Embed Size (px)
Citation preview

Novel etiology of hereditary erythroid disorders
Ph.D. Thesis
Lucie Láníková (born Piterková)
Department of Biology, Faculty of Medicine and Dentistry, Palacky University
Olomouc 2013

Declaration/Prohlášení Hereby I declare that I have written this work on my own under the supervision of Assoc. Prof. Vladimír Divoký, Ph.D., and that all used literature is cited and mentioned in references. Tímto prohlašuji, že předloženou práci jsem napsala samostatně, pod vedením školitele doc. RNDr. Vladimíra Divokého, Ph.D. a s použitím citované literatury. In Olomouc/V Olomouci ……………………………………………………………………..

Abstract/Abstrakt Our study defines new etiology of two distinct congenital erythroid disorders: β-thalassemia and polycythemia and provides novel insights into phenotypic heterogeneity associated with the positional effect of VHL mutations. β-thalassemia is a common hereditary hemoglobin disorder characterized by quantitative reduction of functional β-globin chains. Earlier, we have reported a novel etiology of β-thalassemia caused by insertion of an LINE-1 element into the intron-2 of the β-globin gene, leading to β-globinL1+ allele. The exact mechanism how this intronic insertion of transposable element attenuates human β-globin gene expression was not known. Therefore, we tested several hypotheses which led to the elucidation of the molecular mechanism leading to the thalassemia phenotype due to the LINE-1 insertion. Germline heterozygous von Hippel-Lindau (VHL) gene mutations underlie dominantly inherited familial VHL tumor syndrome comprised of a predisposition for different tumors. However, recessively inherited congenital polycythemia, exemplified by Chuvash polycythemia, has been associated with two separate homozygous 3’ VHL gene mutations (R200W, H191D). We described and characterized a novel homozygous VHL mutation, in exon-2 (c.413C>T):P138L, which is associated in the affected homozygote with congenital polycythemia but not in her, or her heterozygous relatives, with cancer. We also reported a second polycythemic Croatian VHLH191D homozygote and performed several biochemical and molecular tests to better define the phenotype. Tato práce se zabývá studiem dvou různých typů vrozených poruch erytropoezy: β-talasémie a polycytémie a přináší nové informace k pochopení fenotypové heterogenity asociované s různou pozicí mutací ve VHL genu. β-talasémie jsou vrozené chronické anémie, vznikající v důsledku snížení, nebo absence syntézy β-globinového polypeptidového řetězce. Úplně novou etiologií β-talasémie je inserce funkčního retrotransposonu LINE-1 do druhého intronu β-globinového genu. Přesný mechanismus, jakým LINE-1 element může modulovat expresi lidských genů, není znám. V našem případě jsme dokázali, že talasemický fenotyp je důsledek kombinace několika různých defektů na molekulární úrovni. Vrozené heterozygotní mutace von Hipple-Lindau (VHL) genu jsou nejčastěji asociovány s VHL syndromem a různými druhy rakoviny. Zatímco recesivně dědičné homozygotní mutace na 3´konci VHL genu, v exonu 3 (R200W, H191D), jsou příčinou vzniku polycytémie, jejíž nejznámějším příkladem je tzv. Čuvašská polycytémie. U nově diagnos-tikované pacientky s vysokou hladinou hemoglobinu a erytropoetinu jsme objevili a popsali doposud nepublikovanou homozygotní mutaci v druhém exonu VHL genu (c.413C>T):P138L. Jde o první mutaci v druhém exonu VHL genu, která je asociovaná jen s polycytémií a nikoliv s nádorovým onemocněním. Podrobně jsme také popsali druhý případ výskytu VHLH191D mutace v Chorvatsku.

Acknowledgement I would like to thank to Assoc. Prof. Vladimír Divoký, Ph.D. for giving me the opportunity to be a part of his great team, for the excellent ideas he brought to my scientific projects and for time he spent by critically reviewing my results. I appreciate all the help (Jana Kučerová) and the work (Jana Kučerová, Eliška Trojáčková) which was done in order to help me to finish my projects in Olomouc. I would like to thank also to all the other colleagues in Olomouc, especially to Lenka Calábková and Renáta Mojzíková. My special thanks belong to prof. Josef T. Prchal at University of Utah, Division of Hematology and his lab. During my stay in Salt Lake City I gained incredible amount of new knowledge, scientific experience and self-confidence. I wouldn’t be able to finish my work without the love and support of my husband Ondřej and my family (my mom, dad and sister Martina). Thank you. This work was supported in whole or in part, by grants P301/12/1503 (Ministry of Education, Youth and Sport, Czech Republic), NT13587 (Ministry of Health, Czech Republic), by European Structural Funds (project CZ.1.07/2.3.00/20.0164) and by internal student grants of Faculty of Medicine and Dentistry, Palacky University (LF_2012_16 and LF_2013_10).

Contents
I Theoretical Background .......................................................................................................................... 1
1 Introduction ................................................................................................................................................. 1
1.1 The erythropoiesis ............................................................................................................................. 1
1.1.1 Morphology and composition of the erythrocyte ............................................................... 2
1.1.2 Regulation of erythropoiesis ................................................................................................... 3
1.1.3 Regulation of oxygen sensing ................................................................................................. 4
1.2 Clinical manifestation and classification of erythroid disorders ................................................. 5
1.2.1 Anemia ....................................................................................................................................... 6
1.2.2 Polycythemia ............................................................................................................................. 8
1.2.2.1 Primary polycythemia .............................................................................................................. 8
1.2.2.2 Secondary polycythemia .......................................................................................................... 9
1.2.2.3 Congenital disorders of hypoxia sensing ............................................................................... 9
1.3 Mobile elements and human diseases ........................................................................................... 10
1.3.1 Mobile elements ...................................................................................................................... 10
1.3.2 The role of mobile elements in the pathogenesis of human diseases ............................ 11
II Original Research ................................................................................................................................... 13
2 Aims of the Thesis .................................................................................................................................... 13
3 Materials and Methods ............................................................................................................................. 14
3.1 Patient samples ................................................................................................................................. 14
3.2 Mutation screening .......................................................................................................................... 14
3.3 Cell culture ........................................................................................................................................ 15
3.4 Drugs and inhibitors ....................................................................................................................... 15
3.5 Nuclear Run-On Assay ................................................................................................................... 15
3.6 RNA isolation and quantitative RT-PCR .................................................................................... 16
3.6.1 Human β-globin transcripts quantification ........................................................................... 16
3.6.2 Relative ratio of exon-3 to exon-2 of human β-globin primary transcript ...................... 16
3.6.3 Effect of the VHL mutations on expression of HIFs’ target genes .............................. 16
3.7 DNA methylation analysis ............................................................................................................. 17
3.8 In vitro assay of the sensitivity of erythroid progenitors to EPO ............................................. 17
3.8.1 In vitro assay in semisolid medium ........................................................................................ 17

3.8.2 In vitro assay in liquid culture ................................................................................................. 17
3.9 Functional analysis of VHL protein ............................................................................................. 18
4 Results and Discussion ............................................................................................................................ 19
4.1 List of Publications and Meeting Abstracts ................................................................................ 19
4.2 Molecular mechanisms underlying reduced transcription of β-globin gene ............................. 22
4.3 Novel homozygous VHL mutation in exon 2 ........................................................................... 24
4.4 Polycythemia due to Croatian homozygous VHL (c.571C>G:H191D) mutation .............. 26
5 Summary ..................................................................................................................................................... 29
III Supplements and Appendices ......................................................................................................... 30
6 Bibliography ............................................................................................................................................... 30
7 Acronyms and Abbreviations ................................................................................................................. 37
8 Supplements............................................................................................................................................... 39
8.1 List of primers and PCR reaction conditions ............................................................................. 39
8.1.1 List of primers ......................................................................................................................... 39
8.1.2 PCR reaction conditions ....................................................................................................... 40
8.2 List of other Publications and Meetings Abstracts .................................................................... 41
9 Appendices................................................................................................................................................. 43
Original version of Publications .................................................................................................... 43

I Theoretical Background
1 Introduction 1.1 The erythropoiesis Hematopoiesis is dynamic process, where the hematopoietic stem cells (HSC) give rise to all of the different mature blood cell types (myeloid and lymphoid lineage). HSCs are self-renewing and multipotent, i.e. have the potential to develop into all blood cells, but cannot develop to other cell types. Traditionally, the hematopoietic process is divided into primitive and definitive hematopoiesis based on the developmental program and type of blood cells generated. During embryogenesis, the yolk sac derived hematopoiesis occurs in two distinct waves in the extraembryonic blood islands. The first wave produces primitive macrophages and primitive erythrocytes, thus providing the developing embryos with oxygen [1]. The second wave of the yolk sac hematopoiesis is a transient wave of definitive erythroid precursors that enter early embryonic circulation. The long-term definitive hematopoiesis produces multipotent blood cells from the hemogenic endothelium of the embryo that includes the aorta-gonad-mesonephros (AGM) region of the embryo; these cells also seed the yolk sac, and mainly fetal liver. After the birth, the site of adult hematopoiesis, where HSCs undergo differentiation to generate lineage-committed progenitors and self-renewal to maintain a constant supply of HSCs, is bone marrow. The shifting power during the developmental stages of hematopoiesis, also connected with hemoglobin switching, is thought to be regulated predominantly at the transcriptional level by a network of several transcription and chromatin remodeling factors. Erythropoiesis is physiological process of the red blood cells (RBCs, erythrocytes) production. The first step in HSCs differentiation takes common multipotent CFU-GEMM progenitor (colony-forming unit granulocytic, erythroid, megakaryocyte, macrophage), followed by generation of committed erythroid progenitors: “early” BFU-E (burst forming unit – erythroid), and “later” CFU-E (colony forming unit – erythroid) [2]. Already committed erythroid progenitors develop into the morphologically distinguishable erythroid precursors – proerythroblasts and erythroblasts. The final stages of maturation are accompanied by hemoglobin synthesis and nucleus extradition - the reticulocytes and mature erythrocytes are produced. Erythropoiesis generates ~ 2 x 1011 new erythrocytes (1% of the total red cell mass) every day and the same amount is removed every day from the circulation [3]. The mature RBCs transport oxygen from the lungs to the rest of the body and then return carbon dioxide from the body to the lungs.

I Theoretical Background
1.1.1 Morphology and composition of the erythrocyte Normal RBCs have a diameter of 7.5 to 8.7 m, average volume of 90 fl and a surface area approximately 136 m2 [3]. The normal resting shape of the erythrocyte is a flexible biconcave disc. The erythrocyte spends most of its circulatory life span (100- to 120- days) within capillary channels of the microcirculation. The erythrocyte’s membrane has a unique capacity to “tank-tread” – rotate around the red cell contents. This arrangement transmits shocks from wall contact through the membrane to the viscous hemoglobin solution in the interior rather than concentrating the energy of contact in the membrane [3]. Hemoglobin (Hb) is a two-way respiratory carrier, enables RBCs delivering oxygen from the lungs to the tissues and facilitating the return transport of carbon dioxide [4]. The Hb synthesis in erythroid cells is dependent on three distinct processes: synthesis of globins, synthesis of heme and iron intake. All of these processes have to be tightly regulated and coordinated in order to prevent pathological conditions such as anemia, porphyria, hemochromatosis or polycythemia. Normal mammalian Hb contains two pairs of unlike polypeptide chains: one chain of each pair is α or α-like (ζ) and the other is β, or β-like (γ, δ or ε). The α-chains of all human hemoglobins, except for some embryonic hemoglobins, are the same during all developmental stages. The non-α chains include the β-chain of normal adult hemoglobin (Hb A (α2β2)), the γ-chain of fetal hemoglobin (Hb F (α2γ2)), and the δ-chain of hemoglobin A2 (Hb A2 (α2δ2)), the minor component which accounts for 2.5% of the Hb of normal adults [3]. The globin gene switches occur during development: the embryonic to fetal globin switch (from ζ- to α-chain and ε- to γ-chain), which coincides with the transition from embryonic (yolk sac) to definitive (fetal liver) hematopoiesis; and the fetal to adult switch, which occurs at the perinatal period [5]. The α-globin gene cluster is located on chromosomes 16 and β-globin gene cluster on the short arm of chromosome 11. Each globin subunit must form a stable linkage with heme situated on the external surface of the protein so that oxygen in the RBC cytosol can bind reversibly to the heme’s iron atoms [6]. A heme (ferrous protoporphyrin IX) is a chemical compound which serves as a prosthetic group consisting of an iron ion (Fe2+) situated in the center of a large heterocyclic organic ring called a porphyrin (four pyrrolic groups joined together by methine bridges). The erythroid heme biosynthesis pathway involves 8 different enzymes, when first and last three are mitochondrial and the intermediate four are cytosolic [7]. Approximately 85% of heme is synthesized to meet requirements for hemoglobin synthesis, the rest is synthesized in the liver, when is largely required for cytochromes P450 [8]. The iron metabolism and the regulation of heme synthesis are different in hemoglobin-synthesizing as compared with non-erythroid cells, but limiting factor is always the availability of iron. Iron is an element essential for living organism, but can be also toxic due to its capacity to react with oxygen and catalyze the production of reactive oxygen species [9]. Much of the iron in the human cells occurs in heme form, when hemoglobin, which is 0.34% iron in weight, contains approximately 2 g of body iron in men and 1.5 g in women [3]. The heme iron is continuously recycled following phagocytosis and catabolism of senescent RBCs by the macrophages of reticuloendothelial system. Only a small part represents iron absorbed through the gastrointestinal tract, mostly through the duodenum. The amount of iron absorbed is tightly regulated according to body needs. The central regulator of iron homeostasis is the hepatic antimicrobial peptide hepcidin. Ferroportin serves as the receptor for hepcidin and is destroyed when the complex is formed [3]. Ferroportin transports iron across basolateral membrane of enterocytes into blood

I Theoretical Background
circulation. Ferroportin-associated enzyme hephaestin oxidizes the ferrous iron to the ferric form. Once ferric iron is released to the plasma by ferroportin, it is bound by transferrin, which after forming a complex with the transferrin receptor, transports the metal into erythroid cells. Once bound to the receptor, the transferrin-iron complex is internalized and iron is released inside the cells into an acidified vesicle. Within the vesicle, STEAP3 (STEAP family member) effects the reduction of ferric to ferrous iron and another protein DMT-1 (divalent metal transporter 1) induces the release of Fe2+ into the cytosol, where it is taken up by mitochondria for heme synthesis [3]. 1.1.2 Regulation of erythropoiesis Many hormones/cytokines, receptors and transcription factors control the development of RBCs from HSCs, but the primary controlling molecular hub is erythropoietin-receptor (EPOR) signaling pathway, with erythropoietin (EPO) as principal regulating hormone. Among the other growth factors, which positively stimulate erythropoiesis, are stem cell factor, interleukin-3, granulocyte macrophage colony-stimulating factor and trombopoietin. Each maturation stage requires for growth and survival different combinations of cytokines, but the first EPO-dependent stage of erythroid differentiation represents CFU-E. The key lineage-specific transcription factors are GATA-1 (GATA-binding factor 1), KLF-1 (erythroid Krüppel-like factor 1) or NF-E2 (nuclear factor - erythroid-derived 2), that are absolutely necessary for normal erythropoiesis and activate many erythroid specific genes, including those for globins and erythroid specific membrane proteins.
Figure 1. Schematic representation of EPO/EPOR signaling pathway. Conforma-tional changes of the receptor after the EPO binding induce the transphosphorylation of associated JAK2 molecules and phosphorylation of tyrosine residues in the cytoplasmic tail of the receptor, which allows binding and phosphorylation of signaling molecules. Grb2/Sos complex is adaptor which mediates the cross-talk between EPO/EPOR and Ras/Raf/MAPK pathways. Negative regulators (e.g. SOCS, CIS) are transcriptionally induced by the activated JAK2. See text for details.

I Theoretical Background
EPO is heavily glycosylated protein, which is produced primary in kidney, by peritubular interstitial fibroblast [1] in response to O2 tension. EPO production is regulated almost exclusively by hypoxia at the transcription level (described in detail below, chapter 1.1.3). EPO is not stored but secreted immediately [2]. EPO binds to the cognate receptor (EPOR) of erythroid progenitor cells promoting the survival, proliferation and differentiation of these cells. EPOR belongs to the hemopoietin/interferon super-family of receptors, which lack intrinsic catalytic function therefore has to be associated with signal-transducing proteins. EPO ligation on its receptor induces the conformational alteration of the pre-existing receptor dimers allowing associated tyrosine kinase Janus kinase 2 (JAK2) activation [3]. Activated JAK2 phosphorylates multiple key tyrosine residues in the EPOR cytoplasmic domain, thereby provides docking sites for downstream signaling molecules containing Src homology 2 domain. The signaling cascade initiated by EPO binding includes multiple pathways, e.g. the STATs, PI-3K/AKT, Ras/Raf/MAPK (Figure 1). Beside JAK family kinase (which is primary and critical), erythroid cells can facilitate cellular signaling also via Src family kinase members, e.g. Lyn [4], but its precise involvement in erythropoiesis is not fully elucidated. An important feature of EPOR signaling is its temporal activation of down-stream negative regulators, such as CIS (cytokine inducible SH2 protein) or SOCSs proteins (suppressors of cytokine signaling) and phosphatase SHP1 (Src homology region 2 domain-containing phosphatase-1), which represent the classical feedback loop [5]. In addition, LNK (SH2B adaptor protein 3) adaptor molecule, through its SH2 domain, negatively modulates EPOR signaling by attenuating JAK2 activation, and regulating EPO-mediated erythropoiesis [6]. 1.1.3 Regulation of oxygen sensing All nucleated cells in the body sense and respond to conditions of reduced oxygen availability (hypoxia). Under hypoxia, hypoxia-inducible factors (HIFs) regulate the expression of genes that mediate adaptive response. Within any given cell type, HIFs control the expression of hundred genes, including these that promote glucose uptake, facilitate glycolysis, inhibit the Krebs cycle, increase mitochondrial electron-transport chain efficiency, promote iron mobilization, stimulate angiogenesis, regulate apoptosis and increase the synthesis of EPO [1]. Oxygen depending-regulation of HIFs is depicted in the Figure 2. HIF is a heterodimeric transcription factor that consists of an O2-sensitive α-subunit and a constitutively expressed β-subunit, which is insensitive to changes in oxygen tension and is identical to aryl hydrocarbon receptor nuclear translocator (ARNT) [2]. Three HIF α-subunits are known, HIF-1α, HIF-2α and HIF-3α [3]. HIF-1 was first identified in human Hep3B hepatoma cells using DNA sequences that were derived from the 3’-hypoxia enhancer of the EPO gene [4]. HIF-1α has a ubiquitous pattern of expression in tissues, whereas HIF-2α is restricted to certain cell types including endothelial cells, cardiomyocytes, hepatocytes or glial cells [5]. Less is known about HIF-3α which in certain contexts have been shown to be inhibitory [6], whereas HIF-1 and HIF-2 heterodimers are transcriptional activators. In well-oxygenated cells (normoxia), HIF-α-subunits are continuously synthesized and rapidly degraded. The key event regulating oxygen-sensitive turnover is, in case of HIF-1α, hydroxylation of Pro402 or Pro564 (Pro-OH), or both, by the prolyl hydroxylase domain protein 2 (PHD2), which is a dioxygenase that utilizes O2 and α-ketoglutarate as substrates while generating CO2 and succinate as by-products [7]. Hydroxylated HIF-1α interacts with

I Theoretical Background
the von Hippel-Lindau (VHL) tumor-suppressor protein which recruits an ubiquitin E3 ligase. The polyubiquitination of HIF-1α flags the protein for degradation by the 26S proteasome. Factor inhibiting HIF-1 (FIH-1) also uses oxygen to hydroxylate HIF-1α on an asparagine residue 803 (Asn-OH) [7]. HIF-1α containing Asn-OH cannot be bound by the coactivator protein p300, thereby preventing HIF-1α from activating gene transcription [8]. Under hypoxic conditions PHD2 activity is reduced due to substrate limitation, inhibition of the catalytic center, or both [9]. Proline and asparagine hydroxylation reactions are inhibited, and HIF-α (i.e., either HIF-1α or HIF-2α) rapidly accumulates, dimerizes with HIF-1β, recruits p300, binds to hypoxia response elements, and activates the transcription by RNA polymerase II (Pol II) of hundreds of target genes [10]. Thus, HIF-α hydroxylation provides a mechanism for transducing changes in oxygen availability to the nucleus as changes to gene transcription. Although in vitro approaches identified HIF-1 as the transcription factor responsible for the hypoxic induction of EPO, HIF-2 has now emerged as the main regulator of EPO production in vivo [11]. Perturbation of PHD-VHL-HIF pathway leads to the development of benign erythrocytosis/polycythemias that are associated with increased or inappropriately normal serum EPO levels (with respect to hemoglobin levels, described in detail below, chapter 1.2.2.3).
Figure 2. Oxygen sensing, gene expression and adaptive response to hypoxia [adapted from ref. 10]. (Left) Under normoxic conditions, PHD2 protein constitutively hydroxylates HIF-1α on specific prolyl residues, and FIH-1 constitutively hydroxylates HIF-1α on a specific asparaginyl residue. Hydroxylated HIF-1α subunit is recognized by VHL, a component of an E3 ubiquitin ligase complex and targeted for degradation by the proteasome. (Right) Under hypoxic conditions, HIF hydroxylases are inactivated allowing HIF-1α to heterodimerises with HIF-1β, binds DNA, recruits the p300 co-activator to form an active complex leading to transcription of downstream genes. See text for details.
1.2 Clinical manifestation and classification of erythroid disorders Erythroid disorders are traditionally divided into two groups: (1) anemia and (2) polycythemia, which both can be caused by acquired or inherited genetic defects. In general,

I Theoretical Background
anemias are characterized by a decrease and polycythemias by an increase of the red cell mass. 1.2.1 Anemia There are several different ways of classifying anemia. Anemia is defined by the World Health Organization (WHO) as Hb < 120 g/l in women and Hb < 130 g/l in men. Hemoglobin concentration expresses the oxygen-carrying capacity of blood, which is in fact decreased in anemia. Based on determination of the red cell mass, anemia can be classified as relative or absolute. Relative anemia represents the states, where Hb concentration falls as the result of an increase in the plasma volume and red blood cell mass is not influenced. Classification of absolute anemias with decreased red cell mass is very difficult. In general, the anemias are caused by decreased production or increased destruction of red cells. Pathophysiological classification of anemias is listed in Table 1. Since we investigated the novel molecular mechanism of thalassemia, only pathophysiology of this hereditary disorder of globin synthesis will be further discussed. In thalassemia, there are defects in the production of either the α-like (α-thalassemia) or in β-like (β-thalassemia) globin chains resulting in an imbalance between production of globin chains and deleterious effect of the globin subunits that are produced in excess. Less common forms of thalassemia include γ-, γδβ-, δ- and εγδβ- thalassemias [1]. There are 95 different mutations causing α-thalassemia described worldwide [2]. The majority of most common α-thalassemia determinants are due to deletions that remove some, or all, of the α-globin gene cluster. More than 200 β-thalassemia alleles have been described in the database of human hemoglobin variants, which involve mutations affecting any of the stages from transcription to RNA processing and translation of β-globin mRNA [3]. New etiology of β-thalassemia was described by us, when insertion of the full-length transposable element LINE-1 into the intron-2 of the β-globin gene caused severe reduction in β-globin mRNA production. WHO calculations estimated that at least 5.2% of the world population (and over 7% of pregnant women) carry an affected Hb allele [4] and about 60 000 severely affected infants are born every year [5]. In addition, at least 20% of the world population suffers from α-thalassemia [4], which is more frequently and widely distributed than β-thalassemia. Epidemiological studies strongly suggest that in populations in which malaria is (or was) endemic, individuals with mild form of either α-thalassemia or β-thalassemia trait are protected against Plasmodium falciparum infection, which explains the high carrier frequency via natural selection. The α-globin gene is duplicated (α1 and α2) on each copy of chromosome 16, so there are a total of four α-globin genes in a normal genotype (αα/αα). The clinical severity of α-thalassemia relates to the number of genes affected, when clinically significant types are Hb H disease and Hb Bart’s hydrops fetalis [6]. There are three broad clinical phenotypes in patients with β-thalassemia: minor, intermedia and major. These phenotypes are associated with mutations that either reduce (β+-thalassemia) or abolish (β0-thalassemia) expression of β-globin gene. The only forms of treatment for thalassemic patients are regular blood transfusions, iron chelation therapy (to prevent iron overload) and splenectomy (in cases complicated by hypersplenism). In case of β-thalassemia, experimental approaches (pharmaceutical agents e.g. azacitidine and decitabine [7] or somatic gene therapy [8]) are used to increase expression of γ- or β-globin genes in order to restore the balance between globin chains production.
I Theoretical Background
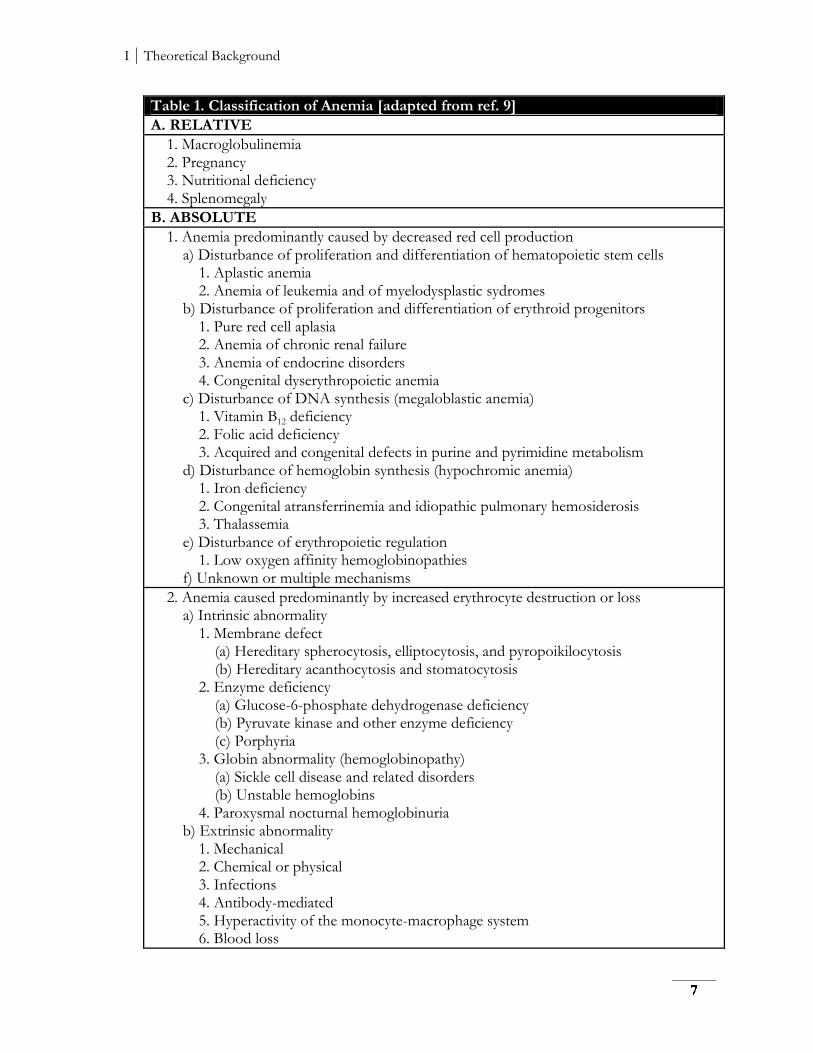
Table 1. Classification of Anemia [adapted from ref. 9]
A. RELATIVE
1. Macroglobulinemia 2. Pregnancy 3. Nutritional deficiency 4. Splenomegaly
B. ABSOLUTE
1. Anemia predominantly caused by decreased red cell production a) Disturbance of proliferation and differentiation of hematopoietic stem cells 1. Aplastic anemia 2. Anemia of leukemia and of myelodysplastic sydromes b) Disturbance of proliferation and differentiation of erythroid progenitors 1. Pure red cell aplasia 2. Anemia of chronic renal failure 3. Anemia of endocrine disorders 4. Congenital dyserythropoietic anemia c) Disturbance of DNA synthesis (megaloblastic anemia) 1. Vitamin B12 deficiency 2. Folic acid deficiency 3. Acquired and congenital defects in purine and pyrimidine metabolism d) Disturbance of hemoglobin synthesis (hypochromic anemia) 1. Iron deficiency 2. Congenital atransferrinemia and idiopathic pulmonary hemosiderosis 3. Thalassemia e) Disturbance of erythropoietic regulation 1. Low oxygen affinity hemoglobinopathies f) Unknown or multiple mechanisms
2. Anemia caused predominantly by increased erythrocyte destruction or loss a) Intrinsic abnormality 1. Membrane defect (a) Hereditary spherocytosis, elliptocytosis, and pyropoikilocytosis (b) Hereditary acanthocytosis and stomatocytosis 2. Enzyme deficiency (a) Glucose-6-phosphate dehydrogenase deficiency (b) Pyruvate kinase and other enzyme deficiency (c) Porphyria 3. Globin abnormality (hemoglobinopathy) (a) Sickle cell disease and related disorders (b) Unstable hemoglobins 4. Paroxysmal nocturnal hemoglobinuria b) Extrinsic abnormality 1. Mechanical 2. Chemical or physical 3. Infections 4. Antibody-mediated 5. Hyperactivity of the monocyte-macrophage system 6. Blood loss

I Theoretical Background
1.2.2 Polycythemia Polycythemia (also known as erythrocytosis) is characterized by an increased red cell blood mass. Polycythemias can be primary or secondary. Primary polycythemias are caused by somatic or germline mutations leading to changes within the erythroid progenitors causing an augmented response to erythropoietin. Secondary polycythemias are caused by either an appropriate or inappropriate increase in the red cell mass as a result of augmented levels of erythropoietin. In some instances, exemplified by Chuvash polycythemia (CP) [1], the first known congenital disorder of hypoxia sensing, erythroid progenitors are in in vitro cultures hypersensitive to EPO; thus, Chuvash polycythemia shares features of both primary and secondary polycythemia. Classification of polycythemias is listed in Table 2.
Table 2. Classification of Polycythemia/Erythrocytosis [adapted from ref. 2]
A. RELATIVE (decreased plasma volume)
1. Dehydration
B. APPARENT (normal plasma and red cell volume)
1. Stress or smoker’s erythrocytosis
C. ABSOLUTE (increased red cell volume)
1. Primary a) Polycythemia vera b) Primary familial and congenital polycythemia 2. Secondary a) Appropriate 1. Altitude 2. Cardiopulmonary disorder 3. Increased hemoglobin affinity for oxygen b) Inappropriate 1. Renal cysts and tumors 2. Hepatoma 3. Cerebellar hemangioblastoma 4. Essential 5. Augmented hypoxia-sensing
1.2.2.1 Primary polycythemia Polycythemia vera (PV) and primary familial and congenital polycythemia (PFCP) are primary polycythemic disorders with erythroid progenitors hypersensitive to EPO. They are caused by somatic (PV) or germ-line (PFCP) mutations that are intrinsic to erythroid progenitors and result in an augmented response to EPO [2]. PV is an acquired clonal hematopoietic stem cell disease characterized by increased production of erythrocytes, granulocytes, platelets and largely polyclonal T lymphocytes [3]. PV erythroid progenitors exhibit disease-specific functional characteristic, i.e. erythropoietin independent colony-formation, a hallmark of PV [4]. Over 95% of PV patients carry a somatic JAK2V617F gain-of-function mutation [5]. JAK2V617F in a large proportion of the PV cases is homozygous due to acquired uniparental disomy on chromosome 9p [6]. Evidence suggests that JAK2V617F is not the disease-initiating mutation and constitutes only part of the

I Theoretical Background
clone [7,8]. Thus, the full mutation landscape and whether and how these alterations contribute to disease initiation and clonal evolution or myelofibrotic transformation are not fully deciphered. PFCP is congenital disorder characterized by an autosomal-dominant mode of inheritance [9]. The distal cytoplasmic region of the EPOR, in association with SHP-1, is required for down-regulation of EPO-mediated activation of JAK2/STAT5 proteins. To date, ten mutations of the EPOR have been convincingly linked to PFCP [10]. All of these mutations result in truncation of the EPOR cytoplasmic carboxyl terminus, leading to loss of its negative regulatory domain and resulting in a gain-of-function of the EPOR. 1.2.2.2 Secondary polycythemia Secondary polycythemia may be subdivided into appropriate and inappropriate, when the former responding normally to tissue hypoxia (i.e. high-altitude polycythemia and hemoglobins with increased affinity for oxygen) and the latter is stimulated by aberrant production of EPO. Polycythemia is often considered a universal, uniform adaptation to hypoxia that would arise in all normal individuals, but high altitude population e.g. Tibetans, are genetically adapted to the environmental stress of high altitude, having normal Hb concentration both at sea level and at high altitude. Several haplotypes have undergone positive selection in Tibetans, including variations at or near the EGLN locus, which encodes PHD2. PHD2D4E and PHD2C127S alterations that originated on the same haplotype about ~6,000 years ago contribute to protection from polycythemia at high altitude, by abrogation of hypoxia-induced HIF-mediated augmentation of erythropoiesis [1]. 1.2.2.3 Congenital disorders of hypoxia sensing Chuvash polycythemia has autosomal-recessive trait and is the first recognized hereditary condition of augmented hypoxia-sensing [2]. The Chuvash people reside in the mid-Volga River region in Russia where CP affects hundreds of individuals (homozygous VHL c.598C>T; VHLR200W mutations), making it the most common congenital polycythemia [3]. Outside of Chuvashia, CP has also been found sporadically in diverse ethnic and racial groups [4,5,6] and a high prevalence of this disorder was reported in the Italian island of Ischia [7]. VHLR200W mutation impairs the interaction of VHL with α-subunit of HIF, thus reducing the rate of ubiquitin-mediated HIF destruction. As a result, the levels of the HIF-1 and HIF-2 heterodimers increase and lead to increased expression of target genes including EPO, vascular endothelial growth factors (VEGF), and plasminogen activator inhibitor (PIA) among others [3]. Clinically, CP patients are predisposed to develop thrombosis, bleeding, cerebrovascular events, and increased mortality independent of the increase in hematocrit [8]. Despite increased expression of HIF-1α and VEGF in the normoxic state, which has been proposed to be related with development of hemangioblastoma and renal cell carcinoma [9], CP patients do not display a predisposition to tumor formation [8]. In contrast, autosomal dominant mutations of the VHL gene cause VHL syndrome [9]. Heterozygotes for these dominant VHL mutations are at increased risk of developing hemangioblastomas, renal cell carcinoma, pheochromocytoma, pancreatic endocrine tumors, and endolymphatic sac tumors when they acquire a somatic mutation in the normal VHL allele in trans [10,11].

