Embed Size (px)
Citation preview

9 J Med Dent SciK. Abe et al.J Med Dent Sci 2017; 64: 9-17
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease. A common characteristic of ALS pathology is cytoplasmic inclusions primarily composed of transactive response DNA-binding protein of 43 kDa (TDP-43). Production of TDP-43 in the central nervous system is strictly regulated, but it is not known whether this is also true in the skin of ALS patients. We found a gradual but significant reduction in epidermal TDP-43 mRNA expression with illness progression in ALS patients with upper-limb onset. However, the immunoblotting analysis revealed more TDP-43 protein in the skin of patients with upper-limb onset than of those with other onsets. There was no correlation between the TDP-43 mRNA expression and protein levels, indicating that the mechanism of TDP-43 autoregulation in the patients’ skin gradually failed. ALS diagnosis depends on clinical signs and electrophysiological findings, making early diagnosis difficult. TDP-43, as quantified by immunoblot analysis of biopsied skin, is a potential new biomarker of ALS.
Key Words: ALS, autoregulation, propagation, skin, TDP‑43
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal motor neuron disease characterized by rapidly progressive muscle weakness, spasticity, atrophy, and paralysis1. Most cases of ALS are sporadic, and about 10% are
of familial form. Both upper and lower motor neurons gradually degenerate, and respiratory muscle failure eventually causes death. Not all voluntary muscles are uniformly affected in ALS; the extraocular muscles and external anal sphincter muscles are selectively spared. In addition, sensory impairment, autonomic neurological symptoms, and skin problems such as decubitus ulcer do not appear until the end stages of the illness. Although this selectivity of affected systems in ALS has been known for some time, its causes have not yet been elucidated.
TDP‑43 is a highly conserved and widely expressed RNA‑binding protein2, 3 that functions as a splicing regulator4. Abnormal cytoplasmic inclusions of TDP‑43 are found in the majority of cases of ALS5, 6, but the role of TDP‑43 in the pathology of ALS has not been elucidated sufficiently. Although TDP‑43 is produced widely throughout the body, including in the pancreas, placenta, lungs, genital tract, spleen, heart, and brain7, the reason its pathology is limited to the nervous system remains unclear. The skin of ALS patients exhibits unique pathological and biochemical alterations in collagen, elastic fibers, and the basal layer8–10. One histochemical study has shown that TDP‑43 signal intensity is higher in the skin of ALS patients than in disease control patients11, suggesting that TDP‑43 production might also be higher. However, to our knowledge, there has not yet been a published biochemical analysis of TDP‑43 in the skin of ALS patients.
Given that TDP‑43 regulates its own production by binding to the 3’UTR12, 13 of its pre‑mRNA to strictly maintain a certain amount of the protein, we considered the possibility that TDP‑43 protein might be abnormally produced through the failure of autoregulation of TDP‑43 in the epidermal cells of ALS patients.
Here, we tested whether the systemic modification of TDP‑43 mRNA or protein and the characteristic pathology seen in the nervous systems of ALS patients were also present in the patients’ skin cells.
Corresponding Author: Takanori Yokota, MD, PhDDepartment of Neurology and Neurological Science, Tokyo Medical and Dental University, 1–5–45 Yushima, Bunkyo‑ku, Tokyo 113–8510, JapanTel: +81‑3‑5803‑4029 Fax: +81‑3‑5803‑4029E‑mail: tak‑[email protected] December 8, 2016;Accepted March 10, 2017
Original Article
TDP-43 in the skin of amyotrophic lateral sclerosis patients
Keisuke Abe1), Takuya Ohkubo1) and Takanori Yokota1)
1) Department of Neurology and Neurological Science, Tokyo Medical and Dental University

10 J Med Dent SciK. Abe et al.
Material and methods
SubjectsThe study included 22 ALS patients (mean age, 64.5
± 9.4 years), 26 patients with neuropathy as disease controls (mean age, 67.1 ± 10.0 years), and 3 ALS autopsy patients within 24 h post‑mortem. All ALS patients had been diagnosed clinically, by using the El Escorial criteria of the World Federation of Neurology, as having ALS14. The diagnoses of the disease control patients were antineutrophil cytoplasmic antibody–associated neuropathy (5), sarcoid neuropathy (3), neuropathy with Sjögren’s syndrome (2), neuropathy with monoclonal gammopathy of undetermined significance (2), multiple mononeuropathy (2), paraneoplastic sensory neuropathy (1), chronic ataxic sensory neuropathy (1), Guillain‑Barré syndrome (1), chronic inflammatory demyelinating polyradiculopathy (1), hereditary neuropathy with liability to pressure palsies (1), neuropathy with cryoglobulinemia (1), diabetic neuropathy (1), vasculitic neuropathy (1), and unclassified neuropathy (4).
