Embed Size (px)
Citation preview

OSTEOPOROSIS PRIMARIA EN PEDIATRÍA:
OSTEOGÉNESIS IMPERFECTASTEOGÉNESIS IMPERFECTADescripción, clasificación y tratamiento Descripción, clasificación y tratamiento
Cristina Tau●Médico Pediatra
● Especialista en Metabolismo Cálcico y Ó[email protected]

Hereditaria
Disminución de masa ósea
Fragilidad ósea
OSTEOGENESIS IMPERFECTA (OI)
Fragilidad ósea
Fracturas
Deformaciones óseas progresivas

- Déficit de crecimiento
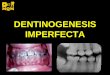
- Formación defectuosa de dientes(Dentinogenesis Imperfecta)
- Sordera
- Macrocefalia
- Escleróticas azules
- Escoliosis
- Tórax en barril
- Hiperlaxitud ligamentaria

OSTEOGENESIS IMPERFECTA
Tipos clásicosSillence
I- Forma LeveII- Extremadamente severa o LetalII- Extremadamente severa o LetalIII- SeveraIV- Moderada a severa

OI TIPO I
•••• Forma leve y mas común
•••• Fracturas prepuberales •••• Fracturas prepuberales
•••• Retraso de crecimiento leve

OI TIPO III OI TIPO IV
•••• Severas no-letales
•••• Fracturas recurrentes
•••• Deformaciones óseas progresivas

OI TIPO IV
OI TIPO III
•••• Deficiencia severa de crecimiento
OI TIPO IV
••••Deformaciones óseas leve a moderadas
•••• Pequeña estatura variable

TIPO II
•••• La forma mas grave
•••• Letal perinatal
Sillence, 1979
•Bebés con piernas cortas
• Huesos largos muy deformados
••••Múltiples fracturas de costillas

TIPOS V–VIII
Continuan la numeración de Sillence Basada en criterios diferentes
● Tipos V y VI se definieron utilizando la
histología ósea. El fenotipo podría estar histología ósea. El fenotipo podría estar
incluído en el tipo III o IV de Sillence
● Tipos VII y VIII. El fenotipo coincide con
tipos II y III de Sillence

El colágeno tipo I es la proteina defectuosa en osteogenesis imperfecta
Se halla entre otros en:
● Huesos
● Tendones
● Ligamentos
● Piel

Colágeno: Triple hélice, 2 cadenas α1α1 , 1 cadena α2.α2.Los Genes que codifican las dos cadenas Genes que codifican las dos cadenas α1(I)α1(I) yy α2(I) α2(I) son son
COL1A1COL1A1 ó ó COL1A2COL1A2, situados en los cromosomas 17 y 7 , situados en los cromosomas 17 y 7 respectivamente respectivamente respectivamente respectivamente

UNIDAD BÁSICA DEL COLÁGENO: TRIPLE HÉLICEUNIDAD BÁSICA DEL COLÁGENO: TRIPLE HÉLICE
Secuencia repetida de tripéptido glicina-x (prolina)-y (hidroxiprolina) La glicina está y (hidroxiprolina)(Gly) - X - Y)n
La glicina está presente en cada
tercera posición de las cadenas. Se
sitúa cada 3 residuos
helicoidales

La mayoría de las OI (90%) se originan por mutaciones heterocigotas,
más de 1500 descriptas, autosómica dominante o de novo en uno de
los genes COL1A1 o COL1A2

Los restantes casos de OI (10%) son autosómico-recesivos (A-R) y se
caracterizan por una elevada heterogeneidad genética.

