Embed Size (px)
Citation preview

LE NEUROBLASTOME BILATERAL :
Expérience du Service d’Hématologie et Oncologie
Pédiatrique de Rabat.
Mémoire de fin de stage.
Service d’Hématologie et Oncologie Pédiatrique (SHOP)
Hôpital d’enfants de Rabat (HER)
Du 02 Novembre 2015 au 29 Avril 2016.
Travail réalisé par : Dr ARRACHIDI Nabila
Encadré par : Pr El Kababri Maria

SOMMAIRE

Introduction .............................................................................................................................. 1
1. Objectif : .......................................................................................................................... 3
2. Matériels et méthodes : ................................................................................................... 3
Résultats .................................................................................................................................... 4
1. Données épidémiologiques : ........................................................................................... 5
2. Données cliniques : ......................................................................................................... 5
2.1. Circonstances de découverte : .................................................................................. 5
3. Données diagnostiques : .................................................................................................. 6
3.1. Imagerie : ................................................................................................................. 6
3.2. Dosages biologiques : .............................................................................................. 8
3.3. Anatomopathologie et biologie de la tumeur : ......................................................... 8
4. Bilan d’extension : .......................................................................................................... 9
5. Classification pré thérapeutique : .................................................................................... 9
6. Données thérapeutiques : ................................................................................................ 9
7. Données évolutives : ..................................................................................................... 10
Discussion ................................................................................................................................ 12
1. Epidémiologie : ............................................................................................................. 13
1.1. Fréquence : ............................................................................................................. 13
1.2. Age et sexe : ........................................................................................................... 13
1.3. Etiologie : ............................................................................................................... 13
2. Diagnostic : ................................................................................................................... 14
2.1. Données cliniques de la forme abdominale bilatérale du neuroblastome : ............ 14
3. Marqueurs biologiques : ................................................................................................ 15
3.1. Catécholamines urinaires : ..................................................................................... 15
3.2. Lactate déshydrogénase (LDH) : ........................................................................... 15
3.3. Férritine : ................................................................................................................ 15
3.4. Neurone specific enolase (NSE) : .......................................................................... 16

4. Apport de l’imagerie : ................................................................................................... 16
4.1. Imagerie de la tumeur primitive : ........................................................................... 16
4.2. Imagerie des métastases : ....................................................................................... 17
4.3. Apport de l’imagerie dans la surveillance : ........................................................... 19
5. Caractéristiques histo-pathologiques : .......................................................................... 19
6. Apport de la cytogénétique et de la biologie moléculaire : ........................................... 21
7. Classifications pronostiques : ........................................................................................ 23
7.1. Classification EVANS : ......................................................................................... 23
7.2. Classification INSS (International Neuroblastoma Staging System): ................... 24
7.3. Classification TNM : .............................................................................................. 24
7.4. Classification de « pediatric oncology group » POG : .......................................... 25
7.5. Classification clinico-biologique pronostique de Brodeur : .................................. 26
8. Prise en charge thérapeutique : ...................................................................................... 26
8.1. La prise en charge du neuroblastome bilatéral en fonction du groupe de risque : . 27
8.2. La prise en charge du neuroblastome bilatéral selon l’INGR (International
Neuroblastoma Group Risk) ............................................................................................. 29
8.3. Le traitement chirurgical ........................................................................................ 29
8.4. Chimiothérapie : ..................................................................................................... 33
8.5. Radiothérapie : ....................................................................................................... 34
8.6. Les agents différenciants : Les rétinoïdes .............................................................. 35
9. Perspectives thérapeutiques : ........................................................................................ 35
Conclusion ............................................................................................................................... 36
Références ............................................................................................................................... 38

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
1
Introduction

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
2
Le neuroblastome est une tumeur maligne dérivée des cellules de la crête neurale. C’est
la tumeur solide extra crânienne la plus fréquente de l’enfant de moins de 5 ans, qui
représente 8 à 10% des cancers de l’enfant et 15% de tous les décès pédiatriques dus au
cancer. Derrière le terme de neuroblastome, se cachent différentes entités dont la présentation
et l’évolution sont extrêmement variables : certaines formes peuvent régresser spontanément,
d’autres guérissent après chirurgie et/ou chimiothérapie et d’autres ont un pronostic plus
sombre malgré des thérapeutiques intensives.
Les neuroblastomes sont en fait probablement plusieurs maladies différentes. Ce qui
donne encore plus d’intérêt à l’étude de cette maladie est le fait que le neuroblastome est la
première tumeur dont les caractéristiques moléculaires ont été utilisées pour guider la prise en
charge thérapeutique, en adaptant le traitement en fonction des caractéristiques propres du
patient et de la tumeur. Dans environ 70% des cas, les neuroblastomes sont abdominaux, dont
45% au niveau de la surrénale et 25% au niveau des ganglions sympathiques. L’atteinte
bilatérale abdominale est rare, très peu de cas sont rapportés dans la littérature. Dans les deux
dernières décennies, des progrès dans le diagnostic et le traitement du neuroblastome ont été
réalisés. Des méthodes d’imagerie médicale ont été développées. Des critères histologiques,
des marqueurs biologiques, génétiques et chromosomiques ont également été définis et ont
permis l’établissement et l’amélioration progressive de classification de groupes de patients.
Des protocoles associant la chimiothérapie à la chirurgie, à la radiothérapie, voire à
l’autogreffe de cellules souches ont été développés avec des stratégies thérapeutiques adaptées
aux groupes de risque.
Nous rapportons dans ce travail l’expérience du service d’hématologie et oncologie
pédiatrique de Rabat dans la prise en charge du neuroblastome bilatéral.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
3
1. Objectif :
Le but de ce travail est d’étudier les caractéristiques cliniques, biologiques,
radiologiques, histologiques, pronostiques et thérapeutiques sur une série de patients suivis
pour neuroblastomes bilatéraux au service d’hématologie et d’oncologie pédiatrique de
l’hôpital d’enfants de Rabat.
2. Matériels et méthodes :
Notre travail est réalisé dans le service d’hématologie et d’oncologie pédiatrique de
l’hôpital d’enfants de Rabat.
Il s’agit d’une étude rétrospective sur une période de quinze ans allant de Janvier 2001 à
Décembre 2015. Le registre informatique du CHOP de l’HER a été alors consulté et un total
de 378 patients suivis pour "Neuroblastome" est enregistré durant cette période. Nous n’avons
tenu compte que de ceux codés «bilatéraux». Seuls 8 patients ont été recensés pour ce travail.
Les critères d’inclusion dans notre étude étaient :
Neuroblastome bilatéral,
Diagnostiqué et pris en charge entre Janvier 2001 et Décembre 2015 à l’hôpital
d’enfants de Rabat,
Quel que soit le stade et l’âge.
Nous avons pu recueillir des informations concernant l’âge des 8 malades, leurs sexes,
le type histologique et le stade de la maladie, les données cliniques, biologiques,
radiologiques, thérapeutiques ainsi que le devenir de ces patients. Les données contenants ces
informations ont été formulées sous forme d’observations.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
4
Résultats