Skin biopsy and tissue processingWe obtained 10 columnar specimens (diameter, 3 mm)
from the skin overlying the biceps brachii in each ALS patient, 10 specimens from the sural nerve area in each disease control patient, and 10 specimens from each of these areas in each ALS autopsy patient. Local anesthesia with 1% procaine was given before skin biopsy. None of the patients had abnormalities (e.g., atrophy, hypertrophy, recent bruising, induration) at the biopsy site. For each set of 10 specimens, 8 were frozen at –80 ℃ and 2 were fixed in 10% formalin solution and embedded in paraffin.
Quantitative real-time PCR assayWe stripped off the epidermis from the dermis by
incubating each specimen for 20 min in phosphate buffered saline (PBS) containing 3.8% ammonium thiocyanate15, 16. Total RNA was extracted from the epidermis by using Isogen reagent for RNA extraction (Nippon Gene, Tokyo, Japan). DNase‑treated RNA (1 μg) was reverse transcribed with SuperScript III by using random hexamers (Life Technologies, Carlsbad, CA). Quantitative real‑time (qRT) PCR analysis was performed with a TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) and a Light Cycler 480 Real‑Time PCR Instrument (Roche Diagnostics, Mannheim, Germany). The primers and probes for the TDP‑43 (Hs00606522_m1) and keratin 10 (KRT10; Hs00166289_m1) genes were designed by TaqMan
Gene Expression Assays (Applied Biosystems, Foster City, CA). KRT10 is expressed mainly in the epidermis16 and was used as an internal marker.
ImmunoblottingAfter the epidermis had been stripped off each
specimen, the epidermis was homogenized with 200 μL of homogenate buffer (10 mM Tris‑HCl, 0.8 M NaCl, 1 mM EGTA, and 1 mM dithiothreitol). Subsequently, 1% Triton X‑100 was added. After incubation for 60 min on ice, the sample was centrifuged at 100,000 × g for 20 min at 20 ℃. All proteins were separated by electrophoresis on a 10% polyacrylamide gel (ATTO Corporation, Tokyo, Japan) and transferred onto polyvinylidene difluoride membranes. The membranes were probed with rabbit polyclonal anti‑TDP‑43 antibody (1:1000, 12892‑1‑AP, Proteintech Group, Inc., Rosemont, IL) and mouse monoclonal anti‑α‑tubulin antibody (1:1000, M175‑3, MBL, Nagoya, Japan), after which the corresponding proteins were detected with an anti‑rabbit secondary antibody (1:1,000, sc‑2020, Santa Cruz Biotechnology, Inc., Dallas, TX) and anti‑mouse secondary antibody (1:1,000, sc‑2020, Santa Cruz Biotechnology). Blots were visualized with SuperSignal West Femto Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA) and detected by using a ChemiDoc System (Bio‑Rad, Hercules, CA). Quantitative analysis of the detected band image was performed by using Image J 1.50e (Wayne Rasband, National Institutes of Health, USA).
ImmunohistochemistryEach paraffin block was sliced into 4‑μm‑thick
sections, which were deparaffinized with xylene and hydrated in ethanol. The sections were then treated with 1% hydrogen peroxide in PBS (pH 7.4) for 60 min, after which they were incubated for 60 min in PBS with 5% goat serum to block non‑specific binding of antibody and probed for 60 min with rabbit polyclonal anti‑TDP‑43 antibody (1:1000, 12892‑1‑AP, Proteintech Group, Inc.). Finally, the sections were stained for immunoreactivity by using ‑an ABC kit (Vector Laboratories, Burlingame, CA). All samples were stained at the same time. Glass slides were selected from each group in such a way that observers were blind to the selection when they examined them under a microscope.
Statistical analysisData are presented as means ± SD. Differences
between means were evaluated with Student’s two‑tailed t‑test. We regarded P < 0.05 as significant. We
11TDP-43 in the skin of amyotrophic lateral sclerosis patients
used the least‑squares method to determine whether the amount of protein or mRNA of TDP‑43 was associated with age, duration of illness or each other. We made a hypothesis model that the two parameters correlated linearly, set up a null hypothesis that it had a significant slope (we defined the slope of regression line as ‘a’), and verified its authenticity by regression analysis.
Standard protocol approvals, registrations, and patient consents
The local ethics committees of Tokyo Medical and Dental University School of Medicine approved our standard protocol and registrations. All patients gave us informed consent for the procedures.