I Theoretical Background
Some patients with VHL syndrome also develop acquired polycythemia due to EPO production by a tumor [9]. Other than VHLR200W germline mutations also cause polycythemia. Some patients with congenital polycythemia have proven to be compound heterozygotes for the Chuvash mutation, a few cases of congenital polycythemia, known to have mutations of only one VHL allele were reported, but lacking an obvious pathophysiological explanation [12]. The mutation in PHD2 (PHD2P317R) was identified in the family, in which heterozygotes for this mutation have mild or borderline polycythemia [13]. The P317R mutation affects a residue that is in very close vicinity of the catalytic domain and impairs binding to both HIF-1α and HIF-2α. Since then, five additional patients with unexplained polycythemia who are heterozygote carriers of different PHD2 mutations have been reported [14]. Almost all patients with PHD2-associated polycythemia have normal EPO level. If the cause of the polycythemia is the haploinsufficiency or dominant negative effect remains unsolved. Study of different families with polycythemia also revealed the presence of heterozygous missense mutation in the coding sequence of HIF-2α. Patients with HIF-2α mutations have typically elevated EPO [14]. There is heterogeneity in the functional defects associated with HIF-2α mutations but all the findings support a critical role of HIF-2α in controlling the expression of human EPO.
1.3 Mobile elements and human diseases 1.3.1 Mobile elements Human genome is flooded with the repetitive sequences capable of moving to new locations, by process known as a ‘transposition’ (excision of the sequences from current genomic location and insertion into a new genomic site). Transposable elements (TEs; also known as “jumping genes”) occupy almost half, 45% [1], of the human genome, making the TE content of our genome one of the highest among mammals, second only to the opossum genome with a reported TE content of 52% [2]. The actual contribution of repeats to mammalian genomes is significantly larger, as the older, ancestral repeats have diverged beyond the current recognition limit [3]. TEs can be separated, based on their mechanism of replication, into two major classes: DNA transposons and retrotransposons. DNA transposons, which move by a ‘cut-and-paste’ mechanism using an encoded transposase gene [4] are currently not mobile in the human genome, they were active during early primate evolution until ~37 million years ago (Myr) [5]. In contrast, retrotransposons duplicate through RNA intermediates that are reverse transcribed and inserted at new genomic locations, referred as ‘copy-and-paste’ mechanism [5]. Retroelements are subdivided into two major groups: those containing long-terminal repeats, LTR retroelements, and all others belong into the category of non-LTR retroelements [2]. LTR retroelements are endogenous retroviruses, which account for ~8% of the human genome but their activity is presently very limited, when the peak of accumulation was estimated ~25 Myr [6]. Non-LTR retrotransposons include autonomous and non-autonomous members. The autonomous long interspersed element-1 (LINE-1 or L1), and its non-autonomous partners, such as ‘SINE-R, VNTR, and Alu’ (SVA) and the short interspersed element (SINE), are the only mobile elements with clear evidence of current retrotranspositional activity in the human genome [2], as indicated by the more than

I Theoretical Background
96 reported cases of de novo insertions that are responsible for genetic disorders and cancer [5,7,8]. SVA and SINEs lack the ability to retrotranspose, but can co-opt L1 machinery for their replication [9]. There are >500,000 L1 copies in the human genome as a result of their continued mobilization activity over the past 150 Myr [5]. Most L1s are inactive due to point mutations, rearrangements, or truncations with only a subset of estimated 80 - 100 elements [8], currently functional in any individual. A substantial fraction of the human genome, >30%, is derived directly or indirectly from L1 retrotransposon activity [8], which makes them the most successful TEs in the human genome by mass. Although L1 transcription and retrotransposition may occur in any cell type at the level of an individual, only events occurred during germ-cell development will be incorporated into the germline lineage and contribute to future generations [3]. However, a growing evidence has indicated that somatic retrotransposition in mammals not only occurs, but is likely to occur at a substantial frequency [10]. The ability of TE to cause diseases via the inactivation of genes by insertional mutagenesis together with the abuse of cellular mechanisms for their own propagation has long been characterized as “parasitic”. But there are also several ways how L1 can generate new alleles. When L1 retrotransposes it can also co-transpose adjacent non-L1 sequences to its new integration site. In this process (designated as ‘3’ transduction’) the presumed L1 polyadenylation signal is bypassed in favor of a downstream endogenous polyadenylation signal, allowing downstream flanking host sequences to ‘‘come along for the ride’’ [11]. Similarly, ‘5’ transduction’ occurs when a cellular promoter, localized upstream to the donor L1, transcribes both 5’ adjacent sequence and L1 which is then subjected to reverse transcription/integration [11]. Some of these L1-generated alleles have survived the test of time, and strongly suggest that L1 may create a platform of potential alleles that can be subjected to the forces of natural selection. Nowadays there is increasing tendency to assume, that TEs are cultivated in the genome for their beneficial possibilities, in particular, to drive genome evolution and alter gene expression [7,12] – key components of plasticity necessary for adaptation and survival. 1.3.2 The role of mobile elements in the pathogenesis of human diseases The fact that many sporadic human diseases have sub-group of unknown etiology suggests a possibility that TE-associated DNA disruption plays crucial role at least in some of them. While the disruption of normal gene function by transposable elements upon integration into exonic regions is obvious, their post-insertional effects on gene expression have not received much attention [1]. Up to date, the known impact on the expression of host genes is via modification of the transcript quality or quantity [2], transcriptional interference [3], or by the control of pathways that affect the mRNA life-cycle [4]. Germline mutations caused by TE insertions have led to several human genetic disorders such as cystic fibrosis, retinitis pigmentosa, hemophilia etc. Recently, the new generation sequencing techniques allow identification of genome and transcriptome aberrations on the level of individuals and help to clarify the extent of the contribution of mobile elements to genetic instability in many human diseases, including different types of cancer. Genetic instability is one of the key features associated with cancer. In contrast to normal cells, the majority of human cancers, and cancer-derived cell lines, support variable, but typically much higher endogenous full-length L1 mRNA expression [5]. The major mechanisms for silencing of TE’s potentially harmful retrotransposing activity is DNA

I Theoretical Background
methylation at the CpG site in the L1 promoter [6] and malignant tumor cells are generally featured by global hypomethylation, including L1 [7]. The hypomethylation of L1 has been reported in urothelial bladder carcinoma, malignant testicular tumors and prostatic adenocarcinoma [8]. The association of retroelement expression with genetic instability in cancer remains largely correlative [9,10], but the observations of augmented mutation rates in the majority of human cancers suggest, that retroelement expression and presumably activity could be one of the forces which accelerate tumor evolution [5]. Another negative fitness consequence arising from the proliferation of TE sequences within mammalian genomes involves the tendency to serve as sources of homology for non-allelic homologous recombinations (NAHR) [11]. Such misrouted recombination events involving TEs represents a viable mechanism for large-scale chromosomal translocation mutations [12]. A much larger fraction of TE-related diseases in humans results from recombination mutations than from insertional mutations [11]. NAHR events have been well recognizes as a major source of DNA damage that leads to either duplications deletion of the sequences between the two participating Alu elements [5]. Numerous examples of Alu/Alu NAHR contributing to cancer have been reported. For example, 23 out of 29 reported recombination events in the BRCA1 gene, which has an unusually high density of Alu elements (41.5% of the gene), involve Alu elements [5]. Interestingly, Alu/Alu cancer-related recombination events do not occur only in germ-lines but also somatically. Among them are recurring duplication of the MYB locus that can contribute to T-cell acute lymphoblastic leukemia or the case of MLL (mixed lineage leukemia), where Alu mediated recombination generates tandem duplications of exons [13,14]. L1 contributes to the instability in human cancers also by creating double strand breaks (DSBs) [15]. DSBs are disruptive forms of DNA damage that are typically corrected prior to its proceeding through the cell cycle.

II Original Research
2 Aims of the Thesis This study is focused on molecular pathophysiology of congenital erythroid disorders with novel etiologies. It was aimed to: 1. describe molecular mechanism, how the presence of active retrotransposon in the non-coding region of β-globin gene attenuates its expression and thus leads to β-thalassemia, 2. investigate the role of novel homozygous VHL mutation in the pathogenesis of polycythemia. Secondary, it was aimed to: 3. characterize the clinical and biological differences between two known positionally close inherited homozygous VHL mutations causing polycythemia: Chuvash R200W and Croatian H191D.

II Original Research
3 Materials and Methods The Materials and Methods section contains only detail information about experiments which were performed by me either at Department of Biology, Faculty of Medicine and Dentistry, Palacky University or at Division of Hematology, School of Medicine, University of Utah. All the other experiments, done by collaborators at different Institutions are presented only in brief and cited accordingly. Each chapter in this section is subdivided into paragraphs and each paragraph is labeled (on the right margin) with the abbreviation of the corresponding project, i.e. L1 (see chapter 4.2), VHL P138L (see chapter 4.3) and VHL H191D (see chapter 4.4).
3.1 Patient samples Peripheral blood from propositus and her mother were obtained by venipuncture. All the samples were obtained with approval of the Institutional Review Board (IRB) committee of Palacky University in Olomouc, Czech Republic. Informed consent for all subjects was provided according to the Declaration of Helsinki. The propositus is a 15-year-old girl of Asian Indian extraction (Punjabi ethnicity), who has been known to be polycythemic from infancy. Her parents are hematologically normal and are of the same ethnicity but not known to be related. Peripheral blood of the propositus and her parents was obtained by venipuncture after obtaining a signed University of Utah’s IRB informed consent. Blood samples from 23 persons were collected from two different families, including two propositi with polycythemia and 21 relatives. Peripheral blood samples were collected in EDTA and/or ACD tubes. Written inform consent was obtained from all participants. The IRB of the University of Utah approved the study.
3.2 Mutation screening Genomic DNA was isolated from whole peripheral blood using phenol-chloroform protocol [1]. To characterize the β-thalassemia mutation, genomic DNA was analyzed by restriction mapping using several restriction endonucleases and Southern blot hybridization (for details see chapter 9, Supplemental Materials and Methods). Granulocyte and mononuclear cell fractions from peripheral blood were isolated according to previously published protocol [2]. Genomic DNA was isolated from granulocytes using Gentra-Puregene Kit (Qiagen, Germantown, MD), and VHL gene’s
L1 VHL P138L VHL H191D L1 VHL P138L VHL H191D

II Original Research
exons were amplified using Hot Star Master Mix (Qiagen). The sequences of primers and reaction conditions are listed in Supplements and Appendices. Sequencing was performed using the standard protocol and the same amplification primers.
3.3 Cell culture
Interspecific hybrids of the patients’ Epstein Barr Virus (EBV) transformed lymphocytes and mouse erythroleukemia (MEL) cells were generated as described elsewhere [3]. From the positive cell clones were further derived two lines – one contained affected chromosome 11 (designated “MEL HBBL1+”) and the second with normal chromosome 11 (designated “MEL HBBwt”). Hybrid mouse/human erythroleukemia cell lines were maintained in humidified atmosphere at 37°C, 5% CO2 in high-glucose Iscove’s Modified Dulbecco’s Media (IMDM) with GlutaMAX (Invitrogen, NY, USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen), 100 U/mL penicillin and 100 μg/mL streptomycin (both Invitrogen). 786-0 cell line (renal cell adenocarcinoma origin) was purchased from ATCC (CRL-1932™, Manassas, VA). Cells were maintained in humidified atmosphere at 37°C, 5% CO2 in high-glucose Dulbecco’s Modified Eagle’s Medium (D-MEM) with GlutaMAX (Invitrogen, NY, USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen), 100 U/mL penicillin and 100 μg/mL streptomycin (both Invitrogen).
3.4 Drugs and inhibitors Hybrid human/mouse cells were treated by 1.8% dimethyl sulfoxide (DMSO, Sigma-Aldrich, St. Louis, MO) for 72 h to induced differentiation. For purpose of different experiments MEL cell lines were treated: 24h with emetine (0.5 μM, Sigma-Aldrich) for nonsense-mediated mRNA decay (NMD) inhibition; in various time points (0, 1, 2, 4, 8 and 12 hours) with actinomycin D (Act-D, 1 μM, Sigma-Aldrich) to block mRNA synthesis. To test the DNA methylation pattern/reactivation of silenced gene expression the hybrid cells were treated with 5-aza-2’-deoxycytidine (DAC, 2 μM or 5 μM, Sigma-Aldrich) and/or trichostatin A (TSA, 100 nM, Sigma-Aldrich) for 24h simultaneously with DMSO or for 24h and 48h before the induction of differentiation by DMSO.
3.5 Nuclear Run-On Assay Nuclei from hybrid MEL HBBL1+and MEL HBBwt cells were isolated with Nuclei EZ Prep Nuclei Isolation Kit (Sigma-Aldrich), according to the manufacturer’s protocol. The total nuclei were split into two equal aliquots and 2x reaction buffer was added (containing 30mM Tris, pH 8; 2,5 mM MgCl2; 150 mM KCl; 20% glycerol; 1mM DTT and 40 U RNasin (Promega, Madison, WI) and reaction was set as described [4]. The in vitro transcription reaction was initiated with the addition of 0.5 mM of each ribonucleotide triphosphates (rATP, rCTP, rGTP, rUTP) (rNTP) (Invitrogen) and incubated for 40 minutes at 30°C. Second aliquot without added rNTPs was incubated at the same
L1 VHL P138L L1
L1

II Original Research
conditions. The rate of transcription (normalized fold increase) was determined as the ratio of nuclear fraction primary β-globin transcript measured in exon-1 (primers HBB ex1 F and R) with added rNTPs to the same fraction basal level (without added rNTPs) after 40 minutes of in vitro transcription. LDHA was used as a reference gene for quantification. The sequences of primers and reaction conditions are listed in Supplements and Appendices.
3.6 RNA isolation and quantitative RT-PCR
3.6.1 Human β-globin transcripts quantification Total RNA from MEL HBBL1+and MEL HBBwt cell lines was isolated with TRI reagent (Roche Applied Science) followed by TURBO DNA-free DNase I treatment (Ambion, Life Technologies, NY). For quantification of human β-globin mRNA by quantitative RT-PCR (qPCR) RNA was reverse-transcribed using a Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Science). cDNA was quantified on Light Cycler 480 (Roche Applied Science). For quantification of aberrantly spliced variants and NMD activation assay, exon-3 primers (HBB ex3 F and R) and UPL (Universal Probe Library, Roche Applied Science) probe #56 were used. For mRNA stability assay and effect of AZA and/or TSA treatment, exon-2 primers (HBB ex2 F and R) were used for β-globin mRNA and primers HBG F a R for γ-globin mRNA quantification. Levels of human β-globin and γ-globin transcripts were normalized to human LDHA (primers hLDHA F and R and UPL probe #47) using efficiency corrected formula for relative expression ratio [5]. The sequences of primers and reaction conditions are listed in Supplements and Appendices. 3.6.2 Relative ratio of exon-3 to exon-2 of human β-globin primary transcript Nuclei’s RNA from MEL HBBL1+ and MEL HBBwt cell lines was isolated with TRI reagent (Roche Applied Science) followed by TURBO DNA-free DNase I treatment (Ambion, Life Technologies, NY). The HBB exon-3/exon-2 ratio of MEL HBBL1+ native primary transcript from the nuclear fraction that was prepared as mentioned above was normalized to the exon-3/exon-2 ratio of the control MEL HBBwt native primary nuclear transcript using formula 2ΔΔCt. The method for calculating ΔΔCt was similar to the method used for the relative quantification and is as follows: (CtMEL HBBL1+ ex2 – CtMEL HBBwt ex2) – (CtMEL HBBL1+ ex3 – CtMEL HBBwt ex3). Exon-2 primers HBB ex 2 F and R, exon-3 primers HBB ex 3 F and R, and exon-3 UPL probe #56 were used for the determination of this ratio. The efficiency of both amplicons was measured using a dilution series standard curve and was very close to 2. The sequences of primers and reaction conditions are listed in Supplements and Appendices. 3.6.3 Effect of the VHL mutations on expression of HIFs’ target genes Total RNA was isolated from granulocytes using TRI reagent solution (Molecular Research Center, Cincinnati, OH) and then treated with DNA-free™ DNase Treatment & Removal Reagents (Ambion, Life Technologies, NY) to remove any contaminating DNA. 500 ng of DNA-free RNA was reverse-transcribed using SuperScript® VILO™ cDNA Synthesis Kit (Invitrogen) according to manufacturer’s instruction protocol. qPCR were performed with specific TaqMan® Gene Expression probes (Applied Biosystems, Carlsbad,
L1 L1
L1
VHL P138L VHL H191D

II Original Research
CA) for following genes: ADM (Hs00181605), TFRC (Hs00951083), NDRG1 (Hs00608387), PDK1 (Hs00176853), SLC2A1 (Hs00892681), VEGF (Hs00900055), BNIP3 (Hs00969291), BNIP3L (Hs00188949), HK1 (Hs00175976). All samples were assayed in triplicates. Data were normalized to HPRT (4333768F) and GAPDH (4333764F) reference genes. The statistical significance of relative expression changes of target mRNA levels normalized to a reference genes was analyzed by the pair-wise fixed reallocation randomization test using the REST© 2009 software [6].
3.7 DNA methylation analysis Bisulfite modification was performed on genomic DNA from MEL HBBL1+ and MEL HBBwt cells using the EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA). The promoter and enhancer region of β-globin gene and the -globinL1+
L1 promoter were amplified with semi-nested PCR. β-globin promoter primers were published elsewhere [7], enhancer region was amplified with HBBenh bis F and R primers for first round, then HBBenh bis F2 and R for semi-nested PCR. L1 promoter was amplified with L1 meth F and R primers for first round, followed by semi-nested round with primers L1 meth F2 and R. The purified products were subcloned into the pCR®2.1-TOPO® plasmid (Invitrogen) and at least 8 positive clones for each region were sequenced (in the case of -globinL1+ promoter and 3' enhancer 10 to 20 clones were sequenced). The sequences of primers and reaction conditions are listed in Supplements and Appendices.
3.8 In vitro assay of the sensitivity of erythroid progenitors to EPO 3.8.1 In vitro assay in semisolid medium In vitro sensitivity of erythroid progenitors to EPO was performed on mononuclear cells isolated from the peripheral blood using Histopaque (Sigma-Aldrich) density gradient centrifugation and plating (2.3×105/mL) to methylcellulose media (MethoCult® H4531; StemCell Technologies, Vancouver, BC) without addition of EPO or with addition of various concentrations of EPO (StemCell Technologies), ranging from 0.015 to 3.0 U/mL. Cell cultures were maintained in humidified atmosphere of 5% CO2 at 37°C for 14 days. Erythroid burst-forming unit colonies (BFU-Es) were scored by standard morphologic criteria. 3.8.2 In vitro assay in liquid culture Expansion of the progenitor cells from the mononuclear cell population was performed based on our published protocol [8]. Briefly, 1x106 cells/mL were cultured in the Stem-Span™ Serum-Free Expansion Medium (StemCell Technologies) containing different cytokine cocktail - day 1-7 (100 ng/mL of fetal liver tyrosine kinase 3 ligand, 100 ng/mL of thrombopoietin, and 100 ng/mL of stem cell factor), day 8-14 (50 ng/mL of stem cell factor, 50 ng/mL of insulin like growth factor-1, and 3 U/mL of EPO), day 15-21 (50 ng/mL of insulin like growth factor-1, and 3 U/mL of EPO). All cytokines were kind gift of Amgen (Thousand Oaks, CA).
VHL P138L VHL H191D L1
VHL P138L VHL H191D VHL H191D

II Original Research
3.9 Functional analysis of VHL protein VHL Human cDNA ORF Clone was obtained from OriGene (RC216151, Rockville, MD). Site-directed mutagenesis was performed using the QuickChange II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) and allele-specific oligonucleotides for VHLP138L and VHLH191D. The sequences of primers and reaction conditions are listed in Supplements and Appendices. Transfection of all mutated, wild type and empty plasmids (which was used as a negative control) was done using Lipofectamine 2000 reagent (Invitrogen). Cells were selected 48 hours after transfection using 1 mg/mL G418 (Invitrogen) and cultured for 21 days. Resistant clones were isolated using 96-well limiting dilution. 30 single clones were picked up for each VHL construct and tested for expression of VHL(Myc-DDK) by TaqMan® Gene Expression assay on demand (Applied Biosystems). To determine the half-life of pVHL, 786-0 stable transfected clones were treated with 200 μM cycloheximide (Sigma-Aldrich) and cells were harvested at different time-points (0, 2, 4, 6, 8 and 10 hours) to ice cold RIPA buffer (Sigma-Aldrich) supplemented with following protease inhibitors: 1 M DTT, 1 M NAF, 10 M β glycerophosphate, 10 g/mL leupeptin, 2 mg/mL aprotinin, 0.1 M Na3VO4 and 0.1 M PMSF. Proteins were electrophoretically resolved on SDS-polyacrylamide gels and electroblotted onto Immobilon® PVDF mebranes (Millipore, Billerica, MA). Membranes were incubated with primary antibodies rabbit anti-human VHL (FL-181, Santa Cruz, Dallas, TX, 1:500) and rabbit anti-human actin (Sigma-Aldrich, 1:1000) at 4°C overnight, washed in PBS with 0.05% Tween 20, and incubated for 1h with the goat anti-rabbit horse radish peroxidase (HRP)-conjugated secondary antibody (Thermo Fisher, Waltham, MA). HRP activity was detected with ECL detection kit (Pierce, Rockford, IL, USA) on Blue Lite Autorad films (BioExpress, UT, USA). Quantitation of pVHL signals was performed using ImageJ software.
VHL P138L VHL H191D

II Original Research
4 Results and Discussion 4.1 List of Publications and Meeting Abstracts This thesis contains data that were presented in publications and on meetings listed below: Publications
Lanikova L, Kucerova J, Indrak K, Divoka M, Issa JP, Papayannopoulou T, Prchal
JT, Divoky V. β-thalassemia due to intronic LINE-1 insertion in the β-globin gene: molecular mechanisms underlying reduced transcript of the β-globinL1 allele. Human Mutation. 2013; doi: 10.1002/humu.22383/epub ahead of print.
Lanikova L, Lorenzo F, Yang CH, Vankayalapati H, Drachtman R, Divoky V, Prchal
JT. Novel homozygous VHL mutation in exon 2 is associated with congenital polycythemia but not with cancer. Blood. 2013;121(19):3918-24.
Piterkova L*, Tomasic Ljubas N*, Huff Ch, Bilic E, Yoon D, Miasnikova GY, Sergueeva AI, Niu X, Nekhai S, Gordeuk V, Prchal JT. Polycythemia due to Croatian homozygous VHL (571C>G:H191D) mutation has a different phenotype than Chuvash polycythemia (VHL 598C>T:R200W). Haematologica. 2013; 98(4):560-7.
Selected Meeting Abstracts
Lanikova L, Prchal JT. VHL mutations associated with congenital polycythemia. - 6th Symposium on Advances in Molecular Hematology, XXVII. Olomoucké
hematologické dny s mezinárodní účastí, May 12-14 (2013), Olomouc.
Piterkova L, Lorenzo F, Vankayalapali H, Drachtman R, Prchal JT. Novel homozygous VHL mutation in exon 2 (413C>T:P138L) is associated with congenital polycythemia, elevated level of RUNX1/AML1 transcript, but not with the cancer. Blood. Nov 2012; 120(21): 2081.
- 54th Annual Meeting of American Society of Hematology, December 8-11 (2012),
Atlanta (Georgia), USA.

II Original Research
Divoka M, Mojzikova R, Piterkova L, Pospisilova P, Partschova M, Horvathova M, Pospisilova D, Laluhova Striezencova Z, Cermak J, Indrak K, Divoky V. Molecular Characterization of Hemoglobinopathies and Red Cell Enzymopathies in the Czech and Slovak Populations: An Update. Blood. Nov 2011; 118(21): 5307.
- 53rd Annual Meeting of American Society of Hematology, December 10-13
(2011), Sand Diego (California), USA.
Divoka M, Partschova M, Mojzikova R, Piterkova L, Horvathova M, Cermak J, Pospisilova D, Indrak K, Divoky V. Molecular characterization of beta-thalassemia and hemoglobin variants in the Czech and Slovak populations: An Update. Haematologica. 2011; 96(s2): 626.
- 17th Annual Meeting of European Society of Hematology, June 9-12 (2011),
London, United Kingdom.
Piterkova L, Kucerova J, Indrak K, Divoky V. Decreased rate of β-globinL1+ allele
transcription due to intronic LINE-1 insertion in the β-globin gene is associated with β-globinL1+ promoter and enhancer hypermethylation which is not reverted by decitabine. Blood. 2010; 116(21): 860.
- 52nd Annual Meeting of American Society of Hematology, December 4-7 (2010),
Orlando (Florida), USA.
Piterkova L, Kucerova J, Indrak K, Divoky V. Intronic LINE-1 insertion in β-globin gene cause β-thalassemia due to aberrant splicing, nonsense-mediated decay and decreased rate of β-globinL1+ allele transcription.
- XXII. Biochemický zjazd, Martin, September 8-12 (2010), Martin, Slovak
Republic.
Piterkova L, Kucerova J, Indrak K, Divoky V. Intronic LINE-1 insertion in the β-globin gene causes β-thalassemia due to aberrant splicing, nonsense-mediated decay and decreased rate of β-globinL1+ allele transcription. Haematologica. 2010; 95(s2): 417-418.
- 16th Annual Meeting of European Society of Hematology, June 10-13 (2010),
Barcelona, Spain.
Piterkova L, Kucerova J, Indrak K, Divoky V. Negativní vliv retrotransposonu LINE-1 na lidský genom: nová molekulární příčina vzniku β-talasémie.
- X. mezioborové setkání mladých vědeckých a výzkumných pracovníků, Sigma-
Aldrich, May 25-28 (2010), Hotel Devět skal. Best oral presentation award.

II Original Research
Piterkova L, Kucerova J, Indrak K, Divoky V. A novel etiology of β-thalassemia: Evidence for LINE-1 retrotransposon contribution to human disease.
- Conference of Medical and Pharmaceutical Schools, November 19-21 (2009),
Hradec Králové.
Piterkova L, Kucerova J, Indrak K, Divoky. Molekulární mechanismus působení L1 elementu na expresi β-globinového genu: nová příčina vzniku β-talasémie. - Science conference of Ph.D. students, September 8-9 (2009), Faculty of Medicine
and Dentistry, Palacky University, Olomouc. 2nd place in Best Oral Presentation Competition.

II Original Research
4.2 Molecular mechanisms underlying reduced transcription of β-globin gene with intronic LINE-1 insertion leading to β+-thalassemia phenotype
β-thalassemia is an inherited disorder of β-globin chain production. The known molecular mechanisms responsible for β-thalassemia include point mutations located in the β-globin gene or its promoter (the majority of the β-thalassemia mutations) and a small number of deletions removing either a part of the β-globin gene or its locus control region - LCR [1]. The point mutations underlying β-thalassemia are classified according to the mechanism by which they affect gene regulation. Mutation affecting transcription can involve either the conserved DNA sequences in the β-globin promoter (e.g. in the TATA or CCAAT boxes) or the stretch of 50 nucleotides in the 5’ UTR. There are over 50 different mutations that affect splice junction dinucleotides (GT at 5’ and AG at 3’) which completely abolish or reduce the efficiency of normal splicing. Several mutations also activate cryptic splice sites, which disturb the normal splice sites during the processing of pre-mRNA. Other RNA processing mutants affect the polyadenylation signal (AATAAA) and the 3’ UTR [2,3,4]. There are rare forms of β-thalassemia, which are associated with mutations independent of the β-globin complex, e.g. GATA 1 [5] or Xeroderma Pigmentosum D genes [6]. We described a mother and daughter of Ukrainian descent with clinical presentation of β+-thalassemia trait. Inexplicably their phenotypes were more severe than that of typical β+-thalassemia heterozygotes. The propositus (25-year-old Caucasian female) had the hemoglobin concentration values 11 to 12 g/dl, MCV of 60-70 fl, and MCH of 19-20 pg. Red cell morphological abnormalities included hypochromic microcytosis, target cells, poikilocytes and 0.3 to 1.5% of reticulocytes. The HbA2 levels were elevated to 5.3% and the HbF levels were slightly just above 1%. No inclusion bodies of precipitated globin chains were detected in the patient’s erythroid cells after brilliant cresyl blue staining. Hematological indices of the propositus’ mother were comparable. No neurological or other systemic defects were found on both patients physical examination. Analysis of the patient’s β-globin genes revealed no obvious mutation except that each was heterozygous for an unexpected rearrangement detected by gene mapping and this finding provided the impetus for further studies. Hybridization of several restriction enzyme digests of propositus’ DNA to an intron-2 (β-IVS-II) probe showed two abnormal fragments, one major and one minor, indicated unreported rearrangement involving an insertion into the β-globin gene. The insertion (named L1β-thal, Figure 3) occurred 98 bp 5’ to the 3’ end of β-IVS-II. The L1 was flanked by a 9-13 bp target site duplication, with the presumptive first 9 bases (T/AAAATAAAA) forming a consensus L1 endonuclease cleavage site [7]. Analysis of the inserted retrotransposon demonstrated its antisense orientation with respect to the β-globin gene. The inserted L1 was full length, displayed 99.5% homology with a consensus sequence of retrotranspositionally-active human L1s (33 nucleotide differences in 6.0 kb [8]), including only 6 bp differences in the 910 bp 5’ UTR containing the L1 promoter (GenBank accession: AF149422.1). It had two open reading frames and an intact 3’ poly (A) track. The β+-thalassemia nature of this mutation was investigated in reticulocytes and in interspecific hybrids of propositus’ chromosome 11 and mouse erythroleukemia cells. Both mRNA and globin-chain data revealed that the L1 insertion leads to reduced production of β-globin transcript and peptide from the mutant locus, i.e. to β+-thalassemia. L1β-thal led to a

II Original Research
dramatic reduction of β-globin transcripts produced by the affected allele, as assessed by RT-PCR using the propositus’ reticulocyte RNA. We were also able to identify abnormally spliced β-globin transcripts by RT-PCR; the sizes of these PCR products suggested the presence of rare messages with unusual combinations of β-globin exons with intronic and/or insertion-derived sequences. All aberrantly spliced β-globin transcripts contain premature translation termination codon, which makes them susceptible to nonsense-mediated mRNA decay (NMD). We proved that NMD participates in reduction of steady-state mRNA level from affected allele. The low mRNA amount could also result from other causes influencing β-globinL1 mRNA stability.
Figure 3. Schematic diagram of the -globinL1 allele. (middle) The three exons of β-globin gene are shown as grey rectangles, introns as black; the 6 kb insertion is depicted as a green rectangle; the β-IVS-II is divided into two segments of different sizes. (up) Structure of the human L1 retrotransposon [adapted from ref. 9]. The full-length human L1 retrotransposon is 6 kb, and contains: 1) a 910-bp 5’ UTR region with bidirectional promoter activity; ASP (antisense promoter) 2) ORF1 region, which encodes a 40-kD a basic RNA-binding protein with a leucine zipper domain (lz); 3) ORF2 region, which encodes a 150-kDa protein with endonuclease (EN) and reverse transcriptase (RT) activities, and a conserved C-terminal zinc knuckle domain (z); and 4) a 3’ UTR region dispensable for retrotransposition activity. The 3’ UTR contains a functional polyadenylation signal, which occasionally may be bypassed in favor of a stronger downstream signal. L1 terminates with a poly(A) sequence (pA), and is flanked by 2-bp to 20-bp target site duplications (TSDs). (below) Schematic diagram of the L1β-thal insertion and β-globin gene breakpoints. The arrows above the nucleotide sequence show the residues at positions 98 and 99 bp 5’ to the 3’ end of β-IVS-II. The L1 insertion was flanked by a 9-13 bp target site duplication (the run of four As, shown by the hatched bracket, could be duplicated or could be a part of the L1-poly(A) tail). Lower steady-state amount of mRNA produced by β-globinL1 allele also resulted from reduced rate of transcription and decreased production of full-length β-globin transcripts. Hypermethylated promoter/enhancer sequences of β-globinL1 allele indicated epigenetic

II Original Research
modification caused by the presence of L1. Treatment with demethylation agent did not lead to restoration of transcription. Histone deacetylase inhibitor partially reactivated the β-globinL1 transcription in spite of permanent β-globinL1 promoter CpG methylation suggesting that decreased rate of transcription from β-globinL1 allele is associated with altered chromatin. In conclusion, we report a combination of several molecular events leading to the β+-thalassemia phenotype due to intronic LINE-1 insertion in the β-globin gene. Although similar effects of intronic L1 sequence on normal mRNA processing have been reported for other host genes (such as altered splicing, hybrid L1/host gene transcripts, truncation of host gene transcripts [reviewed by ref. 10]) we demonstrated these defective transcript-processing mechanisms in a combination with an L1-mediated epigenetic silencing of the host gene expression. Our data suggested that the main effect of intronic L1 insertion on the host gene expression is transcriptional repression due to regional spreading of methylation affecting both 5’ promoter and 3’ enhancer. However, our attempts to reactivate the silenced β-globinL1 expression revealed important consequences: not a demethylation agent, but trichostatin, a potent histone deacetylase inhibitor, led to (partial) restoration of the host gene transcription suggesting that decreased rate of transcription from β-globinL1 allele is associated also with altered chromatin caused either by β-globinL1 promoter-enhancer displacement due to insertion and/or possibly by spreading of repressive marks induced by L1 to the neighboring β-globinL1 gene promoter [11,12]. These results might have therapeutic implications in diseases caused by deleterious effects of retrotransposons on the expression of nearby genes.
4.3 Novel homozygous VHL mutation in exon 2 is associated with congenital polycythemia but not with cancer
Inherited mutations of the VHL gene cause VHL syndrome, an autosomal dominant disorder [13] that demonstrates marked phenotypic variability and age-dependent penetrance. Heterozygotes for such mutations are at increased risk of developing retinal and central nervous system haemangioblastomas, clear cell renal cell carcinoma, phaeochromocytoma, pancreatic islet tumors and endolymphatic sac tumors [14]. Tumors develop from cells that acquire a somatic mutation of the unaffected VHL gene in addition to the germ line mutation on the other allele (two hit model of cancerogenesis) [15]. In rare cases, affected patients may present with polycythemia, a paraneoplastic manifestation of VHL syndrome presumably due to inappropriate production of EPO by the tumor cells. In fact, production of EPO by the tumor has been demonstrated both in the case of hemangioblastoma and renal cell carcinoma; the polycythemia usually resolves after removal of the tumor [16]. There are two known homozygous VHL gene mutations causing polycythemia that are not associated with a VHL cancer syndrome - an R200W mutation endemic in Chuvashia causing the first recognized disorder of augmented hypoxia sensing in normoxia [17], and an H191D mutation of Croatian origin [18]. Both are located in the distal exon-3 of the VHL gene of its C-terminal domain. As different positions of loss-of-function mutations of VHL gene are associated with different type of cancers, it has been proposed that only C-terminal domain VHL mutations would cause polycythemia. However, we contradicted this notion. We report a novel homozygous variant of the VHL gene located in the middle of coding region in exon-2; c.413C>T:P138L. The propositus is a 15 year old Punjabi female with

II Original Research
congenital polycythemia; i.e. elevated EPO 40 mIU/mL, no JAK2 mutations, and hemoglobin 19-20 g/dl. Her parents are VHLP138L heterozygotes and no VHL tumors are reported in the extended family, in contrast to the other VHL P138 residue (P138R [19], P138T [20]) mutations that have been reported in VHL syndrome and renal cancer. pVHL is a negative regulator of hypoxia inducible transcription factors as it degrades α subunits of HIFs. It has been proposed, that an association between mutated pVHL and suppressor of cytokine signaling 1 leads to JAK2 up-regulation and enhanced erythropoiesis [21]. We show that the VHLP138L mutation, which lies in the catalytic HIF-1α peptide ligand-binding region, perturbs pHIF-1α pVHL interaction due to a conformational effect on the W117 and S111 residues lying within 2.8 to 4.3 Å distance from the mutated L138. The effect of this single mutation on overall structure is a shift of 1.9 Å RMSD (root mean square deviation) from the wild-type complex structure (see Figure 4). The accumulation of HIFs and up-regulated transcription of downstream target genes including those for glucose transporter-1 (SLC2A1) and transferrin (TF) were found in the propositus’ granulocytes.
Figure 4. Molecular dynamics simulations study of pVHL-ElonginC-ElonginB complex and interaction with HIF-1α. (i) Superimposition of wild type (grey color) and mutated pVHL (green color) is shown. The wt P138 (in violet) and mutated L138 (in green) sites and the critical active site residues for the HIF-1α peptide (PDB:1LM8) binding region are depicted. The P138L mutations perturbs pHIF-1α interactions with pVHL due to the conformational effect on the W117 and S111 residues (shown in orange) at 2.8 to 4.3 Å distance from mutated L138. (ii) Detail superimposition of wild type and P138L pVHL in the interaction with HIF-1α.

