Embed Size (px)
Citation preview

Ortsgerichtete Mutagenese zur Erstellung von
Sequenz-Funktionsbeziehungen von
Acyltransferasen aus der Polyketidbiosynthese
Masterarbeit
zur Erlangung des akademischen Grades
Master of Science in Biochemie
der Naturwissenschaftlichen Fakultät der
Heinrich-Heine-Universität Düsseldorf
vorgelegt von
Carolin Bisterfeld
Dortmund
2012

Referent: Professor Dr. Jörg Pietruszka
Koreferent: Professor Dr. F. Schulz

Diese Arbeit wurde in der Zeit von April 2012 bis September 2012 unter der Leitung
von Professor Dr. F. Schulz am Max-Planck-Institut für molekulare Physiologie in
Dortmund angefertigt.

Danksagung
Gerne möchte ich all den Personen danken, die diese Arbeit erst möglich gemacht
haben.
Dabei gilt mein größter Dank Herrn Prof. Dr. Frank Schulz, der mir die Möglichkeit
gab, die Arbeit in seinem Institut zu absolvieren und mir dieses faszinierende Thema
überlies, dem ich mich mit viel Freude gewidmet habe. Bei auftretenden Problemen
hast du mich stets unterstützt und mich mit neuen Anregungen und Hilfestellungen
motiviert. Vielen Dank für dein Vertrauen in mich!
Ein großer Dank gilt auch Herrn Prof. Dr. Jörg Pietruszka, der sich bereiterklärt hat
diese Arbeit zu betreuen. Deine Unterstützung und dein Einsatz, auch über die
Entfernung, haben mir sehr geholfen.
Uschi Sundermann danke ich für die Betreuung meiner Arbeit. Du hast mich in
dieses komplexe Thema eingeführt und mir einige Kniffe bei den praktischen
Arbeiten beigebracht. Du hattest stets ein offenes Ohr für mich und durch deine
herzliche und positive Art fühlte ich mich sehr wohl und motiviert. Vielen Dank, du
warst eine fantastische Betreuerin!
Ich möchte auch allen anderen Mitgliedern des Arbeitskreises für die herzliche
Aufnahme und die nette Arbeitsatmosphäre danken. Die Zusammenarbeit mit euch
hat mir sehr viel Spaß gemacht. Ein besonderer Dank gilt hierbei Stephan Klopries,
der mir für die chemischen Arbeiten Platz in seinem Abzug zur Verfügung stellte und
mich bei allen Fragen rund um die Synthese unterstützt hat.
Ein ganz persönliches Dankeschön gilt meinem Freund Tobias, meinen Eltern und
Geschwistern, ohne die mein Studium nicht möglich gewesen wäre. Eure Liebe,
bedingungslose Unterstützung und Fürsorge geben mir stetig neue Kraft und neues
Selbstvertrauen.

Inhaltsverzeichnis | 1
INHALTSVERZEICHNIS
1 EINLEITUNG ....................................................................................................... 6
1.1. Polyketide .................................................................................................. 7
1.1.1. Polyketidsynthasen – Die Biosynthese von Polyketiden ........................ 8
1.1.1.1. Biosynthese von Erythromycin ...................................................... 10
1.1.1.1.1. Spezifität und biotechnologisches Potenzial der
AT-Domäne der Erythromycin-PKS .......................................... 14
2 AUFGABENSTELLUNG ................................................................................... 17
3 ERGEBNISSE UND DISKUSSION ................................................................... 24
3.1. Mutagenese der AT6-Domäne aus DEBS3 ............................................. 24
3.1.1. Wiederherstellung der Wildtyp Aktivität von S. erythraea∆AT6hygR .... 24
3.1.2. Präparation der AT6* Mutanten zur Stereospezifität ........................... 29
3.1.2.1. Sättigungsmutagenese zur Erstellung der
Stereospezifitätsvarianten ............................................................. 29
3.1.3. Restriktionanalyse und Sequenzierung zur Verifizierung der
präparierten Plasmide; Stereospezifität ............................................... 33
3.1.4. Konjugation der AT6* Varianten in S. erythraea∆AT6hygR;
Stereospezifität .................................................................................... 36
3.1.5. Analyse der AT6* Varianten in S. erythraea∆AT6hygR;
Stereospezifität .................................................................................... 36
3.1.5.1. Fermentation in 24-Lochplatten und anschließende
Massenanalyse ............................................................................. 36
3.1.5.2. Fermentation zur präparativen Gewinnung von Erythromycin und
Analyse mittels 1H-NMR ................................................................ 39
3.1.5.3. Fermentation und Aufarbeitung von S. erythraea NRRL-B-24071
(Wildtyp) ........................................................................................ 40
3.1.5.4. Fermentation und Aufarbeitung von erzeugten S. erythraea
Varianten ....................................................................................... 45
3.1.6. Präparation der AT6* Mutanten zur Substratspezifität ......................... 47

Inhaltsverzeichnis | 2
3.1.6.1. Sättigungsmutagenese zur Erstellung der AT6* Varianten;
Substratspezifität ........................................................................... 47
3.1.7. Restriktionsanalyse und Sequenzierung zur Verifizierung der
präparierten Plasmide; Substratspezifität ............................................ 49
3.2. Synthese des SNAC aktivierten rac-Methylmalonat 13 ........................... 50
3.3. Fütterungen mit Malonaten ...................................................................... 53
3.4. Abschließende Diskussion ....................................................................... 54
4 ZUSAMMENFASSUNG UND AUSBLICK ........................................................ 57
4.1. Projekt zur Erweiterung der Stereospezifität ............................................ 58
4.2. Projekt zur Erweiterung der Substratspezifität ......................................... 60
4.3. Fütterung mit Malonaten .......................................................................... 60
5 MATERIAL UND METHODEN .......................................................................... 62
5.1. Mikroorganismen, Plasmide, Oligonukleotide .......................................... 62
5.1.1. Mikroorganismen ................................................................................. 62
5.1.2. Vektoren und Plasmide ....................................................................... 62
5.1.3. Oligonukleotide .................................................................................... 64
5.2. Nährmedien ............................................................................................. 68
5.2.1. Nährmedien für S. erythraea ............................................................... 68
5.2.2. Nährmedien für E. coli ......................................................................... 69
5.3. Allgemeine Lösungen und Puffer ............................................................. 69
5.3.1. Antibiotika ............................................................................................ 69
5.3.2. Lösungen für die Plasmid Isolation ...................................................... 70
5.3.3. Lösungen für die Konjugation .............................................................. 70
5.3.4. Lösungen für die Präparation ultra-kompetenter Zellen ....................... 70
5.3.5. Lösungen für die Gelelektrophorese .................................................... 71
5.3.6. Substrat-Puffer für die Fütterung ......................................................... 71
5.4. Enzyme .................................................................................................... 71
5.5. Allgemeine Methoden der Molekularbiologie ........................................... 72

Inhaltsverzeichnis | 3
5.5.1. Lagerung und Kultivierung der Mikroorganismen ................................ 72
5.5.1.1. Lagerung von E. coli ..................................................................... 72
5.5.1.2. Lagerung von S. erythraea ............................................................ 72
5.5.1.3. Kultivierung von E. coli .................................................................. 72
5.5.1.4. Kultivierung von S. erythraea ........................................................ 72
5.5.1.5. Screening von S. erythraea in 24-Lochplatten des System
Duetz[53] ......................................................................................... 73
5.5.1.6. Extraktion mit Ethylacetat .............................................................. 74
5.5.1.7. Fermentation zur Gewinnung von Erythromycin in präparativen
Mengen ......................................................................................... 74
5.5.1.8. Extraktion und Aufreinigung von Erythromycin aus
Fermentationskulturen .................................................................. 75
5.5.2. Plasmidpräparation .............................................................................. 76
5.5.3. Ethanol-Präzipitation ........................................................................... 77
5.5.4. PCR-Protokolle .................................................................................... 77
5.5.4.1. Kolonie-PCR ................................................................................. 77
5.5.4.2. Zielgerichtete Mutagenese durch overlap extension PCR ............ 78
5.5.4.2.1. DpnI Verdau des Mutterstranges nach der PCR................... 81
5.5.5. Agarose-Gelelektrophorese von DNA ................................................. 81
5.5.5.1. Konzentrationsbestimmung mittels Gelelektrophorese ................. 82
5.5.6. Aufreinigung von DNA mittels Gelelektrophorese................................ 82
5.5.7. Restriktionsanalyse ............................................................................. 83
5.5.7.1. Restriktionsverdau vor der Polymerase Kettenreaktion ................ 83
5.5.7.2. Restriktionsverdau vor der Klonierung .......................................... 83
5.5.7.3. Kontrollverdau ............................................................................... 84
5.5.8. Klonierungen mittels SLIC-MIX ........................................................... 85
5.5.9. Einbringen genetischer Information in E. coli ...................................... 87
5.5.9.1. Präparation von ultra-kompetenten E. coli-Zellen ......................... 87

Inhaltsverzeichnis | 4
5.5.9.2. Transformation durch Hitzeschock ................................................ 88
5.5.10. Einbringen genetischer Information in Actinomyceten mittels
Konjugation ......................................................................................... 88
5.5.10.1. Vorbereitung des E. coli Donorstammes ....................................... 89
5.5.10.2. Vorbereitung des S. erythraea Rezipientenstammes .................... 89
5.5.10.3. Konjugation und Selektion ............................................................ 89
5.5.11. Testung auf antimikrobielle Aktivität .................................................... 90
5.5.12. Sequenzierung .................................................................................... 90
5.5.13. Protokolle zur Mutagenese der AT6-Domäne ..................................... 91
5.5.13.1. Wiederherstellung der Wildtyp Aktivität ......................................... 91
5.5.13.2. Präparation der Stereospezifitäts-Mutanten .................................. 91
5.5.13.3. Präparation der Substratspezifitäts-Mutanten ............................... 93
5.5.13.4. Protokolle zur Konjugation der erzeugten DEBS3AT6*
Expressionsplasmide in S. erythraea∆AT6hygRAT6*;
Stereospezifitätsvarianten ............................................................. 95
5.5.13.5. Screening der Stereospezifitätsmutanten ..................................... 96
5.5.14. Fütterungsexperimente mit Malonaten ................................................ 96
5.6. Allgemeine chemische Methoden ............................................................ 98
5.6.1. Geräte und Chemikalien ...................................................................... 98
5.6.1.1. Geräte ........................................................................................... 98
5.6.1.2. Chemikalien .................................................................................. 98
5.6.2. Chromatographische Verfahren........................................................... 98
5.6.2.1. Säulenchromatographie ................................................................ 98
5.6.2.2. Dünnschichtchromatographie........................................................ 98
5.6.2.3. Präparative Dünnschichtchromatographie .................................... 98
5.6.3. Analytische Verfahren ......................................................................... 99
5.6.3.1. NMR-Spektroskopie ...................................................................... 99
5.6.3.2. Massenspektrometrie .................................................................. 100

Inhaltsverzeichnis | 5
5.6.3.2.1. GC-MS ................................................................................ 100
5.6.3.2.2. LC/ESI-MS zur allgemeinen Analyse der Masse ................ 100
5.6.3.2.3. LC/ESI-MS zur Analyse von Fermentationsprodukten ........ 101
5.6.3.2.4. MS/MS ................................................................................ 101
5.6.3.2.5. Hochauflösende Massen Spektrometrie (HRMS) ............... 102
5.7. Synthese des Substrates rac-Methylmalonyl-SNAC (13) ...................... 102
5.7.1. N-(2-mercaptoethyl)acetamid (SNAC) (14) ....................................... 102
5.7.2. 3-(tert-butoxy)-2-methyl-3-oxopropansäure (11) ............................... 103
5.7.3. tert-Butyl-3-((2-acetamidethyl)thio-2-methyl-3-oxopropanat (12) ....... 104
5.7.4. 3-((2-acetamidethyl)thio)-2-methyl-3-oxopropanoat (13) ................... 105
5.7.5. Erythromycin A (7) ............................................................................. 105
6 ABKÜRZUNGEN UND AKRONYME .............................................................. 106
7 REFERENZEN ................................................................................................ 111
8 ANHANG ......................................................................................................... 114
8.1. NMR-Spektren ....................................................................................... 114
9 EIDESSTATTLICHE ERKLÄRUNG................................................................ 117
10 STRUKTUREN DER SUBSTANZEN .............................................................. 118

Einleitung | 6
1 Einleitung
Viele der heute verwendeten pharmazeutischen Wirkstoffe sind Naturstoffe oder von
diesen abgeleitet. Mit einem Anteil von 50 % der zugelassenen Medikamente sind
sie nicht mehr aus der heutigen Medizin wegzudenken.[1] Diese enorme Bedeutung
der Natur beruht auf der fantastischen strukturellen und funktionellen Vielfalt an
Substanzen, die darüberhinaus bis heute nur in einem geringen Maß erforscht
wurde. Neben dem pharmazeutischen und auch agrarökologischen Interesse, bieten
die hochkomplexen Substanzen zudem eine Herausforderung für die chemische
Synthese.[2] Es ist also nicht verwunderlich, dass ein großes interdisziplinäres
Forschungsinteresse an Naturstoffen und deren Funktionen besteht.
Die Wirkung einiger pflanzlicher Substanzen war bereits früh erkannt. Die Menschen
nutzten dieses Wissen nicht nur zur medizinischen Versorgung, sondern zum
Beispiel auch als Waffen. So wurde bereits in der Steinzeit das Gift aus Tollkirschen
– Tropan (1) – als Pfeilgift verwendet. Eine der ältesten heute erhaltenen
Aufzeichnungen aus dem Jahre 2600 v.Chr. beschreibt die Gewinnung von
Substanzen aus Myrrhe und Schlafmohn, die auch heute noch als Arzneien
verwendet werden.[3] Ein Meilenstein der jüngeren Naturstoff-Geschichte setzte
Alexander Fleming 1928 mit der Entdeckung des Penicillins (2) aus einem
Schimmelpilz, eines der bis heute bekanntesten Antibiotika.[4] Dies war auch der
Beginn der Nutzung von Bakterien als Quelle für neue Wirkstoffe.
Abbildung 1: Strukturen von Tropan (1) und Penicillin (2).
Heute wird kontinuierlich nach neuen Substanzen mit antibiotischen Eigenschaften
gesucht. Neben den terrestrischen Lebewesen, geraten in den letzten Jahren auch
zunehmend marine Organismen als mögliche Ressource für neue Naturstoffe in den
Fokus. Die Zahl der bekannten Naturstoffe ist dabei stetig angestiegen. So wurden
von 2000-2008 300 Substanzen mit antimikrobieller Wirkung und allein 2008 1065
neue marine Verbindungen beschrieben.[5,6]

Einleitung | 7
Die Nutzung eines Naturstoffes als kommerziellen Arzneistoff setzt die mögliche
Bereitstellung in großen Mengen voraus. Dies ist über eine klassische chemische
Synthese aufgrund der komplexen Strukturen meist sehr aufwendig und kostspielig,
und daher häufig nicht umsetzbar. Die Biotechnologie bietet hier die Möglichkeit
durch Nutzung von einzelnen Enzymen oder ganzen Mikroorganismen die Strukturen
zugänglich zu machen.[7] Zusätzlich können durch die Kombination der
Biotechnologie und chemischer Synthese eine Vielzahl von neuen Derivaten erzeugt
werden. Dies ist besonders bezogen auf die ständig zunehmende Resistenz vieler
Bakterien gegenüber den bisher eingesetzten Antibiotika von großem Interesse.
Hierbei gibt es verschiedene Ansätze. Die Partialsynthese, eine Kombination der
Biotechnologie und chemischer Synthese bietet die Möglichkeit eine Vielzahl von
Derivaten bereitzustellen. Dazu werden Fermentationsprodukte durch nachfolgende
chemische oder enzymatische Syntheseschritte in vitro modifiziert.[8] Ein weiterer
Ansatzpunkt ist die Versorgung der produzierenden Organismen mit modifizierten
Vorstufen.
Durch die enormen Fortschritte der Molekularbiologie rücken auch zunehmend
biochemische Methoden in den wissenschaftlichen Fokus. Durch die
kombinatorische Biosynthese sollen die am natürlichen Syntheseweg beteiligten
Enzyme verändert werden, um somit die Bereitstellung von alternativen,
nicht-natürlichen Produkten zu ermöglichen.[9]
1.1. Polyketide
Eine prominente Klasse der Naturstoffe sind die Polyketide. Diese strukturell und
funktionell hoch heterogenen Substanzen werden hauptsächlich von Bakterien,
Pilzen und Pflanzen gebildet.[10] Es sind Sekundärmetabolite, die unterschiedliche
biologische Funktionen erfüllen. Während sie in Pflanzen zum Beispiel als Farbstoff
dienen, setzen Bakterien sie als Abwehr- oder Botenstoffe ein.[11] Diverse biologische
Aktivitäten (antibiotische, fungizide, antivirale, immunsuppressive, etc.) qualifizieren
viele Polyketide als pharmakologische Wirkstoffe.[12,13,14] Zu den Polyketiden gehören
Polyphenole, Makrolide, Polyene, Endiine und Polyether.[15]

Einleitung | 8
Abbildung 2: Strukturen von Polyketiden, die als pharmazeutische Arzneistoffe eingesetzt werden. Reduzierte Polyketide: Rapamycin (3), Lovastatin (5), Monensin A (6), Erythromycin A (7). Aromatische Polyketide: Doxorubicin (4).
Trotz ihrer enormen strukturellen und funktionellen Diversität haben alle Polyketide
einen gemeinsamen Biosyntheseweg, bei dem erstaunlicherweise das
vergleichsweise einfache Molekül Acetat als Baustein dient.[15] Die Umsetzung zum
komplexen Polyketid erfolgt über eine kontrollierte iterative Abfolge von
decarboxylierenden Claisen-Thioester-Kondensationen, die von Polyketidsynthasen
selektiv katalysiert werden.[15]
1.1.1. Polyketidsynthasen – Die Biosynthese von Polyketiden
Im Jahre 1893 beschäftigte sich James N. Collie mit der Synthese eines Polyketids
und prägte dabei auch den Begriff.[16] Seitdem wurde interdisziplinär geforscht um
den Mechanismus der hochspezifischen Umwandlung von Carbonsäuremonomeren
zu komplexen Substanzen zu entschlüsseln. Es wurde begonnen, die für die
Biosynthese verantwortlichen Enzyme durch Klonierung und Isolierung zu
charakterisieren.[17,18] Nach den ersten Erkenntnissen wurden die Polyketidsynthasen
(PKS) nach ihrer Struktur und ihrem Reaktionsmechanismus in drei Klassen
unterschieden.[19] Die Typ-I-PKS bauen reduzierte Polyketide auf. Es sind riesige
Enzymkomplexe, die sich aus mehreren aneinandergereihten katalytisch aktiven
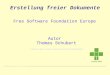
Einleitung | 9
Modulen zusammensetzen.[15] Die Typ-II-PKS sind bisher nur in Prokaryonten
bekannt und sind für die iterative Synthese aromatischer Polyketide verantwortlich.
Sie bestehen aus kleineren Enzymen, die in einem dissoziierbaren Komplex
angeordnet sind und mehrere Kettenverlängerungen katalysieren. [20] Daneben gibt
es noch die Chalconsynthasen, die in eine dritte Klasse – die Typ-III-PKS –
eingeordnet werden.[15]
Der prinzipielle Ablauf der Polyketidbiosynthese steht in Analogie zu der
Fettsäurebiosynthese. Bei beiden werden aktivierte Acyl-Startereinheiten mit
ebenfalls aktivierten Malony-CoA Derivaten aus dem Primärmetabolismus über
decarboxylierende Claisen-Thioester-Kondensationen zu β-Ketoacylpolymeren
umgesetzt. Dabei bleiben sie über eine Thiolbindung mit dem Enzym kovalent
verbunden.[15]
Abbildung 3: Übersicht der Fettsäure- und Polyketidbiosynthese. Der Aufbau der wachsenden Kette während der Fettsäurebiosynthese erfolgt durch eine Reihe von Claisen-Kondensationen, die durch Ketosynthasen (KS) katalysiert werden. Nach jedem Kondensationsschritt wird die Ketofunktion des so initial synthetisierten β-Ketoesters in drei Schritten vollständig reduziert, bevor die nächste Runde der Kettenverlängerung initiiert werden kann. Bei der Synthese aromatischer Polyketide hingegen entfallen die Reduktionsschritte komplett, sodass der β-Ketoester nicht modifiziert wird. Dies resultiert in einem hochreaktiven poly-β-Ketoester, der in weiteren Reaktionen durch Enzyme cyclisiert wird resultierend in aromatischen Strukturen. Bei reduzierten Polyketiden finden einzelne Reduktionsschritte nur optional statt, wodurch es zu einer Vielfalt aus Hydroxyl-, Carbonylgruppen, Doppelbindungen und Methylengruppen kommt. Abbildung in Anlehnung an Hopwood 1997.
[21]

Einleitung | 10
Der β-Ketoester durchläuft bei den Fettsäuresynthasen (FAS) nach jeder
Kettenverlängerung einen obligatorischen reduktiven Zyklus, katalysiert durch drei
Enzyme.[22] Auch bei den Polyketidsynthasen gibt es drei reduzierende Enzyme,
jedoch sind die reduktiven Schritte bei Typ-I-PKS optional und können teilweise oder
vollständig übersprungen werden. Diese Variabilität resultiert in einer deutlich
komplexeren Strukturdiversität, da sowohl β-Ketone, β-Hydroxyl- oder Enoylgruppen
auftreten können.[23]
Nach der für das spezifische Polyketid notwendigen Anzahl an Verlängerungs-Zyklen
werden die Substrate vom Enzymkomplex abgespalten. Es folgen post-PKS
Prozessierungen, die in den endgültigen Strukturen der Polyketide resultieren.[24] Die
optionalen Reduktionsschritte, sowie die Modifikationen der Primärprodukte
ermöglichen die große Zahl von bisher mehr als 7000 natürlichen Polyketiden.
Da diese Arbeit sich mit der Typ-I-PKS der Erythromycin A (7)-Biosynthese in dem
Bodenbakterium Saccharopolyspora erythraea beschäftigt, wird im Folgenden näher
auf dessen Reaktionsmechanismus eingegangen.
1.1.1.1. Biosynthese von Erythromycin
Die Erythromycin-PKS (DEBS) aus dem Bodenbakterium Saccharopolyspora
erythraea katalysiert die Synthese des 6-Desoxyerythronolid [6-dEB (8)], welches
durch post-PKS Modifikationen zum Erythromycin A (7) umgesetzt wird.[25]
Die DEBS ist ein Archetyp der nicht-iterativen Typ-I-PKS, die als erste modulare PKS
durch Sequenzierung der korrespondierenden Gene identifiziert wurde.[26,27] Es ist
ein multimodulares Enzym, welches zu den größten bisher bekannten Proteinen
zählt. Die einzelnen Module sind an einem linearen Fließband angeordnet, wobei die
Übergänge zwischen den einzelnen Enzymen fließend ineinander übergehen.
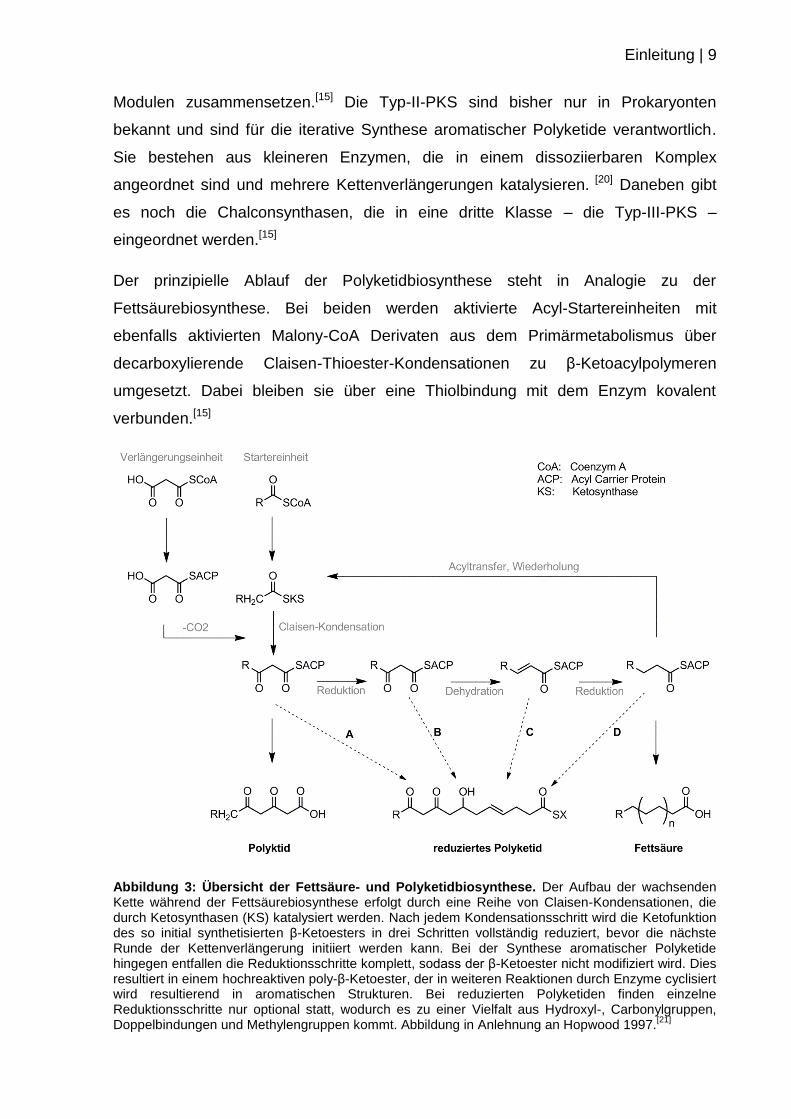
Einleitung | 11
Abbildung 4: Grundmechanismen der Erythromycin A (7) Biosynthese.[26,27]
Abkürzungen: ACP: Acylcarrierprotein, AT: Acyltransferase, DH: Dehydratase, ER: Enoylreduktase, KR: Ketoreduktase, KS: Ketosynthase, TE: Terminale Esterase. Die Biosynthese wird von der Polyketidsynthase DEBS katalysiert. Dieser 2 MDa große Multienzymkomplex besteht aus drei Enzymen, DEBS1, 2 und 3, die nicht kovalent miteinander verknüpft als molekulares Fließband agieren. Jedes dieser Enzyme beinhaltet zwei bis drei Module (schwarze Kästen), die kovalent miteinander verknüpft sind. Als Startereinheit dient Propionyl-CoA, welches mit sechs Äquivalenten Methylmalonyl-CoA als Verlängerungsbausteine verknüpft wird. Ein minimales Modul (vgl. Modul 3) besteht aus drei Domänen, die eine Kettenverlängerung um 2 C-Atome inklusive der Seitenkette (resultierend in der Bildung eines β-Ketoesters) katalysieren: Eine Ketosynthase, welche eine decarboxylierende Claisenkondensation katalysiert, eine Acyltransferase welche den Baustein auswählt und diesen auf das Acylcarrierprotein überträgt. Der β-Ketoester kann dann nachfolgend durch die reduktiven Domänen weiter prozessiert werden. Die Anzahl der reduktiven Domänen des jeweiligen Moduls bestimmt hierbei den Grad der Reduktion der wachsenden Kette. Final spaltet eine Terminale Esterase die Polyketidkette vom Enzym ab und ermöglicht eine Zyklisierung. Durch Post-PKS Prozessierungen wie Glykosylierungen und Methylierungen wird die Biosynthese des biologisch aktiven, komplexen Naturstoffes Erythromycin A (7) komplementiert.

Einleitung | 12
Insgesamt codieren drei Gene (eryAI, eryAII und eryAIII) für jeweils ein Enzym
(DEBS1, 2 und 3), die mit je etwa 350 kDa eine außergewöhnliche Größe
besitzen.[17],[28] Jedes DEBS-Enzym setzt sich aus zwei Modulen zusammen, die je
einen Kettenverlängerungsschritt katalysieren. Jedes Modul besteht aus einer
Ketosynthase (KS), einer Acyltransferase (AT) und einem Acylcarrierprotein (ACP),
sowie einer variierenden Zahl von reduktiven Domänen. Zusätzlich gibt es noch ein
Lademodul (bestehend aus einer AT und einem ACP) und die Terminale Esterase
am Ende der PKS (vgl. Abbildung 4). Durch den iterativen Mechanismus der PKS,
wird jedes Modul nur einmal durchlaufen, sodass vom Aufbau der PKS auf die
korrelierende Struktur des Primärproduktes geschlossen werden kann. Dieser
Zusammenhang wird als Prinzip der Kolinearität bezeichnet.[15]
Die ACP-Domänen der DEBS werden posttranslational durch eine
Phosphodiesterbindung eines konservierten Serin-Restes an Coenzym A aktiviert.
Diese Bindung resultiert in einer Konformationsänderung des ACP (inaktive
apo-Form aktive holo-Form), die durch eine Phosphopantetheinyltransferase
(PPTase) katalysiert wird.[29]
Abbildung 5: Aktivierung des ACP durch Coenzym A katalysiert durch eine PPTase. Der Phosphopantetheinarm wird vereinfacht durch eine Wellenlinie mit der aktiven Thiol-Gruppe am Ende dargestellt.
Die AT-Domäne des Lademoduls ist für die Substratauswahl – hier Propionyl-CoA –
verantwortlich. Durch einen nukleophilen Angriff des Thiolrestes wird das Substrat
auf das ACP übertragen und anschließend auf die Ketosynthase des ersten Moduls
transferiert (vgl. Abbildung 6 A). Ketosynthasen katalysieren die Kettenverlängerung
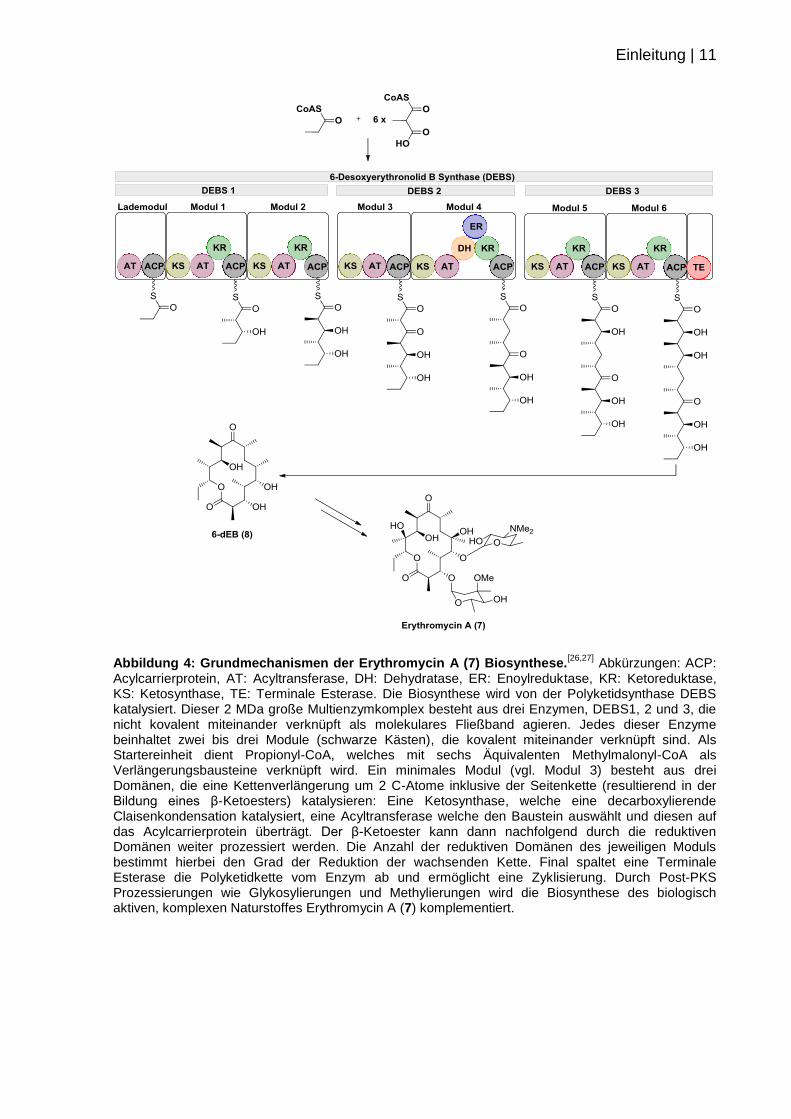
Einleitung | 13
über eine Thio-Claisen-Kondensation, wobei ein β-Ketoester entsteht. Dabei greift
die am ACP kovalent gebundene Verlängerungseinheit Methylmalonyl-CoA die
wachsende Kette an, die zuvor auf die Ketosynthase-Domäne übertragen wurde.
Hierbei liefert die Abspaltung des CO2 die nötige Energie für die Reaktion (vgl.
Abbildung 6 B).
Abbildung 6: Mechanismen der Erythromycin-Biosynthese. A: Wahl der Verlängerungseinheit durch die AT-Domäne und Transfer auf das ACP durch einen nukleophilen Angriff am Beispiel des Moduls 1. B: Kettenverlängerung durch eine carboxylierende Claisen-Kondensation zwischen ACP
und KS am Beispiel des Modul 1.
Die Ketoreduktase (KR), die Dehydratase (DH) und die Enoylreduktase (ER) sind als
optionale reduktive Domänen für die komplexe β-Ketoprozessierungen der Polyketid-
Seitenketten verantwortlich. So wird in den Modulen 1, 2, 5 und 6 jeweils nach der
Kondensation eine Reduktion des β-Carbonyls durch die entsprechende KS
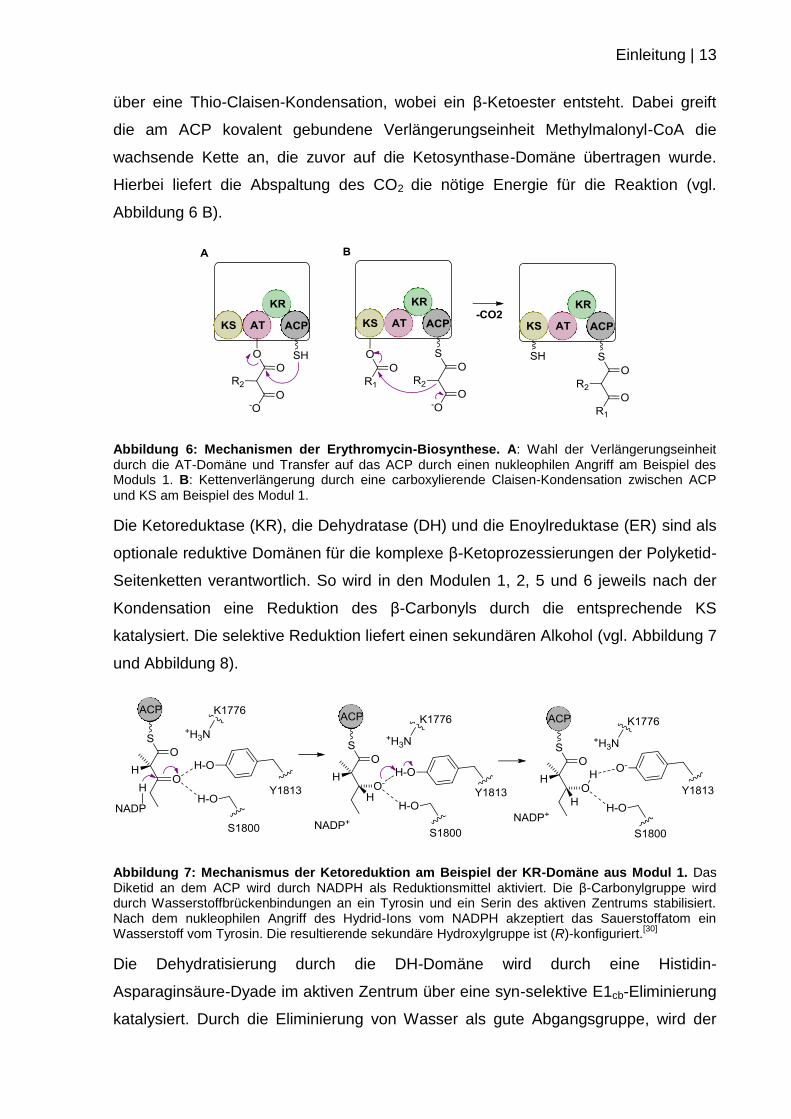
katalysiert. Die selektive Reduktion liefert einen sekundären Alkohol (vgl. Abbildung 7
und Abbildung 8).
Abbildung 7: Mechanismus der Ketoreduktion am Beispiel der KR-Domäne aus Modul 1. Das Diketid an dem ACP wird durch NADPH als Reduktionsmittel aktiviert. Die β-Carbonylgruppe wird durch Wasserstoffbrückenbindungen an ein Tyrosin und ein Serin des aktiven Zentrums stabilisiert. Nach dem nukleophilen Angriff des Hydrid-Ions vom NADPH akzeptiert das Sauerstoffatom ein Wasserstoff vom Tyrosin. Die resultierende sekundäre Hydroxylgruppe ist (R)-konfiguriert.
[30]
Die Dehydratisierung durch die DH-Domäne wird durch eine Histidin-
Asparaginsäure-Dyade im aktiven Zentrum über eine syn-selektive E1cb-Eliminierung
katalysiert. Durch die Eliminierung von Wasser als gute Abgangsgruppe, wird der