Results
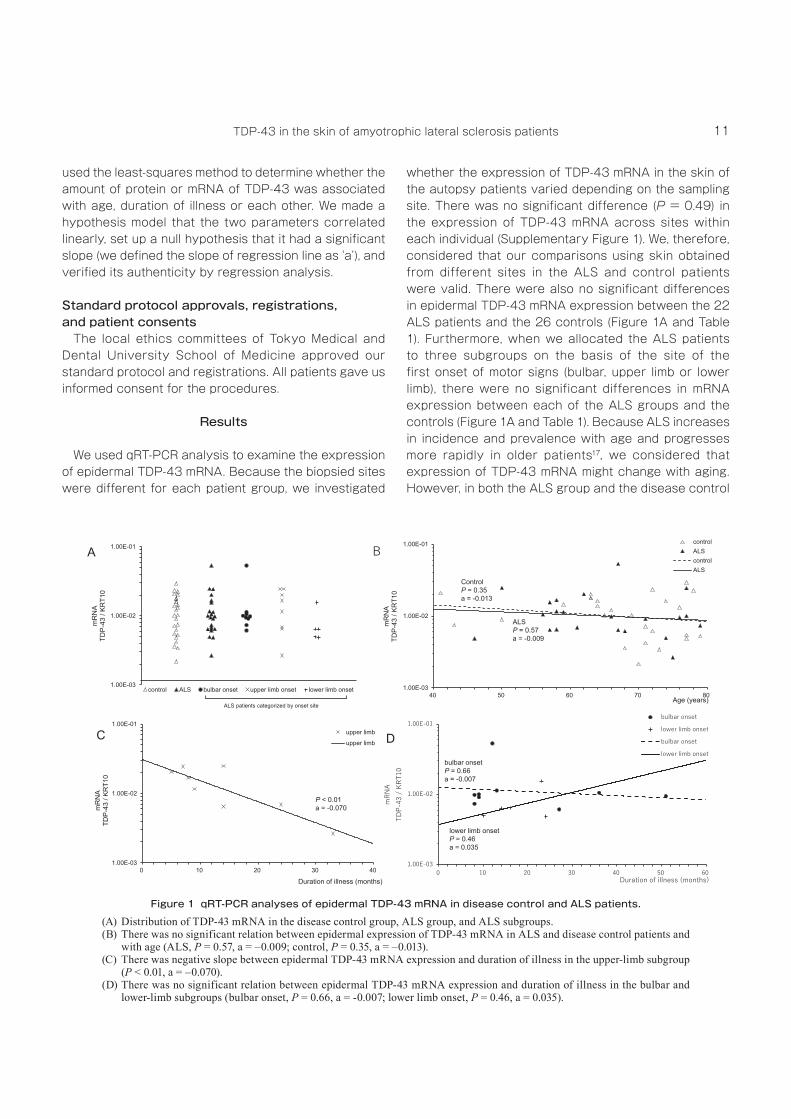
We used qRT‑PCR analysis to examine the expression of epidermal TDP‑43 mRNA. Because the biopsied sites were different for each patient group, we investigated
whether the expression of TDP‑43 mRNA in the skin of the autopsy patients varied depending on the sampling site. There was no significant difference (P = 0.49) in the expression of TDP‑43 mRNA across sites within each individual (Supplementary Figure 1). We, therefore, considered that our comparisons using skin obtained from different sites in the ALS and control patients were valid. There were also no significant differences in epidermal TDP‑43 mRNA expression between the 22 ALS patients and the 26 controls (Figure 1A and Table 1). Furthermore, when we allocated the ALS patients to three subgroups on the basis of the site of the first onset of motor signs (bulbar, upper limb or lower limb), there were no significant differences in mRNA expression between each of the ALS groups and the controls (Figure 1A and Table 1). Because ALS increases in incidence and prevalence with age and progresses more rapidly in older patients17, we considered that expression of TDP‑43 mRNA might change with aging. However, in both the ALS group and the disease control
Figure 1 qRT-PCR analyses of epidermal TDP-43 mRNA in disease control and ALS patients.(A) Distribution of TDP‑43 mRNA in the disease control group, ALS group, and ALS subgroups.(B) There was no significant relation between epidermal expression of TDP‑43 mRNA in ALS and disease control patients and
with age (ALS, P = 0.57, a = –0.009; control, P = 0.35, a = –0.013).(C) There was negative slope between epidermal TDP‑43 mRNA expression and duration of illness in the upper‑limb subgroup
(P < 0.01, a = –0.070).(D) There was no significant relation between epidermal TDP‑43 mRNA expression and duration of illness in the bulbar and
lower‑limb subgroups (bulbar onset, P = 0.66, a = ‑0.007; lower limb onset, P = 0.46, a = 0.035).
12 J Med Dent SciK. Abe et al.
group, there were no significant changes in TDP‑43 mRNA expression as a function of age by regression analysis (ALS: P = 0.57, control: P = 0.35) (Figure 1B). We then examined whether the expression of epidermal TDP‑43 mRNA changed with the progression of illness
in ALS. There was a negative slope between TDP‑43 mRNA expression and disease duration in the upper limb onset subgroup (P < 0.01) (Figure 1C), however, no such relation was observed in the bulbar and lower limb onset subgroup (bulbar onset: P = 0.66, lower limb onset: P = 0.46) (Figure 1D). In this study, since we did not take skin samples before the development of ALS, we can not evaluate whether the expression level of mRNA actually changes with the progression of disease in one patient. We need to gather more skin samples to confirm the relation between them.
