Embed Size (px)
Citation preview

Object-oriented Programming Paradigms for MolecularModeling
AMIT GUPTA, SHAJI CHEMPATH, MARTIN J. SANBORN, LOUIS A. CLARK and RANDALL Q. SNURR*
Department of Chemical Engineering and Center for Catalysis and Surface Science, Northwestern University, Evanston, IL 60208, USA
(Received April 2002; In final form July 2002)
This paper discusses the application of object-orientedprogramming (OOP) design concepts to the developmentof molecular simulation code. A number of newlanguages such as Fortran 90 (F90) have been developedover the last decade that support the OOP designphilosophy. We briefly describe the salient features ofF90 and some basic object-oriented design principles. Asan illustration of the design concepts we implement ageneral interface in F90 for calculating pairwise inter-actions that can be extended easily to any number ofdifferent forcefield models. The ideas presented here areused in the development of a multipurpose simulationcode, named Music. An example of the use of Music forgrand canonical Monte Carlo (GCMC) simulations offlexible sorbate molecules in zeolites is given. Theexample illustrates how OOP allowed existing code formolecular dynamics and GCMC to be easily combined toperform hybrid GCMC simulations with minimal codingeffort.
Keywords: Object-oriented programming; Molecular simulation;Fortran 90; Hybrid Monte Carlo
INTRODUCTION
Computer simulation consists of two levels ofabstraction. The first level involves taking a physicalphenomenon we wish to simulate and, after makingsuitable assumptions and approximations, descri-bing it as a sequence of mathematical steps. Thesecond level then involves communicating themathematical model to the computer using a suitableform of programming abstraction called a program-ming language. Many programming languages havebeen developed since the early days of assembly
language. This development has been spurred by theconstant need to enable the programmer to writeprograms faster with greater program efficiency,understandability, portability and extensibility. Alsoas new programming paradigms were defined, newlanguages had to be developed to support theseparadigms. Structured programming and object-oriented programming (OOP) are two importantparadigms that have shaped the development ofmost modern languages.
In the 1970s and early 1980s structured programming[1] was the dominant programming methodology.Higher-level languages such as Fortran, C and Pascalwhich supported this form of coding became verypopular during that period. The idea behindstructured programming was to divide each pro-gram into a series of smaller tasks, which in turncould be further simplified until the task could beimplemented without further decomposition. Thiswas referred to as the “top-down” design approach,and it worked well since most complex problems areinherently hierarchical. However, each top-downdesign produced a solution unique to that problem,and hence it was difficult to reuse subroutines inother projects without extensive modifications. Also,this approach did not give adequate consideration tothe data being used.
Then in the last one and half decades anotherapproach was developed, where instead of sub-dividing the problem into tasks the design focus wasshifted to the data structure. This came from therealization that a system’s functionality tends tochange more than the data on which it acts. Data andways of structuring it were given primary impor-tance and the code was organized in modules
ISSN 0892-7022 print/ISSN 1029-0435 online q 2003 Taylor & Francis Ltd
DOI: 10.1080/0892702031000065719
*Corresponding author. Tel.: þ1-847-467-2977. Fax: þ1-847-467-1018. E-mail: [email protected]
Molecular Simulation, 2003 Vol. 29 (1), pp. 29–46

