Embed Size (px)
Citation preview

Oligomeric interaction of the PapB transcriptionalregulator with the upstream activating region of piliadhesin gene promoters in Escherichia coli
Yan Xia, Kristina Forsman †, Jana Jass and Bernt EricUhlin *
Department of Microbiology, Umea University, S-90187Umea, Sweden.
Summary
Transcriptional regulation of the pap genes, whichencode fimbrial adhesins in uropathogenic Escheri-chia coli , depends on an upstream activating region.This region contains binding sites for a transcriptionfactor, PapB, which is a member of a growing familyof putative regulatory proteins found in several viru-lence-associated fimbrial gene systems. To assess thenature of the PapB binding sites, we studied differentnaturally occurring variants and a number of in vitroconstructed mutant binding sites. DNase I footprintinganalysis and visualization of the PapB–DNA complexby atomic force microscopy showed that the proteinoccupied a DNA region of more than 50 bp. PurifiedPapB protein was shown to recognize a motif includ-ing a 9 bp repeat sequence containing T/A triplets ata conserved position. PapB binding was affected bydistamycin, and the results were consistent with thepossibility that the binding to DNA occurred throughminor groove interaction. From these analyses andestimation of the relative number of PapB proteins perbinding site, we suggest that PapB binds the DNA inan oligomeric fashion and may function as an architec-tural factor in the transcriptional control of adhesinexpression.
Introduction
Expression of Pap pili in the uropathogenic Escherichiacoli strain J96 depends on transcription of the major papoperon, starting at the papB promoter, and the divergentpapI operon (Baga et al., 1985; Goransson et al., 1989a).Both operons require activation by the cAMP–CRP com-plex bound to a site within an upstream activating sequ-ence (UAS) between the two promoters (Goransson et al.,
1989b; Forsman et al., 1992). The papB gene product sti-mulates pap operon expression and acts as transcriptionalregulator of the pap promoters. When the amount of PapBis increased above a certain level, transcription from thepapB promoter is repressed. Therefore, PapB has an auto-regulatory function (Forsman et al., 1989). There are PapB-like proteins encoded by many fimbrial gene systems. SfaBis a positive regulator influencing the production of the Sfimbriae (Morschhauser et al., 1993). The products of thefanA and fanB genes both show resemblance to PapBand are suggested as acting as transcriptional anti-termi-nators involved in the control of K99 fimbriae production(Roosendaal et al., 1989). Biosynthesis of the E. coli CS31Asurface antigen is negatively controlled by the PapB homo-logue, ClpB, together with LRP (Christine, 1996). In the pefoperon, located on the 90 kb virulence plasmid in Salmo-nella typhimurium, the gene pefB is postulated to encodea PapB-like protein (Friedrich et al., 1993). Evidently, thisfamily of regulatory proteins occurs in several importantpathogens. However, the molecular targets and mode ofaction of the PapB-like proteins are so far largely unknown.
In vitro binding studies have shown that the PapB pro-tein interacts with three sequences (sites 1, 2 and 3):two within the papI–papB intercistronic region and onewithin the papB coding sequence (Forsman et al., 1989).The finding that the three sites vary in their affinity forPapB is consistent with the role of PapB in autoregulation.PapB binding to the high-affinity site (site 1) would activatetranscription, while binding to the low-affinity site, site 2(which overlaps the transcriptional start site), wouldrepress transcription (Forsman et al., 1989). In initial muta-genesis experiments, we found that the deletion of site 3 didnot abolish autoregulation by PapB (unpublished data). Site2 overlaps the promoter, and this sequence is likely to affectthe promoter function per se. The distance between thecentre of PapB binding site 1 and the transcriptional startsite for the papB operon is 260 bp. In contrast, most tran-scriptional activators bind closer to the promoter, aroundposition ¹40 to ¹60 (Collado–Vides et al., 1991). The dis-tance between the transcriptional start site for the papIoperon and the centre of PapB binding site 1 is 71 bp.Here, we report our studies on the PapB–site 1 interaction.We also investigated the in vitro PapB interaction withbinding sites from promoter regions of other pili–adhesinoperons. Based on the results obtained, we proposed
Molecular Microbiology (1998) 30(3), 513–523
Q 1998 Blackwell Science Ltd
Received 8 May, 1998; revised 24 July, 1998; accepted 25 July,1998. †Present address: Astra Hassle AB, Molecular Biology,S-43183 Molndal, Sweden. *For correspondence. E-mail [email protected]; Tel. (90) 785 6731; Fax (90) 772 630.

that PapB could recognize sequences containing 9 bprepeats and interacted with the DNA along the minor grooveas an oligomer.
Results
Binding sites for the PapB protein in different piliregulatory regions
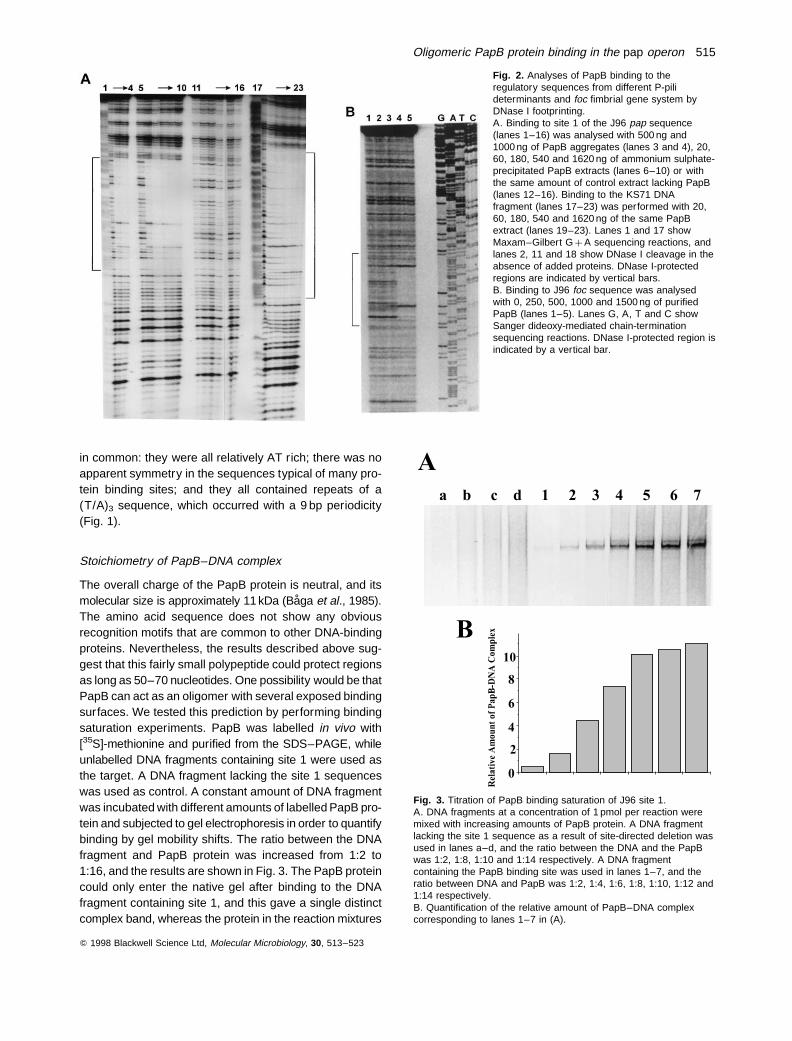
Earlier in vitro binding studies have shown that the PapBprotein can interact with several sequences, two withinthe papI–papB intercistronic region and one within thepapB coding sequence (Forsman et al., 1989). In thisstudy, we aimed at characterizing the mode of binding atthe high-affinity site located in between the two divergentpap promoters, which is referred to as the PapB bindingsite 1. The PapB protein has a tendency to form large, inso-luble aggregates, and we developed a procedure to purifythe protein in soluble form after denaturation–renaturationas described in Experimental procedures. To define thePapB recognition sequence further, we compared thecorresponding regulatory regions of pap genes from afew additional uropathogenic E. coli strains and the focregulatory region of J96 (Fig. 1). These sequences werehighly homologous to PapB binding site 1 of strain J96,
but also showed some interesting differences. The strainC1212 carries two pap gene clusters, denoted pap-17and pap-21, whose DNA sequences differ mainly in thesite 1 region (Blyn et al., 1989). The pap-17 site 1 is iden-tical to that of J96, while the pap-21 locus contains theadditional 9 bp sequence 58-CTGTGTTTG-38 (Fig. 1). TheDNA of a P-pili determinant from strain KS71 has an18 bp insertion (corresponding to two copies of the same9 bp sequence) in its site 1 (Rhen and Vaisanen-Rhen,1987; Fig. 1). We compared the binding of PapB proteinto these sites from J96 and KS71 by examining the pat-terns of protection from DNase I cleavage. The footprintanalysis showed that the 18 bp extra DNA in the KS71site 1 extended the PapB-protected region by 18 nucleo-tides compared with that of J96 (Fig. 2A).
