Embed Size (px)
Citation preview

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 1/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 1
PHARMEUROPA 18.1CONTENTS January 2006
,Pharmeuropa Scientific Notes Online 25
Publication of Supplements 5.5 and 5.6 2
General Information 5
Electronic version of the 5th Edition of the
European Pharmacopoeia 5
CRS: conditions of sale 6
List of codes of groups of experts 11
List of texts published in Supplement 5.5 12
Comments concerning some revised / corrected texts
published in Supplement 5.5 14
Elaboration / Revision of a monograph (Procedure 1) 18
Technical Guide: 4th
Edition - 2005 ( new) 19Pharmeuropa Scientific Notes ( new) 17
Pharmeuropa Bio 21
Proceedings of conferences of the EDQM 22
List of Standard Terms: 5th Edition 23
Proficiency testing studies (PTS): 2006 24
Knowledge database 11
Press releases
• Certification of suitability of monographs of the European
Pharmacopoeia (Istanbul, Turkey, 27-28 October 2005) 13
• Pharmacopoeial Discussion Group (PDG)
(Chicago, USA, 7-11 November 2005) 25
International Conferences 26
Training Sessions on the 5th Edition of the European
Pharmacopoeia: Chemicals
2-3 March 2006, London, UK 27
27-28 April 2006, Chicago, USA 32
Certification of Suitability of theMonographs of the Ph. Eur. 37
List of certificates 37
Scientific Notes 41
Effect of Temperature on Plasma Freezing under Industrial
Conditions 41
Draft Monographs and General Textsfor Comment 47
Allopurinol 47
Bisoprolol hemifumarate 49
Cefamandole nafate 53
Chlortalidone 55
Cimetidine 57
Cimetidine hydrochloride 60
Cisplatin 63
Clonidine hydrochloride 65
Clotrimazole 67
Colony-forming cell assay for human haematopoietic
progenitor cells (2.7.28) 69
Dacarbazine 71Devil’s claw dry extract 73
Diethylcarbamazine citrate 75
Dipyridamole 78Dopamine hydrochloride 80
Dorzolamide hydrochloride 82
Ethambutol hydrochloride 84
Fenoterol hydrobromide 86
Fexofenadine hydrochloride 88
Fluorescein 90
Fluorodopa (18F) injection 92
Fluorouracil 94
Glucagon, human 96
Glycerol monocaprylate 98
Glycerol monocaprylocaprate 100
Human anti-D immunoglobulin 102
Human anti-D immunoglobulin
for intravenous administration 102
Human plasma for fractionation 103
Human prothrombin complex 105
Influenza vaccine (surface antigen,
inactivated, virosome) 106
Isotretinoin 108
Liquorice dry extract, quantified 110
Magnesium citrate, anhydrous 112
Meclozine hydrochloride 112
Molsidomine 115
Moxidectin for veterinary use 117
Norgestimate 121
Nucleated cell count and viability (2.7.29) 122
Paraffin, white soft 125Paraffin, yellow soft 127
Pentaerythrityl tetranitrate, diluted 128
Perindopril tert-butylamine 131
Potassium clavulanate 134
Potassium clavulanate, diluted 137
Rectal preparations 139
Ropivacaine hydrochloride monohydrate 141
Sertraline hydrochloride 143
Sodium acetate trihydrate 146
Sodium fluoride 146
Vaginal preparations 147
Vinpocetine 149
Warfarin sodium 151
Warfarin sodium clathrate 153 Wettability of porous solids including powders (2.9.45) 155
Willow bark dry extract 158
Illustrations of powdered drugs in herbal monographs:
• Calendula flower 161
• Ribwort plantain 161
International Harmonisation 163
Carmellose 163
White petrolatum jelly 164
Yellow petrolatum jelly 165
PDG state of work (November 2005) 167
Projected timetable for publication and implementation
of texts signed off by the PDG (November 2005) 169Contents of the JP Forum (Vol. 14, No. 3) 171
Contents of the USP Forum (Vol. 31, No. 6) 172

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 2/176
2 © PHARMEUROPA Vol. 18, No. 1, January 2006
THE EUROPEAN PHARMACOPOEIA
5th Edition: initial volume 5.0 (2 volumes) + 8 Supplements (5.1 - 5.8)
Supplements 5.6, 5.7, 5.8 published in 2006
The 5 th Edition 5.0 (2 volumes) has been available since June 2004 (for prices and ordering
information please consult http://book.pheur.org). It is comprised of texts that were implemented
on 1 st January 2005 and a cumulative list of reagents.
Publication of Supplements
The supplements are not cumulative and are to be kept for the duration of the 5 th Edition.
Modifications (revisions/corrections) to texts are indicated by a line in the margin .
Supplement 5.1 has been available since September 2004; it is comprised of texts that were
implemented on 1st April 2005.
Supplement 5.2 has been available since December 2004; it is comprised of texts that were
implemented on 1st July 2005.
Supplement 5.3 has been available since June 2005; it is comprised of texts implemented on1st January 2006.
Supplement 5.4 has been available since September 2005; it is comprised of texts that will
be implemented on 1st April 2006 and a cumulative list of reagents.
Supplement 5.5 has been available since December 2005; it is comprised of texts that will be
implemented on 1st July 2006.
Supplement 5.6 will be available in June 2006; it is comprised of texts that will be
implemented on 1st January 2007.
_____________________________________________________________________________
Online access is available to the database giving names of reagents,especially chromatographic columns. The address is:
http://www.pheur.org/knowledge.htm

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 3/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 3

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 4/176
4 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 5/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 5
General Information
General Information
5th EDITION OF THE EUROPEAN PHARMACOPOEIA
ELECTRONIC VERSION1920 New and Revised Monographs and 293 General Texts
With the electronic version of the 5th Edition of the European Pharmacopoeia you can view 1920 monographs,293 general texts (including general monographs and methods of analysis), 2297 reagents, and also have a directinternet link to the most recent catalogue of reference substances, which contains 1730 references.
The electronic format has the following convenient features:
• hierarchical table of contents, subject index and keyword search;
• hyperlinks in the text of a monograph giving access to information on general methods, reagents and reference
substances used in the monograph;
• changed (inserted and deleted) texts indicated in both the HTML version and the Acrobat version;
• direct access from a monograph or a general method to the CRS database on the internet;
• use of a standard internet browser to access the data;
• direct printing of an Acrobat version for each individual monograph;
• internet and intranet versions available.
NEW : direct links to the general notices and the list of general monographs from each text.
The electronic version is available in English, in French and in a bilingual version.
PRICES: SEE THE CATALOGUE ON OUR INTERNET SITE http://book.pheur.org
Our prices are indicated in EUR but we accept payments in national currencies.
FREQUENTLY ASKED QUESTIONS:WHAT IS THE ROLE OF
THE EUROPEAN PHARMACOPOEIA?
The European Pharmacopoeia is the original source of harmonised quality standards for medicines; all of itspublished texts have undergone European harmonisation. These texts are mandatory in 34 European countries* andin the European Union and replace any pre-existing national texts on the same subject.
As laid down in their national legislation, certain member states** may continue to issue a national pharmacopoeiathat republishes all or some of the harmonised European texts (most often General Chapters), translated if necessaryinto the national language. In all cases it is the European text that is implemented and made legally binding in all ofthe member states.
*Austria, Belgium, Bosnia and Herzegovina, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Greece,
Hungary, Iceland, Ireland, Italy, Latvia, Lithuania, Luxembourg, Malta, Netherlands, Norway, Portugal, Romania, Serbia and Montenegro, SlovakRepublic, Slovenia, Spain, Sweden, Switzerland, “The Former Yugoslav Republic of Macedonia”, Turkey, United Kingdom.
**Austria, Bulgaria, Czech Republic, Germany, Greece, Hungary, Portugal, Romania, Spain, Switzerland, United Kingdom.
________________________________________________________________________________

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 6/176
6 © PHARMEUROPA Vol. 18, No. 1, January 2006
General Information
REFERENCE SUBSTANCES, PREPARATIONS ANDSPECTRA OF THE EUROPEAN PHARMACOPOEIA
The reference substances and preparations are selected
and verified batches suitable for use as prescribed in theEuropean Pharmacopoeia. The European PharmacopoeiaCommission does not guarantee their use for purposesother than those prescribed. Each vial supplied contains aquantity sufficient for the prescribed use.
It is recommended that the products be used as soon aspossible.
The stability of the contents of opened vials or ampoulescannot be guaranteed.
It should be noted that no certificates of analysis,expiry dates, nor any data not relevant to the use ofthe products as defined by the Ph. Eur. monograph
are provided with the reference material. The productscomply with the requirements of the monograph and aremonitored regularly.
For complete information, the catalogue can bedownloaded from our website.
CONDITIONS OF SALE
1730 reference substances, reference preparations
and reference spectra are supplied by the Technical
Secretariat of the European Pharmacopoeia Commission.
Prices
PRICE LIST
Prices are identified for each product in the catalogue.
However, please note that prices and package sizes are
subject to change without notice.
The European Directorate for the Quality of Medicines(EDQM) does not operate a discount policy. The sale
prices are exclusive of duties and taxes and are givenin Euros. It is the responsibility of the buyer (or the
recipient of the delivery if different from the buyer) tocontact the national fiscal or customs authorities to paythe duties and taxes. In no event shall the said duties and
taxes be paid by the Council of Europe (EDQM).
In the European Union (EU), there is no VAT identifi-
cation number for organisations with diplomatic status.The Council of Europe (EDQM) therefore has no VATidentification number and is not subject to duties and
taxes.
The goods remain the property of the Council of Europe(EDQM) until the invoice has been paid in full. Catalogue
items are not returnable for exchange or refund.
DELIVERY AND RELATED COSTS
Unless otherwise stated below or specifically agreed with
the customer, the goods are shipped to the buyer on
a DDU (Incoterms 2000) basis, namely, delivered duty
unpaid and insurance included. Where the shipment isidentified below as airport consignment only, the goodsare shipped to the buyer on a CIP (Incoterms 2000) basis,
namely carriage and insurance included.
— The Council of Europe (EDQM) delivers the goods tothe buyer not cleared for import and not unloaded by
any means of transport.
— The Council of Europe (EDQM) bears the cost andrisks of packing, transport to the delivery site and
insurance.
— In no event shall the Council of Europe (EDQM) be
held responsible for any deterioration of the goodsdue to their delayed delivery by the carrier.
— The buyer is responsible for the cost of import
customs clearance, for paying the duties and taxesrequired in the country of import and for unloading
the goods. The buyer shall be entirely responsibleif the goods are held up at customs at the time ofimport into the buyer’s country. In addition, the
buyer is responsible for any risks associated with useof their own carrier.
Where the shipping costs are paid by the customer,
the goods are shipped to the buyer on an EX Works
(Incoterms 2000) basis, with neither carriage norinsurance included. Therefore, the Council ofEurope (EDQM) takes no responsibility in any case of
deterioration or loss of goods.
— In no event shall the Council of Europe (EDQM) beable to provide any assistance.
Delivery charges
( New fees apply as from 1 st September 2005)
The extra charges are applied per shipment. A shipmentcomprises only the reference substances that can beshipped under the same conditions. Consequently, goods
requiring specific packaging (e.g. ice), dangerous goodsor controlled substances will be invoiced separately fromthe rest of the order and extra charges will be incurred.
As one order could include several shipments, theCouncil of Europe (EDQM) advises its customers to re-group their orders by type of shipment so the customerscan better track the progress of a complete order and savemoney in shipping charges.
Where the buyer requests shipping conditions other thanthose recommended in our catalogue, or another carrier,the Council of Europe (EDQM) takes no responsibility inany case of deterioration of the goods or loss of parcel.
Extra charges (postage and packaging) will be applied in
the following cases (for larger quantities, prices are givenon request). Please note that prices are subject to change
without notice.

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 7/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 7
General Information
— Other European countries: 180 EUR per item
— Outside Europe: 250 EUR per item
f) Dangerous goods in excepted quantities: sent bycarrier chosen by EDQM: Isosorbide dinitrate CRS,Glyceryl trinitrate CRS, Oxaliplatin and its impuritiesCRS, Cisplatin CRS, Dichlorodiaminocyclohexane
platinum CRS(Note: for countries inside the EU (with the exceptionof Cyprus and Malta), our shipment is on a “door todoor” basis. For all other countries (and the exceptionsabove), our shipment is by airport consignment only)
— EU: 100 EUR per item
— Other European countries: 125 EUR per item
— Outside Europe: 125 EUR per item
g) Dangerous goods sent by road: carrier chosenby EDQM: Isosorbide 5-mononitrate CRS,
Pentaerythrityl tetranitrate diluted CRS
— EU: 150 EUR per item (“door to door”)
— Other European countries: 180 EUR per item (“doorto door”)
— Outside Europe: cannot be sent
h) Precursors (controlled drugs: sent by carrier chosenby EDQM)
(Note: for countries outside the EU, our shipment is byairport consignment only)
— France: no extra charge, price is inclusive of packagingand postage. At the client’s request, Express Courierdelivery is charged at 18 EUR per shipment
— EU: 18 EUR per shipment
— Other European countries: 160 EUR per shipment
— Outside Europe: 160 EUR per shipment
NB: these extra charges include packaging, shippingand management of permits
i) Psychotropic substances (controlled drugs: sent bycarrier chosen by EDQM)
(Note: for countries outside France, our shipment isby airport consignment only)
— France: no extra charge, price is inclusive of packagingand postage. At the client’s request, Express Courierdelivery is charged at 18 EUR per shipment
— EU (except France): 110 EUR per shipment
— Outside EU: 160 EUR per shipment
NB: these extra charges include packaging, shippingand management of permits
j) Narcotics (controlled drugs: sent by carrier chosen byEDQM)
(Note: for countries outside France, our shipment is byairport consignment only)
— France: 50 EUR per shipment
— EU (except France): 110 EUR per shipment
a) Shipment at room temperature
— France: no extra charge, price is inclusive ofpackaging and postage. At the client’s request, ExpressCourier delivery is charged at 18 EUR per shipment
— EU: 18 EUR per shipment
— Other European countries : 80 EUR per shipment
— Outside Europe: 120 EUR per shipment (note: forIndia, South America and Africa, our shipment is byairport consignment only)
— Shipping costs paid by the customer: 10 EUR pershipment
b) Shipment under ice: sent in cooled freight containersand by express courier
(Note: for all countries inside the EU (with theexception of Cyprus, Estonia, Malta, Poland) ourshipment is on a “door to door” basis. For all othercountries (and the exceptions above), our shipment isby airport consignment only)
— EU: 50 EUR per shipment
— Other European countries: 70 EUR per shipment
— Outside Europe: 120 EUR per shipment
— Shipping costs paid by the customer: 20 EUR per
shipment
c) Shipment under dry ice: sent in cooled freightcontainers (dry ice) and by express courier
(Note: for all countries inside the EU (with theexception of Cyprus, Estonia, Malta, Poland) ourshipment is on a “door to door” basis. For all other
countries (and the exceptions above), our shipment isby airport consignment only)
— EU: 90 EUR per shipment
— Other European countries: 120 EUR per shipment
— Outside Europe: 200 EUR per shipment
— Shipping costs paid by the customer: 55 EUR pershipment
d) Hepatitis C virus BRP, B19 virus DNA for NAT : dry ice+ dangerous goods – from 5 to 100 vials (from 1 to20 sales units). For orders of over 100 vials (20 salesunits): prices on request
(Note: for countries outside France, our shipment is byairport consignment only)
— EU: 250 EUR per shipment
— Other European countries: 250 EUR per shipment
— Outside Europe: 250 EUR per shipment
e) Dangerous goods: sent by carrier chosen by EDQM: Swine erysipelas bacteria, serotype 1 BRP, Swine erysipelas bacteria, serotype 2 BRP, Pertussis toxin BRP, Diphtheria toxin BRP, Bleomycin sulphate CRS, Brucella melitensis BRP, Isosorbide 2-nitrate CRS
(Note: for countries outside France, our shipment is byairport consignment only)
— EU: 150 EUR per item

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 8/176
8 © PHARMEUROPA Vol. 18, No. 1, January 2006
General Information
— Outside EU: 160 EUR per shipment
NB: these extra charges include packaging, shippingand management of permits
k) Reference spectra
— France: no extra charge, price is inclusive of packagingand postage. At the client’s request, Express Courierdelivery is charged at 18 EUR per shipment
— EU: 18 EUR per shipment
— Other European countries: 50 EUR per shipment
— Outside Europe: 50 EUR per shipment
— Shipping costs paid by the customer: 10 EUR per
shipment.
How do I order?
The reference substances, reference preparations andreference spectra are supplied by the EDQM.
ORDER FORM
Please send your order using the CRS order form (seepage xv of the CRS catalogue) or by sending an official
purchase order on company letterhead to the EDQM.The order form may be downloaded from www.pheur.orgunder Reference Substances.
Fax: +33 (0)3 88 41 27 71 – for the attention of CRS Sales
E-mail: [email protected]
Letter: Council of Europe, European Directorate for
the Quality of Medicines, FAO CRS Sales Team,BP907, F-67029 Strasbourg Cedex, France
Customers are financially responsible for duplicate ordersin the following cases:
— confirmation orders that are not clearly marked asbeing a confirmation of an order that has alreadybeen sent to the Council of Europe (EDQM)
— submission of the same order multiple times (i.e., via
fax, e-mail, mail or any combination thereof)
Please note we do not accept orders by telephone.
If you are using any other documentation other than the
official CRS order form please ensure you have included:
— details of the Invoicing/Billing address includingname of company, post code, town, country and
telephone number
— details of the Delivery/Dispatch address (if different)including name of company, post code, town,
country (please note STREET ADDRESS ONLY, noP.O. Boxes)
— contact name, telephone number, fax number ande-mail address: an e-mail address is required for order
confirmation and shipping notification purposes
— VAT number (mandatory within the EuropeanUnion)
— your order reference/purchase order reference
— item order code
— official name of the CRS/BRP as set out in the CRScatalogue
— sales/unit quantity
— name and account number of the carrier (if you wish
to use your own)
If orders are received without the official name of theCRS/BRP and the full item order code (as set out inthe catalogue) the EDQM takes no responsibility for an
incorrect item being dispatched.
Unfortunately, we will not be able to process any orders
received without the above information.
SPECIAL DOCUMENTATION is required for controlled drugs and
chemical precursors of narcotics (Vienna Convention)(see our catalogue for further information).
Payment
Payment can be made by cheque made payable to theCouncil of Europe/EDQM and sent to the above address(see 3.1) or by bank transfer to our bank.
Société Générale, 255, route de Mittelhausbergen, 67200Strasbourg, France
IBAN account number for international transfers:(FR 76) 30003 02360 00550034256 76
National transfers: 30003 02360 00550034256 76
SWIFT: SOGEFRPP
You can also pay by credit card (Visa, Eurocard,
Mastercard or American Express) by writing down thecard number, the expiry date and the card holder’s name,
and including the card holder’s signature. Please note
that we do not accept credit card numbers by telephone.
In all cases, the payment should be net of charge for
the Council of Europe and invoices should be paid within 30 days from the date of invoice. Any other fees,
such as customs duties, taxes, or tariffs are also theresponsibility of the customer.
For certain countries, especially those having strict
monetary regulations, we reserve the right to requirepre-payment for new clients and large orders. In case of
doubt, please contact us at [email protected]. Payment byletter of credit is not accepted.

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 9/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 9
General Information
Payment
I would like to pay now. I will automatically receive an invoice/receipt
I enclose a cheque made payable to Council of Europe/EDQM
I wish to payby credit card
CRS Order Form
Your Order Reference (*) [reference] Date (*)
CONDITIONS OF SALE
We sell on our standard terms of business. For details please see our catalogue.
BILLING ADDRESSYour Client Code / :
Company Name (*):
Invoice Address (*)
City (*)
PostCode (*):
Country (*)
Contact Name (*)
VAT N°(* in Europe)
Tel (*): Fax:
E-mail:
DELIVERY ADDRESS(Please complete if different from invoicing address)
Company Name (*):
Delivery Address (*)
City (*)
PostCode (*):
Country (*)
Contact Name (*)
VAT N°(* in Europe)
Tel (*): Fax:
E-mail:
DELIVERY CHARGES and PRICES
The price should not be regarded as representing the selling price of a commercial product. The prices quoted in our catalogue are exclusive ofduties and taxes. Extra handling charges may be applied. Please see our catalogue for details.
Expiry Date Name Signature
Euro/Mastercard N° American Express N°Visa N°
All items marked with an asterisk (*) are mandatory
Total Goods/€
Reference(*) Item (*) Quantity (1) (*) TotalUnit Price
Council of EuropeEuropean Directorate for the Quality of Medicines
BP 907, 67029 Strasbourg Cedex 1 (France)
http://www.pheur.org
Tel: +33 (0)3 88 41 30 30 Fax: + 33 (0)3 88 41 27 71 E-mail:[email protected]
SIRET: 778860080010 APE Code APE:990Z
VAT N° : Not applicable to Council of Europe- diplomatic privilege.
I will pay on receipt of an invoice
(1) A CRS/BRP may include several individual vials (see sale unit in catalogue), in such instances do not order in terms of total numberof vials

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 10/176
10 © PHARMEUROPA Vol. 18, No. 1, January 2006
General Information
ORDER FORM
CATALOGUE OF
- CHEMICAL REFERENCE SUBSTANCES -
- BIOLOGICAL REFERENCE PREPARATIONS -
- INFRARED REFERENCE SPECTRA - MISCELLANEOUS REAGENTS-
The catalogue of reference substances of the European Directorate for the Quality of Medicines isa publication of the Council of Europe, issued three times a year to include the latest substancesadopted by the European Pharmacopoeia Commission.
This catalogue is free; if you would like to receive subsequent updated versions, please completeand return this form.
RECIPIENT:
Please check the appropriate box: Prof Dr Mr Ms
Surname...................................................................First name...........................................................
Company/Laboratory ..........................................................................................................................
Industrial Laboratory Private Control Laboratory Public Control Laboratory
Department: Invoicing/Purchasing Analytical Laboratory
Address.................................................................................................................................................
..............................................................................................................................................................
City.............................................................Postal Code .........................Country.................................
Tel.............................................................................Fax......................................................................
ADDITIONAL RECIPIENT IN YOUR COMPANY/LABORATORY:
Please check the appropriate box: Prof Dr Mr Ms
Surname...................................................................First name...........................................................
Department: Invoicing/Purchasing Analytical Laboratory
Other service (please describe)...................................................................................
EUROPEAN DIRECTORATE FOR THE QUALITY OF MEDICINES
226, avenue de Colmar - BP 907 - F 67029 Strasbourg Cedex 1, France
Fax +33(0)3 88 41 27 71

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 11/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 11
General Information
1 Microbiology6 Biological substances6B Human blood and blood products7 Antibiotics10A Organic chemistry - synthetic products10B Organic chemistry - synthetic products
10C Organic chemistry - synthetic Products10D Organic chemistry - synthetic Products
11 Organic chemistry - natural products12 Galenical products13A Phytochemistry13B Phytochemistry13H Fatty oils and derivatives14 Radioactive compounds
15 Sera and vaccines15V Veterinary sera and vaccines
LIST OF CODES OF GROUPS OF EXPERTS(November 2004)
BOT Botulinum toxinBSR Bovine serumCEL Cellulose derivativesCRB CarbohydratesCTP Cell therapy productsFRC Functionality-related characteristicsGEL GelatinGTP Gene therapy productsHFA Propellants
HOM HomeopathyICP Inductively coupled plasma spectrometryINC Inorganic chemistry
INH InhalationsLEC Lecithins for pharmaceutical purposesMAB Monoclonal antibodiesMMM Alternative microbiological methodsMYC MycoplasmasP4 Procedure 4POW Powder characterisation techniquesRGN ReagentsST Standard terms
STA Statistics VIT Vitamins WAT Water
Groups of experts
Working parties
________________________________________________________________________________
Consult KNOWLEDGE, the new free database at www.pheur.org (under Tools section) which
will tell you if a substance or a method of analysis is part of the work programme of the
European Pharmacopoeia, its state of work, if its draft text was published in PHARMEUROPA and if so inwhich issue, and in which volume of the European Pharmacopoeia the official obligatory text can be found.
You can also see if a text published in the European Pharmacopoeia is undergoing revision.
Also available:
• the trade name(s) of the reagent(s) such as those used in chromatography columns or biological kits thatwere used at the time of the elaboration of the monograph to carry our certain tests
• downloadable reference chromatograms• technical information explaining how to carry out the described tests
• the list of reference substances used in the monograph• the list of certificates granted.
KNOWLEDGE
KNOWLEDGETHE NEW FREE DATABASE AVAILABLE ATTHE NEW FREE DATABASE AVAILAB LE AT
www pheur orgwww.pheur.orgYOU HAVE A QUESTION ON A SUBSTANCE OR A METHOD OF ANALYSIS YOU HAVE A QUESTION ON A SUBSTANCE OR A METHOD OF ANALYS IS
I N T HE E UR OPE AN P HA RMAC OP OE IAIN THE EUROPEAN PHARMACOPOEIA?

