Embed Size (px)
Citation preview

Peroxidase-Mediated Removal of aPolychlorinated Biphenyl UsingNatural Organic Matter as the SoleCosubstrateL I S A M . C O L O S I , †
D A N I E L J . B U R L I N G A M E , ‡
Q I N G G U O H U A N G , ‡ A N DW A L T E R J . W E B E R . , J R . * , ‡
Departments of Civil and Environmental Engineering and ofChemical Engineering, University of Michigan, Ann Arbor,Michigan 48109
Natural organic matter (NOM) of hydroxylated aromaticcharacter can undergo catalyst-mediated self-couplingreactions to form larger molecular aggregates. Indeed, suchreactions are central to natural humification processes.Nonhydroxylated persistent aromatic contaminants such aspolychlorinated biphenyls (PCBs) are, conversely, inertwith respect to such reactions. It is here demonstratedhowever that significant coincidental coupling and removalof a representative aqueous-phase PCB occurs duringhorseradish peroxidase (HRP)-catalyzed oxidative couplingreactions of a representative aquatic NOM. Experimentswith Suwannee River fulvic acid as a reactive cosubstrateindicate that 2,2′-dichlorobiphenyl (PCB-4) is covalentlyincorporated into aggregating NOM, likely through fortuitouscross-coupling reactions. To develop a better understandingof potential mechanisms by which the observed phe-nomenon occurs, two hydroxylated monomeric cosubstratesof known molecular structure, phenol and 4-methoxyphenol,were investigated as alternative cosubstrates. PCB-4removal appears from these experiments to relate to certainmolecular characteristics of the native cosubstratemolecule (reactivity with HRP, favorability for radicalattack, and hydrophobicity) and its associated phenoxyradical (stability). The findings reveal potential pathwaysby which PCBs, and perhaps other polyaromatic contaminants,may be naturally transformed and detoxified in nature.The results further provide a foundation for developmentof enhanced-humification strategies for remediation of PCB-contaminated environmental systems.
IntroductionBiogeochemical degradation and humification comprise twoclasses of complex counterdirectional processes governingthe ongoing environmental transformation and turnover ofnatural organic matter (NOM) (1). Degradation processeseffect reductions in the sizes of macromolecules to smallerlabile molecules for eventual mineralization, while humifi-cation involves processes in which small organic moleculesare aggregated into macromolecular/supramolecular struc-
tures. These structures may be classified as humic substances.Both classes of transformations can be exploited to achieveengineered remediation of soil and sediment systems thathave been contaminated by organic xenobiotics; however,the humification pathway has generally received much lessattention in this regard than has the degradation/mineral-ization pathway (2).
Oxidative coupling processes constitute a class of reactionscritically involved in NOM humification processes. Thesereactions, generally catalyzed by metal oxides or oxidativeenzymes, have been shown to facilitate incorporation of bothnatural and anthropogenic phenols and anilines into activelyaggregating or “polymerizing” NOM matrixes (2-5). In thissense, humification becomes an especially attractive reme-diation strategy because the coupling of xenobiotic mono-mers can mediate irreversible immobilization and ultimatedetoxification (6-12). It has also been demonstrated thatthe removal of substrate compounds may be enhanced bythe addition of a cosubstrate that is comparatively morereactive toward the selected catalyst than the target xenobioticitself (7, 13-15). This enhancement has been attributed toan increased quantity of nonspecific free radicals generatedduring reactions between the catalyst and the cosubstrateand to subsequent participation of these radicals in cross-coupling reactions between substrates (6, 7). Finally, it hasbeen suggested that phenolic cosubstrates undergoingoxidative coupling may, perhaps by the same mechanism,result in concomitant transformation of inert contaminants.That is, the presence of an active substrate has been shownto engender polymerization of compounds (e.g., polychlo-rinated biphenyls (PCBs), polyaromatic hydrocarbons (PAHs))that, lacking a phenolic or anilinic subunit, may notthemselves serve as active donors for the catalyst (2, 16).
It has become increasingly evident, as our understandingof humification processes has evolved, that the traditionalcharacterization of humic substances as macromolecularpolymers may require reconsideration. It has indeed beensuggested that these materials may be better classified assupramolecular associations of relatively small heterogeneousmolecules (17, 18). This view is at least more consistent withobserved direct polymerizations of humic substances in thepresence of suitable catalysts. In these reactions activatedaromatic constituents of the loosely bound humic structurespresumably undergo the same radical-mediated polymer-izations as do phenolic or anilinic monomers (17, 19-21).That humic constituents may influence the transformationof xenobiotics during oxidative coupling by participating incross-coupling with the latter has in fact been documented(3, 5, 6, 22). Such observations support a hypothesis thatcertain contaminants, even though themselves inert withrespect to a catalyst, may be transformed and incorporatedinto humic substances, presumably through free radicalreactions mediating the oxidative cross-coupling reactionsof cosubstrates.
Motivated by the hypothesis articulated above, we in thispaper describe experiments conducted to evaluate thathypothesis. Specifically, an inert xenobiotic, a PCB, is shownto be transformed effectively during the oxidative couplingreactions between a humic substance serving as the solecosubstrate and horseradish peroxidase (HRP) as a modelcatalyst. In search of an understanding of associated mech-anisms, we also investigated two specific monomeric testcosubstrates of well-known molecule structure, phenol and4-methoxyphenol, and evaluated the manner in whichstructural features of these cosubstrates relate to the facilita-tion of PCB coupling and removal. Because no attempt was
* Corresponding author phone: (734) 763-1464; fax: (734) 9364391; e-mail: [email protected].