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
5
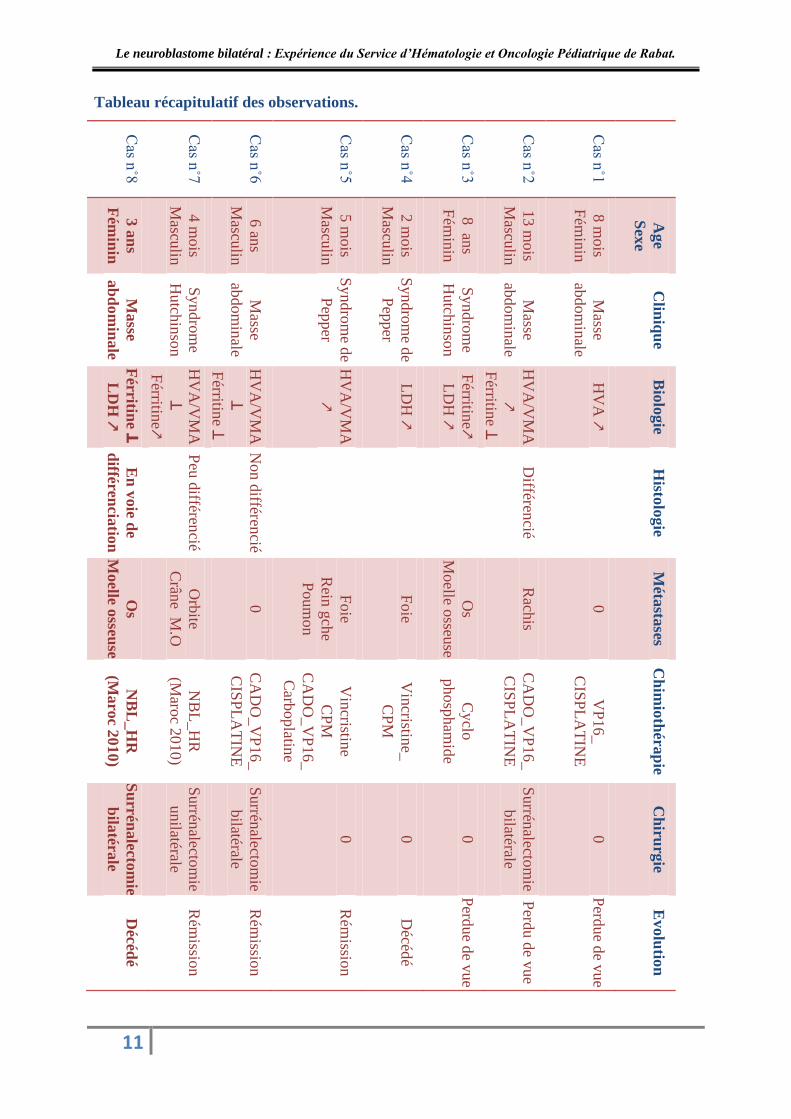
1. Données épidémiologiques :
Notre série est composée de 5 garçons et 3 filles, soit un sexe ratio de 1.66. L’âge
moyen au moment de diagnostic est de 30 mois, soit 2 ans et demi avec des extrêmes allant de
2mois et 8 ans.
La répartition géographique des patients n’a présenté aucune particularité tandis que le
niveau socio économique est bas.
Age Sexe Origine Niveau
socio-économique
Cas n˚1 8 mois Féminin Rabat Bas
Cas n˚2 13 mois Masculin Rabat Bas
Cas n˚3 8 ans Féminin Taza Bas
Cas n˚4 2 mois Masculin Azrou Bas
Cas n˚5 5 mois Masculin Guercif Bas
Cas n˚6 6 ans Masculin Rabat Bas
Cas n˚7 4 mois Masculin Tétouan Bas
Cas n˚8 3 ans Féminin Tétouan bas
2. Données cliniques :
2.1. Circonstances de découverte :
La présentation clinique au moment du diagnostic a été le plus souvent liée à la
présence des métastases.
Le diagnostic a été évoqué devant la découverte d’une masse ou deux masses
abdominales dans 5 cas au total ; isolées dans un cas (cas n˚1), associée à des ADP
périphériques dans 2 cas (cas n˚2 et n˚6), et associée à un syndrome de Hutchinson dans 2 cas
(cas n˚3 et n˚7).
Chez 2 patients (cas n˚4 et n˚5) le diagnostic a été fait devant des signes en faveur d’un
syndrome de Pepper, avec comme symptomatologie initiale une distension abdominale et une
détresse respiratoire mettant en jeu le pronostic vital chez un cas (cas n˚4), et hépatomégalie
chez les 2 cas.
Par ailleurs, un enfant (cas n˚8) a présenté des douleurs abdominales non spécifiques
associées à une anémie et une thrombopénie.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
6
Aucun diagnostic n’a été porté sur une échographie systématique de grossesse.
Symptomatologie clinique :
Détresse vitale Tumeur Métastases
Cas n˚1 2 Masses abdominales _ _
Cas n˚2 Masse abdominale _ _
Cas n˚3 2 Masses abdominales Syndrome de Hutchinson _
Cas n˚4 _ Syndrome de Pepper Détresse respiratoire
Cas n˚5 _ Syndrome de Pepper _
Cas n˚6 Masse abdominale _ _
Cas n˚7 Masse abdominale Syndrome de Hutchinson _
Cas n˚8 Douleur abdominale Bi-cytopénie _
3. Données diagnostiques :
3.1. Imagerie :
Tous les patients de notre série ont bénéficié d’une échographie abdominale et d’une
TDM abdominale ou abdomino-pelvienne à plusieurs reprises pour le diagnostic et pour le
suivi de l’efficacité thérapeutique, ainsi qu’une radiographie thoracique.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
7
Echographie abdominale TDM abdo-pelvienne
Cas n˚1 Masses surrénaliennes
bilatérales hétérogènes Masses surrénaliennes bilatérales :
- droite : 100x74x105mm
- gauche : 96x94x100mm
Calcifications
Absence d’adénopathies profondes
Cas n˚2 Masses surrénaliennes
bilatérales hétérogènes Masses surrénaliennes bilatérales :
- droite : 126x126x120 mm
- gauche : 37x33x20 mm
Calcifications, nécrose
ADP pré-caves et hilaires hépatiques
Cas n˚3 Masses surrénaliennes
bilatérales hétérogènes Masses surrénaliennes bilatérales :
- droite : 19.8x12x10 mm
- gauche : 10x11x9 mm
Calcifications, composante kystique
Absence d’adénopathie
Cas n˚4 - Masses surrénaliennes
bilatérales hétérogènes
- Hépatomégalie
multinodulaire
Masses surrénaliennes bilatérales :
- droite : 32x16.8 mm
- gauche : 43x28 mm
Absence d’adénopathies profondes
Hépatomégalie truffée de nodules
Cas n˚5 Masses surrénaliennes
bilatérales hétérogènes
Hépatomégalie
multinodulaire
Masses surrénaliennes bilatérales :
- droite : 62x46.3x53.1 mm
- gauche : 14.4x9.4x10.6 mm
Hépatomégalie truffée de nodules (29 mm)
Adénopathies lombo-aortiques, inter-aortico-cave et
hilaires
Cas n˚6 Masses surrénaliennes
bilatérales hétérogènes Masses surrénaliennes bilatérales :
- droite : 33x50 mm
- gauche : 13x14x10 cm
Adénopathies rétro-péritonéales latéro-aortiques gauches
Cas n˚7 Masses surrénaliennes
bilatérales hétérogènes Masses surrénaliennes bilatérales :
- droite : 50x20 mm
- gauche : 76x80x62 mm
Calcifications, nécrose
Adénopathies cœlio-mésentériques, pré et latéro
aortiques
Nodule hépatique (7mm), nodules péritonéaux et
pariétaux fessiers.
Cas n˚8 Masses surrénaliennes
bilatérales hétérogènes Masses surrénaliennes bilatérales :
- droite : 84x69x46 mm
- gauche : 110x97 mm
Absence d’adénopathies
Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
8
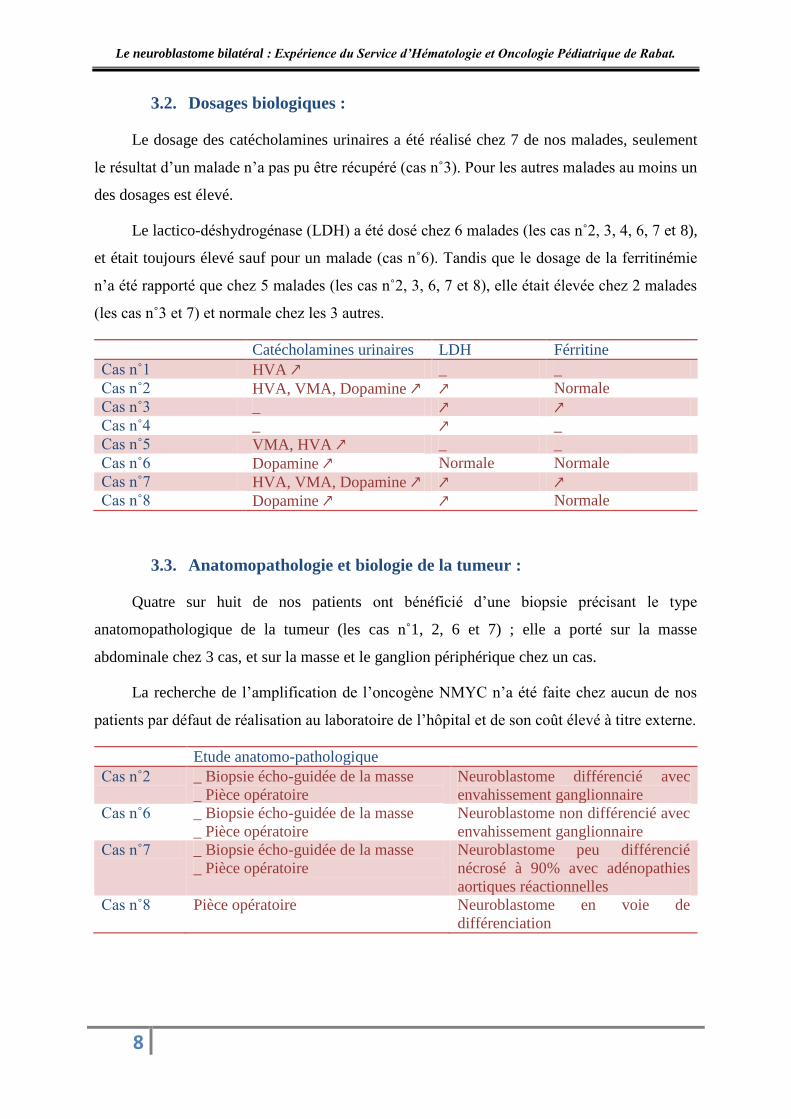
3.2. Dosages biologiques :
Le dosage des catécholamines urinaires a été réalisé chez 7 de nos malades, seulement
le résultat d’un malade n’a pas pu être récupéré (cas n˚3). Pour les autres malades au moins un
des dosages est élevé.
Le lactico-déshydrogénase (LDH) a été dosé chez 6 malades (les cas n˚2, 3, 4, 6, 7 et 8),
et était toujours élevé sauf pour un malade (cas n˚6). Tandis que le dosage de la ferritinémie
n’a été rapporté que chez 5 malades (les cas n˚2, 3, 6, 7 et 8), elle était élevée chez 2 malades
(les cas n˚3 et 7) et normale chez les 3 autres.
Catécholamines urinaires LDH Férritine
Cas n˚1 HVA ↗ _ _
Cas n˚2 HVA, VMA, Dopamine ↗ ↗ Normale
Cas n˚3 _ ↗ ↗ Cas n˚4 _ ↗ _
Cas n˚5 VMA, HVA ↗ _ _
Cas n˚6 Dopamine ↗ Normale Normale
Cas n˚7 HVA, VMA, Dopamine ↗ ↗ ↗ Cas n˚8 Dopamine ↗ ↗ Normale
3.3. Anatomopathologie et biologie de la tumeur :
Quatre sur huit de nos patients ont bénéficié d’une biopsie précisant le type
anatomopathologique de la tumeur (les cas n˚1, 2, 6 et 7) ; elle a porté sur la masse
abdominale chez 3 cas, et sur la masse et le ganglion périphérique chez un cas.
La recherche de l’amplification de l’oncogène NMYC n’a été faite chez aucun de nos
patients par défaut de réalisation au laboratoire de l’hôpital et de son coût élevé à titre externe.
Etude anatomo-pathologique
Cas n˚2 _ Biopsie écho-guidée de la masse
_ Pièce opératoire
Neuroblastome différencié avec
envahissement ganglionnaire
Cas n˚6 _ Biopsie écho-guidée de la masse
_ Pièce opératoire
Neuroblastome non différencié avec
envahissement ganglionnaire
Cas n˚7 _ Biopsie écho-guidée de la masse
_ Pièce opératoire
Neuroblastome peu différencié
nécrosé à 90% avec adénopathies
aortiques réactionnelles
Cas n˚8 Pièce opératoire Neuroblastome en voie de
différenciation