Einleitung | 14
sekundäre Alkohol zu einer Doppelbindung reduziert. Die ER-Domäne ist in der Lage
mit dem Cofaktor NADPH als Reduktionsmittel die Doppelbindung weiter zu
reduzieren, was in einer Methylengruppe resultiert (vgl. Abbildung 8).[31]
Abbildung 8: Überblick über die reduktive Schleife innerhalb einer modularen PKS. A: Viertes Modul der Erythromycin-PKS mit allen reduktiven Domänen. B: Reduktive Schritte der KR-, DH- und
ER-Domänen.
Die Abwesenheit von reduktiven Domänen in Modul 3 (vgl. Abbildung 4) resultiert in
der Ketogruppe an Position 9 des 6-Desoxyerythronolid (8). Im vierten Modul findet
eine vollständige Reduktion der entsprechenden Carbonylgruppe durch die KR-, DH-
und ER-Domäne, was in einer Methylengruppe resultiert.
Durch die Terminale Esterase wird die Abspaltung der kovalent gebundenen
Polyketidkette von dem ACP des sechsten Moduls katalysiert. Dadurch wird ein
Carbanion an C1 gebildet, welches vom Hydroxyl-Sauerstoff am C13 nukleophil
angegriffen wird. Das so entstandene 6-dEB (8) wird durch post-PKS Modifikationen
zum Erythromycin A (7) prozessiert. Dabei entsteht über Hydroxylierungen und
Glycosylierungen mit dem Zucker Mycarose und einem Aminozucker die biologisch
aktive Verbindung. Die Intermediate Erythromycin B bis D entstehen durch
nicht-vollständige Prozessierungen, sie kommen nur zu einem geringen Anteil in
Fermentationen von S. erythraea vor.[32]
1.1.1.1.1. Spezifität und biotechnologisches Potenzial der AT-Domäne der
Erythromycin-PKS
Da diese Arbeit sich speziell mit der AT6-Domäne beschäftigt, soll im Folgenden
näher auf die AT-Domänen eingegangen werden.
Die AT-Domänen der Erythromycin-PKS besitzen mit der ausnahmslosen Auswahl
des Methylmalonyl-CoA als Verlängerungseinheit eine sehr hohe Substratspezifität.
Zugleich weisen sie eine erstaunliche Stereoselektivität auf, da sie

Einleitung | 15
interessanterweise einzig das 2(S)-Enantiomer des Methylmalonyl-CoA
einbauen.[33,34] Die entsprechenden Stereozentren des Erythromycin A (7) sind
jedoch ausschließlich (R)-konfiguriert. Somit muss die -Methylgruppe jeweils nach
der ersten, fünften und sechsten Kondensation selektiv epimerisiert werden. Bisher
ist leider noch nicht bekannt, wodurch diese Epimerisierungen kontrolliert werden.
Innerhalb der PKS werden das ACP, sowie die KS-Domänen als mögliche
Katalysatoren diskutiert.[35,36]
Bei Überlegungen zur Erzeugung von Erythromycin-Derivaten, ein zentrales Ziel der
Wissenschaft zur Evaluierung von Leitstrukturen[37], bietet die Spezifität der
AT-Domänen einen aussichtsreichen Ansatzpunkt. Es wurden verschiedene
Methoden zur Etablierung von allgemeingültigen Strategien angewandt.
Dazu gehört zum Beispiel ein Ansatz der kombinatorischen Biosynthese[9], bei dem
durch Substitution der AT-Domänen mit denen alternativer Spezien neue
Polyketid-Varianten erzeugt werden sollen.[38,39,40] Eine bemerkenswerte Arbeit ist
der Aufbau einer Bibliothek aus 61 Analoga von 6-dEB (8) mitunter durch
Austausche der AT-Domänen zwischen der Erythromycin- und der Rapamycin-PKS
in einem gentechnisch veränderten Wirt.[41] Hierbei wurden die Experimente an einer
vollständigen PKS durchgeführt. Da Hybrid-PKS, die aus diesen domain swapping-
Experimenten resultierten, jedoch häufig eine geringere Produktion des gewünschten
Produktes aufweisen als die entsprechenden Wildtypen, konnten die Derivate nicht in
präparativen Mengen erhalten werden.[42,43]
Diese, sowie zahlreiche weitere Swapping-Experimente führten zur erfolgreichen
Generierung von Hybrid-PKS, jedoch resultierten viele Ansätze auch in
nicht-funktionellen Enzymen.[40] Dies liegt vor allem an der Problematik die Modul-
und Domängrenzen zu erkennen und der strukturellen Integrität der erzeugten
Hybrid-PKS.[12]
Eine neue Strategie der kombinatorischen Biosynthese ist die Einführung von
Punktmutationen. Somit werden einige Problematiken wie die Fehlfaltung von
Proteinen bei Swapping-Experimenten umgangen. Es konnten bereits Erfolge dieses
Ansatzes, zum Beispiel durch die Relaxation der Substratspezifität von
AT-Domänen[44], verzeichnet werden. Eine systematische Anwendung von
Punktmutationen ermöglichte einen Zugang zu einer Reihe von neuen

Einleitung | 16
Polyketid-Derivaten.[45] Schwierigkeiten liegen hierbei im Verständnis der
Enzymchemie und der strukturellen Auswirkungen von Aminosäureaustausche.[20]
Daneben ist es ohne Kristallstruktur schwierig sinnvolle Aminosäurepositionen für
einen Austausch zu definieren. Dies kann teilweise durch die Anwendung von
Homologiemodellen umgangen werden. Problematisch bleibt jedoch die Analyse der
resultierenden Ergebnisse, besonders wenn sie nicht den, auf Basis des Modells
gestützten, Erwartungen entsprechen. In der Arbeitsgruppe wurden bereits
Untersuchungen zu Sequenz-Funktionsbeziehungen der AT6 unternommen. Hierbei
wurden auf der Basis von Alignments 14 Positionen identifiziert, die mit der
Substratspezifität bezüglich des Einbaus von Methylmalonyl-CoA versus Malonyl-
CoA korrelieren. Es konnten drei Varianten erzeugt werden, die
2-Desmethylerythromycin (9) liefern.[46]
Abbildung 9: Struktur des 2-Desmethylerythromycin (9). Ein durch Punktmutation erzeugtes
Erythromycin-Derivat.[46]

Aufgabenstellung | 17
2 Aufgabenstellung
Ein wichtiges strukturelles Element der Polyketide ist ihre Stereochemie.
Erythromycin A (7) besitzt allein 18 Stereozentren und damit theoretisch 218 mögliche
Stereoisomere. Tatsächlich ist aber bisher nur ein einzelnes Isomer gefunden
worden.[47]
Abbildung 10: Erythromycin A (7) mit seinen 18 Stereozentren.
Die Stereochemie übt einen großen Einfluss auf die globale Struktur des Moleküls
aus und prägt dessen Funktion bzw. biologische Wirkung entscheidend.[48] Die Natur
hat hier Wege gefunden, nur eine spezifische Struktur zu synthetisieren. Dies wirft
die Frage nach dem mechanistischen Ursprung der Spezifität auf.[49] Die Chemie
beschäftigt sich bereits seit Jahrzehnten mit der Frage, wie die Konfiguration von
Stereozentren kontrolliert werden kann. In den letzten Jahren geriet dabei auch
zunehmend die Biokatalyse in den wissenschaftlichen Fokus.[50] Es wurden Modelle
entwickelt, die die Stereospezifität von Enzymen erklären sollen. Häufig fehlt jedoch
noch das nötige Verständnis, um den Mechanismus vollständig aufzuklären. Bei den
hochkomplexen Polyketidsynthasen ist die Stereochemie der reduktiven Domänen
bereits recht gut verstanden.[30,31] Die AT-Domänen jedoch sind bisher
vergleichsweise schlecht untersucht. Es ist aktuell noch nicht bekannt, ob die
Konfiguration der Seitenkette allein durch die AT-Domänen bestimmt wird, oder ob
weitere Domänen wie beispielsweise die KS oder das ACP auch eine Rolle dabei
spielen.
Die Arbeitsgruppe Schulz beschäftigt sich seit geraumer Zeit mit der
Substratspezifität von Acyltransferasen. Als Modellsystem wurde die AT6-Domäne
der Erythromycin Biosynthesemaschinerie (DEBS) gewählt. Durch vorrangegangene
Untersuchungen von Struktur-Funktionsbeziehungen konnte ein Bild der
Substraterkennung durch die DEBS AT6-Domäne gewonnen werden. So wurde der

Aufgabenstellung | 18
Einfluss von verschiedenen Resten auf die Enzymaktivität untersucht. Weitergehend
konnte in der Arbeitsgruppe aufgezeigt werden, dass in Folge einer einzelnen
Punktmutation innerhalb dieser AT6-Domäne in Folge einer Punktmutation die
Substratspezifität relaxiert werden kann, wodurch der Einbau des sterisch
anspruchsvolleren Propargylmalonats ermöglicht wurde.[46] Diese Punktmutation an
der Aminosäureposition Val295 belegt, dass diese in direktem Kontakt mit der
Seitenkette des Substrates steht. Diese Aminosäureposition konnte erst mit Hilfe
eines Modells der substratgebundenen AT6-Domäne – durch Kooperation der
Arbeitsgruppe Schulz mit der Arbeitsgruppe für Theoretische Chemie um Dr. Elsa
Sanchez-Garcia vom MPI für Kohlenforschung entworfen – identifiziert werden.
Abbildung 11: Modellstruktur der DEBS AT6 Region. Die Aminosäuren Met235 (gelb) und Val295 (blau), sowie das Substrat (S)-MM-CoA sind gekennzeichnet. Das Modell stammt vom
Kooperationspartner Kenny Bravo-Rodriguez.
Das Modell zeigt, dass die Seitenkette des Malonats auf das Val295 zeigt. Basierend
auf den experimentellen Daten erfolgte eine nähere Auswertung des Modells,
resultierend in einer möglichen Erklärung zum ausschließlichen Einbau des
(S)-Methylmalonats. Ein auf Basis eines Modells getroffenes Ergebnisbild ist zwar
recht suggestiv, jedoch konnte durch erfolgreiche Mutagenese-Experimente die
Qualität der modellierten Struktur bestätigt werden. Die Aminosäure Val295 besitzt
also eine Wirkung auf die Substraterkennung jenseits der offensichtlichen
suggestiven Grafikdarstellung. Eine weitere Deutung des Modells zeigte auf, dass
die Aminosäuren Val295 und Met235, die in der Substratbindetasche gegenüber

Aufgabenstellung | 19
liegen, aller Wahrscheinlichkeit nach ein Einbau des (R)-Methylmalonats
ausschließen. Es wurden Modelle berechnet, bei denen die Substratbindung im
nativen Zustand und bei einem Austausch der Aminosäuren Val295 und Met235
untersucht wurden. Die Berechnungen weisen darauf hin, dass die Variante
Val295Met & Met235Val in der Lage sein könnte das (R)-Methylmalonat einzubauen.
Abbildung 12: Aktives Zentrum der AT6. Wildtyp mit Met235 (gelb), Val295 (blau) und (S)-MMCoA.
Abbildung 13: Aktives Zentrum der AT6. Doppelmutante Met235Val (blau), Val295Met (gelb) und (R)-MMCoA.
Auf Basis dieser Ergebnisse entwickelte sich das Projekt dieser Arbeit. Ziel war es
eine Variante der AT6 zu generieren, die das Substrat (R)-Methylmalonat akzeptiert
und einbaut. Dabei war es sinnvoll sich auf die AT6 zu konzentrieren, da hierzu in
der Arbeitsgruppe bereits Untersuchungen gemacht wurden und daher ein Spektrum
aus verschiedenen Methoden zur Mutation entwickelt worden ist. Zudem wurde das
Aufgabenstellung | 20
Modell auf der Basis der AT6-Domäne entwickelt. Es wurde eine Bibliothek von
Enzymvarianten konstruiert, die auf die Aminosäurepositionen Val295 und Met235
fokussiert ist.
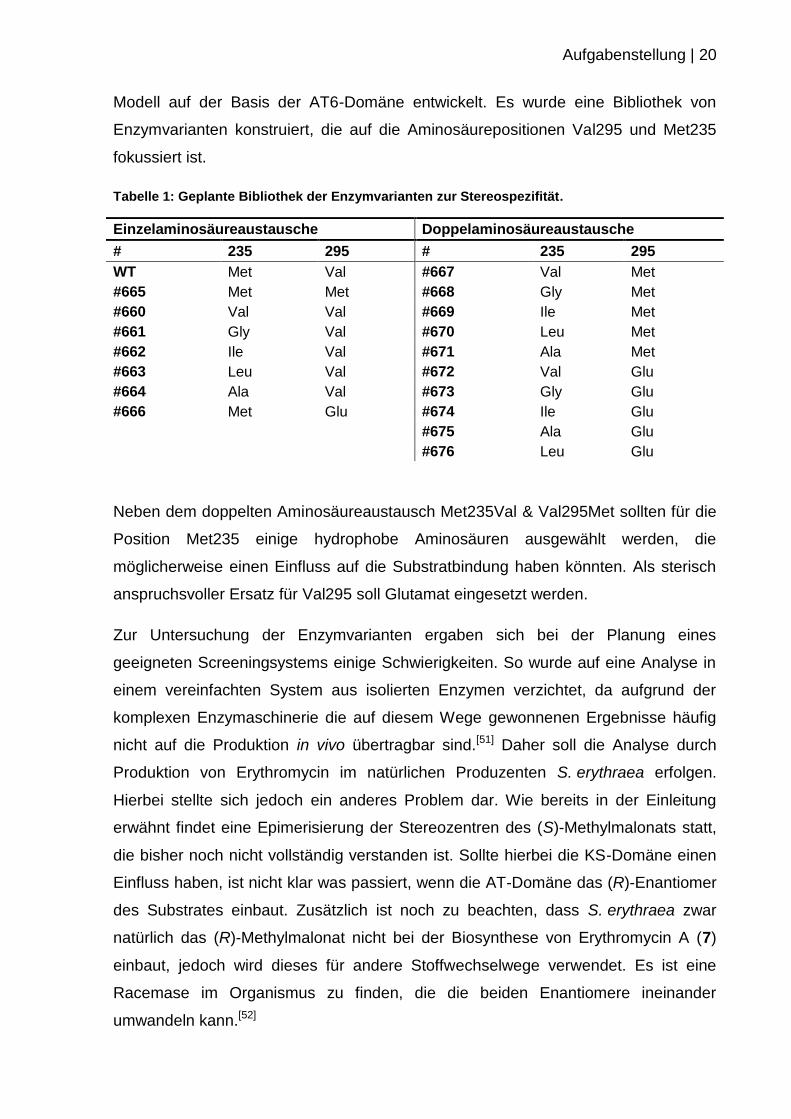
Tabelle 1: Geplante Bibliothek der Enzymvarianten zur Stereospezifität.
Einzelaminosäureaustausche Doppelaminosäureaustausche
# 235 295 # 235 295
WT Met Val #667 Val Met
#665 Met Met #668 Gly Met
#660 Val Val #669 Ile Met
#661 Gly Val #670 Leu Met
#662 Ile Val #671 Ala Met
#663 Leu Val #672 Val Glu
#664 Ala Val #673 Gly Glu
#666 Met Glu #674 Ile Glu
#675 Ala Glu
#676 Leu Glu
Neben dem doppelten Aminosäureaustausch Met235Val & Val295Met sollten für die
Position Met235 einige hydrophobe Aminosäuren ausgewählt werden, die
möglicherweise einen Einfluss auf die Substratbindung haben könnten. Als sterisch
anspruchsvoller Ersatz für Val295 soll Glutamat eingesetzt werden.
Zur Untersuchung der Enzymvarianten ergaben sich bei der Planung eines
geeigneten Screeningsystems einige Schwierigkeiten. So wurde auf eine Analyse in
einem vereinfachten System aus isolierten Enzymen verzichtet, da aufgrund der
komplexen Enzymaschinerie die auf diesem Wege gewonnenen Ergebnisse häufig
nicht auf die Produktion in vivo übertragbar sind.[51] Daher soll die Analyse durch
Produktion von Erythromycin im natürlichen Produzenten S. erythraea erfolgen.
Hierbei stellte sich jedoch ein anderes Problem dar. Wie bereits in der Einleitung
erwähnt findet eine Epimerisierung der Stereozentren des (S)-Methylmalonats statt,
die bisher noch nicht vollständig verstanden ist. Sollte hierbei die KS-Domäne einen
Einfluss haben, ist nicht klar was passiert, wenn die AT-Domäne das (R)-Enantiomer
des Substrates einbaut. Zusätzlich ist noch zu beachten, dass S. erythraea zwar
natürlich das (R)-Methylmalonat nicht bei der Biosynthese von Erythromycin A (7)
einbaut, jedoch wird dieses für andere Stoffwechselwege verwendet. Es ist eine
Racemase im Organismus zu finden, die die beiden Enantiomere ineinander
umwandeln kann.[52]

Aufgabenstellung | 21
Der Einbezug des globalen Kontexts einer intakten PKS in sämtlichen
Untersuchungen hat hierbei den Preis eines Screeningsystems mit einem komplexen
Auslesen der Resultate, da eine Observable mehrere Determinanten enthält. Die
Mutanten der AT6-Domäne werden in vitro durch zielgerichtete Mutagenese erzeugt
und durch einen Vektor und einen Donorstamm mittels intergenetischer Konjugation
in S. erythraea eingebracht. Durch Fermentation sollen präparative Mengen des
putativen Erythromycin-Stereoisomers zugänglich gemacht werden. Bei der
Doppelmutante Val295Met & Met235Val sollte die Aktivität der PKS vollständig
inaktiv sein, außer die Konzentration des in vivo produzierten (R)-Methylmalonats ist
groß genug, um als Substrat zur Verfügung zu stehen. Da dies jedoch als eher
unwahrscheinlich betrachtet wird, soll mit einer Fütterung mit rac-Methylmalonyl-
SNAC (13) komplementiert werden. Der einfachste Nachweis, ob bei einer
Enzymvariante das (R)-Methylmalonat eingebaut wurde resultierend in einer
Konfigurationsänderung des Zielmoleküls, ist durch eine NMR-Analyse des
produzierten Erythromycin-Derivates.
Insgesamt sollen also S. erythraea_AT6* Varianten erzeugt und fermentiert werden.
Erythromycin soll aus den Fermentationskulturen extrahiert und isoliert werden, um
mittels 1H-NMR analysiert zu werden. Zur Vorauswahl geeigneter Kandidaten der
präparativen Fermentation soll eine Kultivierung der S. erythraea_AT6* Varianten in
24-Lochplatten des System Duetz durchgeführt werden. Die Fermentationsprodukte
sollen mittels LC/ESI-MS analysiert werden, um die Produktion von Erythromycin
festzustellen. Das Substrat rac-Methylmalonyl-SNAC (13) zur Fütterung soll dabei
über eine chemische Synthese im Rahmen dieser Arbeit hergestellt werden. Die
Synthese des enantiomerenreinen (R)-MM-SNAC ist in diesem Fall nicht sinnvoll, da
es durch die oben erwähnte Racemase des Organismus epimerisiert werden würde.
Somit kann auf eine deutlich anspruchsvollere enantiomerenreine Synthese
verzichtet werden.

Aufgabenstellung | 22
Abbildung 14: Syntheseplanung des Substrates rac-Methylmalonyl-SNAC (13).
Ausgehend von tert-Butylpropionat (10) soll über eine Carboxylierung mit CO2 die
geschützte Methylmalonsäure 11 hergestellt werden. Im Anschluss soll diese mit
SNAC (14) gekoppelt werden. SNAC (14) ist ein Coenzym A Mimetikum, welches die
Malonsäure aktiviert. Die Vorteile gegenüber Coenzym A sind die
Membrangängigkeit, sowie die deutlich einfachere und preiswertere Synthese. Als
letzten Schritt folgt die Entschützung des Substrates 13.
Ein weiteres Ziel dieser Arbeit ist die Erweiterung der Substratspezifität der
AT6-Domäne. Dazu wurde, aufbauend auf den Ergebnissen von Uschi
Sundermann[45], eine Bibliothek von Enzymvarianten geplant. Durch
Mutageneseexperimente zeigte sich, dass die Aminosäurepositionen Tyr297 und
Ser299 ebenfalls in direktem Kontakt zum akzeptierten Substrat stehen. Durch
spezifische Aminosäureaustausche an diesen beiden Positionen konnte der Einbau
von Propargylmalonat erzielt werden. Erstaunlich ist, dass eine Mutation von nur
einer der beiden Positionen nicht zu diesem Ergebnis führte. Diese Ergebnisse
stimmen ebenfalls mit dem Modell der AT6 überein und bestätigten dessen Wert.[46]
Um eine mögliche Erweiterung der Substratakzeptanz zu erreichen, wurde eine
Bibliothek geplant, die auf diesen Aminosäurepositionen fokussiert ist. Auch hier
sollten die Mutationen durch Sättigungsmutagenese eingebracht werden. Das
Screening sollte über Fermentationen von S. erythraea in 24-Lochplatten des System
Duetz mit Fütterung von Propargylmalonat und anfolgender Analyse der
Erythromycin-Derivate über LC/ESI-MS erfolgen.

Aufgabenstellung | 23
Tabelle 2: Geplante Bibliothek von Enzymvarianten zur Substratspezifität.
Tyr297X Ser299X
Val -
His -
Ala Ala
Gly Gly
Leu Leu
WT WT
Des Weiteren soll eine Untersuchung von S. erythraea zur Aufnahme von nicht
aktivierten Malonaten stattfinden. Dazu sollen der S. erythraea WT (NRRL-B-24071)
und die Mutante S. erythraea∆AT6hygR_pKSSU656 (Val295Ala) fermentiert werden.
Diese AT6 Variante baut neben Methylmalonat auch Propargylmalonat ein und weist
damit eine erweiterte Substratspezifität auf. Aufgrund fehlender Crotonylreduktase-
Aktivität ist Ethylmalonyl-CoA nicht in S. erythraea vorhanden.[40] Um zu testen, ob
diese Mutante auch Ethylmalonat akzeptiert, soll daher eine Fütterung der Kulturen
mit Diethylethylmalonat (15) erfolgen. Zusätzlich soll auch Diethylpropargylmalonat
(16) gefüttert werden, um einen Vergleich der Einbauraten zu der SNAC-aktivierten
Malonsäure zu erhalten und somit eine Kontrolle bezüglich fehlender SNAC-
Aktivierung zu generieren. Eine Analyse der Fermentationsprodukte soll zeigen, ob
die Malonate – auch ohne Aktivierung – eingebaut werden können.
Abbildung 15: Strukturen von Diethylethylmalonat (15) und Diethylpropargylmalonat (16).

Ergebnisse und Diskussion | 24
3 Ergebnisse und Diskussion
3.1. Mutagenese der AT6-Domäne aus DEBS3
Die Mutagenese der AT6-Domäne aus DEBS3 stellt durch die hohe Komplexität des
Enzymes eine große Herausforderung dar. In der Arbeitsgruppe konnten durch
intensive Forschungsarbeit ein Spektrum an gentechnischen Methoden zum
Einbringen von zielgerichteten Mutationen in die PKS etabliert werden. Diese werden
zur Erzeugung der Stereo- und Substratspezifitätsvarianten in diesem Projekt
angewandt. Die erzeugten AT6 Varianten werden nicht heterolog in Wirten, sondern
in S. erythraea, dem Erythromycin-Originalproduzenten exprimiert.
3.1.1. Wiederherstellung der Wildtyp Aktivität von S. erythraea∆AT6hygR
Der Stamm S. erythraea∆AT6hygR, der in dieser Arbeitsgruppe konstruiert wurde, ist
nicht in der Lage Erythromycin zu bilden, da das Enzym DEBS3 durch das Inserieren
der Hygromycin-Kassette an die Position der AT6-Domäne inaktiv ist. Das Plasmid
pKSSU89 wurde ebenfalls in der Arbeitsgruppe konstruiert, um eine schnelle und
gezielte Integration in das Genom der Actinomyceten zu erzielen. Es trägt eine
vollständige DEBS3 kodierende Gensequenz, einen Actinorhodin-Promotor (actII),
eine Apramycin Resistenzkassette sowie eine attP Integrasebindestelle des
Bakteriophagen VWB (für eine gezielte Integration in das Genom von
Actinomyceten).
Abbildung 16: Shuttle-Vektor pKSSU89. Die Aminosäurepositionen Val295 und Met235, sowie die NotI Schnittstellen sind gekennzeichnet.
Wird dieser Vektor (pKSSU89) in den Stamm S. erythraea∆AT6hygR transformiert, so
kann er über die Integrasebindestelle an das Genom binden. Dabei wird der Vektor
in das Genom integriert. Dieser Vorgang ist jedoch nicht dauerhaft stabil und kann
pKSSU89

Ergebnisse und Diskussion | 25
auch wieder rückgängig gemacht werden. Aus diesem Grund muss bei der
Kultivierung der Selektionsdruck mit dem entsprechenden Antibiotikum aufrecht
gehalten werden.
Ist der Vektor in das Genom von S. erythraea∆AT6hygR integriert, so ist der Stamm
wieder in der Lage Erythromycin zu bilden. Das durch den Vektor pKSSU89 kodierte
Enzym DEBS3 übernimmt das Intermediat, welches von DEBS1 und 2 produziert
wurde. Wird der Vektor pKSSU89 innerhalb der AT6 kodierenden Region vor der
Konjugation mutiert (AT6*), können S. erythraea Mutanten erzeugt werden, die einen
Aminosäureaustausch innerhalb der AT6-Domäne aufweisen (S. erythraeaAT6*).
Hierbei ist es notwendig die S. erythraea∆AT6hygR Mutante zu verwenden, um den
natürlichen Produktionsweg von Erythromycin zu unterbinden und so sicherzustellen,
dass die DEBS3 Variante das Intermediat weiter prozessiert.
Zuerst mussten ausreichende Mengen des Vektors pKSSU89 generiert werden.
Dazu wurde die Plasmid-DNA mittels alkalischer Lyse isoliert. Die erhaltene DNA
wurde mittels Kolonie-PCR, sowie Restriktionsanalyse mit dem Enzym NotI
verifiziert.
Abbildung 17: Kolonie-PCR von pKSSU89 tragenden Transformanden.
Um S. erythraeaAT6_pKSSU89 zu generieren – sprich Wildtyp Aktivität
wiederherzustellen – musste der Vektor pKSSU89 zuerst in E. coli
ET12567/pUZ8002 Zellen transformiert werden. Positive Klone (Selektion über
entsprechende Antibiotika) wurden anschließend als Donorstamm für eine
intergenetische Konjugation in S. erythraea∆AT6hygR Zellen eingesetzt. Die
Transkonjuganten wurden durch Überschichtung mit antibiotikahaltigem LB-Agar
identifiziert.
5.0 kb
1.5 kb
1.0 kb
0.5 kb

Ergebnisse und Diskussion | 26
Zur Überprüfung der Erythromycin-Produktion der Stämme S. erythraea
NRRL-B-24071, S. erythraea∆AT6hygR und S. erythraea∆AT6hygR_pKSSU89 sollte
zunächst ein Hemmhoftest durchgeführt werden. Dazu wurden Proben von
Fermentationskulturen der Stämme in ausgestanzte Löcher einer Agarplatte
gegeben. Bacillus subtilis diente hierbei als Referenzstamm.
Abbildung 18: Untersuchung der antimikrobiellen Aktivität durch einen Hemmhoftest unter Verwendung von Bacillus subtilis als Referenzstamm. Die Fermentationskultur des Wildtyps erzeugt eine deutlich sichtbare Wachstumsinhibitionszone, wohingegen bei S. erythraeaΔAT6hyg
R
keine Ausbildung einer Inhibitionszone zu beobachten ist.
Anhand der Abbildung 18 ist zu erkennen, dass der Stamm S. erythraea
NRRL-B-24071 (Wildtyp) einen Hemmhof aufweist. Durch vorhandenes Erythromycin
wird Bacillus subtilis am Wachstum gehemmt. Der Stamm S. erythraea∆AT6hygR
dagegen weist erwartungsgemäß keinen Hemmhof auf, da hier die Produktion von
Erythromycin durch die Hygromycin-Kassette inaktiv ist. Der Stamm
S. erythraea∆AT6hygR_pKSSU89 sollte ebenfalls einen Hemmhof aufweisen, da
durch die Integration der Vektor-DNA die Fähigkeit zur Produktion von Erythromycin
wiederhergestellt sein sollte. Auf der Platte ist jedoch nur ein minimaler Hemmhof
erahnbar. Da gentechnisch veränderte S. erythraea Kulturen häufig eine geringere
Produktion als der Wildtyp aufweisen, wurde eine nähere Analyse durch eine
LC/ESI-MS Messung durchgeführt.
Für die LC/ESI-MS Messungen wurden die Fermentationsprodukte aus den Kulturen
mit Ethylacetat extrahiert. Nach Entfernen des Lösungsmittels wurden die Proben in
Methanol aufgenommen und der Massenanalyse unterzogen. Dabei konnte eindeutig
detektiert werden, dass der S. erythraea Wildtyp NRRL-B-24071 Erythromycin
produziert [vgl. Abbildung 19 (A)], wohingegen die Fermentation der DEBS3
S. ery∆AT6hygR_
pKSSU89
S. ery∆AT6hygR
S. ery WT
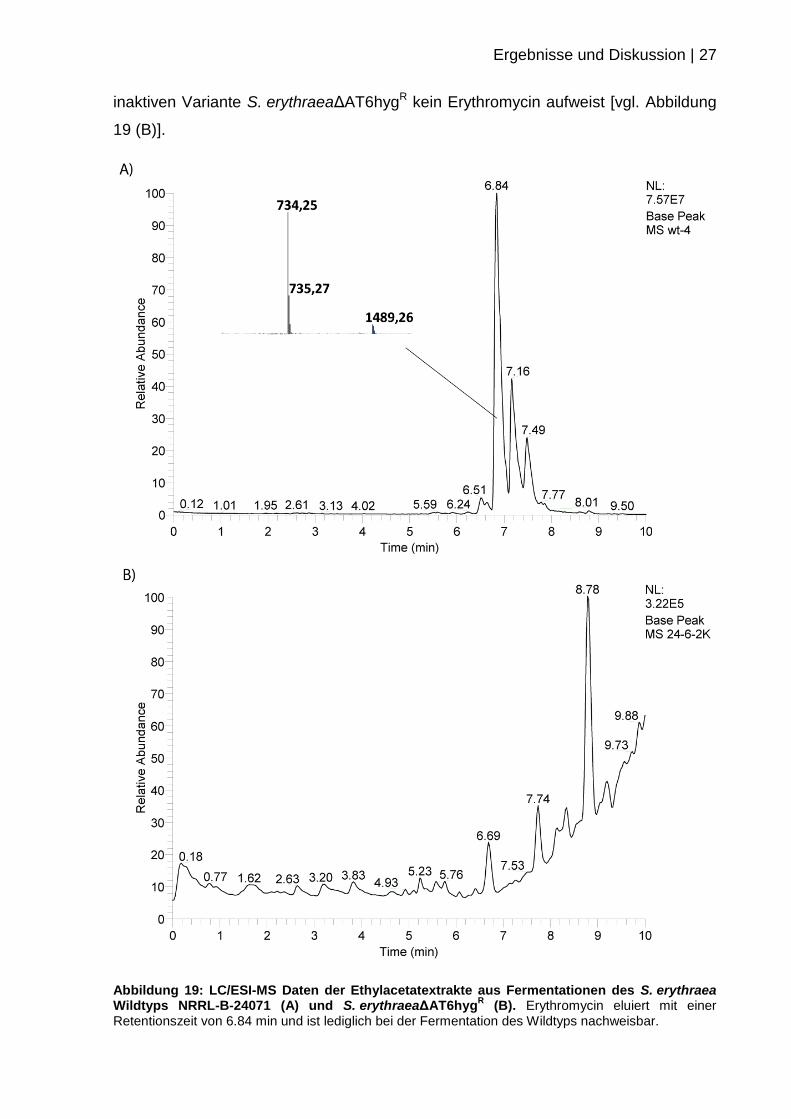
Ergebnisse und Diskussion | 27
inaktiven Variante S. erythraeaΔAT6hygR kein Erythromycin aufweist [vgl. Abbildung
19 (B)].
Abbildung 19: LC/ESI-MS Daten der Ethylacetatextrakte aus Fermentationen des S. erythraea Wildtyps NRRL-B-24071 (A) und S. erythraeaΔAT6hyg
R (B). Erythromycin eluiert mit einer
Retentionszeit von 6.84 min und ist lediglich bei der Fermentation des Wildtyps nachweisbar.
Ergebnisse und Diskussion | 28
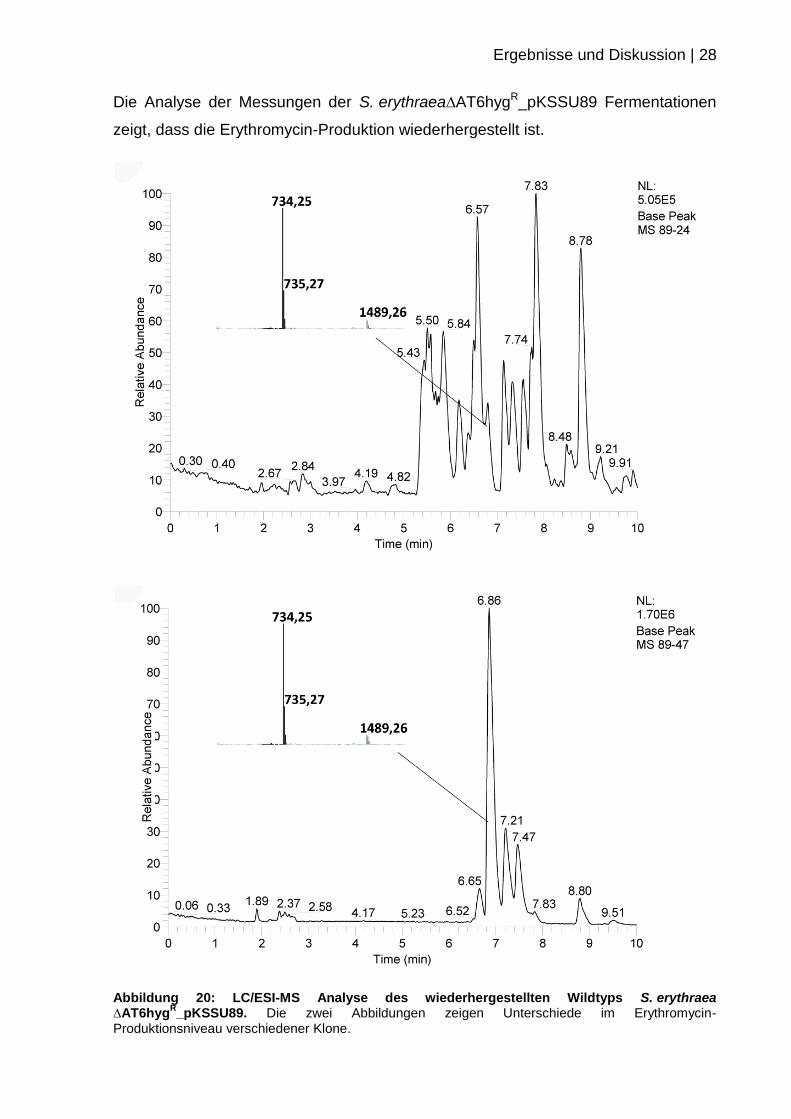
Die Analyse der Messungen der S. erythraea∆AT6hygR_pKSSU89 Fermentationen
zeigt, dass die Erythromycin-Produktion wiederhergestellt ist.
Abbildung 20: LC/ESI-MS Analyse des wiederhergestellten Wildtyps S. erythraea ∆AT6hyg
R_pKSSU89. Die zwei Abbildungen zeigen Unterschiede im Erythromycin-
Produktionsniveau verschiedener Klone.