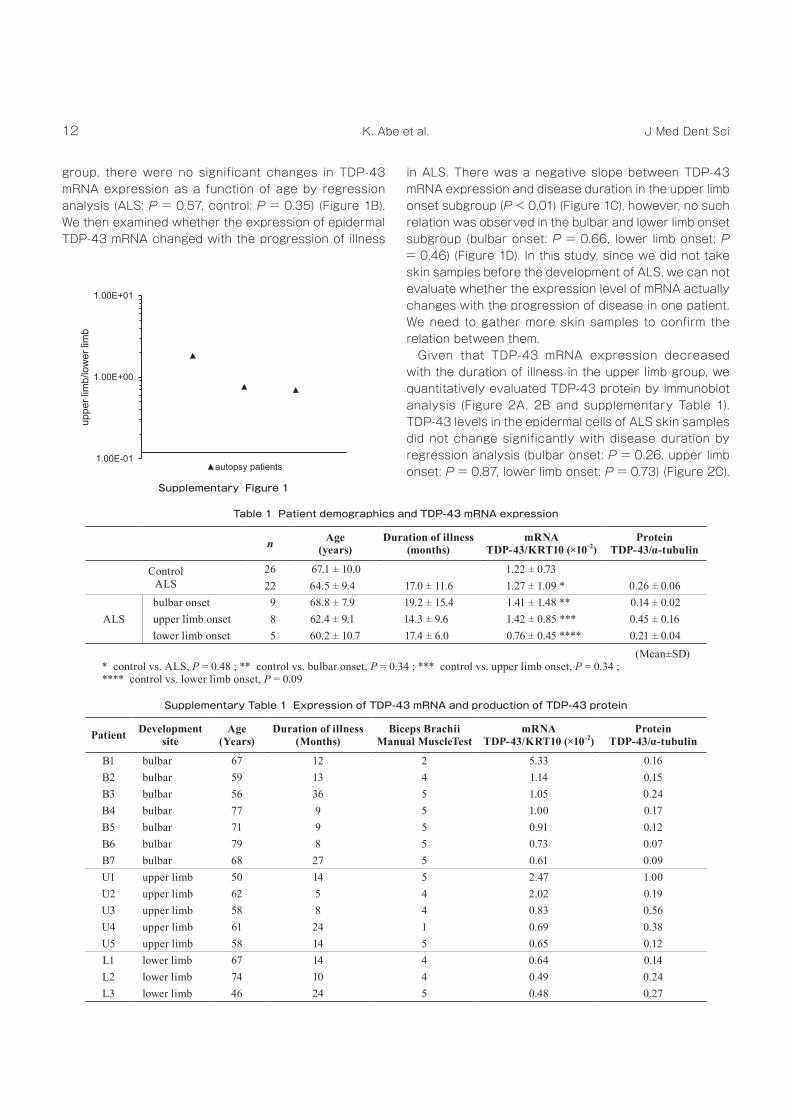
Given that TDP‑43 mRNA expression decreased with the duration of illness in the upper limb group, we quantitatively evaluated TDP‑43 protein by immunoblot analysis (Figure 2A, 2B and supplementary Table 1). TDP‑43 levels in the epidermal cells of ALS skin samples did not change significantly with disease duration by regression analysis (bulbar onset: P = 0.26, upper limb onset: P = 0.87, lower limb onset: P = 0.73) (Figure 2C).
Table 1 Patient demographics and TDP-43 mRNA expression
n Age (years)
Duration of illness(months)
mRNATDP-43/KRT10 (×10-2)
ProteinTDP-43/α-tubulin
ControlALS
26 67.1 ± 10.0 1.22 ± 0.7322 64.5 ± 9.4 17.0 ± 11.6 1.27 ± 1.09 * 0.26 ± 0.06
ALSbulbar onset 9 68.8 ± 7.9 19.2 ± 15.4 1.41 ± 1.48 ** 0.14 ± 0.02 upper limb onset 8 62.4 ± 9.1 14.3 ± 9.6 1.42 ± 0.85 *** 0.45 ± 0.16 lower limb onset 5 60.2 ± 10.7 17.4 ± 6.0 0.76 ± 0.45 **** 0.21 ± 0.04
(Mean±SD)* control vs. ALS, P = 0.48 ; ** control vs. bulbar onset, P = 0.34 ; *** control vs. upper limb onset, P = 0.34 ; **** control vs. lower limb onset, P = 0.09
Supplementary Table 1 Expression of TDP-43 mRNA and production of TDP-43 protein
Patient Development site
Age (Years)
Duration of illness(Months)
Biceps BrachiiManual MuscleTest
mRNA TDP-43/KRT10 (×10-2)
Protein TDP-43/α-tubulin
B1 bulbar 67 12 2 5.33 0.16 B2 bulbar 59 13 4 1.14 0.15 B3 bulbar 56 36 5 1.05 0.24 B4 bulbar 77 9 5 1.00 0.17 B5 bulbar 71 9 5 0.91 0.12 B6 bulbar 79 8 5 0.73 0.07 B7 bulbar 68 27 5 0.61 0.09 U1 upper limb 50 14 5 2.47 1.00 U2 upper limb 62 5 4 2.02 0.19 U3 upper limb 58 8 4 0.83 0.56 U4 upper limb 61 24 1 0.69 0.38 U5 upper limb 58 14 5 0.65 0.12 L1 lower limb 67 14 4 0.64 0.14 L2 lower limb 74 10 4 0.49 0.24 L3 lower limb 46 24 5 0.48 0.27
Supplementary Figure 1
13TDP-43 in the skin of amyotrophic lateral sclerosis patients
However, the upper limb onset group had generally higher TDP‑43 levels than the other groups (P = 0.07) (Figure 2B). TDP‑43 protein levels appeared to increase with increasing TDP‑43 mRNA expression in the upper limb onset group but not statistically significant (P = 0.33) (Figure 2D).