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 12/176
12 © PHARMEUROPA Vol. 18, No. 1, January 2006
General Information
LIST OF TEXTS PUBLISHED IN SUPPLEMENT 5.5
A vertical line in the margin indicates where part of a text has been revised or corrected. A horizontal line in the margin indicates where part of a text has been deleted. It is to be emphasised that these indications, which are not necessarily exhaustive, are given for information and do not form an official part of the texts. Editorial changes are not indicated.
Individual copies of texts will not be supplied.
NEW TEXTS
REVISED TEXTS
GENERAL CHAPTERS
5.1.6. Alternative methods for control of microbiologicalquality
MONOGRAPHS
The monographs below appear for the first time in the European Pharmacopoeia. They will be implemented on1 July 2006 at the latest.
Vaccines for human use
Influenza vaccine (surface antigen, inactivated, preparedin cell cultures) (2149)Influenza vaccine (whole virion, inactivated, prepared incell cultures) (2308)
Radiopharmaceutical preparations
Sodium iodide (123I) solution for radiolabelling (2314)Technetium (99mTc) bicisate injection (2123)
Monographs
-Acetyldigoxin (2168) Artichoke leaf (1866)Chondroitin sulphate sodium (2064)Clobetasol propionate (2127)Danaparoid sodium (2090)Doxazosin mesilate (2125)Etidronate disodium (1778)
Febantel for veterinary use (2176)Fumitory (1869)Iotrolan (1754)Nandrolone decanoate (1992)Pine silvestris oil (1842)Silica, hydrophobic colloidal anhydrous (2208)Thioctic acid (1648)
Venlafaxine hydrochloride (2119)
GENERAL CHAPTERS
2.4.29. Composition of fatty acids in oils rich in omega-3-acids
2.6.15. Prekallikrein activator2.6.21. Nucleic acid amplification techniques2.6.22. Activated coagulation factors2.7.4. Assay of human coagulation factor VIII2.7.11. Assay of human coagulation factor IX2.7.21. Assay of human von Willebrand factor2.7.22. Assay of human coagulation factor XI4. Reagents ( new, revised, corrected )5.10. Control of impurities in substances for
pharmaceutical use
MONOGRAPHSThe monographs below have been technically revised
since their last publication. They will be implemented on1 July 2006 .
General monographs
Substances for pharmaceutical use (2034)
Dosage forms
Capsules (0016)Ear preparations (0652)Liquid preparations for cutaneous application (0927)Liquid preparations for oral use (0672)Rectal preparations (1145)
Semi-solid preparations for cutaneous application (0132)Tablets (0478)
Vaginal preparations (1164)
Vaccines for human use
Pneumococcal polysaccharide conjugate vaccine(adsorbed) (2150)
Monographs
Aluminium hydroxide, hydrated, for adsorption (1664)Beclometasone dipropionate, anhydrous (0654)Beclometasone dipropionate monohydrate (1709)Benzyl alcohol (0256)Centaury (1301)Colestyramine (1775)Digoxin (0079)Dipivefrine hydrochloride (1719)Evening primrose oil, refined (2104)
Formoterol fumarate dihydrate (1724)Gemfibrozil (1694)Glutathione (1670)Human coagulation factor XI (1644)Ketorolac trometamol (1755)Lauroyl macrogolglycerides (1231)Linoleoyl macrogolglycerides (1232)Liquorice root (0277)Macrogol 20 glycerol monostearate (2044)Maltitol, liquid (1236)Methadone hydrochloride (0408)Oleoyl macrogolglycerides (1249)Oxazepam (0778)Pancuronium bromide (0681)
Star anise (1153)Sucrose (0204)Sumatriptan succinate (1573)

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 13/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 13
General Information
CORRECTED TEXTS
SUPPRESSION OF TEXTS
The texts below have been corrected and are republished in their entirety. These corrections are to be taken into account from the publication date of Supplement 5.5.
GENERAL CHAPTERS
2.3.1. Identification reactions of ions and functional
groupsMONOGRAPHS
Dosage forms
Eye preparations (1163)
Monographs
Arachis oil, refined (0263)Carboprost trometamol (1712)Cefradine (0814)Chloroquine sulphate (0545)Desogestrel (1717)
Heparin calcium (0332)Heparin sodium (0333)Ipratropium bromide (0919)Levothyroxine sodium (0401)Norethisterone (0234)Oxytetracycline dihydrate (0199)Oxytetracycline hydrochloride (0198)Propranolol hydrochloride (0568)Sulbactam sodium (2209)Sulfaguanidine (1476)Tetracycline (0211)Tetracycline hydrochloride (0210)all- rac--Tocopherol (0692)all- rac--Tocopheryl acetate (0439)Zinc oxide (0252)
The following text is deleted on 1 April 2006 .
MONOGRAPHS
Monographs
Glucagon (0612)
CERTIFICATION OF SUITABILITY OF MONOGRAPHS OFTHE EUROPEAN PHARMACOPOEIA*
Istanbul, Turkey, 27-28 October 2005
Two days of exchanges and lively discussions confirmedthat the procedure for the certification of monographs ofthe European Pharmacopoeia is a major tool of growingimportance for guaranteeing the quality of substancesfor pharmaceutical use in the context of constantlydeveloping world trade. The procedure also plays animportant role in the implementation of the revisedEuropean Directives (2001/83/EEC as amended by2004/27/EEC and 2001/82/EEC as amended by2004/28/EEC).
Several ideas for future development have becomeapparently necessary in terms of:
— communication between the different partners and
the transparency of the procedure;
— optimisation of the timeline for the assessment ofdossiers;
— reinforcement of the importance of dialogue betweenthe authorities to guarantee the recognition of thecertificates of suitability and of inspections in thelight of the new legislation on inspections of activeprimary ingredients.
The objectives of this conference were particularly
important since they involved reviewing the resultsobtained since the last conference (4 years ago), andsharing points of view and experiences with the principalusers. The proposed programme facilitated dialogue
with users, indeed 12 workshop sessions (coveringthe procedure for renewals and revisions, deficienciesin dossiers, sterile products and inspections) and56 individual consultations (or One-to-One sessions)
were organised, giving each attendee the opportunityto express and exchange views with European authorityrepresentatives and the assessors who evaluate thecertification dossiers.
Over 180 representatives involved in the quality ofmedicines, from 32 countries including Canada, India,China, South Korea, Israel and the United States,participated in this international conference organisedby the European Directorate for the Quality of Medicines(EDQM) of the Council of Europe. The success of theconference is evidence of the dynamism of internationalactivities in the domain of the quality of medicines, andthe importance of co-operation between the differentpartners implicated (EDQM, European Commission,EMEA, national licensing authorities, inspection andindustries).
* The procedure for the certification of suitability of monographs of the European Pharmacopoeia The European directives 2001/83/EEC and 2001/83 EEC amended, on the criteria for the quality, safety and efficacy of medicines on the market,
refer to the specifications of the European Pharmacopoeia to define quality criteria for medicines for human and veterinary use respectively.
Within this legal framework, a supplier of raw materials must provide clients in the pharmaceutical industry with proof that the purity of itsproduct is suitably controlled by the monographs of the European Pharmacopoeia; this is the role of the certificate of suitability. Since thebeginning of the procedure, more than 1860 certificates, including 502 concerning the evaluation of the reduction of the TSE risk, have beengranted by the EDQM following evaluation of dossiers by assessors designated by the various national licensing authorities.
_______________________________________________________________________________________

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 14/176
14 © PHARMEUROPA Vol. 18, No. 1, January 2006
General Information
COMMENTS CONCERNING SOME REVISED/CORRECTED TEXTS PUBLISHED IN SUPPLEMENT 5.5
Here follows information concerning certain technical modifications to some revised/corrected texts adoptedby the European Pharmacopoeia Commission at the June 2005 session. This information completes themodifications indicated by lines in the margin in the supplement. Therefore, the information below is not
necessarily exhaustive.
ANALYTICAL METHODS
2.4.29. Composition of fatty acids in oils rich in omega-3-acids
In the test for system suitability, as in the case of thetest for oligomers in the omega-3-acid ethyl esters 90monograph, the first 3 requirements are consideredsufficient; the 4th requirement is not routinely performedby the producers who proposed the test, and is deleted.
2.6.15. Prekallikrein activator
This general chapter has been revised based on theoutcome of the international collaborative study BSP049organised by the EDQM, to mention that a microtitreplate-based method, which is nowadays the mostfrequently used, may also be used instead of methodsusing autoanalysers, which were more appropriate whena large number of samples had to be analysed.
2.6.21. Nucleic acid amplification techniques
This general chapter has been revised to makeit applicable to new applications such as the test
for mycoplasmas (see revised chapter 2.6.7) anda quantitative test system used to control anti-Dplasma for B19 virus (see monograph Human anti-Dimmunoglobulin (0557)).
2.6.22. Activated coagulation factors
This general chapter has been revised together withgeneral chapters 2.7.11 and 2.7.22 to:
— change protamine sulphate R to being an example ofa suitable substance to neutralise the heparin;
— replace the reference to cephalin R and platelet substitute R, which were obsolete, by a phospholipidpreparation to act as a platelet substitute.
2.7.4. Assay of human coagulation factor VIII
The assay of human coagulation factor VIII using achromogenic substrate was first included in the EuropeanPharmacopoeia in 1993, replacing the two-stage assay,in line with the recommendation of the Scientificand Standardisation Committee of the InternationalSociety on Thrombosis and Haemostasis (SSC ISTH).Commercial kits are used for the assay and thedescription of the method is generic to allow the use ofall currently available kits with acceptable performance.
The revision brings no essential major changes to themethod. Work has been carried out recently to definecritical aspects of the method, particularly with respect
to B domain-deleted factor VIII. Problems encountered with the latter product are best resolved by the useof a B domain-deleted factor VIII reference standardfor routine assay. The experimental work carried outrecently indicates that it is best for these products to haltfactor Xa generation when the factor Xa concentrationhas reached approximately 50 per cent of the maximum(plateau) level. Furthermore, since it has been shown
that, from a statistical point of view, the potency found with independent or serial dilutions is not significantlydifferent, independent dilutions are no longer required.
2.7.11. Assay of human coagulation factor IX
This general chapter has been revised to:
— mention factor IX-deficient plasma as a predilutionmedium, since this is used routinely;
— allow the use of commercial APTT reagents and omitthe reference to cephalin-based reagents, which are nowobsolete.
2.7.21. Assay of human von Willebrand factor
This general chapter has been revised to improve andsupplement the description of ristocetin cofactor activity,and in particular to introduce general statements onquantitative assays. Furthermore, since it has been shownthat, from a statistical point of view, the potency found
with independent or serial dilutions is not significantlydifferent, independent dilutions are no longer required.
2.7.22. Assay of human coagulation factor XI
The general chapter has been revised to:
— mention factor XI-deficient plasma as a predilutionmedium since this is used routinely;
— allow the use of commercial APTT reagents and omitthe reference to cephalin-based reagents, which are nowobsolete.
5.10. Control of impurities in substances forpharmaceutical use
The section on Interpretation of the test for relatedsubstances in the monographs on active substances hasbeen modified to replace the examples by a decision
tree, which better illustrates the interpretation ofmonographs.

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 15/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 15
General Information
VACCINES FOR HUMAN USE
DOSAGE FORMS
GENERAL MONOGRAPHS
Substances for pharmaceutical use (2034)
In the section dealing with related substances, thepossibility of exemptions to the general provisions hasbeen introduced, since it is now seen to be appropriate tomake exceptions in some specific monographs.
The section on residual solvents has been modified tostate explicitly that the content of residual solvents istaken into account for calculation of specific opticalrotation and specific absorbance.
Capsules (0016)
In order to take account of the new harmonised chapteron Disintegration, adaptations have been introduced tothe relevant sections.
Ear preparations (0652)
The test for Deliverable mass or volume has been the
cause of some misunderstanding amongst users: it wasnot a quality control test, and aimed only to ensurethat the filling was such that the labelled dose could be
withdrawn; furthermore, it has been considered to be vague and impractical. It has therefore been replaced byan additional sentence under Production.
Liquid preparations for cutaneous application (0927)Liquid preparations for oral use (0672)
The test for Deliverable mass or volume has been thecause of some misunderstanding amongst users: it wasnot a quality control test, and aimed only to ensurethat the filling was such that the labelled dose could be
withdrawn; furthermore, it has been considered to be vague and impractical. It has therefore been replaced byan additional sentence under Production.
Rectal preparations (1145)
The test for deliverable mass or volume has been thecause of some misunderstanding amongst users: it wasnot a quality control test, and aimed only to ensurethat the filling was such that the labelled dose could be
withdrawn; furthermore, it has been considered to be vague and impractical. It has therefore been replaced byan additional sentence under Production.
Also, in order to take account of the new harmonisedchapters on Dissolution and Disintegration, adaptationshave been introduced to the relevant sections.
Semi-solid preparations for cutaneous application(0132)
The test for deliverable mass or volume has been thecause of some misunderstanding amongst users: it wasnot a quality control test, and aimed only to ensurethat the filling was such that the labelled dose could be
withdrawn; furthermore, it has been considered to be vague and impractical. It has therefore been replaced by
an additional sentence under Production.
Tablets (0478)
This monograph was published in Pharmeuropa 15.2 and 16.2 for enquiry related to uniformity of subdividedtablets and to oral lyophilisates. Due to the commentsreceived, it was necessary to carry out a new enquiryas regards oral lyophilisates ( Pharmeuropa 17.4). Asregards subdivision of tablets, uniformity of mass isnow tested on 30 parts; no 2nd test is required in caseof failure. This revision is the result of the enquiryand the study performed by the OMCLs on the basis of
Pharmeuropa 16.2. This text also takes into account
the new harmonised chapters on Disintegration andDissolution.
Vaginal preparations (1164)
The test for deliverable mass or volume has been thecause of some misunderstanding amongst users: it wasnot a quality control test, and aimed only to ensurethat the filling was such that the labelled dose could be
withdrawn; furthermore, it has been considered to be vague and impractical. It has therefore been replaced byan additional sentence under Production.
Also, in order to take account of the new harmonised
chapters on Dissolution and Disintegration, adaptationshave been introduced to the relevant sections.
Pneumococcal polysaccharide conjugate vaccine(adsorbed) (2150)
The monograph has been revised to clarify that the testfor sterility that is carried out on intermediates (thepneumococcal polysaccharides, the carrier protein andthe monovalent bulk conjugate) uses 10 ml for eachmedium or the equivalent of 100 doses, whichever is
less. Furthermore, it harmonises this monograph withthe monograph on Meningococcal group C conjugate
vaccine (2112).

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 16/176
16 © PHARMEUROPA Vol. 18, No. 1, January 2006
General Information
MONOGRAPHS
Aluminium hydroxide, hydrated, for adsorption (1664)
Based on batch data, the limit for iron has been increasedfrom 10 ppm to 15 ppm.
Beclometasone dipropionate, anhydrous (0654)Beclometasone dipropionate monohydrate (1709)
Following the establishment of beclometasonedipropionate for system suitability CRS andbeclometasone dipropionate for peak identification CRS,the identification of impurities D and M has beenmodified. In addition, relative retentions of the otherdetectable impurities have been deleted according tousual practice.
Benzyl alcohol (0256)
In the test for residue on evaporation, the temperature
of 100 °C indicated for evaporation on a water-bath wastoo low in view of the boiling point of benzyl alcohol(205 °C), therefore the evaporation method has beenchanged.
In the assay, it is now specified that the mixture is heatedon a water-bath.
Digoxin (0079)
This monograph has been revised to replace the TLCtest for related substances by LC and to replace the assay
with an LC test using the same system. For this productof natural origin having a complex impurity profile, it
has not proved possible to apply the general policy forimpurities shown in the monograph on Substances for
Pharmaceutical Use (2034). It has been necessary to givean acceptance criterion equivalent to 0.2 per cent for “anyother impurities”. An acceptance criterion for the sum ofimpurities (specified and unspecified) has been definedand in order to provide a further degree of control, anacceptance criterion for the subtotal of unspecifiedimpurities has been added.
Dipivefrine hydrochloride (1719)
The following changes have been made:
— an increase in the amount of concentrated ammoniasolution used in the mobile phase to improve thesymmetry of the peaks;
— the introduction of a higher upper limit for thesymmetry factor in the assay, i.e. 2.0 for the principalpeak in the chromatogram obtained with referencesolution (c).
Evening primrose oil, refined (2104)
The unsaponifiable matter is largely composed of natural vitamins. It is therefore desirable that the oil is smoothlyrefined so that the unsaponifiable matter remains rather
high. Based on batch data, the limit for unsaponifiablematter has been increased from 2.0 per cent to 2.5 percent.
Gemfibrozil (1694)
Following a study in the EDQM laboratory, method C hasbeen replaced by method F in the test for heavy metals.Furthermore, impurity D has been corrected.
Glutathione (1670)
Following the establishment of the CRS for glutathione,several batches have been tested and it appeared that thelimits for impurity D and for the total of impurities weretoo strict. These 2 limits have therefore been increased.
Human coagulation factor XI (1644)
This monograph has been revised to harmonise thetotal protein test with other monographs. Reference togeneral chapter 2.5.33 is not appropriate since 7 differentmethods are described and it has not been shown that
these 7 methods are equivalent. Since the determinationof nitrogen by sulphuric acid digestion (Kjeldahl method)is the only method that can be performed without usinga reference preparation, this is the method of choice.Indeed, this is a big advantage since results obtainedcan be compared directly. Other methods can also beused provided that they have been validated against thedetermination of nitrogen by the sulphuric acid digestionmethod described in the monograph.
Ketorolac trometamol (1755)
Following a study in the EDQM laboratory, method C hasbeen replaced by method F in the test for heavy metals.
Lauroyl macrogolglycerides (1231)Linoleoyl macrogolglycerides (1232)
By analogy with the revision made to the monograph Stearoyl macrogolglycerides (1268) in 2004, theconditions used for the test for free glycerol have beenmodified.
Macrogol 20 glycerol monostearate (2044)
In the test for hydroxyl value the method has beenchanged: method A yields better results, within the limitsof 65 to 85.
Methadone hydrochloride (0408)
The silver nitrate titration in the assay has been replacedby the acid-base titration, which is more specific as itdetermines the active moiety; in addition, the titrant islow-cost, odourless, and easy to handle. This titration hasbeen shown to be equivalent to the silver nitrate titrationand to the previous perchloric acid titration.
Oleoyl macrogolglycerides (1249)
By analogy with the revision made to the monographstearoyl macrogolglycerides (1268) in 2004, the
conditions used for the test for free glycerol have beenmodified.

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 17/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 17
General Information
Pancuronium bromide (0681)
2 new impurities have been added to the transparencylist. Pancuronium bromide and its impurities cannotbe analysed by LC because of insufficient sensitivity(little or no chromophores). For this reason, the currentTLC method has been optimised to allow detection of
impurities at 0.1 per cent.
Sucrose (0204)
As the amount of lead in the test solution is in practiceextremely low, the system suitability criterion in the testfor lead has been changed.
Sumatriptan succinate (1573)
As it is difficult to obtain samples of the individualimpurities to prepare replacement CRS batches, a methodhas been developed based on the injection of mixtures ofsumatriptan with impurities obtained by evaporation, toidentify the specified impurities in the test for impurity A
and impurity H and in the test for related substances.
NEW : PHARMEUROPA SCIENTIFIC NOTES
For prices and ordering information please consult the catalogue on our internet site http://book.pheur.org
• Few Bicyclic Acetals at Reducing End of Low-Molecular-Weight Heparins: Might they RestrictSpecification of Pharmacopoeia?
• The Control of Impurities in ChlortalidoneUsing a Reversed-Phase Stationary Phase
• Factor VIII Test in Reference Preparations:Compensation for Different Dilutions
• The Control of Impurities in AmitriptylineHydrochloride Using a Reversed-Phase
Hybrid Stationary Phase
• A Precise Colour Determination Method for Tablets -an Application of Instrumental Colour Measurementin the Pharmaceutical Development
• Development of an in vivo Test Procedure for the Easeof Breaking of Scored Tablets
• Chromogenic Assay of Human Coagulation Factor VIII:Statistical Comparison of 2 Working DilutionProcedures
• Impurity Profile of Amino Acids?
• Batch Variability of Bacitracin: HPLC versus MEKC
• Quality Criteria of Homoeopathic Mother Tinctures:Considerations Regarding Suitable Tests forHomoeopathic Monographs
• Instructions for Authors
Available now (English only)
As from Pharmeuropa 17.4, Scientific Notes are principally published in a new publication called Pharmeuropa Scientific Notes.
Articles published in Pharmeuropa Scientific Notes are indexed in the PubMed database of the National Library of Medecine, available on the internet site (www.ncbi.nlm.nih.gov).
The first edition of Pharmeuropa Scientific Notes (Pharmeuropa SN 2005-1) became available in August 2005.
This issue is included in the subscription to Pharmeuropa, and is not available separately.
SCIENTIFIC NOTES 2005-1
________________________________________________________________________________
8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 18/176
18 © PHARMEUROPA Vol. 18, No. 1, January 2006
General Information
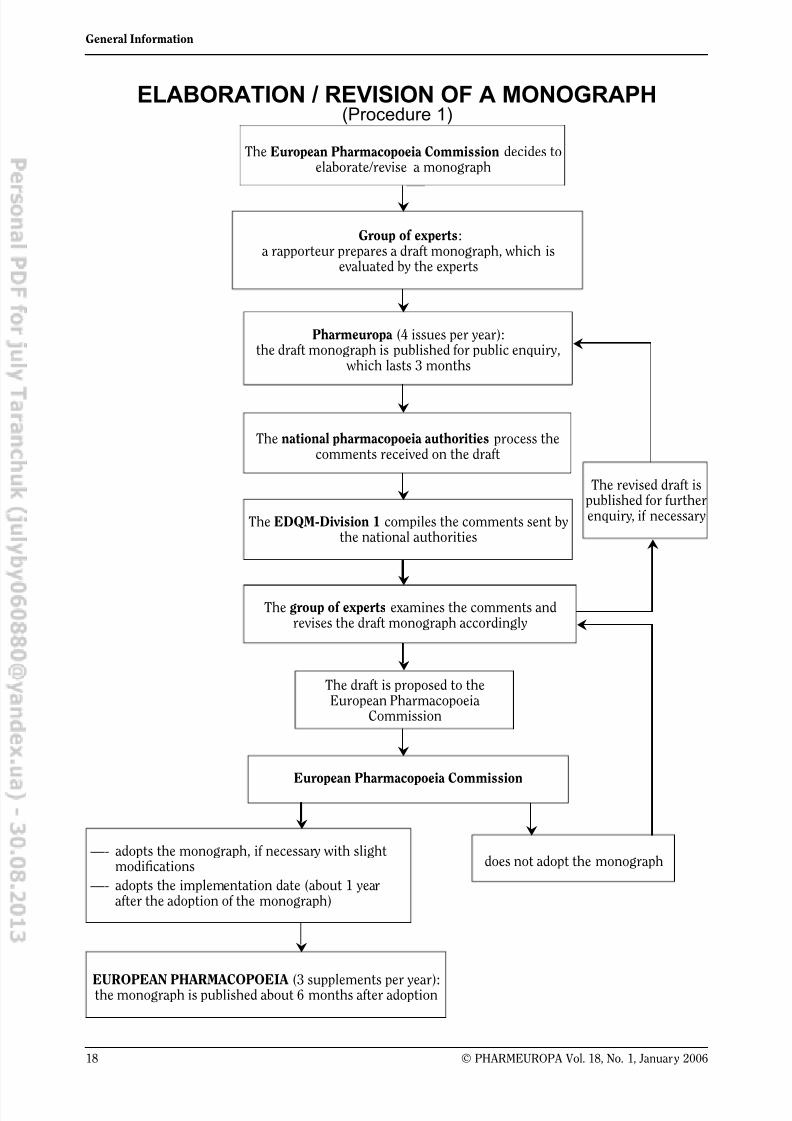
ELABORATION / REVISION OF A MONOGRAPH(Procedure 1)
The European Pharmacopoeia Commission decides toelaborate/revise a monograph
Group of experts:a rapporteur prepares a draft monograph, which is
evaluated by the experts
Pharmeuropa (4 issues per year):the draft monograph is published for public enquiry,
which lasts 3 months
The national pharmacopoeia authorities process thecomments received on the draft
The EDQM-Division 1 compiles the comments sent bythe national authorities
The group of experts examines the comments andrevises the draft monograph accordingly
European Pharmacopoeia Commission
does not adopt the monograph
The revised draft ispublished for furtherenquiry, if necessary
—- adopts the monograph, if necessary with slightmodifications
—- adopts the implementation date (about 1 yearafter the adoption of the monograph)
EUROPEAN PHARMACOPOEIA (3 supplements per year):the monograph is published about 6 months after adoption
The draft is proposed to theEuropean Pharmacopoeia
Commission