II Original Research
We then analyzed the VHLP138L propositus and her heterozygous parents’ erythroid progenitors and found them to be hypersensitive to EPO (a feature of primary polycythemias without EPO independent colonies). Because of reports that RUNX1/AML1 transcript levels are specifically upregulated in erythroid progenitors in polycythemia vera and allegedly responsible for PV EPO hypersensitivity [22,23], we analyzed erythroid RUNX1/AML1 transcripts in the propositus and found them increased. In conclusion, we demonstrated that a homozygous mutation in exon-2 of the VHL gene can also be associated with a polycythemic phenotype rather than VHL syndrome tumors. Further, we report that increased levels of RUNX1/AML1 transcripts are not specific for PV but can be seen in other primary polycythemias. This report provides further evidence for the heterogeneity of clinical phenotypes of VHL mutations resulting in cancers or in augmented erythropoiesis. The molecular basis of these differences remains unclear. We can only speculate that the relatively small VHL peptide comprised of 213 codons yet encoded by a large >11,2 kb VHL gene may have multiple functions, possibly due to interactions with other modifying factors, that await future clarification. We submit that description of families with congenital disorders and unique phenotypes due to VHL mutations provides an attractive opportunity for structure-function relationship analyses of VHL protein and leads to an enhanced understanding of polycythemic disorders and diseases of hypoxia sensing.
4.4 Polycythemia due to Croatian homozygous VHL (c.571C>G:H191D) mutation has a different phenotype than Chuvash polycythemia (VHL c.598C>T:R200W)
The only known endemic polycythemia, Chuvash polycythemia, is a disorder of global augmented hypoxia sensing due to elevated hypoxia inducible transcription factors (HIF-1 and HIF-2). The underlying genetic defect is the homozygosity with respect to a C→T missense mutation in VHL gene, causing an arginine-to-tryptophan change at amino-acid residue 200 (Arg200Trp) [17]. The mutation has also non-erythroid consequences such as increased stroke and low blood pressure, pulmonary hypertension [24,25], and increased PIA [26], which have even greater impact on morbidity and mortality than the initially uncovered erythroid effect. The estimated time of origin of VHLR200W mutation is prior to divergence of the human races approximately 14,000 - 62,000 years ago, suggesting that while those homozygous for this mutation have increased mortality and are selected against, more numerous heterozygotes must have a survival or reproductive advantage yet to be defined [27]. Beside Chuvash polycythemia, there is accumulating evidence of other congenital polycythemias characterized by VHL mutations [16,18,28]. In the study, we report second homozygous polycythemic patient for the VHL c.71C>G (H191D) germ-line mutation, a 5 year old Croatian girl from Herzegovina, a region located in the southern part of Bosnia contiguous to Dalmatia. Herzegovina is the counterpart to the Croatian region and is populated largely by people of Croatian ethnicity along the border. We set up to define the phenotype of this mutation in this and the previously reported homozygote [18] with particular emphasis on a critical comparison with CP. We also pursued a hypothesis that a putative survival advantage of H191D heterozygotes may account for the possible high prevalence of heterozygosity in Croatians and thus set up to

II Original Research
determine its approximate origin in evolution by determining haplotype sharing among affected individuals. We analyzed the two homozygotes (p12 and p18, see Figure 5) by high-density genotyping to examine their relatedness and the VHL haplotype. We detected evidence of a significant relationship between the two individuals (p=4x10-18), with a maximum likelihood estimate of 8th degree relatives (95% C.I. 6th degree - 11th degree) [29], exactly matching the known pedigree (see Figure 5). These individuals share a 16 cM haploid segment on chromosome 11 and a 24 cM haploid IBD (identity by descent) segment on chromosome 3 (see Figure 6). Within the segment on chromosome 3, a 15.6 cM autozygous segment is present in the female (p18), and a 1.6 cM autozygous segment is present in the male (p12). Both autozygous segments contain the VHL mutation, and the smaller segment is diploid IBD. Two other large autozygous segments are present in the male (15.5 cM on chromosome 15 and 10 cM on chromosome 4). The presence of autozygosity at the VHL mutation and elsewhere in the genome is a strong indication that all four of the parents of p12 and p18 are related, sharing a common ancestor between approximately 3 and 6 generations ago. This common ancestor is likely the founder of the VHLH191D mutation. Since the analysis demonstrated a recent origin of this mutation and, thus, the frequency of this mutation among tested families cannot be used in arguments for or against any survival benefit or detriment.
Figure 5. Pedigree of the VHL H191D families available for study with extended family history. Each family is denoted by a family number (F1, F2). The ‘p’ code (p01, p02 and so on) indicates individuals from whom DNA samples have been obtained. The arrow indicates new polycythemic patient. The VHL mutation of the paternal side of F1 must have been inherited from the husband of p01. The husband of p01 was not related to F2 according to the pedigree and the mutation did not segregate among the known relatives. Heterozygote individuals denoted by asterisk as well as the husband of p01 presumably shared the common ancestor, the founder of the mutation.
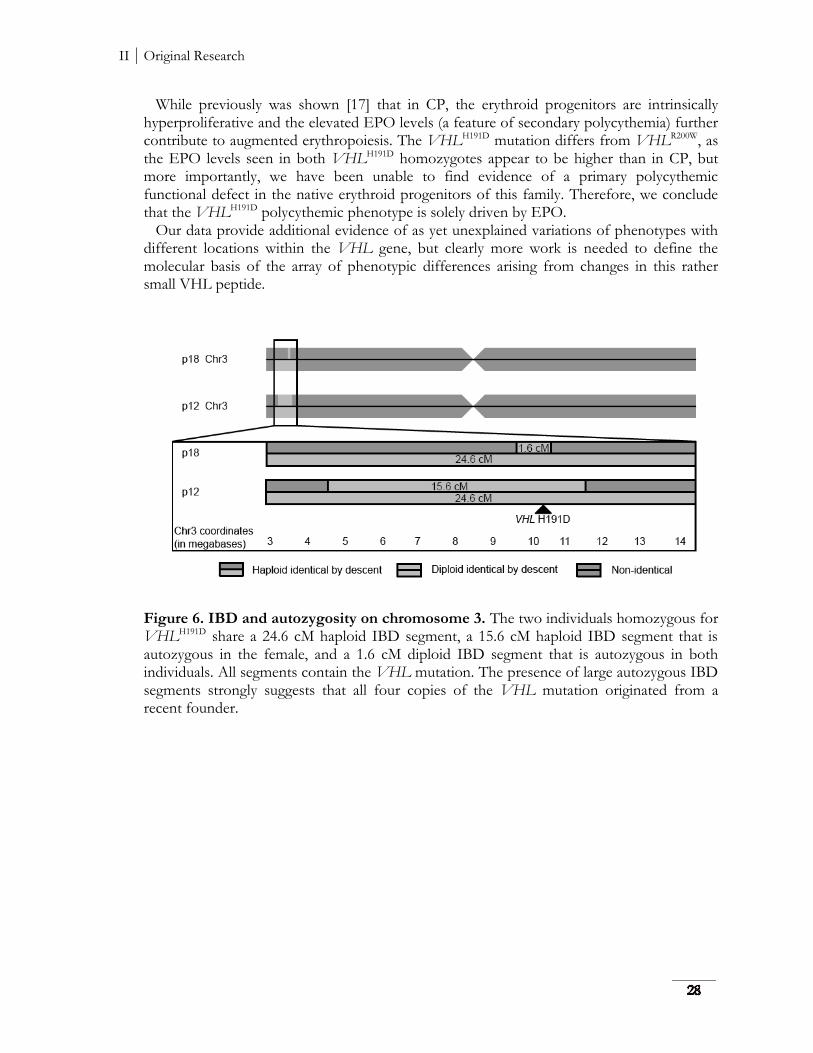
II Original Research
While previously was shown [17] that in CP, the erythroid progenitors are intrinsically hyperproliferative and the elevated EPO levels (a feature of secondary polycythemia) further contribute to augmented erythropoiesis. The VHLH191D mutation differs from VHLR200W, as the EPO levels seen in both VHLH191D homozygotes appear to be higher than in CP, but more importantly, we have been unable to find evidence of a primary polycythemic functional defect in the native erythroid progenitors of this family. Therefore, we conclude that the VHLH191D polycythemic phenotype is solely driven by EPO. Our data provide additional evidence of as yet unexplained variations of phenotypes with different locations within the VHL gene, but clearly more work is needed to define the molecular basis of the array of phenotypic differences arising from changes in this rather small VHL peptide.
Figure 6. IBD and autozygosity on chromosome 3. The two individuals homozygous for VHLH191D share a 24.6 cM haploid IBD segment, a 15.6 cM haploid IBD segment that is autozygous in the female, and a 1.6 cM diploid IBD segment that is autozygous in both individuals. All segments contain the VHL mutation. The presence of large autozygous IBD segments strongly suggests that all four copies of the VHL mutation originated from a recent founder.

II Original Research
5 Summary In 1988, a novel molecular mechanism producing inactivation of gene expression and causing human disease, genomic insertion of a long interspersed nuclear elements (LINE-1, L1), was described [1]. The human genome contains about 5 x 105 copies of these retrotransposons [2]; most of which are randomly 5’ truncated and inactive. However, there are 3000 - 4000 full-length L1 elements in the human genome and roughly 80 - 100 of them are capable of ongoing retrotranspositon [3]. De novo L1 insertions into genes such as factor VIII [1], dystrophin [4] or retinitis pigmentosa [5] demonstrated the mutagenic potential of active L1 elements in the genesis of human disease. Our group has described a family with β-thalassemia due to the insertion of a full-length L1 element into the β-globin gene and shown that this L1 element retained the capacity for high retrotransposition frequency. This was the first example of an intact, functional L1 causing human disease. We now demonstrated that this retrotransposon was inserted in the antisense orientation into the 3’ end of intron-2 of the β-globin gene and led to a severe reduction of β-globin gene expression due to aberrant splicing, nonsense-mediated decay, decreased production of full-length transcripts and epigenetic transcriptional repression of β-globinL1 allele. Although mutations in the human globin genes have been extensively studied for a long time, this kind of molecular etiology of thalassemia has not been previously described. Studies on Chuvash polycythemia and other VHL-associated polycythemias are important because VHL was originally identified as a tumor-suppressor gene mutated in VHL syndrome, where tumors are initiated by biallelic VHL inactivation and are associated with abnormal activation of hypoxic gene response pathways [6]. Whereas, the majority of the VHL syndrome mutations abolish the ability of VHL to polyubiquitinate HIF-α, polycythemic VHL mutations lead to a more modest partial loss of activity [7]. Quantitative difference in the loss of activity could explain the variable phenotype among VHL mutations, but VHL gene may have also other functions, possibly due to interactions with other modifying factors, that can contribute to the onset of diseases and which await future clarification. We described a new homozygous VHL exon-2 mutation, VHLP138L, which is associated in the affected homozygote with congenital polycythemia but not in her, or her relatives, with cancer or other VHL syndrome tumors. This contrasts with reports of other heterozygous VHL mutations encoding the same amino acid residue (VHL P138R, P138T) that have been reported in VHL syndrome and renal cancer. We also show that this mutation is not only associated with elevated EPO levels but also with a hallmark of primary polycythemia, i.e. EPO hypersensitivity. We also provided evidence that polycythemia due to Croatian homozygous VHLH191D mutation has a different phenotype than Chuvash polycythemia.

III Supplements and Appendices
6 Bibliography 1 Introduction 1.1 The erythropoiesis 1.1.1 Morphology and composition of the erythrocyte
[1] Sood R, Liu P. Novel Insights into the Genetic Controls of Primitive and Definitive Hematopoiesis from Zebrafish Models. Adv Hematol. 2012;2012:830703.
[2] Gregory CJ, Eaves AC. Three stages of erythropoietic progenitor cell differentiation distinguished by a number of physical and biologic properties. Blood. 1978; 51:527-37.
[3] Part VI. The Erythrocyte. In Kaushansky K, Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Prchal JT. Williams Hematology. 8th edn. McGraw-Hill Companies, Inc. 2011; 409-865.
[4] Marengo-Rowe AJ. Structure-function relations of human hemoglobins. Proc (Bayl Univ Med Cent). 2006; 19(3):239-45.
[5] Stamatoyannopoulos G. Control of globin gene expression during development and erythroid differentiation. Exp Hematol. 2005; 33(3):259-71.
[6] Forget BG, Bunn HF. Classification of the disorders of hemoglobin. Cold Spring Harb Perspect Med. 2013; 3(2):a011684.
[7] Ponka P. Tissue-specific regulation of iron metabolism and heme synthesis: distinct control mechanisms in erythroid cells. Blood. 1997;89(1):1-25.
[8] Richardson DR, Ponka P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim Biophys Acta. 1997 Mar 14;1331(1):1-40.
[9] Chapter 18. Iron homeostasis. In Beaumont C, Beris P, Beuzard Y, Brugnara C. Disorders of iron homeostasis, erythrocytes, erythropoiesis. 1st edn. European School of Haematology, Handbook. 2006; 392-408.
1.1.2 Regulation of erythropoiesis
[1] Haase VH. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013; 27(1):41-53.
[2] Part VI. The Erythrocyte. In Kaushansky K, Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Prchal JT. Williams Hematology. 8th edn. McGraw-Hill Companies, Inc. 2011; 409-865.
[3] Remy I, Wilson IA, Michnick SW. Erythropoietin Receptor Activation by a Ligand-Induced Conformation Change. Science. 1999; 283(5404):990-993.
[4] Chin H, Arai A, Wakao H, Kamiyama R, Miyasaka N, Miura O. Lyn physically associates with the erythropoietin receptor and may play a role in activation of the Stat5 pathway. Blood. 1998; 91(10):3734-45.
[5] Krebs DL, Hilton DJ. SOCS: physiological suppressors of cytokine signaling. J Cell Sci. 2000;113 (16):2813-9.
[6] Tong W, Zhang J, Lodish HF. Lnk inhibits erythropoiesis and Epo-dependent JAK2 activation and downstream signaling pathways. Blood. 2005;105(12):4604-12.

III Supplements and Appendices
1.1.3 Regulation of oxygen sensing
[1] Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105(2):659-69.
[2] Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995; 92(12): 5510-5514.
[3] Gu YZ, Moran SM, Hogenesch JB, Wartman L, Bradfield CA. Molecular characterization and chromosomal localization of a third α-class hypoxia inducible factor subunit, HIF-3α. Gene Expr. 1998;7(3):205-13.
[4] Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270(3):1230-7.
[5] Wiesener MS, Jürgensen JS, Rosenberger C, Scholze CK, Hörstrup JH, Warnecke C, Mandriota S, Bechmann I, Frei UA, Pugh CW, Ratcliffe PJ, Bachmann S, Maxwell PH, Eckardt KU. Widespread hypoxia inducible expression of HIF-2α in distinct cell populations of different organs. FASEB J. 2003;17(2):271-3.
[6] Makino Y, Cao R, Svensson K, Bertilsson G, Asman M, Tanaka H, Cao Y, Berkenstam A, Poellinger L. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature. 2001; 414:550-554.
[7] Semenza GL. Hypoxia-Inducible Factor 1 (HIF-1) Pathway. Sci STKE. 2007;(407):cm8.
[8] Peet D, Linke S. Regulation of HIF: Asparaginyl hydroxylation. Novartis Found. Symp. 2006; 272:37-49.
[9] Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, Scarpulla RC, Chandel NS. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1(6):409-14.
[10] Semenza GL. Oxygen Sensing, Homeostasis, and Disease. N Engl J Med. 2011; 365(6): 537-547.
[11] Haase VH. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013; 27(1):41-53. 1.2 Clinical manifestation and classification of erythroid disorders 1.2.1 Anemia
[1] Chapter 9. Molecular basis of thalassaemia syndromes. In Beaumont C, Beris P, Beuzard Y, Brugnara C. Disorders of iron homeostasis, erythrocytes, erythropoiesis. 1st edn. European School of Haematology, Handbook. 2006; 210-228.
[2] A Database of Human Hemoglobin Variants and Thalassemias http://globin.cse.psu.edu/hbvar/ menu.html. Assessed February 18, 2013.
[3] Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. The Lancet. 2012; 379(9813):373-383.
[4] Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480-7.
[5] Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115(22):4331-6.
[6] Singer ST. Variable Clinical Phenotypes of α-Thalassemia Syndromes. TheScientificWorldJOURNAL. 2009;9:615-625.
[7] Perrine SP. Fetal Globin Induction—Can It Cure β Thalassemia? Hematology Am Soc Hematol Educ Program. 2005;38-44.
[8] Sadelain M, Rivella S, Lisowski L, Samakoglu S, Rivière I. Globin gene transfer for treatment of the beta-thalassemias and sickle cell disease. Best Pract Res Clin Haematol. 2004;17(3):517-34.
[9] Part VI. The Erythrocyte. In Kaushansky K, Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Prchal JT. Williams Hematology. 8th edn. McGraw-Hill Companies, Inc. 2011; 409-865.
1.2.2 Polycythemia 1.2.2.1 Primary polycythemia
[1] Ang SO, Chen H, Hirota K, et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet. 2002;32(4):614-621.

III Supplements and Appendices
[2] Part VI. The Erythrocyte. In Kaushansky K, Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Prchal JT. Williams Hematology. 8th edn. McGraw-Hill Companies, Inc. 2011; 823-838.
[3] Spivak JL. Polycythemia vera: myths, mechanisms, and management. Blood. 2002;100:4272-90.
[4] Prchal JF, Axelrad AA. Letter: Bone-marrow responses in polycythemia vera. N Engl J Med. 1974; 290:1382.
[5] James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, Garçon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144-8.
[6] Kralovics R, Guan Y, Prchal JT. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp Hematol. 2002;30:229-36.
[7] Kralovics R, Teo SS, Li S, Theocharides A, Buser AS, Tichelli A, Skoda RC. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood. 2006;108:1377-80.
[8] Nussenzveig RH, Swierczek SI, Jelinek J, Gaikwad A, Liu E, Verstovsek S, Prchal JF, Prchal JT. Polycythemia vera is not initiated by JAK2V617F mutation. Exp Hematol. 2007;35, 32-8.
[9] Prchal JT, Crist WM, Goldwasser E, Perrine G, Prchal JF. Autosomal dominant polycythemia. Blood. 1985;66:1208.
[10] Gordeuk VR, Stockton DW, Prchal JT. Congenital polycythemias/erythrocytoses. Haematologica. 2005;90(1):109-16.
1.2.2.2 Secondary polycythemia 1.2.2.3 Congenital disorders of hypoxia sensing
[1] Lorenzo FRV, Huff CD, Myllymäki M, Swierczek S, Salama ME, Semenza G, Gordeuk VR, Jorde L, Koivunen P, Prchal JT. A Novel EGLN1/PHD2 High-Frequency Variant in Tibetans Protects Against Hypoxia-Induced Polycythemia. Blood (ASH Annual Meeting Abstracts) 2012 120: Abstract 2079.
[2] Ang SO, Chen H, Hirota K, et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet. 2002;32(4):614-621.
[3] Liu E, Percy MJ, Amos CI, Guan Y, Shete S, Stockton DW, McMullin MF, Polyakova LA, Ang SO, Pastore YD, Jedlickova K, Lappin TR, Gordeuk V, Prchal JT. The worldwide distribution of the VHL 598C>T mutation indicates a single founding event. Blood. 2004;103(5):1937-40.
[4] Pastore YD, Jelinek J, Ang S, et al. Mutations in the VHL gene in sporadic apparently congenital polycythemia. Blood. 2003;101(4):1591-1595.
[5] Pastore Y, Jedlickova K, Guan Y, et al. Mutations of von Hippel-Lindau tumor-suppressor gene and congenital polycythemia. Am J Hum Genet. 2003;73(2):412-9.
[6] Percy MJ, Beard ME, Carter C, Thein SL. Erythrocytosis and the Chuvash von Hippel-Lindau mutation. Br J Haematol. 2003;123(2):371-2.
[7] Perrotta S, Nobili B, Ferraro M, Migliaccio C, Borriello A, Cucciolla V, Martinelli V, Rossi F, Punzo F, Cirillo P, Parisi G, Zappia V, Rotoli B, Della Ragione F. Von Hippel-Lindau-dependent polycythemia is endemic on the island of Ischia: identification of a novel cluster. Blood. 2006;107(2):514-9.
[8] Gordeuk VR, Sergueeva AI, Miasnikova GY, et al. Congenital disorder of oxygen sensing: association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood. 2004:103(10):3924-32.
[9] Friedrich CA. Genotype-phenotype correlation in von Hippel-Lindau syndrome. Hum Mol Genet. 2001;10 (7):763-767.
[10] Maher ER, Webster AR, Richards FM, Green JS, Crossey PA, Payne SJ, Moore AT. Phenotypic expression in von Hippel-Lindau disease: correlations with germline VHL gene mutations. J Med Genet. 1996;33(4):328-32.
[11] Maher ER. Genetics of phaeochromocytoma. Br Med Bull. 2006;79-80(1):141-151.
[12] Bento MC, Chang KT, Guan Y, Liu E, Caldas G, Gatti RA, Prchal JT. Congenital polycythemia with homozygous and heterozygous mutations of von Hippel-Lindau gene: five new Caucasian patients. Haematologica. 2005;90(1):128-9.

III Supplements and Appendices
[13] Percy MJ, Zhao Q, Flores A, Harrison C, Lappin TR, Maxwell PH, McMullin MF, Lee FS. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci U S A. 2006;103(3):654-9.
[14] Lee FS, Percy MJ. The HIF pathway and erythrocytosis. Annu Rev Pathol. 2011;6:165-92.
[15] Part VI. The Erythrocyte. In Kaushansky K, Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Prchal JT. Williams Hematology. 8th edn. McGraw-Hill Companies, Inc. 2011; 823-838.
1.3 Mobile elements and human diseases 1.3.1 Mobile elements
[1] Lander ES et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860-921.
[2] Belancio VP, Deininger PL, Roy-Engel AM. LINE dancing in the human genome: transposable elements and disease. Genome Med. 2009;1(10):97.
[3] Babushok DV, Kazazian HH Jr. Progress in Understanding the Biology of the Human Mutagen LINE-1. Hum Mutat. 2007;28(6):527-39.
[4] Reznikoff WS. Tn5 as a model for understanding DNA transposition. Molecular Microbiology. 2003;47(5):1199-1206.
[5] Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10(10):691-703.
[6] Mills RE, Bennett EA, Iskow RC, Devine SE. Which transposable elements are active in the human genome? Trends Genet. 2007;23:183-191.
[7] Belancio VP, Hedges DJ, Deininger P. Mammalian non-LTR retrotransposons: for better or worse, in sickness and in health. Genome Res. 2008;18:343-358.
[8] Hancks DC, Kazazian HH Jr.Active human retrotransposons: variation and disease. Current Opinion in Genetics & Development. 2012;22(3):191-203.
[9] Kazazian HH Jr. Mobile elements: drivers of genome evolution. Science. 2004;303(5664):1626-32.
[10] Kazazian HH Jr. Mobile DNA transposition in somatic cells. BMC Biology. 2011;9:62
[11] Han JS, Boeke JD. LINE-1 retrotransposons: modulators of quantity and quality of mammalian gene expression? BioEssays. 2005;27:775-784
[12] Goodier JL, Kazazia HH Jr. Retrotransposons Revisited: The Restraint and Rehabilitation of Parasites. Cell. 2008;135:23-35.
1.3.2 The role of mobile elements in the pathogenesis of human diseases
[1] Kines KJ, Belancio VP. Department of Structural and Cellular Biology, Tulane Expressing genes do not forget their LINEs: transposable elements and gene expression. Front Biosci. 2012;17:1329-1344.
[2] Han JS, Szak ST, Boeke JD. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature. 2004;429(6989):268-74.
[3] Soifer HS, Zaragoza A, Peyvan M, Behlke MA, Rossi JJ. A potential role for RNA interference in controlling the activity of the human LINE-1 retrotransposon. Nucleic Acids Res. 2005;33(3):846-56.
[4] Rangwala SH, Zhang L, Kazazian HH Jr. Many LINE1 elements contribute to the transcriptome of human somatic cells. Genome Biol. 2009;10(9):R100.
[5] Belancio VP, Roy-Engel AM, Deininger PL. All you need to know about retrotransposon and cancer. Semin Cancer Biol. 2010; 20(4): 200-210.
[6] Hata K, Sakaki Y. Identification of critical CpG sites for repression of L1 transcription by DNA methylation. Gene. 1997;189(2):227-34.
[7] Chalitchagorn K, Shuangshoti S, Hourpai N, Kongruttanachok N, Tangkijvanich P, Thongngam D., Voravud N, Sriuranpong V, Mutirangura A. Distinctive pattern of LINE-1 methylation level in normal tissue and the association with carcinogenesis. Oncogene. 2004;23:8841-8846.
[8] Aporntewan C, Phokaew C, Piriyapongsa J, Ngamphiw C, Ittiwut C, Tongsima S, Mutirangura A. Hypomethylation of intragenic LINE-1 represses transcription in cancer cells through AGO2. PLoS One. 2011;6(3):e17934.
[9] Daskalos A, Nikolaidis G, Xinarianos G, Savvari P, Cassidy A, Zakopoulou R, Kotsinas A, Gorgoulis V, Field JK, Liloglou T. Hypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancer. Int J Cancer. 2009;124(1):81-7.

III Supplements and Appendices
[10] Roman-Gomez J, Jimenez-Velasco A, Agirre X, Cervantes F, Sanchez J, Garate L, Barrios M, Castillejo JA, Navarro G, Colomer D, Prosper F, Heiniger A, Torres A. Promoter hypomethylation of the LINE-1 retrotransposable elements activates sense/antisense transcription and marks the progression of chronic myeloid leukemia. Oncogene. 2005;24:7213-7223.
[11] Belancio VP, Hedges DJ, Deininger P. Mammalian non-LTR retrotransposons: for better or worse, in sickness and in health. Genome Res. 2008;18:343-358.
[12] Hedges DJ, Deininger PL. Inviting Instability: Transposable elements, Double-strand breaks, and the Maintenance of Genome Integrity. Mutat Res. 2007; 616(1-2):46-59.
[13] O’Neil J, Tchinda J, Gutierrez A, Moreau L, Maser RS, Wong KK, Li W, McKenna K, Liu XS, Feng B, Neuberg D, Silverman L, DeAngelo DJ, Kutok JL, Rothstein R, DePinho RA, Chin L, Lee C, Look AT. Alu elements mediate MYB gene tandem duplication in human T-ALL. J Exp Med. 2007;204:3059-3066.
[14] Strout MP, Marcucci G, Bloomfield CD, Caligiuri MA. The partial tandem duplication of ALL1 (MLL) is consistently generated by Alu-mediated homologous recombination in acute myeloid leukemia. Proc Natl Acad Sci U S A. 1998;95:2390-2395.
[15] Gasior SL, Wakeman TP, Xu B, Deininger PL. The human LINE-1 retrotransposon creates DNA double-strand breaks. J Mol Biol. 2006;357(5):1383-93
3 Materials and Methods
[1] Green MR, Sambrook J. Molecular Cloning: A Laboratory Manual (Fourth Edition). Oxford University Press, 2012.
[2] Prchal JT, Throckmorton DW, Carroll AJ 3rd, Fuson EW, Gams RA, Prchal JF. A common progenitor progenitor for human myeloid and lymphoid cells. Nature. 1978;274(5671):590-591.
[3] Yagi M, Stamatoyannopoulos G, Papayannopoulou T. Monoclonal antibody 53.6 recognizes a novel proliferation-associated antigen encoded on human chromosome 11. J Immunol. 1987;138(7):2208-2212.
[4] Yang Ch, Feng J, Song W, et al. A mouse model for nonsense mutation bypass therapy shows a dramatic multiday response to geneticin. Proc Natl Acad Sci USA. 2007;4(39):15394-15399.
[5] Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45.
[6] Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30(9):e36.
[7] Mabaera R, Richardson CHA, Johnson K, Hsu M, Fiering S, Lowrey CH. Developmental and
differentiation specific patterns of human β- and -globin promoter DNA methylation. Blood. 2007;110(4):1343-1352.
[8] Gaikwad A, Nussenzveig R, Liu E, Gottshalk S, Chang K, Prchal JT. In vitro expansion of erythroid progenitors from polycythemia vera patients leads to decrease in JAK2 V617F allele. Exp Hematol. 2007;35(4):587-595.
4 Results and Discussion
[1] Stamatoyannopoulos G, Nienhuis AW, Majerus PW, Varmus H. The Molecular Basis of Blood Diseases. Philadelphia, PA: Saunders; 1994.
[2] Part VI. The Erythrocyte. In Kaushansky K, Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Prchal JT. Williams Hematology. 8th edn. McGraw-Hill Companies, Inc. 2011; 409-865.
[3] Chapter 9. Molecular basis of thalassaemia syndromes. In Beaumont C, Beris P, Beuzard Y, Brugnara C. Disorders of iron homeostasis, erythrocytes, erythropoiesis. 1st edn. European School of Haematology, Handbook. 2006; 210-228.
[4] Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. The Lancet. 2012; 379(9813):373-383.
[5] Yu C, Niakan KK, Matsushita M, Stamatoyannopoulos G, Orkin SH, Raskind WH. X-linked thrombocytopenia with thalassemia from a mutation in the amino finger of GATA-1 affecting DNA binding rather than FOG-1 interaction. Blood. 2002;100(6):2040-5.

