Embed Size (px)
Citation preview

NOVEL TARGETS WITHIN THE HEPATITIS C VIRUS NONSTRUCTURAL PROTEIN NS4B AND THEIR INHIBITION
USING DISTINCT CLASSES OF SMALL MOLECULES
A DISSERTATION SUBMITTED TO THE DEPARTMENT OF MICROBIOLOGY AND IMMUNOLOGY
AND THE COMMITTEE ON GRADUATE STUDIES OF STANFORD UNIVERSITY
IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY
Paul David Bryson December 2009

http://creativecommons.org/licenses/by-nc/3.0/us/
This dissertation is online at: http://purl.stanford.edu/bs187wm8852
© 2010 by Paul David Bryson. All Rights Reserved.
Re-distributed by Stanford University under license with the author.
This work is licensed under a Creative Commons Attribution-Noncommercial 3.0 United States License.
ii

I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Jeffrey Glenn, Primary Adviser
I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Harry Greenberg
I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Shoshana Levy
I certify that I have read this dissertation and that, in my opinion, it is fully adequatein scope and quality as a dissertation for the degree of Doctor of Philosophy.
Peter Sarnow
Approved for the Stanford University Committee on Graduate Studies.
Patricia J. Gumport, Vice Provost Graduate Education
This signature page was generated electronically upon submission of this dissertation in electronic format. An original signed hard copy of the signature page is on file inUniversity Archives.
iii

iv
Abstract
Hepatitis C Virus (HCV) is the causative agent of significant liver disease,
including cirrhosis and hepatocellular carcinoma. This virus infects greater than 2% of
the world’s population, and treatment options for these patients are limited; thus, this
virus represents a major public health problem. In this thesis, we aim to enhance our
ability to solve this problem by seeking to unravel the role that NS4B, one of the HCV
nonstructural proteins, plays in the viral life cycle. In focusing on this viral protein, we
not only achieve a better understanding of HCV’s biology, but we also identify
multiple small molecules that may directly lead to better treatment options for those
afflicted with this virus.
Drawing on similarities to other plus-sense RNA viruses, we identify an RNA
binding activity in NS4B, demonstrate that this activity is specific for the 3’ terminus
of the HCV genome template, and characterize the domains of NS4B that are
responsible for this activity. We utilize the microfluidic technology developed for this
assay to perform a high-throughput screen for small molecule inhibitors of this RNA
binding activity, and identify clemizole hydrochloride, among others, as an effective
inhibitor. Cell-based studies show that inhibition of RNA binding through either
pharmacologic or genetic methods inhibits HCV replication. Genetic analysis further
identifies two mutations in the HCV NS3 protein that each can overcome the genetic
disruption of NS4B’s RNA binding domain.
In addition to clemizole, our work identifies a small molecule inhibitor of HCV
that affects a different activity of NS4B, its ability to form membrane-associated foci.

v
We provide genetic, biochemical, and cell biological evidence that NS4B is the target
of this drug, and an in vitro light scattering assay further suggests that it is the second
amphipathic helix of NS4B that is the target. In total, our results demonstrate that two
different activities of NS4B are each a valid pharmacological target for HCV antivirals
and uncover two candidate compounds that have potential for further pharmaceutical
development.

vi
Acknowledgments
I owe deep gratitude to many people who have helped me through the winding
road to this dissertation. First and foremost, I want to thank my PI, Jeffrey Glenn, who
has helped steer me through this path. I have to confess that occasionally—especially
when I got stuck on a project—I would feel somewhat less than “happy.” But
fortunately, you always had a 1-day experiment to turn things around and keep me
moving forward.
I am also grateful for the excellent colleagues I met in Jeff’s group. Thank you
to Menashe and Shirit for getting me started, to Namjoon, Choongho, and Ella for
keeping me going, to Wei for helping me out at all hours of the day, to Rick for
keeping me well hydrated, and to all for making the experience not just long, but
enjoyable. I will miss you guys.
I am very fortunate to have had the opportunity to obtain the level of education
I received at Stanford. My interactions with faculty really solidified my ambitions to
become a faculty member one day. I would like to thank my committee, Harry, Peter
and Shoshana for adding a dose of reality to my zany ideas. Joe Lipsick and James
Nelson were excellent teaching mentors, and I hope to follow in their lead some day.
Upi Singh and John Boothroyd both left me with the desire to explore new fields.
Thanks to all the faculty who put together the teaching curriculum for us M&I
students. And finally, I really appreciate all the staff, especially Julie, Mary Jeanne,
and Wanapa, who allowed me to focus on the science and not the business of being a
graduate student.

vii
On a somewhat more personal note, I am grateful to have entered with a class
of students who ended up being good friends and colleagues. You all have made this
period so much more enjoyable than it could have been. Special thanks to Jeff, Drew
and Drew for providing me with a roof over my head and to others for offering to help
me finish my work here.
Most importantly, I couldn’t have made it this far without the love and support
of my family. Xiaochin, sometimes I think you like science more than I do, and it
thrills me every time I get to explain my latest experiment to you. Mom, thanks for
always believing in me and pushing me to succeed at the next level. Hallie, I really
appreciate the grounding effect you and your family has had for me. And Copland,
thanks for listening to my practice talks—here’s a treat, or “bark bark, bark bark bark
treat!”

viii
Table of Contents ABSTRACT .......................................................................................................................................... IV
ACKNOWLEDGMENTS.................................................................................................................... VI
TABLE OF CONTENTS.................................................................................................................. VIII
LIST OF FIGURES................................................................................................................................X
LIST OF TABLES..................................................................................................................................X
CHAPTER 1. INTRODUCTION...........................................................................................................1
HCV EPIDEMIOLOGY.............................................................................................................................2 HCV THERAPY ......................................................................................................................................2 HCV MOLECULAR BIOLOGY ..................................................................................................................3 HCV NS4B ...........................................................................................................................................5 THEMES UNCOVERED IN THIS RESEARCH ...............................................................................................7
CHAPTER 2. DISCOVERY OF A HEPATITIS C TARGET AND ITS PHARMACOLOGICAL INHIBITORS BY MICROFLUIDIC AFFINITY ANALYSIS ...........................................................9
2.1 INTRODUCTION ..............................................................................................................................10 2.2 RESULTS ........................................................................................................................................12
Microfluidic assay validation: HuD binding to RNA.....................................................................13 NS4B binds HCV RNA and Kd is determined by microfluidics ......................................................16 NS4B specifically binds the 3’ terminus of the (–) viral strand .....................................................17 An ARM in NS4B is essential for RNA binding and HCV replication............................................19 High-throughput screening for inhibitory compounds...................................................................21 Inhibitors of HCV RNA replication................................................................................................25 Clemizole-resistant mutants...........................................................................................................25
2.3 DISCUSSION ...................................................................................................................................29 2.4 METHODS ......................................................................................................................................32
Plasmids.........................................................................................................................................32 In vitro RNA transcription and fluorescent and radioactive labeling............................................33 Device design.................................................................................................................................34 RNA binding assay.........................................................................................................................35 Screening of inhibitory compound library .....................................................................................39 Determination of IC50 for in vitro RNA binding.............................................................................40 Expression and purification of recombinant NS4B........................................................................40 GST pull down assay......................................................................................................................41 RNA Filter Binding Assay..............................................................................................................42 Cell cultures and electroporation ..................................................................................................42 Viability assay................................................................................................................................43 Luciferase assay.............................................................................................................................44 Real-time PCR ...............................................................................................................................45 Selection of resistant mutants ........................................................................................................45 Whole cell RNA electroporation ....................................................................................................46 Statistical analysis .........................................................................................................................46
CHAPTER 3. MUTATIONS IN NS3 COMPENSATE FOR THE DISRUPTION OF AN RNA-BINDING DOMAIN IN NS4B .............................................................................................................47
3.1 INTRODUCTION ..............................................................................................................................48 NS4B contains an RNA-binding activity ........................................................................................48 NS4B interacts with NS3................................................................................................................48
3.2 RESULTS ........................................................................................................................................49

ix
RBD mutations prevent HCV replication.......................................................................................49 RBD mutations do not affect HCV localization .............................................................................53 Primary-site revertants are not observed ......................................................................................55 Secondary-site mutations in NS3 restore HCV replication in KR247AA replicons .......................57
3.3 DISCUSSION ...................................................................................................................................59 METHODS ............................................................................................................................................62
Plasmids.........................................................................................................................................62 Colony formation assay .................................................................................................................63 Infection-transfection.....................................................................................................................63 Picking colonies, RT-PCR, & sequencing......................................................................................64 Luciferase assay.............................................................................................................................64
CHAPTER 4. A SMALL MOLECULE INHIBITS HCV REPLICATION AND DISRUPTS NS4B’S SUBCELLULAR DISTRIBUTION ......................................................................................65
4.1 INTRODUCTION ..............................................................................................................................66 4.2 RESULTS ........................................................................................................................................67
The small molecule, anguizole, inhibits HCV RNA replication. ....................................................67 Mutations conferring resistance to anguizole map to NS4B..........................................................70 Anguizole treatment leads to an altered subcellular distribution pattern of the NS4B protein. ....74 A resistance mutation also alters the subcellular distribution pattern of the NS4B protein. .........77 Anguizole interacts with the second amphipathic helix of NS4B. ..................................................78
4.3 DISCUSSION ...................................................................................................................................82 4.4 MATERIALS AND METHODS...........................................................................................................85
DNA constructs and peptides.........................................................................................................85 Drugs and antibodies.....................................................................................................................86 Cell culture, NS4B-GFP transfections, and fluorescence microscopy...........................................86 Stable luciferase replication assays ...............................................................................................87 Transient luciferase replication assays..........................................................................................88 Analysis of resistance mutants .......................................................................................................89 Dynamic Light Scattering (DLS)....................................................................................................90
CHAPTER 5. CONCLUSIONS ...........................................................................................................92
SUMMARY ...........................................................................................................................................93 SIGNIFICANCE......................................................................................................................................94
REFERENCES ......................................................................................................................................96
APPENDIX A. SUPPLEMENTARY MATERIAL TO CHAPTER 2 ..................................................................97 Calibration curve ...........................................................................................................................97 RNA binding experiment with high ionic strength buffer...............................................................97 ATA inhibits RNA binding by NS4B in a dose-dependent manner.................................................98 Specificity of hits identified in the small molecule screen..............................................................98
APPENDIX B. SUPPLEMENTARY MATERIAL TO CHAPTER 4 ................................................................109 BIBLIOGRAPHY..................................................................................................................................111

x
List of Figures FIGURE 1.1. THE HCV GENOME STRUCTURE. ............................................................................................4 FIGURE 1.2 REPLICATION COMPLEX OF HCV .............................................................................................6 FIGURE 2.1 PROTEIN-RNA INTERACTIONS MEASURED ON MICROFLUIDIC PLATFORM..............................15 FIGURE 2.2 NS4B BINDS SPECIFICALLY TO THE 3’ TERMINUS OF THE HCV NEGATIVE STRAND RNA......18 FIGURE 2.3 IDENTIFICATION OF RNA BINDING DOMAINS WITHIN NS4B. .................................................20 FIGURE 2.4 SMALL-MOLECULE SCREEN REVEALS THAT CLEMIZOLE HYDROCHLORIDE INHIBITS RNA
BINDING BY NS4B AND HCV RNA REPLICATION IN CELL CULTURE. .............................................24 FIGURE 2.5 CLEMIZOLE-RESISTANT MUTANT. ..........................................................................................28 FIGURE 3.1. PUTATIVE RNA BINDING DOMAINS ARE NECESSARY FOR HCV REPLICATION. .....................52 FIGURE 3.2. DISRUPTION OF RNA BINDING DOMAINS DOES NOT AFFECT THE FORMATION OF PUTATIVE
HCV REPLICATION COMPLEXES......................................................................................................54 FIGURE 3.3. REPLICATING COLONIES CONTAINING KR247AA MUTATION CONTAIN SECONDARY-SITE
MUTATIONS.....................................................................................................................................56 FIGURE 3.4. MUTATIONS IN NS3 ENHANCE REPLICATION OF RBD DEFECTIVE MUTANTS. .......................58 FIGURE 3.5. LOCATION OF COMPENSATORY MUTATIONS IN NS3-4A DIMER. ...........................................61 FIGURE 4.1. A SMALL MOLECULE INHIBITS HCV REPLICATION................................................................69 FIGURE 4.2. RESISTANT MUTATIONS MAP TO NS4B. ................................................................................71 FIGURE 4.3. CHARACTERIZATION OF THE H94R RESISTANCE MUTATION. ................................................73 FIGURE 4.4. ANGUIZOLE ALTERS THE SUBCELLULAR DISTRIBUTION OF NS4B-GFP IN TRANSIENTLY
TRANSFECTED CELLS. .....................................................................................................................76 FIGURE 4.5. THE H94R MUTATION ALTERS NS4B-GFP’S SUBCELLULAR DISTRIBUTION. ........................79 FIGURE 4.6. ANGUIZOLE INTERACTS WITH THE SECOND AMPHIPATHIC HELIX (AH2) OF NS4B. ..............81 FIGURE A.1. MICROFLUIDIC BASED RNA BINDING ASSAY. ....................................................................101 FIGURE A.2. SIGNAL TO NOISE RATIO IN OUR RNA BINDING ASSAY.......................................................103 FIGURE A.3. MICROFLUIDICS-BASED ANALYSIS OF RNA BINDING BY ANOTHER HUMAN PROTEIN FROM
THE ELAV-LIKE FAMILY, HUR (ELAV L1). ................................................................................105 FIGURE A.4. BINDING OF NS4B TO HCV RNA BY CONVENTIONAL METHODS.......................................106 FIGURE A.5. ATA INHIBITS RNA BINDING BY NS4B IN A DOSE DEPENDENT MANNER...........................107 FIGURE A.6. CLEMIZOLE INHIBITS HCV REPLICATION BY REAL-TIME PCR ASSAYS. .............................108 FIGURE B.1. SUBCELLULAR DISTRIBUTION PATTERN OF NS4B-GFP VARIES IN ANGUIZOLE-TREATED
CELLS............................................................................................................................................109 FIGURE B.2. DISTRIBUTION PATTERNS OF CELLULAR MARKERS DO NOT VARY WITH ANGUIZOLE
TREATMENT. .................................................................................................................................110
List of Tables TABLE 3.1. PRIMER SEQUENCES…................................................................................................. 63

1
Chapter 1. Introduction

2
HCV epidemiology
Hepatitis C Virus is a serious cause of liver disease. Patients infected with this
virus can develop chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma.
Throughout the world, more than 170 million people are thought to be infected1 and
HCV is responsible for roughly 27% of liver cirrhoses and 25% of hepatocellular
carcinomas worldwide. Based on global mortality estimates, HCV is thus directly
responsible for 366,000 deaths per year2.
In the United States, prevalence is somewhat lower, though HCV is still the
most common bloodborne infection in the US3 and end-stage liver disease caused by
chronic hepatitis C is the most common cause of liver transplantation4. Approximately
1.6% of the American population has been exposed to HCV, and 1.3% is still positive
for HCV RNA5. This infection is clearly a significant public health problem, and
means to treat its pathogenicity are badly needed.
HCV therapy
Current standard of care regimens for HCV include treatment with Pegylated
interferon- and ribavirin. However, this treatment has significant downsides. First, it
is successful in only 54-56% of patients treated6. Second, these treatments are often
associated with significant side effects, and treatment is not recommended for many
patients with various contraindications; as a result, a sustained viral response to
Pegylated interferon- + ribavirin is achieved in as little as 15% of patients who
present with HCV7. Thus, the current treatment for patients is not effective at curing

3
this disease. Compounding the ineffectiveness of the current antiviral therapy is the
fact that no vaccine is yet available for this pathogen. Therefore, there is a great need
for improved antiviral therapies, and a large amount of effort is being put into the
search for such therapies. In order to develop specific antiviral drugs, an
understanding of the viral life cycle has been crucial (discussed in detail below).
HCV molecular biology
HCV is a member of the Hepacivirus genus, in the Flaviviridae family. Its 9.6
kb genome is composed of a single strand of positive-sense RNA. Encoded within this
genome are three structural proteins, Core, E1 and E2, which together form the viral
particle. Downstream of these proteins are the nonstructural proteins, which include
p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B (Figure 1.1). A number of model
systems have been utilized to examine the role of each of these proteins, as well as that
of RNA elements of the genome (reviewed in 8). Of relevance to this dissertation are
the 5’ untranslated region (UTR) of the RNA genome and the proteins NS3, NS4B,
and NS5B.
The 5’ UTR forms a highly ordered structure composed of four domains, I-IV.
These form an internal ribosome entry site (IRES) that allows for the cap-independent
translation of the viral genome. Notably, elements of the 5’UTR that are not necessary
for translation are essential for RNA replication9. As will be discussed in Chapter 2,
this region appears to encode the RNA sequence necessary for efficient binding
between NS4B and HCV RNA.

4
Figure 1.1. The HCV Genome structure.
HCV contain an RNA genome of 9.6kb. The 5’UTR contains four ordered domains,
which form an internal ribosome entry site (IRES). The HCV genome encodes 10
proteins, divided into structural (Core, E1, E2) and nonstructural (p7, NS2, NS3,
NS4A, NS4B, NS5B). Their known functions are shown here (reprinted by permission
from Macmillan Publishers Ltd: Nature Reviews Microbiology (5, 453-463),
copyright 2007).

5
NS3 is a multifunctional protein that contains both a protease domain and a
helicase domain. Its protease activity is a common target of antiviral drugs, some of
which have progressed into Phase IIb clinical trials10, 11. Furthermore, this protein has
been shown to interact genetically with NS4B, which will be discussed in Chapter 3.
NS5B is the RNA-dependent RNA polymerase that is responsible for copying
the HCV genome. As described in Chapter 4, this protein is another major target of
antiviral therapies, and various nucleoside and non-nucleoside inhibitors are in
development12.
HCV NS4B
The main subject of this dissertation is the NS4B viral protein. This protein has
been the subject of less investigation than the other nonstructural proteins.
Nevertheless, several groups have revealed structural and functional insights into this
protein’s role, outlined below.
NS4B was first characterized to be sufficient for the formation of the
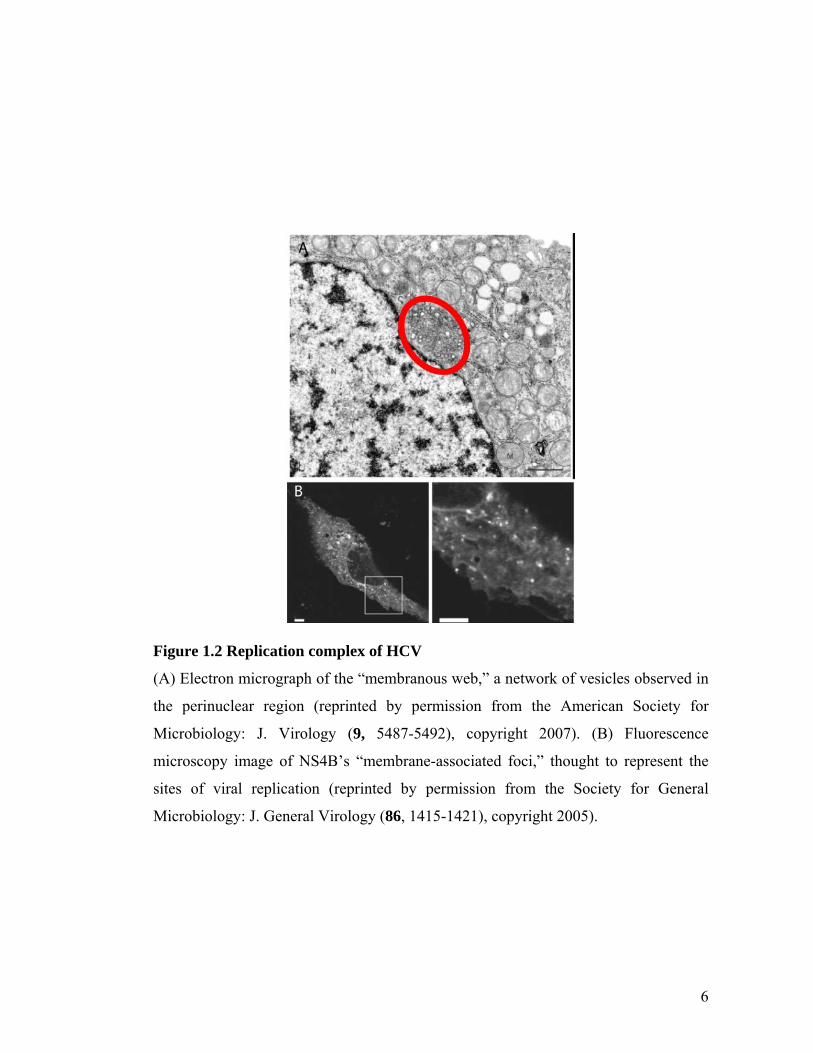
membranous web, a vesicular structure observed in HCV-infected cells13 (Figure
1.2A). All RNA viruses appear to replicate in association with membranes, and this
membranous web is hypothesized to be the site of replication for HCV. As discussed
in Chapter 4, many of the HCV NS proteins, as well as nascent HCV RNA, have been
localized to this web14, 15. In addition, the equivalent of the membranous web is
thought to be visible with light microscopy as membrane-associated foci that adopt an
endoplasmic reticular pattern16 (Figure 1.2B).
6
Figure 1.2 Replication complex of HCV
(A) Electron micrograph of the “membranous web,” a network of vesicles observed in
the perinuclear region (reprinted by permission from the American Society for
Microbiology: J. Virology (9, 5487-5492), copyright 2007). (B) Fluorescence
microscopy image of NS4B’s “membrane-associated foci,” thought to represent the
sites of viral replication (reprinted by permission from the Society for General
Microbiology: J. General Virology (86, 1415-1421), copyright 2005).

7
Structually, NS4B is associated with membranes and is thought to contain four
or five transmembrane domains17. Also, it contains several amphipathic helices, which
play a role in its membrane localization18, 19. These functions will be examined in
Chapter 4.
In addition, NS4B has been described to contain a nucleotide binding motif
that is essential for its GTPase activity20. Also, NS4B has been described to
oligomerize, for which lipid modifications on the C-terminus are important.21
Themes uncovered in this research
This thesis enumerates the cumulative results of several different projects
performed during the author’s dissertation. All three of the chapters are centered on
NS4B’s role in the replication of viral RNA, but they are different in the approach
taken to that role. One project focuses on the cellular biology of how NS4B may be
involved in the formation of the complexes on which replication occurs (Chapter 4).
The other project takes both a biochemical and genetic approach in order to examine
one functional aspect of the NS4B protein, its RNA-binding activity (Chapters 2 and
3). By examining disparate aspects of NS4B’s role in the HCV life cycle, this thesis
aims to elucidate a broader understanding of this protein’s evolution as an essential
part of the HCV genome.
Much of this work was performed in collaboration with others in Jeffrey
Glenn’s lab and beyond. Specifically, Chapter 2 was a collaboration with Shirit Einav
of the Glenn lab and Doron Gerber of the Stephen Quake lab, and this chapter has
been published in Nature Biotechnology22. Paul Bryson, the author of this thesis, was

8
responsible for the developing the RNA-binding hypothesis, identifying potential
RNA-binding domains, designing the RNA-binding experiments, executing some of
the binding experiments, and contributing a portion of the final manuscript. Chapter 3
has not yet been published as a journal article, and all the material presented therein is
primarily the responsibility of this thesis’s author. Chapter 4 was a collaboration with
members of the Genelabs Technologies company and has been submitted for
publication. Paul Bryson was responsible for the design of the project, the execution of
some of the replication assays and resistant mutant identification, and of all the
fluorescence microscopy. Furthermore, he was responsible for the data analysis and
the composition of the manuscript.