† Department of Civil and Environmental Engineering.‡ Department of Chemical Engineering.
Environ. Sci. Technol. 2007, 41, 891-896
10.1021/es061616c CCC: $37.00 2007 American Chemical Society VOL. 41, NO. 3, 2007 / ENVIRONMENTAL SCIENCE & TECHNOLOGY 9 891Published on Web 12/23/2006

made to measure radical species, it remains impossible toascertain that the PCBs react directly with cosubstrateradicals; however, the cosubstrate molecular data are pre-sented as indirect support for their presumed involvement.
After decades of discharge into the natural environment,PCBs have become widely distributed in aquatic sediments,threatening human and environmental health as they maketheir way into both aquatic and terrestrial food chains (23).The findings presented here reveal a potential means bywhich these contaminants can be transformed and detoxifiedin the natural environment. They thus lay the groundworkand foundation for development of enhanced-humificationstrategies for engineered remediation of PCB-contaminatedenvironments.
Experimental SectionMaterials. Extracellular horseradish peroxidase (type I, RZ) 1.3), hydrogen peroxide (29.9%, ACS reagent grade), 2,2′-azinobis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) (98%in diammonium salt form), and radiolabeled phenol (U-14C-phenol in toluene solution) were purchased from SigmaChemical Co. (St. Louis, MO). Cosubstrates including phenol(99%), 4-methoxyphenol (99%), and Suwannee River fulvicacid (SRFA) were obtained from Acros Chemical Co. (MorrisPlains, NJ), Aldrich Chemical Co. (Milwaukee, WI), and theInternational Humic Substances Society (IHSS), respectively.2,2′-Dichlorobiphenyl (PCB-4) (99.35%) was obtained fromNeosyn Laboratories Inc. (New Milford, CT).
Quantifying Apparent Removal of PCB-4 at SelectedCosubstrate/PCB Ratios Over Consecutive Reaction Peri-ods. Reactions mediated by HRP/H2O2 were carried out atroom temperature in 5 mL of a 2.0 µM stock solution ofPCB-4 prepared in a phosphate buffer solution (PBS; 10 mM,pH 7.0). Agitated Teflon-capped 11 mL glass test tubes wereused as completely mixed batch reactors (CMBRs). In initialexperiments designed to determine optimal cosubstrateratios, different volumes of cosubstrate solution and comple-mentary PBS were added, such that the total volume in eachreactor was always 6 mL. In this way, an array of reactorscontaining varying ratios of cosubstrate to PCB was createdin quadruplicate, and each CMBR contained the same finalPCB concentration (1.8 µM).
A 100 mg/L total SRFA stock solution was prepared andthen filtered using 0.45 µm glass fiber filters to yield a solubletotal organic carbon (TOC) concentration of 52.44 mg/L.Because the molecular weight of SRFA is not well character-ized, the SRFA/PCB-4 ratios were then computed on a TOCbasis. Concentrated stock solutions of two test cosubstrates(phenol and 4-methoxyphenol) were prepared and deliveredin methanol to achieve high cosubstrate/PCB-4 ratios withoutsignificant dilution of the PCB concentration. To ensureuniform HRP activity at different ratios, each CMBR receivedthe same total volume of methanol in the form of a stockcosubstrate solution, dilution methanol, or some combina-tion of the two to yield a final 5.5% methanol (v/v) content.
HRP activities for the phenol, 4-methoxyphenol, and SRFAratio experiments were 0.01, 0.01, and 2 U/mL, respectively,where 1 unit (U) of HRP activity is defined as the amountcatalyzing the oxidation of 1 µmol of ABTS/min. The sameexcess concentration of hydrogen peroxide (150 µM) wasused for the reactions with phenol and 4-methoxyphenol,but a more aggressive condition (2 mM) was used with SRFAto compensate for its decreased aromaticity compared tothat of phenol and 4-methoxyphenol and to ensure saturationof the increased HRP dose required in the SRFA experiments.Following addition of the hydrogen peroxide, each CMBRwas agitated gently on a shake table for 30 min prior to theaddition of 96 µL of 1.0 N HCl to terminate the reaction.Following termination, samples were immediately centri-fuged at 2205g for 25 min to remove polymerized reaction
byproducts. Equal volumes of the supernatant and isooctanewere then added to a fresh tube and vortexed vigorously for30 s. Upon separation of the phases, analytical samples weretaken from the isooctane. Extraction recovery for the liquid-liquid extraction was between 89% and 97%.