OSTEOGENESIS IMPERFECTA-Clasificación. Actualmente XV tipos
Tipo Gen Fenotipo
H.Dominante.SILLENCE
I COL1A1-COL1A2 Leve sin deformaciones
II COL1A1-COL1A2 Letal perinatal
III COL1A1-COL1A2 Deformante progresiva
IV COL1A1-COL1A2 Deformante moderada
V IFITM5 Moderada callos V IFITM5 Moderada callos
hipertróficos,histología distinta
VI SERPINF1 Moderada a severa-histología
distinta
VII CRTAP Severa a letal
VIII LEPRE1 Severa a letal
IX PPIB Moderada a a letal
Lindahl et al.2014

OSTEOGENESIS IMPERFECTA-Clasificación
Tipo Gen Fenotipo
X SERPINH1 Severa a letal
XI FKBP10 Deformante progresiva,
Sme de Bruck
XII SP7 moderada
XIII BMP1 Severa,alta masa ósea
XIV TMEM38B Moderada a severa
XV WNT1 Moderada, deformante
progresiva
Inclasificada CREB3L1 SeveraInclasificada CREB3L1 Severa
X-LINKED
Sospecha defecto osteocito PLS3 leve
Lindahl et al.2014

DOS CLASES GENERALES DE MUTACIONES DEL COLÁGENO TIPO I RESULTAN EN OI
● 1) Fallo para sintetizar los productos de un alelo de COL1A1 lleva a haploinsuficiencia
● 2) Mutaciones que resultan en moléculas de colágeno con estructura anormal ● mas frecuente: sustitución de glicine por otro AA en la cadena α1(I) ó α2(I)
● MAS DE 800 MUTACIONES DE GLICINA HELICOIDALES DIFERENTES HAN SIDO IDENTIFICADAS EN OI

● La relación entre genotipo y fenotipo no está
aún bien conocida

RELACIÓN ENTRE EDAD Y Z-SCORES DE DMO ÁREA LUMBAR EN PACIENTES CON MUTACIONES DE HAPLOINSUFICIENCIA EN COL1A1 (HI), Y MUTACIONES EN COL1A1 Y COL1A2 QUE LLEVAN A
SUSTITUCIONES DE GLICINA EN LA HÉLICE DEL COLLAGEN TYPE I
● Pacientes con COL1A1
haploinsuficiencia:
mayor estatura, peso
y densidad mineral ósea
lumbar por área
● Histomorfometría
ósea: ancho de
RAUCH et al, JBMR 2009
lumbar por área ósea: ancho de
corticales 49% mayor en
los pacientes con COL1A1
Haploinsuficiencia
RAUCH et al, JBMR 2009

PACIENTES CON OI NO LETALES CON SUSTITUCIONES DE GLICINA (n 161)
Serina fue el AA sustitutivo más frecuentemente hallado
En la cadena α 1(I) (n 42), los pacientes tenían menor estatura y mas frecuentes del Tipo III , positivos para DI, comparados con los que tenían las sustituciones de
serina en la cadena α 2(I) (n 40). Z-score de Talla: -6 vs -3.4
RAUCH et al, Eur J Hum Genet 2010
RAUCH et al, EUR J HUM GENET, 2010
En la cadena α2(I) la estatura se correlacionó con la localización de la mutación
Entre los pacientes de diferentes familias que tuvieron la misma mutación en la cadena α1(I) ó α2(I), se halló concordancia en ● DI ● Escleróticas azules ● Fracturas y deformaciones al nacimiento

���� GENES COMPROMETIDOS EN OI● COL1A1
● COL1A2
● CRTAP
● FKBP10
● LEPRE1
● PPIB (cyclophilin B)
● SERPINF1
● IFITM5
● SERPINH1● SERPINH1
● SP7
● BMP1
● TMEM38B
● WNT1
● CREB3L1
● PLS3
Los genes más estudiados son COL1A1 y COL1A2

PREVALENCIA DE OI
�������� Estimada en 1 por 15.000 a 20.000 nacidos vivos
Sin embargo, las formas leves son sub-diagnosticadas, y la prevalencia actual puede ser más alta.la prevalencia actual puede ser más alta.
No se han hallado diferencias basadas en la Raza ni Sexo.