The foc genes encode pili of the F1C serotype, and thefoc regulatory region shows homology to that of pap (VanDie et al., 1984; our unpublished data). The putative PapBbinding site was identified by DNase I footprinting in thefoc operon, and the extent of the protected sequencewas similar to that of pap site 1 of J96 (Fig. 2A and B).When comparing the nucleotide sequence in the focDNA with that of the three above-mentioned PapB bindingsites from different pap operons, we found some features
Q 1998 Blackwell Science Ltd, Molecular Microbiology, 30, 513–523
Fig. 1. Comparison of PapB-binding sequences from different pap and foc fimbrial gene systems. The 9 bp sequence motifs containing T/Atriplets are underlined by solid or dotted lines. The conserved nucleotide positions in the repeat sequences from each gene system are shown(shaded) in the corresponding compilation (only the top strand sequence shown) at the bottom.
514 Y. Xia, K. Forsman, J. Jass and B. E. Uhlin
in common: they were all relatively AT rich; there was noapparent symmetry in the sequences typical of many pro-tein binding sites; and they all contained repeats of a(T/A)3 sequence, which occurred with a 9 bp periodicity(Fig. 1).
Stoichiometry of PapB–DNA complex
The overall charge of the PapB protein is neutral, and itsmolecular size is approximately 11 kDa (Baga et al., 1985).The amino acid sequence does not show any obviousrecognition motifs that are common to other DNA-bindingproteins. Nevertheless, the results described above sug-gest that this fairly small polypeptide could protect regionsas long as 50–70 nucleotides. One possibility would be thatPapB can act as an oligomer with several exposed bindingsurfaces. We tested this prediction by performing bindingsaturation experiments. PapB was labelled in vivo with[35S]-methionine and purified from the SDS–PAGE, whileunlabelled DNA fragments containing site 1 were used asthe target. A DNA fragment lacking the site 1 sequenceswas used as control. A constant amount of DNA fragmentwas incubated with different amounts of labelled PapB pro-tein and subjected to gel electrophoresis in order to quantifybinding by gel mobility shifts. The ratio between the DNAfragment and PapB protein was increased from 1:2 to1:16, and the results are shown in Fig. 3. The PapB proteincould only enter the native gel after binding to the DNAfragment containing site 1, and this gave a single distinctcomplex band, whereas the protein in the reaction mixtures
Q 1998 Blackwell Science Ltd, Molecular Microbiology, 30, 513–523
Fig. 2. Analyses of PapB binding to theregulatory sequences from different P-pilideterminants and foc fimbrial gene system byDNase I footprinting.A. Binding to site 1 of the J96 pap sequence(lanes 1–16) was analysed with 500 ng and1000 ng of PapB aggregates (lanes 3 and 4), 20,60, 180, 540 and 1620 ng of ammonium sulphate-precipitated PapB extracts (lanes 6–10) or withthe same amount of control extract lacking PapB(lanes 12–16). Binding to the KS71 DNAfragment (lanes 17–23) was performed with 20,60, 180, 540 and 1620 ng of the same PapBextract (lanes 19–23). Lanes 1 and 17 showMaxam–Gilbert G þ A sequencing reactions, andlanes 2, 11 and 18 show DNase I cleavage in theabsence of added proteins. DNase I-protectedregions are indicated by vertical bars.B. Binding to J96 foc sequence was analysedwith 0, 250, 500, 1000 and 1500 ng of purifiedPapB (lanes 1–5). Lanes G, A, T and C showSanger dideoxy-mediated chain-terminationsequencing reactions. DNase I-protected region isindicated by a vertical bar.
Fig. 3. Titration of PapB binding saturation of J96 site 1.A. DNA fragments at a concentration of 1 pmol per reaction weremixed with increasing amounts of PapB protein. A DNA fragmentlacking the site 1 sequence as a result of site-directed deletion wasused in lanes a–d, and the ratio between the DNA and the PapBwas 1:2, 1:8, 1:10 and 1:14 respectively. A DNA fragmentcontaining the PapB binding site was used in lanes 1–7, and theratio between DNA and PapB was 1:2, 1:4, 1:6, 1:8, 1:10, 1:12 and1:14 respectively.B. Quantification of the relative amount of PapB–DNA complexcorresponding to lanes 1–7 in (A).
Oligomeric PapB protein binding in the pap operon 515
with the control fragment remained at the top of the gel.The binding to site 1 appeared to reach saturation at theratio of 1:8–1:10 between the DNA fragment and the PapBprotein. Similar results were obtained if we kept the amountof the labelled PapB constant while different amounts ofDNA fragment containing site 1 were used (data notshown). Taken together, our results indicated that the J96site 1 can be occupied by 8–10 PapB molecules. In thecase of KS71A, which has two more repeats of the (T/A)3sequence, four more PapB molecules were required tocover the binding site, as judged by the fact that saturationwas reached at a ratio of 1:14 (data not shown).