Once the optimal ratio of cosubstrate to PCB wasdetermined for the SRFA and the monomeric cosubstrates,experiments were conducted using a time-sequenced mul-tiple addition reaction scheme and an initial PCB-4 con-centration of 1.8 µM. In each experiment, an appropriatevolume of phenol, 4-methoxyphenol, or SRFA was added toachieve the predetermined optimal cosubstrate/PCB-4 ratios.Specifically, 24 identical CMBRs were prepared to test eachcosubstrate. Four blank CMBRs each received a volume ofPBS equal to the sum of the HRP + H2O2 doses and servedas the sample for time t ) 0. The remaining reactors weredosed with the enzyme and catalyst and set on the shakertable to react. At preselected 30 min time intervals, sets offour test tubes were “sacrificed” by additions of the acid toterminate the reactions. The remaining test tubes were thenredosed, set back on the shake table, and allowed to reactfor additional 30 min intervals ranging from 60 min to 3 h.A control reaction was performed using this same protocol(HRP + H2O2 + PCB-4) in the absence of any cosubstrate.It should be noted that HRP dosages were selected to ensuresaturation of the enzyme with respect to the cosubstrate attime zero for a subsequent 30 min reaction period, i.e., 2U/mL for SRFA, 0.75 U/mL for phenol, and 0.007 U/mL for4-methoxyphenol. Although an excess concentration ofhydrogen peroxide (150 µM) was used for both monomericcosubstrates, an even more aggressive condition (2 mM) wasused with SRFA.
A Hewlett-Packard 5890 series II gas chromatographequipped with a Zebron ZB-1 column (30 m × 0.25 mm ×0.25 µm; Phenomenex, Torrance, CA) and an electron capturedetector was used to measure concentrations of PCB-4 inthe isooctane samples. Nitrogen was used as the carrier gasat a flow rate of 6.4 mL/min. Injected sample volumes were4 µL, and initial temperatures for the injector, oven, anddetector were 280, 350, and 325 °C, respectively. The retentiontime for PCB-4 was 10.3 min using the following heatingprotocol: from 110 to 200 °C in 8 min and held until 15 min,from 200 to 250 °C in 2 min and held until 20 min. FollowingGC analysis, the difference between the average control(prereaction) and each experimental (postreaction) concen-tration was computed to determine apparent PCB removalin each reacted sample.
Characterizing Adsorptive versus Reactive Removal ofPCB-4. PCB removal via adsorption to precipitated productsof phenol coupling was also evaluated using a methodpreviously described by Weber and Huang (2). Six triplicatesets of test tube reactors received 4 mL each of 1.8 µM PCB-4stock solution and varying dosages of dry preformed polymersolids (adsorbent). The contents of these reactors were mixedon a shaker table for 2.5 h and the reactors then centrifugedto separate solids from the aqueous phase. Aliquots ofsupernatant were then transferred to fresh test tubes, in whichaqueous-phase PCBs were extracted into equal volumes ofisooctane for subsequent GC analysis. The doses of adsorbentwere selected to simulate the amount of precipitatedpolymers formed as a function of time during reaction ofphenol with HRP: 0 mg/L (0 min), 50 mg/L (∼60 min), 100mg/L (∼90 min), 300 mg/L (∼120 min), and 600 mg/L (∼150min). The doses were selected on the basis of a time-courseevaluation of the reaction of radiolabeled phenol (14C-phenol)with HRP. Briefly, 2 L of 1500 µM 14C-phenol stock solutionwas prepared in PBS, stirred continuously, and dosed at 30min intervals with 0.5 U/mL HRP and 150 µM H2O2. Prior toeach dose, 2 mL samples were collected and centrifuged toremove solids. The supernatant was then analyzed in a
892 9 ENVIRONMENTAL SCIENCE & TECHNOLOGY / VOL. 41, NO. 3, 2007

scintillation counter to determine the concentration of 14C-phenol in the aqueous phase. This quantity was thensubtracted from the starting concentration to determine howmuch phenol had been converted to precipitated solids asa function of time.
An identical reaction, using phenol rather than 14C-phenol,was performed to generate the required quantities ofpreformed adsorbent. Following termination of this reaction,the entire contents were centrifuged at 5000 rpm for 20 min.The pellets were then combined into a single batch and thenevaporated to dryness under nitrogen gas. The massesrequired for the doses specified previously were weighedout individually.
Evaluating the Effects of PCB-4 on the Rates of HRPReactions with Test Cosubstrates. Following determinationof optimal cosubstrate/PCB-4 ratios for both test cosub-strates, the effects of the PCB on the initial rate of reactionbetween HRP and each test cosubstrate were evaluated. Tothis end, an approach similar to that described in a previouspaper by Colosi et al. was taken (24). Stock solutions of phenol(1600 µM) and 4-methoxyphenol (500 µM) in PBS wereprepared with and without the addition of PCB-4 (1.8 µM)to achieve the optimal cosubstrate/PCB-4 ratios in thoseCMBRs containing both chemicals. Reactors containing 5mL of stock solution were run in triplicate, both with andwithout PCBs, and then dosed with sufficiently low HRP toensure saturation of the enzyme with respect to the cosub-strate, i.e., 0.75 and 0.01 U/mL for phenol and 4-methox-yphenol, respectively. Reactions were initiated upon additionof 150 µM H2O2 and the CMBRs shaken by hand for 20 s priorto addition of 100 µL of 1.0 N HCl to terminate the reaction.Blank CMBR tests were run in triplicate with and withoutPCB-4 and dosed with equivalent volumes of PBS in placeof H2O2. Immediately following acid addition, all CMBRs werecentrifuged at 2205g for 25 min, and the supernatants fromeach reactor were transferred to amber HPLC vials. An Agilent1100 series high-performance liquid chromatograph equippedwith a Phenomenex C18 column (250 × 2.0 mm, 5 µm particlesize) was used to detect pre- and postreaction concentrationsof phenol (mobile phase 40% reagent-grade acetonitrile(ACN), 60% distilled deionized water (DDI), flow rate 0.3mL/min, tR ) 4.7 min) and 4-methoxyphenol (mobile phase30% ACN, 70% DDI, flow rate 0.4 mL/min, tR ) 3.9 min). Forboth chemicals, concentrations were measured using UVabsorbance with external five-point calibration. Followingmeasurement of the cosubstrate concentration in the blank(S0) and reaction CMBRs (S20), initial reaction rates (r0) weredefined for the first reaction interval according to r0 ) (S0 -S20)/∆t.