Tipo VI (mutación inactiva del gen SERPINF1 (codifica PEDF)
Es la única OI que presenta falta de mineralización del osteoide
No responde al tratamiento con bisfosfonatos
Farber et al. 2014Rauch et al. JCEM 2012
La determinación sérica de PEDF ayuda al diagnóstico pero no provee información sobre la remodelación y mineralización ósea

TRATAMIENTO

Dirigido a
● Prevenir y Controlar los síntomas
OBJETIVO
● Prevenir y Controlar los síntomas
● promover la movilidad independiente
●mejorar la mas ósea y la fuerza muscular

--MédicoMédico
--FisioterapiaFisioterapia y y rehabilitaciónrehabilitación
--OrtopédicoOrtopédico
--De la De la pérdidapérdida de de audiciónaudición--De la De la pérdidapérdida de de audiciónaudición
--De la De la familiafamilia
--De la De la mujermujer embarazada,deembarazada,de la la mujermujer adultaadulta
--ConsejoConsejo genéticogenético: : precisaprecisa el el diagnósticodiagnóstico, , riesgoriesgo de de otrootro embarazoembarazo afectadoafectado
de OI, de OI, diagnósticodiagnóstico prenatalprenatal

- MÉDICO:
● Bisfosfonatos
●Calcio, vitamin D●Calcio, vitamin D

30 Niños con OI severa; Edad: 3-16 años. Pamidronato cíclico e.v. 3 días cada 4-6 meses + Ca yVit D durante 1.3- 5 años↑↑↑↑DMO 42 ±±±± 29% (0.20 a 0.37g/cm²)Z-score DMO: –5.3 ±±±± 1.2→→→→ -3.4 ±±±± 1.5↓↓↓↓ Fracturas: 2.3±±±±2.2→→→→ 0.6±±±±0.5/año
GLORIEUX et al, NEJM 1998GLORIEUX et al, NEJM 1998

●Disminución del Número de fracturas
●Mejoría de la Función del esqueleto
●Aumento de Mineralización ósea
●Aumento de la Fuerza muscular

BIFOSFONATOS UTILIZADOS EN OSTEOGÉNESISIMPERFECTA
-Pamidronato oral
-Olpadronato oral
-Alendronato oral
-Risedronato oral
-Pamidronato ev
-Neridronato ev
-Zoledronato ev

COMPLICACIONES DEL TRATAMIENTO CON BISFOSFONATOS
Luego de varios años de tratamiento, la curación de las osteotomías es lenta pero la reparación de las las osteotomías es lenta pero la reparación de las
fracturas es normal

TRATAMIENTOS EN INVESTIGACIÓN
-Denosumab, Anticuerpo monoclonal anti-RANKL
-Anticuerpo monoclonal anti-esclerostina

DENOSUMAB
Two years’ experience with denosumab forchildren with Osteogenesis imperfecta type VI
Heike Hoyer-Kuhn1, Christian Netzer, Friederike Koerber, Eckhard Schoenau and Oliver Semler
Orphanet Journal of Rare Diseases 2014, 9:145

Evaluation of teriparatide treatment in adultswith osteogenesis imperfectaEric S. Orwoll, Jay Shapiro, Sandra Veith, Ying Wang, Jodi Lapidus, Chaim Vanek, Jan L. Reeder,
Tony M. Keaveny, David C. Lee, Mary A. Mullins, Sandesh C.S. Nagamani, and Brendan Lee.
J Clin Invest 2014, 124(2): 491
OI tipo I mejoría de DMO, no hubo beneficio en OI tipos III y IV

Terapia celular
Terapia Génica
Tratamientos curativos en investigación
Vectores virales para las formas recesivas

CONCLUSIONES
● La Osteogenesis Imperfecta es una enfermedad discapacitante heterogénea causada principalmente por un defecto en los genes COL1A1 ó COL1A2
● Actualmente, investigadores han hallado alguna correlación entre genotipo y fenotipo

CONCLUSIONES II
● En espera del desarrollo de terapias celular y génicas, el tratamiento con bisfosfonatos mejora la calidad de vida de los pacientes, aumentando la calidad de vida de los pacientes, aumentando la movilidad, la masa ósea, y disminuyendo el número de fracturas. El tratamiento es más efectivo durante el período de crecimiento