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 19/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 19
General Information
________________________________________________________________________________
NEW : TECHNICAL GUIDE FOR THEELABORATION OF MONOGRAPHS
4th Edition (2005) available soon on the EDQM internet site
The Technical Guide for the Elaboration of Monographs describes the scientific approach used for the elaboration ofmonographs and the establishment of specifications of the European Pharmacopoeia. The guide also describes howto scientifically elaborate the various sections that must be included in each monograph, for example, definition,characters, the physical and chemical reactions constituting the identification section, purity tests, assay methods
and storage conditions. It is continually being updated.
Available soon as a free download on the EDQM internet site http://www.pheur.org (bilingual version)
Contents
Preface
Introduction by Claude Huriet (France)History and definitions by Povl Riis (Denmark)
ETHICAL DILEMMAS IN RESEARCH
Uses and abuses of biomedical researchby Jan Helge Solbakk (Norway)
Selection and recruitment of participants: European standardsby Herman Nys (Belgium)
Placebo: its action and place in health researchby Andrzej Górski (Poland)
Cancer clinical trials by Maxime Seligmann (France)
Some ethical considerations in industry-sponsored clinicaltrials by Tom Gallacher and N. Sreeharan (United Kingdom)
Women in biomedical research by Outi Leena L. Hovatta(Sweden)
ETHICAL EYE - BIOMEDICAL RESEARCH (2004)
What are the rules and underlying values governing biomedical research in Europe? What form do these values takeand where do they originate? Does biomedical research pose a threat to individuals and their rights? What balanceshould be struck between freedom of research and protection of the individual? All these questions are examined inthis book from a pan-European perspective.
The authors look at various international and European standards, including the Helsinki Declaration of the WorldMedical Association, EU Directive 2001/20 on pharmaceutical research and the Council of Europe’s Convention onHuman Rights and Biomedicine. The last named was signed in Oviedo in 1997 and is the first binding internationaltreaty on the subject, with a special chapter on scientific research on human beings. The Convention establishes acommon minimum level of protection of fundamental rights throughout Europe. It will soon be supplemented by anadditional protocol specifically concerned with biomedical research.
The book contains a glossary and a list of relevant international conventions and treaties, web sites and publications.It is aimed at both specialists and a wider public interested in this subject.
ISBN 92-871-5462-7. Price: 15 EUR (Europe) / US$ 23 (outside Europe), + 10 % postage.
This book is available in French and English from:
Council of Europe Publishing - Sales Unit
Ms Sophie Lobey, F- 67075 Strasbourg Cedex, France.
Tel: +33 (0)3 88 41 25 81 - Fax: +33 (0)3 88 41 39 10
Email: [email protected] - Web site http://book.coe.int
BIOMEDICAL RESEARCH IN EUROPE
Germany: current legislation by Jochen Taupitz (Germany)
Central and eastern Europe: research-related problems fortransition countries by Eugenijus Gefenas (Lithuania)
Italy: some shortcomings of biomedical research by StéphaneBauzon (Italy)
United Kingdom: data protection and confidentiality byMichel Coleman and Vivienne Harpwood (United Kingdom)
EUROPE AND BIOMEDICAL RESEARCH
European law and biomedical research by Peteris Zilgalvis(Council of Europe)
APPENDICES
Selected websites
Draft protocol on biomedical research, Council of Europe
The Helsinki Declaration, World Medical Association

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 20/176
20 © PHARMEUROPA Vol. 18, No. 1, January 2006
General Information
GUIDE TO SAFETY AND QUALITY ASSURANCEFOR ORGANS, TISSUES AND CELLS
2nd Edition (2004)
The purpose of this guide is to provide guidance for all those involved in transplantation to maximise the quality,
and thereby the success rate, of transplants, and to minimise the risks to all involved in this complex procedure. It
includes safety and quality standards for the procurement, preservation, processing and distribution of organs, tissues
and cells of human origin (allogeneic and autologous) used for transplantation purposes. This guide will be regularly
updated, in line with the latest technical advances.
As the European Union Directive on Tissues and Cells (2004/23/EC) was recently adopted, the European Commission
will build on the Council of Europe’s guide when establishing technical standards under the directive. This
co-operation will ensure that the same standards are applied throughout Europe.
ISBN 92-871-5518-6. Price: 13 EUR (Europe) / US$ 20 (outside Europe), + 10 % postage.
The 2nd Edition of the Guide can be obtained in English and French from:
Council of Europe Publishing - Sales UnitMs Sophie Lobey, F-67075 Strasbourg Cedex, France.Tel: +33 (0)3 88 41 25 81 - Fax : +33 (0)3 88 41 39 10E-mail: [email protected] - Web site: http://book.coe.int
Questions and comments on the content should be sent directly to the division in charge:
Council of Europe, Health DivisionMr Karl-Friedrich Bopp, F-67075 Strasbourg Cedex, France.Tel: +33 (0)3 88 41 22 14 - Fax: +33 (0)3 88 41 27 26E-mail: [email protected] of Europe Portal: www.coe.intHealth and Ethics: www.coe.int/T/E/Social_Cohesion/Health
________________________________________________________________________________
VISIT OUR INTERNET SITE http://www.pheur.organd get the news as it happens at the
European Directorate for the Quality of Medicines (EDQM)and the
European Pharmacopoeia
(site is in English only)
access the latest news on official publications
consult the list of adopted monographs and CRS after each session of the Commission
download more than 1500 safety datasheets and about 170 leaflets
find out about the new developments in the procedure for certification of suitability of monographs of theEuropean Pharmacopoeia
access the work programme database of the European Pharmacopoeia (KNOWLEDGE DATABASE), thecertification and the reference substance databases
find official surveys in progress, announcements of conferences and international seminars, etc.
keep up-to-date on the latest technical, scientific and regulatory developments
find out about career opportunities at the EDQM
access the EDQM’s on-line publications ordering service at: http://book.pheur.org
NEW : The EDQM has launched a new system for sending questions to the Public Relations Unit, the ElectronicPublications Helpdesk and Monographs. Any questions should be submitted through the HELPDESK, which is
accessible from the EDQM internet site at http://www.pheur.org/site/page_521.php
Please send us your comments and suggestions to help us develop this site!

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 21/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 21
General Information
PHARMEUROPA BIO
These issues are included in the subscription to Pharmeuropa.
BIOLOGICALS 2003-1
• Collaborative Study for the Establishment of The
European Pharmacopoeia BRP Batch 1 for
Diphtheria Toxin
• Collaborative Study for the Establishment of
European Pharmacopoeia BRP Batch 2 for Inactivated
Poliomyelitis Vaccine for In Vitro D Antigen Assay
• Collaborative Study for the Establishment of A Global
(WHO International / US/Ph. Eur.) Standard for the
Potency Assay of Human Anti-D Immunoglobulin
• Feasibility Study to Evaluate the Correlation Between
Results of a Candidate In Vitro Assay and Established
In Vivo Assays for Potency Determination of Newcastle
Disease Vaccines
Available now (English only)
BIOLOGICALS 2002-1
• Collaborative Study for Establishment of an HPLC-
Method for Batch Consistency Control of Recombinant
Interferon-Alfa-2
• Calibration of European Pharmacopoeia BRP Batch 3/
Mega 2 (US/FDA) Standard for Human Coagulation
Factor VIII Concentrate for Use in the Potency Assay
• Collaborative Study for the Establishment of the
European Pharmacopoeia BRP for Oral Poliomyelitis
Vaccine (OPV) Batch 3 for Use in the Potency Assay
• Establishment of the European Pharmacopoeia BRP for
Hepatitis A Vaccine Type B (Aventis Pasteur) Batch 2
Available now (English only)
For prices and ordering information please consult the catalogue on our internet site http://book.pheur.org
BIOLOGICALS 2003-2
• Collaborative Studies for the Establishment of
Reference Substances for the Microbiological Assay of
Antibiotics
• Collaborative Study for Establishment of a Global
Standard for the Potency Assay of Human Anti-D
Immunoglobulin
• Collaborative Study for Establishment of a European
Pharmacopoeia Biological Reference Preparation (BRP)
For B19 Virus DNA Testing of Plasma Pools by Nucleic
Acid Amplification Technique
BIOLOGICALS 2004-1
• Validation Study to Evaluate the Reproducibility of a
Candidate In Vitro Potency Assay of Newcastle Disease
Vaccines and to Establish the Suitability of a Candidate
Biological Reference Preparation
• Establishment of Batch 4 of the Biological Reference
Preparation (BRP) for Rabies Vaccine (Inactivated) for
Veterinary Use
• Collaborative Study for the Establishment of
Erythropoietin BRP Batch 2
• Somatropin and its Variants: Structural
Characterization and Methods of Analysis
• Capillary Electrophoresis for the Control of Impurities
of rDNA Somatropin
• Collaborative Study to Establish the Low-Molecular-
Mass Heparin for Assay European Pharmacopoeia
Biological Reference Preparation (BSP060)
Available now (English only)
BIOLOGICALS 2005-1
• Collaborative Study to Establish a New Biological
Reference Preparation for Prekallikrein Activator
• Collaborative Study for the Establishment of the Ph.
Eur. BRP Batch 1 for Anti-Vaccinia Immunoglobulin
• Feasibility Study to Develop a Common in vitro
D-Antigen Assay for Inactivated Poliomyelitis Vaccines
• Allergy Vaccines: a Need for Standardisation in Mass
Units of Major Allergen
• Efficacy Demonstration of Tetanus Vaccines by double
antigen ELISA
• International Symposium on Alternatives to Whole Cell
Pertussis Vaccine Potency Assay Available now (English only)
• Collaborative Study for the Validation of Serological
Methods for Potency Testing of Diphtheria Toxoid
Vaccines: Part 1
• Collaborative Study for the Validation of Serological
Methods for Potency Testing of Diphtheria Toxoid
Vaccines: Extended study: Correlation of Serology with
In Vivo Toxin Neutralisation
• Establishment of European Pharmacopoeia Biological
Reference Preparations Batch 2 for rDNA Hepatitis B
vaccine (Method A and B)
• Control of Clostridium Perfringens Vaccines by Means
of an Indirect Competitive ELISA for the Epsilon
Toxin Component – Examination of the Assay by a
Collaborative Study
Available now (English only)

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 22/176
22 © PHARMEUROPA Vol. 18, No. 1, January 2006
General Information
EDQM CONFERENCE PROCEEDINGS
Certification of Suitability of Monographs of theEuropean Pharmacopoeia (CEP) - New Developmentsof the Procedure, How to Apply for a CEP8-9 November 2001, Athens, Greece
The Future Face of the European Pharmacopoeia -Current Concerns in Pharmaceutical Analysis8-9 February 2001, Cannes, France
Herbal Medicinal Products: Quality Evaluation -Contribution of the European Pharmacopoeia16-17 November 2000, Nice, France
Tetanus Vaccine for Human Use22-23 June 2000, Strasbourg, France
Mycoplasma Testing: The Potentialities and Roleof PCR Tests13-14 March 2000, Paris, France
The following free Conference Proceedings are available to download from the EDQM internet site
http://www.pheur.org/site/page_601.php
Biologicals beyond 2000: Challenge for QualityStandards in an Evolving Field27-29 September 1999, Strasbourg, France
General Monographs on Dosage Forms and Pharmaco-Technological Test Methods26-27 October 1998, Seville, Spain
The Vision of the European Pharmacopoeia in the21st Century - The Dynamics of Quality of Medicines inEurope4-7 December 1996, Prague, Czech Republic
Sterility Tests and Efficacy of AntimicrobialPreservation5-6 February 1996, Barcelona, Spain
Certificates of Suitability of Monographs of the
European Pharmacopoeia: Implementation of the 5th
Edition – New Procedures for Revision and Renewal of
Certificates
27-28 October 2005, Istanbul, Turkey
OMCL Information Day: Place and Role of the European
OMCL Network within the Regulatory Framework in
Europe
27 May 2005, Rome, Italy
Alternatives to Whole Cell Pertussis Vaccine Potency
Assay
16 March 2005, Geneva, Switzerland
Quality of Homoeopathic Products in the New European
Legislative Framework
15 February 2005, Strasbourg, France
Serological Potency Tests for Diphtheria and Other
Vaccines
6-7 October 2004, Budapest, Hungary
Quality on the Move: Dynamics of the EuropeanPharmacopoeia
4-6 October 2004, Budapest, Hungary
Process Analytical Technologies International Symposium
3-4 May 2004, Cannes, France
Microbiological Control Methods in the EuropeanPharmacopoeia: Present and Future
5-6 May 2003, Copenhagen, Denmark
Foot and Mouth Disease Vaccines: Current Situation
17-18 March 2003, Strasbourg, France
Standardisation and Quality Control Cell and GeneTherapy Products
24-25 February 2003, Strasbourg, France
Replacement, Reduction and Refinement of the Use of Animals in the Quality Control of Vaccines
7-8 November 2002, Strasbourg, France
Excipients: Classical Requirements and FunctionalityRelated Testing
4-5 April 2002, Brussels, Belgium
******
The following Conference Proceedings can be ordered from the EDQM. For prices and ordering please consult thecatalogue on our internet site http://book.pheur.org
All above proceedings are available in English only and are not included in the subscription to Pharmeuropa.

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 23/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 23
General Information
LIST OF STANDARD TERMS5th EDITION (printed version available)
(27 European languages)
The present list of Standard Terms is a revised list that was drawn up in response to a request from the EuropeanCommission. It covers medicines for both human and veterinary use. These Standard Terms are to be used in
answering the questions in Module 1 (item 1.2 and 1.3) of the EU application form.
The list of Standard Terms is composed of:
— an introduction:• a section of general principles and instructions for the use of Standard Terms,• the summary of the changes (amendments, additions, deletions) performed since the last publication
(December 2002),• the procedure for the addition, deletion or modification of terms in the list of Standard Terms (requests
restricted to licensing authorities);
— 3 lists of standard terms:• list of pharmaceutical forms,• list of routes and/or methods of administration,• list of containers, closures and administration devices.
The 5th
Edition contains translations in 27 European languages: Bulgarian, Croatian, Czech, Danish, Dutch, English,Finnish, French, German, Greek, Hungarian, Icelandic, Italian, Macedonian, Norwegian, Polish, Portuguese, Slovak,Slovenian, Spanish, Swedish and Turkish. 5 new languages have been added compared to the printed versionpublished in December 2002: Lithuanian, Estonian, Latvian, Romanian and Maltese.
The corresponding online version is available only to people who ordered the printed version of the 5th Edition(December 2004).
Price: see the catalogue on our internet site http://book.pheur.org
GUIDE TO THE PREPARATION, USE AND QUALITYASSURANCE OF BLOOD COMPONENTS
11th Edition (2005) of the technical appendix to Recommendation No. R (95) 15This guide contains a compendium of measures designed to ensure the safety, efficacy and quality of bloodcomponents, and is particularly intended for all those working in blood transfusion services. It describes the differentblood components and gives information on their clinical indications and possible side effects.
This guide continues to be the ‘golden standard’ for blood transfusion services and forms the basis for many nationalguidelines in Europe and elsewhere.
During the preparation of this 11th edition, the Council of Europe and the European Commission worked closelytogether to ensure that the requirements under Article 29 of the European Union Directive 2002/98/EC and thoseof this guide were compatible. In particular, the section in Part A on the quality system for blood transfusionestablishments has been completely overhauled.
Where necessary, chapters have also been revised to take into account what can now be achieved with new advancesin technology.
This reference book will be of interest to blood transfusion centres, legislators, health personnel and all those
working in the field of blood transfusion.The European Pharmacopoeia monograph on human plasma for fractionation refers inter alia to therecommendations made in this Guide.
ISBN 92-871-5667-0. Price: 19 EUR (Europe) / US$ 29 (outside Europe), + 10 % postage.
The 11th Edition of the Guide can be obtained in English and French from:Council of Europe Publishing - Sales UnitMs Sophie Lobey, F-67075 Strasbourg Cedex, France.Tel: +33 (0)3 88 41 25 81 - Fax: +33 (0)3 88 41 39 10E-mail: [email protected] - Web site: http://book.coe.int
Questions and comments on the content should be sent directly to the division in charge:Council of Europe, Health DivisionMr Karl-Friedrich Bopp, F-67075 Strasbourg Cedex, France.Tel: +33 (0)3 88 41 22 14 - Fax: +33 (0)3 88 41 27 26
E-mail: [email protected] of Europe Portal: www.coe.intHealth and Ethics: www.coe.int/T/E/Social_Cohesion/Health
________________________________________________________________________________

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 24/176
24 © PHARMEUROPA Vol. 18, No. 1, January 2006
General Information
Please complete and return this form to: Josiane Bourrély, Division IV, EDQM before 10 February 2006
by post: EDQM, BP 907, 67029 Strasbourg Cedex 1, Franceby fax: +33 (0)3 88 41 27 71; by E-mail: [email protected]
REGISTRATION DETAILS
PARTICIPANT DETAILS*(Delivery address) INVOICING DETAILS (if different from participant/delivery details)
First Name First Name
Last Name Last Name
Company/Institution
Company/Institution
Name of Unit/Section (to be mentioned in the attestation of the participant)
Address(No POBoxes)
Address
Postcode Postcode
Town Town
Country Country
VAT No(EU only)
VAT No(EU only)
Tel Tel
Fax Fax
E-mail E-mail
Purchase Order Reference (to be mentioned on the invoice)
* Please note that all related information, documentation or material (e-mails, protocols, samples, reports, attestations of participation) will be
sent to the above-mentioned registered participant at the above-mentioned address.
PTS ref Name of Study Date of Availability
Participation DispatchConditions
Delivery Charges
1
PTS081 Loss on drying (2.2.32) End Feb 06 ❏ YES ❏ NO AmbientTemperature
(a)
PTS082 Dissolution test (extended-release tablets, paddleapparatus, spectrophometric determination)
End April 06 ❏ YES ❏ NO AmbientTemperature
(a)
PTS083 Potentiometric titration (2.2.20) End June 06 ❏ YES ❏ NO AmbientTemperature
(a)
PTS084 Assay and related substances by LC (activesubstance for human and veterinary use, isocratic RP-LC,UV-detection)
End Sept 06 ❏ YES ❏ NO AmbientTemperature
(a)
PTS085 Microbiological assay of antibiotics (2.7.2) End Nov 06 ❏ YES ❏ NO AmbientTemperature
(a)
AREA OF ACTIVITY: ❏ OMCL ❏ Private QC pharmaceutical laboratory ❏ Other ( please specify) __________________
FEESThe amount due per study is 230 Euros for laboratories not belonging to the OMCL Network.1In addition to these costs, extra charges for delivery will be added for each PTS study dispatched. These charges are set out in our Official
Catalogue of Pharmaceutical Reference Substances and Preparations – see Section 2.2: Delivery and Related Costs Prices - Deliverycharges (a).
DELIVERYEach PTS will be shipped either on a DDU or CIP basis (Incoterms 2000) as set out in our Official Catalogue of Pharmaceutical ReferenceSubstances and Preparations – see Section 2.2: Delivery and Related Costs Prices - Delivery charges (a). A copy of our catalogue isavailable on our website www.pheur.org
CANCELLATION AND PAYMENT About 2 weeks before the PTS study becomes available, we will send you an order confirmation by e-mail and an invoice. The cancellation ofan invoiced PTS is only possible if we have been informed within 3 working days of that order confirmation. In all cases the payment shouldbe net of charges for the Council of Europe and paid within 30 days from the date of invoice. Details of how to pay are available on ourwebsite and will be outlined on our invoice.
Date Signature
R E G I S T R A T I O N F O R M : P
r o f i c i e n c y T e s t i n g S t u d
i e s ( P T S )
Y e a r 2 0 0 6 – P h y s i c o - C h e m i c a l S t u d i
e s

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 25/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 25
General Information
NEW : PHARMEUROPA ONLINE
Pharmeuropa, Pharmeuropa Bio and Pharmeuropa Scientific Notes Online are now available as a complementaryservice for subscribers to the printed edition of Pharmeuropa, and will be offered for a trial period without an
additional fee for those who ordered the printed version of Pharmeuropa Vol. 18 (2006). All issues stretching back to volume 10 (1998) are stored as Acrobat PDF-files, and can be searched with a search engine identical to the one usedfor the online versions of the European Pharmacopoeia and the Standard Terms.
A username and a password are required to access Pharmeuropa Online, and instructions for creating these using theEDQM Certificate of Authenticity can be found on the inside-front cover of this issue of Pharmeuropa.
________________________________________________________________________________
The Pharmacopoeial Discussion Group [EuropeanPharmacopoeia (EP), Japanese Pharmacopoeia (JP),United States Pharmacopeia (USP)] met in association
with the Expert Working Groups of the InternationalConference on Harmonisation (ICH). The World HealthOrganisation attended as an observer.
Calcium disodium edetate, Calcium phosphate dibasicdihydrate, Calcium phosphate dibasic anhydrous: theseharmonised monographs were signed off.
Benzyl alcohol, Lactose anhydrous, Methylcellulose:
revisions of these harmonised monographs were signedoff.
Microbial contamination: the PDG has worked onthe harmonisation of 3 important general chapters
with relevance for the ICH Q6A guideline: microbialenumeration methods, tests for specified micro-organisms, and acceptance criteria for pharmaceuticalpreparations. These harmonised chapters were signed off.
Implementation of harmonised texts: to accelerateinter-regional implementation of harmonised texts,the JP has implemented a new procedure for rapidimplementation of harmonised general chapters related
to the Q6A guideline, since full harmonisation is achievedonly when all 3 pharmacopoeias have published andimplemented a monograph/general chapter.
ICH Expert Working Group Q4B (Regulatory Acceptance of Pharmacopoeial Interchangeability): on November9, 2005, the PDG held a joint meeting with Q4B todiscuss the regulatory acceptance of harmonisedmonographs and general chapters, particularly thoseof relevance for the ICH Q6A guideline; the documentdetailing the roles and responsibilities of the PDG andQ4B EWG was discussed and further refinement wasnecessary; 5 packages for harmonised general chaptershave been submitted by the PDG to the Q4B group,including dissolution, extractable volume, particulate
matter in parenterals, residue on ignition/sulphatedash, and sterility test. Examination of the test forresidue on ignition/sulphated ash has been completedby the Q4B group and tests, analytical procedures,and acceptance criteria of the 3 pharmacopoeias willbe recognised as interchangeable by the regulatoryauthorities in the 3 regions once the harmonised text hasbeen published and implemented in all 3 regions. Thepackage for dissolution was submitted to the Q4B groupin August 2005; the PDG is awaiting feedback on thistopic. The USP has revised its text for extractable volumeand the PDG is awaiting feedback from the Q4B group.Particulate matter was discussed at the November 9meeting and Q4B provided a preliminary report outlininga number of issues remaining to be resolved in orderto achieve regulatory interchangeability. A number ofissues remain to be resolved for the sterility test in orderto achieve regulatory interchangeability. Additionally,packages for the PDG harmonised texts on disintegrationand uniformity of dosage units are in preparation forsubmission to Q4B.
Industry Associations: a meeting with industryassociations from the 3 ICH regions was held onNovember 8, 2005, to exchange information on progress
with the current work program and future harmonisation
needs. Industry associations were encouraged to play anactive role in the harmonisation process as importantstakeholders.
Excipients Councils: a meeting was held on November 10,2005, with Tri-PEC (IPEC Americas, IPEC Europe,Japanese Pharmaceutical Excipients Council) to discussthe work program on harmonisation of excipientmonographs. Current issues include the policy forfunctionality-related characteristics, use of additives andprocessing aids in excipients, co-processed excipients,impurities in excipients, and the future of harmonisation.
The PDG will hold its next meeting on 5-8 June, 2006, in Yokohama, Japan.
PHARMACOPOEIAL DISCUSSION GROUP (PDG)
Chicago, USA, 7-10 November 2005

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 26/176
26 © PHARMEUROPA Vol. 18, No. 1, January 2006
International Conferences
International Conferences AGENDA 2006
INTERNATIONAL CONFERENCES & SYMPOSIUM***
TRAINING SESSIONS ON THE 5th EDITIONOF THE EUROPEAN PHARMACOPOEIA
CHEMICALS – NEW PROGRAMME
London, UK, 2-3 March 2006
Chicago, USA, 27-28 April 2006
***The EDQM is pleased to announce a new training programme on the European Pharmacopoeia 5th Edition. Thenew programme aims to provide professionals with an in-depth and up-to-date knowledge of the most importantand practical aspects of the European Pharmacopoeia. New additions for 2006 include practical examples and casestudies, question and answer sessions giving you the opportunity to clarify any issues that may arise and the chanceto meet one-to-one with the speakers who work in a certain area, thus providing you with more meaningful and
worthwhile interactions.
Do not miss these opportunities to meet the EDQM/European Pharmacopoeia.
More information will be available on the EDQM website www.pheur.org and in the next issues of Pharmeuropa.
GENERAL CONDITIONS FOR REGISTRATION AT THE EDQM CONFERENCES
AND TRAINING SESSIONS
HOW TO REGISTER
Register as soon as possible: places are limited.
We recommend using a separate form for each participant. Please fill in the registration form* and return it to theEDQM Public Relations Unit, by post, by fax or by e-mail duly completed with the selected method of payment.
NEW Online Registration: Online conference registration is now possible. To use the online registration form, justclick on the icon ‘EDQM Events - Register online’ and follow the steps described.
REGISTRATION FEE
— A special rate is given to permanent staff of national authorities, university, R&D public centres.
— The registration fee is not subject to VAT and covers attendance at the lectures, working documents, lunches,coffee breaks and the official dinner.
— The registration fee does not include your hotel accommodation costs (see hotel reservation form) and travelexpenses.
METHOD OF PAYMENT
You can make your payment: by bank transfer, by enclosed cheque or by credit card.
The relevant bank transfer information is given on the invoice.
CANCELLATION POLICY
One month before the event 80% of the registration fee will be refunded; no refunds will be given after this date.However, registration may be transferred to another person at any time. In case of no show, the registration fee isdue.
* All information requested on the registration form is necessary and used only for the organisation of the seminar.

