Embed Size (px)
Citation preview

On the Analogy of B-BO and B-Au Chemical Bonding in B11O- and B10Au- Clusters
Hua-Jin Zhai,† Chang-Qing Miao,‡ Si-Dian Li,*,‡ and Lai-Sheng Wang*,†
Institute of Molecular Sciences, Shanxi UniVersity, Taiyuan 030006, People’s Republic of China, Institute ofMaterials Science and Department of Chemistry, Xinzhou Teachers’ UniVersity,Xinzhou 034000, Shanxi, People’s Republic of China, and Department of Chemistry, Brown UniVersity,ProVidence, Rhode Island 02912, United States
ReceiVed: September 11, 2010; ReVised Manuscript ReceiVed: October 15, 2010
During photoelectron spectroscopy experiments, the spectra of B11O- and B10Au- clusters are found to exhibitsimilar patterns except for a systematic spectral shift of ∼0.5 eV, hinting that they possess similar geometricstructures. The electron affinities are measured to be 4.02 ( 0.04 eV for B11O and 3.55 ( 0.02 eV for B10Au.DFT calculations at the B3LYP level show that B11O- and B10Au- adopt similar C1 (1A) ground states,which are based on the quasiplanar B10 cluster interacting with a BO unit and Au, respectively. The B11O-
and B10Au- clusters are thus valent isoelectronic because both BO and Au can be viewed as monovalentunits, forming highly covalent B-BO and B-Au bonds analogous to the B-H bond in B10H-. For B10Au-,we also find a highly symmetric D10h (1A1g) planar molecular wheel as a minimum on the potential energysurface. However, it is 45 kcal/mol above the ground state at the B3LYP level and not viable for experimentalobservation. Natural bond orbital analyses reveal interesting coValent versus ionic B-Au bonding in the C1
B10Au- and D10h B10Au- structures, respectively, providing insight for the design of Dnh MBn molecularwheels.
1. Introduction
Boron possesses interesting chemical bonding properties andhas played essential roles in advancing chemical bondingmodels.1,2 Primarily due to the electron deficiency of boron,delocalized three-center two-electron bonds are common inboranes3 and elemental boron clusters.4-8 Boron also formsinteresting oxides, and the simplest diatomic BO molecule(boronyl) has long been recognized as a σ-radical9-11 with aB≡O triple bond. BO and BO- are valent isoelectronic to CNand CN-/CO, respectively, which are important inorganicligands. However, despite decades of persistent interest in boronoxides,12,13 boronyl chemistry remains relatively unknown.14-20
We are interested in elucidating the electronic structure andchemical bonding of boron oxide clusters using photoelectronspectroscopy (PES) and theoretical calculations.11,21-23 Werecently characterized two boron oxide clusters,22 B3O2
- andB4O3
-, which possess linear B(BO)2- (D∞h, 3Σg) and triangular
B(BO)3- (D3h, 2A2′′) structures, that is, two and three boronyls
bonded to a single B atom. We further showed that the B4O22-
cluster has a linear diboronyl diborene ground state, B2(BO)22-,
which can be viewed as two boronyls bonded to a B2 core andconcurrently features a rare B≡B triple bond.23 Our combinedexperimental and theoretical studies21-23 have stimulated furthertheoretical efforts on boron oxide clusters.24-27 However, noneof the Bm(BO)n
- (m > 2) clusters has been characterizedexperimentally. Among earlier studies relevant to boronylchemistry, gas-phase microwave14 and matrix infrared spec-troscopy15 suggested rather strong B-O linkage in XBO. PESdata revealed a similarity between H3CBO and its isoelectronicH3CCN,16 and thus the B-O linkage in the former was described
as a triple bond. Very recently, the first metal boronyl compoundfeaturing a B≡O triple bond was synthesized in the bulk.18
The bonding properties of Au in alloy clusters represent yetanother interesting aspect in cluster research.28-30 The isolobalanalogy between a gold phosphine unit and a hydrogen atomhas helped the rationalization of the structure and bonding inAu compounds.28 Further analogy between a bare Au atom andH has been discovered recently in gas-phase binary Auclusters.31-35 In systematic studies on Si-Au clusters, SiAu4,SiAun (n ) 2, 3), Si2Aun (n ) 2, 4), and Si3Au3 were shown topossess structures and bonding similar to the silane SiH4, SiHn,Si2Hn (n ) 2, 4), and Si3H3, respectively.31-33 For the B7Au2
-
alloy cluster,34 the B-Au bonding was shown to be covalent,similar to the B-H bonding in B7H2
-.36 The B10Au- cluster inthe current work serves as another example to further test theAu/H analogy in Au alloy clusters. Moreover, stimulated bythe elucidation of the chemical bonding in planar boronclusters,5-7 there is increasing computational interest in planarMBn molecular wheels.37-44 Among these MBn clusters are theD10h B10Au- anion39 and its valent isoelectronic D10h B10Cu-
and D10h B10Zn species.40 However, the viability of all theseD10h molecular wheels has yet to be confirmed experimentally.
Here we report a combined PES and theoretical study ofB11O- and B10Au- clusters. During PES experiments on boronoxide clusters and B-Au alloy clusters, B11O- and B10Au- areobserved to exhibit similar PES patterns except for a slight shiftin binding energies. Theoretical calculations show that B11O-
and B10Au- possess similar C1 (1A) ground-state structures,where the B11O- cluster can be described as B10(BO)-, that is,a boronyl B≡O unit interacting with an aromatic B10
- core.Both the B-BO and B-Au bonds are shown to be highlycovalent, analogous to the B-H bond. The current studyhighlights not only the robustness of the B≡O boronyl groupin boron-rich boron oxide clusters but also the Au/H analogyin B-Au complexes. A highly symmetric D10h B10Au- molec-
* To whom correspondence should be addressed. E-mail: [email protected] (S.-D.L.); [email protected] (L.-S.W.).
† Brown University.‡ Shanxi University and Xinzhou Teachers’ University.
J. Phys. Chem. A 2010, 114, 12155–12161 12155
10.1021/jp108668t 2010 American Chemical SocietyPublished on Web 10/29/2010
ular wheel is shown to be a high-lying minimum 45 kcal/molabove the C1 ground state. The bonding of this cluster iscompared with that of the C1 global minimum, providing insightinto the stability of Dnh-type MBn molecular wheels.
