Embed Size (px)
Citation preview

1OPEN COMPUTING GRIDFOR MOLECULAR SCIENCES
Mathilde Romberg, Emilio Benfenati, and Werner Dubitzky
All substances are poisons; there is none that is not a poison. The right dose differentiatesa poison from a remedy.
Paracelsus (1493–1541)
1.1 INTRODUCTION
The number of chemicals in society is largely increasing, and therewith the risk ofbeing exposed to chemicals increases. Knowledge of possible toxic effects of thesechemicals is vital, as are the measurement and assessment of the effects and relatedrisks. Within the European Union, the Registration, Evaluation, and Authorisation ofChemicals (REACH) legislation [1] places responsibility on the chemical industriesto properly assess the risks associated with their products. It has been estimated thatabout 30,000 new chemicals will be put on the European market in the coming years.The assessment of these chemicals would cost billions of euros and involve the use ofmillions of animals. REACH also aims to ensure that risks from substances of veryhigh concern (SVHC) are properly controlled or that the substances are substituted. Tomatch REACH requirements, fast and reliable methods with reproducible results arecrucial, and regulatory bodies would be able to approve results. Property predictionand modeling will play an important role in this case [2].
Toxicology, the study of harmful interactions between chemicals and biologicalsystems [3], uses more and more computer models. These models are based on alreadyavailable data and help to reduce in vivo testing. Toxicity modeling and its data havemany applications such as characterizing hazards, assessing environmental risks, andidentifying potential lead components in drug discovery. A well-established methodfor toxicity modeling is quantitative structure–activity relationship (QSAR) or quan-titative structure–property relationship (QSPR) [4,5]. On the basis of the availablemeasured and calculated properties or activities and descriptors of compounds, pre-dictive models for a certain property are built, which are then used to predict that
Grid Computing for Bioinformatics and Computational Biology. Edited by E.-G. Talbi and A.Y. Zomaya.Copyright © 2008 John Wiley & Sons, Inc.
1
COPYRIG
HTED M
ATERIAL

2 Open Computing Grid for Molecular Sciences
property for new compounds. An example for a property is the lethal dose (LD50),which is the amount of a substance that kills 50% of the population exposed to it.This property is mainly used to compare the toxicity of different compounds and toclassify them, for example, for hazard warnings.
Classical QSAR models have been based on a very limited number of parameters,which have been measured (such as simple physicochemical properties) or calculated.The model target has been to find a relationship between these parameters and theproperty within a very limited congeneric series of chemicals. These chemicals share acommon skeleton, and a few fragments are linked to it. In more recent years, there hasbeen a significant change in the QSAR scenario: The interest has shifted from theidentification of the relationship between the parameters and the property to a morepractical use, the prediction of the properties of new chemicals. This calls attention tothe predictive power of the model, since previously a model was not verified but wassimply assessed with statistical measurements evaluating the fitting of the calculatedvalues. Meanwhile, the challenge has become to model larger sets of compounds,and in addition the number of calculated chemical descriptors or fragments hasdrastically increased to several thousands. Finally, new more powerful algorithms areused, and these tools also introduce the possibility to extract new knowledge fromthe data instead of simply leading the algorithms toward well-known parametersbased on a priori knowledge or hypotheses.
Classical bioinformatics applications such as data warehousing and data miningare a major part of the model development as a result of the following:
(a) The available data are stored in very different sources such as published journalpapers, spreadsheets, and relational databases in different formats and notationswith different nomenclature and
(b) Relations between the data are mined and used for building predictive modelsof various kinds such as multilinear regression (MLR), partial least squares(PLS), or artificial neural networks (ANNs).
Other applications applied within the process of prediction model development belongto the field of molecular modeling. Calculating certain properties of a molecule on thebasis of its two- and three-dimensional structures provides the basis for the predictionof an endpoint such as LD50. Currently, the model-building and prediction processincludes a variety of steps that a toxicologist or a pharmacologist would performmanually step by step by taking care of data selection, parameter setting, data formatconversions and data transfer between each pair of subsequent steps, and so on.
Pharmaceutical industry and regulatory bodies together with environmental agen-cies are very interested in finding fast, cost-effective, easy, and reliable ways to identifycompounds with respect to their toxicity. The process of determining lead compoundsfor a new drug takes years [6,7] in the laboratory, and in addition about 90% of thepotential drugs entering the preclinical phase fail in further process due to their toxi-city [8,9]. In recent years, pharmaceutical companies along with research initiativeshave investigated modeling and prediction methods together with grid computing to

Grids for Toxicology and Drug Discovery 3
streamline and speed up processes. The prominent interest of industries lies in costreduction, for example, reducing failure rate and using in-house PCs’ idle time torun modeling tasks [10,11]. Software providers offer matching grid solutions [12,13]for the latter. These approaches exploit the embarrassingly parallel1 nature of theapplications and offer sophisticated scheduling mechanisms. They are deployed asin-house systems, that is, they do not span multiple organizations, mainly for securityreasons. Companies do not risk their data and methods being exposed to outsiders.
Publicly funded research projects in bioinformatics investigate data and compu-tational grid methods to integrate huge amounts of data, develop ontologies, modelworkflows, efficiently integrate application software including legacy codes, definestandards, and offer easy-to-use and efficient tools [14–21].
Section 1.2 of this chapter will highlight grid systems in toxicology and drugdiscovery and their main characteristics. Section 1.3 will give an in-depth overviewof the European OpenMolGRID approach, while Section 1.4 will conclude with anoutlook for future developments.
1.2 GRIDS FOR TOXICOLOGY AND DRUG DISCOVERY
Toxicology covers important issues in life and environmental sciences. It is essentialthat the characteristics of a chemical be identified before producing and releasing itinto the environment. In drug discovery, one aim is to exclude toxic, chemically un-stable and biologically inactive compounds from the drug discovery [22,23] early on inthe process. Therefore, models are being developed for predicting which compoundsare liable to fail at a later stage of the process. In this context, QSAR models areone of the most popular methods. Another goal is to identify compounds that wouldbind to a given biological receptor. The area of docking is important to understandbiological processes and find cures that succeed by activating or by inhibiting proteinactions [24]. The docking studies require the modeling of the enzyme (which has tobe known) in addition to the modeling of the small chemical compounds to be studied(ligand). However, these docking studies are more complex and do require a carefultridimensional description of the ligand. This is not always necessary in the case ofQSAR models. For this reason, faster and simpler screening based on easier methodsis often performed by drug companies, and the detailed docking studies are performedonly for a limited number of chemicals. However, grid technologies introduce newpossibilities.
The major objectives for using grid technology [25,26] to support biomathematicsand bioinformatics approaches in drug discovery and related fields are to shorten thetime to solution and reduce its costs. Users of such technology are (computational)biologists, pharmacologists, and chemists, who are usually not computer systemexperts. To bridge this gap, providing a user-friendly system is crucial. It allows
1An application is called embarrassingly parallel if no particular effort is needed to split it into a largenumber of independent problems that can be executed in parallel, and these processes do not need tocommunicate with each other.