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 27/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 27
International Conferences
2006 TRAINING SESSION: LONDON
HOW TO USE THE EUROPEAN PHARMACOPOEIA 5TH EDITION
CHEMICAL PRODUCTS
Duration: 1,5 days, Location: Local Government House, Smith Square,
London SW1P 3HZ, UK
Working language: English
***
NEW PROGRAMME
THURSDAY 2 MARCH 2006
8:45 - 9:15 Registration and Welcome Coffee
Opening remarks and general introduction9:15 - 9:30 Dr Michael Morris, Chair of the European Pharmacopoeia Commission
SESSION 1: GENERAL INTRODUCTION
The European Regulatory System: Interactive and complementary relationship between EU/European
Commission/European Council/EMEA/EDQM/Council of Europe, place of the EDQM and the European
Pharmacopoeia
9:30 - 9:50 Dr Agnès Artiges, Director of the European Directorate for the Quality of Medicines,
EDQM, Council of Europe
9:50 - 10:00 Discussion
SESSION 2: HOW TO FIND YOUR WAY THROUGH THE EUROPEAN PHARMACOPOEIA
- General concepts in the European Pharmacopoeia: theory and rationaleGeneral notice, General monographs and chapters (chromatographic separation techniques, ..)
- Role and status of the European Pharmacopoeia10:00 - 10:45 Mr Peter Castle, Secretary to the European Pharmacopoeia Commission, EDQM, Council
of Europe
10:45 - 11:00 Discussion
11:00 - 11:30 Coffee Break
- A practical approach: Cases studies of specific monographs (active substances and excipients),
use of reference standards and links with the Certificate of suitability of monographs of the
European Pharmacopoeia (CEP)
11:30 - 12:15 Speaker to be confirmed12:15 - 12:30 Discussion
SESSION 3: CURRENT PROGRESS IN THE FIELD OF INTERNATIONAL
HARMONISATION
- Regulatory interchangeability between FDA (US) / EU / *MHLW (J)
12:30 - 12:50 Dr Michael Morris12:50 - 13:00 Discussion
13:00 - 14:00 Lunch break
SESSION 4: HOW TO PARTICIPATE IN THE ELABORATION & REVISION
OF THE EUROPEAN PHARMACOPOEIA
14:00 – 14:20 Mr Peter Castle
14:20 – 14:30 Discussion
SESSION 5: THE EUROPEAN PHARMACOPOEIA PUBLICATIONS AND SERVICES
- Publications: The 5th
Edition of the European Pharmacopoeia: Implementation and the
publication schedule, Pharmeuropa and list of standard terms
14:30 - 14:50 Dr Hans-Joachim Bigalke, Publications Division, EDQM, Council of Europe
14:50 - 15:00 Discussion
15:00 - 15:30 Coffee break
*Ministry of Health, Labour and Welfare
N E W P R O G R A M M E : T R A I N I N G S E S S I O N 5 T H
E D I T
I O N
2 - 3 M a r c h 2 0 0 6 – L o c a t i o n : L
o c a l G o v e r n m e n t H o u s e ,
L o n d o n , U K

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 28/176
28 © PHARMEUROPA Vol. 18, No. 1, January 2006
International Conferences
- European Pharmacopoeia Reference Standards
15:30 - 15:50 Speaker to be confirmed15:50 - 16:00 Discussion
SESSION 6: THE EUROPEAN PHARMACOPOEIA INTERNET SITES, HOW TO ACCESS
THE ONLINE DATABASES AND THE NEW HELPDESK
16:00 - 16:20 Mrs Caroline Larsen Le Tarnec, Public Relations Officer, EDQM, Council of Europe16:20 - 16:30 Discussion
NEW! SESSION 7: ONE TO ONE CONSULTATIONS
16:30 - 17:30
Topics under which a consultation can be arranged: General questions on the EDQM, the Europeanregulatory framework and harmonisation; Technical questions on PhEur monographs and texts;Publications and services; Electronic version of the European Pharmacopoeia
FRIDAY 3 MARCH 2006 (MORNING ONLY)
8:30 - 9:00 Welcome Coffee
During the morning, the electronic/online version of the European Pharmacopoeia will be set up
and demonstrated to participants interested in learning more about this version. The EDQM will
give advice on how to search, retrieve and print documents and illustrate where to find
information such as chromotograms, reference substances and Certificates of suitability.
SESSION 8: GENERAL INTRODUCTION TO THE CERTIFICATION PROCEDURE AND
INSPECTIONS
- General considerations on the Certification procedure and Inspections
Origin, scope, how it works, who is involved9:00 - 9:15 Dr Agnès Artiges9:15 - 9:25 Discussion
- How to apply for a CertificateContent of the dossier for chemical substancesComments on the main deficiencies found in dossiers
9:25 - 11:05 Dr Andrew McMath, Certification Unit, EDQM, Council of Europe10:10 - 10:40 Coffee break
10:55 - 11:05 Discussion
- Revisions and renewals
11:05 - 11:15 Dr Andrew McMath11:15 - 11:25 Discussion11:25 Final addresses and Closure of the meeting
NEW! SESSION 9: ONE TO ONE CONSULTATIONS
11:45 - 12:45
Topics covered: General questions on the EDQM, the European regulatory framework andharmonisation; Technical questions on PhEur monographs and texts; Certification procedure;Publications and services; Electronic version of the European Pharmacopoeia
Who should attend?
This training session should be attended by professionals from industry, in particular persons involved inthe manufacture and control of drug substances/products or the preparation of registration dossiers; frominspectorates, regulatory agencies and academic institutions.
Contact details:
Further information on the conference, accommodation and registration forms will be made available onthe EDQM internet site: http://www.pheur.org. Address: Public Relations Unit, EDQM, 226 avenue deColmar BP 907, F-67029, Strasbourg, Cedex 1:Tel: +33 (0) 3 88 41 30 30 (Dial 4 for Conferences); Fax: + 33 (0) 3 88 41 27 71;Internet: See under ‘HELPDESK’ on our website: http://www.pheur.org/site/page_630.php)
FOR MORE INFORMATION PLEASE VISIT THE WEBSITE: http://www.pheur.org
N E W
P R O G R A M M E : T R A I N
I N G S E S S I O N 5 T H
E D I T
I O N
2 - 3 M a r c
h 2 0 0 6 – L o c a t i o n : L o
c a l G o v e r n m e n t H o u s e , L o n d o n , U K

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 29/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 29
International Conferences
Please complete and send this form to: Caroline Larsen Le Tarnec, Public Relations, EDQM Fax: +33 0)3 88 41
27 71; Internet: Please attach the registration form to our enquiry form see section ‘Helpdesk’ on the website:
http://www.pheur.org/site/page_630.php ) or register online.
REGISTRATION DETAILS
DATE OF REGISTRATION (DD/MM/YY) : _ _ _ _ _ _
REGISTRATION FEE (*See general conditions) 600 €* or 200 €*
ONE TO ONE MEETING Yes, I am interested in reserving a meeting No, I am not interested
Which topic(s)? EU Regulations Monographs, revisions Publications Certification Internet
PARTICIPANT DETAILS Please complete one form per participant
Title (Dr., Mr, Mrs, Ms, …)
First Name
Family Name
Company/Institution
Address forCorrespondence
Postcode
Town
Country
Telephone
Fax
AREA OF ACTIVITY/ Manufacturer of raw material Manufacturer of medicines for human use for veterinary use
OCCUPATION: QA QC R & D Regulatory affairs Regulatory authority: Licensing Pharmacopoeias Inspection OMCL University Other (please specify)……………………
PAYMENT (NEW )Following receipt of your registration form, we will send you an invoice. Please note that we must receive paymentbefore the conference takes place. Details of payment methods will be outlined on the invoice. However, you willbe able to settle your invoice by:
1. PERSONAL OR COMPANY CHEQUE made payable to Council of Europe/EDQM2. BANK TRANSFER3. CREDIT CARD
DETAILS FOR INVOICING PURPOSES (if different from participant details)
Company/Institution
Address
Postcode
Town
Country
VAT Number (EU only)
Contact Name
Telephone
Fax
PO Number/ Reference
CANCELLATION CHARGES: I have read and accept the cancellation terms as stated on our web-site.
Date Signature
FOR MORE INFORMATION PLEASE VISIT REGULARLY THE WEBSITE : http://www.pheur.org
R E G I S T R A T I O N F O R M
:
T R A I N I N G S E S S I O N 5
t h E
D I T I O N
2 - 3 M a r c h 2 0 0 6 –
L o c a t i o n : L o c a l G o v e r n m e n t H o u s e ,
L o n d o n ,
U K

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 30/176
30 © PHARMEUROPA Vol. 18, No. 1, January 2006
International Conferences
2006 TRAINING SESSION: LONDON
EUROPEAN PHARMACOPOEIA 5TH EDITION CHEMICALS2-3 MARCH 2006
ONE TO ONE CONSULTATION QUESTION FORM(Thursday 2 March 2006 from 16:30 – 17:30
AND Friday 3 March 2006 from 11:30 – 12:30)
If you would like to register for a one-to-one consultation (15 minutes) with a member of the EDQMscientific team during the training course, please complete this consultation request form and send itto the EDQM by fax to +33 3 88 41 27 71 or via the Helpdesk on the website:http://www.pheur.org/site/page_630.php)Priority will be given to those who book in advance.
PARTICIPANT DETAILS
Title (Dr., Mr, Mrs, Ms)
First Name
Family Name
Company/Institution
The time slots available for one-to-one consultations will be limited. The EDQM shall do all it can toaccommodate all interested participants.
If it is on a specific application, please mention the reference numberYour question:
Date Signature
FOR MORE INFORMATION ABOUT TRAINING SESSIONS AND CONFERENCES ORGANISED BY THE EDQMPLEASE VISIT REGULARLY THE WEBSITE : http://www.pheur.org
O N E
- T O - O
N E R E S E R V A T I O N
F O R M :
T R A I N I N G S E S S I O N 5 t h E
D I T I O N
2 - 3 M
a r c h 2 0 0 6 – L o c a t i o n
: L o c a l G o v e r n m e n t H o
u s e , L o n d o n , U K

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 31/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 31
International Conferences
HOTEL RESERVATION FORM
2006 TRAINING COURSE LONDON: 2-3 MARCH 2006Location: Local Government House, London, UK
NOVOTEL LONDON WATERLOO HOTEL RESERVATION FORM Fax: +44 (0)207 793 0202
To be filled in and sent by fax before the 15 February 2006
Global reservation: 20 rooms reserved from Wednesday 1st March to Friday 3
rd March included.
Contact reservations: NOVOTEL – LONDON WATERLOO HOTEL: 113 Lambeth Palace Road, London,
SE1 7LS, UK, Contact: Assita Kone Tel +44 (0)207 793 5718
Fax +44 (0)207 793 0202; E-mail : [email protected]
General information: The quotas of rooms reserved will be available until 15 February 2006.
After this date the availability and the negotiated prices will not be guaranteed. The
rooms should be reserved individually by each participant and are available on a
first come, first served basis. Cancellation policy: Any room cancelled 15 days priorto arrival 50% will be charged and 7 days before arrival 100% will be charged on the
credit card provided.
Participant: Surname Forename
Company/ employer Address
City Postal Code
Country E-mail
Tel N° Fax N°
Hotel reservation: Arrival day/hour ……………..... Departure day/hour ……………….....
Please indicate the room and nights required by a circle
SINGLE
OCCUPANCY
DOUBLE
OCCUPANCY
NIGHT
01/03/2006
NIGHT
02/03/2006
NOVOTEL
LONDON
WATERLOO
HOTEL145 GBP* 165 GBP* YES YES
* Taxes and breakfast inclusive
Method of payment: Credit card: Visa EuroCard / MasterCard Amex
Credit card number: Expiry date:
Cardholder: ______________________________________________________
I authorise the NOVOTEL – LONDON WATERLOO HOTEL to charge against my credit card the
amount equivalent to one night in order to block my reservation for the duration of the seminar.
Date: _____________________ Signature: ________________________________________
HOTEL CONFIRMATION RESERVATION N°:___________ Signature:____________________
H O T E L R E G I S T R A T I O N F
O R M :
T R A I N I N G S E S S I
O N 5 t h E
D I T I O N
2 - 3 M A R C H 2 0 0 6 – L
o c a t i o n : L o c a l G o v e r n m e n t
H o u s e , L o n d o n

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 32/176
32 © PHARMEUROPA Vol. 18, No. 1, January 2006
International Conferences
2006 TRAINING SESSION: UNITED STATESTHE EUROPEAN PHARMACOPOEIA 5TH EDITION
CHEMICAL DRUG PRODUCTS AND SUBSTANCESDuration: 2 days, Location: Radisson SAS, 160E Huron Street, Chicago, USA
Working language: English
***
NEW PROGRAMME
THURSDAY 27 APRIL 2006
8:00-8:30 Registration and Welcome Coffee
8:30-8:45 Opening remarks and general introduction
8:45-9:05 European regulations for medicines: How does the system work? Relationship between EU/EMEA and the EDQM of the Council of Europe. Place and roles of the EDQM and the European Pharmacopoeia
9:05-9:25 EDQM, general organisation and process of elaboration & revision of the European Pharmacopoeia. How to submit a new monograph/revision proposal.
9:25-10:10 Pharmacopoeias and international harmonisation process
10:10-10:30 Open discussion
10:30-11:00 Coffee Break
11:00-12:00 How to use the European Pharmacopoeia: General Notices, General chapters, generalmonographs and monographs on dosage forms. Frequently asked questions.
12:00-12:20 Open discussion
12:30-13-20 Lunch Break
13:20-14:20 Cases studies of specific monographs (active substances and excipients), use of reference standards, links with the Certificate of suitability of monographs of the European Pharmacopoeia (CEP)
14:20-15:30 The European Pharmacopoeia Publications (printed and electronic publications) and Reference Standards
15:30-16:30 The European Pharmacopoeia Internet sites, How to access to the online databases and to
the new users’ support: the HELPDESK
16:30-16:45 Open discussion
17:00-18:00 One to One consultations.
Subject to a prior appointment, questions has to be sent prior the session to guarantee an efficient
answer/advice. Topics under which a consultation can be arranged: General questions on the EDQM, the
European regulatory framework and harmonisation; Technical questions on PhEur monographs and texts;
Publications and services; Electronic version of the European Pharmacopoeia
N
E W P R O G R A M M E : T R A
I N I N G S E S S I O N 5 T H
E D
I T I O N
2 7 - 2 8 A p r i l 2 0 0 6 – L o c a t i o n : R a d
i s s o n H o t e l , 1 6 0 E H u r o n
S t . ,
C h i c a g o ,
U S A

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 33/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 33
International Conferences
FRIDAY 28 APRIL 2006
8:00-8:30 Welcome Coffee
8:30-9:30 Presentation of PHARMEUROPA online and PHARMEUROPA Scientific Notes
9:30-10:30 The certification procedure and inspections: General considerations, How to apply fora Certificate
10:30-10:50 Open discussion
10:50-11:10 Coffee break
11:10-12:10 Revisions and renewals in the procedure of certification
12:10-12:30 Open discussion
12:30-13:30 Lunch break
13:30-14:30 General Chapters in the European Pharmacopoeia: How to understand and to usethem in practice. Decisions tree for impurities.
14:30-15:30 How to use the European Pharmacopoeia in your daily laboratory work Case of alternative methods, Test procedures in a monograph, How to perform Specific tests,
How to interpret chromatograms and list of impurities
15:30-15:50 Open discussion
16:00 Final addresses and Closure of the meeting
16:00-17:15 One to One consultations
Topics covered: General questions on the EDQM, the European regulatory framework and
harmonisation; Technical questions on PhEur monographs and texts; Certification procedure;
Publications and services; Electronic version of the European Pharmacopoeia
During coffee breaks, the electronic/online version of the European Pharmacopoeia will be set up
and demonstrated to participants interested in learning more about this version.
Who should attend?This conference should be attended by professionals from industry, in particular persons involved in
the manufacture and control of drug substances/products or the preparation of registration dossiers;
from inspectorates, regulatory agencies and academic institutions.
Contact details:Further information on the training course, accommodation and registration forms will be made available
on the EDQM internet site: http://www.pheur.org
Register at: http://www.pheur.org/site/page_597.php or contact the Public Relations Unit, EDQM;
226 avenue de Colmar, BP907, F-67029, Strasbourg, Cedex 1, FRANCE; Tel: 00 33 3 88 41 30 30 (Dial 4);
Fax: 00 33 3 88 41 27 71;
Email: Via the EDQM Helpdesk - go to: http://www.pheur.org/site/page_521.php
FOR MORE INFORMATION PLEASE VISIT THE WEBSITE : http://www.pheur.org
N E
W P R O G R A M M E : T R A I N I N G S E S S I O N 5 T H
E D I T I O N
2 7 - 2 8 A p r i l
2 0 0 6 – L o c a t i o n : R a d i s s o n H o t e l , 1 6 0 E H u r o n S
t . , C h i c a g o , U S A

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 34/176
34 © PHARMEUROPA Vol. 18, No. 1, January 2006
International Conferences
Please complete and send this form to: Caroline Larsen Le Tarnec, Public Relations, EDQM Fax: +33 0)3 88 41
27 71; Internet: Please attach the registration form to our enquiry form see section ‘Helpdesk’ on the website:http://www.pheur.org/site/page_630.php ) or REGISTER ON-LINE
REGISTRATION DETAILS
DATE OF REGISTRATION (DD/MM/YY) : _ _ _ _ _ _
REGISTRATION FEE (*See general conditions) 950 €* or 400 €*
ONE TO ONE MEETING Yes, I am interested in reserving a meeting No, I am not interested
Which topic(s)? EU Regulations Monographs, revisions Publications Certification Internet
PARTICIPANT DETAILS Please complete one form per participant
Title (Dr., Mr, Mrs, Ms, …)
First Name
Family Name
Company/InstitutionAddress forCorrespondence
Postcode
Town
Country
Telephone
Fax
AREA OF ACTIVITY/ Manufacturer of raw material Manufacturer of medicines for human use for veterinary use
OCCUPATION: QA QC R & D Regulatory affairs
Regulatory authority: Licensing Pharmacopoeias Inspection OMCL University Other (please specify)……………………
PAYMENT (NEW )
Following receipt of your registration form, we will send you an invoice. Please note that we must receive paymentbefore the conference takes place. Details of payment methods will be outlined on the invoice. However, you willbe able to settle your invoice by:
1. PERSONAL OR COMPANY CHEQUE made payable to Council of Europe/EDQM2. BANK TRANSFER3. CREDIT CARD
DETAILS FOR INVOICING PURPOSES (if different from participant details)
Company/Institution
Address
Postcode
Town
CountryVAT Number (EU only)
Contact Name
Telephone
Fax
PO Number/ Reference
CANCELLATION CHARGES: I have read and accept the cancellation terms as stated on our web-site.
Date Signature
FOR MORE INFORMATION PLEASE VISIT REGULARLY THE WEBSITE : http://www.pheur.org
R E G I S T R A T I O N F O R M :
T R A I N I N G S E S S I O N 5 t h E
D I T I O N
2 7 - 2 8 A p r i l 2 0 0 6 – R a d i s s o n H o t e l , 1 6 0 E H u r o n S t . , C h i c a g o , U S A

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 35/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 35
International Conferences
2006 TRAINING SESSION: UNITED STATES
EUROPEAN PHARMACOPOEIA 5TH EDITION CHEMICALS27-28 APRIL 2006
ONE TO ONE CONSULTATION QUESTION FORM
If you would like to register for a one-to-one consultation (15 minutes) with a member of the EDQMscientific team during the training course, please complete this consultation request form and sendit to the EDQM by fax to +33 3 88 41 27 71 or via the Helpdesk on the website:http://www.pheur.org/site/page_630.php)Priority will be given to those who book in advance.
PARTICIPANT DETAILSTitle (Dr., Mr, Mrs, Ms)
First Name
Family Name
Company/Institution
The time slots available for one-to-one consultations will be limited. The EDQM shall do all it can to
accommodate all interested participants.
If it is on a specific application, please mention the reference numberYour question:
Date Signature
FOR MORE INFORMATION ABOUT TRAINING SESSIONS AND CONFERENCES ORGANISED BY THE EDQMPLEASE VISIT REGULARLY THE WEBSITE : http://www.pheur.org
O N E
- T O - O
N E R E S E R V A T I O N
F O R M :
T R A I N I N G S E S S
I O N 5 t h E
D I T I O N
2 7 - 2 8 A p r i l 2 0 0 6 –
L o c a t i o n : R a d
i s s o n H o t e l , 1 6 0 E H u r o
n S t r e e t , C h i c a g o ,
U S A