Oligomeric binding by PapB visualized by atomicforce microscopy
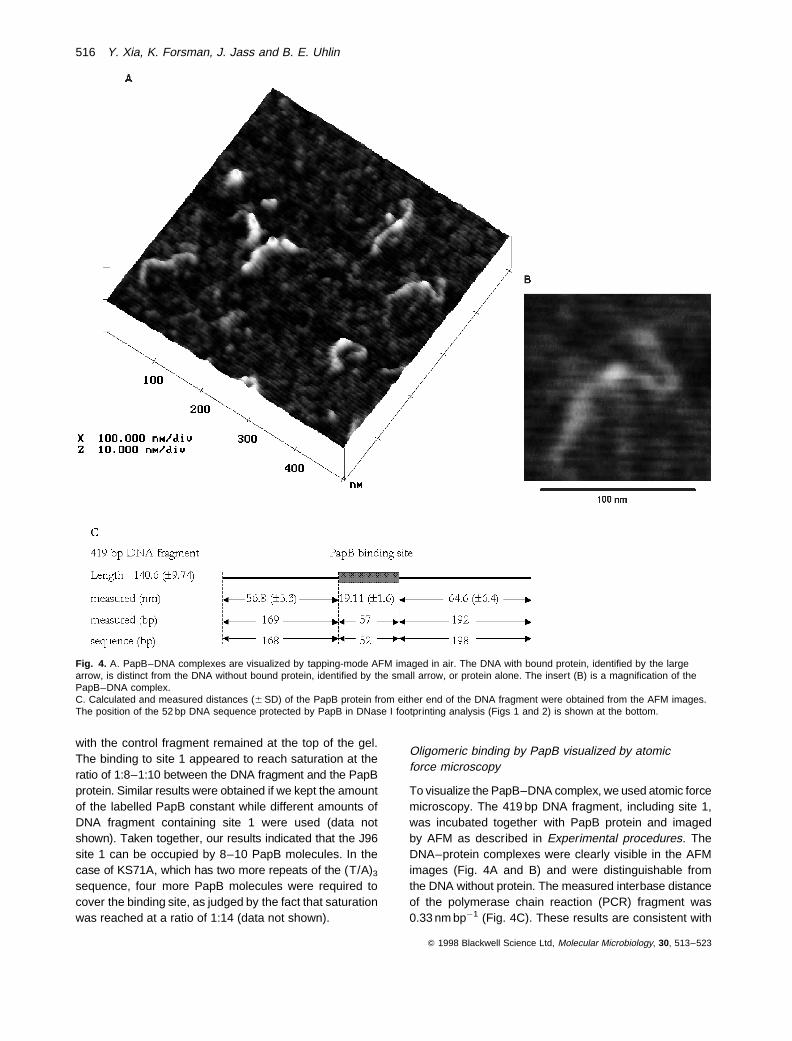
To visualize the PapB–DNA complex, we used atomic forcemicroscopy. The 419 bp DNA fragment, including site 1,was incubated together with PapB protein and imagedby AFM as described in Experimental procedures. TheDNA–protein complexes were clearly visible in the AFMimages (Fig. 4A and B) and were distinguishable fromthe DNA without protein. The measured interbase distanceof the polymerase chain reaction (PCR) fragment was0.33 nm bp¹1 (Fig. 4C). These results are consistent with
Q 1998 Blackwell Science Ltd, Molecular Microbiology, 30, 513–523
Fig. 4. A. PapB–DNA complexes are visualized by tapping-mode AFM imaged in air. The DNA with bound protein, identified by the largearrow, is distinct from the DNA without bound protein, identified by the small arrow, or protein alone. The insert (B) is a magnification of thePapB–DNA complex.C. Calculated and measured distances (6 SD) of the PapB protein from either end of the DNA fragment were obtained from the AFM images.The position of the 52 bp DNA sequence protected by PapB in DNase I footprinting analysis (Figs 1 and 2) is shown at the bottom.
516 Y. Xia, K. Forsman, J. Jass and B. E. Uhlin

the interbase distance reported by others for B-DNA (Gut-hold et al., 1994; Allison et al., 1996). The measured dis-tance of the PapB binding site from either end of the DNAfragment corresponded to the estimated basepair distancedetermined from the sequence. Some apparently randomnon-specific binding was also observed in the images. Asseen in Fig. 4C, the PapB protein was consistently foundapproximately 10 bp from the centre of the fragment andcovered 14% of the DNA length, corresponding to 57 bp(6 5). This suggests that the protein covers the completebinding site 1 identified by the DNase I footprinting data.The results were fully consistent with the findings describedabove, suggesting an oligomeric complex of PapB boundto the DNA.
Competition between PapB and distamycin for DNAbinding
To assess further how the PapB protein might interact withDNA, we challenged the PapB protein–DNA binding reac-tion with distamycin, a minor groove DNA-binding drug(Coll et al., 1987), and the major groove binder methylgreen (Zimmer and Wahnert, 1986). We found that dista-mycin by itself caused some retardation of the DNA frag-ments at concentrations above 10 mM (data not shown).The drug also altered the mobility of the PapB–DNA com-plexes at those concentrations (Fig. 5). In the presence of10–50 mM distamycin, we observed ‘supershifted’ bandscorresponding to complexes presumably containing DNAwith both PapB and distamycin bound. At even higher con-centrations (100–1000 mM), the pattern changed drasti-cally, and evidently the PapB binding was abolished, asjudged by the fact that the DNA fragments migrated aswhen only distamycin was present. Figure 5 also showsresults from the gel mobility shift assay of PapB bindingto the DNA fragment in the presence of methyl green. Incontrast to what was observed with distamycin, there wasno obvious effect on PapB binding obtained with methylgreen up to a concentration of 100 mM, despite the factthat distamycin and methyl green have similar DNA-binding
affinities (Kumar and Muniyappa, 1992). As a comparison,similar experiments were performed with H-NS, which wasconcluded to bind DNA through major groove interactions(Tippner et al., 1994). As expected, we found a significantinfluence on H-NS binding to DNA in the presence of100 mM methyl green (data not shown). Taken together,the results suggest that the PapB protein binding dependson the DNA conformation, and they were also consistentwith the possibility of interaction in the minor groove.