2. Experimental and Computational Methods
2.1. Photoelectron Spectroscopy. The experiments werecarried out using a magnetic-bottle PES apparatus equipped witha laser vaporization cluster source, details of which have beendescribed previously.45,46 Briefly, the B11O- cluster anions wereproduced by laser vaporization of a pure disk target made ofenriched 10B isotope (99.75%) in the presence of a helium carriergas seeded with 0.01% O2. The B10Au- clusters were producedusing an Au/B mixed disk target (10B isotope enriched, 99.75%)and a pure helium carrier gas. Cluster anions from the laservaporization source were analyzed using a time-of-flight massspectrometer. The B11O- and B10Au- clusters of current interestwere each mass-selected and decelerated before being photo-detached. Two detachment photon energies were used in thecurrent experiments: 266 nm (4.661 eV) and 193 nm (6.424eV). Effort was made to choose colder clusters (that is, thosewith long resident times in the nozzle) for photodetachment,which was shown previously to be important for obtaining highquality PES data.47 Photoelectrons were collected at nearly 100%efficiency by the magnetic bottle and analyzed in a 3.5 m longelectron flight tube. The PES spectra were calibrated using theknown spectra of Rh- and Au-, and the energy resolution ofthe apparatus was ∆Ek/Ek ∼ 2.5%, that is, ∼25 meV for 1 eVelectrons.
2.2. Computational Methods. Structural optimizations werecarried out at the hybrid B3LYP level,48 with the augmentedDunning’s all-electron basis set (aug-cc-pvtz)49 for B, O, andH and the Stuttgart relativistic small-core pseudopotential andvalence basis set augmented with two f and one g functions[Stuttgart_rsc_1997_ecp + 2f1g: R(f) ) 0.498, R(f) ) 1.461,R(g) ) 1.218] for Au.50 A variety of initial structures wereoptimized in search of the ground-state structures of B11O- andB10Au- and their neutrals. For the purpose of comparison, thesame set of structures was also calculated for B10H- and B10H.Frequency calculations were done to confirm that all obtainedground-state structures are true minima. Excitation energies ofthe neutrals were calculated with the time-dependent DFT(TDDFT) method51,52 at the ground-state structures of the anions.All calculations were carried out using Gaussian 03.53
3. Experimental Results
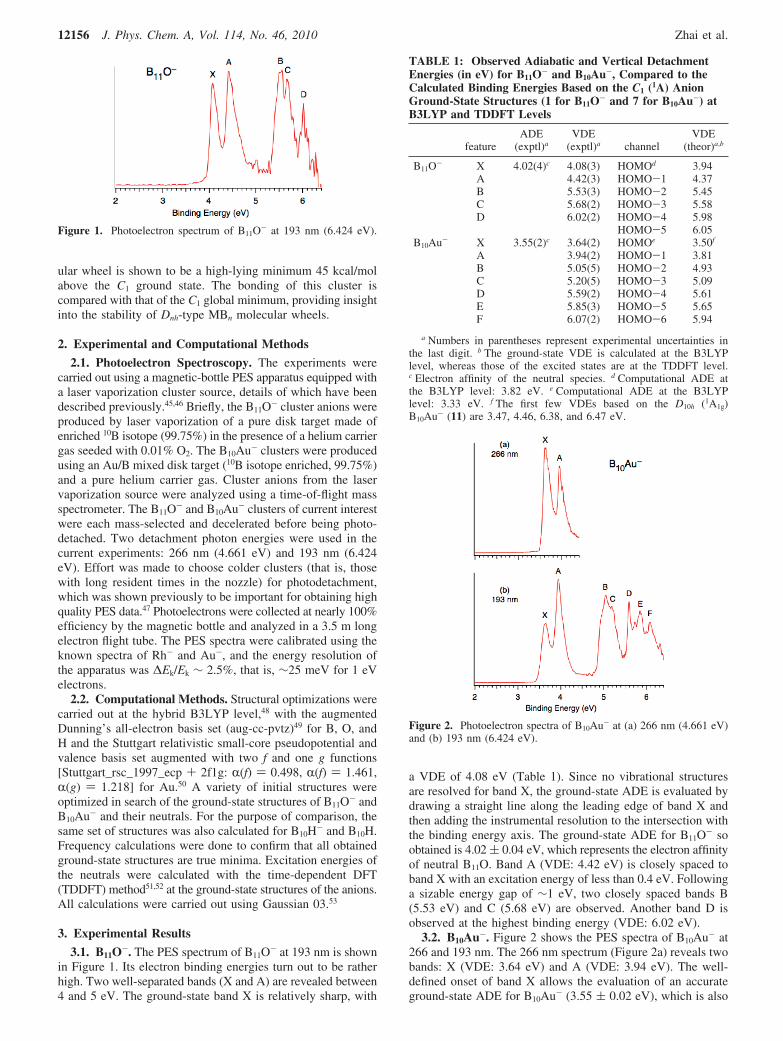
3.1. B11O-. The PES spectrum of B11O- at 193 nm is shownin Figure 1. Its electron binding energies turn out to be ratherhigh. Two well-separated bands (X and A) are revealed between4 and 5 eV. The ground-state band X is relatively sharp, with
a VDE of 4.08 eV (Table 1). Since no vibrational structuresare resolved for band X, the ground-state ADE is evaluated bydrawing a straight line along the leading edge of band X andthen adding the instrumental resolution to the intersection withthe binding energy axis. The ground-state ADE for B11O- soobtained is 4.02 ( 0.04 eV, which represents the electron affinityof neutral B11O. Band A (VDE: 4.42 eV) is closely spaced toband X with an excitation energy of less than 0.4 eV. Followinga sizable energy gap of ∼1 eV, two closely spaced bands B(5.53 eV) and C (5.68 eV) are observed. Another band D isobserved at the highest binding energy (VDE: 6.02 eV).
3.2. B10Au-. Figure 2 shows the PES spectra of B10Au- at266 and 193 nm. The 266 nm spectrum (Figure 2a) reveals twobands: X (VDE: 3.64 eV) and A (VDE: 3.94 eV). The well-defined onset of band X allows the evaluation of an accurateground-state ADE for B10Au- (3.55 ( 0.02 eV), which is also
Figure 1. Photoelectron spectrum of B11O- at 193 nm (6.424 eV).