4 Open Computing Grid for Molecular Sciences
the user to solve the biochemical problem without the knowledge about the detailsof the underlying system. Users require access to their private and publicly availabledata, execution of legacy software, and visualization of results. In cases where usersdevelop their own application software, it should also be easily integratable.
There exist quite a few initiatives, projects, and systems that exploit grid methodsfor chemoinformatics, bioinformatics, and computational biology, and some of whichfocus on applications relevant to this chapter. One of the early grid projects in drugdiscovery is the Virtual Laboratory [27]. In the beginning of this century, the VirtualLaboratory project set up an infrastructure based on the Globus Toolkit 2.4 [28] andthe Nimrod-G resource broker [29], specifically designed for parametric studies. TheVirtual Laboratory environment provides software tools and resource brokers thatfacilitate large-scale molecular studies on geographically distributed computationaland data grid resources. This is used for examining or screening millions of chemicalcompounds (molecules) in the Chemical Data Bank (CDB) to identify those hav-ing potential use in drug design. The DOCK software package [30] is integrated formolecular docking calculations and for access to the CDB, and the data replica cata-logs are provided. The user interface allows us to specify input and output data setsand locations, set parameters for the DOCK software, submit jobs, and retrieve out-put. This command-line interface has recently been replaced by a Web portal withinthe Australian BioGrid initiative [31].
DDGrid [32] is a subproject of the China grid initiative that also focuses ondocking. Its goal is to analyze chemical databases and identify compounds appro-priate for a given biological receptor. It offers access to a variety of databases suchas Specs (chemically available compounds’ structure database), MDL Comprehen-sive Medicinal Chemistry 3D (pharmaceutical compounds), National Cancer Insti-tute Database (NCI)-3D (structures with corresponding 3D models), China NaturalProducts Database, Traditional Chinese Medicinal Database (TCMD), and ZINC-ChemBridge (chemical structures). The user is provided with tools for preprocess-ing of data (Combimark), for visualization, for structure search, and for encryptingand decrypting of data. The core middleware layer is based on the Berkeley OpenInfrastructure for Network Computing (BOINC, [33]), which is a well-establishedbase for the group of “at home” systems. For example, Rosetta@home [34] usesPCs all over the world to model protein structures and interactions to understand dis-eases and find cures. Rosetta@home distinguishes itself from other grid initiatives indrug discovery by using voluntarily donated free CPU cycles to execute the Rosettaprogram. The “users” of these systems have no influence on the application. Theydownload the software that uses the free CPU cycles to run the application set up bya research group, which in this case was David Baker’s group at the University ofWashington in Seattle [35].
Within the UK e-Science program, the e-Science Solutions for the Analysis ofComplex Systems (eXSys) project [36] studies drug discovery from the angle ofinteraction networks. It analyzes protein interaction networks to identify sets of pro-teins in a bacterium that, if they were inhibited, would destroy the bacterium butnot affect its host organism. These proteins qualify as potential drug targets. eXSystackles data access and integration issues by building local data sets for intracellular

Grids for Toxicology and Drug Discovery 5
metabolic or protein interaction networks from heterogeneous resources such as theDatabase of Interacting Proteins (DIP, [37]), the Kyoto Encyclopedia of Genes andGenomes (KEGG, [38]), the Swissprot protein databank [39], and publications. Aproject internal common data format is established in which all data are integrated.The necessary network analysis programs plus their integration as grid services aredeveloped. A graphical user interface allows users to select interaction networks froma local database, analyze them, and visualize the results. myGrid [40] and OGSA-DAI/OGSA-DQP [41] are used for data access and analysis of various data sources.The myGrid infrastructure offers a workbench well suited for bioinformaticians.It includes workflow generation and enactment, as well as a variety of servicesfor data integration such as knowledge annotation and verification of experiments(KAVE), semantic discovery (Feta), and life science identifier (LSID) services for datahandling.
The aspects of workflows for the drug design pipeline are also dealt with in theWide In Silico Docking On Malaria (WISDOM) data challenge [42], a project tochallenge the infrastructure built by the Enabling grids for e-Science (EGEE) project[43]. WISDOM seeks to find new inhibitors for a family of proteins by using in silicodocking on the grid. During the 6-week data challenge in mid-2005, a terabyte of datawas produced using 80 CPU years. These data from over 40 million docked ligandsare now being further analyzed.
While most of the drug-discovery-related grid projects and systems deal withsimulations and modeling of docking ligands to proteins and the identification ofprotein functions, it is also important to identify and optimize lead molecules withthe targeted therapeutic potential. Pharmaceutical companies set up in-house gridsystems to also cover this aspect. Little is published about these grid systems, butinformation can be found in case studies of software vendors [10] and press releases[44–46]. Key to this approach is workflow modeling, semantic Web technologies, anddata management.
The approaches described use a variety of grid middleware or infrastructuresystems, including grid service and pregrid-service versions of the Globus Toolkit,gLite (basis of the EGEE test bed), Web services, network computing (desktop grid),myGrid, and the pregrid-service version of UNICORE. With respect to middleware,all further developments aim at a service-oriented architecture (SOA), whatever typeof resource is being used. The Open grid Services Architecture (OGSA [47]) has beendefined by the Open grid Forum [48] to achieve a standardized grid framework. Fordrug discovery, the topics workflow modeling, application integration, standards fordata structures, metadata and ontologies, and data integration are equally important.
Recently, a series of activities has addressed both the issues of databases of chem-ical compounds used in the world and how to predict their environmental and toxicproperties. The European Commission’s Joint Research Centre is considering thestrategic development of a general system to predict properties of industrial chemi-cals. The U.S. Environmental Protection Agency (EPA), which adopts predictive toolsfor the property predictions of chemicals for decades, is also enlarging its set of tools.The Danish EPA predicted properties of tens of thousands of chemicals using a setof software. All these initiatives show the deep interest in a more powerful approach

