Embed Size (px)
Citation preview

CE
MIL
AL
TU
NA
Y
İS
TA
NB
UL
ÜN
İVE
RS
İTE
Sİ S
AĞ
. BİL
.
EN
ST
.
YÜ
KS
EK
LİS
AN
S
İS
TA
NB
UL
-20
18
Tez kabul edildikten sonra yapılan sabit ciltte
sırt yazısı bu şablona göre yazılacak. Yazılar tek satır
olacak
Cilt sırtı yazıların yönü yukarıdan aşağıya
(sol yandaki gibi) olacak .
Tez, Yüksek Lisans’sa, YÜKSEK LİSANS
TEZİ;
Doktora ise DOKTORA TEZİ ifadesi
kalacak
Adınızı soyadınızı giriniz
Tez Sınavının yapılacağı yılı yazınız


ii
T.C.
İSTANBUL ÜNİVERSİTESİ
SAĞLIK BİLİMLERİ ENSTİTÜSÜ
DANIŞMAN
DOÇ. DR. SELÇUK SÖZER TOKDEMİR
GENETİK ANABİLİM DALI
GENETİK PROGRAMI
İSTANBUL-2018
CEMİL ALTUNAY
POLİSİTEMİA VERA’DA SİTOKİN SİNYAL
YOLAĞININ ETKİSİ
( YÜKSEK LİSANS TEZİ )

iii

iv
BEYAN

v
İTHAF
Aileme ithaf ediyorum

vi
TEŞEKKÜR
Değerli desteklerinden dolayı danışmanım Selçuk Sözer TOKDEMİR‘e;
Ekip arkadaşlarım İldeniz USLU, Hilal HEKİMOĞLU, Can Veysel ŞOROĞLU,
Büşra YAŞA, Selin Fulya TOPRAK ve Cemal Çağıl KOÇANA’ya;
Aziz Sancar Deneysel Tıp Araştırma Enstitüsü Öğretim Üyelerine, çalışanlarına
ve tüm arkadaşlarıma;
Sağlık teknikeri Abdullah YILMAZ ve Biyolog Ayşe ENGİN’e;
Değerli arkadaşlarım Sami YEŞİLKAYA, Emre MURATOĞLU, Ali İsa TAŞ,
Ferid TAŞ, Barış TAŞ, Burak GÜLBOL, Ferdi KURTOĞLU ve Koray REYHAN’a;
Lisans ve Yüksek Lisans arkadaşım Esra KAYNAK’a;
Tuğçe TORUN’a;
Annem Faize ALTUNAY, babam Mehmet Ziya ALTUNAY ve kardeşim Mert
ALTUNAY’a teşekkür ederim.
Bu çalışma, İstanbul Üniversitesi Bilimsel Araştırma Projeleri Birimi tarafından
desteklenmiştir. Proje No: 27338

vii
İÇİNDEKİLER
TEZ ONAYI ................................... ERROR! BOOKMARK NOT DEFINED.
BEYAN .............................................................................................................. İV
İTHAF .................................................................................................................. V
TEŞEKKÜR ....................................................................................................... Vİ
İÇİNDEKİLER ................................................................................................. Vİİ
TABLOLAR LİSTESİ ......................................................................................... X
SEMBOLLER / KISALTMALAR LİSTESİ ................................................... Xİİ
ÖZET ................................................................................................................ XV
ABSTRACT .................................................................................................... XVİ
1. GİRİŞ VE AMAÇ ............................................................................................. 1
2. GENEL BİLGİLER .......................................................................................... 3
2.1. Miyeloproliferatif Neoplaziler ..................................................................... 3
2.1.1. Tarihçe .................................................................................................... 3
2.1.2. Sınıflandırma ........................................................................................... 4
2.1.3. Klasik Miyeloproliferatif Neoplazilerin Genetik Temeli ve Moleküler
Patofizyolojisi ............................................................................................................... 5
2.1.4. JAK2 Gen Mutasyonu ve Mutasyonun MPN Patogenezindeki Rolü ..... 6
2.1.5. “MPN ile sınırlı” Mutasyonlar, JAK2 Kinaz-bağımlı Sitokin Reseptör
Yollarını Aktive Eder .................................................................................................... 7
2.1.6. MPN Başlangıcında Önemli Faktörler .................................................. 10
2.2. Sitokinler .................................................................................................... 11
2.2.1. Kemokinler............................................................................................ 12
2.2.2. IFN ile Uyarılabilen CXCR3 Kemokinleri ........................................... 15
2.2.3. CXCR3 .................................................................................................. 16
2.2.3.1. CXCR3'ün Tanımlanması ve Ekspresyonu ...................................... 16
2.2.3.2. Alternatif Eklemeyle Oluşturulan CXCR3 Varyantlarının Keşfi .... 18
2.2.3.3. CXCR3'ün in vivo İşlevi için Ekleme Varyantlarının Potansiyel
Önemi ........................................................................................................................ 20
2.2.4. Kemokin ile Uyarılan CXCR3 İnternalizasyonu .................................. 21
2.2.4.1. CXCR3 Ligandlarının Hastalık ve Tedavi ile İlişkisi ...................... 22
2.2.5. MPN ile İlişkilendirilen Sitokin ve Kemokinler ................................... 23

viii
3. GEREÇ VE YÖNTEM ................................................................................... 25
3.1. Gereç .......................................................................................................... 25
3.1.1. Hasta Örnekleri ..................................................................................... 25
3.1.2. Deneylerde Kullanılan Kontrol Örnekleri ............................................. 27
3.1.2.1. Sağlıklı Kontrol Örnekleri ................................................................ 28
3.1.3. Kullanılan Kimyasal Maddeler ............................................................. 28
3.1.4. Kullanılan Kitler ................................................................................... 29
3.1.5. Kullanılan Cihazlar ............................................................................... 30
3.1.6. Kullanılan Tampon ve Çözeltiler .......................................................... 31
3.2. Yöntem ....................................................................................................... 32
3.2.1. Mononükleer Hücre İzolasyonu ............................................................ 32
3.2.2. Hücre Dondurma ................................................................................... 33
3.2.3. Hücre Çözme......................................................................................... 34
3.2.4. Hücre Ayrımı ........................................................................................ 35
3.2.4.1. CD34 MicroBead Kit (MACS Separation) ile Hücre Ayrım
Prosedürü................................................................................................................... 35
3.2.4.2. CD34+/-Lineage- Popülasyon Eldesi Prosedürü ............................... 36
3.2.5. Hücrelerden DNA İzolasyonu ............................................................... 38
3.2.6. İki Aşamalı Allel Spesifik Nested PZR ................................................ 38
3.2.7. Agaroz Jelde Görüntüleme .................................................................... 42
3.2.7.1. Agaroz Jelin Hazırlanması ............................................................... 42
3.2.7.2. Jele Yükleme Ve Görüntüleme ........................................................ 42
3.2.8. RNA İzolasyonu .................................................................................... 42
3.2.9. Ters Transkriptaz Polimeraz Zincir Reaksiyonu .................................. 44
3.2.10. Gerçek Zamanlı Kantitatif PZR (q-PCR)............................................ 46
3.2.11. Akım Sitometri Cihazında Antikor Hücre Yüzey Analizi .................. 50
3.2.12. CD34+ ve CD34+Lin- Hücrelere GM-CSF Uygulaması ..................... 50
3.2.13. Ekspresyon Verilerinin İstatiksel Analizi ........................................... 51
4. BULGULAR ................................................................................................... 52
4.1. DNA İzolasyon Sonuçları .......................................................................... 52
4.2. RNA İzolasyon Sonuçları .......................................................................... 53
4.3. İki Aşamalı Nested PZR Sonuçları ............................................................ 55
4.4. Gen İfadesi Sonuçları ................................................................................. 58

ix
4.5. Hücre Yüzey CXCR3 Reseptör Anlatımı .................................................. 60
4.5.1. CD34+ ve CD34+Lin- Hücrelere GM-CSF Uygulaması Sonuçları ....... 62
5. TARTIŞMA .................................................................................................... 64
KAYNAKLAR ................................................................................................... 68
HAM VERİLER ................................................................................................. 83
FORMLAR ......................................................................................................... 84
ETİK KURUL KARARI .................................................................................... 85
PATENT HAKKI İZNİ ...................................................................................... 86
İNTİHAL RAPORU İLK SAYFASI .................................................................. 87
ÖZGEÇMİŞ ........................................................................................................ 88

x
TABLOLAR LİSTESİ
Tablo 2-1: Güncellenmiş (2016) WHO sınıflandırmasına göre MPN’lerin başlıca
alt tipleri (3) ...................................................................................................................... 4
Tablo 3-1: Hastaların Klinik Parametreleri ........................................................ 25
Tablo 3-2 ............................................................................................................. 28
Tablo 3-3: Kullanılan kimyasal maddeler .......................................................... 28
Tablo 3-4: Kullanılan Kitler ............................................................................... 30
Tablo 3-5: Kullanılan cihazlar ............................................................................ 30
Tablo 3-6: Hücre Sayımında Kullanılan Formül ................................................ 33
Tablo 3-7: Allel Spesifik Nested PZR 1. Aşama bileşenleri .............................. 39
Tablo 3-8: İki aşamalı PZR için tasarlanan primer setleri .................................. 39
Tablo 3-9: İki aşamalı PZR’nin 1. aşama koşulları ............................................ 40
Tablo 3-10: Allel Spesifik Nested PZR 2. aşama bileşenleri ............................. 41
Tablo 3-11: Allel spesifik Nested PZR’nin 2. aşama koşulları .......................... 41
Tablo 3-12: Kalıp RNA/Primer Karışımı ........................................................... 44
Tablo 3-13: Reaksiyon Karışımı ......................................................................... 45
Tablo 3-14: cDNA Sentez Koşulları ................................................................... 45
Tablo 3-15: Gen ekspresyon analizleri için PZR bileşenleri .............................. 47
Tablo 3-16: Kantitatif gerçek zamanlı PZR Reaksiyon Bileşenleri ................... 47
Tablo 3-17: CXCR3A, CXCR3B, CXCL9 ve GAPDH Primer Dizileri ............... 49
Tablo 4-1: Mononükleer Hücrelerden İzole Edilen DNA örneklerinin ölçüm
değerleri .......................................................................................................................... 52
Tablo 4-2: Mononükleer Hücrelerden İzole Edilen DNA örneklerinin ölçüm
değerleri .......................................................................................................................... 53
Tablo 4-3: Hastaların Mutasyon Durumları ........................................................ 56

xi
ŞEKİLLER LİSTESİ
Şekil 2-1: JAK kinaz ailesinin homoloji bölgeleri (46) ........................................ 7
Şekil 2-2: MPN hücrelerinde JAK – STAT sinyali (53) ...................................... 8
Şekil 2-3: JAK2V617F ve CALR mutantlarının onkojenik özelliklerinde sitokin
reseptörlerinin rolü (4) .................................................................................................... 10
Şekil 2-4: CXC kemokinlerin genel yapısı (91) ................................................. 13
Şekil 2-5: CXCR3 ligandlarının özgünlüklerine katkıda bulunabilecek
mekanizmalara genel bakış (91) ..................................................................................... 14
Şekil 2-6: Cxcr3 gen yapısına genel bakış (91) .................................................. 18
Şekil 4-1: Alel Spesifik PZR Jel Görüntüsü ....................................................... 58
Şekil 4-2: CXCR3B gen ifadesi sonucu. ............................................................. 59
Şekil 4-3: CXCR3A gen ifadesi sonucu. ............................................................. 59
Şekil 4-4: CXCR9 gen ifadesi sonucu. ................................................................ 60
Şekil 4-5: 22 ve 24 numaları hastaların akımölçer sonuçları. ............................. 61
Şekil 4-6: Akımölçer verilerinin istatistiksel analizi. ......................................... 62

xii
SEMBOLLER / KISALTMALAR LİSTESİ
γδT: Gama delta T hücreleri
γIRE: γ-interferon yanıt elementi
ACKR: Atipik kemokin reseptörleri
Bç: Baz çifti
[Ca2+]i: Hücre içi kalsiyum konsantrasyonları
CAC: Klinik Danışma Komitesi
CALR: Calreticulin
[cAMP]i: Siklik adenosin monofosfat
CD34: (Cluster of differentiation 34, hematopoietic progenitor cell antigen)
cDNA: Komplementer Deoksiribonükleik Asit
CXCL9: C-X-C Motif Chemokine Ligand 9
CXCL10: C-X-C Motif Chemokine Ligand 10
CXCL11: C-X-C Motif Chemokine Ligand 11
CXCL12: C-X-C Motif Chemokine Ligand 12
CXCR3: C-X-C Motif Chemokine Receptor 3
CXCR3A: C-X-C Motif Chemokine Receptor 3 – A izoformu
CXCR3B: C-X-C Motif Chemokine Receptor 3 – B izoformu
DH: Dendritik hücre
dNTP: Deoksinükleotid Trifosfat
DNA: Deoksiribonükleik Asit
DMSO: Dimetil Sülfoksit
DSMZ: Deutsche Sammlung von Mikroorganismen und Zellkulturen
EDTA: Etilen Diamin Tetra Asetik Asit
Epo: Eritropoietin
EpoR: Eritropoietin Reseptörü
ET: Esansiyel Trombositoz
F: Fenilalanin
FACS: Fluorescence Assisted Cell Sorting

xiii
FBS: Fetal Sığır Serumu, Fetal Bovine Serum
FERM: Dört-noktalı-bir (four-point-one) ezrin radixin moesin
G: Guanin
Gαi: G alfa proteinlerinin inhibitör tipi
Gαs: Uyarıcı G alfa protein sinyali
GAG: Glikosaminoglikanlar
G-CSFR: Granülosit Koloni Uyarıcı Faktör Reseptörü
GM-CSF: (Granülosit Makrofaj Koloni Uyarıcı Faktör)
GPCR: G protein-bağlı reseptörlerdir
HEL: Human Erythroleukemia Cell Line
HKK: Hematopoetik Kök Hücre
HUMEC: İnsan mikrovasküler endotelyal hücreleri
IFN-γ: İnterferon γ
(IL)-2: İnterlökin
ILC: Lenfoid hücreler
IMDM: Iscove's Modified Dulbecco's Media
IP-9: IFN-γ ile uyarılabilen protein-9
IP-10: IFN- γ ile uyarılabilen 10 kDa'luk protein
I-TAC: IFN ile uyarılabilen T hücresi α kemoatraktanı
JAK2: Janus Kinaz 2
kDa: Kilodalton
KML: Kronik Miyeloid Lösemi
LC480: LightCycler-480
LPS: Lipopolisakkarit
Mig: IFN-γ ile uyarılabilen monokin
MPH: Miyeloproliferatif Hastalıklar
MPL: Miyeloproliferatif lösemi virüsü
MPNs: Miyeloproliferatif Neoplaziler
mRNA: Mesajcı Ribonükleik Asit
MS: Milattan Sonra

xiv
MUT: Mutant (JAK2V617F pozitif)
NF-κB2: Nükleer faktör kappa B2
NK: Doğal öldürücü hücreler
PBMC: Periferal kan mononükleer hücreleri
PBS: Fosfat Salin Çözeltisi, Phosphate Buffered Saline
Ph: Philadelphia Kromozomu
PHA: Fitohemaglutinin
PI: Propidyum İyodid
PI3K: Fosfatidilinositol 3-kinaz
PMF: Primer Miyelofibroz
PV: Polisitemia Vera
PZR: Polimeraz Zincir Reaksiyonu
qPZR: Kantitatif Polimeraz Zincir Reaksiyonu
RNA: Ribonükleik Asit
SDS: Sodyum Dodesil Sülfat
SET-2: Esansiyel Trombositoz Hücre Soyu
siRNA: Small Interfering Ribonükleik Asit
STAT: Signal transducer and activator of transcription
T: Timin
TAE: Tris-acetate-EDTA
Th1: Yardımcı T hücrelerinde
TPO: Trombopoietin
TYK2: Tirozin Kinaz 2
UV: Ultra Viyole
V: Valin
WHO: World Health Organization
WT: Wild Type (JAK2V617F negatif)

xv
ÖZET
Altunay C. Polisitemia Vera'da Sitokin Sinyal Yolağının Etkisi. İstanbul
Üniversitesi Sağlık Bilimleri Enstitüsü, Genetik ABD. Yüksek Lisans Tezi. İstanbul.
2018.
Klasik Miyeloproliferatif Neoplaziler (MPN), miyeloproliferatif bozukluklar
arasında en sık görülen hastalıklardandır. Tamamen işlevsel olan ve son aşamaya kadar
farklılaşmış kan hücrelerinin aşırı üretimi ile karakterize edilirler. Önceki bir çalışmada
Granülosit-Makrofaj Koloni Uyarıcı Faktörün (GM-CSF) sağlıklı hematopoietik kök
hücrelerde proliferasyon kontrolünden sorumlu CXCR3 ekspresyonunu arttırdığı
gösterilmiştir. Buradan yola çıkarak bu çalışmanın hipotezi; CXCL9-CXCR3 sinyal
yolağının PV progresyonunda etkisi vardır. Bu çalışmada, MPN grubuna dahil olan
Polisitemia vera’da (PV) sitokin sinyal yolağı etkisi araştırılmıştır. İnsan perifer kan
mononükleer hücrelerinde ve kanser kök hücrelerinde CXCL9 kemokini ve bunun
reseptörü olan CXCR3’ün iki izoformunun (CXCR3A ve CXCR3B) gen ifade
seviyeleri, mononükleer hücre yüzeyinde ise CXCR3 reseptör varlığı incelenmiştir.
Çalışmada ekspresyon seviyelerini araştırmak amacıyla kantitatif gerçek zamanlı
polimeraz zincir reaksiyonu, hücre yüzey reseptör durumunun incelenmesi içinse akım
ölçer metotları kullanılmıştır. Mononükleer ve CD34+ hücrelerde ifade seviyelerinin
direkt olarak incelenmesinin yanında bu hücreler için gerçek zamanlı PZR metodu
hücre kültüründe GM-CSF uygulamasının ardından CXCR3 reseptör ekspresyonuna
bakmak amacıyla kullanılmıştır. Mononükleer hücrelerde CXCR3A ekspresyonunun
mRNA düzeyinde hastalarda sağlıklılara göre istatistiksel olarak anlamlı arttığı
bulunmuşur. Hasta ve sağlıklı kontrol grupları arasında CXCR3B ve CXCL9
ekspresyon seviyeleri kıyaslandığında mRNA düzeyinde istatistiksel olarak anlamlı bir
fark olmadığı tespit edilmiştir. Hücre yüzey reseptör durumuna bakıldığında CXCR3
reseptörünün hastalardan elde edilen mononükleer hücre yüzeylerinde sağlıklı gruptan
elde edilenlere göre anlamlı bir azalma olduğu görülmüştür. Bu sonuçlar PV’de
CXCR3A/CXCR3B dengesi ile bu reseptörlere özgün olarak bağlanan kemokinler
CXCL9, CXCL10 ve CXCL11’in inflamasyon ve kanser progresyonu ile ilişkili
olabileceğini düşündürmektedir.
Anahtar Kelimeler : Myeloproliferatif Bozukluklar, Polisitemia Vera, Sitokinler,
CXCR3, CXCL9
Bu çalışma, İstanbul Üniversitesi Bilimsel Araştırma Projeleri Birimi tarafından
desteklenmiştir. Proje No: 27338

xvi
ABSTRACT
Altunay C. Effect of Cytokine Signaling Pathway in Polycythemia Vera.
İstanbul University, Institute of Health Science, Department of Genetics, MSc Thesis.
İstanbul. 2018.
The classical myeloproliferative neoplasms (MPNs) are the most frequent
diseases among myeloproliferative disorders. MPNs are characterized by excessive
production of terminally differentiated blood cells. Granulocyte-Macrophage Colony
Stimulating Factor (GM-CSF) has previously been shown to increase CXCR3
expression responsible for proliferation control in healthy hematopoietic stem cells.
Taken together, the hypothesis of study is that the CXCL9-CXCR3 signaling pathway is
involved in the PV progression. In this study, effect of the cytokine signaling pathway
in Polycythemia vera (PV) was investigated. The aim was to investigate the expression
levels of CXCL9 and its receptor CXCR3, in human peripheral blood mononuclear cells
(PBMC) and cancer stem cells, and finally the CXCR3 on the surface of PBMC. Real-
time polymerase chain reaction (qPCR) was used to investigate expression levels, and
flow cytometry methods were used to examine cell surface receptor presence. In
addition, the qPCR method for these cells has been used to look at CXCR3 expression
after GM-CSF application in cell culture. In mononuclear and CD34+ cells, the mRNA
level of CXCR3A expression was found to be increased in patients. There was no
statistically significant difference in mRNA levels between CXCR3B and CXCL9
expression levels between patients and healthy controls. Given the cell surface receptor
status, the CXCR3 receptor was found to have a significant reduction in mononuclear
cell surfaces obtained from patients. These results may indicate that the
CXCR3A/CXCR3B balance and the chemokines CXCL9, CXCL10 and CXCL11,
which bind specifically to these receptors, may be associated with inflammation and
cancer progression in PV.
Key Words: Myeloproliferative Neoplasms, Polycythemia Vera, Cytokines,
Chemokine CXCL9, CXCR3
The present work was supported by the Research Fund of Istanbul University.
Project No. 27338

1. GİRİŞ VE AMAÇ
Myeloproliferatif neoplaziler (MPN) Miyelofibroz (MF), Polisitemia vera (PV)
ve Esansiyel trombositemi’den (ET) oluşur (1,2). BCR-ABL-negatif MPN'ler olarak da
adlandırılan klasik MPN'ler, miyeloproliferatif bozukluklar arasında en sık görülen
hastalıklardır (3). MPN'ler, tamamen işlevsel olan, son aşamaya kadar farklılaşmış kan
hücrelerinin aşırı üretimi ile karakterize edilir. Bu hematopoietik hücre bozuklukları,
miyeloid soy hücre tiplerinin (≥1) klonal proliferasyonu ile karakterize edilir (1,2,4).
Çoğunlukla Janus kinaz 2 (JAK2), kalretikülin (CALR) veya trombopoietin reseptörü
(MPL) genlerindeki mutasyonlarla ilişkilidir (5-11). Klinik bulgular MPN alt tipine göre
değişebilir. Bunlar arasında polisitemi, anemi, lökositoz, trombositoz, yorgunluk ve
hepatosplenomegali bulunur (6,12,13). Genel olarak, hastalar trombotik ve
tromboembolik olaylarda artmış risk taşırlar ve genel popülasyona göre daha yüksek
mortalite riskine sahiptirler (14-19). MF'ye (PV veya ET olanlar için) veya akut
miyeloid lösemiye ilerlemeler hastalar arasında büyük bir endişe kaynağı olmaya devam
etmektedir (5,20).
MPN'ler, çoğu hasta için genellikle düşük yaşam kalitesine yol açan
hastalıklardır (20-24). Semptomlar kaşıntı, gece terlemeleri, mikrovasküler belirtiler,
splenomegali ve splenomegali ile ilişkili bulguları (örn; karın ağrısı, erken tokluk)
içerebilir ve yorgunluk en şiddetli göstergelerden biridir (12,21-23).
Enflematuvar sitokinlerin, kemokinlerin ve büyüme faktörlerinin birçok farklı
hücresel kaynağı mevcuttur ve bu hücre çeşitleri MPN alt tipine ve oluşan
komplikasyonlara (tromboz ve kemik iliği fibrozisi) bağlı olarak değişmektedir.
ET, PMF ve PV hastalarında, sağlıklı bireylere kıyasla, çeşitli sitokin ve
kemokinlerin plazma seviyelerinde yükselme saptanmıştır (25). Bu bulgu, inflamatuar
bir sürecin MPN'nin fizyopatolojisine dahil olabileceğini, sitokinlerin ve kemokinlerin
otokrin, parakrin ve endokrin olaylarda rol oynayabileceğini ve hatta hematopoietik nişi
etkileyebileceğini göstermektedir. MPN hastaları ile yapılan çalışmalarda sağlıklı
bireylere kıyasla yüksek sitokin/kemokin üretim sıklığı ile karakterize olan işlevsiz
sitokin/kemokin üretimi gösterilmiştir. Ayrıca, sitokin/kemokin seviyeleri ve
hematolojik parametreler arasındaki korelasyon, bağışıklık sistem bozukluğunu,
hematopoietik (kan) hücrelerin üretimini etkileyebileceğini ve bu hücrelerin

2
çoğalmasını desteklediğini göstermektedir. Bulgular ayrıca, MPN hastalarında
sitokinlerin ve kemokinlerin aşırı üretimini ve bunun proinflamatuar bir durumla
sonuçlandığını da vurgulamaktadır (25).
Hastalarda, sağlıklı bireylere kıyasla, çeşitli sitokinlerin plazma seviyelerinde
yükselme saptanmış ve bunların MPN patogenezinde rol oynayabileceği gösterilmiştir.
Önceki bir çalışmada Granülosit-Makrofaj Koloni Uyarıcı Faktörün (GM-CSF) sağlıklı
hematopoietik kök hücrelerde proliferasyon kontrolünden sorumlu CXCR3
ekspresyonunu arttırdığı gösterilmiştir. Bu bilgiler ışığında oluşturmuş olduğumuz
hipotez: CXCL9-CXCR3 sinyal yolağının PV patogenez ve progresyonunda etkili
olduğudur.
Çalışmamızın amaçları PV’de önemli olduğu düşünülen CXCL9 kemokininin ve
onun reseptörü olan CXCR3’ün;
Amaç 1: MPN hasta ve kontrol grup Perifer Mononükleer Hücrelerde (MNH)
CXCL9 ve CXCR3 mRNA anlatım düzeyi,
Amaç 2: Kanser kök hücrelerinde CXCL9 ve CXCR3 mRNA anlatım düzeyi
Amaç 3: CXCR3’ün MNH yüzey anlatım düzeylerinin araştırılmasıdır
Çalışmamızın hedefi, PV prognoz ve tedavi süreçlerinin kolaylaştırılmasına
katkı sağlamaktır. Elde edilen bulgular bilimsel çalışmaları yeni kemokin ve kemokin
reseptörlerini de prognoz ve tadavi süreçlerinde etkili olmaları açısından aday hale
getirebilecektir. PV’de kemokin ve kemokin reseptörlerinin hastalık süreçlerine etkisini
gösteren herhangi bir bilimsel çalışma mevcut olmadığından, çalışmamız özgün bir
çalışmadır. Elde edilen veriler, PV’nin yanında diğer MPN’lere ve farklı kanser
türlerine katı sağlayabilecek potansiyeldedir.