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
9
4. Bilan d’extension :
Tous nos patients ont bénéficié d’un médullogramme au niveau des 2 crêtes, tandis que
seulement 4 cas ont bénéficié d’une scintigraphie osseuse.
Médullogramme Scintigraphie osseuse Autre imagerie
Cas n˚1 Non concluant Non faite Normale
Cas n˚2 Normal Métastases rachis Normale
Cas n˚3 Métastases médullaires _ Métastases os, orbite
Cas n˚4 Normal _ Normale
Cas n˚5 Métastases médullaires _ Normale
Cas n˚6 Normal Normale Normale
Cas n˚7 Métastases médullaires Métastases osseuses Métastases orbite, crâne
Cas n˚8 Métastases médullaires Métastases osseuses Métastases crâne
5. Classification pré thérapeutique :
Au total, 7 de nos malades ont pu être classés après bilan d’extension selon la
classification pré thérapeutique « EVANS » : 4 malades sont de stade IV, 2 malades sont de
stade IV S, et un malade est de stade III.
6. Données thérapeutiques :
Tous nos patients qui ont achevé leurs traitements ont bénéficié d’une chimiothérapie
néo adjuvante de réduction tumorale suivie d’une chirurgie suivie d’une chimiothérapie post
opératoire.
La chimiothérapie initiale a été administrée chez tous nos malades pour réduire le
volume tumoral, rendre la tumeur opérable et/ou pour la régression des métastases. Les
protocoles utilisés ont été étudiés en fonction de l’âge et du stade d’EVANS. Le nombre de
cures et de cycles administré est variable en fonction du protocole et de la réponse au
traitement. La chirurgie n’a été possible que chez 4 de nos malades ; 2 malades ont été perdu
de vue, un autre décédé en début de traitement avant la chirurgie, et un malade présentant un
syndrome de Pepper a bien évolué sous chimiothérapie initiale, n’ayant pas nécessité de
chirurgie et a acheminé par une chimiothérapie palliative à base de Cyclophosphamide.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
10
L’intervention chirurgicale a consisté en une surrénalectomie bilatérale chez 3 de nos
malades ; totale pour la grosse tumeur et partielle pour la plus petite. Et une surrénalectomie
unilatérale partielle chez un seul de nos malades.
La chimiothérapie post opératoire a été administrée chez les quatre malades opérés.
7. Données évolutives :
L’évolution de quatre de nos patients était excellente avec rémission complète de la
tumeur.
Deux de nos huit patients ont été perdus de vue avant la chirurgie. Un patient est décédé
en début de la chimiothérapie initiale, et un autre patient a présenté une rechute tumorale
nécessitant une reprise chirurgicale puis il est décédé.
Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
11
Tableau récapitulatif des observations.
Cas n
˚8
Cas n
˚7
Cas n
˚6
Cas n
˚5
Cas n
˚4
Cas n
˚3
Cas n
˚2
Cas n
˚1
3 a
ns
Fém
inin
4 m
ois
Mascu
lin
6 an
s
Mascu
lin
5 m
ois
Mascu
lin
2 m
ois
Mascu
lin
8 an
s
Fém
inin
13 m
ois
Mascu
lin
8 m
ois
Fém
inin
Age
Sex
e
Masse
ab
dom
inale
Syndro
me
Hutch
inso
n
Masse
abdom
inale
Sy
ndro
me d
e
Pep
per
Syndro
me d
e
Pep
per
Syndro
me
Hutch
inso
n
Masse
abdom
inale
Masse
abdom
inale
Clin
iqu
e
Férritin
e Ʇ
LD
H ↗
HV
A/V
MA
Ʇ
Férritin
e↗
HV
A/V
MA
Ʇ
Férritin
e Ʇ
HV
A/V
MA
↗
LD
H ↗
Férritin
e↗
LD
H ↗
HV
A/V
MA
↗
Férritin
e Ʇ
HV
A ↗
Bio
logie
En
voie d
e
différen
ciatio
n
Peu
différen
cié
Non d
ifférencié
Différen
cié
Histo
logie
Os
Moelle o
sseuse
Orb
ite
Crân
e M.O
0
Foie
Rein
gch
e
Poum
on
Foie
Os
Moelle o
sseuse
Rach
is
0
Méta
stases
NB
L_H
R
(Maro
c 2010
)
NB
L_H
R
(Maro
c 2010
)
CA
DO
_V
P16_
CIS
PL
AT
INE
Vin
cristine
CP
M
CA
DO
_V
P16_
Carb
oplatin
e
Vin
cristine_
CP
M
Cyclo
phosp
ham
ide
CA
DO
_V
P16_
CIS
PL
AT
INE
VP
16_
CIS
PL
AT
INE
Ch
imio
théra
pie
Su
rrénalecto
mie
bila
térale
Surrén
alectom
ie
unilatérale
Surrén
alectom
ie
bilatérale
0
0
0
Surrén
alectom
ie
bilatérale
0
Ch
irurg
ie
Décéd
é
Rém
ission
Rém
ission
Rém
ission
Décéd
é
Perd
ue d
e vue
Perd
u d
e vue
Perd
ue d
e vue
Evolu
tion

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
12
Discussion

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
13
1. Epidémiologie :
1.1. Fréquence :
Le neuroblastome bilatéral est une entité rare, avec une incidence <10% [1,2]. Ceci
rejoint les données de notre série qui montre la rareté de la forme bilatérale du neuroblastome
et l’évalue à 2,11 % de l’ensemble des neuroblastomes.
Le neuroblastome, toutes localisations confondues, se situe au 2ème rang des tumeurs
solides de l’enfant après les tumeurs cérébrales. Au Centre d’Hématologie et d’Oncologie
Pédiatrique (CHOP) de l’Hôpital d’Enfants de Rabat, le neuroblastome représente 9% de tous
les cancers de l’enfant, avec en moyenne 28 nouveaux cas par an. Il occupe la quatrième place
après les leucémies aigues lymphoblastiques, les lymphomes malins non hodgkiniens et la
maladie de hodgkin.
1.2. Age et sexe :
L’âge médian au moment de diagnostic du neuroblastome bilatéral dans notre étude est
de 9 mois et demi, comme est le cas dans l’étude de Kushner et al [3], et plus de la moitié des
cas ont été diagnostiqués avant l’âge d’un an.
Concernant le sexe, le neuroblastome bilatéral présente une prédominance féminine
dans la majorité des articles [4,6,7], tandis que le neuroblastome, toutes localisations
confondues, est légèrement plus fréquent chez les garçons que chez les filles, avec un sexe
ratio de 1,2 [5,8]. Dans notre série on note une légère prédominance masculine avec un sexe
ratio de 1,6.
1.3. Etiologie :
Le Neuroblastome surrénalien bilatéral ou le neuroblastome primaire multiple a été
décrit, dans environ 20% des cas, chez des patients ayant un neuroblastome familial [3]. Dans
notre série, aucun cas de neuroblastome familial n’a été signalé. L'apparition de tumeurs
primaires multiples chez les jeunes enfants a été décrite dans la littérature et pourrait souligner
une prédisposition génétique à cette maladie [9,10]. Maris et ses collègues ont rapporté la
preuve de la présence d'un locus de prédisposition héréditaire du neuroblastome bilatéral sur
le chromosome 16p12-13 [11].
Il y a eu une certaine controverse si ces tumeurs bilatérales sont des métastases
primaires synchrones doubles ou des métastases controlatérales d'une tumeur unilatérale [12]
reflétant ainsi l’origine multicentrique de cette tumeur [13,14]. Cependant, la plupart des cas

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
14
signalés étaient synchrones [7,15] et à ce jour, seulement 2 cas asynchrones ont été rapportés
dans la littérature [14,15].
2. Diagnostic :
2.1. Données cliniques de la forme abdominale bilatérale du
neuroblastome :
En période anténatale, le diagnostic est évoqué par l’échographie du 3e trimestre de
grossesse qui met en évidence une masse abdominale supra rénale bilatérale. Dans notre série
aucun cas n’a été diagnostiqué en anténatal, alors que 5 cas sont rapportés dans la littérature
[4,6,16].
En période post natale, les signes cliniques révélateurs sont extrêmement variables,
pouvant être liés à la tumeur primitive, ou aux métastases, qui sont le plus souvent hépatiques
chez les nourrissons et ostéo-médullaires chez l’enfant de plus d’un an. Les patients avec un
site de développement de la tumeur au niveau abdominal sont susceptibles de développer une
masse abdominale de découverte systématique ou accompagnée de signes de compression
digestive. L’hypertension artérielle est possible chez ces patients, due à une compression de
l’artère rénale par la tumeur ou une hypersécrétion des catécholamines.
Les patients ayant une maladie métastatique, peuvent présenter différents symptômes en
fonction de la localisation métastatique (os, moelle osseuse, orbite, foie…). Très rarement des
syndromes paranéoplasiques spécifiques du neuroblastome sont associés, tel que le syndrome
opso-myoclonique et le syndrome de Kerner-Morisson.
Certains patients ayant une présentation bilatérale du neuroblastome ont d’autres
syndromes associés tels que le syndrome d'hypoventilation congénitale avec aganglionose
colique totale, l’anémie de Fanconi et le syndrome VACTERL ou la microcéphalie.
Dans notre population, les signes cliniques ayant conduit au diagnostic sont dans 7 cas
sur 8 liés aux métastases, ceci rejoint les données de la littérature où la maladie dans sa
localisation bilatérale a généralement été détectée en raison de symptômes liés aux métastases
[4,7]. Parmi 27 cas publiés chez qui les circonstances de découverte ont été décrites, le
diagnostic a été évoqué dans 3 cas devant une masse abdominale et dans 24 cas devant la
présence de métastases [6,14,17]. Deux cas de notre cohorte avaient un syndrome de Pepper
dont un patient avait une détresse respiratoire sévère engageant le pronostic vital.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
15
3. Marqueurs biologiques :
3.1. Catécholamines urinaires :
La plupart des neuroblastomes produisent des quantités importantes de catécholamines
et de leurs métabolites qui sont excrétés dans les urines. Les catécholamines urinaires sont des
examens de routine pour le diagnostic et le suivi de ces tumeurs.
Le dosage des catécholamines donne des indications importantes, en effet la sécrétion
de la dopamine, de l’HVA, et de la VMA, qui sont des marqueurs spécifiques, est directement
proportionnelle à la masse tumorale. Aussi, des valeurs élevées de VMA, HVA, dopamine, et
de noradrénaline, ainsi que l’augmentation des rapports DA/HVA ou DA /VMA constituent
des facteurs de mauvais pronostic. Alors qu’un rapport VMA/HVA supérieur ou égal à 1.5 a
longtemps été considéré comme un facteur de pronostic favorable.
Dans notre série le dosage des catécholamines urinaires a été réalisé chez 7 de patients,
seulement le résultat d’un malade n’a pas pu être récupéré (cas n˚3). Pour les autres malades
au moins un des taux a été élevé.
3.2. Lactate déshydrogénase (LDH) :
Le LDH est un marqueur non spécifique du volume tumoral ou de son renouvellement
rapide. Un taux élevé est observé dans plus de trois quart des cas de neuroblastomes, et est
significativement associé à une survie inférieure dans ces tumeurs. Cependant, sa valeur
pronostique est discutée, approuvée par certains et non retrouvée par d’autres en analyse
multivariée. Ce marqueur a été dosé chez 6 de nos malades et s’est révélée toujours élevé sauf
pour un malade.
3.3. Férritine :
C’est une protéine de transport et du stockage du fer qui est synthétisée en quantité
anormale par certaines cellules cancéreuses. La ferritinémie est élevée si le neuroblastome est
métastatique (stade 4) ou avec une extension locorégionale (stade 3). Sa valeur pronostique
est le plus souvent effacée par sa corrélation avec d’autres facteurs biologiques. Le dosage de
cette protéine n’a été rapporté que chez 5 de nos malades, elle était élevée chez 3 malades et
normale chez les 2 autres. Dans la littérature, la férritine a été dosée chez 2 cas seulement et
elle était élevée [2,16].