Mutations in the PapB binding site that attenuatePapB’s affinity for DNA
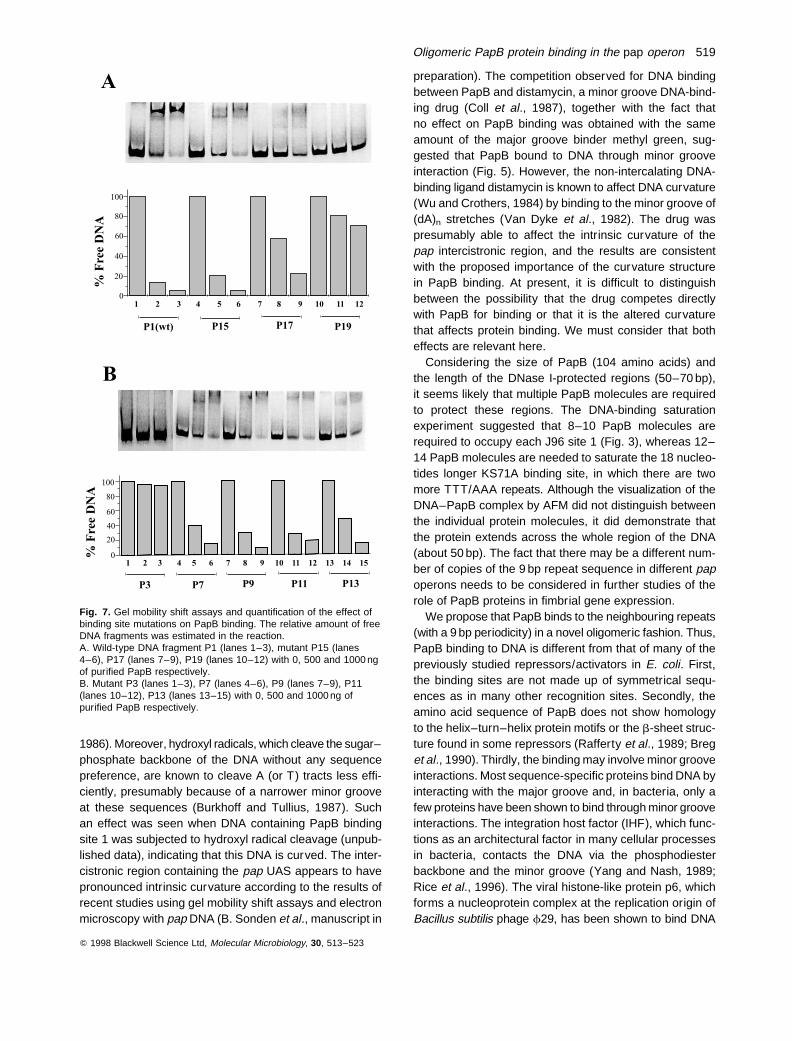
In order to assess further the nucleotide sequence require-ments for of PapB–DNA interactions, we constructed aseries of PapB binding site derivatives from J96 site 1.The mutants included derivatives in which the four con-served TTT/AAA sequences were mutated to CTC/GAGeither one by one or all in combination. Another group ofderivatives was based on ‘G’ insertions in which one G resi-due was inserted such that it altered the distance betweenthe repetitive TTT/AAA sequences (Fig. 6). The PapBprotein had a weaker binding ability for all the mutatedsequences, as shown by gel mobility shift assays (Fig. 7Aand B) and by DNase I footprinting analyses (data notshown). In contrast to the wild-type sequence, there wasusually no clearly distinguishable shifted band correspond-ing to a single PapB–DNA complex in the case of PapBbinding to the mutant sites. The quantitative analysisshowed that more of the DNA fragments containing mutantsite 1 sequences were left unbound in the presence of thesame amount of PapB when compared with the wild-typeDNA fragment (Fig. 7A and B). The PapB protein couldalmost not bind to the site 1 derivatives in which the fourTTT/AAA repeats were all mutated to four CTC/GAG orwhen four ‘G8 residues were inserted between the repeti-tive TTT/AAA sequences. The mutations also affected thein vivo expression as monitored by pap–lacZ fusion con-structs. The level of b-galactosidase expression from aseries of pap(I þ, B þ)A:lacZ plasmids carrying such muta-tions was measured upon growth in the logarithmic phase.All mutations reduced in vivo transcription according tothis analysis, and the resulting levels were between 14%and 42% of the wild-type level (Fig. 6). The lowest levelswere comparable with those of constructs lacking theentire site 1 region (i.e. pKF19) or carrying the papB struc-tural gene deletion (i.e. pHMG15). The in vitro and in vivoresults were thereby consistent and pointed to the impor-tance of the repeated (T/A)3 sequences occurring with9 bp periodicity for PapB binding.
Discussion
We investigated the mode of PapB interaction with site 1
Q 1998 Blackwell Science Ltd, Molecular Microbiology, 30, 513–523
Fig. 5. Gel mobility shift assay of the DNA and PapB complexes inthe presence of distamycin or methyl green at the differentconcentrations indicated.
Oligomeric PapB protein binding in the pap operon 517

in vitro and the role of this interaction in vivo in transcrip-tional activation of the major pap operon. The variablesize resulting from the insertion/deletion of a 9 bp sequ-ence of the PapB-binding sequences among the differentpap operons suggested that PapB interacted with severalrepeated target sequences within these regions. Therepeated nucleotide sequence motif TTT/AAA(N)6TTT/AAA(N)6TTT/AAA(N)6TTT/AAA was found in site 1, andthe results from mutagenesis of the DNA sequence suggestthat PapB binding depends on such repeats. We concludedthat PapB recognized a DNA structure including (T/A)3
repeats with 9 bp periodicity rather than a specific DNAsequence. This hypothesis was supported by the observa-tion that an 18 bp insertion into site 1 extended the PapBfootprints by the same number of nucleotides (i.e. strainKS71; Fig. 2A). The DNase I footprinting analysis ofPapB binding to the foc regulatory region (Fig. 2B) alsoshowed a 9 bp periodicity in terms of (T/A)3 sequences.
The average helical repeat of B-DNA in vitro is estimated
to be 10.5 bp per turn, although there may be differencesin vivo (Rhodes, and Klug, 1980; Bellomy and Records,1990). The helical repeat may be 9 bp per turn whenPapB is bound to site 1, or PapB may form a left-handedhelix around the DNA, similar to that proposed for theMetJ protein of E. coli (Rafferty et al., 1989). The HU pro-tein of E. coli is also suggested to bind to DNA as severaladjacent heterodimers occupying 9 bp each (Bonnefoy andRouviere-Yaniv, 1991).
Sequences that are AT rich seem to have rather specialproperties. For example, the A tracts of the oligonucleotide58-GGAAATTTCC-38 show a gradual compression of theminor groove in the 58 to 38 direction as determined byNMR (Katahira et al., 1990). AT-rich DNA is preferentiallypresent where the minor groove of nucleosomal DNA isplaced on the inside of the nucleosome complex (reviewedby Travers and Klug, 1987). A or T tracts longer than threenucleotides result in curved DNA when positioned on thesame side of the helix (Hagerman, 1985; Koo et al.,
Q 1998 Blackwell Science Ltd, Molecular Microbiology, 30, 513–523
Fig. 6. Outline of pap:lacZ fusions containing mutations in PapB binding site1 or in the papB gene. Transcripts originating from initiation atthe papB promoter are indicated by the horizontal arrow above the map. The sequences corresponding to wild type (pHMG1) and mutant(pXN1–6) variants of site 1 are indicated below with the repeated T triplets underlined and the basepair substitutions or G residue insertionsshown for each mutant. On the left are designations of the corresponding oligonucleotide primers designed for construction of wild-type andmutant DNA fragments for PapB-binding analyses (see Fig. 7). The deletion mutant derivative (pKF19) and the papB insertion mutantderivative (pHMG15) used as controls are shown at the bottom. The b-galactosidase specific activities are given as relative values with theactivity of the wild-type plasmid pHMG1 set to 1.0.