Ergebnisse und Diskussion | 29
Bei der Analyse von mehreren Klonen der Mutante zeigen sich Unterschiede im
Produktionsniveau von Erythromycin. Diese hohe Klon-zu-Klon Variabilität ist typisch
für gentechnisch veränderte S. erythraea Stämme.[46]
3.1.2. Präparation der AT6* Mutanten zur Stereospezifität
Die Präparation der Sterospezifitätsvarianten Bibliothek – fokussiert auf die
Positionen Val295 und Met235 – erfolgte mittels Sättigungsmutagenese. Der Vektor
pKSSU89 trägt die vollständige DEBS3 Gen-Kassette und wird in der PCR als
Template verwendet. Die Enzymvarianten sollten mittels spezifisch definierten
Oligonukleotiden erzeugt werden, um aussagekräftige Datensätze mit einem
praktikablen Screening-Aufwand zu erhalten. Die präparierten PCR-Produkte sollten
in einen E. coli Shuttle-Vektor pKSSU96 (basiert auf pKSSU89 Vektor, AT6 Region
durch eine Hygromycin-Resistenz Kassette ausgetauscht, DEBS3 inaktiv) kloniert
werden. Die resultierenden wiederhergestellten pKSSU89* Plasmide sollten dann in
S. erythraea∆AT6hygR konjugiert werden, um das inaktive DEBS3 zu
komplementieren.
3.1.2.1. Sättigungsmutagenese zur Erstellung der Stereospezifitätsvarianten
Die Präparation der Einzelaminosäureaustausche erfolgte zuerst. Dazu wurden die
entsprechenden Oligos in der zweistufigen overlap extension PCR eingesetzt.
Abbildung 21: Schema zum Arbeitsablauf der zielgerichteten Mutagenese am Beispiel der Mutation Met235Val.
Da nicht immer eine nebenproduktfreie Amplifizierung möglich war, wurde nach
beiden PCR Durchgängen eine Gelaufreinigung durchgeführt.
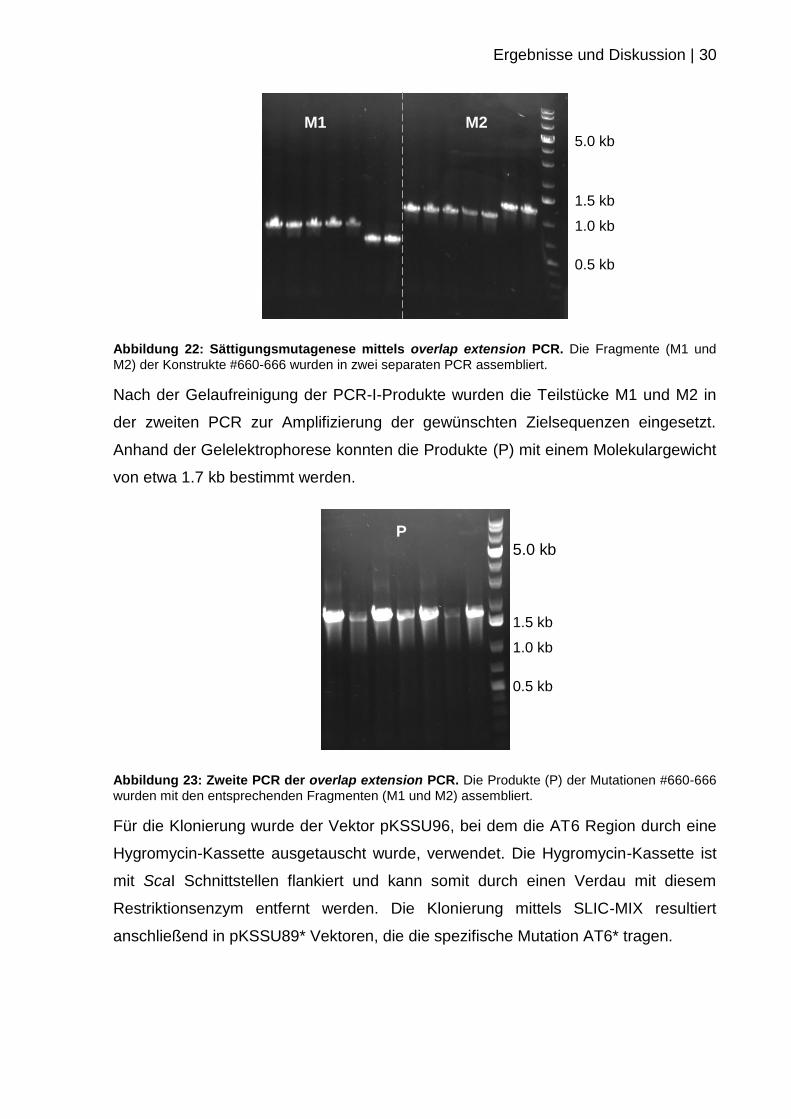
Ergebnisse und Diskussion | 30
Abbildung 22: Sättigungsmutagenese mittels overlap extension PCR. Die Fragmente (M1 und
M2) der Konstrukte #660-666 wurden in zwei separaten PCR assembliert.
Nach der Gelaufreinigung der PCR-I-Produkte wurden die Teilstücke M1 und M2 in
der zweiten PCR zur Amplifizierung der gewünschten Zielsequenzen eingesetzt.
Anhand der Gelelektrophorese konnten die Produkte (P) mit einem Molekulargewicht
von etwa 1.7 kb bestimmt werden.
Abbildung 23: Zweite PCR der overlap extension PCR. Die Produkte (P) der Mutationen #660-666
wurden mit den entsprechenden Fragmenten (M1 und M2) assembliert.
Für die Klonierung wurde der Vektor pKSSU96, bei dem die AT6 Region durch eine
Hygromycin-Kassette ausgetauscht wurde, verwendet. Die Hygromycin-Kassette ist
mit ScaI Schnittstellen flankiert und kann somit durch einen Verdau mit diesem
Restriktionsenzym entfernt werden. Die Klonierung mittels SLIC-MIX resultiert
anschließend in pKSSU89* Vektoren, die die spezifische Mutation AT6* tragen.
M1 M2
5.0 kb
1.5 kb
1.0 kb
0.5 kb
M1
5.0 kb
1.5 kb
1.0 kb
0.5 kb
P

Ergebnisse und Diskussion | 31
Abbildung 24: Vektor pKSSU96 für die Klonierung mittels SLIC-MIX. Die Hygromycin-Kassette (blau), sowie die Schnittstellen der Restriktionsenzyme ScaI und FseI sind gekennzeichnet.
Nach erfolgter Klonierung und Transformation in E. coli OmniMAX™ 2 T1 Zellen
wurde am darauffolgenden Tag zur Identifizierung potenziell richtiger (Insert-
tragender) Klone eine Kolonie-PCR durchgeführt. Hierbei bindet ein Oligonukleotid
spezifisch innerhalb des Inserts, wohingegen das zweite Oligonukleotid an DEBS3
annealt. Somit ist eine Kontrolle des positiven Einbaus des Inserts gewährleistet.
Abbildung 25: Kolonie-PCR zur Identifizierung Insert-tragender Klone (#660-666). Die 1.7 kb
großen PCR Produkte konnten in allen untersuchten Klonen nachgewiesen werden.
Die ausgewählten Klone sind hier meist positiv, wodurch bestätigt wird, dass die
Klonierung mit der SLIC-MIX Methode sehr zuverlässig ist. Die Klone wurden
weitergehend mittels Restriktionsanalyse mit dem FastDigest Enzym NotI und einer
Sequenzierung verifiziert (vgl. 3.1.3).
Die Plasmide wurden über eine Miniplasmidpräparation isoliert und als Template für
die Präparation der Doppelaminosäureaustausche eingesetzt. Dazu wurde erneut die
overlap extension PCR mit den entsprechenden Oligonukleotiden eingesetzt. Es
5.0 kb
1.5 kb
1.0 kb
0.5 kb
pKSSU96
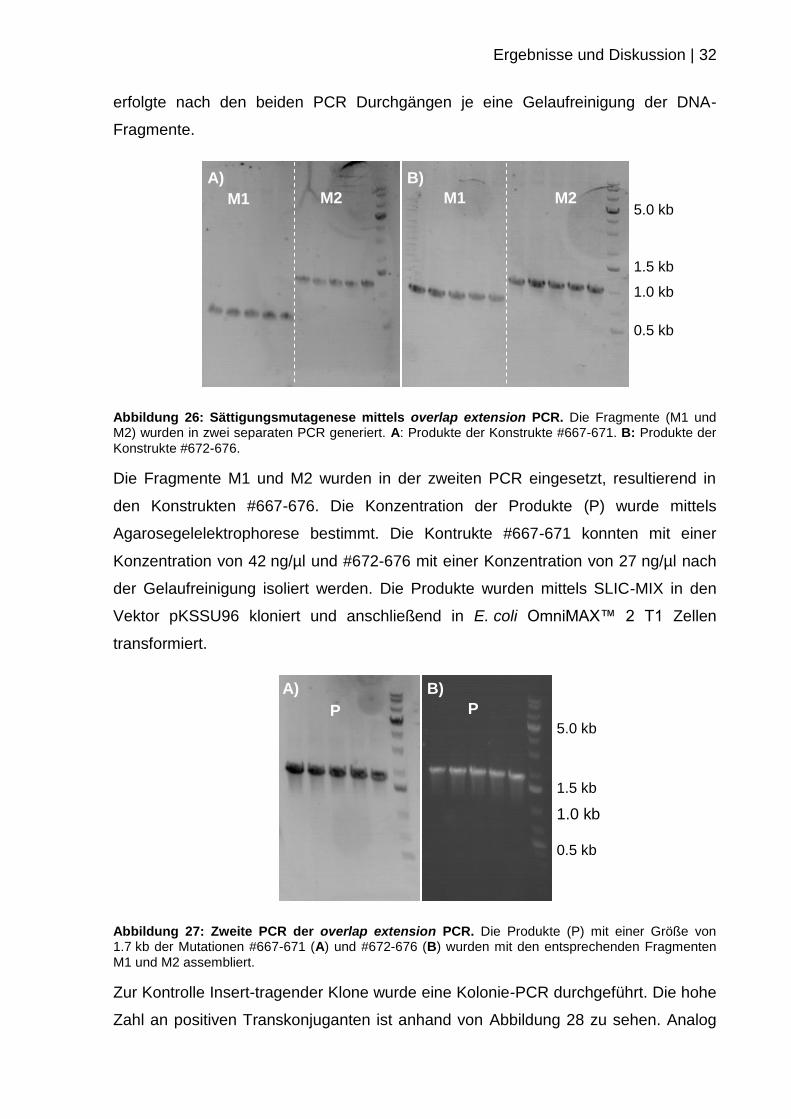
Ergebnisse und Diskussion | 32
erfolgte nach den beiden PCR Durchgängen je eine Gelaufreinigung der DNA-
Fragmente.
Abbildung 26: Sättigungsmutagenese mittels overlap extension PCR. Die Fragmente (M1 und M2) wurden in zwei separaten PCR generiert. A: Produkte der Konstrukte #667-671. B: Produkte der
Konstrukte #672-676.
Die Fragmente M1 und M2 wurden in der zweiten PCR eingesetzt, resultierend in
den Konstrukten #667-676. Die Konzentration der Produkte (P) wurde mittels
Agarosegelelektrophorese bestimmt. Die Kontrukte #667-671 konnten mit einer
Konzentration von 42 ng/µl und #672-676 mit einer Konzentration von 27 ng/µl nach
der Gelaufreinigung isoliert werden. Die Produkte wurden mittels SLIC-MIX in den
Vektor pKSSU96 kloniert und anschließend in E. coli OmniMAX™ 2 T1 Zellen
transformiert.
Abbildung 27: Zweite PCR der overlap extension PCR. Die Produkte (P) mit einer Größe von 1.7 kb der Mutationen #667-671 (A) und #672-676 (B) wurden mit den entsprechenden Fragmenten
M1 und M2 assembliert.
Zur Kontrolle Insert-tragender Klone wurde eine Kolonie-PCR durchgeführt. Die hohe
Zahl an positiven Transkonjuganten ist anhand von Abbildung 28 zu sehen. Analog
A) B)
B) A)
M1 M2 M1 M2
P P
5.0 kb
1.5 kb
1.0 kb
0.5 kb
5.0 kb
1.5 kb
1.0 kb
0.5 kb
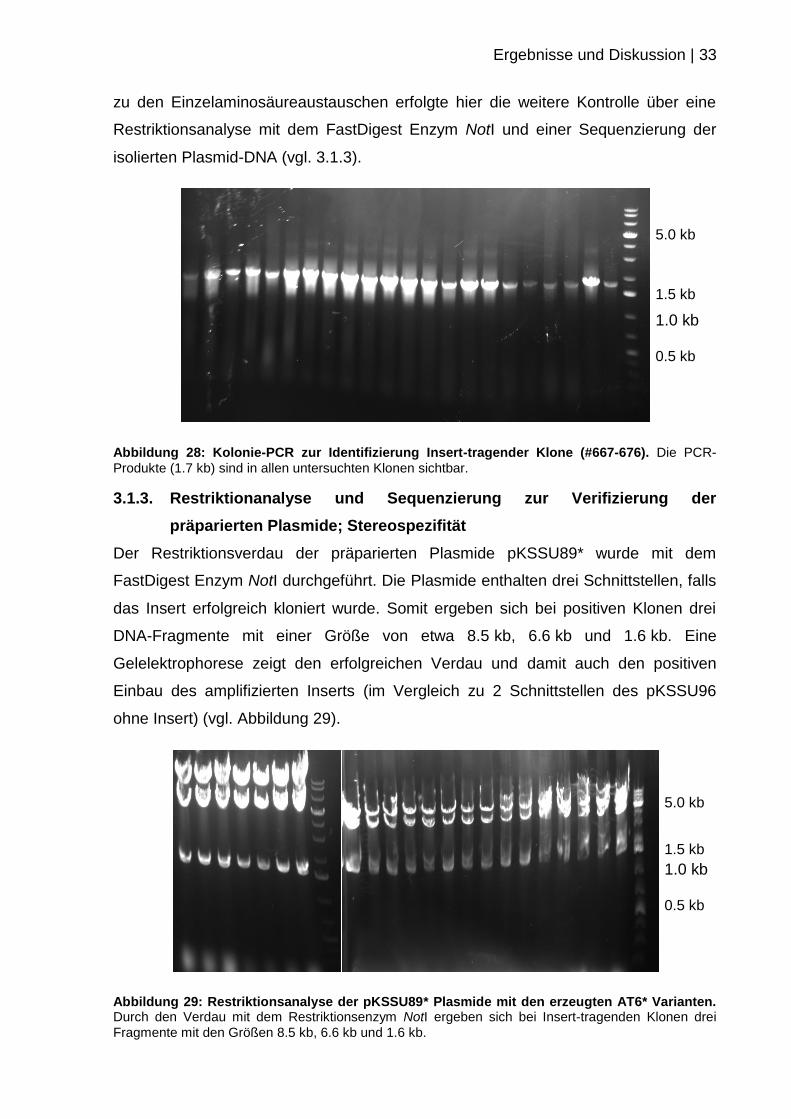
Ergebnisse und Diskussion | 33
zu den Einzelaminosäureaustauschen erfolgte hier die weitere Kontrolle über eine
Restriktionsanalyse mit dem FastDigest Enzym NotI und einer Sequenzierung der
isolierten Plasmid-DNA (vgl. 3.1.3).
Abbildung 28: Kolonie-PCR zur Identifizierung Insert-tragender Klone (#667-676). Die PCR-
Produkte (1.7 kb) sind in allen untersuchten Klonen sichtbar.
3.1.3. Restriktionanalyse und Sequenzierung zur Verifizierung der
präparierten Plasmide; Stereospezifität
Der Restriktionsverdau der präparierten Plasmide pKSSU89* wurde mit dem
FastDigest Enzym NotI durchgeführt. Die Plasmide enthalten drei Schnittstellen, falls
das Insert erfolgreich kloniert wurde. Somit ergeben sich bei positiven Klonen drei
DNA-Fragmente mit einer Größe von etwa 8.5 kb, 6.6 kb und 1.6 kb. Eine
Gelelektrophorese zeigt den erfolgreichen Verdau und damit auch den positiven
Einbau des amplifizierten Inserts (im Vergleich zu 2 Schnittstellen des pKSSU96
ohne Insert) (vgl. Abbildung 29).
Abbildung 29: Restriktionsanalyse der pKSSU89* Plasmide mit den erzeugten AT6* Varianten. Durch den Verdau mit dem Restriktionsenzym NotI ergeben sich bei Insert-tragenden Klonen drei
Fragmente mit den Größen 8.5 kb, 6.6 kb und 1.6 kb.
5.0 kb
1.5 kb
1.0 kb
0.5 kb
5.0 kb
1.5 kb
1.0 kb
0.5 kb

Ergebnisse und Diskussion | 34
Die Sequenzanalyse der präparierten Plasmide pKSSU89* erfolgte über Alignments
(T-Coffee – Multiple Sequence Alignment) mit dem Wildtyp. Die Sequenzvergleiche
sind in der Abbildung 30 und Abbildung 31 aufgeführt und zeigen die
Aminosäureaustausche an den Positionen Met235 und Val295 der AT6-Domäne. Die
Sequenzen zeigen die erfolgreichen Aminosäureaustausche der Mutanten #660-666
(vgl. Abbildung 30). Die Varianten #667-676 mit doppelten Aminosäureaustauschen
wurden mit den Plasmiden der Einzelaminosäureaustausche als Template generiert.
Die Plasmide mit den Konstrukten #667-671 zeigen die geplanten Mutationen,
jedoch weisen die Konstrukte #672-676 an der Position Val295 die Aminosäure
Glutamin statt Glutamat auf. Dies liegt an einer fehlerhaften Bestellung der
Oligonukleotide. Da die Aminosäure Glutamat aus sterischen Gründen gewählt
wurde und Glutamin eine ähnliche Größe aufweist, wurden die Plasmide dennoch für
das im Weiteren folgende Screening über Fermentation verwendet.
# 235 AS # 295 AS
WT ATG Met WT GTG Val
660 GTC Val 665 ATG Met
661 GGC Gly 666 GAG Glu
662 ATC Ile
663 TTG Leu
664 GCC Ala
Abbildung 30: Nukleotidsequenzvergleich der AT6-Domäne von S. erythraea NRRL-B-24071 (Wildtyp) mit den korrespondierenden Sequenzen der Mutanten mit einem Aminosäureaustausch #660-666 im Bereich um Met235 sowie Val295. "***" zeigen die Aminosäureaustausche.

Ergebnisse und Diskussion | 35
# 235 AS 295 AS
WT ATG Met GTG Val
667 GTC Val ATG Met
668 GGC Gly ATG Met
669 ATC Ile ATG Met
670 TTG Leu ATG Met
671 GCC Ala ATG Met
672 GTC Val CAG Gln
673 GGC Gly CAG Gln
674 ATC Ile CAG Gln
675 TTG Leu CAG Gln
676 GCC Ala CAG Gln
Abbildung 31: Nukleotidsequenzvergleich der AT6-Domäne von S. erythraea NRRL-B-24071 (Wildtyp) mit den korrespondierenden Sequenzen der Mutanten mit doppeltem Aminosäureaustausch #667-676 im Bereich um Met235 sowie Val295. "***" zeigen die Aminosäureaustausche.

Ergebnisse und Diskussion | 36
3.1.4. Konjugation der AT6* Varianten in S. erythraea∆AT6hygR;
Stereospezifität
Die Einbringung der heterologen Plasmid-DNA in S. erythraea erfolgte über
intergenetische Konjugation. Die häufig verwendete Transformation ist bei
Actinomyceten meist nicht ohne Weiteres möglich, da es sich um Gram-positive
Bakterien handelt. Die Konjugation liefert hier jedoch zuverlässig eine hohe Zahl von
positiven Transkonjuganten, ist jedoch aufwendiger in der Durchführung als die
üblicherweise eingesetzte Transformation. Es wurde E. coli ET12567/pUZ8002 als
Donorstamm verwendet. Dieser Stamm ist methylierungsnegativ und trägt einen
F-Faktor, der für die Übertragung der DNA notwendig ist. Um eine Integration der
übertragenden DNA in das Genom von S. erythraea zu erreichen, wurden integrative
Vektoren verwendet. Diese enthalten Integrasestellen (basierend auf Phasen-
Integrasen), die an spezifischen Stellen innerhalb des Genoms integrieren können.
Da diese Integration auch umkehrbar ist, womit eine relativ geringe Stabilität
einhergeht, ist es notwendig einen Selektionsdruck mit den entsprechenden
Antibiotika stetig aufrecht zu halten. Nach etwa 7 Tagen konnten positive
Transkonjuganten isoliert werden. Diese wurden auf ABB13-Agarplatten kultiviert, bis
sie eine ausreichende Größe hatten. Im Anschluss wurden die Kolonien in einer
Fermentation eingesetzt oder für kurze Zeit bei Raumtemperatur gelagert.
3.1.5. Analyse der AT6* Varianten in S. erythraea∆AT6hygR; Stereospezifität
3.1.5.1. Fermentation in 24-Lochplatten und anschließende Massenanalyse
Abbildung 32: Kultivierung der Hauptkulturen in 24-Lochplatten des System Duetz.[53]
Ergebnisse und Diskussion | 37
Die Analyse der AT6* Varianten erfolgte durch Fermentation inklusive Fütterung der
Bakterien mit dem SNAC aktivierten rac-Methylmalonat (rac-MM-SNAC) (13) und
anschließender Extraktion von Erythromycin. Es wurden alle Varianten und der
Wildtyp mit und ohne Zugabe des Substrates 13 untersucht. Um die AT6* Varianten
auf eine positive Erythromycin-Produktion zu testen, wurden alle erzeugten Varianten
in 24-Lochplatten des System Duetz[53] kultiviert. Diese Kultivierung der Varianten in
kleinen Medienvolumina ermöglichte die Fermentation einer großen Zahl von
Kulturen in einem praktikablen Zeit- und Arbeitsaufwand. Zusätzlich liefert sie den
Vorteil einer gleichmäßigen Belüftung der Kulturen. Nach einer Kultivierungsdauer
von insgesamt 7 Tagen (2 Tage Vorkultur und 5 Tage Hauptkultur) wurden die
Fermentationsprodukte mit Ethylacetat extrahiert. Die Extrakte wurden nach
Verdampfung des Lösungsmittels in Methanol gelöst und über eine LC/ESI-MS
Messung analysiert. Konnte Erythromycin in den Proben nachgewiesen werden,
erfolgte eine quantitative Analyse der gefundenen Menge im Vergleich zum analog
fermentierten Wildtyp. Um statistisch signifikante Resultate zu bekommen, wurden
jeweils vier Klone jeder Variante parallel fermentiert und analysiert.
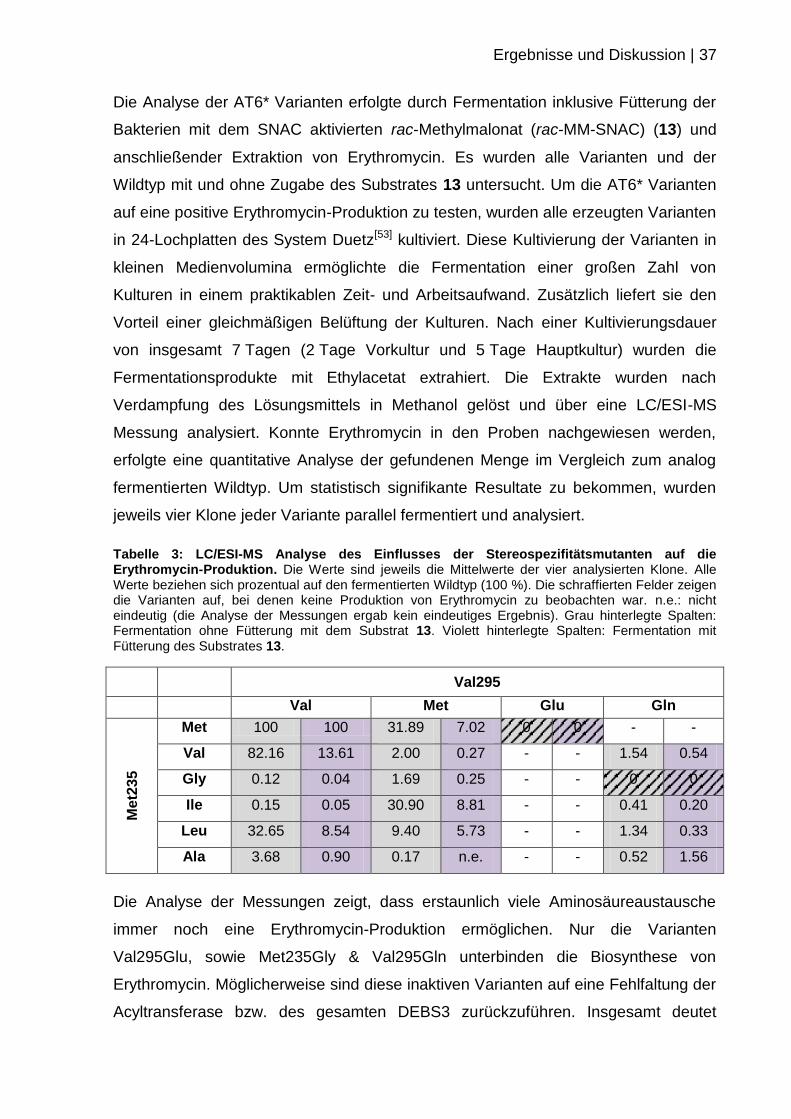
Tabelle 3: LC/ESI-MS Analyse des Einflusses der Stereospezifitätsmutanten auf die Erythromycin-Produktion. Die Werte sind jeweils die Mittelwerte der vier analysierten Klone. Alle Werte beziehen sich prozentual auf den fermentierten Wildtyp (100 %). Die schraffierten Felder zeigen die Varianten auf, bei denen keine Produktion von Erythromycin zu beobachten war. n.e.: nicht eindeutig (die Analyse der Messungen ergab kein eindeutiges Ergebnis). Grau hinterlegte Spalten: Fermentation ohne Fütterung mit dem Substrat 13. Violett hinterlegte Spalten: Fermentation mit Fütterung des Substrates 13.
Val295
Val Met Glu Gln
Me
t23
5
Met 100 100 31.89 7.02 0 0 - -
Val 82.16 13.61 2.00 0.27 - - 1.54 0.54
Gly 0.12 0.04 1.69 0.25 - - 0 0
Ile 0.15 0.05 30.90 8.81 - - 0.41 0.20
Leu 32.65 8.54 9.40 5.73 - - 1.34 0.33
Ala 3.68 0.90 0.17 n.e. - - 0.52 1.56
Die Analyse der Messungen zeigt, dass erstaunlich viele Aminosäureaustausche
immer noch eine Erythromycin-Produktion ermöglichen. Nur die Varianten
Val295Glu, sowie Met235Gly & Val295Gln unterbinden die Biosynthese von
Erythromycin. Möglicherweise sind diese inaktiven Varianten auf eine Fehlfaltung der
Acyltransferase bzw. des gesamten DEBS3 zurückzuführen. Insgesamt deutet

Ergebnisse und Diskussion | 38
dieses Ergebnis mit nur zwei inaktiven bei insgesamt 17 analysierten Varianten auf
eine recht große Toleranz der AT6 in den Positionen 235 und 295 bezogen auf
dessen Aktivität. Auffällig ist auch, dass einige Mutationen in einer sehr geringen
Aktivität gegenüber dem Wildtyp resultieren. Dies zeigt einen negativen Effekt des
Aminosäureaustausches auf das Erythromycin-Produktionsniveau. Indes resultiert
keine der Varianten in einem positiven Einfluss gegenüber dem Wildtyp, was jedoch
nicht überraschend ist, da S. erythraea insgesamt sehr empfindlich auf
gentechnische Manipulation reagiert.[46] Während die Einzelmutante Met235Val noch
eine recht hohe Erythromycin-Produktion gegenüber dem Wildtyp aufweist, ist diese
bei der Mutation Val235Met deutlich geringer. Die Doppelmutante Met235Val &
Val295Met dagegen weist mit 2 % ohne Fütterung und 0.27 % mit Fütterung fast
keine Aktivität mehr auf. Aufgrund der Fütterung zusammen mit der Vermutung der
Akzeptanz von (R)-Methylmalonat dieser Variante entspricht eine geringere Aktivität
mit Fütterung keineswegs den Erwartungen. Dieser Effekt kann auf einer Inaktivität
des Enzyms beruhen oder möglicherweise durch eine komplexe Selektivität des
Enzyms verursacht worden sein. So besteht die Möglichkeit, dass die Doppelmutante
kein (S)-Methylmalonat akzeptiert und ein zusätzlicher Selektivitätsfilter
(beispielsweise die KS-Domäne – bereits in der Aufgabenstellung diskutiert) das
eingebaute (R)-Methylmalonat nicht toleriert. Dieses Ergebnis ist durchaus
interessant und sollte durch weitere Experimente untersucht werden.
Die Varianten mit Glutamin an der Position 295 zeigen durchweg ein vergleichsweise
schlechtes Erythromycin-Produktionsniveau. Scheinbar ist also die Aminosäure
Glutamin an der Position 295 nicht förderlich für die Erythromycin-Produktion.
Welchen Einfluss die Varianten auf die Stereospezifität der AT6 haben ist durch die
LC/ESI-MS Analyse allein nicht zu klären. Dazu muss eine 1H-NMR-Analyse
durchgeführt werden.

Ergebnisse und Diskussion | 39
3.1.5.2. Fermentation zur präparativen Gewinnung von Erythromycin und
Analyse mittels 1H-NMR
Abbildung 33: Fermentation von S. erythraea in 5 L Kolben mit Stahlfeder.
Zur Gewinnung von präparativen Mengen des Erythromycins wurde S. erythraea in
einem Gesamtvolumen von 1 L (0.5 L pro 5 L Kolben) fermentiert. Das produzierte
Erythromycin sollte aus den Kulturen isoliert werden, um eine Analyse der
Konfiguration durchführen zu können. Die Fermentation erfolgte in Glaskolben mit
einer Stahlfeder am Boden und verlief inklusive Vor- und Hauptkultur über 7 Tage. Im
Anschluss wurden die Kulturen eingefroren, um das Erythromycin aus den Zellen
freizusetzen. Die Schwierigkeiten bei der Präparation von Erythromycin aus
Fermentationskulturen bestehen in der großen Anzahl von Fermentationsprodukten
sowie im Etablieren geeigneter reproduzierbarer Methoden. In dieser Arbeit wurde
eine Extraktionskaskade mit anfolgender präparativer Dünnschichtchromatographie
durchgeführt. Durch die Extraktion mit Ethylacetat sollen organische Substanzen aus
der Fermentationskultur isoliert werden. Die folgende Extraktion mit Wasser (pH 4.5)
soll die Separation von z. B. Lipiden ermöglichen. Im dritten Schritt der Extraktion soll
Erythromycin wieder in die organische Phase überführt werden (vgl. Abbildung 34).