3
2. GENEL BİLGİLER
2.1. Miyeloproliferatif Neoplaziler
2.1.1. Tarihçe
Hipokrat ve Galen’e (MS 129–200) göre kan, dört “salgı”dan biri olarak kabul
edilmiştir. Diğer üçü ise balgam, siyah ve sarı safradır. “Plethora” (Yunanca "bolluk")
ise kanın domine ettiği salgıların dengesizliği durumudur (26). Hipokratik düşünce tarzı
yüzyıllar boyunca hüküm sürmüş ve flebotomi (kan alma) genellikle humoral dengeyi
sürdürme çabasıyla uygulanmıştır (27). İsveçli bir bilim adamı olan Robin Fahreus
(1888–1968), bir test tüpünde dört ayrı katmana ayrılan pıhtılaşmış kanın, "hümoral
teori"nin temelini oluşturduğunu ileri sürmüştür (28). Sarı tabakanın serumu temsil
ettiği, yukarıdan aşağıya diğer üç tabakanın ise beyaz kan hücrelerini, kanı (oksijenli
kırmızı kan hücreleri) ve oksijensiz kırmızı kan hücrelerini temsil ettiği düşünülmüştür
(28).
1935 yılında Hirsch, post-Polisitemia vera miyelofibrozu (post-PV
miyelofibroz) tanımlamıştır (29) ve 1939'da Vaughan ve Harrison, Primer miyelofibroz
(PMF), Polisitemia Vera (PV) ve Esansiyel Trombositoz (ET) arasındaki ilişkiyi, ortak
bir atasal hücreden köken almaları açısından ele almışlardır (30). Bu görüş diğer bilim
insanları tarafından da desteklenmiştir (31,32) ve nihayetinde 1951'de Dameshek
tarafından miyeloproliferatif hastalıklar (MPH) konsepti olarak açıklanmıştır (33).
Dameshek, laboratuvar ve klinik ilgi alanları geniş olmasına rağmen, 1951’de
MPH kavramını tanımlamasıyla bilinmektedir (33). Pubmed'e göre, “miyeloproliferatif”
sıfatı ilk kez 1951'de William Dameshek'in “Miyeloproliferatif sendromlar hakkında
bazı spekülasyonlar” başlıklı bilimsel makalesinde kullanılmıştır ve Blood dergisinde
yayımlanmıştır. Dameshek miyeloproliferatif bozukluklar kavramını, kemik iliğinde
hematopoetik öncüllerin aşırı proliferasyonu ve olgun kan hücrelerinin aşırı üretimi ile
karakterize edilen koşullar olarak ortaya koymuştur. 1951'de William Dameshek,
Kronik Myeloid Lösemi’yi (KML), PV’yi, ET’yi, PMF’yi ve Eritrolösemi'yi bir araya
getirerek "miyeloproliferatif bozukluklar" kavramını tanımlamıştır (34). 1960 yılında,
Nowell ve Hungerford KML'deki Philadelphia kromozomunu keşfetmiştir (34,35).
1967'de Fialkow ve arkadaşları, KML'yi bir klonal kök hücre hastalığı olarak belirlemek
için X'e bağlı polimorfizmleri kullanmışlardır (34). 1967'de, PV Çalışma Grubu, PV'nin

4
tarihini incelemek ve büyük ölçekli klinik deneyler yapmak için Louis Wasserman
tarafından bir toplantıya çağrılmıştır (34). 1972'de, Janet Rowley, Philadelphia (Ph)
kromozomunun 9. ve 22. kromozomlar arasında bir karşılıklı translokasyon olduğunu
keşfetmiştir, böylece onkogenik BCR-ABL mutasyonunun karakterizasyonunun yolunu
açmıştır (34). 1996'da Brian Druker İmatinib'i keşfetmiştir (34). 2005 yılında James
Chloé, BCR-ABL-negatif MPH'lerde bir fonksiyon kazandırıcı mutasyon olan JAK2
mutasyonu (JAK2V617F) tanımlamıştır ve PV, ET ve PMF'de KML benzeri bir tedavi
stratejisi ortaya çıkmıştır (34).
2.1.2. Sınıflandırma
Dünya Sağlık Örgütü (WHO), Hematopatoloji Topluluğu ve Avrupa
Hematopatoloji Derneği ile işbirliği içinde, sırasıyla 2001 ve 2008 yıllarında,
Hematopoetik ve Lenfoid Doku Tümörlerinin WHO Sınıflandırmasının üçüncü ve
dördüncü baskılarını yayımlamıştır (2). 2014 baharında, dünyanın dört bir yanından 100
kişilik - patolog, hematolog, onkolog ve genetikçiden oluşan - bir klinik danışma
komitesi (CAC), sınıflandırmanın dördüncü baskısına revizyon önerisinde bulunmuştur.
Bu nedenlerle dördüncü baskı güncellenmektedir ve en son yapılan 2016
sınıflandırması hastalık kategorilerinin önemli bir revizyonu değildir (2).
MPN kategorileri, sınıflandırmanın 2008’deki dördüncü baskısından bu yana
önemli ölçüde değişmemiştir, fakat yeni mutasyonların keşifleri ve var olan bazı
morfolojik özelliklerin daha iyi anlaşılması, hastalıklar için tanı kriterlerini etkilemiştir
(2).
Güncellenmiş (2016) WHO sınıflandırmasına göre MPN’lerin başlıca alt tipleri
Tablo 2-1’de listelenmiştir.
Tablo 2-1: Güncellenmiş (2016) WHO sınıflandırmasına göre MPN’lerin başlıca alt tipleri
(2)
Miyeloproliferatif neoplaziler (MPN)
Kronik miyeloid lösemi (KML), BCR-ABL1+
Kronik nötrofilik lösemi (KNL)

5
Polisitemia vera (PV)
Primer miyelofibroz (PMF)
PMF, prefibrotik / erken evre
PMF, fibrotik evre
Esansiyel trombositemi
Kronik eozinofilik lösemi
Sınıflandırılamayan MPN
2.1.3. Klasik Miyeloproliferatif Neoplazilerin Genetik Temeli ve Moleküler
Patofizyolojisi
BCR-ABL- MPN'ler olarak da adlandırılan klasik MPN'ler, miyeloproliferatif
bozukluklar arasında en sık görülen hastalıklardır (3). MPN'ler, tamamen işlevsel olan,
son aşamaya kadar farklılaşmış kan hücrelerinin aşırı üretimi ile karakterize edilir.
Klasik MPN'ler 3’e ayrılır: PV, ET ve PMF. Tüm MPN’ler, klonal olarak büyüyen ve
hemen hemen tüm miyeloid hücreleri, B hücrelerini ve doğal öldürücü (natural killer,
NK) hücreleri oluşturan, tek bir somatik mutasyona uğramış hematopoetik kök
hücrelerden (HKH) kaynaklanır (36). MPN’de HKK'nin klonal genişlemesine, tek veya
bir dizi hiperplazi eşlik eder. PV sadece fazla eritrosit sayısı ve baskın eritroid soyu ile
değil, aynı zamanda megakaryositik / granülositik soyların değişken hiperplazisi ile de
ilişkilidir. ET, megakaryositik hiperplazi ile birlikte artmış trombosit sayısıyla
karakterize edilirken, PMF, hem kemik iliği fibrozunun (kollajen liflerinin fazlalığı)
hem de megakaryositik hiperplazi varlığıyla tanımlanan klinik ve biyolojik
özellikleriyle daha heterojen bir hastalıktır. Her ne kadar bu üç MPN’nin klinik tabloları
kendi özgün formlarında farklı olsa da hastalık başlangıcında kesin tanı konması çoğu
zaman zorlayıcıdır. Bu durum, Dünya Sağlık Örgütü'nün (WHO) MPN tanı kriterlerinin
2016 revizyonu ile yansıtılmıştır (2).
Somatik mutasyonlar, sadece MPN'lerde değil, aynı zamanda miyeloid
malinitelerin çoğunda da HKK'lerin klonal büyümesinden sorumludur (3). Mikroarray
ve yeni nesil dizileme (YND) kullanılarak yapılan yüksek çözünürlüklü genom

6
analizleri, tüm miyeloid malinitelerde birkaç gen mutasyonunun keşfiyle
sonuçlanmıştır. Bu gen mutasyonlarından nispeten az bir kısmı, tek veya sınırlı sayıda
hastalık ile ilişkili bulunmuştur. Bu nedenle, bu mutasyonlar “MPN ile sınırlı” olanlar,
“MPN ile sınırsız” olanlar ve diğer miyeloid malinitelerde bulunanlar şeklinde
sınıflandırılmıştır.
2.1.4. JAK2 Gen Mutasyonu ve Mutasyonun MPN Patogenezindeki Rolü
2005'ten önce, BCR-ABL- klasik MPN'lerin moleküler patogenezi
bilinmemekteydi. 2005 yılında, JAK2 geninin 14. ekzonunda 1849. nükleotit olan
Guanin’in (G) Timin’e (T) dönüştüğü somatik mutasyonun keşfi büyük bir buluştu. Bu
mutasyon, JAK2 proteininin psödokinaz bölgesinde 617. kodonda valinin fenilalaninle
yer değiştirmesi (JAK2V617F) ile sonuçlanır (Şekil 2-1) (35,37-39). Bu mutasyon
PV'nin %95'inde ve ET’nin ve PMF'nin %50-60'ında olmak üzere MPN'lerin yaklaşık
%70'inde bulunabilmektedir. JAK2V617F mutasyonu sıklıkla, 9. kromozomun kısa kolu
üzerinde heterozigotluk kaybına (9pLOH) yol açan mitotik rekombinasyonun meydana
gelmesinden dolayı heterozigotluktan homozigotluğa geçişe uğrar. JAK2V617F
mutasyonu multipotent hematopoetik bir progenitörde ortaya çıkar, tüm miyeloid
soylarda bulunur ve ayrıca lenfosit hücrelerde, özellikle B, NK ve T hücrelerinde de
hastalıkta saptanabilir. Diğer bazı malin hemopatilerde nadiren bulunabilir (40).
Bununla birlikte, JAK2V617F, yenidoğanlar da dahil olmak üzere, normal
popülasyonda çok düşük seviyede (% 1'den az) tespit edilmiştir (41-43). Yaşlanma ile
ilişkili klonal hematopoezde en sık görülen mutasyonlardan biridir (44).
JAK2 ekson 12 mutasyonları da MPN'lerde bulunmuştur ve JAK2V617F- PV'nin
çoğunluğunda mevcuttur (Şekil 2-1) (45). JAK2 ekson 12 mutasyonlarının hepsi, Src
homolojisi 2 (SH2) ve psödokinaz bölgeleri arasında, Lys 539 civarında, 536 ve 547
amino asitleri arasındaki bir bölgededir.

7
Şekil 2-1: JAK kinaz ailesinin homoloji bölgeleri (46)
V617F mutasyonu JH2 veya psödokinaz bölgesindedir, JAK2'deki ekson 12 mutasyonları JH2
alanına yakındır. (Debra Tyler'ın görselidir.)
2.1.5. “MPN ile sınırlı” Mutasyonlar, JAK2 Kinaz-bağımlı Sitokin Reseptör
Yollarını Aktive Eder
JAK2, iki kinaz bölgesi ile karakterize edilen JAK ailesinin bir üyesidir. İki bölgeden
bir tanesi C terminalinde katalitik olarak aktifken diğer bölge katalitik olarak inaktif
(veya çok zayıf) olan ve kinaz bölgesinin kendi kendine aktivasyonunu önleyen
psödokinazdır (Şekil 2-2) (47). JAK proteinleri, N terminalinde dört-noktalı-bir (four-
point-one) ezrin radixin moesin (FERM)-benzeri bölge ve SH2-benzeri bir bölgeye
sahiptir (Şekil 2-2). JAK'ların sitokin reseptörlerine kovalent olmayan bağlanması
FERM bölgesine bağlıdır. JAK ailesi kinazları, hücre içinde reseptörlere yapısal olarak
bağlı oldukları için, hematopoietik sitokin reseptör familyasının katalitik kısmı olarak
düşünülebilir. Ayrıca, JAK'ların reseptörler ile ilişkisi, hücre yüzeyine uygun
konumlanması için önemlidir. Eritropoietin (EPO) reseptörü (EPOR), miyeloproliferatif
lösemi virüsü (MPL) ve granülosit koloni uyarıcı faktör reseptörü (G-CSFR) gibi
homodimerik reseptörler JAK2’yi kullanırken heterodimerik reseptörler JAK1 ve
JAK2/tirozin kinaz 2 (TYK2) veya JAK3'ü kullanır. Sitokin bağlanması, reseptöre
bağlanan JAK'ları trans-fosforilasyon ile aktive eden reseptörlerin konformasyonundaki
değişiklikleri uyarır (Şekil 2-2). Aktive edilmiş JAK'lar, ardından, başta STAT’lar
olmak üzere, diğer sinyalleme molekülleri için kullanılan reseptörleri fosforile ederler.
Bu yolakta STAT'lar, homodimerizasyon veya heterodimerizasyonlarını ve ardından
hedef genlerin transkripsiyonunu düzenledikleri çekirdeğe geçişlerini indükleyen
JAK'lar tarafından fosforile edilirler (Şekil 2-3). JAK2'nin psödokinaz bölgesinin 2 rolü

8
vardır: bir tanesi kinaz bölgesini inhibe etmektedir, diğeri de sitokine bağımlı
aktivasyonu desteklemektir. V617F, JAK2'yi tam olarak anlaşılmayan bir mekanizma
ile aktive eder, fakat hem yapısal hem de fonksiyonel veriler V617F tarafından
indüklenen birinci konformasyonel değişimin psödokinaz bölgesinin C sarmalını
içerdiğini göstermiştir (48,49). Ekson 12'deki mutasyonların proteinde meydana geldiği
bölge ve aktivasyon mekanizması JAK2V617F mutasyonunkinden farklıdır (50).
JAK2V617F veya JAK2 ekson 12 mutasyonları, interlökin-3 (IL-3) bağımlı hücre
hatlarında ifade edildiğinde, sitokine aşırı duyarlılığı veya sitokin bağımsızlığını
indükler. EPOR gibi homodimerik reseptörlerin varlığı bu biyolojik etkiyi büyük ölçüde
kolaylaştırır (51). JAK2V617F’nin düşük ifade seviyelerinde sinyalizasyonu
indüklemek için bir sitokin reseptörünün bulunması mutlaka gereklidir. JAK2V617F,
STAT'larin ve fosfatidilinositol 3-kinaz (PI3K) ve MAPK yollarının yapısal
aktivasyonunu uyarır. Hücre soylarında JAK2V617F tarafından indüklenen bu sitokin
hipersensitivitesi veya bağımsızlığı, daha önce PV eritroid progenitörlerinde tarif edilen
EPO hipersensitivitesine veya bağımsızlığına benzer (37,52).
Şekil 2-2: MPN hücrelerinde JAK – STAT sinyali (53)
(a) JAK2, bir FERM bölgesi (sitokin reseptör etkileşimi), bir SH2 bölgesi (etkileşimli
proteinlerin katılması), bir psuedokinaz bölgesi ve bir kinaz bölgesi dahil olmak üzere yedi

9
bölgeye (JH7-JH1) sahiptir. En yaygın miyeloproliferatif neoplazi (MPN) mutasyonu,
JAK2V617F, proteinin psödokinaz bölgesinde bulunur. Ekson 12 mutasyonu bu bölgeye bitişik
olarak bulunur. Yaygın akut lenfoblastik lösemi (ALL) mutasyonu R683G, JAK2'nin
psödokinaz bölgesinde de mevcuttur. (b) JAK molekülleri, sitokin reseptörlerinin
dimerizasyonu ve otofosforilasyonu için bir araya gelirler. JAK'lar daha sonra karşılık gelen
dimerleri transfosforile ve aktive edebilirler. JAK2'deki aktive edici mutasyonlar, konstitütif
fosforilasyon ile sonuçlanır. JAK molekülleri, STAT yolu dahil olmak üzere çok sayıda sinyal
yolunu aktive edebilir. JAK2'nin yer değiştirerek çekirdeğe gittiği ve histon 3 üzerinde tirozin
41'in fosforilasyonunu düzenlediği gösterilmiştir, bu da hedef genlerin ve bunların ifadelerinin
düzenlenmesinde kritik olabilir. Kısaltmalar: JH1–7, JAK homoloji bölgeleri 1–7; SH2, Src
homoloji 2; FERM, dört noktalı bir, ezrin, radixin, moesin; JAK2, Janus kinaz 2; STAT, sinyal
transdüserleri ve transkripsiyon aktivatörleri; P, fosforilasyon.
Sonuçta, MPN ile ilişkili başlıca mutasyonlar olan JAK2V617F, JAK2 ekson 12
mutantları, MPLW515L/K ve CALR mutantları, sitokin/reseptör/JAK2 yolaklarını ve
bunların devamındaki sinyalizasyonu aktive eder. Bu aktivasyon JAK2V617F
mutantlarında 3 homodimerik reseptör (EPOR, MPL, G-CSFR) aracılığıyla, CALR
mutantlarında ise esas olarak MPL aracılığıyla sağlanır (Şekil 2-3). Bu durum, bu farklı
mutasyonların neden genelde birbirlerini karşılıklı olarak dışladıklarını açıklayabilir.
Karşılıklı dışlamanın ardındaki mekanizmaları aydınlatmayı amaçlayan çalışmalar
mevcuttur (54). Bu çalışmalarda hücre döngüsü durmasının ve yaşlanmanın bu yanıtta
rol oynadığı öne sürülmüştür. Bu yaklaşıma göre mutant bir hücrenin ek bir mutasyonu
edinmesi, belli sinyal yolaklarını ve yaşlanmayı hiperaktif hale getirir ve aynı
mutasyonu taşıyan hücreler baskın hale gelmektedir. Bununla birlikte, nadiren, bu
mutasyonların 2 tanesi aynı hastada tespit edilebilir, ancak genellikle farklı klonlarda
veya alt klonlarda bulunurlar.

10
Şekil 2-3: JAK2V617F ve CALR mutantlarının onkojenik özelliklerinde sitokin
reseptörlerinin rolü (3)
(A) JAK2V617F, sırasıyla eritrositoz, trombositoz ve nötrofili olmak üzere 3 ana homodimerik
reseptör olan EPOR, MPL ve G-CSFR üzerinden sinyalizasyonu aktive eder. (B) CALR
mutantları esas olarak MPL'yi ve düşük bir seviyede G-CSFR'yi aktive eder, ancak EPOR'u
aktive etmez, bu durum mutantlarla ilişkili trombositozu açıklar.
2.1.6. MPN Başlangıcında Önemli Faktörler
Yaşlanma: Yaşlanan sağlıklı popülasyonda JAK2V617F'nin nispeten daha sık
olduğuna dair kanıtlar artmaktadır ve sıklığının <%0.5 olduğu tahmin edilmektedir (41).
Bu durum, JAK2V617F'nin tek başına klonal hematopoezi meydana getirmek için
yeterli olduğunu ve MPN'de hastalık başlatan bir rolü olduğunu gösterir. Bununla
birlikte, JAK2V617F sık bir mutasyon olup, MPN başlatmak için düşük bir penetransa
sahiptir, bu da JAK2V617F’nin başka faktörlerle birlikte hastalığa yol açabilmesi için
ilişkilendirilmesi gerektiğini düşündürmektedir.
Enflamasyon: Tüm MPN'ler, özellikle de PMF enflamatuar yanıt ile ilişkilidir.
Özellikle, yüksek plazma düzeylerinde var olan enflamatuar sitokinlerin yanı sıra
kortikosid ve ruxolitinib gibi anti-enflamatuar tedavilerle hafifleyen semptomların
varlığı enflemasyonu göstermektedir (55). Bu enflamatuar sitokinler, mutasyona

11
uğramış ve uğramamış hematopoietik hücrelerin yanı sıra mezenkimal stromal hücreler
gibi hematopoetik olmayan hücreler tarafından da sentezlenir (56). Ayrıca, JAK2V617F
progenitörlerinin, JAK2V617F HKK'leri için uygun bir ortam sağlayan nestin+
hücrelerinin apopitotik ölümünü indükleyen IL1β salgıladıkları gösterilmiştir (57).
Bunun yanında, tümör nekroz faktör α’nın (TNFα) JAK2V617F HKK'lere bir rekabet
avantajı sağladığı gösterilmiştir (58).
Yatkınlık: JAK2, özellikle JAK2 46/1 haplotipi, TERT, MECOM, SH2B3,
CHEK2, PINT ve GFI1B gibi genlerdeki yaygın polimorfizmlere karşılık gelen birkaç
yatkınlık alleli, zayıf bir şekilde (2 ila 6 kat) hastalığın gelişimini arttırır (41,59).
Mekanizma halihazırda bilinmemektedir ancak bu yatkınlık varyantları ya DNA hasar
cevabında (CHEK2, TERT, JAK2 46/1 haplotip) ya da JAK2/STAT yolunda rol
oynayan genleri içermektedir (60,61).
2.2. Sitokinler
Sitokinler (Yunanca sito-, hücre; ve -kinos, hareket), hücresel iletişimde yaygın
olarak kullanılan bir sinyal molekülleri kategorisidir. Bunlar proteinler, peptitler veya
glikoproteinlerdir. Hücre sinyalizasyonunda önemli olan, yaygın bulunan, küçük
proteinlerdir (~ 5–20 kDa) (62). Sitokinler hücreler tarafından salınırlar ve diğer
hücrelerin davranışlarını etkilerler. Bazı durumlarda salındığı hücrenin kendisini de
etkileyebilirler. Sitokinler kemokinleri, interferonları, interlökinleri, lenfokinleri, tümör
nekroz faktörlerini kapsarlar, ancak genellikle hormon veya büyüme faktörü değillerdir
(63). Sitokinler geniş bir hücre grubu tarafından üretilirler; makrofajlar, B lenfositler, T
lenfositler ve mast hücreleri, endotel hücreler, fibroblastlar ve çeşitli stromal hücreler
gibi bağışıklık hücreleri de bu gruba dahildir. Bir sitokin birden fazla hücre tipi
tarafından üretilebilir (64). Sitokinler reseptörler yoluyla etki ederler ve özellikle
bağışıklık sisteminde önemlidirler. Humoral ve hücre bazlı bağışıklık yanıtları
arasındaki dengeyi düzenlerler ve belirli hücre popülasyonlarının olgunlaşmasını,
büyümesini ve tepkilerini düzenlerler. Bazı sitokinler, diğer sitokinlerin etkilerini
karmaşık yollarla güçlendirir veya kısıtlar. Tüm bunların yanında sitokinler, hastalıkta
özellikle enfeksiyona, immün yanıtlara, inflamasyona, travmaya, sepsise, kansere ve
reprodüksiyona karşı verilen yanıtlarda önemlidirler (65).

12
2.2.1. Kemokinler
Kemokinler olarak adlandırılan bazı sitokinler belirli hücre tiplerini kimyasal
olarak çekerler (66). Bu kemokinler, bir yaralanma veya enfeksiyon bölgesindeki
hücreler tarafından salınırlar ve hasarı onarmak veya istilacı ile mücadele etmek için
bölgeye başka bağışıklık hücrelerini çağırırlar (67). Kemokinler, iltihaplanma sürecinde
önemli bir rol oynamaktadırlar ve bağışıklık tepkilerini düzenlemeye yardımcı olacak
yeni ilaçlar için umut verici hedeftirler (68).
Kemokinler veya kemotaktik sitokinler düşük moleküler kütleli proteinlerdir (8–
12 kDa) ve zaman-mekan bağımlı bir şekilde lökosit migrasyonunu yönlendiren ayırt
edici bir işleve sahiptirler (69-74). Spesifik lökosit alt tiplerinin kontrollü kemotaksisi,
sadece immün hücre hedeflemesi, embriyogenez ve anjiyogenez dahil olmak üzere
homeostatik süreçlerde değil, aynı zamanda kanser, iltihaplanma ve otoimmünite gibi
patofizyolojik ortamlarda da gereklidir (75-80). Bu nedenle, kemokinler sağlık ve
hastalık sırasında doğuştan gelen ve sonradan kazanılmış bağışıklık olaylarında anahtar
oyunculardır. Biyolojik fonksiyonlar için bağlandıkları reseptörler, esas olarak G alfa
proteinlerinin inhibitör tipini (Gαi) aktive eden, daha sonra adenilat siklazın
inhibisyonuna neden olan ve böylece hücre içi siklik adenosin monofosfat ([cAMP]i)
konsantrasyonlarını azaltan spesifik G protein-bağlı reseptörlerdir (GPCR'ler) (70,78).
Bununla birlikte, G proteininden bağımsız sinyalizasyonu da aktive edebilirler. Bunların
arasında β-arrestin ile ilişkili yolaklar mevcuttur (81). Spesifik GPCR'ler ile etkileşime
ek olarak, kemokin varlığı, aktivite ve reseptör tercihi, glikosaminoglikanlar (GAG'ler),
atipik kemokin reseptörleri (ACKR'ler), gen transkripsiyonu, mRNA stabilitesi,
alternatif gen ekleme, karşılıklı sinerjizm veya antagonizm ile kemokin etkileşimleri ve
post-translasyonel modifikasyonlar da dahil olmak üzere çoklu seviyelerde modüle
edilir (82-85). Bu nedenle, in vivo olarak fonksiyon gösteren son kemokin, çok sayıda
düzenleyici mekanizmanın karmaşık bir sonucudur.
Başlıca biyolojik fonksiyonlara ilişkin olarak, aslında kemokin ailesinin,
sırasıyla, endojen (örn., Sitokinler) veya eksojen (örn., Mikrobiyal ürünler) uyaran
tarafından önceden oluşturulmuş veya önceden indükleme gerektiren, homeostatik ve
enflamatuar proteinlere sınıflandırılabileceği önerilmiştir (86-89). Bununla birlikte, bu
sınıflandırmanın mutlak olmadığı anlaşılmıştır. Çünkü CXCL12 gibi birçok kemokin
hem homeostatik hem de enflamatuar rollerde görev almaktadır.

