Embed Size (px)
Citation preview

Passive Lipoidal Diffusion and Carrier-Mediated Cell Uptake Are BothImportant Mechanisms of Membrane Permeation in DrugDispositionDennis Smith,*,† Per Artursson,‡ Alex Avdeef,§ Li Di,∥ Gerhard F. Ecker,⊥ Bernard Faller,#
J. Brian Houston,¶ Manfred Kansy,□ Edward H. Kerns,■,▽ Stefanie D. Kramer,○ Hans Lennernas,‡
Han van de Waterbeemd,● Kiyohiko Sugano,△ and Bernard Testa▲
†4 The Maltings, Walmer, Kent, CT14 7AR, U.K.‡Department of Pharmacy, Biomedical Centre, Uppsala University, S-752 63 Uppsala, Box 580, Sweden§1732 First Avenue, #102, New York, New York 10128, United States∥Pharmacokinetics, Dynamics and Metabolism, Pfizer Inc., Groton, Connecticut 06340, United States⊥Department of Medicinal Chemistry, University of Vienna, Althanstrasse, 141090 Wien, Austria#Novartis Institutes for Biomedical Research, WSJ-350.3.04, CH-4002 Basel, Switzerland¶School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Oxford Road, Manchester, M13 9PT, U.K.□The Non-Clinical Safety Department, F. Hoffmann-La Roche, CH-4070 Basel, Switzerland■National Center for Advancing Translational Sciences, National Institutes of Health, 9800 Medical Center Drive, Rockville,Maryland 20850, United States○Institute of Pharmaceutical Sciences, ETH, Zurich, Switzerland●14 Rue de la Rasclose, 66690 Saint Andre, France△Research Formulation, Sandwich Laboratories, Ramsgate Road, Sandwich, Kent CT13 9NJ, U.K.▲Department of Pharmacy, University Hospital Lausanne, CH-1011 Lausanne, Switzerland
ABSTRACT: Recently, it has been proposed that drug permeation is essentiallycarrier-mediated only and that passive lipoidal diffusion is negligible. This opposesthe prevailing hypothesis of drug permeation through biological membranes,which integrates the contribution of multiple permeation mechanisms, includingboth carrier-mediated and passive lipoidal diffusion, depending on thecompound’s properties, membrane properties, and solution properties. Theprevailing hypothesis of drug permeation continues to be successful forapplication and prediction in drug development. Proponents of the carrier-mediated only concept argue against passive lipoidal diffusion. However, thearguments are not supported by broad pharmaceutics literature. The carrier-mediated only concept lacks substantial supporting evidence and successfulapplications in drug development.
KEYWORDS: permeation, passive lipoidal diffusion, “carrier-mediated only” concept
■ INTRODUCTION
Absorption, distribution, and elimination of drug molecules andtheir metabolites from the point of dosing, throughout bodytissues, into cells, exposure to biochemical targets, and routes ofclearance involve crossing diverse tissue, cellular, and organellemembranes. The process of membrane crossing is broadlyreferred to as “permeation”, which includes mechanisms of passivelipoidal diffusion, carrier-mediated influx, carrier-mediated efflux,paracellular diffusion, aqueous boundary layer (mucus) diffusion,endocytosis, and others. Owing to the importance of permeationin drug absorption and disposition, these mechanisms have beenand continue to be an active research area. The preponderance of
the data on permeation has led to a widely accepted and appliedprevailing drug permeation paradigm, which embodies thecontribution of all the mechanisms. This paradigm is routinelyapplied with success in drug design to optimize efficacy and safety,predict human PK/PD, select dose, and plan dosing regimens.This review discusses the prevailing permeation hypothesis,
which we conclude is consistent with the experimental data.
Received: November 26, 2013Revised: February 10, 2014Accepted: April 11, 2014Published: April 11, 2014
Review
pubs.acs.org/molecularpharmaceutics
© 2014 American Chemical Society 1727 dx.doi.org/10.1021/mp400713v | Mol. Pharmaceutics 2014, 11, 1727−1738

Importantly, this review also addresses uncertainties amongdrug researchers that result from recent articles1−4 published bya group that assert the carrier-mediated only concept (CMOC)of drug permeation and attempt to invalidate passive lipoidaldiffusion across biological membranes into cells and across celllayer membranes. We conclude that the CMOC lacks sufficientevidence. The CMOC is not a sound scientific principle andlacks experimental evidence. Carrier-mediated transport andpassive lipoidal diffusion permeation mechanisms are comple-mentary, their relative contribution depending on manyphysicochemical and biochemical factors (e.g., concentrationgradient, lipophilicity and hydrogen-bonding capacity of thediffusing compound, transporter affinity, and Km). Theintegrated system follows the laws of thermodynamics.Discussion of divergent hypotheses, based on experimental
data, is certainly desirable. Thus, recent articles5,6 havediscussed the experimental evidence that both passive lipoidaldiffusion and carrier-mediated transport permeation mecha-nisms affect the absorption and disposition of drugs. There isbroad consensus in the pharmaceutics field on the existence,effects, and application of knowledge about both passivelipoidal diffusion and carrier-mediated transport. Clearly, drugresearchers have, throughout the history of drug discovery,utilized physicochemical properties (lipophilicity, hydrogen-bonding capacity, molecular size, ionization/charge), whichhave been correlated to passive lipoidal diffusion permeationrate and not to carrier-mediated permeation rate, to enhancethe success of drug delivery.7−23 In recent years, drug discoveryresearchers have also utilized knowledge about transporteruptake to enhance drug exposure to certain tissues. Forexample, liver specific transporters (OATP1B1 and 1B3)selectively increase liver concentration of their substrates,which minimize the exposure to peripheral tissue and reducetoxicity.24,25
Founded on our experience with detailed experimentation ondrug permeation mechanisms, scientific literature evidence, andapplication of drug permeation mechanisms in drug research,we reiterate and support the roles of both passive lipoidaldiffusion and carrier-mediated transport in the prevailing drugpermeation theory. The purpose of this review is to provideguidance for drug researchers, so that potential drug candidateswill not be overlooked by mistaken evaluations that ignore theimportant effects of passive lipoidal diffusion.Prevailing Drug Permeation Philosophy Restated. The
prevailing theory of drug permeation through biologicalmembranes is as follows:
1. There are multiple permeation mechanisms by whichdrug molecules, in general, cross biological membranesand are absorbed and distributed throughout the body,including passive lipoidal diffusion, carrier-mediated influx,carrier-mediated efflux, paracellular diffusion, aqueousboundary layer (mucus) diffusion, and transcytosis.
2. The rate with which a given compound crosses specificbiological barriers (epithelium, endothelium) varies with(a) the compound’s properties (e.g., lipophilicity, pKa,and transporter parameters [Km and Vmax]), (b) themembrane’s properties (e.g., composition, transporterfunction, tight junction characteristics, and membranepotential), and (c) solution properties (pH, pH gradientacross membrane, and permeant concentration gradientacross membrane).
This theory is based on results from drug disposition inpharmaceutical research and from fundamental investigations asdiscussed below.
Unifying Role of Lipoidal Permeability in DrugDisposition. Overview. Tissues of the body are constructedof cells. Cells are encapsulated by membranes made ofphospholipid bilayers. Proteins are embedded in the cellmembrane and function as sensors for the cell, maintain theintracellular homeostasis, or transport nutrients and othersubstances. Cells are joined, through junction proteins, invarying degrees of tightness.Paracellular permeation occurs when drug molecules diffuse
through the cell junctions. Examples of the role of paracellularpermeation in defining drug permeation include the following:
1. For much of the vasculature of systemic circulation, thejunctions are not tight and allow aqueous diffusion ofeven large drug molecules. Thus, drug molecules incirculation are rapidly exposed to tissue cell surfaces.
2. The capillary vasculature in the brain has very tightjunctions between cells, forming the distinct blood−brainbarrier (BBB), which greatly attenuates the diffusion ofdrug molecules to the surface of the CNS tissue cells.
3. The cells of the upper gastrointestinal tract aremoderately tight and allow a portion of small drugmolecules (typically polar and less than 300 Da) to passthrough the junctions. These drugs can also reach cellsurfaces for many tissues but will have restricted access tothe CNS.
Paracellular diffusion does not account for most drugpermeation processes, such as for larger molecules, exposureto intracellular drug targets, or penetration to CNS drug targetsthrough the BBB. Experimental data also support twoadditional major permeation mechanisms: passive lipoidaldiffusion through the phospholipid membrane and carrier-mediated transport through one of the transporter proteins.The theoretical framework and the experimental evidence formembrane permeability of drugs have been reviewed.5,6
Additional recent data26 from fluorine NMR studies on uptakeof modified nucleosides (L-FMAU) into erythrocytes (bio-logical systems that include transporters) provide clearindication of two different mechanism governing uptake ofL-FMAU in erythrocytes: “facilitated transport via nucleosidetransporter and nonfacilitated diffusion”.Removal of drug and metabolite molecules from the body is
by metabolism, renal clearance, or “biliary excretion”. Again,transport proteins may play an important role for somecompounds. Passive lipoidal permeability has a central role incontrolling these processes, as outlined below.Passive lipoidal permeability is correlated positively with
lipophilicity (e.g., as expressed by the log of the octanol−waterpartition, log P, or the apparent value at a given pH, often7.4, log D) and negatively with the hydrogen-bonding capacityof a molecule (e.g., as expressed by the polar surface area, PSA,the solvatochromic parameters α (H-bond donor) and β(H-bond acceptor), and the molecular hydrogen-bondingpotentials, MHBPs).27−30 If a drug molecule has positive log Dand low PSA, passive lipoidal permeability is high and is a largefraction of the total permeability, owing to the large surface areaof the cell membrane. In this case, any transporter permeability isa low fraction of the total permeability, even if the compound hascarrier-mediated transport, because it is limited by the carrierconcentration and the Km (left diagram in Figure 1). If a drug
Molecular Pharmaceutics Review
dx.doi.org/10.1021/mp400713v | Mol. Pharmaceutics 2014, 11, 1727−17381728

molecule has negative log D and high PSA, passive lipoidalpermeability is low and, if the molecule has carrier-mediatedpermeability, a significant portion of the total permeability maybe carrier-mediated (right diagram in Figure 1). The schematicsshown in Figure 1 illustrate the separation between drugs ofhigh and low lipoidal permeability.The evidence for lipoidal diffusion is provided by many
experiments, and importantly the results are consistent forpassage across any cell type, organ, or species and are notenergy or concentration dependent.Evidence from in Vitro Monolayer Cell Systems. Lipoidal
diffusion is well exemplified by in vitro monolayer cell systemsused to predict or rationalize drug absorption. These measureflux in an apical to basolateral (A−B) direction and vice versa.A highly passive lipoidally permeable drug will show thefollowing:
• a comparatively high rate of apical to basolateral (A−B)flux of a group of compounds
• no effect of concentration on rate constant, up to thelimit of solubility
• near identical A−B and B−A flux regardless ofconcentration
• lack of effect of inhibitors of known transport proteins• similar rates of flux in cell lines selectively cultured for
low and high transporter expression.