Determinations of Pertinent Molecular Descriptors ofCosubstrate Molecules. Correlations between moleculardescriptors and PCB-4 removal efficiency were attemptedfor both monomeric cosubstrates evaluated. Literature valuesfor octanol-water partitioning coefficients (log P) wereemployed to assess their relative hydrophobicity (25).
Electronic parameters utilized in a comparison of mo-lecular reactivity for the selected cosubstrates were computedusing the HyperChem Molecular Modeling System, profes-sional version 7.1 (Hypercube, Inc., Gainesville, Florida) (26).Cosubstrate structures were optimized using the OPLSmolecular mechanics force field and the Polak-Ribiereoptimization algorithm set to a convergence gradient criterionof less than 0.1 kcal/(Å mol). Subsequent quantum optimi-zation was achieved using the ZINDO/1 semiempiricalmethod with the same convergence algorithm and criterion.After optimization, EHOMO (energy of the highest occupiedmolecular orbital), ELUMO (energy of the lowest unoccupiedmolecular orbital), and the HOMO-LUMO gap (H-L gap )ELUMO - EHOMO) were computed for each cosubstrate molecule.Polarizability, an indicator of reactivity that parametrizes
the flexibility of a molecule’s electron cloud, was alsocalculated. Finally, quantum-optimized structures of eachcosubstrate’s respective radical (phenoxy and 4-methoxy)were computed using the ZINDO/1 algorithm in conjunctionwith unrestricted Hartree-Fock (UHF) calculations and thesame convergence criterion. In each case, the total chargewas set to zero with doublet multiplicity. The quantum-optimized structures were used to compute the total energy(Etotal) for each radical.
Results and DiscussionQuantifying Apparent PCB-4 Removal at Selected Cosub-strate/PCB Ratios. Figure 1 depicts apparent removal ofPCB-4 following 30 min reactions of HRP with SRFA, phenol,and 4-methoxyphenol at an array of selected cosubstrate/PCB ratios. Despite dramatic differences in the cosubstratemolecules investigated, their impacts on PCB-4 removal sharea common general trend, as shown in Figure 1. In eachinstance, PCB-4 removal increases with increasing cosub-strate concentration from zero to a maximum and then beginsto decrease with a further increase in cosubstrate concentra-tion. This is consistent with the hypothesis that HRP reactionsgenerate free radicals in their reactions with cosubstratesthat can then nonselectively attack PCB molecules, leadingto their incorporation in precipitable products of the oxidativecoupling reactions. The PCB molecule, because it lacks ahydroxylated aromatic moiety, is not itself a direct HRPsubstrate, such that some cosubstrate must be reacted withthe enzyme to generate nonspecific phenoxy radicals. At lowconcentrations of cosubstrate (corresponding to the lowerratios), rates of PCB-4 removal appear to be limited bysuboptimal rates of radical formation. In contrast, whencosubstrate concentrations are higher than optimal (corre-sponding to higher ratios), interactions between radicals andPCB molecules become increasingly less probable; i.e., thepreponderance of cosubstrate molecules effectively out-competes the PCB. Thus, most efficient PCB removal willoccur at the optimal ratio of cosubstrate to PCB. As indicatedin Figure 1, this ratio is 10× for SRFA, 900× for phenol, and200× for 4-methoxyphenol. It is noteworthy that while eachof the plots in Figure 1 shares the same general trend, eachreaches a different maximum value at dramatically differentoptimal ratios.
Characterizing Apparent PCB-4 Removal over Con-secutive Reaction Periods. Figure 2 depicts the results of
FIGURE 1. Apparent PCB-4 removal following reaction of HRP withthe cosubstrate at an array of selected cosubstrate/PCB ratios.Enzyme concentrations used in conjunction with SRFA (top), phenol(middle), and 4-methoxyphenol (bottom) as the cosubstrate are 2,0.01, and 0.01 U/mL, respectively. Error bars represent 95% confidenceintervals for the mean value (n ) 4).
