Embed Size (px)
DESCRIPTION
Parallel Methods for Nano/Materials Science Applications. (Electronic Structure Calculations). Andrew Canning Computational Research Division LBNL & UC Davis. Outline. Introduction to Nano/Materials science Electronic Structure Calculations (DFT) - PowerPoint PPT Presentation
Citation preview

Parallel Methods for Nano/Materials Science Applications
Andrew Canning
Computational Research Division LBNL & UC Davis
(Electronic Structure Calculations)

Outline
• Introduction to Nano/Materials science • Electronic Structure Calculations (DFT) • Code performance on High Performance
Parallel Computers • New Methods and Applications for
Nanoscience
1991 Silicon surface reconstruction (7x7), Phys. Rev. (Stich, Payne, King-Smith, Lin, Clarke) Meiko I860, 64 processor Computing Surface (Brommer, Needels, Larson, Joannopoulos) Thinking Machines CM2, 16,384 bit processors
2005 1000 atom Molybdenum simulation with Qbox SC05. (F. Gygi et al. ) BlueGene/L, 32,000 processors (LLNL)
1998 FeMn alloys (exchange bias), Gordon Bell prize (Ujfalussy, Stocks, Canning, Y. Wang, Shelton et al.) Cray T3E, 1500 procs. first > 1 Tflop Simulation
Milestones in Parallel Calculations

Electronic Structure Calculations
• Accurate Quantum Mechanical treatment for the electrons• Each electron represented on grid or with some basis functions
(eg. fourier components) • Compute Intensive: Each electron requires 1 million points/basis
(need 100s of electrons) • 70-80% NERSC Materials Science Computer Time
(first-principles electronic structure)
InP quantum dot (highest electron energy state in valence band)

Motivation for Electronic Structure Calculations
• Most Materials Properties Only Understood at a fundamental level from Accurate Electronic Structure (Strength, Cohesion etc)
• Many Properties Purely Electronic eg. Optical Properties (Lasers)
• Complements Experiments • Computer Design Materials at the
nanoscale

Materials Science Methods
• Many Body Quantum Mechanical Approach (Quantum Monte Carlo) 20-30 atoms
• Single Particle QM (Density Functional Theory) No free parameters. 100-1000 atoms
• Empirical QM Models eg. Tight Binding 1000-5000 atoms
• Empirical Classical Potential Methods thousand-million atoms
• Continuum Methods
atoms

Ab initio Method: Density Functional Theory (Kohn 98 Nobel Prize)
),..(),..(}||||
1
2
1{ 11
,,
2NN
Ii Iiji jii
i
rrErrRr
Z
rr
)()(}||||
)(
2
1{ 2 rErV
Rr
Zrd
rr
riiiXC
I I
2
12 |),..(||)(|)( N
ii rrrr
Kohn Sham Equation (65): The many body ground state problem can be mapped onto a single particle problem with the same electron density and a different effective potential (cubic scaling).
Use Local Density Approximation (LDA) for )]([ rVXC (good Si,C)
Many Body Schrodinger Equation (exact but exponential scaling )

Selfconsistent calculation
)()()},(2
1{ 2 rErrV iii
Nii ,..,1}{
2|)(|)( rrN
ii
),( rV
Self
con
sis
ten
cy
N electronsN wave functionslowest N eigenfunctions

Choice of Basis for DFT(LDA)
Increasing basis size M
Gaussian FLAPW Fourier grid
Percentage of eigenpairs M/N
30% 2%Eigensolvers
Direct (scalapack) Iterative

Plane-wave Pseudopotential Method in DFT
)()())}((||||
)(
2
1{ 2 rErrV
Rr
Zrd
rr
rjjjXC
I I
Solve Kohn-Sham Equations self-consistently for electron wavefunctions within the Local Density Appoximation
rkgi
g
jgkj ekCr ).(
, )()( 1. Plane-wave expansion for
2. Replace “frozen” core by a pseudopotential
Different parts of the Hamiltonian calculated in different spaces (fourier and real) 3d FFT used

PARATEC (PARAllel Total Energy Code)
• PARATEC performs first-principles quantum mechanical total energy calculation using pseudopotentials & plane wave basis set
• Written in F90 and MPI• Designed to run on large parallel
machines IBM SP etc. but also runs on PCs
• PARATEC uses all-band CG approach to obtain wavefunctions of electrons
• Generally obtains high percentage of peak on different platforms• Developed with Louie and Cohen’s groups (UCB, LBNL),
Raczkowski