9
Chapter 2. Discovery of a hepatitis C target and its pharmacological inhibitors by microfluidic affinity
analysis

10
2.1 Introduction
Over 150 million people are infected with Hepatitis C Virus (HCV)
worldwide. Unfortunately, many of these individuals are unable to clear their infection
with the current standard of care, which consists of a combination of interferon and
ribavirin23. Moreover, this treatment is associated with significant side effects,
precluding its use by many individuals. Thus, current therapies are inadequate for the
majority of the patients23, and there is a pressing need for new HCV drugs23.
The 9.6-kb, positive, single-stranded RNA HCV genome encodes a 3,000-
amino-acid polyprotein which is proteolytically processed into structural proteins,
which are components of the mature virus, and nonstructural proteins (NS), which are
involved in replicating the viral genome24. Like other positive strand RNA viruses25,
HCV appears to replicate in association with intracellular membrane structures. In the
case of HCV, the structures are termed the membranous web26 and are believed to be
induced by the NS4B protein. NS4B is also required to assemble the other viral NS
proteins within the apparent sites of RNA replication18. It is not known how viral
RNA, especially the negative strand template required for production of progeny
genomes, might be incorporated or maintained at these replication sites.
NS4B and HCV RNA have been shown to colocalize to the membranous
web27, 28, suggesting that NS4B is in intimate contact with viral RNA in the context of
authentic viral RNA replication. The hepatitis A and polio picornaviruses have
proteins termed 2C which are required for replication, bind RNA29, 30, and have an N-
terminal amphipathic helix and a nucleotide binding motif29-31. NS4B contains the

11
same structural features, and both of them are required for HCV replication18, 20. We
thus hypothesized that NS4B may similarly bind RNA, that this interaction might be
critical for the HCV life cycle, and that RNA binding by NS4B could be amenable to
pharmacologic disruption. To test these hypotheses, we sought to establish an in vitro
RNA binding assay in a format enabling simultaneous analysis of multiple conditions,
mutants, and replicates, quantitative dissociation constant (Kd) measurements, and
high-throughput screening for potential pharmacologic inhibitors.
Like the majority of drug targets32, NS4B is a membrane protein. Membrane
proteins are notoriously difficult to express and characterize biochemically, especially
in the quantities required for pharmaceutical screening. Moreover, solubilization by
detergents can alter their natural membrane associated topology. To ameliorate these
problems, we used microfluidic tools to perform binding assays with nanoliter protein
consumption. This enabled use of an in vitro cell lysate expression system, which was
supplemented with microsomal membranes to create more natural folding conditions,
under which the best NS4B topology data available to date has been obtained33. The
reduced yield relative to conventional expression methods is offset by low sample
consumption.
Previous microfluidic tools to measure drug interactions have been limited to
enzymatic targets which can catalyze formation of a fluorescent substrate34. In this
case we directly measured binding constants by using mechanical trapping of
molecular interactions (MITOMI), a microfluidic affinity assay that has previously
been used to measure interactions between transcription factors and DNA35. We have
extended the previous work by showing that MITOMI can be used both to measure

12
binding constants of membrane protein-RNA interactions and to measure inhibition of
such interactions by small molecules in a high throughput screen. The latter point was
particularly surprising in that the elastomer used to fabricate the device is known to
have limitations in chemical compatibility36, 37; here we show that this does not
prevent its use in a drug screen nor does it prevent discovery of a small molecule with
the desired pharmacological properties. Taken together, the results of this paper reveal
a novel HCV target and are the first demonstration that microfluidic technology can be
used to discover a new pharmaceutical, thereby successfully consummating more than
a decade of effort to apply microfluidic tools to drug discovery38, 39.
2.2 Results
We validated the use of this platform for RNA binding by studying two human
proteins from the embryonic lethal abnormal visual system (ELAV) family, the RNA
binding activity of which has been previously well-characterized40-42. We then applied
this methodology to study RNA interactions with the transmembrane HCV NS4B
protein. We used this platform to (i) test the hypothesis that HCV NS4B binds RNA,
(ii) determine the Kd for this interaction, (iii) study the substrate specificity of this
binding, (iv) determine the amino acids within NS4B required for RNA binding, and
(v) screen a compound library for pharmacologic inhibitors of NS4B RNA binding
and HCV replication.

13
Microfluidic assay validation: HuD binding to RNA
HuD is a host cell protein from the ELAV-like family with well characterized
RNA binding activity. It is a cytoplasmic protein that contains 3 conserved RNA
recognition motifs through which it binds AU-rich elements (ARE) in the 3’ UTR (un-
translated region) of genes such as those encoding cytokines and proto-oncogenes.
Binding was tested against two Cy3-labeled RNA probes: AU3 and a known AU3
mutant to which HuD binding is impaired41, 42 (Figure 2.1). RNA binding experiments
with MITOMI were performed essentially as described by Maerkl and Quake35, except
that in this case RNA was used instead of DNA (Figure A.1). Briefly, we spotted a
microarray of target RNA sequences labeled with Cy3 onto an epoxy-coated slide.
These arrays were used to program the microfluidic devices by aligning each spot in
the array to a unit cell in the device. After bonding the microfluidic device to a
microarray it was subjected to surface patterning that resulted in a circular area coated
with biotinylated anti-histidine antibodies within each unit cell. The device was then
loaded with in vitro transcription/translation mixture containing DNA templates
coding for HuD fused in frame with a C-terminal V5-6 histidine tag (HuD-V5-his) or
Gus protein fused in frame with a C–terminal 6 histidine tag (Gus-his). Bodipy-labeled
tRNALys was added for protein labeling. Each unit cell was then isolated by the control
of three micromechanical valves followed by an incubation to allow protein synthesis,
binding of the synthesized protein to the surface biotinylated anti-his antibodies,
solvation of target RNA, and equilibration of proteins and target RNA. MITOMI was
then performed by actuation of a “button” membrane to trap surface-bound complexes
while expelling any solution phase molecules. After a brief wash to remove untrapped

14
unbound material, the trapped molecules and expressed protein were subsequently
detected with an array scanner. The ratio of bound RNA to expressed protein was
calculated for each data point by measuring the median signal of Cy3 to median signal
of bodipy.
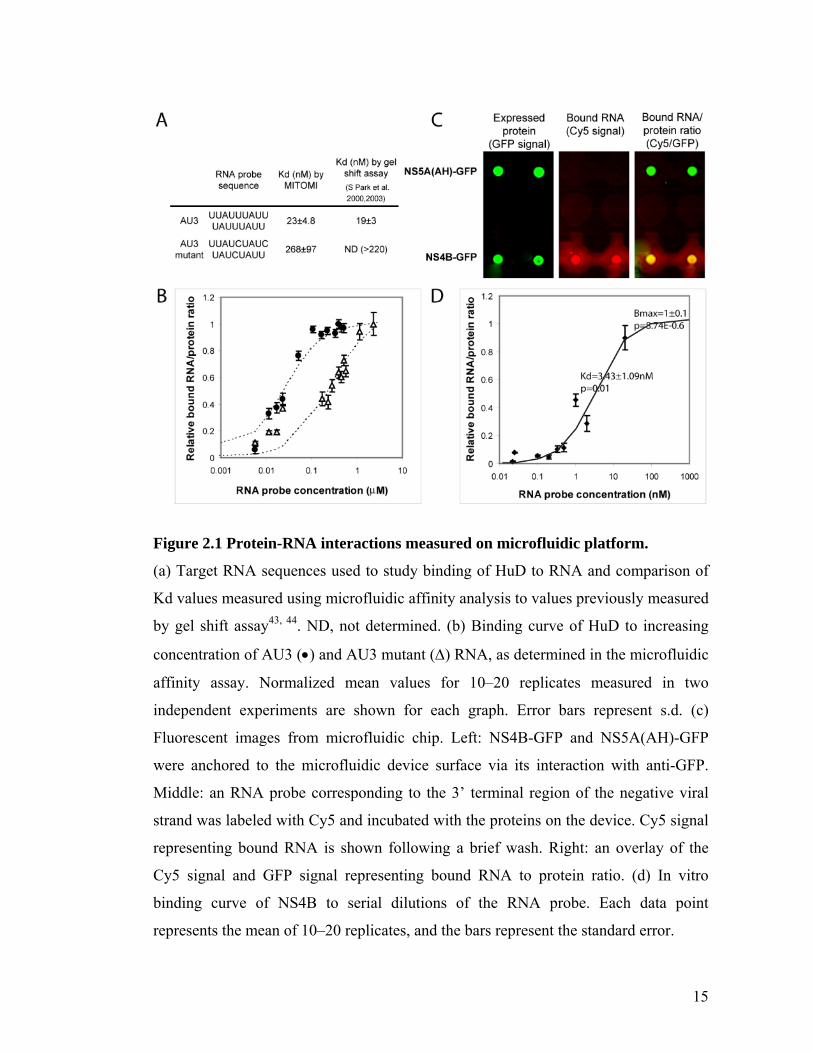
15
Figure 2.1 Protein-RNA interactions measured on microfluidic platform.
(a) Target RNA sequences used to study binding of HuD to RNA and comparison of
Kd values measured using microfluidic affinity analysis to values previously measured
by gel shift assay43, 44. ND, not determined. (b) Binding curve of HuD to increasing
concentration of AU3 () and AU3 mutant () RNA, as determined in the microfluidic
affinity assay. Normalized mean values for 10–20 replicates measured in two
independent experiments are shown for each graph. Error bars represent s.d. (c)
Fluorescent images from microfluidic chip. Left: NS4B-GFP and NS5A(AH)-GFP
were anchored to the microfluidic device surface via its interaction with anti-GFP.
Middle: an RNA probe corresponding to the 3’ terminal region of the negative viral
strand was labeled with Cy5 and incubated with the proteins on the device. Cy5 signal
representing bound RNA is shown following a brief wash. Right: an overlay of the
Cy5 signal and GFP signal representing bound RNA to protein ratio. (d) In vitro
binding curve of NS4B to serial dilutions of the RNA probe. Each data point
represents the mean of 10–20 replicates, and the bars represent the standard error.

16
The assay detected strong binding of HuD to the AU3 RNA probe; background
binding by Gus-his was 7-16 fold lower than the HuD signal (Figure 2.1A). This
background level did not increase with RNA probe concentration and was subtracted
from all chambers (Figure A.2). The binding affinity of HuD to the AU3 probe was
much greater than to the AU3 mutant probe: the Kd for the AU3 binding was
determined to be 23+5 nM and for the AU3 mutant 268+95 nM (Figure 2.1A). These
values agree with previous measurements in a gel shift assay41, 42 and serve to validate
the MITOMI microfluidic affinity assay for RNA-protein interactions. RNA binding
analysis of another protein from the ELAV-like family, HuR, is shown in Figure A.3.
NS4B binds HCV RNA and Kd is determined by microfluidics
We then tested whether HCV NS4B binds RNA. Since NS4B is important in
viral RNA replication, and initiation of positive-strand RNA synthesis is likely to start
at the 3’ terminus of the negative-strand RNA, we first tested binding of NS4B to this
region, using a probe designated 3’ negative terminus. A fusion of the amphipathic
helix (AH) of NS5A to the N-terminus of GFP18 was used as a negative control. This
protein also binds to membranes and can thus anchor the microsomal membranes to
the device surface via the interaction of GFP with anti-GFP (Figure 2.1C). The Kd of
NS4B binding to the 3’ terminus of the HCV negative strand RNA was measured at
3.43+1 nM (Figure 2.1D). To the best of our knowledge this is the first report that
HCV NS4B binds RNA. Conventional GST pull down assays and RNA filter binding
assays using recombinant forms of purified NS4B expressed in E. coli confirmed this

17
finding (Figure A.4), although these were less convenient and amenable to the types of
analyses and high throughput format that we sought to perform.
NS4B specifically binds the 3’ terminus of the (–) viral strand
We measured the substrate specificity of the observed NS4B-HCV RNA
interaction with three additional HCV probes (Figure 2.2a). The probes designated 5’
UTR pos and 3’ UTR pos correspond to the 5’ UTR and 3’ UTR sequences of the
positive viral strand, respectively, and 5’ negative terminus corresponds to the 5’
terminus of the negative strand. The RNA binding experiment was repeated and
binding of NS4B to equimolar concentrations (3 nM) of the various probes was
compared. Whereas NS4B binds all four HCV probes, its apparent affinity to the 3’
negative terminus probe is 5- to 12-fold greater than to the other three HCV RNA
regions tested or to an unrelated delta virus RNA genome sequence45, 46 (see Figure
2.2B). These findings suggest that it is the RNA sequence, and likely the secondary
RNA structure, that determines the specificity of NS4B binding to the 3’ terminus of
the negative strand.
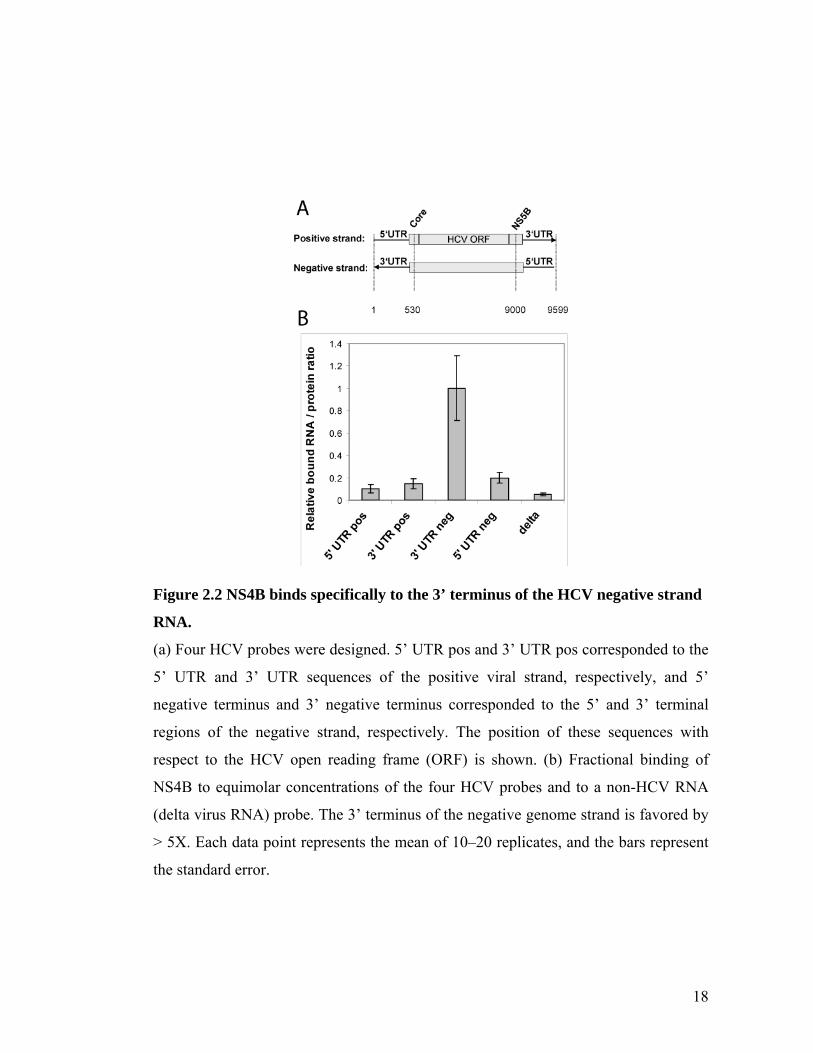
18
Figure 2.2 NS4B binds specifically to the 3’ terminus of the HCV negative strand
RNA.
(a) Four HCV probes were designed. 5’ UTR pos and 3’ UTR pos corresponded to the
5’ UTR and 3’ UTR sequences of the positive viral strand, respectively, and 5’
negative terminus and 3’ negative terminus corresponded to the 5’ and 3’ terminal
regions of the negative strand, respectively. The position of these sequences with
respect to the HCV open reading frame (ORF) is shown. (b) Fractional binding of
NS4B to equimolar concentrations of the four HCV probes and to a non-HCV RNA
(delta virus RNA) probe. The 3’ terminus of the negative genome strand is favored by
> 5X. Each data point represents the mean of 10–20 replicates, and the bars represent
the standard error.

19
An ARM in NS4B is essential for RNA binding and HCV replication
Various structural motifs responsible for the interaction between proteins and
RNA have been reported47. One of these is the arginine rich motif, (ARM). ARMs
were originally defined as short (10 to 20 amino acids) arginine-rich sequences found
in viral, bacteriophage, and ribosomal proteins (Figure 2.3a)30, 47, 48. There is little
identity between ARM sequences, other than the preponderance of arginine residues.
Subsequently, ARM-like motifs, consisting of longer sequences containing fewer
arginines were identified30, 48. Inspection of the primary sequence of NS4B reveals the
presence of multiple positively-charged amino acids (see Figure 2.3B). The majority
of these arginine residues are within the last 71 amino acids, in the C-terminal region
of NS4B (Figure 2.3B, C). This region is predicted to form a cytoplasmic segment
based on empirical topology studies using glycosylation markers33. Elements of this
region conform with previously described ARM-like motifs.
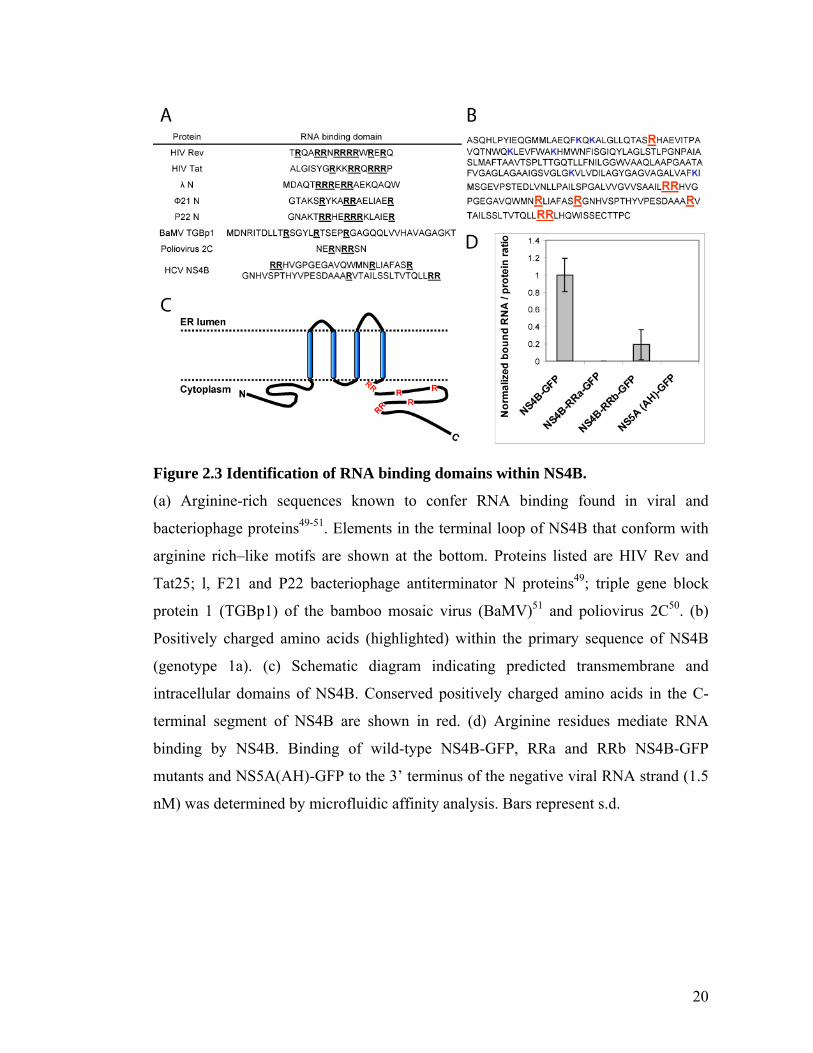
20
Figure 2.3 Identification of RNA binding domains within NS4B.
(a) Arginine-rich sequences known to confer RNA binding found in viral and
bacteriophage proteins49-51. Elements in the terminal loop of NS4B that conform with
arginine rich–like motifs are shown at the bottom. Proteins listed are HIV Rev and
Tat25; l, F21 and P22 bacteriophage antiterminator N proteins49; triple gene block
protein 1 (TGBp1) of the bamboo mosaic virus (BaMV)51 and poliovirus 2C50. (b)
Positively charged amino acids (highlighted) within the primary sequence of NS4B
(genotype 1a). (c) Schematic diagram indicating predicted transmembrane and
intracellular domains of NS4B. Conserved positively charged amino acids in the C-
terminal segment of NS4B are shown in red. (d) Arginine residues mediate RNA
binding by NS4B. Binding of wild-type NS4B-GFP, RRa and RRb NS4B-GFP
mutants and NS5A(AH)-GFP to the 3’ terminus of the negative viral RNA strand (1.5
nM) was determined by microfluidic affinity analysis. Bars represent s.d.

21
Using a series of point mutations, we tested if RNA binding by NS4B is
mediated by some of its positively-charged residues. A substitution of Arg-Arg in
positions 192-193 with Ala-Ala was termed “RRa mutant” and a similar substitution
in positions 247-248 was designated “RRb mutant”. While the RRb mutant decreased
binding by NS4B to the 3’ negative terminus probe by 5 fold, RNA binding activity to
the RRa mutant was completely eliminated at the same probe concentration (1.5nM;
Figure 2.3D). These results suggest that the tested arginine residues mediate RNA
binding by NS4B in vitro. To test the hypothesis that these positively charged residues
are also important for HCV replication, we introduced these mutations into high
efficiency subgenomic HCV replicons and assayed them in standard replicon colony
formation assays (data not shown). The various mutations significantly impaired HCV
RNA replication and the degree of replication inhibition correlated with the degree of
NS4B RNA binding impairment (Bryson P et al, manuscript in preparation). These
results suggest that efficient binding of HCV RNA is required for viral replication in
vitro. Alignment of the sequences of natural HCV isolates currently available in
databases reveals that these positively-charged residues are highly conserved across all
HCV genotypes. This conservation suggests that there is a requirement for the RNA
binding residues for productive viral infection in vivo.
High-throughput screening for inhibitory compounds
We screened a compound library for small molecules that could inhibit the
RNA-NS4B interaction. As shown in Figure 2.4A, 1280 compounds from a small
molecular library were spotted on epoxy-coated slides as a microarray. The array was

22
allowed to dry, and was then aligned and bonded to a microfluidic device. The rest of
the assay was performed as before, except that the device was loaded with NS4B-GFP
followed by Cy5-labeled 3’ terminus negative RNA probe. In the primary screen, the
compounds were spotted at a concentration of ~1mM. The entire library was screened
in duplicate using only two microfluidic devices. Out of 1280 compounds, 104 were
found to have an inhibitory effect (>90% inhibition) on RNA binding by NS4B. In
addition, there were 110 compounds for which there was a significant discrepancy
between the two tested replicates or to which one or two of the measurements were
disrupted due to technical reasons.
The 214 compounds (104+110) identified in the primary screen were subjected
to a secondary screen (Figure 2.4A). This was done in a similar manner except that a
smaller device was used, the spotted compound concentration was 10 fold lower than
in the primary screen and 5 replicates were spotted for each compound. 18 compounds
were confirmed to significantly inhibit RNA binding by NS4B out of the 214
compounds tested (Figure 2.4B). Most of the identified compounds did not inhibit
binding of HuR protein to its own 4A target RNA sequence (Figure A.3) nor did they
inhibit HuR binding to its previously described HCV RNA target: the 3’ terminus of
the negative viral strand52, suggesting that these hits are specific. This data and
additional data supporting the validity of the screen are presented in Appendix A and
Figure A.5.
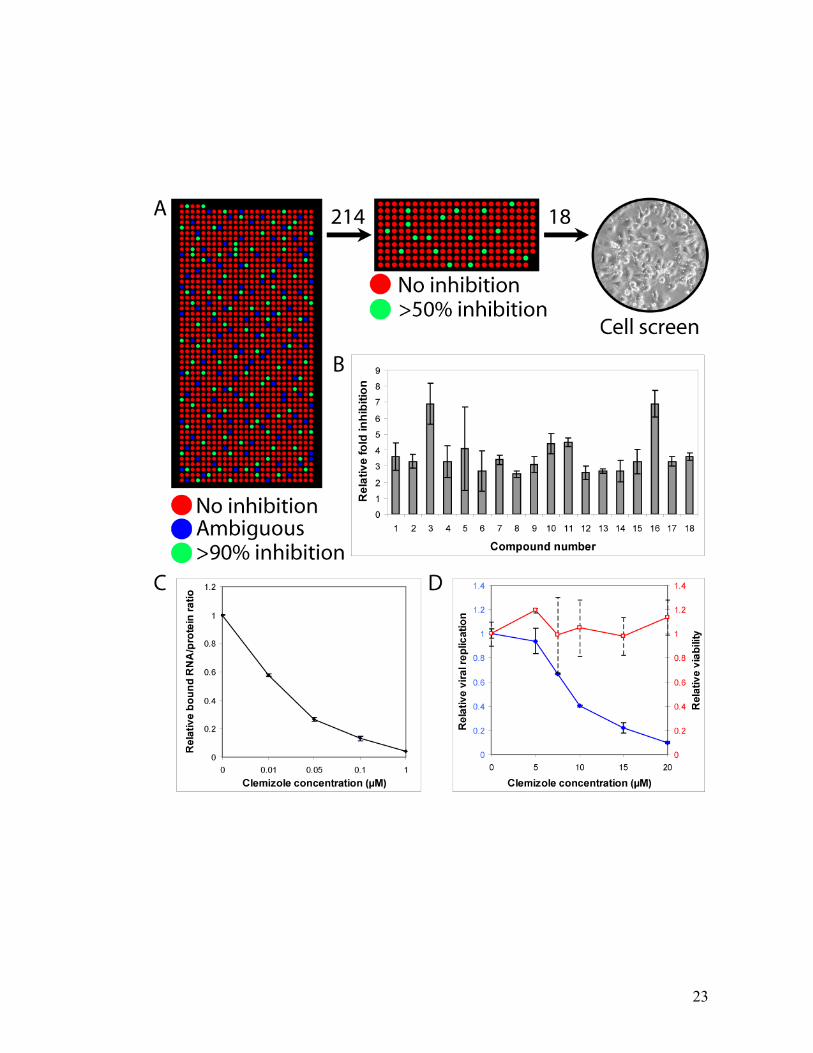
23

24
Figure 2.4 Small-molecule screen reveals that clemizole hydrochloride inhibits
RNA binding by NS4B and HCV RNA replication in cell culture.
(a) The first screen represented a low-stringency measurement of inhibition of 1,280
compounds where the latter were categorized as having high (green), ambiguous
(blue) or no (red) inhibition. Based on the initial screen, 214 compounds were then
measured again with higher stringency and with a greater number of replicates and the
best 18 inhibitors were tested for their ability to inhibit HCV replication via a cellular
assay. (b) In vitro inhibition of NS4B-RNA binding by the top 18 small molecules. (c)
HCV luciferase reporter–linked cellular assay showing that clemizole inhibits HCV
replication (left axis, blue ) with no measurable toxicity to the cell as measured by
alamarBlue (right axis, red ). (d) In vitro NS4B-RNA binding: inhibition curve of
clemizole. Each data point represents the mean of 10–20 replicates and the bars
represent the standard error.