VOL. 41, NO. 3, 2007 / ENVIRONMENTAL SCIENCE & TECHNOLOGY 9 893
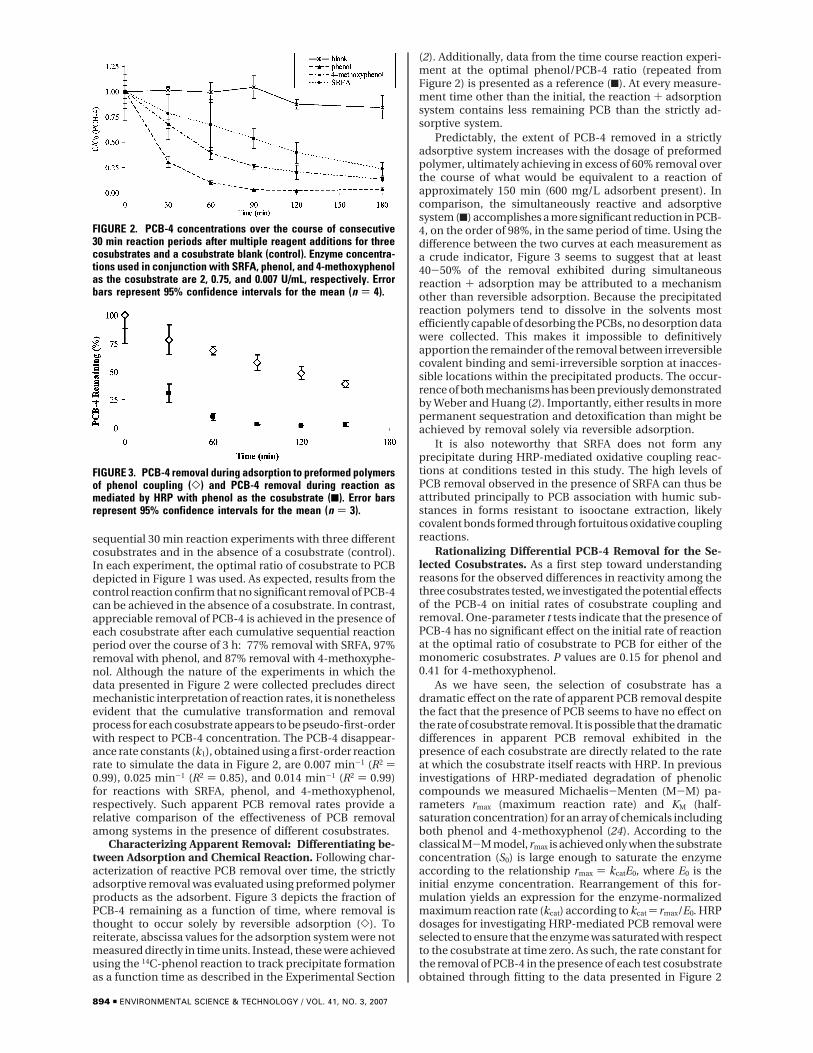
sequential 30 min reaction experiments with three differentcosubstrates and in the absence of a cosubstrate (control).In each experiment, the optimal ratio of cosubstrate to PCBdepicted in Figure 1 was used. As expected, results from thecontrol reaction confirm that no significant removal of PCB-4can be achieved in the absence of a cosubstrate. In contrast,appreciable removal of PCB-4 is achieved in the presence ofeach cosubstrate after each cumulative sequential reactionperiod over the course of 3 h: 77% removal with SRFA, 97%removal with phenol, and 87% removal with 4-methoxyphe-nol. Although the nature of the experiments in which thedata presented in Figure 2 were collected precludes directmechanistic interpretation of reaction rates, it is nonethelessevident that the cumulative transformation and removalprocess for each cosubstrate appears to be pseudo-first-orderwith respect to PCB-4 concentration. The PCB-4 disappear-ance rate constants (k1), obtained using a first-order reactionrate to simulate the data in Figure 2, are 0.007 min-1 (R2 )0.99), 0.025 min-1 (R2 ) 0.85), and 0.014 min-1 (R2 ) 0.99)for reactions with SRFA, phenol, and 4-methoxyphenol,respectively. Such apparent PCB removal rates provide arelative comparison of the effectiveness of PCB removalamong systems in the presence of different cosubstrates.
Characterizing Apparent Removal: Differentiating be-tween Adsorption and Chemical Reaction. Following char-acterization of reactive PCB removal over time, the strictlyadsorptive removal was evaluated using preformed polymerproducts as the adsorbent. Figure 3 depicts the fraction ofPCB-4 remaining as a function of time, where removal isthought to occur solely by reversible adsorption (]). Toreiterate, abscissa values for the adsorption system were notmeasured directly in time units. Instead, these were achievedusing the 14C-phenol reaction to track precipitate formationas a function time as described in the Experimental Section
(2). Additionally, data from the time course reaction experi-ment at the optimal phenol/PCB-4 ratio (repeated fromFigure 2) is presented as a reference (9). At every measure-ment time other than the initial, the reaction + adsorptionsystem contains less remaining PCB than the strictly ad-sorptive system.
Predictably, the extent of PCB-4 removed in a strictlyadsorptive system increases with the dosage of preformedpolymer, ultimately achieving in excess of 60% removal overthe course of what would be equivalent to a reaction ofapproximately 150 min (600 mg/L adsorbent present). Incomparison, the simultaneously reactive and adsorptivesystem (9) accomplishes a more significant reduction in PCB-4, on the order of 98%, in the same period of time. Using thedifference between the two curves at each measurement asa crude indicator, Figure 3 seems to suggest that at least40-50% of the removal exhibited during simultaneousreaction + adsorption may be attributed to a mechanismother than reversible adsorption. Because the precipitatedreaction polymers tend to dissolve in the solvents mostefficiently capable of desorbing the PCBs, no desorption datawere collected. This makes it impossible to definitivelyapportion the remainder of the removal between irreversiblecovalent binding and semi-irreversible sorption at inacces-sible locations within the precipitated products. The occur-rence of both mechanisms has been previously demonstratedby Weber and Huang (2). Importantly, either results in morepermanent sequestration and detoxification than might beachieved by removal solely via reversible adsorption.