518 Y. Xia, K. Forsman, J. Jass and B. E. Uhlin
1986). Moreover, hydroxyl radicals, which cleave the sugar–phosphate backbone of the DNA without any sequencepreference, are known to cleave A (or T) tracts less effi-ciently, presumably because of a narrower minor grooveat these sequences (Burkhoff and Tullius, 1987). Suchan effect was seen when DNA containing PapB bindingsite 1 was subjected to hydroxyl radical cleavage (unpub-lished data), indicating that this DNA is curved. The inter-cistronic region containing the pap UAS appears to havepronounced intrinsic curvature according to the results ofrecent studies using gel mobility shift assays and electronmicroscopy with pap DNA (B. Sonden et al., manuscript in
preparation). The competition observed for DNA bindingbetween PapB and distamycin, a minor groove DNA-bind-ing drug (Coll et al., 1987), together with the fact thatno effect on PapB binding was obtained with the sameamount of the major groove binder methyl green, sug-gested that PapB bound to DNA through minor grooveinteraction (Fig. 5). However, the non-intercalating DNA-binding ligand distamycin is known to affect DNA curvature(Wu and Crothers, 1984) by binding to the minor groove of(dA)n stretches (Van Dyke et al., 1982). The drug waspresumably able to affect the intrinsic curvature of thepap intercistronic region, and the results are consistentwith the proposed importance of the curvature structurein PapB binding. At present, it is difficult to distinguishbetween the possibility that the drug competes directlywith PapB for binding or that it is the altered curvaturethat affects protein binding. We must consider that botheffects are relevant here.
Considering the size of PapB (104 amino acids) andthe length of the DNase I-protected regions (50–70 bp),it seems likely that multiple PapB molecules are requiredto protect these regions. The DNA-binding saturationexperiment suggested that 8–10 PapB molecules arerequired to occupy each J96 site 1 (Fig. 3), whereas 12–14 PapB molecules are needed to saturate the 18 nucleo-tides longer KS71A binding site, in which there are twomore TTT/AAA repeats. Although the visualization of theDNA–PapB complex by AFM did not distinguish betweenthe individual protein molecules, it did demonstrate thatthe protein extends across the whole region of the DNA(about 50 bp). The fact that there may be a different num-ber of copies of the 9 bp repeat sequence in different papoperons needs to be considered in further studies of therole of PapB proteins in fimbrial gene expression.
We propose that PapB binds to the neighbouring repeats(with a 9 bp periodicity) in a novel oligomeric fashion. Thus,PapB binding to DNA is different from that of many of thepreviously studied repressors/activators in E. coli. First,the binding sites are not made up of symmetrical sequ-ences as in many other recognition sites. Secondly, theamino acid sequence of PapB does not show homologyto the helix–turn–helix protein motifs or the b-sheet struc-ture found in some repressors (Rafferty et al., 1989; Breget al., 1990). Thirdly, the binding may involve minor grooveinteractions. Most sequence-specific proteins bind DNA byinteracting with the major groove and, in bacteria, only afew proteins have been shown to bind through minor grooveinteractions. The integration host factor (IHF), which func-tions as an architectural factor in many cellular processesin bacteria, contacts the DNA via the phosphodiesterbackbone and the minor groove (Yang and Nash, 1989;Rice et al., 1996). The viral histone-like protein p6, whichforms a nucleoprotein complex at the replication origin ofBacillus subtilis phage f29, has been shown to bind DNA
Q 1998 Blackwell Science Ltd, Molecular Microbiology, 30, 513–523
Fig. 7. Gel mobility shift assays and quantification of the effect ofbinding site mutations on PapB binding. The relative amount of freeDNA fragments was estimated in the reaction.A. Wild-type DNA fragment P1 (lanes 1–3), mutant P15 (lanes4–6), P17 (lanes 7–9), P19 (lanes 10–12) with 0, 500 and 1000 ngof purified PapB respectively.B. Mutant P3 (lanes 1–3), P7 (lanes 4–6), P9 (lanes 7–9), P11(lanes 10–12), P13 (lanes 13–15) with 0, 500 and 1000 ng ofpurified PapB respectively.
Oligomeric PapB protein binding in the pap operon 519

in a multimeric fashion through minor groove interaction(Serrano et al., 1990; 1994; Freire et al., 1994). Anotherexample is the PurR repressor binding to a 16 bp operatorsite that was characterized by crystal structure analysis(Schumacher et al., 1994). In eukaryotic cells, the TATAbox-binding protein (TBP) recognizes its AT-rich DNA tar-get via minor groove interactions, inducing a conforma-tional change in the double helix (Patikoglou, 1997). Ourearlier findings led us to propose that PapB may activatetranscription by counteracting repression caused by thehistone-like protein H-NS (Forsman et al., 1992). The pre-sent results suggest that the oligomeric protein interactionat the UAS, thereby presumably stabilizing a suitableDNA conformation, could be an important part of suchan anti-repression.
Experimental procedures
Bacterial strains and culture conditions
Escherichia coli strains used in this study were J96 (Hull et al.,1981), JM103 (Messing et al., 1981), MC1029 (Casadabanand Cohen, 1980) and BL21(DE3) (Studier and Moffat,1986). Cells were grown in L-broth (Bertani, 1951) at 378C.Growth occurred on plates with tryptone yeast agar (TYS;Swedish Labfab). Antibiotics used in selective media werecarbenicillin (100 mg ml¹1) and kanamycin (50 mg ml¹1).
Plasmids
The PapB-overproducing plasmid pKF10 has been describedearlier (Forsman et al., 1989). A cosmid clone pBSN50 fromstrain J96 was used as the source of foc DNA (unpublishedconstruction). The source of pap DNA from strain KS71was the plasmid pKTH3025 (Rhen et al., 1985). A series ofpap–lac operon fusion plasmids was based on the vectorpRZ5202 (Reznikoff and McClure, 1986). The derivatives,pHMG1 (Goransson and Uhlin, 1984) and pHMG15 (Bagaet al., 1985), have been described previously. Plasmid pKF19was obtained by deleting the entire PapB binding site 1(52 bp) in pHMG1. Plasmid pHMG102, which carries an SphI(position 583)–XhoI (linker insertion) pap fragment insertedinto the corresponding sites of pUC18, was used for subcloningof in vitro constructed mutations. The DNA fragments contain-ing different mutations in site 1 were created using the PCR-based method of overlapping extension (Horton et al., 1993).The two common primers used in all these PCR reactionswere: no. 740 58-CCCAAGCTTAATCCGTTACCGCCAGCG-CCT-38; and no. 1140 58-CCCAAGCTTTAAACGATCTTTT-AACCCACAAAAC-38. The sequences corresponding to theprimers used for introducing the individual mutations wereas listed in Fig. 6. These PCR fragments were digested withCsp 45I–Nsi I and subcloned into plasmid pHMG102. Theresulting plasmids were then cleaved with SphI–ApaI andligated with the pap(I þ, B þ) A:lacZ fusion plasmid pHMG1cleaved with the same enzymes. The resulting pap–lacZfusion plasmids pXN1, 2, 3, 4, 5 and 6 carried the variousPapB binding site 1 mutations, as outlined in Fig. 6. To con-struct the His-tagged PapB-overproducing plasmid pYN1,
the DNA fragment carrying the papB gene was amplified byPCR with plasmid pHMG1 as template and using primersb1: 58-GCCCGAATTCGGATCCATGGCGCATCATGAAGT-CAT-38; and b3: 58-GCCCCTGCAGAAGCTTTTATTAGTC-AAATGCCGACGA-38. The PCR product was then digestedwith BamHI–HindIII and cloned into the same sites of plasmidpQE 30 (QIAGEN).