13
Kemokinler yapısal olarak CXC, CC, C veya CX3C ligandları şeklinde
sınıflandırılır. Bu sınıflandırma, salgılanan olgun proteinin NH2-terminal dizisinde
bulunan korunmuş Sistein (Cys) rezidülerinin sayısına ve pozisyonuna bağlı olarak
yapılmıştır (73,78,90). CXC kemokinler NH2-terminal Cys rezidülerinin arasında
rastgele ("X") bir amino asit içerirler (Şekil 2-4). Kemokin reseptörlerinin
sınıflandırılması, etkileşime girdiği kemokin alt ailesine göre yapılır. Örneğin CC
kemokin reseptörler (CCR'ler) CC kemokinleriyle, CXC kemokin reseptörler
(CXCR'ler) CXC kemokinleri ile etkileşime girerler (78). Spesifik bir kemokin,
komplementer alt sınıfından bir veya daha fazla reseptörü tanıyabilir ve bunun tersi de
mümkündür. Bu durum kemokin ağında olağanüstü bir karışıklığın var olmasına neden
olur. Son birkaç yılda, bir kemokin reseptörünün hücre içi sinyal yollarından tercihli
olarak birkaçını aktifleştirebileceğinin kanıtlanması, durumu daha da karmaşık bir hale
getirmiştir (81). Bu durum muhtemelen ilgili reseptöre ve liganda değil, aynı zamanda
çalışılan hücre tipine veya dokusuna da bağlıdır.
Şekil 2-4: CXC kemokinlerin genel yapısı (91)

14
Kemokinler, karşılıklı olarak 30s, 40s ve 50s ilmeklerine bağlanan üç antiparalel iplikçik
(pembe) ve bir COOH-terminal α-sarmal (turuncu) içerir. Esnek NH2-terminal alanını sırasıyla
bir N ilmiği ve 310 sarmal takip eder. Salgılanmış olgun proteinin 3D yapısı, dört korunmuş
Cys rezidüsü (mavi) tarafından oluşturulan iki disülfid köprüsü ile stabilize edilir (91).
İnsan CXC kemokinlerinden yedi tanesi (CXCL1–3 ve CXCL5-8) korunmuş bir
Glu-Leu-Arg (“ELR”) amino asit motifi içerir (73,78,90). ELR motifi olmayan CXC
kemokinlerinin çoğu CXCR3 ile etkileşime girer (73). Bu CXCR3 ligandları için, bir
yandan trombositle ilgili agonistler CXCL4 ve CXCL4L1 ve diğer yandan başlıca
indükleyici olarak interferon γ (IFN-γ)'yı paylaşan CXCL9, CXCL10 ve CXCL11
kemokinleri şeklinde ayrım yapılabilir (92). Tek bir reseptör ve indükleyici
paylaşmalarına rağmen, ortaya çıkan kanıtlar IFN ile indüklenen CXCR3 ligandlarının
in vivo olarak benzer olmayan rolleri olduğuna işaret etmektedir (93). CXCR3
ligandlarının bu özgünlüklerine katkıda bulunabilecek belli başlı mekanizmaların
olduğu düşünülmektedir (Şekil 2-5).
Şekil 2-5: CXCR3 ligandlarının özgünlüklerine katkıda bulunabilecek mekanizmalara
genel bakış (91)
CXCL9, CXCL10 ve CXCL11, ortak reseptör olarak CXCR3'ü ve baskın indükleyici olarak
IFN-γ'yı paylaşan yapısal olarak ilişkili kemokinlerdir. Yapısal ve işlevsel benzerliklere rağmen,
ortaya çıkan kanıtlar CXCL9, CXCL10 ve CXCL11 için in vivo olarak birbilerine benzemeyen
rolleri olduğuna işaret etmektedir. IFN ile indüklenebilir CXCR3 ligandlarının özgünlükleri pek
çok durumdan köken alabilir. Bunlar; bu ligandlarının spesifik indükleyicilere yanıt olarak
spesifik hücre tiplerinde salgılanmaları (A, B), spesifik CXCR3 etkileşim özellikleri ve farklı
CXCR3 izoformlarının varlığı (C), ilgili başlıca sinyal kaskadları (D), T hücre polarizasyonu

15
üzerindeki etkileri (E), CCR antagonizmi (F), ACKR etkileşimleri (G), posttranslasyonel
işlemler (H) ve GAG bağlama özellikleri (I). ACKR, atipik kemokin reseptörü; CCR, CC
kemokin reseptörü; CXCR, CXC kemokin reseptörü; GAG, glikozaminoglikan; GRK, G
protein-bağlı reseptör kinazlar; HUMEC, insan mikrovasküler endotel hücresi; IFN, interferon;
LPS, lipopolisakkarit; PAD, peptidlarginin deiminaz; PBMC, periferik kan mononükleer hücre;
PG, peptidoglikan; STAT, sinyal dönüştürücü ve transkripsiyon aktivatörleri; TNF, tümör
nekroz faktörü.
2.2.2. IFN ile Uyarılabilen CXCR3 Kemokinleri
1985 yılında, IFN-γ aracılı inflamatuar yanıtı çözmek için yapılan bir çalışmada,
trombosit kaynaklı proteinlerle yüksek homolojiye sahip protein kodlayan bir gen fark
edilmiştir (94). Proteinin moleküler kütlesi yaklaşık 10 kDa ve protein "IFN- γ ile
uyarılabilen 10 kDa'luk protein" (IP-10) olarak adlandırılmıştır. Beş yıl sonra, 1990'da,
IFN-γ tarafından seçici olarak indüklenen ve başka bir trombosit faktör-4-benzeri
proteini kodlayan bir mRNA tarif edilmiştir (95). Bu molekül IFN-α, IFN-β ve
lipopolisakkarit (LPS) dahil olmak üzere başka hiçbir makrofaj aktivatörü tarafından
indüklenmeyen bir moleküldür. Yazarlar, molekülün “IFN-γ ile uyarılabilen monokin”
(Mig) olarak adlandırılması gerektiğini öne sürdüler. IP-10 ve Mig'in, benzer proteinler
oldukları, 4. Kromozomun q21.1 kolu üzerinde yer alan genleriyle ve başlangıç
kodonlarının 16 kb'den daha az bir mesafede olmalarıyla ortaya çıkarılmıştır (96).
Yapılan çalışmalar IP-10’un ve Mig’in, korunmuş bir ELR amino asit motifi içermeyen
ve NH2-terminal dizisinde rastgele bir rezidü ("X") ile ayrılan iki korunmuş Cys
rezidüsü içeren kemotaktik sitokinler veya kemokinler olduğunu ortaya çıkarmıştır. Her
ikisi de bu iki kemokin için seçici bir reseptör olarak rapor edilen CXCR3 üzerinden
etki ederler (97). Daha sonra, iki araştırma grubu uyarılmış astrositler ve
keratinositlerde üçüncü bir ELR negatif, IFN ile uyarılabilen CXC kemokini
tanımlamışlardır (98,99). Bu protein IP-10 ve Mig ile güçlü bir şekilde ilişkilidir ve
CXCR3 için daha yüksek bir afinite göstermektedir. Bu üçüncü IFN ile ilişkili CXCR3
ligandı, ilk yayınlarda "IFN-γ ile uyarılabilen protein-9 (IP-9)" veya "IFN ile
uyarılabilen T hücresi α kemoatraktanı" (I-TAC) olarak isimlendirildi ve karşılık gelen
gen yine 4q21'de bulundu (99,100). Kurulan yeni sistematik kemokin nomenklatüründe,
Mig, IP-10 ve I-TAC/IP-9, sırasıyla CXCL9, CXCL10 ve CXCL11 olarak yeniden
adlandırılmıştır (90), ve genel olarak IFN ile uyarılabilen CXCR3 ligandları olarak
adlandırılmaktadırlar.

16
IFN ile uyarılabilen CXCR3 kemokinleri, amino asit sekanslarında yaklaşık
%40 homoloji gösterirler ve insan mikrovasküler endotelyal hücreleri (HUMEC),
keratinositler ve fibroblastlar dahil olmak üzere çeşitli hücreler tarafından üretilirler
(Şekil 2-5) (98,99,101-103). Ek olarak, CXCL9 ve CXCL11 yaygın olarak periferal kan
mononükleer hücreleri (PBMC'ler) ve daha spesifik olarak makrofajlar (CXCL9) ve
astrositler (CXCL11) tarafından salgılanırlar (95). Ağırlıklı olarak CXCL10 üreten
lökositler T hücreleri ve monositlerdir (102,104,105). Farklı temel hücresel kökenlere
ek olarak, özgün promotörler IFN ile uyarılabilen CXCR3 kemokinlerinin
ekspresyonlarını kontrol ederler (Şekil 2-5). Cxcl9 promotörünün bir γ-interferon yanıt
elementi (γIRE) ve bir nükleer faktör kappa B2 (NF-κB2) bölgesi vardır ve CXCL9
protein ifadesi tam olarak IFN-γ'ya bağlıdır (106-108). Cxcl10 ve Cxcl11
promotörlerinin her ikisi de IFN-γ tarafından indüklendiklerinden ve bir interferon yanıt
elemanı (IRSE) ve bir NF-κB1 bölgesi içerdiklerinden belli bir benzerlik derecesi
gösterirler (108-111). Cxcl10 promotöründeki IRSE, genin IFN-α’ya ve IFN-β'ya
duyarlılığına aracılık eder. Bu nedenle hem Tip I hem de Tip II IFN'ler CXCL10
ekspresyonunun güçlü uyaranlarıdır. Dikkat çekici bir şekilde, CXCL11, IFN-β ve IFN-
γ tarafından uyarılır, ancak IFN-α ile uyarılmaz (112). IFN ile uyarılabilen üç CXCR3
ligandı için, ilgili IFN'ler tarafından indüklenen gen transkripsiyonu, fibroblastlarda ve
endotel hücrelerde TNF-α ve IL-1β varlığında kuvvetli bir şekilde artmaktadır (113).
Şaşırtıcı bir şekilde, bakteriyel lipopolisakkaritler (LPS) ve peptidoglikanlar aynı
zamanda fibroblastlarda ve endotel hücrelerde üç CXCR3 ligandını sinerjistik olarak
indüklemelerine rağmen, bu ajanlar, CXCR3 ligandlarının lökositler tarafından IFN
uyarımıyla üretimlerini inhibe etmiştir (101-103). Ek olarak, tek hücre seviyesinde,
endotel hücreler, CXCR3 ligandlarının fibroblast ve lökositlerden daha iyi
üreticileridirler (101-103).
2.2.3. CXCR3
2.2.3.1. CXCR3'ün Tanımlanması ve Ekspresyonu
İnsan kemokin reseptörü CXCR3, ilk kez 1996 yılında tanımlanmıştır (97).
Reseptör orijinal olarak "monositler veya granülositler tarafından ifade edilmeyen ilk
lenfosit kemokin reseptörü" olarak tarif edilmiştir (97). CXCR3’e karşılık gelen gen iki
yıl sonra bulunmuştur ve X kromozomu üzerinde, q13.1 bölgesinde yer almaktadır
(Şekil 2-6) (114). Gen, yaklaşık 41 kDa'lık bir moleküler kütleye sahip 368 amino

17
asitlik bir membran molekülünü kodlar (97). CXCR3, yedi transmembran sarmalı içeren
A sınıfı bir GPCR'dir. Reseptör, ağırlıklı olarak aktive edilmiş T hücreleri üzerinde
eksprese edilir. CXCR3, düzenleyici T hücreleri, CD4 pozitif ve CD8 efektör ve hafıza
T hücreleri üzerinde tespit edilmiştir, Th2 hücreleri ile karşılaştırıldığında yardımcı T
(Th)1 hücrelerinde daha yüksek seviyelerde olduğu tespit edilmiştir (97,114-127).
Dendritik hücre (DH) aracılı T hücresi aktivasyonu, başlangıçta CXCR3 negatif olan T
lenfositler üzerinde CXCR3'ü indükler. Ayrıca interlökin (IL)-2, hücre kültürlerinde,
fitohemaglutinin (PHA) varlığında ya da yokluğunda, CXCR3'ü yüksek verimle
arttırabilir ve toplam kültürün yaklaşık % 95’inde CXCR3 pozitifliğine neden olabilir
(114). Lökositlerin diğer alt tipleri, örneğin, doğuştan gelen lenfoid hücreler (ILC'ler),
Gama delta T (γδT) hücreleri, doğal öldürücü (NK) hücreler, NKT hücreleri, spesifik B
lenfositleri ve DH'lerin kendileri de fonksiyonel CXCR3'ü ifade edebilirler (115,128-
133). Ayrıca, CXCR3 ekspresyonu, bağışıklık sistemi ile ilgili olmayan çeşitli
hücrelerde gösterilmiştir. Bu hücreler fibroblastlar, endotel ve epitel hücreler, astrositler
ve düz kas hücreleridir (133,134). Son zamanlarda, eozinofiller ve iltihaplı ortamdaki
nötrofiller üzerinde de CXCR3 bulunmuştur (135-137). Bu durum, CXCR3'ün
granülositler üzerinde bulunmadığının tam olarak doğru olmadığını göstermektedir.

18
Şekil 2-6: Cxcr3 gen yapısına genel bakış (91)
Cxcr3, kromozom X üzerinde q13.1 bölgesinde bulunur ve üç ekson ile bir intron içerir.
Alternatif ekleme, üç farklı CXC kemokin reseptörü 3 (CXCR3) proteinini kodlayan, yapısal ve
fonksiyonel olarak farklı üç mRNA üretir. Standart CXCR3A 368 amino asit içerir. Dört NH2-
terminal rezidüsü, Cxcr3'ün 1. eksonu tarafından kodlanır ve kalan tüm amino asitler, 3. ekson
tarafından kodlanır. CXCR3B (415 amino asit), 2. ekson tarafından kodlanan 51 amino asitlik
özgün bir NH2 terminal ucu içerir. Hem CXCR3A hem de CXCR3B, yedi transmembran
bölgesi içerir. Önemli ölçüde kısaltılmış olan CXCR3-alt (267 amino asit), transkripsiyonel
ekson atlamadan kaynaklanır ve sadece dört veya beş transmembran bölge içerir.
2.2.3.2. Alternatif Eklemeyle Oluşturulan CXCR3 Varyantlarının Keşfi
Başlangıçta tarif edilen 368 amino asitlik CXCR3 proteininin adı daha sonra
CXCR3A olarak değiştirilmiştir ve Cxcr3 geninin alternatif eklemelerinden
kaynaklanan iki diğer CXCR3 izoformu keşfedilmiştir (Şekil 2-6). Birkaç çalışmada,
özgün sinyal kaskadlarının ve ekspresyon paternlerinin her bir CXCR3 varyant
oluşumuna dayandırılabileceği iddia etmiştir (138,139). Gerçekten de, ligandların
varlığından bağımsız olarak, CXCR3 varyantlarının, spesifik hücre tiplerinde farklı

19
şekillerde eksprese edilebildiği ve kısmen farklı sinyal transdüksiyon yollarını aktive
edebileceğine dair kanıtlar mevcuttur (138-142). Bu da, alternatif gen ekleme işleminin,
CXCR3'ün ve in vivo ligandlarının duruma özgü görevlerinin ve özelliklerinin
belirlenmesinde rol oynayabileceğini düşündürmektedir. En çok bulunan form olan
CXCR3A, kemotaksiyi ve kalsiyum mobilizasyonunu uyarmak için CXCL9, CXCL10
ve CXCL11 ile etkileşir. CXCL11 ve CXCL10, inhibe edici Gα proteinlerinin (Gαi)
aktivasyonunu, β arrestin-1 ve β arrestin-2 alımını ve ERK1/2 fosforilasyonunu uyarır
(138,141-143). CXCR3A'nın CXCL9 ile etkileşmesi üzerine verilen yanıt genel olarak
CXCL10 ve CXCL11 ile etkileşmesi üzerine verilen yanıttan daha zayıf olmasına
rağmen, HEK293T hücre transfektanlarında, üç ligandın tamamı reseptör
internalizasyonunu etkili bir şekilde uyarabilmektedir (138). Gαi proteinlerine bağlanma
durumu, CXCR3A aktivasyonunun, adenil siklaz aktivitesinin inhibisyonuna ve
ardından endojen [cAMP]i konsantrasyonunun azalmasına neden olduğu anlamına
gelmektedir. Bu sinyal yolağı en sonunda hücre içi kalsiyum konsantrasyonlarında
([Ca2+]i), hücre proliferasyonunda ve migrasyonla ilişkili hücresel tepkilerin
başlatılmasında artışa neden olur (97,114). Cxcr3'ün 2. eksonunun 5′ ucundaki bir
alternatif ekleme, ekspresyonu daha az olan 415 amino asitlik CXCR3B'yi üretir. Bu
ikinci CXCR3 varyantı, CXCR3A'nın dört NH2-terminal rezidüsünün yerini alan
özgün, 51 amino asitlik bir NH2-terminal kuyruğu içerir. mRNA seviyesinde CXCR3A
ve CXCR3B kalp, böbrek, karaciğer ve iskelet kası dokularında bulunurken, CXCR3A
plasentada da bulunur (139). Bağışıklık hücreleri esas olarak CXCR3A'yı eksprese etse
de genellikle düşük seviyelerde CXCR3B ekspresyonu görülür (92). Ayrıca, endotel
hücreleri CXCR3B'yi seçici olarak eksprese edebilirler. IFN ile uyarılabilen üç CXCR3
kemokin ligandına ek olarak iki trombosit kökenli kemokin olan CXCL4 ve CXCL4L1
de CXCR3A ve CXCR3B'ye bağlanırlar (139,144).
IFN ile uyarılabilen CXCR3 agonistleri arasında CXCL10, CXCR3B için
bağlanma afinitesi en yüksek olan kemokindir (139). IFN ile uyarılabilen tüm CXCR3
ligandları, CXCR3A için CXCR3B'den daha yüksek bir afinite gösterirler. Üstelik,
CXCR3B ile oluşan kemokin sinyali, kalsiyum hareketi ile ilişkili değildir. CXCR3A
transfektanlarına kıyasla, p21 mRNA seviyelerinin CXCR3B ile transfekte edilen
hücrelerde on kat daha yüksek olduğu gösterilmiştir (139). Siklin bağımlı kinaz
inhibitörü p21, DNA hasarı sonucunda hücre döngüsünün durdurulmasında önemli bir
role sahiptir (145,146) ve p21 ekspresyonunun uyarılmasının, uyarıcı G alfa (Gαs)

20
protein sinyalinin başlamasından ve ardından [cAMP]i artışından kaynaklanan
antiproliferatif tepkide yer alan mekanizmaların bir parçası olduğu düşünülmektedir
(147). Bu nedenle, kemokin reseptörlerinin ve CXCR3A'nın aksine, CXCR3B'nin,
mikrovasküler endotel hücrelerinde ligand etkileşimi üzerine Gαs proteinlerine
bağlanabileceği ve [cAMP]i'nin aslında bu CXCR3 varyantına bağlı olduğunu açıkladığı
öne sürülmüştür. Ayrıca, Gα proteinine bağlanmadaki bu farklılıkların, CXCR3A ve
CXCR3B ile indüklenen zıt hücresel tepkileri açıkladığı ileri sürülmüştür (139). Bu
hipotezi destekleyen kanıtlar, CXCR3B aktivasyonunun antiproliferatif bir yanıt
başlattığı ve hücre göçünü olumsuz yönde etkilediği gözlemleriyle sağlanmıştır. Ayrıca
CXCR3B'nin, CXCR3 ligandlarının antianjiyogenik etkilerinden sorumlu reseptör
olduğuna inanılmaktadır (139). Bununla birlikte, CXCR3B ile transfekte edilmiş
HEK293T hücrelerinde herhangi bir Gαs uyarımı gözlenememiştir ve farelerde
CXCR3B formu mevcut değildir. Ancak CXCR3 ligandları ve özellikle CXCL4L1 bu
hayvanlarda antianjiyogenik aktiviteyi etkili bir şekilde korumaktadır (138,144,148).
2.2.3.3. CXCR3'ün in vivo İşlevi için Ekleme Varyantlarının Potansiyel Önemi
Kullanılan deney modeline bağlı olarak, CXCR3 varyantlarına odaklanan özgün
çalışmalarda çelişkili sonuçlar elde edilmiştir. Aslında, CXCR3 ekleme varyantına bağlı
bir mekanizmadan ziyade hücreye bağlı bir mekanizmanın, ligand uyarımı üzerine
aktive edilen Gα protein (Gαi veya Gαs) tipi için belirleyici olabileceği öne sürülmüştür.
Bu, en azından kısmen de olsa, sadece bir izoformu bulunan ve başlangıçta klasik bir
Gαi-bağlı kemokin reseptörü olarak kabul edilen murin CXCR3'ün anjiyogenez
inhibisyonu gibi etkilere sebep olduğunu açıklayabilmektedir (144). Farelerde
CXCR3/Gαi sinyali göz önüne alındığında, Gαi proteini Gαi2’ye karşılık gelir, Gαi3 ise
bu hayvan modelinde inhibitör etki gösterir (149). İnsandaki Cxcr3 için farelerde
karşılık gelen gende alternatif ekleme olmadığı gerçeği, insan kemokin ağı ve murin
karşılığı arasındaki potansiyel önemli farkların altını çizen sayısız örneklerden biridir.
İzoforma spesifik antikorlarla yapılan çalışmaların sınırlı sayıda olması
nedeniyle, CXCR3 varyantlarının sağlık ve hastalık durumlarında IFN ile ilişkili
CXCR3 kemokin ağına kesin katkısı büyük ölçüde bilinmemektedir. İltihaplı bir
hücresel ortamda, genellikle belirli bir kemokin reseptörü ve bunun ligandları bulunur
ve hatta tek bir hücre tarafından birlikte sentezlenebilirler. CXCR3A ve CXCR3B'nin T
hücreleri üzerinde birlikte ifade edildiğini bildirilmiştir (139). Bununla birlikte,

21
CXCR3B’nin, CXCR3A'nın aksine, HUMEC'ler üzerinde eksprese edildiği ve CXCR3
ligandlarının CXCR3B aracılığıyla bu mikrovasküler endotelyal hücrelerin büyümesini
inhibe ettiği gösterilmiştir. Aksine, insan mezengial hücreleri esas olarak CXCR3B'yi
değil, CXCR3A'yı ifade eder (139). Over kanseri hastalarında üç CXCR3 varyantının da
farklı ekspresyonları bildirilmiştir. En yüksek CXCR3B ekspresyonu normal dokuda
(CXCR3B ekspresyonu; normal > endometriozis > kanser dokusu) görülmüştür.
CXCR3A ekspresyonu ise endometrioziste ve kanser dokusunda normal dokudan daha
yüksektir (150). Prostat kanseri örneklerinde CXCR3A'nın mRNA seviyelerinin artmış
ve CXCR3B'nin mRNA seviyelerinin azalmış olduğu bulunmuştur. Üstelik, bu değişmiş
ekspresyon düzeylerinin, kanser hücrelerinin bozulmuş bir göç ve invazyon davranışına
sebep olduğu ve CXCR3A ekspresyon artışı ile CXCR3B ekspresyon azalışının nihai
sonucunun tümör progresyonu ve metastaz olduğu ortaya çıkarılmıştır (151).
2.2.4. Kemokin ile Uyarılan CXCR3 İnternalizasyonu
CXC kemokin reseptörü 3, kemokin agonistlerinin varlığında daha da artan
yapısal internalizasyon gösterir (152). CXCL11, T hücreleri ve uyarılmış endotel
hücreleri arasındaki temastan sonra, CXCR3 internalizasyonunun uyarılmasından
sorumlu olan baskın IFN ile uyarılabilen CXCR3 ligandı olarak bulunmuştur (153).
Ayrıca, CXCR3 ile transfekte edilmiş HEK hücrelerinin kullanıldığı bir çalışmada,
CXCL11'in CXCR3 internalizasyonunu destekleyen ana kemokin olduğu iddia
edilmiştir (154). Bu veriler, CXCR3 internalizasyonunun üstün bir indükleyicisi olarak
hareket ederek CXCL11'in, diğer iki IFN ile uyarılabilen CXCR3 ligandları ve
trombosit türevli CXCR3 agonistleri CXCL4 ve CXCL4L1 için reseptörün
mevcudiyetini azalttığı anlamına gelebilmektedir. Üstelik, CXCL9’un ve CXCL11'in,
sırasıyla β-arrestin alımını ve reseptör internalizasyonunu başlatma eğilimli ligandlar
oldukları iddia edilmiştir. Aksine, transfekte edilmiş HEK hücreleri üzerindeki özgün
CXCR3 varyantlarının internalizasyon özellikleri arasında farka bakıldığında,
CXCL11’in CXCR3A ve CXCR3B'nin internalizasyonunu daha az uyardığı
görülmüştür (138). CXCL10, CXCR3A internalizasyonunu 10 dakika içinde %40
oranında başlatırken CXCL9 ile uyarılan CXCR3A internalizasyonunun üç kat daha
yavaş olduğu ortaya çıkarılmıştır (138). CXCR3 internalizasyonunu uyarmak için IFN
ile uyarılabilen CXCR3 ligandlarının potansiyelleri ile ilgili yapılan iki çalışmada elde
edilen çelişkili sonuçlar, ilk çalışmanın yazarlarının CXCR3 varyantları arasında fark

22
gözetmeksizin sadece β-arrestine bağımlı CXCR3 internalizasyonunu dikkate alırken,
diğerleri, reseptörün internalizasyonunun β-arrestin'den bağımsız bir şekilde
gerçekleşebileceğini göstermişlerdir (138,152,154,155). CXCR3B internalizasonunu
ise, CXCL9 veya CXCL10 orta derecede uyarmaktadır (138,155). Özetle, çeşitli
çalışmalardan elde edilen farklı sonuçlar, CXCR3 internalizasyon özelliklerinin
muhtemelen kullanılan deney düzeneğine bağlı olduğunu göstermektedir. Ayrıca
internalizasyon, alternatif CXCR3 ekleme varyantları için farklı şekillerde ortaya
çıkmaktadır. Bununla birlikte, CXCR3 izoformlarının in vivo fizyolojik özellikleri şu
anda büyük ölçüde bilinmemektedir. Önceki çalışmalar, bu farklı reseptör varyantlarının
özgün rolleri olduğu fikrini desteklerken, CXCR3 izoformları ile hücrelerin transfekte
edilmesi yapaydır ve in vivo durumu gösterdiği kesin değildir, bu da, endojen olarak
ifade edilen üç CXCR3 varyantı arasında ayrım yapabilen oldukça spesifik antikorlarla
çalışmalara ihtiyaç olduğunu göstermektedir.
2.2.4.1. CXCR3 Ligandlarının Hastalık ve Tedavi ile İlişkisi
T hücrelerinin ve NK hücrelerinin inflamatuar yapıları ve kemotaktik
aktiviteleri, CXCL9’un, CXCL10’un ve CXCL11'in inflamasyon ve otoimmünitede
anahtar rolleri olabileceği anlamına gelmekedir. Ayrıca, CXCL10’un ilk olarak in vitro
ve daha sonra in vivo olarak antianjiyogenik etkileri gösterilmiştir (156,157). Bu durum
ise IFN ile uyarılabilen CXCR3 ligandlarının biyolojik fonksiyonlarının, CXCR3 ifade
eden lökositlerin göçünü yönlendirmeye yönelik fonksiyonlarının ötesine uzandığı
anlamına gelir. Bununla birlikte, IFN ile uyarılabilen ve trombosit türevli diğer iki
CXCR3 agonisti CXCL4 ve CXCL4L1'in anjiyostatik özellikleri gösterilmiştir
(144,148,158-161). CXCR3 nötralize edici antikorların eklenmesinin, insan endotel
hücrelerinin göçünü engellediği ve fare korneasında CXCL4L1 ile indüklenen
antianjiyogenik aktiviteyi inhibe ettiği bulunmuştur (144,162). Üstelik, anti-CXCR3
antikorları, tümör büyümesinin inhibisyonunu önlemiştir ve CXCL4L1'in CXCR3 -/-
farelerde tümör büyümesi üzerinde hiçbir etkisi görülmemiştir (144). Bu sonuçlar, en
azından kemokinler ekzojen olarak (dışardan) eklendiğinde, CXCR3 ligandlarının
anjiyostatik etkilerinin CXCR3'e bağlı bir olgu olduğunu gösterebilmektedir.
CXCR3A'nın, ligandlarının anti-anjiyogenik etkilerine aracılık eden bir reseptör
olmadığını gösteren belirtiler de vardır. Bu durum, CXCR3 reseptör varyantlarının ya