III Supplements and Appendices
[6] Viprakasit V, Gibbons RJ, Broughton BC, Tolmie JL, Brown D, Lunt P, Winter RM, Marinoni S, Stefanini M, Brueton L, Lehmann AR, Higgs DR. Mutations in the general transcription factor TFIIH result in beta-thalassaemia in individuals with trichothiodystrophy. Hum Mol Genet. 2001;10(24):2797-802.
[7] Nienhuis AW, Anderson WF. Isolation and translation of hemoglobin messenger RNA from thalassemia, sickle cell anemia, and normal human reticulocytes. J Clin Invest. 1971;50:2458-2460.
[8] Feng Q, Moran JV, Kazazian HH Jr, Boeke JD. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 1996;87:905-916.
[9] Kimberland ML, Divoky V, Prchal J, Schwahn U, Berger W, Kazazian HH Jr. Full-length human L1 insertions retain the capacity for high frequency retrotransposition in cultured cells. Hum Mol Genet. 1999;8:1557-1560.
[10] Babushok DV, Kazazian HH Jr. Progress in Understanding the Biology of the Human Mutagen LINE-1. Hum Mutat. 2007;28(6):527-39.
[11] Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10(10):691-703.
[12] Garcia-Perez JL, Morell M, Scheys JO, et al. Epigenetic silencing of engineered L1 retrotransposition events in human embryonic carcinoma cells. Nature. 2010;466(7307):769-73.
[13] Montoya-Durango DE, Liu Y, Teneng I, et al. Epigenetic control of mammalian LINE-1 retrotransposon by retinoblastoma proteins. Mutat Res. 2009; 665(1-2):20-8.
[14] Bader HL, Hsu T. Systemic VHL gene functions and the VHL disease. FEBS Lett. 2012;586(11):1562-1569.
[15] Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet. 2011;19(6):617-623.
[16] Gordeuk VR, Stockton DW, Prchal JT. Congenital polycythemias/erythrocytoses. Haematologica. 2005; 90:109-116.
[17] Pastore YD, Jelinek J, Ang S, et al. Mutations in the VHL gene in sporadic apparently congenital polycythemia. Blood. 2003;101(4):1591-1595.
[18] Ang SO, Chen H, Hirota K, et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet. 2002;32(4):614-621.
[19] Pastore Y, Jedlickova K, Guan Y, et al. Mutations of von Hippel-Lindau tumor-suppressor gene and congenital polycythemia. Am J Hum Genet. 2003;73(2):412-9.
[20] Leonardi E, Martella M, Tosatto SC, Murgia A. Identification and in silico analysis of novel von Hippel-Lindau (VHL) gene variants from a large population. Ann Hum Genet. 2011;75(4):483-96.
[21] Gnarra JR, Tory K, Weng Y, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet. 1994;7(1):85-90.
[22] Russell RC, Sufan RI, Zhou B, et al. Loss of JAK2 regulation via a heterodimeric VHL-SOCS1 E3 ubiquitin ligase underlies Chuvash polycythemia. Nat Med. 2011;17(7):845-853.
[23] Mutschler M, Magin AS, Buerge M, et al. NF-E2 overexpression delays erythroid maturation and increases erythrocyte production. Br J Haematol. 2009;146(2):203-217.
[24] Wang W, Schwemmers S, Hexner EO, Pahl HL. AML1 is overexpressed in patients with myeloproliferative neoplasms and mediates JAK2V617F-independent overexpression of NF-E2. Blood. 2010;116(2):254-266.
[25] Bushuev VI, Miasnikova GY, Sergueeva AI. Endothelin-1, vascular endothelial growth factor and systolic pulmonary artery pressure in patients with Chuvash polycythemia. Haematologica. 2006;91(6):744-9.
[26] Gordeuk VR, Prchal JT. Vascular complications in Chuvash polycythemia. Semin Thromb Hemost. 2006;32(3):289-94.
[27] Gordeuk VR, Sergueeva AI, Miasnikova GY, et al. Congenital disorder of oxygen sensing: association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood. 2004:103(10):3924-32.
[28] Liu E, Percy MJ, Amos CI, et al. The worldwide distribution of the VHL 598C>T mutation indicates a single founding event. Blood. 2004;103(5):1937-40.
[29] Lee FS, Percy MJ. The HIF Pathway and Erythrocytosis. Annu. Rev. Pathol. Mech. Dis. 2011;6:165-92.
[30] Huff CD, Witherspoon DJ, Simonson TS, et al. Maximum-likelihood estimation of recent shared ancestry (ERSA). Genome Res. 2011;21(5):768-774.

III Supplements and Appendices
5 Summary
[1] Kazazian HH Jr, Wong C, Youssoufian H, Scott AF, Phillips DG, Antonarakis SE. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature. 1988;332:164-166.
[2] Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10(10):691-703.
[3] Hancks DC, Kazazian HH Jr.Active human retrotransposons: variation and disease. Current Opinion in Genetics & Development. 2012;22(3):191-203.
[4] Narita N, Nishio H, Kitoh Y, et al. Insertion of a 5’ truncated L1 element into the 3’ end of exon 44 of the dystrophin gene resulted in skipping of the exon during splicing in a case of Duchenne muscular dystrophy. J Clin Invest. 1993;91:1862-1867.
[5] Schwahn U, Lenzner S, Dong J, et al. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat Genet. 1998;19:327-332.
[6] Kim WY, Kaelin WG. Role of VHL Gene Mutation in Human Cancer. JCO. 2004;22(24): 4991-5004.
[7] Lee FS, Percy MJ. The HIF Pathway and Erythrocytosis. Annu. Rev. Pathol. Mech. Dis. 2011;6:165-92.

III Supplements and Appendices
7 Acronyms and Abbreviations ACD citrate-dextrose solution Act-D actinomycin D ADM adrenomedullin ARNT aryl hydrocarbon receptor nuclear translocator Asn asparagine ATP adenosine triphosphate BFU-E burst forming unit - erythroid BNIP3 BCL2/adenovirus E1B 19kDa interacting protein 3 BNIP3L BCL2/adenovirus E1B 19kDa interacting protein 3 like BRCA1 breast cancer 1 cDNA complementary deoxyribonucleic acid CFU-E colony forming unit - erythroid CFU-GEMM colony-forming unit granulocytic, erythroid, megakaryocyte, macrophage CIS cytokine inducible SH2 protein CP Chuvash polycythemia CTP cytosine triphosphate DAC decitabine: 2’-deoxy-5-azacytidine D-MEM Dulbecco’s Modified Eagle Medium DMSO dimethyl sulfoxide DMT-1 divalent metal transporter 1 DNA deoxyribonucleic acid DSBs double strand breaks DTT dithiothreitol EDTA ethylene-diamine-tetraacetic acid EBV Epstein-Barr virus EGLN egl nine homolog 1 EPO erythropoietin EPOR erythropoietin receptor FIH-1 factor inhibiting hypoxia inducible factor 1 GAPDH glyceraldehyde 3-phosphate dehydrogenase GATA-1 GATA-binding factor 1 GTP guanine triphosphate Grb2 growth factor receptor-bound protein 2 Hb hemoglobin HBB beta-globin HBG gamma-globin HIF hypoxia inducible factor HK1 hexokinase 1 HPRT hypoxanthine-guanine phosphoribosyl transferase HSC hematopoietic stem cell IBD identity by descent IVS intervening sequence JAK2 Janus kinase 2

III Supplements and Appendices
KCl potassium chloride KLF-1 erythroid Krüppel-like factor 1 L1/ LINE-1 long interspersed element-1 LCR locus control regions LDHA lactate dehydrogenase A LNK/SH2B src homology 2 B adaptor protein 3 LTR long terminal repeats MAPK mitogen-activated protein kinase MCH mean corpuscular hemoglobin MCV mean corpuscular volume MEL murine erythroleukemia cell line MLL mixed lineage leukemia Myr million year NAF sodium fluoride NAHR non-allelic homologous recombination NDRG1 N-myc downstream regulated 1 NF-E2 nuclear factor - erythroid-derived 2 NMD nonsense-mediated decay NTPs nucleotide triphosphate ORF open reading frame PDK1 pyruvate dehydrogenase kinase 1 PFCP primary familial and congenital polycythemia PHD2 prolyl hydroxylase protein 2 PI-3K/AKT phosphoinositide 3-kinase/protein kinase B PIA plasminogen activator inhibitor-1 Pol II polymerase II PMSF phenylmethylsulfonyl fluoride Pro proline PV polycythemia vera RBCs red blood cells REST relative expression software tool RMSD root-mean-square deviation RNA ribonucleic acid RT-PCR/PCR reverse transcription polymerase chain reaction RUNX1/AML runt-related transcription factor 1/acute myeloid leukemia SH2 src homology 2 SHP1 src homology region 2 domain-containing phosphatase-1 SINE-R short interspersed elements (‘R’ indicating a sequence of retroviral origin) SLC2A1 solute carrier family 2 (facilitated glucose transporter), member 1 SOCS suppressor of cytokine signaling Sos son of sevenless STATs/STAT 5 signal transducer and activator of transcription 5 STEAP3 6-transmembrane epithelial antigen of prostate family member 3 SVA SINE-R VNTR Alu TE transposable element TFRC transferrin receptor protein TSA trichostatin TSD tandem side duplication UPL universal probe library UTP uracil triphosphate UTR untranslated region VEGF vascular endothelial growth factor VHL von Hippel-Lindau gene/protein VNTR variable number tandem repeat

III Supplements and Appendices
8 Supplements 8.1 List of primers and PCR reaction conditions 8.1.1 List of primers Primer name Sequence =========================================== VHL exon 1 F 5’ CGAAGACTACGGAGGTCGAC VHL exon 1 R 5’ GGCTTCAGACCGTGCTATCG VHL exon 2 F 5’ GTGTGGCTCTTTAACAACC VHL exon 2 R 5’ CTGTACTTACCACAACAACC VHL exon 3 F 5’ TCCTTGTACTGAGACCCTAG VHL exon 3 R 5’ AGCTGAGATGAAACAGTCTA HBB ex1 F 5’ TGAGGAGAAGTCTGCCGTT HBB ex1 R 5’ GGGCCTCACCACCAACTT HBB ex2 F 5’ CAAGGGCACCTTTGCCACA HBB ex2 R 5’ CCTGAAGTTCTCAGGATCCACG HBB ex3 F 5’ ACAAGTATCACTAAGCTCGCTTTCT HBB ex3 R 5’ TAGTTGGACTTAGGGAACAAAGG hLDHA F 5’ CTGTCATGGGTGGGTCCTT hLDHA R 5’ GCAACATTCATTCCACTCCA HBG F 5’ GGCAACCTGTCCTCTGCCTC HBG R 5’ GAAATGGATTGCCAAAACGG HBBenh bis F 5’ TTTGATTTTATTTAGTTTTTTTGTTTAGAG HBBenh bis R 5’ CCAACAAACCTCTAATCTCTTCCTA HBBenh bis F2 5’ TTTTAGTTGTTTTTATGAATGTTTTT L1 meth F 5’ TTGAGTTAGGTGTGGGATATAGTTT L1 meth F2 5’ GTTTTTTAGGTGAGGTAATGTTT L1 meth R 5’ ATATAAACATAATTAACAAAAAAACCTAAC VHL P138L_FW 5’ CTGAATTATTTGTGCTATCTCTCAATGTTGA VHL P138L_RV 5’ TCAACATTGAGAGATAGCACAAATAATTCAG VHL H191D_FW 5’ GAAGATCTGGAAGACGACCCAAATGTGCAGA VHL H191D_RV 5’ TCTGCACATTTGGGTCGTCTTCCAGATCTTC
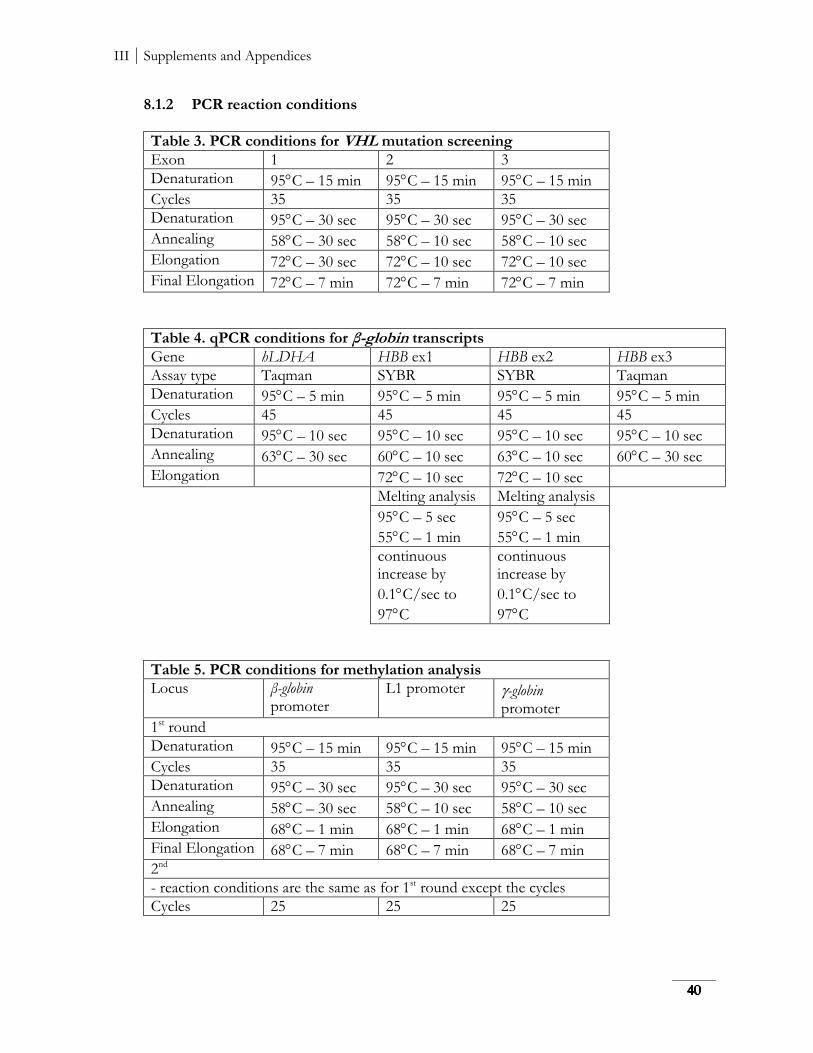
III Supplements and Appendices
8.1.2 PCR reaction conditions
Table 3. PCR conditions for VHL mutation screening
Exon 1 2 3
Denaturation 95C – 15 min 95C – 15 min 95C – 15 min
Cycles 35 35 35
Denaturation 95C – 30 sec 95C – 30 sec 95C – 30 sec
Annealing 58C – 30 sec 58C – 10 sec 58C – 10 sec
Elongation 72C – 30 sec 72C – 10 sec 72C – 10 sec
Final Elongation 72C – 7 min 72C – 7 min 72C – 7 min
Table 4. qPCR conditions for β-globin transcripts
Gene hLDHA HBB ex1 HBB ex2 HBB ex3
Assay type Taqman SYBR SYBR Taqman
Denaturation 95C – 5 min 95C – 5 min 95C – 5 min 95C – 5 min
Cycles 45 45 45 45
Denaturation 95C – 10 sec 95C – 10 sec 95C – 10 sec 95C – 10 sec
Annealing 63C – 30 sec 60C – 10 sec 63C – 10 sec 60C – 30 sec
Elongation 72C – 10 sec 72C – 10 sec
Melting analysis Melting analysis
95C – 5 sec
55C – 1 min
95C – 5 sec
55C – 1 min
continuous increase by
0.1C/sec to
97C
continuous increase by
0.1C/sec to
97C
Table 5. PCR conditions for methylation analysis
Locus β-globin promoter
L1 promoter -globin promoter
1st round
Denaturation 95C – 15 min 95C – 15 min 95C – 15 min
Cycles 35 35 35
Denaturation 95C – 30 sec 95C – 30 sec 95C – 30 sec
Annealing 58C – 30 sec 58C – 10 sec 58C – 10 sec
Elongation 68C – 1 min 68C – 1 min 68C – 1 min
Final Elongation 68C – 7 min 68C – 7 min 68C – 7 min
2nd
- reaction conditions are the same as for 1st round except the cycles
Cycles 25 25 25

III Supplements and Appendices
Table 6. PCR conditions for site mutagenesis
Mutation VHLP138L VHLH191D
Denaturation 95C – 30 sec 95C – 30 sec
Cycles 16 16
Denaturation 95C – 30 sec 95C – 30 sec
Annealing 55C – 1 min 60C – 1 min
Elongation 68C – 6 min 68C – 6 min
8.2 List of other Publications and Meetings Abstracts Publications
Ye Z, Liu CF, Lanikova L, Dowey SN, He C, Huang X, Brodsky RA, Spivak JL, Prchal JT, Cheng L. Differential sensitivity to JAK inhibitory drugs by isogenic human erythroblasts and hematopoietic progenitors generated from patient iPSCs. Stem Cells. 2013; accepted.
Lorenzo FR, Yang CH, Lanikova L, Burtos L, Prchal JT. Novel Compound VHL Heterozygosity (VHL T124A/L188V) Associated With Congenital Polycythemia. Br J Haematol. 2013; doi: 10.1111/bjh.12431/epub ahead of print.
Prchal JT, Swierczek SI, Piterkova L, Jelinek J, Parker CJ, Cairns B. Reliable assessment of X-chromosome inactivation via DNA or RNA based assays remains controversial. Blood; e-Letter, Jun 2012.
Swierczek SI, Piterkova L, Jelinek J, Agarwal N, Hammoud S, Wilson A, Hickman K, Parker CHJ, Cairns B, Prchal JT. Methylation of AR locus does not always reflect X chromosome inactivation state. Blood. 2012; 119(13): e100 - e109.
Meeting Abstracts
Piterkova L*, Tian L*, Wang L, Ye Z, Cheng L, Wheeler DA, Hakonarson H, Prchal JT. Whole Genome Sequencing of Four CD34+-Derived iPSC Polycythemia Vera Clones From a Single Female. Blood. Nov 2012; 120(21): 705. - 54th Annual Meeting of American Society of Hematology, December 8-11 (2012),
Atlanta (Georgia), USA.
Wang L, Swierczek SI, Piterkova L, Hickman K, Wheeler DA, Prchal JT. Whole Exome Sequencing of Polycythemia Vera Reveals Novel Recurrent Somatic and Germline Variation. Blood. Nov 2012; 120(21): 1755. - 54th Annual Meeting of American Society of Hematology, December 8-11 (2012),
Atlanta (Georgia), USA.

III Supplements and Appendices
Ye Z, Piterkova L, Liu C, Dowey S, Chou BK, Huang X, Spivak J, Swierczek S, Moliterno A, Prchal JT, Cheng L. Distinct Induced Pluripotent Stem Cell Clones with Somatic Mutations Prepared From PV Patients. Blood. Nov 2011; 118(21): 2826. - 53rd Annual Meeting of American Society of Hematology, December 10-13
(2011), San Diego (California), USA.

III Supplements and Appendices
9 Appendices
Original version of Publications
Lanikova L, Kucerova J, Indrak K, Divoka M, Issa JP, Papayannopoulou T, Prchal
JT, Divoky V. β-thalassemia due to intronic LINE-1 insertion in the β-globin gene: molecular mechanisms underlying reduced transcript of the β-globinL1 allele. Human Mutation. 2013; doi: 10.1002/humu.22383/epub ahead of print.
Lanikova L, Lorenzo F, Yang CH, Vankayalapati H, Drachtman R, Divoky V, Prchal
JT. Novel homozygous VHL mutation in exon 2 is associated with congenital polycythemia but not with cancer. Blood. 2013;121(19):3918-24.
Piterkova L*, Tomasic Ljubas N*, Huff Ch, Bilic E, Yoon D, Miasnikova GY, Sergueeva AI, Niu X, Nekhai S, Gordeuk V, Prchal JT. Polycythemia due to Croatian homozygous VHL (571C>G:H191D) mutation has a different phenotype than Chuvash polycythemia (VHL 598C>T:R200W). Haematologica. 2013; 98(4):560-7.

BRIEF REPORTOFFICIAL JOURNAL
www.hgvs.org
β-Thalassemia Due to Intronic LINE-1 Insertion in theβ-Globin Gene (HBB): Molecular Mechanisms UnderlyingReduced Transcript Levels of the β-GlobinL1 Allele
Lucie Lanikova,1,2 Jana Kucerova,1,3 Karel Indrak,3 Martina Divoka,3 Jean-Pierre Issa,4 Thalia Papayannopoulou,5
Josef T. Prchal,2 and Vladimir Divoky1,3 ∗
1Department of Biology, Faculty of Medicine and Dentistry, Palacky University, Olomouc, Czech Republic; 2Departments of Medicine, Pathologyand Genetics, University of Utah and Medical Service, George E. Wahlen Department of Veterans Affairs Medical Center, Salt Lake City, Utah;3Department of Hemato-Oncology, Faculty of Medicine and Dentistry and University Hospital, Olomouc, Czech Republic; 4Fels Institute, TempleUniversity School of Medicine, Philadelphia, Pennsylvania; 5Department of Medicine/Division of Hematology, School of Medicine, University ofWashington, Seattle, Washington
Communicated by Haig H. KazazianReceived 15 April 2013; accepted revised manuscript 10 July 2013.Published online 22 July 2013 in Wiley Online Library (www.wiley.com/humanmutation). DOI: 10.1002/humu.22383
ABSTRACT: We describe the molecular etiology ofβ+-thalassemia that is caused by the insertion of the full-length transposable element LINE-1 (L1) into the intron-2 of the β-globin gene (HBB). The transcript level of theaffected β-globin gene was severely reduced. The remain-ing transcripts consisted of full-length, correctly processedβ-globin mRNA and a minute amount of three aberrantlyspliced transcripts with a decreased half-life due to activa-tion of the nonsense-mediated decay pathway. The lowersteady-state amount of mRNA produced by the β-globinL1
allele also resulted from a reduced rate of transcriptionand decreased production of full-length β-globin primarytranscripts. The promoter and enhancer sequences of theβ-globinL1 allele were hypermethylated; however, treat-ment with a demethylating agent did not restore theimpaired transcription. A histone deacetylase inhibitorpartially reactivated the β-globinL1 transcription despitepermanent β-globinL1 promoter CpG methylation. Thisresult indicates that the decreased rate of transcriptionfrom the β-globinL1 allele is associated with an alteredchromatin structure. Therefore, the molecular defectcaused by intronic L1 insertion in the β-globin gene rep-resents a novel etiology of β-thalassemia.Hum Mutat 00:1–5, 2013. C© 2013 Wiley Periodicals, Inc.
KEY WORDS: β-thalassemia; β-globin; HBB; LINE-1;epigenetic repression
Additional Supporting Information may be found in the online version of this article.∗Correspondence to: Vladimir Divoky, Department of Biology, Faculty of Medicine
and Dentistry, Palacky University, Hnevotinska 3, CZ-77515 Olomouc, Czech Republic.
E-mail: [email protected]
Contract grant sponsors: Czech Ministry of Health (NT/13587, NT/11208); EU
(CZ.1.07/2.3.00/30.0041); and Palacky University (LF_2013_004, LF_2013_010).
We have previously reported that the insertion of a full-lengthLINE-1 (L1) element (GenBank accession: AF149422.1) in the an-tisense orientation into the 3′-end of intron-2 of the β-globin gene(HBB; MIM #141900; GenBank accession: NM 000518.4) causesβ+-thalassemia in a mother and daughter of Ukrainian descent[Divoky et al., 1996]. The β-thalassemia trait in the propositus andher mother was diagnosed in the laboratory of the late Dr. Huisman(Augusta, GA) [Indrak et al., 1992]. We have shown that this L1element retained the capacity for high-frequency retrotranspositionin cultured cells [Kimberland et al., 1999]. Here, we analyzed themolecular mechanisms that are responsible for the severe reductionin β-globin gene expression in the presence of the L1 insertion andcause the β+-thalassemia phenotype.
Theβ+-thalassemia nature of this mutation was investigated in thereticulocytes from the patient and in the interspecific hybrids createdfrom the fusion of transformed lymphocytes of the propositus withmouse erythroleukemia cells (MEL) containing either the affectedor normal chromosome 11 [Papayannopoulou et al., 1986]. The cellline containing the mutant chromosome 11 from the propositus wasdesignated as “MEL HBBL1+,” and the control cell line with a normalchromosome 11 was designated as “MEL HBBwt.” Blood samplesfrom the propositus and her mother were obtained with the approvalof the institutional review board committees of Palacky University inOlomouc and the Institute of Hematology and Blood Transfusion inPrague, Czech Republic. Informed consent was obtained as per theDeclaration of Helsinki. The details of the experimental proceduresand the sequences of the primers used for this study are found inthe Supp. Methods and Supp. Tables.
The propositus (25-year-old Caucasian female) had microcyticanemia (Hb 11.3 g/dL, red blood cells 5.5 × 1012/L, mean corpuscu-lar volume 63 fL, and mean corpuscular hemoglobin 20 pg) with anormal reticulocyte count. Her HbA2 was increased to 5.3% and theHbF was just above 1%. No inclusion bodies of precipitated globinchains were detected in her erythroid cells after brilliant cresyl bluestaining. Hematological indices of the patient’s mother were compa-rable; neither patient had neurological or other systemic abnormal-ities. Southern blot and sequencing analyses revealed heterozygosityfor the full-length retrotransposon (L1, i.e., L1β-thal) inserted in theantisense orientation into the 3′-end of intron-2 (β-IVS-II) of theβ-globin gene (hereafter referred to as β-globinL1; Supp. Fig. S1)deposited into the HBVar database (http://www.lovd.nl/HBB). The
C© 2013 WILEY PERIODICALS, INC.