6 Open Computing Grid for Molecular Sciences
capable to cope with a problem that involves many programs, databases, and resources,which would surely benefit from an integrated strategy supported by grid.
The following section will detail these characteristics taking the OpenMolGRIDsystem as an example.
1.3 EXAMPLE OpenMolGRID
The Open Computing grid for Molecular Sciences and Engineering (OpenMolGRID,[49]) system has been developed to support the lead component identification indrug discovery and designed in an open manner to be of immediate use for otherapplications in biochemistry and pharmacology. The objectives of this project are to
� Develop tools that permit end users to securely and seamlessly access, integrate,and use relevant globally distributed data resources;
� Develop tools that permit end users to securely and seamlessly access, use, andschedule globally distributed computational methods and tools; and
� Provide foundations and design principles for developing and constructing next-generation molecular engineering systems.
The selected underlying grid middleware UNICORE [50] offers well-designed in-terfaces for application software integration both on the user client side and on theexecution server side. It provides data access and data transfer together with work-flow modeling, execution, and monitoring. To facilitate the development of predictionmodel and prediction workflows, the OpenMolGRID project developed abstractionlayers to easily integrate application software and to access different publicly avail-able relevant databases (e.g., ECOTOX [51], NTP [52]), and built a data warehousefrom that data [53]. It includes automated workflow support that simplifies the task ofthe user by including support steps such as data conversion and data transfer into theworkflow, by automatically assigning appropriate execution servers, and by exploitingparallelism [54,55].
The general architecture underlying the OpenMolGRID system is depicted inFig. 1.1. The abstract resource interfaces are the key to flexibility providing a com-mon interface accessible by the UNICORE server and a resource-specific interfaceon the resource side. Each resource has an XML resource description attached toinform the server and the client about input and output characteristics and behavior.The challenges addressed in the OpenMolGRID project are as follows:
� Molecular design and engineering are computationally very demanding, andthey generates huge amounts of data.
� Data from a variety of different sources need to be integrated using data ware-housing techniques, and the data need to be accessible seamlessly.
� The scientific workflows involve heterogeneous data, compute, and applicationresources.

Example OpenMolGRID 7
Figure 1.1. OpenMolGRID high-level architecture.
� The scientific workflows are fairly complex and involve multiple dependentsteps.
These challenges become obvious when analyzing, for example, the QSAR model-building process: First, relevant experimental (toxicity) data are searched from one ormultiple sources, which requires that all sources be accessible through the system andthat the relevant data can be extracted. The structural information is then extracted anda 3D structure is generated for each molecule. Application software such as MOL-GEO [56] is used to accomplish this task. In the next step, the generated moleculargeometry is optimized using the molecular modeling software for example, MOPAC[57]. These applications process a single structure at a time so that the tasks can easilybe distributed. This is also true for the next step in the workflow, the calculation of des-criptors, for example, using the molecular descriptor analyzer (MDA) module fromthe Codessa [58] software package. The results from all structures serve as input tomodel development software—for example, the best multilinear regression (BMLR)analysis module of Codessa. The following sections will describe the OpenMolGRIDsolutions in detail.
1.3.1 Data Management
Besides storage of data, the major challenge in data management is the access toevery kind of data source containing data relevant to drug discovery, toxicology,pharmacology, and, of course, the interpretation and integration of these data.

8 Open Computing Grid for Molecular Sciences
1.3.1.1 Access to Data Sources
The abstraction layer for data sources is realized as a set of metadata and a server-sidewrapper application called database access tool that encapsulates the communicationwith the underlying database system. The output data are sent to the client in an XMLformat that is designed for easy automatic processing. The metadata file containsinformation on the database layout and semantic information—for example, databasename, access restrictions, information about the database intended for the user, tablenames, field names, and types.
The XML structure defined to transfer the data from the data source to the userclient or as input to other applications is a list of elements. This facilitates an easytransformation to other formats, for instance, application software input formats, andfor easy extraction of certain fields, for example, the structure of a chemical compound.
1.3.1.2 Data Warehouse
Predictive QSAR/QSPR modeling requires the handling and management of chem-ical structure and property data, along with data relating to molecular descriptors.Often these data must be retrieved from public or private data repositories as wellas integrated and formatted so that it is amenable to data mining methods such aslinear regression methods, artificial neural networks, and decision tree algorithms.Data warehousing [59] provides the data integration and formatting functionality re-quired by data mining applications. It is employed to integrate, cleanse, normalize,and consolidate data from different sources and to map them onto “ready-to-use” datastructures (e.g., by denormalizing relational database tables). Within the OpenMol-GRID system, a grid-enabled data warehouse for molecular engineering environmentshas been developed [53]. Its main purpose is to provide integrated and consolidateddata originating from selected data resources relevant to molecular engineering. Thefollowing data resources have been integrated:
� National Toxicology Program database, which provides information regardingpotentially toxic chemicals to health regulatory and research agencies, the sci-entific and medical communities, and the public NTP [52]
� ECOTOX (ecotoxicology) databases Aquire and Terretox, which providechemical toxicity information for aquatic and terrestrial life, respectively [51]
� Multidrug resistance (MDR) data set, proprietary� G-protein-coupled receptor (GPCR) data set, proprietary
The databases integrated in the data warehouse are harvested from the sources men-tioned above and are mapped into the warehouse and its physical repository. A de-tailed view of the OpenMolGRID data warehouse and its relation to the Web and otherOpenMolGRID components is depicted in Fig. 1.2. The warehouse processes followthe typical extract, transform, and load scheme (also known as ETL). According toreflect updates in the underlying databases, the warehousing process is performedperiodically. The extract component transfers the database from its public Web siteas single or multiple files (depending on the database) to the data warehouse. Each

Example OpenMolGRID 9
Figure 1.2. OpenMolGRID data warehouse and related components.
database has its own implementation-specific format from which the data are extractedand mapped into the data transformation environment. Within this environment, thedata are denormalized from relational databases, cleansed (inconsistent entries areremoved), enriched, and standardized based on the requirements of the molecularengineering environments. New fields are computed to facilitate different types ofanalyses. For example, the log inverse of the measured dosage of a chemical’s tox-icity is often more useful for some calculations and models than that of the toxicityvalue itself. By providing this value within the warehouse’s data structures, the userdoes not have to perform this calculation and can focus on the more intricate aspectsof the modeling task at hand. Data normalization may involve, for example, missingvalue imputation, mean centering, or alignment to canonical units. In addition, com-plex data transformations (descriptor calculations) are integrated into the warehouseagain to offer values the users would have to calculate otherwise.
The transformed data are then loaded into the data warehouse’s physical datastorage, a PostgreSQL relational database. Client access to data in the OpenMolGRIDdata warehouse is enabled via the generic database access tool mentioned above.Inputs and outputs are encapsulated in an OpenMolGRID-specific XML syntax anddata are easily identifiable due to being associated with generic data types definedespecially for OpenMolGRID’s data needs. These data types are used throughout allapplications in the OpenMolGRID system.
The data warehouse’s transformation environment includes the calculation of cer-tain descriptor values as mentioned. Specialized software is required to perform thesecalculations and they are expensive to compute, especially if there are a large numberof chemicals and several representations of the same chemical. From a data ware-house perspective, these descriptor calculations are complex data transformations.