around it. By combining the data structure with itsintrinsic functionality, these modules were “self-sufficient” entities. These modules could be just“plugged” into new programs, thus improvingreusability and reducing coding time. Also, sincethese modules were designed to emulate real worldobjects they also made the code easier to understandand maintain. These modules were called objects andthe programming approach was referred to as object-oriented programming, often abbreviated as OOP. Thatsome of the most popular languages such as Cþþ ,Java and Fortran 90 (F90) today support this codingmethodology is a testament to its advantages.
OOP with F90 has found widespread applicationin the simulation community over the last few years[2]. The principal advantage of F90 over the otherOOP languages is that it is fully backwardscompatible with Fortran 77 (F77), allowing program-mers to use the vast amount of F77 code that has beendeveloped over the years. Recently, Morieira andMidkiff [3] analyzed the features of F90 with aprogram for calculating the electrostatic potential ina shielded microstrip structure. They compared theperformance of F90 with High Performance Fortran(HPF) and Cþþ . They found that while HPF even ona single node was faster than F90 for some systems,F90 was faster than Cþþ for every system. Akin [4]illustrates a number of object-oriented features in F90that are important for engineering computation, andCarr [5] discusses how objects can streamline codedevelopment by implementing a module for per-forming vector operations in F90. These and a host ofother papers from the numerical computing com-munity [6–9] are indicative that although thiscommunity has its share of die-hard F77 users, F90has made significant headway in the last few years.However, OOP in F90 has not yet made significantinroads in molecular modeling.
This paper is organized as follows. In the nextsection we review the basics of OOP. We thendescribe some of the salient features of F90 and itsdifferences from F77. We then discuss some of thetypical challenges encountered while writing mole-cular modeling programs and discuss how OOP canbe used to generate elegant solutions to some of theseproblems. We then include source code for someobjects that are small and may be potentially usefulfor molecular modeling. Finally, we present the useof our Music code for developing a new type ofgrand canonical Monte Carlo (GCMC) technique forsimulation of adsorption in zeolites.
BASIC TENETS OF OOP
As discussed in the previous section one of the keyfeatures of any higher level language is its ability toabstract data. Programming languages that support
OOP go further by providing features such asencapsulation, inheritance and polymorphism,which are described below. These features make iteasier to reuse and extend code while minimizing theprobability of introducing bugs. To understand howthese features work it is useful to divide program-mers into two groups. One group is concerned withproviding fundamental subroutines or librarieswhich form the building blocks of larger appli-cations. The second group of programmers then arethose who use these fundamental libraries in theirapplications.
Encapsulation refers to hiding information that thecreator of the object does not want the user to haveaccess to. The user can only access the properties ofany object by function calls provided by the creator.This has a number of advantages. The user sees amuch cleaner interface by which the object can beused rather than having to worry about theunnecessary implementation details. The list offunctions provided by the creator to access the objectis referred to as the Application Programming Interface(API ). The more important benefit is that if thecreator of the object module wants to change the datarepresentation of the module it can be done withoutworrying about breaking the code that uses theobject. All that the creator has to ensure is that thefunctions in the API still work correctly.
OOP tries to give the programmer the power tocreate objects that correspond closely to objects in thereal-world problem domain. Just as objects in the realworld are related by an “is-a” type of relationship(e.g. an apple “is-a” fruit), so are objects in OOP. Thiskind of relationship is implemented in OOP byinheritance. Given two objects P and C, if C inheritsfrom P then it automatically gets all the datastructures and functionality of P. In addition, theobject C can further add to what it inherits, therebyincreasing its own functionality. Thus, inheritance isa way of taking a general sort of an object andtailoring to fit specific needs. For instance, everysquare matrix “is a” matrix, so every function that ispossible on a matrix type object is also possible on asquare matrix object. In addition, the functioneigenvalue is only valid for a square matrix. So, themodule square_matrix could use inheritance toget all the functionality for the module matrix andin addition define its own function for calculatingeigenvalues. In OOP terminology module matrixwould be referred as the base class and modulesquare_matrix as the derived class. Inheritancefurther emphasizes the data-centric nature of OOPby enabling the user to create hierarchies of datatypes.
Closely related to inheritance is the concept ofpolymorphism. Polymorphism enables the program-mer to provide a generic interface to a number ofrelated subroutines. Polymorphism comes in two
A. GUPTA et al.30

flavors—static polymorphism also known as over-loading and dynamic or run-time polymorphism. Staticpolymorphism is used to resolve calls to subroutineswith the same names on the basis of parameterspassed to the subroutine. For instance, there are anumber of ways of specifying parameters tocalculate the area of a triangle. Imagine writing tworoutines for calculating the area of a triangle asfollows:
Then getArea(3.0, 4.0, 5.0) calls the firstfunction and getArea(3.0, 4.0) calls the secondfunction, thus providing a more uniform andintuitive interface to the function.
Dynamic polymorphism involves resolving callsto functions related by an inheritance hierarchy atrun-time as opposed to the compile-time resolutionprovided by static polymorphism. By virtue ofinheritance a “parent” parameter object can bereplaced by anyone of its “child” objects in afunction or subroutine call. Inheritance guaranteesthat a derived class object will have at least the samefunctionality as its base class if not more. Thisimplies that different types of objects related byinheritance can now be passed to the same routine.In OOP since the functional implementations areclosely tied to the objects, this in turn implies thatdepending on the object passed to the routine at run-time different functions may have to be called. Thisability of a routine to take different types of objects asparameters and resolve the function calls at run-timeis referred to as dynamic polymorphism. This sort ofpolymorphism, while not directly supported in F90,can be simulated with the use of pointers asdescribed by Decyk et al. [8,9].
Object-oriented program design requires theprogrammer to think about the problem at a more
fundamental level to identify relevant objects andthen define the relationships among them. Languagefeatures such as inheritance and polymorphism thenfacilitate translation of these ideas into computercode. Admittedly OOP ideas take some getting usedto. There are a number of excellent papers and books[2,9,10] available to provide a deeper understandingof these concepts and their applications.
FEATURES OF FORTRAN 90
Fortran 90 was designed by the Fortran committeewith some of the following goals in mind asdescribed by Reid [11].
. Provide greater expressive power;
. Enhance safety;
. Provide extra fundamental features (such asdynamic storage);
. Exploit modern hardware better;
. Improve portability between different machinearchitectures.
In this section, we give a very brief overview ofsome of the key differences between F77 and F90,and we start building up some objects of use inmolecular simulations. For more details about F90please refer to Refs. [9,12].
F90 makes a number of ornamental changes toimprove the source code formatting. F90 supportsfree-formatting, which disables any special signifi-cance that was attached to columns in F77. An “&” atthe end of a line indicates that the line is continued.The comment character “!” can be placed anywhereon a line with the compiler ignoring anything thatfollows. We use this improved formatting in the codelisted in this paper.
User-defined Type Definitions
One of the biggest improvements over F77 is theability to abstract data into user-defined types (alsoknown as derived types). For instance, we canassociate a number of characteristics to the terms“atoms” and “molecules”. Abstractions of theseterms can be defined using the Type keyword asshown below:
PARADIGMS FOR MOLECULAR MODELLING 31