insertion occurred at 98 bp 5′- to the 3′-end of β-IVS-II and wasflanked by a 9–13-bp target site duplication [Divoky et al., 1996;Kimberland et al., 1999].
The β-globin transcripts from affected and unaffected β-globinalleles of the propositus were distinguishable by a silent codon 2polymorphism [Antonarakis et al., 1985; Orkin et al., 1982]. Thispolymorphism allowed the quantification of the transcript amountand the determination of the β+-thalassemia phenotype (Fig. 1A).The propositus was a codon 2 CAT/CAC heterozygote, whereas themother of the propositus, who also carried the L1 insertion, was acodon CAC homozygote. Thus, the L1-containing allele was derivedfrom framework 1 [Antonarakis et al., 1985] with a codon 2 CACsequence that creates a recognition site for the BsiHKAI enzyme.
To assess whether the β+-thalassemia is due to abnormal splic-ing of the β-globin message, reticulocyte and MEL HBBL1+ mR-NAs were analyzed by RT-PCR using several primers. Sequencingof the RT-PCR products revealed both correctly processed tran-scripts as well as three rare messages with unusual sequences pro-duced by the β-globinL1 allele (Fig. 1B). All of the aberrantly splicedβ-globin transcripts contained a premature translation terminationcodon, which makes them susceptible to nonsense-mediated mRNAdecay (NMD). To determine whether the NMD pathway degradesthe β-globinL1 mRNA and thus is responsible for the reduction inthe steady-state mRNA level produced by the mutant allele, we cul-tivated the induced MEL hybrids cells overnight with the NMDinhibitor emetine [Yang et al., 2007]. Quantitative real-time PCR(qPCR) showed a 2.4-fold increase (P < 0.05 vs. control) in the ac-cumulation of transcripts from the β-globinL1 allele in the presenceof emetine (Fig. 1C), whereas no increase in the amount of wild-typeβ-globinL1 transcripts was observed under the same conditions. Tofurther confirm that the low stability of the β-globinL1 transcriptswas due to NMD, transcription in the MEL HBBL1+ and MEL HBBwt
hybrids was blocked with actinomycin D in the presence and absenceof emetine. Indeed, the decreased stability of the β-globinL1 mRNAwas mainly due to NMD. The half-life of the β-globinL1 mRNA wasrestored almost to normal when emetine was used (Supp. Fig. S2).
In addition to a decrease in RNA stability, a decreased rate of tran-scription could be responsible for the reduced amount of β-globinL1
mRNA. This possibility was examined using a nuclear run-on assay[Yang et al., 2007]. The increase in the β-globin nuclear fractionprimary transcript level during 40 min of in vitro transcription inthe presence or absence of ribonucleotide triphosphates (rNTPs, seeSupp. Methods for experimental details) was measured. The datashowed that the β-globinL1 allele has a transcription rate that isreduced 4.7-fold versus the wild type (Fig. 1D).
Previous studies have demonstrated that L1 insertions disruptthe formation of the full-length transcript of the target gene by pro-ducing prematurely polyadenylated mRNA. These truncated tran-scripts form because of the use of the L1 polyadenylation signal(s).Additionally, the release of RNApol-II from the L1 sequence causesinefficient elongation [Han et al., 2004]. We measured the rela-tive amount of the β-globinL1 primary transcript using qPCR withprimers flanking the insertion (Fig. 1E). The nuclear fraction RNAwas collected from MEL HBBwt and MEL HBBL1+ hybrids, and therelative amount of wild-type and β-globinL1 primary transcripts wasmeasured at exon-2 and exon-3 and expressed as the exon-3/exon-2 ratio normalized to the control as described [Pfaffl, 2001] (seeSupp. Methods). The ratio was 0.805, which suggests that 19.5% ofthe MEL HBBL1+ primary transcript is not extended beyond the L1insertion. The truncated transcripts could result from either pre-mature polyadenylation or incomplete elongation.
Retrotransposons are considered to be epigenetic mediators ofgene expression in mammalian cells [Whitelaw and Martin, 2001].
We examined whether the decreased rate of β-globinL1 allele tran-scription is associated with the β-globinL1 promoter and 3′-enhancermethylation that could be reversed by a demethylating agent. Thelocations of the CpG dinucleotides (CpGs) within the β-globinL1
promoter and enhancer and also within the L1 promoter are de-picted in Figure 2A. The results from bisulfite sequencing (Fig. 2B)show that the β-globinL1 promoter sequence was hypermethylated(65% Me-CpG vs. 35% Me-CpG in the control MEL HBBwt cellline). The 3′-enhancer sequence was fully methylated in compari-son with the control sequences. Treatment of the induced hybridMEL cells with decitabine (5-aza-2′-deoxycytidine, DAC, 2 μM)for 24 hr did not change the methylation status of the β-globinL1
promoter, nor did it lead to an increase in expression of β-globinL1
mRNA (Fig. 2B and C). Different treatment conditions (treatmenttime 48 hr or 5 μM DAC) gave similar results. Expression of theγ -globin gene, which was used as a treatment control, was increasedafter DAC exposure as expected [Mabaera et al., 2008] (Fig. 2C).The partial demethylation of the β-globinL1 erythroid-specific 3′-enhancer sequence [Boehringer et al., 1987] (Fig. 2B) suggested thatinsensitivity to DAC under these assay conditions was a feature ofthe β-globinL1 and L1 promoters. Even the more stringent DAC-treatment conditions [Qin et al., 2009] did not reactivate β-globinL1
expression (data not shown).The histone deacetylation of the surrounding chromatin was im-
plicated in the inactivation of L1 host genes [Garcia-Perez et al.,2010]. To test whether transcriptional silencing of the β-globinL1 al-lele is related to repressive chromatin modifications such as deacety-lation, we treated induced MEL HBBL1+ cells with trichostatin A(TSA), a histone-deacetylase (HDAC) inhibitor (Fig. 2D). The TSA-induced reactivation of developmentally silenced γ -globin expres-sion in the somatic cell hybrids was previously documented [Swanket al., 2003], and therefore, it was used as an internal treatment con-trol (Fig. 2E). Although exposure of the induced MEL HBBL1+ cells toTSA (alone or in combination with DAC) did not change the denselymethylated profile of the β-globinL1 promoter sequence (Fig. 2D),this treatment partially restoredβ-globinL1 expression (Fig. 2E). Thisresult suggests that histone deacetylation is (in part) responsible forthe β-globinL1 silencing that could be relieved by TSA.
L1β -thal displays characteristics of an active retrotransposon [Kim-berland et al., 1999]. Therefore, it is possible that transcripts origi-nating from the L1β -thal promoter could interfere with the β-globingene that is transcribed in the opposite orientation to this L1[Whitelaw and Martin, 2001]. The ε-globin gene silencing by a tran-scribed Alu element provides precedence for in cis transcriptionalinterference by neighboring transcripts from the antisense strand inglobin gene regulation [Wu et al., 1990]. Thus, we assayed for thepresence of L1β-thal-specific transcripts in patient mRNA that wasisolated from harvested BFU-E colonies and in mRNA from the in-terspecific hybrids (see Supp. Methods and Supp. Tables for details).No L1β-thal-specific transcripts could be detected in the BFU-Es fromthe patients. Several L1 transcripts were detected in the induced MELHBBL1+ cells; however, their sequence analyses revealed other thanL1β-thal origin (data not shown). Correspondingly, bisulfite sequenc-ing revealed a highly methylated pattern along the L1β-thal promoterregion (Fig. 2B and D), suggesting that the absence of endogenousL1 transcripts is a consequence of the L1β-thal promoter hypermethy-lation. We also investigated whether the L1β-thal insertion splits theHBB gene’s transcript, that is, the “gene-breaking” model [Wheelanet al., 2005]. We identified neither upstream HBB-specific tran-scripts that terminate in the major antisense polyadenylation signal[Han et al., 2004] nor evidence for L1 antisense promoter-derivedHBB-specific transcripts (see Supp. Methods and Supp. Tables fordetails).
2 HUMAN MUTATION, Vol. 00, No. 0, 1–5, 2013
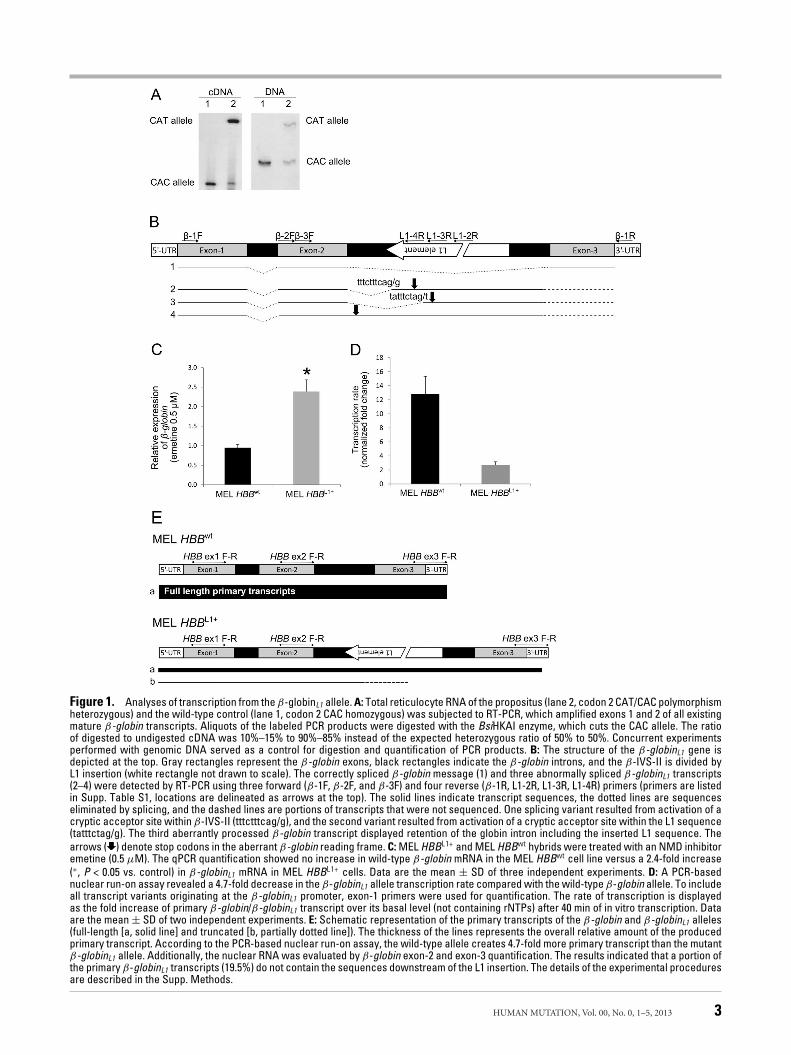
Figure 1. Analyses of transcription from the β-globinL1 allele. A: Total reticulocyte RNA of the propositus (lane 2, codon 2 CAT/CAC polymorphismheterozygous) and the wild-type control (lane 1, codon 2 CAC homozygous) was subjected to RT-PCR, which amplified exons 1 and 2 of all existingmature β-globin transcripts. Aliquots of the labeled PCR products were digested with the BsiHKAI enzyme, which cuts the CAC allele. The ratioof digested to undigested cDNA was 10%–15% to 90%–85% instead of the expected heterozygous ratio of 50% to 50%. Concurrent experimentsperformed with genomic DNA served as a control for digestion and quantification of PCR products. B: The structure of the β-globinL1 gene isdepicted at the top. Gray rectangles represent the β-globin exons, black rectangles indicate the β-globin introns, and the β-IVS-II is divided byL1 insertion (white rectangle not drawn to scale). The correctly spliced β-globin message (1) and three abnormally spliced β-globinL1 transcripts(2–4) were detected by RT-PCR using three forward (β-1F, β-2F, and β-3F) and four reverse (β-1R, L1-2R, L1-3R, L1-4R) primers (primers are listedin Supp. Table S1, locations are delineated as arrows at the top). The solid lines indicate transcript sequences, the dotted lines are sequenceseliminated by splicing, and the dashed lines are portions of transcripts that were not sequenced. One splicing variant resulted from activation of acryptic acceptor site within β-IVS-II (tttctttcag/g), and the second variant resulted from activation of a cryptic acceptor site within the L1 sequence(tatttctag/g). The third aberrantly processed β-globin transcript displayed retention of the globin intron including the inserted L1 sequence. Thearrows ( ) denote stop codons in the aberrant β-globin reading frame. C: MEL HBBL1+ and MEL HBBwt hybrids were treated with an NMD inhibitoremetine (0.5 μM). The qPCR quantification showed no increase in wild-type β-globin mRNA in the MEL HBBwt cell line versus a 2.4-fold increase(∗, P < 0.05 vs. control) in β-globinL1 mRNA in MEL HBBL1+ cells. Data are the mean ± SD of three independent experiments. D: A PCR-basednuclear run-on assay revealed a 4.7-fold decrease in the β-globinL1 allele transcription rate compared with the wild-type β-globin allele. To includeall transcript variants originating at the β-globinL1 promoter, exon-1 primers were used for quantification. The rate of transcription is displayedas the fold increase of primary β-globin/β-globinL1 transcript over its basal level (not containing rNTPs) after 40 min of in vitro transcription. Dataare the mean ± SD of two independent experiments. E: Schematic representation of the primary transcripts of the β-globin and β-globinL1 alleles(full-length [a, solid line] and truncated [b, partially dotted line]). The thickness of the lines represents the overall relative amount of the producedprimary transcript. According to the PCR-based nuclear run-on assay, the wild-type allele creates 4.7-fold more primary transcript than the mutantβ-globinL1 allele. Additionally, the nuclear RNA was evaluated by β-globin exon-2 and exon-3 quantification. The results indicated that a portion ofthe primary β-globinL1 transcripts (19.5%) do not contain the sequences downstream of the L1 insertion. The details of the experimental proceduresare described in the Supp. Methods.
HUMAN MUTATION, Vol. 00, No. 0, 1–5, 2013 3
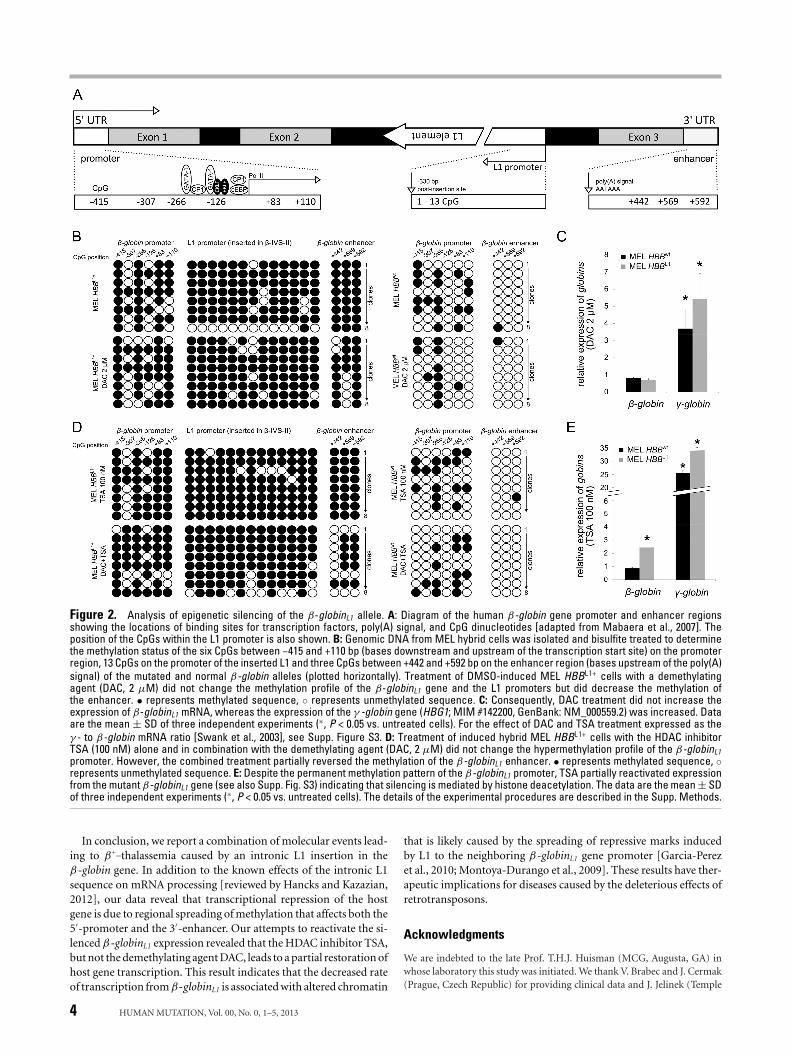
Figure 2. Analysis of epigenetic silencing of the β-globinL1 allele. A: Diagram of the human β-globin gene promoter and enhancer regionsshowing the locations of binding sites for transcription factors, poly(A) signal, and CpG dinucleotides [adapted from Mabaera et al., 2007]. Theposition of the CpGs within the L1 promoter is also shown. B: Genomic DNA from MEL hybrid cells was isolated and bisulfite treated to determinethe methylation status of the six CpGs between −415 and +110 bp (bases downstream and upstream of the transcription start site) on the promoterregion, 13 CpGs on the promoter of the inserted L1 and three CpGs between +442 and +592 bp on the enhancer region (bases upstream of the poly(A)signal) of the mutated and normal β-globin alleles (plotted horizontally). Treatment of DMSO-induced MEL HBBL1+ cells with a demethylatingagent (DAC, 2 μM) did not change the methylation profile of the β-globinL1 gene and the L1 promoters but did decrease the methylation ofthe enhancer. • represents methylated sequence, ◦ represents unmethylated sequence. C: Consequently, DAC treatment did not increase theexpression of β-globinL1 mRNA, whereas the expression of the γ -globin gene (HBG1; MIM #142200, GenBank: NM_000559.2) was increased. Dataare the mean ± SD of three independent experiments (∗, P < 0.05 vs. untreated cells). For the effect of DAC and TSA treatment expressed as theγ - to β-globin mRNA ratio [Swank et al., 2003], see Supp. Figure S3. D: Treatment of induced hybrid MEL HBBL1+ cells with the HDAC inhibitorTSA (100 nM) alone and in combination with the demethylating agent (DAC, 2 μM) did not change the hypermethylation profile of the β-globinL1promoter. However, the combined treatment partially reversed the methylation of the β-globinL1 enhancer. • represents methylated sequence, ◦represents unmethylated sequence. E: Despite the permanent methylation pattern of the β-globinL1 promoter, TSA partially reactivated expressionfrom the mutant β-globinL1 gene (see also Supp. Fig. S3) indicating that silencing is mediated by histone deacetylation. The data are the mean ± SDof three independent experiments (∗, P < 0.05 vs. untreated cells). The details of the experimental procedures are described in the Supp. Methods.
In conclusion, we report a combination of molecular events lead-ing to β+–thalassemia caused by an intronic L1 insertion in theβ-globin gene. In addition to the known effects of the intronic L1sequence on mRNA processing [reviewed by Hancks and Kazazian,2012], our data reveal that transcriptional repression of the hostgene is due to regional spreading of methylation that affects both the5′-promoter and the 3′-enhancer. Our attempts to reactivate the si-lenced β-globinL1 expression revealed that the HDAC inhibitor TSA,but not the demethylating agent DAC, leads to a partial restoration ofhost gene transcription. This result indicates that the decreased rateof transcription from β-globinL1 is associated with altered chromatin
that is likely caused by the spreading of repressive marks inducedby L1 to the neighboring β-globinL1 gene promoter [Garcia-Perezet al., 2010; Montoya-Durango et al., 2009]. These results have ther-apeutic implications for diseases caused by the deleterious effects ofretrotransposons.
Acknowledgments
We are indebted to the late Prof. T.H.J. Huisman (MCG, Augusta, GA) inwhose laboratory this study was initiated. We thank V. Brabec and J. Cermak(Prague, Czech Republic) for providing clinical data and J. Jelinek (Temple
4 HUMAN MUTATION, Vol. 00, No. 0, 1–5, 2013

University, Philadelphia, PA) for preliminary analyses. We also thank themembers of the V. Divoky lab and M. Mrug (UAB, Birmingham, AL) forvaluable discussions.
Disclosure Statement: The authors declare no conflicts of interest.
References
Antonarakis SE, Kazazian HH Jr, Orkin SH. 1985. DNA polymorphism and molecularpathology of the human globin gene clusters. Hum Genet 69:1–14.
Boehringer RR, Hammer RE, Brinster RL, Palmiter RD, Townes TM. 1987. Two 3′
sequences direct adult erythroid-specific expression of human β-globin genes intransgenic mice. Proc Natl Acad Sci USA 84:7056–7060.
Divoky V, Indrak K, Mrug M, Brabec V, Huisman THJ, Prchal JT. 1996. A novelmechanism of β-thalassemia. The insertion of L1 retrotransposable element intoβ globin IVSII. Blood 88:148a (abstract).
Garcia-Perez JL, Morell M, Scheys JO, Kulpa DA, Carter CC, Hammer GD, Collins KL,O’Shea KS, Menendez P, Moran JV. 2010. Epigenetic silencing of engineered L1retrotranspo-sition events in human embryonic carcinoma cells. Nature 4669:769–773.
Han JS, Szak ST, Boeke JD. 2004. Transcriptional disruption by the L1 retrotransposonand implications for mammalian transcriptomes. Nature 429:268–274.
Hancks DC, Kazazian HH Jr. 2012. Active human retrotransposons: variation anddisease. Curr Opin Genet Dev 22:191–203.
Indrak K, Brabec V, Indrakova J, Chrobak L, Sakalova A, Jarosova M, Cermak J, Fei YJ,Kutlar F, Gu YC, Baysal E, Huisman THJ, et al. 1992. Molecular characterizationof β-thalassemia in Czechoslovakia. Hum Genet 88:399–404.
Kimberland ML, Divoky V, Prchal JT, Schwahn U, Berger W, Kazazian HH Jr. 1999.Full-length human L1 insertions retain the capacity for high frequency retrotrans-position in cultured cells. Hum Mol Genet 8:1557–1560.
Mabaera R, Greene MR, Richardson CA, Conine SJ, Kozul CD, Lowrey CH. 2008.Neither DNA hypomethylation nor changes in the kinetics of erythroid differen-tiation explain 5-azacytidine’s ability to induce human fetal hemoglobin. Blood111:411–420.
Mabaera R, Richardson CA, Johnson K, Hsu M, Fiering S, Lowrey CH. 2007.Developmental- and differentiation-specific patterns of human gamma- and be-taglobin promoter DNA methylation. Blood 110:1343–1352.
Montoya-Durango DE, Liu Y, Teneng I, Kalbfleisch T, Lacy ME, Steffen MC, RamosKS. 2009. Epigenetic control of mammalian LINE-1 retrotransposon by retinoblas-toma proteins. Mutat Res 665:20–28.
Orkin SH, Kazazian HH Jr, Antonarakis SE, Goff SC, Boehm CD, Sexton JP, Waber PG,Giardina PJ. 1982. Linkage of β-thalassaemia mutations and β-globin gene poly-morphisms with DNA polymorphisms in human β-globin gene cluster. Nature296:627–631.
Papayannopoulou T, Brice M, Stamatoyannopoulos G. 1986. Analysis of humanhemoglobin switching in MEL x human fetal erythroid cell hybrids. Cell 46:469–476.
Pfaffl MW. 2001. A new mathematical model for relative quantification in real-timeRT-PCR. Nucleic Acids Res 29:e45.
Qin T, Jelinek J, Si J, Shu J, Issa JP. 2009. Mechanisms of resistance to 5-aza-2′-deoxycytidi-ne in human cancer cell lines. Blood 113:659–667.
Swank RA, Skarpidi E, Papayannopoulou T, Stamatoyannopoulos G. 2003. Thehistone deacetylase inhibitor, trichostatin A, reactivates the developmen-tally silenced gamma globin expression in somatic cell hybrids and inducesgamma gene expression in adult BFUe cultures. Blood Cells Mol Dis 30:254–257.
Wheelan SJ, Aizawa Y, Han JS, Boeke JD. 2005. Gene-breaking: a newparadigm for human retrotransposon-mediated gene evolution. Genome Res 15:1073–1078.
Whitelaw E, Martin DI. 2001. Retrotransposons as epigenetic mediators of phenotypicvariation in mammals. Nat Genet 27:361–365.
Wu J, Grindlay GJ, Bushel P, Mendelsohn L, Allan M. 1990. Negative regulation ofthe human epsilon-globin gene by transcriptional interference: role of an Alurepetitive element. Mol Cell Biol 10:1209–1216.
Yang C, Feng J, Song W, Wang J, Tsai B, Zhang Y, Scaringe WA, Hill KA, MargaritisP, High KA, Sommer SS. 2007. A mouse model for nonsense mutation bypasstherapy shows a dramatic multiday response to geneticin. Proc Natl Acad Sci USA104:15394–15399.
HUMAN MUTATION, Vol. 00, No. 0, 1–5, 2013 5

1
SUPPLEMENTAL METHODS, TABLES AND FIGURES
Databases
The mutation has been deposited in the LOVD database and HBvar database:
http://lovd.bx.psu.edu/variants.php?select_db=HBB&action=view&view=0002122%2C0001811%2C0
http://globin.bx.psu.edu/cgi-bin/hbvar/query_vars3?mode=output&display_format=page&i=2892
Diagnosis of -thalassemia
The HbA2 levels were measured by cation exchange high-performance liquid chromatography
(HPLC) and the HbF levels by alkali denaturation and HPLC (Kutlar, et al., 1991). Analysis of
the -globin genes of the patient revealed no obvious mutation except that they were
heterozygous for an unexpected rearrangement that was detected by gene mapping (not shown).
RNA quantification by primer extension of RT-PCR products
Total reticulocyte RNA from the propositus and the control was subjected to RT-PCR with
primers -F and -Ex2-R (Supp. Table S1). After 19 cycles of PCR, the amplified products were
labeled with one cycle of primer extension using a [-32
P] ATP end-labeled primer -Ex2-R
(95°C, 2 min; 58°C, 2 min; 72°C, 8 min). Aliquots of the PCR products were then digested for 2
h at 65°C with BsiHKAI (New England Biolabs, Beverly, MA), analyzed on an 8% non-
denaturing polyacrylamide gel, and visualized by autoradiography. The intensities of the bands
were quantified using a PhosphorImager (Molecular Dynamics).
Preparation of human/mouse cell hybrids
Interspecific hybrids of Epstein Barr Virus (EBV) transformed lymphocytes from the propositus
and mouse erythroleukemia (MEL) cells were generated. The fusions were done in the presence
of polyethylene glycol (PEG, 1500, 50 % w/v) as previously described (Papayannopoulou, et al,
1996). After a period of two to three weeks, clonal outgrowths were evident in many of the
wells. Hybrid cells from the clonal outgrowths that contained human chromosome 11 were
selected based on reactivity to antibody 53/6, which recognizes an antigen encoded by
chromosome 11 (Yagi, et al., 1987). Two lines were derived from the positive cell clones: one
containing the affected chromosome 11 (designated “MEL HBBL1+
”) and one with the normal
chromosome 11 (designated “MEL HBBwt
”). The hybrids were expanded and cultured in IMDM

2
supplemented with 10% bovine calf serum (Invitrogen, Life Technologies, NY) in a humidified
incubator at 37°C and 5% CO2 and were induced to differentiate by adding 1.8% dimethyl
sulfoxide (DMSO, Sigma-Aldrich, St. Louis, MO) for 72 h.
The hybrid human/mouse cells were treated under various conditions for different
experiments as follows: for 24 h with emetine (0.5 μM, Sigma-Aldrich, St. Louis, MO) for
nonsense-mediated mRNA decay (NMD) inhibition; for various times (1 - 12 h) with
actinomycin D (Act-D, 1 μM, Sigma-Aldrich, St. Louis, MO) to block mRNA synthesis; and for
24 h simultaneously with DMSO or for 24 h and 48 h before the induction of differentiation by
DMSO with 5-aza-2'-deoxycytidine (DAC, 2 μM or 5 μM, Sigma-Aldrich, St. Louis, MO) and/or
trichostatin A (TSA, 100 nM, Sigma-Aldrich, St. Louis, MO) to test the DNA methylation
pattern/reactivation of silenced gene expression.
Subcloning and sequencing of alternatively spliced transcripts
RNA was isolated from the reticulocytes from the propositus and from the in vitro cultured
human/mouse cell hybrids. The -globin transcripts were analyzed by RT-PCR and sequencing
of the subcloned PCR products. The primer pairs used to amplify and sequence the transcripts
were: -1F and -1R, for first round PCR; -2F with L1-4R, -3F with L1-4R, -2F with L1-2R
and -3F with L1-3R for nested PCR. The sequences of the PCR primers are listed in Supp.
Table S1.
Human -globin transcript quantification by real-time qPCR
Total RNA from the MEL HBBL1+
and MEL HBBwt
cell lines was isolated with TRI reagent
(Roche Applied Science) followed by TURBO DNA-free DNase I treatment (Ambion, Life
Technologies, NY). For quantification of the human -globin mRNA by qPCR, RNA was
reverse-transcribed using a Transcriptor First Strand cDNA Synthesis Kit (Roche Applied
Science). cDNA was quantified on a Light Cycler 480 (Roche Applied Science). For
quantification of the aberrantly spliced variants and the NMD activation assay, exon-3 primers
(HBB ex3 F and R) and UPL (Universal Probe Library, Roche Applied Science) probe #56 were
used. For the mRNA stability assay and the effect of AZA and/or TSA treatment, exon-2 primers
(HBB ex2 F and R) were used for -globin mRNA and primers HBG F a R for γ-globin mRNA
quantification. The levels of the human -globin and γ-globin transcripts were normalized to

3
human LDHA (OMIM 150000, GenBank accession: NM_005566.3) (primers LDHA F and R and
UPL probe #47) using an efficiency corrected formula for the relative expression ratio (Pfaffl, et
al., 2001). The sequences of the primers and the reaction conditions are listed in Supp. Tables S1
and S2.
PCR-based Nuclear Run-On Assay
Nuclei from the hybrid MEL HBBL1+
and MEL HBBwt
cells were isolated with the Nuclei EZ
Prep Nuclei Isolation Kit (Sigma-Aldrich, St. Louis, MO), according to the manufacturer’s
protocol. The total nuclei were split into two equal aliquots and 2x reaction buffer was added
(containing 30 mM Tris, pH 8; 2,5 mM MgCl2; 150 mM KCl; 20% glycerol; 1 mM DTT and 40
U RNasin; Promega, Madison, WI), and the reaction was performed, as previously described
(Yang, et al., 2007). The in vitro transcription reaction was initiated by the addition of 0.5 mM of
each ribonucleotide triphosphate (rATP, rCTP, rGTP, rUTP) (rNTP) (Invitrogen, Life
Technologies, NY) and incubated for 40 min at 30°C. The second aliquot, which did not contain
rNTPs, was incubated under the same conditions. The rate of transcription (normalized fold
increase) was determined as the ratio of the nuclear fraction primary -globin transcript measured
in exon-1 (primers HBB ex1 F and R) with added rNTPs to the same fraction at basal level
(without added rNTPs) after 40 min of in vitro transcription. LDHA was used as a reference gene
for quantification. The sequences of primers are listed in Supp. Table S1.
Relative ratio of exon-3 to exon-2 of the human -globin primary transcript
The HBB exon-3/exon-2 ratio of MEL HBBL1+
native primary transcript from the nuclear fraction
that was prepared as mentioned above was normalized to the exon-3/exon-2 ratio of the control
MEL HBBwt
native primary nuclear transcript using formula 2Ct
. The method for calculating
Ct was similar to the method used for the relative quantification and is as follows: (CtMEL
HBBL1+ ex2 – CtMEL HBBwt ex2) – (CtMEL HBBL1+ ex3 – CtMEL HBBwt ex3). Exon-2 primers HBB ex 2 F and
R, exon-3 primers HBB ex 3 F and R, and exon-3 UPL probe #56 were used for the determination
of this ratio. The efficiency of both amplicons was measured using a dilution series standard
curve and was very close to 2. The sequences of PCR primers are listed in Supp. Table S1.

4
DNA methylation analysis
Bisulfite modification was performed on the genomic DNA from MEL HBBL1+
and MEL HBBwt
hybrids cells using the EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA). The
promoter and enhancer regions of the -globin gene and the L1-thal promoter were amplified
using semi-nested PCR. The -globin promoter primers were published elsewhere (Mabaera, et
al., 2007); the enhancer region was amplified with HBBenh bis F and R primers for the first
round, and then HBBenh bis F2 and R were used for semi-nested PCR. The L1 promoter was
amplified with L1 meth F and R primers for the first round, followed by primers L1 meth F2 and
R for the semi-nested round. The purified products were subcloned, and at least 8 positive clones
for each region were sequenced. In the case of the -globinL1 promoter and 3' enhancer, 10 to 20
clones were sequenced. The sequences of the PCR primers are listed in Supp. Table S1.
L1-specific RT-PCR and sequencing
In vitro cultures of BFU-E colonies were performed as described (Horvathova, et al., 2012).
Total RNA from reticulocytes and from harvested BFU-Es was treated twice with DNAse I (10
U/1 g of RNA) at 37C for 1 hour and then re-extracted using phenol-chloroform and
precipitated with ethanol. Overall, 5 pmols of the L1-specific primer were annealed (in 45C for
30 min) to 1 g of total RNA (isolated from reticulocytes and from BFU-E colonies) in 1x
hybridization buffer (1 M NaCl; 10 mM Tris-HCl pH 7.5; 1 mM EDTA; 0.2% SDS) and were
extended with reverse transcriptase (Superscript II, Invitrogen, Life Technologies, NY)
according to manufacturer’s instructions. This cDNA template was amplified by semi-nested
PCR using the same antisense primer as for reverse transcription and two sense primers (L1 F 1
for the first round and L1 F 2 for the second round). DNA sequencing was performed in both
directions. The sequences of the primers are listed in Supp. Table S1.
Gene-breaking RT-PCR and sequencing
Total RNA from MEL HBBL1+
and MEL HBBwt
cell lines was isolated with TRI reagent
followed by TURBO DNA-free DNase I treatment. Transcripts terminating in L1 major
antisense polyadenylation site were searched by using 0.12 μM L1-polyT chimera primer and
reverse transcriptase (Superscript II, Invitrogen, Life Technologies, NY) (Wheelan, et al., 2005);
the cDNA synthesis was performed with 3 μg total RNA. 10 μL of the cDNA synthesis solution

5
was used for PCR amplification with HotStarTaq Master Mix Kit (Qiagen, QIAGEN Valencia,
CA) and 0.5 μM primers for the first round: NRO B1 F and MAPS L1 R and second round:
NRO B2 F and MAPS L1 R. In searching for transcripts starting in the L1 antisense promoter
GeneRacerTM
Oligo dT primer was used (GeneRacerTM
Kit, Invitrogen, Life Technologies, NY)
and ASP L1 F primer (Mätlik, et al., 2006) for the first round, and ASP L1 F and HBB ex3 R
primers for the second round. The PCR products (major bands) were purified on agarose gels,
cloned by using a TOPO TA Cloning (GeneRacerTM
Kit, Invitrogen, Life Technologies, NY)
and sequenced. The sequences of the primers are listed in Supp. Table S1.
Kutlar A, Huisman THJ. 1991. The detection of hemoglobinopathies. In: Hommes FA, ed.
Techniques in Diagnostic Human Biochemical Genetics: A Laboratory Manual. New
York, NY: Wiley-Liss. 519-560.
Horvathova M, Kapralova K, Zidova Z, Dolezal D, Pospisilova D, Divoky V. 2012.
Erythropoietin-driven signaling ameliorates the survival defect of DMT1-mutant erythroid
progenitors and erythroblasts. Haematologica 97(10):1480-1488.
Mabaera R, Richardson CHA, Johnson K, , Hsu M, Fiering S, Lowrey CH. 2007. Developmental-
and differentiation-specific patterns of human - and -globin promoter DNA
methylation. Blood 110(4):1343-1352.
Mätlik K, Redik K, Speek M. 2006. L1 Antisense Promoter Drives Tissue-Specific Transcription
of Human Genes. J Biomed Biotechnol 2006: 71753.
Papayannopoulou T, Brice M, Stamatoyannopoulos G. 1996. Analysis of human hemoglobin
switching in MEL x human fetal erythroid cell hybrids. Cell 46(3):469-476.
Pfaffl MW. 2001. A new mathematical model for relative quantification in
real-time RT-PCR. Nucleic Acids Res 29(9):e45.
Wheelan SJ, Aizawa Y, Han JS, Boeke JD. 2005. Gene-breaking: A new paradigm for human
retrotransposon-mediated gene evolution. Genome Res 15:1073–1078.
Yagi M, Stamatoyannopoulos G, Papayannopoulou T. 1987. Monoclonal antibody 53.6
recognizes a novel proliferation-associated antigen encoded on human chromosome 11. J
Immunol 138(7):2208-2212.