10 Open Computing Grid for Molecular Sciences
Therefore, the most frequently used molecular descriptors are calculated for eachmolecular structure in the data warehouse. Besides the traditional molecular descrip-tor types, a physicochemical parameter, the log P value (octanol–water partition co-efficient), is also calculated for the compounds. Essentially, the descriptor calculationprocedure amounts to virtualization of parts of the data warehouse’s data transforma-tion processes. This virtualization functionality is realized by the development of thecommand-line interface for UNICORE.
1.3.1.3 A Data Type for Toxicity
A major challenge is that many data resources contain inconsistencies by way ofthe same data (types) represented in different records (the idea of a record variesfrom source to source). For example, supposing we have decided that the stan-dardized data unit for a particular dosage field is milligrams per kilogram (mg/kg),there may be variations in the style, a source represents this. Some records maycontain Mg/kg (or some other variation), thereby causing inconsistencies with thestandard realized in the OpenMolGRID warehouse. The taxonomy step in the pro-cess flow described enables any number of substitutions to be defined to ensurethat consistency is maintained within, and between, data resources entering thewarehouse. Characteristic of many data sources is the idea that each data fieldcontains a value from a set of allowable values. This can be problematic when anumber of resources are being integrated into a data warehouse. For example, adosage field can have several measurement units associated with it—for example,g/kg, mg/kg, or �g/kg—which have to be aligned to be usable for data mining. Inthe absence of a data warehouse, this must be performed manually, but in theOpenMolGRID data warehouse, anautomated mechanism is required. The mech-anism was developed based on canonical units or primitives. Measurement unitscan be broken down into several categories—for example, length, weight, and time.Each of these categories has an associated base unit, the unit primitive—for exam-ple, kilograms for weight. For the conversion between various forms of the samemeasurement category, scaling factors (which can be more complex mathematicalformulations) are defined, in both directions, to enable dynamic conversion from oneunit to another.
1.3.1.4 Data Storage
Besides the data warehouse that contains the cleaned and transformed data fromavailable data sources, space to store (intermediate) results is required. A relationaldatabase has been set up to support the complete molecular engineering process. Itis capable of handling all data generated in the OpenMolGRID system (molecules,descriptors, models, experimental property values, predicted property values, etc.).It is set up as a read/write store, while the data warehouse is read-only from the endusers’ perspective.
Very important for a data store for molecular sciences is a structure and substructuresearch capability that has been developed. This function is necessary for identifyingthe best subset of data (chemical compounds) to be used for further analysis and isfundamental in chemical and related communities. The substructure search is realized

Example OpenMolGRID 11
Figure 1.3. Application interface.
as a two-part process with two different queries. The first query aims to select asubset of structures that may contain the substructure. This is performed using afingerprinting approach, which significantly accelerates the search. Fingerprints of thestructures are matched against the fingerprint of the substructure, and those structuresthat cannot possibly match are removed from the set. The second query is performedwithin the matching subset to select structures that actually contain the full chemicalsubstructure. The comparison is computationally expensive, which makes it importantto use the first step to reduce the input set for the second query and thereby make theoverall process more efficient.
1.3.2 Application Integration
Similar to the integration of data sources through metadata and access software(database access tool), all kinds of applications can be integrated into the systemas shown in Fig. 1.3. The abstract interface is realized as a wrapper to existing soft-ware modules. It provides the description of the application (its metadata) and theinput/output (I/O) data format conversion routines from the standard data format tothe proprietary and vice versa. The metadata also define the interface and I/O formatinformation used by clients. As a result, a well-defined application on the server sidecan be addressed on the user client side by an application-specific interface as shownin the following section.
1.3.3 User Interface
OpenMolGRID, being based on the UNICORE grid middleware [60], includes theUNICORE graphical client (see Fig. 1.4), which is shown here with the detailedworkflow for model building (for the description of the coarse-grained workflow,see introduction to Section 1.3). It is a Java application offering job creation andmonitoring for complex multistep and multisite jobs. Jobs are composed of subjobs,tasks, and dependencies reflecting temporal and data dependencies. Jobs are repre-sented by acyclic directed graphs with tasks and subjobs as vertices and dependencies
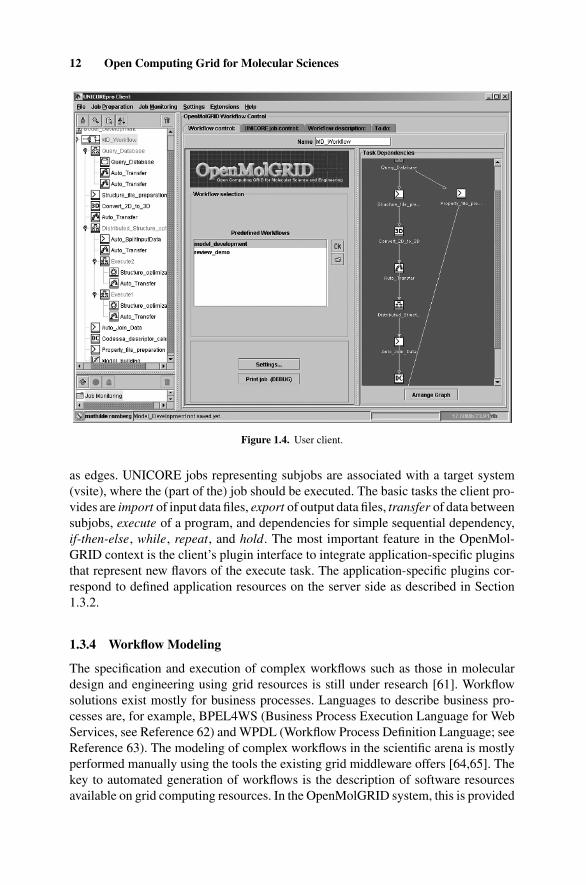
12 Open Computing Grid for Molecular Sciences
Figure 1.4. User client.
as edges. UNICORE jobs representing subjobs are associated with a target system(vsite), where the (part of the) job should be executed. The basic tasks the client pro-vides are import of input data files, export of output data files, transfer of data betweensubjobs, execute of a program, and dependencies for simple sequential dependency,if-then-else, while, repeat, and hold. The most important feature in the OpenMol-GRID context is the client’s plugin interface to integrate application-specific pluginsthat represent new flavors of the execute task. The application-specific plugins cor-respond to defined application resources on the server side as described in Section1.3.2.
1.3.4 Workflow Modeling
The specification and execution of complex workflows such as those in moleculardesign and engineering using grid resources is still under research [61]. Workflowsolutions exist mostly for business processes. Languages to describe business pro-cesses are, for example, BPEL4WS (Business Process Execution Language for WebServices, see Reference 62) and WPDL (Workflow Process Definition Language; seeReference 63). The modeling of complex workflows in the scientific arena is mostlyperformed manually using the tools the existing grid middleware offers [64,65]. Thekey to automated generation of workflows is the description of software resourcesavailable on grid computing resources. In the OpenMolGRID system, this is provided