There is only small variation in in vitro permeation rates ofdrugs between various cell layer permeation experiments inwhich large variation exists in transporters.31 The same is truein permeation rates of drugs between Caco-2 (from humancolon carcinoma) and MDCK (from canine kidney),32,33 wherethe following observations were made (Figure 2):
• There is a very close correlation between physicochem-ical systems (including PAMPA and liposomes) and cellbased systems.
• It does not require energy.• It happens in all organs, species, and cell types.
Typical MCDK35 and averaged Caco-236 results areillustrated in Table 1 for a series of beta adrenoceptor blockers.Permeation rate increases with increasing lipophilicity, asobserved in many experiments,21 and clearly indicated forCaco-2 in Table 1, which is consistent with passive diffusionthrough the lipophilic core of the bilayer membrane. The highpassive lipoidally permeable compounds (lipophilic, with lowpolar surface area such as bufarolol, alprenolol, and
propranolol) show identical high rates of transport in eitherdirection across the MDR1-MDCKII cells and are notinfluenced by the presence of a transporter inhibitor. Lesspassive lipoidally permeable compounds, such as labetalol,show different rates A−B and B−A (due to different heights ofthe energy barrier), and are markedly affected by the presenceof the transporter inhibitor. The transport of atenolol, a lowmolecular weight drug of low lipophilicity, is based onparacellular permeation. The permeation rate is related toionization constant pKa and pH according to the “pH-partitiontheory”37 and to intrinsic lipophilicity (log P), with permeationrate increasing in relation to increased concentration of acompound’s neutral species (to which log P refers), which isthe form that permeates the bilayer membrane while thecharged form has very low permeability. log D is a particularlyuseful term in explaining the interplay between intrinsiclipophilicity (log P) and pKa. In Table 1, the drugs havesimilar pKa, but the degree of ionization means that their log Dat pH 7.4 is approximately 2 log units lower than their intrinsiclipophilicity (log P). Other experiments show that permeationrate is well correlated between a biological membrane (e.g.,Caco-2), an artificial membrane (e.g., PAMPA), and octanol/water partition coefficient.21,38
Figure 1. Schematic relating to drugs and their dependence on passive lipoidal diffusion and carrier-mediated transport to cross membranes. Drugmolecules with balanced lipophilicity and relatively low hydrogen-bonding capacity rapidly cross membranes by lipoidal diffusion (white doublearrows), owing to the high cell surface area, at high rates compared to minimal transporter impact (gray unidirectional arrows). Drug molecules withgreater hydrogen-bonding capacity and lower lipophilicity cross membranes by lipoidal diffusion less readily and may, therefore, be influenced bytransporters for which they have substantial affinity and transport rate.
Figure 2. Caco-2 and MDCK intrinsic permeability, log P0 (unchargedform), values of 79 drugs thought to cross cell layers predominantly bypassive lipoidal diffusion. The expression of drug-transporting proteinsin the Caco-2 cell line differ substantially from laboratory tolaboratory.34 Furthermore, the expression of drug-transportingproteins in Caco-2 and other cell lines differs substantially.31 In spiteof that, the two cell models indicate nearly the same intrinsicpermeability, with r2 = 0.90, slope = 1 and intercept ∼0.33
Molecular Pharmaceutics Review
dx.doi.org/10.1021/mp400713v | Mol. Pharmaceutics 2014, 11, 1727−17381729
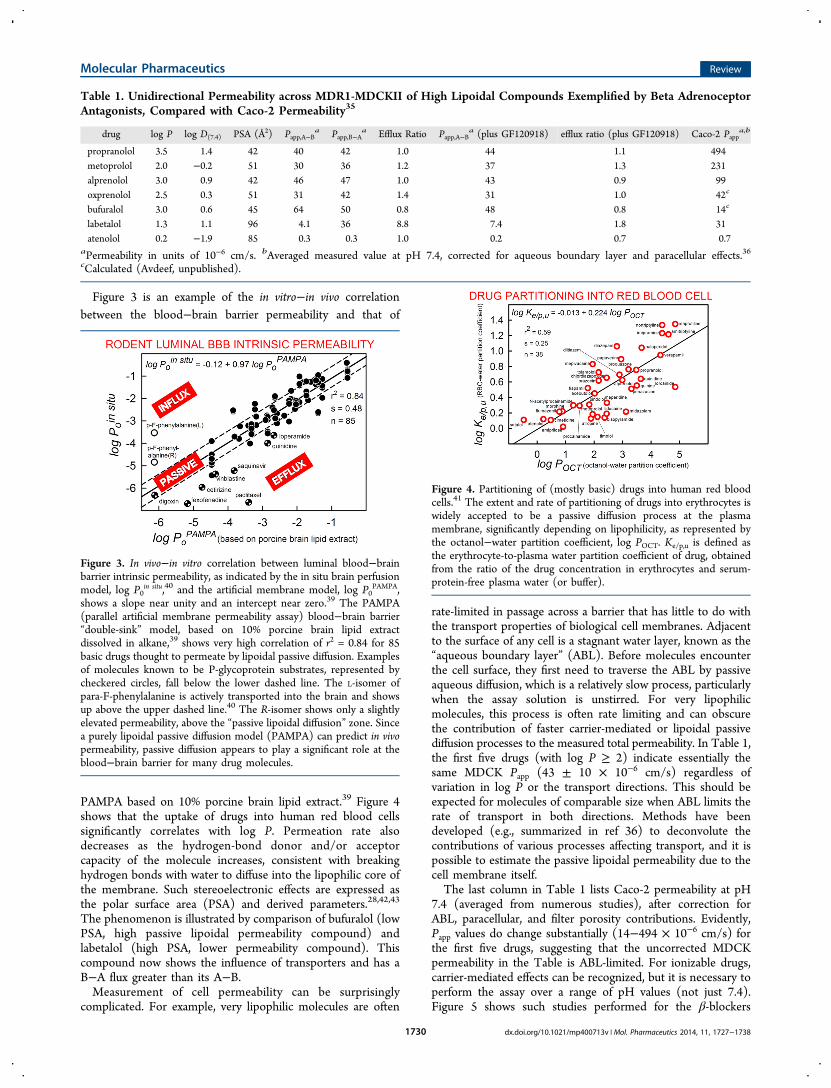
Figure 3 is an example of the in vitro−in vivo correlationbetween the blood−brain barrier permeability and that of
PAMPA based on 10% porcine brain lipid extract.39 Figure 4shows that the uptake of drugs into human red blood cellssignificantly correlates with log P. Permeation rate alsodecreases as the hydrogen-bond donor and/or acceptorcapacity of the molecule increases, consistent with breakinghydrogen bonds with water to diffuse into the lipophilic core ofthe membrane. Such stereoelectronic effects are expressed asthe polar surface area (PSA) and derived parameters.28,42,43
The phenomenon is illustrated by comparison of bufuralol (lowPSA, high passive lipoidal permeability compound) andlabetalol (high PSA, lower permeability compound). Thiscompound now shows the influence of transporters and has aB−A flux greater than its A−B.Measurement of cell permeability can be surprisingly
complicated. For example, very lipophilic molecules are often
rate-limited in passage across a barrier that has little to do withthe transport properties of biological cell membranes. Adjacentto the surface of any cell is a stagnant water layer, known as the“aqueous boundary layer” (ABL). Before molecules encounterthe cell surface, they first need to traverse the ABL by passiveaqueous diffusion, which is a relatively slow process, particularlywhen the assay solution is unstirred. For very lipophilicmolecules, this process is often rate limiting and can obscurethe contribution of faster carrier-mediated or lipoidal passivediffusion processes to the measured total permeability. In Table 1,the first five drugs (with log P ≥ 2) indicate essentially thesame MDCK Papp (43 ± 10 × 10−6 cm/s) regardless ofvariation in log P or the transport directions. This should beexpected for molecules of comparable size when ABL limits therate of transport in both directions. Methods have beendeveloped (e.g., summarized in ref 36) to deconvolute thecontributions of various processes affecting transport, and it ispossible to estimate the passive lipoidal permeability due to thecell membrane itself.The last column in Table 1 lists Caco-2 permeability at pH
7.4 (averaged from numerous studies), after correction forABL, paracellular, and filter porosity contributions. Evidently,Papp values do change substantially (14−494 × 10−6 cm/s) forthe first five drugs, suggesting that the uncorrected MDCKpermeability in the Table is ABL-limited. For ionizable drugs,carrier-mediated effects can be recognized, but it is necessary toperform the assay over a range of pH values (not just 7.4).Figure 5 shows such studies performed for the β-blockers
Table 1. Unidirectional Permeability across MDR1-MDCKII of High Lipoidal Compounds Exemplified by Beta AdrenoceptorAntagonists, Compared with Caco-2 Permeability35
drug log P log D(7.4) PSA (Å2) Papp,A−Ba Papp,B−A
a Efflux Ratio Papp,A−Ba (plus GF120918) efflux ratio (plus GF120918) Caco-2 Papp
a,b
propranolol 3.5 1.4 42 40 42 1.0 44 1.1 494metoprolol 2.0 −0.2 51 30 36 1.2 37 1.3 231alprenolol 3.0 0.9 42 46 47 1.0 43 0.9 99oxprenolol 2.5 0.3 51 31 42 1.4 31 1.0 42c
bufuralol 3.0 0.6 45 64 50 0.8 48 0.8 14c
labetalol 1.3 1.1 96 4.1 36 8.8 7.4 1.8 31atenolol 0.2 −1.9 85 0.3 0.3 1.0 0.2 0.7 0.7
aPermeability in units of 10−6 cm/s. bAveraged measured value at pH 7.4, corrected for aqueous boundary layer and paracellular effects.36cCalculated (Avdeef, unpublished).