6
Yang Ch, Feng J, Song W, et al. 2007. A mouse model for nonsense mutation bypass therapy
shows a dramatic multiday response to geneticin. Proc Natl Acad Sci USA 4(39):15394-
15399.
Supp. Figure S1. Schematic diagram of the L1-thal insertion and -globin gene breakpoints.
(A and B) The inverse PCR strategy used to clone the 5’ and 3’ breakpoints. (A) An AccI
genomic fragment (1.1 kb) containing the 5’ junction of the -globin IVS-II with the insertion
sequence was self-ligated in the AccI site and then linearized in the BamHI site. Primers -Ex2-R
and -IVS-II-F were used to amplify the junction sequence. (B) A PstI genomic fragment (1.6
kb) containing the 3’ junction of the insertion with -globin IVS-II sequence was ligated in the
PstI site and then linearized in the EcoRI site. Primers -Ex3-F and -Ex3-R were used to
amplify the linear fragments containing the junction. (C) Sequence of the breakpoints. The
insertion occurred at 98 bp 5´ to the 3´ end of -IVS-II. The arrows above the nucleotide
sequence show the residues at positions 98 and 99 bp 5’ to the 3’ end of -globin IVS-II. The L1
insertion was flanked by a 9-13 bp target site duplication (the run of four As, shown by the
hatched bracket, could be duplicated or could be a part of the L1-poly(A) tail) (Kimberland, et
al., 1999). The L1-poly(A) tail length is 107(A); the GenBank sequence database (accession
AF149422.1) shows only 27(A).
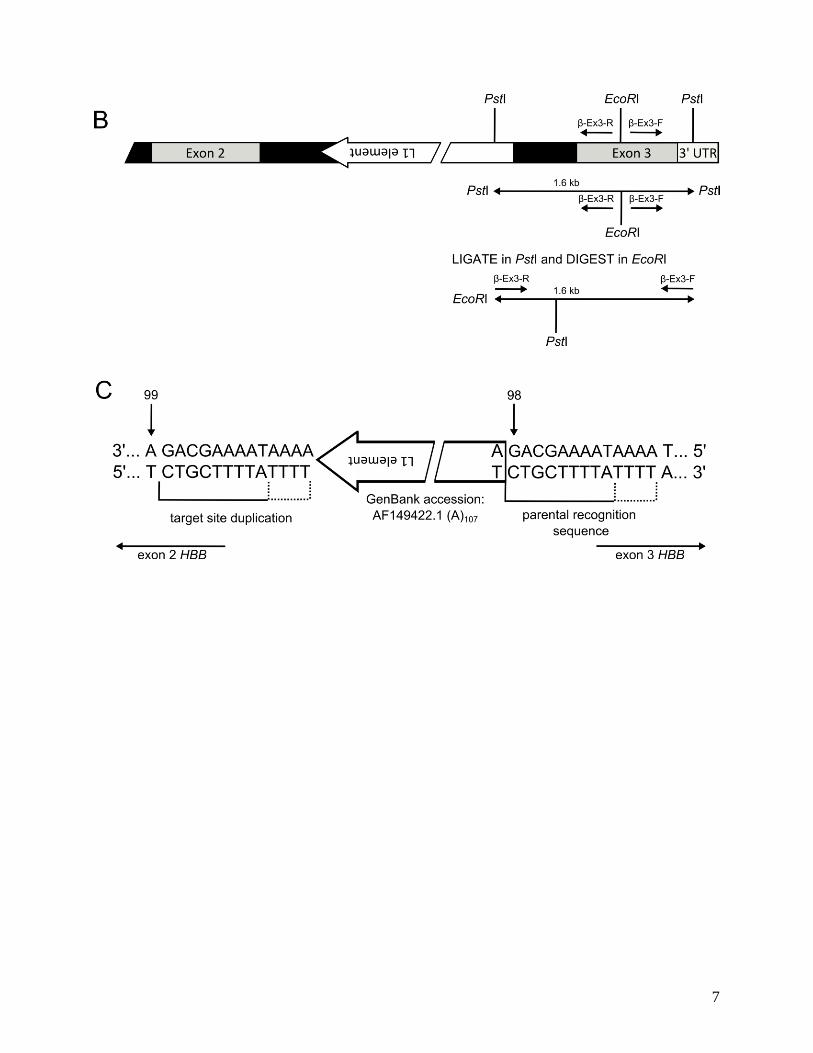
7
8
Supp. Figure S2. The half-life of -globin and -globinL1 mRNA was determined after blockage
of transcription for various times (1 – 12 h) with actinomycin D (ActD, 1 μM) in induced hybrid
MEL cells. The half-life was calculated (Dölken, et al., 2008) from the relative amount of
transcript present at each time point during ActD treatment. The half-life of the wild type -
globin mRNA in the control MEL HBBwt
hybrid cell line was 11.71 h. The half-life of total
mRNA from -globinL1 allele, measured in MEL HBBL1+
cells, was only 4.31 h. To test whether
the low stability of -globinL1 transcripts is solely a consequence of NMD, we also measured the
mRNA half-life in the presence of emetine (0.5 μM). The stability of the -globinL1 mRNA was
restored almost to normal levels when emetine was used. The data are the mean ± SD of three
independent experiments.
9
Supp. Figure S3. The ratio of γ- to β-globin mRNA in hybrid MEL cells treated with DAC (2
M) and TSA (100 nM) at day 3 of 1.8% DMSO-induced differentiation. The γ/β-globin
transcript ratio of MEL HBBL1+
cells was normalized to the γ/β-globin transcript ratio of control
cells MEL HBBwt
using the formula 2Ct
where Ct was calculated as: (Ct– Ctβ)MEL HBBwt –
(Ct– Ctβ)MEL HBBL1+. Data are the mean ± SD of three independent experiments. The increased
γ/β-globin mRNA ratio in DMSO-induced and DAC-treated MEL HBBL1+
cells is consistent with
the activation of γ-globin in the presence of persistent suppression of β-globin expression due to
the L1 insertion. TSA treatment resulted in an equal γ/β-globin transcript ratio in MEL HBBwt
and
MEL HBBL1+
cells.
Dölken L, Ruzsics Z, Rädle B, et al. 2008. High-resolution gene expression profiling for
simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14(9):1959-
72.
Kimberland ML, Divoky V, Prchal JT, Schwahn U, Berger W, Kazazian HH Jr. 1999. Full-
lenght human L1 insertions retain the capacity for high frequency retrotransposition in
cultured cells. Hum Mol Genet 8(8):1557-1560.
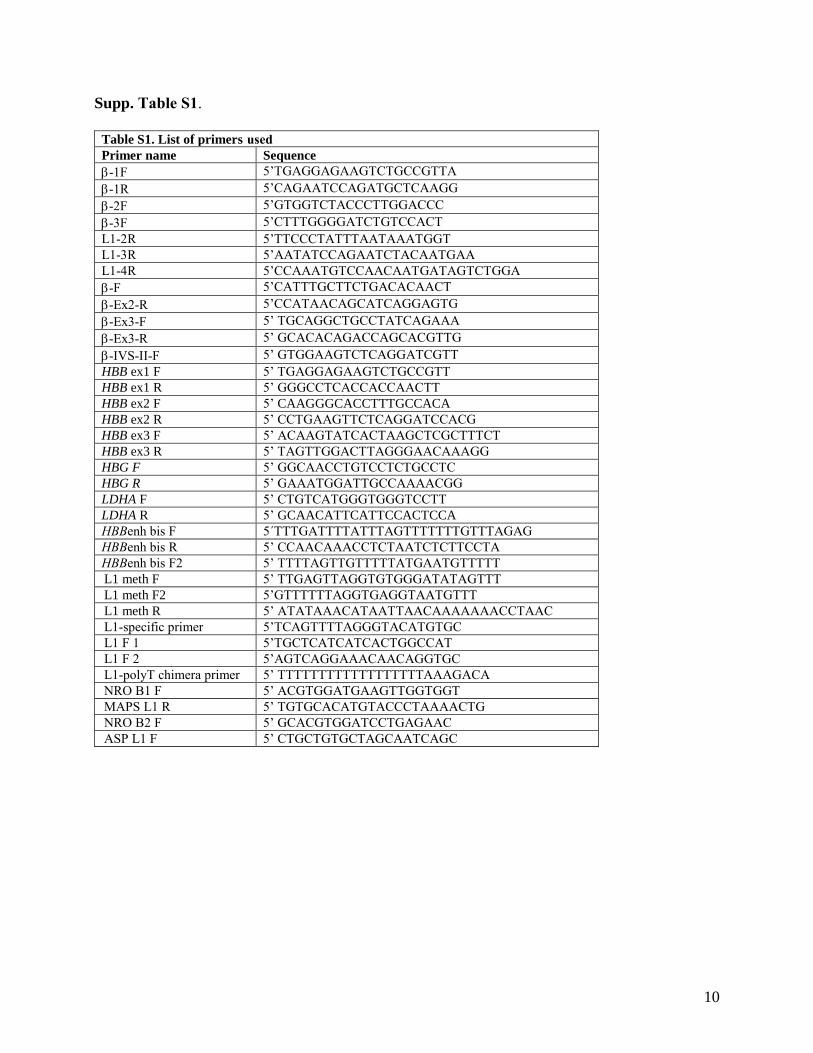
10
Supp. Table S1.
Table S1. List of primers used Primer name Sequence
-1F 5’TGAGGAGAAGTCTGCCGTTA
-1R 5’CAGAATCCAGATGCTCAAGG
-2F 5’GTGGTCTACCCTTGGACCC
-3F 5’CTTTGGGGATCTGTCCACT
L1-2R 5’TTCCCTATTTAATAAATGGT
L1-3R 5’AATATCCAGAATCTACAATGAA
L1-4R 5’CCAAATGTCCAACAATGATAGTCTGGA
-F 5’CATTTGCTTCTGACACAACT
-Ex2-R 5’CCATAACAGCATCAGGAGTG
-Ex3-F 5’ TGCAGGCTGCCTATCAGAAA
-Ex3-R 5’ GCACACAGACCAGCACGTTG
-IVS-II-F 5’ GTGGAAGTCTCAGGATCGTT
HBB ex1 F 5’ TGAGGAGAAGTCTGCCGTT
HBB ex1 R 5’ GGGCCTCACCACCAACTT
HBB ex2 F 5’ CAAGGGCACCTTTGCCACA
HBB ex2 R 5’ CCTGAAGTTCTCAGGATCCACG
HBB ex3 F 5’ ACAAGTATCACTAAGCTCGCTTTCT
HBB ex3 R 5’ TAGTTGGACTTAGGGAACAAAGG
HBG F 5’ GGCAACCTGTCCTCTGCCTC
HBG R 5’ GAAATGGATTGCCAAAACGG
LDHA F 5’ CTGTCATGGGTGGGTCCTT
LDHA R 5’ GCAACATTCATTCCACTCCA
HBBenh bis F 5´TTTGATTTTATTTAGTTTTTTTGTTTAGAG
HBBenh bis R 5’ CCAACAAACCTCTAATCTCTTCCTA
HBBenh bis F2 5’ TTTTAGTTGTTTTTATGAATGTTTTT
L1 meth F 5’ TTGAGTTAGGTGTGGGATATAGTTT
L1 meth F2 5’GTTTTTTAGGTGAGGTAATGTTT
L1 meth R 5’ ATATAAACATAATTAACAAAAAAACCTAAC
L1-specific primer 5’TCAGTTTTAGGGTACATGTGC
L1 F 1 5’TGCTCATCATCACTGGCCAT
L1 F 2 5’AGTCAGGAAACAACAGGTGC
L1-polyT chimera primer 5’ TTTTTTTTTTTTTTTTTTAAAGACA
NRO B1 F 5’ ACGTGGATGAAGTTGGTGGT
MAPS L1 R 5’ TGTGCACATGTACCCTAAAACTG
NRO B2 F 5’ GCACGTGGATCCTGAGAAC
ASP L1 F 5’ CTGCTGTGCTAGCAATCAGC
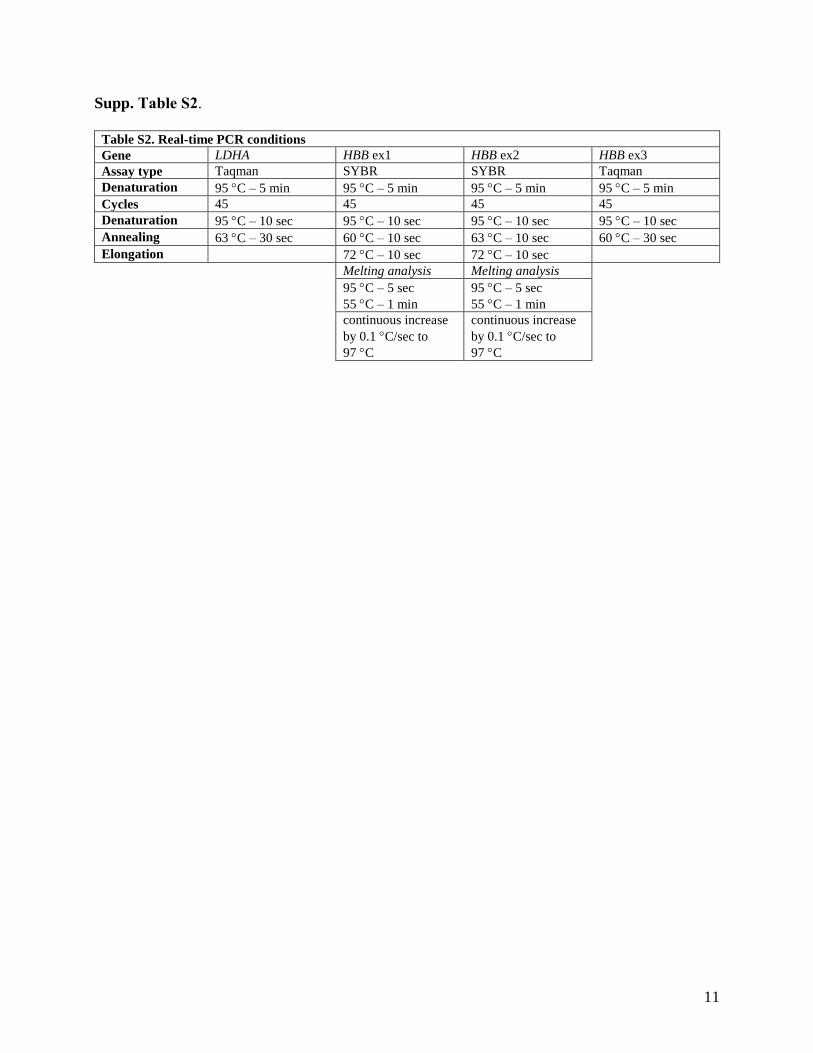
11
Supp. Table S2.
Table S2. Real-time PCR conditions
Gene LDHA HBB ex1 HBB ex2 HBB ex3
Assay type Taqman SYBR SYBR Taqman
Denaturation 95 C – 5 min 95 C – 5 min 95 C – 5 min 95 C – 5 min
Cycles 45 45 45 45
Denaturation 95 C – 10 sec 95 C – 10 sec 95 C – 10 sec 95 C – 10 sec
Annealing 63 C – 30 sec 60 C – 10 sec 63 C – 10 sec 60 C – 30 sec
Elongation 72 C – 10 sec 72 C – 10 sec
Melting analysis Melting analysis
95 C – 5 sec
55 C – 1 min
95 C – 5 sec
55 C – 1 min
continuous increase
by 0.1 C/sec to
97 C
continuous increase
by 0.1 C/sec to
97 C

Regular Article
RED CELLS, IRON, AND ERYTHROPOIESIS
Novel homozygous VHL mutation in exon 2 is associated with congenitalpolycythemia but not with cancerLucie Lanikova,1,2 Felipe Lorenzo,1 Chunzhang Yang,3 Hari Vankayalapati,4 Richard Drachtman,5 Vladimir Divoky,2
and Josef T. Prchal1
1Division of Hematology, University of Utah and VA Health Care, Salt Lake City, UT; 2Department of Biology, Faculty of Medicine and Dentistry,
Palacky University, Olomouc, Czech Republic; 3Surgical Neurology Branch, National Institute of Neurological Disorders and Stroke, National Institutes of
Health, Bethesda, MD; 4Huntsman Cancer Institute, Salt Lake City, UT; and 5Cancer Institute of New Jersey, Robert Wood Johnson Medical School, New
Brunswick, NJ
Key Points
• We describe a novelhomozygous mutation in exon2 of the VHL gene causingcongenital polycythemia.
• We demonstrate the VHLP138L
effect on the augmentation oferythropoiesis, along withstructural and functionalstudies of this mutation.
Germline von Hippel–Lindau (VHL) gene mutations underlie dominantly inherited
familial VHL tumor syndrome comprising a predisposition for renal cell carcinoma,
pheochromocytoma/paraganglioma, cerebral hemangioblastoma, and endolymphatic
sac tumors. However, recessively inherited congenital polycythemia, exemplified by
Chuvash polycythemia, has been associated with 2 separate 39 VHL gene mutations in
exon 3. It was proposed that different positions of loss-of-function VHL mutations are
associated with VHL syndrome cancer predisposition and only C-terminal domain-
encoding VHL mutations would cause polycythemia. However, now we describe a
new homozygous VHL exon 2 mutation of the VHL gene:(c.413C>T):P138L, which is
associated in the affected homozygote with congenital polycythemia but not in her, or
her-heterozygous relatives, with cancer or other VHL syndrome tumors. We show that
VHLP138L has perturbed interaction with hypoxia-inducible transcription factor (HIF)1a.
Further, VHLP138L protein has decreased stability in vitro. Similarly to what was reported
in Chuvash polycythemia and some other instances of HIFs upregulation, VHLP138L erythroid progenitors are hypersensitive to
erythropoietin. Interestingly, the level of RUNX1/AML1 and NF-E2 transcripts that are specifically upregulated in acquired
polycythemia vera were also upregulated in VHLP138L granulocytes. (Blood. 2013;121(19):3918-3924)
Introduction
The von Hippel-Lindau (VHL) tumor suppressor gene encodesa multifunctional protein that interacts with diverse partners andpromotes degradation of hypoxia-inducible transcription factors(HIFs) by facilitating their ubiquitinization and eventual proteasomaldegradation. Germline dominantly inherited mutations in VHL pre-dispose patients to highly vascularized malignant tumors, includingrenal cell carcinoma of the clear-cell type, hemangioblastoma, andpheochromocytoma/paraganglioma.1,2 Less frequently, VHL muta-tions have been associated with benign tumors, including those of theinner ear (endolymphatic sac tumor), pancreas (pancreatic cysts,serous cystadenoma, and pancreatic neuroendocrine tumors), andtestes (epididymal cystadenomas).2 The incidence of VHL disease isthought to be about 1 in 36 000 births with an estimated de novomutation rate of 4.4 3 1026 gametes per generation.3 VHL tumorsgenerally develop after age of 124 and have more than 90%penetrance in the highest age classes (96% at 51–60 years of age,99% at 61–70 years of age).3
Polycythemia (also known as erythrocytosis) is characterizedby an increased red cell blood mass. Polycythemias can be primaryor secondary. Primary polycythemias are caused by somatic orgermline mutations leading to changes within the erythroid progenitors
causing an augmented response to erythropoietin (EPO). Second-ary polycythemias are caused by either an appropriate or inap-propriate increase in the red cell mass as a result of augmentedlevels of EPO. In Chuvash polycythemia, the first known con-genital disorder of hypoxia-sensing,5 erythroid progenitors inin vitro cultures are hypersensitive to EPO; thus, Chuvashpolycythemia shares features of both primary and secondarypolycythemia.6
Because different positions of loss-of-function mutations of theVHL gene are associated with different type of cancers, it has beenproposed that only C-terminal domain–encoding VHL mutationswould cause polycythemia.7 There are 2 known homozygous VHLgene mutations causing polycythemia that are not associated withany VHL syndrome malignant or benign tumors; these are theVHLR200W mutation, constituting the disorder of augmented hypoxia-sensing in normoxia (ie, Chuvash polycythemia5) and the VHLH191D
mutation found in a Croatian subject.8 Both are located in the distalportion of exon 3 of the VHL gene in its C-terminal domain. In ad-dition, there are several reports of compound heterozygotes asso-ciated with congenital polycythemia, 3 in combination with VHLR200W
mutation, and others employing different exon 3 VHL missense
Submitted November 26, 2012; accepted March 18, 2013. Prepublished online
as Blood First Edition paper, March 28, 2013; DOI 10.1182/blood-2012-11-
469296.
The publication costs of this article were defrayed in part by page charge
payment. Therefore, and solely to indicate this fact, this article is hereby
marked “advertisement” in accordance with 18 USC section 1734.
© 2013 by The American Society of Hematology
3918 BLOOD, 9 MAY 2013 x VOLUME 121, NUMBER 19
For personal use only. at UNIV OF UTAH on June 3, 2013. bloodjournal.hematologylibrary.orgFrom

mutations (Table 1).9-13 It has been suggested that the genomicconfiguration of the 39 region of VHL exon 3 gene has a specificerythropoiesis-promoting effect independent of EPO by JanusKinase-2 (JAK2) hyperactivation, because JAK2 is the crucial com-ponent of intracellular activation of EPO/EPO receptor signaling.14
Compatible with this report, to date no congenital polycythemia as-sociated with VHL homozygous or compound heterozygous muta-tions outside of VHL exon 3 have been described. We now challengethis premise by reporting a family with congenital polycythemia witha new homozygous VHLmutation in exon 2 along with structural andfunctional studies of the mutation (Figure 1).
Materials and methods
Patient samples
The propositus is a 15-year-old girl of Asian Indian extraction (Punjabiethnicity), who has been known to be polycythemic from infancy. Herparents are hematologically normal and are of the same ethnicity but notknown to be related. Peripheral blood of the propositus and her parents wasobtained by venipuncture after obtaining a signed Institutional ReviewBoard informed consent in accordance with the Declaration of Helsinki.This study received approval from Institutional Review Board protocol#17665; molecular biology of polycythemia and thrombocytosis. Gran-ulocyte and mononuclear cell fractions were isolated according to apreviously published protocol.15
Mutation screening
Genomic DNA was isolated from granulocytes using Gentra-Puregene Kit(Qiagen, Germantown, MD), and VHL gene was amplified using Hot Star
Master Mix (Qiagen) and the following primers: VHL_exon1_F 59CGAAGACTACGGAGGTC GAC; VHL_exon1_R 59GGCTTCAGACCGTGCTATCG; VHL_exon2_F 59GTGTGGCTCTTTAACAACC; VHL_exon2_R59CTGTACTTACCACAACAACC; VHL_exon3_F 59TCCTTGTACTGAGACCCTAG; and VHL_exon3_R 59AGCTGAGATGAAACAGTCTA. Se-quencing was performed using the same amplification primers and protocolestablished by the University of Utah, Core DNA Sequencing Facility.
In vitro assay of erythroid progenitors’ sensitivity to EPO
In vitro sensitivity of erythroid progenitors to EPO was performed on mono-nuclear cells isolated from the peripheral blood using Histopaque (Sigma-Aldrich, St. Louis, MO) density gradient centrifugation and plating (2.3 3105/mL) to methylcellulose media (MethoCult H4531; StemCell Technol-ogies, Vancouver, BC) with addition of various concentrations of EPO(StemCell Technologies), ranging from 0.015 to 3.0 U/mL. Cell cultureswere maintained in humidified atmosphere of 5% CO2 at 37°C for 14 days.Erythroid burst-forming unit colonies (BFU-Es) were scored by standardmorphologic criteria. The number of BFU-Es grown in individual con-centrations of EPO was expressed as a percentage of maximum vs the con-centration of EPO. Maximum growth (100%) represents the highest colonynumber grown in culture. The assay was carried out on erythroid progen-itors from a VHLP138L-homozygous patient (n 5 1), from patient with het-erozygous gain-of-function HIF2aM535V mutation (n 5 1), and 8 healthycontrols (n 5 8). Results for healthy controls are pooled and T bars des-ignate standard deviations.
Quantitative analysis of HIF target genes expression
RNA was isolated from the patient’s granulocytes using TRI reagent andresidual DNA was removed by DNA-free DNase Treatment & RemovalReagents (Ambion, Life Technologies, NY). Gene expression experimentswere performed on a FastPCR 7500 instrument using TaqMan Gene Ex-pression assays TFRC (Hs00951083), SLC2A1 (Hs00892681), HK1(Hs00175976), RUNX1 (Hs00231079), NF-E2 (Hs00232351), and reference
Table 1. Homozygous and compound heterozygous VHL mutations in individuals with erythrocytosis/polycythemia reported in theliterature
VHL genotype Ethnicity Notes
Compound heterozygous VHL genotype
235 C→T/586 C→G R79C/L188V Caucasian9
376 G→A/548 C→T D126N/S183L British10
598 C→T/574 C→T R200W/P192A American (white)11
598 C→T/562 C→G R200W/L188V American (white)11
598 C→T/388 G→C R200W/V130L American (white)8
Homozygous VHL genotype
598 C→T/598 C→T R200W/R200W Chuvash,5 Italian,12 Danish,11 German,13
Turkish,13 American (white)11Frequent thrombotic complications
571 C→G/571 C→G H191D/H191D Croatian11,27
413 C→T/413 C→T P138L/P138L Punjabi
Figure 1. The schematic structure of the VHL gene. Sequencing of the second exon of the VHL gene revealed c.413C.T:P138L homozygous VHL mutation in the
propositus, which was inherited from her parents, both VHLP138L heterozygous. UTR, untranslated region.
BLOOD, 9 MAY 2013 x VOLUME 121, NUMBER 19 NOVEL HOMOZYGOUS POLYCYTHEMIC VHL MUTATION 3919
For personal use only. at UNIV OF UTAH on June 3, 2013. bloodjournal.hematologylibrary.orgFrom
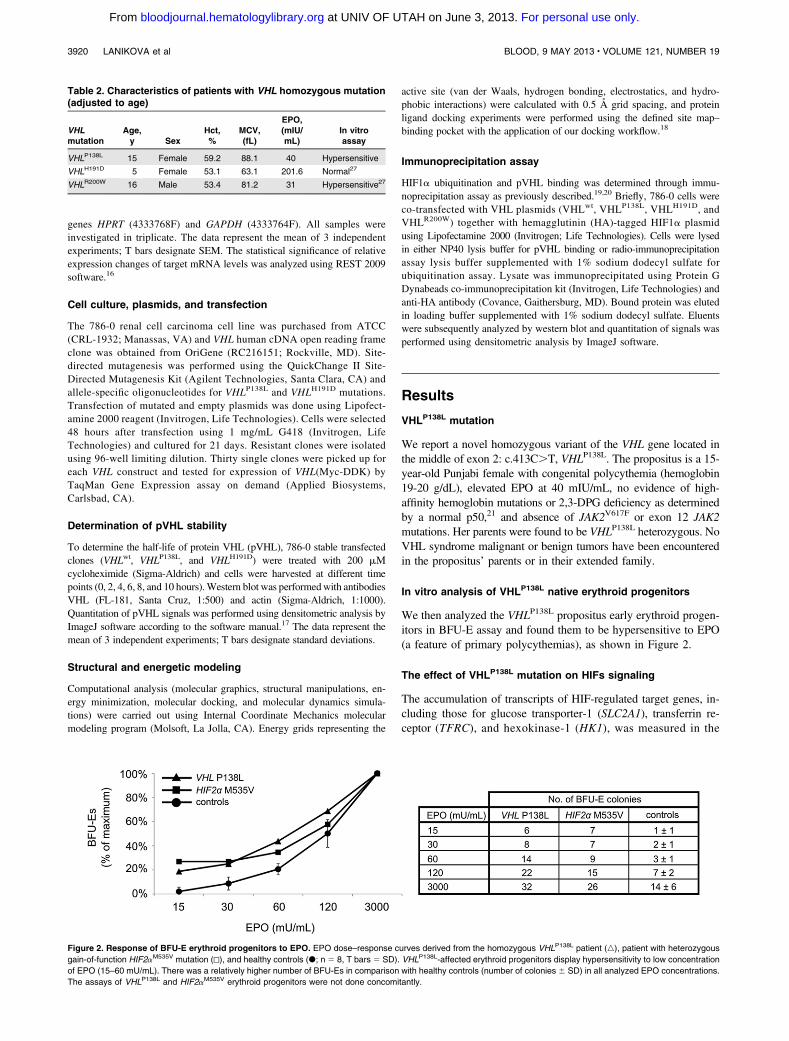
genes HPRT (4333768F) and GAPDH (4333764F). All samples wereinvestigated in triplicate. The data represent the mean of 3 independentexperiments; T bars designate SEM. The statistical significance of relativeexpression changes of target mRNA levels was analyzed using REST 2009software.16
Cell culture, plasmids, and transfection
The 786-0 renal cell carcinoma cell line was purchased from ATCC(CRL-1932; Manassas, VA) and VHL human cDNA open reading frameclone was obtained from OriGene (RC216151; Rockville, MD). Site-directed mutagenesis was performed using the QuickChange II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) andallele-specific oligonucleotides for VHLP138L and VHLH191D mutations.Transfection of mutated and empty plasmids was done using Lipofect-amine 2000 reagent (Invitrogen, Life Technologies). Cells were selected48 hours after transfection using 1 mg/mL G418 (Invitrogen, LifeTechnologies) and cultured for 21 days. Resistant clones were isolatedusing 96-well limiting dilution. Thirty single clones were picked up foreach VHL construct and tested for expression of VHL(Myc-DDK) byTaqMan Gene Expression assay on demand (Applied Biosystems,Carlsbad, CA).
Determination of pVHL stability
To determine the half-life of protein VHL (pVHL), 786-0 stable transfectedclones (VHLwt, VHLP138L, and VHLH191D) were treated with 200 mMcycloheximide (Sigma-Aldrich) and cells were harvested at different timepoints (0, 2, 4, 6, 8, and 10 hours).Western blot was performedwith antibodiesVHL (FL-181, Santa Cruz, 1:500) and actin (Sigma-Aldrich, 1:1000).Quantitation of pVHL signals was performed using densitometric analysis byImageJ software according to the software manual.17 The data represent themean of 3 independent experiments; T bars designate standard deviations.
Structural and energetic modeling
Computational analysis (molecular graphics, structural manipulations, en-ergy minimization, molecular docking, and molecular dynamics simula-tions) were carried out using Internal Coordinate Mechanics molecularmodeling program (Molsoft, La Jolla, CA). Energy grids representing the
active site (van der Waals, hydrogen bonding, electrostatics, and hydro-phobic interactions) were calculated with 0.5 A grid spacing, and proteinligand docking experiments were performed using the defined site map–binding pocket with the application of our docking workflow.18
Immunoprecipitation assay
HIF1a ubiquitination and pVHL binding was determined through immu-noprecipitation assay as previously described.19,20 Briefly, 786-0 cells wereco-transfected with VHL plasmids (VHLwt, VHLP138L, VHLH191D, andVHLR200W) together with hemagglutinin (HA)-tagged HIF1a plasmidusing Lipofectamine 2000 (Invitrogen; Life Technologies). Cells were lysedin either NP40 lysis buffer for pVHL binding or radio-immunoprecipitationassay lysis buffer supplemented with 1% sodium dodecyl sulfate forubiquitination assay. Lysate was immunoprecipitated using Protein GDynabeads co-immunoprecipitation kit (Invitrogen, Life Technologies) andanti-HA antibody (Covance, Gaithersburg, MD). Bound protein was elutedin loading buffer supplemented with 1% sodium dodecyl sulfate. Eluentswere subsequently analyzed by western blot and quantitation of signals wasperformed using densitometric analysis by ImageJ software.
Results
VHLP138L mutation
We report a novel homozygous variant of the VHL gene located inthe middle of exon 2: c.413C.T, VHLP138L. The propositus is a 15-year-old Punjabi female with congenital polycythemia (hemoglobin19-20 g/dL), elevated EPO at 40 mIU/mL, no evidence of high-affinity hemoglobin mutations or 2,3-DPG deficiency as determinedby a normal p50,21 and absence of JAK2V617F or exon 12 JAK2mutations. Her parents were found to be VHLP138L heterozygous. NoVHL syndrome malignant or benign tumors have been encounteredin the propositus’ parents or in their extended family.
In vitro analysis of VHLP138L native erythroid progenitors
We then analyzed the VHLP138L propositus early erythroid progen-itors in BFU-E assay and found them to be hypersensitive to EPO(a feature of primary polycythemias), as shown in Figure 2.
The effect of VHLP138L mutation on HIFs signaling
The accumulation of transcripts of HIF-regulated target genes, in-cluding those for glucose transporter-1 (SLC2A1), transferrin re-ceptor (TFRC), and hexokinase-1 (HK1), was measured in the
Table 2. Characteristics of patients with VHL homozygous mutation(adjusted to age)
VHLmutation
Age,y Sex
Hct,%
MCV,(fL)
EPO,(mIU/mL)
In vitroassay
VHLP138L 15 Female 59.2 88.1 40 Hypersensitive
VHLH191D 5 Female 53.1 63.1 201.6 Normal27
VHLR200W 16 Male 53.4 81.2 31 Hypersensitive27
Figure 2. Response of BFU-E erythroid progenitors to EPO. EPO dose–response curves derived from the homozygous VHLP138L patient (n), patient with heterozygous
gain-of-function HIF2aM535V mutation (N), and healthy controls (d; n 5 8, T bars 5 SD). VHLP138L-affected erythroid progenitors display hypersensitivity to low concentration
of EPO (15–60 mU/mL). There was a relatively higher number of BFU-Es in comparison with healthy controls (number of colonies 6 SD) in all analyzed EPO concentrations.
The assays of VHLP138L and HIF2aM535V erythroid progenitors were not done concomitantly.
3920 LANIKOVA et al BLOOD, 9 MAY 2013 x VOLUME 121, NUMBER 19
For personal use only. at UNIV OF UTAH on June 3, 2013. bloodjournal.hematologylibrary.orgFrom
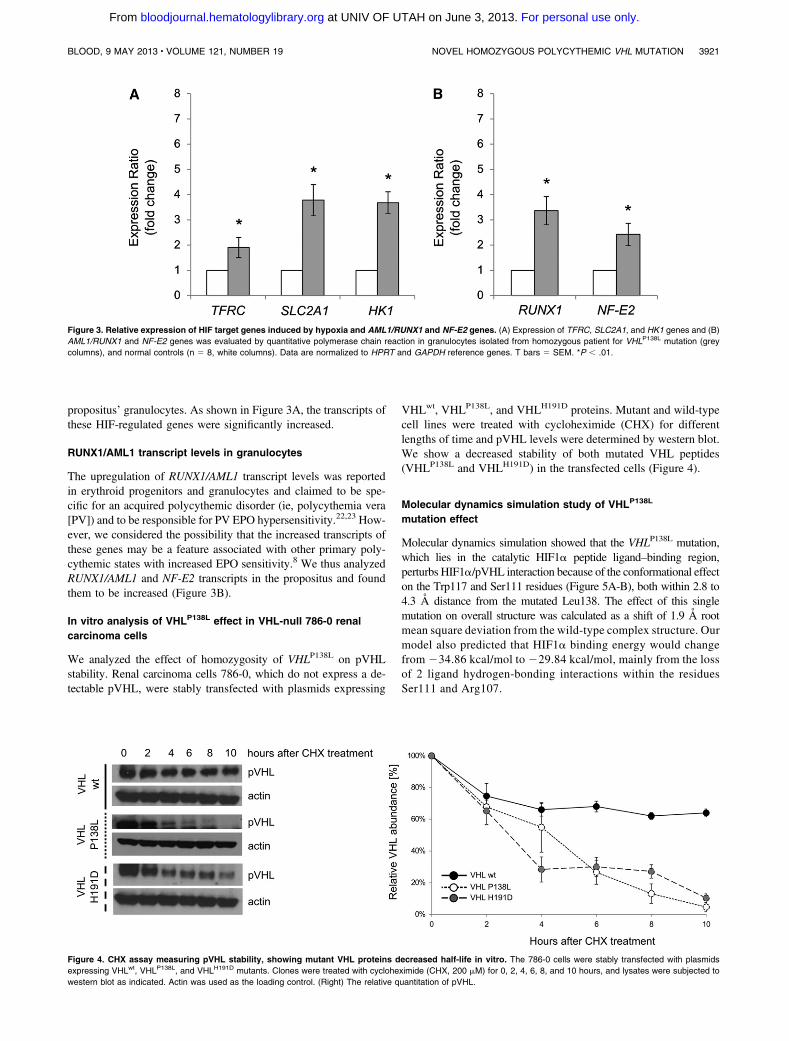
propositus’ granulocytes. As shown in Figure 3A, the transcripts ofthese HIF-regulated genes were significantly increased.
RUNX1/AML1 transcript levels in granulocytes
The upregulation of RUNX1/AML1 transcript levels was reportedin erythroid progenitors and granulocytes and claimed to be spe-cific for an acquired polycythemic disorder (ie, polycythemia vera[PV]) and to be responsible for PV EPO hypersensitivity.22,23 How-ever, we considered the possibility that the increased transcripts ofthese genes may be a feature associated with other primary poly-cythemic states with increased EPO sensitivity.8 We thus analyzedRUNX1/AML1 and NF-E2 transcripts in the propositus and foundthem to be increased (Figure 3B).
In vitro analysis of VHLP138L effect in VHL-null 786-0 renal
carcinoma cells
We analyzed the effect of homozygosity of VHLP138L on pVHLstability. Renal carcinoma cells 786-0, which do not express a de-tectable pVHL, were stably transfected with plasmids expressing
VHLwt, VHLP138L, and VHLH191D proteins. Mutant and wild-typecell lines were treated with cycloheximide (CHX) for differentlengths of time and pVHL levels were determined by western blot.We show a decreased stability of both mutated VHL peptides(VHLP138L and VHLH191D) in the transfected cells (Figure 4).
Molecular dynamics simulation study of VHLP138L
mutation effect
Molecular dynamics simulation showed that the VHLP138L mutation,which lies in the catalytic HIF1a peptide ligand–binding region,perturbs HIF1a/pVHL interaction because of the conformational effecton the Trp117 and Ser111 residues (Figure 5A-B), both within 2.8 to4.3 A distance from the mutated Leu138. The effect of this singlemutation on overall structure was calculated as a shift of 1.9 A rootmean square deviation from the wild-type complex structure. Ourmodel also predicted that HIF1a binding energy would changefrom 234.86 kcal/mol to229.84 kcal/mol, mainly from the lossof 2 ligand hydrogen-bonding interactions within the residuesSer111 and Arg107.
Figure 3. Relative expression of HIF target genes induced by hypoxia and AML1/RUNX1 and NF-E2 genes. (A) Expression of TFRC, SLC2A1, and HK1 genes and (B)
AML1/RUNX1 and NF-E2 genes was evaluated by quantitative polymerase chain reaction in granulocytes isolated from homozygous patient for VHLP138L mutation (grey
columns), and normal controls (n 5 8, white columns). Data are normalized to HPRT and GAPDH reference genes. T bars 5 SEM. *P , .01.
Figure 4. CHX assay measuring pVHL stability, showing mutant VHL proteins decreased half-life in vitro. The 786-0 cells were stably transfected with plasmids
expressing VHLwt, VHLP138L, and VHLH191D mutants. Clones were treated with cycloheximide (CHX, 200 mM) for 0, 2, 4, 6, 8, and 10 hours, and lysates were subjected to
western blot as indicated. Actin was used as the loading control. (Right) The relative quantitation of pVHL.
BLOOD, 9 MAY 2013 x VOLUME 121, NUMBER 19 NOVEL HOMOZYGOUS POLYCYTHEMIC VHL MUTATION 3921
For personal use only. at UNIV OF UTAH on June 3, 2013. bloodjournal.hematologylibrary.orgFrom
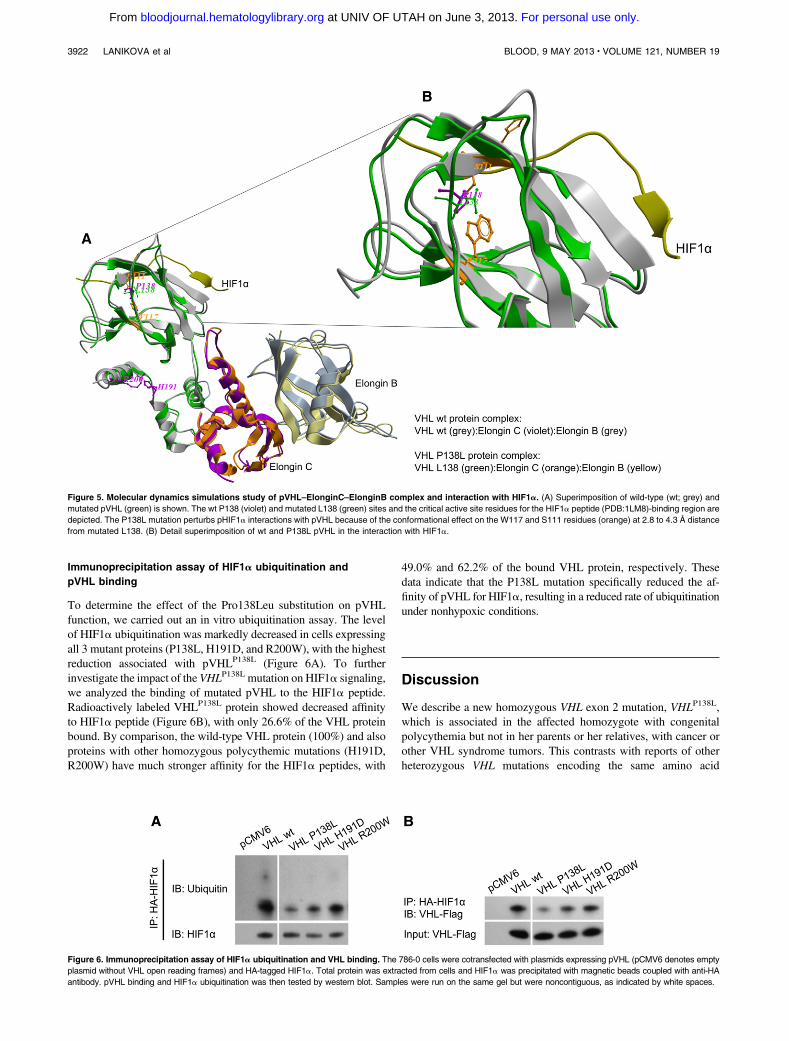
Immunoprecipitation assay of HIF1a ubiquitination and
pVHL binding
To determine the effect of the Pro138Leu substitution on pVHLfunction, we carried out an in vitro ubiquitination assay. The levelof HIF1a ubiquitination was markedly decreased in cells expressingall 3 mutant proteins (P138L, H191D, and R200W), with the highestreduction associated with pVHLP138L (Figure 6A). To furtherinvestigate the impact of the VHLP138L mutation on HIF1a signaling,we analyzed the binding of mutated pVHL to the HIF1a peptide.Radioactively labeled VHLP138L protein showed decreased affinityto HIF1a peptide (Figure 6B), with only 26.6% of the VHL proteinbound. By comparison, the wild-type VHL protein (100%) and alsoproteins with other homozygous polycythemic mutations (H191D,R200W) have much stronger affinity for the HIF1a peptides, with
49.0% and 62.2% of the bound VHL protein, respectively. Thesedata indicate that the P138L mutation specifically reduced the af-finity of pVHL for HIF1a, resulting in a reduced rate of ubiquitinationunder nonhypoxic conditions.
Discussion
We describe a new homozygous VHL exon 2 mutation, VHLP138L,which is associated in the affected homozygote with congenitalpolycythemia but not in her parents or her relatives, with cancer orother VHL syndrome tumors. This contrasts with reports of otherheterozygous VHL mutations encoding the same amino acid
Figure 5. Molecular dynamics simulations study of pVHL–ElonginC–ElonginB complex and interaction with HIF1a. (A) Superimposition of wild-type (wt; grey) and
mutated pVHL (green) is shown. The wt P138 (violet) and mutated L138 (green) sites and the critical active site residues for the HIF1a peptide (PDB:1LM8)-binding region are
depicted. The P138L mutation perturbs pHIF1a interactions with pVHL because of the conformational effect on the W117 and S111 residues (orange) at 2.8 to 4.3 A distance
from mutated L138. (B) Detail superimposition of wt and P138L pVHL in the interaction with HIF1a.
Figure 6. Immunoprecipitation assay of HIF1a ubiquitination and VHL binding. The 786-0 cells were cotransfected with plasmids expressing pVHL (pCMV6 denotes empty
plasmid without VHL open reading frames) and HA-tagged HIF1a. Total protein was extracted from cells and HIF1a was precipitated with magnetic beads coupled with anti-HA
antibody. pVHL binding and HIF1a ubiquitination was then tested by western blot. Samples were run on the same gel but were noncontiguous, as indicated by white spaces.
3922 LANIKOVA et al BLOOD, 9 MAY 2013 x VOLUME 121, NUMBER 19
For personal use only. at UNIV OF UTAH on June 3, 2013. bloodjournal.hematologylibrary.orgFrom