25
Inhibitors of HCV RNA replication
We measured the in vivo antiviral effect on HCV RNA replication of the
inhibitory compounds identified in the above screen. Following electroporation with a
full-length HCV RNA genome harboring a Luciferase reporter gene53, Huh7.5 cells
were grown in the presence of increasing concentrations of these compounds.
Luciferase assays were performed at 72hr. In parallel, the viability of cells in the
presence of the compounds was assessed by an Alamar Blue-based assay. 6 of the
compounds showed some antiviral effect above that solely attributable to cellular
toxicity. One of these, clemizole hydrochloride (1-p-Chlorobenzyl-2-(1-pyrrolidinyl)
methylbenzimidazole hydrochloride), an H1 histamine receptor antagonist, was found
to significantly inhibit HCV replication. A tenfold decrease in viral replication was
measured at 20µM concentration of the drug, with an EC50 of ~8 µM (Figure 2.4C). At
these concentrations there was no measurable cellular toxicity (Figure 2.4C). Similar
results were obtained by real-time PCR assays performed in clemizole-treated Huh7.5
cells infected with the infectious HCV clone (J6/JFH) 54 (Figure A.6). The in vitro
half-maximal inhibitory concentration (IC50) of clemizole for RNA binding by NS4B
is ~ 24 1 nM (Figure 2.4D).
Clemizole-resistant mutants
The mechanism of action of clemizole’s antiviral activity was further
substantiated by selecting for clemizole resistant HCV mutants. Established HCV
replicon-harboring cells and Huh7 cells electroporated de novo with a genotype 1b
subgenomic HCV replicon (Bart 79I)55 were passaged in the presence of the drug,

26
yielding ~60 colonies that were able to grow in the presence of the compound. 11
individual colonies were successfully expanded, passaged 5-10 times and the HCV
RNA replicating in the cells was subjected to sequence analysis. In addition, RNA
from a pool of clemizole-resistant colonies was isolated and subjected to a similar
analysis. 3 colonies were found to harbor replicons with mutations that mapped to the
NS4B region and 6 colonies were found to harbor replicons with mutations that
mapped to the negative strand 3’ terminus RNA region. In addition there was one
colony with mutations that mapped to both NS4B and the negative strand 3’ terminus,
and one where we have yet to find the location of the mutation conferring resistance to
clemizole. No such mutations were identified in 10 replicon colonies that were
passaged in parallel in the absence of the drug.
Two of the clemizole-resistant NS4B mutants were characterized in detail. The
first, W55R is depicted in Figure 2.5A. It involves the substitution of an arginine for
the tryptophan at amino acid 55 within a predicted cytoplasmically-oriented segment
of NS4B. This mutation is sufficient to confer a clemizole-resistant phenotype in cells:
Huh7.5 cells transfected with either whole cell RNA extracted from the W55R mutant
cells (Figure 2.5B) or with in vitro-transcribed J6/JFH RNA encoding this point
mutation and a linked luciferase reporter gene (Figure 2.5C) were unaffected by 10µM
clemizole. EC50 of clemizole on the W55R mutant J6/JFH RNA was ~18 µM (2.25
times the EC50 on the wild type RNA). Similar to other HCV mutants resistant to an
NS3 protease inhibitor56, the absolute level of replication of the W55R mutant was
lower than that of the wild type genome, suggesting that the drug-resistant mutation
comes at the cost of impaired replication fitness.
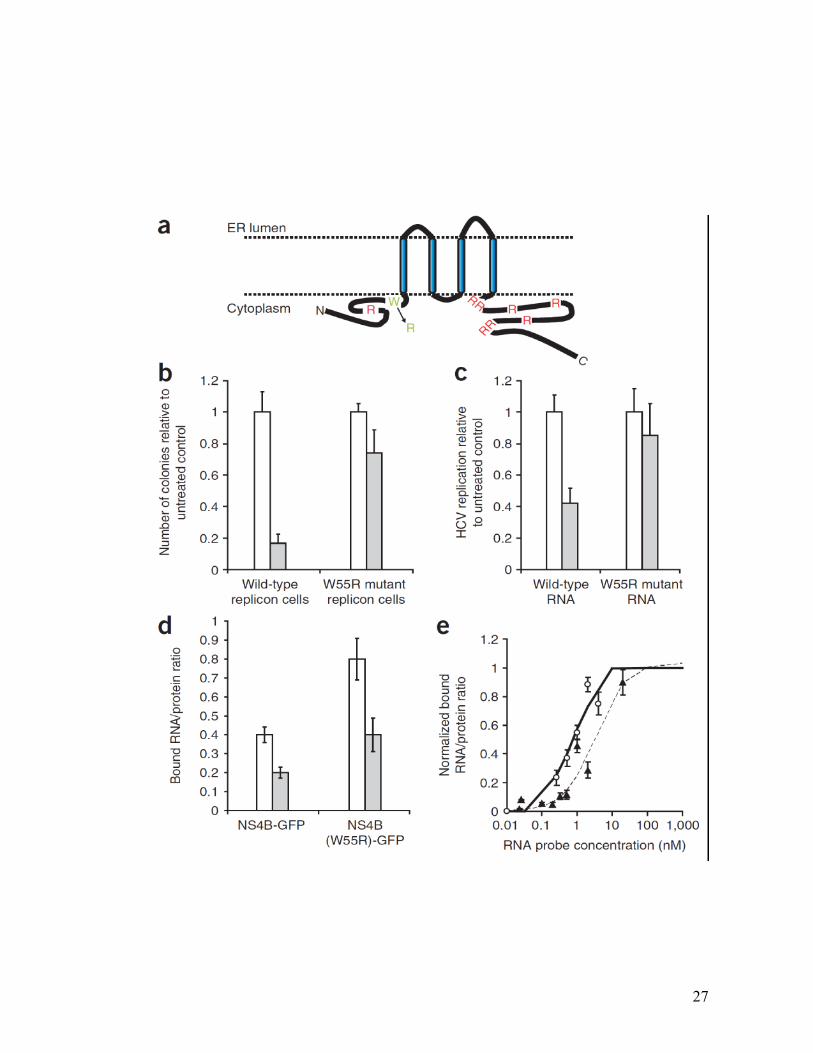
27

28
Figure 2.5 Clemizole-resistant mutant.
Replicon cells were passaged in the presence of clemizole and individual colonies
were isolated, propagated and the HCV genomes harbored within were subjected to
sequence analysis. (a) Schematic diagram indicating predicted transmembrane and
intracellular segments of NS4B. Conserved positively charged amino acids are shown
in red. The clemizole-resistant mutation, W55R, is shown in green. (b) HCV
replication in Huh7.5 cells electroporated with 50 g of whole-cell RNA extracted
from cells harboring either wild type or the W55R mutant clone, followed by growth
in the absence (white bars) or presence (gray bars) of 10 M clemizole. Results
represent relative numbers of colonies obtained compared to each corresponding
untreated control. (c) HCV replication assays initiated by electroporation of in vitro
transcribed luciferase reporter– linked wild-type or W55R mutant HCV RNA
genomes performed in the absence (white bars) or presence (gray bars) of 10 M
clemizole. Results represent replication level of each genome relative to its untreated
level. (d) HCV RNA binding of wild-type NS4B and the W55R NS4B mutant as
measured in vitro by microfluidics in the presence of 10 nM clemizole (gray bars) and
摧 Ѝ� � �摧 �m m ce (white bars). (e) In vitro binding curves of W55R NS4B mutant
(solid line,) and wild-type NS4B (broken line, ) to serial dilutions of the RNA
probe. Each data point represents the mean of 10–20 replicates, and the bars represent
the standard error.

29
We also introduced this mutation into the NS4B-GFP vector and tested it for
its RNA binding activity using the in vitro microfluidic assay. Although both mutant
and wild type NS4B proteins experienced ~ a 2 fold reduction of RNA binding in the
presence of 10nM clemizole, because the baseline RNA binding of the mutant is
higher, the residual amount of RNA bound by the mutant in the presence of clemizole
was comparable to that bound by the wild type in the absence of clemizole (Figure
2.5D). Furthermore, this mutant demonstrates greater apparent affinity to the viral
RNA with a Kd of 0.75nM (vs 3.4 nM for wild type NS4B) (Figure 2.5E).
The second clemizole-resistant mutation, termed R214Q, was identified in a
resistant colony as well as in pooled resistant cells. It involves the substitution of a
glutamine for the arginine at amino acid 214 within the cytoplasmic C-terminal
segment of NS4B. Similar results to the first mutation were obtained in cellular and in
vitro analyses done on this mutation, with an EC50 of 40.3µM (~5 times higher than
the EC50 on the wild type RNA) in the luciferase reporter linked replication assay and
a Kd of 0.6nM in the in vitro binding assay (data not shown). Presumably both of these
mutations alter the conformation of NS4B so as to increase its affinity for the viral
RNA. Indeed the Kds measured by the in vitro RNA binding assay reflect this. Taken
together, the above data provides compelling genetic and biochemical evidence for
clemizole’s mechanism of action.
2.3 Discussion
The above results demonstrate that HCV NS4B binds RNA and that this
binding is specific for the 3’ terminus of the negative strand of the viral genome.

30
Because the in vivo specificity of this interaction has not yet been determined
quantitatively, one should use the usual caution in extrapolating the numerical value of
our in vitro results. We also showed that an arginine rich–like motif in NS4B is
essential for RNA binding and for HCV replication. Clemizole hydrochloride was
found to have a substantial inhibitory effect on HCV RNA replication mediated by its
suppression of NS4B’s RNA binding.
Because NS4B is associated with membranes18 and is known to induce
membrane replication sites26, its RNA binding activity offers a mechanism to
incorporate the viral genome into the HCV replication compartment. This may
facilitate the initiation of synthesis of nascent positive strand from the membrane
anchored negative strand. NS4B may also act by recruiting the polymerase complex to
the HCV RNA, via its interaction with the NS5B polymerase or other components of
the replication complex57.
The in vitro IC50 of clemizole for RNA binding by NS4B is ~ 24 ± 1nM
(Figure 2.4d). It is possible that poor cellular permeability accounts for the ~400 fold
difference between the IC50 measured for in vitro RNA binding by NS4B and the EC50
measured for the antiviral effect in cells. We hypothesize that improved drug delivery
and optimization of the compound following structure-activity relationship (SAR)
analysis are expected to improve its potency as an antiviral agent, and that the
microfluidic platform can facilitate this process.
This study also demonstrates the utility of a microfluidic approach. The
microfluidic affinity assay has several important advantages over currently used

31
methods which make it a promising and general tool for drug discovery, especially
against membrane bound targets. First, protein synthesis by mammalian reticulocyte
lysates in the presence of microsomal membranes provides the natural conditions
required for protein folding. Second, the microfluidic assay eliminates the need for a
high level of protein expression and purification, a problem faced by currently used
methods. Third, this assay is capable of detecting transient and low affinity
interactions35. Fourth, the microfluidic device is capable of making many parallel
measurements, and therefore can be used for high throughput screening. Finally, we
have shown in spite of known chemical compatibility issues with the elastomer used to
fabricate the device, one can successfully screen and discover small molecules with
desired pharmacological properties.
As with HIV and tuberculosis, effective pharmacologic control of HCV will
likely best be achieved by a cocktail of multiple drugs against independent virus-
specific targets. Combination of even a moderate NS4B inhibitor with other emerging
anti-HCV agents represents an attractive paradigm for increasing current virologic
response rates. Although we hypothesize that more potent inhibitors than clemizole
can be obtained, because clemizole has already been extensively used in humans
(albeit for a different indication) it may find immediate use as a critical component of
next generation anti-HCV strategies.

32
2.4 Methods
Plasmids
Standard recombinant DNA technology was used to construct and purify all
plasmids. All regions that were amplified by PCR were analyzed by automated DNA
sequencing. Plasmid DNAs were prepared from large-scale bacterial cultures and
purified by a Maxiprep kit (Marligen Biosciences). Restriction enzymes were
purchased from New England Biolabs.
The open reading frames (ORF) of HuR and HuD were obtained from the
ORFeome library of cDNA clones (Open biosystems). These ORFs were inserted into
the expression vector pcDNA-Dest 40 vector (Invitrogen) by the use of gateway
technology (Invitrogen) allowing addition of a C-terminal V5-his tag.
The plasmid pcDNA-NS4B-GFP encoding NS4B of genotype 1a fused in
frame with a C-terminal GFP was previously described 18. The mutations RRa, RRb
and W55R were introduced into this plasmid by site-directed mutagenesis (using the
QuikChange kit (Stratagene)). The plasmid NS5A(AH)GFP was constructed as
previously reported . Gus-his vector was obtained from Roche.
The plasmids pcDNA3.1-5’ UTR pos which encodes the 5’ UTR of the
positive viral strand, was generated by amplification of the 5’ UTR positive sequence
from the Bart79I plasmid58 with primers containing EcoRV restriction sites, digestion
with EcoRV and ligation into the corresponding site in pcDNA3.1 (Invitrogen). The
plasmid pcDNA3.1- 3’ negative terminus which encodes the 3’ terminal region of the

33
negative RNA strand was generated the same way except that the EcoRV-flanked
insert was ligated in an inverse orientation.
The plasmids pcDNA3.1 – 3’ UTR pos and 5’ negative terminus were
similarly generated except that the inserted gene was flanked by HindIII and Xho1
restriction sites.
The vector encoding the delta virus genomic RNA sequence was cloned by
inserting NheI flanked HDV sequence into a pSPT19 vector (Roche Diagnostics) cut
with XbaI45, 59.
The plasmid FL-J6/JFH-5’C19Rluc2AUbi that consists of the full-length HCV
genome and expresses Renilla luciferase was a gift from Dr. Charles M. Rice53. The
W55R mutation was introduced into this plasmid by site-directed mutagenesis (using
the QuikChange kit (Stratagene)).
In vitro RNA transcription and fluorescent and radioactive labeling
Plasmid DNA of the 5’ and 3’ terminal regions of the negative and positive
viral strands were linearized with XbaI. The plasmid DNA of the delta genomic
sequence was linearized with XbaI. Linearized plasmids were then treated with
proteinase K, followed by phenol-chloroform extraction and precipitation with
ethanol. The DNA was resuspended in RNasefree water to a final concentration of 1
µg/µl. 4 µgs of DNA was used as a template for transcription with the T7 MEGAscript
(Ambion) according to the manufacturer’s protocol. The template DNA was digested
by the addition of 5 U of RQ1 DNase (Ambion) and a 15-min incubation at 37°C. The
unincorporated ribonucleotides were removed by size exclusion with a Micro Bio-

34
Spin P-30 column (Bio-Rad), and the transcribed RNA was extracted with phenol-
chloroform, followed by precipitation in ethanol. The RNA pellet was washed with
70% ethanol and resuspended in H2O. Determination of the RNA concentration was
performed by measurement of the optical density at 260nm. The integrity of the RNA
and its concentration were confirmed by 1% agarose gel electrophoresis and ethidium
bromide staining.
The RNA sequences were labeled with Cy5 by using Label IT kit (Mirus)
according to the manufacturer protocol followed by purification on a microspin
column (Mirus) and ethanol precipitation. The number of fluorescent labels per RNA
molecule was determined by measuring the spectrophotometric absorbance of the
nucleic-dye conjugate at 260nm and the λMAX for Cy5 (650nm). This was proportional
to the probes length and was used to adjust our binding experiments results.
Cy3-labeled RNA probes used to study RNA binding by HuR and HuD were
purchased from IDT.
Radioactive labeling of RNA probes with 32P was done as previously
described.60
Device design
Device fabrication and design was done essentially as described35. In brief, a
“control” layer, which harbors all channels required to actuate the valves, is situated
on top of a “flow” layer, which contains the network of channels being controlled. All
biological assays and fluid manipulations are performed on the flow layer. A valve is
created where a control channel crosses a flow channel. The resulting thin membrane

35
in the junction between the two channels can be deflected by hydraulic
actuation.Using multiplexed valve systems allows a large number of elastomeric
microvalves to perform complex fluidic manipulations within these devices.
Introduction of fluid into these devices is accomplished through steel pins inserted into
holes punched through the silicone. Computer-controlled external solenoid valves
allow actuation of multiplexors, which in turn allow complex addressing of a large
number of microvalves. Each unit cell is controlled by three micromechanical valves
as well as a“button” membrane (Supplementary Figure 2.1). The “button membrane”
of each unit cell masks a circular area within the flow channel between two
“sandwich” valves. Aligning and bonding a microarray to the device, allows
positioning of the spotted material (RNA probes or inhibitory compounds) within the
“RNA chamber” of each unit cell. This chamber can then be opened to the flow
channel by the control of a “neck valve”. The used devices had either 640 or 2400 unit
cells.
RNA binding assay
The approach used was a modification of the previously described method for
protein-DNA interactions35 (Supplementary Figure 2.1 online).
1. For soluble proteins:
A. RNA arraying and device alignment.First, dilution series of Cy3-labeled
target RNA sequences (IDT) were spotted as a microarray onto epoxy coated glass
substrates slide (CEL Associates) by OmniGrid Micro (GeneMachines) microarrayer
using a CMP3B pin (TeleChem International, Inc.) with column and row pitches of

36
680 mm and 320 mm, respectively. Each sample solution contained 1% BSA in dH2O
to prevent covalent linkage of the target RNA to the epoxy functional groups as well
as for visualization during alignment. After spotting the arrays were quality controlled
on a GenePix4000b (Molecular Devices). The arrays were then aligned to a
microfluidic device by hand on a SMZ1500 (Nikon) stereoscope and bonded overnight
in the dark on a heated plate at 40ºC.
B. Surface chemistry (Supplementary Figure 2.1c online). All devices were
driven between 10 and 15 psi in the control line and between 4 and 6 psi for the flow
line. For the initial surface derivatization steps the chamber valves remained closed to
prevent liquid from entering the chambers containing the spotted RNA targets. First,
all accessible surface area was derivatized by flowing a solution of biotinylated BSA
(Pierce) resuspended to 2 mg/mL in dH2O for 30 min through all channels, followed
by a 10 min PBS wash. Next a 500 μg/mL Neutravidin (Pierce) solution in PBS was
flown for 20 min, followed by a 10 min PBS wash. Next, the”button” membrane was
closed and the PBS wash continued for an additional 5 min. Then all remaining
accessible surface area except for the circular area of 60 μm masked by the button was
passivated with the same biotinylated solution as above for 30 min, followed by a 10
min PBS wash. Finally a 1:1 solution of biotinylated-6-histidine antibody (Qiagen) in
2% BSA in PBS was loaded for 2-5 min, after which the ”button” membrane was
opened and flow continued for 20 min allowing specific functionalization of the
previously masked circular area. This was again followed by a 10 min PBS wash
completing the surface derivatization procedure.

37
C. Protein synthesis and MITOMI. Next a standard 25 μL TNT T7 coupled
wheat germ extract mixture (Promega) was prepared and spiked with 1 μL
tRNALys−bodipy−fl (Promega) and 2 μL of plasmid DNA template coding for the
appropriate protein (HuR, HuD or Gus). The mixture was immediately loaded into the
device and flushed for 5 min, after which the chamber valves were opened allowing
for dead end filling of the chambers with wheat germ extract. The chamber valves
were again closed and flushing continued for an additional 5 min. Next the segregation
valves separating each unit cell were closed followed by opening of the chamber
valves allowing for equilibration of the unit cell by diffusion. The entire device was
heated to 30ºC on a temperature controlled microscope stage and incubated for up to
90 min to complete protein synthesis, binding of protein to the surface bound
biotinylated-6-histidine antibody, solvation of target RNA, and equilibration of
proteins and target RNA. MITOMI was then performed by closing the ”button”
membrane as well as the chamber valves allowing trapping surface-bound complexes
while expelling any solution phase molecules. Radial closure prevents solvent pockets
from forming between the two interfaces and effectively creates zero dead volume
while preserving the equilibrium concentrations of the molecular interactions to be
detected. This was followed by a 5 min PBS wash to remove untrapped unbound
material. The device was then imaged on a modified arrayWoRxe (AppliedPrecision)
microarray scanner to detect protein synthesis and the trapped molecules.
D. Image and Data Analysis. All images were analyzed with GenePix3.0
(Molecular Devices). For each experiment an image was taken after MITOMI and the
final PBS wash. This was used to determine the concentration of surface bound

38
protein (FITC channel) as well as surface bound target RNA (Cy3 channel). Each unit
cell generated a single data point that consisted of the ratio of median signal of Cy3 to
median signal of bodipy; thus, representing the ratio of surface bound RNA to surface
bound expressed protein. Data from multiple data points were averaged and their
standard deviations calculated. Results were then normalized to 1. Between 10-20
replicates were included for each probe tested. A control protein, Gus-his (Roche) was
included in each experiment and subjected to the same analysis. When present,
detected signal with Gus-his was used for background subtraction.
2. For membrane-associated proteins.
Detection of RNA binding by NS4B was done similarly to the described above
except for several modifications. NS4B was expressed off the microfluidic device by
using coupled transcription/translation rabbit reticulocyte system (Promega). 2 μL
canine microsomal membranes (Promega) were added to the reaction mixture to allow
appropriate protein folding. An NS4B-GFP construct was used thus eliminating the
need for tRNALys−bodipy−fl in the reaction mixture. A biotinylated anti-GFP antibody
(Abcam) was used to specifically functionalize the protected circular area defined by
the button. The protein was anchored to the chip via its interaction with the surface
bound anti-GFP antibodies. The surface bound protein and the Cy5-labeled HCV RNA
probes were loaded into the chip and allowed to incubate for 5-20 min in a 50mM
Hepes KOH (pH 6.8) buffer. Next, MITOMI was performed followed by a brief wash
in Hepes KOH (pH 6.8) buffer. The device was then scanned and results quantified as
described above. NS4B-GFP mutants and NS5A(AH)-GFP were assayed the same
way. Signal detected with NS5A(AH)-GFP was used for background subtraction.

39
Screening of inhibitory compound library
The 1280 compounds of the Lopac library (Sigma) solubilized in Dimethyl
sulfoxide (DMSO) were spotted onto epoxy coated glass substrates slide (CEL
Associates) by OmniGrid Micro (GeneMachines) microarrayer using a CMP3B pin
(TeleChem International, Inc.) as a microarray. For the primary screen compounds
were spotted at a high concentration (~1mM) in duplicates. The array was allowed to
dry, and was then aligned and bonded to a microfluidic device. Two large devices
(2400 unit cells per device) were used for the primary screen. The rest of the
procedure was done similarly to the described above (RNA binding assay). In brief,
the device was subjected to surface patterning that resulted in a circular area coated
with biotinylated anti-GFP antibodies within each unit cell. Next, NS4B-GFP
expressed off chip using coupled transcription/translation rabbit reticulocyte system
(Promega) in the presence of microsomal membranes (Promega) was loaded into the
chip and bound to the surface biotinylated anti-GFP antibodies. Cy5-labeled 3’ neg
terminus RNA probe was then loaded at a concentration of 1.5nM. Each unit cell was
then isolated followed by a 30 min incubation to allow binding of the protein to
surface biotinylated anti-GFP antibodies, solvation of library compounds, and
equilibration of proteins and target RNA. Next, MITOMI was performed trapping
surface-bound complexes while expelling any solution phase molecules. After a brief
wash to remove untrapped unbound material the device was scanned and results
analyzed. The ratio of bound RNA to expressed protein was calculated for each data
point by measuring the median signal of Cy3 to median signal of bodipy. Results were
normalized to signal measured in unit cells containing no inhibitory compound.