CONCLUSIONES III
● La Fisioterapia, la psicoterapia, la rehabilitación, y las cirugías ortopédicas correctoras son complementos en el tratamiento de los complementos en el tratamiento de los pacientes con OI

RAQUITISMOS HEREDITARIOS
-Hipofosfatémico familiar
-Hipofosfatémico con hipercalciuria
-Pseudocarencial por déficit de 1α Hidroxilasa -Pseudocarencial por déficit de 1α Hidroxilasa
-Resistente Tipo II por alteración del receptor de vitamina D (VDR)

ROL DEL FÓSFORO
Importante en múltiples procesos biológicos:
• Mantenimiento de la integridad de membranas celulares y ácidos nucleicos
• Metabolismo celular: ATP• Metabolismo celular: ATP
• Regulación de procesos subcelulares: fosforilación enzimática
• Homeostasis del estado ácido-base
• Mineralización ósea

FÓSFORO
85% Huesos y dientes
14% Otros tejidos14% Otros tejidos
1% Líquido extracelular

REGULADORES DE LA HOMEOSTASIS DEL FÓSFORO
Fósforo de la dieta
PTH
Calcitriol
Fosfatoninas: FGF23, FGF-7, FRP-4, MEPE

ESQUELETO, RIÑON Y PARATIROIDES EN LA REGULACIÓN DE FGF23
Pettifor et al, Calcified Tissue Int 2012

HOMEOSTASIS DEL FÓSFORO EN LA NIÑEZ
Intestino: absorción pasiva, 1,25 (OH)₂D
Riñon: Co-transportadores de Na-PO₄
NaP-INaP-I
NaP-IIa y IIc (Regulados por PTH, FGF23)
NaP-III

CAUSAS DE HIPOFOSFATEMIA EN NIÑOS
Pettifor et al, Calcif Tissue Int 2012

RAQUITISMO HIPOFOSFATÉMICO
-LIGADO AL CROMOSOMA X: PHEX (gene regulador de P con homología a endopeptidasa) mutación inactivante. Aumento de FGF23 circulante o fosfatonina
-AUTOSÓMICO DOMINANTE: (ADHR)-AUTOSÓMICO DOMINANTE: (ADHR)Mutación del gene FGF-23 (aumento de actividad biológica) Aumento en suero
����Disminuye la expresión del co-transporter tipo IIa y IIc Sodio-Fosfato de la células tubulares renales
���� Disminuye la expresión de la 25OHD-1αααα-hidroxilasa renal

INCIDENCIA
1/20.000 individuos

INTRODUCCIÓN
El Raquitismo hipofosfatémico familiar (RHF) es un raquitismo hereditario caracterizado por hipofosfatemia por pérdida renal de fósforo,hipofosfatemia por pérdida renal de fósforo,y pequeña talla

RAQUITISMO HIPOFOSFATÉMICO FAMILIAR
••••Hipofosfatemia
•••• Deformaciones progresivas de miembros inferiores (genu varum, genu valgum, coxa vara)(genu varum, genu valgum, coxa vara)
•••• Pequeña talla
•••• Alteraciones dentarias (retraso de aparición, abcesos)

FISIOPATOLOGÍA
-Mutaciones con pérdida de función del gen regulador del fósforo con homología a endopeptidasa en cromosoma X (PHEX)
-PHEX codifica endopeptidasa de membrana -PHEX codifica endopeptidasa de membrana que se expresa en huesos y dientes
-Pérdida de 86-87% en casos familiares-Pérdida de 57-72% en casos esporádicos

FISIOPATOLOGÍA
-Defecto de mineralización ósea y aumento de FGF23
-FGF23 no es sustrato del PHEX. Causa de elevación de FGF23 no es clara, sin embargo ambos son productos del osteocito

FISIOPATOLOGÍA
-Desorden de la reabsorción tubular de fósforo
-Conversión defectuosa de 1,25(OH)2D-Conversión defectuosa de 1,25(OH)2D
-Defecto del osteoblasto

Actualmente el tratamiento utilizado
consiste en sales de fósforo y calcitriol

CLÍNICA
Pequeña tallaDeformaciones de miembros inferiores
Genu varumGenu varumGenu valgum
Dolicocefalia Abcesos dentarios

RADIOGRAFÍAS
Raquitismo en muñecas, rodillas y tobillos.