Ergebnisse und Diskussion | 40
Abbildung 34: Schema für die Isolation von Erythromycin aus einer S. erythraea Fermentation über Extraktion mit Ethylacetat (pH 9.5), sowie Wasser (pH 4.5).
Die anschließende präparative Dünnschichtchromatographie ermöglichte eine
schnelle Separation der extrahierten Substanzen. Schwierigkeiten ergaben sich, da
Erythromycin nicht UV-aktiv ist und somit ohne Anfärben nicht sichtbar ist. Dieses
Problem wurde gelöst, indem ein schmaler Streifen der DC-Platte mit einer
Kaliumpermanganatlösung angefärbt wurde, was nur zu einem geringen Verlust des
Produktes führte und die Position der Substanzen anzeigte.
3.1.5.3. Fermentation und Aufarbeitung von S. erythraea NRRL-B-24071
(Wildtyp)
Zur Etablierung der Methoden wurde der S. erythraea Wildtyp fermentiert und
aufgearbeitet. Zur Analyse wurden nach den Aufarbeitungsschritten LC/ESI-MS
Messungen durchgeführt. Die Analyse der organischen Phase aus dem zweiten
Extraktionsschritt ergab, dass hier nach einmaliger Extraktion mit leicht saurem
Wasser (pH 4.5) noch Erythromycin vorhanden war. Durch eine viermalige Extraktion
der EtOAc-Phase mit Wasser konnte der Großteil des Erythromycins in die wässrige
Phase überführt werden.
Nach dem dritten Extraktionsschritt, wurde der Extraktionsrückstand der
Fermentation auf eine präparative DC-Platte aufgetragen. Der Rf-Wert des als
Ergebnisse und Diskussion | 41
Referenz aufgetragenen käuflichen Erythromycins betrug 0.66. Das Erythromycin
aus dem Fermentationsrückstand konnte auf einem Bereich mit einem Rf-Wert von
0.14-0.66 detektiert werden. Dieser breite Bereich deutet darauf hin, dass das
Erythromycin wahrscheinlich teilweise protoniert vorlag. Aufgrund der großen Fläche
des Produktes wurde er in drei Fraktionen unterteilt, um eine möglichst reine Probe
zu gewinnen. Das Kieselgel wurde von der DC-Platte gekratzt und mit einem 2:1
Gemisch aus Chloroform und Methanol extrahiert. Nach Entfernen des
Lösungsmittels wurden die Proben erneut durch eine LC/ESI-MS Messung
analysiert. Die drei Fraktionen nach der DC wiesen alle Erythromycin auf. Dies zeigt,
dass es möglich ist Erythromycin durch diese Methode zu isolieren und bis zu einem
gewissen Grad aufzureinigen. Die LC/ESI-MS Messungen zeigten neben den
Erythromycin Derivaten B und C, die nicht durch die verwendeten Methoden zu
separieren sind, zu einem geringen Anteil noch weitere Verunreinigungen.
Erythromycin eluiert mit einer Retentionszeit von 6.8 min und weist ein Masse zu
Ladungsverhältnis von 734.74 (m/z = M+H+) auf. Die reinste Fraktion wurde zur
näheren Analyse einer MS/MS Analyse unterzogen. Das Fragmentierungsmuster
verifizierte das Resultat.
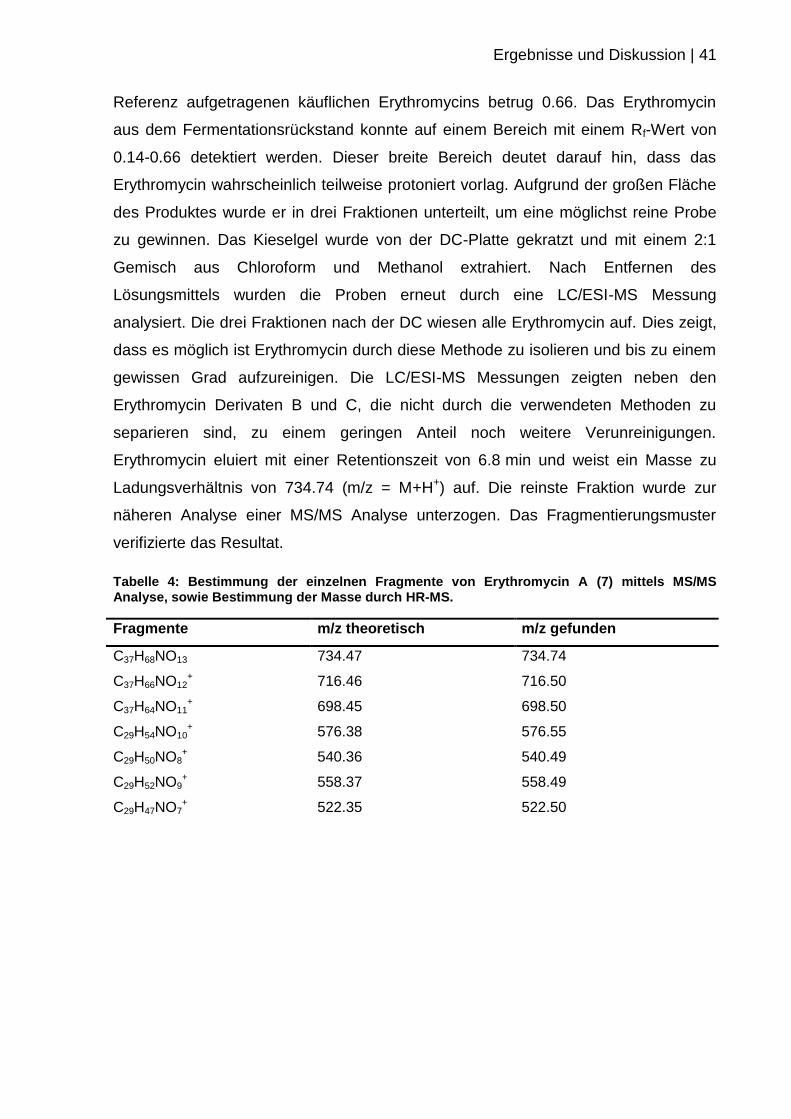
Tabelle 4: Bestimmung der einzelnen Fragmente von Erythromycin A (7) mittels MS/MS Analyse, sowie Bestimmung der Masse durch HR-MS.
Fragmente m/z theoretisch m/z gefunden
C37H68NO13 734.47 734.74
C37H66NO12+ 716.46 716.50
C37H64NO11+ 698.45 698.50
C29H54NO10+ 576.38 576.55
C29H50NO8+ 540.36 540.49
C29H52NO9+ 558.37 558.49
C29H47NO7+ 522.35 522.50

Ergebnisse und Diskussion | 42
Abbildung 35: Fragmentierung von Erythromycin A (7).
Die Fraktion der Wildtypfermentation, die neben Erythromycin am wenigsten
Verunreinigungen aufwies wurde mittels 1H-NMR untersucht. Als Referenz wurde ein
Spektrum von kommerziell erhältlichem Erythromycin aufgenommen. Anhand einer
1H-NMR Auswertung der Literatur wurde das Signal des H-2 identifiziert.[54] Sowohl
im NMR-Spektrum der Referenz, als auch im Spektrum des Erythromycins aus der
Wildtypfermentation konnte das Signal bei 2.84 ppm detektiert werden. Zur Analyse
der Konfiguration an der Position C2 muss vor allem dieses Signal betrachtet
werden. Da die Aufspaltung der Signale für eine Auswertung der Multiplizitäten nicht
ausreichend war, wurden die Spektren übereinandergelegt, um eine Analyse der
Signale durchführen zu können. Die übereinstimmenden Signale deuten darauf hin,
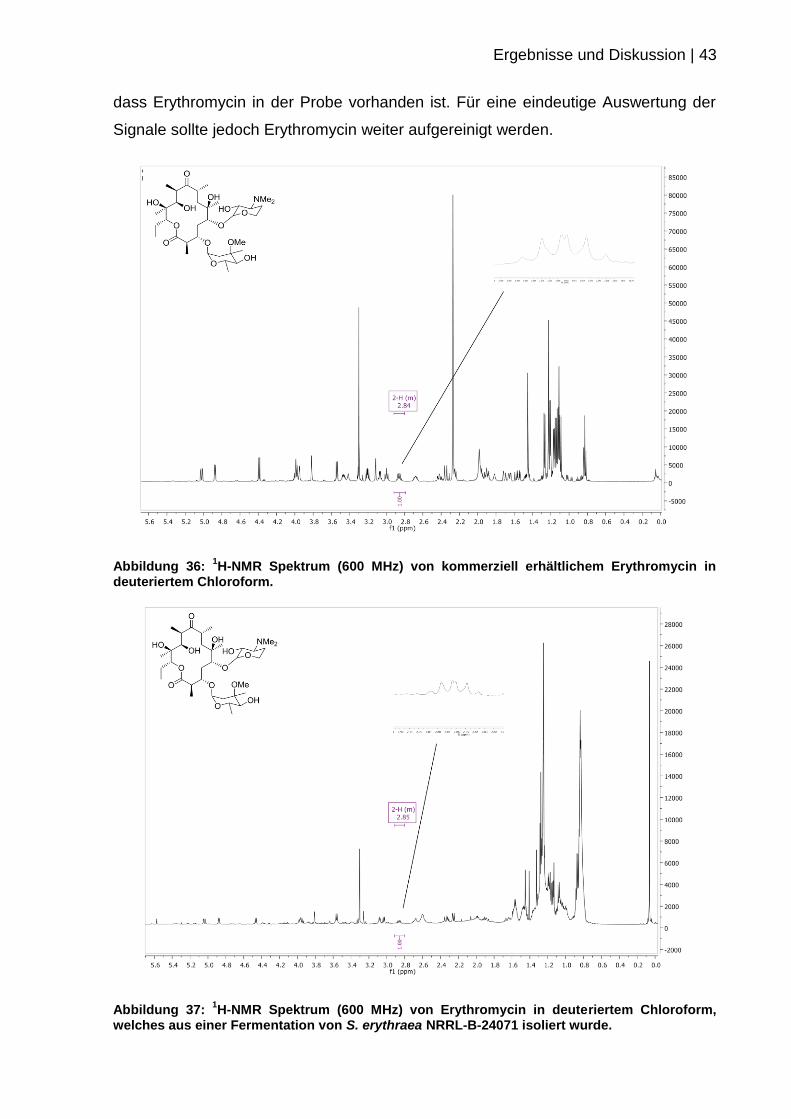
Ergebnisse und Diskussion | 43
dass Erythromycin in der Probe vorhanden ist. Für eine eindeutige Auswertung der
Signale sollte jedoch Erythromycin weiter aufgereinigt werden.
Abbildung 36: 1H-NMR Spektrum (600 MHz) von kommerziell erhältlichem Erythromycin in
deuteriertem Chloroform.
Abbildung 37: 1H-NMR Spektrum (600 MHz) von Erythromycin in deuteriertem Chloroform,
welches aus einer Fermentation von S. erythraea NRRL-B-24071 isoliert wurde.

Ergebnisse und Diskussion | 44
Abbildung 38: Übereinandergelegte 1H-NMR Spektren von kommerziell erhältlichem und aus
einer Fermentation von S. erythraea NRRL-B-24071 isoliertem Erythromycin. Grün: käufliches
Erythromycin. Schwarz: Aus der Fermentation isoliertes Erythromycin.
Um die Reproduzierbarkeit der Aufarbeitungsmethoden zu bestätigen wurde der
Wildtyp erneut fermentiert und mittels LC/ESI-MS Messungen analysiert. Erneut
konnte Erythromycin in den Fraktionen nachgewiesen werden. Die Massen der
erhaltenen Fraktionen wurden bestimmt und sind in folgender Tabelle dargestellt.
Tabelle 5: Massen der Fraktionen bei der Extraktion und Reinigung des Erythromycins aus Fermentationskulturen. Angaben in [mg].
WT1 WT2
EtOAc-Phase 54.6 65.2
Spot1 9.5 11.6
Spot2 20.7 22.2
Spot3 50.3 51.5
Ein Vergleich der Fraktionen der Wildtypfermentationen zeigt durch die große
Ähnlichkeit der Massen die Reproduzierbarkeit der hier angewandten Methoden.
Dies ist notwendig um die Resultate der Fermentationen von S. erythraea Kulturen
der AT6* Varianten auswerten zu können.
Ergebnisse und Diskussion | 45
3.1.5.4. Fermentation und Aufarbeitung von erzeugten S. erythraea Varianten
Die aufwendige Fermentation im präparativen Maßstab erfolgte aus Zeitgründen
nicht mit allen Varianten. Es wurden die Mutationen Met235Val & Val295Met, sowie
die Einzelaminosäureaustausche Met235Val und Val295Met ausgewählt. Die
Aufreinigung des Erythromycins erfolgte analog zum S. erythraea Wildtyp über
Extraktion gefolgt von einer präparativen Dünnschichtchromatographie.
Die erhaltenen Massen der Proben der jeweiligen Fermentation wurden bestimmt
und sind in Tabelle 6 dargestellt. Es wurden Proben nach der Extraktionskaskade
(EtOAc-Phase), sowie nach der DC (Spot 1-3) vermessen. In allen Proben konnte
durch eine LC/ESI-MS Messung Erythromycin nachgewiesen werden.
Tabelle 6: Massen der Fraktionen bei der Extraktion und Reinigung des Erythromycins aus Fermentationskulturen. Angaben in [mg]. Grau hinterlegte Spalten: Fermentation ohne Fütterung mit dem Substrat 13, Violett hinterlegte Spalten: Fermentation mit Fütterung des Substrates 13.
WT1 WT2
Met235Val & Val295Met
Met235Val & Val295Met
Val295Met Met235Val
EtOAc-Phase 54.6 65.2 34.23 52.6 n.b. 70.4
Spot1 9.5 11.6 28.2 9.0 30.9 8.6
Spot2 20.7 22.2 39.3 27.7 32.8 35.2
Spot3 50.3 51.5 32.8 10.5 40.4 13.9
Bei der Betrachtung der erhaltenen Massen wird deutlich, dass die drei Spots nach
der präparativen DC (Spot 1, 2 und 3) zusammen eine größere Masse innehaben,
als nach der letzten Extraktion. Dies ist wahrscheinlich auf das durch Methanol
gelöste Kieselgel zurückzuführen. Um eine genaue Analyse durchführen zu können,
muss das Kieselgel aus den Proben entfernt werden.
Ein Vergleich der Massen der Fermentationen der Mutante Met235Val & Val295Met
mit und ohne Fütterung mit rac-Methylmalonyl-SNAC (13) kann Rückschlüsse auf
den Einbau des gefütterten Substrates zulassen. Die Massen der Spots der
Fermentation mit Fütterung sind geringer, als die ohne Zuführung des Substrates 13.
Dies entspricht den Ergebnissen aus den LC/ESI-MS Analysen, jedoch nicht den
Erwartungen, dass das Erythromycin-Produktionsniveau mit Fütterung größer ist.
Auch ist hier die Masse der Fraktionen der Fermentationen der Varianten Val295Met
und Met235Val höher, als bei der Doppelmutante. Eine Analyse der S. erythraea AT6
Varianten mittels 1H-NMR ist aktuell in Arbeit. Die Analyse der Spektren, speziell des
Signals des H-2 (wahrscheinlich etwa bei 2.84 ppm) kann Aufschluss über die

Ergebnisse und Diskussion | 46
Stereospezifität der Mutanten liefern. Hier ist es sehr spannend, welche Resultate
sich aus der Analyse der Spektren ergeben. Sollte das C2 Stereozentrum des
Erythromycin A (7) (R)-konfiguriert sein (also Wildtyp-Zustand) so wäre dies
zusammen mit dem geringen Produktionsniveau ein weiterer Hinweis für die bereits
erwähnte Hypothese, dass die KS-Domäne das von der AT-Domäne eingebaute
(R)-Methylmalonat nicht akzeptiert und die Biosynthese damit abgebrochen wird.

Ergebnisse und Diskussion | 47
3.1.6. Präparation der AT6* Mutanten zur Substratspezifität
Die Varianten zur Untersuchung einer erweiterten Substratspezifität basieren auf
Ergebnissen von Uschi Sundermann.[46] In einer Analyse von 14 Aminosäuren der
AT6 wurden die Positionen 297 und 299 als vielversprechend identifiziert, da
Punktmutationen an diesen Positionen zu einer erweiterten Substratspezifität führten.
Die erstellte Bibliothek aus 23 Enzymvarianten ist auf diese Positionen fokussiert.
Die Präparation der Substratspezifitätsvarianten verlief analog zu denen der
Stereospezifität. Die Mutanten sollten mittels overlap extension PCR amplifiziert, in
den Shuttle-Vektor pKSSU96 kloniert und in den DEBS3 inaktiven Stamm
S. erythraeaΔAT6hygR konjugiert werden.
3.1.6.1. Sättigungsmutagenese zur Erstellung der AT6* Varianten;
Substratspezifität
Die Erstellung der Substratspezfitätsbibliothek mittels overlap extension PCR wurde
mit dem Vektor pKSSU89 als Template durchgeführt. Die Amplifizierung der
Doppelmutanten erfolgte hierbei nicht auf Basis von zuvor präparierten
Einzelmutanten, sondern ebenfalls mit dem Vektor pKSSU89 als Template für die
PCR indem entsprechende Oligonukleotide mit zweifachen Codonaustauschen
verwendet wurden. Die erste PCR ergab die beiden Fragmente der Konstrukte (M1
und M2).
Abbildung 39: Sättigungsmutagenese mittels overlap extension PCR. Die Fragmente (M1 und
M2) wurden in zwei separaten PCR generiert.
Um die Nebenprodukte der PCR zu entfernen, wurde im Anschluss eine
Gelaufreinigung durchgeführt. Die beiden Fragmente wurden in der zweiten PCR zu
den entsprechenden Produkten verknüpft. Die resultierenden Konstrukte haben ein
Molekulargewicht von etwa 1.7 kb.
M1 M1 M1 M2 M2 M2
5.0 kb
1.5 kb
1.0 kb
0.5 kb
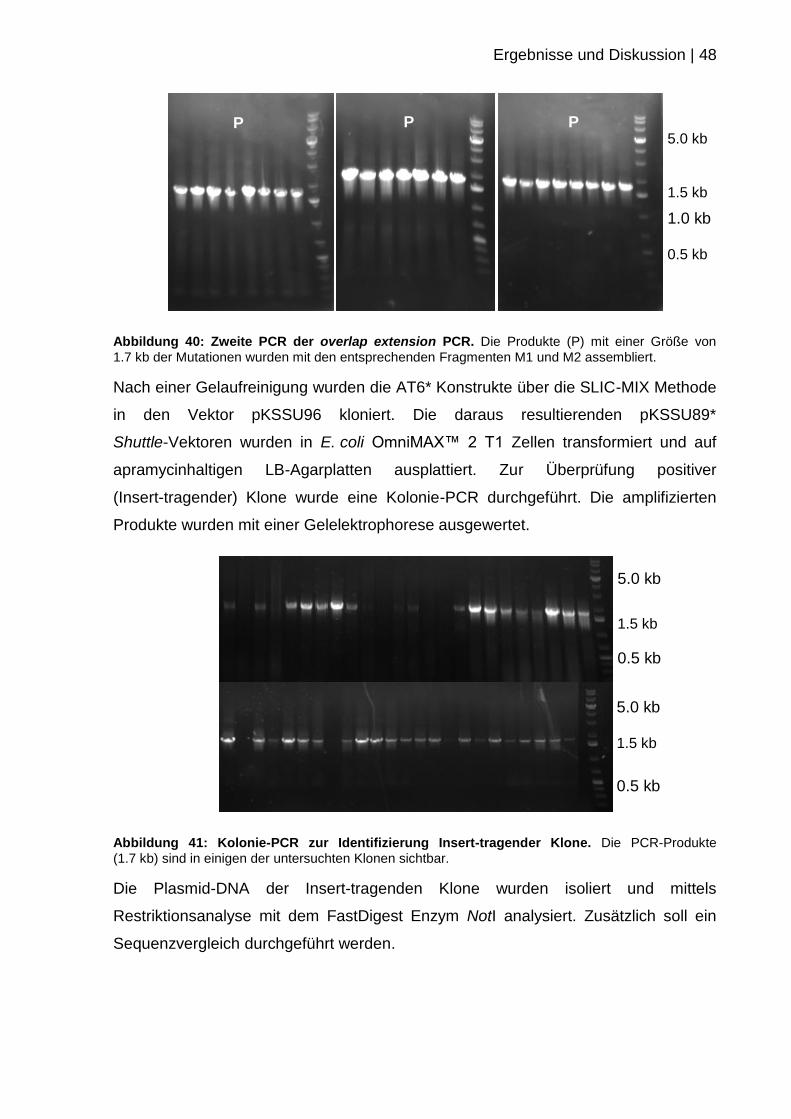
Ergebnisse und Diskussion | 48
Abbildung 40: Zweite PCR der overlap extension PCR. Die Produkte (P) mit einer Größe von
1.7 kb der Mutationen wurden mit den entsprechenden Fragmenten M1 und M2 assembliert.
Nach einer Gelaufreinigung wurden die AT6* Konstrukte über die SLIC-MIX Methode
in den Vektor pKSSU96 kloniert. Die daraus resultierenden pKSSU89*
Shuttle-Vektoren wurden in E. coli OmniMAX™ 2 T1 Zellen transformiert und auf
apramycinhaltigen LB-Agarplatten ausplattiert. Zur Überprüfung positiver
(Insert-tragender) Klone wurde eine Kolonie-PCR durchgeführt. Die amplifizierten
Produkte wurden mit einer Gelelektrophorese ausgewertet.
Abbildung 41: Kolonie-PCR zur Identifizierung Insert-tragender Klone. Die PCR-Produkte
(1.7 kb) sind in einigen der untersuchten Klonen sichtbar.
Die Plasmid-DNA der Insert-tragenden Klone wurden isoliert und mittels
Restriktionsanalyse mit dem FastDigest Enzym NotI analysiert. Zusätzlich soll ein
Sequenzvergleich durchgeführt werden.
P P P
5.0 kb
1.5 kb
1.0 kb
0.5 kb
5.0 kb
1.5 kb
0.5 kb
5.0 kb
1.5 kb
0.5 kb

Ergebnisse und Diskussion | 49
3.1.7. Restriktionsanalyse und Sequenzierung zur Verifizierung der
präparierten Plasmide; Substratspezifität
Der Kontrollverdau mit dem FastDigest Enzym NotI soll in drei Fragmenten mit den
Größen 8.5 kb, 6.6 kb und 1.6 kb resultieren.
Abbildung 42: Restriktionsanalyse der pKSSU89* Plasmide mit den erzeugten AT6* Varianten zur Substratspezifität. Durch den Verdau mit dem Restriktionsenzym NotI ergeben sich bei Insert-
tragenden Klonen drei Fragmente mit den Größen 8.5 kb, 6.6 kb und 1.6 kb.
Die Restriktionsanalyse zeigt, dass die untersuchte Plasmid-DNA das Insert tragen.
Eine Sequenzanalyse zur endgültigen Verifizierung der geplanten Mutationen der
Plasmide ist aktuell in Arbeit.
Die Konjugation in S. erythraea∆AT6hygR und Screening durch Fermentation fand
aus Zeitgründen nicht statt.
5.0 kb
1.5 kb
1.0 kb
0.5 kb

Ergebnisse und Diskussion | 50
3.2. Synthese des SNAC aktivierten rac-Methylmalonat 13
Die Synthese des SNAC aktivierten rac-Methylmalonats (rac-MM-SNAC) (13)
erfolgte weitestgehend nach einer in der Arbeitsgruppe entwickelten dreistufigen
Syntheseroute. Lediglich der erste Schritt der Synthese, die Präparation des
geschützten rac-Methylmalonats (11) wurde von der bekannten Route abgewandelt.
Das Coenzym A Mimetikum SNAC (14) wurde vorab über eine Acetylierung von
Cysteaminhydrochlorid (17) selbst hergestellt.
Abbildung 43: Synthese des Coenzym A Mimetikums SNAC (14). i):2.5 Äq. NaHCO3, 1.0 Äq.
KOH, 1.0 Äq. Ac2O, H2O, RT, 2 h.
Die Acetylierung erfolgte nach einer Aktivierung des Cysteaminhydrochlorids (17) mit
Natriumhydrogencarbonat und Kaliumhydroxid. Nach Zugabe von
Essigsäureanhydrid konnte SNAC (14) nach der Aufarbeitung in einer moderaten
Ausbeute von 64 % isoliert werden.
Die dreistufige Synthese des rac-MM-SNAC (13) wurde ausgehend von
tert-Butylpropionat (10) durchgeführt. Durch Einleiten von Kohlenstoffdioxid in die
Lösung konnte nach Deprotonierung durch NaHMDS die Säure 11 generiert werden.
Abbildung 44: Synthese der Verbindung 11. i): NaHMDS, THF, -78 °C-RT, 1 h. ii) CO2, 14 h.
Das Produkt 11 konnte durch Extraktion in sehr reiner Form jedoch nur mit einer
geringen Ausbeute von 32 % isoliert werden. Eine weitere Aufreinigung war nicht
notwendig. Zur Erzeugung des CO2 Gases wurde Trockeneis bei Raumtemperatur in
einen Kolben gefüllt und in die Reaktionslösung geleitet. Die Ausbeute konnte gering
verbessert werden, indem das naszierende Gas durch ein mit Molekularsieb gefülltes
Gefäß geführt wurde. Die dennoch mäßige Ausbeute wurde wegen der praktisch
einfach durchführbaren, sowie wenig zeitaufwendigen Synthesevorschrift toleriert.
Ein Vergleich mit der in der Arbeitsgruppe entwickelten Synthese des
tert-Butylmethylmalonats (11) basierend auf einer Methylierung von

Ergebnisse und Diskussion | 51
tert-Butylhydrogenmalonat (18) zeigt die Vorteile der Carboxylierungsreaktion
(Abbildung 44 und Abbildung 45).
Abbildung 45: In der Arbeitsgruppe durchgeführte Synthese des tert-Butylmethylmalonats (11). i) 2.1 Äq. LDA, MeI, THF, -78 °C-RT, Schutzgasatmosphäre, 16 h. Die Ausbeuten lagen durchschnittlich bei 35-70 %.
Die Durchführung der Synthese ist durch die benötigte Schutzgasatmosphäre sehr
aufwendig. Zusätzlich muss das Substrat vor Verwendung säulenchromatographisch
aufgereinigt werden, da es in käuflicher Form nicht die benötigte Reinheit hat. LDA
muss ausgehend von Diisopropylamin, welches zuvor frisch destilliert werden sollte,
am Tag der Synthese hergestellt werden. Des Weiteren nimmt die Bildung des
dialkylierten Malonates bei der Hochskalierung als unerwünschtes Nebenprodukt zu
und lässt sich nur schwer vom monoalkylierten Produkt trennen, so dass die erzielten
Ausbeuten bei Ansätzen von größer 500 mg stark sanken.[55] Durch die
Carboxylierung ausgehend von tert-Butylpropionat (10) kann diese sehr
zeitaufwendige Synthese, sowie der Einsatz des sehr giftigen Methyliodids
umgangen werden. Diese Vorteile amortisieren die geringere Ausbeute der
Carboxylierung von 32 % gegenüber den durchschnittlich erreichten Werten der
Methylierung von 35-70 % Ausbeute.
Im zweiten Schritt wurde die Methylmalonsäre 11 mit SNAC (14) gekoppelt um somit
eine zellwandpermeable, tert-Butyl-geschützte, Thioester-aktivierte rac-
Methylmalonsäure zu erhalten. Die Kupplung erfolgte nach einer Aktivierung der
Carbonsäure 11 durch CDI und DMAP. Durch die Bildung des hydrolysempfindlichen
gemischten Anhydrids durch CDI war die Durchführung der Reaktion unter
Schutzgas notwendig.
Abbildung 46: Synthese der Verbindung 12. Kupplung der tert-Methylmalonsäure (11) mit SNAC (14). i): 1.2 Äq. CDI, 0.3 Äq. DMAP, THF, 0 °C, 2 h. ii): 1.5 Äq. SNAC (14), RT, 16 h.
Nach einer säulenchromatographischen Aufreinigung konnte das Produkt 12 in
kristalliner Form mit einer zufriedenstellenden Ausbeute von 74 % isoliert werden.

Ergebnisse und Diskussion | 52
Die Entschützung als finalen Schritt der Synthese des Substrates 13 erfolgte am Tag
der Fütterung, da die Substanz 13 säure-, base-, und temperaturlabil ist und nur in
verdünnten wässrigen Natriumcarbonat-Puffer hinreichend stabil ist um
Fütterungsexperimente durchzuführen.
Abbildung 47: Synthese des rac-MM-SNAC (13). Entschützung des tert-Butyl-MM-SNAC (12).
i): 2.25 Äq. TiCl4, DCM, 0 °C-RT, 6.5 h.
Das Produkt 13 wurde in Substrat-Puffer (ein Glucose-Natriumcarbonat-Puffer, pH 8)
aufgenommen und nach Entfernen des Lösungsmittels und des ausgefallenen
Titandioxids, sowie einer Sterilfiltration direkt der Fermentation zugeführt.
Insgesamt konnte das Substrat (13) mit einer Gesamtausbeute von 24 % über drei
Stufen synthetisiert werden. Es konnte eine Lagerstabilität von tert-
Butylmethylmalonat-SNAC (12) bei -20 °C für einige Wochen beobachtet werden. Da
das Endprodukt 13 jedoch sehr instabil war, wurde der letzte Schritt erst am Tage
der Fütterung durchgeführt.

Ergebnisse und Diskussion | 53
3.3. Fütterungen mit Malonaten
In der Arbeitsgruppe konnte eine AT6* Variante generiert werden, die eine erweiterte
Substratspezifität aufweist. So konnte der Einbau von SNAC aktiviertem
Propargylmalonat nachgewiesen werden. Die Mutante basiert auf einem
Aminosäureaustausch an der Position Val295 (S. erythraea∆AT6hygR_pKSSU656
Val295Ala).[46] Da Mutanten der ersten Generation häufig ein sehr breites
Substratspektrum aufweisen, sollte nach der Bildung weiterer Erythromycin-Derivate
durch diese Mutante gescreent werden. Möglicherweise kann die Variante auch
Ethylmalonat einbauen, jedoch konnten Stassi und Katz et al. nachweisen, dass
S. erythraea keine Crotonylreduktase besitzt und damit kein Ethylmalonyl-CoA
herstellen kann.[40] Daher muss Ethylmalonat der Fermentation zugeführt werden. In
swapping-Experimenten konnte bereits aufgezeigt werden, dass eine Fütterung mit
Diethylethylmalonat (15) zur Synthese eines Erythromycin-Derivates führt. Hierbei
scheint eine Aktivierung mit SNAC (14) nicht notwendig zu sein.
Aufgrund dieser Resultate wurde eine Fermentation der Variante S. erythraea
∆AT6hygR_pKSSU656 (Val295Ala) mit Fütterung von Diethylethylmalonat (15)
durchgeführt. Um einen Vergleich der Aufnahmekapazität in die Zellen zu erhalten,
sollte auch eine Fütterung mit Diethylpropargylmalonat (16) stattfinden.
Eine Analyse der LC/ESI-MS Messungen ergab, dass Propargylmalonat eingebaut
wurde, jedoch konnte das entsprechende Erythromycinderivat nur in sehr geringen
Mengen nachgewiesen werden. Die Mengen sind mit einem Faktor 2 geringer als mit
einer SNAC-Aktivierung.
Die Masse des 2-Ethylerythromycin-Derivat konnte nur im Rauschen detektiert
werden. Es ist möglich, dass ein Einbau stattfindet, die Kapazität der
Zellwandpermeabilität jedoch zu gering ist um eindeutige Aussagen treffen zu
können. Hier sollte eine Fütterung mit SNAC-aktiviertem Ethylmalonat durchgeführt
werden um eindeutige Ergebnisse zu erhalten.

Ergebnisse und Diskussion | 54
3.4. Abschließende Diskussion
Die Präparation von 17 S. erythraeaAT6* Varianten zur Untersuchung der
Stereospezifität basierend auf den Aminosäurepositionen Met235 und Val295 konnte
erfolgreich durchgeführt werden. Die dafür verwendete overlap extension PCR
lieferte zuverlässig die geplanten Mutanten. Die Abweichung der Varianten
pKSSU672-676 mit der Aminosäure Glutamin, statt des geplanten Glutamats, liegt
an einer fehlerhaften Bestellung der hierfür eingesetzten Oligonukleotide. Da
Glutamin einen ähnlichen sterischen Anspruch wie Glutamat besitzt wurden diese
Varianten jedoch weiter verwendet. Alle erzeugten AT6* Konstrukte konnten
erfolgreich mittels SLIC-MIX kloniert werden. Diese Klonierungsmethode ist äußerst
zuverlässig, was durch die positiven Klone der Kolonie-PCR bestätigt wurde. Die
durch intergenetische Konjugation erhaltenen S. erythraea∆AT6hygR_pKSSU89*
Varianten wurden in 24-Lochplatten des System Duetz kultiviert und mit dem
synthetisierten Substrat rac-Methylmalonyl-SNAC (13) gefüttert. Die Synthese konnte
erfolgreich über drei Stufen mit einer Gesamtausbeute von 24 % durchgeführt
werden. Der sogenannte Flaschenhals dieser Syntheseroute ist die Carboxylierung
mit Kohlenstoffdioxid mit einer Ausbeute von 33 %. Durch eine Optimierung dieser
Reaktion könnte die Gesamtausbeute deutlich gesteigert werden.
Eine LC/ESI-MS Analyse der Fermentationsprodukte zeigte, dass die Positionen
Met235 und Val295 eine erstaunliche Toleranz aufweisen. Nur zwei der 17
Mutationen führten hier zu einer vollständigen Unterdrückung der Erythromycin-
Produktion. Die Analyse der LC/ESI-MS Daten ermöglichte jedoch keinen
Rückschluss auf das eingebaute Substrat. Hierzu muss eine 1H-NMR-Analyse der
erhaltenen Fermentationsprodukte erfolgen. Bei der Fermentation zur Gewinnung
von präparativen Mengen des Erythromycins mussten erst einige Hürden wie eine
optimale Extraktion und gute Aufreinigung überwunden werden. Durch den Einsatz
einer dreistufigen Extraktionskaskade und einer präparativen
Dünnschichtchromatographie ist es gelungen Erythromycin aus einer S. erythraea
NRRL-B-24071 (Wildtyp) Kultur zu isolieren. Eine 1H-NMR-Messung sowie ein
Vergleich mit käuflich erworbenem Erythromycin bestätigten den Befund. Als
abschließender Aufreinigungsschritt steht hier nun die Entfernung des in der Probe
befindlichen Kieselgels an. Die Menge und Reinheit des isolierten Produktes ist
jedoch für eine genaue Analyse der Multiplizitäten der NMR-Signale nicht
ausreichend gewesen.

Ergebnisse und Diskussion | 55
Die Analyse der fermentierten S. erythraea∆AT6hygR_pKSSU89* Varianten mittels
LC/ESI-MS ergab, das auch hier Erythromycin isoliert werden konnte. Die erhaltenen
Massen an Erythromycin resultierten in einer Hypothese über die
Stereospezifitätskontrolle der Acyltransferase. Das niedrige Erythromycin-
Produktionsniveau der Fermentation mit Zugabe des Substrates 13 impliziert die
Hypothese, dass möglicherweise die Acyltransferase das (R)-Methylmalonat
akzeptiert, die Ketosynthase das Zwischenprodukt jedoch nicht toleriert und es somit
zu einem Abbruch der Biosynthese kommt. Dies setzt voraus, dass die Ketosynthase
einen Einfluss auf die Stereokontrolle der Seitenketten des Polyketids hat. Um diese
hochspekulative Hypothese genauer zu untersuchen, sollten weitere Experimente,
vielleicht eine Untersuchung in einem vereinfachten System, stattfinden.
Eine nähere Analyse der Stereochemie des isolierten Erythromycins mittels 1H-NMR
steht noch aus. Die hieraus erhaltenen Ergebnisse könnten zu einer Identifikation der
Aminosäuren führen, die einen Einfluss auf die Selektivität der Acyltransferase
haben.
Die Präparation der AT6* Varianten basierend auf Tyr297 und Ser299 erfolgte bis zur
Klonierung in den Vektor pKSSU96. Sobald die Sequenzanalyse der erzeugten
pKSSU89* Plasmide abgeschlossen ist, sollten diese über intergenetische
Konjugation in S. erythraea∆AT6hygR eingebracht werden, um entsprechende
S. erythraea∆AT6hygR_pKSSU89* Varianten zu erzeugen. Diese sollten
anschließend durch ein Screening in 24-Lochplatten des System Duetz inklusive
Fütterung von SNAC aktiviertem Propargylmalonat untersucht werden.
Das Fütterungsexperiment mit nicht aktivierten Malonaten lieferte zwar ein
Erythromycinderivat mit einem Progargylrest an der C2-Position (nachgewiesen
durch eine Analyse der LC/ESI-MS Daten), jedoch war die Menge mit einem Faktor 2
geringer als es bei der Fütterung mit SNAC-aktiviertem Propargylmalonat der Fall
war.[46] Dies zeigt, dass auch bei weiteren Fütterungsexperimenten eine
SNAC-Aktivierung der Substrate durchgeführt werden sollte, um eindeutige
Ergebnisse zu erhalten. Die bei der Analyse der LC/ESI-MS Spektren gefundenen
Spuren an 2-Ethylerythromycin deuten darauf hin, dass die untersuchte Mutante
S. erythraea∆AT6hygR_pKSSU656 (Val295Ala) auch Ethylmalonat als Substrat
einbauen kann. Durch eine Fütterung mit SNAC-aktiviertem Ethylmalonat könnte
dieser Verdacht bestätigt werden.