TABLE 1: Observed Adiabatic and Vertical DetachmentEnergies (in eV) for B11O- and B10Au-, Compared to theCalculated Binding Energies Based on the C1 (1A) AnionGround-State Structures (1 for B11O- and 7 for B10Au-) atB3LYP and TDDFT Levels
featureADE
(exptl)aVDE
(exptl)a channelVDE
(theor)a,b
B11O- X 4.02(4)c 4.08(3) HOMOd 3.94A 4.42(3) HOMO-1 4.37B 5.53(3) HOMO-2 5.45C 5.68(2) HOMO-3 5.58D 6.02(2) HOMO-4 5.98
HOMO-5 6.05B10Au- X 3.55(2)c 3.64(2) HOMOe 3.50f
A 3.94(2) HOMO-1 3.81B 5.05(5) HOMO-2 4.93C 5.20(5) HOMO-3 5.09D 5.59(2) HOMO-4 5.61E 5.85(3) HOMO-5 5.65F 6.07(2) HOMO-6 5.94
a Numbers in parentheses represent experimental uncertainties inthe last digit. b The ground-state VDE is calculated at the B3LYPlevel, whereas those of the excited states are at the TDDFT level.c Electron affinity of the neutral species. d Computational ADE atthe B3LYP level: 3.82 eV. e Computational ADE at the B3LYPlevel: 3.33 eV. f The first few VDEs based on the D10h (1A1g)B10Au- (11) are 3.47, 4.46, 6.38, and 6.47 eV.
Figure 2. Photoelectron spectra of B10Au- at (a) 266 nm (4.661 eV)and (b) 193 nm (6.424 eV).
12156 J. Phys. Chem. A, Vol. 114, No. 46, 2010 Zhai et al.

the electron affinity of B10Au neutral. The relative intensitiesof bands X and A seem to show photon-energy dependence.Such photon-energy-dependent intensity changes can in principlegive valuable information about the nature of the electrons beingdetached.54,55 However, in the current case, the detachmentchannels for the X and A bands are both from delocalizedorbitals of the B10 ring as will be shown later, and no suchinformation can be extracted from the photon-energy-dependentintensity changes.
The overall PES pattern of B10Au- at 193 nm (Figure 2b) issurprisingly similar to that of B11O- (Figure 1), except for asystematic shift to lower binding energies (∼0.5 eV; see Table1). Again, an energy gap (∼1.1 eV) is observed between bandsA and B. Bands B (5.05 eV) and C (5.20 eV) partially overlapwith each other. The higher binding energy portion of the 193nm spectrum shows numerous features beyond 5.5 eV (Figure2b), all appearing to be relatively sharp. Three major bands arelabeled: D (5.59 eV), E (5.85 eV), and F (6.07 eV).
4. Theoretical Results
Structural searches for the B11O- and B10Au- clusters canbe demanding computationally. Fortunately, the PES similari-ties between B11O- and B10Au- (Figures 1 and 2) stronglyhint that the two species are structurally similar and,therefore, should both possess a B10 moiety. Our structuralsearches are thus largely based on the B10 unit, with the extraBO unit and Au attached to different possible sites. Selectedoptimized structures for B11O- (1-5) and B10Au- (7-12),along with the ground-state structures of their neutrals (6and 13), are shown in Figure 3.
Briefly, structures 1 (C1, 1A), 2 (Cs, 1A′), and 3 (C2, 1A) ofB11O- can be viewed as the BO unit being attached terminallyto three possible peripheral sites of the quasiplanar B10
- cluster6
in an in-plane fashion. The BO unit prefers a corner site,resulting in anion ground state 1. Structures 2 and 3 are first-order saddle points, ∼40 kcal/mol higher than structure 1.Structure 5 (C2V, 1A1) can be considered as the BO unit beingattached perpendicular to the plane, which is a third-order saddlepoint, ∼86 kcal/mol above structure 1. Structure 4 (C2V, 1A1) isbased on the B11
- moiety6 with the O atom attached to aperipheral site. It is a second-order saddle point, ∼74 kcal/molabove structure 1. The neutral state 6 (C1, 2A) is located as atrue minimum, which corresponds to the anion ground state 1.
Selected structures for B10Au- (7-12) and B10Au (13) areshown in Figure 3b. Their ground states are 7 (C1, 1A) and 13(C1, 2A), respectively. Among these optimized structures, 7, 9,10, 12, and 13 correspond to structures 1, 3, 4, 5, and 6 forB11O-/B11O, respectively. Note that structures 8, 9, 10, and 12are not minima. Also located for B10Au- is a highly symmetricwheel structure: 11 (D10h, 1A1g), which is a true minimumsubstantially higher in energy (by 45 kcal/mol). To aid theelucidation of chemical bonding in B11O- and B10Au-, a similarset of structures are also optimized for B10H- (14-18) and B10H(19), as shown in Figure 4.
5. Comparison between Experiment and Theory
The well-resolved PES spectra of B11O- and B10Au- (Figures1 and 2) provide electronic and structural information, whichhelps the search for the global minimum structures throughcomparison between experiment and theory. As shown in Table1, the calculated ground-state ADE and VDE for B11O- (1) are3.82 and 3.94 eV, respectively, as compared to the experimentalvalues of 4.02 and 4.08 eV. For B10Au- (7) the calculatedground-state ADE (3.33 eV) and VDE (3.50 eV) may be
compared with the experimental values of 3.55 and 3.64 eV,respectively. It is seen that B3LYP consistently underestimatesthe ground-state ADE by ∼0.2 eV and VDE by ∼0.15 eV. Forthe numerous excited states, TDDFT calculations reproduce theexperimental VDE values within ∼0.1 eV for the majority ofPES bands (Table 1).
The simulated PES spectra for B11O- and B10Au- are shownin Figure 5, both in excellent agreement with the experimentaldata. In particular, the small X-A energy gap and the relativelylarge A-B energy gap, which are characteristic of the experi-
Figure 3. Optimized structures for (a) B11O- (1-5) and B11O (6) and(b) B10Au- (7-12) and B10Au (13). The lowest vibrational frequency(νmin) and relative energy (∆E) are labeled under each structure. Thenumber of imaginary frequencies is shown in parentheses.