It is also noteworthy that SRFA does not form anyprecipitate during HRP-mediated oxidative coupling reac-tions at conditions tested in this study. The high levels ofPCB removal observed in the presence of SRFA can thus beattributed principally to PCB association with humic sub-stances in forms resistant to isooctane extraction, likelycovalent bonds formed through fortuitous oxidative couplingreactions.
Rationalizing Differential PCB-4 Removal for the Se-lected Cosubstrates. As a first step toward understandingreasons for the observed differences in reactivity among thethree cosubstrates tested, we investigated the potential effectsof the PCB-4 on initial rates of cosubstrate coupling andremoval. One-parameter t tests indicate that the presence ofPCB-4 has no significant effect on the initial rate of reactionat the optimal ratio of cosubstrate to PCB for either of themonomeric cosubstrates. P values are 0.15 for phenol and0.41 for 4-methoxyphenol.
As we have seen, the selection of cosubstrate has adramatic effect on the rate of apparent PCB removal despitethe fact that the presence of PCB seems to have no effect onthe rate of cosubstrate removal. It is possible that the dramaticdifferences in apparent PCB removal exhibited in thepresence of each cosubstrate are directly related to the rateat which the cosubstrate itself reacts with HRP. In previousinvestigations of HRP-mediated degradation of phenoliccompounds we measured Michaelis-Menten (M-M) pa-rameters rmax (maximum reaction rate) and KM (half-saturation concentration) for an array of chemicals includingboth phenol and 4-methoxyphenol (24). According to theclassical M-M model, rmax is achieved only when the substrateconcentration (S0) is large enough to saturate the enzymeaccording to the relationship rmax ) kcatE0, where E0 is theinitial enzyme concentration. Rearrangement of this for-mulation yields an expression for the enzyme-normalizedmaximum reaction rate (kcat) according to kcat ) rmax/E0. HRPdosages for investigating HRP-mediated PCB removal wereselected to ensure that the enzyme was saturated with respectto the cosubstrate at time zero. As such, the rate constant forthe removal of PCB-4 in the presence of each test cosubstrateobtained through fitting to the data presented in Figure 2
FIGURE 2. PCB-4 concentrations over the course of consecutive30 min reaction periods after multiple reagent additions for threecosubstrates and a cosubstrate blank (control). Enzyme concentra-tions used in conjunction with SRFA, phenol, and 4-methoxyphenolas the cosubstrate are 2, 0.75, and 0.007 U/mL, respectively. Errorbars represent 95% confidence intervals for the mean (n ) 4).
FIGURE 3. PCB-4 removal during adsorption to preformed polymersof phenol coupling (]) and PCB-4 removal during reaction asmediated by HRP with phenol as the cosubstrate (9). Error barsrepresent 95% confidence intervals for the mean (n ) 3).
894 9 ENVIRONMENTAL SCIENCE & TECHNOLOGY / VOL. 41, NO. 3, 2007

can be further normalized using the appropriate enzymeconcentration. Table 1 presents the normalized apparentrate of PCB-4 degradation, kcat values measured for degrada-tion of the cosubstrate in the absence of PCB-4, and selectedmolecular descriptors of the evaluated cosubstrates.
The cosubstrate 4-methoxyphenol, which reacts morequickly with HRP than phenol, also mediates more effectiveremoval of PCB-4. The data presented in Table 1 are thus inaccord with our conjecture that the apparent rate of PCBremoval is dependent on the rate of reaction between HRPand the cosubstrate. Consistent with the hypothesizedmechanism by which HRP reactions enable cross-couplingand ultimately mediate removal of PCB-4, it seems likelythat those cosubstrates degraded most efficiently by theenzyme generate a larger quantity of radicals per unit oftime, enabling faster polymerization of the PCB, which itselfhas little effect on the reaction rate.
While each cosubstrate’s ability to generate radicalsappears to be associated with its optimal rate of PCB removal,the ratio at which the PCB removal exhibits optimal ratesseems to depend on the electronic character of a cosubstrate’smolecular structure. It seems reasonable that characteristicsfacilitating radical attack might improve the probability thatthe cosubstrate is preferentially polymerized over the PCB,effectively reducing the rate at which the PCB will be removedfrom solution. Of the molecular descriptors in Table 1, theH-L gap and polarizability are related to the overallfavorability of radical attack toward a molecule of a givencosubstrate. As indicated, phenol exhibits a higher H-L gapthan 4-methoxyphenol. This suggests that phenol moleculesare more stable and less excitable than molecules of4-methoxyphenol (14, 27). Similarly, the difference in po-larizability between the two cosubstrates indicates that theelectron cloud surrounding phenol is less flexible than thatof 4-methoxyphenol. On the basis of these criterion com-parisons, 4-methoxyphenol is in both instances a better targetfor radical attack, consistent with Figure 1 in which theoptimal ratio for phenol (900×) is significantly higher thanthe optimal ratio for 4-methoxyphenol (200×). At ratios higherthan that shown to be optimal for PCB removal in Figure 1(bottom), the inherent favorability of 4-methoxyphenolradical attack makes it such that PCB polymerization becomesincreasingly improbable. In contrast, a much higher con-centration of the phenol molecules may be present duringthe reaction because they are unable to compete as effectivelywith the PCB. Extrapolated, this trend can be used to inferthe relative favorability of radical attack toward the PCB inthe presence of the SRFA as the cosubstrate. As indicated inFigure 1, the observed optimal ratio of SRFA to PCB-4 (10×)is an order of magnitude smaller than even that of 4-meth-oxyphenol. This suggests that SRFA is even more optimallysuited for attack by radical species, scavenging as it doesradicals that might otherwise mediate the incorporation ofa PCB molecule. This is consistent with the fact that itmediates slower PCB-4 removal than phenol and 4-meth-oxyphenol, as well as with previous investigations in whichNOM was found to be a good radical scavenger (3, 29, 30).