Figure 3. In vivo−in vitro correlation between luminal blood−brainbarrier intrinsic permeability, as indicated by the in situ brain perfusionmodel, log P0
in situ,40 and the artificial membrane model, log P0PAMPA,
shows a slope near unity and an intercept near zero.39 The PAMPA(parallel artificial membrane permeability assay) blood−brain barrier“double-sink” model, based on 10% porcine brain lipid extractdissolved in alkane,39 shows very high correlation of r2 = 0.84 for 85basic drugs thought to permeate by lipoidal passive diffusion. Examplesof molecules known to be P-glycoprotein substrates, represented bycheckered circles, fall below the lower dashed line. The L-isomer ofpara-F-phenylalanine is actively transported into the brain and showsup above the upper dashed line.40 The R-isomer shows only a slightlyelevated permeability, above the “passive lipoidal diffusion” zone. Sincea purely lipoidal passive diffusion model (PAMPA) can predict in vivopermeability, passive diffusion appears to play a significant role at theblood−brain barrier for many drug molecules.
Figure 4. Partitioning of (mostly basic) drugs into human red bloodcells.41 The extent and rate of partitioning of drugs into erythrocytes iswidely accepted to be a passive diffusion process at the plasmamembrane, significantly depending on lipophilicity, as represented bythe octanol−water partition coefficient, log POCT. Ke/p,u is defined asthe erythrocyte-to-plasma water partition coefficient of drug, obtainedfrom the ratio of the drug concentration in erythrocytes and serum-protein-free plasma water (or buffer).
Molecular Pharmaceutics Review
dx.doi.org/10.1021/mp400713v | Mol. Pharmaceutics 2014, 11, 1727−17381730
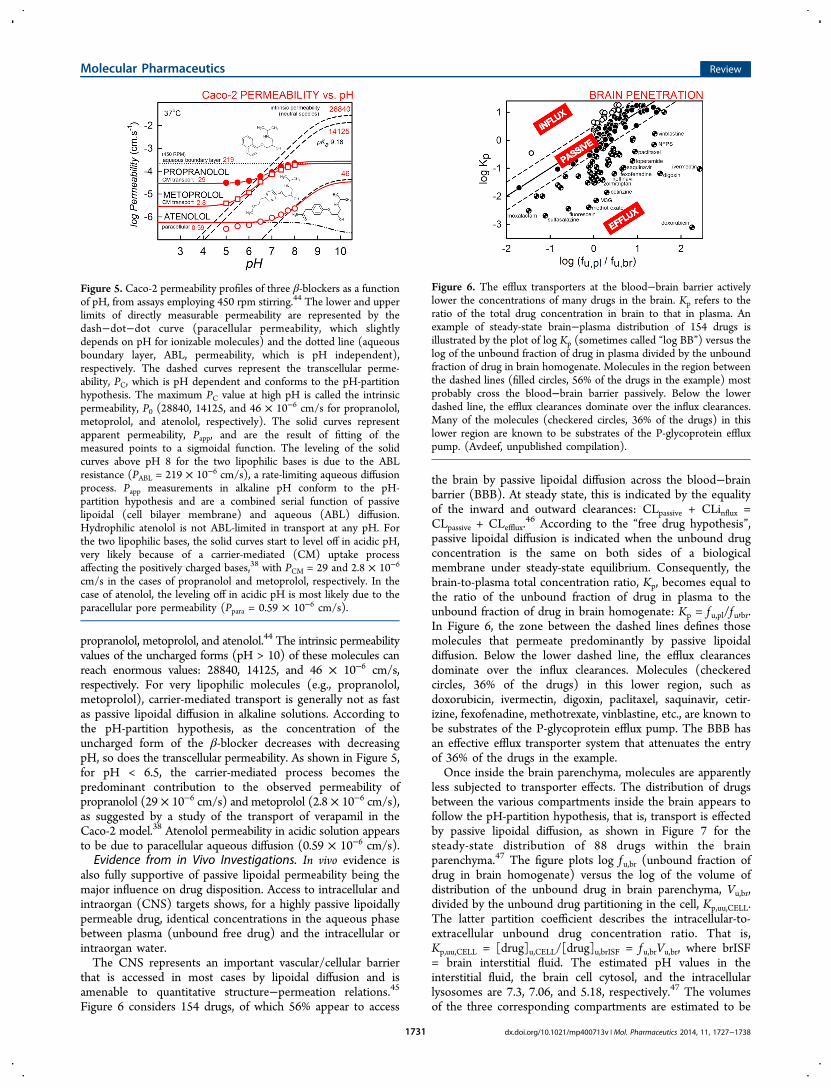
propranolol, metoprolol, and atenolol.44 The intrinsic permeabilityvalues of the uncharged forms (pH > 10) of these molecules canreach enormous values: 28840, 14125, and 46 × 10−6 cm/s,respectively. For very lipophilic molecules (e.g., propranolol,metoprolol), carrier-mediated transport is generally not as fastas passive lipoidal diffusion in alkaline solutions. According tothe pH-partition hypothesis, as the concentration of theuncharged form of the β-blocker decreases with decreasingpH, so does the transcellular permeability. As shown in Figure 5,for pH < 6.5, the carrier-mediated process becomes thepredominant contribution to the observed permeability ofpropranolol (29 × 10−6 cm/s) and metoprolol (2.8 × 10−6 cm/s),as suggested by a study of the transport of verapamil in theCaco-2 model.38 Atenolol permeability in acidic solution appearsto be due to paracellular aqueous diffusion (0.59 × 10−6 cm/s).Evidence from in Vivo Investigations. In vivo evidence is
also fully supportive of passive lipoidal permeability being themajor influence on drug disposition. Access to intracellular andintraorgan (CNS) targets shows, for a highly passive lipoidallypermeable drug, identical concentrations in the aqueous phasebetween plasma (unbound free drug) and the intracellular orintraorgan water.The CNS represents an important vascular/cellular barrier
that is accessed in most cases by lipoidal diffusion and isamenable to quantitative structure−permeation relations.45
Figure 6 considers 154 drugs, of which 56% appear to access
the brain by passive lipoidal diffusion across the blood−brainbarrier (BBB). At steady state, this is indicated by the equalityof the inward and outward clearances: CLpassive + CLinflux =CLpassive + CLefflux.
46 According to the “free drug hypothesis”,passive lipoidal diffusion is indicated when the unbound drugconcentration is the same on both sides of a biologicalmembrane under steady-state equilibrium. Consequently, thebrain-to-plasma total concentration ratio, Kp, becomes equal tothe ratio of the unbound fraction of drug in plasma to theunbound fraction of drug in brain homogenate: Kp = f u,pl/fu,br.In Figure 6, the zone between the dashed lines defines thosemolecules that permeate predominantly by passive lipoidaldiffusion. Below the lower dashed line, the efflux clearancesdominate over the influx clearances. Molecules (checkeredcircles, 36% of the drugs) in this lower region, such asdoxorubicin, ivermectin, digoxin, paclitaxel, saquinavir, cetir-izine, fexofenadine, methotrexate, vinblastine, etc., are known tobe substrates of the P-glycoprotein efflux pump. The BBB hasan effective efflux transporter system that attenuates the entryof 36% of the drugs in the example.Once inside the brain parenchyma, molecules are apparently
less subjected to transporter effects. The distribution of drugsbetween the various compartments inside the brain appears tofollow the pH-partition hypothesis, that is, transport is effectedby passive lipoidal diffusion, as shown in Figure 7 for thesteady-state distribution of 88 drugs within the brainparenchyma.47 The figure plots log f u,br (unbound fraction ofdrug in brain homogenate) versus the log of the volume ofdistribution of the unbound drug in brain parenchyma, Vu,br,divided by the unbound drug partitioning in the cell, Kp,uu,CELL.The latter partition coefficient describes the intracellular-to-extracellular unbound drug concentration ratio. That is,Kp,uu,CELL = [drug]u,CELL/[drug]u,brISF = f u,brVu,br, where brISF= brain interstitial fluid. The estimated pH values in theinterstitial fluid, the brain cell cytosol, and the intracellularlysosomes are 7.3, 7.06, and 5.18, respectively.47 The volumesof the three corresponding compartments are estimated to be
Figure 5. Caco-2 permeability profiles of three β-blockers as a functionof pH, from assays employing 450 rpm stirring.44 The lower and upperlimits of directly measurable permeability are represented by thedash−dot−dot curve (paracellular permeability, which slightlydepends on pH for ionizable molecules) and the dotted line (aqueousboundary layer, ABL, permeability, which is pH independent),respectively. The dashed curves represent the transcellular perme-ability, PC, which is pH dependent and conforms to the pH-partitionhypothesis. The maximum PC value at high pH is called the intrinsicpermeability, P0 (28840, 14125, and 46 × 10−6 cm/s for propranolol,metoprolol, and atenolol, respectively). The solid curves representapparent permeability, Papp, and are the result of fitting of themeasured points to a sigmoidal function. The leveling of the solidcurves above pH 8 for the two lipophilic bases is due to the ABLresistance (PABL = 219 × 10−6 cm/s), a rate-limiting aqueous diffusionprocess. Papp measurements in alkaline pH conform to the pH-partition hypothesis and are a combined serial function of passivelipoidal (cell bilayer membrane) and aqueous (ABL) diffusion.Hydrophilic atenolol is not ABL-limited in transport at any pH. Forthe two lipophilic bases, the solid curves start to level off in acidic pH,very likely because of a carrier-mediated (CM) uptake processaffecting the positively charged bases,38 with PCM = 29 and 2.8 × 10−6
cm/s in the cases of propranolol and metoprolol, respectively. In thecase of atenolol, the leveling off in acidic pH is most likely due to theparacellular pore permeability (Ppara = 0.59 × 10−6 cm/s).