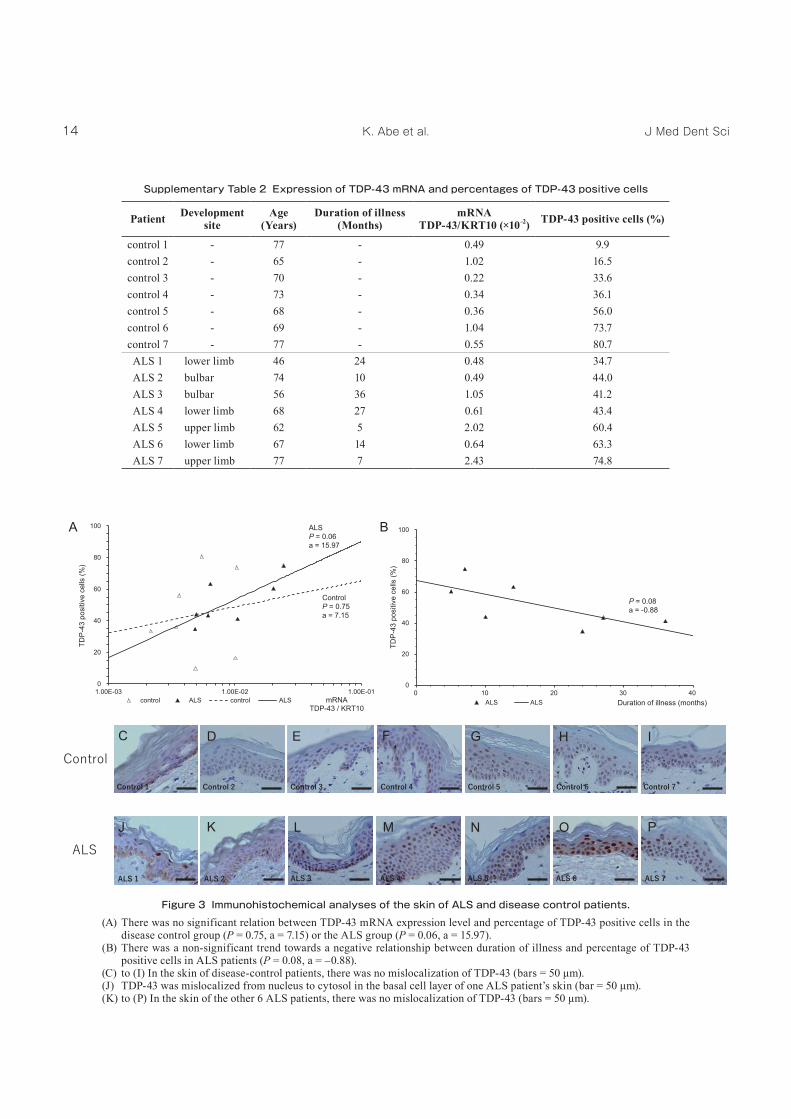
Although TDP‑43 is normally localized to the nucleus, in the central nervous systems of ALS patients, it is mislocalized from the nucleus to the cytosol and forms round or skein‑like inclusions5, 6. Because these inclusions contain insoluble TDP‑43, both the loss of functional TDP‑43 and the toxicity of insoluble TDP‑43 cause motor neuron death5, 6. We expected that a reduction in the level of functional TDP‑43 might also induce TDP‑43 mislocalization within the cells of non‑neural tissues such as the skin. We found no significant difference in the percentage of epidermal cells with strong TDP‑43 intensity (“TDP‑43 positive cells”) between the ALS and disease control groups (P = 0.40) (Supplementary
Table 2). However, the percentage of TDP‑43 positive cells in ALS patients skin was generally varied with the expression of TDP‑43 mRNA (P = 0.06) (Figure 3A) and the duration of illness (P = 0.08) by regression analysis (Figure 3B). Some TDP‑43 was localized in the nuclei of epidermal cells in the control patients (Figure 3C to 3I) and most of the ALS patients (Figure 3K to 3P). However, only in one case did we find TDP‑43 mislocalization in the basal cell layer of the skin of an ALS patient (Figure 3J). The presence of TDP‑43 positive inclusions has been reported in the cytosol of fibroblasts from the skin of patients with sporadic ALS18. The increase in the endoplasmic reticulum (ER) chaperone GRP‑78 and the macroautophagic marker LC3 in the skin of patients with TDP‑43A315T has revealed that ER stress activates autophagy of mutant TDP‑4319. In another report, the function of the ubiquitin‑proteasome system was altered in primary skin fibroblasts from patients with familial ALS20. These findings support the hypothesis that
Figure 2 Immunoblot analyses of TDP-43 in the skin of ALS patients.(A) TDP‑43 levels in the epidermis were increased in some of the patients in the upper‑limb‑onset subgroup (patients U1, U3 and U4).(B) Quantitative analyses of TDP‑43 by using Image J 1.50e.(C) There was no significant relation between TDP‑43 protein level and duration of illness in ALS patients (bulbar onset, P = 0.26, a =
0.003; upper limb onset, P = 0.87, a = 0.005; lower limb onset, P = 0.73, a = 0.004).(D) There was positive slope between TDP‑43 protein level and TDP‑43 mRNA expression in ALS patients but not statistically
significant (P = 0.33, a = 0.291).