Example OpenMolGRID 13
by the application’s abstraction layer. These descriptions can be used for automatedapplication identification and inclusion in multistep workflows. The following para-graphs will describe the solution for automated workflow specification and processingdeveloped within the OpenMolGRID project.
Workflow Specification. Workflows consist of task or process elements with logi-cal, temporal, and data dependencies. Tasks may also be independent of other tasksin a workflow. As described in Section 1.3.3, OpenMolGRID uses the UNICOREclient that offers graphical workflow specification to build up complex jobs. Existingworkflow description languages do not match the UNICORE model with respect toapplication software resources. As these play the most important role within the au-tomatic job generation, a workflow specification language has been developed thatenables a high-level definition of various scientific processes containing sufficientinformation for the automatic generation of complete UNICORE jobs. This includesthe tasks and their dependencies, as well as the necessary resources. XML has beenselected as a specification language for the workflow. A core element in a workflowis the task, which is described by
� A string, the task’s function to be fulfilled by an application resource and sup-ported by a client plugin
� A string, the UNICORE task identifier in the job tree� An integer, the unique numerical task identification within the workflow� A boolean, the export flag specifying whether result files are to be exported to
the user’s workstation� A boolean, the split flag specifying whether the task is data parallel and can be
distributed onto several execution systems� A string, the identifier of an application that is capable of splitting the input data
for this task into n chunks (splittertask)� A string, the identifier of an application that is capable of joining the n result
files into one file (joinerTask)� A set of options to feed the application with parameter settings
A set of resources is specified for a task requesting runtime, number of nodes, numberof processors, and memory. The target system for the execution of all tasks within aUNICORE (sub)job can be specified by usite and vsite.
The following shows the XML workflow specification for the model developmentprocess:
<?xml version=“1.0” ?><!– *** Model Multi-Drug resistance on OpenMolGRID data warehouse data ***−− >
<workflow xmlns=“http://www.openmolgrid.org/namespaces/2004/WorkflowDescription” xmlns:rd=“http://www.openmolgrid.org/namespaces/2004/SimpleResources” >
<group type=“subjob” identifier=“Query Database” id=“1” >

14 Open Computing Grid for Molecular Sciences
<!– wrapper group to allow easy datasource selection –>
<task name=“DataBaseRequest” identifier=“Query Database” id=“11”export=“false” split=“false” >
<option name=“query” value=“SELECT chemical.moldw id,chemical.structuretype, chemical.fileformat,chemical.molecularstructure,property.propertyid,property.propertyname,property.loginverse FROM (chemical LEFT JOIN property ONchemical.moldw id=property.moldw id) WHERE chemical.molecularstructure!=“ and property.propertyname like ‘Multi-Drug%”’ />
</task>
<task name=“DataBaseRequestToPLF” identifier=“Property filepreparation” id=“3” export=“false” split=“false” />
<task name=“DataBaseRequestToSLF” identifier=“Structure filepreparation” id=“2” export=“false” split=“false” />
<resourceRequest><rd:node usite=“Ulster OMG” vsite=“MOLDW” />
</resourceRequest></group>
<task name=“2Dto3Dconversion” identifier=“Convert 2D to 3D” id=“21”export=“false” split=“false” />
<task name=“SemiempiricalCalculation” identifier=“Structure optimization”id=“25” export=“false” split=“true” splitterTask=“SplitStructureList”joinerTask=“JoinStructureLists” >
<option name=“keywords” value=“AM1 PRECISE 1SCF NOINTER” /></task>
<task name=“DescriptorCalculation” identifier=“Codessa descriptorcalculation” id=“29” export=“false” split=“false” />
<task name=“ModelBuilding” identifier=“Model building” id=“40”export=“false” split=“false”><resourceRequest>
<rd:resources runTime=“3600” /></resourceRequest>
</task>
<dependency pred=“11” succ=“2” /><!– db request to structure extract –>
<dependency pred=“11” succ=“3” /><!– db request to property extract –>
<dependency pred=“3” succ=“40” /><!– property extract to model building –>
<dependency pred=“2” succ=“21” /><!– struct extract to 2d to 3d –>
<dependency pred=“21” succ=“25” /><!– 2d to 3d to semiempirical –>
<dependency pred=“25” succ=“29” /><!– semiempirical to descriptor calc –>
<dependency pred=“3” succ=“40” /><!– property extract to model building –>
<dependency pred=“29” succ=“40” /><!– descriptor calc to Modelbuilding –>
<resourceRequest><rd:node usite = “TartuOMG” vsite = “VSite1”/ >
</resourceRequest></workflow>

Example OpenMolGRID 15
Workflow Processing. A workflow specified as described serves as input to theMetaPlugin, a special plugin to the UNICORE client. The MetaPlugin parses theXML workflow, creates the corresponding UNICORE job, and assigns target systemsand resources to it. These tasks include a number of sophisticated actions:
� Subjobs are introduced into the job wherever necessary, for example, whenrequested applications are not available on the same target system.
� Transfer tasks are introduced into the job to transmit data from one target systemto another, which is the execution system of a subjob.
� Data conversion tasks are added between two tasks where the output format(specified in XML according to the application metadata) of one task does notmatch the input format of the successor task.
� Splitter and transfer tasks are added to the workflow as predecessor tasks of asplitable task for input data preparation.
� Subjobs are created around split tasks for each selected target system and atransfer task to transfer the output data back to the superordinate subjob.
� Joiner tasks are added to join the output data of split tasks.� The directed acyclic graph of dependencies between all tasks (the Explicit ones
from the workflow specification and the automatically generated ones) is set up.
The MetaPlugin uses the resource information provided by the target system (vsite),the metadata of the applications, and information about the plugins available to theclient. A resource information provider component has been developed to support theMetaPlugin in resource selection. It returns the client plugin handling the function, thetarget systems offering the corresponding application, and the I/O formats. Currently,the MetaPlugin does resource selection at a basic level, but a more sophisticatedresource broker component can easily be added. The main advantage of this mecha-nism is that a user who wants to do model building can name the coarse-grained tasksand their dependencies in an XML workflow, thereby avoiding the tedious job of thestep-by-step preparation of the complex workflow of the corresponding UNICOREjob. The latter would demand detailed knowledge about, for example, I/O formatsfor the inclusion of data conversion tasks and the manual splitting and distribution oftasks onto appropriate target systems. The automatic UNICORE job creating givesthe flexibility to the system to adapt to the actual grid layout and resource availabilityand helps to avoid human errors.
Figure 1.4 shows on the left-hand side the UNICORE job the MetaPlugin generatedfrom the workflow detailed. All tasks starting with “Auto ” have been added as are thegroups “Execute1” and “Execute2,” where the semiempirical calculation is distributedamong systems offering the necessary application software. “Auto Transfers” areused, for example, to transfer data from the database to the systems where the structureconversion and the model development are to be executed. The “Auto SplitInputData”and “Auto Join Data” tasks have been included to partition the input data for thesemiempirical calculation to allow for its distributed execution and to join the resultfiles after the execution is completed.