Now we can use these types just as we would use aprimitive data type to define our variables. Todeclare an array of three molecules and then accessits individual fields we use the % symbol as in:
Also note that we can use a derived type for thedefinition of fields of another derived type. In thiscase we used AtomParams in MoleculeParams. Thus,we can start to build a hierarchy of data structureswith relationships that reflect the order in thephysical problem space.
Modules
In F77 the basic building blocks of a program werefunctions and subroutines. F90 adds another build-ing block called a module. A large part of the OOPfunctionality of F90 comes from the use of modulessince they provide a way to couple the data and itsassociated functionality into one programming unit.We will use the skeleton implementation of a three-dimensional vector object given in Appendix 1 toillustrate a number of features of F90 in general andmodules in particular.
A module is defined using the keyword Module.Modules can be broken up into two major partsseparated by the Contains keyword. The sectionpreceding this keyword primarily describes the datatype definitions and the interface to the routinesdefined in the module. The routines are then definedafter the Contains keyword.
Modules provide a mechanism for supportingencapsulation in F90 through the keywords Public,Private and Use, Only. Just as we can build a hierarchyof user-defined type definitions, it is possible to builda hierarchy of modules. The Use keyword providesaccess to the variables and routines defined inanother module. As discussed before it is a goodpractice to hide the implementation details from theusers of modules. The keywords Private and Publiccan be used to change the visibility of the variablesand routines defined in a module. It is also goodprogramming to “use” only what is needed from a
module. This list is specified with the Only keyword.This makes the code more maintainable by immedia-tely conveying to the reader the source of variablesand routines used in a module.
In addition to data encapsulation, modules provideexplicit interfaces to the routines defined inside them.This means that the compiler has complete infor-mation regarding the type of parameters being passedbetween routines, enabling it to do parameter type-checking. Another advantage is when passing arraysas arguments there is no longer any need to pass theirdimensions. This can be seen in the subroutinevector3D_init1 in Appendix 1 where the dimen-sion of the variable arr is not specified. Explicitinterfaces thus help to reduce the number ofarguments and make the code less prone to insidiouserrors arising from parameter type incompatibility.
The explicit interface provided inside modules isalso useful in supporting overloading. By usingthe interface keyword a generic name can be assig-ned to routines with different parameters definedin a module. Thus, a call to vector3D_init willbe resolved to either vector3D_init1 orvector3D_init2 depending on whether thesecond argument is an array or a number. In additionto overloading subroutines and functions, it is alsopossible to overload operators as shown with thevector3D_add method. The Optional and Presentkeywords provide another mechanism for designingroutines with flexible argument lists. The Optionalkeyword makes the argument optional to theroutine. To check whether the optional argumentwas passed, the keyword Present is used asillustrated in the routine vector3D_display.
Arrays, Pointers and Dynamic Memory
A much sought after feature in F77 was the ability todynamically allocate memory. This feature wasadded to F90. The dynamic memory is managed byusing the keywords Allocate, Allocatable and Deal-locate. The dynamic allocation is done as shownbelow:
A. GUPTA et al.32

Allocate creates memory on the heap and it staysfor the length of the program. It is good program-ming practice to return the memory to the operatingsystem once we are done with it. This is done byusing the Deallocate command.
Allocatable arrays make for more flexible codewhile minimizing memory waste.
Often we want to have allocatable fields in an abs-tract data type. For instance, in moleculeParamswe may want to make the atom array an allocatabletype to reduce memory waste.
Unfortunately, this is illegal in F90 since Allocatableis not an allowed attribute for fields in the userdefined types (this will be fixed in Fortran 2000).Instead we have to use the pointer attribute. Pointervariables hold the location of memory as theirvalue. Depending on the type of the pointer, thisvalue could refer to the location of an integer, anarray or a derived data type. Another way tothink about pointers is as aliases to a particularvariable, e.g.
The variable ptr is said to point to a or beAssociated with the variable a. To dissociate thepointer we use Nullify(ptr). To find out if apointer is associated we use the keyword Associatedas in
Pointers can be used to solve the problem ofhaving dynamically allocatable fields in derived data
types as shown below. This piece of code willinitialize the number of molecules and number ofatoms as specified by the user.
Pointers also allow us to implement polymor-phism in F90 [9] as we shall see later.
In the style of Matlab w [13], F90 providesfunctionality so that arrays may be used in wholeexpressions with scalars as in,
In addition, subarrays of larger arrays called“sections” [11] may be used as arrays. Forinstance,
Not only is this more succinct but it is also morecomputationally efficient than writing Do loops sincethe optimization is better [11].
Portability
With the proliferation of a large number ofarchitectures and operating systems it is difficult toensure portability especially with regard to numericoperations. Towards this end, F90 provides a way tospecify the precision and range of numbers thatremains constant across platforms. This is done
PARADIGMS FOR MOLECULAR MODELLING 33