residue (VHL P138R, P138T) that have been reported in VHLsyndrome and renal cancer.24-26 We also show that this mutation isnot only associated with elevated EPO levels but also witha hallmark of primary polycythemia (ie, EPO hypersensitivity).
This novel, polycythemia-associated germline homozygosityfor the hypomorphic VHLP138L allele, albeit present in a differentdomain of the VHL protein, has a similar erythropoiesis-stimulatingeffect to the Chuvash VHLR200W mutation in regard to EPO levelsand BFU-Es EPO hypersensitivity (Table 2). It has been suggestedthat these features are due to a conformational change of SOCS1’sbinding groove encoded by VHL gene’s exon 3. SOCS1 is a neg-ative regulator of JAK2, and it has been suggested that the im-paired interaction of VHLR200W and VHLH191D with SOCS1 is acause of EPO hypersensitivity.9 However, this putative mechanismis contradicted by our data of EPO-hypersensitive VHLP138L ery-throid progenitors (Figure 2) and also by the fact that VHLH191D
native erythroid progenitors are not EPO hypersensitive,27 eventhough VHLH191D is located in the SOCS1’s binding groove.9
Further, our data reveal that some HIF2a gain-of-function muta-tion (c.1603A.G:M535V)28 also have EPO-hypersensitive ery-throid progenitors. We conclude that the molecular mechanisms ofEPO-hypersensitive stimulation of erythroid proliferation (a featureof primary polycythemia) remain unexplained and await furtherstudies.
We show that the loss of function of the VHLP138L mutation (aswell as VHLH191D) is at least in part from decreased stability of VHLprotein (Figure 4). Similar protein instability was previously shownfor the Chuvash VHLR200W mutation.29 Further, our computermodeling of the pVHLP138L structure predicted significant interfer-ence with binding of the a subunit of the HIF1 peptide, which wasconfirmed by HIF1a ubiquitination and a pVHL-binding assay(Figure 6). These functional abnormalities would be expected toprolong the half-life of HIF1 or HIF2, essential transcriptional factorsregulating hypoxia sensing. Indeed, this assumption is directlyconfirmed by the enhanced expression of HIF-regulated genesSLC2A1, TFRC, and HK-1 (Figure 3A). These data suggest that theimpaired stability, together with reduced affinity for the HIFa subunitof the polycythemic pVHLmutants, are common features resultingin the delayed ubiquitinization and degradation of HIFs.30
We also demonstrate that granulocytes from the VHLP138L
subject have increased transcripts of RUNX1 and NF-E2 genes(Figure 3B). It was reported that increased transcription of NF-E2is specific for the acquired primary polycythemic disorder, PV.22,23
Our results contradict this assumption. The nonspecificity ofincreased transcripts of these genes in PV is further supported by
our unpublished data in the study of HIF2a germline gain-of-function mutation. These data suggest that the activation of RUNX1and NF-E2 genes is likely secondary to augmented erythropoiesisin polycythemia and not a candidate “driver” of PV augmentederythropoiesis. It remains to be shown if the increased transcriptsof these genes in PV may be due to augmentation of the hypoxiasensing pathway, also possibly present in PV.
This report provides further evidence for the heterogeneity ofclinical phenotypes of VHL mutations resulting in cancers1 or inaugmented erythropoiesis. The molecular basis of these differencesremains unclear at this time. One can only speculate that therelatively small VHL peptide comprising 213 codons yet encoded bya large .11.2-kb VHL gene may have multiple functions, possiblyfrom interactions with other modifying factors, which await futureclarification. We submit that descriptions of families with congenitaldisorders and unique phenotypes resulting from VHL mutationsprovide an attractive opportunity for structure-function relationshipanalyses of VHL protein and lead to an enhanced understanding ofpolycythemic disorders and diseases of hypoxia sensing.
Acknowledgments
This work was supported by the Czech Science Foundation (projectP301/12/1503), the European Structural Funds (project CZ.1.07/2.3.00/20.0164) and by grant LF_2012_016 (L.L. and V.D.).
Authorship
Contribution: L.L. designed the research, performed the research,analyzed data, and wrote the paper; F.L. performed the research andreviewed the paper; C.Y. performed the immunoprecipitation assayand wrote the paper; H.V. performed molecular dynamics simulationsstudy and wrote the paper; R.D. recruited study subjects and reviewedthe manuscript; V.D. analyzed data and reviewed the manuscript; andJ.T.P. conceived the project, designed the study, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no compet-ing financial interests.
Correspondence: Josef T. Prchal, Division of Hematology, 30N1900E, 5C402 SOM, University of Utah, Salt Lake City, UT84132; e-mail: [email protected].
References
1. Bader HL, Hsu T. Systemic VHL gene functionsand the VHL disease. FEBS Lett. 2012;586(11):1562-1569.
2. Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: a clinical and scientific review.Eur J Hum Genet. 2011;19(6):617-623.
3. Maher ER, Iselius L, Yates JR, et al. Von Hippel-Lindau disease: a genetic study. J Med Genet.1991;28(7):443-447.
4. Nichols K, Zelley K. Hereditary CancerPredisposition Program at the Cancer Center. VonHippel-Lindau (VHL) syndrome. http://www.chop.edu/service/oncology/our-programs/hereditary-cancer-predisposition-program/genetic-syndromes-with-cancer-risks/von-hippel-lindau-syndrome.html. Accessed February 18, 2013.
5. Ang SO, Chen H, Hirota K, et al. Disruptionof oxygen homeostasis underlies congenital
Chuvash polycythemia. Nat Genet. 2002;32(4):614-621.
6. Prchal JT. Clinical manifestations andclassification of erythrocyte disorders. In:Kaushansky KLM, Kipps TJ, Beutler E, SeligsohnU, Prchal JT, eds. Williams Hematology.New York, NY: McGraw Hill; 2010:455-463.
7. Richards FM. Molecular pathology of von Hippel-Lindau disease and the VHL tumour suppressorgene. Expert Rev Mol Med. 2001;2001:1-27.
8. Pastore YD, Jelinek J, Ang S, et al. Mutations inthe VHL gene in sporadic apparently congenitalpolycythemia. Blood. 2003;101(4):1591-1595.
9. Bento MC, Chang KT, Guan Y, Liu E, Caldas G,Gatti RA, Prchal JT. Congenital polycythemia withhomozygous and heterozygous mutations of vonHippel-Lindau gene: five new Caucasian patients.Haematologica. 2005;90(1):128-129.
10. Bond J, Gale DP, Connor T, et al. Dysregulationof the HIF pathway due to VHL mutationcausing severe erythrocytosis and pulmonaryarterial hypertension. Blood. 2011;117(13):3699-3701.
11. Pastore Y, Jedlickova K, Guan Y, et al. Mutationsof von Hippel-Lindau tumor-suppressor gene andcongenital polycythemia. Am J Hum Genet. 2003;73(2):412-419.
12. Perrotta S, Nobili B, Ferraro M, et al. Von Hippel-Lindau-dependent polycythemia is endemic onthe island of Ischia: identification of a novelcluster. Blood. 2006;107(2):514-519.
13. Cario H, Schwarz K, Jorch N, et al. Mutations inthe von Hippel-Lindau (VHL) tumor suppressorgene and VHL-haplotype analysis in patients withpresumable congenital erythrocytosis.Haematologica. 2005;90(1):19-24.
BLOOD, 9 MAY 2013 x VOLUME 121, NUMBER 19 NOVEL HOMOZYGOUS POLYCYTHEMIC VHL MUTATION 3923
For personal use only. at UNIV OF UTAH on June 3, 2013. bloodjournal.hematologylibrary.orgFrom

14. Russell RC, Sufan RI, Zhou B, et al. Loss of JAK2regulation via a heterodimeric VHL-SOCS1 E3ubiquitin ligase underlies Chuvash polycythemia.Nat Med. 2011;17(7):845-853.
15. Prchal JT, Throckmorton DW, Carroll AJ III,Fuson EW, Gams RA, Prchal JF. A commonprogenitor for human myeloid and lymphoid cells.Nature. 1978;274(5671):590-591.
16. Pfaffl MW, Horgan GW, Dempfle L. Relativeexpression software tool (REST) for group-wisecomparison and statistical analysis of relativeexpression results in real-time PCR. Nucleic AcidsRes. 2002;30(9):e36.
17. ImageJ User Guide. IJ1.46. http://rsbweb.nih.gov/ij/docs/guide/index.html. Accessed February 18,2013.
18. Abagyan R, Totrov M, Kuznetsov D. ICM—A newmethod for protein modeling anddesign–applications to docking and structureprediction from the distorted native conformation.J Comput Chem. 1994;15(5):488-506.
19. Zhuang Z, Yang C, Lorenzo F, et al. SomaticHIF2A gain-of-function mutations inparaganglioma with polycythemia. N Engl J Med.2012;367(10):922-930.
20. Yang C, Sun MG, Matro J, et al. Novel HIF2Amutations disrupt oxygen sensing leadingto polycythemia, paragangliomas andsomatostatinomas. Blood. 2013;121(13):2563-2566.
21. Beutler E. The “ascorbate” effect on 2,3-DPG isknown to be due to oxalate. Vox Sang. 2004;86(3):199; author reply 200.
22. Mutschler M, Magin AS, Buerge M, et al. NF-E2overexpression delays erythroid maturation andincreases erythrocyte production. Br J Haematol.2009;146(2):203-217.
23. Wang W, Schwemmers S, Hexner EO, PahlHL. AML1 is overexpressed in patientswith myeloproliferative neoplasms andmediates JAK2V617F-independentoverexpression of NF-E2. Blood. 2010;116(2):254-266.
24. Leonardi E, Martella M, Tosatto SC, Murgia A.Identification and in silico analysis of novel vonHippel-Lindau (VHL) gene variants from a largepopulation. Ann Hum Genet. 2011;75(4):483-496.
25. Gnarra JR, Tory K, Weng Y, et al. Mutations of theVHL tumour suppressor gene in renal carcinoma.Nat Genet. 1994;7(1):85-90.
26. Young AC, Craven RA, Cohen D, et al. Analysisof VHL gene alterations and their relationship toclinical parameters in sporadic conventional renalcell carcinoma. Clin Cancer Res. 2009;15(24):7582-7592.
27. Ljubas Tomasic N, Piterkova L, Huff C, et al.Polycythemia due to Croatian homozygous VHL(571C.G:H191D) mutation has a differentphenotype than Chuvash polycythemia (VHL598C.T:R200W). Haematologica. 2013;98(4):560-567.
28. Prchal JT. Secondary polycythemia(erythrocytosis). In: Kaushansky KLM, Kipps TJ,Beutler E, Seligsohn U, Prchal JT, eds. WilliamsHematology. New York, NY: McGraw Hill; 2010:823-839
29. Hickey MM, Lam JC, Bezman NA, Rathmell WK,Simon MC. von Hippel-Lindau mutationin mice recapitulates Chuvash polycythemiavia hypoxia-inducible factor-2alphasignaling and splenic erythropoiesis.J Clin Invest. 2007;117(12):3879-3889.
30. Semenza GL. Hypoxia-inducible factors inphysiology and medicine. Cell. 2012;148(3):399-408.
3924 LANIKOVA et al BLOOD, 9 MAY 2013 x VOLUME 121, NUMBER 19
For personal use only. at UNIV OF UTAH on June 3, 2013. bloodjournal.hematologylibrary.orgFrom

560
ARTICLES
haematologica | 2013; 98(4)
Red Cell Disorders
Introduction
Von Hippel-Lindau (VHL) is a tumor-suppressor gene,mutations of which have long been recognized to predisposeto renal cancer, pheochromocytoma and other cancers.1
Notably, mutations clustering at different positions appear tofavor certain tumor types.2-4 VHL is a negative regulator ofhypoxia inducible transcription factors (HIF). Two mutationslocated at the 3' portion of the VHL coding region cause poly-cythemia but not VHL syndrome cancers. The most frequentcause of congenital polycythemia is homozygosity for thehypomorphic VHL 598C>T (R200W) mutation leading to areduced rate of ubiquitination of a subunits of HIF-1 and HIF-2, the principal mechanism underlying Chuvash poly-cythemia.5 This disease is endemic in the ChuvashAutonomous Republic of the Russia Federation6 and in theItalian island of Ischia,7 and is sporadic worldwide; it is asso-ciated with decreased survival of homozygotes partly due tocerebral vascular events and systemic thrombosis. Othercommon manifestations are varicose veins and vertebralhemangiomas.8 Haplotype analyses demonstrated that themutation likely arose from a single founder 14,000 – 62,000
years ago, indicating a survival advantage of heterozygotes,9
which may in part be due to protection from anemia.10 Inaddition, compound heterozygosity for R200W and otherVHLmutations has been reported in a few patients with con-genital polycythemia.11-14
HIF are master transcription factors that determine cellularresponses by oxygen-dependent destruction of a subunits.VHL is a substrate-recognition component of an E3 ubiquitin-protein ligase complex that, under normoxic conditions, ubiq-uitinates HIF1a and HIF2a and targets them for proteasomaldegradation.15 Disruption of the interaction between the a-subunits of HIF and VHL protein causes accumulation of HIFand altered transcription of downstream target genes includ-ing those for glucose transporter-1 (SLC2A1), vascularendothelial growth factor (VEGF), transferrin (TF) and ery-thropoietin (EPO).5
Eight years ago we described the first example of ahomozygous germ-line VHL mutation other than the VHLR200W mutation, i.e. the 571C>G (H191D) mutation in apolycythemic boy from a region in the south of Croatia calledDalmatia.16 This Croatian VHL mutation is positioned in thesame structural region as the Chuvash polycythemia muta-
©2013 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2012.070508*NLT and LP contributed equally to this manuscript.Manuscript received on May 23, 2012. Manuscript accepted on November 28, 2012.Correspondence: [email protected] or [email protected]
Mutations of VHL (a negative regulator of hypoxia-inducible factors) have position-dependent distinct cancer phe-notypes. Only two known inherited homozygous VHL mutations exist and they cause polycythemia: ChuvashR200W and Croatian H191D. We report a second polycythemic Croatian H191D homozygote distantly related tothe first propositus. Three generations of both families were genotyped for analysis of shared ancestry.Biochemical and molecular tests were performed to better define their phenotypes, with an emphasis on a com-parison with Chuvash polycythemia. The VHL H191D mutation did not segregate in the family defined by theknown common ancestors of the two subjects, suggesting a high prevalence in Croatians, but haplotype analysisindicated an undocumented common ancestor ~six generations ago as the founder of this mutation. We show thaterythropoietin levels in homozygous VHL H191D individuals are higher than in VHL R200W patients of similarages, and their native erythroid progenitors, unlike Chuvash R200W, are not hypersensitive to erythropoietin. Thisobservation contrasts with a report suggesting that polycythemia in VHL R200W and H191D homozygotes is dueto the loss of JAK2 regulation from VHL R200W and H191D binding to SOCS1. In conclusion, our studies furtherdefine the hematologic phenotype of VHL H191D and provide additional evidence for phenotypic heterogeneityassociated with the positional effects of VHL mutations.
The phenotype of polycythemia due to Croatian homozygous VHL (571C>G:H191D) mutation is different from that of Chuvash polycythemia (VHL 598C>T:R200W) Nikica Ljubas Tomasic,1* Lucie Piterkova,2,3* Chad Huff,4 Ernest Bilic,5 Donghoon Yoon,2 Galina Y. Miasnikova,6
Adelina I. Sergueeva,7 Xiaomei Niu,8 Sergei Nekhai,8 Victor Gordeuk,9 and Josef T. Prchal2
1Division of Neonatology, University Hospital Merkur, Zagreb, Croatia; 2Division of Hematology, University of Utah, SaltLake City, UT, USA; 3Department of Biology, Faculty of Medicine and Dentistry, Palacky University, Olomouc, CzechRepublic; 4Department of Human Genetics, University of Utah, Salt Lake City, UT, USA; 5Department of Pediatrics,Division of Hematology, University Hospital Center, Zagreb, Croatia; 6Chuvash Republic Clinical Hospital No. 1,Cheboksary, Russia; 7Cheboksary Children’s Hospital, Cheboksary, Russia; 8Center for Sickle Cell Disease andDepartment of Medicine, Howard University, Washington, DC, USA; and 9Sickle Cell Center and Department ofMedicine, University of Illinois at Chicago, Chicago, IL, USA
ABSTRACT

tion that leads to a modest partial loss of VHL activitysince the affected residue is distant from the functionalVHL domain.17A high level of erythropoietin, due to increased HIF, was
assumed to be a major cause of polycythemia in patientswith these VHL R200W and H191D mutations, which arehallmarks of secondary polycythemic disorders. However,Chuvash polycythemia R200W erythroid progenitors arealso hypersensitive to erythropoietin,5,18 a hallmark of pri-mary polycythemic disorders, by an as of yet unknownunderlying mechanism. Recently, Russell and colleagueshypothesized that the mutated VHL R200W and H191Dregions bind more avidly to suppressor of cytokine signal-ing 1 (SOCS1), a potent negative regulator of erythro-poiesis.19 They proposed that this abnormal associationbetween the VHL protein and SOCS1 hinders Janus kinase2 (JAK2) degradation leading to JAK2 up-regulation, whichcan potentially explain the erythroid hypersensitivity toerythropoietin observed in Chuvash polycythemia, andthey predicted that this erythroid hypersensitivity to ery-thropoietin would also be observed in the Croatian VHLH191D mutation.19In this study, we report another homozygous patient for
the VHL 571C>G (H191D) germ-line mutation, a 5-yearold Croatian girl from Herzegovina, a region located in thesouthern part of Bosnia contiguous to Dalmatia.Herzegovina is the counterpart to the Croatian region andis populated largely by people of Croatian ethnicity alongthe border. We set out to define the phenotype of thismutation in this and the previously reported homozy-gote16 with particular emphasis on a critical comparisonwith Chuvash polycythemia. We also pursued a hypothe-sis that a putative survival advantage of H191D heterozy-gotes may account for the possible high prevalence of het-erozygosity in Croatians and thus set out to determine itsapproximate origin in evolution by determining haplotypesharing among affected individuals.
Design and Methods
Blood samples from 23 persons were collected from two differ-ent families, including two propositi with polycythemia and 21relatives (Figure 1). Peripheral blood samples were collected intotubes containing EDTA and/or ACD. Written informed consentwas obtained from all participants. The Institutional Review Boardof the University of Utah approved the study.
Mutation analysis of the VHL geneGenomic DNA was isolated from granulocytes using the TRI
reagent (Molecular Research Center, Cincinnati, OH, USA). Forgenotyping, 100 ng of patients’ genomic DNA was used.Polymerase chain reactions (PCR) were performed using theHotStar Taq Master Mix Kit (QIAGEN, Germantown, MD, USA)and the following primers: VHL F agttgttggcaaagcctctt, VHL Rcaaaagctgagatgaaacagtg. As the MslI endonuclease abolishes arestriction site of the VHL 571C>G mutation, the PCR productwas then purified with a QIAquick PCR Purification Kit (QIA-GEN) and subjected to restriction with MslI enzyme (NewEngland Biolabs, Beverly, MA, USA) according to the manufactur-ers’ instructions. Cleavage products were evaluated on 2%agarose gels. PCR-direct sequencing was performed to confirm themutation screening, using a standard protocol and the same ampli-fication primers.
Analysis of recent shared ancestry The two individuals homozygous for VHL H191D were geno-
typed using the Illumina HumanOmni1-Quad BeadChip at theChildren's Hospital of Philadelphia, Center for Applied Genomics.Beagle 3.220 was used to phase and impute missing genotypes,with the phase two release of 30 HapMap CEU trios as a refer-ence.21 GERMLINE 1.4.122 inferred the locations and extents ofidentity by descent (IBD) segments (parameters err_het = 2,err_hom = 1, and min_m = 1cM, with marker positions given onthe HapMap r22 genetic map). ERSA 1.0 was applied to theGERMLINE output to test for evidence of recent shared ancestry.23
Phenotypes associated with Croatian and Chuvash VHL mutations
haematologica | 2013; 98(4) 561
Figure 1. Pedigree with genotyping. (A) Pedigree of the family available for study with extended family history. Each family is denoted by afamily number (F1, F2). The “p” code (p01, p02 and so on) indicates individuals from whom DNA samples have been obtained. The arrowindicates a new polycythemic patient. The VHL mutation of the paternal side of F1 must have been inherited from the husband of p01. Thehusband of p01 was not related to F2 according to the pedigree and the mutation did not segregate among the known relatives.Heterozygote individuals denoted by an asterisk as well as the husband of p01 presumably shared the common ancestor, the founder ofthe mutation. (B) Examples of sequences of the third exon of the VHL gene in an unaffected subject (p01), in a heterozygote subject (p02),and in a homozygote (p18) patient.
A B