40
Greater than 90% inhibition was defined as the cutoff for inhibition in the primary
screen. 104 compounds which were above this cutoff and additional 110 yielding
ambiguous results were subjected to a secondary screen. This was performed
similarly, except that two smaller devices (640 unit cells per each) were used, the
spotted compound concentration was 10 fold lower than in the primary screen and 5
replicates were spotted for each compound. Inhibition greater than 2.5 fold was
considered significant. 18 compounds identified in this screen were further analyzed
for their antiviral effect on HCV RNA replication.
Determination of IC50 for in vitro RNA binding
For an accurate measurement of IC50s, serial dilutions of the inhibitory
compound were loaded onto the microfluidic device by continuous flow while
maintaining a steady concentration of the compound in the flow channel. This helped
to avoid expected losses of the spotted compounds from incomplete solubilization
and/or binding of the compound to PDMS. The experiment was performed essentially
as described in RNA binding assay for transmembrane proteins except that the
expressed protein and the Cy5-labeled HCV RNA probes were incubated in the device
in the presence of the inhibitory compounds or their absence. IC50s were measured as
described in the Statistical Analysis section below.
Expression and purification of recombinant NS4B
GST-NS4B and GST were expressed in E.coli BL21 and purified as described
elsewhere 20. NS4B was fused in frame with an N-terminal 6his-MISTIC protein
(membrane-integrating sequence for translation of IM protein constructs)61. Overnight

41
cultures of E.Coli transformed with mistic-NS4B plasmid were diluted 1:100 in 400ml
of fresh medium and grown at 37ºC to an OD of 0.8. Isopropyl--D-
thiogalactopyranoside (IPTG) (Invitrogen) was then added to a final concentration of
0.1mM. After 3 hours growth at 37ºC cells were pelleted and resuspended in 30ml
lysis buffer (50mM NaCl, 50mM Tris HCL (pH8), 100mM Imidazole, 10mM
Decylmaltoside, Complete EDTA free protease inhibitors (Roche Applied Science)).
Cells were lysed by one cycle in a French Press at a pressure of 10,000 psi for 1
minute, followed by centrifugation at 12,000xg for 5 minutes at 4ºC. The supernatant
was loaded on nickel column (Amersham). Following washes, protein was eluted in a
buffer containing 400mM Imidazole. Glycerol was added at a final concentration of
20% and samples were stored at -20ºC. Purification was monitored by SDS-PAGE.
Total protein concentration was measured using the RC-DC assay (Bio-Rad). NS4B-
mistic was identified by Western blot analyses using monoclonal antibodies against 6-
his (Santa Cruz Biotechnology) and NS4B (Virostat). 6his-mistic was expressed and
purified in a similar manner.
GST pull down assay
Similar to a previously described strategy 62, 1g of purified GST-NS4B or
GST was incubated for an hour at 37ºC in a 50l reaction mixture containing 32P-
labeled in vitro transcribed HCV RNA (corresponding to the 3’ terminus of the
negative viral strand) and binding buffer (10mM DTT, 10mM Na HEPES (pH 7.4),
33mM NaCl, 0.1mM EDTA, 10mM MgCl2). 50l of tRNA pre-coated glutathione-
agarose beads (Sigma) were then added, followed by 1 hour incubation at 4ºC to allow

42
binding of GST to the beads. The beads were then washed three times in binding
buffer and bound RNA was measured by liquid scintillation counting of sample
aliquots. Control incubations with an RNA probe prepared in the absence of T7 RNA
polymerase were used for background subtraction.
RNA Filter Binding Assay
Assays were performed essentially as described 60. Briefly, various
concentrations of mistic-NS4B protein or mistic control were incubated for 1 hour at
30°C with 3.3 nM 32P-labeled in vitro transcribed HCV RNA probe in binding buffer
(50 mM HEPES pH 7.0, 50 mM NaCl, 1 mM MgCl2, 10 ng/µl tRNA, and 0.2mM
Decylmaltoside) in a final volume of 40 µl. Membranes were pre-soaked in the
binding buffer and assembled in a dot blot apparatus (Schleicher & Schull) from top to
bottom as follows: nitrocellulose (Biorad), Hybond N+ (Amersham Biosciences),
Whatman 3mm filter paper. The binding reactions were loaded onto the dot-plot
apparatus and filtered through the membranes. After washing, the membranes were
air-dried and visualized by Phospho-imaging. Results represent percentage of bound
RNA calculated by dividing the signal detected in the nitrocellulose membrane by the
sum of the signals detected in the nitrocellulose and the Hybond membranes.
Cell cultures and electroporation
Huh-7.5 cells were maintained in Dulbecco’s modified minimal essential
medium (Gibco) supplemented with 1% L-glutamine (Gibco), 1% penicillin, 1%
streptomycin (Gibco), 1X nonessential amino acids (Gibco) and 10% fetal bovine
serum (Omega Scientific). Cell lines were passaged twice weekly after treatment with

43
0.05% trypsin–0.02% EDTA and seeding at a dilution of 1:5. Subconfluent Huh-7.5
cells were trypsinized and collected by centrifugation at 700 X g for 5 min. The cells
were then washed three times in ice-cold RNase-free PBS (BioWhittaker) and
resuspended at 1.5*107 cells/ml in PBS. Wild type or mutant FL-J6/JFH-
5’C19Rluc2AUbi RNA for electroporation was generated by in vitro transcription of
XbaI-linearized DNA templates using the T7 MEGAscript kit (Ambion), followed by
purification, essentially as described above (In vitro RNA transcription and fluorescent
labeling). 5 µg of RNA was mixed with 400µl of washed Huh-7.5 cells in a 2mm-gap
cuvette (BTX) and immediately pulsed (0.82 kV, five 99 µs pulses) with a BTX-830
electroporator. After a 10 min recovery at room temperature, pulsed cells were diluted
into 10ml of prewarmed growth medium. Cells from several electroporations were
pooled to a common a stock and seeded in 6 well plates (5*105 cells per well). After
24 hr, medium was replaced and cells were grown in the presence of serial dilutions of
the various inhibitory compounds (Sigma) identified in the screen. 17 commercially
available compounds, out of the 18 identified, were analyzed. Untreated cells were
used as a negative control for water soluble compounds. For compounds solubilized in
DMSO, untreated cells were grown in the presence of corresponding concentrations of
the solvent as a negative control. Medium was changed daily. After 72hr of treatment
cells were subjected to an alamar blue based viability assay and luciferase assay.
Viability assay
Following 72 hrs of treatment cells were incubated for 3 hrs at 37ºC in the
presence of 10% Alamar Blue reagent (TREK Diagnostic Systems). Plates were then

44
scanned and fluorescence was detected by using FLEXstation II 384 (Molecular
Devices, Inc.). Depending on the inhibitory compound’s solvent, water or DMSO,
signal was normalized relatively to untreated samples or samples grown in the
presence of DMSO, respectively.
Luciferase assay
Viral RNA replication was determined using Renilla luciferase assay
(Promega). The same samples subjected to the viability assay described above were
analyzed in this assay. According to the manufacturer protocol, cells were washed
with ice cold PBS and scraped off the plate into 250 µl of ice-cold Renilla lysis buffer.
20 µl of the cell lysates were then loaded onto 96 well plates. 100 µl of the Renilla
luciferase assay buffer containing the assay substrate were injected and luciferase
activity was measured using a Berthold LB 96 V luminometer. As above, signal was
normalized relative to untreated samples or samples grown in the presence of the
corresponding concentration of DMSO.
Luciferase activity detected in samples treated with 100 u/ml Interferon alpha
B2 (PBL biomedical labs) was used as a positive control, demonstrating three log
reduction at 72hr treatment. The experiment was repeated four times, each time with
triplicates. IC50s were measured by fitting data to a three parameter logistic curve
using the formula Y=a+(b-a)/(1+10^(X-c)) (BioDataFit, Chang Bioscience, Inc).

45
Real-time PCR
5x104 Huh7.5 ells were infected with cell culture-grown HCV titered at
1.4x104 TCID50/ml, as described 54. 2 hours after infection, cells were washed three
times in culture medium. Cells were then treated daily with various concentrations of
clemizole. After 72 hours samples were subjected to the viability assay described
above, following which TRIzol Reagent (Invitrogen) was added and total cell RNA
was extracted in triplicates according to the manufacturer’s instructions. Reverse
transcription was then performed using random hexamers and Superscript II reverse
transcriptase (Invitrogen, Carlsbad, CA). Real-time PCR was performed on the
resulting cDNA to quantify the amounts of HCV and actin RNA (in separate
reactions) in each sample. Standards were made using an in vitro-transcribed HCV
RNA and human actin standard (Applied Biosystems, Foster City, CA). HCV was
quantified using primers AGAGCCATAGTGGTCT and
CCAAATCTCCAGGCATTGAGC and probe 6-carboxyfluorescein-
CACCGGAATTGCCAGGACGACCGG-6-carboxytetramethylrhodamine. Actin was
quantified using beta-actin control reagents (Applied Biosystems) according to the
manufacturer’s instructions. HCV RNA level was adjusted to actin level and
normalized relative to untreated samples.
Selection of resistant mutants
Established HCV replicons-harboring cells and Huh7 cells electroporated de
novo with a genotype 1b subgenomic HCV replicon (Bart 79I) 55 were passaged in the

46
presence of neomycin and increasing concentration of clemizole (1-16 µM). Colonies
that were able to grow in the presence of the compound were isolated and propagated
for 5-10 passages. Eleven colonies (out of ~60) survived the passages and were
subjected to sequence analysis, as previously described 20.
Whole cell RNA electroporation
Whole cell RNA was extracted from clemizole-resistant replicon clones and
from untreated replicon cells using TRIzol reagent (Invitrogen). Equal amounts of
whole cell RNA (50µg) were electroporated into Huh7.5 cells as described above.
Cells were grown under G418 selection in the presence or absence of clemizole for 3
weeks. The number of colonies was determined using Image J (NIH) following
fixation and staining with Crystal violet.
Statistical analysis
Dissociation equilibrium constants were determined by fitting data to the
equation describing equilibrium binding; Y = a*X/(b+X) (a and b represent maximum
binding and Kd, respectively) by nonlinear least squares regression fitting (BioDataFit,
Chang Bioscience). IC50s were measured by fitting data to a three parameter logistic
curve using the formula Y=a+(b-a)/(1+10^(X-c)) (a, b and c represent minimum
binding, maximum binding and logEC50, respectively)(BioDataFit, Chang Bioscience,
Inc).

47
Chapter 3. Mutations in NS3 compensate for the disruption of an RNA-binding domain in NS4B

48
3.1 Introduction
NS4B contains an RNA-binding activity
As described in Chapter 2, NS4B harbors the ability to bind HCV RNA.
Molecular determinants of this interaction were identified in both the protein and RNA
partners. The 3’ terminus of the negative-sense HCV RNA genome was shown to bind
to NS4B with higher affinity than the other regions of the genome (Figure 2.2).
Meanwhile, two putative RNA-binding domains (RBDs) were identified in the NS4B
sequence, RR192 and RR247 (Figure 2.3). When both arginines of either of these two
domains were mutated to alanines, binding was inhibited. In the case of RR192,
binding was almost completely abrogated, while for RR247, binding was inhibited
roughly 5-fold (Figure 2.3D). Therefore, these two domains are important for RNA
binding in vitro.
In addition, clemizole hydrochloride was identified as an inhibitor of this RNA
binding activity (Figure 2.4). We determined that this compound not only inhibited
RNA binding in vitro, but also impaired replication in cells. These data support our
hypothesis that RNA binding is important for HCV replication in cells, a hypothesis
we will further explore in this chapter.
NS4B interacts with NS3
As described in Chapter 1, HCV replication takes place in a replication
complex. All the viral nonstructural proteins have been localized to this complex, and
it is likely that these proteins physically interact with each other in order to achieve

49
efficient viral replication. In support of this idea, numerous protein-protein
interactions have been observed among the HCV NS proteins63, 64. Because these
proteins are so intimately involved with one another, it is likely that defects in one
nonstructural protein can be suppressed by mutations in another. In fact, NS3 has been
shown to perform such a role in the context of a defective NS4B; a number of amino
acid substitutions in the former have been identified that compensate for defects in the
latter65.
In this chapter, we hypothesize that the putative RBDs are important for HCV
replication in cell culture. We further hypothesize that defects in these RBDs can be
suppressed by secondary-site mutations in the virus. To test these hypotheses, we
examined the replication capacity of HCV replicons containing arginine to alanine
mutations at the RBDs. We also isolated revertant colonies, identified secondary-site
mutations and tested them for suppression of the KR247AA defect.
3.2 Results
RBD mutations prevent HCV replication
To test whether NS4B’s putative RNA-binding domains are important for
HCV replication, we constructed HCV replicons that contained one of the following
mutations: two consecutive arginines at amino acid 192-193 of the NS4B sequence to
two alanines (RR192AA) or a lysine-arginine sequence at amino acid 247-248 to two
alanines (KR247AA)a. Along with a wild-type and polymerase-defective mutant
a Note that the replicon studied is genotype 1b, not 1a as was tested in the in vitro experiments of Chapter 2. Thus, the 247th amino acid is a lysine, not an arginine.

50
construct, these constructs were electroporated into Huh7 cells, and propagated for
three weeks under G418 selection. The resulting colonies were stained and counted
(Figure 3.1).
Wild-type RNA gave many more colonies than both RBD mutants, yielding a
mean of 3330 colonies per g of RNA electroporated (N = 3, SEM = 450). For the
RR192AA mutant, only 4 colonies were observed over 5 experiments, for a mean of
0.8 colonies / g RNA. For the KR247AA mutant, the observed mean was 72 colonies
/ g RNA (N = 3, SEM = 8), roughly 2% of wild-type. No colonies were observed for
the polymerase-defective mutant. These data are consistent with the clemizole
inhibition experiments of Chapter 2; there, the pharmacological disruption of RNA
binding also led to an inhibition of viral replication.
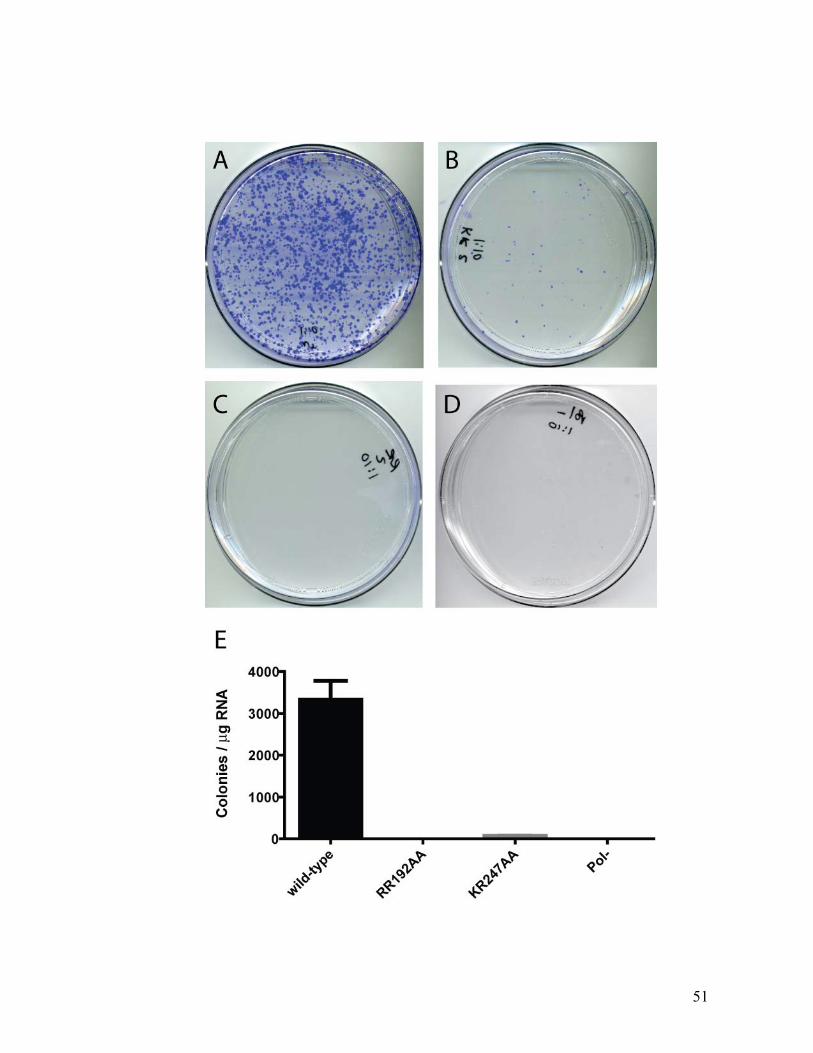
51

52
Figure 3.1. Putative RNA binding domains are necessary for HCV replication.
Colony formation assays were performed with HCV replicons containing either (A)
wild-type, (B) RR192AA, (C) KR247AA, or (D) polymerase defective sequence. The
number of colonies was counted for each sample, and the results are displayed in (E).
Error bars represent SEM.

53
RBD mutations do not affect HCV localization
One possibility for the reduction in viral replication is that the genetic
disruption of the RBD impairs the expression or proper folding of NS4B and that this
inhibits replication. To test this possibility, we monitored the subcellular distribution
of the HCV nonstructural protein replicase complex. Because antibodies for NS4B are
not readily available, we chose to follow the distribution of another nonstructural
protein, NS5A. NS4B’s proper localization has been shown to be essential for the
membrane-associated foci distribution pattern of NS5A18; therefore, we hypothesized
that if NS4B was improperly folded, NS5A’s localization pattern would be disrupted.
To test this hypothesis, we infected Huh7 cells with a vaccinia virus expressing
the T7 polymerase, then transfected these cells with replicon constructs driven by a T7
promoter that contained a wild-type, RR192AA, or KR247AA HCV replicon. After 4
hours, these cells were fixed and subjected to immunofluorescence analysis. As seen
in Figure 3.2, NS5A’s pattern of subcellular distribution in each of the mutant samples
was reticular, with a predominance of foci in the perinuclear region. This localization
pattern matches that which we observed and has been reported elsewhere for wild-
type; therefore, it is likely that these mutations do not affect NS4B’s subcellular
localization.
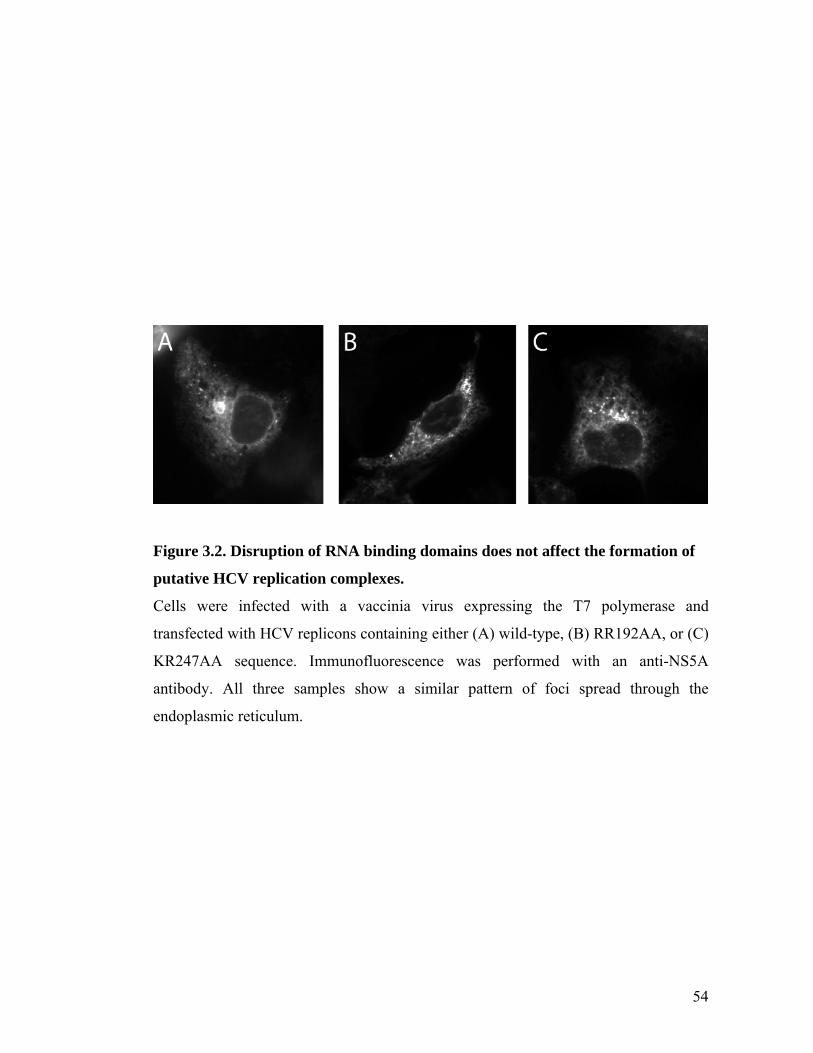
54
Figure 3.2. Disruption of RNA binding domains does not affect the formation of
putative HCV replication complexes.
Cells were infected with a vaccinia virus expressing the T7 polymerase and
transfected with HCV replicons containing either (A) wild-type, (B) RR192AA, or (C)
KR247AA sequence. Immunofluorescence was performed with an anti-NS5A
antibody. All three samples show a similar pattern of foci spread through the
endoplasmic reticulum.

55
Primary-site revertants are not observed
In addition to staining and counting colonies from the electroporations, some
colonies were isolated and clonally expanded, in order to determine the mechanism by
which they overcame the RBD defect. RNA was harvested from these clones and was
reverse-transcribed, amplified and sequenced. Notably, no HCV sequence was
detected in the 3 isolated RR192AA colonies. Of the 20 colonies isolated from the
KR247AA samples, all were HCV positive. However, none of the clones had reverted
to the original KR sequence at position 247 of the NS4B region. Instead, some clones
had accumulated one or more mutations in other parts of the HCV genome. In others,
we did not detect any mutations in the genome.
All the mutations observed in the revertant clones are identified in Figure 3.3.
Figure 3.3A highlights the location of all observed mutations in the HCV genome,
while Figure 3.3B lists the type and location of the substitution in the full-length HCV
polyprotein. In addition, Figure 3.3B lists whether or not these mutations suppressed
the KR247AA defect in a transient transfection assay (described below). Those that
did suppress the defect are highlighted in red in Figure 3.3A.

56
Figure 3.3. Replicating colonies containing KR247AA mutation contain
secondary-site mutations.
RNA harvested from cells replicating under G418 selection was reverse-transcribed,
amplified, and sequenced. No reversions to KR at the RBD were observed. (A)
Twelve other substitutions were observed throughout the genome (*). Two of these
mutations, Q1112R and S1200Y, were confirmed to restore replication to wild-type
levels or above in a luciferase assay (indicated in red). (B) All the observed mutations
are listed, including in which clone they were found, whether they improved
replication in KR247AA replicons, and whether they also improved replication in
wild-type replicons. n.d. = not determined.

57
Secondary-site mutations in NS3 restore HCV replication in
KR247AA replicons
Having identified a dozen mutations that arose under G418 selection, we
assayed each of these mutations to determine whether it could suppress the original
KR247AA defect in NS4B. We generated constructs containing the KR247AA
mutation and one of each of the additional observed secondary-site mutations. These
constructs were generated in a luciferase reporter vector, which was used to test the
replication capacity of the constructs in transient transfection assays.
The majority of the constructs did not improve replication levels above that of
the KR247AA alone mutant (Figure 3.3B). However, two of the mutations in NS3 did
enhance replication levels, Q1112R and S1200Y. To our surprise, the KR247AA
Q1112R mutant replicated 3-fold more efficiently than the wild-type replicon (Figure
3.4). Meanwhile, the KR247AA S1200Y mutant replicated at virtually the same level
as the wild-type. Therefore, these mutations in the N-terminal region of NS3 suppress
the defect of the KR247AA mutation in NS4B.
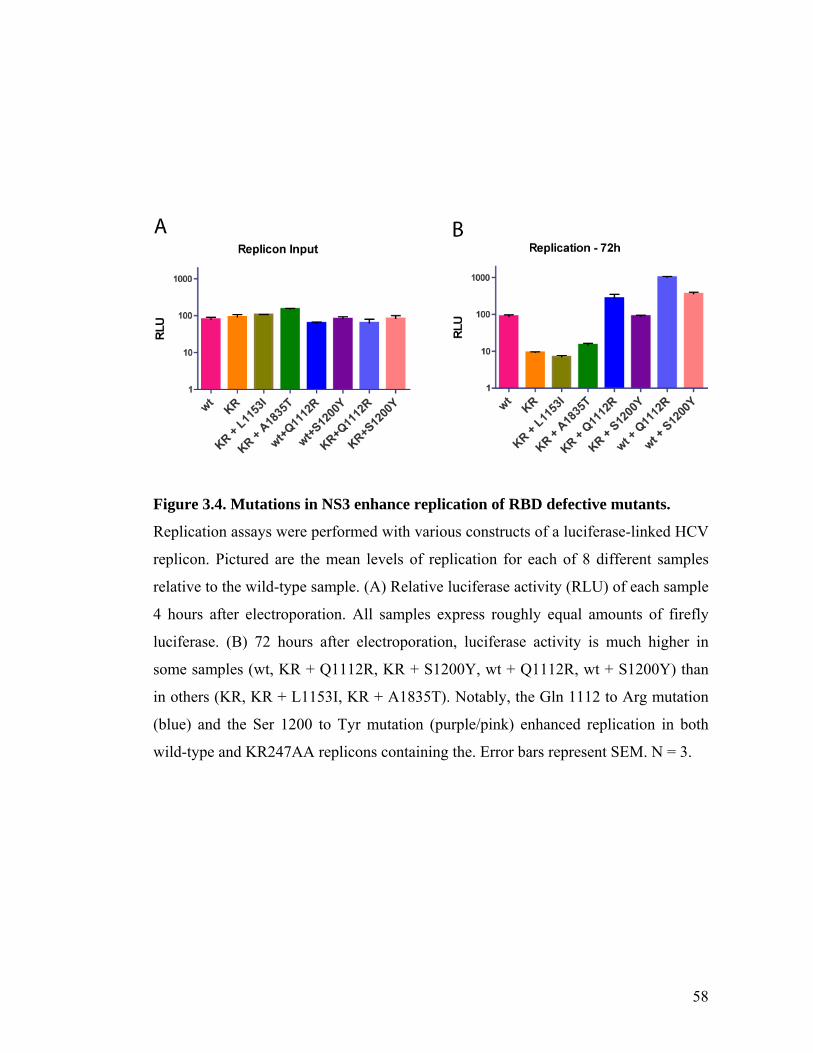
58
Figure 3.4. Mutations in NS3 enhance replication of RBD defective mutants.
Replication assays were performed with various constructs of a luciferase-linked HCV
replicon. Pictured are the mean levels of replication for each of 8 different samples
relative to the wild-type sample. (A) Relative luciferase activity (RLU) of each sample
4 hours after electroporation. All samples express roughly equal amounts of firefly
luciferase. (B) 72 hours after electroporation, luciferase activity is much higher in
some samples (wt, KR + Q1112R, KR + S1200Y, wt + Q1112R, wt + S1200Y) than
in others (KR, KR + L1153I, KR + A1835T). Notably, the Gln 1112 to Arg mutation
(blue) and the Ser 1200 to Tyr mutation (purple/pink) enhanced replication in both
wild-type and KR247AA replicons containing the. Error bars represent SEM. N = 3.