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 36/176
36 © PHARMEUROPA Vol. 18, No. 1, January 2006
International Conferences
HOTEL RESERVATION FORM
2006 TRAINING COURSE CHICAGO: 27-28 APRIL 2006Location: Radisson Hotel & Suites Hotel, Chicago, USA
To be filled in and sent by fax before 27 MARCH 2006 Fax: +1-312-757-5158
Global reservation: 40 rooms reserved from Wednesday 26th
April to Friday 28th
April included.
Contact reservations: RADISSON HOTEL & SUITES: 160E Huron St., Chicago, IL 60611
Contact: Reservations and indicate you are part of the EDQM/European Pharmacopoeia training course;
Tel: +1-312-787-2900; Fax: +1-312-757-5158; E-mail: [email protected]
General information: The quotas of rooms reserved will be available until 27 March 2006.
After this date the availability and the negotiated prices will not be guaranteed. The rooms should be reserved
individually by each participant and are available on a first come, first served basis. Cancellation policy: Before26 April 2006 at no further cost, after this date and in case of no show, one night will be charged to your credit
card.
Participant:Family name: First name:
Address:
City: Postcode:
State/Country:
Tel: Fax:
E-mail:
Hotel reservation:
Arrival day/hour: Departure day/hour:
Please tick the box indicating the type of room you wish to reserve:
❏ Standard Single Room at 149USD per night* ❏ Standard Double Room at 149USD per night*
❏ Single Corner Suite at 189USD per night* ❏ Double Corner Suite at 189USD per night
*Note: Room rates are subject to state and local taxes and excludes breakfast
Please tick the box(es) indicating number of nights accommodation you require:
❏ Yes Night 26 April ❏ Yes Night 27 April ❏ Yes, Night 28 April
Method of payment:
Credit card:❏
Visa❏
EuroCard/ MasterCard ❏
Amex❏
Diners Card
Credit card number: Expiry date:
Cardholder : ______________________________________________________
I authorise the RADISSON HOTEL & SUITES to charge against my credit card the amount equivalent to one
night in order to block my reservation for the duration of the training session.
HOTEL CONFIRMATION RESERVATION N°: ____________ Signature: ______________________
H O T E L R E G I S T R A T I O N F O R M :
T R A I N I N G S E S S I
O N 5 t h E
D I T I O N
2 7 - 2 8 A P R I L 2 0 0 6 – L o c a t i o n : R a d i s s o n H o t e l , 1 6 0 E H
u r o n S t . C h i c a g o ,
U S A

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 37/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 37
Certification of suitability
Certification of Suitabilityof Monographs of the Ph. Eur.
CERTIFICATION SECTION OF THE EDQM’SINTERNET SITE
Visit the certification section of the EDQM’s internet site(www.pheur.org) and find:
— warnings/news;— answers to frequently asked questions;— general information on the certification procedure;— daily updated list of certificates granted.
LIST OF CERTIFICATES
Granted certificates
A daily updated list (more than 1800 certificates) isavailable online in the certification section of our internetsite (www.pheur.org). The database can be searched bysubstance name, certificate number or holder name. TSEcertificates can also be searched selectively. To print thefull list of certificates: select “certificate number” in thedatabase and search for “CEP”.
Voided or suspended certificates
A list of voided certificates is available online in thecertification section of our internet site (www.pheur.org)and is also published in Pharmeuropa. This list includes:
— the certificates not renewed after the 5-year validityperiod (expired certificates);
— the certificates withdrawn at the request of theapplicant (production stopped, site closed etc.);
— the certificates suspended for a defined periodor withdrawn by EDQM (for GMP deficiencies orinsufficient information no longer in line withregulatory requirements). They are identified by anasterisk.
Warning: in the online search of the database, nodifference is made between suspended, expired or
withdrawn certificates.
LIST OF CERTIFICATES
Chemical purity and microbiological qualityThese certificates have not been renewed / have been withdrawn.
* suspended, withdrawn or not renewed by EDQM following an inspection ** withdrawn by CEP Holder after suspension by EDQM as a result of an inspection *** CEP restored
Substance Certificate Holder/Detenteur
Acetylcysteine R0-CEP 1997-071-Rev 00 Sterling SNIFF Italia; I 06073 Solomeo Di
Corciano (Perugia)
Acetylsalicylic acid (Crystals-
Powder-Granules 7017)
R0-CEP 2000-013-Rev 00
***
Alta Laboratories Ltd ; IND Maharashtra
Allopurinol R0-CEP 1997-016-Rev 00 Siegfried Cms Ag/Ltd; CH 4800 Zofingen
Alprazolam R0-CEP 1998-111-Rev 02 Degussa AG; D 01445 Radebeul
Amiloride Hydrochloride R0-CEP 1995-045-Rev 02 Siegfried Cms Ag/Ltd; CH 4800 Zofingen
Amoxicillin Sodium R1-CEP 1996-096-Rev 00 Ribbon SRL Pharmaceutical and Chemical;
I 20145 Milano
Amoxicillin Sodium R1-CEP 1998-002-Rev 01 Sandoz Industrial Products S.A.; E 08520
Barcelona
Amoxicillin trihydrate R0-CEP 1993-001-Rev 00 Flamma SpA; I 24125 Bergamo
Amoxicillin trihydrate; Compacted R0-CEP 1995-032-Rev 02 Gist-Brocades BV; NL 2600 MA Delft
Amoxicillin trihydrate R0-CEP 1998-127-Rev 00 Smithkline Beecham; FR 35380 Plélan le Grand
Amoxillin Sodium sterile R1-CEP 1996-096-Rev 00 Ribbon Srl Pharmaceutical and Chemical I-20145
Milano
Ampicillin Sodium R0-CEP 1995-004-Rev 01 Gist Brocades B.v; NL 2600 AK Delft
Ampicillin Sodium; Sterile R0-CEP 1998-132-Rev 00 Gist-Brocades BV; NL 2600 MA Delft
Ampicillin Trihydrate R0-CEP 1994-010-Rev 00 Gist-Brocades B V; NL 2600 MA Delft
Ampicillin Trihydrate R0-CEP 1992-001-Rev 02 Biochemie SA; E 08400 Granollers - Barcelona
Ampicillin, Anhydrous R0-CEP 1994-009-Rev 00 Gist-Brocades B V; NL 2600 MA Delft
Aspartic acid R0-CEP 2000-295-Rev 02 Bim Sifram Group; FR 75010 Paris
Ascorbic acid R0-CEP 1995-019-Rev 01 Pliva D D Zagreb; CRO 10000 Zagreb
Ascorbic acid R0-CEP 1997-035 Rev 02 Merck Kgaa D 64271 Darmstadt
Atenolol R0-CEP 1998-033 Rev 02 Teva Pharmaceutical Industrie Ltd; IL 49131 Petah
Tiqva
Beclometasone Dipropionate R1-CEP 1992-013-Rev 00 Hoechst Marion Roussel; F 92800 Puteaux
Benzylpenicillin potassium R1-CEP 1992-003-Rev 00 Gist-Brocades BV; NL 2600 MA Delft
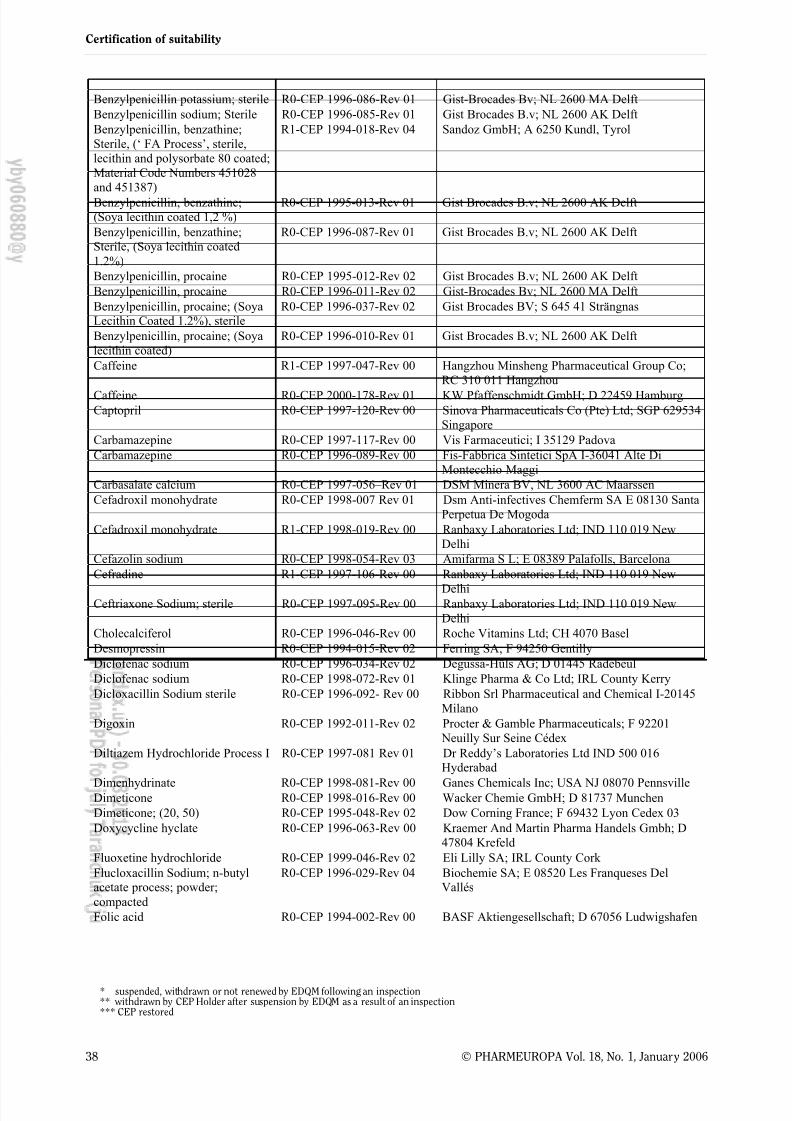
8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 38/176
38 © PHARMEUROPA Vol. 18, No. 1, January 2006
Certification of suitability
* suspended, withdrawn or not renewed by EDQM following an inspection ** withdrawn by CEP Holder after suspension by EDQM as a result of an inspection *** CEP restored
Benzylpenicillin potassium; sterile R0-CEP 1996-086-Rev 01 Gist-Brocades Bv; NL 2600 MA Delft
Benzylpenicillin sodium; Sterile R0-CEP 1996-085-Rev 01 Gist Brocades B.v; NL 2600 AK Delft
Benzylpenicillin, benzathine;
Sterile, (‘ FA Process’, sterile,
lecithin and polysorbate 80 coated;
Material Code Numbers 451028
and 451387)
R1-CEP 1994-018-Rev 04 Sandoz GmbH; A 6250 Kundl, Tyrol
Benzylpenicillin, benzathine;
(Soya lecithin coated 1,2 %)
R0-CEP 1995-013-Rev 01 Gist Brocades B.v; NL 2600 AK Delft
Benzylpenicillin, benzathine;Sterile, (Soya lecithin coated
1.2%)
R0-CEP 1996-087-Rev 01 Gist Brocades B.v; NL 2600 AK Delft
Benzylpenicillin, procaine R0-CEP 1995-012-Rev 02 Gist Brocades B.v; NL 2600 AK Delft
Benzylpenicillin, procaine R0-CEP 1996-011-Rev 02 Gist-Brocades Bv; NL 2600 MA Delft
Benzylpenicillin, procaine; (SoyaLecithin Coated 1.2%), sterile
R0-CEP 1996-037-Rev 02 Gist Brocades BV; S 645 41 Strängnas
Benzylpenicillin, procaine; (Soya
lecithin coated)
R0-CEP 1996-010-Rev 01 Gist Brocades B.v; NL 2600 AK Delft
Caffeine R1-CEP 1997-047-Rev 00 Hangzhou Minsheng Pharmaceutical Group Co;
RC 310 011 Hangzhou
Caffeine R0-CEP 2000-178-Rev 01 KW Pfaffenschmidt GmbH; D 22459 HamburgCaptopril R0-CEP 1997-120-Rev 00 Sinova Pharmaceuticals Co (Pte) Ltd; SGP 629534
Singapore
Carbamazepine R0-CEP 1997-117-Rev 00 Vis Farmaceutici; I 35129 Padova
Carbamazepine R0-CEP 1996-089-Rev 00 Fis-Fabbrica Sintetici SpA I-36041 Alte Di
Montecchio Maggi
Carbasalate calcium R0-CEP 1997-056–Rev 01 DSM Minera BV, NL 3600 AC Maarssen
Cefadroxil monohydrate R0-CEP 1998-007 Rev 01 Dsm Anti-infectives Chemferm SA E 08130 Santa
Perpetua De Mogoda
Cefadroxil monohydrate R1-CEP 1998-019-Rev 00 Ranbaxy Laboratories Ltd; IND 110 019 New
Delhi
Cefazolin sodium R0-CEP 1998-054-Rev 03 Amifarma S L; E 08389 Palafolls, Barcelona
Cefradine R1-CEP 1997-106-Rev 00 Ranbaxy Laboratories Ltd; IND 110 019 New
DelhiCeftriaxone Sodium; sterile R0-CEP 1997-095-Rev 00 Ranbaxy Laboratories Ltd; IND 110 019 New
Delhi
Cholecalciferol R0-CEP 1996-046-Rev 00 Roche Vitamins Ltd; CH 4070 Basel
Desmopressin R0-CEP 1994-015-Rev 02 Ferring SA; F 94250 Gentilly
Diclofenac sodium R0-CEP 1996-034-Rev 02 Degussa-Hüls AG; D 01445 Radebeul
Diclofenac sodium R0-CEP 1998-072-Rev 01 Klinge Pharma & Co Ltd; IRL County Kerry
Dicloxacillin Sodium sterile R0-CEP 1996-092- Rev 00 Ribbon Srl Pharmaceutical and Chemical I-20145
Milano
Digoxin R0-CEP 1992-011-Rev 02 Procter & Gamble Pharmaceuticals; F 92201
Neuilly Sur Seine Cédex
Diltiazem Hydrochloride Process I R0-CEP 1997-081 Rev 01 Dr Reddy’s Laboratories Ltd IND 500 016Hyderabad
Dimenhydrinate R0-CEP 1998-081-Rev 00 Ganes Chemicals Inc; USA NJ 08070 PennsvilleDimeticone R0-CEP 1998-016-Rev 00 Wacker Chemie GmbH; D 81737 Munchen
Dimeticone; (20, 50) R0-CEP 1995-048-Rev 02 Dow Corning France; F 69432 Lyon Cedex 03
Doxycycline hyclate R0-CEP 1996-063-Rev 00 Kraemer And Martin Pharma Handels Gmbh; D
47804 Krefeld
Fluoxetine hydrochloride R0-CEP 1999-046-Rev 02 Eli Lilly SA; IRL County Cork
Flucloxacillin Sodium; n-butylacetate process; powder;
compacted
R0-CEP 1996-029-Rev 04 Biochemie SA; E 08520 Les Franqueses DelVallés
Folic acid R0-CEP 1994-002-Rev 00 BASF Aktiengesellschaft; D 67056 Ludwigshafen
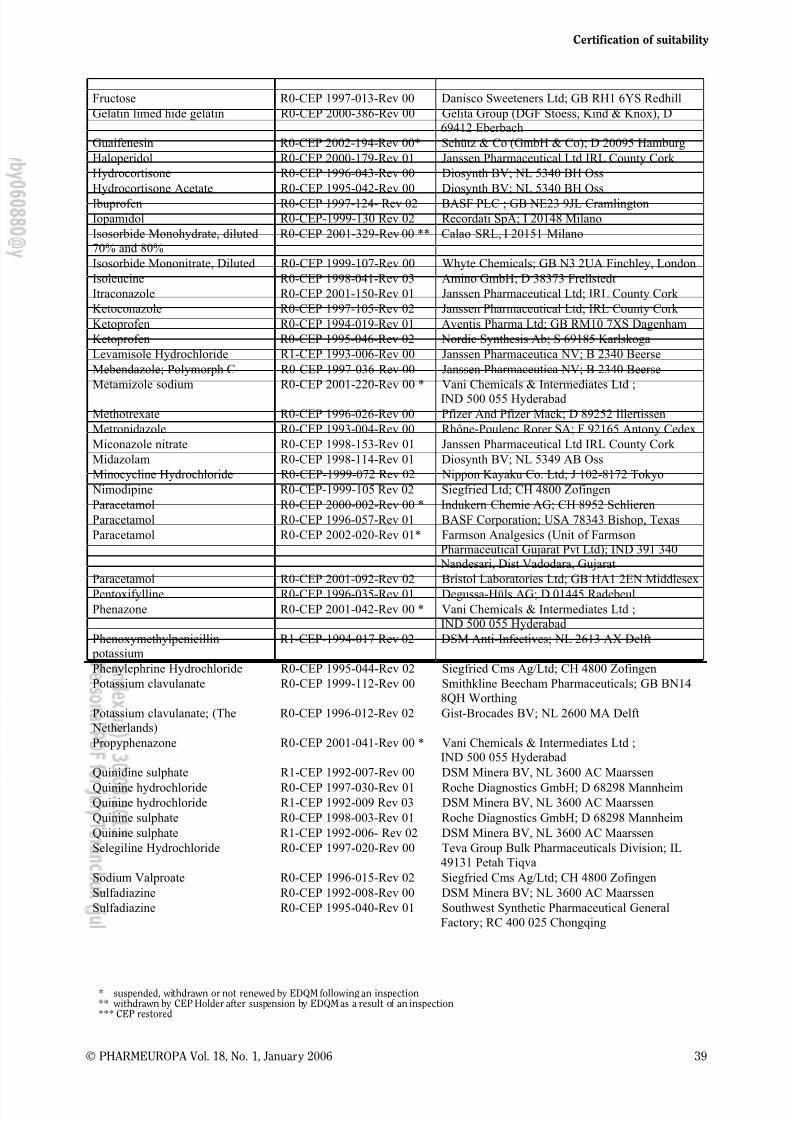
8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 39/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 39
Certification of suitability
* suspended, withdrawn or not renewed by EDQM following an inspection ** withdrawn by CEP Holder after suspension by EDQM as a result of an inspection *** CEP restored
Fructose R0-CEP 1997-013-Rev 00 Danisco Sweeteners Ltd; GB RH1 6YS Redhill
Gelatin limed hide gelatin R0-CEP 2000-386-Rev 00 Gelita Group (DGF Stoess, Kind & Knox), D69412 Eberbach
Guaifenesin R0-CEP 2002-194-Rev 00* Schütz & Co (GmbH & Co); D 20095 Hamburg
Haloperidol R0-CEP 2000-179-Rev 01 Janssen Pharmaceutical Ltd IRL County Cork
Hydrocortisone R0-CEP 1996-043-Rev 00 Diosynth BV; NL 5340 BH Oss
Hydrocortisone Acetate R0-CEP 1995-042-Rev 00 Diosynth BV; NL 5340 BH Oss
Ibuprofen R0-CEP 1997-124- Rev 02 BASF PLC ; GB NE23 9JL Cramlington
Iopamidol R0-CEP-1999-130 Rev 02 Recordati SpA; I 20148 Milano
Isosorbide Monohydrate, diluted
70% and 80%
R0-CEP 2001-329-Rev 00 ** Calao SRL, I 20151 Milano
Isosorbide Mononitrate, Diluted R0-CEP 1999-107-Rev 00 Whyte Chemicals; GB N3 2UA Finchley, London
Isoleucine R0-CEP 1998-041-Rev 03 Amino GmbH; D 38373 Frellstedt
Itraconazole R0-CEP 2001-150-Rev 01 Janssen Pharmaceutical Ltd; IRL County Cork
Ketoconazole R0-CEP 1997-105-Rev 02 Janssen Pharmaceutical Ltd; IRL County Cork
Ketoprofen R0-CEP 1994-019-Rev 01 Aventis Pharma Ltd; GB RM10 7XS Dagenham
Ketoprofen R0-CEP 1995-046-Rev 02 Nordic Synthesis Ab; S 69185 Karlskoga
Levamisole Hydrochloride R1-CEP 1993-006-Rev 00 Janssen Pharmaceutica NV; B 2340 Beerse
Mebendazole; Polymorph C R0-CEP 1997-036-Rev 00 Janssen Pharmaceutica NV; B 2340 Beerse
Metamizole sodium R0-CEP 2001-220-Rev 00 * Vani Chemicals & Intermediates Ltd ;
IND 500 055 Hyderabad
Methotrexate R0-CEP 1996-026-Rev 00 Pfizer And Pfizer Mack; D 89252 Illertissen
Metronidazole R0-CEP 1993-004-Rev 00 Rhône-Poulenc Rorer SA; F 92165 Antony Cedex
Miconazole nitrate R0-CEP 1998-153-Rev 01 Janssen Pharmaceutical Ltd IRL County Cork
Midazolam R0-CEP 1998-114-Rev 01 Diosynth BV; NL 5349 AB Oss
Minocycline Hydrochloride R0-CEP-1999-072 Rev 02 Nippon Kayaku Co. Ltd; J 102-8172 Tokyo
Nimodipine R0-CEP-1999-105 Rev 02 Siegfried Ltd; CH 4800 Zofingen
Paracetamol R0-CEP 2000-002-Rev 00 * Indukern Chemie AG; CH 8952 Schlieren
Paracetamol R0-CEP 1996-057-Rev 01 BASF Corporation; USA 78343 Bishop, Texas
Paracetamol R0-CEP 2002-020-Rev 01* Farmson Analgesics (Unit of Farmson
Pharmaceutical Gujarat Pvt Ltd); IND 391 340
Nandesari, Dist Vadodara, Gujarat
Paracetamol R0-CEP 2001-092-Rev 02 Bristol Laboratories Ltd; GB HA1 2EN Middlesex
Pentoxifylline R0-CEP 1996-035-Rev 01 Degussa-Hüls AG; D 01445 Radebeul
Phenazone R0-CEP 2001-042-Rev 00 * Vani Chemicals & Intermediates Ltd ;
IND 500 055 Hyderabad
Phenoxymethylpenicillin
potassium
R1-CEP-1994-017 Rev 02 DSM Anti-Infectives; NL 2613 AX Delft
Phenylephrine Hydrochloride R0-CEP 1995-044-Rev 02 Siegfried Cms Ag/Ltd; CH 4800 Zofingen
Potassium clavulanate R0-CEP 1999-112-Rev 00 Smithkline Beecham Pharmaceuticals; GB BN14
8QH Worthing
Potassium clavulanate; (The
Netherlands)
R0-CEP 1996-012-Rev 02 Gist-Brocades BV; NL 2600 MA Delft
Propyphenazone R0-CEP 2001-041-Rev 00 * Vani Chemicals & Intermediates Ltd ;IND 500 055 Hyderabad
Quinidine sulphate R1-CEP 1992-007-Rev 00 DSM Minera BV, NL 3600 AC Maarssen
Quinine hydrochloride R0-CEP 1997-030-Rev 01 Roche Diagnostics GmbH; D 68298 MannheimQuinine hydrochloride R1-CEP 1992-009 Rev 03 DSM Minera BV, NL 3600 AC Maarssen
Quinine sulphate R0-CEP 1998-003-Rev 01 Roche Diagnostics GmbH; D 68298 Mannheim
Quinine sulphate R1-CEP 1992-006- Rev 02 DSM Minera BV, NL 3600 AC Maarssen
Selegiline Hydrochloride R0-CEP 1997-020-Rev 00 Teva Group Bulk Pharmaceuticals Division; IL
49131 Petah Tiqva
Sodium Valproate R0-CEP 1996-015-Rev 02 Siegfried Cms Ag/Ltd; CH 4800 Zofingen
Sulfadiazine R0-CEP 1992-008-Rev 00 DSM Minera BV; NL 3600 AC Maarssen
Sulfadiazine R0-CEP 1995-040-Rev 01 Southwest Synthetic Pharmaceutical General
Factory; RC 400 025 Chongqing
8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 40/176
40 © PHARMEUROPA Vol. 18, No. 1, January 2006
Certification of suitability
Transmissible spongiform encephalopathy risk productsThe following certificates have been withdrawn.
* suspended, withdrawn or not renewed by EDQM following an inspection ** withdrawn by CEP Holder after suspension by EDQM as a result of an inspection *** CEP restored
Sulfamethoxazole R0-CEP 1995-026-Rev 02 Shionogi & Co Ltd; J 541-0045 Osaka
Tamoxifen Citrate R0-CEP 1997-024-Rev 01 Siegfried Cms Ag/Ltd; CH 4800 Zofingen
Terfenadine R0-CEP 1997-003-Rev 00 Ranbaxy Laboratories Ltd; IND 144 533 Toansa
Terfenadine R0-CEP 1995-037-Rev 01 Avik Pharmaceutical Ltd; IND 396 195 Vapi
Theophylline R1-CEP 1998-018-Rev 00 * KW Pfannenschmidt GmbH; D 22459 Hamburg
Trimethoprim R0-CEP 1999-132-Rev 02 Welding GmbH & Co KG; D 20354 Hamburg
Trimethoprim R1-CEP 1994-017-Rev 02 Welding GmbH & Co KG; D 20354 HamburgVinblastine Sulphate R1-CEP 1992-005-Rev 00 Eli Lilly SA - Irish Branch; IRL County Cork
Substance Certificate Holder/Detenteur
Brain Heart Infusion R0-CEP 2000-267-Rev 00 BD Diagnostic Systems ; USA 21152 Sparks
Gelatin, Alkaline-Processed
Chromium-Tanned Hide Gelatin-
SPA
R0-CEP 2000-113-Rev 02 Croda Colloids Ltd; GB WA8 8UB Widnes,
Cheshire
Gelatin, Acid bone gelatin R0-CEP 2000-227-Rev 03 Croda Colloids Ltd; GB WA8 8UB Widnes,
Cheshire
Gelatin, Limed bone gelatin R0-CEP 2000-228-Rev 03 Croda Colloids Ltd; GB WA8 8UB Widnes,
Cheshire
Gelatin limed hide gelatin R0-CEP 2000-386-Rev 00 Gelita Group (DGF Stoess, Kind & Knox), D-
69412 Eberbach
Gelatin US Enzymatic BoneGelatin Hydrolysate
R0-CEP 2002-153-Rev 01 Gelita Group; D 69412 Eberbach
Gelatin acid bone gelatin,
American origin
R0-CEP 2000-139-Rev 05 PB Gelatins; B 1800 Vilvoorde
Gelatin acid bone gelatin,
American origin
R0-CEP-2000-026-Rev 03 Rousselot SAS; F 92641 Boulogne Billancourt
Cedex
Gelatin acid bone gelatin,including NaOH treatment
R0-CEP-2001-411-Rev 02 Rousselot SAS; F 92641 Boulogne BillancourtCedex
Gelatin; / Acid bone gelatin;
(French [European] origin)
R0-CEP 2000-030-Rev 00 Skw Biosystems; F 92641 Boulogne Billancourt
Cedex