PARATEC: Code Details
• Code written in F90 and MPI (~50,000 lines) • 33% 3D FFT, 33% BLAS3, 33% Hand coded F90 • Global Communications in 3D FFT (Transpose)• Parallel 3D FFT handwritten, minimize comms. reduce
latency (written on top of vendor supplied 1D complex FFT )
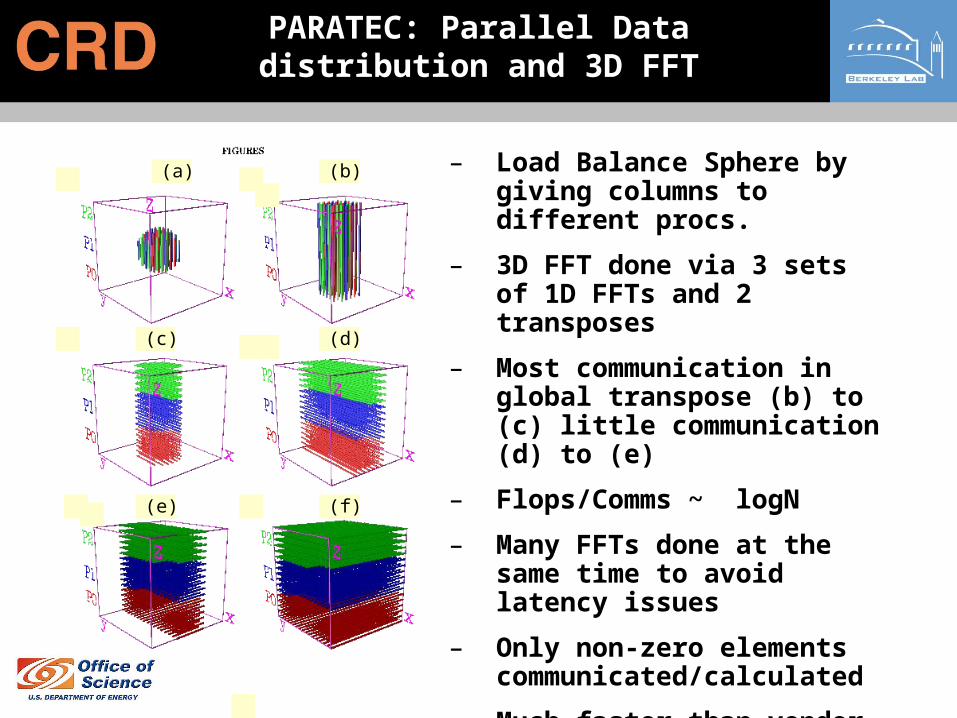
– Load Balance Sphere by giving columns to different procs.
– 3D FFT done via 3 sets of 1D FFTs and 2 transposes
– Most communication in global transpose (b) to (c) little communication (d) to (e)
– Flops/Comms ~ logN
– Many FFTs done at the same time to avoid latency issues
– Only non-zero elements communicated/calculated
– Much faster than vendor supplied 3D-FFT
PARATEC: Parallel Data distribution and 3D FFT
(a) (b)
(e)
(c)
(f)
(d)
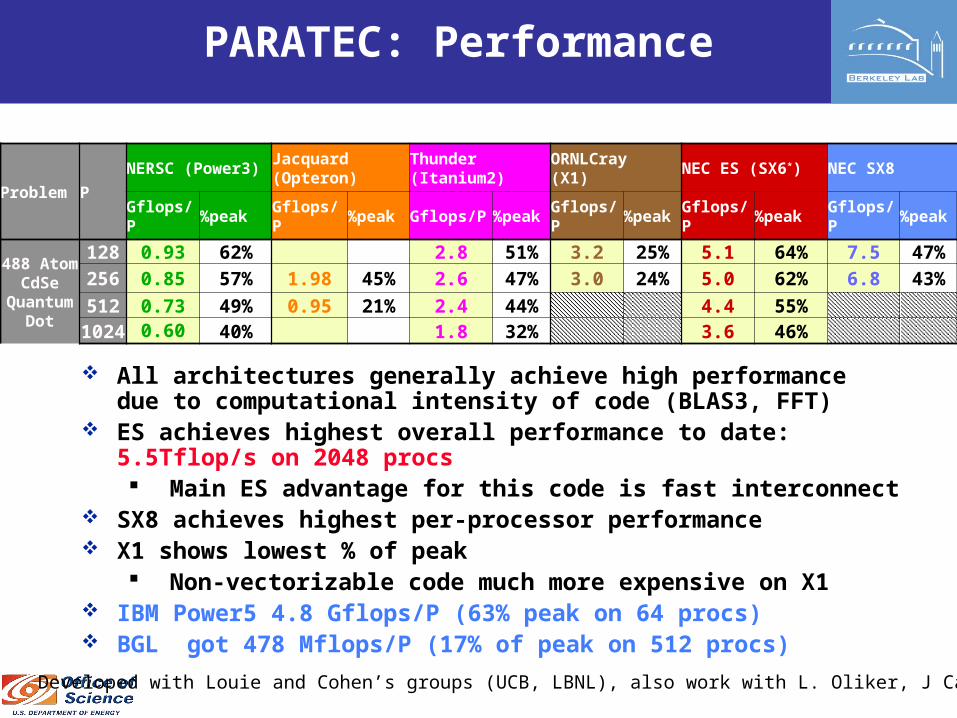
PARATEC: Performance
All architectures generally achieve high performance due to computational intensity of code (BLAS3, FFT)
ES achieves highest overall performance to date: 5.5Tflop/s on 2048 procs Main ES advantage for this code is fast interconnect
SX8 achieves highest per-processor performance X1 shows lowest % of peak