Figure 6. The efflux transporters at the blood−brain barrier activelylower the concentrations of many drugs in the brain. Kp refers to theratio of the total drug concentration in brain to that in plasma. Anexample of steady-state brain−plasma distribution of 154 drugs isillustrated by the plot of log Kp (sometimes called “log BB”) versus thelog of the unbound fraction of drug in plasma divided by the unboundfraction of drug in brain homogenate. Molecules in the region betweenthe dashed lines (filled circles, 56% of the drugs in the example) mostprobably cross the blood−brain barrier passively. Below the lowerdashed line, the efflux clearances dominate over the influx clearances.Many of the molecules (checkered circles, 36% of the drugs) in thislower region are known to be substrates of the P-glycoprotein effluxpump. (Avdeef, unpublished compilation).
Molecular Pharmaceutics Review
dx.doi.org/10.1021/mp400713v | Mol. Pharmaceutics 2014, 11, 1727−17381731

0.2, 0.79, and 0.01 mL per gram of brain tissue, respectively.For intrabrain distribution by passive lipoidal diffusion,Kp,uu,CELL can be simply calculated by taking into account thepKa of the drug and the pH and volumes of the three braincompartments, as demonstrated elegantly. Examples of passivedistribution are shown in Figure 7 by filled circles (87% of the
drugs in the example) bounded by the two dashed lines. Thereappear to be net efflux processes for NFPS, sertraline,sumatriptan, and trifluoperazine (checkered circles, 5% of thedrugs), driving compounds from brain cells into the braininterstitial fluid. Examples of net clearance from the interstitialfluid into the brain cells are shown by molecules represented byunfilled circles (8% of the drugs), several of which bearpermanent positive charge.47 (Moxalactam is likely to be in thepassive lipoidal diffusion group, but it appears as an outlier sinceits measured f u,br > 1, the physically meaningful upper limit.)It is now widely accepted that CNS activity is determined by
the unbound brain drug concentration, Cu,br, and not the totalconcentration in the brain, Cbr
TOT. For those molecules that aresubstrates of receptors accessible from the interstitial fluid, theunbound concentration in the interstitial fluid, Cu,brISF, is bestcorrelated with receptor occupancy. This was experimentallydemonstrated for 18 molecules, where more accurate predictionof the biophase drug concentration was achieved by using Cu,brrather than Cbr
TOT.48 To a lesser degree of accuracy, theunbound plasma concentration, Cu,pl, could be used to predictthe biophase concentration in the brain.Considerable amounts of human data are available. This is
exemplified by studying sedating (highly passive lipoidalpermeable) and nonsedating (lower passive lipoidally perme-able, influenced by efflux transporters) H1 receptor antagonists(antihistamines).49 Chorpheniramine, in contrast to the non-sedating antihistamine ebastine, when studied with PETscanning, had a Kd calculated from CNS receptor occupancy(7 nM) and unbound plasma concentrations matching thatmeasured in in vitro receptor experiments (3−4 nM). Thereceptor occupancy of ebastine was much lower than its plasmaconcentration would suggest. Ebastine rapidly forms thezwitterionic (low passive lipoidal permeability) active metab-olite cavebastine and is partially excluded from the brain due tothis and the activity of efflux transporters.
The clearance of highly lipid-permeable drugs mirrors all theabove. Even if the drug is a possible substrate of a transporter,clearance is invariably metabolic. This is evidenced by thefollowing:
1. A normally negligible excretion or clearance of alipophilic compound in urine. Even though the unbounddrug is filtered at the glomerulus and could beconcentrated in the renal tubule by transporterinvolvement, passive lipoidal diffusion means the drugis passively reabsorbed as the urine is concentrated,resulting in negligible urinary excretion of all highlylipoidally permeable drugs.
2. When urinary pH is altered, compelling proof of lipoidalpermeability is shown for drugs with a positive log Pvalue (i.e., whose neutral form is lipophilic) and anegative log D value (i.e., whose ionized form ishydrophilic).50−52 Thus, the basic drug amphetamine(log P 1.8, pKa 9.94, TPSA 26 Å2) is poorly excreted(3−7%) in alkaline urine and markedly excreted (60%)in acidic urine. This behavior is due to the drugundergoing renal filtration, followed by passive reabsorp-tion of the lipophilic neutral form, or lack of reabsorptionof the hydrophilic cationic form. Conversely, acetylsali-cylic acid (log P 0.90, pKa 3.50, TPSA 64 Å2) showsgreater excretion at a urinary pH of 7 (3%) comparedto pH 5.5 (1%). Memantine, a basic drug (log P 2.1,pKa 10.2, TPSA 26 Å2) has been examined at urinary pHvalues of 5 and pH 8 and shows a 7−10-fold higher renalclearance at acidic compared to basic urinary pH.53 AtpH 5 unbound renal clearance values (6 mL/min/kg)exceed glomerular filtration rate (2 mL/min/kg),indicating some active tubular secretion in addition toglomerular filtration, illustrating how transport proteinscan exist alongside passive diffusion processes. It isdifficult to conceive of a system able to explain the effectsof pH on urinary excretion and clearance other thanpassive lipoidal diffusion.
3. The rapid transfer of drugs in both directions across thesinusoidal membrane results in intracellular hepatocyteconcentrations approximating unbound plasma concen-trations. Although not direct evidence, this probablyexplains the utility of the well-stirred liver model and itsrobust use in in vitro to in vivo predictions of clearancefor highly lipid permeable drugs.
Thus, for an oral, high lipoidal permeability drug, only duringthe absorption process will a concentration gradient exist in thebody and free drug concentrations will be identical betweenplasma water, extracellular water, and total body water. Theabsence of concentration gradient indicates the passivediffusion nature of the process.When a drug has moderate or low passive lipoidal
permeability, behavior different from that outlined aboveoccurs in a predictable manner. As the rate of passive lipoidaldiffusion is lower, the influence of transporter proteins canbecome apparent. Transport gradients will now exist across theaqueous compartments of the body. The evidence for thepassive diffusion of highly permeable drugs now is reflected inalmost an inversion of all the statements above for drugs ofmoderate to low lipid permeability. These concepts allow therational understanding of drug disposition based on compoundcharacteristics.
Figure 7. Many CNS drugs distribute inside the brain by passivelipoidal diffusion. Examples are shown by filled circles (87% of thedrugs in the example) bounded by the two dashed lines. There appearto be net efflux processes (checkered circles, 5% of the drugs), drivingcompounds from brain cells into the brain interstitial fluid. Examplesof net clearance from the interstitial fluid into the brain cells are shownby molecules represented by unfilled circles (8% of the drugs).47
Molecular Pharmaceutics Review
dx.doi.org/10.1021/mp400713v | Mol. Pharmaceutics 2014, 11, 1727−17381732

Transdermal Medicines: A Case for Passive Permeation.Skin was originally considered an essentially impermeablebarrier whose function was restricted to protecting animalorganisms from their environment. Since years, it is now knownthat the skin also represents a potential portal of entry by whichmany chemicals may gain access to the systemic circulation.54
This route of administration of systemically acting drugsprovides strong evidence for the determining involvement ofpassive permeation processes in their transdermal permeation.During skin absorption, drugs have to pass a complex
multilayer structure before reaching the blood.55,56 Structurally,the skin can be viewed as a series of layers, the three majordivisions being epidermis, dermis, and subcutis or hypodermis.The latter lies below the dermis and the vascular system, and istherefore not relevant to percutaneous penetration. The dermis(2 mm) is essentially an acellular collagen-based connectivetissue which supports the many blood vessels, lymphaticchannels, and nerves of the skin. Hair follicles and sweat glandsoriginate in this layer and open directly onto the skin surface.The avascular epidermis consists of stacked layers of cellswhose thickness varies between about 0.1 mm (eyelids) and0.8 mm (palms, soles).57 The outermost layer of the epidermis,namely, the stratum corneum, is composed of 10−15 layers offlat keratin-filled cells, closely packed in a nonpolar lipid matrixcomposed of ceramides, cholesterol, and fatty acids, but largelydevoid of phospholipids.58,59 In contrast and to the best of ourknowledge, there is no influx transporter in the stratumcorneum.The transdermal administration of systemically acting topical
medications relies on specific formulations (e.g., hydrogels,creams, ointments, patches, or disks), from which drugs exitdown a concentration gradient. Given the absence oftransporters, the main rate-determining step in skin penetrationis then diffusion of the solute across the stratum corneum,which represents the principal penetration barrier of theskin.57,60 Under conditions of normal passive diffusion, itappears that the intercellular route predominates over thetranscellular route.61−63 Moreover, an important property ofthe stratum corneum is its ability to become heavily hydrated,modifying also the permeation profile of chemicals.57,64
A direct in vivo proof that transcutaneous medicines areindeed absorbed through the stratum corneum and able toreach their target tissues or organs is found in the mere fact thatthey were cleared for marketing. Table 2 (data taken mainlyfrom the Canadian Drug Bank65) offers a short list oftransdermally active medicines to illustrate the variety oftissues and organs they can target (muscles and joints, sexhormone receptors, central receptors, etc.). Also noteworthy isthe fact that basic, acidic, and neutral compounds coexist in thislist, as is the wide range covered by log P (1−4.1) and pKa
values (4−10). Indirect evidence obtained from the statisticalanalysis of in vivo and in vitro skin permeation studies haverevealed excellent correlations with molecular descriptors suchas molecular weight and volume, log P, log D, and H-bondingparameters.66−71
Why Debate the Role of Passive Lipoidal Perme-ability? There are now authors who doubt the role of passivelipoidal diffusion in drug permeation. It is possible their view iscolored by a growing interest in transporters as drugs becomeless lipid-permeable to conform to target active site character-istics (see above). Some of these authors state that carriermediation is the only existing form of permeation, at theexclusion of lipoidal permeation (CMO concept). Whilehealthy debate is warranted, the group most advocating theCMOC has made statements to discredit passive lipoidaldiffusion and its foundational evidence. This is contrary to theconclusion of many, including the authors. Passive lipoidaldiffusion exists alongside carrier-mediated transport, and thepathways are defined by physicochemical characteristics. Theproponents of the CMOC state that “...evidence and reasoningwe describe may be seen as circumstantial...”.1 The statementsagainst passive lipoidal diffusion have been strongly refuted,5,6
and a brief recounting follows.A Debate on the CMOC or an Integrated System Defined
by Lipoidal Diffusion. The major points made in support of theCMOC are followed by a response. Readers will find greaterdetail in the articles.5,6 Arguments 1−13 are from Dobson andKell;1 argument 14 is from Dobson et al.;2 argument 15 is fromKell et al.:3
1. Lipophilic cations are charged and cannot crossmembranes owing to Born charging. Response: Drugmolecule ions are in equilibrium with neutral nonionizeddrug molecules, which have much higher lipophilicityand much higher passive diffusion permeation rate.According to the pH-partition theory, permeation ratevaries with solution pH and a compound’s pKa such thatan increasing ratio of nonionized/ionized formscorrelates with increasing permeation rate. The termlog D is a descriptor incorporating the effects oflipophilicity and ionization.6
2. The mass ratio of protein:lipid in vivo (1/1 to 3/1)affects the transport properties of lipids. Artificialmembranes do not model biological membranes, owingto the high protein content in vivo. Response: Theseratios include the cytoplasmic and exoplasmic portions ofmembrane protein mass, not just the relevant trans-membrane fraction. The lipid:protein molar ratio isestimated as 40:1, making lipid an important portion ofthe membrane exposed to drug molecules.6 A furtherrefined consideration would take into account the
Table 2. Representative Transdermal Medicines
drug therapeutic class major indication formulation MW log P pKa
diclofenac NSAID locomotor pain hydrogel, cream 296.1 4.5 4.117β-estradiol estrogenic agonist hormone replacement therapy gel, patch, disk 272.4 4.0 10.3fentanyl μ-opioid agonist narcotic analgesia patch 336.5 4.1 8.8glycol salicylate presumably a prodrug of salicylic acid locomotor pain hydrogel, cream 182.2 2.1nicotine nicotinic agonist nicotine withdrawal disk, patch 162.2 1.2 8.8nitroglycerine vasodilator angina pectoris disk, patch 227.1 1.2scopolamine muscarinic antagonist motion sickness disk 303.4 0.98 6.9selegiline MAO-B inhibitor Parkinson’s disease patch 187.3 2.7 8.7testosterone androgenic agonist hormone replacement therapy patch, hydrogel 288.4 3.3
Molecular Pharmaceutics Review
dx.doi.org/10.1021/mp400713v | Mol. Pharmaceutics 2014, 11, 1727−17381733

relative cross-sectional area at the membrane surface of the40 phospholipid molecules to one typical protein. Thelipid surface area would still be significantly greater thanthat of the transporter protein.