using the Selected_real_kind, Selected_int_kind and
Kind keywords. The usage is illustrated below:
The result of intrinsic functions Selected_real_kindand Selected_int_kind is a kind type that meets, orminimally exceeds, the requirements specified by theprecision and range [12]. This mechanism ensuresprogram portability across platforms since differentarchitectures have different precisions for primitivedata types.
MOLECULAR MODELING WITH OOP
Two important aspects of molecular modeling thatare constantly evolving are move types and force-fields. Examples of move types include moleculardynamics integration, Monte Carlo translation of amolecule, etc. New types of moves are developed toaccelerate the speed of the simulation, whether it isto achieve equilibrium more quickly or to lower theratio of CPU time spent per unit of physical timewhile tracking the system dynamics. While theability to sample a phase space more efficientlyincreases the precision of the simulation result, theaccuracy of the simulation is determined by theforcefield and the parameters employed. Conse-quently, new and improved forcefields are alsoconstantly being developed.
As computer simulations become more reliablethere is an increased demand on the researcher tocater to a wider array of problems. This oftenrequires the ability to deal with many different kindsof forcefields, and as the problems get more complex,greater functionality is also required to incorporatedifferent kinds of move types into one simulation.The integration of various forcefields and movetypes brings with it new challenges for datarepresentation. The different move types oftenrequire different forms of representation for thecurrent configuration of the system. For instance, it isusually most convenient to work in Cartesiancoordinates for molecular dynamics simulationswhereas generalized coordinates are preferred fordoing Monte Carlo simulations of molecules withconstraints. This is also true for various termscomprising a forcefield. While terms for pairwiseinteractions require Cartesian distances, terms
involving intramolecular interactions work with
various forms of generalized coordinates.
The problem then is to find a “clean,” easilyextensible approach to represent the data in thesimulation, making the programs more flexible andensuring faster code development. The OOP para-digm lends itself naturally to such an approach byenabling the programmer to cast the data objects asmore direct representations of real world objects.Recently, a number of groups [14–16] have describedthe design of some basic objects that can be used asbuilding blocks for various types of molecularsimulations. They illustrate how relatively straight-forward it is to extend the existing building blocks todesign objects for new simulations. The idea then isto distribute these objects to other groups who cancustomize them according to their needs. Huber et al.[14] and Sadus [15] implement the objects usingCþþ , whereas Kofke et al. [16] use Java as theirlanguage of choice. While F90 does not support OOPin the full traditional sense of the word, it does lenditself to a data-centric programming style. Weillustrate the design and implementation of OOPobjects in F90 by implementing a number offorcefields describing pairwise interactions.
Since F90 does not have an OOP syntax, we definea few programming style rules that enable us toenforce the OOP paradigm more effectively. Theserules are given below:
1. All routines in a module start with the name ofthe module to tie the function more strongly withthe module name.
2. Each module has defined within it a user defineddata type that is representative of the data in themodule.
3. All the public routines in the module take as theirfirst parameter a variable of this representativedata type. This enables a close coupling with thedata object and the functionality that is providedby the routine.
4. Each module has an init routine to initialize thedata fields of the variable of the representativedata type.
5. Each module has a display routine to report thevalues of data fields.
6. Each module has a cleanup routine to take care ofdeallocating memory if necessary.
A. GUPTA et al.34

A molecular simulation program can be viewed asconsisting of two distinct parts, the initialization andthe actual run. The initialization is responsible forsetting up the initial configuration, the forcefieldparameters and the parameters for the differentmove types. One of the difficulties of designing ageneral purpose simulation code is that parametersand initialization for the different forcefields andmove types vary widely. OOP allows one to providea generic interface that is used by any driver programto initialize different move types and forcefields.
We have used the ideas discussed above in thedevelopment of the Multipurpose simulation code(Music). All the forcefield calculations (estimation ofenergies and forces resulting from interparticleinteractions) are wrapped into a forcefieldmodule. Similarly, all the calculations associatedwith move types (molecular dynamics integration,Monte Carlo insertions etc.) are wrapped under themoves module.
The interface for forcefield is defined so that itreceives positions of all atoms in the system andreturns the energies and forces on particles ofinterest. The calculation of forces is optional becauseit is not required during Monte Carlo simulations.Also it is not necessary to perform calculations on allmolecules every time. During a Monte Carloinsertion move it is only necessary to calculate theinteraction energy of the newly inserted moleculewith rest of the system, whereas during an MDsimulation we need the forces on all particles duringeach step of the integration. The forcefield objectcontains all information regarding intermolecularand intramolecular interactions. The intermolecularinteractions are usually calculated using pairwisepotentials like Lennard Jones potentials. The pair-model module which does this is described later inthe section.
The interface for moves is defined so that it takes aparticular configuration of the system and changes itto a different one. It returns the energies of old andnew configurations. For example a Monte Carlotranslation move will pick one of the molecules andtranslate it into a different position and then returnthe old and new energies. This new trial configu-ration can be accepted or rejected by the main callingprogram based on the Monte Carlo acceptancecriterion [17].
The actual run usually consists of a number ofiterations where the phase space is sampled byperturbing the configuration according to movetypes derived from statistical mechanics. Thisprocess can be abstracted as illustrated in Fig. 1.Now if we can define a generic interface that can beused by all move types to “communicate” with anytype of forcefield then we can quickly create newsimulations by “plugging” together move types andforcefields as we desire.
F90 enables us to create such generic interfaces. Toillustrate the methodology we implement an inter-face that is used to initialize various models forcalculating pairwise atomic interactions. This objectwill be used by the forcefieldmodule. The object isimplemented in the module called PairModels andconsists of two main routines, PairModels_initand PairModels_getinteraction (Appendix 2),as well as display and cleanup routines.
The responsibility for initializing parametersand getting interactions is then passed on to therelevant potential model object as specified inPairModel_init. This makes sense since eachtype of potential model object “knows” whatparameters it needs and how to use them. TheLennard-Jones potential model object called lj isshown in Appendix 3. This scheme enables us tospecify the kind of potential model we want to use asan input parameter string without changing anycode. Also, to add another pairwise potential model,all we need to do is modify the modulePairModels, making it easy to extend the code.These changes will not affect other objects dealingwith move types or intramolecular potentials.
The module PairModels presented here canbe downloaded from our website [18]. Along withthe source code in this paper the site has also anumber of other potentially useful modules fordownload.
HYBRID GCMC USING MUSIC
In this section an example of the use of Music forstudying adsorption in zeolites is given. This examplehighlights how object oriented programming facili-tated the incorporation of new capabilities into thecode with minimal programming effort. Zeolites arecrystalline microporous materials with cavities andpores about 3–12 A in diameter. They are used forseparation and catalysis applications. In theseprocesses, adsorption and diffusion of guest mole-cules within the zeolite pores play an important role.
FIGURE 1 The anatomy of a simulation run.
PARADIGMS FOR MOLECULAR MODELLING 35