Ergebnisse und Diskussion | 56
Die durchgeführten Experimente zeigen, dass eine Mutagenese von Polyketid-
Synthasen sehr aufwendig ist. Durch den komplexen Biosyntheseweg ist eine
Deutung der erhaltenen Ergebnisse des Screenings oft sehr vielschichtig und bedarf
weiterer Analysen.

Zusammenfassung und Ausblick | 57
4 Zusammenfassung und Ausblick
Erythromycin ist ein Makrolid-Antibiotikum einer der größten Naturstoffklassen – den
Polyketiden. Diese strukturell und funktionell hoch heterogenen Substanzen werden
von Pilzen, Pflanzen und Bakterien synthetisiert. Erythromycin gehört zu den
reduzierten Polyketiden und wird von einer Typ-I-Polyketid Synthase generiert. Die
6-Desoxyerythronolid B Synthase (DEBS) aus dem Bodenbakterium
Saccharopolyspora erythraea bildet einen Enzymkomplex aus drei nicht kovalent
verknüpften Enzymen und ist für die Biosynthese des Erythromycins verantwortlich.
Abbildung 48: Grundmechanismen der Erythromycin A (7) Biosynthese.[26,27]
Dieser 1.8 MDa große Multienzymkomplex (Monomer) besteht aus drei Enzymen, DEBS1, 2 und 3, die nicht kovalent miteinander verknüpft als molekulares Fließband agieren. Jedes dieser Enzyme beinhaltet zwei bis drei Module (schwarze Kästen), die kovalent miteinander verknüpft sind. Ein minimales Modul (vgl. Modul 3) besteht aus drei Domänen, die eine Kettenverlängerung um 2 C-Atome inklusive der Seitenkette (resultierend in der Bildung eines β-Ketoesters) katalysieren: Eine Ketosynthase (KS), die eine decarboxylierende Claisenkondensation katalysiert, eine Acyltransferase (AT), die den Baustein auswählt und diesen auf das Acylcarrierprotein (ACP) überträgt. Der β-Ketoester kann dann nachfolgend durch die reduktiven Domänen weiter prozessiert werden. Eine vollständige reduktive Schleife besteht aus einer Ketoreduktase (KR), einer Dehydratase (DH) und einer Enoylreduktase (ER). Die Anzahl der reduktiven Domänen des jeweiligen Moduls bestimmt hierbei den Grad der Reduktion der wachsenden Kette. Final spaltet eine Terminale Esterase (TE) die Polyketidkette vom Enzym ab und ermöglicht eine Zyklisierung. Durch Post-PKS Prozessierungen wie Glykosylierungen und Methylierungen wird die Biosynthese des biologisch aktiven, komplexen Naturstoffes Erythromycin A (7) komplementiert.
Der funktionale Komplex der Polyketidsynthase liegt als Dimer mit einer Größe von
etwa 3.5 MDa vor und ist eines der größten bekannten Proteine. Es stellt damit im
Hinblick auf enzyme engineering Strategien eine Herausforderung dar. Im Rahmen
dieser Arbeit wird die Strategie der Punktmutation einer spezifischen Domäne
angewandt. Dieser Weg der kombinatorischen Biosynthese führte bereits in früheren
Experimenten zu einigen Erfolgen.[44,45,46] Eine zusätzliche Herausforderung stellte
hier die Verwendung von S. erythraea als Erythromycin-Originalproduzent dar.

Zusammenfassung und Ausblick | 58
Dieses Gram-positive Bakterium ist genetisch nur schwer zu manipulieren und stellt
besondere Ansprüche an eine produktive Fermentation.
Um eine Diversitätserhöhung von Erythromycin zu erreichen wurden anhand der
Acyltransferase aus Modul 6 mittels Sättigungsmutagenese Bibliotheken präpariert,
die zu einer Erweiterung der Stereo- bzw. Substratspezifität führen sollten.
4.1. Projekt zur Erweiterung der Stereospezifität
Eine Analyse eines Modells der DEBS AT6-Domäne durch die Arbeitsgruppe von
Dr. Elsa Sanchez Garcia (Theoretische Chemie) des Max-Planck-Instituts für
Kohlenforschung (Mülheim) zeigte zwei Aminosäurepositionen auf, die scheinbar für
den selektiven Einbau des (S)-Methylmalonyl-CoA verantwortlich sind – Met235 und
Val295.
Abbildung 49: Aktives Zentrum der AT6-Domäne. A: Wildtyp mit Met235 (gelb), Val295 (blau) und (S)-MMCoA. B: Doppelmutante Met235Val (blau), Val295Met (gelb) und (R)-MMCoA. Das Modell stammt vom Kooperationspartner Kenny Bravo-Rodriguez.
Die geplanten Mutationen konnten über eine overlap extension PCR präpariert
werden. Eine anschließende Klonierung mittels SLIC-MIX resultierte in pKSSU89*
Plasmiden, die die gewünschten Mutationen innerhalb der AT6-Domäne trugen. Die
Plasmid-DNA wurden mittels Restriktionsanalyse und Sequenzvergleich verifiziert.
Nach einer intergenetischen Konjugation in S. erythraea∆AT6hygR, ein Stamm bei
dem DEBS3 durch das Inserieren einer Hygromycin-Kassette inaktiviert wurde,
wurden die Varianten über Kultivierung inklusive Fütterung mit rac-Methylmalonyl-
SNAC (13) in Lochplatten des System Duetz[53] und folgender LC/ESI-MS Analyse
der Fermentationsextrakte gescreent. Dazu wurde das Substrat rac-Methylmalonyl-
SNAC (13) über eine dreistufige chemische Synthese hergestellt. Diese
Syntheseroute ermöglichte eine schnelle Präparation der Verbindung. Der Einsatz
einer Carboxylierung mit Kohlenstoffdioxid ermöglichte zudem eine wenig
A B

Zusammenfassung und Ausblick | 59
aufwendige Reaktion, die eine mühsame Methylierungsreaktion sowie den Einsatz
von giftigem Methyliodid umgeht (vgl. 3.2). Da die Substanz 13 säure-, base-, und
temperaturlabil ist und nur in verdünnten wässrigen Natriumcarbonat-Puffer
hinreichend stabil ist, wurde der letzte Schritt der Synthese am Tag des
Fütterungsexperiments durchgeführt.
Abbildung 50: Prinzipieller Ablauf des Screenings auf Erythromycin-Produktion der Stereoselektivitätsbibliothek.
Es konnte aufgezeigt werden, dass eine große Toleranz gegenüber den eingeführten
Mutationen der zwei entsprechenden Aminosäurepositionen besteht. Eine
1H-NMR-Analyse sollte weitere Auskünfte über die Konfiguration des C2
Stereozentrums von Erythromycin geben. Hierzu musste zuerst eine Methodik zur
Aufarbeitung und Isolation des Erythromycins aus Fermentationskulturen geschaffen
werden. Dies gelang über eine dreistufige Extraktionskaskade, sowie einer
präparativen Dünnschichtchromatographie. Mittels LC/ESI-MS sowie 1H-NMR-
Analyse konnte nachgewiesen werden, dass es gelungen war Erythromycin aus
einer S. erythraea NRRL-B-24071 (Wildtyp) Fermentation zu isolieren. In den
fermentierten Varianten der Mutationen Met235Val & Val295Met, sowie den
Einzelaminosäureaustauschen Met235Val und Val295Met konnte ebenfalls durch
eine LC/ESI-MS Analyse Erythromycin nachgewiesen werden, jedoch ist für eine
Analyse der Konfiguration der Stereozentren eine 1H-NMR-Analyse notwendig. Diese
Analyse kann Aufschluss darüber geben, ob die eingebrachten Mutationen zu einer
Erweiterung der Stereoselektivität der AT6-Domäne geführt haben. Die bisher
erzielten Ergebnisse des Screenings stellen eine Quelle für Spekulationen dar. So
könnte die geringe Erythromycin-Produktivität der Doppelmutante Met235Val &
Val295Met darauf hindeuten, dass neben der Acyltransferase auch die Ketosynthase
für die Stereoselektivität der Polyketid-Seitenketten verantwortlich sein könnte. Zur
näheren Untersuchung sollten weitere Experimente durchgeführt werden.
Shuttle-Vektor
DEBS3*-kodierend
S. erythraea∆AT6hygR
Screening der Mutanten

Zusammenfassung und Ausblick | 60
4.2. Projekt zur Erweiterung der Substratspezifität
Die Bibliothek zur Substratspezifität fokussiert auf den Aminosäurepositionen Tyr297
und Ser299 wurde basierend auf Erkenntnissen geplant, die durch vorrangegangene
zielgerichtete Mutagenese-Experimente erlangt wurden.[46] Durch overlap extension
PCR und Klonierung mittels SLIC-MIX wurden die pKSSU89* Plasmide mit den
entsprechenden Mutationen in Analogie zu den Stereospezifitätsvarianten (vgl.
Abschnitt 4.1) konstruiert. Die Plasmide wurden mittels Restriktionsverdau durch das
FastDigest Enzym NotI auf das Vorhandensein des Inserts (Klonierungserfolg)
kontrolliert. Eine Sequenzanalyse zur Verifizierung der Plasmid-DNA wird aktuell
durchgeführt. Sind die geplanten Mutationen vorhanden, sollte die Plasmid-DNA über
intergenetische Konjugation in das Genom von S. erythraea∆AT6hygR eingebracht
werden. Die resultierenden S. erythraea∆AT6hygR_pKSSU89* Varianten sollten zur
Analyse in Lochplatten des System Duetz fermentiert werden. Dabei sollte mit SNAC
aktivierten Malonatderivaten, wie Propargylmalonat gefüttert werden, um auf eine
erweiterte Substratspezifität zu testen. Die Fermentationsextrakte können mittels
LC/ESI-MS Messungen untersucht werden, da eine Akzeptanz von z.B.
Propargylmalonat zu einem Erythromycin-Derivat mit einer veränderten Masse führt.
Es bleibt abzuwarten, welche Ergebnisse sich aus den noch folgenden Experimenten
ergeben.
4.3. Fütterung mit Malonaten
Die Variante S. erythraea∆AT6hygR_pKSSU656 (Val295Ala) zeigt bereits eine
erweiterte Substratakzeptanz, da sie neben Methylmalonat auch Propargylmalonat
einbaut.[46] Eine Untersuchung mit Diethylethylmalonat (15) sollte aufzeigen, ob auch
dieses Derivat als Substrat akzeptiert wird. Hierbei konnte jedoch festgestellt werden,
dass der Einbau nicht SNAC aktivierter Substanzen zu gering ist, um eindeutige
Aussagen treffen zu können. Da nur Spuren des 2-Ethylerythromycins in den
Fermentationsextrakten gefunden wurden, sollte eine Fütterung der Mutante mit
SNAC-aktiviertem Ethylmalonat durchgeführt werden, um die Ergebnisse zu
verifizieren. Hierbei könnte auch die Fütterung weiterer Malonatderivate auf die
Akzeptanz durch die Val295Ala Mutante der AT6-Domäne getestet werden.
Insgesamt beschäftigte sich diese Arbeit mit der aufwendigen genetischen
Manipulation der Acyltranferase aus Modul 6 der Erythromycin-Polyketidsynthase.
Bei dem Versuch eine Änderung der Stereoselektivität zu erzielen ergaben sich

Zusammenfassung und Ausblick | 61
sowohl Schwierigkeiten bei der Auswahl eines geeigneten Sceeningsystems, als
auch bei der Auswertung der Ergebnisse. Da die endgültigen 1H-NMR-Analysen der
untersuchten AT6* Varinaten noch nicht vorliegen, bleibt abzuwarten welche
Rückschlüsse gezogen werden können. Möglicherweise ist es notwendig weitere
Untersuchungen zum Verständnis der Stereokontrolle der Polyketidsynthase zu
machen. Ebenso sind auch bei den Projekten zur erweiterten Substratspezifität noch
weitere Experimente durchzuführen, um eine Aussage über die Auswirkung der
eingebrachten Mutationen treffen zu können.
Die Ergebnisse zeigen, dass die Manipulation eines der größten bekannten Enzyme,
der Erythromycin-Polyketidsynthase (DEBS) eine komplexe Aufgabe ist. Die in dieser
Arbeit erlangten Erkenntnisse sind jedoch ein weiterer Schritt auf dem Weg diese
Enzyme zu verstehen und nutzen zu können. Zudem können sie Ansatzpunkt für das
Konstruieren weiterer aussichtsreicher Experimente dienen.
Material und Methoden | 62
5 Material und Methoden
5.1. Mikroorganismen, Plasmide, Oligonukleotide
5.1.1. Mikroorganismen
Tabelle 7: In den Experimenten eingesetzte Stämme.
Stamm Genotyp/Phenotyp Herkunft
OmniMAX™ 2 T1
Phage-Resistant
F′ {proAB+ lacIq lacZΔM15 Tn10(TetR) Δ(ccdAB)} mcrA Δ(mrr-hsdRMS-mcrBC) φ80(lacZ)ΔM15 Δ(lacZYA-argF) U169 endA1 recA1 supE44 thi-1 gyrA96 relA1 tonA panD
Invitrogen
E. coli BL21Gold(DE3) B F- omp T hsdSB(rB-mB
-) dcm+ Tetr gal λ(DE3) endA Hte
Stratagene
E. coli ET12567/pUZ8002 F−, dam-13::Tn9, dcm-6 hsdM, hsdR, zjj- 202::Tn10, recF143, galK2, galT22, ara-14, lacY1, xyl15, leuB6, thi1, tonA31, rpsL136, his64, tsx78, mtlI, glnV44
AK Leadlay
Saccharopolyspora erythraea
NRRL-B-24072
n.a. ARS Culture Col.
Bacillus subtilis DSM-10 DSMZ
Saccharopolyspora erythraea
ΔAT6hygR
NRRL-B-24071 mit Austausch der
AT6-Domäne innerhalb von eryAIII
gegen ΩhygR
AK Schulz
5.1.2. Vektoren und Plasmide
Tabelle 8: In dieser Arbeit verwendete und konstruierte Plasmide.
Plasmid kb Basierend auf Resistenz Herkunft
pKSSU89 16.9 pKSSU40, DEBS3 kodierend unter
Kontrolle von PactIII
Apramycin AK Schulz
pKSSU96 16.7 Austausch der AT6 Kassette innerhalb von
pKSSU89 gegen Ωhyg
Apramycin,
Hygromycin
AK Schulz
pKSSU660 16.9 pKSSU89 mit AS Austausch: Met235Val Apramycin diese Arbeit
pKSSU661 16.9 pKSSU89 mit AS Austausch: Met235Gly Apramycin diese Arbeit
pKSSU662 16.9 pKSSU89 mit AS Austausch: Met235Ile Apramycin diese Arbeit
pKSSU663 16.9 pKSSU89 mit AS Austausch: Met235Leu Apramycin diese Arbeit
pKSSU664 16.9 pKSSU89 mit AS Austausch: Met235Ala Apramycin diese Arbeit
pKSSU665 16.9 pKSSU89 mit AS Austausch: Val295Met Apramycin diese Arbeit
pKSSU666 16.9 pKSSU89 mit AS Austausch: Val295Glu Apramycin diese Arbeit

Material und Methoden | 63
Plasmid kb Basierend auf Resistenz Herkunft
pKSSU667 16.9 pKSSU89 mit AS Austausch: Met235Val
und Val295Met
Apramycin diese Arbeit
pKSSU668 16.9 pKSSU89 mit AS Austausch: Met235Gly
und Val295Met
Apramycin diese Arbeit
pKSSU669 16.9 pKSSU89 mit AS Austausch: Met235Ile und
Val295Met
Apramycin diese Arbeit
pKSSU670 16.9 pKSSU89 mit AS Austausch: Met235Leu
und Val295Met
Apramycin diese Arbeit
pKSSU671 16.9 pKSSU89 mit AS Austausch: Met235Ala
und Val295Met
Apramycin diese Arbeit
pKSSU672 16.9 pKSSU89 mit AS Austausch: Met235Val
und Val295Gln
Apramycin diese Arbeit
pKSSU673 16.9 pKSSU89 mit AS Austausch: Met235Gly
und Val295Gln
Apramycin diese Arbeit
pKSSU674 16.9 pKSSU89 mit AS Austausch: Met235Ile und
Val295Gln
Apramycin diese Arbeit
pKSSU675 16.9 pKSSU89 mit AS Austausch: Met235Leu
und Val295Gln
Apramycin diese Arbeit
pKSSU676 16.9 pKSSU89 mit AS Austausch: Met235Ala
und Val295Gln
Apramycin diese Arbeit
pKSSU687 16.9 pKSSU89 mit AS Austausch: Tyr297Val Apramycin diese Arbeit
pKSSU688 16.9 pKSSU89 mit AS Austausch: Tyr297Val und
Ser299Ala
Apramycin diese Arbeit
pKSSU689 16.9 pKSSU89 mit AS Austausch: Tyr297Val und
Ser299Gly
Apramycin diese Arbeit
pKSSU690 16.9 pKSSU89 mit AS Austausch: Tyr297Val und
Ser299Leu
Apramycin diese Arbeit
pKSSU691 16.9 pKSSU89 mit AS Austausch: Tyr297His Apramycin diese Arbeit
pKSSU692 16.9 pKSSU89 mit AS Austausch: Tyr297His und
Ser299Ala
Apramycin diese Arbeit
pKSSU693 16.9 pKSSU89 mit AS Austausch: Tyr297His und
Ser299Gly
Apramycin diese Arbeit
pKSSU694 16.9 pKSSU89 mit AS Austausch: Tyr297His und
Ser299Leu
Apramycin diese Arbeit
pKSSU695 16.9 pKSSU89 mit AS Austausch: Tyr297Ala Apramycin diese Arbeit
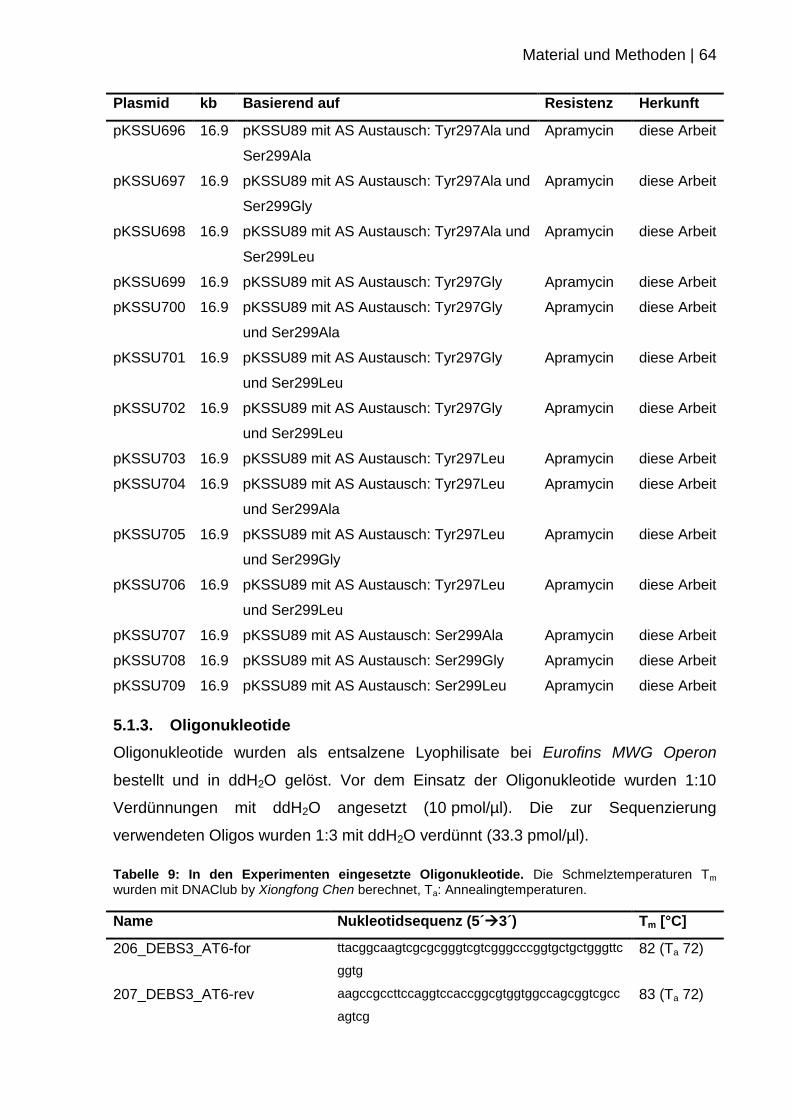
Material und Methoden | 64
Plasmid kb Basierend auf Resistenz Herkunft
pKSSU696 16.9 pKSSU89 mit AS Austausch: Tyr297Ala und
Ser299Ala
Apramycin diese Arbeit
pKSSU697 16.9 pKSSU89 mit AS Austausch: Tyr297Ala und
Ser299Gly
Apramycin diese Arbeit
pKSSU698 16.9 pKSSU89 mit AS Austausch: Tyr297Ala und
Ser299Leu
Apramycin diese Arbeit
pKSSU699 16.9 pKSSU89 mit AS Austausch: Tyr297Gly Apramycin diese Arbeit
pKSSU700 16.9 pKSSU89 mit AS Austausch: Tyr297Gly
und Ser299Ala
Apramycin diese Arbeit
pKSSU701 16.9 pKSSU89 mit AS Austausch: Tyr297Gly
und Ser299Leu
Apramycin diese Arbeit
pKSSU702 16.9 pKSSU89 mit AS Austausch: Tyr297Gly
und Ser299Leu
Apramycin diese Arbeit
pKSSU703 16.9 pKSSU89 mit AS Austausch: Tyr297Leu Apramycin diese Arbeit
pKSSU704 16.9 pKSSU89 mit AS Austausch: Tyr297Leu
und Ser299Ala
Apramycin diese Arbeit
pKSSU705 16.9 pKSSU89 mit AS Austausch: Tyr297Leu
und Ser299Gly
Apramycin diese Arbeit
pKSSU706 16.9 pKSSU89 mit AS Austausch: Tyr297Leu
und Ser299Leu
Apramycin diese Arbeit
pKSSU707 16.9 pKSSU89 mit AS Austausch: Ser299Ala Apramycin diese Arbeit
pKSSU708 16.9 pKSSU89 mit AS Austausch: Ser299Gly Apramycin diese Arbeit
pKSSU709 16.9 pKSSU89 mit AS Austausch: Ser299Leu Apramycin diese Arbeit
5.1.3. Oligonukleotide
Oligonukleotide wurden als entsalzene Lyophilisate bei Eurofins MWG Operon
bestellt und in ddH2O gelöst. Vor dem Einsatz der Oligonukleotide wurden 1:10
Verdünnungen mit ddH2O angesetzt (10 pmol/µl). Die zur Sequenzierung
verwendeten Oligos wurden 1:3 mit ddH2O verdünnt (33.3 pmol/µl).
Tabelle 9: In den Experimenten eingesetzte Oligonukleotide. Die Schmelztemperaturen Tm wurden mit DNAClub by Xiongfong Chen berechnet, Ta: Annealingtemperaturen.
Name Nukleotidsequenz (5´3´) Tm [°C]
206_DEBS3_AT6-for ttacggcaagtcgcgcgggtcgtcgggcccggtgctgctgggttc
ggtg
82 (Ta 72)
207_DEBS3_AT6-rev aagccgccttccaggtccaccggcgtggtggccagcggtcgcc
agtcg
83 (Ta 72)

Material und Methoden | 65
Name Nukleotidsequenz (5´3´) Tm [°C]
213_hygSca-100_for atcacgggcgcggatgtggccgtgg 69
184_DEBS3-seq19-rev acagccgcgccagcgacacc 63
451_Sonde_AT6_fwd tggcaggcgaccacgaacagctccgcggg 73
665_Asn336_seq tgcgctggcgggcaaggg 62
932_Seq_Met235_DEBS3_fwd tcgacgtcgtacagccggtgttg 57
847_Met235Val_fwd ctggcgggcaagggcggcGTCgtctcgttggcggctccc 79
848_Met235Val_rev gggagccgccaacgagacGACgccgcccttgcccgccag 79
849_Met235Gly_fwd ctggcgggcaagggcggcGGCgtctcgttggcggctccc 81
850_Met235Gly_rev gggagccgccaacgagacGCCgccgcccttgcccgccag 81
851_Met235Ile_fwd ctggcgggcaagggcggcATCgtctcgttggcggctccc 79
852_Met235Ile_rev gggagccgccaacgagacGATgccgcccttgcccgccag 79
853_Met235Leu_fwd ctggcgggcaagggcggcTTGgtctcgttggcggctccc 79
854_Met235Leu_rev gggagccgccaacgagacCAAgccgcccttgcccgccag 79
855_Met235Ala_fwd ctggcgggcaagggcggcGCCgtctcgttggcggctccc 81
856_Met235Ala_rev gggagccgccaacgagacGGCgccgcccttgcccgccag 81
857_Val295Met_fwd cgcgccaagacgctcccgATGgactacgcctcgcactcc 74
858_Val295Met_rev ggagtgcgaggcgtagtcCATcgggagcgtcttggcgcg 74
859_Val295Glu_fwd cgcgccaagacgctcccgGAGgactacgcctcgcactcc 75
860_Val295Glu_rev ggagtgcgaggcgtagtcCTCcgggagcgtcttggcgcg 75
877_Val295Gln_fwd cgcgccaagacgctcccgCAGgactacgcctcgcactcc 75
878_Val295Gln_rev ggagtgcgaggcgtagtcCTGcgggagcgtcttggcgcg 75
956_Val297_fwd ccaagacgctcccggtggacGTGgcctcgcactcccgccac
gtcgaggag
83
957_Val297_Ala299_fwd ccaagacgctcccggtggacGTGgccGCCcactcccgcca
cgtcgaggag
84
958_Val297_Gly299_fwd ccaagacgctcccggtggacGTGgccGGCcactcccgcca
cgtcgaggag
84
959_Val297_Leu299_fwd ccaagacgctcccggtggacGTGgccTTGcactcccgcca
cgtcgaggag
82
960_His297_fwd ccaagacgctcccggtggacCACgccTCGcactcccgcca
cgtcgaggag
83
961_His297_Ala299_fwd ccaagacgctcccggtggacCACgccGCCcactcccgcca
cgtcgaggag
84
962_His297_Gly299_fwd ccaagacgctcccggtggacCACgccGGCcactcccgcca
cgtcgaggag
84
963_His297_Leu299_fwd ccaagacgctcccggtggacCACgccTTGcactcccgcca
cgtcgaggag
82

Material und Methoden | 66
Name Nukleotidsequenz (5´3´) Tm [°C]
964_Ala297_fwd ccaagacgctcccggtggacGCCgccTCGcactcccgcca
cgtcgaggag
83
965_Ala297_Ala299_fwd ccaagacgctcccggtggacGCCgccGCCcactcccgcca
cgtcgaggag
85
966_Ala297_Gly299_fwd ccaagacgctcccggtggacGCCgccGGCcactcccgcc
acgtcgaggag
85
967_Ala297_Leu299_fwd ccaagacgctcccggtggacGCCgccTTGcactcccgcca
CgtCgaggag
81
968_Gly297_fwd ccaagacgctcccggtggacGGCgccTCGcactcccgcca
cgtcgaggag
83
969_Gly297_Ala299_fwd ccaagacgctcccggtggacGGCgccGCCcactcccgcc
acgtcgaggag
85
970_Gly297_Gly299_fwd ccaagacgctcccggtggacGGCgccGGCcactcccgcc
acgtcgaggag
85
971_Gly297_Leu299_fwd ccaagacgctcccggtggacGGCgccTTGcactcccgcca
cgtcgaggag
82
972_Leu297_fwd ccaagacgctcccggtggacTTGgccTCGcactcccgcca
cgtcgaggag
82
973_Leu297_Ala299_fwd ccaagacgctcccggtggacTTGgccGCCcactcccgcca
cgtcgaggag
83
974_Leu297_Gly299_fwd ccaagacgctcccggtggacTTGgccGGCcactcccgcca
cgtcgaggag
83
975_Leu297_Leu299_fwd ccaagacgctcccggtggacTTGgccTTGcactcccgcca
cgtcgaggag
81
976_Ala299_fwd ccaagacgctcccggtggactacgccGCCcactcccgccac
gtcgaggag
82
977_Gly299_fwd ccaagacgctcccggtggactacgccGGCcactcccgccac
gtcgaggag
82
978_Leu299_fwd ccaagacgctcccggtggactacgccTTGcactcccgccacg
tcgaggag
80
1002_Val297_rev ctcctcgacgtggcgggagtgcgaggcCACgtccaccggga
gcgtcttgg
83
1003_Val297_Ala299_rev ctcctcgacgtggcgggagtgGGCggcCACgtccaccggg
agcgtcttgg
84
1004_Val297_Gly299_rev ctcctcgacgtggcgggagtgGCCggcCACgtccaccggg
agcgtcttgg
84
1005_Val297_Leu299_rev ctcctcgacgtggcgggagtgCAAggcCACgtccaccggg
agcgtcttgg
82
1006_His297_rev ctcctcgacgtggcgggagtgcgaggcGTGgtccaccggga
gcgtcttgg
83