Non-vectorizable code much more expensive on X1 IBM Power5 4.8 Gflops/P (63% peak on 64 procs) BGL got 478 Mflops/P (17% of peak on 512 procs)
Problem PNERSC (Power3)
Jacquard (Opteron)
Thunder (Itanium2)
ORNLCray (X1) NEC ES (SX6*) NEC SX8
Gflops/P %peak Gflops/P %peak Gflops/P %peak Gflops/P %peak Gflops/P %peak Gflops/P %peak
488 AtomCdSe
Quantum
Dot
128 0.93 62% 2.8 51% 3.2 25% 5.1 64% 7.5 47%256 0.85 57% 1.98 45% 2.6 47% 3.0 24% 5.0 62% 6.8 43%512 0.73 49% 0.95 21% 2.4 44% 4.4 55%1024
0.60 40% 1.8 32% 3.6 46%
Developed with Louie and Cohen’s groups (UCB, LBNL), also work with L. Oliker, J Carter
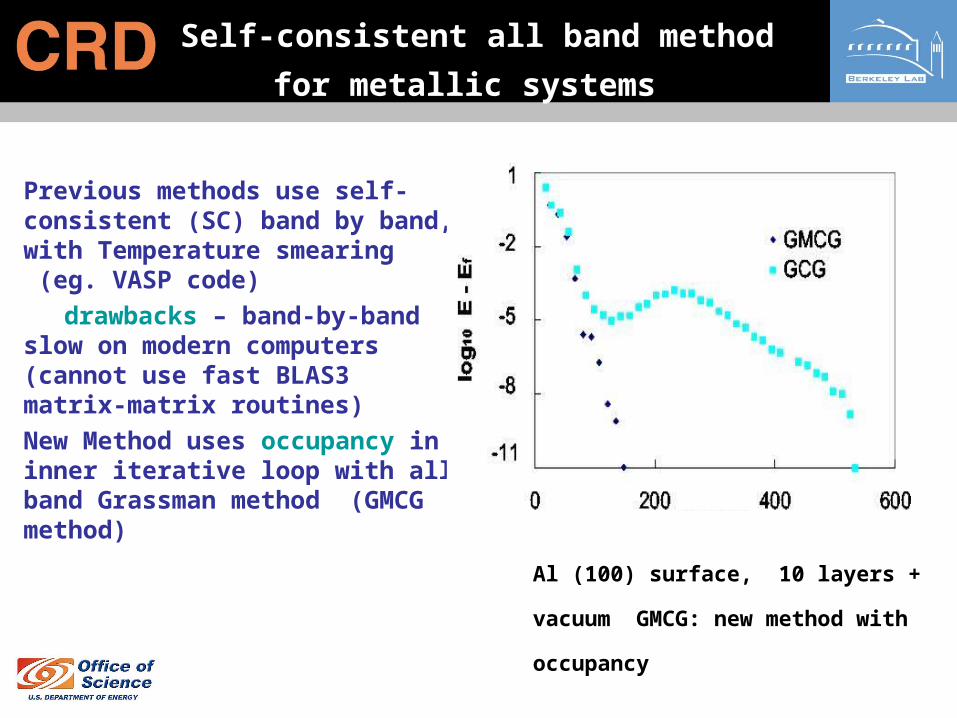
Self-consistent all band method for
metallic systems
• Previous methods use self-consistent (SC) band by band, with Temperature smearing (eg. VASP code)
drawbacks – band-by-band slow on modern computers (cannot use fast BLAS3 matrix-matrix routines)
• New Method uses occupancy in inner iterative loop with all band Grassman method (GMCG method)
Al (100) surface, 10 layers + vacuum
GMCG: new method with occupancy