23
da alternatif reseptörler aracılığıyla alternatif sinyal yollarının anti-anjiyogenik
aktivitede yer aldığını göstermektedir.
Anjiyogenezi düzenlediğine inanıldığı gerçeği, bu kemokinlerin ayrıca tümör
biyolojisi ve hematolojik malinitelerde de rol oynayabileceğini düşündürmektedir (92).
Genel olarak, çeşitli in vivo hastalık modellerinde, durum-bağımlı olan farklı roller IFN
ile uyarılabilen CXCR3 ligandlarına dayandırılmıştır (92,93).
2.2.5. MPN ile İlişkilendirilen Sitokin ve Kemokinler
MPN ile kemokinlerin arasındaki bazı ilişkileri belirten literatür çalışmaları
mevcuttur. Bir çalışmada CCL3’ün MPN’ye neden olabileceği gösterilmiştir (163). Bu
çalışmadaki hastalarda lösemi gelişme riskinin arttığı gösterilmiştir. Bu durumda kemik
iliği stromasından elde edilen CCL3, çeşitli proinflamatuar sitokinlerin üretilmesi ve
lösemi öncesi hücrelerin proliferasyonunu arttırma kapasitesine sahip olan monositlerin
kemik iliğinden sızmasını uyararak lösemi gelişimine neden olabilir.
Ph- MPN'ler (PV, ET ve PMF), birçok inflamatuar sitokinlerin (IL1, IL2, IL6,
IL8, IL12, TNFα ve IFNγ), birkaç büyüme faktörünün (GM-CSF, G-CSF, HGF, PDGF
ve EGF) ve anjiyogenik faktörlerin (VEGF) artmış plazma seviyeleri ile sitokin
üretimindeki önemli değişiklikler ile karakterize edilirler (164). Ayrıca deregülasyonlar
da IL4 ve IL10 gibi anti-enflamatuar sitokinlerle ilgilidir. MPN'ler arasında sitokin
seviyeleride ve profillerinde farklılıklar vardır, ancak bu hastalıklar içinde belirli bir
süreklilik söz konusu değildir. Bazı sitokinler, PMF'de PV'ye göre (IL1, IL1RA, IL2-
Ra, EGF ve IL10) aşırı eksprese edilir. Aksine, bazıları PV'de PMF’e göre (IL7, IFNγ,
GM-CSF, MIP-la, IP-10 ve MIG) aşırı eksprese edilir ve ET; IL6, IL8, IL12, IFNγ,
GM-CSF ve HGF için PV'den daha yüksek ekspresyon seviyelerine sahiptir.
Teknik olarak, farklı çalışmalar arasındaki yöntemlerin standardizasyonu
olmadığından, bu sonuçları birbirleriyle karşılaştırmak zordur. Aynı teknolojilerin
kullanılması ile büyük bir MFP, PV ve ET kohortunun araştırılması, bu farklılıkları
açıklığa kavuşturabilecek ve her bir hastalık için spesifik profil varlıklarını daha iyi
tanımlayabilecektir.
IFN-γ/CXC kemokin sinyallemesi, IFN-γ'nın, IFN-γ heterodimerik reseptöre
bağlanmasından başlayarak, Janus Kinaz (JAK) -STAT yolağını aktive eder ve bu da
STAT1 aktivasyonuna yol açar. Bunu, çekirdeğe STAT1 translokasyonu ve hemen

24
ardından kemokin (CXCL9, CXCL10) transkripsiyonu takip eder. Üretilip hücreden
salınan kemokinler, özgün reseptörleri CXCR3’e bağlanarak belli biyolojik süreçleri
başlatırlar.
Sonuç olarak, yaşlanma ile birlikte IL6 ve interferon-gama ile uyarılabilen
kemokinlerin (CXCL9 ve CXCL10) varlığının arttığı ve aksine IL2, EGF ve EGFR
varlığının azaldığı gösterilmiştir (165,166).
25
3. GEREÇ VE YÖNTEM
3.1. Gereç
3.1.1. Hasta Örnekleri
Çalışmada, İstanbul Üniversitesi, İstanbul Tıp Fakültesi Hematoloji Bölümü’nde
Miyeloproliferatif Neoplazi tanısı almış 33 hastadan flebetomi yolu ile elde edilen
periferik kan mononükleer hücreleri kullanıldı. Hastalara gerekli bilgilendirme
yapılarak, gönüllü onam formu okutuldu ve onayları alındı. Çalışmaya dahil edilen
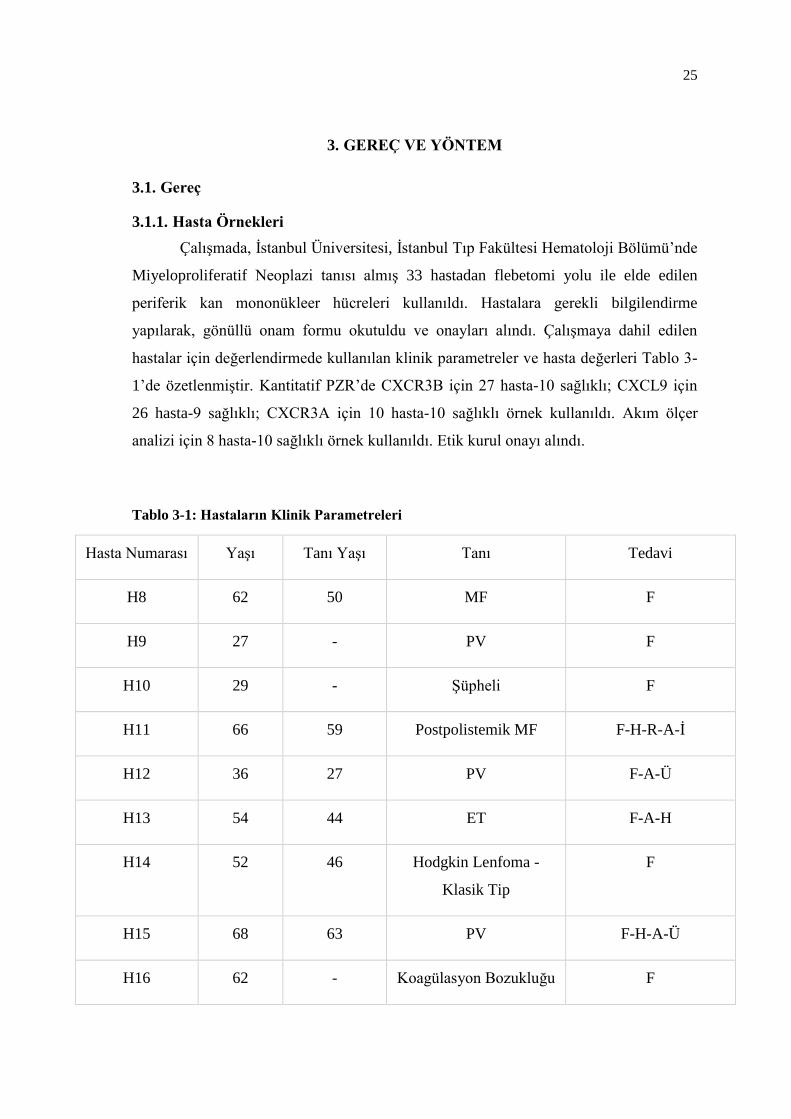
hastalar için değerlendirmede kullanılan klinik parametreler ve hasta değerleri Tablo 3-
1’de özetlenmiştir. Kantitatif PZR’de CXCR3B için 27 hasta-10 sağlıklı; CXCL9 için
26 hasta-9 sağlıklı; CXCR3A için 10 hasta-10 sağlıklı örnek kullanıldı. Akım ölçer
analizi için 8 hasta-10 sağlıklı örnek kullanıldı. Etik kurul onayı alındı.
Tablo 3-1: Hastaların Klinik Parametreleri
Hasta Numarası Yaşı Tanı Yaşı Tanı Tedavi
H8 62 50 MF F
H9 27 - PV F
H10 29 - Şüpheli F
H11 66 59 Postpolistemik MF F-H-R-A-İ
H12 36 27 PV F-A-Ü
H13 54 44 ET F-A-H
H14 52 46 Hodgkin Lenfoma -
Klasik Tip
F
H15 68 63 PV F-H-A-Ü
H16 62 - Koagülasyon Bozukluğu F

26
(Biyopsi yok)
H17 31 27 PV F-A
H18 48 41 PV F-H-A-L-At-S
H19 83 75 PV F-H-C-Ü
H20 32 - PV F
H21 56 43 MPN (PV,ET) F-A-Ü
H22 70 66 ET F-A-H-Ü
H23 53 35 PV F-H-V
H24 63 54 Hairy Cell Lösemi F-A-C
H25 39 - PV F
H26 59 45 ET F-H-T-Ü-A
H28 56 48 PV F-H-Ü
H29 92 64 PV F-H-A-Ü
H34 58 -- PV F
H35 71 - PV F
H36 71 63 Multipl Miyelom F-Ko-Cl
H37 55 49 PV F-A
H38 69 52 PV F-A-Ü-H
H39 48 41 PV F-H-A
H43 65 54 PV F-H-A-Ü

27
H46 55 45 ET F-H-A
H54 55 53 ET F-A
H56 55 55 Hairy Cell Lösemi F-A
H57 - - - F
Tabloda Kullanılan Kısaltmalar: F: Flebetomi, H: Hidroksiüre, T: Tromboredüktin, Ü: Ürikoliz,
A: Asetilsalisilik Asit, R: Ruxolitinib, L: Larcadip, At: Ator, S: Saneloc, C: Coraspin, Ko:
Kordexa, Cl: Clexane
3.1.2. Deneylerde Kullanılan Kontrol Örnekleri
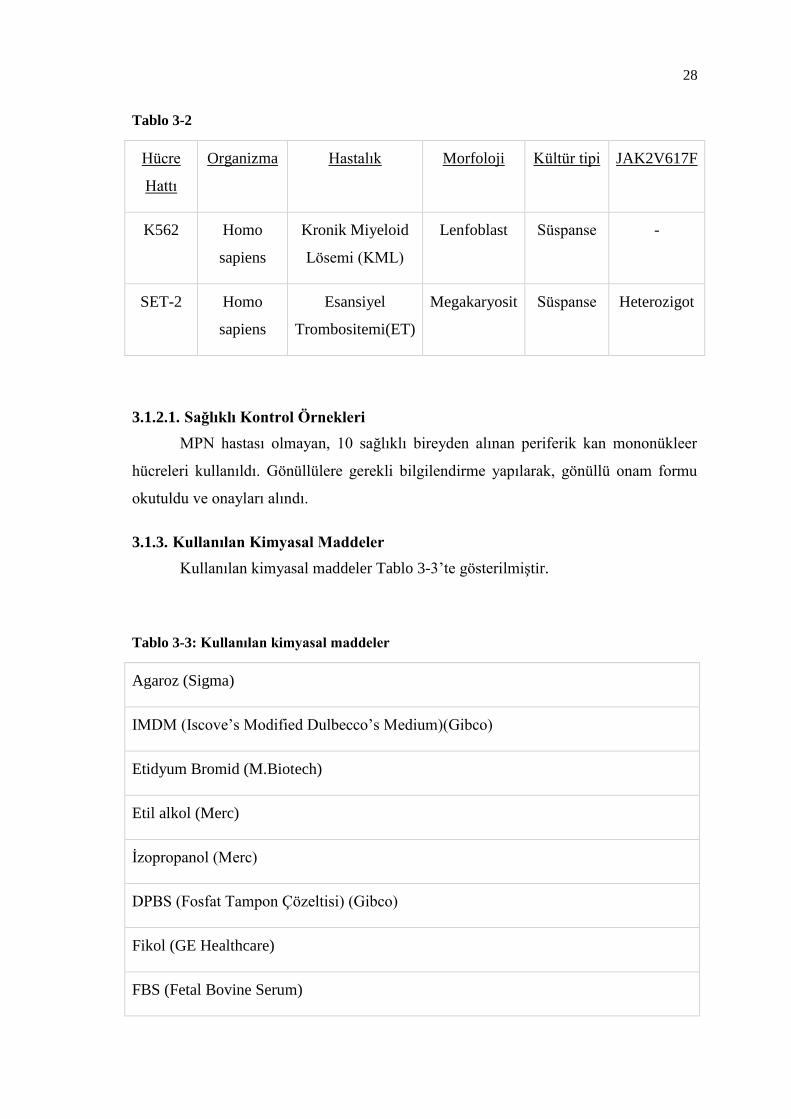
MPN’lerde JAK2V617F mutasyon karakterinin allel düzeyinde belirlenmesi
amacıyla deneylerde kontrol olarak 2 farklı hücre soyu kullanıldı. Bunlar JAK2V617F
mutasyonunu homozigot formda taşıyan HEL (İnsan eritrolösemi hücre soyu) ve
heterozigot formda taşıyan SET-2 (İnsan esansiyel tromboz hücre soyu) hücre
soylarıdır. Ayrıca JAK2V617F mutasyonunu taşımadığı bilinen K562 (İnsan kronik
miyeloid lösemi hücre soyu) hücre soyu da kontrol olarak kullanıldı (Tablo 3-2).
HEL ve SET-2 hücre soyları DSMZ (Leibniz Enstitüsü Alman Mikroorganizma
ve Hücre Kültürü Koleksiyonu) firmasından temin edildi. K562 hücre soyu ise İstanbul
Üniversitesi Deneysel Tıp Araştırma Enstitüsü Genetik Ana Bilim Dalı’ndan Prof. Dr.
Duran ÜSTEK’ten temin edilmiştir.
28
Tablo 3-2
Hücre
Hattı
Organizma Hastalık Morfoloji Kültür tipi JAK2V617F
K562 Homo
sapiens
Kronik Miyeloid
Lösemi (KML)
Lenfoblast Süspanse -
SET-2 Homo
sapiens
Esansiyel
Trombositemi(ET)
Megakaryosit Süspanse Heterozigot
3.1.2.1. Sağlıklı Kontrol Örnekleri
MPN hastası olmayan, 10 sağlıklı bireyden alınan periferik kan mononükleer
hücreleri kullanıldı. Gönüllülere gerekli bilgilendirme yapılarak, gönüllü onam formu
okutuldu ve onayları alındı.
3.1.3. Kullanılan Kimyasal Maddeler
Kullanılan kimyasal maddeler Tablo 3-3’te gösterilmiştir.
Tablo 3-3: Kullanılan kimyasal maddeler
Agaroz (Sigma)
IMDM (Iscove’s Modified Dulbecco’s Medium)(Gibco)
Etidyum Bromid (M.Biotech)
Etil alkol (Merc)
İzopropanol (Merc)
DPBS (Fosfat Tampon Çözeltisi) (Gibco)
Fikol (GE Healthcare)
FBS (Fetal Bovine Serum)

29
Tris baz (BioChemika)
Etilen diamin tetra asetik asit (EDTA) (Gibco)
Steril, DNA/RNA nükleaz içermeyen su
Ultra Saf Su (Biochrom)
50 bp Ladder (BioRad)
Propidium Iodide
2 mL CD34 MicroBeads, insan (MACS)
2 mL FcR Blocking Reagent, insan (MACS)
2 mL Diamond Lin Biotin-Antibody Cocktail, insan (MACS)
2×2 mL Anti-Biotin MicroBeads (MACS)
2 mL CD34 Diamond MicroBeads, insan (MACS)
Universal Probe Library qPCR Probe 27 (Roche)
Universal Probe Library qPCR Probe 4 (Roche)
Universal Probe Library qPCR Probe 60 (Roche)
SYBR Green Probe (BIOLINE)
CD34 PE (8G12) Antikor (BD Pharming)
3.1.4. Kullanılan Kitler
Kullanılan kitler Tablo 3-4’te gösterilmiştir.

30
Tablo 3-4: Kullanılan Kitler
CD34 MicroBead Kit, insan (MACS)
Diamond CD34 Isolation Kit, insan (MACS)
Quick gDNA Micro Prep (Zymo Research D3025)
RNA Purification Kit (Jena Bioscience) (PP-210L)
SCRIPT cDNA Synthesis Kit (Jena Bioscience) (PCR-511S)
Sigma PZR Mix
3.1.5. Kullanılan Cihazlar
Kullanılan cihazlar Tablo 3-5’te gösterilmiştir.
Tablo 3-5: Kullanılan cihazlar
Buzdolabı ve Derin Dondurucular (+4°C, -20°C, -80°C) (Samsung, Haier,Haier)
Buz Makinası (Cornelius)
Jel görüntüleme sistemi (UVP)
Otoklav (Kermanlar)
Vorteks (Kermanlar)
Otomatik Pipetler (Eppendorf)
PZR Aleti (Thermo-Cycler) (Bio-Rad T100)
Elektroforez Aleti (Bio-Rad)
Güç Kaynağı (E-C Apparatus Corporation)

31
Distile Su Cihazı (Millipore)
Masaüstü Mini Santrifüj (Beckman Coulter)
Soğutmalı Santrifüj (Beckman Coulter)
Su Banyosu (Memmert)
Laminer Akış Kabini (NUAIRE)
Hücre Ayırım Ünitesi ( FACSAria Akım Sitometri Cihazı, BD Bioscience)
Nanodrop NanoDrop Technologies
Mikroskop (Olympus)
Mikrodalga Fırın (Arçelik)
Gerçek Zamanlı Kantitatif PZR Cihazı (LightCycler 480 II - Roche)
3.1.6. Kullanılan Tampon ve Çözeltiler
% 70’lik Etil Alkol
70 ml Etil alkol
Steril distile su ile 100ml’ye tamamlandı.
50 X TAE (Tris-Asetik asit-EDTA)
242 g Tris baz
57,1 g Glasiyal asetik asit
100 ml EDTA (0,5M)
Distile su ile 1 lt’ye tamamlandı ve otoklav ile steril edildi.
MACS Solüsyonu
500 ml PBS
% 0.5 (7.5 M) BSA

32
2Mm (0.5M) EDTA
Filtrelendi.
Propidoum Iodide Solüsyonu
10μl Propidoum iodide
10 ml’ ye MACS solüsyonu ile tamamlandı.
Hücre Dondurma Solüsyonu
%80 FBS (Fetal Bovine Serum)
%20 DMSO ile tamamlandı.
20 dakika buzda bekletildi.
Hücre Dondurma Solüsyonu 2
%80 IMDM
%20 FBS (Fetal Bovine Serum) ile tamamlanır. 0,20 μm membranlı filtreden
geçirildi.
Hücre Çözdürme Solüsyonu
%2 FBS (Fetal Bovine Serum)
IMDM ile tamamlandı. 0,20 μm membranlı filtreden geçirildi.
3.2. Yöntem
3.2.1. Mononükleer Hücre İzolasyonu
İstanbul Üniversitesi, İstanbul Tıp Fakültesi Hematoloji Bölümü’nde toplanarak
laboratuvarımıza getirilen MPN tanısı almış hastaların flebetomi uygulanarak toplanan
periferal kanları ile kontrol olarak kullanılan 10 sağlıklı erişkinden elde edilen perifer
kanlar, fikol üzerine yayıldı, fikol gradient santrifüj yöntemi ile ayrıştırıldı ve fikol
içindeki periferal kan mononükleer hücreleri, pastör pipet ile toplandı. Toplanan bu
periferal kan mononükleer hücreleri daha sonra yüzey belirteçlerine göre izole edilerek
DNA ve RNA izolasyonunda kullanıldı, kalan kısmı ise hücre dondurma protokolüne
uygun olarak dondurulup sonraki çalışmalarda kullanıldı.
1. Kabin çamaşır suyuyla silindi, tüm malzemeler alkolle silinerek kabine
alındı. Kan torbası ve filtre hortumu çamaşır suyuyla silindi.

33
2. Kan torbasındaki kan steril şişelerde PBSs ile 1:1 oranında sulandırıldı.
3. 15 mL fikol 50 mL’lik falkonlara dağıtıldı ve ardından üzerlerine 35 mL
kan (kan+PBS) yavaşça eklendi.
4. Falkonlar santrifüjde 2000 rpm’de 30 dakika 15°C’de hızlanma ve
yavaşlama ivmesi en az olacak şekilde santrifüj edildi.
5. Fikol-gradient santrifüj tekniği ile tüpün ağzından dibine doğru sırasıyla
plazma, mononükleer hücre, fikol, granülosit fazları oluşur. Ayrımı net
bir şekilde santrüfüj sonrasında görülen mononükleer hücreler 15 ml
MACS içerisine toplandı. Serum ve granülositler gerektiği kadar başka
çalışmalarda kullanılmak üzere saklandı.
6. Santrifüj sonrasında süpernatantı dikkatli bir şekilde atıldıktan sonra
pellet 10 ml MACS ile çözüldü ve tüm tüpler birleştirildi.
7. Bu tüp 2000 rpm’de 15 dakika santrifüj edildi.
8. Santrifüj sonrasında süpernatant atıldı, pellet 10 ml IMDM ile çözüldü.
9. IMDM içerisindeki hücrelerden pipet 10 μl çekilerek 90 μl IMDM’e
eklendi ve 10’luk dilüsyonlar hazırlandı. Seyreltilen bu hücrelerden 10 μl
çekilip 10 μl trypan blue ile karıştırıldıktan sonra hemositometre ile
sayıldı (Tablo 3-6).
Tablo 3-6: Hücre Sayımında Kullanılan Formül
Hücre sayısı/ml = (Dört Karedeki Hücre Sayısı Ortalaması) x 104 x (Dilusyon Oranı)
3.2.2. Hücre Dondurma
Mononükleer hücre izolasyonundan sonra kalan hücre sayısına göre hücre
dondurma solüsyonları 1:1 oranda hazırlanır.
1. Hücre dondurma solüsyonu 1, hazırlandıktan sonra 20 dakika buzda
bekletildi.

34
2. Hücreler 2000 rpm’de 15 dakika santrifüj edildi, pellet hücre dondurma
solüsyonu 2 ile çözüldü ve hücre dondurma solüsyonu 1 damla damla
eklendi.
3. 2 mL ve 5 mL dondurma tüplerine alındı. 1 gece -80°’de bekletildi ve
ertesi gün uzun süre saklamak amacıyla sıvı azot tankına aktarıldı
3.2.3. Hücre Çözme
Sağlıklı kontrol kanlarından elde edilen mononükleer hücreler dondurulmadan
direkt olarak deneylerde kullanıldı. Bu nedenle bu grup için hücre çözme işlemi
uygulanmadı.
Hasta kanından mononükleer hücre izolasyonundan sonra mononükleer
hücrelerin kalan kısmı sonraki çalışmalarda kullanılmak üzere donduruldu. Hücrelerin
çalışmalarda kullanılması gerektiğinde hücreler azot tankından alındı ve hücre
çözdürme için aşağıdaki protokol takip edildi.
1. Hücreler sıvı azottan alındı ve buza yerleştirildi.
2. 37°C su banyosunda olabildiğince hızlı bir şekilde eritildi.
3. Dondurma tüpünde bulunan hücreler 15 mL’lik falkona aktarıldı.
4. Bu tüpteki hacmin %10’u kadar Dnaz eklendi (200 μl)
5. 37°C su banyosunda 90 saniye inkübe edildi.
6. Filtrelenmiş %2 oranında FBS içeren 10 ml IMDM damla damla eklendi.
7. 1200 rpm’de 7 dakika santrifüj edildi.
8. Pellete 140 μl Dnaz eklendi. (hacmin %7’si kadar)
9. 10 mL FBS’li IMDM tekrar damla damla eklendi. 1500 rpm/ 6 dakika
santrifüj edildi.
10. Pellete 80 μl DNaz eklendi. (hacmin %4’ü kadar)
11. 100 milyon hücreye 5 mL olacak şekilde MACS eklendi.
12. Hücreler sayıldıktan sonra sonraki deneysel aşamalarda kullanıldı.

35
3.2.4. Hücre Ayrımı
Kök hücre ayrımı için iki farklı popülasyon hedeflenmiştir. Bunlar CD34+/- ve
CD34+/-Lineage- popülasyonlarıdır. Bu popülasyon periferik kan mononükleer
hücrelerinden elde edilmiştir.
3.2.4.1. CD34 MicroBead Kit (MACS Separation) ile Hücre Ayrım Prosedürü
İlk olarak, CD34+ hücreler ‘CD34 MicroBeads’ ile manyetik olarak işaretlenir.
Daha sonra, hücre süspansiyonu bir ‘MACS Ayırıcı’nın manyetik alanına yerleştirilen
‘MACS® Kolona’ yüklenir. Manyetik olarak işaretlenmiş CD34+ hücreler, kolontun
içerisinde tutunurlar. Kolonu manyetik alandan çıkardıktan sonra, manyetik olarak
tutunmuş olan CD34+ hücreler pozitif seçilmiş hücreler olarak saflaştırılır.
1. 5 mL MACS tampon çözültisindeki hücreler 1700 rpm’de 10 dakika
santrifüj edildi ve üst sıvı tamamen çekildi.
2. Pellete 300 μl MACS eklendi.
3. 100 μl FcR Blocking Reagent eklendi.
4. 100 μl CD34 MicroBeads eklendi.
5. İyice karıştırıldı ve +4°C’de 30 dakika inkübe edildi.
6. Hücrelere 5 mL MACS eklendi ve 1700 rpm’de 10 dakika santrifüj
edildi. Üst svı tamamen çekildi.
7. Hücreler 500 μl MACS ile yeniden çözüldüler.
8. Manyetik Ayrıma geçildi; kite özgü kolon, ‘MACS Ayırıcı’daki
manyetik alana yerleştirildi ve kolonun altına CD34- hücreleri toplayacak
olan tüp yerleştirildi.
9. Kolon 3 mL MACS ile yıkandı.
10. Hücreler kolona yavaşça bırakıldı ve CD34- hücreler toplanmış oldu.
11. 3 mL MACS yıkama yapmak amacıyla kolona 3 defa daha uygulandı.
12. Kolon manyetik alandan çıkarılarak CD34+ hücreleri toplamak amacıyla
2. bir tüpe yerleştirildi.

36
13. Kolona 5 mL MACS eklendi. CD34+ hücreleri kolondan çıkarmak için
kolonun paketinden çıkan piston sıkıca ve yavaş bir şekilde kolonun
ağzından itibaren bastırıldı.
Manyetik ayrımdan sonra saflık analizi yapıldı. Bunun için bir miktar hücre
alınıp CD34 PE Antikor ile boyandıktan sonra akım hücre ölçer cihazında
analiz edildi.
3.2.4.2. CD34+/-Lineage- Popülasyon Eldesi Prosedürü
Diamond CD34 İzolasyon Kiti, perifer kan mononükleer hücrelerinden
(PBMC'ler) ve kemik iliği mononükleer hücrelerinden son derece saf CD34+ kök
hücrelerinin izolasyonu için geliştirilmiştir. Kök hücrelerin izolasyonu, iki aşamalı bir
prosedürde gerçekleştirilir. İlk olarak, Lin+ hücreleri (CD2, CD3, CD11b, CD14, CD15,
CD16, CD19, CD56, CD61 ve CD235a (Glikopin A)), spesifik antijenlere ve Anti-
Biotin MicroBeads'e karşı biyotin-konjuge antikorların bir kokteyli ile manyetik olarak
etiketlenir. Hücreler ‘ayırıcı’nın manyetik alanına yerleştirilen bir kolonu üzerinde
manyetik olarak ayrılırlar, etiketlenmemiş Lin- hücreler akıp geçerken manyetik olarak
etiketlenmiş Lin+ hücreler kolon içinde kalır. İkinci adımda, Lin- kök hücreler doğrudan
‘CD34 Diamond MicroBeads’ ile etiketlenir. Ardından bir manyetik ayırma daha yapılır
ve CD34+ kök hücreler, kolonun manyetik alandan çıkarılmasından sonra elde edilir.
Prosedür, elde edilen PBMC sayısı belirlendikten sonra uygulandı.
1. 200 g’de 10 dakika santrüfüjden sonra üst sıvı tamamen çekildi.
2. 200 μl MACS ile yeniden sulandırıldı.
3. 50 μl ‘Diamond Lin Biotin-Antibody Coctail’ eklendi.
4. İyice karıştırıldı ve 10 dakika +4°C’de bekletildi.
5. 3 mL MACS eklenerek hücreler yıkandı ve 300 g’de 10 dakika santrifüj
edildi. Üst sıvı tamamen çekildi.
6. 400 μl MACS ile pellet yeniden sulandırıldı.
7. 100 μl ‘AntiBiotin Microbeads’ eklendi.
8. İyice karıştırıldı ve 15 dakika +4°C’de bekletildi.