Material und Methoden | 67
Name Nukleotidsequenz (5´3´) Tm [°C]
1007_His297_Ala299_rev ctcctcgacgtggcgggagtgGGCggcGTGgtccaccggg
agcgtcttgg
84
1008_His297_Gly299_rev ctcctcgacgtggcgggagtgGCCggcGTGgtccaccggg
agcgtcttgg
84
1009_His297_Leu299_rev ctcctcgacgtggcgggagtgCAAggcGTGgtccaccggg
agcgtcttgg
82
1010_Ala297_rev ctcctcgacgtggcgggagtgcgaggcGGCgtccaccggga
gcgtcttgg
83
1011_Ala297_Ala299_rev ctcctcgacgtggcgggagtgGGCggcGGCgtccaccggg
agcgtcttgg
85
1012_Ala297_Gly299_rev ctcctcgacgtggcgggagtgGCCggcGGCgtccaccggg
agcgtcttgg
85
1013_Ala297_Leu299_rev ctcctcgacgtggcgggagtgCAAggcGGCgtccaccggg
agcgtcttgg
81
1014_Gly297_rev ctcctcgacgtggcgggagtgcgaggcGCCgtccaccggga
gcgtcttgg
83
1015_Gly297_Ala299_rev ctcctcgacgtggcgggagtgGGCggcGCCgtccaccggg
agcgtcttgg
85
1016_Gly297_Gly299_rev ctcctcgacgtggcgggagtgGCCggcGCCgtccaccggg
agcgtcttgg
85
1017_Gly297_Leu299_rev ctcctcgacgtggcgggagtgCAAggcGCCgtccaccggg
agcgtcttgg
82
1018_Leu297_rev ctcctcgacgtggcgggagtgcgaggcCAAgtccaccggga
gcgtcttgg
82
1019_Leu297_Ala299_rev ctcctcgacgtggcgggagtgGGCggcCAAgtccaccggg
agcgtcttgg
83
1020_Leu297_Gly299_rev ctcctcgacgtggcgggagtgGCCggcCAAgtccaccggg
agcgtcttgg
83
1021_Leu297_Leu299_rev ctcctcgacgtggcgggagtgCAAggcCAAgtccaccggga
gcgtcttgg
81
1022_Ala299_rev ctcctcgacgtggcgggagtgGGCggcgtagtccaccggga
gcgtcttgg
82
1023_Gly299_rev ctcctcgacgtggcgggagtgGCCggcgtagtccaccggga
gcgtcttgg
82
1024_Leu299_rev ctcctcgacgtggcgggagtgCAAggcgtagtccaccgggag
cgtcttgg
80
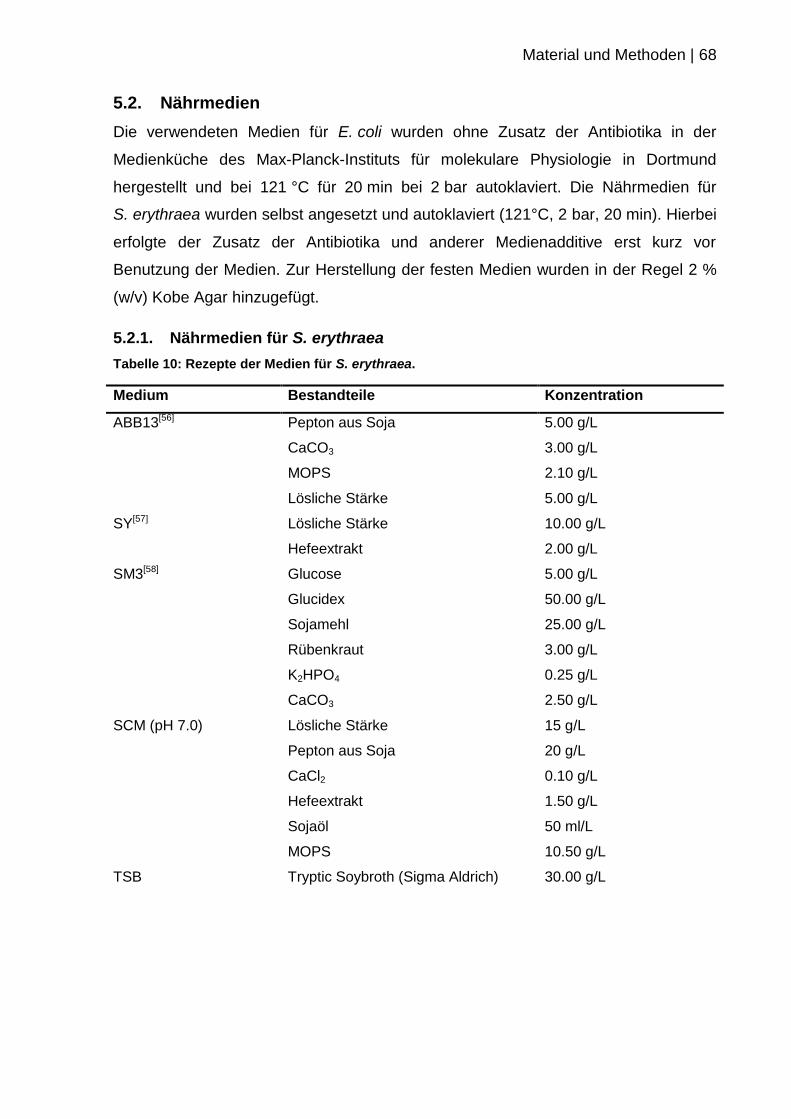
Material und Methoden | 68
5.2. Nährmedien
Die verwendeten Medien für E. coli wurden ohne Zusatz der Antibiotika in der
Medienküche des Max-Planck-Instituts für molekulare Physiologie in Dortmund
hergestellt und bei 121 °C für 20 min bei 2 bar autoklaviert. Die Nährmedien für
S. erythraea wurden selbst angesetzt und autoklaviert (121°C, 2 bar, 20 min). Hierbei
erfolgte der Zusatz der Antibiotika und anderer Medienadditive erst kurz vor
Benutzung der Medien. Zur Herstellung der festen Medien wurden in der Regel 2 %
(w/v) Kobe Agar hinzugefügt.
5.2.1. Nährmedien für S. erythraea
Tabelle 10: Rezepte der Medien für S. erythraea.
Medium Bestandteile Konzentration
ABB13[56] Pepton aus Soja 5.00 g/L
CaCO3 3.00 g/L
MOPS 2.10 g/L
Lösliche Stärke 5.00 g/L
SY[57] Lösliche Stärke 10.00 g/L
Hefeextrakt 2.00 g/L
SM3[58] Glucose 5.00 g/L
Glucidex 50.00 g/L
Sojamehl 25.00 g/L
Rübenkraut 3.00 g/L
K2HPO4 0.25 g/L
CaCO3 2.50 g/L
SCM (pH 7.0) Lösliche Stärke 15 g/L
Pepton aus Soja 20 g/L
CaCl2 0.10 g/L
Hefeextrakt 1.50 g/L
Sojaöl 50 ml/L
MOPS 10.50 g/L
TSB Tryptic Soybroth (Sigma Aldrich) 30.00 g/L
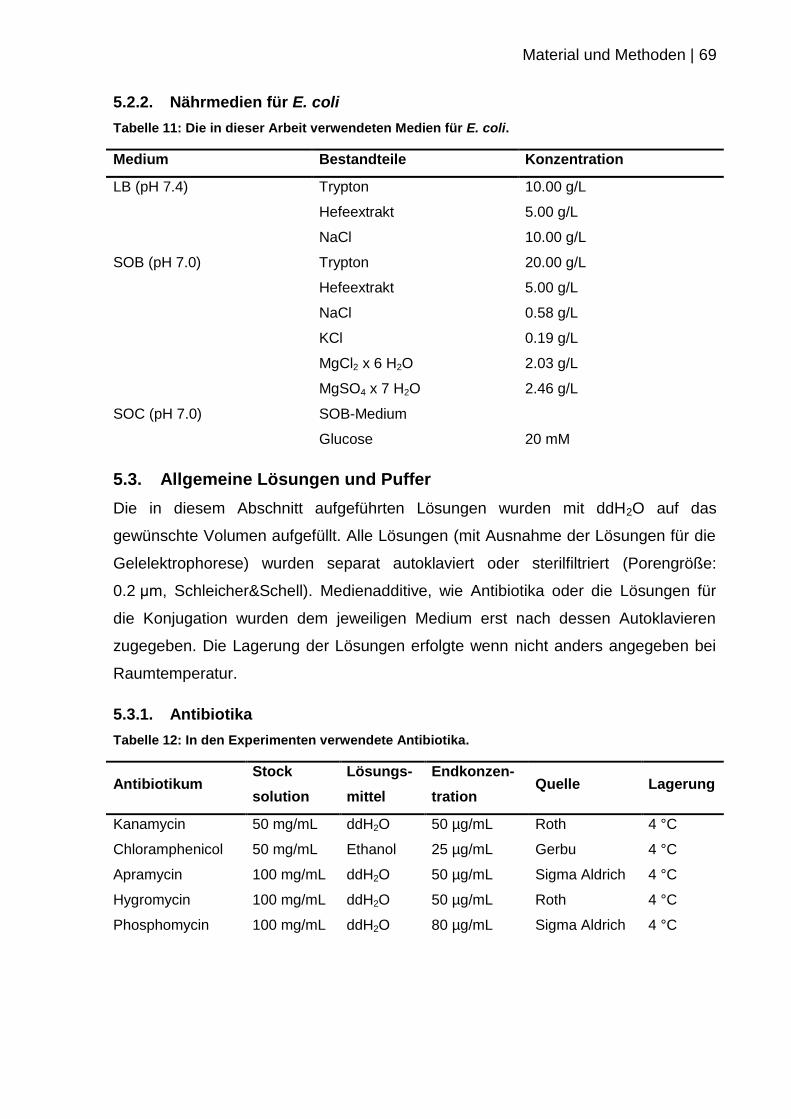
Material und Methoden | 69
5.2.2. Nährmedien für E. coli
Tabelle 11: Die in dieser Arbeit verwendeten Medien für E. coli.
Medium Bestandteile Konzentration
LB (pH 7.4) Trypton 10.00 g/L
Hefeextrakt 5.00 g/L
NaCl 10.00 g/L
SOB (pH 7.0) Trypton 20.00 g/L
Hefeextrakt 5.00 g/L
NaCl 0.58 g/L
KCl 0.19 g/L
MgCl2 x 6 H2O 2.03 g/L
MgSO4 x 7 H2O 2.46 g/L
SOC (pH 7.0) SOB-Medium
Glucose 20 mM
5.3. Allgemeine Lösungen und Puffer
Die in diesem Abschnitt aufgeführten Lösungen wurden mit ddH2O auf das
gewünschte Volumen aufgefüllt. Alle Lösungen (mit Ausnahme der Lösungen für die
Gelelektrophorese) wurden separat autoklaviert oder sterilfiltriert (Porengröße:
0.2 μm, Schleicher&Schell). Medienadditive, wie Antibiotika oder die Lösungen für
die Konjugation wurden dem jeweiligen Medium erst nach dessen Autoklavieren
zugegeben. Die Lagerung der Lösungen erfolgte wenn nicht anders angegeben bei
Raumtemperatur.
5.3.1. Antibiotika
Tabelle 12: In den Experimenten verwendete Antibiotika.
Antibiotikum Stock
solution
Lösungs-
mittel
Endkonzen-
tration Quelle Lagerung
Kanamycin 50 mg/mL ddH2O 50 µg/mL Roth 4 °C
Chloramphenicol 50 mg/mL Ethanol 25 µg/mL Gerbu 4 °C
Apramycin 100 mg/mL ddH2O 50 µg/mL Sigma Aldrich 4 °C
Hygromycin 100 mg/mL ddH2O 50 µg/mL Roth 4 °C
Phosphomycin 100 mg/mL ddH2O 80 µg/mL Sigma Aldrich 4 °C

Material und Methoden | 70
5.3.2. Lösungen für die Plasmid Isolation
Tabelle 13: Eingesetzte Lösungen zur Plasmid-Isolation.
Lösung Bestandteile Konzentration Anmerkung
MP1 Tris pH 8 25 mM bei 4 °C lagern
EDTA 10 mM
Glucose 50 mM
RNAse 200 µg/mL
MP2 NaOH 0.2 M
SDS 1 %
MP3 NaOAc 3 M pH 4.8 – 5.2
MP4 Lithiumchlorid 5 M
5.3.3. Lösungen für die Konjugation
Tabelle 14: Bei der Konjugation verwendete Lösungen.
Lösung Stammlösung Pro Liter Medium Anmerkung
Thiamin 1 % 1 mL Lagerung bei 4 °C
FeSO4 1.2 % 1 mL Zugabe einiger Tropfen HCl,
Lagerung bei 4 °C
MgCl2 10 mM 10 mL
5.3.4. Lösungen für die Präparation ultra-kompetenter Zellen
Tabelle 15: Bei der Präparation von ultra-kompetenten Zellen verwendete Lösung.
Lösung Bestandteile Konzentration Anmerkung
TB-Puffer PIPES 10 mM pH 6.7, Lagerung bei 4 °C
CaCl2 15 mM
KCl 250 mM
MnCl2 55 mM
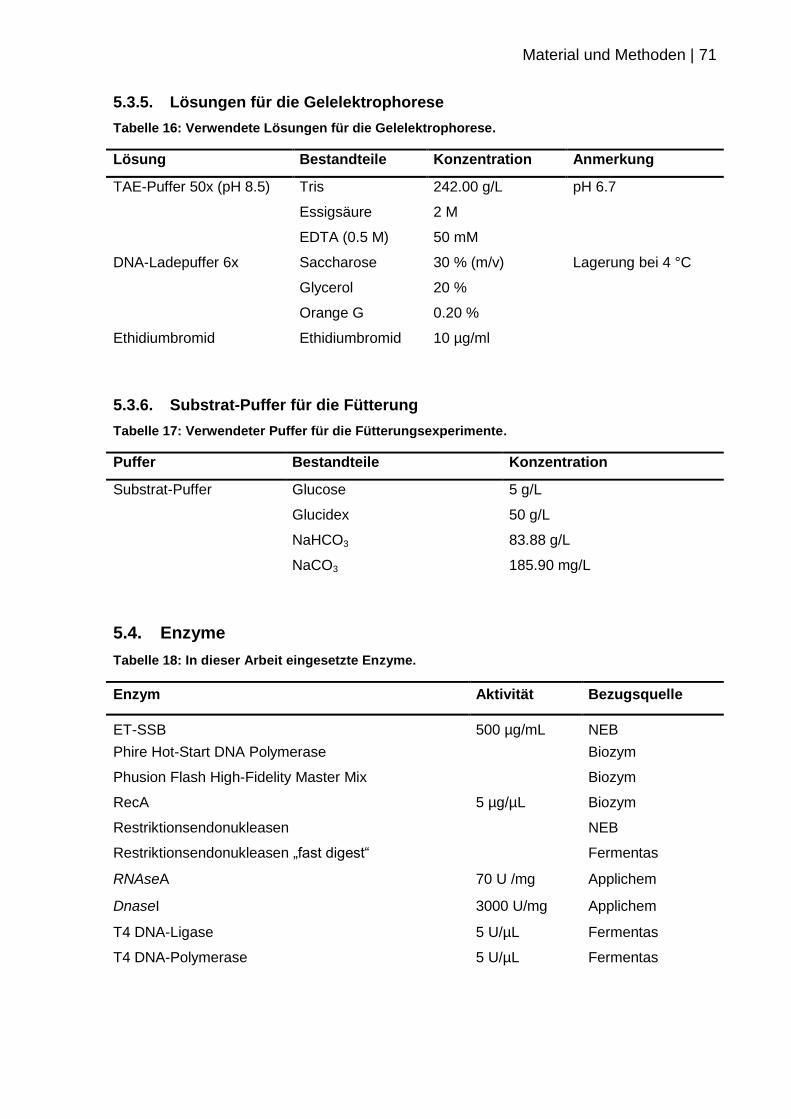
Material und Methoden | 71
5.3.5. Lösungen für die Gelelektrophorese
Tabelle 16: Verwendete Lösungen für die Gelelektrophorese.
Lösung Bestandteile Konzentration Anmerkung
TAE-Puffer 50x (pH 8.5) Tris 242.00 g/L pH 6.7
Essigsäure 2 M
EDTA (0.5 M) 50 mM
DNA-Ladepuffer 6x Saccharose 30 % (m/v) Lagerung bei 4 °C
Glycerol 20 %
Orange G 0.20 %
Ethidiumbromid Ethidiumbromid 10 µg/ml
5.3.6. Substrat-Puffer für die Fütterung
Tabelle 17: Verwendeter Puffer für die Fütterungsexperimente.
Puffer Bestandteile Konzentration
Substrat-Puffer Glucose 5 g/L
Glucidex 50 g/L
NaHCO3 83.88 g/L
NaCO3 185.90 mg/L
5.4. Enzyme
Tabelle 18: In dieser Arbeit eingesetzte Enzyme.
Enzym Aktivität Bezugsquelle
ET-SSB 500 µg/mL NEB
Phire Hot-Start DNA Polymerase Biozym
Phusion Flash High-Fidelity Master Mix Biozym
RecA 5 µg/µL Biozym
Restriktionsendonukleasen NEB
Restriktionsendonukleasen „fast digest“ Fermentas
RNAseA 70 U /mg Applichem
DnaseI 3000 U/mg Applichem
T4 DNA-Ligase 5 U/µL Fermentas
T4 DNA-Polymerase 5 U/µL Fermentas

Material und Methoden | 72
5.5. Allgemeine Methoden der Molekularbiologie
5.5.1. Lagerung und Kultivierung der Mikroorganismen
Im Vorfeld dieser Arbeit wurden die optimalen Wachstumsbedingungen zur
Kultivierung, sowie die Fermentationsbedingungen von S. erythraea NRRL-B-24071
in der Arbeitsgruppe untersucht und optimiert. Die resultierenden Protokolle wurden
für diese Arbeit übernommen. Sie werden in den folgenden Abschnitten geschildert.
5.5.1.1. Lagerung von E. coli
Die langfristige Lagerung von E. coli Zellen erfolgte als Glycerolgefrierkulturen. Dazu
wurden bewachsene Kulturen in LB-Medium mit einer 80 %-igen Glycerollösung
versetzt, sodass die Endkonzentration 20 % Glycerol betrug. Die Stammlösungen
wurden bei -80 °C gelagert. Für ein bis zwei Tage wurden E. coli-Zellen in
LB-Medium oder auf LB-Agar Platten bei 4 °C gelagert.
5.5.1.2. Lagerung von S. erythraea
Die Lagerung der Stammkultur erfolgte bei -80 °C als Glycerolgefrierkultur. Für den
regelmäßigen Gebrauch wurde S. erythraea auf ABB13-Agar Platten ausplattiert und
bei 30 °C bis zur Sporenbildung kultiviert. Die bewachsenen Platten wurden bei
Raumtemperatur gelagert. Für längere Lagerzeiten wurde eine Übernachtkultur in
TSB erzeugt und mit dem gleichen Volumen einer 2 M Sorbitollösung vermengt. Die
Sorbitolstammlösungen wurden bei -80 °C aufbewahrt.
5.5.1.3. Kultivierung von E. coli
Die Kultivierung von E. coli Zellen erfolgte bei 30 °C und 180 rpm in LB-Medium
unter Zusatz der entsprechenden Antibiotika. (Apramycin bei E. coli_pKSSU89 oder
bei auf dem Vektor pKSSU89 basierenden Konstrukten, Apramycin und Hygromycin
bei E. coli_pKSSU96).
5.5.1.4. Kultivierung von S. erythraea
Bei der Kultivierung von S. erythraea ist eine hohe Belüftungsrate der Kulturen zu
beachten. Dazu wurden die verwendeten Glaskolben mit Stahlfedern am Boden
ausgestattet. Bei Verwendung von 24-Lochplatten wurden einige Glasperlen (3-4) in
jedes Loch gegeben.
Die Vorkultur von S. erythraea wurde bei 30 °C und 180 rpm für zwei Tage kultiviert.
Als Medium diente bei der Vorkultur das TSB-Medium, dagegen erfolgte die

Material und Methoden | 73
Erythromycin-Produktion in der Regel in SM3-Medium. In Ausnahmefällen wurde das
SCM-Medium für die Fermentation verwendet. Für die Vorkultur wurde mit einer
Pipettenspitze ein Stück aus einer bewachsenen ABB13-Agarplatte ausgeschnitten
(ca. 0.5 cm2) und in einen Kolben gegeben. Gegebenenfalls wurde das
entsprechende Antibiotikum (Resistenz durch integrierten Vektor vermittelt)
zugegeben. Die Hauptkultur wurde mit 1/20 Volumen der Vorkultur ohne Zusatz
eines Antibiotikums angeimpft und bei 30 °C und 180 rpm für fünf Tage geschüttelt.
5.5.1.5. Screening von S. erythraea in 24-Lochplatten des System Duetz[53]
Abbildung 51: Kultivierung der Hauptkulturen in 24-Lochplatten des System Duetz.
Die Fermentationen für die initialen Screens der S. erythraea AT6* Mutanten wurden
in 24-Lochplatten (Leervolumen 5 mL) des System Duetz[53] kultiviert. Je vier
einzelne Klone wurden pro Mutante eingesetzt. Als Referenz diente die parallele
Kultivierung des S. erythraea Wildtyps (NRRL-B-24071). Für die Vorkultur wurden je
2 mL TSB mit Apramycin in ein Loch überführt und mit einem ca. 0.5 cm2 großen
Stück von einer ABB13 Agarplatte der jeweiligen Transkonjugante versetzt. Die
24-Lochplatten wurden mit einem luftdurchlässigen Verschlussfilm für Zellkulturen
verschlossen und in den Halterungen des System Duetz befestigt. Die Vorkultur
wurde für zwei Tage bei 30 °C und 180 rpm kultiviert. Anschließend wurde die
Hauptkultur in frischen Lochplatten angesetzt. Hierfür wurden 3 mL SM3-Medium mit
1/10 Volumen der Vorkultur angeimpft. Dazu wurden je 300 µL einer 100 mM Lösung
(Endkonzentration 10 mM) der SNAC aktivierten rac-Methylmalonsäure (13)
gegeben. Als Referenz diente hier die analoge Kultivierung der Klone ohne Zusatz

Material und Methoden | 74
des Substrates 13. Die Platten wurden mit dem Deckel des System Duetz
verschlossen, befestigt und für fünf Tage bei 30 °C und 180 rpm fermentiert (vgl.
Abbildung 51). Zur Erstellung von Stammlösungen wurde der Rest der Vorkulturen
mit 80 % Glycerol (Endkonzentration 20 %) versetzt und bei -80 °C gelagert. Im
Anschluss an die Kultivierung wurden die einzelnen Kulturen in 15 mL Röhrchen
überführt und für eine Nacht bei -20 °C eingefroren, um durch eine Öffnung der
Zellen Erythromycin freizusetzen.
5.5.1.6. Extraktion mit Ethylacetat
Die Kulturen wurden in 15 mL Reaktionsgefäße überführt und nachfolgend mit dem
zweifachen Volumen Ethylacetat versetzt und über Nacht bei 19 °C und in der Regel
180 rpm geschüttelt. Es folgte die Zentrifugation bei 4 °C, 4000 g für 10 min.
Anschließend wurde die organische Phase in Reagenzgläser überführt und in einer
Vakuumzentrifuge bei 37 °C bis zur Trockne eingeengt. Die Rückstände wurden in
500 µl Methanol resuspendiert und für 30 min bei 13200 g zentrifugiert. Der
Überstand wurde in Probenröhrchen pipettiert und mittels LC/ESI-MS analysiert.
5.5.1.7. Fermentation zur Gewinnung von Erythromycin in präparativen Mengen
Abbildung 52: Fermentation von S. erythraea in 5 L Kolben mit Stahlfeder.
Zur NMR-Analyse von Erythromycin ist die Präparation in größeren Mengen
notwendig. Dazu wurden Kulturen in einem Gesamtvolumen von 1 L fermentiert. Für
die Vorkultur wurden 30 mL TSB-Medium (enthielt Apramycin, außer beim WT) in

Material und Methoden | 75
einem 250 mL Kolben mit 1.5 mL Flüssig-Glycerolstammlösung oder einem 1 cm2
großem Stück aus einer bewachsenen ABB13-Agarplatte angeimpft und bei 30 °C
und 180 rpm kultiviert bis die Kultur dicht bewachsen war (etwa 1-2 Tage). Die
Kulturen wurden mit weiteren 30 mL TSB-Medium verdünnt, auf zwei 250 mL Kolben
aufgeteilt und unter den gleichen Bedingungen für 24 h weiter geschüttelt. Die
Hauptkulturen aus 1 L SM3-Medium wurden mit 50 mL Vorkultur angeimpft und in
zwei 5 L Kolben mit Spirale für 5 Tage bei 30 °C und 180 rpm fermentiert. Da eine
sehr hohe Schaumentwicklung zu beobachten war, wurde meist am zweiten Tag der
Fermentation tropfenweise Antischaum (Antifoam Y-30, Sigma Aldrich) zu den
Kulturen gegeben, bis kein Schaum mehr vorhanden war (etwa 0.5 ml).
Für die Fütterung mit der SNAC aktivierten Methylmalonsäure (13) wurde beim
Ansetzen der Hauptkulturen der erzeugten S. erythraea AT6* Klone 100 mL einer
100 mM Lösung des Substrates in Substrat-Puffer hinzugegeben.
Nach dem Ablauf der fünftägigen Fermentation wurden diese in Glasflaschen
überführt und bei -80 °C für eine Nacht tiefgefroren, um die Zellen zu öffnen.
5.5.1.8. Extraktion und Aufreinigung von Erythromycin aus Fermentations-
kulturen
Zur Isolierung und Aufreinigung von Erythromycin wurden die tiefgefrorenen Kulturen
bei 4 °C über Nacht aufgetaut. Im Anschluss wurden die Zellen und festen
Medienbestandteile durch Zentrifugation bei 4 °C, 4000 g für 20 min (Sorvall
Evolution RC) sedimentiert. Der Überstand wurde filtriert und das Filtrat mit
Ethylacetat extrahiert. Dabei wurde ein Protokoll zur Extraktion von
Erythromycin A (7) aus einer S. erythraea Fermentationskultur von Beran et al.[59]
angewandt. Das Protokoll wurde zur Erhöhung der Ausbeute wie im Folgenden
geschildert optimiert. Nachdem das Erythromycin A (7) mit EtOAc aus der
Fermentationskultur extrahiert wurde, erfolgte die Extraktion der EtOAc-Phase mit
angesäuertem Wasser (pH 4.5). Hierbei wurde nicht einmal, sondern viermal
extrahiert. Das resultierende Protokoll ist in Abbildung 53 schematisch dargestellt.
Zur Analyse wurden von den eingeengten organischen Phasen Proben genommen
und mittels LC/ESI-MS vermessen (im Schema mit einem "*" gekennzeichnet). Zur
weiteren Aufreinigung wurde der erhaltene Feststoff einer präparativen DC (vgl.
5.6.2.3) unterzogen.

Material und Methoden | 76
Abbildung 53: Schema für die Isolation von Erythromycin aus einer S. erythraea Fermentation."*": Von diesen Phasen wurden nach Entfernen des Lösungsmittels Proben für eine
LC/ESI-MS Messung genommen.
5.5.2. Plasmidpräparation
Die Plasmidpräparation aus E. coli Zellen erfolgte ausgehend von einer
Übernachtkultur, die zuvor bei 30 °C und 180 rpm kultiviert worden ist. Es wurden je
1.5 mL der Zellsuspensionen für 2 min (4 °C, 13200 rpm) zentrifugiert. Der
Überstand wurde verworfen und das Zellsediment wurde dem folgenden Protokoll
unterzogen:
(Die Zusammensetzung der Lösungen MP1-MP4 sind in Tabelle 13 beschrieben)
Resuspendieren der Zellen in 50 µL MP1
Zelllyse durch Zugabe von 100 µL MP2, durch Invertieren mischen, Inkubieren
(max. 5 min) bis die Lösung klar ist
Zugabe von 75 µL MP3, mischen durch Invertieren
Zugabe von 225 µL MP4, mischen durch Invertieren
Sedimentieren von Proteinen, genomischer DNA und Zellbestandteile durch
Zentrifugation (10 min, 4 °C, 13200 rpm)

Material und Methoden | 77
Überstand abnehmen und zum Fällen der Plasmid-DNA mit 900 µL absolutem
Ethanol versetzen, durch Invertieren mischen
Pelletieren der Plasmid-DNA durch Zentrifugation für 15 min (4 °C,
13200 rpm)
Überstand wird verworfen, das Sediment wird zum Waschen mit 300 µL
70 %-igem Ethanol versetzt und 2-3 min (4 °C, 13200 rpm) zentrifugiert
Überstand wird verworfen, erneut kurz zentrifugiert und der restliche
Überstand entfernt
Die gefällte und pelletierte DNA wird bei RT getrocknet
Lösen der Plasmid-DNA in 40 µL ddH2O
5.5.3. Ethanol-Präzipitation
Zur Fällung und Reinigung von Plasmid-DNA wurden die wässrigen DNA-Lösungen
nach folgendem Protokoll behandelt:
Zugabe von 1/10 Probenvolumen MP3 (vgl. Tabelle 13)
Zugabe von eiskaltem, absolutem Ethanol (2.5-faches Volumen)
Mischen durch Invertieren
Zentrifugation bei 4 °C, 13200 g für 10 min
Der Überstand wird verworfen
Die sedimentierte DNA wird mit 70 %-igem Ethanol gewaschen und im
Anschluss bei RT getrocknet
Resuspension der DNA im ½ Volumen ddH2O
Die Konzentration wird mittels Gelelektrophorese bestimmt
5.5.4. PCR-Protokolle
5.5.4.1. Kolonie-PCR
Die Kolonie-PCR diente der Identifizierung möglicher positiver Transkonjuganten
(Insert-tragender Klone). Von bewachsenen Agarplatten wurden einzelne Kolonien
mit Hilfe einer Pipettenspitze partiell von der Agarplatte abgestrichen und in
PCR-Gefäße, die mit 5 µL ddH2O gefüllt waren, überführt. Es folgte die Inkubation
bei 99 °C für 10 min. 1 µL der Zellsuspension wurde dann nachfolgend in die
eigentliche PCR eingesetzt. Das Pipettierschema eines PCR-Ansatzes ist in Tabelle
19 aufgeführt.

Material und Methoden | 78
Tabelle 19: Pipettierschema für die Kolonie-PCR.
Komponente Menge
Betain (5 M) 2 µL
206_DEBS3_AT6-for (10 µM) 1 µL
207_DEBS3_AT6-rev (10 µM) 1 µL
Zellsuspension 1 µL
2x Mastermix Phire 5 µL
Das verwendete Temperaturprofil der PCR kann der Tabelle 20 entnommen werden.
Da die Schmelztemperatur Kann der Oligonukleotide >72 °C ist, konnte auf einen
separaten Annealing-Schritt verzichtet werden.
Tabelle 20: Temperaturprofil der Kolonie-PCR.
Schritt Temperatur Zeit
Denaturierung 99 °C 3 min
Denaturierung 99 °C 50 sec
Annealing/Elongation 72 °C 1 min
Finale Elongation 72 °C 1 min
Ende 4 °C ∞
Im Anschluss wurden 2 µL DNA-Ladepuffer zu den PCR-Produkten gegeben, die
gesamte Probe in ein 1 %-iges Agarosegel pipettiert und zusammen mit dem
Längenstandard (GeneRulerTM 1 kb Plus DNA Ladder, Fermentas) einer
Gelelektrophorese unterzogen.
5.5.4.2. Zielgerichtete Mutagenese durch overlap extension PCR
Die einzelnen Aminosäureaustausche innerhalb der DEBS AT6-Domäne wurden
mittels Sättigungsmutagenese mit spezifischen Oligonukleotiden durchgeführt.
Dieses Verfahren besteht aus zwei PCR Schritten (vgl. Abbildung 54).
25x

Material und Methoden | 79
Abbildung 54: Schema zum Arbeitsablauf der zielgerichteten Mutagenese am Beispiel der Mutation Met235Val.
Da die Klonierung mittels SLIC-MIX (vgl. Kapitel 5.5.8) erfolgen sollte, mussten die
entsprechenden Sequenzen mit endständigen, komplementären Sequenzen
flankierend zum Zielvektor pKSSU96 versehen werden. Als Template diente das
mittels EcoRV linearisierte Plasmid pKSSU89. Die eingesetzten Oligonukleotide
waren durch die benötigten komplementären Sequenzen vergleichsweise lang. In der
ersten PCR wurden in getrennten Ansätzen separat je zwei Fragmente erzeugt, die
einen überlappenden Bereich von etwa 15 Nukleotiden links und rechts der
einzufügenden Mutation besaßen. Das Pipettierschema, sowie das eingesetzte
Temperaturprofil werden in Tabelle 19 und Tabelle 20 aufgeführt.
Tabelle 21: Pipettierschema der zielgerichteten Mutagenese, PCR Schritt I.
Komponente Menge
Fragment 1 Fragment 2
Betain (5 M) Betain (5 M) 2 µL
206_DEBS3_AT6-for (10 µM) 207_DEBS3_AT6-rev (10 µM) 1 µL
Oligo_rev (10 µM) Oligo_fwd (10 µM) 1 µL
Linearisierte Vektor DNA
(ca. 50 ng/µL)
Linearisierte Vektor DNA
(ca. 50 ng/µL)
1 µL
Mastermix Phusion 2x Mastermix Phusion 2x 5 µL

Material und Methoden | 80
Tabelle 22: Temperaturprofil zur zielgerichteten Mutagenese, PCR Schritt I.
Schritt Temperatur Zeit
Denaturierung 99 °C 3 min
Denaturierung 99 °C 15 sec
Annealing 60 °C 1 sec
Elongation 72 °C 50 sec
Finale Elongation 72 °C 1 min
Ende 4 °C ∞
Zur Überprüfung der PCR wurde im Anschluss mit 3 µL des PCR-Produktes eine
Gelelektrophorese durchgeführt. Bei erfolgreicher PCR, wurde der Ansatz mit
insgesamt 30 µL Volumen wiederholt. Nach erneuter Kontrolle mittels
Gelelektrophorese wurden die DNA-Fragmente gelaufgereinigt.
In einer zweiten PCR wurden die beiden gereinigten Fragmente durch den Einsatz
der beiden äußeren Oligos assembliert. Der verwendete Ansatz wird in Tabelle 23
dargestellt. Das verwendete Temperaturprofil ist analog zur ersten PCR (Tabelle 22).
Tabelle 23: Pipettierschema der zweiten PCR zielgerichteten Mutagenese, overlap extension PCR, Schritt II.
Komponente Menge
Betain (5 M) 2 µL
206_DEBS3_AT6-for (10 µM) 1 µL
207_DEBS3_AT6-rev (10 µM) 1 µL
Fragment 1 0.5 µL
Fragment 2 0.5 µL
Mastermix Phusion 2x 5 µL
Auch hierbei wurde zunächst der Erfolg der PCR mittels Gelelektrophorese (mit 3 µL
PCR-Produkt) überprüft und im Anschluss der Ansatz auf 30 µL hochskaliert. Nach
erneuter Gelelektrophorese wurden die PCR-Produkte gegebenenfalls einem DpnI-
Verdau unterzogen und dann gelaufgereinigt. Die Konzentration der isolierten und
gereinigten DNA wurde durch ein Agarose-Gel bestimmt.
25x

Material und Methoden | 81
5.5.4.2.1. DpnI Verdau des Mutterstranges nach der PCR
Die Template-DNA aus E. coli liegt methyliert vor. Durch Verdau mit dem
Restriktionsenzym DpnI wird diese methylierte DNA abgebaut. So kann nach
erfolgter PCR sichergestellt werden, dass die parentale DNA abgebaut wird. Der
dafür pipettierte Ansatz kann der Tabelle 24 entnommen werden.
Tabelle 24: Verdau der PCR-Produkte durch das FastDigest Enzym DpnI von Fermentas.
Komponente Volumen
PCR-Produkte 27 µL
FastDigest-Puffer 10x 3 µL
DpnI 1 µL
Der Ansatz wurde für 30 min bei 37 °C inkubiert. Anschließend wurde Ladepuffer
hinzugefügt und die gesamte Probe einer Gelaufreinigung unterzogen.
5.5.5. Agarose-Gelelektrophorese von DNA
Die Gelelektrophorese wurde mit 1 %-igen Agarosegelen durchgeführt. Zur
Herstellung der Gele wurde 1 % (w/v) Agarose in 1x TAE-Puffer angesetzt und in der
Mikrowelle aufgekocht bis alles gelöst war. Die abgekühlte, aber flüssige
Agaroselösung wurde mit Ethidiumbromid (etwa 15 µL einer 10 mg/mL Lösung auf
200 mL Agaroselösung) versetzt und in eine Gelform gegossen. Das ausgehärtete
Gel wurde in eine Gelkammer gelegt und mit TAE-Puffer befüllt bis das Gel
vollständig mit Flüssigkeit bedeckt war. Die zu untersuchenden DNA-Proben
(1-15 µL) wurden mit 1/6 Volumen DNA-Ladepuffer versetzt und in die Geltaschen
gegeben. Zusätzlich wurde der Längenstandard GeneRulerTM 1 kb Plus DNA Ladder
(Fermentas, vgl. Abbildung 55), der zuvor mit Ladepuffer und Wasser (1:3) verdünnt
wurde, aufgetragen.

Material und Methoden | 82
Abbildung 55: Längenstandard für die Agarosegele: GeneRulerTM 1 kb Plus DNA Ladder.
Die Auftrennung der DNA-Fragmente wurde bei 120 V für etwa 26 min durchgeführt.
5.5.5.1. Konzentrationsbestimmung mittels Gelelektrophorese
Zur Konzentrationsbestimmung der DNA-Proben nach der Ethanol-Fällung oder
Gelaufreinigung wurden je 1 µL der vorliegenden Proben und 1 µL einer 1:10
Verdünnung mit ddH2O in ein Gel eingetragen. Daneben erfolgte die Auftragung des
Längenstandards in Volumina von 1 µL sowie 5 µL. Nach erfolgter Gelelektrophorese
wurde die Intensität der DNA-Banden verglichen und mittels der angegebenen
DNA-Mengen des Längenstandards die Konzentration abgeschätzt.
Eine Berechnung der Konzentration wird hier beispielhaft gezeigt: Ist die Intensität
einer Bande der aufgetragenen Probe mit der der 1500 bp DNA-Bande des
Längenstandards vergleichbar, so ist bei 1 µL aufgetragener Probe die Konzentration
gleich 80/3 ng/µL (1:3 Verdünnung des Standards). Bei 5 µl aufgetragener Probe
wird zusätzlich mit dem Faktor fünf multipliziert (80/3*5 ng/µL).
5.5.6. Aufreinigung von DNA mittels Gelelektrophorese
Zur nebenproduktfreien Isolierung der Ziel-DNA nach einer PCR wurde eine
Gelaufreinigung durchgeführt. Dabei wurde die DNA-Probe entsprechend ihres
Volumens auf mehrere Taschen aufgeteilt. Nach erfolgter Gelelektrophorese konnten
die Banden durch das Interkalieren des Ethidiumbromids in die DNA-Fragmente
unter UV-Licht detektiert und ausgeschnitten werden. Hierbei ist wichtig, dass die
Dokumentation des Gels erst nach Ausschneiden der Gelstücke erfolgt, damit die

Material und Methoden | 83
DNA nicht unnötig geschädigt wird. Das Gelstück wurde in ein
1.5 mL-Reaktionsgefäß überführt und die DNA mittels Gel Extraktions Kit von
PEQLAB Biotechnologie GmbH isoliert. Dabei wurde das Kurzprotokoll von PEQLAB
befolgt. Der optionale Waschschritt mit Binding Buffer wird nicht durchgeführt.
5.5.7. Restriktionsanalyse
5.5.7.1. Restriktionsverdau vor der Polymerase Kettenreaktion
Um die PCR zu erleichtern und somit eine erhöhte Effizienz zu erzielen, wurden die
eingesetzten Vektoren zuvor linearisiert. Hierzu wurden FastDigest Enzyme
verwendet. Für die Linearisierung von pKSSU89 wurde EcoRV eingesetzt. Der
Reaktionsansatz kann der folgenden Tabelle entnommen werden. Er wurde bei
37 °C für 30 min inkubiert.
Tabelle 25: Ansatz für den Restriktionsverdau.
Komponente Volumen
DNA (Plasmid) 20 µL
Puffer 10x (FD) 5 µL
Restriktionsendonuklease (FD) 3 µL
ddH2O 22 µL
5.5.7.2. Restriktionsverdau vor der Klonierung
Abbildung 56: Vektor pKSSU96. Die Hygromycin-Kassette (blau), sowie die Schnittstellen der Restriktionsenzyme ScaI und FseI sind gekennzeichnet.
Vor der Klonierung wurde der Vektor pKSSU96 linearisiert, indem die Hygromycin-
Kassette herausgeschnitten wurde. Dazu wurde das FastDigest Enzym ScaI
pKSSU96

Material und Methoden | 84
verwendet. Wegen der enormen Größe des Vektors (ca. 16.8 kb) ist eine
Gelaufreinigung mit einer Ausbeute von nahezu 0 ng/µL nicht sinnvoll. Daher wird
zusätzlich ein FseI Verdau durchgeführt. Die FseI-Schnittstellen liegen innerhalb der
Hygromycin-Kassette. Damit wird sichergestellt, dass der Vektor nahezu vollständig
linearisiert vorliegt. Der Verdau mit ScaI erfolgt für 45 min bei 37 °C. Der dafür
pipettierte Ansatz kann der Tabelle 26 entnommen werden.
Tabelle 26: ScaI Verdau des Vektors pKSSU96.
Komponente Volumen
DNA (Plasmid) 20 µL
Puffer 10x (FD) 5 µL
ScaI (FD) 3 µL
ddH2O 22 µL
Für den Verdau mit FseI wurde die DNA präzipitiert. Der Verdau erfolgte für 30 min
bei 37 °C. In Tabelle 27 ist der hierfür verwendete Ansatz aufgeführt.
Tabelle 27: FseI Verdau des Vektors pKSSU96.
Komponente Volumen
DNA (Plasmid) 25 µL
Puffer 4 5 µL
FseI 1 µL
BSA 0.4 µL
ddH2O 18.6 µL
Nach erneuter Ethanolpräzipitation wurde die DNA resuspendiert und die
Konzentration über eine Agarose-Gelelektrophorese ermittelt.
5.5.7.3. Kontrollverdau
Die Verifizierung von Konstrukten wurde durch einen Verdau mit einem
Restriktionsenzym überprüft. Hier wurde NotI, ein FastDigest Enzym von Fermentas,
eingesetzt. In der Ziel-DNA-Sequenz bestehend aus Vektor inklusive Insert waren
drei Schnittstellen für NotI vorhanden. Davon liegt eine innerhalb der Insert-DNA und
zwei liegen im Vektorrückgrat. Dementsprechend gibt es bei positivem Einbau des
Inserts drei DNA-Fragmente.