Figure 4. Optimized structures for (a) B10H- (14-18) and B10H (19).The lowest vibrational frequency (νmin) and relative energy (∆E) arelabeled under each structure. The number of imaginary frequencies isshown in parentheses.
Bonding in B11O- and B10Au- Clusters J. Phys. Chem. A, Vol. 114, No. 46, 2010 12157
mental PES spectra, are perfectly borne out in the TDDFTcalculations. The agreement between experiment and theory isremarkable, lending considerable credence to the identifiedground-state structures for both B11O- and B10Au- and theirneutrals.
6. Discussion
6.1. Structural Similarities between B11O-, B10Au-, andB10H-. The similarity in the PES spectra of B11O- and B10Au-
is surprising at first glance, suggesting that these two clustersmay possess similar structures. The key to the structuralconnection between B11O- and B10Au- is the occurrence of theB10 unit and the peripheral BO group in B11O- (Figure 3a).Both BO and Au can be viewed as monovalent species, whichare bonded to the same peripheral site on the B10 unit (Figure3). It is noted that the B11O- (1) ground-state structure,B10(BO)-, differs substantially from that of the bare B11
-
cluster,6 indicating the predominant role of the BO group ingoverning the structures of boron-rich oxide clusters. Thestructures of B11O- and B10Au- (Figure 3) may be comparedto that of B10H- (Figure 4). The three anion species possesssimilar global minimum structures (1, 7, and 14), all involvinga B10 structural unit, with BO, Au, and H bonded to the sameperipheral corner site. The similar structures for B10X- (X )BO, Au, H) underlie the electronic and structural robustness ofthe doubly (σ- and π-) aromatic B10 cluster as an inorganicbuilding block.6,8
6.2. Chemical Bonding in B10X- (X ) H, Au, BO). TheB10
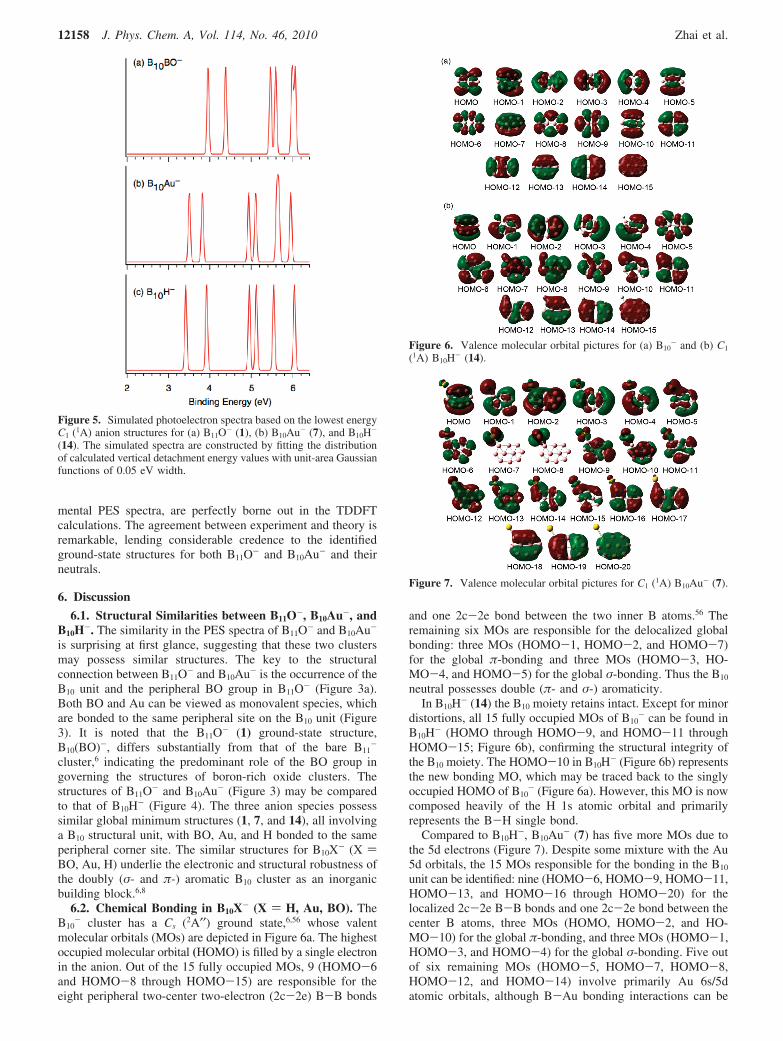
- cluster has a Cs (2A′′) ground state,6,56 whose valentmolecular orbitals (MOs) are depicted in Figure 6a. The highestoccupied molecular orbital (HOMO) is filled by a single electronin the anion. Out of the 15 fully occupied MOs, 9 (HOMO-6and HOMO-8 through HOMO-15) are responsible for theeight peripheral two-center two-electron (2c-2e) B-B bonds
and one 2c-2e bond between the two inner B atoms.56 Theremaining six MOs are responsible for the delocalized globalbonding: three MOs (HOMO-1, HOMO-2, and HOMO-7)for the global π-bonding and three MOs (HOMO-3, HO-MO-4, and HOMO-5) for the global σ-bonding. Thus the B10
neutral possesses double (π- and σ-) aromaticity.In B10H- (14) the B10 moiety retains intact. Except for minor
distortions, all 15 fully occupied MOs of B10- can be found in
B10H- (HOMO through HOMO-9, and HOMO-11 throughHOMO-15; Figure 6b), confirming the structural integrity ofthe B10 moiety. The HOMO-10 in B10H- (Figure 6b) representsthe new bonding MO, which may be traced back to the singlyoccupied HOMO of B10
- (Figure 6a). However, this MO is nowcomposed heavily of the H 1s atomic orbital and primarilyrepresents the B-H single bond.