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
16
3.4. Neurone specific enolase (NSE) :
La NSE est une enzyme de la glycolyse non spécifique des neuroblastomes. Son intérêt
est plutôt pronostique, car son taux augmente avec le stade et a un impact négatif sur la survie.
Cependant, il n’a pas de valeur indépendamment des autres facteurs pronostiques.
4. Apport de l’imagerie :
4.1. Imagerie de la tumeur primitive :
Elle a pour but de localiser anatomiquement la tumeur, de préciser ses mensurations et
ses rapports avec les vaisseaux et les organes de voisinage, permettant ainsi d’évaluer son
opérabilité.
Echographie abdominale :
L’échographie est l’examen de première intention quand la tumeur est abdominale ou
pelvienne. C’est un examen simple, ne nécessitant pas de prémédication, anodin et répétable,
son caractère non invasif le rend utile dans la surveillance sous traitement.
Tomodensitométrie abdominale ou abdomino-pelvienne :
Le scanner permet de localiser avec précision la tumeur primitive et donne une vision
globale de l’espace anatomique concerné. Il permet la mensuration de la tumeur, établit ses
rapports avec les organes de voisinage et les vaisseaux, évalue l’existence de kystes et de
calcifications et recherche les adénopathies satellites. Aussi, cet examen peut visualiser une
extension intra rachidienne de la tumeur à travers un foramen intravertébral, tumeur dite «en
sablier».
On distingue trois stades d’extension loco régionales identifiables au scanner :
Tableau 1 : Stades d’extension locorégionales observables au scanner (IGR 2004)
Stades Particularités anatomiques tumorales
I Tumeur nettement séparée des structures de voisinage, en particulier des
vaisseaux, par un liseré graisseux.
II Tumeur volumineuse et/ou située au contact des axes vasculaires qu’elle
refoule sans qu’il soit possible de savoir s’il s’agit d’un simple contact ou
d’une adhérence.
II Tumeur englobant un ou plusieurs axes vasculaires non résécables, tel que
l’aorte et ses principales branches collatérales : tronc cœliaque et les artères
mésentériques supérieures et inférieures.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
17
Imagerie par résonance magnétique (IRM) :
L’IRM est complémentaire du couple échographie-scanner. Elle est surtout très
performante pour visualiser les tumeurs en sablier, et est donc l’examen de choix pour ces
tumeurs. Au niveau rétro péritonéal, cet examen permet un excellent bilan quel que soit le
volume de la masse, il évalue au mieux les adénopathies, les rapports avec les vaisseaux,
l’extension intrarachidienne ou l’infiltration médiastinale postérieure éventuelle et il permet
également de dépister des lésions vertébrales dans le champ de l’examen. Malheureusement,
cet examen n’a été réalisé chez aucun de nos malades à cause de son coût élevé.
L’analyse du scanner ou de l’IRM permet de décider de l’opérabilité de la tumeur au
terme d’une discussion impliquant oncologues, radiologues et chirurgiens. Une tumeur est
opérable quand une exérèse complète peut être faite sans risque de complication ni sacrifice
d’organe.
La radiologie conventionnelle :
- La radiographie thoracique reste systématique, c’est le premier examen réalisé en cas
de point d’appel thoracique. Par ailleurs, il peut montrer un élargissement d’un espace inter
pédiculaire sur le rachis évocateur d’une extension en sablier, ou bien une atteinte osseuse
ostéolytique. Il peut montrer des métastases pulmonaires qui sont exceptionnelles lors du
diagnostic.
- L’abdomen sans préparation n’est pas indispensable au diagnostic. Lorsqu’il est
réalisé, on peut retrouver un syndrome de masse opaque refoulant les clartés digestives et
contenant de fines calcifications punctiformes. Il peut aussi montrer des modifications
osseuses liées aux extensions intrarachidiennes et foraminales.
4.2. Imagerie des métastases :
Près de 60% des patients atteints de neuroblastome sont porteurs de métastases au
moment de diagnostic. Leur siège est médullaire, osseux ou les deux, dans 82% des cas.
Métastases osseuses :
La scintigraphie à la MIBG (méta-iodo-benzyl-guanidine) est un examen à la fois
sensible et spécifique. Quand la tumeur fixe la MIBG, ce qui est le cas dans 95% des
neuroblastomes, elle permet d’établir une cartographie de la maladie. Ainsi cet examen
permet de confirmer la localisation principale de la tumeur primitive et de détecter

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
18
d’éventuelles localisations secondaires ostéomédullaires et osseuses. La spécificité de cette
technique approche 100 %.
La scintigraphie au diphosphonate marquée au technétium 99m : Ce n’est que lorsque la
tumeur primitive ne fixe pas la MIBG qu’une scintigraphie au technétium 99 est pratiquée à
la recherche de métastases osseuses. Cependant cet examen n’est pas spécifique de la maladie,
il est moins sensible et permet d’évaluer moins finement la réponse au traitement. Dans notre
série un seul malade a bénéficié de la scintigraphie à la MIBG et trois autres ont bénéficié de
la scintigraphie au technétium 99m.
Les radiographies du squelette ont un rôle limité et ne sont pas nécessaires excepté pour
préciser des lésions douteuses trouvées sur la scintigraphie osseuse.
Métastases médullaires :
Les métastases médullaires sont recherchées par les myélogrammes et les biopsies
médullaires qui sont prélevés dans des sites différents, deux sites différents doivent être
évaluables avec deux techniques différentes. Tous nos malades ont bénéficié d’un
médullogramme au niveau des 2 crêtes alors que seulement trois d’entre eux ont bénéficié
d’une biopsie ostéo-médullaire complémentaire. Ainsi l’évaluation de l’extension
ostéomédullaire est fondée sur les prélèvements de moelle et la scintigraphie à la MIBG,
technique de référence permettant une évaluation globale de la maladie.
Autres métastases :
La radiographie thoracique : on se contente du cliché standard de face des poumons, à
la recherche d’éventuelles localisations pulmonaires ou pleurales. Or les métastases
pulmonaires sont exceptionnelles.
L’échographie et le scanner abdominal : permettent en même temps que le bilan de la
tumeur primitive de rechercher l’existence de métastases hépatiques.
Le scanner cérébral : est utilisé pour étudier les atteintes osseuses et épidurales du crâne,
aussi il est nécessaire en cas de localisation métastatique orbitaire.
Dans notre série un seul des deux malades présentant le syndrome de Hutchinson a
bénéficié d’une TDM crânio-cérébro-orbitaire, faute de moyens.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
19
4.3. Apport de l’imagerie dans la surveillance :
La tumeur primitive
Pour les localisations abdominopelviennes, l’évaluation de la réduction de volume
tumoral sous chimiothérapie est réalisée par échographie. La fréquence des contrôles est
dictée par les protocoles. L’imagerie postopératoire doit être réalisée environ à un mois de
l’intervention, avec la même technique d’imagerie qu’en préopératoire, à la recherche d’un
reliquat tumoral.
Les localisations secondaires
La surveillance des localisations secondaires ostéomédullaires repose sur la
scintigraphie à MIBG si elle est disponible, sinon sur l’IRM. Celle des localisations
hépatiques repose sur l’échographie.
5. Caractéristiques histo-pathologiques :
Le diagnostic de neuroblastome est fortement suspecté sur l’ensemble des arguments
cliniques, biologiques et radiologiques précédemment décrits. Il doit être confirmé par une
analyse histologique d’une biopsie ou par la présence de cellules tumorales typiques dans la
moelle osseuse (aspiration ou biopsie). Microscopiquement, la tumeur se compose de
neuroblastes qui sont des petites cellules rondes indifférenciées et volontiers groupés en
rosettes, il existe des formes plus différenciées prenant un aspect ganglioneural, et l’on parle
alors de ganglioneuroblastome. A un degré de plus, la tumeur a un aspect de ganglioneurome
avec des cellules ganglioneurales matures et des éléments schwanniens fusiformes sans
contingent neuroblastique. On distingue alors :
- Les neuroblastomes (à stroma pauvre), composés à plus de 50% de neuroblastes et de
substance fibrillaire (indifférenciés, peu différenciés, ou en voie de différenciation).
- Les ganglioneuroblastomes (à stroma riche) composés de plus de 50% de tissu
stromal ganglioneuromateux (nodulaires, mélangés, ou borderline).
- Et les ganglioneuromes bénins composés exclusivement d'éléments matures (cellules
ganglionnaires et cellules de Schwann).
La forme particulière du neuroblastome in situ est connue depuis longtemps des
pathologistes et représenterait un simple retard à la régression spontanée des neuroblastes
fœtaux. Il existe des classifications anatomopathologiques pour le neuroblastome, et aucune
ne peut être proposée sur un prélèvement cytologique. Elles ne sont applicables que sur une