Self-consistent all band method for metals
i
iini
/|}V2
2
1{|min f
i
}{i
)(Vout r
P
ote
nti
al M
ixin
g
Vou
t
Vin
)()()(*
rrrii
iif
KS - DFT
i
iini|}V2
2
1{|min

The Quantization Condition of Quantum-well The Quantization Condition of Quantum-well States in Cu/Co(100)States in Cu/Co(100)
Copper substrate
Cobalt layerCopper Wedge
0
54 Å
4 m
m sc
an
d
photon beam in
electronsout
• Theoretical investigation of Quantum Well states in Cu films using our codes (PARATEC, PEtot) to compare with experiments at the ALS (E. Rotenberg, Y.Z. Wu, Z.Q. Qiu)
• New computational methods for metallic systems used in the calculations.
•Lead to an understanding of surface effects on the Quantum Well States. Improves on simple Phase Accumulation Model used previously
-1.5
-1.0
-0.5
0.0
E-E
F (e
V)
20151050Cu thickness(ML)
Difference between theory and experiment improved by taking surface effects into account
QW states in Copper Wedge

Computational challenges (larger nanostructures)
•Ab initio method PARATEC
atoms
moleculesnanostructures bulk
size1-100 atoms
1000-10^6 atoms
Infinite(1-10 atomsin a unit cell)
methodAb initio Method PARATEC
Challenge forcomputationalnanoscience.
Ab initioelementsand reliability
New methodologyand algorithm(ESCAN)
Even largerSupercomputer
)( 3NO
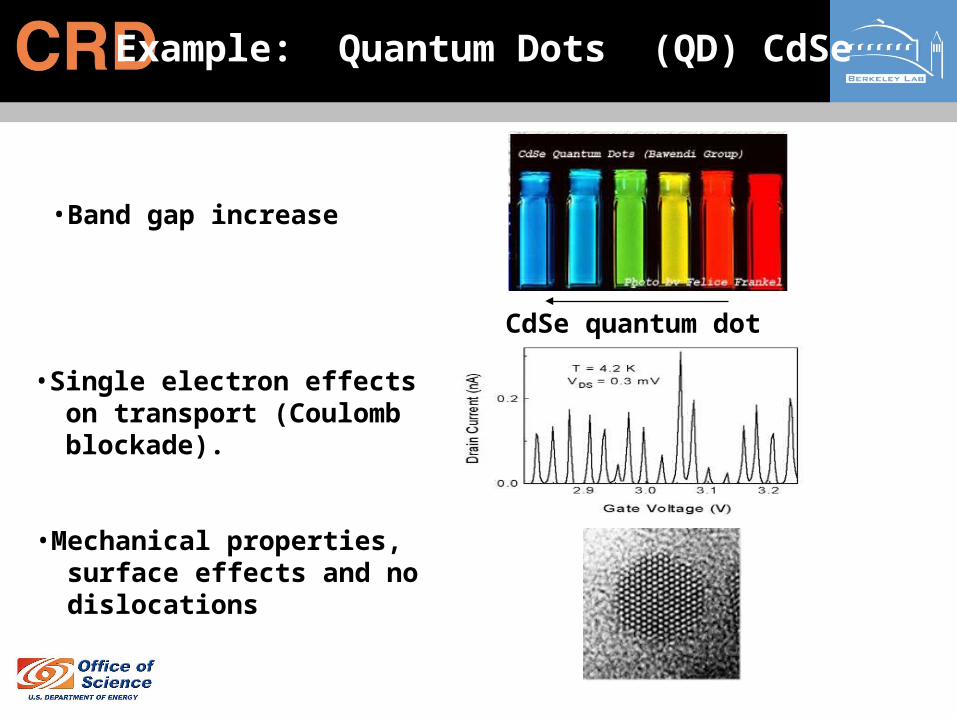
Example: Quantum Dots (QD) CdSe
•Band gap increase
CdSe quantum dot (size)
•Single electron effects on transport (Coulomb blockade).
•Mechanical properties, surface effects and no dislocations
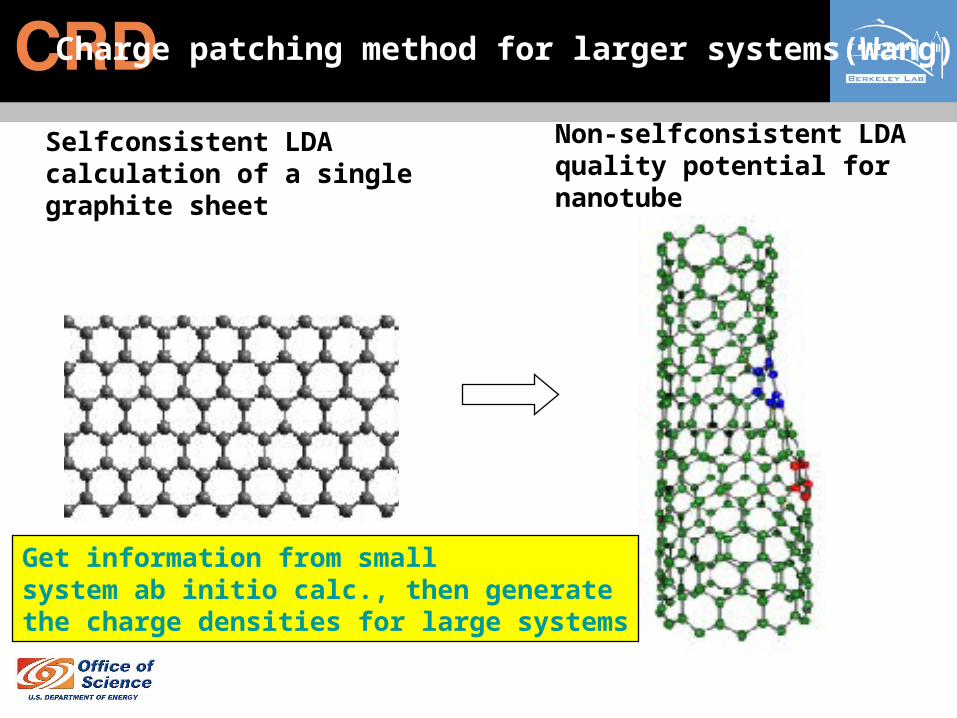
Charge patching method for larger systems(Wang)