GCMC simulations can be used to study theadsorption of the guest molecules in the zeolitecavities [19]. The regular GCMC procedures areapplicable only when the guest molecule beingadsorbed is considered rigid. We have used a methodcalled hybrid GCMC in which regular GCMC iscombined with the hybrid Monte Carlo method totake care of the flexibility of the guest molecule.
Hybrid GCMC
The hybrid Monte Carlo (HMC) method wasintroduced by Duane et al. [20] in 1987. A detaileddescription is given in Ref. [21]. This method isusually used for simulating an NVT ensemble, but inthe current work we have used HMC combined withGCMC to simulate a grand canonical ensemble.HMC uses overall system moves instead of the usualtranslation and rotation moves of individual mole-cules for exploring the configurations of the system.In these hybrid moves, the particles in the simulationvolume are given velocities taken from a Maxwell–Boltzmann distribution and then integrated for aspecific amount of time (t ) using a specificintegration step (Dt ). The resulting configuration isaccepted or rejected based on a Metropolis accep-tance criterion [21]. The hybrid GCMC simulationthus consists of HMC moves, insertion moves anddeletion moves.
Prior to the hybrid GCMC simulation a library ofconfigurations of a single guest molecule in an idealgas phase is generated by doing HMC at the sametemperature as the main simulation. For insertion ofa molecule into the system during hybrid GCMC,one of the configurations is selected from the libraryand then inserted into the zeolite. The Eulerianangles of orientation for the molecule are selectedrandomly during each insertion. The insertions intothe simulation volume can be done randomly or withbias towards the low energy regions of the zeolite[22]. It can be shown that this scheme leads to thesame acceptance criterion for insertion as in regularGCMC.
PAccðN ! N þ 1Þ ¼ min 1;fV
ðN þ 1ÞkTexpð2DV=kTÞ
� �
where DV denotes the potential energy of inter-action between the whole system and the sorbatemolecule being inserted and f is the fugacity of thesorbate at the given pressure.
Combining MD and GCMC
Using the Music framework, it was extremely easy todevelop the hybrid GCMC code. The integrationcode (MD code) and the GCMC code had alreadybeen developed. The different move types used for
these simulations were already developed as movetype objects and placed in the modules. See Fig. 2 foran arrangement of the modules in Music. Theindividual move types are wrapped under themoves module which provides a common interfacefor accessing all move types. This common interfacemakes the process of calling individual move typeseasier. The main calling programs (top level of Fig. 2)need not know much about the interface for theindividual move types. During an MD simulationthe main calling program calls the moves modulerepeatedly and moves subsequently calls theintegrate module. During a GCMC simulationmoves is called repeatedly and moves calls theinsert, delete, translate or rotate modules.All of these move types access forcefield whichcontains details about the interaction betweenmolecules to calculate the energies and accelerations.To create a hybrid GCMC simulation a main drivermodule called hybridgc was written, which callsintegrate, insert and delete move types.Since all of these modules were well testedpreviously and their API was well defined, thecreation of hybridgc and testing became very easy.The flexibility added by defining the move types andplacing them under the common interface calledmoves can be explained with the help of an example.Suppose one of the developers in the Musicdevelopers group wants to do a hybrid GCMCsimulation. If the developer also wants to include aMonte Carlo translation move in the simulation inaddition to the insert, delete and integratemoves, all that needs to be done is to add a few linesof code in the hybridgc module which makes a callto translate through the moves module. Themodule translate was probably developed bysome other developer interested in regular GCMC.However since the module is placed under thecommon interface moves, the hybrid GCMC deve-loper can also access it and use it. This makes codesharing among developers working on differentmove types easier.