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
20
pièce d’exérèse chirurgicale, avant toute chimiothérapie. La classification histopronostique de
Shimada (Shimada 1984) reste la référence. Elle est basée sur l'âge de l'enfant et sur 3 critères
histologiques : la richesse du stroma, le grade de différenciation et l'index mitotique et
caryorrhectique (MKI).
Critères histologiques de mauvais pronostic (Shimada):
- Neuroblastome à stroma riche nodulaire
- Neuroblastome à stroma pauvre avec :
Âge > 5 ans
Histologie indifférenciée après 1,5 an
MKI > 100 après 1,5 an
MKI > 200 avant 1,5 an
Actuellement, on utilise la classification INPC (International Neuroblastoma Pathologic
Classification) où les critères morphologiques ont été standardisés. Celle-ci est largement
inspirée de la classification Shimada.
Tableau 2 : Classification INPC

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
21
En combinant cette classification histologique (différenciation) avec l’âge, le MKI et la
présence de calcifications, on peut distinguer des groupes histopronostiques :
- Les formes de pronostic défavorable :
Stroma riche nodulaire,
Stroma pauvre et :
Âge > 5ans,
Histologie indifférenciée et âge entre 1.5 et 5ans,
Histologie différenciée et MKI >100,
Âge <1.5 ans et MKI >200.
- Les formes de pronostic favorable :
Stroma riche, sans nodule
Stroma pauvre et :
âge <1.5 ans et MKI <200,
âge entre 1.5 et 5ans,
histologie différenciée et MKI <100.
Dans les séries précédentes de neuroblastomes bilatéraux, les tumeurs sont d’histologie
le plus souvent favorable selon la classification de Shimada [6,18,12,15]. Dans notre série 4
malades ont été opérés, et les types anatomopathologiques retrouvés après examen des pièces
opératoires sont : des neuroblastomes bien différenciés chez 2 cas, un neuroblastome peu
différencié chez un cas, et des neuroblastomes en voie de différentiation chez un cas lors de sa
première chirurgie, puis un ganglioneuroblastome mélangé à stroma riche lors de sa 2e
chirurgie.
6. Apport de la cytogénétique et de la biologie moléculaire :
Le comportement agressif des tumeurs à histologie défavorable est fortement corrélé
avec l’amplification de l’oncogène NMYC, premier oncogène relevant dans le
neuroblastome. Le NMYC est un proto-oncogène localisé sur le bras court du chromosome 2,
qui, dans le neuroblastome, peut être amplifié jusqu’à plusieurs centaines de copies de gène
par cellule. L’amplification de cet oncogène de plus de 10 copies est fortement associée à une
histologie défavorable, à un stade avancé de la pathologie, à un comportement tumoral
agressif et à un risque élevé de rechute. Le statut de l’oncogène NMYC est devenu un facteur
pronostique essentiel, décisionnel pour le traitement. Et une perspective d’avenir consistera à