DNA techniques
Plasmid isolation, gel electrophoresis, transformation, amplifi-cation of DNA by PCR and DNA labelling were performedusing standard procedures (Maniatis et al., 1982). Restrictionendonuclease digestions, DNA ligation reactions and DNAsequencing with Sequenase were performed under the con-ditions recommended by the manufacturers (New EnglandBiolabs, Pharmacia, United States Biochemical Corporation).
b-Galactosidase assay
To measure the b-galactosidase specific activity, we used themethod described by Miller (1972).
Purification of PapB protein
Cell lysate from strain JM103/pKF10 was heated at 1008C for5 min in SDS sample buffer and loaded onto 15% preparativeSDS–PAGE. The PapB protein was extracted by cutting thecorresponding band, washing twice with 1 mM dithiothreitol(DTT) for 15 min and eluting with elution buffer (0.05 M Tris-Cl, pH 7.9, 0.1% SDS, 0.1 mM EDTA, 5 mM DTT, 150 mMNaCl) at 48C for 48 h. The eluate was then precipitated byfour or five volumes of cold acetone with 1 mM DTT for30 min at ¹808C. After centrifugation at 10 000 r.p.m. for20 min, the pellet was washed several times with 80% ace-tone, 1 mM DTT and air dried. It was then dissolved in thedenaturation buffer (6 M guanidine hydrochloride, 50 mMHEPES–NaOH, pH 7.5, 150 mM NaCl, 1 mM DTT, 0.1 mMEDTA, 20% glycerol) and kept at room temperature for 15–20 min. This was finally diluted 50 times or more into thesame buffer without guanidine hydrochloride and kept at48C for 24–48 h. The renatured protein was dialysed againstSB buffer (10 mM Tris-HCl, pH 8.0, 1 mM NH4Cl, 0.1 mMEDTA, 1 mM DTT, 50% glycerol) and kept at ¹208C.
The His-tagged PapB was purified from strain BL21(DE3)/pYN1 through a Ni-NTA column (Qiagen). Bacteria from a400 ml culture were collected and resuspended in 15 ml of bind-ing buffer (5 mM imidazole, 500 mM NaCl, 20 mM Tris-Cl,pH 7.9). After sonication, the supernatant was applied to thecolumn, which had been pre-equilibrated with binding buffer.The column was then washed with 10 volumes of binding bufferand 6 volumes of washing buffer (60 mM imidazole, 500 mMNaCl, 20 mM Tris-Cl, pH 7.9). The His-tagged PapB was finallyeluted with elution buffer (500 mM imidazole, 500 mM NaCl,20 mM Tris-Cl, pH 7.9) at a flow rate of 10 ml h¹1.
Analyses of protein–DNA interactions
Gel mobility shift assays and DNase I footprinting analyseswere performed as described previously (Forsman et al.,
Q 1998 Blackwell Science Ltd, Molecular Microbiology, 30, 513–523
520 Y. Xia, K. Forsman, J. Jass and B. E. Uhlin

1989; Goransson et al., 1989b). DNA fragments for gel mobil-ity shift assays were obtained by PCR amplification usingplasmid pHMG1 as the template. The primers no. 1140 58-CCCAAGCTTTAAACGATCTTTTAACCCACAAAAC-38 andno. 740 58-CCCAAGCTTAATCCGTTACCGCCAGCGCCT-38 were used to obtain a 419-bp-long fragment containingsite 1. To obtain the 250-bp-long fragments, including mutantsites (Fig. 7), we used the primers corresponding to sequencesshown in Fig. 6 (p1–p19) in combination with primer no. 1140.The PCR products were purified and 32P end- labelled with T4polynucleotide kinase. Purified PapB protein (500–1000 ng)was mixed with labelled DNA fragments (5000–10 000 c.p.m.)in the presence of 0.5 mg of poly-(dI–dC) and 50 mM KCl inbuffer B (25 mM HEPES, pH 7.5, 0.1 mM EDTA, 5 mM DTT,10% glycerol) in a final volume of 10 ml. The reaction mixtureswere incubated at 258C for 15 min and then immediatelyloaded onto 8% polyacrylamide–bis gel for electrophoresis.In the competition experiment, purified PapB protein wasincubated with the end-labelled 419 bp DNA fragment aloneor in the presence of the indicated amount of distamycin ormethyl green as described above. The DNA fragment usedin the DNase I protection assay for the foc regulatory regionwas obtained by PCR amplification using an unlabelled primeroligo1: 58-TGCCTGGTAATCCGTTACCGC-38 and 32P end-labelled primer oligo7: 58-TTGACGATCTTTTGATCTGTA-38,with plasmid pBSN50 as the template. The DNA fragment forthe analysis of binding to KS71 DNA was obtained by end-labelling in the Csp 45I site and subsequent Nsi I cleavage ofplasmid pKTH3025 (Rhen et al., 1985). The fragment usedin the DNase I protection assay for the pap regulatory regionwas obtained by cleaving the M13 clone containing the PapBbinding site with Csp 45I, end-labelled with Klenow andrecleaved with HindIII. The footprinting reactions were carriedout as follows. A 1 ml sample of 32P end-labelled DNA fragment(1 ng, 20 000–50 000 c.p.m.) was mixed with 1 mg of poly-(dI–dC) in buffer B with 50 mM KCl (final concentration). PapBprotein (20–1620 ng) was added, and the mixtures were incu-bated at 258C for 15 min. DNase I digestions were carried outafter the addition of 100 ng of DNase I and MgCl2 to a final con-centration of 5 mM. After 2 min, the reactions were stopped bythe addition of 12 ml of stop mix (0.25 M EDTA, 1.5 M NaCl,1.5 mg ml¹1 oyster glycogen). The DNA was extracted with1:1 (v/v) phenol–chloroform, ethanol precipitated and ana-lysed on 8% polyacrylamide–urea gels.