37
9. 5 mL MACS eklenerek hücreler yıkandı ve 300 g’de 10 dakika santrifüj
edildi. Üst sıvı tamamen çekildi.
10. 1000 μl MACS’ta yeniden sulandırıldıç
11. Manyetik ayrıma geçildi. Manyetik ayrım esnasında sonraki adımlara
geçmeden önce daima kolonun boşalması beklendi.
12. Kolon manyetik alana yerleştirildi (MACS ayırıcıya).
13. Kolon 2 mL MACS ile yıkandı.
14. Kolona hücre süspansiyonu uygulandı.
15. ‘İşaretlenmemiş’ hücreler toplandı ve kolon 2 mL MACS ile yıkandı.
Akan tüm sıvı toplandı; bunlar ‘işaretlenmemiş’ Lin- hücrelerdir. İki defa
MACS eklenerek yıkama adımları gerçekleştirildi. Yeni MACS sadece
kolon boşaldıktan sonra eklendi.
16. CD34+ kök hücrelerin manyetik işaretlenmesine geçildi.
17. 300 g’de 10 dakika santrifüj edildi. Üst sıvı tamamen çekildi.
18. 200 μl MACS’ta yeniden sulandırıldı.
19. 50 μl ‘CD34 Diamond MicroBeads’ eklendi.
20. İyice karıştırıldı ve +4°C’de 30 dakika bekletildi.
21. 2,5 mL MACS eklenerek hücreler yıkandı ve 300 g’de 10 dakika
santrifüj edildi. Üst sıvı tamamen çekildi.
22. 500 μl MACS ile pellet yeniden sulandırıldı.
23. ‘Manyetik Ayrım: CD34+ kök hücrelerin pozitif seçilimi’ne geçildi
24. Kolon manyetik alana yerleştirildi.
25. Kolon 500 μl MACS geçirilerek hazırlandı.
26. Kolona hücre süspansiyonu uygulandı.
27. Kolon 3 defa 500 μl MACS ile yıkandı. (Kolon boşaldıkça eklendi.)
28. Kolon ayırıcıdan çıkarıldı ve uygun bir toplama tüpüne yerleştirildi.

38
29. 1 mL MACS kolona eklendi. Pistonun kolon ağzından itibaren sıkıca
itilmesiyle manyetik olarak işaretlenmiş hücreler elde edildi.
3.2.5. Hücrelerden DNA İzolasyonu
Sağlıklı bireylerden ve MPN hastalarından toplanan periferal kanlardan izole
edilen mononükleer hücrelerde ve iki farklı kit kullanılarak, hücre ayırıcı ile ayrımı
sağlanmış CD34+, CD34- hücreleri için DNA izolasyonu yapıldı.
1. Hücre süspansiyonları masaüstü mini santrifüj kullanılarak 3 000 g’de 5
dakika santrifüj edildi, üst sıvı mikropipet ile dikkatlice çekilerek
hücrelerin pellet haline gelmesi sağlandı.
2. Pellet olarak saklanmış örnekler üzerine 200 μl ‘Lysis Buffer’ eklendi ve
4-6 saniye vortekslenip 5-10 dakika oda sıcaklığında bekletildi.
3. Zymo Spin kolona aktarıldıktan sonra 10.000 g’de 1 dakika santrifüj
edildi ve yıkama aşamasına geçildi.
Yıkama Aşaması;
1. Spin kolon üzerine 200 μl ‘’Pre-Wash Buffer’’ eklendi ve 10.000
g’de 1 dakika santrifüj edildi.
2. 500 μl ‘’g-DNA Wash Buffer’’ spin kolona eklendi tekrar 10.000
g’de 1 dakika santrifüj edildi.
3. Kolon, RNaz-DNaz içermeyen 1,5 ml ependorflara alındı ve 10 μl
‘’Elution Buffer’' eklendi.
4. 2-5 dakika oda sıcaklığında bekletildi ve en yüksek hızda 30 saniye
santrifüj edildi.
DNA’nın Saklanması
Uzun süreli saklama için -20°C’de, kısa süre içinde kullanılacağında
+4°C’de muhafaza edildi.
3.2.6. İki Aşamalı Allel Spesifik Nested PZR
İki aşamalı veya iç içe polimeraz zincir reaksiyonu (allel spesifik PZR), spesifik
olmayan bağlanmayı azaltarak istenmeyen ürünlerin oluşmasını engelleyen bir
polimeraz zincir reaksiyonu modifikasyonudur. İki faklı primer seti gereklidir. İlk

39
primer seti ile mutasyonu içeren bölge çoğaltılır. İlk aşamadan sonra jelde 521 bazlık
DNA parçaları görülür. İlk aşama sonunda mutant ve yabani tipteki JAK2 geni ayırt
edilememektedir. PZR işlemi 2. aşamasında mutasyon bölgesine özgü farklı primer seti
kullanılır. Bu şekilde sadece istenilen bölgenin çoğaltılması sağlanarak spesifik bantlar
elde edilir.
İki aşamalı allel spesifik nested PZR’de uygulanan protokol şu şekildedir:
1. Sigma PZR karışımı, primerler ve kalıp DNA -20oC’ de saklandı.
Çalışma sırasında tüm bileşenler buz içerisinde tutuldu.
2. Allel spesifik nested PZR 1. aşamasında Tablo 3-7’de belirtilen
miktarlarla homojen bir karışım hazırlanıp DNaz/RNaz içermeyen PZR
tüplerine 18’er μl dağıtıldı.
Ortak karışımda bulunanlar: steril su, Sigma PZR karışımı ve
primerlerdir.
Tablo 3-7: Allel Spesifik Nested PZR 1. Aşama bileşenleri
Bileşen Miktar
Sigma PZR Mix 10 μl
Primer 1 (Forward) 1 μl
Primer 2 (Reverse) 1 μl
Kalıp DNA 2 μl
H2O 20 μl’ye tamamalandı
3. Allel spesifik nested PZR’nin 1. aşamasında P1 ve P2 primerleri, ikinci
aşamasında ise P3, P4, P5 ve P6 primerleri kullanıldı (Tablo 3-8).
Tablo 3-8: İki aşamalı PZR için tasarlanan primer setleri

40
Primer Dizisi Ürün
1.
AŞAMA
PRİMER
SETİ
P1
(Forvard)
5’-GATCTCCATATTCCAGGCTTACACA-3’
453
bç P2
(Reverse)
5’-TATTGTTTGGGCATTGTAACCTTCT-3’
2.
AŞAMA
PRİMER
SETİ
Mutant
Spesifik
Primerler
P3
(Forvard)
5’-CCTCAGAACGTTGATGGCA-3’
279
bç P4
(Reverse)
5’-ATTGCTTTCCTTTTTCACAAGA-3’
Yabani
Tip
Spesifik
Primerler
P5
(Forvard)
5’-
AGCATTTGGTTTTAAATTATGGAGTATATG-
3’
229
bç
P6
(Reverse)
5’-GTTTTACTTACTCTCGTCTCCACAAAA-3’
4. 2 μl kalıp DNA, negatif kontrol tüpü haricindeki tüm tüplere dağıtıldı.
Negatif kontrol tüpüne de DNA yerine 2 μl steril su eklendi.
5. Allel spesifik nested PZR’nin 1. Aşaması için reaksiyon uygun
programda başlatıldı. (Tablo 3-9).
Tablo 3-9: İki aşamalı PZR’nin 1. aşama koşulları
94oC 1 dakika
95 oC 30 saniye
60 oC 30 saniye 35 Döngü

41
72 oC 30 saniye
72 oC 2 dakika
6. PZR’nin 1. aşamasından sonra elde edilen ürün, 1:40 oranında
seyreltilerek PZR’nin 2. aşamasında kullanıldı.
7. Tablo 3-10’da belirtilen miktarlarda olmak üzere yine DNA haricinde bir
karışım hazırlandı ve 18’er μl steril tüplere dağıtıldı ve seyreltilen
DNA’lardan 2’şer μl karışıma dağıtıldı. Negatif kontrol tüplerine 2 μl
steril su eklendi.
Tablo 3-10: Allel Spesifik Nested PZR 2. aşama bileşenleri
Bileşen Miktar (20 μl)
Sigma PZR Mix 10 μl
Primer 3 (Forward) 0,8 μl
Primer 4 (Reverse) 0,8 μl
Primer 5 (Forward) 0,8 μl
Primer 6 (Reverse) 0,8 μl
Kalıp DNA
(1. aşama PZR ürünü 1:40 seyreltik)
2 μl
H2O 20 μl’ye tamamlandı
8. 2. aşama uygun koşullarda gerçekleştirildi (Tablo 3-11).
Tablo 3-11: Allel spesifik Nested PZR’nin 2. aşama koşulları

42
94 oC 1 dakika
95 oC 30 saniye
59 oC 25 saniye 35 Döngü
72 oC 25 saniye
72 oC 2 dakika
3.2.7. Agaroz Jelde Görüntüleme
Reaksiyon sonrasında örnekler agaroz jelde yürütüldü ve görüntüleri alındı. Jele
yüklenen örneğin mutant alleli (JAK2V617F) taşıması durumunda 279 bç uzunluğunda
bant görülür, yabanil tip allel taşıması durumunda ise 229 bç uzunluğunda bant görülür.
3.2.7.1. Agaroz Jelin Hazırlanması
1 gram agaroz, 50 mL 1X TAE tamponuna eklenerek %2’ lik jel hazırlandı.
Agarozun TAE içerisinde tam olarak çözünebilmesi için karışım mikrodalga fırında
ısıtıldı. Tam olarak çözündükten sonra şişeye 2,5 μl etidyum bromür ilave edilerek
karıştırıldı. Karışım tarakların bulunduğu jel kasedine döküldü ve donup jel haline
gelmesi için bekletildi.
3.2.7.2. Jele Yükleme Ve Görüntüleme
Agaroz jel, kasette donduktan sonra taraklar çıkartıldı. Kasedin içindeki donmuş
jel yürütme tankına yerleştirildi ve kasede yürütme tamponu olan 1X TAE eklendi.
Nested pzr ürünlerinden 2’şer μl alındı ve jel yükleme tamponundan 6,5’er μl
eklendikten sonra jelde oluşan kuyulara yüklendi. Marker da yüklendikten sonra 45
dakika 65 voltta yürütüldü ve ardından görüntü U.V. ışık altında elde edildi.
3.2.8. RNA İzolasyonu
Sağlıklı kontrollerden ve MPN hastalarından alınan perifer kan örneklerinden
izole edilen MNC’lerden, CD34+ ve CD34- hücrelerden RNA izolasyonu yapılması
amacıyla ‘Jena Bioscience - Total RNA Purification Kit’ kullanıldı. Prosedür şu
şekildedir:

43
A. Örnek Hazırlama ve Hücre Lizizi
1. Hücre pelletine 500 μl Liziz Tamponu (2-Merkaptoetanol (ME)
eklenmiş) eklendi.
2. 10 saniye vorteklenerek ile örnek liziz edildi.
B. Kolon Aktivasyonu
1. Kolonlar 2 mL’lik toplama tüplerine yerleştirildiler.
2. Kolon içine 100 μl ‘Activation Buffer’ eklendi.
3. 10 000 g’de 30 saniye santrifüj edildi.
4. Altta toplanan sıvı atıldı.
5. Hemen sonraki adıma geçildi.
C. Kolona Örnek Yüklenmesi
1. Hazırlanan lizata 300 μl İzopropanol eklendi ve vortekslendi.
2. Karışım direkt olarak kolon içine aktarıldı.
3. 10 000 g’de 30 saniye santrüfüj edildi.
4. Alltaki sıvı atıldı.
D. Birinci Kolon Yıkaması
1. Kolona 700 μl ‘Primary Washing Buffer (ethanol eklenmiş)’
uygulandı.
2. 10 000 g’de 30 saniye santrifüj edildi.
3. Alttaki sıvı atıldı.
E. İkinci Kolon Yıkaması
1. Kolon içine 700 μl ‘Secondary Washing Buffer (ethanol eklenmiş)’
uygulandı.
2. 10 000 g’de 30 saniye santrifüj edildi.
3. Alttaki sıvı atıldı.
4. Kolon, etabolü uzaklaştırmak için, 10 000g’de 2 dakika yeniden
santrifüj edildi.

44
F. RNA’nın Elde Edilmesi
1. Kolon, Dnaz/Rnaz içermeyen bir mikrosantrifüj tüpüne yerleştirildi.
2. Kolon membranının merkezine 40 μl ‘Elution Buffer’ eklendi.
3. Oda sıcaklığında 1 dakika bekletildi.
4. RNA’yı elde etmek için 10 000 g’de 1 dakika santrifüj edildi.
5. Elde edilen RNA -20 ya da -80°C’de saklandı.
3.2.9. Ters Transkriptaz Polimeraz Zincir Reaksiyonu
Sağlıklı ve hasta MNC’lerinden ve CD34+ ve CD34- kök hücrelerinden izole
edilen RNA örneklerinden komplementer DNA (cDNA) elde edildi. cDNA elde etmek
amacıyla “Script cDNA Sentez Kiti (Jena Bioscience)” kullanılarak cDNA sentezi
yapıldı ve şu protokol izlendi:
1. DNaz/RNaz içermeyen steril PZR tüplerine Tablo 3-12’de belirtilen
maddeler, belirtilen miktarlarda eklenerek RNA/primer karışımı
hazırlandı. Tüm aşamalar buz üzerinde gerçekleştirildi.
2. Karışım, 70oC’ de 5 dakika inkübe edildikten sonra hemen buza alındı.
3. İnkübasyon esnasında, Tablo 3-13’de belirtilen bileşenler ile örnek
sayısına göre bir reaksiyon karışımı hazırlandı.
4. İnkübasyonun ardından primer/kalıp RNA karışımına, esnasında Tablo
3-13’e göre hazırlanmış olan reaksiyon karışımından 10’ar μl eklendi.
Toplamda 20 μl’lik bir karışım elde edildi.
Tablo 3-12: Kalıp RNA/Primer Karışımı
Bileşen Bileşenin Stok
Konsantrasyonu
Reaksiyon
Konsantrasyonu
Miktar
(10 μl)
H2O 10 μl’ye
tamamlanır.
Primer: Oligo- - 50 pmol (300ng) 0,5 μl

45
dT15-25 Random
Hexamer
50 pmol (100ng) 0,5 μl
Kalıp RNA - 55 ng X μl
Tablo 3-13: Reaksiyon Karışımı
Bileşen Bileşenin Stok
Konsantrasyonu
Reaksiyon
Konsantrasyonu
Miktar
(10 μl)
H2O 10 μl’ye
tamamlanır.
SCRIPT-RT
Tampon
Solüsyonu
5X 1X 4 μl
dNTP Karışımı Her biri için 10 mM Her biri için 500
μM
1 μl
DTT Solüsyonu 100 mM 5 mM 1 μl
RNaz inhibitörü 40 ünite/μl 40 ünite 1 μl
SCRIPT Ters
Transkriptaz
200 ünite/μl 100 ünite 0,5 μl
5. Bileşenleri belirtilen miktarlarda içeren PZR tüpleri, PZR cihazına
yerleştirildi ve Tablo 3-14’te belirtilen koşullarda reaksiyon uygulandı.
Tablo 3-14: cDNA Sentez Koşulları
+42oC 10 dakika

46
+50oC 50 dakika
+70oC 10 dakika
+4oC ∞
6. cDNA, direkt sonraki aşama için kullanıldı veya -20oC’de kısa süreli
saklandı.
3.2.10. Gerçek Zamanlı Kantitatif PZR (q-PCR)
CXCR3A, CXCR3B ve CXCL9 genlerinin Polisitemia Vera prognozunda etkili
olabileceği düşünüldüğünden, bu genlerin olası ekspresyon değişimlerinin hastalıkla
ilişkisini incelemek amacıyla ekspresyon analizleri yapıldı.
Gerçek zamanlı kantitatif PZR ile karşılaştırmalı ölçümler yapıldı. Prob olarak
önce Universal Probe Library (UPL) daha sonra SYBR Green kullanıldı. UPL problar,
5' ve 3' uçlarından sırasıyla Fluorescein (FAM) ve koyu bir boya olan ‘quencher’
(TAMRA) boyası ile işaretlenirler. qPCR problarının hibridizasyonunu gerektiren
özgünlüğü ve erime sıcaklığını (Tm) korumak için, ‘Kilitli Nükleik Asitler’ (LNA) her
bir UPL probunun dizisine dahil edilirler. LNA'lar, standart DNA nükleotidlerine
kıyasla yüksek bağlanma kuvvetlerine sahip DNA nükleotid analoglarıdır. 3’ ucunda
bulunan floresan yaymayan ve baskılayıcı olan TAMRA molekülü, 5’ ucunda bulunan
ve florasan ışıma yapabilen FAM boyasının sinyal oluşturmasına engel olmaktadır. Bu
sayede prob ışıma yapamamaktadır. Taq polimeraz, primerlerin bağlanmasıyla yeni
zincir oluşmaya başladığı zaman, 5'→3' yönünde uzama işlemini yaparken, ekzonükleaz
aktivitesi ile probu keser. FAM molekülünün TAMRA molekülünden ayrılması ile
floresan ışıma gerçekleşir. Amplifikasyon ürünü çoğaldıkça her döngüde floresan ışıma
miktarı da artarak devam eder. SYBR Green, DNA molekülleri arasına eklenerek çift
iplikçikli DNA moleküllerine bağlanan, yaygın olarak kullanılan bir floresan boyadır.
Kantitatif PZR'de kullanılır çünkü floresan ölçümü, DNA’nın ne kadar çoğaldığını
kısmi veya kesin olarak belirlemek için her amplifikasyon döngüsünün sonunda
yapılabilir.

47
Kantitatif PZR’de ekspresyon analizi amacıyla CD34+, CD34- ve mononükleer
hücrelerden elde edilen cDNA’lar kullanıldı. Ekspresyon analizleri, CXCR3A,
CXCR3B ve CXCL9 genlerinden her birine özgü tasarlanmış primerler,
UniversalProbeLibrary (UPL) problar (PZR karışımı olarak ‘Roche Light Cycler® 480
Probes Master (2x conc.)’ ve ayrıca SYBR Green kullanılarak yapılmıştır.
Elde edilen bulgular SYBR ile yapılan deneylerden elde edilmiştir.
qPCR uygulamasında izlenen adımlar şu şekildedir:
PZR için gerekli içeren bir reaksiyon karışımı, örnek miktarının %10’u
kadar fazla olacak şekilde hazırlandı (Tablo 3-15).
Ortak karışımda; prob (UPL veya SYBR Green), Roche Light Cycler®
480 Probes Master (UPL Prob için), primerler ve steril su kullanıldı.
Bileşenlerin konsantrasyonu Tablo 3-16’te gösterilmektedir.
Tablo 3-15: Gen ekspresyon analizleri için PZR bileşenleri
Bileşenler UPL SYBR
Roche Light Cycler® 480 Probes Master
Forward Primer 8 μl 7 μl
Reverse Primer Probes Master var Probes Master yok
Probe (UPL veya SYBR Green)
Su
cDNA 2 μl 3 μl
Tablo 3-16: Kantitatif gerçek zamanlı PZR Reaksiyon Bileşenleri
İçerik Konsantrasyon
(UPL)
Konsantrasyon
(SYBR)
Miktar
(UPL)
Miktar
(SYBR)
Son
Konsantrasyon
Son
Konsantrasyon

48
(UPL) (SYBR)
Roche
Light
Cycler®
480
Probes
Master
2X
-
5 μl
-
1X
-
Forward
Primer
100 μM 10 μM 0,8 μl 0,4 μl 200 nM 400 nM
Reverse
Primer
100 μM 10 μM 0,8 μl 0,4 μl 200 nM 400 nM
Prob
(UPL
veya
SYBR)
20 U 2X 0,2 μl 5 μl 0,04 U 1X
dH₂O - - 1,2 μl 1,2 μl - -
Toplam
Miktar
- - 8 μl 7 μl - -
Ekspresyon analizi için CXCR3A, CXCR3B ve CXCL9 genlerinin yanında
referans gen olarak GAPDH kullanıldı.
CXCR3A geni için, ENST00000373693.3 kodlu transkript varyantı kullanıldı.
Varyantın boyu 1624 bp olup 368 amino asitlik bir proteini kodlamaktadır. CXCR3A
gen dizisi referans alınarak sipariş edilen primerler kullanıldı. CXCR3A için sadece
SYBR Green probu kullanılarak ekspresyon sonuçları elde edildi.
CXCR3B geni için, ENST00000373691.4 kodlu transkript varyantı kullanıldı.
Varyantın boyu 1861 bp olup 415 amino asitlik bir proteini kodlamaktadır. Amplikon
boyutu 111 nükleotittir. CXCR3B gen dizisi referans alınarak sipariş edilen primerler

49
kullanıldı. SYBR Green ve 27 numaralı UPL prob kullanılarak ekspresyon sonuçları
elde edildi.
CXCL9 geni için, ENST00000264888.5 kodlu transkript varyantı kullanıldı.
Varyantın boyu 2740 bp olup 125 amino asitlik bir proteini kodlamaktadır. Amplikon
boyutu 73 nükleotittir. CXCL9 gen dizisi referans alınarak sipariş edilen primerler
kullanıldı. SYBR Green ve 4 numaralı UPL prob kullanılarak ekspresyon sonuçları elde
edildi.
GAPDH geni için, ENST00000229239.9 kodlu transkript varyantı kullanıldı.
Varyantın boyu 1875 bp olup 335 amino asitlik bir proteini kodlamaktadır. Amplikon
boyutu 66 nükleotittir. CXCL9 gen dizisi referans alınarak sipariş edilen primerler
kullanıldı. SYBR Green ve 60 numaralı UPL prob kullanılarak ekspresyon sonuçları
elde edildi.
CXCR3A, CXCR3B, CXCL9 ve GAPDH primer dizileri Tablo 3-17’ te
gösterilmektedir.
Tablo 3-17: CXCR3A, CXCR3B, CXCL9 ve GAPDH Primer Dizileri
Primerler Primer Dizileri
CXCR3A İleri Primer 5’ ACCCAGCAGCCAGAGCACC 3’
CXCR3A Geri Primer 5’ TCATAGGAAGAGCTGAAGTTCTCCA 3’
CXCR3B İleri Primer 5’ CAACCACAAGCACCAAAGC 3’
CXCR3B Geri Primer 5’ TCTTCTGCGTGATCCCATC 3’
CXCL9 İleri Primer 5’ CCTTAAACAATTTGCCCCAAG 3’
CXCL9 Geri Primer 5’ TTGAACTCCATTCTTCAGTGTAGC 3’
GAPDH İleri Primer 5’ AGCCACATCGCTCAGACAC 3’
GAPDH Geri Primer 5’ GCCCAATACGACCAAATCC 3’

50
3.2.11. Akım Sitometri Cihazında Antikor Hücre Yüzey Analizi
Sağlıklı kontrollerden ve hastalardan elde edilen MNC, CD34+ ve CD34- hücre
gruplarının yüzeylerinde CXCR3 reseptörünün durumu incelendi. İşlem için her bir
tüpte 50 000 - 100 000 hücre kullanıldı. Hücreler pellet haline getirildikten sonraki
adımlar şu şekildedir;
1. Pelletlere 500’er μl MACS eklendi ve santrifüj edilerek hücreler yıkandı.
Santrifüj işlemi 1800 rpm’de 5 dakika uygulandı.
2. Üst sıvı döküldü yeniden 500’er μl MACS ile sulandırılma işlemi
yapıldı.
3. Her bir hücre örneği için CXCR3 boyası içermeyen bir ‘boyasız’ tüp elde
edildi. Her bir boyasız hücre tipi için birer tane de CXCR3 boyalı tüp
hazırlandı.
4. Hücrelere CXCR3 boyası eklendikten sonra buzda ve karanlıkta olacak
şekilde 20 dakika bekletildi.
5. Ardından tüplere 500’er μl daha MACS eklenerek 1800 rpm’de 5 dakika
santrifüj edildi.
6. Üst sıvı atıldıktan sonra PI tamponundan 500’er μl eklendikten sonra
akım sitometri cihazında okutma işlemi gerçekleştirildi.
7. Cihazda ana hücre grubu belirlendikten sonra PI ile canlı-ölü hücrelerin
ayrımı sağlandı. Canlı hücrelerin bulunduğu bölge seçildikten sonra
CXCR3 pozitifliği için sonuçlar elde edildi.
3.2.12. CD34+ ve CD34+Lin- Hücrelere GM-CSF Uygulaması
Literatür taramalarında GM-CSF’in (Granülosit-Makrofaj Koloni Stimulasyon
Faktörü) CD34+ hücrelerde CXCR3 ekspresyonunu arttırdığı bilinmektedir (167). PV
hastalarında elde edilen CD34+ ve CD34+Lin- hücreler 2 gün boyunca GM-CSF ile
muamele edilmiştir. Uygulanan GM-CSF konsantrasyonu 10 ng/ml’dir. Besi yeri olarak
IMDM-%20 FBS kullanılmıştır. 6-kutulu tabağın her bir kuyusuna 3 mL besiyeri içinde
100.000-150.000 hücre eklenecek şekilde uygulandı. 2 gün sonunda toplanan
hücrelerden sırasıyla RNA izolasyonu, cDNA sentezi ve kantitatif PZR işlemleri
uygulandı. Bu işlem ayrıca hasta MNC ve kordon kanı hücreleri için uygulandı.

51
3.2.13. Ekspresyon Verilerinin İstatiksel Analizi
Ekspresyon analizi amacıyla uygulanan qPCR’de her bir örnek için triplike
çalışma gerçekleştirildi. PZR verimi 2 olarak belirlendi.
CXCR3A, CXCR3B ve CXCL9 gen ekspresyon düzeyleri GAPDH referans geni
kullanılarak araştırıldı. 2-ΔΔCt= 2-(CtHasta–CtReferans)-(CtSağlıklı-CtReferans) yöntemi ile hesaplandı
(168). Grafikleri oluşturma ve istatiksel analizler için GraphPad Prism 5.0 yazılımı
kullanıldı. Sağlıklı ve hasta örneklerin karşılaştırmalı mRNA düzeylerini incelemek için
Mann-Whitney U testi uydulandı. Mann-Whitney U testi, aynı popülasyondan gelen iki
örneği karşılaştırmak için kullanılan ve iki örneğin eşit olup olmadığını test etmek için
kullanılan, parametrik olmayan bir testtir.
Standart sapma (SD) değeri hesaplanırken “Mean ± SEM” yöntemi kıllanıldı.
Grafiklerde standart sapmanın ortalamaya yakın olması, ortalamadan sapma riskinin
düşük olduğu anlamına gelmektedir.