59
Finally, we sought to determine whether these two mutations in NS3 enhance
HCV replication only in the context of the KR247AA mutation or whether they also
enhance replication in an otherwise wild-type replicon. We generated two constructs
in a wild-type backbone, one containing the Q1112R mutation and the other
containing the S1200Y mutation, and tested them in the transient transfection assay.
As shown in Figure 3.4, these mutations also enhanced the replication capacity of
wild-type replicons. The introduction of the Q1112R mutation to a wild-type replicon
enhanced replication by 10-fold, while the introduction of this mutation to a
KR247AA replicon increased replication by 30-fold. The S1200Y mutation enhanced
wild-type replication by 3.6-fold, while it enhanced KR247AA replication by 9.6-fold.
Thus, these mutations enhance HCV replication in a general manner, and they increase
replication more dramatically in the context of the KR247AA mutants.
3.3 Discussion
In this chapter, we have shown that the domains important for RNA binding in
vitro are also important for HCV replication in cells. Furthermore, one of these
domains, RR192, was found to be essential for replication, while the other could be
complemented. These results correlate with our previous in vitro binding data, in
which RR192 was essential for RNA binding, while KR247 appeared to play a
secondary role. We also determined that the formation of the replication complex does
not appear to be affected in either of these mutants, and that revertant colonies did not
contain a primary-site reversion. Finally, two mutations in the NS3 region of the HCV
genome were found to suppress the KR247AA defect.

60
These two mutations, Q1112R and S1200Y, are both located in the protease
domain of NS3. In their study of NS4B:NS3 interactions, Paredes et al.65 found that
the exact same mutation Q1112R could compensate for a more general genetic defect
in NS4Bb. Moreover, they found several additional mutations within the NS3 protease
domain, P1115R, E1202G, and M1205K. As described in that paper, and as shown in
Figure 3.5, all these mutations cluster together in the same region of the quaternary
protein structure. This region is distal to the protease active site and may be important
in interacting with the NS4B protein. In agreement with their data, the S1200Y
mutation is also located in this region, supporting the hypothesis that these mutations
might suppress the NS4B defect through a common mechanism.
As demonstrated by Figure 3.4, each of these two mutations causes a
significant increase in viral replication in wild-type viruses. This suggests that these
mutations may allow for more efficient replication in general. Such an improvement in
efficiency may compensate for the moderate impairment conferred by the KR247AA
mutation. Interestingly, these data do not agree with the report in which Q1112R was
originally identified65. While our luciferase assay showed an increase in replication of
greater than 10-fold (Figure 3.4), Paredes et al. found an increase of less than 1.5-fold.
This disparity may be partly due to minor differences in the HCV replicons used, to
differences in the cell lines used, or perhaps differences in the sensitivities of the assay
used in each case.
b To impair NS4B, the authors created a hybrid replicon containing a genotype 1a NS4B within the context of a genotype 1b backbone.

61
Figure 3.5. Location of compensatory mutations in NS3-4A dimer.
A ribbon diagram of the 3-D structure, as obtained from the protein data bank
(accession number 1CU1), of an NS3-4A dimer is shown. On the upper monomer
(green), the amino acids comprising the protease active site and the compensatory
mutations identified in this manuscript are shown in red. On the lower monomer
(blue), the same amino acids, as well as those previously identified to be
compensatory in the NS3 protease domain are shown in red. These mutations cluster
together in one region of the protein that is not part of the protease active site.
Underlined amino acids indicate those found in this study. The image was generated
with 3D Molecule Viewer of the Vector NTI suite (Invitrogen).

62
In addition, data from patient isolates show that these two substitutions,
Q1112R and S1200Y, are not commonly found in vivo66. Position 1112 is not highly
conserved in patients, and proline, glutamine, leucine, serine, alanine, histidine, and in
one case, arginine, have been observed at that location. In contrast, position 1200 is
highly conserved, with 71% of isolates containing a serine at that location, and 99%
containing either a serine, threonine, or alanine. Tyrosine has not been observed at that
position. Based on these data, it is likely that any enhancement of replication observed
in vitro by these mutations is counterbalanced by a selective disadvantage in vivo. In
the context of developing antiviral drugs that can achieve the same effect as the
KR247AA mutation, such an observation is encouraging in that these Q1112R and
S1200Y resistant mutants may not evolve in infected patients.
While the KR domain has been shown to be important for RNA binding in
vitro, it is possible that this domain may serve another function in the context of viral
replication. Therefore, future experiments will be targeted towards testing NS4B’s
RNA-binding activity in cells and determining the importance of these RBDs to such
an activity. To accomplish this, infection-transfection experiments may be performed,
as in Figure 3.2, in combination with biochemical RNA:protein interaction assays.
Methods
Plasmids
For the colony formation assays, the Bart79I plasmid20 was used. For the
luciferase assays, the Bart79ILuc* (Chapter 4) was used. Cloning of all mutations in

63
these plasmids was performed using the Quikchange method (Stratagene). Primers for
each of these mutations are listed in Table 3.1.
Primer name Primer Sequence (5’ to 3’) RR192AA AGCGATACTGGCTGCGCACGTGGGCCCA KR247AA TCAGCTGCTGGCGGCGCTTCACCAGTGGA Q1112R CGTCGGCTGGCGAGCGCCCCCCG P1142S GGCATGCCGATGTCATTTCGGTGCGCC L1153I CAGGGGGAGCCTAATCTCCCCCAGG R1157K GAGCCTACTCTCCCCCAAGCCCGTCTC S1200Y CTTTGTACCCGTCGAGTATATGGAAACCACTATGC A1835T GCGCCGGCATCACTGGAGCGGCT S2466N CATCTCGCAGCGCAAACCTGCGGCAGAAG Q2750H GCGGGGACCCATGAGGACGAGGC
Table 3.1. Primer sequences
Colony formation assay
Colony formation assays were performed as described20, using a minimum of
three separate electroporations for each of the wild-type, RR192AA, KR247AA, or
pol- RNA. Plates were harvested 20 days post-electroporation and stained with 0.1%
crystal violet.
Infection-transfection
Infection-transfection assays were performed as described18. 2 g of Bart79I
plasmid was transfected after infection with vaccinia virus at a MOI of 5. Coverslips
were fixed at 4 hours post-transfection and immunostaining was performed as
described in Chapter 4, using a monoclonal NS5A antibody67 (1:1000).

64
Picking colonies, RT-PCR, & sequencing
Colonies growing under G418 selection were picked using sterile cloning disks
and expanded under G418 selection until growth had exceeded a 6-well dish. Cellular
RNA was harvested with Trizol according to the manufacturer’s instructions
(Invitrogen). HCV replicon RNA was reverse-transcribed and amplified with
Superscript II (Invitrogen), using gene-specific primers. The PCR products were
directly sequenced and compared to wild type sequence. Of the 20 colonies isolated,
all were sequenced in the NS4B and 5’UTR regions. For the remainder of the HCV
genome, approximately 4 clones were sequenced for each region.
Luciferase assay
Luciferase assays were performed as described in Chapter 4. 5 g of plasmid
DNA was electroporated into Huh7.5 cells. Plasmids used contained wild-type, the
KR247AA mutation, or a combination of the KR247AA mutation and one of Q1112R,
P1142S, L1153I, R1157K, S1200Y, A1835T, S2466N, Q2750H, or wild-type and one
of Q1112R or S1200Y. Cells were harvested at 4 hours, 72 hours, and 96 hours post-
electroporation. At the time of harvest, an alamarBlue© viability assay was performed
according to the manufacturer’s recommendations (Invitrogen). Reported luciferase
values are normalized to each samples viability value.

65
Chapter 4. A small molecule inhibits HCV replication and disrupts NS4B’s subcellular distribution

66
4.1 Introduction
Worldwide, 170 million people are infected with Hepatitis C Virus (HCV), a
(+) sense, single-stranded RNA virus1. These individuals experience significant
morbidity related to their infection, and mortality rates are high. Moreover, treatment
options for this disease are inadequate for the majority of patients. The standard of
care regimen, which relies on pegylated interferon- plus ribavirin, is successful in
only about 50% of patients; moreover, it is associated with significant side effects8.
Therefore, better antiviral therapies are needed to address this public health problem.
As a better picture of the molecular biology of HCV has emerged, a number of
specific antivirals have been identified, and several pharmaceuticals that target the
viral nonstructural proteins NS3 and NS5B have progressed into phase II clinical
trials12. Notably, resistant mutants have already been observed for both these types of
compounds68; therefore, it is likely that a combination of antivirals will provide the
best sustained response in patients.
One potential target for a complementary antiviral is the HCV nonstructural 4B
(NS4B) protein. Several functions have been ascribed to this protein, including the
ability to rearrange cellular membranes into a so-called membranous web, visualizable
through electron microscopy and believed to represent the viral replication platform13,
and to form membrane-associated foci (MAF), believed to reflect, in part, the light
microscopic equivalent of the membranous web16. Furthermore, NS4B can bind and
hydrolyze GTP20, bind to HCV RNA22, and interact physically with other

67
nonstructural components of the HCV replication complex63. Any one of these
functions represents a potential target for pharmacological disruption.
Recently, Chunduru et al. have identified 23 small molecules that bind to
NS4B with a dissociation constant (KD) to NS4B of 5 M or less69. Of those leads, 4
molecules were shown to inhibit HCV replication with an EC50 of 1 M or less, as
measured by an ELISA for NS5A protein levels. Here, we focus on one of these
compounds, which we designate “anguizole.” We show that this compound indeed
inhibits HCV RNA replication with little toxicity to host cells. Furthermore, we
provide genetic and biochemical evidence that NS4B is the target of this drug, and we
demonstrate that its mechanism of action proceeds through the disruption of MAF
formation. Finally, our in vitro dynamic light scattering assay suggests that the second
N-terminal amphipathic helix (AH2) of NS4B interacts with this compound, and we
identify a mutation in the N-terminal region that confers resistance to the drug.
4.2 Results
The small molecule, anguizole, inhibits HCV RNA replication.
Chunduru et al.69 have recently performed a large-scale screen for small
molecules that could bind to recombinant NS4B. This assay was based on monitoring
changes in intrinsic protein fluorescence of recombinant NS4B as an indirect measure
of candidate ligand binding. One of the compounds identified in the screen, 7-
[chloro(difluoro)methyl]-5-furan-2-yl-N-(thiophen-2-ylmethyl)pyrazolo[1,5-
a]pyrimidine-2-carboxamide, is shown in Figure 4.1A, and is hereon referred to as

68
“anguizole”. In the screen, this compound was shown to inhibit HCV protein
expression, as detemined by an ELISA-based HCV replication assay. Initially, we
sought to confirm that anguizole does indeed possess specific anti-HCV activity in the
HCV replicon system.
Luciferase-linked HCV replicons were treated with various concentrations of
anguizole, and luciferase activity was assayed as a measure of HCV replication. A
representative experiment is shown in Figure 4.1b. Treatment with anguizole
significantly inhibited viral RNA replication in both genotype 1b and 1a replicons.
The mean 50% effective concentration (EC50) of the compound in genotype 1b was
310 nM and in genotype 1a was 560 nM. Anguizole appeared to have no significant
cytotoxic effect at the concentrations studied, with a mean 50% cytotoxicity
concentration (CC50) of > 50 M, as measured by the cell proliferation reagent, WST-
1(Roche). Notably, we were unable to determine the EC50 for genotype 2a replicons,
suggesting that this compound’s effect on viral replication may be genotype-specific.
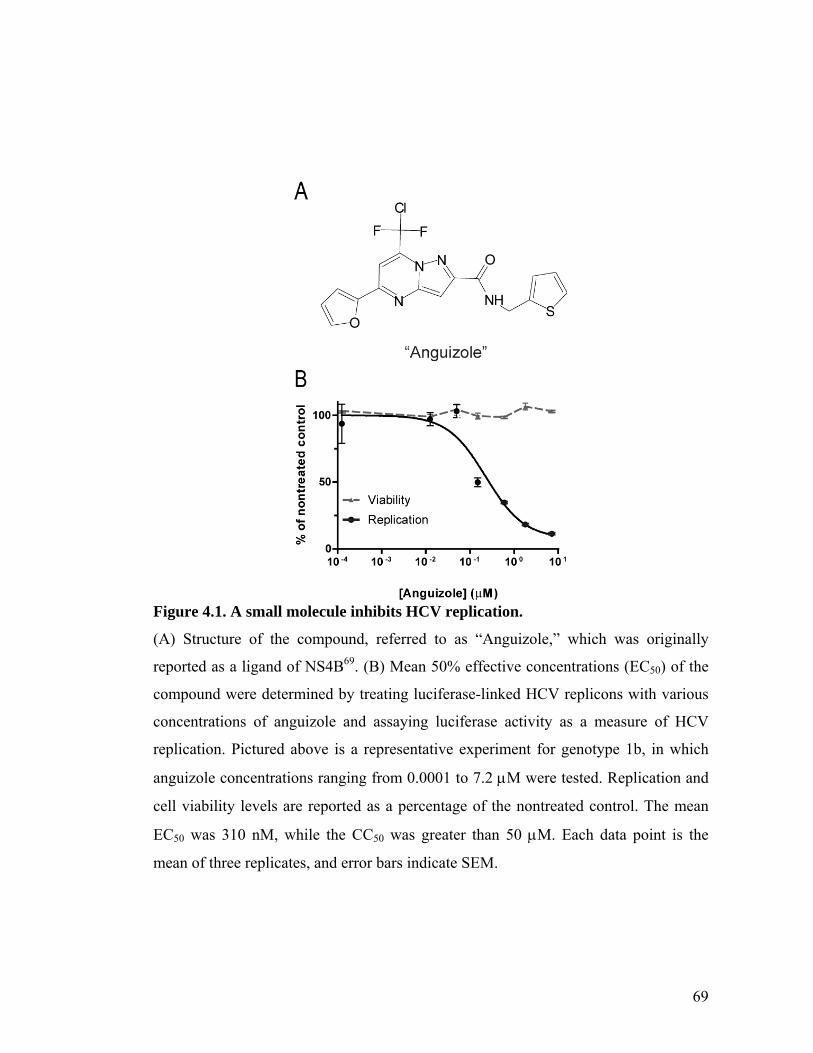
69
Figure 4.1. A small molecule inhibits HCV replication.
(A) Structure of the compound, referred to as “Anguizole,” which was originally
reported as a ligand of NS4B69. (B) Mean 50% effective concentrations (EC50) of the
compound were determined by treating luciferase-linked HCV replicons with various
concentrations of anguizole and assaying luciferase activity as a measure of HCV
replication. Pictured above is a representative experiment for genotype 1b, in which
anguizole concentrations ranging from 0.0001 to 7.2 M were tested. Replication and
cell viability levels are reported as a percentage of the nontreated control. The mean
EC50 was 310 nM, while the CC50 was greater than 50 M. Each data point is the
mean of three replicates, and error bars indicate SEM.

70
Mutations conferring resistance to anguizole map to NS4B.
In order to determine anguizole’s mechanism of action, we first sought to
obtain genetic evidence that anguizole interacts with NS4B. Therefore, we selected for
anguizole-resistant mutants. Replicon cells were grown in the presence of the drug at
either 5- or 10-fold the EC50, and resistant colonies were isolated and propagated.
RNA was extracted from 18 colonies, and the NS4B coding region was reverse-
transcribed, amplified, and sequenced. Several mutations within NS4B were identified
in the resistant colonies (Figure 4.2A), while no NS4B mutations were observed in
untreated replicon cells grown in parallel.
Present in 14 of the 18 isolated clones, the most commonly selected mutation
conferring anguizole resistance was a substitution of histidine to either arginine
(H94R) or asparagine in position 94 of the NS4B amino acid sequence, corresponding
to position 1805 of the HCV polyprotein. To confirm that it was responsible for the
phenotypic resistance to anguizole, the H94R mutation was cloned into a firefly
luciferase-linked HCV reporter construct and electroporated into Huh7.5 cells. The
cells were then dosed with various concentrations of anguizole solubilized in DMSO,
and cells were treated with DMSO alone as a negative control. Luciferase expression
levels were measured after 5 days of treatment. As shown in Figure 4.3A, the replicon
harboring the H94R mutation was indeed much less sensitive to anguizole treatment
(EC50 = 7.5 M, 95% CI = 3.2-17.4 M), relative to the wild-type replicon (EC50 =
0.20 M, 95% CI = 0.08-0.52 M).

71
Figure 4.2. Resistant mutations map to NS4B.
Colony formation assays were performed at either 5- or 10 times the EC50 for this
compound. Replicating colonies were isolated and cDNA from these colonies was
sequenced. (A) Several point mutations in the genotype 1b NS4B coding region were
identified in resistant colonies. Colonies containing such mutations and the stringency
of selection are indicated. At the top are listed the wild-type amino acid found at the
indicated location in the NS4B polypeptide sequence. Observed mutations are listed
below. Highlighted is the most common mutation observed, a mutation of NS4B’s 94th
amino acid—corresponding to the HCV polyprotein’s 1805th amino acid—from a
histidine to either an arginine or an asparagine. (B) A predicted topology of the NS4B
protein, as modified from17. The location of the H94 mutation is indicated. Two
predicted N-terminal amphipathic helices are shown (AH1 and AH2), as are the four
predicted transmembrane domains (TM1-4).

72
To evaluate the replication capacity of the H94R NS4B mutant replicon,
nontreated WT and H94R replicons were monitored as a function of time over the
course of 6 days post-electroporation (Figure 4.3B). Replication kinetics of the
luciferase-linked H94R replicon were similar to those of the wild-type replicon;
however, the efficiency of replication was roughly two-fold lower for the mutant.
Therefore, the H94R mutation appears to carry a significant fitness cost in the
replication of the virus.
Taken together, these findings suggest that anguizole does not merely target
NS4B in vitro, but also likely in the context of viral replication in cells; furthermore,
its antiviral activity appears to depend on the sequence of NS4B.
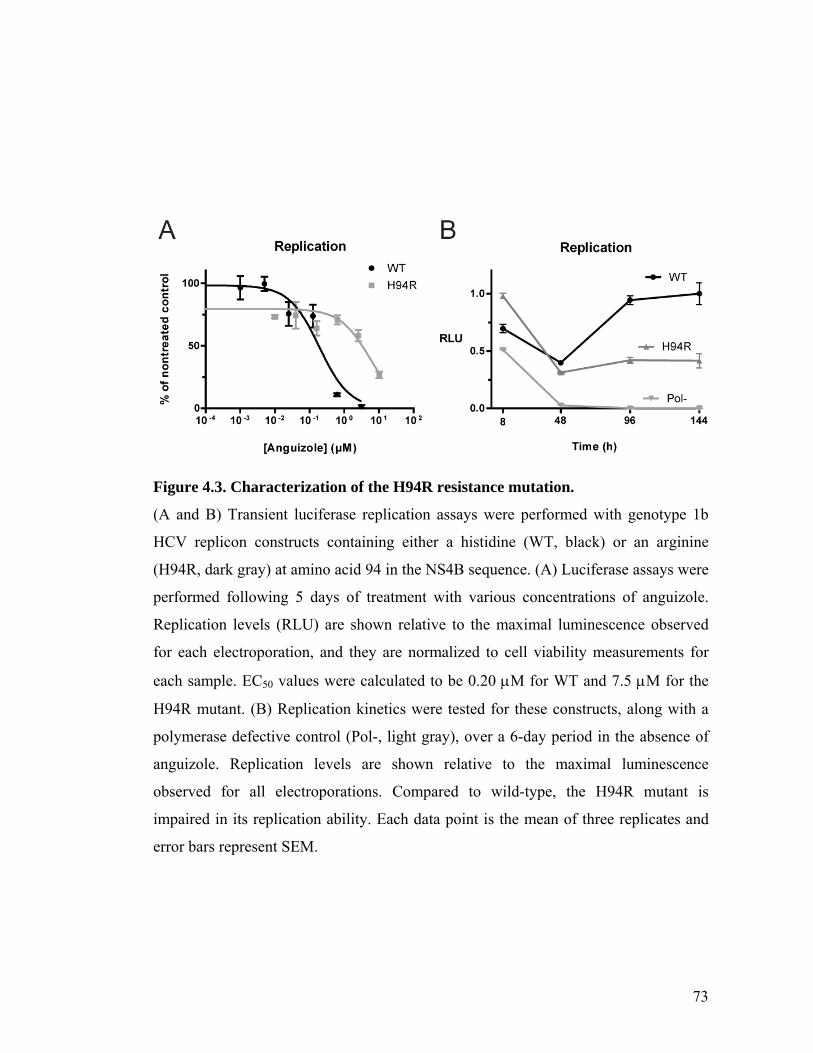
73
Figure 4.3. Characterization of the H94R resistance mutation.
(A and B) Transient luciferase replication assays were performed with genotype 1b
HCV replicon constructs containing either a histidine (WT, black) or an arginine
(H94R, dark gray) at amino acid 94 in the NS4B sequence. (A) Luciferase assays were
performed following 5 days of treatment with various concentrations of anguizole.
Replication levels (RLU) are shown relative to the maximal luminescence observed
for each electroporation, and they are normalized to cell viability measurements for
each sample. EC50 values were calculated to be 0.20 M for WT and 7.5 M for the
H94R mutant. (B) Replication kinetics were tested for these constructs, along with a
polymerase defective control (Pol-, light gray), over a 6-day period in the absence of
anguizole. Replication levels are shown relative to the maximal luminescence
observed for all electroporations. Compared to wild-type, the H94R mutant is
impaired in its replication ability. Each data point is the mean of three replicates and
error bars represent SEM.

74
Anguizole treatment leads to an altered subcellular distribution
pattern of the NS4B protein.
We next sought to determine the mechanism by which anguizole affects NS4B.
Since NS4B alone has been shown to be sufficient for assembling the membrane-
associated foci (MAF) that are believed to correlate to the sites of viral replication16,
we hypothesized that anguizole might alter the integrity of the MAF.
To test this hypothesis, a plasmid expressing NS4B fused in frame with a C-
terminal green fluorescent protein (GFP) was transfected into Huh7.5 cells. As shown
in Figure 4.4, cells treated with 5 M anguizole for 48 hours displayed a NS4B
distribution pattern distinct from the MAF visible in non-treated cells. Rather than
appearing as small foci (Figure 4.4A), a significant amount of the NS4B proteins in
these treated cells appeared to form elongated snake-shaped structures (Figure 4.4B &
4C). As shown in the insets, these “snakes” were both longer and shaped differently
than the MAF observed in nontreated cells, suggesting that anguizole treatment was
altering the formation of NS4B MAF.
Notably, there was significant variation in the extent of the snake phenotype. A
pattern resembling a network of extremely elongated snakes (Figure 4.4C) was visible
in rare cells, while moderately elongated snakes (Figure 4.4B) were visible in roughly
one fifth of the cells expressing NS4B. In the remaining cells in which NS4B-GFP
was expressed, the distribution pattern of NS4B-GFP either adopted a diffuse
cytoplasmic pattern (Figure B.1A) or displayed a pattern of small, bright foci (Figure
B.1A) unlike the MAF observed in untreated cells.
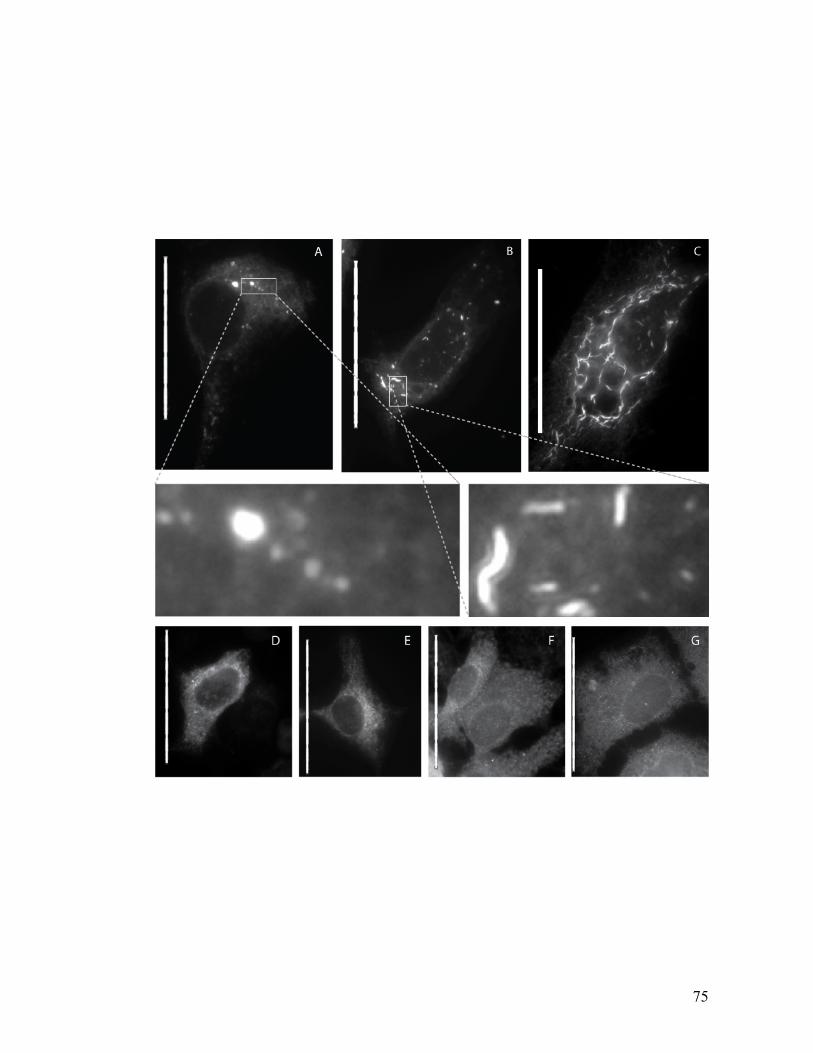
75

76
Figure 4.4. Anguizole alters the subcellular distribution of NS4B-GFP in
transiently transfected cells.
Huh7.5 cells were cultured in the absence (A, D, F) or presence (B, C, E, G) of 5 M
anguizole, either alone (F, G) or following transfection with NS4B-GFP (A, B, C) or
GFP-NS5A (D, E). (A) In nontreated cells, NS4B-GFP displays an ER-associated
pattern of localization, along with several membrane-associated foci (MAF) of varying
size throughout the cytoplasm. (B and C) In the presence of anguizole, NS4B-GFP
appears to form elongated, curved structures (“snakes”), most commonly observed to
be small and dispersed (B), but occasionally observed to be dramatically long (C).
(Insets of A and B) Zoomed-in images highlight the differences in shape and length
between MAF and snakes. (D and E) The distribution pattern of a GFP-NS5A fusion
protein is equivalent in untreated (D) and treated (E) samples. (F and G) The pattern of
calnexin staining is equivalent in untreated (F) and treated cells (G). Scale bars
represent 50 m.