AFM imaging of DNA–protein complexes
For atomic force microscopy (AFM) imaging, the protein–DNA binding reaction was modified to exclude glycerol andpoly-(dI–dC). Approximately 250–500 ng of purified (His-tagged) PapB was added to 30 ng of PCR-amplified 419 bpDNA fragment in binding buffer B and incubated for 20 minat room temperature. An aliquot of the binding mixture(10 ml) was diluted with 20 ml of ultrapure (Millipore) waterand immediately placed onto a silanized mica surface. Thesamples were incubated at 378C for 2 min to disperse andseparate the DNA (Li et al., 1992), gently rinsed with ultrapurewater, blotted around the edges and dried with nitrogen gas.The silanized mica was prepared according to Lyubchenkoet al. (1993) by placing freshly cleaved mica into a 2 l desiccatorcontaining a drop of 3-aminopropyltriethoxy silane (APTES)
for 2–4 h. The APTES-treated mica was kept in a nitrogenatmosphere in the desiccator for no more than 1 week beforeuse. The samples were imaged in air with a Nanoscope IIIMultiMode AFM (Digital Instruments) in tapping mode. Stan-dard silicon cantilevers (Digital Instruments) with an approxi-mate tip radius of 20–40 nm were used. Typical scanparameters were: scan rate 1.9–3 Hz; drive frequency 310–350 kHz; and humidity 10–15%. DNA measurements wereobtained using public domain software UTHSCSA ImageTool 1.27. The average (n ¼ 9) measured DNA length in nmand the known fragment size (419 bp) was used to calculatethe interbase distance.
Test of PapB-binding saturation
Strain JM103/pKF10 was grown in 10 ml of MOPS (–Met) tomid-log phase and induced with IPTG (final concentration1 mM) for 1.5 h. Bacteria (5 ml) were labelled with 200 ml of[35S]-Met for 15 min at 378C. After centrifugation, the pelletwas washed with 5 ml of cold MOPS and dissolved in 0.5 mlof SDS sample buffer. The 35S-labelled PapB was purifiedfrom the gel as described above and incubated with unlabelledDNA fragment [with (419 bp) or without (367 bp) binding site 1]at different ratios in buffer B in the binding saturation test. Thebinding was monitored by gel electrophoresis as describedabove for gel mobility shift assays.
Acknowledgements
We thank Dr Gertrud Puu at FOA, Umea, Sweden for provid-ing the AFM. This work was supported by grants from theSwedish Natural Science Research Council, the SwedishMedical Research Council and the Goran Gustafsson Foun-dation for Research in Natural Science and Medicine.
References
Allison, D.P., Kerper, P.S., Doktycz, M.J., Spain, J.A., Mod-rich, P., Larimer, F.W., et al. (1996) Direct atomic forcemicroscope images of EcoRI endonuclease site specificallybound to plasmid DNA molecules. Proc Natl Acad Sci USA93: 8826–8829.
Baga, M., Goransson, M., Normark, S., and Uhlin, B.E.(1985) Transcriptional activation of a Pap pilus virulenceoperon from uropathogenic Escherichia coli. EMBO J 4:3887–3893.
Bellomy, G.R., and Records, Jr, M.T. (1990) Stable DNA loopsin vivo and in vitro : roles in gene regulation at a distanceand in biophysical characterization of DNA. Prog NucleicAcids Res Mol Biol 39: 81–128.
Bertani, G. (1951) Studies on lysogenesis I. The mode ofphage liberation by lysogenic Escherichia coli. J Bacteriol62: 293–300.
Blyn, L.B., Braaten, B.A., White-Ziegler, C.A., Rolfson, D.H.,and Low, D.A. (1989) Phase variation of pyelonephritis-associated pili in Escherichia coli : evidence for transcrip-tional regulation. EMBO J 8: 613–620.
Bonnefoy, E., and Rouviere-Yaniv, J. (1991) HU and IHF, twohomologous histone-like proteins of Escherichia coli, formdifferent protein–DNA complexes with short DNA frag-ments. EMBO J 10: 687–696.
Q 1998 Blackwell Science Ltd, Molecular Microbiology, 30, 513–523
Oligomeric PapB protein binding in the pap operon 521

Breg, J.N., Van Opheusden, J.H.J., Burgering, M.J.M.,Boelens, R., and Kaptein, R. (1990) Structure of Arc repres-sor in solution: evidence for a family of beta-sheet DNA-binding proteins. Nature 346: 586–589.
Burkhoff, A.M., and Tullius, T.D. (1987) The unusual confor-mation adopted by the adenine tracts in kinetoplast DNA.Cell 48: 935–943.
Casadaban, M.J., and Cohen, S.N. (1980) Analysis of genecontrol signals by DNA fusion and cloning in Escherichiacoli. J Mol Biol 138: 179–207.
Christine, M. (1996) The clp (CS31A) operon is negativelycontrolled by Lrp, ClpB, and L-alanine at the transcriptionallevel. Mol Microbiol 21: 281–292.
Coll, M., Frederick, C., Wang, A.H., and Rich, A. (1987) Abifurcated hydrogen-bonded conformation in the d(A.T)base pairs of the DNA dodecamer d(CGCAAATTTGCG)and its complex with distamycin. Proc Natl Acad Sci USA84: 8385–8389.
Collado-Vides, J., Magasanik, B., and Gralla, J.D. (1991) Con-trol site location and transcriptional regulation in Escheri-chia coli. Microbiol Rev 55: 371–394.
Forsman, K., Goransson, M., and Uhlin, B.E. (1989) Auto-regulation and multiple DNA interactions by a transcriptionalregulatory protein in E. coli pili biogenesis. EMBO J 8:1271–1277.
Forsman, K., Sonden, B., Goransson, M., and Uhlin, B.E.(1992) Antirepression function in Escherichia coli for thecAMP–cAMP receptor protein transcriptional activator.Proc Natl Acad Sci USA 89: 9880–9884.
Freire, R., Salas, M., and Hermoso, J.M. (1994) A new pro-tein domain for binding to DNA through the minor groove.EMBO J 13: 4353–4360.
Friedrich, M.J., Kinsey, N.E., Vila, J., and Kadner, R.J.(1993) Nucleotide sequence of a 13.9 kb segment of the90 kb virulence plasmid of Salmonella typhimurium: thepresence of fimbrial biosynthesis genes. Mol Microbiol 8:543–558.
Goransson, M., and Uhlin, B.E. (1984) Environmental tem-perature regulates transcription of a virulence pili operonin E. coli. EMBO J 3: 2885–2888.
Goransson, M., Forsman, K., and Uhlin, B.E. (1989a) Regu-latory genes in the thermoregulation of Escherichia coli piligene transcription. Genes Dev 3: 123–130.
Goransson, M., Forsman, K., Nilsson, P., and Uhlin, B.E.(1989b) Upstream activating sequences that are sharedby two divergently transcribed operons mediate cAMP–CRP regulation of pilus–adhesin in Escherichia coli. MolMicrobiol 3: 1557–1565.
Guthold, M., Bezanilla, M., Erie, D.A., Jenkins, B., Hansma,H.G., and Bustamante, C. (1994) Following the assemblyof RNA polymerase–DNA complexes in aqueous solutionswith the scanning force microscope. Proc Natl Acad SciUSA 91: 12927–12931.