52
4. BULGULAR
4.1. DNA İzolasyon Sonuçları
Bu çalışmada toplam 31 hasta ve 10 kontrol kullanılmış ve bunların önce perifer
kan hücrelerinden mononükleer hücreler izole edilmiştir. Sonra kanser kök hücre
izolasyonları yapılmıştır. Sonra da GM-CSF uyarımı yapılmıştır. Hasta kanlarından elde
edilen MNC’lerden DNA izolasyonu yapılmıştır. NanoDrop ile yapılan DNA ölçümleri
Tablo 4-1’de göserilmiştir. Örneklerin ng/μl, 260/280 ve 260/230 değerleri tabloda
belirtilmiştir.
Tablo 4-1: Mononükleer Hücrelerden İzole Edilen DNA örneklerinin ölçüm değerleri
Örnek ng/μl 260/280 260/230
H8 77,8 1,88 0,60
H10 51,0 1,93 1,15
H11 31,8 1,82 0,88
H15 31,0 2,13 0,51
H17 122,7 1,87 1,88
H22 100,4 1,88 1,72
H23 311,9 1,87 1,31
H24 49,6 1,87 1,42
H25 252,6 1,86 2,05
H26 196,6 1,89 1,82
H28 104,3 1,83 0,32
H29 42,1 1,68 0,07
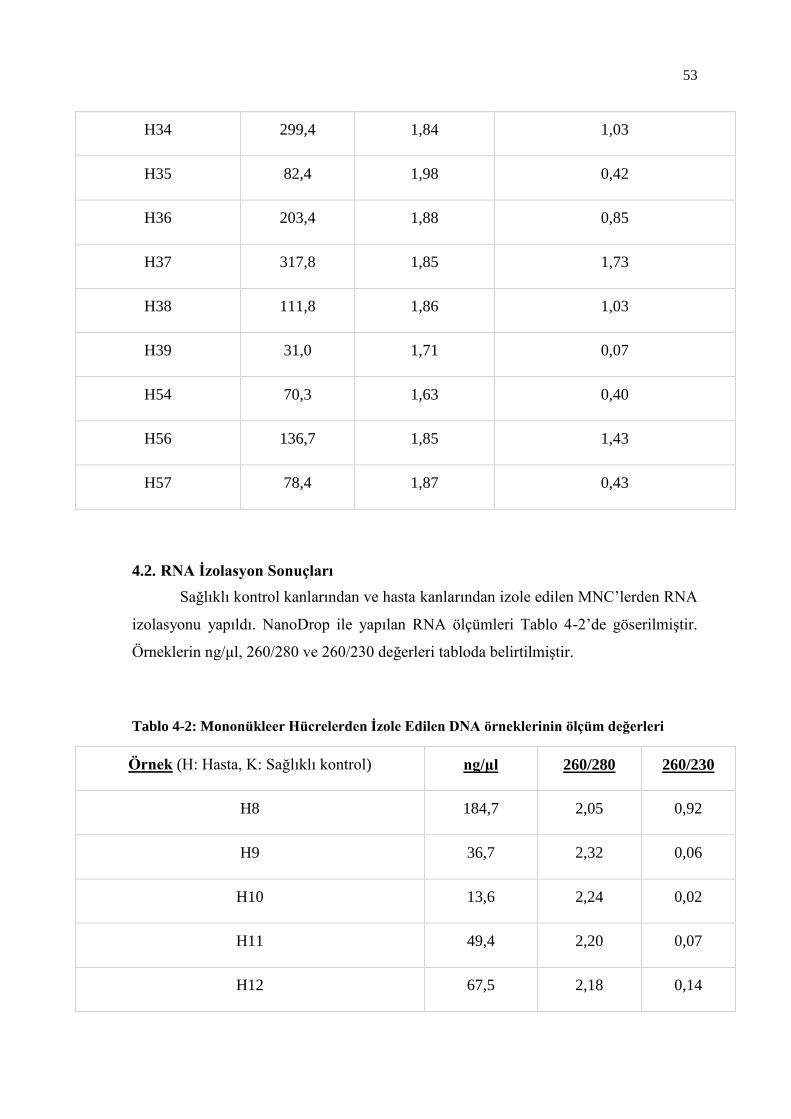
53
H34 299,4 1,84 1,03
H35 82,4 1,98 0,42
H36 203,4 1,88 0,85
H37 317,8 1,85 1,73
H38 111,8 1,86 1,03
H39 31,0 1,71 0,07
H54 70,3 1,63 0,40
H56 136,7 1,85 1,43
H57 78,4 1,87 0,43
4.2. RNA İzolasyon Sonuçları
Sağlıklı kontrol kanlarından ve hasta kanlarından izole edilen MNC’lerden RNA
izolasyonu yapıldı. NanoDrop ile yapılan RNA ölçümleri Tablo 4-2’de göserilmiştir.
Örneklerin ng/μl, 260/280 ve 260/230 değerleri tabloda belirtilmiştir.
Tablo 4-2: Mononükleer Hücrelerden İzole Edilen DNA örneklerinin ölçüm değerleri
Örnek (H: Hasta, K: Sağlıklı kontrol) ng/μl 260/280 260/230
H8 184,7 2,05 0,92
H9 36,7 2,32 0,06
H10 13,6 2,24 0,02
H11 49,4 2,20 0,07
H12 67,5 2,18 0,14

54
H13 40,5 1,99 0,07
H14 35,7 2,02 0,35
H15 43,8 2,14 0,08
H16 22,2 2,16 0,04
H17 27,4 2,11 0,06
H18 36,8 2,02 0,14
H19 48,9 2,00 0,07
H20 63,8 2,00 0,70
H21 49,7 2,03 0,31
H22 213,1 1,92 0,86
H23 14,7 2,00 0,08
H24 127,8 2,00 0,20
H25 57,9 2,00 0,18
H26 39,0 2,08 0,11
H28 48,5 2,05 0,22
H29 96,6 2,08 0,23
H34 17,0 2,17 0,04
H35 27,5 2,01 0,25
H36 113,6 2,08 0,25
H37 57,3 2,10 0,10

55
H38 73,8 1,97 0,49
H39 56,8 2,25 0,09
H43 73,6 2,03 1,26
H46 68,1 2,06 0,88
H54 44,8 1,76 0,30
H56 35,5 2,11 0,25
H57 40,0 2,18 0,09
K1 98,8 1,90 0,21
K2 520,4 2,08 0,59
K3 92,0 2,07 0,14
K4 163,6 2,09 0,32
K5 274,8 2,06 1,08
K6 103,9 1,95 0,23
K7 93,5 2,02 0,45
K8 78,8 2,06 0,19
K9 82,5 2,01 0,57
K10 198,9 2,03 0,32
4.3. İki Aşamalı Nested PZR Sonuçları
Hastaların mutasyon durumlarının tespit edilmesi için yapılan iki aşamalı allel
spesifik PZR sonuçları Tablo 4-3’te verilmiştir. Jel elektroforezi (Şekil 4-1) 229 ve 279
bç'lik Nested PZR ürün boyutunu göstermektedir. Çalışmada kullanılan hasta
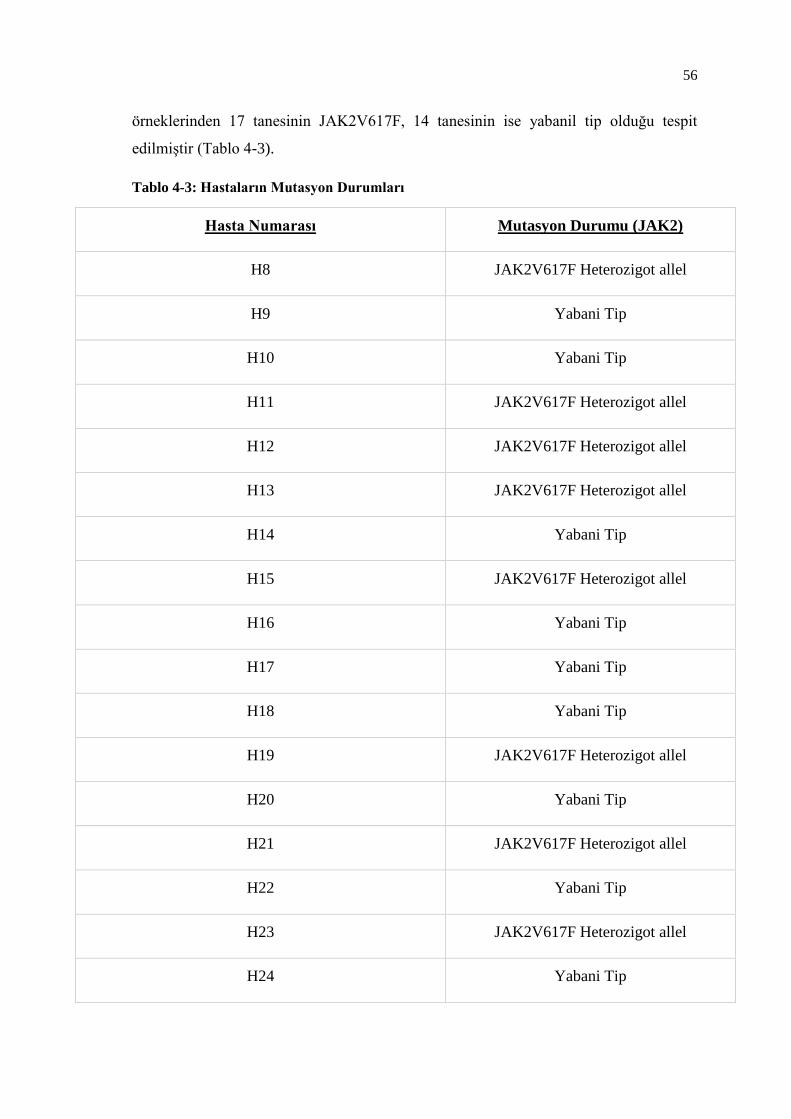
56
örneklerinden 17 tanesinin JAK2V617F, 14 tanesinin ise yabanil tip olduğu tespit
edilmiştir (Tablo 4-3).
Tablo 4-3: Hastaların Mutasyon Durumları
Hasta Numarası Mutasyon Durumu (JAK2)
H8 JAK2V617F Heterozigot allel
H9 Yabani Tip
H10 Yabani Tip
H11 JAK2V617F Heterozigot allel
H12 JAK2V617F Heterozigot allel
H13 JAK2V617F Heterozigot allel
H14 Yabani Tip
H15 JAK2V617F Heterozigot allel
H16 Yabani Tip
H17 Yabani Tip
H18 Yabani Tip
H19 JAK2V617F Heterozigot allel
H20 Yabani Tip
H21 JAK2V617F Heterozigot allel
H22 Yabani Tip
H23 JAK2V617F Heterozigot allel
H24 Yabani Tip

57
H25 Yabani Tip
H26 JAK2V617F Heterozigot allel
H28 JAK2V617F Heterozigot allel
H29 JAK2V617F Heterozigot allel
H34 Yabani Tip
H35 JAK2V617F Heterozigot allel
H36 Yabani Tip
H37 Yabani Tip
H38 JAK2V617F Heterozigot allel
H39 JAK2V617F Heterozigot allel
H43 JAK2V617F Heterozigot allel
H46 JAK2V617F Heterozigot allel
H54 JAK2V617F Heterozigot allel
H56 Yabani Tip
H57 -

58
Şekil 4-1: Alel Spesifik PZR Jel Görüntüsü
Bu deneyde pozitif mutant kontrol (JAK2V617F mutant) olarak H15 MNC’den, JAK2
pozitif yabanil tip kontrol olarak K562 hücre hattından elde edilen DNA örnekleri
kullanıldı. 279 bç’lik bandın varlığı mutasyon varlığını göstermektedir. 229 bç’lik
fragman yabanil tipi ifade etmektedir. H48, H49 ve H54 MNC’lerde 279 bç’lik parça
olduğunda bu hastaların JAK2V617F açısında mutant oldukları tespit edilmiştir. H52 ve
H53 MNC’lerde yabanil tip JAK2 alleline sahip bireylerdir. Kısaltmalar: MNC:
mononükleer hücre; H: hasta; bç: baz çifti
4.4. Gen İfadesi Sonuçları
Yapılan gen ifadesi çalışmaları öncelikle hasta ve kontrol toplam mononükleer
hücrelerinde gerçekleştirilmiştir. Literatürde belirtildiği üzere CXCR3’ün toplam MNC
ve izole edilmiş lenfosit gruplarının ekspresyonları arasında rölatif bir fark
olmadığından (169) lenfosit izolasyonu yapılmayıp CXCR3 ifadesi toplam MNC’de
tespit edilmiştir.
Yapılan CXCR3 gen ifade analizlerinde mononükleer hücrelerden elde edilen
RNA’lar kullanılarak CXCR3B geni için: 27 hasta, 10 kontrol (Şekil 4-2); CXCR3A geni
için: 10 hasta, 10 kontrol (Şekil 4-3) ve CXCL9 geni için ise 26 hasta 9 kontrol (Şekil 4-
4) kullanılarak mononükleer hücrelerle gerçekleştirilmiştir. Sonuçlar istatistiksel
anlamlılık bakımından P değerleriyle birlikte gösterilmiştir. Grafiklerde standart
sapmanın ortalamaya yakın olması, ortalamadan sapma riskinin düşük olduğu anlamına
gelmektedir.
Ardından kanser kök hücrelerinde CXCR3 ve CXCL9 gen ifadesi çalışmaları
gerçekleştirilmiştir. Toplam 4 hastadan elde edilen CD34+ ve CD34+/Lin- gruplarına ait
hücreler kullanılmıştır. Bu çalışmada CXCL9 ve CXCR3 anlatım seviyesi tespit
edilememiştir.

59
Şekil 4-2: CXCR3B gen ifadesi sonucu.
Hasta ve kontrol sütunları için “ortalama ± standart sapma” değerleri sırasıyla "8.366 ± 0.5493
N=27" ve "9.067 ± 0.8859 N=10" şeklindedir. P=0,51’dir.
Şekil 4-3: CXCR3A gen ifadesi sonucu.
Hasta ve kontrol sütunları için “ortalama ± standart sapma” değerleri sırasıyla "7.029 ± 0.4493
N=10" ve "5.088 ± 0.7116 N=10" şeklindedir. P =0,03’tür.
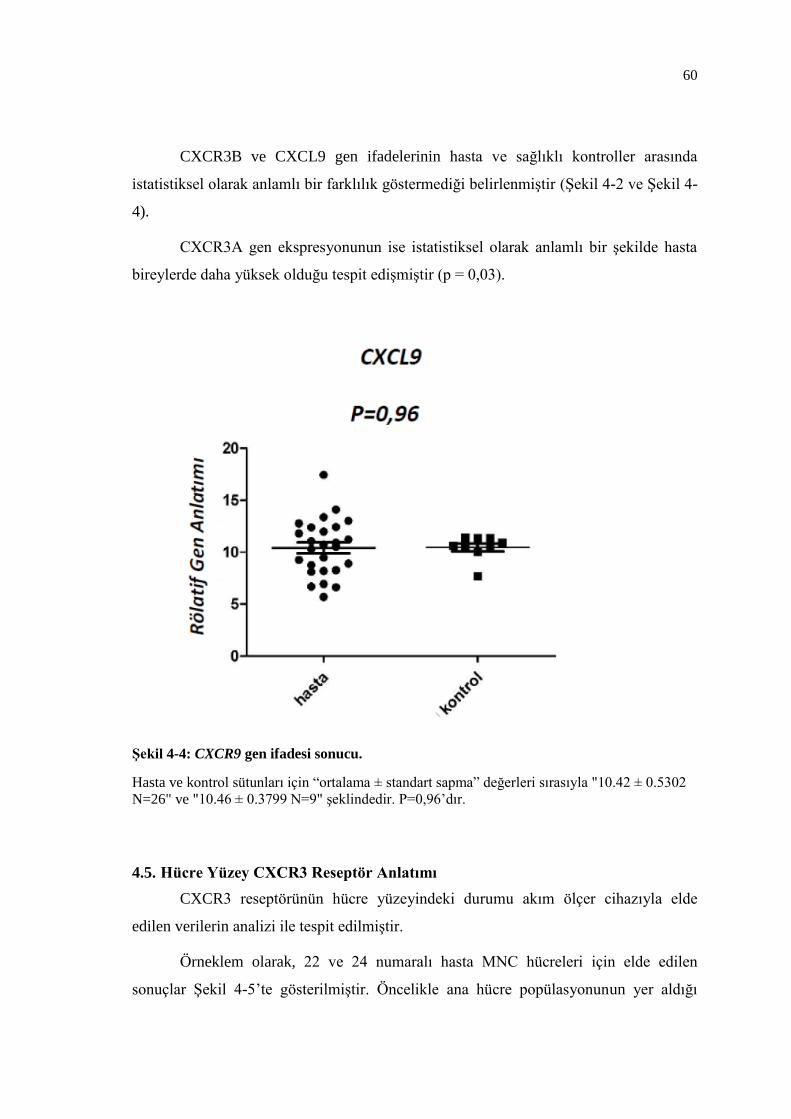
60
CXCR3B ve CXCL9 gen ifadelerinin hasta ve sağlıklı kontroller arasında
istatistiksel olarak anlamlı bir farklılık göstermediği belirlenmiştir (Şekil 4-2 ve Şekil 4-
4).
CXCR3A gen ekspresyonunun ise istatistiksel olarak anlamlı bir şekilde hasta
bireylerde daha yüksek olduğu tespit edişmiştir (p = 0,03).
Şekil 4-4: CXCR9 gen ifadesi sonucu.
Hasta ve kontrol sütunları için “ortalama ± standart sapma” değerleri sırasıyla "10.42 ± 0.5302
N=26" ve "10.46 ± 0.3799 N=9" şeklindedir. P=0,96’dır.
4.5. Hücre Yüzey CXCR3 Reseptör Anlatımı
CXCR3 reseptörünün hücre yüzeyindeki durumu akım ölçer cihazıyla elde
edilen verilerin analizi ile tespit edilmiştir.
Örneklem olarak, 22 ve 24 numaralı hasta MNC hücreleri için elde edilen
sonuçlar Şekil 4-5’te gösterilmiştir. Öncelikle ana hücre popülasyonunun yer aldığı
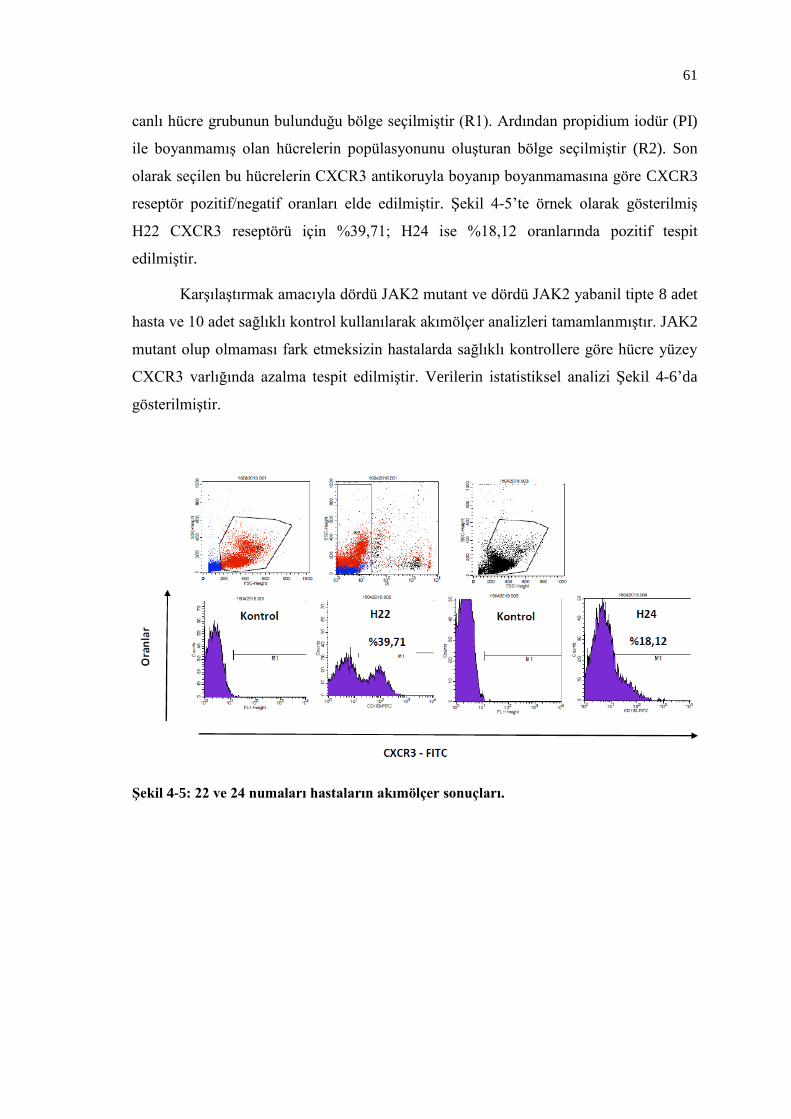
61
canlı hücre grubunun bulunduğu bölge seçilmiştir (R1). Ardından propidium iodür (PI)
ile boyanmamış olan hücrelerin popülasyonunu oluşturan bölge seçilmiştir (R2). Son
olarak seçilen bu hücrelerin CXCR3 antikoruyla boyanıp boyanmamasına göre CXCR3
reseptör pozitif/negatif oranları elde edilmiştir. Şekil 4-5’te örnek olarak gösterilmiş
H22 CXCR3 reseptörü için %39,71; H24 ise %18,12 oranlarında pozitif tespit
edilmiştir.
Karşılaştırmak amacıyla dördü JAK2 mutant ve dördü JAK2 yabanil tipte 8 adet
hasta ve 10 adet sağlıklı kontrol kullanılarak akımölçer analizleri tamamlanmıştır. JAK2
mutant olup olmaması fark etmeksizin hastalarda sağlıklı kontrollere göre hücre yüzey
CXCR3 varlığında azalma tespit edilmiştir. Verilerin istatistiksel analizi Şekil 4-6’da
gösterilmiştir.
Şekil 4-5: 22 ve 24 numaları hastaların akımölçer sonuçları.

62
Şekil 4-6: Akımölçer verilerinin istatistiksel analizi.
Hasta ve kontrol sütunları için “ortalama ± standart sapma” değerleri sırasıyla "19.33 ± 5.394
N=8" ve "36.78 ± 3.351 N=10" şeklindedir. P=0,01’dir.
4.5.1. CD34+ ve CD34+Lin- Hücrelere GM-CSF Uygulaması Sonuçları
Amaç CD34+ hücrelerde normal şartlarda var olmayan CXCR3 gen ifadesinin
PV hastalarında GM-CSF uygulaması ile artıp artmadığını incelemekti. Perifer kandan
elde edilen MNC’lerden CD34+ ve CD34+Lin- hücre popülasyonları GM-CSF ile
muamele edildikten sonra RNA izolasyonları ve kantitatif PZR işlemi gerçekleştirildi.
Bu deneyler 4 hastada ve sağlıklı kontrol olarak kordon kanı kullanılarak
gerçekleştirildi. Yapılan çalışmalarda CD34+ ve CD34+Lin- kök hücre
popülasyonlarında CXCR3 anlatımı tespit edilememiştir. GM-CSF uygulamasının da bu
anlatımı değiştirmediği görülmüştür.
Tüm bu çalışmalara ek olarak kullanılmış olan hücre izolasyon ve dondurma
tekniklerinin bir artefaktı sonucu CXCR3 gen anlatımında bir farklılık olabileceği test
edilmiştir. Bu çalışma için yeni izole edilmiş ve dondurulmuş hücrelerle yapılan deney

63
sonuçları karşılaştırılmış ve taze veya dondurulmuş hücrelerden elde edilen CXCR3 ve
CXCL9 gen anlatım sonuçları arasında herhangi bir fark tespit edilmemiştir.
Ayrıca, kantitatif PZR deneyleri iki tip prob ile ayrı reaksiyonlar halinde
gerçekleştirilmiştir. İlk deneyler UPL problar kullanılarak gerçekleştirilmiştir. UPL
probla yapılan deneylerde hiçbir hücre tipi için (MNC, CX34+/-) Ct değeri elde
edilememiştir. SYBR Green ile uygulanan deneylerde ise hasta ve sağlıklı MNC’ler için
Ct değeri elde edilebilmiş ve gen ekspresyon analizleri gerçekleştirilmiştir. Çift
iplikçikli DNA'nın arasına eklenen SYBR Green I gibi boyalar, daha kullanışlıdır. Her
bir hedef için sentezlenmesi gereken spesifik probların (UPL prob) aksine, tespit
araçları olarak boyaların (SYBR Green) kullanımı ve gen ifadesi tespiti daha kolaydır.