3. Correlations of drug uptake with log P and Caco-2permeation can be weak. Response: For drugspermeating predominantly by passive lipoidal diffusion,the apparent Caco-2 permeability coefficient, Papp, can be(and often is) affected by the aqueous boundary layer,filter, paracellular, and lipoidal (transcellular) perme-ability, as well as the solution pH, as illustrated by theexamples in Figure 5. log P cannot be directly comparedto log Papp. The Caco-2 intrinsic permeability, log P0, isthe rational term to compare to log P. P0 is easy todeduce from Papp,
36 but this is seldom done, which oftenleads to “weak” correlation, as “apple seeds are comparedto whole watermelons.” Caco-2 cells from 10 differentlaboratories were compared in terms of transportermRNA levels of 72 drug and nutrient transporters, and17 other targets. It was concluded that “Caco-2 cells fromdifferent laboratories produce different results even whenusing standard protocols for transport studies. Thedifferences may be due to transporter expression asshown for e.g. PepT1 and MDR1 which in turn isdetermined by the culture conditions. Although themajority of the laboratories used similar cultureconditions, absolute expression of genes was variableindicating that even small differences in cultureconditions have a significant impact on gene expression,although the overall expression patterns were similar”.34
Therefore, it is not astonishing that results of Caco-2cell based permeabilities, when correlated with octanollog P/D values, sometimes show differences incorrelations. This is mainly due to the origin andcomposition of the analyzed data set (ratio activelyversus passively [lipoidal diffusion] transported com-pounds) and use of partition coefficients (log P) or pH-dependent distribution coefficients (log D). Interestingly,correlations of transport studies performed with differentcell lines (e.g., Caco-2/MDCK) commonly used inabsorption prediction, with presumably different trans-porterexpression levels, often give excellent correlations,32
further supporting the coexistence of active and passivetransport in biological systems.
4. Transport across model artificial membranes is stated tooccur via pore defects or dissolution in the lipid mixturethat are not seen in vivo. Response: The studies citedare computational simulations (so-called moleculardynamics) of Na+ and Cl− ion (non-drug-like) transportunder unusual conditions. No convincing experimentalevidence for the relevance of pores has been reported.Other experiments indicate the unimportance of pores.Membrane resistance excluding pore diffusion is usuallydetermined by conductivity measurements. Otherwisefunction of, e.g., ion channels could not be determined.6
5. A dominant role for carrier-mediated transport (andagainst passive diffusion) is inferred from the hundreds ofpublications on drug transporters. Response: A largenumber of papers have been published in recent years ontransporters. These result from the recent intenseresearch on transporters. However, it is a logical fallacyand a sleight of hand to state that this is evidence of therate and extent of dominance of carrier-mediated
permeation over passive lipoidal diffusion. (An analogywould be to state that newspapers contain a predom-inance of articles about bad events (e.g., fires, wars,violence, accidents), therefore, bad events dominate goodevents in the world.) Thus, the large number of citationsof publications on transporter research is misleading,because the research they report or review was notundertaken nor concluded by the publication authors asevidence that supports CMOC, as is implied (“There isconsiderable and increasing evidence that drugs get intocells more or less solely by hitchhiking on carriersnormally used for the transport of nutrients andintermediary metabolites”.3
6. Selected small molecules, urea and glycerol, which crossBLM, permeate to some extent in vivo via transporters,except in yeast because glycerol is an osmolyte.Response: Urea and glycerol are more hydrophilicthan typical drugs that permeate membranes, thus, theyare not good models of permeants on which to supporttheories.
7. In liposomes the rate of transfer of nonelectrolytesdepends on MW rather than log P. Response: Liposomescorrelate well with the permeation behaviors usuallyobserved in artificial and biological membranes.18,36,72−74
Molecular weight is partly correlated with lipophilicityand hydrogen-bonding capacity, and as molecular weightincreases in drugs normally so does hydrogen bonding.Molecular weight is therefore a hybrid term expected toshow a relationship to lipoidal membrane permeation.
8. In vitro models of diffusion rates across membranes arenot based on large sample numbers and validated withcompounds not used in the method development.Response: This is out of date information.36
9. The flux across in vitro PAMPA membranes can be pooreven when human absorption is good (e.g., cephalexin,tiacrilast). Response: This is out of date information andalso is misleading. PAMPA membranes serve to modelpassive diffusion, whereas cephalexin and a number ofother molecules are carrier-mediation transported, asextensively compiled.36 The present authors claim thatpassive and active transport processes coexist. PAMPAhas been described to only account for passivemembrane permeation processes. Therefore, it is notastonishing that actively transported compounds like,e.g., cephalexin cannot be correctly predicted regardinghuman absorption by methods exclusively focusing onpassive transport.
10. Activities of anesthetics were previously thought to becontrolled by passive diffusion and correlated to log P,but are now thought to be protein-binding related.Response: There is common agreement that drugmolecules and anesthetics might interact with proteins,but this is misleading in the context of the discussion,which is around drug transport and not mechanism ofaction. A recent publication on anesthetics hassummarized thus: “The molecular mechanism of generalanesthesia is still a controversial issue. Direct effect bylinking of anesthetics to proteins and indirect action onthe lipid membrane properties are the two hypotheses inconflict.”75
11. Many molecules (e.g., ethanol) have relatively specificreceptors, so they may have similar protein binding(unidentified) that effects membrane permeation.
Molecular Pharmaceutics Review
dx.doi.org/10.1021/mp400713v | Mol. Pharmaceutics 2014, 11, 1727−17381734

Response: This is an assumption and generalizationawaiting to be proven by experimental data, but whichcurrently does not rule out transport by passive (lipoidaldiffusion) mechanism.
12. Carrier-mediated drug uptake is observed where it hasbeen studied. (Presumably this circumstantially indicatesthat transporters will be found for all drugs.) Response:Carrier-mediated drug uptake may be observed, but itmay not account for 100% of the transport. InMichaelis−Menten analysis, the nonsaturable termusually is related to the passive diffusion contribution.