Compared to B10H-, B10Au- (7) has five more MOs due tothe 5d electrons (Figure 7). Despite some mixture with the Au5d orbitals, the 15 MOs responsible for the bonding in the B10
unit can be identified: nine (HOMO-6, HOMO-9, HOMO-11,HOMO-13, and HOMO-16 through HOMO-20) for thelocalized 2c-2e B-B bonds and one 2c-2e bond between thecenter B atoms, three MOs (HOMO, HOMO-2, and HO-MO-10) for the global π-bonding, and three MOs (HOMO-1,HOMO-3, and HOMO-4) for the global σ-bonding. Five outof six remaining MOs (HOMO-5, HOMO-7, HOMO-8,HOMO-12, and HOMO-14) involve primarily Au 6s/5datomic orbitals, although B-Au bonding interactions can be
Figure 5. Simulated photoelectron spectra based on the lowest energyC1 (1A) anion structures for (a) B11O- (1), (b) B10Au- (7), and B10H-
(14). The simulated spectra are constructed by fitting the distributionof calculated vertical detachment energy values with unit-area Gaussianfunctions of 0.05 eV width.
Figure 6. Valence molecular orbital pictures for (a) B10- and (b) C1
(1A) B10H- (14).
Figure 7. Valence molecular orbital pictures for C1 (1A) B10Au- (7).
12158 J. Phys. Chem. A, Vol. 114, No. 46, 2010 Zhai et al.

seen in HOMO-5, HOMO-12, and HOMO-14. HOMO-15is the key MO which is responsible for the B-Au σ-bonding.This MO is equivalent to the B-H σ-bonding orbital in B10H-
(14, HOMO-10; Figure 6b).B10(BO)- (1) possesses 40 valence electrons, which occupy
20 MOs as shown in Figure 8. Again, 15 of these MOs can beeasily identified for bonding in the B10 unit: the localizedperipheral 2c-2e B-B bonds and one 2c-2e bond betweenthe two-center B atoms (HOMO-5, HOMO-6, HOMO-8,HOMO-10, HOMO-14 through HOMO-18), delocalizedglobal π-bonding (HOMO, HOMO-2, and HOMO-7), anddelocalized global σ-bonding (HOMO-1, HOMO-3, andHOMO-4). Among the remaining MOs, HOMO-9, HO-MO-11, and HOMO-12 are responsible for B≡O bonding inthe boronyl group, whereas HOMO-19 is the O 2s lone pair.The key bonding MO (HOMO-13) is primarily responsiblefor the B-BO single σ-bond, which links the B10 core and theperipheral BO group. Indeed, HOMO-13 is nearly identical tothe corresponding HOMO-10 of B10H- (Figure 6b) andHOMO-15 of B10Au- (Figure 7), all of which stem from theσ-bonding interaction between the singly occupied B10
- HOMOand the H, Au, or BO radicals.
Thus, B10X- (X ) H, Au, BO) anions can be considered ascovalent complexes between a doubly (σ- and π-) aromatic B10
-
core and a monovalent σ-radical, which are linked through asingle B-X σ-bond. The calculated B-X bond distances at theB3LYP level are 1.184 Å for B10H- (14), 2.029 Å for B10Au-
(7), and 1.628 Å for B10(BO)- (1), which are all typical valuesfor single B-X bonds.22,35,36 Other than the B-X bond, bothB10 and X in B10X- maintain their structural and chemicalintegrity. For example, the BO group in B10(BO)- (1) retainsits triple bond character (HOMO-9, HOMO-11, and HO-MO-12; Figure 8), and the B≡O bond distance (1.215 Å atthe B3LYP level) matches that of free BO (1.203 Å).11 As seenfrom Figures 6-8, the top few occupied MOs (e.g., HOMOthrough HOMO-4) are similar for B10(BO)-, B10Au-, andB10H-, which underlies the similarity of the PES spectra forB11O- and B10Au-. The negative charge in the B10X- anions islocated on the B10 unit (see the HOMOs of B10X-). Therefore,the B-X covalent bond is similar in the ground states of B10X-
and B10X. The B-X bond distances are elongated only veryslightly in the neutrals relative to their anions: 0.004 Å for B10H,0.028 Å for B10Au, and 0.010 Å for B11O.
6.3. On the Viability of the Hypercoordinate D10h B10Au-
Molecular Wheel. The quest of planar hypercoordinate mo-lecular wheels is primarily stimulated by the discovery of pureboron molecular wheels.5-8 Hypercoordinate molecular wheelscontaining a boron ring such as D6h CB6
2-, D7h CB7-, C2V
(effectively D8h) CB8, D9h AlB9, and D9h FeB9- have been
suggested computationally.37,41-43 However, recent joint experi-mental and theoretical studies have shown that D6h CB6
2-, D7h
CB7-, and D8h CB8 are highly unstable and that carbon avoids
the hypercoordinate central position in these species.38 Thehighest coordination number in a planar environment observedexperimentally to date is eight in the B9
- molecular wheel.5 Asimilar molecular wheel with a nonacoordinate Al has beenproposed for AlB9.43 There has been interest to design evenlarger boron rings with a center metal atom, such as the D10h
B10Au-.39
Our structural searches indeed find a true D10h (1A1g)minimum for B10Au- (11, Figure 3). However, at the B3LYPlevel it is 45 kcal/mol above the C1 (1A) ground state (7), andit is not viable experimentally. The simulated PES spectrumbased on the D10h B10Au- anion is shown in Figure 9, where itis compared with the 193 nm experimental PES spectrum.Because of the high symmetry of the D10h isomer, its simulatedPES spectrum only contains two peaks in the binding energyregime of 3-6.3 eV. This spectral pattern completely disagreeswith the observed PES spectra for B10Au-, confirming theB3LYP result that the D10h isomer is much higher in energy.