FIGURE 2 The hierarchy of move type modules. Dotted lines:call tree for a GCMC simulation. Dashed lines: call tree for an MDsimulation. Solid lines: call tree for hybrid GCMC simulation.
A. GUPTA et al.36
The potential for a decrease in computationalspeed may make some researchers cautious aboutadopting OOP. However, with improving compilers,differences in speed between F77 and F90 arebecoming quite small. To quantify this for our case,we performed MD simulations of butane in thezeolite silicalite, using 64 butane molecules (asdescribed below) in 16 unit cells of zeolite withperiodic boundary conditions. Comparing our older,highly-optimized F77 code against the new Musiccode, we found that the OOP code was 2.6 timesslower. We believe that there is room for optimi-zation of the Music code, which would reduce thisfactor, and again improved compilers will also help.The reduced time in writing and maintaining thecode, however, fully compensates for any increasedrun-time in our experience.
Effect of Sorbate Flexibility on Adsorption
Using hybrid GCMC we calculated the adsorptionisotherms of butane in the zeolite silicalite at 300 K.The butane molecule was modeled using a unitedatom model with bond stretching, bond bending andbond torsion potentials. The forcefield parameterswere the same as the ones used by Macedonia andMaginn [23]. In addition to those parameters (whichkept bond lengths fixed), we also included the bondstretching potential energy using the Morse equation[24]. We also performed regular GCMC simulationsof completely rigid butane in silicalite for compari-son. One might expect to see larger number ofmolecules adsorbed in the silicalite with the flexiblebutane model compared to the rigid model, since a
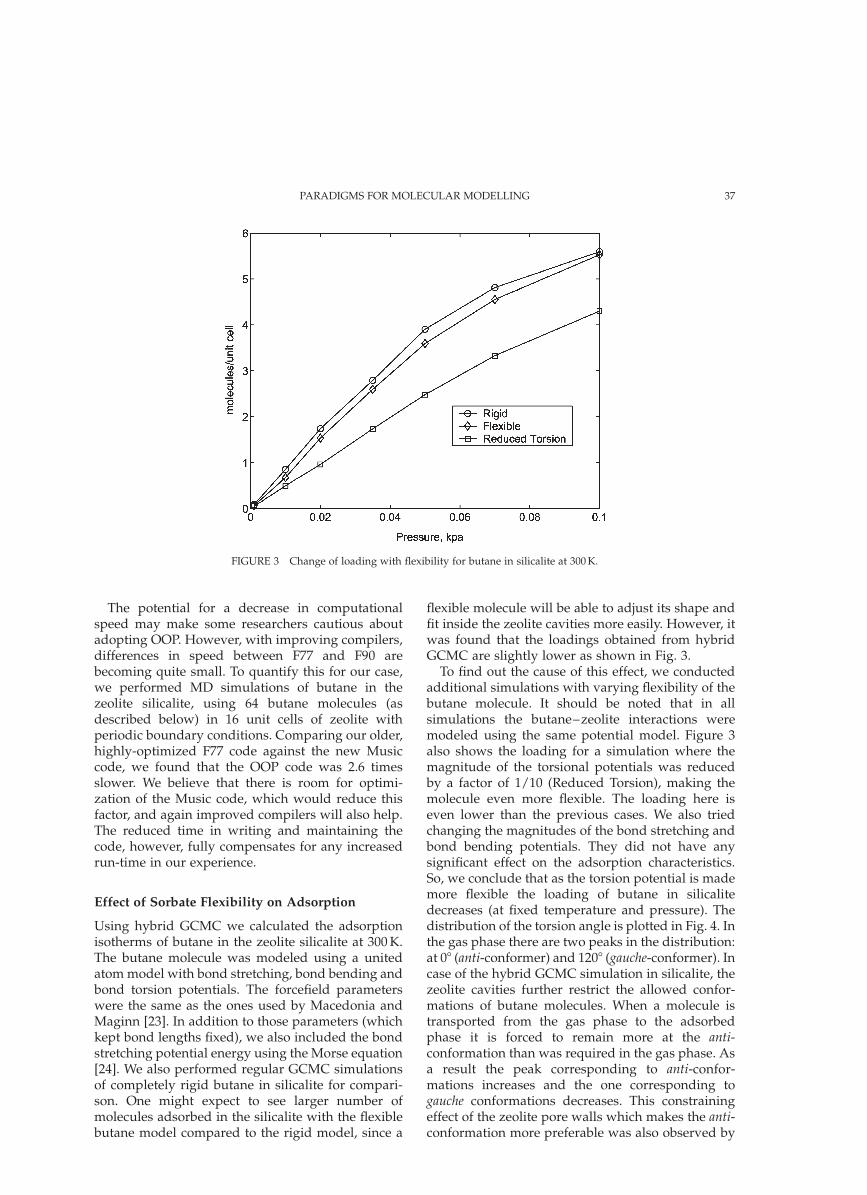
flexible molecule will be able to adjust its shape andfit inside the zeolite cavities more easily. However, itwas found that the loadings obtained from hybridGCMC are slightly lower as shown in Fig. 3.
To find out the cause of this effect, we conductedadditional simulations with varying flexibility of thebutane molecule. It should be noted that in allsimulations the butane–zeolite interactions weremodeled using the same potential model. Figure 3also shows the loading for a simulation where themagnitude of the torsional potentials was reducedby a factor of 1/10 (Reduced Torsion), making themolecule even more flexible. The loading here iseven lower than the previous cases. We also triedchanging the magnitudes of the bond stretching andbond bending potentials. They did not have anysignificant effect on the adsorption characteristics.So, we conclude that as the torsion potential is mademore flexible the loading of butane in silicalitedecreases (at fixed temperature and pressure). Thedistribution of the torsion angle is plotted in Fig. 4. Inthe gas phase there are two peaks in the distribution:at 08 (anti-conformer) and 1208 (gauche-conformer). Incase of the hybrid GCMC simulation in silicalite, thezeolite cavities further restrict the allowed confor-mations of butane molecules. When a molecule istransported from the gas phase to the adsorbedphase it is forced to remain more at the anti-conformation than was required in the gas phase. Asa result the peak corresponding to anti-confor-mations increases and the one corresponding togauche conformations decreases. This constrainingeffect of the zeolite pore walls which makes the anti-conformation more preferable was also observed by