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 41/176
Effect of temperature on plasma freezing
Scientific NotesThe Secretariat of the European Pharmacopoeia recalls that articles reflect the authors’ opinions. In order that all opinions are taken into account, concerned users and readers of Pharmeuropa are invited to send their commentsto the Secretariat. Readers are reminded that details regarding contributions made to Pharmeuropa are cited onthe rear cover of this issue.
M.I. Bravo, S. Grancha, J.I. Jorquera
The European Pharmacopoeia monograph on Human plasma for fractionation does not define the freezing processtime but does define the freezing temperature ( 30 °C or below). Initial freezing conditions are crucial for the quality of plasma. These conditions were intended to preserve labile proteins such as fVIII, but they can also be considered favourable for the plasma quality in general.
This study evaluates the way the industrial plasma freezing affects labile coagulation factors. We have studied the
freezing of plasma in industrial-size chambers at temperatures close to
30 °C,
25 °C and
20 °C, and the possibledifferences between performing the freezing process in a chamber or in a freezer, in order to elucidate whether or not these parameters affect the quality of plasma.
For this study, plasma bottles were frozen in industrial chambers set at 30 °C, 25 °C and 20 °C, and in a freezer set at 20 °C. The freezing rates were followed by means of probes in plasma control bottles. From this plasma,coagulation factors (fVIII, fIX and fibrinogen) were analysed before and after freezing, and cryoprecipitate was obtained in some cases. Statistically significant differences exist in fVIII:C recovery in thawed plasma between freezing at 30 °C and at 20 °C (n = 11; 85.4 ± 4.3 % versus 74.6 ± 6.0 % (chamber) or 79.3 ± 6.3 % (freezer)). There is nodifference between 30 °C and 25 °C, or between freezing at 20 °C in a chamber or in a freezer. No significant loss of activity in thawed plasma is observed for fIX and fibrinogen at 25 °C or 20 °C versus 30 °C. The fVIII and vWF recovery in cryoprecipitates does not show differences (464.2 IU fVIII/ml at 30 °C, 446.7 IU fVIII/ml at 25 °C, and 475.8 IU fVIII/ml at 20 °C).
The results obtained from this study support that plasma might also be frozen at 25 °C or below without any impact onits quality, and that sporadic and short term deviations, from 30 °C or below up to 25 °C, in the currently required
freezing temperature, would not have an effect on the labile factors recovery.
Plasma, freezing, coagulation factor, cryoprecipitate.
Currently, the European Pharmacopoeia monograph onHuman plasma for fractionation [1] defines: ‘When obtainedby plasmapheresis, plasma intended for the recovery of proteins that are labile in plasma is frozen by coolingrapidly at 30 °C or below as soon as possible and at thelatest within 24 h of collection.’ The monograph on Humanplasma for fractionation does not define the freezing processtime but does define the freezing temperature (‘ 30 °Cor below’); in the last revision, the concept ‘in a chamber’was added in order to avoid measuring the temperature inthe plasma. But this last proposal may be open to different interpretations because it does not explicitly indicate thecore plasma temperature that plasma should reach before it is stored at 20 °C (storage conditions).
Once frozen, the monograph defines the plasma storagetemperature at 20 °C or below. The DGTI multicentertrial initiated by the section ‘Blood Plasma Constituents’determined that the stability of frozen fresh plasma (FFP)during 2 and 3 years of storage at 20 °C, 25 °C, 30 °C,and 40 °C shows no detectable protein changes in theFFP [2].
Initial freezing conditions are crucial for the quality of plasma. Early studies [3], assessing the preservation of labile
proteins such as coagulation factor VIII (fVIII), show that thefreezing conditions should be such that at a low surroundingtemperature (e.g. 40 °C in those experiments) freezing tothe core of the plasma container is reached as rapidly aspossible. The study of Åkerblom et al . [4], found a highlysignificant difference of fVIII activity comparing freezing at 20 °C with freezing within 40 minutes at 40 °C. Thissuggests that the freezing should proceed in a short time at temperatures below the theoretical eutectic point ( 23 °Cfor plasma) [5, 6]. These freezing conditions were shown toincrease the yield of labile products such as fVIII, but they
can also be considered favourable for the plasma quality ingeneral.This study evaluates the way the industrial plasma freezingaffects labile coagulation factors. When a chamber is set at atemperature of 30 °C or below, it might happen that thistemperature temporarily rises slightly over the target duringthe day due to opening, loading of the equipment etc. Thereis no scientific data available stating that a difference existsbetween the plasma freezing performed at temperaturesbelow 30 °C or at temperatures slightly higher than 30 °C. For this reason, we have also studied the plasmafreezing in industrial-size chambers at temperatures close to 30 °C, 25 °C and 20 °C. Finally, possible differencesbetween performing the freezing process in a chamber or
M.I. Bravo. (Corresponding author: e-mail: [email protected]), Instituto Grifols, S.A. Pol. Levante. Can Guasch, 2. 08150 Parets del Vallès,Barcelona, SpainS. Grancha. Instituto Grifols, S.A. Pol. Levante. Can Guasch, 2. 08150 Parets del Vallès, Barcelona, Spain J.I. Jorquera. Instituto Grifols, S.A. Pol. Levante. Can Guasch, 2. 08150 Parets del Vallès, Barcelona, Spain
© Pharmeuropa Vol 18, No. 1, January 2006 41

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 42/176
Effect of temperature on plasma freezing
in a freezer were studied, to elucidate whether or not theincrease in temperature occurring when loading a small-sizefreezer affects the plasma quality.
Plasmapheresis plasma for fractionation was collected inbottles of approximately 800 ml of plasma volume containingsodium citrate as anticoagulant. All plasma was introducedinto a chamber at 30 °C or below immediately aftercollection (within 30 min). The freezing process in theplasmapheresis centres takes less than 24 h for the plasmato reach 30 °C or below.The bottles of plasma were thawed in a water bath at 37 °C, the content was homogenised by gentle stirring and,immediately after, plasma was assayed without additionalfreezing for fVIII and factor IX (fIX) activity, and fibrinogen(clottable protein). Aliquots of all samples were separatedand immediately frozen at 70 °C or below. Occasionally,these aliquots were used to confirm results. No apparent fVIII loss happened because of the additional freezing of these samples.
Plasma was then frozen under different study conditions( 30 °C as control, 25 °C and 20 °C). Two independent runs of freezing at 25 °C (n = 11 bottles each) wereperformed on different days. Once the core plasmatemperature was obtained (as measured with thermocoupleprobes in control plasma units), the plasma bottles weremaintained at this temperature for a period of 6-30 h, andthen changed to 70 °C or below until tested (within5 days).Some bottles (n = 11) were used to assay the coagulationfactors content and others (n = 6) for cryoprecipitatepreparation.In order to assay fVIII, fIX and fibrinogen recovery at thedifferent freezing temperatures, plasma bottles stored at
70 °C or below were thawed again as described beforeand immediately assayed for the factors content. In thiscase, aliquots of all thawed bottles were also separated andimmediately frozen at 70 °C or below. Occasionally, thesealiquots were used to confirm the results.
FVIII activity was determined by a chromogenic
method in an automatic system with S-2765 substrate(N-a-Z-D-Arg-Gly-Arg-pNA) from Chromogenix; fIX activityby a coagulation method based in the determination of activated partial thromboplastin time, and the fibrinogen(clottable protein) by the von Clauss coagulation methodin an automatic system. Von Willebrand factor ristocetincofactor (vWF:RCo) was assayed by monitoring platelet aggregation using BC von Willebrand Reagent (Behring).
The plasma temperature was monitored by means of probesplaced in the middle of control plasma bottles, whichwere connected to a recorder (KM1242 from Comark, orTestostor® 171 from Logger).
Bottles (coming from room temperature: 23 ± 3 °C, afterbeing thawed in the water bath at 37 °C) were uniformlydistributed into the freezer ( 20 °C) or on the chambershelves ( 30 °C, 25 °C and 20 °C; Figure 1). The bottlesentered the chambers in 2 groups and the freezer in 4 groupsto minimise the impact on surrounding temperature; theevolution of plasma freezing was followed up in each groupby means of a probe placed in the center of a bottle.
Freezing at 30 °C was performed in a chamber with avolume of 2388 m3. The study at 25 °C and 20 °C wascarried out in a chamber with a volume of 87.52 m3. Anadditional study at 20 °C was performed in a freezer witha volume of 0.5 m3. At the time of this study, the chambers
Figure 1 – Photograph of the general set up of bottles and probes in the chambers
42 © Pharmeuropa Vol 18, No. 1, January 2006
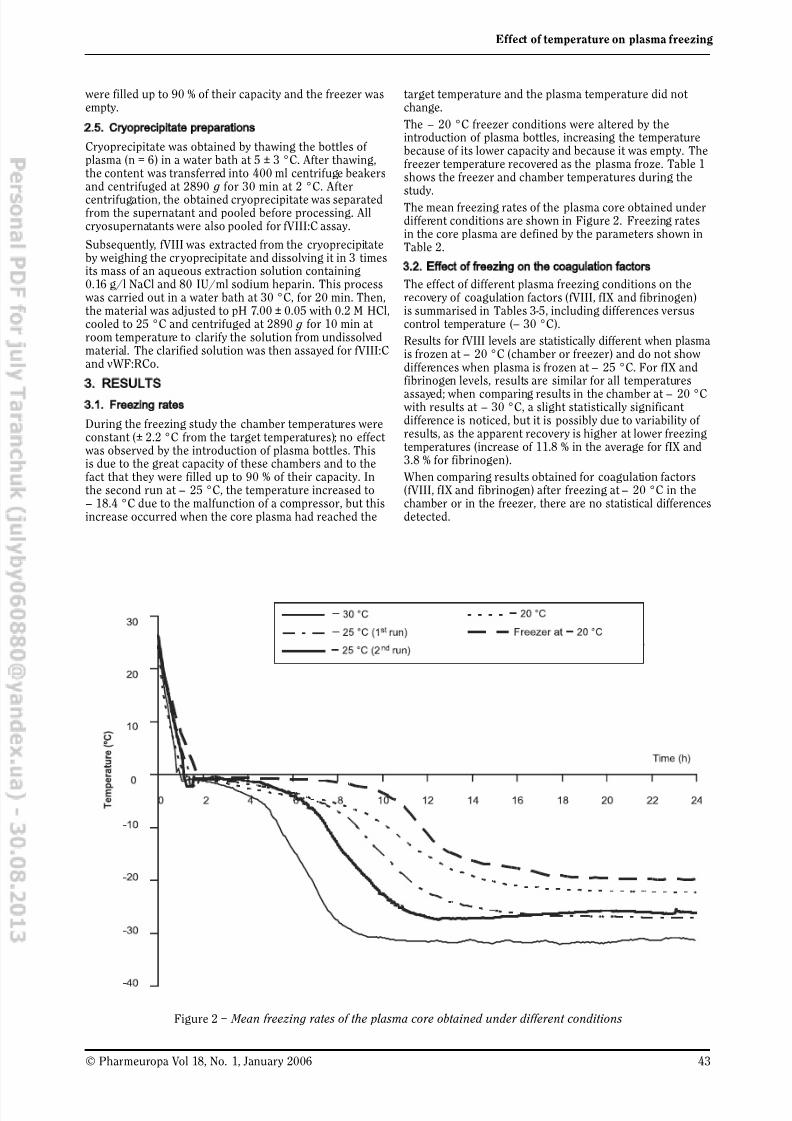
8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 43/176
Effect of temperature on plasma freezing
were filled up to 90 % of their capacity and the freezer wasempty.
Cryoprecipitate was obtained by thawing the bottles of plasma (n = 6) in a water bath at 5 ± 3 °C. After thawing,the content was transferred into 400 ml centrifuge beakersand centrifuged at 2890 g for 30 min at 2 °C. After
centrifugation, the obtained cryoprecipitate was separatedfrom the supernatant and pooled before processing. Allcryosupernatants were also pooled for fVIII:C assay.
Subsequently, fVIII was extracted from the cryoprecipitateby weighing the cryoprecipitate and dissolving it in 3 timesits mass of an aqueous extraction solution containing0.16 g/l NaCl and 80 IU/ml sodium heparin. This processwas carried out in a water bath at 30 °C, for 20 min. Then,the material was adjusted to pH 7.00 ± 0.05 with 0.2 M HCl,cooled to 25 °C and centrifuged at 2890 g for 10 min at room temperature to clarify the solution from undissolvedmaterial. The clarified solution was then assayed for fVIII:Cand vWF:RCo.
During the freezing study the chamber temperatures wereconstant (± 2.2 °C from the target temperatures); no effect was observed by the introduction of plasma bottles. Thisis due to the great capacity of these chambers and to thefact that they were filled up to 90 % of their capacity. Inthe second run at 25 °C, the temperature increased to 18.4 °C due to the malfunction of a compressor, but thisincrease occurred when the core plasma had reached the
target temperature and the plasma temperature did not change.
The 20 °C freezer conditions were altered by theintroduction of plasma bottles, increasing the temperaturebecause of its lower capacity and because it was empty. Thefreezer temperature recovered as the plasma froze. Table 1shows the freezer and chamber temperatures during the
study.The mean freezing rates of the plasma core obtained underdifferent conditions are shown in Figure 2. Freezing ratesin the core plasma are defined by the parameters shown inTable 2.
The effect of different plasma freezing conditions on therecovery of coagulation factors (fVIII, fIX and fibrinogen)is summarised in Tables 3-5, including differences versuscontrol temperature ( 30 °C).Results for fVIII levels are statistically different when plasmais frozen at 20 °C (chamber or freezer) and do not showdifferences when plasma is frozen at 25 °C. For fIX andfibrinogen levels, results are similar for all temperatures
assayed; when comparing results in the chamber at 20 °Cwith results at 30 °C, a slight statistically significant difference is noticed, but it is possibly due to variability of results, as the apparent recovery is higher at lower freezingtemperatures (increase of 11.8 % in the average for fIX and3.8 % for fibrinogen).When comparing results obtained for coagulation factors(fVIII, fIX and fibrinogen) after freezing at 20 °C in thechamber or in the freezer, there are no statistical differencesdetected.
Figure 2 – Mean freezing rates of the plasma core obtained under different conditions
© Pharmeuropa Vol 18, No. 1, January 2006 43
8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 44/176
Effect of temperature on plasma freezing
Cryoprecipitate was obtained from plasma frozen at different temperatures; the results are shown in Table 6. These resultsshow that fVIII and vWF recoveries in the cryoprecipitateare comparable for all temperatures assayed.
Freezing of plasma in a chamber at 30 °C or below is theusual process currently performed to meet the EuropeanPharmacopoeia’s requirements on plasma for fractionationintended for the production of labile coagulation factors.
As observed in the study, freezing of plasma in bottles (of approximately 800 ml) at a temperature of 30 °C wascomplete after 9 h. This period was prolonged up to 11-14 hat 25 °C, 14 h at 20 °C in the chamber and up to 20 hin the study performed at 20 °C in the freezer. Under allconditions studied, the freezing curves have similar profilesbut different maximum speeds (see Table 2).
This study was aimed at analysing whether or not thesedifferences in temperature during plasma freezing,involving longer freezing times and different final plasma
temperatures, affect coagulation factors (fVIII, fIX andfibrinogen) and the cryoprecipitate production. From theresults of fVIII recovery obtained after plasma freezing underthe studied conditions, it is observed that, although thedifference in fVIII recovery is not highly relevant (between85.5 % and 74.6 %), a statistically significant differenceexists between freezing at 30 °C and at 20 °C. Bycontrast, this difference does not exist when comparing theresults obtained at 30 °C with those obtained at 25 °C
(two separate runs). FIX and fibrinogen activity recoveriesare not decreased because of freezing at 25 °C or 20 °C.The results obtained for fVIII recovery in plasma (85.4 %at 30 °C; 85.5-83.8 % at 25 °C and 74.6-79.3 % at 20°C) are similar to those obtained by Åkerblom et al . [4]when freezing plasma in bags (200 ml) at temperatures of 20 °C and 40 °C, obtaining fVIII recoveries of 80 % and86 %, respectively.FVIII and vWF recoveries in the cryoprecipitates obtainedfrom plasma frozen at 30 °C, 25 °C or 20 °C donot show relevant differences. The fVIII yields obtained(475.8-446.7 IU fVIII/l plasma) are similar to those obtainedby Farrugia et al . [3] (425 IU fVIII/kg plasma) when freezingplasma quickly at 70 °C.
When the plasma freezing is performed at 25 °C, therecovery of labile coagulation factors in thawed plasmais identical to that observed when the plasma is frozenat 30 °C. The recovery of fVIII and vWF activities incryoprecipitates obtained from plasma frozen at 30 °C, 25 °C or 20 °C is also equivalent.
The results obtained from this study support that plasmamight also be frozen at 25 °C or below without any impact on its quality, and that sporadic and short term deviations,from 30 °C or below, up to 25 °C, in the currentlyrequired freezing temperature, would not have an effect onthe labile factors recovery.
Table 1 – Summary of freezer and chamber temperatures during the study
Chamber at
- 30 °C
A* B*
- 25 °C** (1st run) - 25°C**(2ndrun) - 20 °CFreezer at - 20 °C
Mean - 30.5 °C - 30.3 °C - 25.7 °C - 26.5 °C - 21.2 °C - 17.9 °C
Maximum - 27.9 °C - 28.7 °C - 24.8 °C - 25.8 °C - 20.3 °C - 11.1 °C
Minimum - 31.5 °C - 31.3 °C - 26.4 °C - 27.2 °C - 21.7 °C - 21.0 °C
* The A probe is placed at the top of the entrance door, and the B probe is placed at the inner part of the chamber; for this study the plasma wasplaced at the inner part of the chamber.
** The temperatures after the malfunction of the compressor are not taken into account.
Table 2 – Freezing rate parameters in the core plasma
Chamber at
Freezing rateparameters - 30 °C - 25 ° C (1st run) - 25 ° C (2nd run) - 20 °C Freezer at - 20 °C
Time to reach 0 °C(minutes)
60 80 70 75 105
Time to reach targettemperature (hours)
9 14 11 14 20
Maximum freezingrate* (°C/hour)
- 8.6 - 4.1 - 5.5 - 3.2 - 4.8
*Obtained by calculation from the linear part (gradient) of the freezing curve.
44 © Pharmeuropa Vol 18, No. 1, January 2006
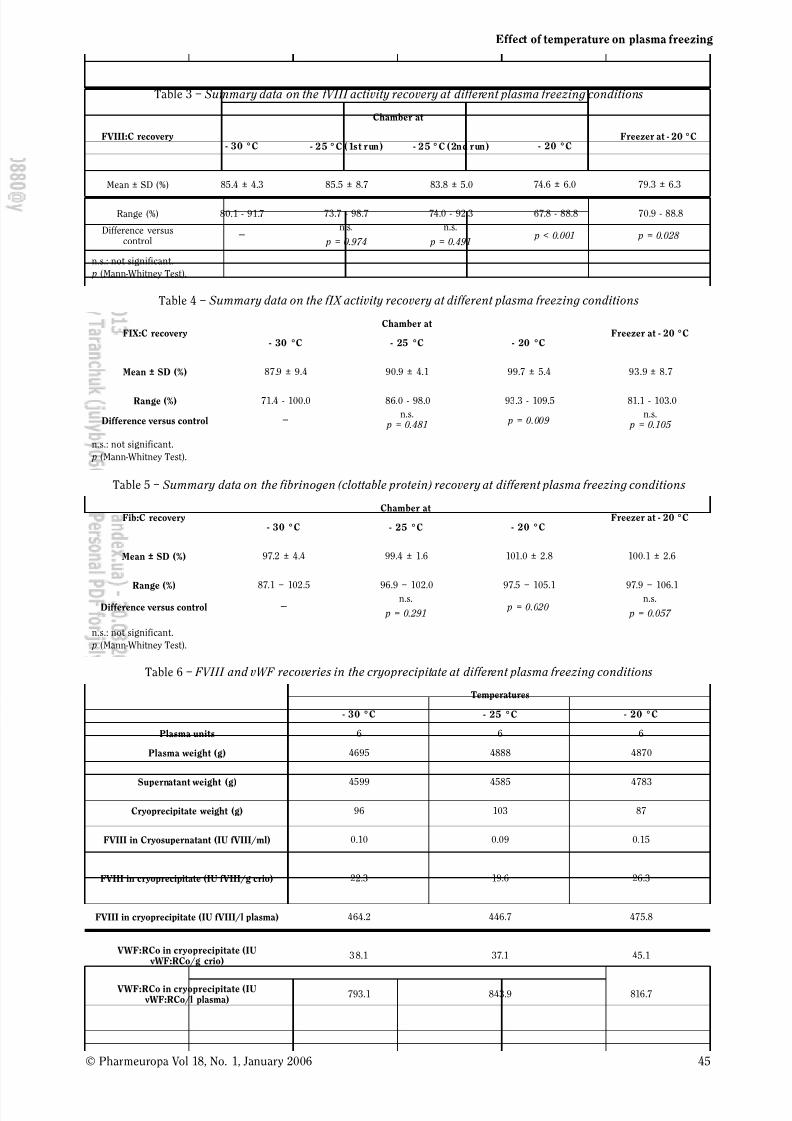
8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 45/176
Effect of temperature on plasma freezing
Table 3 – Summary data on the fVIII activity recovery at different plasma freezing conditions
Chamber at
FVIII:C recovery- 30 °C - 25 ° C (1st run) - 25 ° C (2nd run) - 20 °C
Freezer at - 20 °C
Mean ± SD (%) 85.4 ± 4.3 85.5 ± 8.7 83.8 ± 5.0 74.6 ± 6.0 79.3 ± 6.3
Range (%) 80.1 - 91.7 73.7 - 98.7 74.0 - 92.3 67.8 - 88.8 70.9 - 88.8
Difference versuscontrol
--- n.s.
p = 0.974
n.s.
p = 0.491 p < 0.001 p = 0.028
n.s.: not significant. p (Mann-Whitney Test).
Table 4 – Summary data on the fIX activity recovery at different plasma freezing conditions
Chamber atFIX:C recovery
- 30 °C - 25 °C - 20 °CFreezer at - 20 °C
Mean ± SD (%) 87.9 ± 9.4 90.9 ± 4.1 99.7 ± 5.4 93.9 ± 8.7
Range (%) 71.4 - 100.0 86.0 - 98.0 93.3 - 109.5 81.1 - 103.0
Difference versus control --- n.s. p = 0.481 p = 0.009
n.s. p = 0.105
n.s.: not significant. p (Mann-Whitney Test).
Table 5 – Summary data on the fibrinogen (clottable protein) recovery at different plasma freezing conditions
Chamber atFib:C recovery
- 30 °C - 25 °C - 20 °CFreezer at - 20 °C
Mean ± SD (%) 97.2 ± 4.4 99.4 ± 1.6 101.0 ± 2.8 100.1 ± 2.6
Range (%) 87.1 – 102.5 96.9 – 102.0 97.5 – 105.1 97.9 – 106.1Difference versus control ---
n.s.
p = 0.291 p = 0.020
n.s.
p = 0.057
n.s.: not significant. p (Mann-Whitney Test).
Table 6 – FVIII and vWF recoveries in the cryoprecipitate at different plasma freezing conditions
Temperatures
- 30 °C - 25 °C - 20 °C
Plasma units 6 6 6
Plasma weight (g) 4695 4888 4870
Supernatant weight (g) 4599 4585 4783
Cryoprecipitate weight (g) 96 103 87
FVIII in Cryosupernatant (IU fVIII/ml) 0.10 0.09 0.15
FVIII in cryoprecipitate (IU fVIII/g crio) 22.3 19.6 26.3
FVIII in cryoprecipitate (IU fVIII/l plasma) 464.2 446.7 475.8
VWF:RCo in cryoprecipitate (IUvWF:RCo/g crio)
38.1 37.1 45.1
VWF:RCo in cryoprecipitate (IUvWF:RCo/l plasma)
793.1 843.9 816.7
© Pharmeuropa Vol 18, No. 1, January 2006 45