Why is the D10h (1A1g) B10Au- (11) minimum so much higherin energy relative to the C1 (1A) B10Au- (7) ground state? TheD10h B10 ring is intrinsically electron deficient and would requireelectron donation from the center atom in the molecular wheel.39
NBO analyses reveal that the Au center, which has the naturalelectronic configuration of 6s0.455d9.62, carries a positive netatomic charge of q(Au) ) +0.90 |e| in D10h B10Au-. However,as a highly electronegative metal, it is energetically not favorablefor Au to donate its 6s1 valence electron. MO analysis alsoreveals that the Au center in D10h B10Au- contributes very littleto the “disk delocalization”,5 which would be critical to stabilizethe molecular wheel. On the other hand, in the C1 (1A) B10Au-
(7) and C1 (2A) B10Au (13) structures, the integrity and double(σ- and π-) aromaticity of the B10 unit are maintained, and theB-Au bonding is primarily covalent: q(Au) )+0.05 and +0.28|e| in 7 and 13, respectively. The B-Au covalent bond energy,which is anticipated to be ∼3.7 eV,57 provides additionalstabilization to the C1 ground-state structures. Similar covalentbonding also enhances the stability of the C1 ground-statestructures for B11O- and B10H-. NBO analysis shows that q(BO)) +0.10 |e|, q(BO) ) +0.14 |e|, q(H) ) +0.02 |e|, and q(H) )+0.05 |e| for 1, 6, 14, and 19, respectively. The bonding analysesof the C1 versus D10h structures of B10Au- should be valuablefor the design of hypercoordinate Dnh MBn molecular wheels.
7. Conclusions
In conclusion, we report a combined photoelectron spectros-copy and density functional theory study on B11O- and B10Au-
Figure 8. Valence molecular orbital pictures for C1 (1A) B11O- (1).
Figure 9. Simulated photoelectron spectrum (solid line) based on theD10h (1A1g) B10Au- (11) anion structure, as compared with the 193 nmexperimental data (dashed line). The simulated spectrum is constructedby fitting the distribution of calculated vertical detachment energy valueswith unit-area Gaussian functions of 0.05 eV width.
Bonding in B11O- and B10Au- Clusters J. Phys. Chem. A, Vol. 114, No. 46, 2010 12159

clusters, which give similar PES spectra. DFT calculations showthat B11O- and B10Au- possess similar C1 (1A) structures, whichare based on the quasiplanar B10 unit with BO and Au bondedto the same peripheral corner site. B11O- and B10Au- are thusvalent isoelectronic, consistent with their similar PES spectra.The B-BO and B-Au bonding are shown to be highly covalent,analogous to the B-H bonding in B10H-. A highly symmetricD10h (1A1g) molecular wheel is located as a minimum for B10Au-,45 kcal/mol above the ground state. The current results reinforcethe importance of boronyl as a structural unit and the hydrogenanalogy of Au, and shed light on the design of Dnh-type MBn
molecular wheels.
Acknowledgment. This work was supported by the NationalScience Foundation (DMR-0904034 to L.-S.W) and the NationalNatural Science Foundation of China (No. 20873117 to S.-D.L).
References and Notes
(1) Greenwood, N. N.; Earnshaw, A. Chemistry of the Elements, 2nded.; Butterworth-Heinemann: Oxford, 1997.
(2) Cotton, F. A.; Wilkinson, G.; Murrillo, C. A.; Bochmann, M.AdVanced Inorganic Chemistry, 6th ed.; John Wiley & Sons: New York,1999.
(3) (a) Lipscomb, W. N. Boron Hydrides; Benjamin: New York, 1963.(b) Lipscomb, W. N. Science 1977, 196, 1047.
(4) (a) Hanley, L.; Whitten, J. L.; Anderson, S. L. J. Phys. Chem. 1988,92, 5803. (b) Kato, H.; Yamashita, K.; Morokuma, K. Chem. Phys. Lett.1992, 190, 361. (c) Martin, J. M. L.; Francois, J. P.; Gijbels, R. Chem.Phys. Lett. 1992, 189, 529. (d) Kawai, R.; Weare, J. H. Chem. Phys. Lett.1992, 191, 311. (e) Boustani, I. Int. J. Quantum Chem. 1994, 52, 1081. (f)Ricca, A.; Bauschlicher, C. W., Jr. Chem. Phys. 1996, 208, 233. (g)Boustani, I. Phys. ReV. B 1997, 55, 16426. (h) Gu, F. L.; Yang, X. M.;Tang, A. C.; Jiao, H. J.; Schleyer, P. v. R. J. Comput. Chem. 1998, 19,203. (i) Fowler, J. E.; Ugalde, J. M. J. Phys. Chem. A 2000, 104, 397. (j)Aihara, J. I.; Kanno, H.; Ishida, T. J. Am. Chem. Soc. 2005, 127, 13324.(k) Oger, E.; Crawford, N. R. M.; Kelting, R.; Weis, P.; Kappes, M. M.;Ahlrichs, R. Angew. Chem., Int. Ed. 2007, 46, 8503. (l) Szwacki, N. G.;Sadrzadeh, A.; Yakobson, B. I. Phys. ReV. Lett. 2007, 98, 166804. (m)Tang, H.; Ismail-Beigi, S. Phys. ReV. Lett. 2007, 99, 115501.
(5) Zhai, H. J.; Alexandrova, A. N.; Birch, K. A.; Boldyrev, A. I.;Wang, L. S. Angew. Chem., Int. Ed. 2003, 42, 6004.
(6) Zhai, H. J.; Kiran, B.; Li, J.; Wang, L. S. Nat. Mater. 2003, 2,827.
(7) (a) Kiran, B.; Bulusu, S.; Zhai, H. J.; Yoo, S.; Zeng, X. C.; Wang,L. S. Proc. Natl. Acad. Sci. U.S.A. 2003, 102, 961. (b) Sergeeva, A. P;Zubarev, D. Yu.; Zhai, H. J.; Boldyrev, A. I.; Wang, L. S. J. Am. Chem.Soc. 2008, 130, 7244. (c) Huang, W.; Sergeeva, A. P.; Zhai, H. J.; Averkiev,B. B.; Wang, L. S.; Boldyrev, A. I. Nat. Chem. 2010, 2, 202.
(8) For a review on all-boron aromatic clusters, see: Alexandrova,A. N.; Boldyrev, A. I.; Zhai, H. J.; Wang, L. S. Coord. Chem. ReV. 2006,250, 2811.
(9) Huber, K. P.; Herzberg, G. Constants of Diatomic Molecules; VanNostrand Reinhold: New York, 1979.
(10) Pyykko, P. Mol. Phys. 1989, 67, 871.(11) Zhai, H. J.; Wang, L. M.; Li, S. D.; Wang, L. S. J. Phys. Chem. A
2007, 111, 1030.(12) Bauer, S. H. Chem. ReV. 1996, 96, 1907.(13) (a) Weltner, W., Jr.; Warn, J. R. W. J. Chem. Phys. 1962, 37, 292.