77
To test the specificity of anguizole’s interaction with NS4B, we examined the
effect of anguizole treatment on another viral protein and on subcellular structures in
Huh7.5 cells. First, when we treated cells expressing an NS5A-GFP fusion protein, no
snake-like structures were observed (Figure 4.4E). Also, when markers of the ER,
actin filaments, microtubules, or mitochondria were visualized, no differences
between the cellular structure of treated and nontreated cells were observed, both in
cells transfected with NS4B-GFP and in those not transfected (Figure 4.4G, B.2).
Therefore, we hypothesized that the observed effect is likely the result of a specific
interaction between anguizole and the NS4B protein.
A resistance mutation also alters the subcellular distribution
pattern of the NS4B protein.
Having identified a mutation in NS4B that confers resistance to anguizole
treatment, we hypothesized that this mutation may also affect NS4B-mediated MAF
formation. To test this hypothesis, we characterized the subcellular distribution of
H94R NS4B-GFP, both in the presence and absence of anguizole.
Transient transfections were performed with a construct of NS4B-GFP
carrying the H94R mutation, as described for Figure 4.4. In the absence of anguizole,
cells expressing the H94R NS4B-GFP mutant displayed a distribution pattern
significantly different from the wild-type MAF pattern (Figure 4.5A vs. Figure 4.4A).
Notably, this pattern was strikingly similar to the snake-like pattern seen in wild-type
cells treated with anguizole, with the exception that the H94R snakes tended to be
longer and more dramatic than those observed in the wild-type samples. These results

78
suggest that the H94R mutation itself is sufficient to disrupt the process of NS4B
MAF formation.
In contrast, cells treated with 5 M anguizole displayed a different distribution
of H94R NS4B-GFP (Figure 4.5B). In these cells, no snake patterns were detected,
and many large foci were present throughout the cytoplasm. These foci are
reminiscent of MAF, but their expression pattern is distinct from that of wild-type
NS4B-GFP in that H94R foci tend to be somewhat larger and present in higher
numbers than wild-type NS4B foci.
Anguizole interacts with the second amphipathic helix of NS4B.
Previously, our laboratory identified an N-terminal amphipathic helix (AH1) in
NS4B that is essential for wild-type distribution of NS4B throughout the cell18. We
have also identified a second, smaller amphipathic helix (AH2), consisting of amino
acids 43 – 65 of the NS4B protein. Recently, we have found that upon addition to a
monodisperse population of lipid vesicles, a synthetic peptide comprising this AH2
dramatically alters the size distribution of the vesicles by inducing massive vesicle
aggregation (N.J. Cho, H. Dvory-Sobol, C.H. Lee, S.J. Cho, P.D. Bryson, C. Frank,
and J.S. Glenn, manuscript in preparation). We hypothesized that the AH2 represents a
target for anguizole and that addition of anguizole might disrupt AH2’s lipid vesicle
aggregating activity.
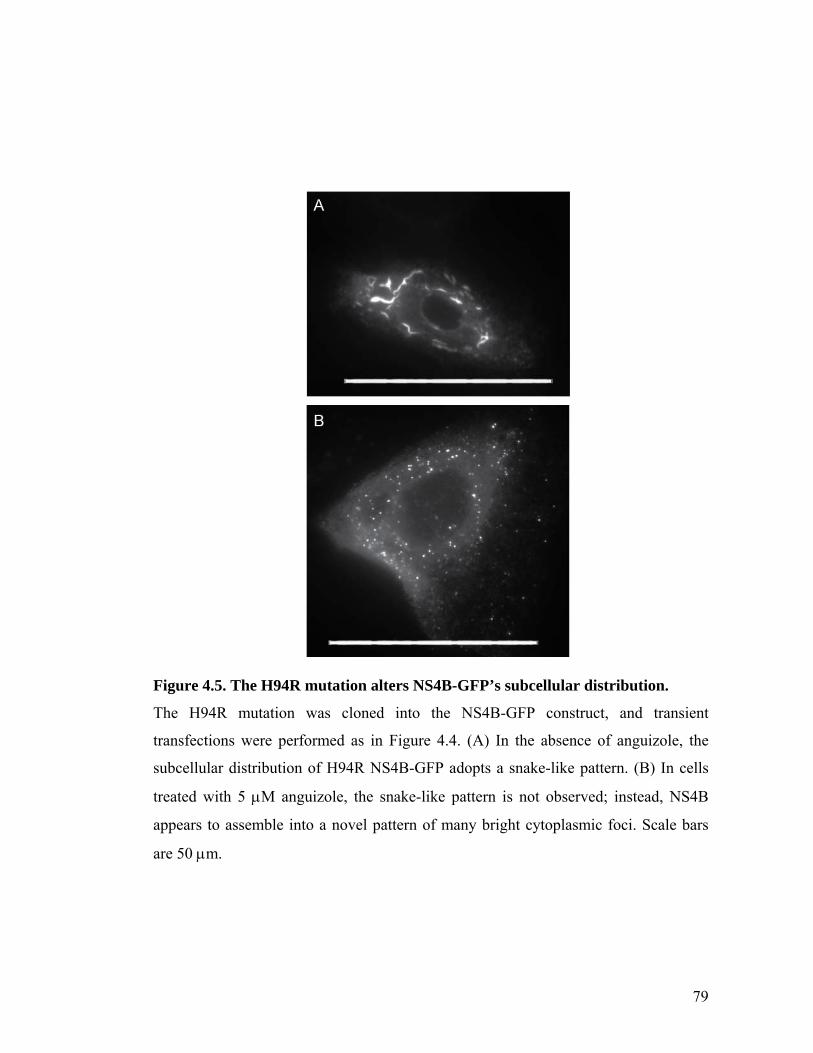
79
Figure 4.5. The H94R mutation alters NS4B-GFP’s subcellular distribution.
The H94R mutation was cloned into the NS4B-GFP construct, and transient
transfections were performed as in Figure 4.4. (A) In the absence of anguizole, the
subcellular distribution of H94R NS4B-GFP adopts a snake-like pattern. (B) In cells
treated with 5 M anguizole, the snake-like pattern is not observed; instead, NS4B
appears to assemble into a novel pattern of many bright cytoplasmic foci. Scale bars
are 50 m.

80
To test this hypothesis, dynamic light scattering (DLS) measurements were
performed with POPC vesicles and AH2 peptide in the presence or absence of
anguizole. Clemizole hydrochloride—which has been recently shown to inhibit HCV
RNA:NS4B binding, for which a C-terminal domain mediating RNA binding is
essential22—was used as a negative control. As shown in Figure 4.6, the addition of
AH2 peptide to the POPC vesicles radically affected the effective diameter of vesicles
in solution, changing the 88 nm diameter untreated vesicles to aggregates with a mean
effective diameter of 5377 nm (Fig 6B vs. 6A). While clemizole exerted no significant
effect on AH2’s vesicle aggregating activity (Figure 4.6D), anguizole dramatically
inhibited AH2’s ability to aggregate vesicles (Figure 4.6C). Specifically, vesicles
incubated with AH2 and anguizole adopted a mean effective diameter of 126 nm,
comparable to what is observed for vesicles without AH2 (88 nm, Fig 6E). These
results suggest that anguizole specifically interacts with the AH2 segment of NS4B.
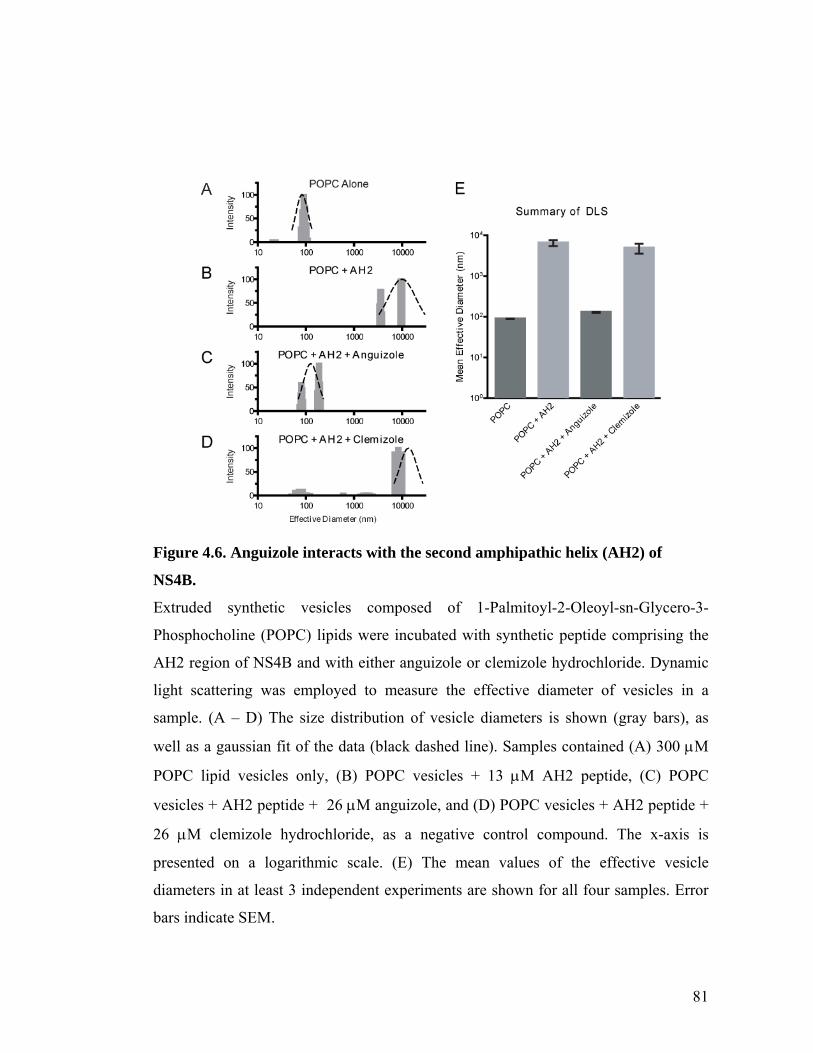
81
Figure 4.6. Anguizole interacts with the second amphipathic helix (AH2) of
NS4B.
Extruded synthetic vesicles composed of 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-
Phosphocholine (POPC) lipids were incubated with synthetic peptide comprising the
AH2 region of NS4B and with either anguizole or clemizole hydrochloride. Dynamic
light scattering was employed to measure the effective diameter of vesicles in a
sample. (A – D) The size distribution of vesicle diameters is shown (gray bars), as
well as a gaussian fit of the data (black dashed line). Samples contained (A) 300 M
POPC lipid vesicles only, (B) POPC vesicles + 13 M AH2 peptide, (C) POPC
vesicles + AH2 peptide + 26 M anguizole, and (D) POPC vesicles + AH2 peptide +
26 M clemizole hydrochloride, as a negative control compound. The x-axis is
presented on a logarithmic scale. (E) The mean values of the effective vesicle
diameters in at least 3 independent experiments are shown for all four samples. Error
bars indicate SEM.

82
4.3 Discussion
In this study, we present evidence concerning the mechanism of a novel means
of inhibiting HCV replication. Anguizole, the small molecule studied here, is a
member of a previously uncharacterized class of inhibitors for this pathogen, and we
demonstrate that mutations in the viral NS4B coding region are sufficient to confer
resistance to this compound. Furthermore, we show that anguizole can disrupt the
formation of NS4B membrane-associated foci and that it also disrupts the interaction
of a specific peptide segment of NS4B with lipid vesicles, suggesting that anguizole
can directly bind this region of the protein. Thus, this compound employs a novel
mechanism of action for inhibiting HCV replication.
In addition to the data from the original screen that identified anguizole 69,
three pieces of evidence presented in this study suggest that anguizole’s inhibition of
viral replication is a result of directly targeting the viral NS4B protein. First, the
mutation of a single amino acid in the NS4B coding region is sufficient to
dramatically reduce the virus’ susceptibility to drug treatment (Figure 4.3). Second,
treatment with anguizole significantly alters the subcellular distribution pattern of
NS4B (Figure 4.4), but not that of another viral protein or cellular proteins. Third,
anguizole specifically blocks the function of a portion of the NS4B polypeptide, the
amphipathic helix comprising amino acids 43 – 65. Taken together, these data point to
NS4B as the target with which anguizole interacts. Because NS4B induces the
formation of membranous viral replication structures (the “membranous web”)13 and
is required for the proper localization of other nonstructural viral components18, 63, it is

83
logical that the disruption of functional NS4B would lead to a reduction in viral
replication. We propose that this is what happens upon either the addition of anguizole
or the mutation of NS4B histidine 94 (Figure 4.4C and Figure 4.5A).
Our in vitro dynamic light scattering experiments (Figure 4.6) suggest that
anguizole interacts directly with the second amphipathic helix of NS4B (amino acids
43-65). Interestingly, to date our resistant mutant analysis (Figure 4.2) has not
identified any mutations that map within this region of NS4B. The most common
mutation, which was found to be phenotypically resistant to anguizole, was located at
the 94th amino acid in the NS4B sequence. We interpret these potentially conflicting
results as follows: (1) mutations in the AH2 region are very poorly tolerated by the
virus; (2) mutations at position 94, in contrast, are well tolerated; and (3) the H94R
mutation, in particular, might cause a conformational change in the NS4B protein, one
which counteracts anguizole’s ability to inhibit HCV replication. In support of point
(2), we note that position 94 in NS4B is not strictly conserved in patient isolates. In
addition to histidine, glutamine (1a), asparagine (1b), serine (1b), and threonine (2a)
have each been observed in reference sequences of the indicated genotype66.
Furthermore, we found that the H94R mutation alone is sufficient to drastically alter
NS4B’s subcellular distribution (Figure 4.5A), supporting point (3). The nature of
such a conformational change is unclear, but one possibility is that it partially blocks
anguizole’s binding site on the AH2. Structural studies may help to further clarify the
molecular interactions occurring in this situation.
In addition to identifying a novel mechanism of HCV inhibition, our results
have enhanced our understanding of the biological processes that occur during HCV

84
replication. For example, we have shown that anguizole can disrupt both NS4B
AH2:lipid interactions in vitro (Figure 4.6) and the formation of MAF in cell culture
(Figure 4.4). These results are consistent with the hypothesis that NS4B’s AH2 is a
key determinant for the formation of MAF, a correlate of HCV replication complexes.
In addition, we have shown that the MAF formation process can be altered both
pharmacologically and genetically, resulting in the cytoplasmic distribution of
elongated snake-like structures. Previous work has shown that NS4B can physically
interact with other nonstructural components of the virus, including itself21, 63, and we
hypothesize that these snakes represent assemblies of NS4B proteins that have lost
their ability to form the proper protein:protein or protein:lipid interactions. Future
studies may reveal more insight into these interactions and, more generally, the
processes involved in replication complex formation.
To the best of our knowledge, anguizole is the first pharmacological compound
shown to affect NS4B subcellular distribution. Because targeting multiple stages of
the viral life cycle is likely to be essential for effective HCV therapy70, anguizole is
thus representative of an important new potential class of HCV inhibitors. Not only is
such a class likely to be complementary to inhibitors that target other viral proteins71,
72, but it may also complement inhibitors that target other functions of the same NS4B
protein. In support of such an outcome, our results show that clemizole hydrochloride
does not inhibit AH2’s vesicle-altering properties while anguizole does (Figure 4.6).
Similarly, anguizole does not inhibit NS4B RNA binding, while clemizole does (S.
Einav, unpublished data). Development of such compounds may lead to an efficacious
antiviral cocktail and help manage this disease for the millions of people it affects.

85
4.4 Materials and Methods
DNA constructs and peptides
Standard recombinant DNA technology was used to clone all constructs. The
plasmid pEF-NS4B-GFP was cloned previously20. To generate the H94R mutation in
this construct, site-directed PCR mutagenesis was performed as described20. The first
round of amplification used the primer sets [pEF-6879F, 5’-
GGCCAAGATCTGCACACTGGTATT-3’, to H94R_R, 5’-
TAAACAGGAGGGTACGTTGGGTGGTGAGCGG-3’] and [H94R_F, 5’-
CCGCTCACCACCCAACGTACCCTCCTGTTTA-3’, to pEF-1783R, 5’-
AGGCTGATCAGCGGGTTTA-3’]. The second round of PCR amplification was
achieved using the primer set pEF-6879F to pEF-1783R.
The Bart-Luciferase plasmid, Bart79ILuc*, was cloned from the Bart79I
parent20, an HCV genotype 1b Con1 sequence containing the adaptive mutation
S2204I, and the pGL3-Basic parent (Promega) as follows. A NotI site was introduced
after the 15th amino acid of Core in Bart79I using PCR mutagenesis. The luciferase
gene was PCR amplified from the pGA3Basic plasmid using the primers NotLucS,
GAATGCGGCCGCAATGGAAGACGCCAAAAACATAAAG, and LucDraAS,
CGATTTAAATTACACGGCGATCTTTCCGCCC. The PCR product was ligated
into the Bart79I plasmid following restriction digestions of both components with
NotI and DraI. In addition, the ScaI restriction site in the Luciferase coding region was
removed by introducing a silent mutation into the site by PCR mutagenesis.

86
A peptide comprising the second amphipathic helix region of the genotype 1b
NS4B protein was synthesized (Anaspec). This peptide, Pep4BAH2, contained the
sequence, H-WRTLEAFWAKHMWNFISGIQYLA-NH2.
Drugs and antibodies
7-[chloro(difluoro)methyl]-5-furan-2-yl-N-(thiophen-2-
ylmethyl)pyrazolo[1,5-a]pyrimidine-2-carboxamide (Chemical Block
A2828/0119446), which we refer to here as anguizole, was solubilized in DMSO, and
aliquots of 10mM stocks were maintained at -20° C. Immediately prior to use, these
stocks were diluted to 200-fold the desired final concentration, and this solution was
added directly to the dishes.
Reagents used for fluorescence microscopy included the following antibodies:
rabbit anti-calnexin (Stressgen SPA-860) rabbit anti-PDI (Stressgen SPA-890), mouse
anti--tubulin (Sigma T9026), and Alexa Fluor® 594 conjugated secondary antibodies
(Invitrogen A11012, A21044). Actin filaments and mitochondria were visualized with
phalloidin-TRITC (Sigma P1951) and the Mitotracker dye (Invitrogen M7512),
respectively.
Cell culture, NS4B-GFP transfections, and fluorescence
microscopy
Cells of the human hepatoma cell line, Huh7.5, were cultured in monolayers as
described73, with media consisting of DMEM (Mediatech) supplemented with 1% L-
glutamine (Mediatech), 1% penicillin, 1% streptomycin (Mediatech), and 10% fetal

87
bovine serum (Omega Scientific). Transfections of pEF-NS4B-GFP constructs were
performed with Lipofectamine-2000 (Invitrogen), according to the manufacturer’s
instructions. Following transfection, the cells were cultured in standard media
containing 5 M anguizole and 0.5% (70 mM) DMSO. Controls were cultured in
media containing 0.5% DMSO. Unless noted otherwise, the cells were treated for 48
hours, fixed in 4% formaldehyde, and mounted with Prolong® Gold antifade reagent
with DAPI (Invitrogen). Slides were visualized under a Nikon E600 fluorescence
microscope and images were captured with a SPOT digital camera and Openlab image
acquisition software (Improvision).
Immunofluorescence was performed as above and as described74, with the
following variations: following fixation, cells were permeabilized with 0.2% saponin,
and subcellular structures were visualized using the primary antibodies, rabbit anti-
calnexin (1:1000), rabbit anti-PDI (1:2000), and mouse anti--tubulin (1:1000),
followed by the appropriate secondary antibody (1:600). Actin was visualized with
TRITC-conjugated phalloidin (50 g/ml), and mitochondria were visualized with the
Mitotracker mitochondrial dye (100 nM), according to the manufacturer’s instructions.
Stable luciferase replication assays
The ET cell line was established by stably transfecting Huh7 cells with RNA
transcripts harboring a firefly luciferase-ubiquitin-neomycin phosphotransferase
fusion protein and EMCV IRES-driven genotype 1b Con1 NS3-5B polyprotein
containing the cell culture adaptive mutations E1202G, T1280I, and K1846T75. Cells
were plated at 0.5-1.0 x104 cells/well in 96-well plates and incubated for 24 hrs. Then,

88
anguizole was added to the cells to achieve 10 final concentrations ranging from 0.097
to 50 μM. Luciferase activity was measured 48 hours later by adding a lysis buffer and
the substrate (Promega E2661 and E2620). Under the same conditions, cytotoxicity of
the compounds was determined using the cell proliferation reagent, WST-1 (Roche),
according to the manufacturer’s instructions. Percent inhibition of replication was
determined relative to a no compound control. EC50 values were calculated using the
equation: % inhibition = 100%/[1 + 10(log EC50 – log (I)) * b)], where b is Hill’s coefficient.
Transient luciferase replication assays
Bart-Luciferase RNA was generated by linearizing the Bart79ILuc* vector
with the ScaI restriction enzyme (NEB). Subsequently, in vitro transcription was
performed with the Megascript T7 kit according to the manufacturer’s instructions
(Ambion).
In vitro-transcribed Bart-Luciferase RNA was transfected into Huh7.5 cells by
electroporation, as previously described76. Briefly, 5 g of RNA was transfected into 6
x 106 cells with five 99 µs pulses at 0.82kV over 1.1 seconds, using a BTX-830
electroporator. Cells were seeded into 6-well plates at 3 x 105 cells/well. For EC50
measurements, serial dilutions of anguizole were added to each well to achieve six
final concentration of anguizole ranging from 0.001 to 3.125 M (WT) or 0.01 to
10.24 M (H94R), while the percentage of DMSO in each well remained constant, at
0.5%. Luciferase activity was measured after 5 days of treatment.
For comparing replication kinetics between the wild-type and H94R replicons,
Bart-Luciferase RNA from the wild-type or mutant constructs was electroporated into

89
Huh7.5 cells as above. Bart-Luciferase RNA of a construct containing an NS5B
polymerase lethal mutation20 served as a negative control. Cells were seeded into 6-
well plates at either 5-, 3-, or 1.5 x 105 cells/well. Cells were lysed at 48 hour
timepoints throughout a 6-day experiment.
Prior to lysis of each sample, cell viability was measured with the
alamarBlue® reagent (TREK Diagnostic Systems), according to the manufacturer’s
recommendations.
Relative levels of replication were measured using the Luciferase Assay
System (Promega). Briefly, cells were lysed in 150 l Cell Culture Lysis Buffer,
scraped from the well, transferred into microcentrifuge tubes, and centrifuged at
16,000 x g for 1 minute. 20 l of the resulting supernatant was mixed with 100 µl of
the Firefly luciferase assay buffer containing the assay substrate and luminescence was
measured using a Berthold LB 96 V luminometer.
In all cases, replication levels were normalized to viability measurements. EC50
values were determined by fitting normalized replication data points to the equation
Y=Bottom + (Top-Bottom) / [1+10^(X-Log[EC50])], using the Graphpad Prism
software (GraphPad Software).
Analysis of resistance mutants
Mutations conferring resistance to anguizole treatment were identified as
previously described77. Briefly, ET cells stably harboring a subgenomic genotype 1b
HCV replicon were cultured in 10 cm dishes with standard Huh7.5 media containing
either 1.5 M or 3 M of anguizole and 1 mg/mL G418. An initial round of cell death

90
was followed by proliferation of replicon cells in the presence of the compound. RNA
was extracted from the cells with Trizol (Invitrogen), according to the manufacturer’s
protocol. RNA was then electroporated into 4 x 106 cells of the human hepatoma cell
line, Huh-Lunet, with a BioRad Gene Pulser at 270V, 960 uF and infinite resistance.
Transfected cells were plated in 25 cm2 dishes and selected with 1 mg/mL G418 and
1.5 or 3 M of anguizole until colonies proliferated. Colonies were picked and
propogated until growth had exceeded a T25 flask. Cells were then trypsinized and
lysed in Trizol. RNA was isolated according to the manufacturer’s instructions. HCV
replicon RNA was reverse-transcribed and amplified with Superscript II (Invitrogen),
using a primer specific to the HCV 3’UTR, and NS4B was amplified using gene-
specific primers. The PCR product was cloned, sequenced, and compared to wild type
NS4B sequence.
Dynamic Light Scattering (DLS)
Unilamellar light vesicles were prepared using the extrusion method, as
previously described78. Vesicle size distribution was determined by quasi-elastic
dynamic light scattering (DLS) as previously described 79. Briefly, DLS
measurements were performed by a 90Plus particle size analyzer from Brookhaven
Instrument Corp. 79 and results were analyzed by digital autocorrelator software
(Brookhaven Instruments Corp.). All measurements were taken at a scattering angle of
90, where the reflection effect is minimized. The hydrodynamic diameter of the
vesicles was determined via non-negatively constrained least squares fitting79. The
lipid and peptide concentrations for DLS measurements were 300 M and 13 M,

91
respectively. For inhibitor experiments, either anguizole or clemizole was first
incubated with the peptide at a 2:1 molar ratio. Then, the mixture was added to the
lipid vesicles. All of the experiments were thermostatically controlled at 25 C.