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
22
rechercher l’amplification de cet oncogène dans le sérum des patients, comme cela a déjà été
prouvé.
Le contenu en ADN des cellules malignes joue un rôle pronostique surtout chez les
enfants pendant leur première année de vie. Une hyperploïdie, le plus souvent une triploïdie
est essentiellement observée dans les neuroblastomes survenant chez les enfants de moins
d’un an et d’évolution favorable, au contraire, la diploïdie des neuroblastomes à une
diminution de la survie et de la survie sans événement. Ce facteur pronostic est en partie
corrélé à l’amplification de NMYC.
Les anomalies génétiques telles que les pertes et les gains de certaines régions
chromosomiques sont des manifestations fréquentes dans les tumeurs malignes.
Tableau 3 : Anomalies chromosomiques récurrentes observées dans les neuroblastomes
Anomalie
chromosomique
Locus impliqué Corrélation avec
l’amplification NMYC
Signification pronostique
globale
17q+ 17q21-qter oui (directe) Défavorable
1p- 1p36 oui (directe) Défavorable
11q- 11q23 oui (inverse) Défavorable
14q- 14q23-qter oui (inverse) Incertaine
Les neurotrophines jouent un rôle critique dans le développement du système nerveux
sympathique, notamment le nerve growth factor (NGF) et le brain derived neutrophic factor
(BDNF). Ils se lient à la cellule par des récepteurs «Trk» de la famille des tyrosines kinases.
Or l’expression de ces récepteurs aux neurotrophines «Trk» est perturbée dans le
neuroblastome. Un niveau d’expression élevé de TrkA, récepteur du facteur de croissance
nerveux (NGF), est corrélé à l’absence d’amplification de NMYC et à un pronostic favorable,
alors que l’expression de TrkB, récépteur d’une autre neurotrophine, BDNF, est liée à
l’amplification de NMYC et à un pronostic défavorable.
CD44 est une molécule impliquée dans les interactions intercellulaires et entre cellule et
matrice extracellulaire. L’absence d’expression de CD44 est associée à un pronostic
défavorable alors que son expression permet de définir un sous groupe de patients de
pronostic favorable, même parmi les stades 3 et 4. Son expression est inversement corrélée à
l’amplification de NMYC.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
23
Dans notre série, la recherche de l’amplification de l’oncogène NMYC n’a été faite chez
aucun de nos malades, par défaut de réalisation au laboratoire et de son coût élevé. Alors que
dans les cas de neuroblastomes bilatéraux rapportés dans la littérature, 6 cas sur 30
documentés ont une amplification de cet oncogène, et sont tous décédés [4,7,19], y compris
les tumeurs stade IV S. Dans une étude multicentrique impliquant 29 cas IVS, seulement 4
avaient une amplification MYCN et ont tous survécu [20] ; ceci peut s’expliquer par le petit
nombre de ces tumeurs amplifiant MYCN et par la biologie particulière de la tumeur dans ce
groupe d'âge [7].
7. Classifications pronostiques :
De nombreuses classifications des neuroblastomes existent sans que l’on puisse
démontrer la supériorité de l’une par rapport à l’autre. Elles sont utiles aux cliniciens et leur
permettent un langage commun. Leur objectif est de définir, de la façon la plus précise
possible, des groupes pronostiques. Ces groupes permettent d’adapter l’intensité thérapeutique
au risque de la maladie.
7.1. Classification EVANS :
C’est la plus ancienne. Il s’agit d’une classification anatomique applicable au moment
du diagnostic permettant une évaluation pronostique en fonction de l’extension de la maladie.
STADES DEFINITIONS
1 Tumeur limitée à l’organe ou à la structure d’origine.
2 Tumeur étendue en dehors de son organe ou de sa structure d’origine, sans dépasser
la ligne médiane.
Les ganglions régionaux homolatéraux peuvent être envahis.
3 Tumeur étendue par continuité au delà de la ligne médiane.
Les ganglions régionaux envahis peuvent être bilatéraux.
4 Tumeur envahissant le squelette, les organes, les tissus mou, les ganglions à distance.
Autre que les stades 4S.
4 S Tumeur stade 1 ou 2 avec métastases limitées au foie, à la peau, à la moelle osseuse.
Sans atteinte radiologique du squelette.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
24
7.2. Classification INSS (International Neuroblastoma Staging System):
C’est la classification la plus utilisée, dérivée de la classification d’EVANS. Il s’agit
d’une classification chirurgicale, elle n’est donc applicable qu’aux patients opérés d’emblée.
STADES DEFINITIONS
Stade 1 Tumeur localisée, exérèse macroscopiquement complète avec ou sans résidu
microscopique, d’éventuels ganglions collés à la tumeur enlevée en bloc peuvent être
positifs. Les ganglions homolatéraux doivent être histologiquement négatifs.
Stade 2A Tumeur localisée, exérèse macroscopiquement incomplète (selon le chirurgien ou
l'imagerie post opératoire), ganglions homolatéraux non adhérents négatifs en
histologie.
Stade 2B
Tumeur localisée avec exérèse complète ou macroscopiquement incomplète, ganglions
homolatéraux non adhérents positifs, ganglions controlatéraux négatifs.
Stade 3 Tumeur non résécable franchissant la ligne médiane (bord externe de la vertèbre du
coté opposé à la tumeur), avec ou sans envahissement ganglionnaire régionale; ou
tumeur unilatérale localisée avec envahissement des ganglions controlatéraux; ou
tumeur médiane avec extension bilatérale par infiltration (non résécable), ou avec
envahissement ganglionnaire.
Stade 4 Quelle que soit la tumeur primitive avec extension aux ganglions à distance, à la moelle
osseuse, à l’os, au foie, au tissu cutané et/ou d’autres organes (sauf stade 4S).
Stade 4S Age < 1 an, tumeur primitive localisée (définie dans les stades 1, 2A, ou 2B), avec
dissémination limitée à la peau, au foie, et/ou à la moelle osseuse (mais avec
infiltration < 10%, et scintigraphie MIBG négative au niveau du squelette), sans lésion
osseuse radiologique.
7.3. Classification TNM :
La classification TNM est la plus adaptée puisqu’elle comporte, d’une part une
classification par stades cliniques (CS), qui permet de juger de la situation pré thérapeutique,
et d’autre part d’une classification post-chirurgicale (PS) par stades pathologiques, qui permet
d’adapter les traitements locaux postopératoires. Dans cette classification, T définit la tumeur
primitive, N les ganglions lymphatiques régionaux et M les métastases.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
25
A. Classification TNM (CS: clinical staging)
Stades T N M
CS1 T1<5 cm N0 M0
CS2 T2 5 < T <10 cm N0 M0
CS3 T< 10 cm N1 M0
CS4 T> 10 cm N1 ou N0 M0
CS4S Tout T Tout N M1
CS5 Idem classification d’EVANS
T MULTIFOCALE
Tout N
Tout M
B. Classification TNM post-chirurgicale (PS)
Stade Qualité d’exérèse Métastases
PS1 Exérèse complète de la tumeur sans ganglion envahis PM0
PS2 Exérèse complète de la tumeur et des ganglions envahis PM0
PS3 A Résidu microscopique de la tumeur et/ou des ganglions PM0
PS3 B Résidu macroscopique de la tumeur et/ou des ganglions PM0
PS3 C Simple biopsie de la tumeur et/ou des ganglions PM0
PS4 Tout type d’exérèse PM1
7.4. Classification de « pediatric oncology group » POG :
Cette classification prend en considération la présence d’ADP régionales métastatiques ;
- Stade A : résection complète de la tumeur primitive avec résidu microscopique. ADP
régionales homolatérales non adhérentes sont histologiquement indemnes. ADP adhérentes à
la surface ou dans la tumeur primitive pouvant être envahis. Si la tumeur primitive est
abdominale ou pelvienne, le foie est histologiquement indemne.
- Stade B : résection incomplète de la tumeur primitive, les ADP et le foie sont
histologiquement indemnes.
- Stade C : résection complète ou incomplète de la tumeur primitive. Les ADP non
adhérentes à la tumeur primitive sont envahies. Le foie est histologiquement indemne.
- Stade D : métastases à distance (ADP, foie, peau, moelle osseuse, os…)
- Stade Ds : enfant<1 an avec stade IVS.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
26
7.5. Classification clinico-biologique pronostique de Brodeur :
Cette classification s’appuie sur des critères pronostiques cliniques (âge, stade au
diagnostic), biologiques (catécholamines urinaires), histologiques (Shimada, CD44) ainsi que
cytogénétiques (ploïdie, perte d’hétérozygotie 1p36) et moléculaires (amplification de
NMYC, expression de TrkA). Cette la classification utilisée aux Etats-unis pour la prise en
charge thérapeutique.
Dans notre cohorte, nos patients ont été classés selon la classification d’EVANS, et 25%
des neuroblastomes surrénaliens bilatéraux étaient stade IVS, alors que ce stade a représenté
50% des cas rapportés dans la littérature [4,7,15,16,17]. Il y a donc, dans les séries de
neuroblastomes bilatéraux une proportion plus importante de stades IVS que dans la
population générale des neuroblastomes où leur fréquence est d’environ 10%. Concernant les
autres stades, notre série comporte 4 cas stade IV (50%), dans la littérature 14 cas identiques
ont été rapportés (20,5%). Le nombre de stades III est identique dans notre série et dans les
cas publiés, un seul cas a été rapporté aussi bien dans notre étude que dans la littérature. Les
stades I et II n’ont pas été signalés dans notre série, alors que dans la littérature, 11 cas de
stade I (16,1%) et 2 cas de stade II (2,9%) ont été rapportés [4,7,15,16,17].
8. Prise en charge thérapeutique :
Les thérapies actuelles utilisées dans le neuroblastome sont : la chirurgie, la
chimiothérapie, la radiothérapie et la greffe des cellules souches hématopoïétiques. La notion
d’opérabilité est un élément majeur dans le choix da la stratégie thérapeutique du
neuroblastome.
L’extrême hétérogénéité des neuroblastomes conduit à une stratégie thérapeutique
reposant sur la stratification selon l’évaluation pronostique, ainsi une prise en charge au profit
de deux stratégies (tableau A) :
Dans le cas des tumeurs à pronostic favorable : l’allégement de la thérapie est préconisé
avec l’utilisation parfois seule de la chirurgie.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
27
Dans le cas de tumeurs agressives : la tendance est à l’intensification de la prise en
charge avec l’utilisation de la chirurgie combinée à la chimiothérapie, la radiothérapie et
parfois la greffe des cellules souches autologues. En se basant sur l’INSS, l’INPC et
l’identification de nombreuses caractéristiques biologiques et génétiques comme des facteurs
de risque et prédicteurs de résultats, un système thérapeutique basé sur le risque a été
développé pour déterminer le traitement. Et les nouveaux protocoles actuels catégorisent les
patients à faible risque, à risque intermédiaire et à haut risque en se basant sur leurs facteurs
pronostiques (tableau B).
Tableau A: Les facteurs pronostiques dans le neuroblastome
Abréviations: LDH=Lactate dehydrogenase; NSE=neurone-specific enolase FL=full-length
transcript; MRP=multidrug-related protein.
8.1. La prise en charge du neuroblastome bilatéral en fonction du groupe
de risque :
Cette classification en trois groupes de risque va permettre de planifier une prise en
charge bien ciblée associant selon le besoin la chirurgie seule ou la chirurgie associée à la
chimiothérapie et/ou à la radiothérapie. On distingue trois groupes de risque : groupe à faible
risque, groupe à risque intermédiaire et groupe à haut risque.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
28
Tableau B: Children’s Oncology Group (COG): Neuroblastoma risk-group schema.
Le groupe à faible risque se résume, comme décrit dans le tableau ci-dessus, en tout
neuroblastome localisé (stade 1 ou 2) avec une biologie et une histologie favorable (c.à.d. sans
amplification de l’oncogène NMYC, pas d’hyperploïdie et bien sur un type histologique peu
ou en voie de différenciation) ou les enfants avec un stade 4S avec une biologie et une
histologie favorable. Ce groupe nécessite une thérapie minime, et la majorité des enfants
peuvent être traités par chirurgie seule. La chirurgie est la première ligne thérapeutique pour
la maladie localisée quand la masse est opérable et dont l’objectif est la résection totale de la
masse tumorale sans laisser de résidu. Une faible dose de chimiothérapie assure un excellent
pronostic pour les tumeurs localisées de faible risque inopérables d’emblée.
Le groupe à risque intermédiaire est constitué de patients de tout âge avec un stade 3
ou les patients de moins de 18 mois avec un stade 4 et qui ont une biologie favorable. La prise
en charge dans ce deuxième groupe consiste en une chimiothérapie à dose modérée ayant
pour objectif de réduire la taille de la tumeur et de la rendre ainsi opérable. Vincristine,
Cyclophosphamide, Etoposide, Ifosfamide, Doxorubicine, Cisplatine et Carboplatine sont les
drogues les plus utilisées dans la majorité des protocoles thérapeutiques et qui ont donné,
selon le Paediatric Oncology Group, d’excellents résultats dans les neuroblastomes à risque
intermédiaire sans amplification de l’oncogène NMYC, et avec hyperploïdie. Les résultats
étaient moins favorables dans les cas avec diploïdie et amplification de l’oncogène.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
29
Le groupe à haut risque comprend les tumeurs localisées avec des facteurs biologiques
défavorables et les neuroblastomes de stade 4 après 18 mois. La stratégie thérapeutique est
complexe, faisant intervenir une phase d’induction par chimiothérapie intensive et chirurgie
de la tumeur primitive, une consolidation par un traitement myélo-ablatif (chimiothérapie à
haute dose puis autogreffe de moelle osseuse), puis un traitement de la maladie résiduelle
minime par un traitement d’entretien (acide 13-cis rétinoïque).
8.2. La prise en charge du neuroblastome bilatéral selon l’INGR
(International Neuroblastoma Group Risk)
Un nouveau système de classification pré thérapeutique basé sur 13 facteurs
pronostiques, cliniques, biologiques, génétiques et histologiques a été récemment développé
par l’INGR afin d’élaborer des protocoles de prétraitement en fonction de risque, applicables
dans le monde entier (tableau C).
Pour les patients à faible risque, l’excision chirurgicale de la tumeur est généralement
curative et permet d’éviter les risques liés à la chimiothérapie. Les patients à risque
intermédiaire sont généralement traités par la chirurgie et la chimiothérapie. Des études visant
à minimiser les régimes thérapeutiques pour ces groupes de patients sont en cours [11].
Le mauvais pronostic des patients à haut risque justifie un régime thérapeutique
beaucoup plus intense, comprenant l’association de la chimiothérapie suivie d’une excision
chirurgicale complète (si possible), une radiothérapie pour atteindre le contrôle local, un
traitement myélo-ablatif avec greffe de la moelle osseuse, et une thérapie biologique [11].
8.3. Le traitement chirurgical
La chirurgie est un élément clé dans le traitement des neuroblastomes. C’est l’élément
capital du traitement des neuroblastomes localisés, car elle peut être le seul traitement
nécessaire. Les critères d’opérabilité ont été définis grâce à un important travail de
coopération entre radiologues et chirurgiens pédiatres. Dans les tumeurs inopérables
initialement, la chirurgie sera faite après réduction du volume tumoral par chimiothérapie néo
adjuvante qui permet d’augmenter les possibilités d’exérèse complète et limiter les
complications chirurgicales. Dans les tumeurs métastatiques, la place de la chirurgie est
controversée. Elle sera pratiquée secondairement après obtention d’une bonne régression des
métastases. Dans les neuroblastomes IVS où la chirurgie n’a qu’un rôle marginal, on peut
comprendre qu’une tentative d’exérèse ne soit recommandée qu’en absence de risques
opératoires majeurs.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
30
Dans la localisation surrénalienne bilatérale, en plus du risque vasculaire, il existe un
risque de défaillance surrénalienne si une chirurgie radicale est effectuée, raison pour laquelle
plusieurs études plaident contre une approche chirurgicale agressive [4].
Dans notre série tous les malades ont reçu une chimiothérapie première, contribuant à
réduire les tumeurs surrénaliennes bilatérales chez tous les patients, à rendre opérable un cas,
et à la mise en rémission des métastases dans les autres cas. Quatre de nos patients (sur les
six qui ont poursuivis le traitement) ont eu une chirurgie surrénalienne : une surrénalectomie
unilatérale et 3 surrénalectomies bilatérales subtotales avec une surrénalectomie totale de la
plus grosse tumeur et énucléation ou résection partielle de la plus petite tumeur, donc une
chirurgie conservatrice visant à préserver la fonction surrénalienne et éviter au malade la
morbidité d’une insuffisance surrénalienne, ceci rejoint la littérature qui suggère que la
préservation de la fonction surrénalienne est convenable et que la surrénalectomie bilatérale
totale doit être limitée aux cas avec glandes irrécupérables, alors que la surrénalectomie
unilatérale avec énucléation controlatérale ou résection partielle est une option acceptable
[18,12].
Pour Pagès et al [4], la stratégie thérapeutique pour ce groupe particulier de tumeurs
pourrait être similaire à celle utilisée pour le neuroblastome unilatérale, à l'exception de la
chirurgie. Cependant, la faible incidence de rechute et le risque d'insuffisance surrénalienne si
la chirurgie radicale est effectuée, plaident contre une approche chirurgicale agressive. Pour
cet auteur le traitement des tumeurs bilatérales avec la même stratégie thérapeutique des
tumeurs unilatérales a donné des résultats similaires en termes de survie. Hiyama et al [18]
recommandent l’excision chirurgicale conservatrice, car la plupart des neuroblastomes
multifocaux ont un faible potentiel de malignité et ont généralement de bons pronostics. La
surrénalectomie bilatérale totale n’est pas requise et il faut tenter de préserver la fonction
surrénalienne, avec un minimum d’envahissement.
Nos deux malades stades IVS avec un syndrome de Pepper ont été traités initialement
par une chimiothérapie seule même en absence de signes de menace chez un cas. Alors que
dans la littérature l’abstention initiale est une attitude thérapeutique employée classiquement
dans les neuroblastomes IVS non menaçants, étant donné la possibilité de régression
spontanée, et la chimiothérapie s’emploie secondairement en cas de progression tumorale ou
de non opérabilité avant d’effectuer le geste chirurgicale. Les modalités chirurgicales utilisées
dans le neuroblastome stade 4S (unilatéral ou bilatéral), surtout chez les nourrissons, ne se
sont pas clairement standardisées. Comme le stade 4S peut régresser spontanément, nous