16 Open Computing Grid for Molecular Sciences
1.3.5 Experience
One of the most noteworthy outcomes of the OpenMolGRID project is that it pavesthe way for standardization of model-building and prediction processes. Within Open-MolGRID, we checked that results obtained with the automatic process are equivalentto those obtained by manual modeling, both for the chemical descriptor calculationand for the QSAR results. This shows that OpenMolGRID has practical potentialas a regular tool for QSAR modeling. It is important to note the advantages in thisdirection offered by the OpenMolGRID approach for models for regulatory purposes.Indeed, in the case of scientific applications the automatic simplified process is surelyconvenient and appealing, and it speeds up the application. But for regulatory pur-poses, it becomes necessary to get the same result independently of the user. So farin most of the cases QSAR models, except those within the classical approach with afew simple parameters, require manual steps that produce variable results due to op-timization differences and the lack of sufficient details. The availability of automaticQSAR modeling tools would surely cover the need for more reproducible results,which is a requirement for results to be used within a regulatory context: The imple-mentation of a candidate protocol for QSAR modeling as a workflow would achievereproducibility, easy models, and suitability for regulatory purposes [66].
Mazzatorta et al. [67] obtained stable and thoroughly validated QSARs using theOpenMolGRID system, and Maran et al. [68] detailed the use of OpenMolGRID forthe development of QSAR models for the prediction of HIV-1 protease inhibitionability of potential inhibitors. They pointed out that building the model is accom-plishable within 1 h using the system instead of 1 day because of automation of theworkflow and parallel execution of tasks. This shows that the objective to shorten thetime to solution has been achieved. Especially the automatic distribution of a taskonto the available systems and the automated output/input format conversion accountfor this.
During the development of the system, application integration has proved to beeasy because the application software itself does not need to be adapted, only awrapper implementing the abstract interface has to be developed. The data ware-house’s transformation process has been significantly improved by the provision of acommand-line interface and a queuing component to the UNICORE system. The datawarehouse uses these components to submit descriptor calculation to grid resourcesand retrieve the output. Indirectly, this also speeds up the user’s workflows becausevalues that every user requires are already calculated up-front and provided in thedata warehouse.
The current lack of an XML editor to generate the workflows makes it difficultfor the toxicologists to prepare their own workflows. These have to be prepared bysomeone familiar with XML and the workflow schema which can easily be coveredfor standard workflows that are prepared initially and made available to everyone—for example, the model development process, but not for, for example, experimentalworkflows.
A set of open issues has arisen from data handling. Standard formats should beused wherever possible, but, for example, there is not yet an established standard

Summary and Outlook 17
for globally unique identifiers for molecular descriptors. How to store the predictivemodels has not yet been resolved because PMML (predictive model markup language,[69]) is not sufficient, and it cannot be used to describe PLS (partial least square) orPCR (principal components regression) models. An extension to PMML could be asolution. For a molecular structure, multiple conformations can exist, and the handlingof these multiple data including their storage, selection, and processing has not yetbeen resolved in chemoinformatics. These topics will, among others, be dealt with inthe EC-funded project Chemomentum [70].
1.4 SUMMARY AND OUTLOOK
QSAR modeling, in silico modeling, is a prominent method in toxicology and phar-macology. Important is the quality of the models; for example, the REACH legislationrequires a clear estimation of the quality of a model, its accuracy, before it can beused for REACH purposes. The European Chemical Bureau is coordinating an actionon QSAR [71] in support of prediction modeling for regulatory purposes. Withindrug discovery, it is vital to have significant models for the prediction of ADMET(absorption, distribution, metabolism, excretion, and toxicity) properties [72]. Themathematical modeling is supported by grid computing because it helps speed up theprocess of model building through parallelization and user-friendliness. Pharmaceuti-cal companies save further costs with in-house grids using their idle desktop PC cyclesinstead of investing in additional compute power. The emerging service-orientedarchitectured grid systems allow interoperability and thereby support for true globalsharing of all kinds of resources (CPU cycles, data, knowledge, applications, etc.).This enables collaboration and may lead to synergy in achieving better and quickerresults. Data and knowledge management are key to interoperability. The data fromdifferent sources need to be interpretable, requiring ontologies to be further developedto enable “understanding.” The Semantic grid [73] offers the necessary framework.It can be used to automate the integration of data from different sources and theirtransformation into knowledge. The CombeChem UK e-Science project [74] demon-strates the advantages of Semantic grid for data from chemistry. On the basis of achemistry ontology and RDF (Resource Description Framework) graphs using XMLdata descriptions, it provides a flexible data structure for data integration and knowl-edge management. Drug development and toxicology are going to gain from the smartlaboratory that has developed. An aspect to be covered is data privacy and security,especially for company-owned data or medical records. These data would not be al-lowed to leave the source, but it may be allowed to mine the data locally and transferonly the results. Therefore, distributed data mining is another topic needing furtherresearch [75]. In addition to security issues, it may not be feasible to transfer data toan execution server because of their sheer volume.
In the future, grid computing will further impact procedures in toxicology andpharmacology. Having standard procedures will help regulatory bodies in their deci-sions. High-quality prediction models will reduce the amount of in vitro and in vivotestings. Nanotoxicology [76], the research on toxic effects of nanoparticles (particles