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 46/176
Effect of temperature on plasma freezing
[1] Human plasma for fractionation, monograph 0853. Ph.Eur. Suppl 5.3. Strasbourg, France: Council of Europe2005.
[2] Kotitschke R, Morfeld F, Kirchmaier CM et al . Stabilityof fresh frozen plasma: results of 36-month storage at 20 °C, 25 °C, 30 °C and 40 °C. Multicenter
Study of the Section ’Blood Plasma Constituents’ of the DGTI. Infusion Therapy and Transfusion Medicine2000; 27:174-80.
[3] Farrugia A, Prowse C. Studies on the procurement of blood coagulation factor VIII: effects of plasma freezing
rate and storage conditions on cryoprecipitate quality. J Clin Pathol 1985; 38 :433-7.
[4] Åkerblom O, Bremme K, Dackland AL et al . Freezingtechnique and quality of fresh frozen plasma. Infusionstherapie 1992; 19:283-7.
[5] Carlebjörk G, Blombäck M, Philstedt P. Freezing of plasma and recovery of factor VIII. Transfusion 1986;
26:159-62.[6] Farrugia A, Hill R, Douglas S et al . Factor VIII/von
Willebrand factor levels in plasma frozen to 30 degreesC in air or halogenated hydrocarbons. Thromb Res1992; 68(1):97-102.
46 © Pharmeuropa Vol 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 47/176
Allopurinol
Draft monographs andgeneral texts for comment
IMPORTANT NOTICE
This section contains proposals for new and revised monographs and general texts, intended for inclusion in the European Pharmacopoeia and submitted for public comment. You can comment through the appropriate national authority at the address listed on the back cover page of the present issue. Readers from countries not members of the European Pharmacopoeia Commission should send their comments directly to the European Directorate for the Quality of Medicines. In order to facilitate the work of the Secretariats of the National Authorities and the European Directoratefor the Quality of Medicines who collect the comments, please mention in any correspondence the PA/PH referencenumber indicated at the beginning of each text. Comments that propose modifications of limits should be supported by analytical data obtained on a significant number of batches. Proposed changes of methodology should be supported by experimental results of a comparative trial of the method published in Pharmeuropa for comment and the proposed alternative. Only comments sent before 31 March 2006 will be considered before the final version is prepared.
It is stressed that these proposals have not been adopted by the European Pharmacopoeia Commission and must not be regarded as official standards.
In the case of proposals for revision, text to be deleted is crossed out and replacements or additions are underlined.
In certain cases, draft monographs require, for appropriate checking, the use of a reference material that is not yet commercially available; in exceptional circumstances, we will try to make the necessary substance available; please submit enquiries to the European Directorate for the Quality of Medicines.
Reference: PA/PH/Exp. 10A/T (05) 85 ANP
NOTE ON THE MONOGRAPH
Hydrazine can be used as starting material in the synthesisof allopurinol and a test has been introduced to limit any residue which could be present. A proposal was published
in Pharmeuropa 16.2. This new proposal includes an LC method that can limit hydrazine to a lower content.XXXX:0576
ALLOPURINOL
Allopurinolum
C5H4N4O M r 136.1
DEFINITION
1,5-Dihydro-4 H -pyrazolo[3,4-d ]pyrimidin-4-one.
Content : 98.0 97.0 per cent to 102.0 per cent (driedsubstance).
CHARACTERS
Appearance : white or almost white powder.
Solubility : very slightly soluble in water and in ethanol(96 per cent). It dissolves in dilute solutions of alkalihydroxides.
IDENTIFICATION First identification : B.
Second identification: A, C, D.
A. Dissolve 10 mg in 1 ml of a 4 g/l solution of sodiumhydroxide R and dilute to 100.0 ml with a 10.3 g/lsolution of hydrochloric acid R. Dilute 10.0 ml of this solution to 100.0 ml with a 10.3 g/l solution of hydrochloric acid R. Examined between 220 nm and350 nm ( 2.2.25 ), the solution shows an absorptionmaximum at 250 nm and an absorption minimum at 231 nm. The ratio of the absorbance measured at theabsorption minimum at 231 nm to that measured at theabsorption maximum at 250 nm is 0.52 to 0.62.
B. Infrared absorption spectrophotometry ( 2.2.24). Pr epar ati on : discs.
Comparison : allopurinol CRS .C. Dissolve 0.3 g in 2.5 ml of dilute sodium hydroxide
solution R and add 50 ml of water R. Add slowly andwith shaking 5 ml of silver nitrate solution R1. A whiteprecipitate is formed which does not dissolve on theaddition of 5 ml of ammonia R.
D. Thin-layer chromatography ( 2.2.27 ).
Test solution. Dissolve 20 mg of the substance to beexamined in concentrated ammonia R and dilute to10 ml with the same solvent.
Reference solution. Dissolve 20 mg of allopurinol CRS in concentrated ammonia R and dilute to 10 ml with thesame solvent.
Plate : TLC silica gel F 254 plate R.
Mobile phase : anhydrous ethanol R, methylenechloride R (40:60 V/V ).
Application : 10 μl.
Development : over 2/3 of the plate.
Drying : in air.
Detection : examine in ultraviolet light at 254 nm.
Re sults : the principal spot in the chromatogram obtainedwith the test solution is similar in position and size tothe principal spot in the chromatogram obtained withthe reference solution.
© PHARMEUROPA Vol. 18, No. 1, January 2006 47

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 48/176
Allopurinol
TESTS
Related substances. Liquid chromatography ( 2.2.29). Pr epar e the solutions immedi at ely befor e use. Use freshly prepared solutions, and store and inject them at 8 °C,using a cooled autosampler.
Test solution (a). Dissolve 25.0 mg of the substance to beexamined in 2.5 ml of a 4 g/l solution of sodium hydroxide R
and dilute immediately to 50.0 ml with the mobile phase.Test solution (b). Dissolve 20.0 mg of the substance to beexamined in 5.0 ml of a 4 g/l solution of sodium hydroxide Rand dilute immediately to 250.0 ml with the mobile phase.
Reference solution (a). Dilute 2.0 ml of test solution (a)to 100.0 ml with the mobile phase. Dilute 5.0 ml of thissolution to 100.0 ml with the mobile phase.
Reference solution (b). Dissolve 5.0 mg of allopurinol impurity A CRS , 5.0 mg of allopurinol impurity B CRS and5.0 mg of allopurinol impurity C CRS in 5.0 ml of a 4 g/lsolution of sodium hydroxide R and dilute immediately to100.0 ml with the mobile phase. Dilute 1.0 ml of this solutionto 100.0 ml with the mobile phase.
Reference solution (c). Dissolve 20.0 mg of allopurinol CRS in 5.0 ml of a 4 g/l solution of sodium hydroxide R anddilute immediately to 250.0 ml with the mobile phase.
Column :
— size : l = 0.25 m, Ø = 4.6 mm;
— stationary phase : octadecylsilyl silica gel for chromatography R (5 μm)(1).
Mobile phase : 1.25 g/l solution of potassium dihydrogen phosphate R.
Flow rate : 1.4 ml/min.
Detection : spectrophotometer at 230 nm.
Injection : 20 μl of test solution (a) and reference solutions (a)
and (b). Run time : twice the retention time of allopurinol.
Elution order : impurity A, impurity B, impurity C,allopurinol.
Retention time : allopurinol = about 10 min.
System suitability : reference solution (b):
— resolution : minimum 1.1 between the peaks due toimpurities B and C.
Limits :
— impurity A : not more than twice the area of the principalpeak in the chromatogram obtained with referencesolution (a) (0.2 per cent);
— impurity B : not more than the area of the principal peakin the chromatogram obtained with reference solution (a)(0.1 per cent);
— impurity C : not more than the area of the correspondingpeak in the chromatogram obtained with referencesolution (b) (0.1 per cent);
— unspecified impurities : for each impurity, not morethan the area of the principal peak in the chromatogramobtained with reference solution (a) (0.10 per cent);
— total of impurities other than A, B and C : not more than3 times the area of the principal peak in the chromatogramobtained with reference solution (a) (0.3 per cent) ;
— disregard limit : 0.5 times the area of the principal peakin the chromatogram obtained with reference solution (a)
(0.05 per cent).
Impurities D and E. Liquid chromatography ( 2.2.29). P r epar e the soluti ons immediat ely befor e use. Use freshly prepared solutions, and store and inject them at 8 °C,using a cooled autosampler.
Solution A : 1.25 g/l solution of potassium dihydrogen phosphate R.
Test solution. Dissolve 50.0 mg of the substance to be
examined in 5.0 ml of a 4 g/l solution of sodium hydroxide Rand dilute immediately to 100.0 ml with solution A.
Reference solution. Dissolve 5.0 mg of allopurinol impurity D CRS and 5.0 mg of allopurinol impurity E CRS in 5.0 ml of a 4 g/l solution of sodium hydroxide R anddilute immediately to 100.0 ml with solution A. Dilute 1.0 mlof this solution to 100.0 ml with solution A.
Column :— size : l = 0.05 m, Ø = 4.6 mm;
— stationary phase : base-deactivated octadecylsilyl silica gel for chromatography R (3 μm)(2).
Mobile phase : methanol R, 1.25 g/l solution of potassiumdihydrogen phosphate R (10:90 V/V ).
Flow rate : 2 ml/min. Detection : spectrophotometer at 230 nm.
Injection : 20 μl.
Run time : 1.5 times the retention time of impurity E.
Retention times : impurity D = about 3.6 min;impurity E = about 4.5 min.
System suitability : reference solution:— resolution : minimum 2.0 between the peaks due to
impurities D and E.
Limits :
— impurity D : not more than the area of the correspondingpeak in the chromatogram obtained with the referencesolution (0.1 per cent),
— impurity E : not more than the area of the correspondingpeak in the chromatogram obtained with the referencesolution (0.1 per cent).
Impurity F. Liquid chromatography ( 2.2.29).
Under the following conditions, any hydrazine in the samplereacts with benzaldehyde to give benzalazine.
Solvent mixture. Mix equal volumes of dilute sodiumhydroxide solution R and methanol R.
Solution A. Dissolve 2.0 g of benzaldehyde R in the solvent mixture and dilute to 50.0 ml with the solvent mixture.
Test solution. Dissolve 250.0 mg of the substance tobe examined in 5 ml of the solvent mixture. Add 4 mlof solution A, mix and allow to stand for 2.5 h at roomtemperature. Add 5.0 ml of hexane R and shake for 1 min.Allow the layers to separate and use the upper layer.
Reference solution. Dissolve 10.0 mg of hydrazine sulphate R in the solvent mixture by sonicating for about 2 min and dilute to 50.0 ml with the solvent mixture. Dilute1.0 ml to 20.0 ml with the solvent mixture. Dilute 1.0 ml of this solution to 20.0 ml with the solvent mixture. To 5.0 mlof the solution obtained, add 4 ml of solution A, mix andallow to stand for 2.5 h at room temperature. Add 5.0 ml of hexane R and shake for 1 min. Allow the layers to separateand use the upper layer.
Blank solution. To 5 ml of the solvent mixture add 4 mlof solution A, mix and allow to stand for 2.5 h at roomtemperature. Add 5.0 ml of hexane R and shake for 1 min.
Allow the layers to separate and use the upper layer.
(1) Hypersil ODS is suitable.(2) Hypersil BDS C18 is suitable.
48 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 49/176
Bisoprolol hemifumarate
Column :
— size : l = 0.250 m, Ø = 4.0 mm;
— stationary phase : cyanosilyl silica gel for chromatography R (5 μm) with a pore size of 100 nm(3) ;
— temperature : 30 °C.
Mobile phase : 2-propanol R, hexane R (5:95 V/V ).
Flow rate : 1.5 ml/min.
Detection : spectrophotometer at 310 nm.
Injection : 20 μl.
Relative retention with reference to benzaldehyde (retentiontime = about 2.8 min): benzalazine = about 0.8.
System suitability : reference solution:
— resolution : minimum 2 between the peaks due tobenzalazine and benzaldehyde;
— signal-to-noise-ratio : minimum 20 for the peak due tobenzalazine.
Limit :
— impurity F : the area of the peak due to benzalazine inthe chromatogram obtained with the test solution isnot more than the area of the corresponding peak inthe chromatogram obtained with the reference solution(2.5 ppm).
Heavy metals ( 2.4.8 ): maximum 20 ppm.
1.0 g complies with test C. Prepare the reference solutionusing 2 ml of lead standard solution (10 ppm Pb) R.
Loss on drying ( 2.2.32 ): maximum 0.5 per cent, determinedon 1.000 g by drying in an oven at 100-105 °C.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAYLiquid chromatography ( 2.2.29) as described in the test forrelated substances with the following modification.
Injection : test solution (b) and reference solution (c).
Calculate the percentage content of C5H4N4O from thedeclared content of allopurinol CRS .
IMPURITIES
Specified impurities: A, B, C, D, E, F.
A. R1 = NH2, R2 = H: 5-amino-1 H -pyrazole-4-carboxamide,
B. R1 = NH2, R2 = CHO: 5-(formylamino)-1 H -pyrazole-4-carboxamide,
D. R1 = O-C2H5, R2 = H: ethyl 5-amino-1 H -pyrazole-4-carboxylate,
E. R1 = O-C2H5, R2 = CHO: ethyl 5-(formylamino)-1 H -pyrazole-4-carboxylate,
C. 5-(4 H -1,2,4-triazol-4-yl)-1 H -pyrazole-4-carboxamide,
F. H2N-NH2 : diazane (hydrazine).
Reference: PA/PH/Exp. 10B/T (01) 67 ANP
NOTE ON THE MONOGRAPH
Related substances tests A and B correspond to different impurity profiles. As indicated in general chapter 5.10.Control of impurities in substances for pharmaceutical use, only the test or tests relevant for the known impurity
profile need to be carried out. XXXX:1710
BISOPROLOL HEMIFUMARATE
Bisoprololi hemifumaras
C20H33NO6 M r 383.5
DEFINITION
( RS )-1-[4-[[2-(1-Methylethoxy)ethoxy]methyl]phenoxy]-3-[(1-methylethyl)amino]propan-2-ol hemifumarate.
Content : 99.0 per cent to 101.0 per cent (anhydroussubstance).
CHARACTERS
Appearance : white or almost white powder.
Solubility : very soluble in water, freely soluble in methanol.
It shows polymorphism.
IDENTIFICATIONInfrared absorption spectrophotometry ( 2.2.24).
Comparison : bisoprolol hemifumarate CRS .
If the spectra obtained in the solid state show differences,dissolve the substance to be examined and the referencesubstance separately in methanol R, evaporate to drynessand record new spectra using the residues.
TESTS
Related substances.
A. Impurities A, B, C, D, E and F. Liquid chromatography( 2.2.29).
Test solution. Dissolve 25 mg of the substance to be
examined in mobile phase A and dilute to 25.0 ml withmobile phase A.
(3) Lichrospher 100 CN is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 49
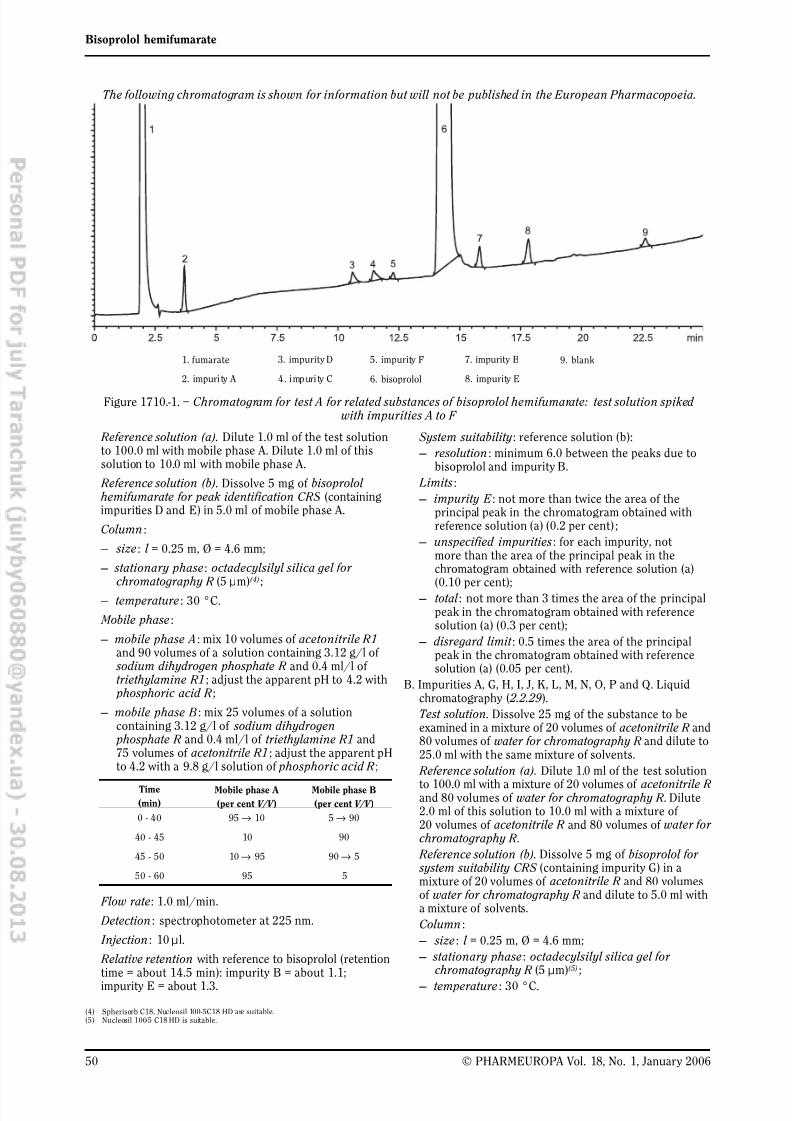
8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 50/176
Bisoprolol hemifumarate
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. fumarate 3. impurity D 5. impurity F 7. impurity B 9. blank
2. impurity A 4 . impurity C 6. bisoprolol 8. impurity E
Figure 1710.-1. – Chromatogram for test A for related substances of bisoprolol hemifumarate: test solution spiked with impurities A to F
Reference solution (a). Dilute 1.0 ml of the test solutionto 100.0 ml with mobile phase A. Dilute 1.0 ml of thissolution to 10.0 ml with mobile phase A.
Reference solution (b). Dissolve 5 mg of bisoprolol hemifumarate for peak identification CRS (containingimpurities D and E) in 5.0 ml of mobile phase A.
Column :
— size : l = 0.25 m, Ø = 4.6 mm;
— stationary phase : octadecylsilyl silica gel for chromatography R (5 μm)(4) ;
— temperature : 30 °C.
Mobile phase :
— mobile phase A : mix 10 volumes of acetonitrile R1and 90 volumes of a solution containing 3.12 g/l of sodium dihydrogen phosphate R and 0.4 ml/l of triethylamine R1 ; adjust the apparent pH to 4.2 with phosphoric acid R ;
— mobile phase B : mix 25 volumes of a solutioncontaining 3.12 g/l of sodium dihydrogen phosphate R and 0.4 ml/l of triethylamine R1 and75 volumes of acetonitrile R1 ; adjust the apparent pHto 4.2 with a 9.8 g/l solution of phosphoric acid R ;
Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 - 40 95 → 10 5 → 90
40 - 45 10 90
45 - 50 10 → 95 90 → 5
50 - 60 95 5
Flow rate : 1.0 ml/min.
Detection : spectrophotometer at 225 nm.
Injection : 10 μl.
Relative retention with reference to bisoprolol (retentiontime = about 14.5 min): impurity B = about 1.1;
impurity E = about 1.3.
System suitability : reference solution (b):— resolution : minimum 6.0 between the peaks due to
bisoprolol and impurity B. Limits :— impurity E : not more than twice the area of the
principal peak in the chromatogram obtained withreference solution (a) (0.2 per cent) ;
— unspecified impurities : for each impurity, not more than the area of the principal peak in thechromatogram obtained with reference solution (a)(0.10 per cent);
— total : not more than 3 times the area of the principalpeak in the chromatogram obtained with referencesolution (a) (0.3 per cent);
— disregard limit : 0.5 times the area of the principalpeak in the chromatogram obtained with referencesolution (a) (0.05 per cent).
B. Impurities A, G, H, I, J, K, L, M, N, O, P and Q. Liquidchromatography ( 2.2.29).Test solution. Dissolve 25 mg of the substance to beexamined in a mixture of 20 volumes of acetonitrile R and80 volumes of water for chromatography R and dilute to25.0 ml with the same mixture of solvents. Reference solution (a). Dilute 1.0 ml of the test solution
to 100.0 ml with a mixture of 20 volumes of acetonitrile Rand 80 volumes of water for chromatography R. Dilute2.0 ml of this solution to 10.0 ml with a mixture of 20 volumes of acetonitrile R and 80 volumes of water for chromatography R. Reference solution (b). Dissolve 5 mg of bisoprolol for system suitability CRS (containing impurity G) in amixture of 20 volumes of acetonitrile R and 80 volumesof water for chromatography R and dilute to 5.0 ml witha mixture of solvents.Column :— size : l = 0.25 m, Ø = 4.6 mm;— stationary phase : octadecylsilyl silica gel for
chromatography R (5 μm)(5) ;
— temperature : 30 °C.
(4) Spherisorb C18, Nucleosil 100-5C18 HD are suitable.(5) Nucleosil 100-5 C18 HD is suitable.
50 © PHARMEUROPA Vol. 18, No. 1, January 2006
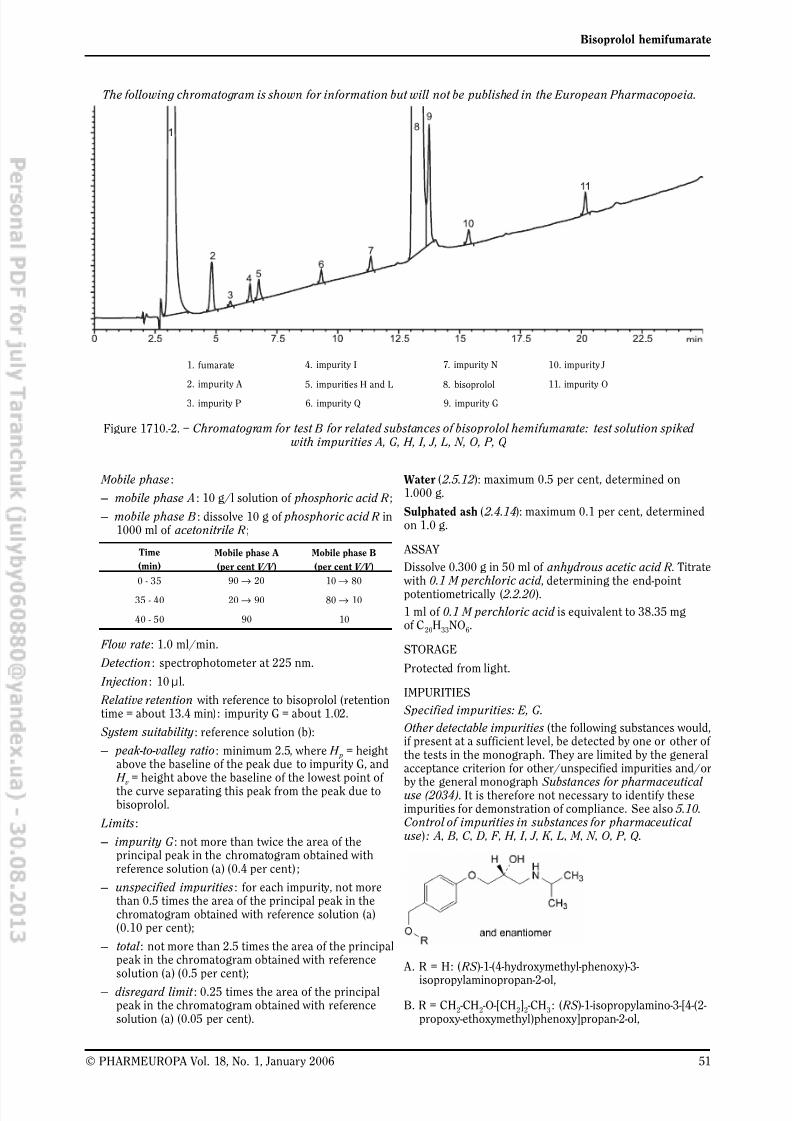
8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 51/176
Bisoprolol hemifumarate
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. fumarate 4. impurity I 7. impurity N 10. impurity J
2. impurity A 5. impurities H and L 8. bisoprolol 11. impurity O
3. impurity P 6. impurity Q 9. impurity G
Figure 1710.-2. – Chromatogram for test B for related substances of bisoprolol hemifumarate: test solution spiked with impurities A, G, H, I, J, L, N, O, P, Q
Mobile phase :
— mobile phase A : 10 g/l solution of phosphoric acid R ;
— mobile phase B : dissolve 10 g of phosphoric acid R in1000 ml of acetonitrile R ;
Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 - 35 90 → 20 10 → 80
35 - 40 20 → 90 80 → 10
40 - 50 90 10
Flow rate : 1.0 ml/min.
Detection : spectrophotometer at 225 nm.
Injection : 10 μl.
Relative retention with reference to bisoprolol (retentiontime = about 13.4 min) : impurity G = about 1.02.
System suitability : reference solution (b):
— peak-to-valley ratio : minimum 2.5, where H p = height above the baseline of the peak due to impurity G, and H
v
= height above the baseline of the lowest point of the curve separating this peak from the peak due tobisoprolol.
Limits :
— impurity G : not more than twice the area of theprincipal peak in the chromatogram obtained withreference solution (a) (0.4 per cent) ;
— unspecified impurities : for each impurity, not morethan 0.5 times the area of the principal peak in thechromatogram obtained with reference solution (a)(0.10 per cent);
— total : not more than 2.5 times the area of the principalpeak in the chromatogram obtained with referencesolution (a) (0.5 per cent);
— disregard limit : 0.25 times the area of the principalpeak in the chromatogram obtained with referencesolution (a) (0.05 per cent).
Water ( 2.5.12 ): maximum 0.5 per cent, determined on1.000 g.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAY
Dissolve 0.300 g in 50 ml of anhydrous acetic acid R. Titratewith 0.1 M perchloric acid , determining the end-point potentiometrically ( 2.2.20).1 ml of 0.1 M perchloric acid is equivalent to 38.35 mgof C20H33NO6.
STORAGE
Protected from light.
IMPURITIES
Specified impurities: E, G.
Other detectable impurities (the following substances would,if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the generalacceptance criterion for other/unspecified impurities and/or
by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify theseimpurities for demonstration of compliance. See also 5.10.Control of impurities in substances for pharmaceutical use) : A, B, C, D, F, H, I, J, K, L, M, N, O, P, Q.
A. R = H: ( RS )-1-(4-hydroxymethyl-phenoxy)-3-
isopropylaminopropan-2-ol,B. R = CH2-CH2-O-[CH2]2-CH3 : ( RS )-1-isopropylamino-3-[4-(2-
propoxy-ethoxymethyl)phenoxy]propan-2-ol,
© PHARMEUROPA Vol. 18, No. 1, January 2006 51
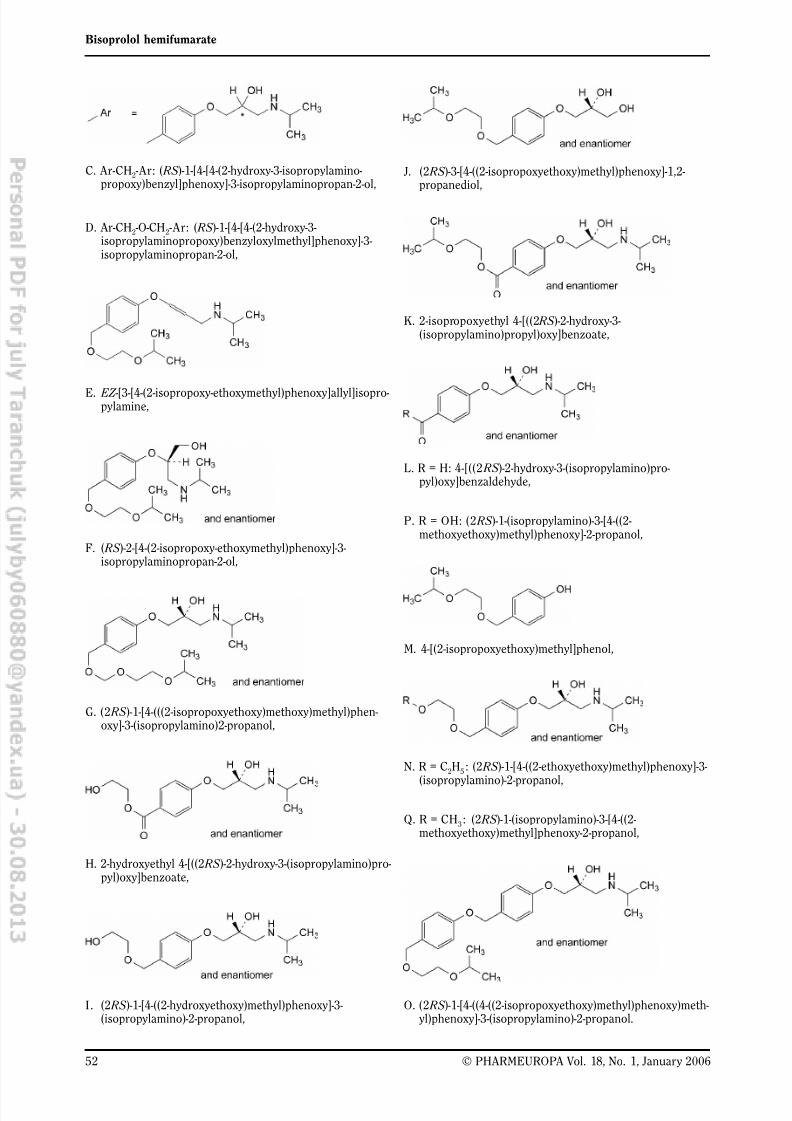
8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 52/176
Bisoprolol hemifumarate
C. Ar-CH2-Ar: ( RS )-1-[4-[4-(2-hydroxy-3-isopropylamino-propoxy)benzyl]phenoxy]-3-isopropylaminopropan-2-ol,
D. Ar-CH2-O-CH2-Ar: ( RS )-1-[4-[4-(2-hydroxy-3-isopropylaminopropoxy)benzyloxylmethyl]phenoxy]-3-isopropylaminopropan-2-ol,
E. EZ -[3-[4-(2-isopropoxy-ethoxymethyl)phenoxy]allyl]isopro-pylamine,
F. ( RS )-2-[4-(2-isopropoxy-ethoxymethyl)phenoxy]-3-isopropylaminopropan-2-ol,
G. (2 RS )-1-[4-(((2-isopropoxyethoxy)methoxy)methyl)phen-oxy]-3-(isopropylamino)2-propanol,
H. 2-hydroxyethyl 4-[((2 RS )-2-hydroxy-3-(isopropylamino)pro-pyl)oxy]benzoate,
I . (2 RS )-1-[4-((2-hydroxyethoxy)methyl)phenoxy]-3-(isopropylamino)-2-propanol,
J. (2 RS )-3-[4-((2-isopropoxyethoxy)methyl)phenoxy]-1,2-propanediol,
K. 2-isopropoxyethyl 4-[((2 RS )-2-hydroxy-3-(isopropylamino)propyl)oxy]benzoate,
L. R = H: 4-[((2 RS )-2-hydroxy-3-(isopropylamino)pro-pyl)oxy]benzaldehyde,
P. R = OH: (2 RS )-1-(isopropylamino)-3-[4-((2-methoxyethoxy)methyl)phenoxy]-2-propanol,
M. 4-[(2-isopropoxyethoxy)methyl]phenol,
N. R = C2H5 : (2 RS )-1-[4-((2-ethoxyethoxy)methyl)phenoxy]-3-
(isopropylamino)-2-propanol,
Q. R = CH3 : (2 RS )-1-(isopropylamino)-3-[4-((2-methoxyethoxy)methyl]phenoxy-2-propanol,
O. (2 RS )-1-[4-((4-((2-isopropoxyethoxy)methyl)phenoxy)meth-yl)phenoxy]-3-(isopropylamino)-2-propanol.
52 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 53/176
Cefamandole nafate
Reference: PA/PH/Exp. 7/T (05) 61 ANP
NOTE ON THE MONOGRAPH
The expression of content has been revised in order toreflect the fact that cefamandole is not to be considered as an impurity but as a participant to the activity of the substance. A new acceptance range for specific optical
rotation has been included. Readers are kindly requested to send information onthe relative retentions of impurities A, C, D and E to theSecretariat.
XXXX:1402
CEFAMANDOLE NAFATE
Cefamandoli nafas
DEFINITIONSodium Mixture of sodium (6 R,7 R)-7-[[(2 R)-2-(formyloxy)-2-phenylacetyl]amino]-3-[[(1-methyl-1 H -tetrazol-5-yl)sulphanyl]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate and(6 R,7 R)-7-[[(2 R)-2-hydroxy-2-phenylacetyl]amino]-3-[[(1-methyl-1 H -tetrazol-5-yl)sulphanyl]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate (cefamandole), withsodium carbonate. Semi-synthetic product derived from afermentation product.
Content :
— cefamandole (C18H18N6O5S2) : 84.0 per cent to 93.0 percent (anhydrous and sodium carbonat e-free substance),
— cefamandole nafate (C19H17N6NaO6S2): 93.0 per cent to102.0 per cent (anhydrous and sodium carbonate-freesubstance), for the sum of the content of cefamandolenafate and cefamandole expressed as cefamandole nafate ;
— cefamandole (C18H18N6O5S2): maximum 9.5 per cent
(anhydrous and sodium carbonate-free substance);— sodium carbonate (Na2CO3): 4.8 per cent to 6.4 per cent.
CHARACTERS
Appearance : white or almost white powder.
Solubility : freely soluble in water, sparingly soluble inmethanol.
IDENTIFICATIONA. Infrared absorption spectrophotometry ( 2.2.24).
Preparation : discs.
Comparison : cefamandole nafate CRS .
B. It gives reaction (a) of sodium ( 2.3.1).
TESTSSolution S. Dissolve 2.5 g in carbon dioxide-free water Rand dilute to 25 ml with the same solvent.
Appearance of solution. Solution S is clear ( 2.2.1) and itsabsorbance ( 2.2.25 ) at 475 nm is maximum 0.03.
pH : 6.0 to 8.0 for solution S, measured after 30 min.
Specific optical rotation ( 2.2.7 ) : 25.0 to 33.0 35.0 to45.0 (anhydrous and sodium carbonate-free substance).Dissolve 1.00 g in acetate buffer solution pH 4.7 R anddilute to 10.0 ml with the same solvent.
Related substances. Liquid chromatography ( 2.2.29). Prepare the solutions immediately before use.
Solvent mixture. Mix 18 volumes of acetonitrile R and75 volumes of a 10 per cent V/V solution of triethylamine Rpreviously adjusted to pH 2.5 with phosphoric acid R.
Test solution. Dissolve 0.100 g of the substance to beexamined in the solvent mixture and dilute to 10.0 ml withthe solvent mixture.
Reference solution (a). Dilute 1 ml of the test solution to10 ml with the solvent mixture, then heat at 60 °C for 30 min.
Reference solution (b). Dilute 1.0 ml of the test solution to100.0 ml with the solvent mixture.
Column :— size : l = 0.25 m, Ø = 4.6 mm;— stationary phase: octadecylsilyl silica gel for
chromatography R (5 μm).
Mobile phase :— triethylamine phosphate solution : dissolve 2.0 g of
sodium pentanesulphonate R in 350 ml of water R,add 40 ml of triethylamine R, adjust to pH 2.5 with phosphoric acid R and dilute to 700 ml with water R ;
— mobile phase A: mix 1 volume of triethylamine phosphatesolution and 2 volumes of water R ;
— mobile phase B : mix equal volumes of triethylaminephosphate solution, methanol R and acetonitrile R ;
Time(min)
Mobile phase A(per cent V/V )
Mobile phase B(per cent V/V )
0 - 1 100 0
1 - 35 100 → 0 0 → 100
35 - 45 0 100
45 - 50 0 → 100 100 → 0
Flow rate : 1.5 ml/min.
Detection : spectrophotometer at 254 nm.
Injection : 20 μl loop injector.
Relative retention with reference to cefamandole nafate(retention time = about 24 min) : cefamandole = about 0.8.System suitability : reference solution (a):— resolution : minimum 5.0 between the peaks due to
cefamandole and cefamandole nafate.
Limits :— any impurity : for each impurity, not more than the area
of the principal peak in the chromatogram obtained withreference solution (b) (1.0 per cent) ;
— total : not more than 5 times the area of the principal peakin the chromatogram obtained with reference solution (b)(5.0 per cent);
— disregard limit : 0.1 times the area of the principal peakin the chromatogram obtained with reference solution (b)(0.1 per cent).
Cefamandole free acid : maximum 9.5 per cent (anhydrousand sodium carbonat e-free substance), determined by liquidchromat ography as described under Assay.
2-Ethylhexanoic acid ( 2.4.28 ): maximum 0.3 per cent m/m.
© PHARMEUROPA Vol. 18, No. 1, January 2006 53