Selfconsistent LDAcalculation of a single graphite sheet
Non-selfconsistent LDAquality potential for nanotube
Get information from smallsystem ab initio calc., then generatethe charge densities for large systems
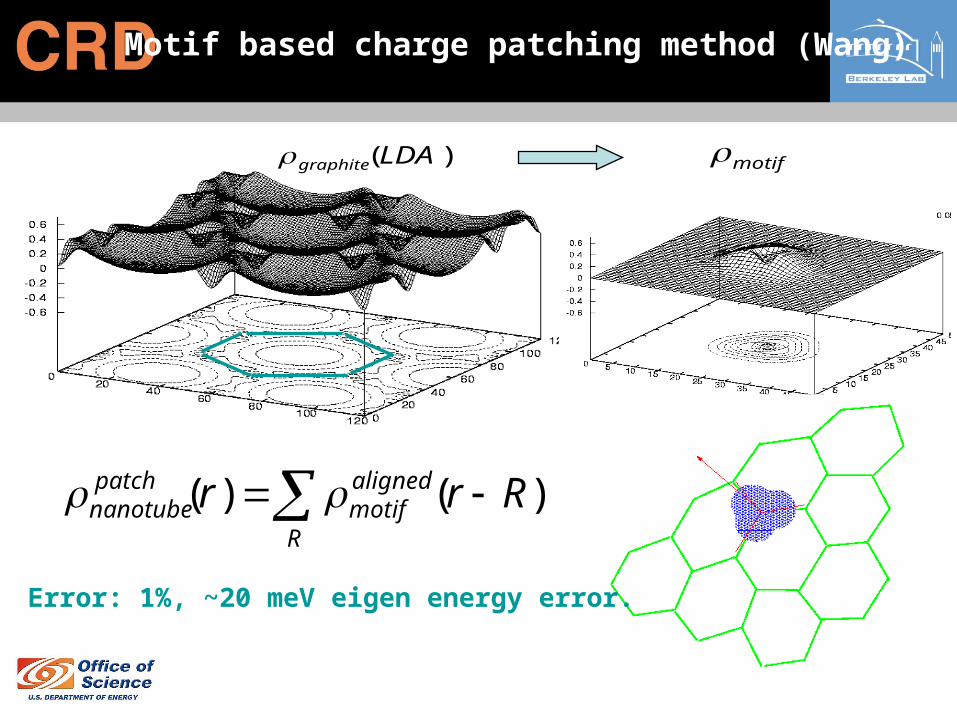
)(LDAgraphite motif
Motif based charge patching method (Wang)
)()( RrrR
alignedmotif
patchnanotube
Error: 1%, ~20 meV eigen energy error.
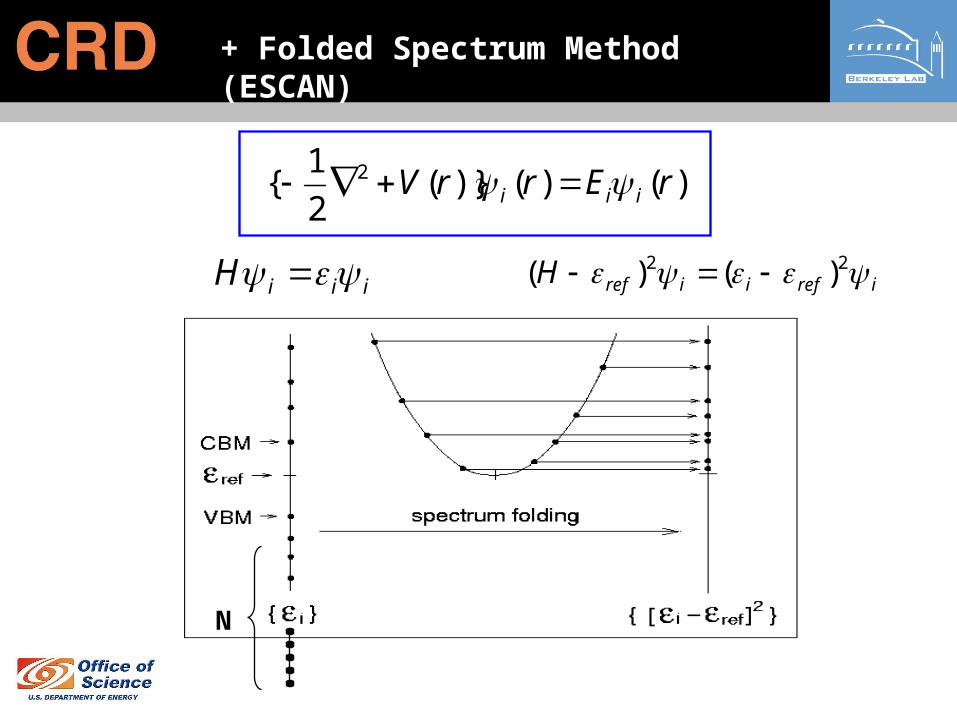
+ Folded Spectrum Method (ESCAN)
iiiH irefiirefH 22 )()(
N
)()()}(2
1{ 2 rErrV iii
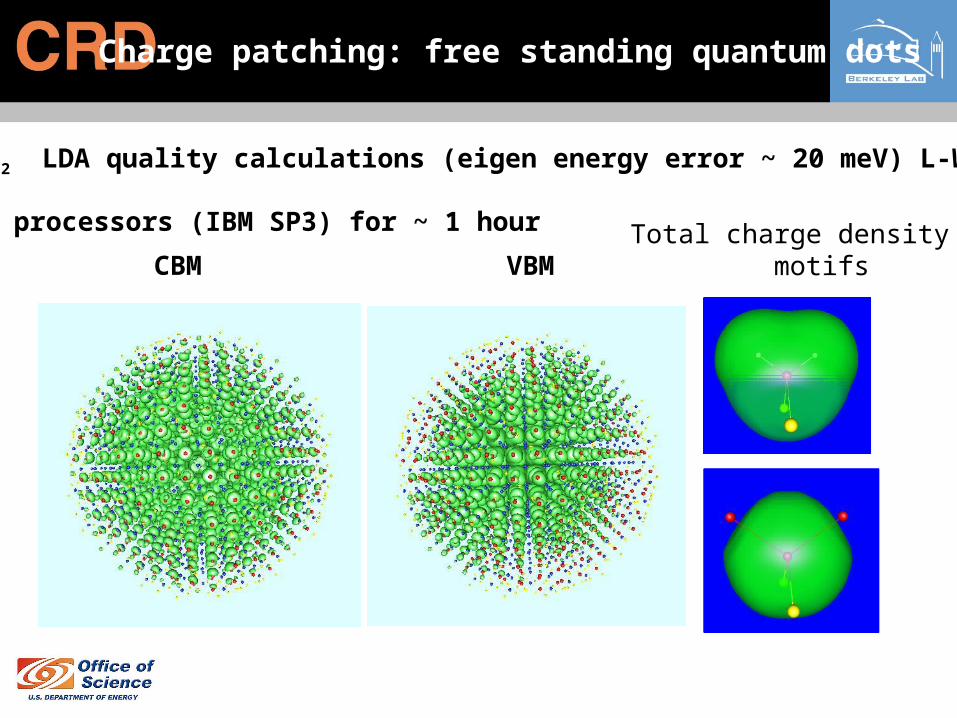
Charge patching: free standing quantum dots
In675P652 LDA quality calculations (eigen energy error ~ 20 meV) L-W Wang
CBM VBM
64 processors (IBM SP3) for ~ 1 hour Total charge density motifs
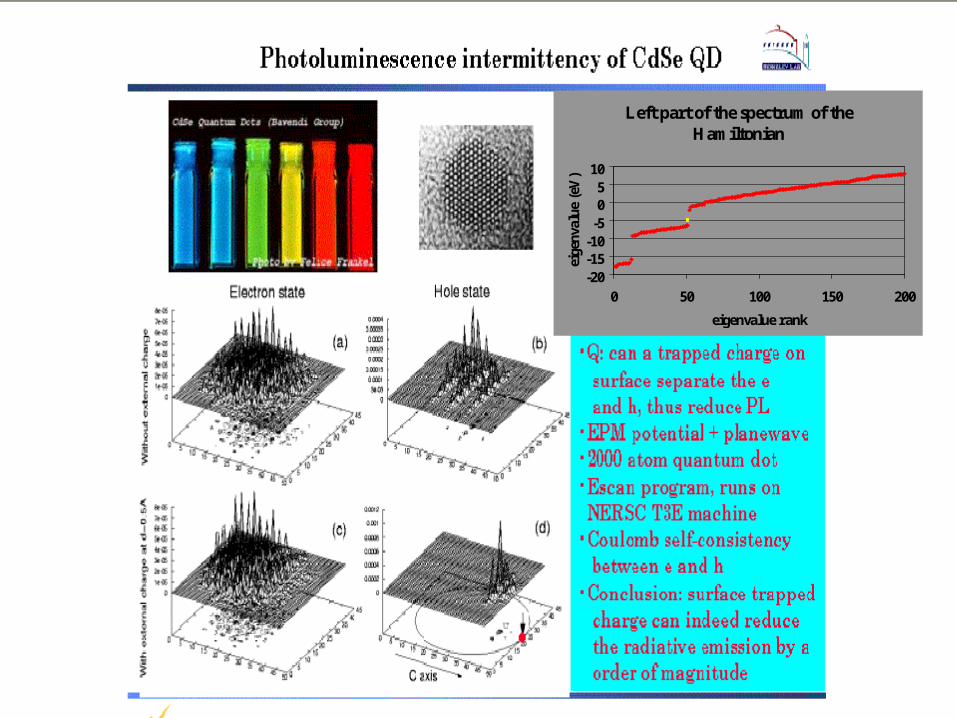