(b) Sommer, A.; White, D.; Linevsky, M. J.; Mann, D. E. J. Chem. Phys.1963, 38, 87. (c) Ruscic, B. M.; Curtiss, L. A.; Berkowitz, J. J. Chem.Phys. 1984, 80, 3962. (d) Doyle, R. J., Jr. J. Am. Chem. Soc. 1988, 110,4120. (e) Hanley, L.; Anderson, S. L. J. Chem. Phys. 1988, 89, 2848. (f)Burkholder, T. R.; Andrews, L. J. Chem. Phys. 1991, 95, 8697. (g)Nemukhin, A. V.; Weinhold, F. J. Chem. Phys. 1993, 98, 1329. (h) Peiris,D.; Lapicki, A.; Anderson, S. L.; Napora, R.; Linder, D.; Page, M. J. Phys.Chem. A 1997, 101, 9935.
(14) Kawashima, Y.; Endo, Y.; Kawaguchi, K.; Hirota, E. Chem. Phys.Lett. 1987, 135, 441.
(15) (a) Lory, E. R.; Porter, R. F. J. Am. Chem. Soc. 1971, 93, 6301.(b) Snelson, A. High Temp. Sci. 1972, 4, 141. (c) Snelson, A. High Temp.Sci. 1972, 4, 318. (d) Andrews, L.; Burkholder, T. R. J. Phys. Chem. 1991,95, 8554. (e) Lanzisera, D. V.; Andrews, L. J. Phys. Chem. A 1997, 101,1482. (f) Bettinger, H. F. Organometallics 2007, 26, 6263.
(16) Bock, H.; Cederbaum, L.; von Niessen, W.; Paetzold, P.; Rosmus,P.; Solouki, B. Angew. Chem., Int. Ed. Engl. 1989, 28, 88.
(17) (a) McAnoy, A. M.; Dua, S.; Schroder, D.; Bowie, J. H.; Schwarz,H. J. Phys. Chem. A 2003, 107, 1181. (b) McAnoy, A. M.; Dua, S.;Schroder, D.; Bowie, J. H.; Schwarz, H. J. Phys. Chem. A 2004, 108, 2426.
(18) Braunschweig, H.; Radacki, K.; Schneider, A. Science 2010, 328,345.
(19) (a) Ehlers, A. W.; Baerends, E. J.; Bickelhaupt, F. M.; Radius, U.Chem.sEur. J. 1998, 4, 210. (b) Li, S. D.; Miao, C. Q.; Guo, J. C.; Ren,G. M. J. Comput. Chem. 2005, 26, 799. (c) Ren, G. M.; Li, S. D.; Miao,C. Q. J. Mol. Struct.: THEOCHEM 2006, 770, 193. (d) Drummond, M. L.;Meunier, V.; Sumpter, B. G. J. Phys. Chem. A. 2007, 111, 6539.
(20) Zhou, M.; Jiang, L.; Xu, Q. Chem.sEur. J. 2004, 10, 5817.(21) Zubarev, D. Yu.; Boldyrev, A. I.; Li, J.; Zhai, H. J.; Wang, L. S.
J. Phys. Chem. A 2007, 111, 1648.(22) Zhai, H. J.; Li, S. D.; Wang, L. S. J. Am. Chem. Soc. 2007, 129,
9254.(23) Li, S. D.; Zhai, H. J.; Wang, L. S. J. Am. Chem. Soc. 2008, 130,
2573.(24) Yao, W. Z.; Guo, J. C.; Lu, H. G.; Li, S. D. J. Phys. Chem. A
2009, 113, 2561.(25) Shao, C. B.; Jin, L.; Fu, L. J.; Ding, Y. H. Theor. Chem. Acc. 2009,
124, 161.(26) Ducati, L. C.; Takagi, N.; Frenking, G. J. Phys. Chem. A 2009,
113, 11693.(27) (a) Nguyen, M. T.; Matus, M. H.; Ngan, V. T.; Grant, D. J.; Dixon,
D. A. J. Phys. Chem. A 2009, 113, 4895. (b) Tai, T. B.; Nguyen, M. T.Chem. Phys. Lett. 2009, 483, 35. (c) Tai, T. B.; Nguyen, M. T.; Dixon,D. A. J. Phys. Chem. A 2010, 114, 2893.
(28) For selected reviews, see: (a) Hall, K. P.; Mingos, D. M. P. Prog.Inorg. Chem. 1984, 32, 237. (b) Jones, P. G. Gold Bull. 1981, 14, 102;1981, 14, 159; 1983, 16, 114; 1986, 19, 46. (c) Whetten, R. L.; Shafigullin,M. N.; Khoury, J. T.; Schaaff, T. G.; Vezmar, I.; Alvarez, M. M.; Wilkinson,A. Acc. Chem. Res. 1999, 32, 397.
(29) (a) Zhai, H. J.; Burgel, C.; Bonacic-Koutecky, V.; Wang, L. S.J. Am. Chem. Soc. 2008, 130, 9156. (b) Wang, X. B.; Wang, Y. L.; Yang,J.; Xing, X. P.; Li, J.; Wang, L. S. J. Am. Chem. Soc. 2009, 131, 16368.
(30) Wang, L. S. Phys. Chem. Chem. Phys. 2010, 12, 8694.(31) Kiran, B.; Li, X.; Zhai, H. J.; Cui, L. F.; Wang, L. S. Angew. Chem.,
Int. Ed. 2004, 43, 2125.(32) Li, X.; Kiran, B.; Wang, L. S. J. Phys. Chem. A 2005, 109, 4366.(33) Kiran, B.; Li, X.; Zhai, H. J.; Wang, L. S. J. Chem. Phys. 2006,
125, 133204.(34) Zhai, H. J.; Wang, L. S.; Zubarev, D. Yu.; Boldyrev, A. I. J. Phys.
Chem. A 2006, 110, 1689.(35) Zubarev, D. Yu.; Li, J.; Wang, L. S.; Boldyrev, A. I. Inorg. Chem.