18 Open Computing Grid for Molecular Sciences
of nanometer size), will need computational and prediction models and procedures fordetermining physicochemical parameters and effects and for risk assessment. Drugdevelopment will extend its use of computational methods and knowledge exploitationto find cures. The systems, biology approach to simulate all processes in a biologicalsystem—for example, a cell—is used to further understand the way the system worksand can be influenced, which may improve drug discovery [77].
ACKNOWLEDGMENTS
This work has partially been funded by the European Commission under Grants IST-2001-37238 andFP6-2005-IST-5-033437.
REFERENCES
1. EU Chemicals Legislation—REACH (Registration, Evaluation and Authorisation of Chemicals).http://ec.europa.eu/enterprise/reach/index en.htm.
2. M.T.D. Cronin, J.S. Jaworska, J.D. Walker, M.H.I. Comber, C.D. Watts, A.P. Worth, Use of QSARsin international decision-making frameworks to predict health effects of chemical substances,Environmental Health Perspectives 111(10), 1391–1401, 2003.
3. J. Timbrell, Introduction to Toxicology, third edition, Taylor & Francis, Boca Raton, FL, 2002.
4. C. Hansch, A. Leo, Exploring QSAR: Fundamentals and Applications in Chemistry and Biology,Vol. 1, American Chemical Society, Washington, DC, Chapters 6 and 11, 1995.
5. J.D. McKinney, A. Richard, C. Waller, M.C. Newman, F. Gerberick, The practice of structure activityrelationships (SAR) in toxicology, Toxicological Sciences 56(1), 8–17, 2000.
6. American Association for the Advancement of Science. http://www.aaas.org/international/africa/gbdi/mod1c.html.
7. PPD Corporate Information. http://www.ppdi.com/corporate/faq/about drug development/home.htm.
8. UK Department of Trade and Industry (DTI), call for research proposals. http://www.technologyprogramme.org.uk/site/DTISpring06/Spring06Docs/BioscienceHealthcare06.pdf.
9. J.A. DiMasi, Risks in new drug development: Approval success rates for investigational drugs,Clinical Pharmacology & Therapeutics 69(5), 297–307, 2001.
10. Novartis Grid MP case study. http://www.ud.com/resources/files/cs novartis.pdf.
11. ADMEToxGrid project. http://www.lpds.sztaki.hu/index.php?loadprojects/current/comgenex.php.
12. United Devices Grid MP on Intel Architecture. http://www.ud.com/resources/files/wp intel ud.pdf.
13. Platform Computing products for life sciences.http://www.platform.com/Life.Sciences/CustomersCaseStudies.htm.
14. North Carolina bioportal. http://www.ncbiogrid.org.
15. Workflow management for bioinformatics (pegasys). http://bioinformatics.ucb.ca/pegasys.
16. Cancer biomedical informatics grid (cagrid). https://cabig.nci.nih.gov/workspaces/Architecture/caGrid.
17. BioPAUA. http://www.biopaua.lncc.br/ENGL/index.php.
18. Open bioinformatics grid. http://www.obigrid.org.
19. Asia Pacific biogrid initiative. http://www.apbionet.org/apbiogrid.
20. Genegrid—virtual bioinformatics laboratory. http://www.qub.ac.uk/escience/dev/article.php, sectionprojects.
21. OpenMolGrid project. http://www.openmolgrid.org/.

References 19
22. Drug Discovery Process, graphics by Novartis.http://www.nibr.novartis.com/images/OurScience/drug.discovery graph.jpg.
23. Genelabs’ Drug Discovery Process. http://www.genelabs.com/research/discoveryProcess.html.
24. E.A. Lunney, Computing in drug discovery: The design phase, IEEE Computing in Science andEngineering 3(5), 105–108, 2001.
25. I. Foster, C. Kesselman (Eds.), The Grid: Blueprint for a New Computing Infrastructure, MorganKaufmann Publishers, San Francisco, CA, 1998.
26. I. Foster, C. Kesselman (Eds.), The Grid 2: Blueprint for a New Computing Infrastructure, secondedition, Morgan Kaufmann Publishers, San Francisco, CA, 2004.
27. R. Buyya, K. Branson, J. Giddy, D. Abramson, The virtual laboratory: enabling molecular modellingfor drug design on the World Wide Grid, Concurrency and Computation: Practice and Experience15(1), 1–25, 2003.
28. Globus Toolkit 2.4 Overview. http://www.globus.org/toolkit/docs/2.4/overview.html.
29. R. Buyya, D. Abramson, J. Giddy, Nimrod/G: An architecture for a resource management andscheduling system in a global computational grid, in: Proceedings of the HPC ASIA 2000, Beijing,China, IEEE Computer Society Press, Los Alamitos, CA, 2000.
30. B. Shoichet, D. Bodian, I. Kuntz, Molecular docking using shape descriptors, Journal of Computa-tional Chemistry 13(3), 380–397, 1992.
31. H. Gibbins, K. Nadiminti, B. Beeson, R. Chhabra, B. Smith, R. Buyya, The Australian BioGrid Portal:Empowering the Molecular Docking Research Community. Technical Report, GRIDS-TR-2005-9,Grid Computing and Distributed Systems Laboratory, University of Melbourne, Australia, June 13,2005.
32. W. Zhang, J. Shen, Drug Discovery Grid (DDGrid), Second Grid@Asia Workshop, Shanghai, China,February 20–22, 2006.
33. Berkeley Open Infrastructure for Network Computing (BOINC). http://boinc.berkeley.edu/.
34. O. Schueler-Furman, C. Wang, P. Bradley, K. Misura, D. Baker, Progress in modeling of proteinstructures and interactions, Science 310, 638–642, 2005.
35. The Baker Laboratory. http://depts.washington.edu/bakerpg/.
36. e-Science Solutions for the Analysis of Complex Systems (eXSys).http://www.neresc.ac.uk/projects/eXSys.
37. Database of Interacting Proteins (DIP). http://dip.doe-mbi.ucla.edu/.
38. KEGG: Kyoto Encyclopedia of Genes and Genomes. http://www.genome.ad.jp/kegg/.
39. UniProtKB/Swiss-Prot Protein Knowledgebase. http://www.ebi.ac.uk/swissprot/.
40. R. Stevens, A. Robinson, C.A. Goble, my Grid: personalised bioinformatics on the information grid,Bioinformatics 19(Suppl. 1), i302–i304, 2003.
41. Open Grid Services Architecture—Data Access and Integration. http://www.ogsadai.org.uk/.
42. Initiative for grid-enabled drug discovery against neglected and emergent diseases.http://wisdom.eu-egee.fr/.
43. Enabling Grids for e-Science. http://www.eu-egee.org/.
44. Will LION and IBM succeed with the drug research Grid solution?http://www.gridtoday.com/02/1111/100708.html.
45. Platform partners with Matrix Science to accelerate drug discovery for pharmaceutical companies.http://www.hoise.com/primeur/02/articles/weekly/AE-PR-12-02-38.html.
46. Output of e-Science project helps GSK speed up drug discovery.http://www.rcuk.ac.uk/escience/news/ahm9.asp.
47. The Open Grid Services Architecture, Version 1.0. 2005. Global Grid Forum,http://www.gridforum.org/documents/GFD.30.pdf.
48. The Open Grid Forum (former Global Grid Forum and Enterprise Grid Alliance). http://www.ogf.org.