LABORATORIO
���� calcemia (Ca) ���� fosfatemia (P) ���� fosfatasas alcalinas (FAL) ���� 25-hidroxivitamina D (25OHD) ���� 25-hidroxivitamina D (25OHD) � paratohormona (PTH)���� creatinina, ���� calciuria (UCa) de 24 hs���� reabsorción tubular de fósforo (RTP)

Evolución clínica
Radiológica
LaboratorioLaboratorio

LABORATORIO
HIPOFOSFATEMIA
Todos los niños tienen hipofosfatemia
La reabsorción tubular de fósforo está La reabsorción tubular de fósforo está disminuída (v.0-95%)

VALORES DE FÓSFORO EN LA NIÑEZ
Neo-3 meses: 4.5-8.3 mg/dl
Niños 1 a 2 años: 4.5-5.8 mg/dl
Niños: 3.5-5.5 mg/dl
Adolescentes: 2.5-4.5 mg/dl

REABSORCIÓN TUBULAR DE FÓSFORO EN LA NIÑEZ
RTP: 1-uP x sCreat x100 = 80-95%sP x uCreat
Como la RTP depende de P sérico, Walton y Bijvoet crearon un Nomograma que no es aplicable en Niños: un Nomograma que no es aplicable en Niños:
TmPi/GFR
Alon y Hellerstein tabla aplicable en niños

LABORATORIO
Las fosfatasas alcalinas están elevadas en todos
los niños.

RESULTADOS LABORATORIO
Los pacientes tienen Normocalcemia,
PTH normal e hipocalciuria

TRATAMIENTO
Sales de fósforo: 5 dosis/día
Calcitriol: c/12 horas

MONITOREO DEL TRATAMIENTO
CADA 4 MESES:
-Curva de Crecimiento-Corrección de las deformaciones-Actividad de FAL
CADA 6 MESES:CADA 6 MESES:
-PTH
ANUAL:
-Ecografía Renal-Radiología

CONCLUSIONES
-En la actualidad no hay un tratamiento completamente satisfactorio para el RHF
-El tratamiento debe iniciarse temprano para que sea efectivo

CONCLUSIONES II
����Los pacientes con RHF tratados con sales de fósforo y calcitriol mantienen un buen crecimiento, mejoran las deformaciones óseas, los parámetros bioquímicos y los signos radiológicos de la parámetros bioquímicos y los signos radiológicos de la enfermedad
����Se requiere un cuidadoso seguimiento. El monitoreo periódico permite detectar y corregir las posibles complicaciones de hiperparatiroidismo y/ó hipercalciuria

RECOMENDACIONES DURANTE ELTRATAMIENTO
-Hipercalciuria: Disminuir ó suspender el Calcitriol
-Hiperparatiroidismo: Suspender el tratamiento hasta la estabilización

RAQUITISMO HIPOFOSFATÉMICO
[1082] Efficacy and Safety of a Human Monoclonal Anti-FGF23 Antibody (KRN23) in
Cumulative 4-Month Dose Escalation (KRN23-INT-001) and 12-Month Long-Term Extension
Study (KRN23-INT-002) in Adult Subjects with X-Linked Hypophosphatemia (XLH)
Thomas Carpenter, Yale University School of Medicine, USA
Conclusions: Blocking FGF23 activity by SC administration of KRN23 every 28 days for
an extended period of up to 16 months demonstrated a favorable safety profile and
increased serum Pi, TmP/GFR and 1,25(OH)2D.
ASBMR 2014