Material und Methoden | 85
Abbildung 57: Shuttle Vektor pKSSU89*. Die Aminosäurepositionen Val295 und Met235, sowie die NotI Schnittstellen sind gekennzeichnet. Die Fragmente nach einem Restriktionsverdau mit NotI haben ein Molekulargewicht von etwa 9.9, 5.2 und 1.6 kb.
Der pipettierte Ansatz kann der folgenden Tabelle entnommen werden. Die Zugabe
des Restriktionsenzyms erfolgte zuletzt und im Anschluss wurde der Ansatz für
15 min bei 37 °C inkubiert.
Tabelle 28: Kontrollverdau mit dem FastDigest Enzym NotI von Fermentas.
Komponente Volumen
DNA 4 µL
Puffer 10x 1 µL
NotI 1 µL
ddH2O 4 µL
Der verdauten Plasmid-DNA wurden 2 µL DNA-Ladepuffer hinzugefügt und die
gesamte Probe wurde auf ein 1%-iges Agarosegel aufgetragen und einer
Gelelektrophorese unterzogen.
5.5.8. Klonierungen mittels SLIC-MIX
Der in dieser Arbeit verwendete SLIC-MIX zur Klonierung basiert auf dem von Li und
Elledge[60] entwickeltem sequence and ligation independent cloning (SLIC).
Entscheidend hierbei sind der Einsatz von Vektor und Insert in äquimolaren Mengen,
sowie der Zusatz von T4-DNA Polymerase, die durch die Abwesenheit von
Nukleotiden ihre 3´-5´-Endonukleaseaktivität ausübt. Es entstehen kurze
einzelsträngige DNA Fragmente mit überhängenden Enden. Um eine erfolgreiche
Verknüpfung der beiden Fragmente zu ermöglichen wird das Insert mit flankierenden
pKSSU89*

Material und Methoden | 86
Sequenzen (mindestens 15 nt lang) amplifiziert (vgl. 5.5.4.2), welche komplementär
zu den Enden des linearisierten Vektors sind. Nach Transformation in E. coli werden
die Doppelstrangbrüche (Nicks) geschlossen.
Die Weiterentwicklung der SLIC Methode in der Arbeitsgruppe[61] resultiert in einer
Erhöhung der Klonierungseffizienz durch den Einsatz von ET-SSB – einem
Einzelstrang stabilisierenden Protein – während der T4-DNA Polymerase
Behandlung. Die Methode dient vor allem dem Klonieren von großen und GC-reichen
PCR-Produkten. Der zuvor linearisierte Vektor und das mittels PCR amplifizierte
Zielgen wurden separat in äquimolaren Mengen für 60 min bei 22 °C mit
T4 DNA-Polymerase inkubiert. Es wurden in der Regel 500 ng Vektor-DNA
eingesetzt. Der vollständige Reaktionsansatz kann dem folgenden Pipettierschema
(vgl. Tabelle 29) entnommen werden.
Tabelle 29: Ansatz der SLIC-MIX Klonierung.
Lösung Vektor Insert
DNA 500 ng Äquimolar
10x T4-DNA Polymerase Puffer 1 µL 1 µL
BSA (100x) 0.10 µL 0.10 µL
ET-SSB (1:10) 1.60 µL 0.80 µL
T4-DNA Polymerase (Fermentas) 0.33 µL 0.10 µL
ddH2O Ad 10 µL Ad 10 µL
Durch Zugabe von dCTP (1/10 Volumina, 10 mM) wurde der Abbau des
Doppelstranges gestoppt. Anschließend wurde in einem zweiten Schritt jeweils 1 µL
beider behandelter DNA-Fragmente in ein neues Reaktionsgefäß pipettiert. Die
in vitro Verknüpfung von Insert und Vektor wird durch das Enzym Rekombinase A
(RecA) katalysiert. Der Ansatz wird für 45 min bei 37 °C inkubiert.
Tabelle 30: Pipettierschema für den Schritt 2 der SLIC MIX-Klonierung.
Lösung Mengenangabe
Vektor-DNA 1 µL
Insert-DNA 1 µL
10x T4 DNA Ligase Puffer (Fermentas) 0.5 µL
RecA (1:25) 0.1 µL
ddH2O Ad 5 µL

Material und Methoden | 87
Die Klonierung mittels SLIC-MIX ist in Abbildung 58 schematisch dargestellt.
Abbildung 58: Schematische Darstellung der SLIC-MIX Methode[61]
Abkürzungen: Nukleotide (nt), Einzelstrangbindendes Protein (SSB), T4 DNA Polymerase (T4 DNA Pol), Rekombinase A (Rec A), Replikationsursprung E. coli (colE1ori), Apramycin Resistenzkassette [aac(3)IV], Origin des Plasmidtransfers (orIT RK2), Integrase des Bakteriophagen φ BT1(IntB1).
Es wurden 4 µL des Klonierungsansatzes in 50 µL chemisch kompetente
OmniMAXTM 2T1R Zellen transformiert (vgl. 5.5.9). Die Zellen wurden auf
Apramycin-haltige LB-Agarplatten ausgestrichen und über Nacht bei 37 °C inkubiert.
5.5.9. Einbringen genetischer Information in E. coli
5.5.9.1. Präparation von ultra-kompetenten E. coli-Zellen
Zur Steigerung der Transformationseffizienz wurden ultra-kompetente E. coli-Zellen
nach einem literaturbekannten Protokoll nach Inoue et al[62] präpariert. Dazu wurde
1 µL der entsprechenden Zellen mit ddH2O verdünnt (1:10) und auf einer
LB-Agarplatte mit entsprechendem Antibiotikum ausgestrichen. Die Platte wurde
über Nacht bei 37 °C inkubiert.

Material und Methoden | 88
Es wurden 10-12 große Kolonien gepickt und in 250 mL SOB-Medium in
einem 1 L Kolben bei 19 °C und 120 rpm kultiviert, bis eine OD600 von 0.5
erreicht worden ist (etwa 24-36 h)
Der Kolben wurde für 10 min auf Eis aufbewahrt und anschließend wurde die
Kultur in sterile Zentrifugengefäße überführt
Zentrifugation bei 4000 rpm und 4 °C für 10 min pelletierte die Zellen
Der Überstand wurde verworfen und das Sediment behutsam in 20 mL
TB-Puffer (eiskalt) und 1.4 mL DMSO (wurde für eine Nacht bei -20 °C
gelagert) resuspendiert
Die Zelllösung wurde aliquotiert (50-100 µL), in flüssigem Stickstoff
schockgefroren und bei -80 °C gelagert
5.5.9.2. Transformation durch Hitzeschock
Die Transformation durch Hitzeschock erfolgte mit chemisch kompetenten E. coli
Zellen. Dazu wurde folgendes Protokoll befolgt:
Ein 50 µL Aliquot der ultra-kompetenten Zellen wird auf Eis aufgetaut
4-5 µL des zu transformierenden Plasmids werden hinzugegeben
15 min auf Eis inkubieren
Die Zellen werden für 1 min bei 42 °C einem Hitzeschock ausgesetzt
(Thermomixer, Eppendorf)
Anschließend werden die Zellen direkt auf Eis gegeben und für 2-5 min
inkubiert
Es werden 200 µL SOC-Medium hinzugegeben
Zur Regeneration der Zellen werden sie für mindestens 45 min bei 37 °C
inkubiert (Thermomixer, Eppendorf)
Die Zellsuspension wird auf antiobiotikabeinhaltende LB-Platten (Resistenz
durch transformiertes Plasmid vermittelt) ausgestrichen und über Nacht bei
37 °C kultiviert
5.5.10. Einbringen genetischer Information in Actinomyceten mittels
Konjugation
Ein Transfer der Plasmid-DNA aus einem E. coli-Donorstamm in einen
Actinomyceten als Rezipient kann über intergenetische Konjugation erfolgen. In
dieser Arbeit wurde als Donor der methylierungsdefiziente Stamm E. coli ET12567
eingesetzt. Dieser enthält das Plasmid pUZ8002, ein sich selbst nicht

Material und Methoden | 89
transferierendes RP4 Derivat, wodurch der Stamm zum DNA-Transfer fähig ist. Als
Rezipent diente S. erythraea∆AT6hygR.
5.5.10.1. Vorbereitung des E. coli Donorstammes
Nach erfolgter Transformation des zu übertragenden Plasmids in E. coli
ET12567/pUZ8002 und Inkubation der Zellen bei 37 °C auf antiobiotikabeinhaltenden
LB-Platten über Nacht wurde je eine Kolonie von der Agarplatte gepickt und damit
eine 2 mL LB-Medium Übernachtkultur angeimpft. Das Medium enthielt die
entsprechenden Antibiotika (hier: Apramycin – Resistenz durch zu transformierendes
Plasmid vermittelt, Chloramphenicol und Kanamycin – Resistenz durch pUZ8002
vermittelt). Die Kultur wurde über Nacht bei 30 °C und 180 rpm geschüttelt.
Anschließend wurde die bewachsene Flüssigkultur mit frischem LB-Medium (enthielt
die entsprechenden Antibiotika) in einem Verhältnis von 1:5 verdünnt und für weitere
3 h bei 30 °C und 180 rpm inkubiert. Die Zellsuspension wurde zentrifugiert (5 min,
4000 g, RT) und die pelletierten Zellen zweimal mit etwa 5 mL TSB-Medium
gewaschen, bevor sie in etwa 125 µL TSB-Medium resuspendiert wurden.
5.5.10.2. Vorbereitung des S. erythraea Rezipientenstammes
Es wurde die Sorbitolstammlösung von S. erythraea∆AT6hygR verwendet. Pro
Konjugationsansatz wurden 2 mL dieser Myceliumlösung eingesetzt. Die Lösungen
wurden bei RT aufgetaut und zentrifugiert (5 min, 4000 g, RT). Nach zweimaligem
Waschen der Zellpellets mit TSB-Medium wurden die Zellen in TSB-Medium
resuspendiert. Dabei wurden je Konjugationsansatz 200 µL Myceliumlösung
verwendet.
5.5.10.3. Konjugation und Selektion
Es wurden je 125 µL E. coli Kultur und 200 µL Myceliumlösung für einen
Konjugationsansatz gemischt (gründliches Auf- und Abpipettieren). Die Suspension
wurde tropfenweise auf sehr trockene SY-Agarplatten (komplettiert mit Thiamin,
FeSO4 und MgCl2) pipettiert. Nach einer kurzen Antrocknungszeit (etwa 5 min) im
laminaren Luftstrom der Sterilbank wurde die Zellsuspension gründlich mit einem
Einwegspatel auf der Agarplatte verteilt. Im Anschluss wurden die Platten für weitere
15-20 min getrocknet. Als Negativkontrolle wurden auf einer Platte einzig 200 µL
Mycelium ausplattiert und in gleicher Weise wie die Konjugationsansätze behandelt.
Die Platten wurden bei 30 °C für etwa 16-18 h inkubiert, bevor sie mit Soft-Agar
bestehend aus TSB-Medium und SY-Agar (4:1), sowie mit den Antibiotika Apramycin

Material und Methoden | 90
und Phosphomycin (zur Abtötung der E. coli-Zellen) komplementiert, überschichtet
wurden. Nach kurzer Antrocknungszeit wurden die Platten bei 30 °C weiter inkubiert,
bis kleine Kolonien sichtbar wurden (etwa 5 Tage). Die Kolonien wurden mit einem
Zahnstocher auf ABB13-Platten (enthielt Phosphomycin und Apramycin) übertragen.
Nach einer weiteren Inkubationsphase bei 30 °C für etwa 5 Tage wurden die Platten
bei Raumtemperatur gelagert.
5.5.11. Testung auf antimikrobielle Aktivität
Zur Bestimmung der antimikrobiellen Aktivität wurde der Hemmhof-Test mit dem
Gram-positiven Bakterium Bacillus subtilis (DSM10) durchgeführt. Hierzu wurden
200 µL einer B. subtilis Sporensuspension zu 200 mL abgekühltem, flüssigen
LB-Agar pipettiert und in Petrischalen gegossen. Mit Hilfe einer umgekehrten 50 µL
Pipettenspitze wurden Löcher im Agar erzeugt. In diese Löcher wurden 50 µL
Fermentationssuspension pipettiert und über Nacht bei 37 °C inkubiert. Die vom
Wachstum ausgesparten Hemmhöfe erlauben eine Beurteilung der antimikrobiellen
Aktivität der zu testenden Substanzen.
5.5.12. Sequenzierung
Um die korrekte Amplifikation der gewünschten Mutationen zu bestätigen, wurden
nach der Plasmidpräparation Sequenzierungen bei StarSEQ GmbH in Auftrag
gegeben. Dazu wurden je 4 µL Plasmid-DNA, 2 µL Betain (5 M) und 1 µL des
entsprechenden Oligos (10 µM) zusammen pipettiert und eingeschickt. Die
verwendeten Oligos für die jeweiligen Plasmide sind in Tabelle 31 aufgeführt.
Tabelle 31: Verwendete Oligonukleotide für die Sequenzierung der entsprechenden Vektoren.
Vektor Oligo 235 Oligo 295 Oligo 297 und 299
pKSSU660-676 451_Sonde_AT6_fwd bzw.
932_Seq_Met235_DEBS3_fwd
665_Asn336_seq -
pKSSU687-709 - - 665_Asn336_seq
Die erhaltenen Ergebnisse wurden über Alignments mit T-Coffee – Multiple
Sequence Alignment ausgewertet.
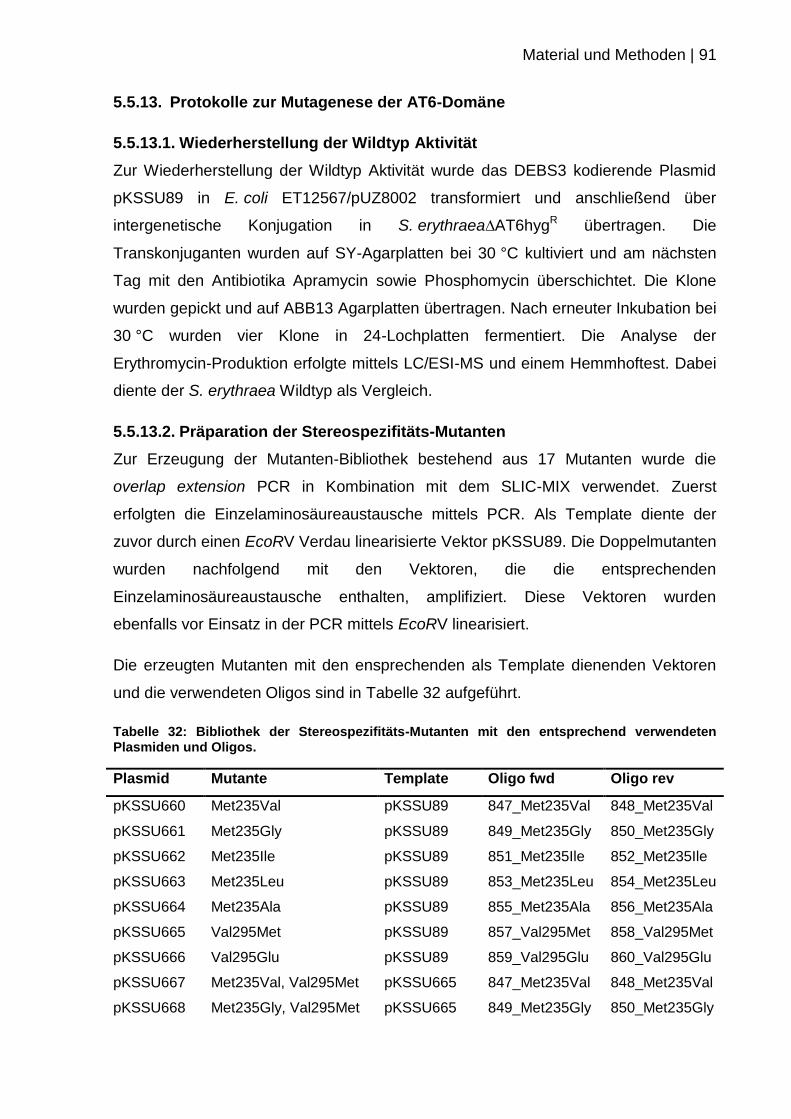
Material und Methoden | 91
5.5.13. Protokolle zur Mutagenese der AT6-Domäne
5.5.13.1. Wiederherstellung der Wildtyp Aktivität
Zur Wiederherstellung der Wildtyp Aktivität wurde das DEBS3 kodierende Plasmid
pKSSU89 in E. coli ET12567/pUZ8002 transformiert und anschließend über
intergenetische Konjugation in S. erythraea∆AT6hygR übertragen. Die
Transkonjuganten wurden auf SY-Agarplatten bei 30 °C kultiviert und am nächsten
Tag mit den Antibiotika Apramycin sowie Phosphomycin überschichtet. Die Klone
wurden gepickt und auf ABB13 Agarplatten übertragen. Nach erneuter Inkubation bei
30 °C wurden vier Klone in 24-Lochplatten fermentiert. Die Analyse der
Erythromycin-Produktion erfolgte mittels LC/ESI-MS und einem Hemmhoftest. Dabei
diente der S. erythraea Wildtyp als Vergleich.
5.5.13.2. Präparation der Stereospezifitäts-Mutanten
Zur Erzeugung der Mutanten-Bibliothek bestehend aus 17 Mutanten wurde die
overlap extension PCR in Kombination mit dem SLIC-MIX verwendet. Zuerst
erfolgten die Einzelaminosäureaustausche mittels PCR. Als Template diente der
zuvor durch einen EcoRV Verdau linearisierte Vektor pKSSU89. Die Doppelmutanten
wurden nachfolgend mit den Vektoren, die die entsprechenden
Einzelaminosäureaustausche enthalten, amplifiziert. Diese Vektoren wurden
ebenfalls vor Einsatz in der PCR mittels EcoRV linearisiert.
Die erzeugten Mutanten mit den ensprechenden als Template dienenden Vektoren
und die verwendeten Oligos sind in Tabelle 32 aufgeführt.
Tabelle 32: Bibliothek der Stereospezifitäts-Mutanten mit den entsprechend verwendeten Plasmiden und Oligos.
Plasmid Mutante Template Oligo fwd Oligo rev
pKSSU660 Met235Val pKSSU89 847_Met235Val 848_Met235Val
pKSSU661 Met235Gly pKSSU89 849_Met235Gly 850_Met235Gly
pKSSU662 Met235Ile pKSSU89 851_Met235Ile 852_Met235Ile
pKSSU663 Met235Leu pKSSU89 853_Met235Leu 854_Met235Leu
pKSSU664 Met235Ala pKSSU89 855_Met235Ala 856_Met235Ala
pKSSU665 Val295Met pKSSU89 857_Val295Met 858_Val295Met
pKSSU666 Val295Glu pKSSU89 859_Val295Glu 860_Val295Glu
pKSSU667 Met235Val, Val295Met pKSSU665 847_Met235Val 848_Met235Val
pKSSU668 Met235Gly, Val295Met pKSSU665 849_Met235Gly 850_Met235Gly

Material und Methoden | 92
Plasmid Mutante Template Oligo fwd Oligo rev
pKSSU669 Met235Ile, Val295Met pKSSU665 851_Met235Ile 852_Met235Ile
pKSSU670 Met235Leu, Val295Met pKSSU665 853_Met235Leu 854_Met235Leu
pKSSU671 Met235Ala, Val295Met pKSSU665 855_Met235Ala 856_Met235Ala
pKSSU672 Met235Val, Val295Gln pKSSU660 877_Val295Gln 878_Val295Gln
pKSSU673 Met235Gly, Val295Gln pKSSU661 877_Val295Gln 878_Val295Gln
pKSSU674 Met235Ile, Val295Gln pKSSU662 877_Val295Gln 878_Val295Gln
pKSSU675 Met235Leu, Val295Gln pKSSU663 877_Val295Gln 878_Val295Gln
pKSSU676 Met235Ala, Val295Gln pKSSU664 877_Val295Gln 878_Val295Gln
Die Ansätze der Einzelaminosäureaustausche (pKSSU660-666) erfolgten
notwendigerweise zuerst. Nach der ersten PCR wurden die Produkte einem
DpnI-Verdau unterzogen, wodurch die Template-DNA zerstört werden sollte. Die
Produkte wurden über eine Gelelektrophorese analysiert und im Anschluss erfolgte
die Gelaufreinigung der Fragmente. Die zweite PCR, in der jeweils die zueinander
gehörigen Fragmente eingesetzt wurden, resultierte in je einer Ziel-DNA-Sequenz
(AT6*). Diese PCR-Produkte wurden mittels Gelelektrophorese überprüft und erneut
gelaufgereinigt.
Für die Doppelaminosäureaustausche (pKSSU667-676) wurden die klonierten
Vektoren der Einzelaminosäureaustausche als Template verwendet (vgl. Tabelle 32).
Das Protokoll der overlap extension PCR erfolgte analog zu dem der
Einzelmutanten, mit Ausnahme des DpnI Verdaus nach der ersten PCR. Die
DNA-Fragmente wurden hier direkt gelaufgereinigt und für die zweite PCR
verwendet.
Von allen Ziel-DNA Sequenzen wurde die Konzentration mittels
Agarose-Gelelektrophorese anhand des Größenstandards berechnet.
Die gereinigte AT6* Ziel-DNA mit den zum Vektor komplementären Enden wurde
über die SLIC-MIX-Methode in den Vektor pKSSU96 integriert. Dieser wurde dafür
mit den Restriktionsenzymen ScaI und FseI geschnitten. Es folgte eine
Ethanolfällung und Bestimmung der Konzentration der Vektor-DNA. Die linearisierte
DNA und die AT6*-DNA-Sequenzen wurden in äquimolaren Mengen im SLIC-MIX
eingesetzt. Da der Vektor mit ca. 1.6 kb etwa 10x größer ist als das zu inserierende
Insert mit 1.7 kb wurde 500 ng Vektor und 50 ng Insert – sprich äquimolare Mengen

Material und Methoden | 93
– verwendet. Nach der Ligation von Vektor und Insert durch die Rekombinase A im
dritten Schritt des SLIC-MIX wurden je 4 µL der neuen Vektor-DNA in kompetente
E coli OmniMAX™ 2 T1 transformiert. Die Zellen wurden auf LB-Platten mit
Apramycin ausplattiert und über Nacht bei 37 °C inkubiert.
Am darauffolgenden Tag wurden die Klone mit einer Kolonie-PCR überprüft. Dafür
wurden in der Regel 3-8 Klone jeder Platte zur Hälfte abgestrichen. Hierfür wurden
die Oligos mit den Nummern 206 und 207 verwendet. Die erfolgreiche Klonierung
wurde mittels einer Agarose-Gelelektrophorese überprüft. Wurde das Insert
amplifiziert, konnte eine Bande bei etwa 1.7 kb beobachtet werden. Mit den positiven
Klonen wurden Übernachtkulturen in LB-Medium (komplettiert mit Apramycin)
angeimpft.
5.5.13.3. Präparation der Substratspezifitäts-Mutanten
Die Mutagenese zur Erzeugung der Substratspezifitäts-Mutanten erfolgte auch hier
mittels zielgerichteter PCR. Alle Mutanten wurden mit dem Plasmid pKSSU89 als
Template erzeugt. Der Vektor wurde vor dem Einsatz in der PCR durch das Enzym
EcoRV linearisiert. Die verwendeten Oligos enthielten – falls nötig – zwei Mutationen.
Somit konnten alle Mutanten parallel präpariert werden. Die für die jeweilige Mutante
verwendeten Oligos sind in Tabelle 33 aufgeführt.
Tabelle 33: Bibliothek der Substratspezifitäts-Mutanten mit den entsprechend verwendeten Plasmiden und Oligos.
Plasmid Mutante Template Oligo fwd Oligo rev
pKSSU687 Tyr297Val pKSSU89 956_Val297 1002_Val297
pKSSU688 Tyr297Val,
Ser299Ala
pKSSU89 957_Val297_Ala299 1003_Val297_Ala299
pKSSU689 Tyr297Val,
Ser299Gly
pKSSU89 958_Val297_Gly299 1004_Val297_Gly299
pKSSU690 Tyr297Val,
Ser299Leu
pKSSU89 959_Val297_Leu299 1005_Val297_Leu299
pKSSU691 Tyr297His pKSSU89 960_His297 1006_His297
pKSSU692 Tyr297His,
Ser299Ala
pKSSU89 961_His297_Ala299 1007_His297_Ala299
pKSSU693 Tyr297His,
Ser299Gly
pKSSU89 962_His297_Gly299 1008_His297_Gly299
pKSSU694 Tyr297His,
Ser299Leu
pKSSU89 963_His297_Leu299 1009_His297_Leu299

Material und Methoden | 94
pKSSU695 Tyr297Ala pKSSU89 964Ala297 1010_Ala297
pKSSU696 Tyr297Ala,
Ser299Ala
pKSSU89 965_Ala297_Ala299 1011_Ala297_Ala299
pKSSU697 Tyr297Ala,
Ser299Gly
pKSSU89 966_Ala297_Gly299 1012_Ala297_Gly299
pKSSU698 Tyr297Ala,
Ser299Leu
pKSSU89 967_Ala297_Leu299 1013_Ala297_Leu299
pKSSU699 Tyr297Gly pKSSU89 968_Gly297 1014_Gly297
pKSSU700 Tyr297Gly,
Ser299Ala
pKSSU89 969_Gly297_Ala299 1015_Gly297_Ala299
pKSSU701 Tyr297Gly,
Ser299Leu
pKSSU89 970_Gly297_Gly299 1016_Gly297_Gly299
pKSSU702 Tyr297Gly,
Ser299Leu
pKSSU89 971_Gly297_Leu299 1017_Gly297_Leu299
pKSSU703 Tyr297Leu pKSSU89 972_Leu297 1018_Leu297
pKSSU704 Tyr297Leu,
Ser299Ala
pKSSU89 973_Leu297_Ala299 1019_Leu297_Ala299
pKSSU705 Tyr297Leu,
Ser299Gly
pKSSU89 974_Leu297_Gly299 1020_Leu297_Gly299
pKSSU706 Tyr297Leu,
Ser299Leu
pKSSU89 975_Leu297_Leu299 1021_Leu297_Leu299
pKSSU707 Ser299Ala pKSSU89 976_Ala299 1022_Ala299
pKSSU708 Ser299Gly pKSSU89 977_Gly299 1023_Gly299
pKSSU709 Ser299Leu pKSSU89 978_Leu299 1024_Leu299
In der ersten PCR wurden zwei Fragmente erzeugt, die einen überlappenden
Bereich in der Mitte enthalten. Nach einer Agarose-Gelelektrophorese zur Kontrolle
der PCR und Gelaufreinigung der Fragmente, wurden diese in einer zweiten PCR
zusammengefügt. Die Ziel-DNA AT6* wurde auch mittels Gelelektrophorese
überprüft und gelaufgereinigt. Die Konzentration der DNA wurde anhand eines
Größenstandards durch eine Agarose-Gelelektrophorese bestimmt.
Die Klonierung erfolgte – analog zur Präparation der Stereospezifitätsvarianten – in
den Vektor pKSSU96. Auch hier wurde dafür die SLIC-MIX Methode verwendet. Der
Vektor wurde zuvor durch einen ScaI und FseI Verdau geschnitten. Im ersten Schritt
der Klonierung wurden Vektor und Insert in äquimolaren Mengen eingesetzt und

Material und Methoden | 95
parallel, jedoch getrennt voneinander mit der T4-Polymerase behandelt. Nach
Stoppen dieser Reaktion durch Zugabe von dCTP wurden Vektor und Insert im
folgenden Schritt durch Einsatz des Enzyms Recombinase A ligiert.
Nach der Klonierung erfolgte die Transformation der Plasmide pKSSU89* in
kompetente E coli OmniMAX™ 2 T1 Zellen. Die transformierten Zellen wurden auf
LB-Platten ausgestrichen und über Nacht bei 37 °C inkubiert.
Am darauffolgenden Tag wurden 1-4 Klone zur Hälfte abgestrichen und in eine
Kolonie-PCR (Oligos 206 & 207) eingesetzt. Klone, bei denen durch die PCR das
Insert nachgewiesen werden konnte, wurden in LB-Medium mit entsprechendem
Antibiotikum kultiviert.
Nach Inkubation über Nacht wurden die Kulturen zentrifugiert und die Plasmide über
eine Plasmidpräparation isoliert. Durch einen Kontrollverdau mit NotI wurde das
Vorhandensein des gewünschten Plasmides pKSSU89* kontrolliert. Die positiv
getesteten Plasmide wurden bei -20 °C gelagert.
5.5.13.4. Protokolle zur Konjugation der erzeugten DEBS3AT6* Expressions-
plasmide in S. erythraea∆AT6hygRAT6*; Stereospezifitätsvarianten
Die Konjugation in S. erythraea∆AT6hygR erfolgte über einen intergenetischen
Plasmidtransfer mit E. coli ET12567/pUZ8002 als Donorstamm.
Zunächst wurde die Vektor-DNA (pKSSU660-672) aus den Übernachtkulturen
(E. coli OmniMAX™ 2 T1) über eine Miniplasmidpräparation isoliert. Es wurden 4 µL
der isolierten DNA für einen Kontrollverdau mit NotI verwendet, um die erfolgreiche
Klonierung erneut zu überprüfen. Die Plasmide einer der positiv getesteten Klone
wurden und in E. coli ET12567/pUZ8002 Zellen transformiert. Die transformierten
Zellen wurden auf Apramycin beinhaltenden LB-Agarplatten ausgestrichen und über
Nacht bei 37 °C inkubiert. Am darauffolgenden Tag wurde ein Klon gepickt und in
einer Übernachtkultur (LB-Medium mit entsprechendem Antibotikum) kultiviert. Die
Übernachtkultur wurde am nächsten Tag 1:5 verdünnt und nochmal für 5 h kultiviert.
Nach der Wachstumsphase wurden die E. coli ET12567 Zellen, die das gewünschte
Plasmid beinhalten, zentrifugiert und zusammen mit vorbereiteten
S. erythraea∆AT6hygR Zellen auf SY-Agarplatten konjugiert. Als Negativkontrolle
diente eine Agarplatte auf denen nur S. erythraea Zellen ausplattiert wurden. Nach
Inkubation über Nacht wurden die Platten mit TSB:SY-Agar (4:1) überschichtet und

Material und Methoden | 96
für etwa 5 Tage weiter inkubiert bis Kolonien sichtbar wurden. Wenn die
Negativkontrolle ohne Bewuchs war, wurden die Kolonien auf ABB13-Agarplatten
übertragen und für weitere 5 Tage inkubiert.
5.5.13.5. Screening der Stereospezifitätsmutanten
Die erzeugten S. erythraeaAT6* Varianten wurden einem Screening unterzogen.
Dazu wurden je vier Klone einer Mutante von der jeweiligen ABB13-Agarplatte
ausgewählt und in 24-Lochplatten des System Duetz fermentiert. Die Vorkulturen
wurden mit je einem Agarstück angeimpft (etwa 0.5 cm2). Die Hauptkulturen erfolgten
anschließend parallel mit und ohne Zugabe des Substrates (13). Der Wildtyp wurde
als Kontrolle ebenfalls mit und ohne Zugabe des synthetischen Bausteins 13
fermentiert. Im Anschluss an die Fermentation wurden die Kulturen mit Ethylacetat
extrahiert und mittels LC/ESI-MS analysiert.
Anhand der Daten des Screenings in Lochplatten wurden Mutanten bestimmt, die für
ein Screening im 1 L-Maßstab sinnvoll erschienen. Es wurden die Zellen mit den
Vektoren pKSSU667, pKSSU665 und pKSSU660 im 1 L-Maßstab mit Fütterung des
Substrates 13 fermentiert. Als Vergleichswert wurde die Mutante pKSSU667
ebenfalls ohne zusätzliche Substratzugabe fermentiert. Als Kontrolle der
Fermentationen diente die Kultivierung des Wildtyps ohne Fütterung.
Die Extraktion des Erythromycins erfolgte im Anschluss an die Fermentation nach
dem in 5.5.1.6 aufgeführten Protokoll. Die weitere Aufreinigung wurde mittels
präparativer DC durchgeführt (vgl. 5.6.2.3). Die gewonnene Probe wurde mittels
LC/ESI-MS und 1H-NMR analysiert.
5.5.14. Fütterungsexperimente mit Malonaten
Die Kultivierung der S. erythraea Kulturen entspricht dem Protokoll aus Kapitel
5.5.1.4. Die Fütterung wurde mit käuflich erworbenem Diethylpropargylmalonat (16)
und Diethylethylmalonat (15) durchgeführt. Es wurde die Mutante S. erythraea
∆AT6_pKSSU656 (Val295Ala) und als Referenz der S. erythraea Wildtyp
(NRRL-B-24071) untersucht. Die Kulturen wurden in 50 mL Kolben mit Spirale
angesetzt. Es wurden Kulturen vom Wildtyp und von je vier Klonen der Mutante
angesetzt. In der Vorkultur der Mutante wurde das TSB-Medium mit dem
Antibiotikum Apramycin (Resistenz vermittelt über das Plasmid) versetzt. Die
Fütterung mit den Malonaten erfolgte in drei Endkonzentrationen (10, 15 und

Material und Methoden | 97
20 mM). Zur Kontrolle wurde jeder Klon und der Wildtyp ohne Substratzugabe
kultiviert. Nach der Fermentation wurden die Lösungen in 15 mL Gefäße überführt
und bei -20 °C über Nacht eingefroren. Die Extraktion erfolgte nach dem in Kapitel
5.5.1.5 aufgeführten Protokoll. Die Proben wurden mittels LC/ESI-MS und MS/MS
analysiert.