20 Open Computing Grid for Molecular Sciences
49. M. Romberg (Ed.), OpenMolGRID—Open Computing Grid for Molecular Science and Engineering,NIC series No. 29, Forschungszentrum Julich, ISBN 3-00-016007-8, July 2005.
50. UNICORE—Uniform Interface to Computing Resources. http://unicore.sourceforge.net.
51. ECOTOXicology Database. http://www.epa.gov/ecotox.
52. National Toxicology Program. http://ntp-server.niehs.nih.gov.
53. W. Dubitzky, D. McCourt, M. Galushka, M. Romberg, B. Schuller, Grid-enabled data warehousingfor molecular engineering, Parallel Computing 30(9–10), 1019–1035, 2004.
54. B. Schuller, M. Romberg, L. Kirtchakova, Application Driven Grid Developments in the Open-MolGRID Project, in: P.M.A. Sloot, A.G. Hoekstra, T. Priol, A. Reinefeld, and M. Bubak (Eds.),Advances in Grid Computing—EGC 2005, LNCS 3470: February 23–29, 2005.
55. S. Sild, U. Maran, M. Romberg, B. Schuller, E. Benfenati, OpenMolGRID: Using automatedworkflows in GRID computing environment. in: P.M.A. Sloot, A.G. Hoekstra, T. Priol, A. Reinefeld,and M. Bubak (Eds.), Advances in Grid Computing—EGC 2005, LNCS 3470, pp. 464–473, February2005.
56. E.V. Gordeeva, A.R. Katritzky, Rapid conversion of molecular graphs to three-dimensional represen-tation using the MOLGEO program, Journal of Chemical Information and Computer Sciences 33,102–111, 1993.
57. J.J. Stewart, MOPAC: A semiempirical molecular orbital program, Journal of Computer-AidedMolecular Design 4, 1–45, 1990.
58. COmprehensive DEscriptors for Structural and Statistical Analysis (Codessa) QSPR/QSAR Software,http://www.codessa-pro.com/index.htm.
59. L. Moss, A. Adelman, Data warehousing methodology, Journal of Data Warehousing 5, 23–31, 2000.
60. D. Erwin (Ed.), UNICORE Plus Final Report—Uniform Interface to Computing Recources, JointProject Report for the BMBF Project UNICORE Plus Grant Number: 01 IR 001 A-D, ISBN3-00-011592-7. Available at http://www.unicore.org/documents/UNICOREPlus-Final-Report.pdf.
61. Open Grid Forum (OGF) Research Group on Workflow Management (WFM-RG).https://forge.gridforum.org/projects/wfm-rg/.
62. Business Process Execution Language for Web Services. Version 1.0. July 31, 2002. http://www-106.ibm.com/developerworks/library/ws-bpel1/.
63. M. zur Muehlen, J. Becker, WPDL: State-of-the-art and directions of a meta-language for workflowprocesses, in: Lothar Bading, Boris Pettkoff, August-Wilhelm Scheer, Siegfried Wendt (Eds.),Proceedings of the 1st Know-Tech Forum, Potsdam, 1999.
64. D. Hull, K. Wolstencroft, R. Stevens, C. Goble, M.R. Pocock, P. Li, T. Oinn, Taverna: A tool for build-ing and running workflows of services, Nucleic Acids Research 34(Web Server issue):W729–W732,doi:10.1093/nar/gkl320, 2006.
65. S. Majithia, M.S. Shields, I.J. Taylor, I. Wang, Triana: A graphical web service composition andexecution toolkit, In Proceedings of the IEEE International Conference on Web Services (ICWS ’04),IEEE Computer Society, Los Alamitos, CA, pp. 514–524, 2004.
66. E. Benfenati, Modelling Aquatic Toxicity with Advanced Computational Techniques: Procedures toStandardize Data and Compare Models, in: J.A. López, E. Benfenati, and W. Dubitzky (Eds.), Procee-dings of International Symposium Knowledge Exploration in Life Science Informatics 2004 (KELSI2004), Lecture Notes in Computer Science, LNCS 3303, Springer, New York, pp. 235–248, 2004.
67. P. Mazzatorta, M. Smiesko, E. Lo Piparo, E. Benfenati, QSAR model for predicting pesticide aquatictoxicity, Journal of Chemical Information Model, 45, 1767–774, 2005.
68. U. Maran, S. Sild, I. Kahn, K. Takkis, Mining of the chemical information in GRID environment,Future Generation Computer Systems, in press, Corrected Proof, Available online 5 July 2006.
69. Predictive Model Markup Language. http://www.dmg.org/pmml-v3-1.html.
70. Chemomentum (Grid Services based Environment to enable Innovative Research).http://www.chemomentum.org.

References 21
71. Computational Toxicology (including QSARs) Joint Research Centre (JRC) Action no1321 of theEuropean Chemicals Bureau (ECB). http://ecb.jrc.it/QSAR/.
72. I.V. Tetko, P. Bruneau, H.-W. Mewes, D.C. Rohrer, G.I. Poda, Can we estimate the accuracy ofADME-Tox predictions? Drug Discovery Today, 11(15–16), 700–707, 2006.
73. D. De Roure, N.R. Jennings, N.R. Shadbolt, The Semantic Grid: A Future e-Science Infrastructure,in: F. Berman, A.J.G. Hey, and G. Fox (Eds.), Grid Computing: Making the Global Infrastructure aReality, John Wiley & Sons, Hoboken, NJ, 2003, pp. 437–470.
74. K.R. Taylor, J.W. Essex, J.G. Frey, H.R. Mills, G. Hughes, E.J. Zaluska, The semantic grid andchemistry: experiences with combechem, Journal of Web Semantics 4(2), 84–101, 2006.
75. D.B. Skillicorn, Distributed Data-Intensive Computation and the Datacentric Grid, White Paper,School of Computing, Queen’s University, Kingston, Canada, 2003.
76. A. Bassan, S. Eisenreich, B. Sokull-Kluettgen, A. Worth, The role of ECB in the risk assessment ofnanomaterials. Is there a future for “computational nanotoxicology”? Poster, The 12th InternationalWorkshop on QSAR in Environmental Toxicology.http://ecb.jrc.it/DOCUMENTS/QSAR/INFORMATION SOURCES/PRESENTATIONS/Bassan Lyon 0605 poster.pdf.
77. E.C. Butcher, Can cell systems biology rescue drug discovery? Nature Reviews Drug Discovery 4(6),461–467, 2005.

22