13. Drugs can concentrate in specific tissues beyond thestoichiometry of internal binding sites. This phenomen-on absolutely requires an active uptake process.Response: This can be due to pH gradients betweenintracellular and extracellular compartments as describedfor, e.g., basic amines and safety relevant lysosomeaccumulation (phospholipidosis).76
14. Biophysical forces in drug−lipid membrane interactions(e.g., lipophilicity, hydrogen bonding) are no differentfrom drug−protein interaction. Thus, physicochemicalproperties and the rule of 5 need not be evidence ofpassive diffusion.77 Response: Of course biophysicalforces apply to both carriers and bilayers. However, thephysical property differences between the rate limitingbarriers for a particular drug in carriers and bilayers candictate the predominant route of transport.19 A secondargument is that ligand−protein recognition is domi-nated by highly selective stereoelectronic features farmore than by global (molecular) physicochemicalproperties.78−80
15. The notion of passive lipoidal permeation is traced backto artificial membrane systems, which are not successfulpredictors of membrane permeation. Response: On thecontrary, artificial membrane models have been success-ful predictors of passive lipoidal permeation.11,14,17−20,36
Accurate Understanding of Drug Permeation Mech-anisms is Important for Drug Development Success. Amajor value of the prevailing permeation hypothesis is that itguides drug research and development, providing a reliablefoundation for lead selection, candidate optimization, preclin-ical studies, and product development. Therefore, it isimperative that accurate permeation theories are implemented.Translation of a discovery therapeutic “hit” to a clinicalcandidate, in part, requires accurate prediction of drug exposureto the target, which is enabled, along with models of other drugproperties (e.g., solubility, metabolism), by an accuratepermeation model. The prevailing permeation hypothesis hasfacilitated success in drug design, interpretation of pharmaco-kinetics, and planning clinical dose and dosing regimens.Permeability is a component of many in vivo pharmacokinetic
processes in drug absorption and disposition, including thefollowing examples in which permeation rate can be rate-determining:
• absorption: permeation across the gastrointestinalepithelial membranes to reach blood capillaries
• hepatic clearance: permeation across the sinusoidal andcanalicular hepatocyte cell membrane
• renal clearance: permeation across the nephron tubules• CNS exposure: permeation across blood−brain barrier• efficacy: permeation across the cell membrane for
intracellular targets
Permeability can also affect in vitro cell-based assays in drugdiscovery because molecules must cross cell membranes toreach intracellular targets.Many of the rates for the preceding permeability processes
are measured using in vitro and in vivo protocols during drugdiscovery and development to observe the rates of permeationof an experimental compound in compound screening for leadoptimization and in detailed characterization for preclinicalstudies predicting human pharmacokinetics and safety. The insilico, in vitro, and in vivo tools that have been developed usingthe prevailing drug permeation hypothesis provide reliablepredictions of drug disposition in vitro, in preclinical species andin human. Drug researchers regularly apply these tools and haveachieved notable progress and success in drug design andoptimization.81
Major New Evidence Is Needed To Support theCMOC. There is insufficient evidence supporting CMOC tolead to its significant application in drug development, and theprevailing permeation hypothesis continues to be widelyapplied. A fresh collection of experiments initiated based onthe unique claims of CMOC has not been reported. Com-mentaries on CMOC have mainly focused on reinterpretationof data from earlier experiments, not excluding elements ofconfirmation bias.82 Only one research paper83 with resultsfrom experiments that are based on CMOC found previouslyunknown linkage of carriers to permeability of selectedcompounds that are toxic to yeast. These carriers have not,to our knowledge, been validated and characterized with thetype of rigor expected of any new target or transporter in drugresearch or for publication.Since its first description in 2008, the CMOC has not been
the basis for new advances in drug delivery and disposition.There have been no original research articles, attributed to afoundation of CMOC, in which carriers were demonstrated tocompletely explain the permeation of clinical drugs that werepreviously thought to permeate mainly by passive lipoidaldiffusion. Of the many in silico tools used widely in drugdisposition prediction, CMOC has not been embraced forincorporation. Considerable experimental evidence and peerreview concurrence would be needed to gain acceptance forCMOC, in accordance with the usual practices for permeationresearch. These include the following:
1. For the large number of drugs that are currentlyconcluded to primarily permeate via passive diffusion,the CMOC should provide data that
a. Identify the specific transporter(s), whether knownor new, that transport each. Note that the CMOCpostulates the existence of a large number ofunidentified transporters to explain transportacross any lipoidal barrier. Specific evidence,however, is still missing.
b. Account quantitatively for the known permeationrates and in vivo disposition effects. Indeed, theCMOC proponents note that many approveddrugs interact with a transporter,4 but no data areprovided to characterize the rate and extent ofthese interactions and effects on in vivo disposition.For each tissue to which the drug is known to haveexposure, proof is needed that the purportedtransporter(s) are in sufficient concentration toproduce the observed level of drug exposure.
Molecular Pharmaceutics Review
dx.doi.org/10.1021/mp400713v | Mol. Pharmaceutics 2014, 11, 1727−17381735

c. For cases where the drug is known to permeatetranscellularly (i.e., across both membranes, suchas the gastrointestinal epithelium, blood−brainbarrier, Caco-2), CMOC-based investigationsshould provide evidence for both the apical andbasolateral transporters for each drug andmetabolites. Furthermore, where the drug isknown to permeate transcellularly in bothdirections (i.e., apical-to-basolateral and baso-lateral-to-apical, such as in Caco-2), CMOCproponents should identify the transporters inboth apical and basolateral membranes that allowtransport in both directions for that membrane.
2. Discover major new influx transporters whose substratesare lipophilic compounds.
a. These carriers should recognize and transportcompounds having a physicochemical profile(lipophilicity, MW, hydrogen-bonding capacity)currently linked to passive lipoidal diffusion. Thisdemand is in line with the argument of CMOCproponents that new, yet unidentified, transporterswill transport lipophilic compounds that satisfy therule of 5, rules 1 to 4.
b. There is a clear need to clone the genes for thepurported new transporters, express them, andstudy their characteristics in an in vitro cell line.These transporters should demonstrate the follow-ing common transporter characteristics, except inlimited cases:
i. Display saturation of permeation rate withincreasing drug concentration, since it is farfrom sufficient to speculate, on behalf ofCMOC, that there are so many transportersfor every compound that permeation ratedoes not saturate. Note however that a fewtransporters (e.g., PEPT1) that have veryhigh capacity are limited cases that do notreadily saturate.
ii. Show inhibition of permeation rates by aninhibitor.
iii. Show stereospecific differences in perme-ation rates.
c. Exhibit variations of permeation rate with pH thatare consistent with the pH-partition theory.
d. Show that the transporters’ permeation rates followthe observed concentration gradient dependence.
3. Discover and characterize new high-flux transporters inCaco-2, MDCK, 2/4/1A, and other cell lines commonlyused for in vitro cell layer permeation experiments. Thisdemand is warranted by the speculations of CMOCproponents that many new significant transporters willbe discovered in these highly studied cell lines.
4. Discover a mechanism that stops or slows down passivelipoidal diffusion in biological membranes. This demandis justified by the fact that passive lipoidal diffusion hasbeen demonstrated in liposomes in vitro, and these are amodel of the observed passive lipoidal permeation ofbiological membranes, as discussed above.
■ CONCLUSIONDebates on important scientific points are necessary andvaluable for advancing science. The current debate, in our view,
has contained some unusual elements. First, we agree with thesignificant role of carrier-mediated transport in drug perme-ation, however, we disagree with the attempt to invalidate thewell-established scientific process of passive lipoidal diffusion,which is done, seemingly, to support the new CMO concept.The arguments against passive lipoidal diffusion are frequentlyillogical, and they cherry-pick the data and overlook the largebody of data that support the major contribution of passivelipoidal diffusion in drug permeation. Second, insufficientexperiments have been planned, conducted, and reported thatare based on the necessary outcomes of CMOC, which woulddifferentiate it from the prevailing permeation theory. Third, wedo not think that CMOC is widely accepted and supported, ashas been implied by the statement, “There is burgeoningevidence for the carrier-mediated view of drug uptake...”.4 Thereferences that are listed in support of this view are reports oftransporter research or literature reviews by authors who do notconclude that their research supports CMOC. Until significantdirect evidence is provided that supports CMOC, drugresearchers should withhold decisions based on CMOC thatcould affect drug discovery and development.Expansion of investigation on drug permeation is greatly
encouraged. The prevailing drug permeation hypothesis, whichincorporates several parallel active and passive permeationmechanisms (i.e., passive lipoidal diffusion, carrier-mediatedinflux, carrier-mediated efflux, paracellular diffusion, mucusresistance, endocytosis, transcytosis), whose rates are deter-mined by the properties of the compound and the membrane,is consistent with the experimental evidence and providessuccessful predictions for drug discovery and development.CMOC has not produced new evidence and predictions thatwarrant changing drug permeation theories, nor theirapplication to drug research.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] Address▽21705 Mobley Farm Drive, Laytonsville, MD 20882, UnitedStates.NotesThe authors declare no competing financial interest.