More subtle than the inference regarding differences inoptimal ratios across cosubstrates, Figure 1 also indicatesthat the optimal extent of reactive removal could be related
to the stability of each cosubstrate’s associated radical. Allthings being equal, a stable radical species will be longer-lived, increasing its opportunity to interact with a moleculeof PCB. The 4-methoxy radical (ZINDO-computed Etotal )-52703 kcal/mol) is more stable and ostensibly more likelyto promote PCB polymerization than the phenoxy radical(ZINDO-computed Etotal ) -37083 kcal/mol). This is con-sistent with a decreased extent of PCB removal for phenol(30%, middle of Figure 1) relative to 4-methoxyphenol (37%,bottom of Figure 1) at their respective optimal ratios. Again,this trend may be extrapolated to make inferences about thestructure of radicals formed during reaction of HRP withSRFA as the cosubstrate. The extent of PCB removal for SRFA(29%, top of Figure 1) is somewhat smaller but not signifi-cantly different from that for phenol. This perhaps indicatesthat electron-donating aliphatic substituents of SRFA radicalsmake them structurally more similar to phenoxy than4-methoxy radicals, but less stable than either.
Finally, it seems possible that the differences in hydro-phobicity between the test cosubstrates may affect the relativeeffectiveness of adsorptive removal as the values of theiroctanol-water partitioning coefficients (P) are slightly dif-ferent. As indicated in Table 1, 4-methoxyphenol (log P )1.58) is slightly more hydrophobic than phenol (log P ) 1.46).By extension, polymers formed from the oxidative couplingof 4-methoxyphenol might more readily adsorb the highlyhydrophobic PCB-4 molecules, contributing to the greaterapparent removal as indicated by the enzyme-normalizedrate constants presented in Table 1. Significantly, as notedearlier, no precipitation of polymers was observed duringreactions with the SRFA. This implies that PCB removal inthe case of this natural cosubstrate resulted principally fromits covalent incorporation into the SRFA matrix.
AcknowledgmentsWe thank Jennifer Gehle for her diligent laboratory assistanceand Tom Yavaraski for important guidance with respect tothe analytical procedures and methodologies. This researchwas financed in large measure by Research Grant P42ES04911-14 from the National Institute of Environmental HealthSciences. L.M.C. appreciates the support provided by aNational Science Foundation Graduate Research Fellowship.
Literature Cited(1) Hedges, J. I. In Humic Substances and Their Role in the
Environment; Frimmel, F. H., Christman, R. F., Eds.; John Wiley& Sons: New York, 1988.
(2) Weber, W. J., Jr.; Huang, Q. Inclusion of persistent organicpollutants in humification processes. Direct chemical incor-poration of phenanthrene via oxidative coupling. Environ. Sci.Technol. 2003, 37, 4221-4227.
(3) Park, J. W.; Dec, J.; Kim, J. E.; Bollag, J. M. Effect of humicconstituents on the transformation of chlorinated phenols andanilines in the presence of oxidoreductive enzymes or birnessite.Environ. Sci. Technol. 1999, 33, 2028-2034.
(4) Weber, E. J.; Spidle, D. L.; Thorn, K. A. Covalent binding ofaniline to humic substances. 1. Kinetic studies. Environ. Sci.Technol. 1996, 30, 2755-2763.
(5) Huang, Q.; Weber, W. J., Jr. Interactions of soil-derived dissolvedorganic matter with phenol in peroxidase-catalyzed oxidativecoupling reactions. Environ. Sci. Technol. 2004, 38, 338-344.
(6) Bollag, J.-M. Decontaminating soil with enzymes. Environ. Sci.Technol. 1992, 26, 1876-1881.
(7) Roper, J. C.; Sarkar, J. M.; Dec, J.; Bollag, J. M. Enhancedenzymatic removal of chlorophenols in the presence of co-substrates. Water Res. 1995, 29, 2720-2724.
(8) Park, J. W.; Dec, J.; Kim, J. E.; Bollag, J. M. Transformation ofchlorinated phenols and anilines in the presence of humic acid.J. Environ. Qual. 2000, 29, 214-220.
(9) Hatcher, P. G.; Bortiatynski, J. M.; Minard, R. D.; Dec, J.; Bollag,J. M. Use of high resolution 13C NMR to examine the enzymaticcovalent binding of 13C-labeled 2,4-dichlorophenol to humicsubstances. Environ. Sci. Technol. 1993, 27, 2096-2103.