14 J Med Dent SciK. Abe et al.
Supplementary Table 2 Expression of TDP-43 mRNA and percentages of TDP-43 positive cells
Patient Development site
Age(Years)
Duration of illness(Months)
mRNA TDP-43/KRT10 (×10-2) TDP-43 positive cells (%)
control 1 ‑ 77 ‑ 0.49 9.9 control 2 ‑ 65 ‑ 1.02 16.5 control 3 ‑ 70 ‑ 0.22 33.6 control 4 ‑ 73 ‑ 0.34 36.1 control 5 ‑ 68 ‑ 0.36 56.0 control 6 ‑ 69 ‑ 1.04 73.7 control 7 ‑ 77 ‑ 0.55 80.7 ALS 1 lower limb 46 24 0.48 34.7 ALS 2 bulbar 74 10 0.49 44.0 ALS 3 bulbar 56 36 1.05 41.2 ALS 4 lower limb 68 27 0.61 43.4 ALS 5 upper limb 62 5 2.02 60.4 ALS 6 lower limb 67 14 0.64 63.3 ALS 7 upper limb 77 7 2.43 74.8
Figure 3 Immunohistochemical analyses of the skin of ALS and disease control patients.(A) There was no significant relation between TDP‑43 mRNA expression level and percentage of TDP‑43 positive cells in the
disease control group (P = 0.75, a = 7.15) or the ALS group (P = 0.06, a = 15.97).(B) There was a non‑significant trend towards a negative relationship between duration of illness and percentage of TDP‑43
positive cells in ALS patients (P = 0.08, a = –0.88).(C) to (I) In the skin of disease-control patients, there was no mislocalization of TDP-43 (bars = 50 μm).(J) TDP-43 was mislocalized from nucleus to cytosol in the basal cell layer of one ALS patient’s skin (bar = 50 μm).(K) to (P) In the skin of the other 6 ALS patients, there was no mislocalization of TDP-43 (bars = 50 μm).

15TDP-43 in the skin of amyotrophic lateral sclerosis patients
alterations in the amount or quality of TDP‑43 occur in the skin of ALS patients.
Discussion
We investigated whether the failure of TDP‑43 regulation was related to the progression of ALS. TDP‑43 strictly regulates its own expression12, 13. Whereas homozygous TDP‑43 knockout (KO) mice die early in embryonic development, heterozygous KO mice show signs of muscle weakness but non‑variant production of TDP‑43. Compensatory mechanisms in heterozygous KO mice might increase TDP‑43 production by enhancing the stability of the mRNA from the normal allele21. On the other hand, overproduction of human TDP‑43 in response to infection with adeno‑associated virus in the spinal cords of cynomolgus monkeys causes progressive motor weakness and muscle atrophy with cytoplasmic inclusions, which apparently recapitulate ALS22. Furthermore, TDP‑43 mRNA expression is upregulated by 150% in the frontal cortex of patients with frontotemporal lobar degeneration (FTLD) and motor neuron disease23, 24, suggesting that severe cell toxicity is caused by the failure of TDP‑43 autoregulation. There have been no previous reports of increased TDP‑43 production in the skin or other non‑neural tissues of ALS patients. We found here that TDP‑43 in the skin of the upper‑limb onset group of ALS patients was increased at varying stages of the disease, but without any signs of disease in the skin itself. ALS is considered to be caused by motor nerve degeneration without pathological abnormalities of the weakened muscle and skin. Furthermore, the excessive TDP‑43 protein binds its pre‑mRNA and maintains a certain amount of the protein12, 13. However, our findings revealed that epidermal TDP‑43 protein increased regardless of the expression level of TDP‑43 mRNA in some cases with upper limb onset (Figure 2B and Supplementary Table 1: U1, U3 and U4) and suggest that potential abnormalities of TDP‑43 autoregulation exist in the epidermal cells of ALS patients. Furthermore, the gradual reduction in TDP‑43 mRNA expression with the progression of ALS indicates that dysregulation of TDP‑43 in the early stage of ALS is associated with the development of the disease.