FIGURE 3 Change of loading with flexibility for butane in silicalite at 300 K.
PARADIGMS FOR MOLECULAR MODELLING 37
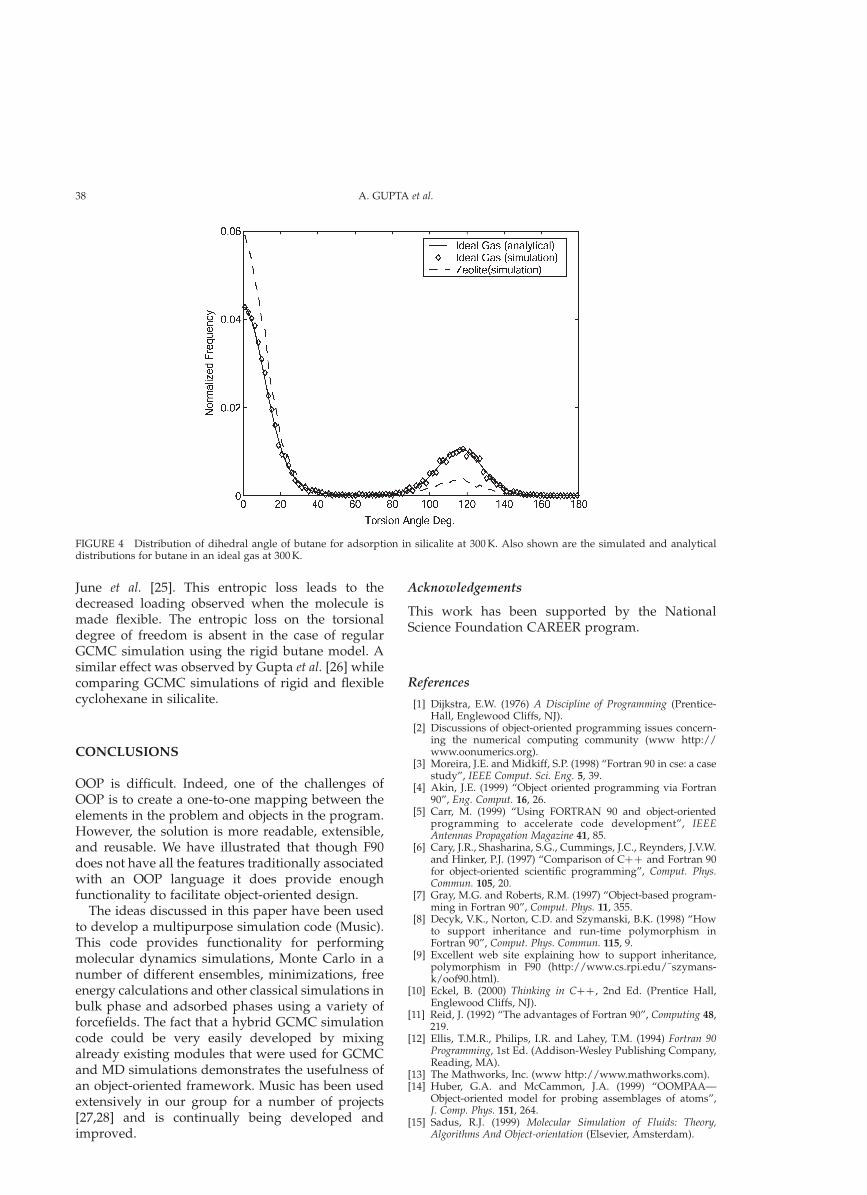
June et al. [25]. This entropic loss leads to thedecreased loading observed when the molecule ismade flexible. The entropic loss on the torsionaldegree of freedom is absent in the case of regularGCMC simulation using the rigid butane model. Asimilar effect was observed by Gupta et al. [26] whilecomparing GCMC simulations of rigid and flexiblecyclohexane in silicalite.
CONCLUSIONS
OOP is difficult. Indeed, one of the challenges ofOOP is to create a one-to-one mapping between theelements in the problem and objects in the program.However, the solution is more readable, extensible,and reusable. We have illustrated that though F90does not have all the features traditionally associatedwith an OOP language it does provide enoughfunctionality to facilitate object-oriented design.
The ideas discussed in this paper have been usedto develop a multipurpose simulation code (Music).This code provides functionality for performingmolecular dynamics simulations, Monte Carlo in anumber of different ensembles, minimizations, freeenergy calculations and other classical simulations inbulk phase and adsorbed phases using a variety offorcefields. The fact that a hybrid GCMC simulationcode could be very easily developed by mixingalready existing modules that were used for GCMCand MD simulations demonstrates the usefulness ofan object-oriented framework. Music has been usedextensively in our group for a number of projects[27,28] and is continually being developed andimproved.
Acknowledgements
This work has been supported by the NationalScience Foundation CAREER program.
References
[1] Dijkstra, E.W. (1976) A Discipline of Programming (Prentice-Hall, Englewood Cliffs, NJ).
[2] Discussions of object-oriented programming issues concern-ing the numerical computing community (www http://www.oonumerics.org).
[3] Moreira, J.E. and Midkiff, S.P. (1998) “Fortran 90 in cse: a casestudy”, IEEE Comput. Sci. Eng. 5, 39.
[4] Akin, J.E. (1999) “Object oriented programming via Fortran90”, Eng. Comput. 16, 26.
[5] Carr, M. (1999) “Using FORTRAN 90 and object-orientedprogramming to accelerate code development”, IEEEAntennas Propagation Magazine 41, 85.
[6] Cary, J.R., Shasharina, S.G., Cummings, J.C., Reynders, J.V.W.and Hinker, P.J. (1997) “Comparison of Cþþ and Fortran 90for object-oriented scientific programming”, Comput. Phys.Commun. 105, 20.
[7] Gray, M.G. and Roberts, R.M. (1997) “Object-based program-ming in Fortran 90”, Comput. Phys. 11, 355.
[8] Decyk, V.K., Norton, C.D. and Szymanski, B.K. (1998) “Howto support inheritance and run-time polymorphism inFortran 90”, Comput. Phys. Commun. 115, 9.
[9] Excellent web site explaining how to support inheritance,polymorphism in F90 (http://www.cs.rpi.edu/~szymans-k/oof90.html).
[10] Eckel, B. (2000) Thinking in Cþþ , 2nd Ed. (Prentice Hall,Englewood Cliffs, NJ).
[11] Reid, J. (1992) “The advantages of Fortran 90”, Computing 48,219.
[12] Ellis, T.M.R., Philips, I.R. and Lahey, T.M. (1994) Fortran 90Programming, 1st Ed. (Addison-Wesley Publishing Company,Reading, MA).
[13] The Mathworks, Inc. (www http://www.mathworks.com).[14] Huber, G.A. and McCammon, J.A. (1999) “OOMPAA—
Object-oriented model for probing assemblages of atoms”,J. Comp. Phys. 151, 264.
[15] Sadus, R.J. (1999) Molecular Simulation of Fluids: Theory,Algorithms And Object-orientation (Elsevier, Amsterdam).
FIGURE 4 Distribution of dihedral angle of butane for adsorption in silicalite at 300 K. Also shown are the simulated and analyticaldistributions for butane in an ideal gas at 300 K.
A. GUPTA et al.38