2006, 45, 5269.(36) Alexandrova, A. N.; Koyle, E.; Boldyrev, A. I. J. Mol. Model. 2006,
12, 569.(37) (a) Exner, K.; Schleyer, P. v. R. Science 2000, 290, 1937. (b) Wang,
Z. X.; Schleyer, P. v. R. Science 2001, 292, 2465.(38) (a) Wang, L. M.; Huang, W.; Averkiev, B. B.; Boldyrev, A. I.;
Wang, L. S. Angew. Chem., Int. Ed. 2007, 46, 4550. (b) Averkiev, B. B;Zubarev, D. Yu.; Wang, L. M.; Huang, W.; Wang, L. S.; Boldyrev, A. I.J. Am. Chem. Soc. 2008, 130, 9248. (c) Averkiev, B. B.; Wang, L. M.;Huang, W.; Wang, L. S.; Boldyrev, A. I. Phys. Chem. Chem. Phys. 2009,11, 9840.
(39) Miao, C. Q.; Guo, J. C.; Li, S. D. Sci. China, Ser. B 2009, 52, 900.(40) Pu, Z. F.; Ito, K.; Schleyer, P. v. R.; Li, Q. S. Inorg. Chem. 2009,
48, 10679.(41) Minyaev, R. M.; Gribanova, T. N.; Starikov, A. G.; Minkin, V. I.
MendeleeV Commun. 2001, 11, 213.(42) Ito, K.; Pu., Z. F.; Li, Q. S.; Schleyer, P. v. R. Inorg. Chem. 2008,
47, 10906.(43) (a) Averkiev, B. B.; Boldyrev, A. I. Russ. J. Gen. Chem. 2008, 78,
769. (b) Guo, J. C.; Yao, W. Z.; Li, Z.; Li, S. D. Sci. China, Ser. B 2009,52, 566.
(44) (a) Erhardt, S.; Frenking, G.; Chen, Z. F.; Schleyer, P. v. R. Angew.Chem., Int. Ed. 2005, 44, 1078. (b) Islas, R.; Heine, T.; Ito, K.; Schleyer,P. v. R.; Merino, G. J. Am. Chem. Soc. 2007, 129, 14767. (c) Yang, Z.;Xiong, S. J. J. Chem. Phys. 2008, 128, 184310. (d) Luo, Q. Sci. China,Ser. B 2008, 51, 607. (e) Wu, Q. Y.; Tang, Y. P.; Zhang, X. H. Sci. China,Ser. B, Chem. 2009, 52, 288.
(45) Wang, L. S.; Cheng, H. S.; Fan, J. J. Chem. Phys. 1995, 102, 9480.(46) Wang, L. S.; Wu, H. In AdVances in Metal and Semiconductor
Clusters, Vol. 4, Cluster Materials; Duncan, M. A., Ed.; JAI Press:Greenwich, CT, 1998; pp 299-343.
(47) (a) Wang, L. S.; Li, X. In Clusters and Nanostructure Interfaces;Jena, P., Khanna, S. N., Rao, B. K., Eds.; World Scientific: River’s Edge,NJ, 2000; pp 293-300. (b) Akola, J.; Manninen, M.; Hakkinen, H.;Landman, U.; Li, X.; Wang, L. S. Phys. ReV. B 1999, 60, R11297. (c) Wang,L. S.; Li, X.; Zhang, H. F. Chem. Phys. 2000, 262, 53. (d) Zhai, H. J.;Wang, L. S.; Alexandrova, A. N.; Boldyrev, A. I. J. Chem. Phys. 2002,117, 7917.
(48) (a) Becke, A. D. J. Chem. Phys. 1993, 98, 5648. (b) Lee, C.; Yang,W.; Parr, R. G. Phys. ReV. B 1988, 37, 785.
(49) Kendall, R. A.; Dunning, T. H.; Harrison, R. J. J. Chem. Phys.1992, 96, 6796.
12160 J. Phys. Chem. A, Vol. 114, No. 46, 2010 Zhai et al.

(50) (a) Schuchardt, K. L.; Didier, B. T.; Elsethagen, T.; Sun, L.;Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T. L. J. Chem. Inf. Model. 2007,47, 1045. (b) Martin, J. M. L.; Sundermann, A. J. Chem. Phys. 2001, 114,3408.
(51) (a) Casida, M. E.; Jamorski, C.; Casida, K. C.; Salahub, D. R.J. Chem. Phys. 1998, 108, 4439. (b) Bauernschmitt, R.; Ahlrichs, R. Chem.Phys. Lett. 1996, 256, 454.
(52) (a) Li, X.; Kiran, B.; Li, J.; Zhai, H. J.; Wang, L. S. Angew. Chem.,Int. Ed. 2002, 41, 4786. (b) Li, J.; Li, X.; Zhai, H. J.; Wang, L. S. Science2003, 299, 864.
(53) Frisch, M. J.; Trucks, G. M.; Schlegel, H. B.; Scuseria, G. E.; Robb,M. A.; Cheeseman, J. R.; Montgomery, J. A.; Vreven, T.; Kudin, K. N.;Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.;Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.;Nakatsuji, H.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian,H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann,R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.;Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.;
Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.;Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz,J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.;Liu; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.;Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe,M.; Gill, P. M. W.; Johnson, B. G.; Chen, W.; Wang, M. W.; Gonzales,C.; Pople, J. A. Gaussian 03, revision A.1; Gaussian, Inc.: Pittsburgh, PA,2003.
(54) (a) Liu, S. R.; Zhai, H. J.; Wang, L. S. Phys. ReV. B 2002, 65,113401. (b) Liu, S. R.; Zhai, H. J.; Wang, L. S. J. Chem. Phys. 2002, 117,9758.
(55) Zhai, H. J.; Wang, B.; Huang, X.; Wang, L. S. J. Phys. Chem. A2009, 113, 3866.
(56) Zubarev, D. Yu.; Boldyrev, A. I. J. Comput. Chem. 2007, 28, 251.(57) (a) Gingerich, K. A. J. Chem. Phys. 1971, 54, 2646. (b) Barysz,
M.; Urban, M. AdV. Quantum Chem. 1997, 28, 257.
JP108668T
Bonding in B11O- and B10Au- Clusters J. Phys. Chem. A, Vol. 114, No. 46, 2010 12161





