TABLE 1. Observed Constants for Apparent PCB-4 andCosubstrate Removal as Well as Selected MolecularCharacteristics for the Monomeric Cosubstrates Evaluated
Cosubstrateln(kPCB)
(s-1)ln(kcat)(s-1)
EHOMO(eV)
ELUMO(eV)
H-L gap(eV)
polariz-ability (A3)
log P(25)
phenol -8.74 7 -8.18 7.85 16.03 11.07 1.464-methoxy-
phenol-5.09 10 -7.09 7.62 14.71 13.54 1.58
VOL. 41, NO. 3, 2007 / ENVIRONMENTAL SCIENCE & TECHNOLOGY 9 895

(10) Naidja, A.; Huang, P. M.; Bollag, J. M. Comparison of reactionproducts from the transformation of catechol catalyzed bybirnessite or tyrosinate. Soil Sci. Soc. Am. J. 1998, 62, 188-195.
(11) Bhandari, A.; Xu, F. Impact of peroxidase addition on thesorption-desorption behavior of phenolic contaminants insurface soils. Environ. Sci. Technol. 2001, 35, 3163-3168.
(12) Dec, J.; Bollag, J. M. Phenoloxidase-mediated interactions ofphenols and anilines with humic materials. J. Environ. Qual.2000, 29, 665-676.
(13) Klibanov, A. M.; Alberti, B. N.; Morris, E. D.; Felshin, L. M. J.Appl. Biochem. 1980, 2, 414-421.
(14) Alberti, B.; Klibanov, A. M. Enzymatic removal of dissolvedaromatics from industrial aqueous effluents. Biotechnol. Bioeng.Symp. 1981, 11, 373-379.
(15) Berry, D. F.; Boyd, S. A. Oxidative coupling of phenols andanilines by peroxidase: structure-activity relationship. Soil Sci.Soc. Am. J. 1984, 48, 565-569.
(16) Klibanov, A. M.; Tu, T. M.; Scott, K. P. Peroxidase-catalyzedremoval of phenols from coal-conversion waters. Science 1983,221, 259-261.
(17) Piccolo, A.; Conte, P.; Tagliatesta, P. Increased conformationalrigidity of humic substances by oxidative biomimetic catalysts.Biomacromolecules 2005, 6, 351-358.
(18) Conte, P.; Piccolo, A. Conformational arrangement of dissolvedhumic substances: Influence of solution composition onassociation of humic molecules. Environ. Sci. Technol. 1999,33, 1682-1690.
(19) Weber, W. J., Jr.; Huang, Q.; Pinto, R. A. Reduction of disinfectionbyproduct formation by molecular reconfiguration of the fulvicconstituents of natural background organic material. Environ.Sci. Technol. 2005, 39, 6446-6452.
(20) Smejkalova, D.; Piccolo, A. Rates of oxidative coupling of humicmonomers catalyzed by a biomimetic iron-porphyrin. Environ.Sci. Technol. 2006, 40, 1644-1649.
(21) Sutton, R.; Sposito, G. Molecular structure in soil humicsubstances: The new view. Environ. Sci. Technol. 2005, 39,9009-9015.
(22) Huang, Q.; Selig, H.; Weber, W. J., Jr. Peroxidase-catalyzedoxidative coupling of phenols in the presence of geosorbents:Rates of non-extractable product formation. Environ. Sci.Technol. 2002, 36, 596-602.
(23) Yamashita, N.; Kannan, K.; Imagawa, T.; Villenueve, D. L.;Hashimoto, S.; Miyazaki, A.; Giesy, J. P. Vertical profiles ofpolychlorinated dibenzo-p-dioxins, dibenzofurans, napthalenes,biphenyls, polycyclic aromatic hydrocarbons, and alkylphenolsin a sediment core from Tokyo Bay, Japan. Environ. Sci. Technol.2000, 34, 3560-3567.
(24) Colosi, L. M.; Huang, Q.; Weber, W. J., Jr. Quantitative structure-activity relationship based quantification of the impacts ofenzyme-substrate binding on rates of peroxidase-mediatedreactions of estrogenic phenolic chemicals. J. Am. Chem. Soc.2006, 128, 4041-4047.
(25) Perry, R. H., Green, D. W., Maloney, J. O., Eds. Perry’s ChemicalEngineer’s Handbook, 7th ed.; McGraw-Hill: New York, 1997.
(26) HyperChem Reference Manual; Hypercube, Inc.: Gainesville,FL, 1996.
(27) DeProft, F.; Geerlings, P. Conceptual and computational DFTin the study of aromaticity. Chem. Rev. 2001, 101, 1451-1464.
(28) Huang, Q.; Weber, W. J., Jr. Transformation and removal ofbisphenol A from aqueous phase via peroxidase-mediatedoxidative coupling reactions: Efficacy, products, and pathways.Environ. Sci. Technol. 2005, 39, 6029-6036.
(29) Lindsey, M. E.; Tarr, M. A. Inhibition of hydroxyl radical reactionwith aromatics by dissolved natural organic matter. Environ.Sci. Technol. 2000, 34, 444-449.
(30) Lindsey, M. E.; Tarr, M. A. Quantitation of hydroxyl radical duringFenton oxidation following a single addition of iron andperoxide. Chemosphere 2000, 41, 409-417.
Received for review July 7, 2006. Revised manuscript receivedOctober 19, 2006. Accepted November 2, 2006.
ES061616C
896 9 ENVIRONMENTAL SCIENCE & TECHNOLOGY / VOL. 41, NO. 3, 2007





