64
5. TARTIŞMA
Lenfosit trafiğinin temel itici gücü olan kemokinler, lenfosit yüzeylerinde
bulunan spesifik reseptörler aracılığıyla sinyallerini iletirler (170). MPN hastalarında,
sağlıklı bireylere kıyasla, çeşitli sitokin ve kemokinlerin plazma seviyelerinde yükselme
saptanmıştır. Bu bulgu, inflamatuar bir sürecin MPN'nin fizyopatolojisine dahil
olabileceğini, kemokinlerin rol oynayabileceğini ve hatta hematopoietik nişi
etkileyebileceğini göstermektedir. Çalışmamızda, hasta ve sağlıklı kontrol
örneklerinden elde edilen periferal kan mononükleer hücrelerde (PBMC) CXCR3A gen
ifadesi kıyaslandığında istatistiksel olarak anlamlı bir artış tespit edilmiştir (p=0,03).
Bunun yanında CXCR3B izoformu ve CXCL9 için gen ifadelerinde hasta ve sağlıklılar
arasında anlamlı bir farkın mevcut olmadığı sonucu elde edilmiştir (p değerleri sorasıyla
0,96 ve 0,51).
Mevcut literatüre göre, CXCR3A hücre proliferasyonu ve kemotaksiden
sorumluyken CXCR3B’nin büyümeyi inhibe ettiği gösterilmiştir (171-173). Ortamı
tümör büyümesi lehine korumak için, CXCR3A ve CXCR3B'nin ifadesi PBMC ve
tümör hücrelerinde farklı şekillerde düzenlenmektedir (174).
CXCR3A ekspresyonunun hastalarda artması ve CXCR3B ekspresyonunun
değişmemesi PV’de kemokin aracılı göç sinyalinin hatalı olmasına, PV-PBMC'nin aşırı
derecede artmış kemotaksisine neden olabilir. CXCR3A varyantının, tümör bölgesine
sitotoksik T hücresi/Doğal öldürücü hücreler (NK)/Doğal öldürücü T hücrelerinin daha
fazla migrasyonunu sağlamak için kemokinler ile daha fazla etkileşerek uyarılmak üzere
ekspresyon artışı gösterdiği kanıtlanmıştır (175).
CXCR3 ekspresyon profilindeki değişiklikler, kanser gelişiminin başlangıç
aşamaları veya metastatik hastalığa ilerlemesi ile ilişkili olabilir. CXCR3A, prostat,
yumurtalık, meme ve böbrek kanserinde tümör ilerlemesiyle ilişkilendirilmiştir
(140,151,176). Ayrıca, CXCL9, CXCL10 ve CXCL11'in, seçici olarak CXCR3A'yı
eksprese eden insan mast hücre hattı (HMC) üzerinde proliferasyonu uyardığı
gösterilmiştir (177,178).
PV’de inflamasyon sıklıkla görülen bir komplikasyondur. Kemokinlerin ve
reseptörlerinin tümör mikroçevresindeki malin transformasyondaki rolü hala

65
tartışmalıdır. Kronik inflamasyon, proliferasyona uğrayan hücrelerde, neoplastik
transformasyona yol açan DNA hasarına neden olur (179-181). İnflamasyon, kanserde
CXCR3’ün artmış ekspresyonuna; promotör demetilasyonunu veya intron CpG
bölgelerinin demetilasyonunu uyararak neden olabilir (182-184). Bu kalıcı
inflamasyonun ve CXCL10’un sürekli salgılanmasının, yapısal CXCR3A-kemokin
(CXCL10) sinyallemesi yoluyla malign neoplastik transformasyonu destekleyen epitel
hücrelerde CXCR3A ifadesini arttırabileceği gösterilmiştir (185). Bu nedenle
çalışmamızda CXCL9’dan sonra CXCL10’un da PV’deki durumunu araştırmak yeni
kapılar açabilir.
PV’de dikkat çeken önemli semptomlardan bir diğeri kendini ciltte gösterir.
Özellikle sıcak bir duşun ardından olmak üzere hastalarda rahatsız edici düzeyde bir
kaşıntı semptomu görülmektedir. Yapılan araştırmalarda, sedef hastalığına sahip
bireylerde IL-29’un CXCR3A taşıyan immün hücrelerin sedef hastalıklı deriye
gitmesine katkıda bulunduğu kanıtlanmıştır. IL-29, insan cildindeki hedef hücreler olan
keratinositlerde ve melanositlerde CXCR3A’nın ligandları CXCL9, CXCL10 ve
CXCL11'in ekspresyonunu uyarmaktadır. CXCL9 uyarımı oldukça zayıfken, CXCL10
indüksiyonu biraz daha kuvvetlidir, IL-29 ile muamele edilmiş keratinositlerde
CXCL11'in ekspresyon artışı en kuvvetli şekilde gözlenmiştir. Fare derisine enjekte
edildiğinde, IL-29’un mürin karşılığı, T-hücrelerinin lokal infıltrasyonuna, T-hücresinin
sitokin ekspresyonuna ve indüklenen iltihaplanma sonucunda deri kalınlaşmasına neden
olmuştur. Sedef hastalığına sahip bireylerin ciltlerinde CXCR3A ligand ekspresyonu,
CXCL11 kan plazma düzeylerinde anlamlı artış ile ve artmış CXCL10 seviyeleri ile
ilişkiliyken, IL-29 ile en düşük uyarımlı ligandın (CXCL9) kan konsantrasyonları
değiştirilmemiştir. Bu nedenle CXCL11’in kandaki seviyesi, IL-29 ve IFN-γ’nın lokal
aktivitesi için bir biyobelirteç olabilir (186). CXCL9’un çalışmamızda da değişmediği
sonucuyla paralel olmasıyla beraber, bu bilgiler birlikte düşünüldüğünde, çalışmamızda
CXCR3A ekspresyonunun hastalarda sağlıklı bireylere göre artmış olması ve CXCL9
ekspresyonunun değişmemiş olması CXCL11 ve CXCL10 ligandlarının ilişkili
olabileceğine ve araştırılmaları gerektiğine işaret etmektedir.
PV’de diğer bir semptom hepatomegalidir. Hepatomegali ile hepatik fibroz
arasındaki ilişki önceki çalışmalarda kanıtlanmıştır (187). CXCR3; fibroblastlar, epitel
ve endotel hücreler, düz kas hücreleri, aktive olmuş T lenfositleri ve dendritik hücreler

66
üzerinde ifade edilir. Bu hücreler ya CXCR3A’yı ya varyantı olan CXCR3B’yi ya da
her ikisinin dengeli bir kombinasyonunu ifade eder (92). CXCL4-CXCR3 yolağının
uyarılması, karaciğer fibrogenezine katkıda bulunan kemokinlerin de kaynağı olan
karaciğer hücrelerinin proliferasyonunu ve aktivasyonunu artırır (188). CXCL4
shRNA'sının, karaciğer hücrelerinde fibroz ile ilişkili proteinlerin (CXCR3, EGFR,
JAK2, STAT3 ve Collagen IV) ekspresyonunu anlamlı ölçüde azalttığı bulunmuştur
(189). Bu durum, hastalarda CXCR3A ekspresyon artışının PV hastalarında
hepatomegaliye katkı sağlıyor olabileceğini göstermektedir.
Hücre yüzey reseptör ekspresyonu sonuçlarımız ile CXCR3A ve CXCR3B
mRNA seviyesindeki ekspresyon sonuçlarımız arasında ters bir korelasyon tespit
edilmiştir. Yani CXCR3A mRNA seviyesini hastalarda artmış durumuda tespit edilmiş
ancak CXCR3 hücre yüzey reseptörünün hastalarda azalmış durumda olduğu sonucunu
elde edilmiştir. mRNA translasyon sonucu proteine çevrildiği için, genellikle mRNA ve
protein seviyeleri arasında bir tür korelasyon olduğu varsayılır. Genellikle mRNA
düzeyi ve protein seviyesi arasında tespit edilen zayıf korelasyonların nedenleri
mevcuttur ve bunlar karşılıklı olarak uyumlu olmayabilir. Bu ters korelasyonun
nedenleri arasında, henüz yeterince tanımlanamamış olan ve mRNA'yı proteine
çevirmeyle ilişkili çok sayıda, karmaşık ve çeşitli post-transkripsiyonel mekanizmaların
varlığı düşünülebilir. Ayrıca, proteinler in vivo yarı ömürlerinde büyük ölçüde farklılık
gösterebilirler. Ubikuitin-proteazom sistemi (UPS), ökaryotik hücrelerde hücre içi
proteinlerin bozulmasından sorumlu ana hücresel mekanizmadır ve proliferasyon, hücre
döngüsü kontrolü, transkripsiyonel düzenleme ve stres tepkisi dahil olmak üzere
hücresel süreçlerin düzenlenmesinde anahtar rol oynar (190). Ayrıca, hücre yüzey
reseptörleri bir şekilde kesime uğrayarak seruma karışmış olabilir. Son olarak hem
protein hem de mRNA deneylerinde teknik olarak net bir sonuç elde etme yeteneğimizi
sınırlandıran önemli zorluklar mevcutttur (191,192).
Nitekim yapılan deneylerde yaşamış olduğumuz teknik sorunlar arasında UPL
probun sonuç vermemesi gelmekteydi. UPL probun çalışmamasının sebebi primerler ile
probun birbirlerine karışmalarının kolaylığına sebep olması olabilir. Bu durum UPL
probların dezavantajlarından birisidir (193).
Çalışmamızda, CXCR3 mRNA ve protein düzeylerinde ekspresyon
seviyelerinin ters çıkmasının daha olası bir sebebi reseptör internalizasyonudur.

67
GPCR'nin internalizasyonu ve geri dönüşüm mekanizmaları hakkında elde edilen
bilgiler son zamanlarda artmıştır. İnternalizasyon bir kez gerçekleştiğinde, bir GPCR
için iki durumdan biri mümkündür; ligandın ayrılması ve fonksiyonel reseptörün
plazma membranına geri dönüşü veya bozunması gerçekleşebilir. Bir transfeksiyon
sistemiyle CXCR3'ün CXCL11 ile indüklenmiş internalizasyonunun meydana geldiği
bulunmuştur (194). PBMC’ler ile yapılan bir çalışmada CXCL11'in taze izole edilmiş
hücreler üzerindeki etkinliğinin CXCL9 ve CXCL10’a göre daha yüksek olduğu
belirgin bir şekilde tespit edilmiştir. Yani, CXCL11 CXCR3'ün internalizasyonunu
önemli bir şekilde uyarmıştır (152). Bu durum, çalışma sonuçlarımıza göre, CXCL11
ekspresyonunun PV’de artmış olabileceği şüphesini doğurmaktadır.
CXCR3B ifadesinin azalması tümör ilişkili-anjiyogenezde azalmayla ve tümöre
bağımlı immünite ile ilişkilidir. CXCR3B, anti-migrasyon ve anti-anjiyogenez
durumlarını sağladığı için her iki izoformu değil de CXCR3A'nın biyobelirteç olarak
hedeflenmesinin tercih edilebileceği ifade edilmiştir (151).
Yeni izole edilmiş kök hücrelerde CXCR3 mesajcı RNA’sının çok düşük
seviyelerde ifade edildiği önceki çalışmalarda gösterilmiştir. Ancak bu ifadenin GM-
CSF tarafından büyük ölçüde arttığı gerçek zamanlı kantitatif polimeraz zincir
reaksiyonu tekniği ile gösterilmiştir (167). Çalışmamızda GM-CSF uygulanan kök
hücrelerde CXCR3 ifadesinde değişiklik gerçekleşmemiştir. Bunun sebebinin son
tüketim tarihi geçmiş GM-CFS kullanımı olduğu düşünülmüştür.
Bu çalışma PV hastalığında CXCR3A/CXCR3B dengesinin bozulmuş olduğunu
gösteren literatürdeki ilk çalışmadır. Çalışma ayrıca bu reseptörlere özgün olarak
bağlanan kemokinlerden CXCL10 ve CXCL11’in de PV progresyonunda etkili
olabileceğine işaret etmektedir.
PV’de CXCR3A/CXCR3B dengesi, inflamasyon ve kanser progresyonu ile
ilişkili olabilir. PV’de artmış CXCR3A ifadesinin ve azalmış CXCR3B ifadesinin
altında yatan mekanizma ile kanser gelişimi ve progresyonundaki rollerini anlamak için
ilave çalışmalar gerekmektedir.

68
KAYNAKLAR
1. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, ve
ark. The 2008 revision of the World Health Organization (WHO) classification
of myeloid neoplasms and acute leukemia: rationale and important changes.
Blood 2009; 114(5): 937-951.
2. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, ve ark.
The 2016 revision to the World Health Organization (WHO) classification of
myeloid neoplasms and acute leukemia. Blood 2016: blood-2016-2003-643544.
3. Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of
classical myeloproliferative neoplasms. Blood 2017; 129(6): 667-679.
4. Geyer J, Orazi A. Myeloproliferative neoplasms (BCR‐ABL1 negative) and
myelodysplastic/myeloproliferative neoplasms: current diagnostic principles and
upcoming updates. International journal of laboratory hematology 2016;
38(S1): 12-19.
5. Kaplan JB, Stein BL, McMahon B, Giles FJ, Platanias LC. Evolving therapeutic
strategies for the classic Philadelphia-negative myeloproliferative neoplasms.
EBioMedicine 2016; 3: 17-25.
6. Geyer HL, Mesa RA. Therapy for myeloproliferative neoplasms: when, which
agent, and how? Blood 2014; 124(24): 3529-3537.
7. Tefferi A. Mutations galore in myeloproliferative neoplasms: would the real
Spartacus please stand up? : Nature Publishing Group; 2011.
8. Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD,
ve ark. Somatic mutations of calreticulin in myeloproliferative neoplasms. New
England Journal of Medicine 2013; 369(25): 2379-2390.
9. Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, ve ark.
Somatic CALR mutations in myeloproliferative neoplasms with nonmutated
JAK2. New England Journal of Medicine 2013; 369(25): 2391-2405.
10. Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, ve ark.
MPLW515L is a novel somatic activating mutation in myelofibrosis with
myeloid metaplasia. PLoS medicine 2006; 3(7): e270.
11. Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, ve ark.
MPL515 mutations in myeloproliferative and other myeloid disorders: a study of
1182 patients. Blood 2006; 108(10): 3472-3476.
12. Emanuel RM, Dueck AC, Geyer HL, Kiladjian J-J, Slot S, Zweegman S, ve ark.
Myeloproliferative neoplasm (MPN) symptom assessment form total symptom
score: prospective international assessment of an abbreviated symptom burden
scoring system among patients with MPNs. Journal of clinical oncology 2012;
30(33): 4098.
13. Geyer HL, Scherber RM, Dueck AC, Kiladjian J-J, Xiao Z, Slot S, ve ark.
Distinct clustering of symptomatic burden among myeloproliferative neoplasm

69
patients: retrospective assessment in 1470 patients. Blood 2014; 123(24): 3803-
3810.
14. Barbui T, Thiele J, Passamonti F, Rumi E, Boveri E, Ruggeri M, ve ark.
Survival and disease progression in essential thrombocythemia are significantly
influenced by accurate morphologic diagnosis: an international study. Journal of
Clinical Oncology 2011; 29(23): 3179-3184.
15. Tefferi A, Rumi E, Finazzi G, Gisslinger H, Vannucchi A, Rodeghiero F, ve ark.
Survival and prognosis among 1545 patients with contemporary polycythemia
vera: an international study. Leukemia 2013; 27(9): 1874.
16. Gangat N, Caramazza D, Vaidya R, George G, Begna K, Schwager S, ve ark.
DIPSS-plus: A refined dynamic international prognostic scoring system (DIPSS)
for primary myelofibrosis that incorporates karyotype, platelet count and
transfusion status. Am Soc Hematology; 2010.
17. Passamonti F, Cervantes F, Vannucchi AM, Morra E, Rumi E, Pereira A, ve ark.
A dynamic prognostic model to predict survival in primary myelofibrosis: a
study by the IWG-MRT (International Working Group for Myeloproliferative
Neoplasms Research and Treatment). Blood 2010; 115(9): 1703-1708.
18. Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, ve ark.
New prognostic scoring system for primary myelofibrosis based on a study of
the International Working Group for Myelofibrosis Research and Treatment.
Blood 2009; 113(13): 2895-2901.
19. Kaifie A, Kirschner M, Wolf D, Maintz C, Hänel M, Gattermann N, ve ark.
Bleeding, thrombosis, and anticoagulation in myeloproliferative neoplasms
(MPN): analysis from the German SAL-MPN-registry. Journal of hematology &
oncology 2016; 9(1): 18.
20. Mesa R, Miller CB, Thyne M, Mangan J, Goldberger S, Fazal S, ve ark.
Myeloproliferative neoplasms (MPNs) have a significant impact on patients’
overall health and productivity: the MPN Landmark survey. BMC cancer 2016;
16(1): 167.
21. Scherber R, Dueck AC, Johansson P, Barbui T, Barosi G, Vannucchi AM, ve
ark. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-
SAF): international prospective validation and reliability trial in 402 patients.
Blood 2011: blood-2011-2001-328955.
22. Johansson P, Mesa R, Scherber R, Abelsson J, Samuelsson J, Birgegård G, ve
ark. Association between quality of life and clinical parameters in patients with
myeloproliferative neoplasms. Leukemia & lymphoma 2012; 53(3): 441-444.
23. Abelsson J, Andréasson B, Samuelsson J, Hultcrantz M, Ejerblad E, Johansson
B, ve ark. Patients with polycythemia vera have worst impairment of quality of
life among patients with newly diagnosed myeloproliferative neoplasms.
Leukemia & lymphoma 2013; 54(10): 2226-2230.
24. Mesa RA, Niblack J, Wadleigh M, Verstovsek S, Camoriano J, Barnes S, ve ark.
The burden of fatigue and quality of life in myeloproliferative disorders
(MPDs). Cancer 2007; 109(1): 68-76.

70
25. da Costa Cacemiro M, Cominal JG, Tognon R, de Souza Nunes N, Simões BP,
de Figueiredo-Pontes LL, ve ark. Philadelphia-negative myeloproliferative
neoplasms as disorders marked by cytokine modulation. Hematology,
Transfusion and Cell Therapy 2018.
26. Cruse JM. History of medicine: the metamorphosis of scientific medicine in the
ever-present past. Am J Med Sci 1999; 318(3): 171-180.
27. Sigerist HE. A History Of Medicine Volume 2. 1961.
28. FAHREUS R. The suspension-stability of the blood. Acta Med Scand. 1921; 55:
1-228.
29. Hirsch E. Generalized osteosclerosis with chronic polycythemia vera. Arch
Pathol 1935; 19: 91-97.
30. Vaughan JM, Harrison C. Leuco‐erythroblastic anæmia and myelosclerosis. The
Journal of Pathology 1939; 48(2): 339-352.
31. Rosenthal N, ERF LA. Clinical observations on osteopetrosis and myelofibrosis.
Archives of Internal Medicine 1943; 71(6): 793-813.
32. Heller EL, Lewisohn MG, Palin WE. Aleukemic myelosis: chronic nonleukemic
myelosis, agnogenic myeloid metaplasia, osteosclerosis, leuko-erythroblastic
anemia, and synonymous designations. The American journal of pathology
1947; 23(3): 327.
33. Dameshek W. Some speculations on the myeloproliferative syndromes. Blood
1951; 6(4): 372-375.
34. Tefferi A. The history of myeloproliferative disorders: before and after
Dameshek. Leukemia 2008; 22(1): 3.
35. Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR, ve ark. A
gain-of-function mutation of JAK2 in myeloproliferative disorders. New
England Journal of Medicine 2005; 352(17): 1779-1790.
36. Spivak JL. The chronic myeloproliferative disorders: clonality and clinical
heterogeneity. Paper presented at: Seminars in hematology2004.
37. James C, Ugo V, Le Couédic J-P, Staerk J, Delhommeau F, Lacout C, ve ark. A
unique clonal JAK2 mutation leading to constitutive signalling causes
polycythaemia vera. Nature 2005; 434(7037): 1144.
38. Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, ve ark.
Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential
thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer cell
2005; 7(4): 387-397.
39. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, ve ark.
Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative
disorders. The Lancet 2005; 365(9464): 1054-1061.
40. Levine RL, Loriaux M, Huntly BJ, Loh ML, Beran M, Stoffregen E, ve ark. The
JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia
and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic
lymphocytic leukemia. Blood 2005; 106(10): 3377-3379.

71
41. Hinds DA, Barnholt KE, Mesa RA, Kiefer AK, Do CB, Eriksson N, ve ark.
Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and
myeloproliferative neoplasms. Blood 2016; 128(8): 1121-1128.
42. Hirsch P, Mamez A, Belhocine R, Lapusan S, Tang R, Suner L, ve ark. Clonal
history of a cord blood donor cell leukemia with prenatal somatic JAK2 V617F
mutation. Leukemia 2016; 30(8): 1756.
43. McKerrell T, Park N, Moreno T, Grove CS, Ponstingl H, Stephens J, ve ark.
Leukemia-associated somatic mutations drive distinct patterns of age-related
clonal hemopoiesis. Cell reports 2015; 10(8): 1239-1245.
44. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, ve
ark. Clonal hematopoiesis of indeterminate potential and its distinction from
myelodysplastic syndromes. Blood 2015; 126(1): 9-16.
45. Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, ve ark. JAK2
exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. New
England Journal of Medicine 2007; 356(5): 459-468.
46. Levine RL, Gilliland DG. Myeloproliferative disorders. Blood 2008; 112(6):
2190-2198.
47. Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological
malignancies. Oncogene 2013; 32(21): 2601.
48. Bandaranayake RM, Ungureanu D, Shan Y, Shaw DE, Silvennoinen O, Hubbard
SR. Crystal structures of the JAK2 pseudokinase domain and the pathogenic
mutant V617F. Nature Structural and Molecular Biology 2012; 19(8): 754.
49. Shan Y, Gnanasambandan K, Ungureanu D, Kim ET, Hammarén H, Yamashita
K, ve ark. Molecular basis for pseudokinase-dependent autoinhibition of JAK2
tyrosine kinase. Nature Structural and Molecular Biology 2014; 21(7): 579.
50. Leroy E, Dusa A, Colau D, Motamedi A, Cahu X, Mouton C, ve ark.
Uncoupling JAK2 V617F activation from cytokine-induced signalling by
modulation of JH2 αC helix. Biochemical Journal 2016; 473(11): 1579-1591.
51. Lu X, Levine R, Tong W, Wernig G, Pikman Y, Zarnegar S, ve ark. Expression
of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated
transformation. Proceedings of the National Academy of Sciences of the United
States of America 2005; 102(52): 18962-18967.
52. Ugo V, Marzac C, Teyssandier I, Larbret F, Lécluse Y, Debili N, ve ark.
Multiple signaling pathways are involved in erythropoietin-independent
differentiation of erythroid progenitors in polycythemia vera. Experimental
hematology 2004; 32(2): 179-187.
53. LaFave LM, Levine RL. JAK2 the future: therapeutic strategies for JAK-
dependent malignancies. Trends in pharmacological sciences 2012; 33(11): 574-
582.
54. Cisowski J, Sayin V, Liu M, Karlsson C, Bergo M. Oncogene-induced
senescence underlies the mutual exclusive nature of oncogenic KRAS and
BRAF. Oncogene 2016; 35(10): 1328.

72
55. Tefferi A. Pathogenesis of myelofibrosis with myeloid metaplasia. Journal of
Clinical Oncology 2005; 23(33): 8520-8530.
56. Plo I. p53 at the crossroads of MPN treatment. Blood 2014; 124(5): 668-669.
57. Arranz L, Sánchez-Aguilera A, Martín-Pérez D, Isern J, Langa X, Tzankov A,
ve ark. Neuropathy of haematopoietic stem cell niche is essential for
myeloproliferative neoplasms. Nature 2014; 512(7512): 78.
58. Fleischman AG, Aichberger KJ, Luty SB, Bumm TG, Petersen CL, Doratotaj S,
ve ark. TNFα facilitates clonal expansion of JAK2V617F positive cells in
myeloproliferative neoplasms. Blood 2011; 118(24): 6392-6398.
59. Tapper W, Jones AV, Kralovics R, Harutyunyan AS, Zoi K, Leung W, ve ark.
Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to
myeloproliferative neoplasms. Nature communications 2015; 6: 6691.
60. Saliba J, Saint-Martin C, Di Stefano A, Lenglet G, Marty C, Keren B, ve ark.
Germline duplication of ATG2B and GSKIP predisposes to familial myeloid
malignancies. Nature genetics 2015; 47(10): 1131.
61. Harutyunyan AS, Giambruno R, Krendl C, Stukalov A, Klampfl T, Berg T, ve
ark. Germline RBBP6 mutations in familial myeloproliferative neoplasms.
Blood 2016; 127(3): 362-365.
62. Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines
and the expanding diversity of effector T cell lineages. Annual review of
immunology 2007; 25: 821-852.
63. Watford WT, Moriguchi M, Morinobu A, O'Shea JJ. The biology of IL-12:
coordinating innate and adaptive immune responses. Cytokine Growth Factor
Rev 2003; 14(5): 361-368.
64. Lackie J. A dictionary of biomedicine. Oxford University Press; 2010.
65. Schulte W, Bernhagen J, Bucala R. Cytokines in sepsis: potent
immunoregulators and potential therapeutic targets--an updated view. Mediators
of inflammation 2013; 2013: 165974.
66. Graves DT, Jiang Y. Chemokines, a family of chemotactic cytokines. Critical
reviews in oral biology and medicine : an official publication of the American
Association of Oral Biologists 1995; 6(2): 109-118.
67. Stow JL, Murray RZ. Intracellular trafficking and secretion of inflammatory
cytokines. Cytokine Growth Factor Rev 2013; 24(3): 227-239.
68. Van der Meide PH, Schellekens H. Cytokines and the immune response.
Biotherapy (Dordrecht, Netherlands) 1996; 8(3-4): 243-249.
69. Luster AD. Chemokines—chemotactic cytokines that mediate inflammation.
New England Journal of Medicine 1998; 338(7): 436-445.
70. Locati M, Massimo, Murphy M, Philip M. Chemokines and chemokine
receptors: biology and clinical relevance in inflammation and AIDS. Annual
review of medicine 1999; 50(1): 425-440.
71. Thelen M. Dancing to the tune of chemokines. Nature immunology 2001; 2(2):
129.

73
72. Thelen M, Stein JV. How chemokines invite leukocytes to dance. Nature
immunology 2008; 9(9): 953.
73. Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity 2012;
36(5): 705-716.
74. Blanchet X, Langer M, Weber C, Koenen RR, von Hundelshausen P. Touch of
chemokines. Frontiers in immunology 2012; 3: 175.
75. Romagnani P, Lasagni L, Annunziato F, Serio M, Romagnani S. CXC
chemokines: the regulatory link between inflammation and angiogenesis. Trends
in immunology 2004; 25(4): 201-209.
76. Russo RC, Garcia CC, Teixeira MM, Amaral FA. The CXCL8/IL-8 chemokine
family and its receptors in inflammatory diseases. Expert review of clinical
immunology 2014; 10(5): 593-619.
77. Keeley EC, Mehrad B, Strieter RM. Chemokines as mediators of tumor
angiogenesis and neovascularization. Experimental cell research 2011; 317(5):
685-690.
78. Bachelerie F, Ben-Baruch A, Burkhardt A, Combadiere C, Farber J, Graham G,
ve ark. Update on the extended family of chemokine receptors and introducing a
new nomenclature for atypical chemokine receptors. Pharmacol Rev 2013;
66(1): 71P-79.
79. Corsiero E, Nerviani A, Bombardieri M, Pitzalis C. Ectopic lymphoid structures:
powerhouse of autoimmunity. Frontiers in immunology 2016; 7: 430.
80. Opdenakker G, Proost P, Van Damme J. Microbiomic and posttranslational
modifications as preludes to autoimmune diseases. Trends in molecular
medicine 2016; 22(9): 746-757.
81. Steen A, Larsen O, Thiele S, Rosenkilde MM. Biased and g protein-independent
signaling of chemokine receptors. Frontiers in immunology 2014; 5: 277.
82. Mortier A, Van Damme J, Proost P. Overview of the mechanisms regulating
chemokine activity and availability. Immunology letters 2012; 145(1-2): 2-9.
83. Mortier A, Gouwy M, Van Damme J, Proost P. Effect of posttranslational
processing on the in vitro and in vivo activity of chemokines. Experimental cell
research 2011; 317(5): 642-654.
84. Moelants EA, Mortier A, Van Damme J, Proost P. In vivo regulation of
chemokine activity by post‐translational modification. Immunology & Cell
Biology 2013; 91(6): 402-407.
85. Metzemaekers M, Van Damme J, Mortier A, Proost P. Regulation of chemokine
activity–a focus on the role of dipeptidyl peptidase IV/CD26. Frontiers in
immunology 2016; 7: 483.
86. Allen SJ, Crown SE, Handel TM. Chemokine: receptor structure, interactions,
and antagonism. Annu. Rev. Immunol. 2007; 25: 787-820.
87. Moser B. Chemokines: role in immune cell traffic. European cytokine network
2003; 14(4): 204-210.

74
88. Moser B, Willimann K. Chemokines: role in inflammation and immune
surveillance. Annals of the rheumatic diseases 2004; 63(suppl 2): ii84-ii89.
89. Mantovani A. The chemokine system: redundancy for robust outputs.
Immunology today 1999; 20(6): 254-257.
90. Bacon K, Baggiolini M, Broxmeyer H, Horuk R, Lindley I, Mantovani A, ve
ark. Chemokine/chemokine receptor nomenclature. J. Leukoc. Biol 2001; 70:
465-466.
91. Metzemaekers M, Vanheule V, Janssens R, Struyf S, Proost P. Overview of the
mechanisms that may contribute to the non-redundant activities of interferon-
inducible CXCR3 ligands. Frontiers in Immunology 2017; 8: 1970.
92. Van Raemdonck K, Van den Steen PE, Liekens S, Van Damme J, Struyf S.
CXCR3 ligands in disease and therapy. Cytokine & growth factor reviews 2015;
26(3): 311-327.
93. Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and
antagonistic functions. Immunology & Cell Biology 2011; 89(2): 207-215.
94. Luster AD, Unkeless JC, Ravetch JV. γ-Interferon transcriptionally regulates an
early-response gene containing homology to platelet proteins. Nature 1985;
315(6021): 672.
95. Farber JM. A macrophage mRNA selectively induced by gamma-interferon
encodes a member of the platelet factor 4 family of cytokines. Proceedings of
the National Academy of Sciences 1990; 87(14): 5238-5242.
96. Lee H-H, Farber J. Localization of the gene for the human MIG cytokine on
chromosome 4q21 adjacent to INP10 reveals a chemokine “mini-cluster”.
Cytogenetic and Genome Research 1996; 74(4): 255-258.
97. Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, ve ark.
Chemokine receptor specific for IP10 and mig: structure, function, and
expression in activated T-lymphocytes. Journal of Experimental Medicine 1996;
184(3): 963-969.
98. Tensen CP, Flier J, van der Raaij-Helmer EM, Sampat-Sardjoepersad S, van der
Schors RC, Leurs R, ve ark. Human IP-9: A Keratinocyte-Derived High Affinity
CXC-Chemokine Ligand for the IP-10/Mig Receptor (CXCR3) 1. Journal of
investigative dermatology 1999; 112(5): 716-722.
99. Cole KE, Strick CA, Paradis TJ, Ogborne KT, Loetscher M, Gladue RP, ve ark.
Interferon–inducible T cell alpha chemoattractant (I-TAC): a novel Non-ELR
CXC Chemokine with potent activity on activated T cells through selective high
affinity binding to CXCR3. Journal of Experimental Medicine 1998; 187(12):
2009-2021.
100. Erdel M, Theurl M, Meyer M, Duba H-C, Utermann G, Werner-Felmayer G.
High-resolution mapping of the human 4q21 and the mouse 5E3 SCYB
chemokine cluster by fiber-fluorescence in situ hybridization. Immunogenetics
2001; 53(7): 611-615.
101. Loos T, Dekeyzer L, Struyf S, Schutyser E, Gijsbers K, Gouwy M, ve ark. TLR
ligands and cytokines induce CXCR3 ligands in endothelial cells: enhanced
CXCL9 in autoimmune arthritis. Laboratory Investigation 2006; 86(9): 902.