92
Chapter 5. Conclusions

93
Summary
The goal of this work was to better characterize the HCV viral life cycle and
identify small molecule inhibitors that could modulate this life cycle. Towards this
goal, we focused on understanding one viral protein, NS4B, and the multiple roles it
plays in HCV replication. We undertook a multi-pronged approach towards these
goals and focused on two activities, NS4B’s binding of viral RNA and its formation of
membrane-association foci.
In Chapter 2, we identified NS4B as an RNA binding protein. We showed that
this RNA binding activity is specific for the 3’ terminus of the negative-sense HCV
genome, and we determined that two domains in the C-terminus of NS4B contribute to
this activity. In addition, our high-throughput screen revealed several compounds that
could inhibit this activity, and one of these, clemizole hydrochloride, was shown to
also inhibit HCV replication. Finally, we identified a mutation in NS4B that conferred
resistance to clemizole and enhanced NS4B’s RNA-binding activity.
In Chapter 3, we further characterized NS4B’s RNA-binding domains. We
showed that each of these domains is important for HCV replication, and our
replication data correlated with our in vitro binding data. We demonstrated that the
subcellular distribution of NS5A in cells harboring RBD-mutated HCV replicons is
unchanged from that of cells containing wild-type replicons. Interestingly, we identify
two mutations in the protease domain of NS3 that suppress the defect in one of these
RNA binding domains.

94
In Chapter 4, we characterize the mechanism of inhibition of another HCV
antiviral, termed “anguizole.” We show that this compound targets NS4B and that it
can disrupt NS4B’s ability to form membrane-associated foci. Furthermore, we
identify resistant mutations in NS4B and utilize an in vitro light scattering assay to
show that the second amphipathic helix of NS4B is the likely target of the drug.
Significance
This dissertation has uncovered several new features of HCV biology, which
should be helpful in better understanding how this virus propagates. First, we have
shown that the NS4B protein can bind HCV RNA, and that this activity of NS4B is
important for viral replication. One can imagine many different ways in which an
RNA-binding activity may be important to the viral life cycle (discussed in Chapter
2.3), and future studies may uncover the exact stage at which this RNA-binding
activity becomes important. Though other proteins in the virus have been found to
bind RNA, our work was the first to show that the small molecule clemizole
hydrochloride can interrupt this protein:RNA interaction. This compound represents a
good lead towards a potential new class of HCV antivirals.
Our discovery of suppressive mutations in NS3 is consistent with the idea that
all the nonstructural proteins work in intimate association in order to replicate the viral
genome. How these two mutations may enhance HCV replication is unclear, but based
on the previous discovery of multiple mutations present in that location, it is likely that
that region of NS3 is important in interacting with NS4B. Perhaps small molecules can
be designed in the future to block NS4B from interacting with this region of NS3.

95
Other future studies might investigate the effect of these mutations on NS4B RNA
binding either in vitro, or in a cell-based assay.
The characterization of anguizole is another example of how many new
pathways exist for developing antiviral drugs. This compound is the first to be
reported to affect NS4B’s membrane associated foci formation, and likely will lead to
many more compounds that can target this activity. Furthermore, the experiments
presented in this thesis exemplify how useful small molecules can be in the study of
HCV biology. Using this compound, we identified regions of the protein that are a
likely target of the drug and show that these regions may also be involved in
membrane-associate foci formation. Future studies will likely examine the mechanism
by which revertants can overcome the anguizole inhibition. Structural studies may also
clarify how NS4B, anguizole, and lipid vesicles all interact with one another.
By elucidating the details of HCV’s life cycle and establishing new types of
small molecules as potential HCV antivirals, we hope that the new avenues of research
developed here will help lead to new classes of drugs to combat HCV infections.
Thus, we hope this thesis will contribute to making a significant difference in the lives
of millions of individuals around the world who currently must confront the ravages of
hepatitis C.

96
References

97
Appendix A. Supplementary material to Chapter 2
Calibration curve To confirm adequate solubilization of the dried spotted RNA probes and to
exclude the possibility that a large fraction of the solvents stick to the
polydimethylsiloxane (PDMS) composing the microfluidic device, we generated a
calibration curve. Known concentrations of soluble labeled RNA probes were loaded
into the device by continuous flow and mean values of Cy3 signal from multiple unit
cells were averaged. Based on this curve, we calculated the actual concentration of
target RNA sequences used in our assay. On average the actual assayed concentration
was 0.55±0.02 of the spotted one. Of note, a similar factor (0.57) was calculated based
on the ratio between a typical print volume and unit cell volume. All the presented
data were adjusted according to this factor.
RNA binding experiment with high ionic strength buffer The ionic strength of the buffer used in an RNA binding experiment may have
an effect on the protein-RNA interaction. To confirm that the high affinity interactions
detected in our RNA binding assay are not a result of enhancement of electrostatic
interactions due to a low ionic strength buffer we performed RNA binding
experiments of NS4B by microfluidics using high (150mM) ionic strength conditions.
RNA binding by NS4B in the presence of PBS (137 mM NaCl, 2.7 mM KCl, 10 mM
Na2HPO4, 2 mM KH2PO4) or 150mM Hepes was similar to that detected with 50mM
Hepes buffer, commonly used for RNA binding assays by others (1, 2). The Kd of
NS4B binding to the 3’ terminus of the HCV negative strand RNA in the high ionic

98
strength conditions was measured at 6.6+3.3 nM (vs. a Kd of 3.4+1.0 nM in the
presence of low (50mM) ionic strength conditions). These results suggest that there is
only a small electrostatic component to the observed RNA-binding.
ATA inhibits RNA binding by NS4B in a dose-dependent manner Prior to the library screen we wished to determine whether HCV RNA binding
by NS4B can be inhibited pharmacologically. Aurintricarboxilic acid (ATA) is a
compound known to inhibit interactions of proteins with nucleic acids 3. We thus
hypothesized that ATA may similarly inhibit binding of NS4B to HCV RNA. To test
this hypothesis, the NS4B RNA binding assay was repeated in the presence of
increasing concentrations of ATA. Indeed, ATA was found to inhibit binding of HCV
RNA by NS4B in a dose-dependent manner, with an IC 50 of 0.49±0.01μM (p value –
0.0003) (Figure A.5). To our surprise, having not previously realized that ATA was
one of the compounds included in the LOPAC library, ATA was one of the 18
compounds identified in the screen. Since we had previously shown that this
compound inhibits the RNA binding activity of NS4B, this turned to be a useful
internal control for the validity of our screen.
Specificity of hits identified in the small molecule screen To determine the specificity of the hits identified in our small molecule screen
we chose the HuR protein (one of the human RNA binding protein used to validate our
RNA binding assay). Interestingly, other than binding to its target RNA sequence, 4A
(Figure A.3), this protein has been previously shown by others 4 to bind the 3’
terminus of the negative HCV strand. Binding of HuR to the consensus 4A RNA
sequence was tested in the presence of the inhibitory molecules shown to inhibit RNA

99
binding by NS4B. No inhibitory effect on RNA binding was detected with the
majority of the hits including clemizole at a concentration of 0.1mM. Similarly,
0.1mM of the identified compounds didn’t have an inhibitory effect on binding of
HuR to the 3’ terminus of the negative HCV RNA strand. In contrast to the other hits,
ATA, known as a non-specific inhibitor of protein-nucleic acids interactions,
significantly inhibited HuR binding to both 4A and 3’ terminus of the negative HCV
strand. These results suggest that the identified hits including clemizole are indeed
specific to RNA binding by NS4B.
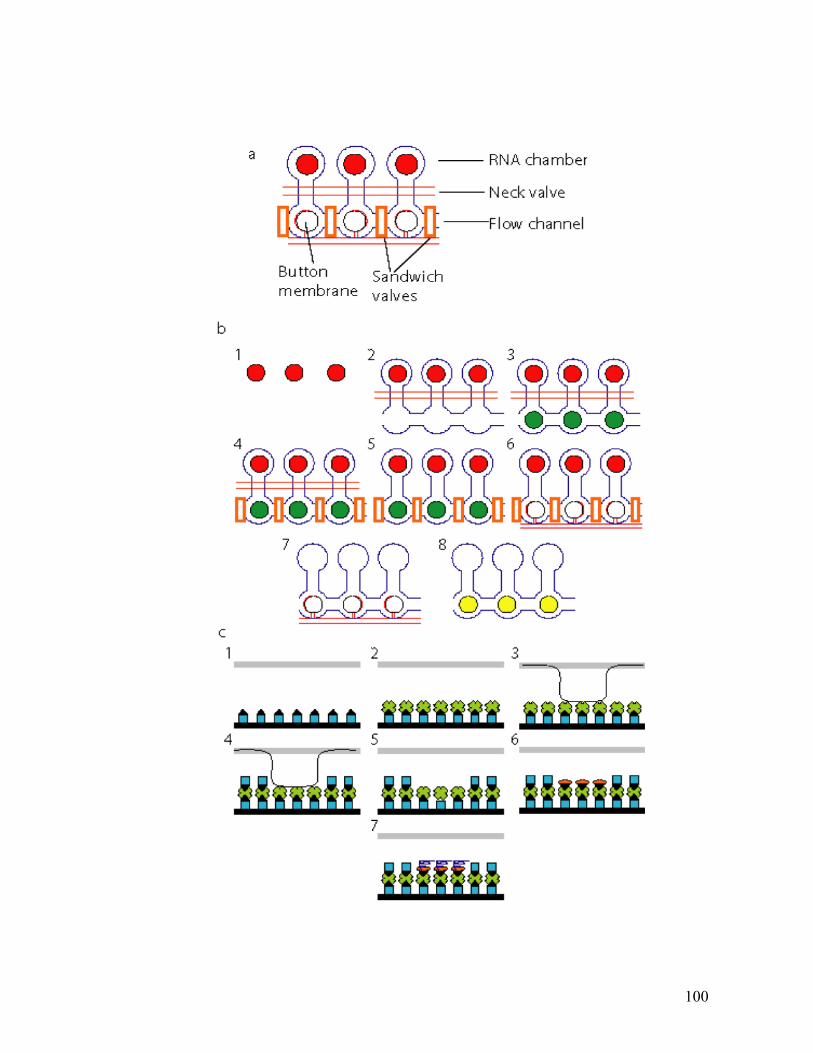
100

101
Figure A.1. Microfluidic based RNA binding assay.
3 individual unit cells (out of hundreds/thousands per microfluidic device) are shown
in this scheme.
a- Compartments and micromechanical valves. A valve is created where a control
channel crosses a flow channel. The resulting thin membrane in the junction between
the two channels can be deflected by hydraulic actuation.Using multiplexed valve
systems allows a large number of elastomeric microvalves to perform complex fluidic
manipulations within these devices.
b- Experimental protocol. ( ) represents Cy3 labeled RNA probe. ( ) represents
surface bound bodipy-labeled protein. 1) target RNA sequences labeled with Cy3 were
spotted onto an epoxy-coated slide as a microarray 2) The microfluidic device was
aligned and bonded to the slide allowing contact of each spot in the array with a unit
cell in the device. The device was subjected to surface patterning that resulted in a
circular area coated with biotinylated anti-histidine antibodies within each unit cell
(see c). The “neck valve” separating the flow channels from the RNA chamber
remained closed during this process. 3) The device was then loaded with in vitro
transcription/ translation mixture containing DNA template encoding a his-labeled
protein. Bodipy-labeled tRNALys was added for protein labeling. 4) Each unit cell
was then isolated by the control of two “sandwich” micromechanical valves. 5) the
“neck valve” was opened forming a single compartment by combining the flow
channel compartment with the RNA chamber. This was followed by an incubation to
allow protein synthesis, binding of the synthesized protein to the surface biotinylated
anti-his antibodies, solvation of target RNA, and equilibration of proteins and target
RNA. 6) MITOMI was then performed by actuation of a “button” membrane trapping
surface-bound complexes while expelling any solution phase molecules. 7) The
“sandwich” valves were opened followed by a brief wash to remove untrapped
unbound material. 8) the “button” membrane was opened and the device was scanned
by an array scanner. Trapped RNA molecules and expressed protein were detected.
The ratio of bound RNA to expressed protein was calculated for each data point by
measuring the median signal of Cy3 to median signal of bodipy (represented by ).

102
c- Surface patterning. 1)Accessible surface area was derivatized by flowing a
solution of biotinylated BSA ( ) through all flow channels 2) a Neutravidin solution
( ) was loaded 3) The “button” membrane was activated 4) all remaining accessible
surface area except for a circular area of 60 μm masked by the button was passivated
with biotinylated solution ( ) 5) the “button” membrane was opened 6)a solution of
biotinylated-antibody ( ) was loaded allowing specific functionalization of the
previously masked circular area 7) In vitro expressed protein ( ) was loaded into the
device and bound to the biotinylated antibody coating the discrete circular area. Each
of the described steps was followed by a PBS wash.
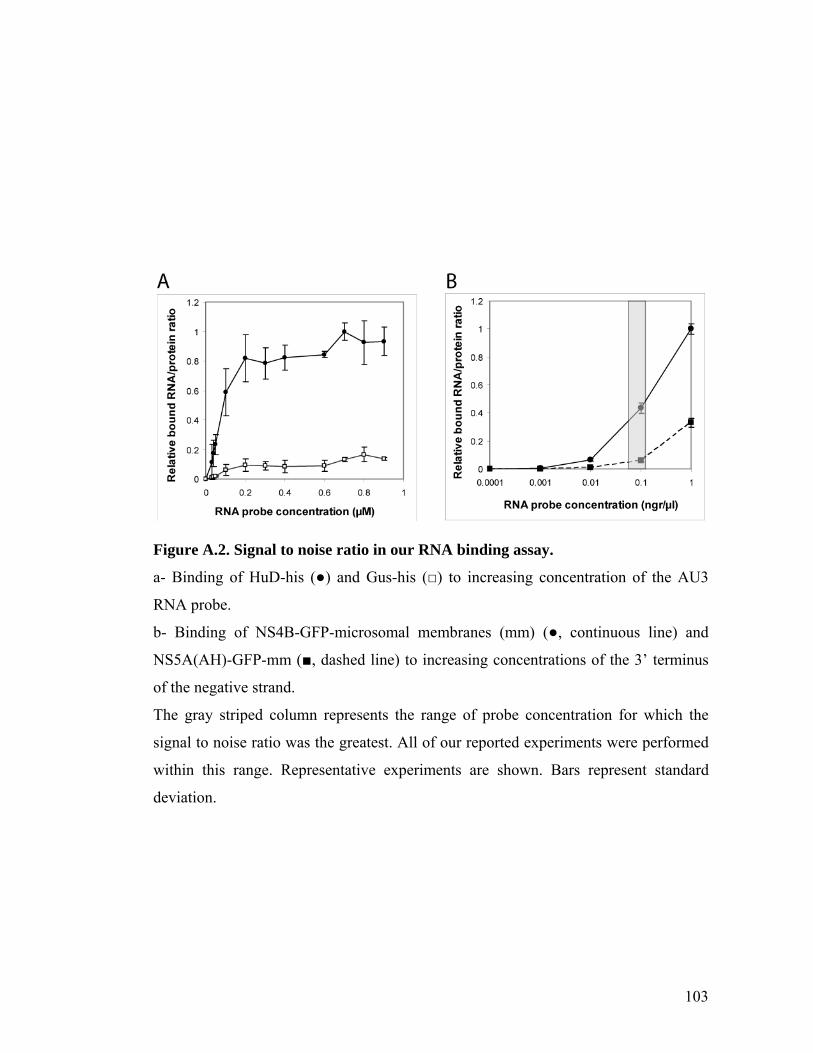
103
Figure A.2. Signal to noise ratio in our RNA binding assay.
a- Binding of HuD-his (●) and Gus-his (□) to increasing concentration of the AU3
RNA probe.
b- Binding of NS4B-GFP-microsomal membranes (mm) (●, continuous line) and
NS5A(AH)-GFP-mm (■, dashed line) to increasing concentrations of the 3’ terminus
of the negative strand.
The gray striped column represents the range of probe concentration for which the
signal to noise ratio was the greatest. All of our reported experiments were performed
within this range. Representative experiments are shown. Bars represent standard
deviation.
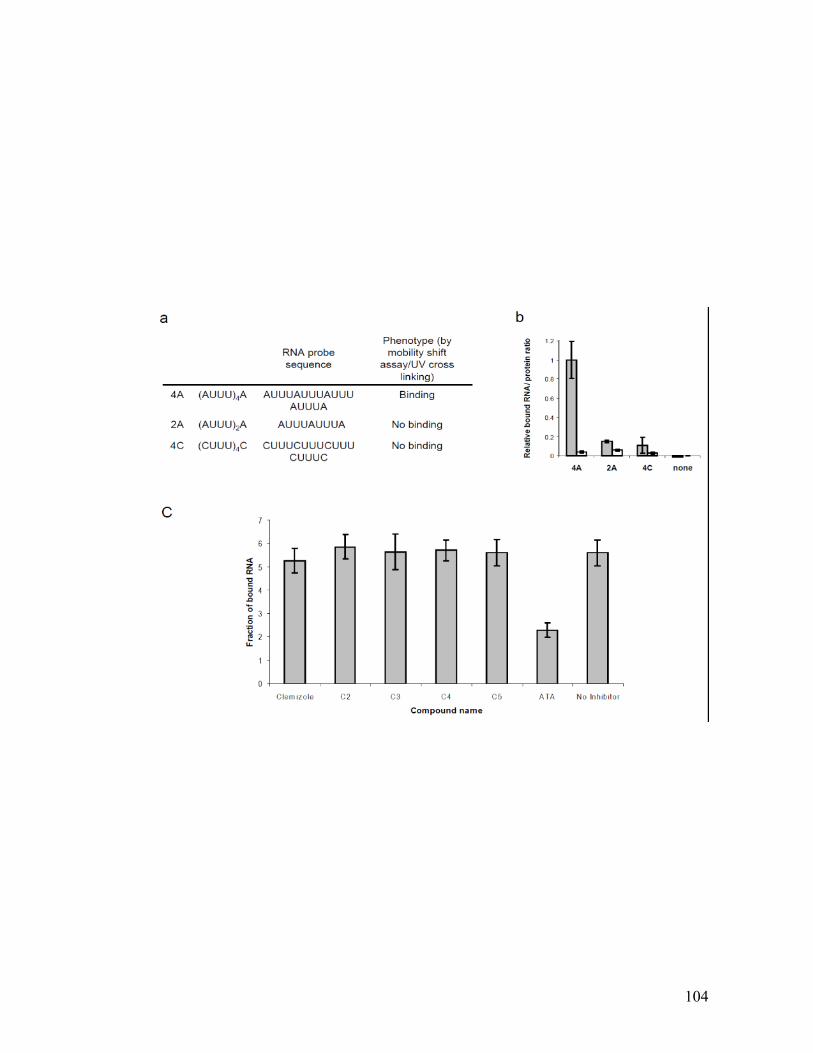
104