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
31
suggérons que la surrénalectomie bilatérale dans la maladie 4S doit être évitée afin de
prévenir l'insuffisance surrénalienne potentielle [19,2,6].
Nos malades stade IV ont été traités par chimiothérapie première jusqu’à la rémission
des métastases, suivie de la chirurgie. Ceci rejoint la littérature où l’exérèse de la tumeur
primitive ne s’envisage qu’après avoir obtenu le contrôle des métastases. La balance
bénéfice/risque doit être évaluée en fonction de l’âge de l’enfant et de la présence d’une
amplification NMYC dans la tumeur. Hsu et al proposent que la résection complète ne soit
pas réalisée à tout prix si la tumeur amplifie NMYC [22]. Aussi le risque de récidive locale
augmente en présence de NMYC amplifié et la radiothérapie du lit tumoral a été proposée
pour prévenir la récidive locale.
Donc dans les cas publiés des neuroblastomes bilatéraux, l’évolution des stades IV
apparait beaucoup plus liée à la biologie de la tumeur qu’à la bilatéralité de l’exérèse
[4,13,16] et l'excision bilatérale doit être évitée, sauf dans le cas des tumeurs amplifiant
NMYC [4]. Il a été montré que la majorité des enfants avec un neuroblastome localisé de
stades INSS 1 et 2 peuvent guérir par la chirurgie seule, et que la qualité de l’exérèse
chirurgicale n’influence pas la survie. Ceci est vrai même pour les formes bilatérales.
Chez les enfants porteurs d’un neuroblastome plus avancé (même non métastatique)
INSS 3 ou stade III d’Evans, toutes localisations comprises, le bénéfice d’une exérèse large a
été évoqué. Dans les formes bilatérales, notre malade ainsi que le seul cas stade 3 publié dans
la littérature [18], ont subi une chirurgie conservatrice avec surrénalectomie bilatérale
subtotale avec de bons résultats (les deux cas sont vivants), donc la qualité de l’exérèse ne
semblait pas avoir d’influence significative sur la survie.
Dans notre série 2 de nos 8 cas sont décédés, un cas stade 4S avec un syndrome de
Pepper compliqué de détresse respiratoire, et l’autre cas avait plus d’un an avec une maladie
stade 4. Un seul cas seulement a rechuté, il s’agit d’une récidive locale au niveau de la
surrénale reséquée partiellement, nécessitant une surrénalectomie totale de la glande restante
avec développement d’une insuffisance surrénalienne et la mise en route d’un traitement
hormonal substitutif, ce cas est décédé.
Dans la littérature il a été souligné que les neuroblastomes multifocales ont
généralement des caractéristiques biologiques favorables et un bon pronostic [18]. Toutefois,
un nombre considérable de cas rapportés de tumeurs surrénaliennes bilatérales sont décédés
de la maladie [13,19], et certains avaient des métastases généralisées et particulièrement

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
32
agressives [19]. Pederivia et al ont conclu que le pronostic de ce groupe particulier de
neuroblastomes est incertain et que la mortalité est élevée [7]. Alors que Pagès et al ont
montré de bonnes survies globales (79% à 5 ans) concluant que la majorité des
neuroblastomes bilatéraux ont un pronostic favorable, sauf ceux qui présentent des facteurs de
risque, tels que l’amplification de NMYC [4] et que ces derniers sont comparables aux cas de
neuroblastome unilatéral à haut risque avec les mêmes caractéristiques de mauvais pronostic.
Le pronostic des neuroblastomes surrénaliens bilatéraux avec un syndrome de Pepper est
favorable du fait de l’âge inférieur à un an, de l’absence des métastases osseuses et d’une
absence d’une amplification du gène NMYC dans la majorité des cas [6]. Dans les cas
rapportés dans la littérature, plusieurs nourrissons avec maladie stade 4S sont décédés
rapidement après le diagnostic [4,19], ils avaient tous un syndrome de Pepper avec détresse
respiratoire. Ceci rejoint notre cas avec syndrome de Pepper compliqué de détresse
respiratoire, qui est décédé un mois après le diagnostic. Dans les précédentes séries la rechute
est peu fréquente, en effet 5 cas seulement ont rechuté parmi les cas publiés de
neuroblastomes bilatéraux, 3 d’entre eux sont décédé [2,4]. La majorité des enfants décédés
avaient des tumeurs amplifiant le NMYC [4,7,19]. Malheureusement dans notre population,
on n’a pas pu réaliser cet examen puisqu’on n’a pas cette facilité dans notre institution, aussi à
cause de son coût élevé et le manque de moyens des familles.
La chirurgie radicale dans le neuroblastome surrénalien bilatéral est susceptible de
produire des séquelles fonctionnelles importantes, notamment l’insuffisance surrénalienne.
Un seul cas dans notre série a développé une insuffisance surrénalienne après une
surrénalectomie bilatérale totale effectuée en 2 temps et aucun de nos cas ayant subi une
surrénalectomie bilatérale subtotale n’a eu cette morbidité. Dans la littérature, un cas sur les 4
ayant subi une surrénalectomie bilatérale totale a eu une insuffisance surrénalienne [23]. Les 3
autres sont décédés quelques semaines après le diagnostic [7,13]. Et 2 cas seulement sur les
13 qui ont eu une exérèse bilatérale subtotale ont développé cette insuffisance séquellaire
nécessitant un traitement hormonal substitutif à long terme, ces 2 patients sont vivants [4].
Au total, nos résultats suggèrent que les neuroblastomes bilatéraux ont un bon pronostic,
et nos données montrent une excellente survie des enfants traités pour un neuroblastome
bilatéral métastatique en soulignant le risque de séquelles à long terme après une chirurgie
radicale. L’efficacité de la stratégie reposant sur une chimiothérapie préopératoire suggère
que la question de l’opérabilité doit être considérée très attentivement au diagnostic, plutôt
que d’envisager une chirurgie d’emblé aux résultats parfois aléatoires et avec un risque de

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
33
séquelles potentiellement lourdes. Considérant l’excellent pronostic des enfants en rémission
complète ou partielle et la faible incidence de rechute, laisser un résidu postopératoire parait
acceptable lorsqu’une morbidité importante semble nécessaire pour réussir une exérèse
complète. Et l’objectif de la chirurgie dans le neuroblastome surrénalien bilatéral est de tenter
de préserver la fonction surrénalienne en utilisant une chirurgie conservatrice.
8.4. Chimiothérapie :
La chimiothérapie initiale reste capitale dans la stratégie de traitement du
neuroblastome. En effet il s’agit d’une tumeur chimio sensible et 60% des patients ont des
métastases au moment du diagnostic. Elle concerne tous les patients porteurs d’un
neuroblastome métastatique ou localisé inextirpable. A doses conventionnelles, elle permet
l’amélioration des formes localisées et des formes métastatiques.
Les drogues ayant montré leur efficacité sont : la Vincristine, la Cyclophosphamide,
l’Ifosfamide, le Cisplatine, le Carboplatine, la Doxorubicine et les épipodophyllotoxines
(Etoposide et Téniposide). La majorité des protocoles de chimiothérapie en cours dans le
monde associent ces substances et consistent en l’administration toutes les trois semaines
d’une combinaison de deux ou trois de ces substances. Les principales associations utilisées
actuellement sont : CO (Cyclophosphamide et Vincristine), CADO (Cyclophosphamide,
Vincristine, Doxorubicine), Etoposide-Carboplatine, et Etoposide-Cisplatine.
Pour les tumeurs inopérables d’emblée, la chimiothérapie permet de réduire la taille et
ainsi d’en faciliter l’ablation complète dans un second temps. Et dans le cas des tumeurs
métastatiques, la chirurgie est pratiquée secondairement, après qu’une régression des
métastases a été obtenue par la chimiothérapie d’induction.
Dans la plupart des protocoles, notamment français, le traitement des stades IV des
enfants âgés de plus de 1 an ou ayant une tumeur amplifiant NMYC consiste en une
chimiothérapie conventionnelle d’induction, suivie d’une intensification thérapeutique par
chimiothérapie à haute dose et greffe de cellules souches hématopoïétiques autologues. Cette
attitude thérapeutique permet d’augmenter la survie des patients, notamment lorsque le
NMYC est amplifié. Une fois le traitement d’induction réalisé et si l’enfant est en rémission
au niveau des métastases, le traitement de la tumeur primitive par chirurgie plus ou moins
radiothérapie est envisagé. Dans notre série aucun patient n’a reçu d’intensification
thérapeutique, alors que 4 cas dans la littérature des neuroblastomes bilatéraux ont reçu ce
schéma thérapeutique [4,21].