Left part of the spectrum of the Hamiltonian
-20-15-10-505
10
0 50 100 150 200
eigenvalue rank
eige
nval
ue (
eV)
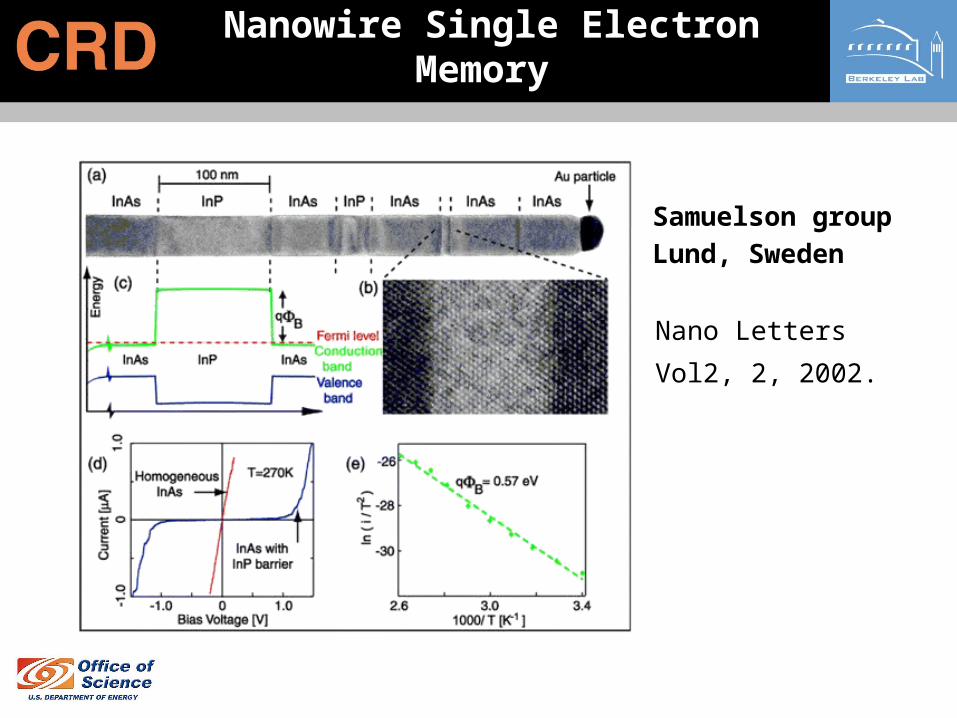
Nanowire Single Electron Memory
Samuelson group
Lund, Sweden
Nano Letters Vol2, 2,
2002.
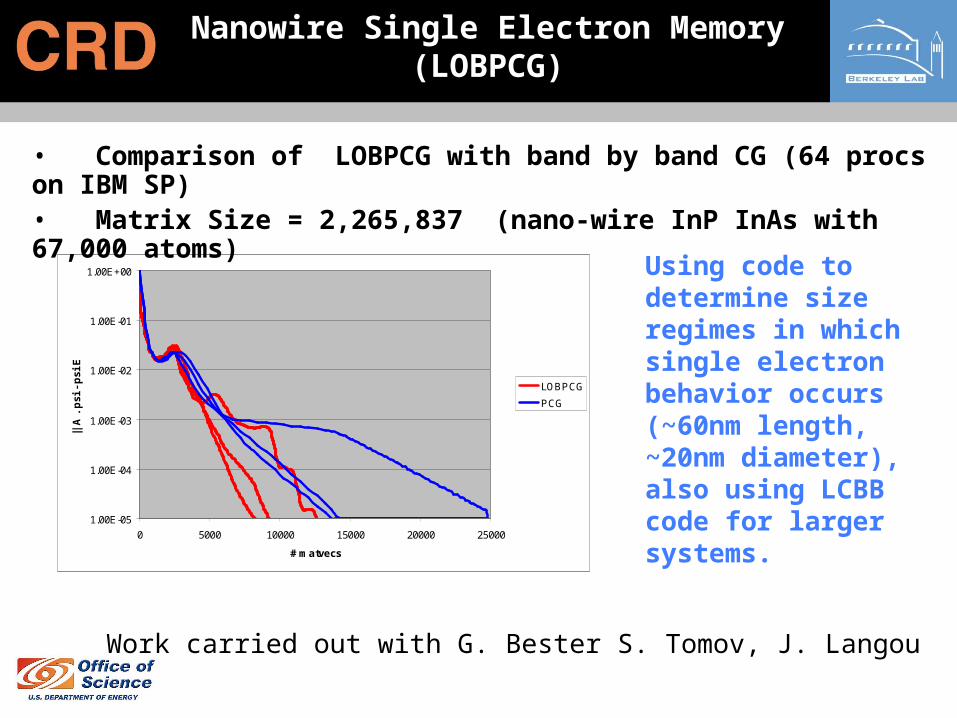
Nanowire Single Electron Memory (LOBPCG)
1.00E-05
1.00E-04
1.00E-03
1.00E-02
1.00E-01
1.00E+00
0 5000 10000 15000 20000 25000
# matvecs
|| A
. p
si -
psi
E |
|
LOBPCG
PCG
• Comparison of LOBPCG with band by band CG (64 procs on IBM SP) • Matrix Size = 2,265,837 (nano-wire InP InAs with 67,000 atoms)
Using code to determine size regimes in which single electron behavior occurs (~60nm length, ~20nm diameter), also using LCBB code for larger systems.
Work carried out with G. Bester S. Tomov, J. Langou