Material und Methoden | 98
5.6. Allgemeine chemische Methoden
5.6.1. Geräte und Chemikalien
5.6.1.1. Geräte
Für Reaktionen unter Schutzgasatmosphäre wurden nach den üblichen Verfahren
(Schlenk)-Kolben verwendet, die vor Benutzung im Trockenschrank bei 110 °C
ausgeheizt und unter Hochvakuum getrocknet wurden. Als Schutzgas wurde hier
Argongas verwendet.
5.6.1.2. Chemikalien
Die verwendeten Chemikalien stammten entweder aus Schenkungen oder wurden
bei den Firmen Sigma Aldrich, Alfa Aesar und VWR käuflich erworben. Die
Lösungsmittel wurden i.A. in trockener Form käuflich erworben. Wasserfreies DCM
wurde mit CaH2 unter Argonatmosphäre frisch destilliert.
5.6.2. Chromatographische Verfahren
5.6.2.1. Säulenchromatographie
Die Säulenchromatographie erfolgte über Kieselgel der Firma Acros mit einer
Partikelgröße von 35-70 µm. Die Beladung der Säule erfolgte stets mit einer
Suspension aus Kieselgel und dem verwendeten Lösungsmittelgemisch. Das
verwendete Lösungsmittelgemisch ist bei den entsprechenden Versuchsvorschriften
angegeben.
5.6.2.2. Dünnschichtchromatographie
Für die Dünnschichtchromatographie wurden ausschließlich Aluminium-Fertigfolien
Silicagel 60 mit Fluoreszenzindikator F-254 der Firma Merck eingesetzt. Die
Detektion erfolgte über UV-Absorbtion oder durch Entwicklung mit einem Heizluftfön
nach Behandlung mit einer Permanganatlösung (2 mL 5 % NaOH, 6.6 g K2CO3, 1 g
KmnO4, 100 mL H2O).
5.6.2.3. Präparative Dünnschichtchromatographie
Die präparative Dünnschichtchromatographie wurde auf 20x20 cm PLC Platten, mit
einer 2 mm Kieselgel 60 Schicht, einer Konzentrierungszone (20x4 cm) und einem
Fluoreszenzindikator F-254 von Merck durchgeführt. Das verwendete Protokoll ist
einer literaturbekannten Vorschrift[59] angelehnt.

Material und Methoden | 99
Die extrahierten Feststoffe wurden in dem kleinstmöglichen Volumen (etwa 100 µL
für ca. 50 mg) Chloroform:MeOH 2:1 gelöst. Es wurde eine möglichst dünne Linie
innerhalb der Konzentrierungszone aufgetragen. Zusätzlich wurde als Referenz
käuflich erworbenes Erythromycin aufgetragen. Die Platte wurde getrocknet, damit
das aufgetragene Lösungsmittel vollständig verdampft war bevor die DC-Platte in die
DC-Kammer gestellt wurde. Das verwendete Laufmittel war ein Gemisch aus
Chloroform, Methanol und Ammoniaklösung in einem Volumenverhältnis von
(90:10:1). Zur Detektion des nicht UV-aktiven Erythromycins wurde ein 1-2 cm breiter
Streifen am Rand der DC-Platte mit einer Permanganatlösung (vgl. 5.6.2.2)
angefärbt und mittels Heizluftfön (max. 50 °C) entwickelt. Auf der Höhe des/der
sichtbaren Flächen wurde das Kieselgel mit Hilfe eines Spatels von der Platte
gekratzt und in ein 15 bzw. 50 mL Röhrchen überführt. Die abgekratzten
Kieselgelproben wurden mit dem zweifachen Volumen eines Chloroform:Methanol-
Gemisches (2:1) versetzt und über Nacht bei 19 °C und 150 rpm geschüttelt.
Anschließend wurde die Suspension filtriert und das Lösungsmittel am
Rotationsverdampfer eingeengt. Zur Analyse wurde eine Probe entnommen.
5.6.3. Analytische Verfahren
5.6.3.1. NMR-Spektroskopie
Alle Messungen wurden in deuterierten Lösungsmittel aufgenommen. Als interner
Standard dienten die Restprotonen der deuterierten Lösungsmittel CDCl3-d1
( H = 7.26 ppm, singulett, C = 77.0 ppm, triplett), MeOD-d4 ( H = 3.31 ppm, quintett,
H = 4.84 ppm, singulett, C = 49.05 ppm, septett), D2O-d2 ( H = 4.79 ppm, singulett).
Die Messungen der 1H-NMR-Spektren und der 13C-NMR-Spektren erfolgten auf
einem Varian Mercury 400 Spektrometer (Messfrequenz 400 MHz bzw. 101 MHz)
oder an einem Bruker 600 (600 MHz). Die 1D und 2D-NOESY-Spektren wurden an
einem Bruker 600 (600 MHz) Spektrometer aufgenommen.
Chemische Verschiebungen wurden in ppm und die Größe der
Kopplungskonstanten J in Hz angegeben. Die Zuordnung der Signale erfolgte falls
nötig anhand der Kopplungskonstanten, DEPT-, COSY-, HSQC-, TOCSY-, HMBC-
Spektren. Für die Beschreibung der Multiplizität der Signale wurden die gängigen
Abkürzungen verwendet: s (Singulett), d (Duplett), t (Triplett), q (Quartett), quin
(Quintett), m (Multiplett), mc (zentriertes Mutiplett).

Material und Methoden | 100
5.6.3.2. Massenspektrometrie
5.6.3.2.1. GC-MS
Die aufgeführten Massen wurden mittels GC-MS bestimmt. Es wurde ein Hewlett
Packard 6890 Serie GC-System mit einem Hewlett Packard 5973 Mass Selective
Detector eingesetzt. Mit einer HP19091M-102 HP-5 (325°C max.) Trace Analysis
Säule. Für die Analyse wurden 2 Methoden verwendet:
Tabelle 34: Systemeinstellungen der Methoden bei der Massenspektrometrie.
# Methode Druck Flussrate
1 DB_50_S 2.100 bar 105 mL/min
2 DB_100_S 2.499 bar 105 mL/min
Methode 1 beginnt mit 50 °C für 2 min unter anschließendem Erhitzen mit einem
Temperaturgradienten von 40°C/min für 5.75 min auf 300 °C und einer weiteren
Verweilzeit von 14 min bei 300°C.
Methode 2 beginnt mit 100 °C für 1 min unter anschließendem Erhitzen mit einem
Temperaturgradienten von 40°C/min für 5 min auf 300 °C und einer weiteren
Verweilzeit von 11 min bei 300 °C.
5.6.3.2.2. LC/ESI-MS zur allgemeinen Analyse der Masse
Für die allgemeine Analyse der Massen wurde eine LC/ESI-MS Messung an einem
Hewlett Packard Series 1100/Finnigan LCQ Advantage MAX System mit einer
CC125/4 Nucleodur C18 Gravity (Macherey & Nagel) 3 μm Chromatographiesäule
eingesetzt. Die Detektion erfolgte bei 280 nm, die Flussrate Betrug 1.0 ml/min.
Eluent A: H2O + 0.1 % HCOOH; Eluent B: Acetonitril + 0.1 % HCOOH.
Verwendeter Gradient: 0-1 min: 90 % A/10 % B
1-10 min: 0 % A/100 % B
10-12 min: 0 % A/100 % B
12-12.1 min: 90 % A/10 % B
12.1-15 min: 90 % A/10 % B

Material und Methoden | 101
5.6.3.2.3. LC/ESI-MS zur Analyse von Fermentationsprodukten
Die Untersuchung von Fermenationsextrakten wurde an einer HPLC mit
anschließendem Massenspektrometer durchgeführt. Dabei diente das Accela
HPLC-System (bestehend aus Pumpe, Autosampler, Säulenofen, PDA Detektor und
UV-Detektion bei 210 nm und 254 nm) der Separation der Extraktionsprodukte.
Durch die Kopplung der HPLC mit einem Orbitrap Massenspektrometer (ausgestattet
mit einer LTQ XL linear ion trap; Thermo Electron Corporation Deutschland) konnten
die Massen der einzelnen Bestandteile gemessen werden. Es wurden nur
Lösungsmittel mit HPLC-Grad von Chromasolv, Deutschland eingesetzt. Bis zu 15 µL
der jeweiligen Probe wurden mittels Autosamlpler (T= 10 °C) bei einer Flussrate von
500 µL/min auf eine CC12514 Nucleodur C18 Gravity Säule (3 µm, Macherey-Nagel
Deutschland) aufgetragen.
Der eingesetzte lineare Gradient kann dem folgenden Schema entnommen werden
(Eluent A: doppeltdeionisiertes Wasser mit 0.1 % Ameisensäure; Eluent B: Acetonitril
mit 0.1 % Ameisensäure):
Start 80 % Eluent A/20 % Eluent B
Innerhalb von 10 min zu 0 % Eluent A/100 % Eluent B
Waschen für 5 min 0 % Eluent A/100 % Eluent B
Reäquilibrierung innerhalb von 5 min 80 % Eluent A/20 % Eluent B
Die anschließende Analyse der Masse erfolgte über EI im positiven
Ionisationsmodus. Hier wurde eine Spannung von 4 kV verwendet. Die Kappilare
wurde auf eine Spannung von 18 V bei einer Temperatur von 275 °C eingestellt. Die
aufgenommenen Spektren erfolgten bei einem Massen-zu-Ladung Verhältnis (m/z)
von 200 bis 2000.
5.6.3.2.4. MS/MS
In einigen Fällen wurde zusätzlich eine Fragmentierung der Moleküle analysiert.
Nach einem Scan von 200-2000 erfolgte die Fragmentierung bei 35 % über die
collisoin induced dissociation (CID).

Material und Methoden | 102
5.6.3.2.5. Hochauflösende Massen Spektrometrie (HRMS)
Die hochaufgelösten Massenspektren wurden mit LTQ Orbtitrap gekoppelt mit einem
Accela HPLC-System (Säule Hypersil Gold, Länge 50 mm, Innendurchmesser 1 mm,
Partikelgröße 1.9 µm, Ionisierungsmethode: Elektrospray Ionisation) aufgenommen.
Dabei wurden Wellenlängen im Bereich von 200-600 nm gemessen und Massen im
m/z-Bereich von 150-2000 detektiert.
5.7. Synthese des Substrates rac-Methylmalonyl-SNAC (13)
5.7.1. N-(2-mercaptoethyl)acetamid (SNAC) (14)
Nach einer Synthese von Hughes and Keatinge-Clay [63] wurden 11.4 g
Cysteaminhydrochlorid (17) (100 mmol) in 500 mL H2O gelöst. Nacheinander wurden
25.2 g (300 mmol, 3 Äq.) Natriumhydrogencarbonat und 5.6 g (100 mmol, 1 Äq.)
KOH zugegeben. Nachdem sich alles gelöst hat wurden 10 mL (100 mmol, 1 Äq.)
Essigsäureanhydrid zugetropft. Der Reaktionsansatz wird 2 h bei Raumtemperatur
gerührt bevor er mit HCl auf pH 5-6 angesäuert wurde. Es wurde mit EtOAc
extrahiert, über NaSO4 getrocknet und das Lösungsmittel am Rotationsverdampfer
entfernt. Es konnten 7.56 g (63.4 mmol, 64 %) des Rohprodukts 14 als gelbliches Öl
isoliert werden, die ohne weitere Aufreinigung eingesetzt wurden.
Rf-Wert: 0.42 (DCM:MeOH 9:1).
HRMS (GC-MS): 120.04730 [M+H+], berechnet 120.04776 [M+H+].
GC-MS: tR = 5,329 min; DB_50_S; m/z 119 [M+], m/z 102 [M-C2H5+H+].
1H-NMR: (400 MHz, CDCl3-d1) = 2.00 (s, 3 H, 4-H), 2.66 (m, 2 H, 1-H), 3.41 (m, 2
H, 2-H), 6.05 (s, 1 H, SH).
13C-NMR: (101 MHz, CDCl3-d1) = 23.3 (C4, CH3), 24.7 (C1, CH2), 42.6 (C2, CH2),
170.39 (C3, C=O).

Material und Methoden | 103
5.7.2. 3-(tert-butoxy)-2-methyl-3-oxopropansäure (11)
Unter Argonatmosphäre wurden 300 mL trockenes THF auf -78 °C temperiert und
10 g (76.9 mmol) tert-Butylpropionat (10) darin gelöst. Dazu wurden 85 mL NaHMDS
(1 M in THF) zugetropft und für 1 h gerührt. Der Reaktionsansatz wird langsam auf
Raumtemperatur erwärmt, wobei die Einleitung von CO2-Gas (Leitung über
Molekularsieb zur Trocknung) gestartet wird. Überschüssiges Gas wird über einen
mit Öl gefüllten Blasenzähler ausgeleitet. Der Reaktionsansatz wurde für 18 h bei RT
gerührt. Nach Zugabe von ges. NH4Cl-Lösung wird THF unter reduziertem Druck
entfernt und NaHCO3 wird zur Reaktionsmischung gegeben. Es wird mit EtOAc (2x)
extrahiert. Die wässrigen Phasen werden vereint und mit konzentrierter HCl
angesäuert (pH 1). Nach erneuter Extraktion mit EtOAc (3x) werden die organischen
Phasen vereint, über NaSO4 getrocknet und das Lösungsmittel unter Vakuum
entfernt. Das Produkt wird ohne weitere Aufarbeitung eingesetzt. Es konnten 4.19 g
(24.1 mmol, 32 %) der Malonsäure 11 isoliert werden.
Rf-Wert:0.48 (CHCl3:MeOH 9:1).
HRMS (GC-MS): 175.09634 C8H15O4 [M+H+], 197.07835 C8H15O4Na [M+Na+],
berechnet: 175.09649 C8H15O4 [M+H+], 197.07843 C8H15O4Na [M+Na+].
GC-MS: DB_100_S, m/z: 229 M+H+, m/z:.200., m/z: 155., m/z: 126.
1H-NMR: (400 MHz, CDCl3-d1) = 1.43 (d, 3J4-H,2-H = 7.3 Hz, 3 H, 4-H). 1.47 (s, 9 H,
tBu), 3.38 (q, 3J2-H, 4-H = 7.3 Hz, 1 H, 2-H).
13C-NMR: (101 MHz, CDCl3-d1) = 14.0 (C4, CH3), 28.09 (C6, tBu), 46.86 (C2, CH),
82.71 (C5, Cq-tBu), 169.79 (C1, COOH), 175.54 (C3, COOR).

Material und Methoden | 104
5.7.3. tert-Butyl-3-((2-acetamidethyl)thio-2-methyl-3-oxopropanat (12)
Unter Schutzgasatmosphäre wurden 6.1 g (35.02 mmol) der Malonsäure 11 in
100 mL THF gelöst. Der Reaktionsansatz wurde auf 0 °C gekühlt und mit 6.8 g
(41.94 mmol, 1.2 Äq.) CDI, sowie 1.28 g (10.48 mmol, 0.3 Äq.) DMAP versetzt. Es
wurde für zwei Stunden bei 0 °C gerührt und anschließend 6.24 g (52.35 mmol,
1.5 Äq.) SNAC (14) zugegeben. Nach weiteren 16 h Rühren bei RT wurde THF unter
reduziertem Druck entfernt und das Rohprodukt 12 wurde säulenchromatographisch
aufgereinigt (DCM:MeOH 100:0 bis 99:1). Es konnten 7.09 g (25.75 mmol, 74 %) des
Produktes isoliert werden.
Rf-Wert: 0.6 (DCM:MeOH 9:1).
HRMS (LC/ESI-MS): 276.12654 C12H22O4NS [M+H+], 298.10841 C12H22O4NSNa
[M+Na+], berechnet: 276.12641 C12H22O4NS [M+H+], 298.10835 C12H22O4NSNa
[M+Na+].
LC/ESI-MS: tR = 7.31 min; pos. Mode; m/z : 275.92 [M+H+].
1H-NMR: (400 MHz, CDCl3-d1) = 1.34 (d, 3J4-H,2-H = 7.2 Hz, 3 H, 4-H), 1.39 (s, 9 H,
tBu), 1.90 (s, 3 H, 4´-H), 2.92- 3.08 (m, 2 H, 1´-H), 3.45 – 3.30 (m, 2 H, 2´-H), 3.51 (q,
3J2-H,4-H = 7.2 Hz, 1 H, 2-H).
13C-NMR: (101 MHz, CDCl3-d1) = 14.23 (C4, CH3), 23.3 (C4´, CH3), 28.07 (C6,
tBu), 28.95 (C1´, CH2), 39.68 (C2, CH), 55.52 (C2´, CH2), 82.4 (C5, Cq-tBu), 168.6
(C3, COOR), 170.52 (C3´, C=O), 197.02 (C1, C=O).

Material und Methoden | 105
5.7.4. 3-((2-acetamidethyl)thio)-2-methyl-3-oxopropanoat (13)
Unter Argonatmosphäre wurden 300 mL trockenes DCM auf 0 °C gekühlt. Darin
wurden 3 g (10.9 mmol) der tBu geschützten SNAC-gekoppelten Malonsäure 12
gelöst. Nach Zugabe von 2.7 mL (24.6 mmol, 2.25 Äq.) Titan(IV)chlorid wurde für
0.5 h bei 0 °C und für weitere 4-6 h (DC-Kontrolle) bei Raumtemperatur gerührt. Der
Reaktionsansatz wurde nach vollständigem Umsatz mit 109 mL Substrat-Puffer
(Endkonzentration 100 mM, vgl. 0) gequencht. DCM wurde unter reduziertem Druck
bei RT entfernt. Die wässrige Lösung wurde 10 min bei 4 °C, 4000 g zentrifugiert.
Der Überstand wurde abgenommen und sterilfiltriert. Die Lösung wurde am gleichen
Tag der Fermentation beigefügt.
Rf-Wert: 0.1 (DCM:MeOH 9:1).
5.7.5. Erythromycin A (7)
Erythromycin A (7) wurde aus Fermentationskulturen von S. erythraea über eine
Extraktionskaskade extrahiert (vgl. 5.5.1.8). Nach Aufreinigung durch eine
präparative DC wurde die Probe mittels LC/ESI-MS und 1H-NMR analysiert.
Rf-Wert: 0.66.
LC/ESI-MS: tR = 6.8 min; pos. Mode; m/z : 734.74 [M+H+].

Abkürzungen und Akronyme | 106
6 Abkürzungen und Akronyme
6-DEB 6-Desoxyerythronolid
A
A Adenin, Adenosin
ACP Acyl Carrier Protein
Apr Apramycin
Äq. Äquivalente
AS Aminosäure
AT Acyltransferase
ATP Adenosintriphosphat
B
bp Basenpaar
BLAST Basic Logical Alignment Search Tool
B. subtilis Bacillus subtilis
bzw. beziehungsweise
C
C Cytosin
ca. circa
Cam Chloramphenicol
CDI Carbonyldiimidazol
CoA Coenzym A
COSY correlated spectroscopy
D
D Dalton
DC Dünnschichtchromatographie
DCM Dichlormethan
dCTP Desoxycytosintriphosphat
DEBS 6-Desoxyerythronolid B Synthase
DH Dehydratase

Abkürzungen und Akronyme | 107
ddH2O Doppelt destilliertes Wasser
DMAP 4-N,N-Dimethylaminopyridin
DMSO Dimethylsulfoxid
DNA Desoxyribonukleinsäure
Dnase Desoxyribonuklease
E
E. coli Escherichia coli
EI Elektronenstoß-Ionisation
ER Enoylreduktase
Ery Erythromycin
ESI Elektronenspray-Ionisation
et al. und andere
EtOAc Ethylacetat
ET-SSB Einzelstrangstabilisierendes Protein
F
FAS Fettsäurebiosynthese
fwd Vorwärts (forward)
G
G Guanin
GC Guanin Cytosin
GC-MS Gaschromatographie gekoppelt mit einer Massenspektrometrie
H
h Stunden
HMBC heteronuclear multiple bond correlation
HPLC high performance liquid chromatography
HRMS Hochauflösende Massen Spektrometrie
HSQC heteronuclear single quantum coherence
hyg Hygromycin

Abkürzungen und Akronyme | 108
J
J Kopplungskonstante
K
Kann Kanamycin
kb Kilobasen
KR Ketoreduktase
KS Ketosynthase
L
LB Lysogeny Broth
LC liquid chromatography
LDA Lithiumdiisopropylamid
M
MeOH Methanol
min Minute
MMCoA Methylmalonyl-CoA
MPI Max-Planck-Institut
MS Massenspektrometrie
m/z Masse- zu Ladungsverhätnis
N
n.a. nicht angegeben
NADPH Nicotinsäureamid-Adenin-Dinukleotidphosphat
NaHMDS Natrium-bis(trimethylsilyl)amid
n.b. nicht bestimmt
n.e. nicht eindeutig
NMR nuclear magnetic resonance
NRRL ARS Culture Collection
nt Nukleotide
O
OD Optische Dichte

Abkürzungen und Akronyme | 109
o/n over night
Ori Replikationsursprung
P
PCR Polymerasekettenreaktion
Phm Phosphomycin
PIPES Piperazin-1,4-bis(2-ethansulfonsäure)
PK Polyketid
PKS Polyketid Synthase
ppm parts per million
PPTase Phosphopantetheinyltransferase
R
R Resistenz
rac Racemat
RecA Rekombinase A
rev Rückwärts (revers)
Rf Retentionsfaktor
Rt Retentionszeit
RT Raumtemperatur
S
s Sekunde
S. ery Saccharopolyspora erythraea
S. erythraea Saccharopolyspora erythraea
SLIC Sequence and ligation independent cloning
SNAC S-Nitroso-N-acetylcystein
SOB Super Optimal broth
SOC Super Optimal broth with Catabolite
T
t Zeit
T Thymin, Thymidin
Ta Annealingtemperatur

Abkürzungen und Akronyme | 110
TAE Tris-Acetat-EDTA
tBu tert.-Butyl
TE Terminale Esterase
tert tertiär
THF Tetrahydrofuran
Tm Schmelztemperatur
Tris Tris(hydroxymethyl)-aminomethan
U
UV Ultra-Violettstrahlung
V
v.Chr. vor Christus
vgl. vergleiche
W
w/v Gewicht/Volumen
WT Wildtyp
Darüber hinaus verwendete Abkürzungen sind IUPAC- bzw. SI-konform. Die
Aminosäuren wurden nach dem international üblichen Ein- bzw. Dreibuchstabencode
abgekürzt (IUPAC-IUB Comission of Biochemical Nomenclature, Pure Appl. Chem.
1982, 54, 1517 und 1525).

Referenzen | 111
7 Referenzen
[1] D. J. Newman, G.M. Cragg, J.Nat.Prod. 2012,75, 311-335.
[2] P. M. Dewick, Medicinical Natural Products – A Biosynthetic Approach, 3 rd ed., John Wiley & Sons Ldt, Chichester, 2009.
[3] B. Jessel, O. Tschimpke, M. Walser, HOFFMANN UND CAMPE VERLAG, 2009, Produktivkraft Natur.
[4] A. Fleming, Br J. Exp. Pathol. 1929, 10, 226–236.
[5] M. Saleem, M. Nazir, M. S. Ali, H. Hussain, Y. S. Lee, N. Riaz, A. Jabbar, Nat. Prot. Rep. 2010, 238-254.
[6] J. W. Blunt, B. R. Copp, M. H. G. Munro, P. T. Northcote, M. R. Prinsep, Nat. Prod. Rep. 2010, 165-237.
[7] K. Buchholz, V. Kasche, U. T. Bornscheuer, Biocatalysts and Enzyme Technology, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005.
[8] C. Cuevas, M. Pérez, M. J. Martín, J. L. Chicharro, C. Fernández-Rivas, M. Flores, A. Francesch, P. Gallego, M. Zarzuelo, F. de la Calle, J. García, C. Polanco, I. Rodríguez, I. Manzanares, Org. Lett. 2000, 2, 2545-2548.
[9] U. Sundermann, S. Kushnir, F. Schulz, Nachrichten aus der Chemie, 2011, 59, 297-318.
[10] J. Clardy, C. Walsh, Nature 2004, 432, 829-837.
[11] C. R. Hutchinson, Proc. Natl. Acad. Sci. U S A 1999, 96, 3336-3338.
[12] K. J. Weissman, P. F. Leadlay, Nat. Rev. Microbio.l 2005, 3, 925-936.
[13] C. Khosla, Chem. Rev. 1997, 97, 2577-2590.
[14] S. G. Van Lanen, B. Shen, Curr. Opin. Drug Discov. Devel. 2008, 11, 186-195.
[15] C. Hertweck, Ange. Chem. Int. Edit. 2009, 48, 4688-4716.
[16] J. N. Collie, J. Chem. Soc. Transactions 1893, 63, 329-337.
[17] J. Staunton, K. J. Weissman, Nat. Prod. Rep. 2001, 18, 380-416.
[18] R. Bentley, J. W. Bennett, Annu. Rev. Microbiol. 1999, 53, 411-446.
[19] B. Shen, Curr. Opin. Chem. Biol. 2003, 7, 285-295.
[20] U. Sundermann, S. Kushnir, F. Schulz, Nachr. Chem. 2011, 59, 29-35.
[21] D. A. Hopwood, Chem. Rev. 1997, 97, 2465-2498.
[22] K. Magnuson, S. Jackowski, C. O. Rock, J. E. Cronan, Jr., Microbiol. Rev. 1993, 57, 522-542.
[23] M. A. Fischbach, C. T. Walsh, Chem. Rev. 2006, 106, 3468-3496.

Referenzen | 112
[24] U. Rix, C. Fischer, L. L. Remsing, J. Rohr, Nat. Prod. Rep. 2002, 19, 542-580.
[25] J. Staunton, B. Wilkinson, Chem. Rev. 1997, 97, 2611-2630.
[26] J. Cortes, S. F. Haydock, G. A. Roberts, D. J. Bevitt, P. F. Leadlay, Nature 1990, 348, 176-178.
[27] S. Donadio, M. Staver, J. McAlpine, S. Swanson, L. Katz, Science 1991, 252, 675-679.
[28] S. Donadio, L. Katz, Gene 1992, 111, 51-60.
[29] K. J. Weissman, Vol. 51 (Eds.: W. Wohlleben, T. Spellig, B. Müller-Tiemann), Springer Berlin Heidelberg, 2005, pp. 43-78.
[30] Keatinge-Clay, A.T.; Stroud, R.M., Structure 2006, 14, 737-748.
[31] X. Guo, T. Liu, C. R. Valenzano, Z. Deng, D. E. Cane, J. AM. CHEM. SOC. 2010, 132, 14694–14696.
[32] C. Olano, C. Mendez, J. A. Salas, Nat. Prod. Rep. 2010, 27, 571-616.
[33] C. Khosla, J. Org. Chem. 2009, 74, 6416-6420.
[34] C. Khosla, R. S. Gokhale, J. R. Jacobsen, D. E. Cane, Annu. Rev. Biochem. 1999, 68, 219-253.
[35] L. Katz, G. W. Ashley, Chem. Rev. 2005, 105, 499-528.
[36] D. H. Kwan, F. Schulz, Molecules 2011, 16, 6092-6115
[37] K. J. Weissman, in Methods in Enzymology, Vol. Volume 459 (Ed.: A. H. David), Academic Press, 2009, pp. 3-16.
[38] M. Oliynyk, M. J. B. Brown, J. Cortés, J. Staunton, P. F. Leadlay, Chem. Biol. 1996, 3, 833-839.
[39] J. Lau, H. Fu, D. E. Cane, C. Khosla, Biochem. 1999, 38, 1643-1651.
[40] D. L. Stassi, S. J. Kakavas, K. A. Reynolds, G. Gunawardana, S. Swanson, D. Zeidner, M. Jackson, H. Liu, A. Buko, L. Katz, Proc. Natl. Acad. Sci. U S A 1998, 95, 7305-7309.
[41] R. McDaniel, A. Thamchaipenet, C. Gustafsson, H. Fu, M. Betlach, M. Betlach, G. Ashley, Proc. Natl. Acad. Sci. U S A 1999, 96, 1846-1851.
[42] L. B. Pickens, Y. Tang, Y.-H. Chooi, Annu. Rev. Chem. Biomol. 2011, 2, 211-236.
[43] L J. Staunton, B. Wilkinson, Curr. Opin. Chem. Biol. 2001, 5, 159-164.
[44] C. D. Reeves, S. Murli, G. W. Ashley, M. Piagentini, C. R. Hutchinson, R. McDaniel, Biochemistry 2001, 40, 15464-15470.
[45] S. Kushnir, U. Sundermann, S. Yahiaoui, A. Brockmeyer, P. Janning, F. Schulz, Minimally invasive Mutagenesis gives rise to a Biosynthetic Polyketide Library, Angew Chem Int Ed Engl, in press.
[46] U. Sundermann, Dissertation, TU Dortmund 2012.
[47] D. H. Kwan, F. Schulz, Molecules 2011, 16, 6092-6115.

Referenzen | 113
[48] H. J. Roth, C. E. Müller, G. Folkers, Stereochemie und Arzneistoffe, Wissenschaftliche Verlagsgesellschaft, 1998.
[49] J. Weyer, Angew. Chem, 86, 17, 604-611.
[50] M. T. Reetz, Angew. Chem. 2011, 123, 144-182.
[51] D. H. Kwan, M. Tosin, N. Schläger, F. Schulz, P. F. Leadlay, Org. Biomol. Chem., 2011, 9, 2053-2056.
[52] M. Oliynyk, M. Samborskyy, J. B. Lester, T. Mironenko, N. Scott, S. Dickens, S. F. Haydock, P.F. Leadlay, Nat. Biotechnol. 2007, 25, 447-453.
[53] W. A. Duetz, L. Ruedi, R. Hermann, K. O'Connor, J. Buchs, B. Witholt, Appl. Environ. Microbiol. 2000, 66, 2641-2646.
[54] A. M. El-Brondkly, H. I. Abd-Alla, M. Shaaban, K. A. Shaaban, Indian J. Pharm Science 2008, 70, 310-319.
[55] S. Klopries, Masterarbeit, TU Dortmund 2010.
[56] N. B. Fitzgerald, R. S. English, J. S. Lampel, T. J. Vanden Boom, Appl. Environ. Microbiol. 1998, 64, 1580-1583.
[57] T. Kieser, M. J. Bibb, M. J. Buttner, K. F. Chater, D. A. Hopwood, Practical streptomyces genetics, John Innes Foundation, 2000.
[58] A. Ranganathan, M. Timoney, M. Bycroft, J. Cortés, I. P. Thomas, B. Wilkinson, L. Kellenberger, U. Hanefeld, I. S. Galloway, J. Staunton, P. F. Leadlay, ChemBiol 1999, 6, 731-741.
[59] M. Beran, V. Přikrylová, P. Sedemera, J. Novák, J. Zima, M. Blumauerová, J. Chrom. 1991, 558, 265-272.
[60] M. Z. Li, S. J. Elledge, Nat. Meth. 2007, 4, 251-256.
[61] S. Kushnir, U. Sundermann, F. Schulz, Asian Journal of Biotechnologie, 2012, under revision.
[62] H. Inoue, H. Nojima, H. Okayama, Gene 1990, 96, 23-28.
[63] J. Hughes, A. Keatinge-Clay, Chemistry & Biology 2011, 18, 165-176.

Anhang | 114
8 Anhang
8.1. NMR-Spektren
Abbildung 59: 1H-NMR Spektrum der Verbindung 11.
Abbildung 60: 13
C-NMR Spektrum der Verbindung 11.
11
11

Anhang | 115
Abbildung 61: 1H-NMR Spektrum der Verbindung 12.
Abbildung 62: 13
C-NMR Spektrum der Verbindung 12.
12
12

Anhang | 116
Abbildung 63: 1H-NMR Spektrum der Verbindung 14.
Abbildung 64: 13
C-NMR Spektrum der Verbindung 14.
14
14

Eidesstattliche Erklärung | 117
9 Eidesstattliche Erklärung
Hiermit versichere ich an Eides statt, dass ich die vorliegende Arbeit selbständig und
nur mit den angegebenen Hilfsmitteln angefertigt habe.
Hamm, den 9/20/2012
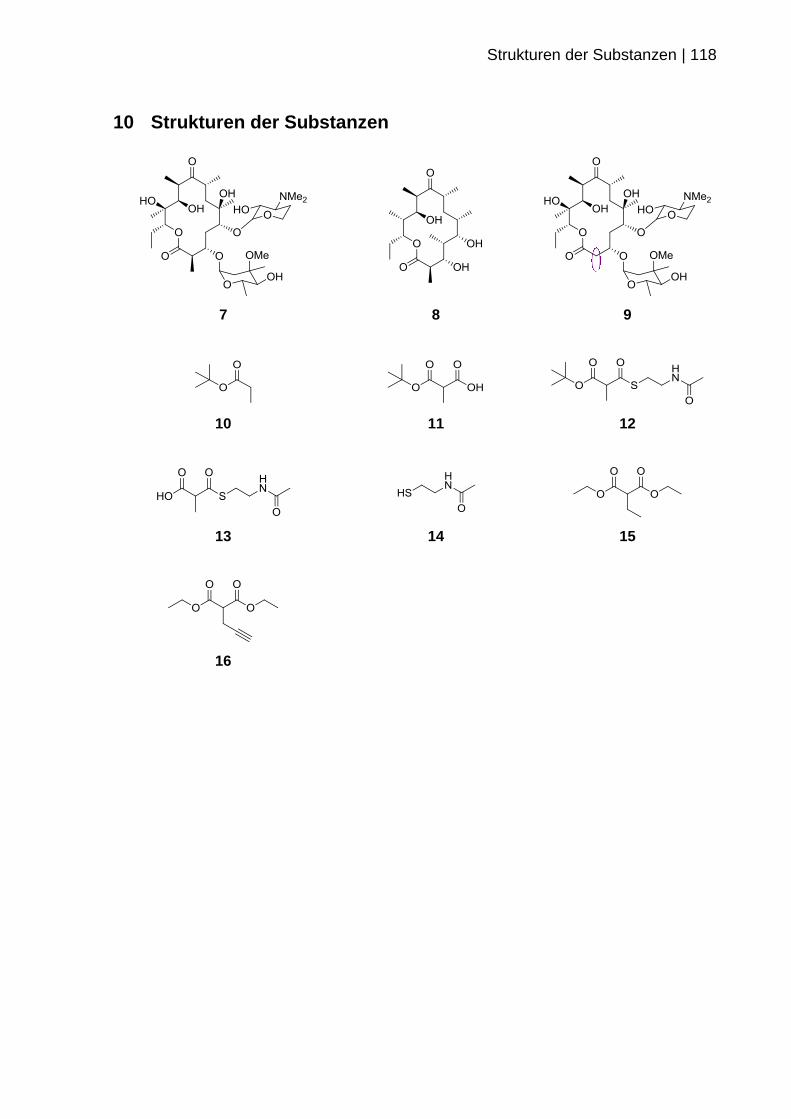
Strukturen der Substanzen | 118
10 Strukturen der Substanzen
7 8 9
10 11 12
13 14 15
16