[16] Kofke, D.A. and Mihalick, B.C. (2002) “Web-based techno-logies for teaching and using molecular simulation”, FluidPhase Equilibria 194–197, 327.
[17] Allen, M.P. and Tildesley, D.J. (1987) Computer Simulation ofLiquids (Oxford University Press, Oxford).
[18] Web site for obtaining source code given in this paper andother potentially useful modules (www http://zeolites.cqe.northwestern.edu/Music/music.html).
[19] Fuchs, A. and Cheetham, A. (2001) “Adsorption of guestmolecules in zeolitic materials: computational aspects”,J. Phys. Chem. B 105, 7375.
[20] Duane, S., Kennedy, A.D., Pendleton, B.J. and Roweth, D.(1987) “Hybrid Monte Carlo”, Phys. Lett. B 195, 216.
[21] Mehlig, B., Heermann, D. and Forrest, B. (1992) “HybridMonte Carlo method for condensed-matter systems”, Phys.Rev. B 45, 679.
[22] Snurr, R.Q., Bell, A.T. and Theodorou, D.N. (1993) “Predictionof adsorption of aromatic hydrocarbons in silicalite fromgrand canonical Monte Carlo simulations with biasedinsertions”, J. Phys. Chem. 97, 13742.
[23] Macedonia, M.D. and Maginn, E.J. (1999) “A biased grandcanonical Monte Carlo method for simulating adsorptionusing all-atom and branched united atom models”, Mol. Phys.96, 1375.
[24] Demontis, P., Suffritti, G.B. and Tilocca, A. (1996) “Diffusionand vibrational relaxation of a diatomic molecule in the porenetwork of a pure silica zeolite: a molecular dynamics study”,J. Chem. Phys. 105, 5586.
[25] June, R.L., Bell, A.T. and Theodorou, D.N. (1990) “Predictionof low occupancy sorption of alkanes in silicalite”, J. Phys.Chem. 94, 1508.
[26] Gupta, A., Clark, L.A. and Snurr, R.Q. (2000) “Grandcanonical Monte Carlo simulations of non-rigid molecules:siting and segregation in silicalite zeolite”, Langmuir 16, 3910.
[27] Gupta, A. and Snurr, R.Q. “A study of pore blockagein silicalite zeolite using minimum energy path simulations”,In Preparation.
[28] Gupta, A. and Snurr, R.Q. “A study of pore blockage insilicalite zeolite using free energy perturbation calculations”,In Preparation.
APPENDIX 1
PARADIGMS FOR MOLECULAR MODELLING 39

A. GUPTA et al.40

PARADIGMS FOR MOLECULAR MODELLING 41

APPENDIX 2
A. GUPTA et al.42

PARADIGMS FOR MOLECULAR MODELLING 43

APPENDIX 3
A. GUPTA et al.44

PARADIGMS FOR MOLECULAR MODELLING 45

A. GUPTA et al.46






