Future Directions
• O(N) based methods (exploit locality) gives sparse matrix problem
• Excited state calculations • Transport Calculations
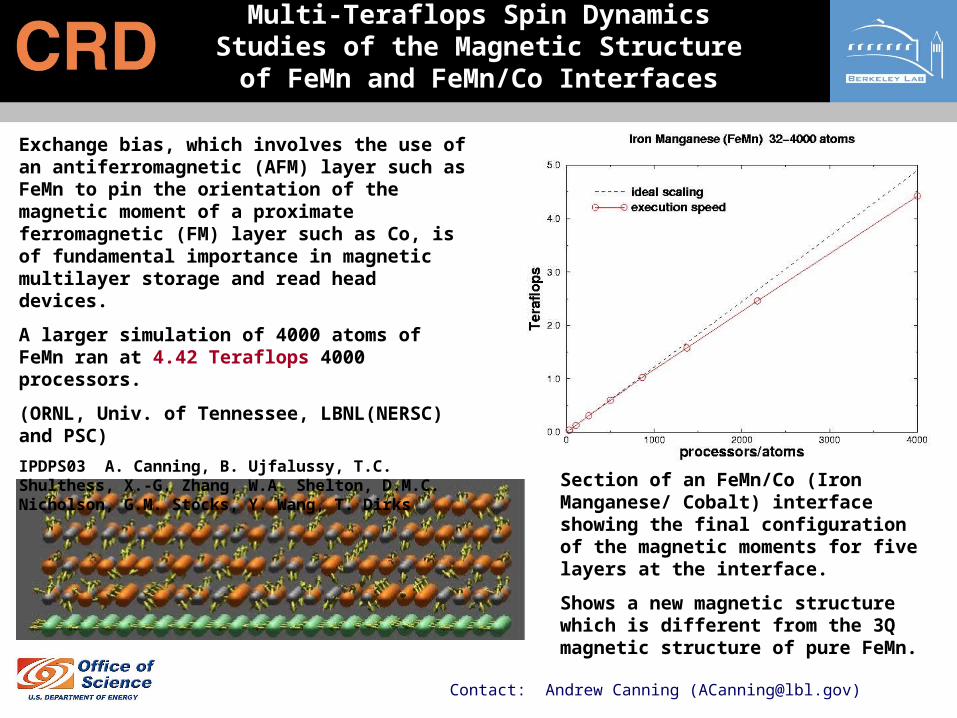
Multi-Teraflops Spin Dynamics Studies of the Magnetic Structure of FeMn and
FeMn/Co Interfaces
Section of an FeMn/Co (Iron Manganese/ Cobalt) interface showing the final configuration of the magnetic moments for five layers at the interface.
Shows a new magnetic structure which is different from the 3Q magnetic structure of pure FeMn.
Exchange bias, which involves the use of an antiferromagnetic (AFM) layer such as FeMn to pin the orientation of the magnetic moment of a proximate ferromagnetic (FM) layer such as Co, is of fundamental importance in magnetic multilayer storage and read head devices.
A larger simulation of 4000 atoms of FeMn ran at 4.42 Teraflops 4000 processors.
(ORNL, Univ. of Tennessee, LBNL(NERSC) and PSC)
IPDPS03 A. Canning, B. Ujfalussy, T.C. Shulthess, X.-G. Zhang, W.A. Shelton, D.M.C. Nicholson, G.M. Stocks, Y. Wang, T. Dirks
Contact: Andrew Canning ([email protected])
Conclusion
First principlecalculation
New algorithmmethodology
Large scalesupercomputers
Accurate Nanostructures simulations
+ +





