■ REFERENCES(1) Dobson, P. D.; Kell, D. B. Carrier-mediated cellular uptake ofpharmaceutical drugs: an exception or the rule? Nat. Rev. DrugDiscovery 2008, 7, 205−220.(2) Dobson, P. D.; Lanthaler, K.; Oliver, S. G.; Kell, D. B.Implications of the dominant role of transporters in drug uptake bycells. Curr. Top. Med. Chem. 2009, 9, 163−181.(3) Kell, D. B.; Dobson, P. D.; Oliver, S. G. Pharmaceutical drugtransport: the issues and the implications that it is essentially carrier-mediated only. Drug Discovery Today 2011, 16, 704−714.(4) Kell, D. B.; Dobson, P. D.; Bilsland, E.; Oliver, S. G. Thepromiscuous binding of pharmaceutical drugs and their transporter-mediated uptake into cells: what we (need to) know and how we cando so. Drug Discovery Today 2013, 18, 218−239.(5) Sugano, K.; Kansy, M.; Artursson, P.; Avdeef, A.; Bendels, S.; Di,L.; Ecker, G. F.; Faller, B.; Fischer, H.; Gerebtzoff, G.; Lennernas, H.;Senner, F. Coexistence of passive and carrier-mediated processes indrug transport. Nat. Rev. Drug Discovery 2010, 9, 597−614.(6) Di, L.; Artursson, P.; Avdeef, A.; Ecker, G. F.; Faller, B.; Fischer,H.; Houston, J. B.; Kansy, M.; Kerns, E. H.; Kramer, S. D.; Lennernas,H.; Sugano, K. Evidence-based approach to assess passive diffusion
Molecular Pharmaceutics Review
dx.doi.org/10.1021/mp400713v | Mol. Pharmaceutics 2014, 11, 1727−17381736

and carrier-mediated drug transport. Drug Discovery Today 2012, 17,905−912.(7) Overton, E. Ueber die allgemeinen osmotischen Eigenschaftender Zelle, ihre vermutlichen Ursachen und ihre Bedeutung fur diePhysiologie. Vierteljahrsschr. Naturforsch. Ges. Zurich 1899, 44, 88−135.(8) Overton, E. Studien uber die Aufnahme der Anilinfarben durchdie lebende. Zell. Jahrb. Wiss. Bot. 1900, 34, 669−701.(9) Collander, R. The permeability of plant protoplasts tononelectrolytes. Trans. Faraday Soc. 1938, 33, 985−990.(10) Davson, H.; Danielli, J. F. The Permeability of NaturalMembranes; Cambridge Univ. Press: London, 1943; pp 80−117.(11) Mueller, P.; Rudin, D. O.; Tien, H. T.; Westcott, W. C.Reconstitution of cell membrane structure in vitro and its trans-formation into an excitable system. Nature 1962, 194, 979−980.(12) Stein, W. D. The Movement of Molecules across Cell Membranes;Academic Press: New York, 1967; pp 65−125.(13) Gallucci, E.; Micelli, S.; Lippe, C. Non-electrolyte permeabilityacross thin-lipid membranes. Arch. Int. Physiol. Biochim. 1971, 79,881−887.(14) Gutknecht, J.; Tosteson, D. C. Diffusion of weak acids acrosslipid membranes: effects of chemical reactions in the unstirred layers.Science 1973, 182, 1258−1261.(15) Rapaport, S. I.; Ohno, K.; Pettigrew, K. D. Drug entry into thebrain. Brain Res. 1979, 172, 354−359.(16) Barry, P. H.; Diamond, J. M. Effects of the unstirred layers onmembrane phenomena. Physiol. Rev. 1984, 64, 763−872.(17) Miyoshi, H.; Maeda, H.; Tokutake, N.; Fujita, T. Quantitativeanalysis of partition behavior of substituted phenols from aqueousphase into liposomes made of lecithin and various lipids. Bull. Chem.Soc. Jpn. 1987, 60, 4357−4362.(18) Bangham, A. D. Surrogate cells or Trojan horses. BioEssays1995, 1081−1088.(19) Xiang, T.-X.; Anderson, B. D. Influence of a transmembraneprotein on the permeability of small molecules across lipidmembranes. J. Membr. Biol. 2000, 173, 187−201.(20) Missner, A.; Pohl, P. 110 years of the Meyer-Overton rule:predicting membrane permeability of gases and other smallcompounds. ChemPhysChem 2009, 10, 1405−1414.(21) Liu, X.; Testa, B.; Fahr, A. Lipophilicity and its relationship withdrug passive permeation. Pharm. Res. 2011, 28, 962−977.(22) Djukic, M.; Munz, M.; Sorgel, F.; Holzgrabe, U.; Eiffert, H.;Nau, R. Overton’s rule helps to estimate the penetration of anti-infectives into patients’ cerebrospinal fluid. Antimicrob. AgentsChemother. 2012, 56, 979−988.(23) Jin, B.; Higashiyama, R.-I.; Nakata, Y.-I.; Yonezawa, J.-I.; Xu, S.;Miyake, M.; Takahashi, S.-I.; Kikuchi, K.; Yazawa, K.-I.; Mizobuchi, S.;Niimi, S.; Kitayama, M.; Koshimoto, C.; Matsukawa, K.; Kasai, M.;Edashige, K. Rapid movement of water and cryoprotectants in pigexpanded blastocysts via channel processes: its relevance to theirhigher tolerance to cryopreservation. Biol. Reprod. 2013, 89, 1−12.(24) Oballa, R. M.; Belair, L.; Black, W. C.; Bleasby, K.; Chan, C. C.;Desroches, C.; Du, X.; Gordon, R.; Guay, J.; Guiral, S.; Hafey, M. J.;Hamelin, E.; Huang, Z.; Kennedy, B.; Lachance, N.; Landry, F.; Li, C.S.; Mancini, J.; Normandin, D.; Pocai, A.; Powell, D. A.; Ramtohul, Y.K.; Skorey, K.; Sørensen, D.; Sturkenboom, W.; Styhler, A.;Waddleton, D. M.; Wang, H.; Wong, S.; Xu, L.; Zhang, L.Development of a liver-targeted stearoyl-CoA desaturase (SCD)inhibitor (MK-8245) to establish a therapeutic window for thetreatment of diabetes and dyslipidemia. J. Med. Chem. 2011, 54, 5082−5096.(25) Pfefferkorn, J. A.; Guzman-Perez, A.; Litchfield, J.; Aiello, R.;Treadway, J. L.; Pettersen, J.; Minich, M. L.; Filipski, K. J.; Jones, C. S.;Tu, M.; Aspnes, G.; Risley, H.; Bian, J.; Stevens, B. D.; Bourassa, P.;D’Aquila, T.; Baker, L.; Barucci, N.; Robertson, A. S.; Bourbonais, F.;Derksen, D. R.; MacDougall, M.; Cabrera, O.; Chen, J.; Lapworth, A.L.; Landro, J. A.; Zavadoski, W. J.; Atkinson, K.; Haddish-Berhane, N.;Tan, B.; Yao, L.; Kosa, R. E.; Varma, M. V.; Feng, B.; Duignan, D. B.;El-Kattan, A.; Murdande, S.; Liu, S.; Ammirati, M.; Knafels, J.; DaSilva-
Jardine, P.; Sweet, L.; Liras, S.; Rolph, T. P. Discovery of (S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)-nicotinic acid as a hepatoselective glucokinase activator clinicalcandidate for treating type 2 diabetes mellitus. J. Med. Chem. 2012,55, 1318−1333.(26) Xu, A. S. L.; Chu, C. K.; London, R. E. F-NMR study of theuptake of 2′-fluoro-5-methyl-β-L arabinofuranosyluracil in erythro-cytes. evidence of transport by facilitated and nonfacilitated pathways.Biochem. Pharmacol. 1998, 55, 1611−1619.(27) Tsai, R. S.; El Tayar, N.; Carrupt, P. A.; Testa, B.Physicochemical properties and transport behaviour of piribedil.Considerations on its membrane-crossing potential. Int. J. Pharmaceut.1992, 80, 39−49.(28) Abraham, M. H. Scales of solute hydrogen-bonding: theirconstruction and application to physicochemical and biochemicalprocesses. Chem. Soc. Rev. 1993, 22, 73−83.(29) Cruciani, G.; Crivori, P.; Carrupt, P. A.; Testa, B. Molecularfields in quantitative structure-permeation relationships: the VolSurfapproach. J. Mol. Struct. (THEOCHEM) 2000, 503, 17−30.(30) Rey, S.; Caron, G.; Ermondi, G.; Gaillard, P.; Pagliara, A.;Carrupt, P. A.; Testa, B. Development of molecular hydrogen bondingpotentials (MHBPs) and their application to structure-permeationrelations. J. Mol. Graphics Modelling 2001, 19, 521−535.(31) Ahlin, G.; Hilgendorf, C.; Karlsson, J.; Al-Khalili Szigyarto, C.;Uhlen, M.; Artursson, P. Endogenous gene and protein expression ofdrug-transporting proteins in cell lines routinely used in drug discoveryprograms. Drug Metab. Dispos. 2009, 37, 2275−2283 (also in Figure2).(32) Irvine, J. D.; Takahashi, L.; Lockhart, K.; Cheong, J.; Tolan, J.W.; Selick, H. E.; Grove, J. R. MDCK (Madin−Darby canine kidney)cells: a tool for membrane permeability screening. J. Pharm. Sci. 1999,88, 28−33.(33) Avdeef, A.; Tam, K. Y. How well can the Caco-2/MDCKmodels predict effective human jejunal permeability? J. Med. Chem.2010, 53, 3566−3584 . (also in Figure 2).(34) Hayeshi, R.; Hilgendorf, C.; Artursson, P.; Augustijns, P.;Brodin, B.; Dehertogh, P.; Fisher, K.; Fossati, L.; Hovenkamp, E.;Korjamo, T.; Masungi, C.; Maubon, N.; Mols, R.; Mullertz, A.;Monkkonen, J.; O’Driscol, C.; Oppers-Tiemissen, H. M.; Ragnarsson,E. G.; Rooseboom, M.; Ungell, A. L. Comparison of drug transportergene expression and functionality in Caco-2 cells from 10 differentlaboratories. Eur. J. Pharm. Sci. 2008, 35, 383−396 (also in Figure 2).(35) Doan, K. M.; Humphreys, J. E.; Webster, L. O.; Wring, S. A.;Shampine, L. J.; Serabjit-Singh, C. J.; Adkinson, K. K.; Polli, J. W.Permeability and P-glycoprotein-mediated efflux differentiate centralnervous system (CNS) and non-CNS marketed drugs. J. Pharmacol.Exp. Ther. 2002, 303, 1029−1037 (also in Table 1).(36) Avdeef, A. Absorption and Drug Development, 2nd ed.; Wiley-Interscience: Hoboken, NJ, 2012 (also in Table 1).(37) Hogben, C. A.; Tocco, D. J.; Brodie, B. B.; Schanker, L. S. Onthe mechanism of intestinal absorption of drugs. J. Pharmacol. Exp.Ther. 1959, 125, 275−82.(38) Avdeef, A.; Artursson, P.; Neuhoff, S.; Lazarova, L.; Grasjo, J.;Tavelin, S. Caco-2 permeability of weakly basic drugs predicted withthe double-sink PAMPA pKa flux method. Eur. J. Pharm. Sci. 2005, 24,333−349 (also in Figure 5).(39) Tsinman, O.; Tsinman, K.; Sun, N.; Avdeef, A. Physicochemicalselectivity of the BBB microenvironment governing passive diffusion −matching with a porcine brain lipid extract artificial membranepermeability model. Pharm. Res. 2011, 28, 337−363 (also in Figure 3).(40) Dagenais, C.; Avdeef, A.; Tsinman, O.; Dudley, A.; Beliveau, R.P-glycoprotein deficient mouse in situ blood-brain barrier permeabilityand its prediction using an in combo PAMPA model. Eur. J. Pharm. Sci.2009, 38, 121−137 (also in Figure 3).(41) Hinderling, P. H. Red blood cells: a neglected compartment inpharmacokinetics and pharmacodynamics. Pharmacol. Rev. 1997, 49,279−295 (also in Figure 4).(42) van de Waterbeemd, H.; Kansy, M. Hydrogen-bonding capacityand brain penetration. Chimia 1992, 46, 299−303.