We have two hypotheses regarding the increase in TDP‑43 production in epidermal cells. One is that abnormal TDP‑43 propagates from neurons to epidermal cells. In our previous review of ALS pathology, we suggested that TDP‑43 pathology could spread by contiguous and non‑contiguous propagation25. Motor signs in ALS patients usually occur in one or two highly localized sites and
spread contiguously. Interestingly, TDP‑43 has a prion‑like domain in the C‑terminal region26, 27, and misfolded TDP‑43 is propagated among neighboring neurons28. Misfolded forms of TDP‑43 have prion‑like properties such as easy aggregation via the insoluble “seeds” that have been extracted from FTLD brains and contiguous progression in a self‑templating manner28. Because TDP‑43 is transmitted and taken up by the axon terminal in vitro29, it can be released into the intercellular space and thus might be incorporated into recipient organs other than the nervous system. Although we took skin samples from over the biceps of all of our ALS patients, our finding that TDP‑43 levels were increased only in the upper‑limb onset group suggests that pathological TDP‑43 can migrate between epidermal cells and the nervous system at an early stage of ALS.
The other hypothesis regarding the increase in TDP‑43 production in epidermal cells is the “multiple hits” hypothesis. About 30% of sporadic ALS patients exhibit a non‑contiguous spread of clinical signs30. Indeed, Sekiguchi et al31. confirmed by needle electromyography that lower motor neuron failures in ALS progress non‑contiguously. The mechanism of remote progression in ALS is unclear. Given that levels of TDP‑43 and its fragments (35 and 25 kDa) in exosomal fractions of cerebrospinal fluid are elevated in ALS patients with frontotemporal dementia32, pathologic TDP‑43 could be transferred remotely via exosomes, although this has not been demonstrated in vivo. In the non‑upper limb onset groups in our study, there were two patients in which the level of epidermal TDP‑43 in the upper limb was increased without upper limb weakness (Figure 2A and Supplementary Table 1: B3 and L3). Although there have been no reports of increased levels of TDP‑43 in the sera of ALS patients, these cases suggest that abnormal TDP‑43 accumulated at the site of development of ALS can be remotely transferred to the skin of the upper limb via the sera or by the other unknown remote progression mechanism.
Wils et al. reported that the level of the C‑terminal 25‑kDa fragment increases with disease progression33. This suggests that TDP‑43 fragments with highly aggregative properties are generated when TDP‑43 pathology occurs. Here, despite our finding of increased TDP‑43 production in some ALS patients, mislocalization of TDP‑43 was not observed in the cytosol (Figure 3K to 3P), except in one patient (Figure 3J). Furthermore, immunoblot analysis did not reveal the C‑terminal 25‑kDa fragments. This is because epidermal cells are exfoliated from the surface of the skin before they can accumulate TDP‑43 fragments and cytosolic inclusions during the

16 J Med Dent SciK. Abe et al.
cell cycle (no more than about 4 weeks). Alternatively, the increase in TDP‑43 production might precede other TDP‑43 pathologies such as fragmentation of TDP‑43 and the generation of cytosolic inclusions.
There are two advantages of evaluating TDP‑43 from skin biopsies: we can obtain fresh samples without post‑mortem changes, and we can evaluate how TDP‑43 production changes with the progression of illness in each patient. Although we found no significant differences in TDP‑43 mRNA expression between the ALS and control groups, epidermal TDP‑43 in first onset site, as assessed by immunoblot, could be a suitable biomarker for ALS.
In summary, to our knowledge, this is the first study to have biochemically analyzed TDP‑43 in the skin of ALS patients. Our findings suggest that the deterioration in autoregulation of TDP‑43 transcription reflect an unknown pathological mechanism of ALS.
References1. Matus S, Valenzuela V, Medinas D et al. ER dysfunction
and protein folding stress in ALS. Int J Cell Biol. 2013; 2013: 674751.
2. Ou SH, Wu F, Harrich D et al. Cloning and Characterization of a Novel Cellular Protein, TDP‑43, That Binds to Human Immunodeficiency Virus Type 1 TAR DNA Sequence Motifs. J Virol. 1995; 69: 3584–3596.
3. Brundin P, Melki R and Kopito R. Prion‑like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol. 2010; 11: 301–307.
4. Buratti E and Baralle FE. Characterization and Functional Implications of the RNA Binding Properties of Nuclear Factor TDP‑43, a Novel Splicing Regulator of CFTR Exon 9. J Biol Chem. 2001; 276: 36337–36343.
5. Arai T, Hasegawa H, Akiyama H et al. TDP‑43 is a component of ubiquitin‑positive tau‑negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006; 351: 602–611.
6. Neumann M, Sampathu DM, Kwong LK et al. Ubiquitinated TDP‑43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006; 314: 130–133.
7. Buratti E, Dörk T, Zuccato E et al. Nuclear factor TDP‑43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001; 20,1774–1784.
8. Ono S, Imai T, Aso A et al. Alterations of skin glycosaminoglycans in patients with ALS. Neurology. 1998; 51: 399–404.
9. Ono S, Toyokura Y, Mannen T et al. Amyotrophic lateral sclerosis: histological, histochemical, and ultrastructural abnormalities of skin. Neurology. 1986; 36: 948–956.