In vitro assay of the sensitivity of erythroidprogenitors to erythropoietinIn vitro sensitivity of erythroid progenitors to erythropoietin was
determined on mononuclear cells isolated from the peripheralblood using Histopaque (Sigma-Aldrich, St. Louis, MO, USA) den-sity gradient centrifugation and plating (2.3×105/mL) on methylcel-lulose media (MethoCult® H4531; StemCell Technologies,Vancouver, BC, Canada) without addition of erythropoietin orwith addition of various concentrations of erythropoietin(StemCell Technologies), ranging from 0.015 to 3.0 U/mL. Cell cul-tures were maintained in a humidified atmosphere of 5% CO2 at37°C for 14 days. Erythroid burst-forming unit colonies (BFU-E)were scored by standard morphological criteria. The assay wascarried out on erythroid progenitors from one homozygous andthree heterozygous VHL H191D individuals, two wild-type rela-tives and a polycythemia vera patient as a positive control. Theprotocol and interpretation of these assays were identical to thoseused for homozygous and heterozygous native progenitors withthe Chuvash polycythemia VHL R200W mutation.5
In vitro expansion of human erythroid progenitorsin liquid cultureThe progenitor cells were expanded from the mononuclear
cell population using our published protocol.24 Briefly, 1x106
cells/mL were cultured in StemSpan™ Serum-Free ExpansionMedium (StemCell Technologies) containing different cytokinecocktails from day 1-7 (100 ng/mL of fetal liver tyrosine kinase 3ligand, 100 ng/mL of thrombopoietin, and 100 ng/mL of stemcell factor), day 8-14 (50 ng/mL of stem cell factor, 50 ng/mL ofinsulin-like growth factor-1, and 3 U/mL of erythropoietin), andday 15-21 (50 ng/mL of insulin-like growth factor-1, and 3 U/mLof erythropoietin). All cytokines were a kind gift from Amgen(Thousand Oaks, CA, USA).
Real-time polymerase chain reaction assayTotal RNA was isolated from granulocytes using TRI reagent
solution (Molecular Research Center, Cincinnati, OH, USA) andthen treated with DNA-free™ DNase Treatment & RemovalReagents (Ambion, Life Technologies, NY, USA) to remove anycontaminating DNA. Five hundred nanograms of DNA-free RNAwere reverse-transcribed using the SuperScript® VILO™ cDNASynthesis Kit (Invitrogen, Life Technologies, NY, USA) accordingto the manufacturer’s instruction protocol. Quantitative PCR wereperformed with specific TaqMan® Gene Expression probes(Applied Biosystems, Carlsbad, CA, USA) for the following genes:ADM (Hs00181605), TFRC (Hs00951083), NDRG1 (Hs00608387),PDK1 (Hs00176853), SLC2A1 (Hs00892681), VEGF (Hs00900055),BNIP3 (Hs00969291), BNIP3L (Hs00188949), and HK1(Hs00175976). All samples were assayed in triplicate. Data werenormalized to HPRT (4333768F) and GAPDH (4333764F) referencegenes. The statistical significance of relative expression changes oftarget mRNA levels normalized to a reference genes was analyzedby the pair-wise fixed reallocation randomization test using REST©
2009 software.25
Biochemical studiesThe complete blood count was performed by an automated
analyzer (Sysmex XT 2000i, Sysmex Corporation, Kobe, Hyogo,Japan). The concentration of erythropoietin in the serum wasdetermined by an enzyme-linked immunosorbent assay (R&DSystems, Minneapolis, MN, USA).
Statistical analysis Analyses were performed with Stata 10.1 (StatCorp., College
Station, TX, USA).
Results
Patients’ characteristicsThe proposita is a 5-year old girl who was referred to
the University Hospital of Zagreb due to failure to thriveat the age of 2 years. Routine blood analysis at the time ofthe current study revealed an erythrocyte count of8.42x1012/L and hemoglobin concentration of 14.5 g/dL.She was iron deficient (ferritin 6.3 mmol/L) and her ery-thropoietin was increased (202 mIU/mL; normal range, 4-28). Her weight and height were below the 5th percentilefor age and gender and psychomotor development wasdelayed, but she was otherwise normal. She experiencesheadaches about twice weekly, on average. Besides ade-quate hydration and daily acetylsalicylic acid (1 mg/kg),she receives no other treatment. Her parents were born inthe same part of the country and deny being related. The previously reported homozygous patient was diag-
nosed with polycythemia and elevated erythropoietin atthe age of 12 years. He is now 26 years old and has occa-sional headaches and malaise, and tires easily. He is phle-botomized once or twice a month depending on symp-toms and hematologic findings. He has been iron deficientbecause of the regular phlebotomies to reduce hemoglo-bin concentration.We found no individuals among the heterozygotes who
had evidence or a history of polycythemia. Neitherhomozygote had evidence of varicose veins which arecommon in young Chuvash polycythemia patients andvirtually invariable after the age of 18 years.8
Analysis of the VHL H191D mutation and mapping of the VHL locusAmong 23 members (12 female, 11 male) of the families
of the two propositi, we found 10 heterozygotes (6female, 4 male; Figure 1) for the VHL H191D mutation.The father of the proposita is distantly related to the firstVHL H191D homozygote polycythemic subject but geno-typing revealed that the VHL mutation in family F1 musthave come from the husband of p01 who was not relatedto family F2 (Figure 1). Thus, the VHL H191D mutationdoes not segregate among the known relatives betweenthese two families, and no evidence suggests that themutation originated from one of two known commonancestors depicted in Figure 1A.We analyzed the two homozygotes (p12 and p18) by
high-density genotyping to examine their relatedness andthe VHL haplotype. We detected evidence of a significantrelationship between the two individuals (P=4x10-18), witha maximum likelihood estimate of 8th degree relatives(95% CI 6th degree - 11th degree)23, exactly matching theknown pedigree (Figure 1). These individuals share a 16cM haploid segment on chromosome 11 and a 24 cM hap-loid IBD segment on chromosome 3 (identical copies of agene segregating from a common ancestor within thedefined pedigree, Figure 2). Within the segment on chro-mosome 3, a 15.6 cM autozygous segment is present inthe female (p18), and a 1.6 cM autozygous segment ispresent in the male (p12). Both autozygous segments con-tain the VHLmutation, and the smaller segment is diploidIBD (Figure 2). Two other large autozygous segments arepresent in the male (15.5 cM on chromosome 15 and 10cM on chromosome 4). The presence of autozygosity atthe VHL mutation and elsewhere in the genome is astrong indication that all four of the parents of p12 and
N.L. Tomasic et al.
562 haematologica | 2013; 98(4)
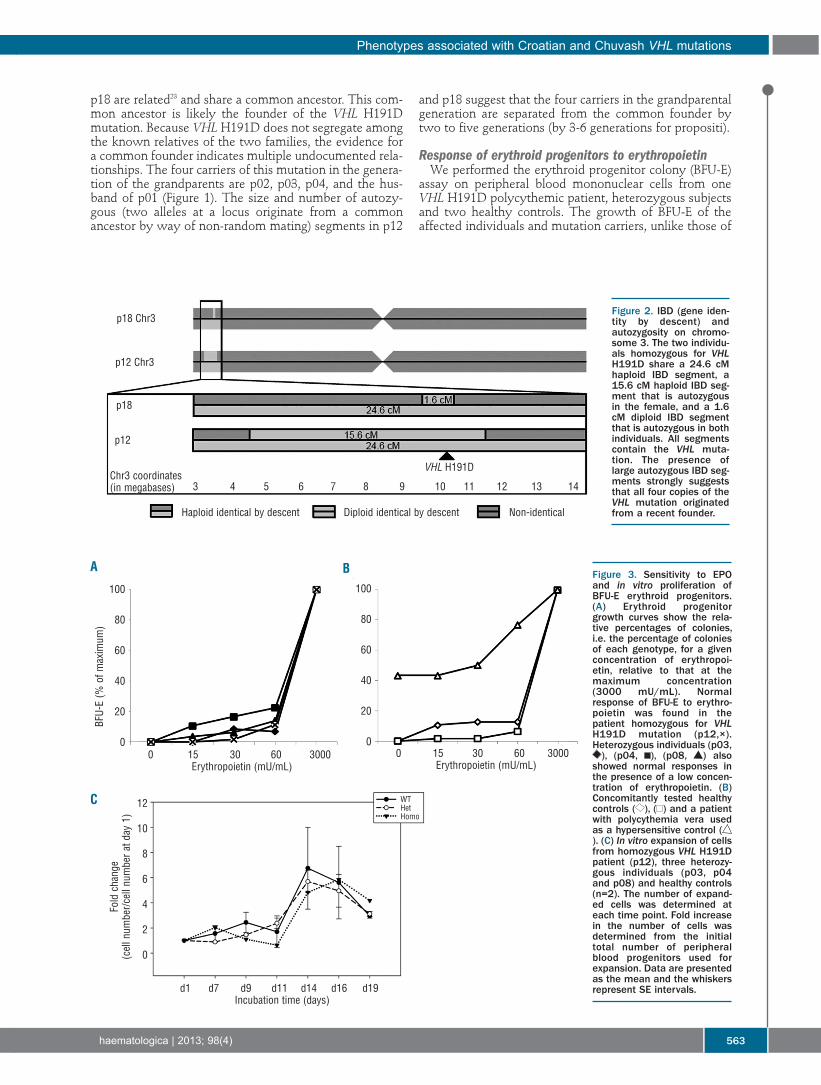
p18 are related23 and share a common ancestor. This com-mon ancestor is likely the founder of the VHL H191Dmutation. Because VHL H191D does not segregate amongthe known relatives of the two families, the evidence fora common founder indicates multiple undocumented rela-tionships. The four carriers of this mutation in the genera-tion of the grandparents are p02, p03, p04, and the hus-band of p01 (Figure 1). The size and number of autozy-gous (two alleles at a locus originate from a commonancestor by way of non-random mating) segments in p12
and p18 suggest that the four carriers in the grandparentalgeneration are separated from the common founder bytwo to five generations (by 3-6 generations for propositi).
Response of erythroid progenitors to erythropoietin We performed the erythroid progenitor colony (BFU-E)
assay on peripheral blood mononuclear cells from oneVHL H191D polycythemic patient, heterozygous subjectsand two healthy controls. The growth of BFU-E of theaffected individuals and mutation carriers, unlike those of
Phenotypes associated with Croatian and Chuvash VHL mutations
haematologica | 2013; 98(4) 563
Figure 2. IBD (gene iden-tity by descent) andautozygosity on chromo-some 3. The two individu-als homozygous for VHLH191D share a 24.6 cMhaploid IBD segment, a15.6 cM haploid IBD seg-ment that is autozygousin the female, and a 1.6cM diploid IBD segmentthat is autozygous in bothindividuals. All segmentscontain the VHL muta-tion. The presence oflarge autozygous IBD seg-ments strongly suggeststhat all four copies of theVHL mutation originatedfrom a recent founder.
Figure 3. Sensitivity to EPOand in vitro proliferation ofBFU-E erythroid progenitors.(A) Erythroid progenitorgrowth curves show the rela-tive percentages of colonies,i.e. the percentage of coloniesof each genotype, for a givenconcentration of erythropoi-etin, relative to that at themaximum concentration(3000 mU/mL). Normalresponse of BFU-E to erythro-poietin was found in thepatient homozygous for VHLH191D mutation (p12,×).Heterozygous individuals (p03,), (p04, ), (p08, ) also
showed normal responses inthe presence of a low concen-tration of erythropoietin. (B)Concomitantly tested healthycontrols ( ), ( ) and a patientwith polycythemia vera usedas a hypersensitive control (). (C) In vitro expansion of cellsfrom homozygous VHL H191Dpatient (p12), three heterozy-gous individuals (p03, p04and p08) and healthy controls(n=2). The number of expand-ed cells was determined ateach time point. Fold increasein the number of cells wasdetermined from the initialtotal number of peripheralblood progenitors used forexpansion. Data are presentedas the mean and the whiskersrepresent SE intervals.
A B
C
100
80
60
40
20
0
100
80
60
40
20
0
BFU-
E (%
of m
axim
um)
Fold cha
nge
(cell n
umbe
r/cell n
umbe
r at d
ay 1)
12
10
8
6
4
2
0
0 15 30 60 3000Erythropoietin (mU/mL)
Chr3 coordinates(in megabases)
p12
p18
p12 Chr3
p18 Chr3
VHL H191D
3 4 5 6 7 8 9 10 11 12 13 14
Haploid identical by descent Diploid identical by descent Non-identical
d1 d7 d9 d11 d14 d16 d19Incubation time (days)
0 15 30 60 3000Erythropoietin (mU/mL)
WTHetHomo
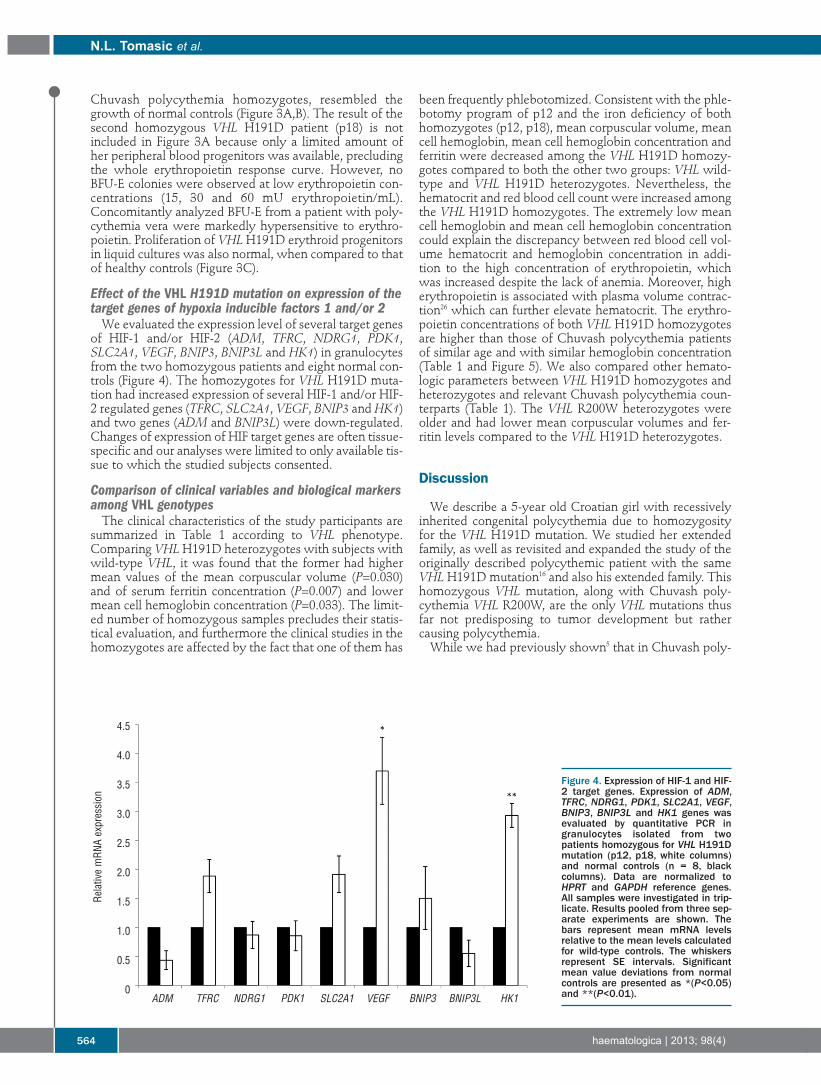
Chuvash polycythemia homozygotes, resembled thegrowth of normal controls (Figure 3A,B). The result of thesecond homozygous VHL H191D patient (p18) is notincluded in Figure 3A because only a limited amount ofher peripheral blood progenitors was available, precludingthe whole erythropoietin response curve. However, noBFU-E colonies were observed at low erythropoietin con-centrations (15, 30 and 60 mU erythropoietin/mL).Concomitantly analyzed BFU-E from a patient with poly-cythemia vera were markedly hypersensitive to erythro-poietin. Proliferation of VHLH191D erythroid progenitorsin liquid cultures was also normal, when compared to thatof healthy controls (Figure 3C).
Effect of the VHL H191D mutation on expression of thetarget genes of hypoxia inducible factors 1 and/or 2We evaluated the expression level of several target genes
of HIF-1 and/or HIF-2 (ADM, TFRC, NDRG1, PDK1,SLC2A1, VEGF, BNIP3, BNIP3L and HK1) in granulocytesfrom the two homozygous patients and eight normal con-trols (Figure 4). The homozygotes for VHL H191D muta-tion had increased expression of several HIF-1 and/or HIF-2 regulated genes (TFRC, SLC2A1, VEGF, BNIP3 and HK1)and two genes (ADM and BNIP3L) were down-regulated.Changes of expression of HIF target genes are often tissue-specific and our analyses were limited to only available tis-sue to which the studied subjects consented.
Comparison of clinical variables and biological markersamong VHL genotypesThe clinical characteristics of the study participants are
summarized in Table 1 according to VHL phenotype.Comparing VHLH191D heterozygotes with subjects withwild-type VHL, it was found that the former had highermean values of the mean corpuscular volume (P=0.030)and of serum ferritin concentration (P=0.007) and lowermean cell hemoglobin concentration (P=0.033). The limit-ed number of homozygous samples precludes their statis-tical evaluation, and furthermore the clinical studies in thehomozygotes are affected by the fact that one of them has
been frequently phlebotomized. Consistent with the phle-botomy program of p12 and the iron deficiency of bothhomozygotes (p12, p18), mean corpuscular volume, meancell hemoglobin, mean cell hemoglobin concentration andferritin were decreased among the VHL H191D homozy-gotes compared to both the other two groups: VHL wild-type and VHL H191D heterozygotes. Nevertheless, thehematocrit and red blood cell count were increased amongthe VHL H191D homozygotes. The extremely low meancell hemoglobin and mean cell hemoglobin concentrationcould explain the discrepancy between red blood cell vol-ume hematocrit and hemoglobin concentration in addi-tion to the high concentration of erythropoietin, whichwas increased despite the lack of anemia. Moreover, higherythropoietin is associated with plasma volume contrac-tion26 which can further elevate hematocrit. The erythro-poietin concentrations of both VHL H191D homozygotesare higher than those of Chuvash polycythemia patientsof similar age and with similar hemoglobin concentration(Table 1 and Figure 5). We also compared other hemato-logic parameters between VHL H191D homozygotes andheterozygotes and relevant Chuvash polycythemia coun-terparts (Table 1). The VHL R200W heterozygotes wereolder and had lower mean corpuscular volumes and fer-ritin levels compared to the VHL H191D heterozygotes.
Discussion
We describe a 5-year old Croatian girl with recessivelyinherited congenital polycythemia due to homozygosityfor the VHL H191D mutation. We studied her extendedfamily, as well as revisited and expanded the study of theoriginally described polycythemic patient with the sameVHL H191D mutation16 and also his extended family. Thishomozygous VHL mutation, along with Chuvash poly-cythemia VHL R200W, are the only VHL mutations thusfar not predisposing to tumor development but rathercausing polycythemia. While we had previously shown5 that in Chuvash poly-
N.L. Tomasic et al.
564 haematologica | 2013; 98(4)
Figure 4. Expression of HIF-1 and HIF-2 target genes. Expression of ADM,TFRC, NDRG1, PDK1, SLC2A1, VEGF,BNIP3, BNIP3L and HK1 genes wasevaluated by quantitative PCR ingranulocytes isolated from twopatients homozygous for VHL H191Dmutation (p12, p18, white columns)and normal controls (n = 8, blackcolumns). Data are normalized toHPRT and GAPDH reference genes.All samples were investigated in trip-licate. Results pooled from three sep-arate experiments are shown. Thebars represent mean mRNA levelsrelative to the mean levels calculatedfor wild-type controls. The whiskersrepresent SE intervals. Significantmean value deviations from normalcontrols are presented as *(P<0.05)and **(P<0.01).ADM TFRC NDRG1 PDK1 SLC2A1 VEGF BNIP3 BNIP3L HK1
4.5
4.0
3.5
3.0
2.5
2.0
1.5
1.0
0.5
0
Relativ
e mRN
A ex
pres
sion
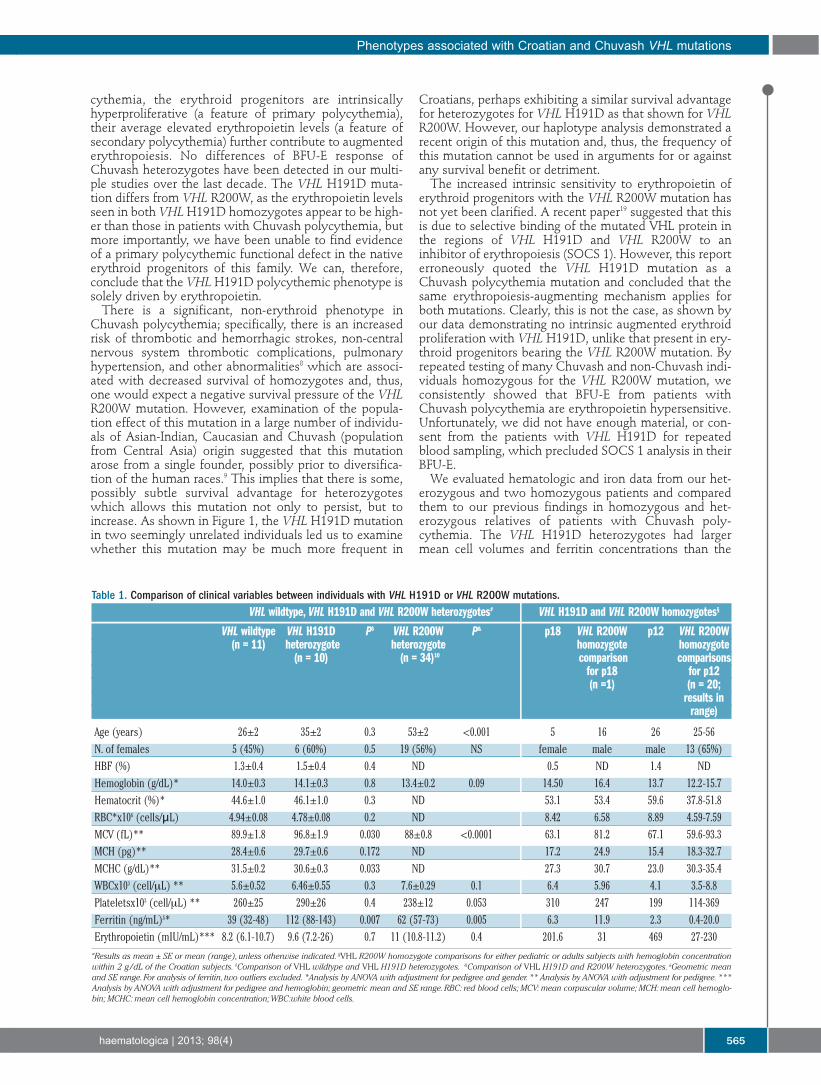
cythemia, the erythroid progenitors are intrinsicallyhyperproliferative (a feature of primary polycythemia),their average elevated erythropoietin levels (a feature ofsecondary polycythemia) further contribute to augmentederythropoiesis. No differences of BFU-E response ofChuvash heterozygotes have been detected in our multi-ple studies over the last decade. The VHL H191D muta-tion differs from VHL R200W, as the erythropoietin levelsseen in both VHLH191D homozygotes appear to be high-er than those in patients with Chuvash polycythemia, butmore importantly, we have been unable to find evidenceof a primary polycythemic functional defect in the nativeerythroid progenitors of this family. We can, therefore,conclude that the VHLH191D polycythemic phenotype issolely driven by erythropoietin. There is a significant, non-erythroid phenotype in
Chuvash polycythemia; specifically, there is an increasedrisk of thrombotic and hemorrhagic strokes, non-centralnervous system thrombotic complications, pulmonaryhypertension, and other abnormalities8 which are associ-ated with decreased survival of homozygotes and, thus,one would expect a negative survival pressure of the VHLR200W mutation. However, examination of the popula-tion effect of this mutation in a large number of individu-als of Asian-Indian, Caucasian and Chuvash (populationfrom Central Asia) origin suggested that this mutationarose from a single founder, possibly prior to diversifica-tion of the human races.9 This implies that there is some,possibly subtle survival advantage for heterozygoteswhich allows this mutation not only to persist, but toincrease. As shown in Figure 1, the VHL H191D mutationin two seemingly unrelated individuals led us to examinewhether this mutation may be much more frequent in
Croatians, perhaps exhibiting a similar survival advantagefor heterozygotes for VHL H191D as that shown for VHLR200W. However, our haplotype analysis demonstrated arecent origin of this mutation and, thus, the frequency ofthis mutation cannot be used in arguments for or againstany survival benefit or detriment.The increased intrinsic sensitivity to erythropoietin of
erythroid progenitors with the VHL R200W mutation hasnot yet been clarified. A recent paper19 suggested that thisis due to selective binding of the mutated VHL protein inthe regions of VHL H191D and VHL R200W to aninhibitor of erythropoiesis (SOCS 1). However, this reporterroneously quoted the VHL H191D mutation as aChuvash polycythemia mutation and concluded that thesame erythropoiesis-augmenting mechanism applies forboth mutations. Clearly, this is not the case, as shown byour data demonstrating no intrinsic augmented erythroidproliferation with VHL H191D, unlike that present in ery-throid progenitors bearing the VHL R200W mutation. Byrepeated testing of many Chuvash and non-Chuvash indi-viduals homozygous for the VHL R200W mutation, weconsistently showed that BFU-E from patients withChuvash polycythemia are erythropoietin hypersensitive.Unfortunately, we did not have enough material, or con-sent from the patients with VHL H191D for repeatedblood sampling, which precluded SOCS 1 analysis in theirBFU-E.We evaluated hematologic and iron data from our het-
erozygous and two homozygous patients and comparedthem to our previous findings in homozygous and het-erozygous relatives of patients with Chuvash poly-cythemia. The VHL H191D heterozygotes had largermean cell volumes and ferritin concentrations than the
Phenotypes associated with Croatian and Chuvash VHL mutations
haematologica | 2013; 98(4) 565
Table 1. Comparison of clinical variables between individuals with VHL H191D or VHL R200W mutations.VHL wildtype, VHL H191D and VHL R200W heterozygotes# VHL H191D and VHL R200W homozygotes§
VHL wildtype VHL H191D P$ VHL R200W P& p18 VHL R200W p12 VHL R200W(n = 11) heterozygote heterozygote homozygote homozygote
(n = 10) (n = 34)10 comparison comparisonsfor p18 for p12(n =1) (n = 20;
results in range)
Age (years) 26±2 35±2 0.3 53±2 <0.001 5 16 26 25-56N. of females 5 (45%) 6 (60%) 0.5 19 (56%) NS female male male 13 (65%)HBF (%) 1.3±0.4 1.5±0.4 0.4 ND 0.5 ND 1.4 NDHemoglobin (g/dL)* 14.0±0.3 14.1±0.3 0.8 13.4±0.2 0.09 14.50 16.4 13.7 12.2-15.7Hematocrit (%)* 44.6±1.0 46.1±1.0 0.3 ND 53.1 53.4 59.6 37.8-51.8RBC*x106 (cells/μL) 4.94±0.08 4.78±0.08 0.2 ND 8.42 6.58 8.89 4.59-7.59MCV (fL)** 89.9±1.8 96.8±1.9 0.030 88±0.8 <0.0001 63.1 81.2 67.1 59.6-93.3MCH (pg)** 28.4±0.6 29.7±0.6 0.172 ND 17.2 24.9 15.4 18.3-32.7MCHC (g/dL)** 31.5±0.2 30.6±0.3 0.033 ND 27.3 30.7 23.0 30.3-35.4WBCx103 (cell/mL) ** 5.6±0.52 6.46±0.55 0.3 7.6±0.29 0.1 6.4 5.96 4.1 3.5-8.8Plateletsx103 (cell/mL) ** 260±25 290±26 0.4 238±12 0.053 310 247 199 114-369Ferritin (ng/mL)£* 39 (32-48) 112 (88-143) 0.007 62 (57-73) 0.005 6.3 11.9 2.3 0.4-20.0Erythropoietin (mIU/mL)*** 8.2 (6.1-10.7) 9.6 (7.2-26) 0.7 11 (10.8-11.2) 0.4 201.6 31 469 27-230#Results as mean ± SE or mean (range), unless otherwise indicated. §VHL R200W homozygote comparisons for either pediatric or adults subjects with hemoglobin concentrationwithin 2 g/dL of the Croatian subjects. $Comparison of VHL wildtype and VHL H191D heterozygotes. &Comparison of VHL H191D and R200W heterozygotes. £Geometric meanand SE range. For analysis of ferritin, two outliers excluded. *Analysis by ANOVA with adjustment for pedigree and gender. ** Analysis by ANOVA with adjustment for pedigree. ***Analysis by ANOVA with adjustment for pedigree and hemoglobin; geometric mean and SE range. RBC: red blood cells; MCV: mean corpuscular volume; MCH: mean cell hemoglo-bin; MCHC: mean cell hemoglobin concentration; WBC:white blood cells.
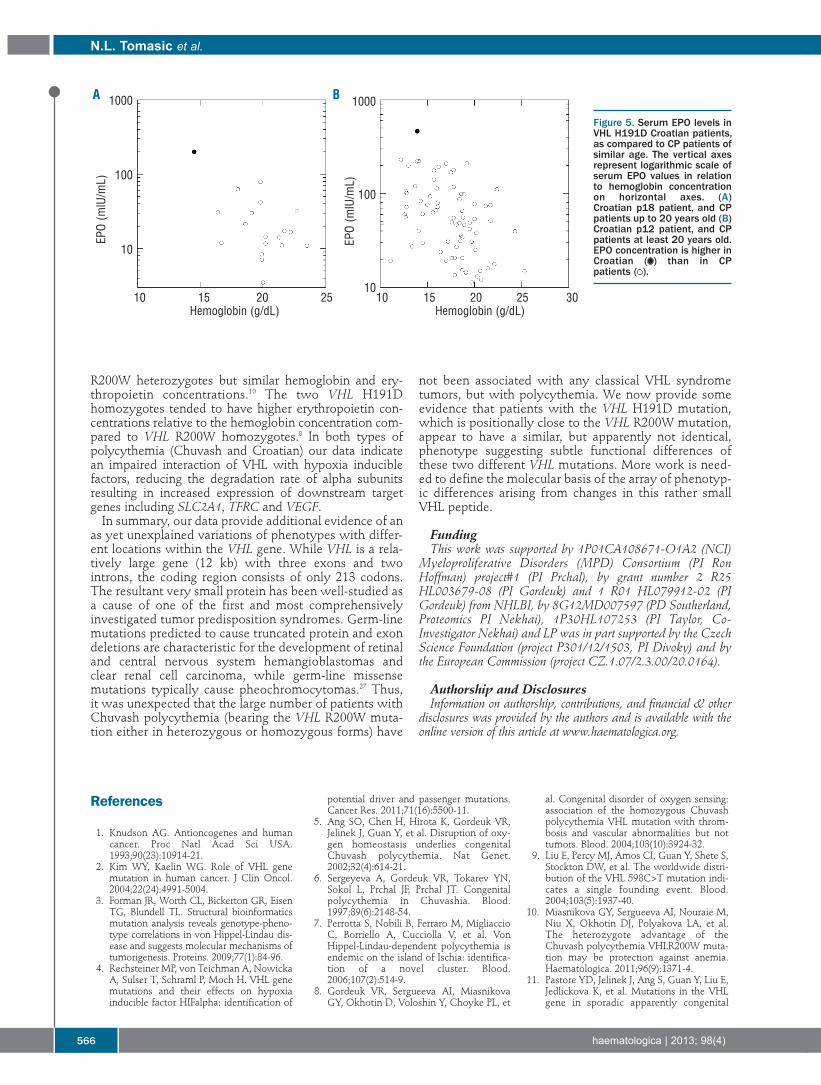
R200W heterozygotes but similar hemoglobin and ery-thropoietin concentrations.10 The two VHL H191Dhomozygotes tended to have higher erythropoietin con-centrations relative to the hemoglobin concentration com-pared to VHL R200W homozygotes.8 In both types ofpolycythemia (Chuvash and Croatian) our data indicatean impaired interaction of VHL with hypoxia induciblefactors, reducing the degradation rate of alpha subunitsresulting in increased expression of downstream targetgenes including SLC2A1, TFRC and VEGF.In summary, our data provide additional evidence of an
as yet unexplained variations of phenotypes with differ-ent locations within the VHL gene. While VHL is a rela-tively large gene (12 kb) with three exons and twointrons, the coding region consists of only 213 codons.The resultant very small protein has been well-studied asa cause of one of the first and most comprehensivelyinvestigated tumor predisposition syndromes. Germ-linemutations predicted to cause truncated protein and exondeletions are characteristic for the development of retinaland central nervous system hemangioblastomas andclear renal cell carcinoma, while germ-line missensemutations typically cause pheochromocytomas.27 Thus,it was unexpected that the large number of patients withChuvash polycythemia (bearing the VHL R200W muta-tion either in heterozygous or homozygous forms) have
not been associated with any classical VHL syndrometumors, but with polycythemia. We now provide someevidence that patients with the VHL H191D mutation,which is positionally close to the VHL R200W mutation,appear to have a similar, but apparently not identical,phenotype suggesting subtle functional differences ofthese two different VHL mutations. More work is need-ed to define the molecular basis of the array of phenotyp-ic differences arising from changes in this rather smallVHL peptide.
Funding This work was supported by 1P01CA108671-O1A2 (NCI)
Myeloproliferative Disorders (MPD) Consortium (PI RonHoffman) project#1 (PI Prchal), by grant number 2 R25HL003679-08 (PI Gordeuk) and 1 R01 HL079912-02 (PIGordeuk) from NHLBI, by 8G12MD007597 (PD Southerland,Proteomics PI Nekhai), 1P30HL107253 (PI Taylor, Co-Investigator Nekhai) and LP was in part supported by the CzechScience Foundation (project P301/12/1503, PI Divoky) and bythe European Commission (project CZ.1.07/2.3.00/20.0164).
Authorship and DisclosuresInformation on authorship, contributions, and financial & other
disclosures was provided by the authors and is available with theonline version of this article at www.haematologica.org.
N.L. Tomasic et al.
566 haematologica | 2013; 98(4)
Figure 5. Serum EPO levels inVHL H191D Croatian patients,as compared to CP patients ofsimilar age. The vertical axesrepresent logarithmic scale ofserum EPO values in relationto hemoglobin concentrationon horizontal axes. (A)Croatian p18 patient, and CPpatients up to 20 years old (B)Croatian p12 patient, and CPpatients at least 20 years old.EPO concentration is higher inCroatian ( ) than in CPpatients ( ).
A B1000
100
10
1000
100
1010 15 20 25
Hemoglobin (g/dL)10 15 20 25 30
Hemoglobin (g/dL)
EPO (m
lU/m
L)
EPO (m
lU/m
L)
References
1. Knudson AG. Antioncogenes and humancancer. Proc Natl Acad Sci USA.1993;90(23):10914-21.
2. Kim WY, Kaelin WG. Role of VHL genemutation in human cancer. J Clin Oncol.2004;22(24):4991-5004.
3. Forman JR, Worth CL, Bickerton GR, EisenTG, Blundell TL. Structural bioinformaticsmutation analysis reveals genotype-pheno-type correlations in von Hippel-Lindau dis-ease and suggests molecular mechanisms oftumorigenesis. Proteins. 2009;77(1):84-96.
4. Rechsteiner MP, von Teichman A, NowickaA, Sulser T, Schraml P, Moch H. VHL genemutations and their effects on hypoxiainducible factor HIFalpha: identification of
potential driver and passenger mutations.Cancer Res. 2011;71(16):5500-11.
5. Ang SO, Chen H, Hirota K, Gordeuk VR,Jelinek J, Guan Y, et al. Disruption of oxy-gen homeostasis underlies congenitalChuvash polycythemia. Nat Genet.2002;32(4):614-21.
6. Sergeyeva A, Gordeuk VR, Tokarev YN,Sokol L, Prchal JF, Prchal JT. Congenitalpolycythemia in Chuvashia. Blood.1997;89(6):2148-54.
7. Perrotta S, Nobili B, Ferraro M, MigliaccioC, Borriello A, Cucciolla V, et al. VonHippel-Lindau-dependent polycythemia isendemic on the island of Ischia: identifica-tion of a novel cluster. Blood.2006;107(2):514-9.
8. Gordeuk VR, Sergueeva AI, MiasnikovaGY, Okhotin D, Voloshin Y, Choyke PL, et
al. Congenital disorder of oxygen sensing:association of the homozygous Chuvashpolycythemia VHL mutation with throm-bosis and vascular abnormalities but nottumors. Blood. 2004;103(10):3924-32.
9. Liu E, Percy MJ, Amos CI, Guan Y, Shete S,Stockton DW, et al. The worldwide distri-bution of the VHL 598C>T mutation indi-cates a single founding event. Blood.2004;103(5):1937-40.
10. Miasnikova GY, Sergueeva AI, Nouraie M,Niu X, Okhotin DJ, Polyakova LA, et al.The heterozygote advantage of theChuvash polycythemia VHLR200W muta-tion may be protection against anemia.Haematologica. 2011;96(9):1371-4.
11. Pastore YD, Jelinek J, Ang S, Guan Y, Liu E,Jedlickova K, et al. Mutations in the VHLgene in sporadic apparently congenital

polycythemia. Blood. 2003;101(4):1591-5.12. Cario H, Schwarz K, Jorch N, Kyank U,
Petrides PE, Schneider DT, et al. Mutationsin the von Hippel-Lindau (VHL) tumor sup-pressor gene and VHL-haplotype analysisin patients with presumable congenital ery-throcytosis. Haematologica. 2005;90(1):19-24.
13. Percy MJ, Beard ME, Carter C, Thein SL.Erythrocytosis and the Chuvash vonHippel-Lindau mutation. Br J Haematol.2003;123(2):371-2.
14. Bento MC, Chang KT, Guan Y, Liu E, CaldasG, Gatti RA, et al. Congenital polycythemiawith homozygous and heterozygous muta-tions of von Hippel-Lindau gene: five newCaucasian patients. Haematologica. 2005;90(1):128-9.
15. Safran M, Kaelin WG Jr. HIF hydroxylationand the mammalian oxygen-sensing path-way. J Clin Invest. 2003;111(6):779-83.
16. Pastore Y, Jedlickova K, Guan Y, Liu E,Fahner J, Hasle H, et al. Mutations of vonHippel-Lindau tumor-suppressor gene andcongenital polycythemia. Am J HumGenet. 2003;73(2):412-9.
17. Lee FS, Percy MJ. The HIF pathway anderythrocytosis. Annu Rev Pathol.2011;6:165-92.
18. Hickey MM, Lam JC, Bezman NA,Rathmell WK, Simon MC. von Hippel-Lindau mutation in mice recapitulatesChuvash polycythemia via hypoxia-inducible factor-2alpha signaling andsplenic erythropoiesis. J Clin Invest.2007;117(12):3879-89.
19. Russell RC, Sufan RI, Zhou B, Heir P, BundaS, Sybingco SS, et al. Loss of JAK2 regula-tion via a heterodimeric VHL-SOCS1 E3ubiquitin ligase underlies Chuvash poly-cythemia. Nat Med. 2011;17(7):845-53.
20. Browning SR, Browning BL. High-resolu-tion detection of identity by descent inunrelated individuals. Am J Hum Genet.2010;86(4):526-39.
21. Frazer KA, Ballinger DG, Cox DR, HindsDA, Stuve LL, Gibbs RA, et al. A secondgeneration human haplotype map of over3.1 million SNPs. Nature. 2007;449(7164):851-61.
22. Gusev A, Lowe JK, Stoffel M, Daly MJ,Altshuler D, Breslow JL, et al. Whole popula-
tion, genome-wide mapping of hidden relat-edness. Genome Res. 2009;19(2):318-26.
23. Huff CD, Witherspoon DJ, Simonson TS,Xing J, Watkins WS, Zhang Y, et al.Maximum-likelihood estimation of recentshared ancestry (ERSA). Genome Res.2011;21(5):768-74.
24. Gaikwad A, Nussenzveig R, Liu E,Gottshalk S, Chang K, Prchal JT. In vitroexpansion of erythroid progenitors frompolycythemia vera patients leads todecrease in JAK2 V617F allele. ExpHematol. 2007;35(4):587-95.
25. Pfaffl MW, Horgan GW, Dempfle L.Relative expression software tool (REST)for group-wise comparison and statisticalanalysis of relative expression results inreal-time PCR. Nucleic Acids Res. 2002;30(9):e36.
26. Spivak JL GP, Ludwig H. Anemia manage-ment in oncology and hematology.Oncologist. 2009;14 (Suppl 1):43-56.
27. Maher ER, Neumann HP, Richard S. vonHippel-Lindau disease: a clinical and scien-tific review. Eur J Hum Genet. 2011;19(6):617-23.
Phenotypes associated with Croatian and Chuvash VHL mutations
haematologica | 2013; 98(4) 567
















![Von Hippel Lindau Disease [VHL]: Magnetic Resonance ... · Kata kunci: penyakit Von Hippel Lindau, hemangioblastoma, karsinoma sel ginjal, kista ginjal. ABSTRACT In this case, we](https://img.pdfslide.net/doc/110x75/5e56f6c31708e23e51691672/von-hippel-lindau-disease-vhl-magnetic-resonance-kata-kunci-penyakit-von.jpg)










![Von Hippel Lindau Disease [VHL]: Magnetic Resonance Imaging](https://img.pdfslide.net/doc/110x75/620636d38c2f7b17300580b0/von-hippel-lindau-disease-vhl-magnetic-resonance-imaging.jpg)