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
34
Dans les stades IVS, les nourrissons avec un syndrome de Pepper menacés par des
signes de compression mettant en jeu le pronostic vital, sont traités par chimiothérapie parfois
associée à de la radiothérapie hépatique, dans notre série et dans la littérature [4,6,19]. Ceci
est l’attitude thérapeutique de référence. Dans les autres stades IVS, la chimiothérapie s’est
employée secondairement, en cas de progression tumorale ou de non opérabilité avant
d’effectuer le geste chirurgical [4,6,7,12].
Dans notre série la chimiothérapie conventionnelle d’induction a réduit le volume
tumoral chez tous nos patients qui ont achevé leur traitement, ce qui a permis une chirurgie
satisfaisante avec un minimum de risque, elle a rendu opérable un cas et a contribué à la mise
en rémission des métastases indispensable à la chirurgie chez 3 de nos cas stade IV. Le
protocole utilisé est une alternance CADO et VP16-Carboplatine ou VP16-Cisplatine, et vu
qu’on avait 2 neuroblastomes haut risque, le protocole Haut Risque Neuroblastome Maroc-
2010 a été utilisé chez ces 2 cas prouvant ainsi son efficacité. Ce protocole comprend 5 cures :
Cisplatine-Etoposide ; vincristine-Cyclophosphamide-Adriamycine ; Ifosfamide-Etoposide ;
Carboplatine-Etoposide et Cisplatine-Etoposide. Notre cas stade IVS avec un syndrome de
Pepper non menaçant a été traité par une chimiothérapie conventionnelle en première
intention sans abstention thérapeutique première ; deux cas similaires sont rapportés dans la
littérature [15,17].
8.5. Radiothérapie :
Radiothérapie externe
Le neuroblastome est une tumeur radiosensible. Cependant la place de la radiothérapie
dans la prise en charge actuelle des patients est à définir. Le risque de séquelles chez l’enfant
de moins de 5 ans (grande majorité traités pour neuroblastome) conditionne, pour une large
part, les discussions autour du rapport bénéfice/risque de cet outil thérapeutique. Longtemps
utilisée pour améliorer le pronostic des formes localisées, l’utilisation de chimiothérapies,
plus efficaces, en a limité considérablement les indications. En revanche, la radiothérapie
garde un rôle important dans la prise en charge palliative, en particulier des patients avec
métastases osseuses douloureuses, elle permet un contrôle rapide de la douleur.
Radiothérapie ciblée par iode 131-méta-iodo-benzyl-guanidine :
La MIBG marquée avec de l’iode 131 peut constituer un moyen d’administrer une
irradiation ciblée. Différentes études ont montré un taux de réponse de 30 à 40%. Ce

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
35
traitement a une efficacité démontrée comme traitement antalgique dans les situations
palliatives, il stabilise également la progression tumorale pendant un temps donné.
Dans notre étude, aucun de nos patients n’a eu un traitement par radiothérapie. Dans les
séries précédentes de neuroblastomes bilatéraux, des cas avec des syndromes de Pepper qui
ont reçu une irradiation hépatique de décompression sont rapportés [4,6,19], aussi 2 cas avec
une tumeur de stade IV ont eu de la radiothérapie au niveau du résidu chirurgical, sont
signalés dans la littérature [4,13].
8.6. Les agents différenciants : Les rétinoïdes
In vitro, l’acide 13-cis rétinoïque diminue la prolifération des neuroblastes, induit une
différentiation, diminue l’expression de NMYC et augmente l’expression de leur récepteur. In
vivo, l’étude de CCG a montré que, quels que soient les traitements reçus antérieurement et le
statut biologique de la tumeur, les patients ayant reçu un traitement d’entretien par l’acide
rétinoïque avaient une probabilité de survie 15% supérieure à ceux qui n’avaient pas reçu ce
traitement. Les résultats de cette étude ont conduit la majorité des équipes dans le monde à
administrer ce traitement aux patients à haut risque. Le Fenrétinide induit une apoptose des
lignées tumorales supérieure à celle des autres rétinoïdes. Cependant, sa place dans le
traitement des neuroblastomes reste à déterminer.
9. Perspectives thérapeutiques :
Le neuroblastome est diagnostiqué dans plus de la moitié des cas au stade métastatique
et 60% des enfants rechutent et décèdent. Par conséquent il est nécessaire d’identifier des
marqueurs plus spécifiques et plus sensibles pour permettre une prise en charge plus précoce
de ces patients mais également de développer de nouvelles thérapies pour les patients
diagnostiqués tardivement et pour lesquels aucun traitement efficace n’est proposé. Les
thérapies anti-angiogéniques ciblent l’angiogénèse tumorale, étape préliminaire à la
dissémination métastatique. Ces thérapies innovantes agissant à un stade tardif du
développement tumoral sont intéressantes chez ces patients diagnostiqués au stade IV et
peuvent représenter une des thérapies futures dans la prise en charge de ces patients.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
36
Conclusion

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
37
Le neuroblastome bilatéral est une entité rare, diagnostiqué autour d’un âge médian de 9
mois et révélé généralement par des symptômes liés aux métastases. La localisation bilatérale
serait liée à une prédisposition génétique et est caractérisée par un pronostic plutôt favorable.
La chirurgie conserve une place centrale dans le traitement du neuroblastome bilatéral,
et l’objectif de l’intervention reste une exérèse complète, à mettre en balance avec les risques
de complications et de séquelles post opératoires. La décision doit être prise dans le contexte
global des autres éléments pronostiques, et l’exérèse complète n’est pas toujours
indispensable. Il est actuellement admis que :
1. Une chimiothérapie première permet d’améliorer l’opérabilité de certaines tumeurs
présentant des facteurs de risque chirurgicaux.
2. Une chirurgie d’emblée expose à la survenue des complications qui peuvent être
évités par une chimiothérapie néo-adjuvante.
3. Une exérèse incomplète est éventuellement préférable à une résection complète avec
des complications.
L’acte chirurgical est toujours soumis à une évaluation bénéfice/risque, et le principal
risque dans la localisation bilatérale de cette tumeur, autre que le risque vasculaire, est le
risque de défaillance surrénalienne si une chirurgie radicale est effectuée ainsi plusieurs
études plaident contre une approche chirurgicale agressive.
Notre travail sur les neuroblastomes surrénaliens bilatéraux montre qu’une exérèse
incomplète permet d’obtenir une survie excellente tout en limitant le risque d’insuffisance
surrénalienne. Et surtout la reconnaissance que la localisation bilatérale est une localisation
favorable.

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
38
Références

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
39
[1]. Nickerson HJ. Favorable biology and outcome of stage IV-S neuroblastoma with
supportive care or minimal therapy: a Children’s Cancer Group study. J Clin Oncol
2000; 18:477–486
[2]. Lo Cunsolo C. Molecular alterations in a case of bilateral adrenal neuroblastoma. Med
Pediatr Oncol 2001; 36:491–493
[3]. Kushner BH. Familial Neuroblastoma : Case Reports, Literature Review and Etiologic
Considerations. Cancer, 1986; 57:1887–1893.
[4]. Perle Maliszewicz Pagès. Bilateral adrenal neuroblastoma. Pediatr Blood Cancer
2009; 52 :196-202
[5]. Brodeur GM. Neuroblastoma. Biological insights into a clinical enigma. Nat Rev
Cancer 2003;3:203-216.
[6]. Kerdudo C. Neuroblastome surrénalien bilatéral et syndrome de pepper : à propos de
quatre observations Arch Pediatr 2004 ; 11(12) :1450-6
[7]. Pederiva F. Bilateral adrenal neuroblastoma is different. Eur J Pediart Surg 2007; 17
:393-396
[8]. Grosfeld JL. Risk based management of childhood solid tumors. J Am Coll Surg
1999; 189:407-425.
[9]. Brodeur GM. Revision of the international criteria for neuroblastoma diagnosis,
staging and reponse to treatment J Clin Oncol 1993 ;11 :1466-1477
[10]. Knudson AG. Mutation and cancer : neuroblastoma and pheochromocytoma Am J
Hum Genet 1972 ; 24 :514-522
[11]. Barrie SR. Neuroblastoma (chapter 31) Pediatr Surg 2012 ;7 :441-458
[12]. Ishiguro Y. Bilateral adrenal neuroblastoma Eur J Pediatr Surg 1994;4 :37-39
[13]. Kramer SA. Bilateral adrenal neuroblastoma Cancer 1980 ;45(8):2208-12
[14]. Suzuki H. Metachronous bilateral adrenal neuroblastoma Cancer 1985 ;56(6) :1490-2
[15]. Kwan Jong-lee. Heterochronous bilateral adrenal neuroblastoma : stage 4S in early
infancy following resection of stage I lesion in the neonatal period Pediatr Surg Int
2012; 28:59-62

Le neuroblastome bilatéral : Expérience du Service d’Hématologie et Oncologie Pédiatrique de Rabat.
40
[16]. Gupta R. Stage 4S bilateral adrenal neuroblastoma in new born APSP J Case Rep
2014;5(1):9
[17]. Monsaingeon M. Comparative values of catecholamines and metabolites for the
diagnosis of neuroblastoma Eur J Pediatr 2003; 62:397-402
[18]. Hiyama E. Multifocal neuroblastoma : biologic behavior and surgical aspects. Cancer
2000 ; 88(8) :1955-63
[19]. Zaizen Y. Bialteral adrenal neuroblastoma Eur J Pediatr Surg 1997 ;7(5) :304-7
[20]. Tonini GP. MYCN oncogene amplification in neuroblastma is associated with worse
prognosis, exept in stage 4s : the italian experience with 295 children J Clin Oncol
1997 ;15 :85-93
[21]. Yanai T. A rare case of bilateral stage IV adrenal neuroblastoma with multiple skin
metastases in neonate : diagnosis, management, and outcome J Pediatr Surg
2004;39(12) :1782-3
[22]. Brouard J. Neonatal Pepper’s syndrome with bilateral adrenal tumor Arch Fr Pediatr
1990 ;47:543
[23]. Schleiermacher G. Treatment of stage 4s neuroblastoma –report of 10
years’experience of the French Society of Paediatric Oncology (SFOP) Br J Cancer
2003 ;89(3) :470-6.





