Hagerman, P.J. (1985) Sequence dependence of the curva-ture of DNA: a test of the phasing hypothesis. Biochemistry24: 7033–7037.
Horton, R.M., Ho, S.N., Pullen, J.K., Hunt, H.D., Cai, Z., andPease, L.R. (1993) Gene splicing by overlap extension.Methods Enzymol 217: 270–279.
Hull, R.A., Gill, R.E., Hsu, P., Minshew, B.H., and Falkow,S. (1981) Construction and expression of recombinant
plasmids encoding type 1 or D-mannose-resistant pili froma urinary tract infection Escherichia coli isolate. InfectImmun 33: 933–938.
Katahira, M., Sugeta, H., and Kyogoku, Y. (1990) A new modelfor the bending of DNAs containing the oligo(dA) tractsbased on NMR observations. Nucleic Acids Res 18:613–618.
Koo, H.S., Wu, H.M., and Crothers, D.M. (1986) DNA bend-ing at adenine thymine tracts. Nature 326: 501–506.
Kumar, K.A., and Muniyappa, K. (1992) Use of structure-directed DNA ligands to probe the binding of recA proteinto narrow and wide grooves of DNA and on its ability topromote homologous pairing. J Biol Chem 267: 24824–24832.
Li, M.-Q., Hansma, H.G., Vesenka, J., Kelderman, G., andHansma, P.K. (1992) Atomic force microscopy of uncoatedplasmid DNA: nanometer resolution with only nanogramamounts of sample. J Biomol Struct Dynamics 10: 607–616.
Lyubchenko, Y., Shlyakhtenko, L., Harrington, R., Oden, P.,and Lindsa, S. (1993) Atomic force microscopy of longDNA: imaging in air and under water. Proc Natl Acad SciUSA 90: 2137–2140.
Maniatis, T., Fritsch, E.F., and Sambrook, J. (1982) MolecularCloning: a Laboratory Manual. Cold Spring Harbor, NY:Cold Spring Harbor Laboratory Press.
Messing, J., Crea, R., and Seeburg, P.H. (1981) A system forshotgun DNA sequencing. Nucleic Acids Res 9: 309–321.
Miller, J.H. (1972) Experiments in Molecular Genetics. ColdSpring Harbor, NY: Cold Spring Harbor Laboratory Press.
Morschhauser, J., Uhlin, B.E., and Hacker, J. (1993) Tran-scriptional analysis and regulation of the sfa determinantcoding for S fimbriae of pathogenic Escherichia coli strains.Mol Gen Genet 238: 97–105.
Patikoglou, G. (1997) Eukaryotic transcription factor–DNAcomplexes. Annu Rev Biophys Biomol Struct 26: 289–325.
Rafferty, J.B.M., Somers, W.S., Saint-Girons, I., and Philips,S.E.V. (1989) Three-dimensional crystal structures ofEscherichia coli met repressor with and without corepres-sor. Nature 341: 705–710.
Reznikoff, W.S., and McClure, W.R. (1986) E. coli promoters.In Maximizing Gene Expression. Reznikoff, W.S., and Gold,L. (eds). Boston: Butterworths, pp. 1–33.
Rhen, M., Vaisanen-Rhen, V., Pere, A., and Korhonen, T.K.(1985) Complementation and regulatory interactionbetween two cloned fimbrial gene clusters of Escherichiacoli strain KS71. Mol Gen Genet 200: 60–64.
Rhen, M., and Vaisanen-Rhen, V. (1987) Nucleotide sequ-ence analysis of a P fimbrial regulatory element of the uro-pathogenic Escherichia coli strain KS71A (O4:K12). MicrobPathogen 3: 387–391.
Rhodes, D., and Klug, A. (1980) Structure of nucleosomecore particles of chromatin. Nature 286: 573–578.
Rice, P.A., Yang, S.W., Mizuuchi, K., and Nash, H.A. (1996)Crystal structure of an IHF–DNA complex: a protein-induced DNA U-turn. Cell 87: 1295–1306.
Roosendaal, B., Damoiseaux, J., Jordi, W., and de Graaf,F.K. (1989) Transcriptional organization of the DNA regioncontrolling expression of the K99 gene cluster. Mol GenGenet 215: 250–256.
Schumacher, M.A., Choi, K.Y., Zalkin, H., and Brennan, R.G.
Q 1998 Blackwell Science Ltd, Molecular Microbiology, 30, 513–523
522 Y. Xia, K. Forsman, J. Jass and B. E. Uhlin

(1994) Crystal structure of LacI member, PurR, bound toDNA: minor groove binding by alpha helices. Science266: 763–770.
Serrano, M., Gutierrez, C., Freire, R., Bravo, A., Salas, M.,and Hermoso, J.M. (1994) Phage f29 protein p6: a viralhistone-like protein. Biochimie 76: 981–991.
Serrano, M., Salas, M., and Hermoso, J.M. (1990) A novelnucleoprotein complex at a replication origin. Science248: 1012–1016.
Studier, F.W., and Moffat, B.A. (1986) Use of bacteriophageT7 RNA polymerase to direct selective high-level expres-sion of cloned genes. J Mol Biol 189: 113–130.
Tippner, D., Afflerbach, H., Bradaczek, C., and Wagner, R.(1994) Evidence for a regulatory function of the histone-like Escherichia coli protein H-NS in ribosomal RNA synth-esis. Mol Microbiol 11: 589–604.
Travers, A., and Klug, A. (1987) Nucleoprotein complexes:DNA wrapping and writhing. Nature 327: 280–281.
Van Die, I., Van Geffen, B., Hoekstra, W., and Bergmans, H.
(1984) Type 1C fimbriae of a uropathogenic Escherichiacoli strain. Cloning and characterization of the genesinvolved in the expression of the 1C antigen and nucleotidesequence of the subunit gene. Gene 34: 187–196.
Van Dyke, M.W., Hertzberg, R.P., and Dervan, P.B. (1982)Map of distamycin, netropsin, and actinomycin bindingsites on heterogeneous DNA: DNA cleavage-inhibition pat-terns with methidiumpropyl-EDTA.Fe(II). Proc Natl AcadSci USA 79: 5470–5474.
Wu, H.M., and Crothers, D.M. (1984) The locus of sequencedirected and protein induced DNA bending. Nature 308:509–513.
Yang, C.C., and Nash, H.A. (1989) The interaction of E. coliIHF protein with its specific binding sites. Cell 57: 869–880.
Zimmer, C., and Wahnert, L. (1986) Nonintercalating DNA-binding ligands: specificity of the interaction and their useas tools in biophysical, biochemical and biological investi-gations of the genetic material. Prog Biophys Mol Biol47: 31–112.
Q 1998 Blackwell Science Ltd, Molecular Microbiology, 30, 513–523
Oligomeric PapB protein binding in the pap operon 523





