10. Ono S and Yamauchi M. Elastin cross‑linking in the skin from patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 1994; 57: 94–96.
11. Suzuki M, Mikami H, Watanabe T et al. Increased expression of TDP‑43 in the skin of amyotrophic lateral sclerosis. Acta Neurol Scand. 2010; 122: 367–372.
12. Ayala YM, De Conti L, Avendaño‑Vázquez SE et al. TDP‑43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011; 30: 277–288.
13. Polymenidou M, Lagier‑Tourenne C, Hutt KR et al. Long pre‑mRNA deletion and RNA misplacing contribute to neuronal vulnerability from loss of TDP‑43. Nat Neurosci. 2011; 14: 459–468.
14. Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci. 1994; 124: Supple 96–107.
15. Clemmensen A, Thomassen M, Clemmensen O et al. Extraction of high‑quality epidermal RNA after ammonium thiocyanate‑induced dermo‑epidermal separation of 4 mm human skin biopsies. Exp Dermatol. 2009; 18: 979–984.
16. Trost A, Bauer JW, Lanschützer C et al. Rapid, high‑quality and epidermal‑specific isolation of RNA from human skin. Exp Dermatol. 2007; 16: 185–190.
17. Atsuta N, Watanabe H, Ito M et al. Age at onset influences on wide‑ranged clinical features of sporadic amyotrophic lateral sclerosis. J Neurol Sci. 2009; 276: 163–169.
18. Paré B, Touzel‑Deschênes L, Lamontagne R et al. Early detection of structural abnormalities and cytoplasmic accumulation of TDP‑43 in tissue‑ engineered skins derived from ALS patients. Acta Neuropathol Commun. 2015; 3: 5.
19. Wang X, Zhou S, Ding X et al. Activation of ER Stress and Autophagy Induced by TDP‑43 A315T as Pathogenic Mechanism and the Corresponding Histological Changes in Skin as Potential Biomarker for ALS with the Mutation. Int J Biol Sci. 2015; 11: 1140–1149.
20. Yang S, Zhang KY, Kariawasam R et al. Evaluation of Skin Fibroblasts from Amyotrophic Lateral Sclerosis Patients for the Rapid Study of Pathological Features. Neurotox Res. 2015; 28: 138–146.
21. Kraemer BC, Schuck T, Wheeler JM et al. Loss of murine TDP‑43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010; 119: 409–419.
22. Uchida A, Sasaguri H, Kimura N et al. Non‑human primate model of amyotrophic lateral sclerosis with cytoplasmic mislocalization of TDP‑43. Brain. 2012; 135: 833–846.
23. Mishra M, Paunesku T, Woloschak GE et al. Gene expression analysis of frontotemporal lobar degeneration of the motor neuron disease type with ubiquitinated inclusions. Acta Neuropathol. 2007; 114: 81–94.

17TDP-43 in the skin of amyotrophic lateral sclerosis patients
24. Gitcho MA, Bigio EH, Mishra M et al. TARDBP 3’‑UTR variant in autopsy‑confirmed frontotemporal lobar degeneration with TDP‑43 proteinopathy. Acta Neuropathol. 2009; 118: 633–645.
25. Kanouchi T, Ohkubo T and Yokota T. Can regional spreading of amyotrophic lateral sclerosis motor symptoms be explained by prion‑like propagation? J Neurol Neurosurg Psychiatry. 2012; 83: 739–745.
26. Goldschmidt L, Teng PK, Riek R et al. Identifying the amylome, proteins capable of forming amyloid‑like fibrils. Proc Natl Acad Sci USA. 2010; 107: 3487–3492.
27. Kim HJ, Kim NC, Wang YD et al. Mutations in prion‑like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013; 495: 467–473.
28. Nonaka T, Masuda‑Suzukake M, Arai T et al. Prion‑like Properties of Pathological TDP‑43 Aggregates from Diseased Brains. Cell Rep. 2013; 4: 124–134.
29. Feiler MS, Strobel B, Freischmidt A et al. TDP‑43 is intercellularly transmitted across axon terminals. J Cell Biol. 2015; 211: 897–911.
30. Fujimura‑Kiyono C, Kimura F, Ishida S et al. Onset and spreading patterns of lower motor neuron involvements predict survival in sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2011; 82: 1244–1249.
31. Sekiguchi T, Kanouchi T, Shibuya K et al. Spreading of amyotrophic lateral sclerosis lesions—multifocal hits and local propagation? J Neurol Neurosurg Psychiatry. 2014; 85: 85–91.
32. Ding X, Ma M, Teng J et al. Exposure to ALS‑FTD‑CSF generates TDP‑43 aggregates in glioblastoma cells through exosomes and TNTs‑like structure. Oncotarget. 2015; 6: 24178–24191.
33. Wils H, Kleinberger G, Janssens J et al. TDP‑43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA. 2010; 107: 3858–3863.





