105
Figure A.3. Microfluidics-based analysis of RNA binding by another human
protein from the ELAV-like family, HuR (ELAV L1).
a. Target RNA sequences used to study binding of HuR to RNA and the phenotype
demonstrated by conventional RNA binding methods 5.
b. HuR binds RNA by microfluidics. A microarray of Cy3-labeled target RNA
sequences was used to program a microfluidic device, and binding of bodiby-labeled
proteins expressed on the device to the RNA sequences was assayed. Results represent
the ratio of bound RNA (median Cy3 signal) to expressed protein (median bodipy
signal). Normalized mean values for 10-20 replicates measured in two independent
experiments are shown. Error bars represent standard deviation. The gray bars
represent binding of HuR-his and the clear bars that of Gus-his, used as a negative
control.
c. HuR RNA binding is not affected by the 5 most active compounds, but is affected
by ATA. We tested binding of HUR to its 4A RNA target in the presence and absence
of NS4B RNA binding inhibitors. Data represent mean value of 10-20 replicates and
bars represent standard deviation.
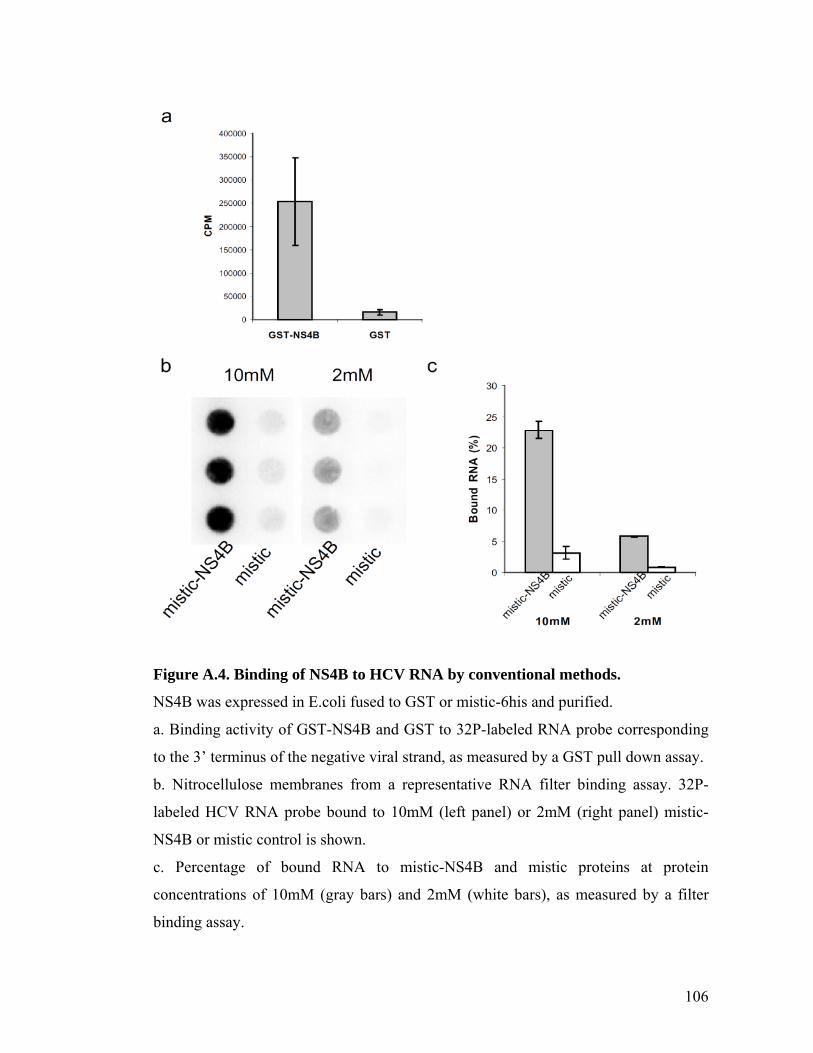
106
Figure A.4. Binding of NS4B to HCV RNA by conventional methods.
NS4B was expressed in E.coli fused to GST or mistic-6his and purified.
a. Binding activity of GST-NS4B and GST to 32P-labeled RNA probe corresponding
to the 3’ terminus of the negative viral strand, as measured by a GST pull down assay.
b. Nitrocellulose membranes from a representative RNA filter binding assay. 32P-
labeled HCV RNA probe bound to 10mM (left panel) or 2mM (right panel) mistic-
NS4B or mistic control is shown.
c. Percentage of bound RNA to mistic-NS4B and mistic proteins at protein
concentrations of 10mM (gray bars) and 2mM (white bars), as measured by a filter
binding assay.
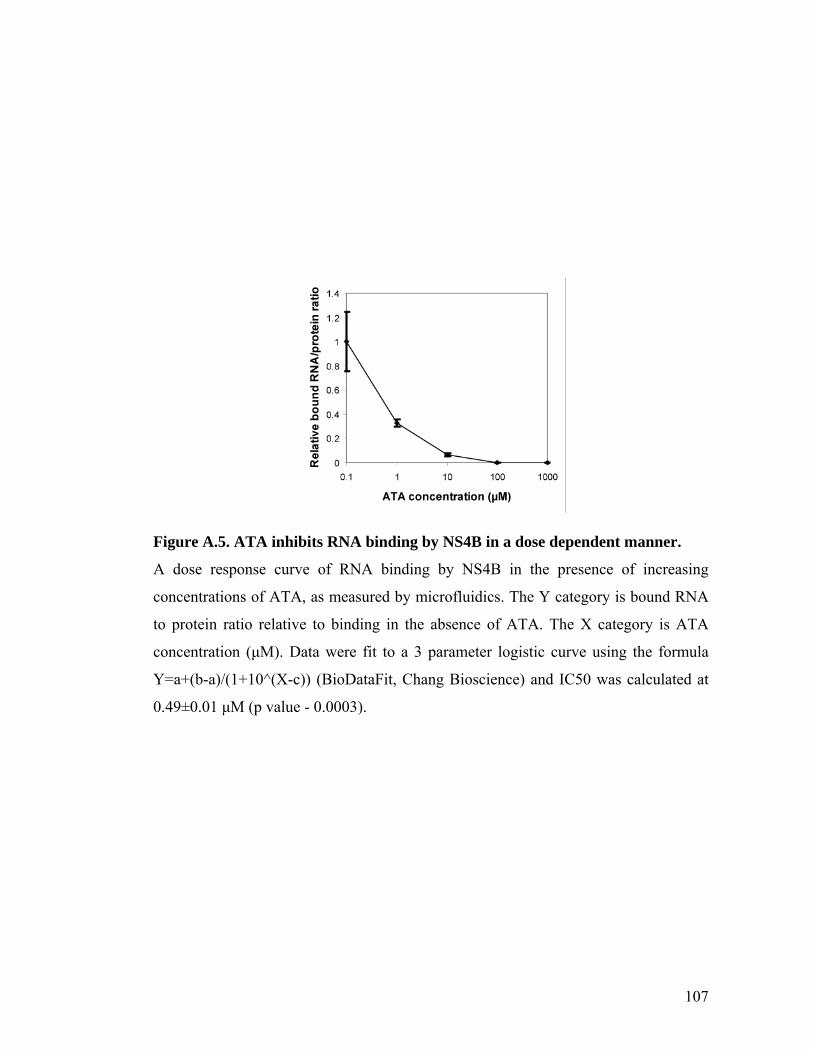
107
Figure A.5. ATA inhibits RNA binding by NS4B in a dose dependent manner.
A dose response curve of RNA binding by NS4B in the presence of increasing
concentrations of ATA, as measured by microfluidics. The Y category is bound RNA
to protein ratio relative to binding in the absence of ATA. The X category is ATA
concentration (μM). Data were fit to a 3 parameter logistic curve using the formula
Y=a+(b-a)/(1+10^(X-c)) (BioDataFit, Chang Bioscience) and IC50 was calculated at
0.49±0.01 μM (p value - 0.0003).
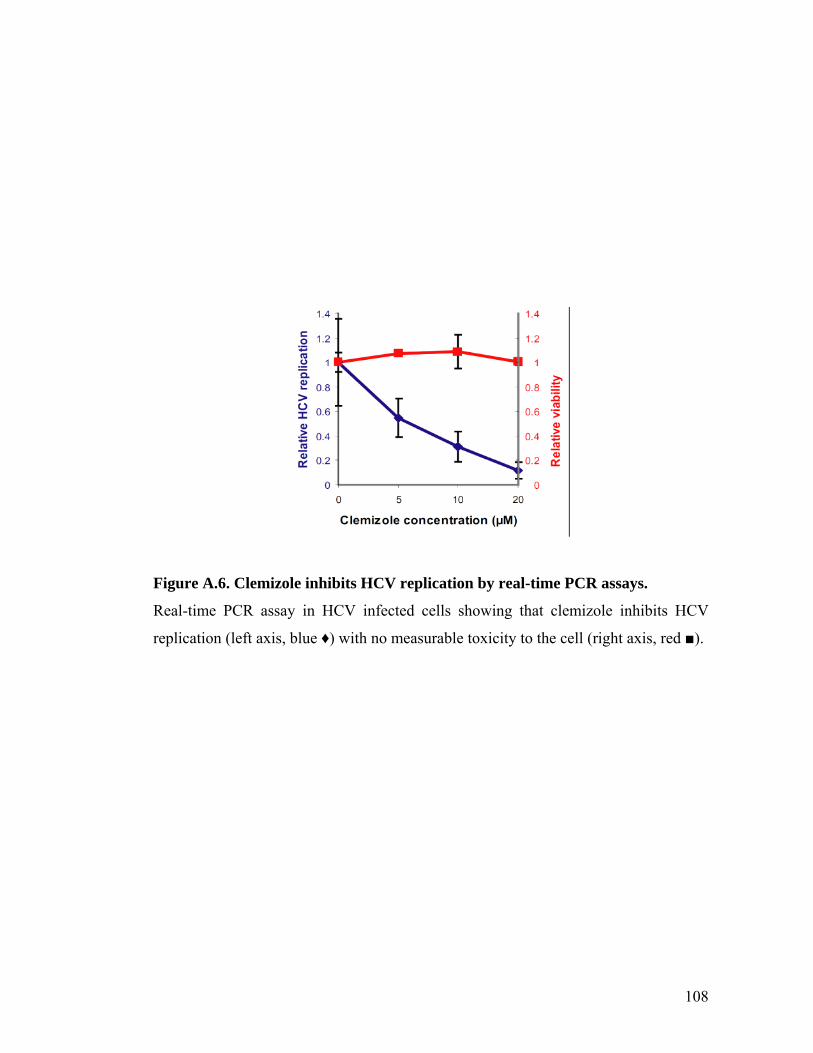
108
Figure A.6. Clemizole inhibits HCV replication by real-time PCR assays.
Real-time PCR assay in HCV infected cells showing that clemizole inhibits HCV
replication (left axis, blue ♦) with no measurable toxicity to the cell (right axis, red ■).
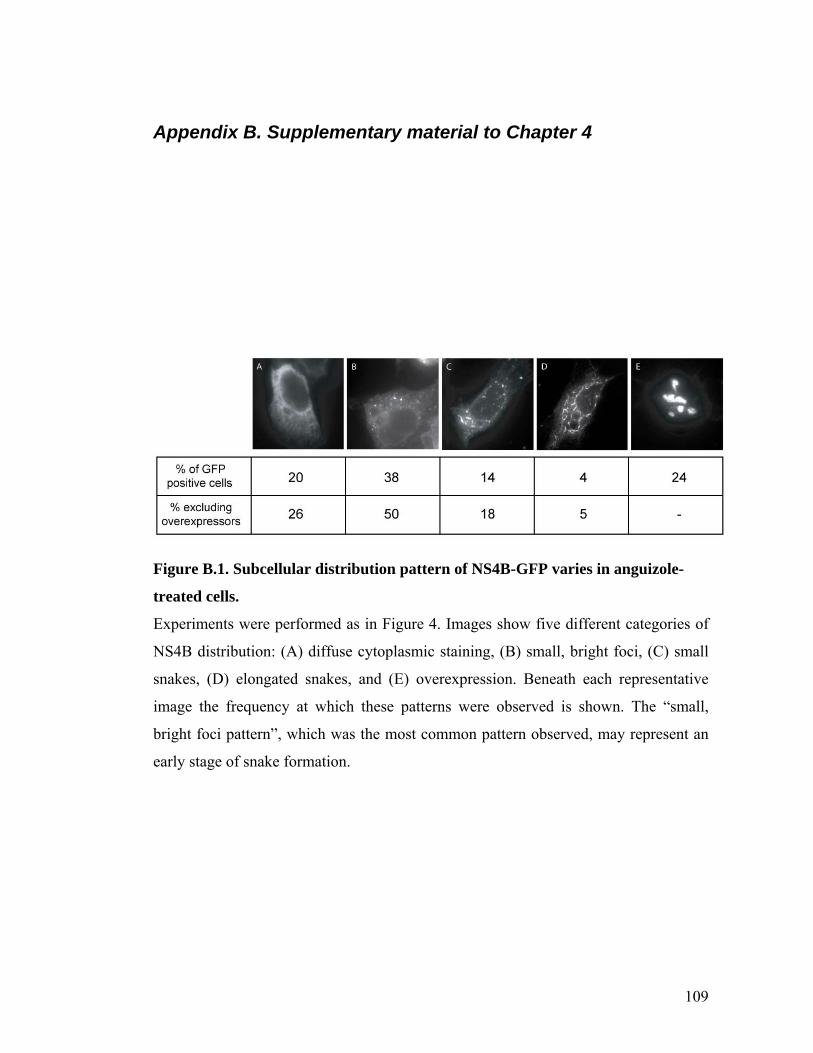
109
Appendix B. Supplementary material to Chapter 4
Figure B.1. Subcellular distribution pattern of NS4B-GFP varies in anguizole-
treated cells.
Experiments were performed as in Figure 4. Images show five different categories of
NS4B distribution: (A) diffuse cytoplasmic staining, (B) small, bright foci, (C) small
snakes, (D) elongated snakes, and (E) overexpression. Beneath each representative
image the frequency at which these patterns were observed is shown. The “small,
bright foci pattern”, which was the most common pattern observed, may represent an
early stage of snake formation.
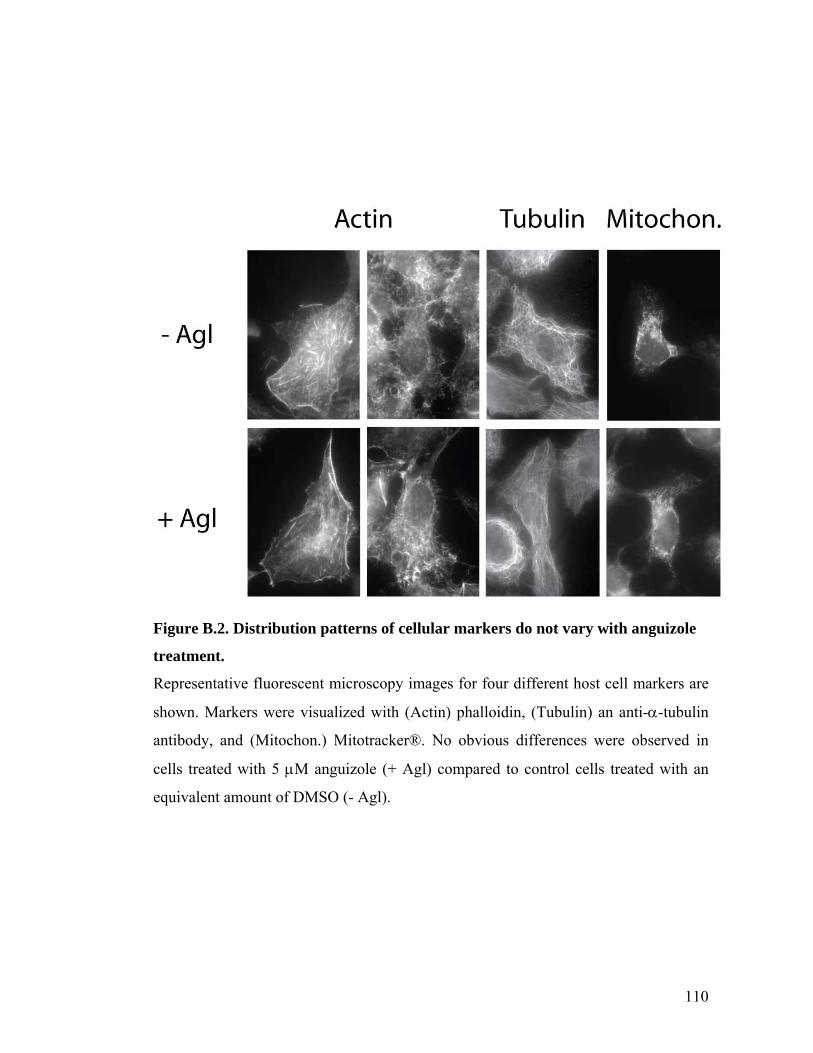
110
Figure B.2. Distribution patterns of cellular markers do not vary with anguizole
treatment.
Representative fluorescent microscopy images for four different host cell markers are
shown. Markers were visualized with (Actin) phalloidin, (Tubulin) an anti--tubulin
antibody, and (Mitochon.) Mitotracker®. No obvious differences were observed in
cells treated with 5 M anguizole (+ Agl) compared to control cells treated with an
equivalent amount of DMSO (- Agl).

111
Bibliography
1. Pawlotsky, J.M. Pathophysiology of hepatitis C virus infection and related liver disease. Trends Microbiol 12, 96-102 (2004).
2. Perz, J.F., Armstrong, G.L., Farrington, L.A., Hutin, Y.J. & Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol 45, 529-38 (2006).
3. Rustgi, V.K. The epidemiology of hepatitis C infection in the United States. J Gastroenterol 42, 513-21 (2007).
4. Afdhal, N.H. The natural history of hepatitis C. Semin Liver Dis 24 Suppl 2, 3-8 (2004).
5. Armstrong, G.L. et al. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 144, 705-14 (2006).
6. Feld, J.J. & Hoofnagle, J.H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 436, 967-72 (2005).
7. Strader, D.B. Understudied populations with hepatitis C. Hepatology 36, S226-36 (2002).
8. Moradpour, D., Penin, F. & Rice, C.M. Replication of hepatitis C virus. Nat Rev Microbiol 5, 453-63 (2007).
9. Friebe, P., Lohmann, V., Krieger, N. & Bartenschlager, R. Sequences in the 5' nontranslated region of hepatitis C virus required for RNA replication. J Virol 75, 12047-57 (2001).
10. Sarrazin, C. et al. SCH 503034, a novel hepatitis C virus protease inhibitor, plus pegylated interferon alpha-2b for genotype 1 nonresponders. Gastroenterology 132, 1270-8 (2007).
11. Reesink, H.W. et al. Rapid decline of viral RNA in hepatitis C patients treated with VX-950: a phase Ib, placebo-controlled, randomized study. Gastroenterology 131, 997-1002 (2006).
12. Manns, M.P. et al. The way forward in HCV treatment--finding the right path. Nat Rev Drug Discov 6, 991-1000 (2007).
13. Egger, D. et al. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 76, 5974-84 (2002).
14. El-Hage, N. & Luo, G. Replication of hepatitis C virus RNA occurs in a membrane-bound replication complex containing nonstructural viral proteins and RNA. J Gen Virol 84, 2761-2769 (2003).
15. Gosert, R. et al. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J Virol 77, 5487-92 (2003).
16. Gretton, S.N., Taylor, A.I. & McLauchlan, J. Mobility of the hepatitis C virus NS4B protein on the endoplasmic reticulum membrane and membrane-associated foci. J Gen Virol 86, 1415-21 (2005).

112
17. Lundin, M., Lindstrom, H., Gronwall, C. & Persson, M.A. Dual topology of the processed hepatitis C virus protein NS4B is influenced by the NS5A protein. J Gen Virol 87, 3263-72 (2006).
18. Elazar, M., Liu, P., Rice, C.M. & Glenn, J.S. An N-terminal amphipathic helix in hepatitis C virus (HCV) NS4B mediates membrane association, correct localization of replication complex proteins, and HCV RNA replication. J Virol 78, 11393-400 (2004).
19. Gouttenoire, J. et al. Identification of a novel determinant for membrane association in hepatitis C virus nonstructural protein 4B. J Virol 83, 6257-68 (2009).
20. Einav, S., Elazar, M., Danieli, T. & Glenn, J.S. A nucleotide binding motif in hepatitis C virus (HCV) NS4B mediates HCV RNA replication. J Virol 78, 11288-95 (2004).
21. Yu, G.Y., Lee, K.J., Gao, L. & Lai, M.M. Palmitoylation and polymerization of hepatitis C virus NS4B protein. J Virol 80, 6013-23 (2006).
22. Einav, S. et al. Discovery of a hepatitis C target and its pharmacological inhibitors by microfluidic affinity analysis. Nat Biotech 26, 1019-1027 (2008).
23. Liang, T.J., Rehermann, B., Seeff, L.B. & Hoofnagle, J.H. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann Intern Med 132, 296-305 (2000).
24. Reed, K.E. & Rice, C.M. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Current topics in microbiology and immunology 242, 55-84 (2000).
25. Rice, C.M. Flaviviridae: The viruses and their replication. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields Virology. (Lippincott-Raven Publications, Philadelphia, Pa., 1996).
26. Egger, D. et al. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 76, 5974-5984 (2002).
27. Gosert, R. et al. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J Virol 77, 5487-5492 (2003).
28. El-Hage, N. & Luo, G. Replication of hepatitis C virus RNA occurs in a membrane-bound replication complex containing nonstructural viral proteins and RNA. J Gen Virol 84, 2761-2769 (2003).
29. Kusov, Y.Y., Probst, C., Jecht, M., Jost, P.D. & Gauss-Müller, V. Membrane association and RNA binding of recombinant hepatitis A virus protein 2C. Arch Virol 143, 931-944 (1998).
30. Rodríguez, P.L. & Carrasco, L. Poliovirus protein 2C contains two regions involved in RNA binding activity. J Biol Chem 270, 10105-10112 (1995).
31. Echeverri, A.C. & Dasgupta, A. Amino terminal regions of poliovirus 2C protein mediate membrane binding. Virology 208, 540-553 (1995).
32. Overington JP, A.-L.B., Hopkins AL. How many drug targets are there? Nat Rev Drug Discov 5, 993-6 (2006).

113
33. Lundin, M., Monné, M., Widell, A., Von Heijne, G. & Persson, M.A.A. Topology of the membrane-associated hepatitis C virus protein NS4B. J Virol 77, 5428-5438 (2003).
34. Hadd AG, R.D., Halliwell JW, Jacobson SC, Ramsey JM. Microchip device for performing enzyme assays. Anal Chem 69, 3407-12 (1997).
35. Maerkl, S.J. & Quake, S.R. A systems approach to measuring the binding energy landscapes of transcription factors. Science 315, 233-237 (2007).
36. Toepke, M.W. & Beebe, D.J. PDMS absorption of small molecules and consequences in microfluidic applications. Lab on a chip 6, 1484-1486 (2006).
37. Lee, J.N., Park, C. & Whitesides, G.M. Solvent compatibility of poly(dimethylsiloxane)-based microfluidic devices. Analytical chemistry 75, 6544-6554 (2003).
38. Whitesides, G.M. The origins and the future of microfluidics. Nature 442, 368-373 (2006).
39. Kang, L., Chung, B.G., Langer, R. & Khademhosseini, A. Microfluidics for drug discovery and development: from target selection to product lifecycle management. Drug discovery today 13, 1-13 (2008).
40. Myer, V.E., Fan, X.C. & Steitz, J.A. Identification of HuR as a protein implicated in AUUUA-mediated mRNA decay. Embo J 16, 2130-2139 (1997).
41. Park, S., Myszka, D.G., Yu, M., Littler, S.J. & Laird-Offringa, I.A. HuD RNA recognition motifs play distinct roles in the formation of a stable complex with AU-rich RNA. Molecular and cellular biology 20, 4765-4772 (2000).
42. Park-Lee, S., Kim, S. & Laird-Offringa, I.A. Characterization of the interaction between neuronal RNA-binding protein HuD and AU-rich RNA. J Biol Chem 278, 39801-39808 (2003).
43. Park, S., Myszka, D.G., Yu, M., Littler, S.J. & Laird-Offringa, I.A. HuD RNA recognition motifs play distinct roles in the formation of a stable complex with AU-rich RNA. Mol Cell Biol 20, 4765-72 (2000).
44. Park-Lee, S., Kim, S. & Laird-Offringa, I.A. Characterization of the interaction between neuronal RNA-binding protein HuD and AU-rich RNA. J Biol Chem 278, 39801-8 (2003).
45. Glenn, J.S., Taylor, J.M. & White, J.M. In vitro-synthesized hepatitis delta virus RNA initiates genome replication in cultured cells. J Virol 64, 3104-7 (1990).
46. Glenn, J.S., Watson, J.A., Havel, C.M. & White, J.M. Identification of a prenylation site in delta virus large antigen. Science 256, 1331-3 (1992).
47. Burd, C.G. & Dreyfuss, G. Conserved structures and diversity of functions of RNA-binding proteins. Science 265, 615-621 (1994).
48. Wung, C.H. et al. Identification of the RNA-binding sites of the triple gene block protein 1 of bamboo mosaic potexvirus. J Gen Virol 80 ( Pt 5), 1119-1126 (1999).
49. Burd, C.G. & Dreyfuss, G. Conserved structures and diversity of functions of RNA-binding proteins. Science 265, 615-21 (1994).
50. Rodriguez, P.L. & Carrasco, L. Poliovirus protein 2C contains two regions involved in RNA binding activity. J Biol Chem 270, 10105-12 (1995).

114
51. Wung, C.H. et al. Identification of the RNA-binding sites of the triple gene block protein 1 of bamboo mosaic potexvirus. J Gen Virol 80 ( Pt 5), 1119-26 (1999).
52. Spångberg, K., Wiklund, L. & Schwartz, S. HuR, a protein implicated in oncogene and growth factor mRNA decay, binds to the 3' ends of hepatitis C virus RNA of both polarities. Virology 274, 378-390 (2000).
53. Tscherne, D.M. et al. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J Virol 80, 1734-1741 (2006).
54. Lindenbach, B.D. et al. Complete replication of hepatitis C virus in cell culture. Science 309, 623-626 (2005).
55. Elazar, M. et al. Amphipathic helix-dependent localization of NS5A mediates hepatitis C virus RNA replication. J Virol 77, 6055-6061 (2003).
56. Tong, X. et al. Identification and analysis of fitness of resistance mutations against the HCV protease inhibitor SCH 503034. Antiviral Res 70, 28-38 (2006).
57. Dimitrova, M., Imbert, I., Kieny, M.P. & Schuster, C. Protein-protein interactions between hepatitis C virus nonstructural proteins. J Virol 77, 5401-5414 (2003).
58. Blight, K.J., Kolykhalov, A.A. & Rice, C.M. Efficient initiation of HCV RNA replication in cell culture. Science 290, 1972-4 (2000).
59. Glenn, J.S., Watson, J.A., Havel, C.M. & White, J.M. Identification of a prenylation site in delta virus large antigen. Science 256, 1331-1333 (1992).
60. Huang, L. et al. Hepatitis C virus nonstructural protein 5A (NS5A) is an RNA-binding protein. J Biol Chem 280, 36417-36428 (2005).
61. Roosild, T.P. et al. NMR structure of Mistic, a membrane-integrating protein for membrane protein expression. Science 307, 1317-1321 (2005).
62. Schilders, G., Raijmakers, R., Raats, J.M.H. & Pruijn, G.J.M. MPP6 is an exosome-associated RNA-binding protein involved in 5.8S rRNA maturation. Nucleic Acids Res 33, 6795-6804 (2005).
63. Gao, L., Aizaki, H., He, J.W. & Lai, M.M. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J Virol 78, 3480-8 (2004).
64. Dimitrova, M., Imbert, I., Kieny, M.P. & Schuster, C. Protein-protein interactions between hepatitis C virus nonstructural proteins. J Virol 77, 5401-14 (2003).
65. Paredes, A.M. & Blight, K.J. A genetic interaction between hepatitis C virus NS4B and NS3 is important for RNA replication. J Virol 82, 10671-83 (2008).
66. Kuiken, C., Yusim, K., Boykin, L. & Richardson, R. The Los Alamos hepatitis C sequence database. Bioinformatics 21, 379-84 (2005).
67. Elazar, M. et al. Amphipathic helix-dependent localization of NS5A mediates hepatitis C virus RNA replication. J Virol 77, 6055-61 (2003).
68. Mo, H. et al. Mutations conferring resistance to a hepatitis C virus (HCV) RNA-dependent RNA polymerase inhibitor alone or in combination with an

115
HCV serine protease inhibitor in vitro. Antimicrob Agents Chemother 49, 4305-14 (2005).
69. Chunduru, S.K. et al. 1686949-A2: (VIROPHARMA INC (VIRO-Non-standard) CHUNDURU S K (CHUN-Individual) BENATATOS C A (BENA-Individual) NITZ T J (NITZ-Individual) BAILEY T R (BAIL-Individual), 2005).
70. De Francesco, R. & Migliaccio, G. Challenges and successes in developing new therapies for hepatitis C. Nature 436, 953-60 (2005).
71. Lin, C., Kwong, A.D. & Perni, R.B. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3.4A serine protease. Infect Disord Drug Targets 6, 3-16 (2006).
72. Malcolm, B.A. et al. SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells. Antimicrob Agents Chemother 50, 1013-20 (2006).
73. Sklan, E.H. et al. TBC1D20 is a Rab1 GTPase-activating protein that mediates hepatitis C virus replication. J Biol Chem 282, 36354-61 (2007).
74. Matto, M., Rice, C.M., Aroeti, B. & Glenn, J.S. Hepatitis C virus core protein associates with detergent-resistant membranes distinct from classical plasma membrane rafts. J Virol 78, 12047-53 (2004).
75. Lohmann, V., Hoffmann, S., Herian, U., Penin, F. & Bartenschlager, R. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J Virol 77, 3007-19 (2003).
76. Lindenbach, B.D. et al. Complete replication of hepatitis C virus in cell culture. Science 309, 623-6 (2005).
77. Trozzi, C. et al. In vitro selection and characterization of hepatitis C virus serine protease variants resistant to an active-site peptide inhibitor. J Virol 77, 3669-79 (2003).
78. Cho, N.J., Kanazawa, K.K., Glenn, J.S. & Frank, C.W. Employing two different quartz crystal microbalance models to study changes in viscoelastic behavior upon transformation of lipid vesicles to a bilayer on a gold surface. Anal Chem 79, 7027-35 (2007).
79. Kolchens, S., Ramaswami, V., Birgenheier, J., Nett, L. & O'Brien, D.F. Quasi-elastic light scattering determination of the size distribution of extruded vesicles. Chem Phys Lipids 65, 1-10 (1993).











![Hepatitis B virus and hepatitis C virus play different ... · alcoholic cirrhosis, hepatitis viruses, tobacco and metabolic diseases[4]. Hepatitis viruses, including hepatitis B virus](https://img.pdfslide.net/doc/110x75/60e46cab5bd9101a6f539e91/hepatitis-b-virus-and-hepatitis-c-virus-play-different-alcoholic-cirrhosis.jpg)

