Molecular Pharmaceutics Review
dx.doi.org/10.1021/mp400713v | Mol. Pharmaceutics 2014, 11, 1727−17381737

(43) Pagliara, A.; Reist, M.; Geinoz, S.; Carrupt, P. A.; Testa, B.Evaluation and prediction of drug permeation. J. Pharm. Pharmacol.1999, 51, 1339−1357.(44) Neuhoff, S.; Ungell, A.-L.; Zamora, I.; Artursson, P. pH-dependent bidirectional transport of weakly basic drugs across Caco-2monolayers: implications for drug−drug interactions. Pharm. Res.2003, 20, 1141−1148 . (also in Fig 5).(45) Ooms, F.; Weber, P.; Carrupt, P. A.; Testa, B. A simple model topredict blood-brain barrier permeation from 3D-molecular fields.Biochim. Biophys. Acta 2002, 1587, 118−125.(46) Hammarlund-Udenaes, M.; Friden, M.; Syvanen, S.; Gupta, A.On the rate and extent of drug delivery to the brain. Pharm. Res. 2008,25, 1737−1750.(47) Friden, M.; Bergstrom, F.; Wan, H.; Rehngren, M.; Ahlin, G.;Hammarlund-Udenaes, M.; Bredberget, U. Measurement of unbounddrug exposure in brain: modeling of pH partitioning explains divergingresults between the brain slice and brain homogenate methods. DrugMetab. Dispos. 2011, 39, 353−362 (also in Figure 7).(48) Liu, X.; Vilenski, O.; Kwan, J.; Apparsundaram, S.; Weikert, R.Unbound brain concentration determines receptor occupancy: acorrelation of drug concentration and brain serotonin and dopaminere-uptake transporter occupancy for eighteen compounds. Drug Metab.Dispos. 2009, 37, 1548−155.(49) Tagawa, M.; Kano, M.; Okamura, N.; Higuchi, M.; Matsuda, M.;Mizuki, Y.; Arai, H.; Iwata, R.; Komemushi, T. F. S.; Ido, T.; Itoh, M.;Sasaki, H.; Watanabe, T.; Yanai, K. Neuroimaging of histamine H1-receptor occupancy in human brain by positron emission tomography(PET): a comparative study of ebastine, a second-generationantihistamine, and (+)-chlorpheniramine, a classical antihistamine.Br. J. Clin. Pharmacol. 2001, 52, 501−509.(50) Beckett, A. H.; Rowland, M. Urinary excretion ofmethylamphetamine in man. Nature 1965, 206, 1260−1261.(51) Davis, J. M.; Kopin, I. J.; Lemberger, L.; Axelrod, J. Effects ofurinary pH on amphetamine metabolism. Ann. N.Y. Acad. Sci. 1971,179, 493−501.(52) Cham, B. E.; Dykman, J. H.; Bochner, F. Urinary excretion ofaspirin. Br. J. Clin. Pharmacol. 1982, 14, 562−564.(53) Freudenthaler, S.; Merneke, I.; Schreb, K.-H.; Boakye, E.;Gundert-Remy, U.; Gleiter, C. H. Influence of urine pH and urinaryflow on the renal excretion of memantine. Br. J. Clin. Pharmacol. 1998,46, 1365−2125.(54) Kalia, Y. N.; Merino, V.; Guy, R. H. Transdermal drug delivery.Clin. Asp. Dermatol. Ther. 1998, 16, 289−299.(55) Flynn, G. L. Physicochemical Determinants of Skin Absorption.In Principles of Route-to-Route Extrapolation for Risk Assessment; Gerrity,T. R., Henry, C. J., Eds.; Elsevier: Amsterdam, 1990; pp 93−127.(56) Flynn, G. L. Cutaneous and Transdermal Delivery: Processesand Systems of Delivery. In Modern Pharmaceutics; Banker, G. S.,Rhodes, C. T., Eds.; Dekker: New York, 1996; pp 239−299.(57) Ritschel, W. A.; Hussain, A. S. The principles of permeation ofsubstances across the skin. Methods Find. Exp. Clin. Pharmacol. 1988,10, 39−56.(58) Hadgraft, J.; Pugh, W. J. The selection and design of topical andtransdermal agents: a review. J. Invest. Dermatol. 1998, 3, 131−135.(59) Lampe, M. A.; Williams, M. L.; Elias, P. M. Human epidermallipids: characterization and modulations during differentiation. J. LipidRes. 1983, 24, 131−140.(60) Scheuplein, R. J.; Blank, I. H. Permeability of the skin.Pharmacol. Rev. 1971, 51, 702−747.(61) Albery, W. J.; Hadgraft, J. Percutaneous absorption: in vivoexperiments. J. Pharm. Pharmacol. 1979, 31, 140−147.(62) Heisig, M.; Lieckfeldt, R.; Wittum, G.; Mazurkevitch, G.; Lee, G.Non steady-state descriptions of drug permeation through stratumcorneum. I. The biphasic brick-and-mortar model. Pharm. Res. 1996,13, 421−426.(63) Nemanic, M. K.; Elias, P. M. In situ precipitation: a novelcytcohemical technique for visualization of permeability pathways inmammalian stratum corneum. J. Histochem. Cytochem. 1980, 28, 573−578.
(64) Mouritsen, O. G.; Jorgensen, K. A new look at lipid-membranestructure in relation to drug research. Pharm. Res. 1998, 15, 1507−1519.(65) www.drugbank.ca.(66) Roberts, M. S.; Pugh, W. J.; Hadgraft, J. Epidermal permeability:penetrant structure relationships. 2. The effect of H-bonding groups inpenetrants on their diffusion through the stratum corneum. Int. J.Pharm. 1996, 132, 23−32.(67) Pugh, W. J.; Roberts, M. S.; Hadgraft, J. Epidermal permeability- penetrant structure relationships. 3. The effect of hydrogen bondinginteractions and molecular size on diffusion across the stratumcorneum. Int. J. Pharm. 1996, 138, 149−165.(68) Abraham, M. H.; Martins, F.; Mitchell, R. C. Algorithms for Skinpermeability using hydrogen bond descriptors: the problem ofsteroids. J. Pharm. Pharmacol. 1997, 49, 858−865.(69) Cronin, M. T. D.; Dearden, J. C.; Moss, G. P.; Murray-Dickson,G. Investigation of the mechanism of flux across human skin in vitroby quantitative structure-permeability relationships. Eur. J. Pharm. Sci.1999, 7, 325−330.(70) Frash, H. F.; Landsittel, D. P. Regarding the sources of dataanalyzed with quantitative structure-skin permeability relationshipmethods. J. Med. Chem. 2002, 45, 399−403.(71) Geinoz, S.; Guy, R. H.; Testa, B.; Carrupt, P. A. Quantitativestructure-permeation relationships (QSPeRs) to predict skin perme-ation: a critical evaluation. Pharm. Res. 2004, 21, 83−92.(72) Avdeef, A.; Box, K. J.; Comer, J. E. A.; Hibbert, C.; Tam, K. Y.pH-metric log P. 10. Determination of vesicle membrane − waterpartition coefficients of ionizable drugs. Pharm. Res. 1997, 15, 208−214.(73) Marbella, L. E.; Cho, H. S.; Spence, M. M. Observing thetranslocation of a mitochondria-penetrating peptide with solid-stateNMR. Biochim. Biophys. Acta Biomembr. 2013, 1828, 1674−1682.(74) Eyer, K.; Paech, F.; Schuler, F.; Kuhn, P.; Kissner, R.; Belli, S.;Dittrich, P. S.; Kramer, S. D. A liposomal fluorescence assay to studypermeation kinetics of drug-like weak bases across the lipid bilayer. J.Controlled Release 2013, 173, 102−109.(75) Reigada, R. Atomistic study of lipid membranes containingchloroform: looking for a lipid-mediated mechanism of anesthesia.PLoS One 2013, 8 (1), 52631 DOI: 10.1371/journal.pone.0052631.(76) Fischer, H.; Atzpodien, E. A.; Csato, M.; Doessegger, L.; Lenz,B.; Schmitt, G.; Singer, T. In silico assay for assessing phospholipidosispotential of small drug-like molecules: training, validation, andrefinement using several datasets. J. Med. Chem. 2012, 55, 126−39.(77) Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J.Experimental and computational approaches to estimate solubility andpermeability in drug discovery and development settings. Adv. DrugDelivery Rev. 2001, 46, 3−26.(78) Testa, B.; Vistoli, G.; Pedretti, A. A Fresh Look at MolecularStructure and Properties. In Drug Properties: Measurement andPrediction; Mannhold, R., Ed.; Wiley-VCH: Weinheim, 2007; pp 3−24.(79) Testa, B.; Vistoli, G.; Pedretti, A.; Caldwell, J. Organicstereochemistry. Part 5. Stereoselectivity in molecular and clinicalpharmacology. Helv. Chim. Acta 2013, 96, 747−798.(80) Vistoli, G.; Pedretti, A.; Testa, B. Assessing drug-likeness −What are we missing? Drug Discovery Today 2008, 13, 285−293.(81) Kola, I.; Landis, J. Can the pharmaceutical industry reduceattrition rates? Nat. Rev. Drug Discovery 2004, 3, 711−715.(82) Lord, C. G.; Ross, L.; Lepper, M. R. Biased assimilation andattitude polarization: the effects of prior theories on subsequentlyconsidered evidence. J. Pers. Soc. Psychol. 1979, 37, 2098−2109.(83) Lanthaler, K.; Bilsland, E.; Dobson, P.D.; Moss, H. J.; Pir, P.;Kell, D. B.; Oliver, S. G. Genome wide assessment of the carriersinvolved in the cellular uptake of drugs: a model system in yeast. BMCBiol. 2011, 9, 70.
Molecular Pharmaceutics Review
dx.doi.org/10.1021/mp400713v | Mol. Pharmaceutics 2014, 11, 1727−17381738





![arXiv:1205.4220v2 [cs.MA] 5 May 2013 · 3. Distributed Optimization via Diffusion Strategies. 4. Adaptive Diffusion Strategies. 5. Performance of Steepest-Descent Diffusion Strategies](https://img.pdfslide.net/doc/110x75/602e1f84e58e05019f17db5f/arxiv12054220v2-csma-5-may-2013-3-distributed-optimization-via-diiusion.jpg)