75
102. Proost P, Vynckier AK, Mahieu F, Put W, Grillet B, Struyf S, ve ark. Microbial
Toll‐like receptor ligands differentially regulate CXCL10/IP‐10 expression in
fibroblasts and mononuclear leukocytes in synergy with IFN‐γ and provide a
mechanism for enhanced synovial chemokine levels in septic arthritis. European
journal of immunology 2003; 33(11): 3146-3153.
103. Proost P, Verpoest S, Borne D, Van K, Schutyser E, Struyf S, ve ark. Synergistic
induction of CXCL9 and CXCL11 by Toll‐like receptor ligands and interferon‐γ
in fibroblasts correlates with elevated levels of CXCR3 ligands in septic arthritis
synovial fluids. Journal of leukocyte biology 2004; 75(5): 777-784.
104. Kaplan G, Luster AD, Hancock G, Cohn ZA. The expression of a gamma
interferon-induced protein (IP-10) in delayed immune responses in human skin.
Journal of Experimental Medicine 1987; 166(4): 1098-1108.
105. Zipfel P, Bialonski A, Skerka C. Induction of members of the IL-8/NAP-1 gene
family in human T lymphocytes is suppressed by cyclosporin A. Biochemical
and biophysical research communications 1991; 181(1): 179-183.
106. Wright TM, Farber J. 5'regulatory region of a novel cytokine gene mediates
selective activation by interferon gamma. Journal of Experimental Medicine
1991; 173(2): 417-422.
107. Wong P, Severns CW, Guyer NB, Wright TM. A unique palindromic element
mediates gamma interferon induction of mig gene expression. Molecular and
cellular biology 1994; 14(2): 914-922.
108. Ohmori Y, Hamilton TA. IL-4-induced STAT6 suppresses IFN-gamma-
stimulated STAT1-dependent transcription in mouse macrophages. The Journal
of immunology 1997; 159(11): 5474-5482.
109. Tensen CP, Flier J, Rampersad SS, Sampat-Sardjoepersad S, Scheper RJ,
Boorsma DM, ve ark. Genomic organization, sequence and transcriptional
regulation of the human CXCL 111 gene. Biochimica et Biophysica Acta (BBA)-
Gene Structure and Expression 1999; 1446(1-2): 167-172.
110. Ohmori Y, Hamilton TA. Cooperative interaction between interferon (IFN)
stimulus response element and kappa B sequence motifs controls IFN gamma-
and lipopolysaccharide-stimulated transcription from the murine IP-10
promoter. Journal of Biological Chemistry 1993; 268(9): 6677-6688.
111. Majumder S, Zhou LZ-H, Chaturvedi P, Babcock G, Aras S, Ransohoff RM.
p48/STAT-1α-containing complexes play a predominant role in induction of
IFN-γ-inducible protein, 10 kDa (IP-10) by IFN-γ alone or in synergy with TNF-
α. The Journal of Immunology 1998; 161(9): 4736-4744.
112. Rani MS, Foster GR, Leung S, Leaman D, Stark GR, Ransohoff RM.
Characterization of β-R1, a gene that is selectively induced by interferon β (IFN-
β) compared with IFN-α. Journal of Biological Chemistry 1996; 271(37):
22878-22884.
113. Proost P, Struyf S, Loos T, Gouwy M, Schutyser E, Conings R, ve ark.
Coexpression and interaction of CXCL10 and CD26 in mesenchymal cells by
synergising inflammatory cytokines: CXCL8 and CXCL10 are discriminative

76
markers for autoimmune arthropathies. Arthritis research & therapy 2006; 8(4):
R107.
114. Loetscher M, Loetscher P, Brass N, Meese E, Moser B. Lymphocyte‐specific
chemokine receptor CXCR3: regulation, chemokine binding and gene
localization. European journal of immunology 1998; 28(11): 3696-3705.
115. Sebastiani S, Allavena P, Albanesi C, Nasorri F, Bianchi G, Traidl C, ve ark.
Chemokine receptor expression and function in CD4+ T lymphocytes with
regulatory activity. The Journal of Immunology 2001; 166(2): 996-1002.
116. Wacleche VS, Goulet J-P, Gosselin A, Monteiro P, Soudeyns H, Fromentin R,
ve ark. New insights into the heterogeneity of Th17 subsets contributing to HIV-
1 persistence during antiretroviral therapy. Retrovirology 2016; 13(1): 59.
117. Abboud G, Desai P, Dastmalchi F, Stanfield J, Tahiliani V, Hutchinson TE, ve
ark. Tissue-specific programming of memory CD8 T cell subsets impacts
protection against lethal respiratory virus infection. Journal of Experimental
Medicine 2016; 213(13): 2897-2911.
118. Inngjerdingen M, Damaj B, Maghazachi AA. Expression and regulation of
chemokine receptors in human natural killer cells. Blood 2001; 97(2): 367-375.
119. Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, ve ark. The
chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with
certain inflammatory reactions. The Journal of clinical investigation 1998;
101(4): 746-754.
120. Suga H, Sugaya M, Miyagaki T, Ohmatsu H, Okochi H, Sato S. CXCR3
deficiency prolongs Th1-type contact hypersensitivity. The Journal of
Immunology 2013; 190(12): 6059-6070.
121. Hasegawa H, Inoue A, Kohno M, Lei J, Miyazaki T, Yoshie O, ve ark.
Therapeutic effect of CXCR3-expressing regulatory T cells on liver, lung and
intestinal damages in a murine acute GVHD model. Gene therapy 2008; 15(3):
171.
122. Mohan K, Cordeiro E, Vaci M, McMaster C, Issekutz TB. CXCR3 is required
for migration to dermal inflammation by normal and in vivo activated T cells:
differential requirements by CD4 and CD8 memory subsets. European journal
of immunology 2005; 35(6): 1702-1711.
123. Hickman HD, Reynoso GV, Ngudiankama BF, Cush SS, Gibbs J, Bennink JR,
ve ark. CXCR3 chemokine receptor enables local CD8+ T cell migration for the
destruction of virus-infected cells. Immunity 2015; 42(3): 524-537.
124. Groom JR, Luster AD. CXCR3 in T cell function. Experimental cell research
2011; 317(5): 620-631.
125. Rabin RL, Park MK, Liao F, Swofford R, Stephany D, Farber JM. Chemokine
receptor responses on T cells are achieved through regulation of both receptor
expression and signaling. The Journal of Immunology 1999; 162(7): 3840-3850.
126. Yamamoto J, Adachi Y, Onoue Y, Adachi YS, Okabe Y, Itazawa T, ve ark.
Differential expression of the chemokine receptors by the Th1‐and Th2‐type
effect or populations within circulating CD4+ T cells. Journal of leukocyte
biology 2000; 68(4): 568-574.

77
127. Kim CH, Rott L, Kunkel EJ, Genovese MC, Andrew DP, Wu L, ve ark. Rules of
chemokine receptor association with T cell polarization in vivo. The Journal of
clinical investigation 2001; 108(9): 1331-1339.
128. Patil RS, Shah SU, Shrikhande SV, Goel M, Dikshit RP, Chiplunkar SV. IL17
producing γδT cells induce angiogenesis and are associated with poor survival in
gallbladder cancer patients. International journal of cancer 2016; 139(4): 869-
881.
129. Poggi A, Carosio R, Fenoglio D, Brenci S, Murdaca G, Setti M, ve ark.
Migration of Vδ1 and Vδ2 T cells in response to CXCR3 and CXCR4 ligands in
healthy donors and HIV-1–infected patients: competition by HIV-1 Tat. Blood
2004; 103(6): 2205-2213.
130. Poggi A, Zancolli M, Catellani S, Borsellino G, Battistini L, Zocchi M.
Migratory Pathways of γδ T Cells and Response to CXCR3 and CXCR4
Ligands. Annals of the New York Academy of Sciences 2007; 1107(1): 68-78.
131. Graves CL, Li J, LaPato M, Shapiro MR, Glover SC, Wallet MA, ve ark.
Intestinal epithelial cell regulation of adaptive immune dysfunction in human
type 1 diabetes. Frontiers in immunology 2017; 7: 679.
132. Muehlinghaus G, Cigliano L, Huehn S, Peddinghaus A, Leyendeckers H, Hauser
AE, ve ark. Regulation of CXCR3 and CXCR4 expression during terminal
differentiation of memory B cells into plasma cells. Blood 2005; 105(10): 3965-
3971.
133. García-López MÁ, Sánchez-Madrid F, Rodríguez-Frade JM, Mellado M,
Acevedo A, García MI, ve ark. CXCR3 chemokine receptor distribution in
normal and inflamed tissues: expression on activated lymphocytes, endothelial
cells, and dendritic cells. Laboratory investigation 2001; 81(3): 409.
134. Goldberg S, Van Der Meer P, Hesselgesser J, Jaffer S, Kolson D, Albright A, ve
ark. CXCR3 expression in human central nervous system diseases.
Neuropathology and applied neurobiology 2001; 27(2): 127-138.
135. Jinquan T, Jing C, Jacobi HH, Reimert CM, Millner A, Quan S, ve ark. CXCR3
expression and activation of eosinophils: role of IFN-γ-inducible protein-10 and
monokine induced by IFN-γ. The Journal of Immunology 2000; 165(3): 1548-
1556.
136. Hartl D, Krauss-Etschmann S, Koller B, Hordijk PL, Kuijpers TW, Hoffmann F,
ve ark. Infiltrated neutrophils acquire novel chemokine receptor expression and
chemokine responsiveness in chronic inflammatory lung diseases. The Journal
of Immunology 2008; 181(11): 8053-8067.
137. Ichikawa A, Kuba K, Morita M, Chida S, Tezuka H, Hara H, ve ark. CXCL10-
CXCR3 enhances the development of neutrophil-mediated fulminant lung injury
of viral and nonviral origin. American journal of respiratory and critical care
medicine 2013; 187(1): 65-77.
138. Berchiche YA, Sakmar TP. CXC chemokine receptor 3 alternative splice
variants selectively activate different signaling pathways. Molecular
pharmacology 2016: mol. 116.105502.

78
139. Lasagni L, Francalanci M, Annunziato F, Lazzeri E, Giannini S, Cosmi L, ve
ark. An alternatively spliced variant of CXCR3 mediates the inhibition of
endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as
functional receptor for platelet factor 4. Journal of Experimental Medicine 2003;
197(11): 1537-1549.
140. Furuya M, Yoneyama T, Miyagi E, Tanaka R, Nagahama K, Miyagi Y, ve ark.
Differential expression patterns of CXCR3 variants and corresponding CXC
chemokines in clear cell ovarian cancers and endometriosis. Gynecologic
oncology 2011; 122(3): 648-655.
141. Korniejewska A, McKnight AJ, Johnson Z, Watson ML, Ward SG. Expression
and agonist responsiveness of CXCR3 variants in human T lymphocytes.
Immunology 2011; 132(4): 503-515.
142. Ehlert JE, Addison CA, Burdick MD, Kunkel SL, Strieter RM. Identification
and partial characterization of a variant of human CXCR3 generated by
posttranscriptional exon skipping. The Journal of Immunology 2004; 173(10):
6234-6240.
143. Zohar Y, Wildbaum G, Novak R, Salzman AL, Thelen M, Alon R, ve ark.
CXCL11-dependent induction of FOXP3-negative regulatory T cells suppresses
autoimmune encephalomyelitis. The Journal of clinical investigation 2014;
124(5): 2009-2022.
144. Struyf S, Salogni L, Burdick MD, Vandercappellen J, Gouwy M, Noppen S, ve
ark. Angiostatic and chemotactic activities of the CXC chemokine CXCL4L1
(platelet factor-4 variant) are mediated by CXCR3. Blood 2011; 117(2): 480-
488.
145. Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated
G1 arrest in human cancer cells. Cancer research 1995; 55(22): 5187-5190.
146. Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown J, ve ark.
Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science
1998; 282(5393): 1497-1501.
147. Lee T-H, Chuang L-Y, Hung W-C. Induction of p21 WAF1 expression via Sp1-
binding sites by tamoxifen in estrogen receptor-negative lung cancer cells.
Oncogene 2000; 19(33): 3766.
148. Struyf S, Burdick MD, Proost P, Van Damme J, Strieter RM. Platelets release
CXCL4L1, a nonallelic variant of the chemokine platelet factor-4/CXCL4 and
potent inhibitor of angiogenesis. Circulation research 2004; 95(9): 855-857.
149. Thompson BD, Jin Y, Wu KH, Colvin RA, Luster AD, Birnbaumer L, ve ark.
Inhibition of Gαi2 activation by Gαi3 in CXCR3-mediated signaling. Journal of
Biological Chemistry 2007; 282(13): 9547-9555.
150. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, ve ark. Specific
recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege
and predicts reduced survival. Nature medicine 2004; 10(9): 942.
151. Wu Q, Dhir R, Wells A. Altered CXCR3 isoform expression regulates prostate
cancer cell migration and invasion. Molecular cancer 2012; 11(1): 3.

79
152. Meiser A, Mueller A, Wise EL, McDonagh EM, Petit SJ, Saran N, ve ark. The
chemokine receptor CXCR3 is degraded following internalization and is
replenished at the cell surface by de novo synthesis of receptor. The Journal of
Immunology 2008; 180(10): 6713-6724.
153. Sauty A, Colvin RA, Wagner L, Rochat S, Spertini F, Luster AD. CXCR3
internalization following T cell-endothelial cell contact: preferential role of IFN-
inducible T cell α chemoattractant (CXCL11). The Journal of Immunology
2001; 167(12): 7084-7093.
154. Rajagopal S, Bassoni DL, Campbell JJ, Gerard NP, Gerard C, Wehrman TS.
Biased agonism as a mechanism for differential signaling by chemokine
receptors. Journal of Biological Chemistry 2013; 288(49): 35039-35048.
155. Smith JS, Alagesan P, Desai NK, Pack TF, Wu J-H, Inoue A, ve ark. CXC motif
chemokine receptor 3 splice variants differentially activate beta-arrestins to
regulate downstream signaling pathways. Molecular pharmacology 2017; 92(2):
136-150.
156. Angiolillo AL, Sgadari C, Taub DD, Liao F, Farber JM, Maheshwari S, ve ark.
Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in
vivo. Journal of Experimental Medicine 1995; 182(1): 155-162.
157. Strieter R, Kunkel S, Arenberg D, Burdick M, Polverini P. Interferon γ-
Inducible Protein-10 (IP-10), a Member of the CXC Chemokine Family, Is an
Inhibitor of Angiogenesis. Biochemical and biophysical research
communications 1995; 210(1): 51-57.
158. Proost P, Schutyser E, Menten P, Struyf S, Wuyts A, Opdenakker G, ve ark.
Amino-terminal truncation of CXCR3 agonists impairs receptor signaling and
lymphocyte chemotaxis, while preserving antiangiogenic properties. Blood
2001; 98(13): 3554-3561.
159. Maione TE, Gray GS, Petro J, Hunt AJ, Donner AL, Bauer SI, ve ark. Inhibition
of angiogenesis by recombinant human platelet factor-4 and related peptides.
Science 1990; 247(4938): 77-79.
160. Hensbergen PJ, Wijnands PGB, Schreurs MW, Scheper RJ, Willemze R, Tensen
CP. The CXCR3 targeting chemokine CXCL11 has potent antitumor activity in
vivo involving attraction of CD8+ T lymphocytes but not inhibition of
angiogenesis. Journal of Immunotherapy 2005; 28(4): 343-351.
161. Addison CL, Arenberg DA, Morris SB, Xue Y-Y, Burdick MD, Mulligan MS,
ve ark. The CXC chemokine, monokine induced by interferon-gamma, inhibits
non-small cell lung carcinoma tumor growth and metastasis. Human gene
therapy 2000; 11(2): 247-261.
162. Romagnani P, Annunziato F, Lasagni L, Lazzeri E, Beltrame C, Francalanci M,
ve ark. Cell cycle–dependent expression of CXC chemokine receptor 3 by
endothelial cells mediates angiostatic activity. The Journal of clinical
investigation 2001; 107(1): 53-63.
163. Aoki Y, Niihori T, Inoue S, Matsubara Y. Recent advances in RASopathies.
Journal of human genetics 2016; 61(1): 33-39.

80
164. Mondet J, Hussein K, Mossuz P. Circulating cytokine levels as markers of
inflammation in philadelphia negative myeloproliferative neoplasms: diagnostic
and prognostic interest. Mediators of inflammation 2015; 2015.
165. Fulop T, Larbi A, Douziech N, Levesque I, Varin A, Herbein G. Cytokine
receptor signalling and aging. Mechanisms of ageing and development 2006;
127(6): 526-537.
166. Shurin GV, Yurkovetsky ZR, Chatta GS, Tourkova IL, Shurin MR, Lokshin AE.
Dynamic alteration of soluble serum biomarkers in healthy aging. Cytokine
2007; 39(2): 123-129.
167. Jinquan T, Quan S, Jacobi HH, Jing C, Millner A, Jensen B, ve ark. CXC
chemokine receptor 3 expression on CD34+ hematopoietic progenitors from
human cord blood induced by granulocyte-macrophage colony-stimulating
factor: chemotaxis and adhesion induced by its ligands, interferon γ–inducible
protein 10 and monokine induced by interferon γ. Blood 2000; 96(4): 1230-
1238.
168. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-
time quantitative PCR and the 2− ΔΔCT method. methods 2001; 25(4): 402-408.
169. Callahan MK, Williams KA, Kivisäkk P, Pearce D, Stins MF, Ransohoff RM.
CXCR3 marks CD4+ memory T lymphocytes that are competent to migrate
across a human brain microvascular endothelial cell layer. Journal of
neuroimmunology 2004; 153(1): 150-157.
170. Springer TA. Traffic signals for lymphocyte recirculation and leukocyte
emigration: the multistep paradigm. Cell 1994; 76(2): 301-314.
171. Baggiolini M. Chemokines and leukocyte traffic. Nature 1998; 392(6676): 565.
172. Glaser J, Gonzalez R, Perreau VM, Cotman CW, Keirstead HS. Neutralization
of the chemokine CXCL10 enhances tissue sparing and angiogenesis following
spinal cord injury. Journal of neuroscience research 2004; 77(5): 701-708.
173. Kelsen SG, Aksoy MO, Yang Y, Shahabuddin S, Litvin J, Safadi F, ve ark. The
chemokine receptor CXCR3 and its splice variant are expressed in human
airway epithelial cells. American Journal of Physiology-Lung Cellular and
Molecular Physiology 2004; 287(3): L584-L591.
174. Chakraborty K, Bose A, Pal S, Chattopadhyay U, Baral R. Interferon-α2b
Restores the Impaired Chemotactic Activity of Peripheral Blood Mononuclear
Cells from Head and Neck Squamous Cell Carcinoma Patients by Modulating
CXC Receptor Ligand Interaction. Journal of Interferon & Cytokine Research
2007; 28(8): 487-500.
175. Chakraborty K, Bose A, Pal S, Sarkar K, Goswami S, Ghosh D, ve ark. Neem
leaf glycoprotein restores the impaired chemotactic activity of peripheral blood
mononuclear cells from head and neck squamous cell carcinoma patients by
maintaining CXCR3/CXCL10 balance. International immunopharmacology
2008; 8(2): 330-340.
176. Datta D, Contreras AG, Grimm M, Waaga-Gasser AM, Briscoe DM, Pal S.
Calcineurin inhibitors modulate CXCR3 splice variant expression and mediate

81
renal cancer progression. Journal of the American Society of Nephrology 2008;
19(12): 2437-2446.
177. Romagnani P, Beltrame C, Annunziato F, Lasagni L, Luconi M, Galli G, ve ark.
Role for interactions between IP-10/Mig and CXCR3 in proliferative
glomerulonephritis. Journal of the American Society of Nephrology 1999;
10(12): 2518-2526.
178. Romagnani P, Lazzeri E, Lasagni L, Mavilia C, Beltrame C, Francalanci M, ve
ark. IP-10 and Mig production by glomerular cells in human proliferative
glomerulonephritis and regulation by nitric oxide. Journal of the American
Society of Nephrology 2002; 13(1): 53-64.
179. Lu H, Ouyang W, Huang C. Inflammation, a key event in cancer development.
Molecular cancer research 2006; 4(4): 221-233.
180. Perwez Hussain S, Harris CC. Inflammation and cancer: an ancient link with
novel potentials. International journal of cancer 2007; 121(11): 2373-2380.
181. Maeda H, Akaike T. Nitric oxide and oxygen radicals in infection,
inflammation, and cancer. BIOCHEMISTRY C/C OF BIOKHIMIIA 1998; 63:
854-865.
182. Ma B, Khazali A, Wells A. CXCR3 in carcinoma progression. Histology and
histopathology 2015; 30(7): 781.
183. Lleo A, Zhang W, Zhao M, Tan Y, Bernuzzi F, Zhu B, ve ark. DNA methylation
profiling of the X chromosome reveals an aberrant demethylation on CXCR3
promoter in primary biliary cirrhosis. Clinical epigenetics 2015; 7(1): 61.
184. Kumar DSS, Wells A. CXCR3 epigenome switches splice variants in prostate
cancer. AACR; 2013.
185. Urra S, Fischer MC, Martínez JR, Véliz L, Orellana P, Solar A, ve ark.
Differential expression profile of CXCR3 splicing variants is associated with
thyroid neoplasia. Potential role in papillary thyroid carcinoma oncogenesis?
Oncotarget 2018; 9(2): 2445.
186. Witte E, Kokolakis G, Witte K, Warszawska K, Friedrich M, Christou D, ve ark.
Interleukin-29 induces epithelial production of CXCR3A ligands and T-cell
infiltration. Journal of Molecular Medicine 2016; 94(4): 391-400.
187. Walters JH, Mc GI. The mechanism of malarial hepatomegaly and its
relationship to hepatic fibrosis. Transactions of the Royal Society of Tropical
Medicine and Hygiene 1960; 54: 135-145.
188. Wasmuth H, Weiskirchen R. Pathogenesis of liver fibrosis: modulation of
stellate cells by chemokines. Zeitschrift fur Gastroenterologie 2010; 48(1): 38-
45.
189. Li J, Liu B, Shi Y, Xie K-L, Yin H-F, Yan L-n, ve ark. CXCL4 Contributes to
the Pathogenesis of Chronic Liver Allograft Dysfunction. Journal of
immunology research 2016; 2016.
190. Leestemaker Y, de Jong A, Witting KF, Penning R, Schuurman K, Rodenko B,
ve ark. Proteasome Activation by Small Molecules. Cell chemical biology 2017;
24(6): 725-736.e727.

82
191. Baldi P, Long AD. A Bayesian framework for the analysis of microarray
expression data: regularized t-test and statistical inferences of gene changes.
Bioinformatics 2001; 17(6): 509-519.
192. SZALLASI Z. Genetic network analysis in light of massively parallel biological
data acquisition. Biocomputing'99: World Scientific; 1999:5-16.
193. Lauriat T. Universal Probe Library From Roche. 2006; 26 Jun,
https://www.biocompare.com/Product-Reviews/40920-Universal-Probe-Library-
From-Roche/.
194. Colvin RA, Campanella GS, Sun J, Luster AD. Intracellular domains of CXCR3
that mediate CXCL9, CXCL10, and CXCL11 function. Journal of Biological
Chemistry 2004; 279(29): 30219-30227.

83
HAM VERİLER

84
FORMLAR

85
ETİK KURUL KARAR

86
PATENT HAKKI İZNİ

87
İNTİHAL RAPORU İLK SAYFASI
88
ÖZGEÇMİŞ
Kişisel Bilgiler
Adı Cemil Soyadı Altunay
Doğ.Yeri Antakya Doğ.Tar. 27.07.1992
Uyruğu T.C. TC Kim No 19697052728
Email [email protected] Tel +90 531 765 48 02
Eğitim Düzeyi
Mezun Olduğu Kurumun Adı Mez. Yılı
Doktora
Yük.Lis.
Lisans Gebze Teknik Üniversitesi 2015
Lise Selim Nevzat Şahin Anadolu Lisesi 2010
İş Deneyimi (Sondan geçmişe doğru sıralayın)
Görevi Kurum Süre (Yıl - Yıl)
1
. Stajyer Berlin Teknik Üniversitei 3 ay (2014)
2
. Stajyer
Max Delbrück Center for Molecular
Medicine 2 ay (2013)
3
. -
89
Yabancı
Dilleri
Okuduğunu
Anlama* Konuşma* Yazma*
YÖKDİL
Puanı
(Diğer)
Puanı
İngilizce İyi İyi İyi 90,00
*Çok iyi, iyi, orta, zayıf olarak değerlendirin
Sayısal Eşit Ağırlık Sözel
ALES Puanı 85,89623 85,29008 72,07582
(Diğer)
Puanı
Bilgisayar Bilgisi
Program Kullanma becerisi
Microsoft Office İyi
Yayınları/Tebligleri Sertifikaları/Ödülleri
- Basic Management Skills Symposium 07.2016
Basic Management Science
Training Programs: 1) Social Engineering I-II 2) Problem Solving Techniques 3)
Time Management 4) 9 Easy Rules for Successful in Business 5) Basic Management
Skills
-ITU BIOTECH ''16 International Congress on Biotechnology and Life Science
07.05.2016
Istanbul Technical University, Istanbul, Turkey
-5th Course in Next Generation Sequencing 04-06.05.2016
Europen School of Genetic Medicine - Istanbul University, Istanbul, Turkey
- Laboratory Processes Course in Biobanking 05.2016

90
Istanbul University - 05.2016
-İÜKÖK Stem Cell Symposium 02.04.2016-03.04.2016
Istanbul University - 02.04.2016-03.04.2016 (16 Hours)
-IX. IUGEN Molecular Biology and Genetics Students’ Winter School 24-
26.02.2012
Istanbul University, Istanbul, Turkey
Özel İlgi Alanları (Hobileri):

91





![T1 FH Tanisma ve Bilgilendirme.ppt [Uyumluluk Modu] · “bir komedi dizisi yapacağım, içerisinde şöyle karakterler olacak, şöyle olacak, ... (Patent alınabilir) Yani aynı](https://img.pdfslide.net/doc/110x75/5cc92fba88c99348378bd8db/t1-fh-tanisma-ve-uyumluluk-modu-bir-komedi-dizisi-yapacagim-icerisinde.jpg)






