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 54/176
Cefamandole nafate
Heavy metals ( 2.4.8 ): maximum 20 ppm.
1.0 g complies with test C. Prepare the reference solutionusing 2 ml of lead standard solution (10 ppm Pb) R.
Water ( 2.5.12 ): maximum 2.0 per cent, determined on0.500 g.
Bacterial endotoxins ( 2.6.14): less than 0.15 IU/mg, if
intended for use in the manufacture of parenteral dosageforms without a further appropriate procedure for theremoval of bacterial endotoxins.
ASSAY
Cefamandole Cefamandole nafate. Liquid chromatography( 2.2.29). Prepare the solutions immediately before use.
Test solution. Dissolve 50.0 mg of the substance to beexamined in the mobile phase and dilute to 100.0 ml withthe mobile phase.
Reference solution (a). Dissolve 50.0 mg of cefamandolenafate CRS in the mobile phase and dilute to 100.0 ml with
the mobile phase. Reference solution (b). Dilute 1 ml of the test solution to10 ml with the mobile phase, then heat at 60 °C for 30 min.
Column :
— size : l = 0.25 m, Ø = 4.6 mm;
— stationary phase : octadecylsilyl silica gel for chromatography R (5 μm).
Mobile phase : mix 25 volumes of acetonitrile R and75 volumes of a 10 per cent V/V solution of triethylamine Rpreviously adjusted to pH 2.5 with phosphoric acid R.
Flow rate : 1.0 ml/min.
Detection : spectrophotometer at 254 nm.
Injection : 20 μl loop injector.
System suitability :
— resolution : minimum 7.0 between the 2 principal peaks inthe chromatogram obtained with reference solution (b) ;
— repeatability : maximum relative standard deviationof 0.8 per cent after a series of single injections of aminimum 3 freshly prepared reference solutions (a).
Calculat e the percentage content of cefamandole(C18H18N6O5S2) from the sum of the contents of cefamandolenafate and cefamandole free acid using the declared cont ent s
of cef amand ol e nafat e CRS . 1 mg of cefamandole nafat e isequivalent to 0.9025 mg of cefamandole.Calculate the percentage content of cefamandole nafatefrom the sum of the content of cefamandole nafate andcefamandole expressed as cefamandole nafate using thedeclared content of cefamandole nafate CRS .
Sodium carbonate. Dissolve 0.500 g in 50 ml of water R.Titrate with 0.1 M hydrochloric acid , determining theend-point potentiometrically ( 2.2.20).
1 ml of 0.1 M hydrochloric acid is equivalent to 10.6 mgof Na2CO3.
STORAGE
In an airtight container, protected from light. If thesubstance is sterile, store in a sterile, airtight, tamper-proof container.
LABELLING
The label states:
— that the substance contains sodium carbonate;
— where applicable, that the substance is free from bacterialendotoxins.
IMPURITIES
A. (6 R,7 R)-7-[[(2 R)-2-(formyloxy)-2-phenylacetyl]amino]-3-methyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-
carboxylic acid (formylmandeloyl-7-amino-desacetoxy-cephalosporanic acid),
B. R = H : (6 R,7 R)-7-[[(2 R)-2-hydroxy-2-phenylacetyl]amino]-3-[[(1-methyl-1 H -t etrazol-5-yl)sulphanyl]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct -2-ene-2-carboxylic acid(cefamandole), deleted,
C. (6 R,7 R)-7-[[(2 R)-2-(acetyloxy)-2-phenylacetyl]amino]-3-[[(1-methyl-1 H -tetrazol-5-yl)sulphanyl]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid(O-acetylcefamandole),
D. 1-methyl-1 H -tetrazole-5-thiol,
E. (6 R,7 R)-7-[[(2 R)-2-(formyloxy)-2-phenylacetyl]amino]-3-[(acetyloxy)methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid (formylmandeloyl-7-ACA).
54 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 55/176
Chlortalidone
Reference: PA/PH/Exp. 10B/T (05) 99 ANP
NOTE ON THE MONOGRAPH
The TLC test for related substances has been replaced by an LC test which allows improved control of impurities.The same chromatographic method is proposed for theassay, as practical difficulties in the titration have beenreported by users. More details on the development work is presented in Pharmeuropa Scientific Notes 2005-1. Only the first identification has been kept as the substance isnot used in pharmacies. The appearance of solution test has been suppressed because no formulations for injectionexist on the market. The test for optical rotation has beendeleted as no data on the optical rotation of the singleenantiomer are available.
XXXX:0546
CHLORTALIDONE
Chlortalidonum
C14H11ClN2O4S M r 338.8
DEFINITION2-Chloro-5-[(1 RS )-1-hydroxy-3-oxo-2,3-dihydro-1 H -isoindol-1-yl]benzenesulphonamide.Content : 98.0 per cent to 102.0 per cent (dried substance).
CHARACTERS Appearance : white or yellowish-white powder.Solubility : practically very slightly soluble in water, soluble
in acetone and in methanol, slightly soluble in ethanol(96 per cent), practically insoluble in methylene chloride. It dissolves in dilute solutions of alkali hydroxides. Melting point : about 220 °C, with decomposition. It showspolymorphism.
IDENTIFICATION First i d entifi cati on: B.
Second id entific ation: A, C, D, E.
A. Dissolve 50.0 mg in ethanol (96 per c ent) R and diluteto 50.0 ml with the same solvent. Dilute 10.0 ml of this solution t o 100.0 ml with ethanol (96 per c ent) R.Examined between 230 nm and 340 nm ( 2.2.25 ), thesolution shows 2 absorption maxima, at 275 nm and
284 nm. The ratio of the absorbance measured at theabsorption maximum at 284 nm to that measured at theabsorption maximum at 275 nm is 0.73 t o 0.88.
B. Infrared absorption spectrophotometry ( 2.2.24). P r epar ati on : discs of pot assium br omi de R.Comparison : chlortalidone CRS .If the spectra obtained show differences, dissolve thesubstance to be examined and the reference substanceseparately in methanol R, evaporate to dryness andrecord new spectra using the residues.
C. Thin-layer chromatography ( 2.2.27 ).T est soluti on. Dissolve 10 mg of the substance t o beexamined in ac etone R and dilute t o 10 ml with the samesolvent. Refer ence solution ( a). Dissolve 10 mg of chl ort ali done CRS in ac et one R and dilute to 10 ml withthe same solvent.
Refer enc e soluti on (b). Dissolve 10 mg of chl ort ali doneCRS and 10 mg of hy dr ochl or othiazid eCRS in acet one R and dilut e to 10 ml with the same solvent.
Plate : TLC si li c a gel GF 25 4 plate R.
M obil e phase : water R, ethyl acetate R (1.5:98.5 V/V ).
Applic ation : 5 μl.
Devel opment : over a path of 10 cm. Drying : in air.
Det ection : examine in ultraviolet light at 254 nm.
Syst em suitability : ref erence solution (b):
— the chromat ogram obtained shows 2 clearly separat edspots.
Results : the principal spot in the chromatogram obtainedwith the test solution is similar in position and size tothe principal spot in the chromat ogram obtained withref erence solution (a).
D. Dissolve about 10 mg in 1 ml of sulphuric acid R. Anintense yellow colour develops.
E. It complies with the test f or optical rotation (see Tests).
TESTS
Appearance of solution. The solution is clear ( 2.2.1) andnot more intensely coloured than int ensity 6 of the range of ref erence solutions of the most appropriate colour ( 2.2.2, Method II ).
Dissolve 1.0 g in dilut e sodium hy dr oxid e soluti on R anddilut e t o 10 ml with the same solvent.
Acidity. Dissolve 1.0 g with heating in a mixture of 25 mlof acetone R and 25 ml of carbon dioxide-free water R.Cool. Titrate with 0.1 M sodium hydroxide, determining theend-point potentiometrically ( 2.2.20). Not more than 0.75 mlof 0.1 M sodium hydroxide is required.
Optical rotation ( 2.2.7 ) : 0.15° t o + 0.15°Dissolve 0.20 g in methanol R and dilute to 20 ml with thesame solvent.
Related substances. Examine by thin-layer chromatography( 2.2.27 ), using as the coating substance a suitable silica gelwith a fluorescent indicator having an optimal int ensity at 254 nm.
T est soluti on. Dissolve 0.2 g of the substance t o be examinedin a mixture of 1 volume of water R and 4 volumes of acetone R and dilut e t o 5 ml with the same mixture of solvents.
Ref er ence soluti on ( a). Dissolve 4 mg of chl ort alid oneimpurity B CRS and 4 mg of chl ort ali done CRS in a mixture
of 1 volume of wat er R and 4 volumes of ac etone R anddilut e t o 10 ml with the same mixture of solvents.
Ref er ence soluti on ( b). Dilute 1 ml of the test solution t o200 ml with a mixture of 1 volume of water R and 4 volumesof ac etone R.
Apply separat ely t o the plat e 5 μl of each solution.Develop over a path of 15 cm using a mixture of 5 volumesof toluene R, 10 volumes of xyl ene R, 20 volumes of conc entr ated ammonia R, 30 volumes of dioxan R and30 volumes of 2-pr opanol R. Allow the plat e to dry in acurrent of warm air and examine in ultraviolet light at 254 nm. In the chromatogram obtained with the t est solution: any spot corresponding to impurity B is not moreint ense than the corresponding spot in the chromatogram
obtained with ref erence solution (a) (1 per cent); andany spot, apart from the principal spot and the spot corresponding to impurity B, is not more intense thanthe spot in the chromat ogram obtained with ref erence
© PHARMEUROPA Vol. 18, No. 1, January 2006 55

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 56/176
Chlortalidone
solution (b) (0.5 per cent). The t est is not valid unless thechromatogram obtained with ref erence solution (a) shows2 clearly separat ed spots.
Related substances. Liquid chromatography ( 2.2.29).
Solvent mixture : a 2 g/l solution of sodium hydroxide R,mobile phase A, mobile phase B (2:48:50 V/V/V ).
Test solution. Dissolve 50.0 mg of the substance to beexamined in t he solvent mixture and dilute to 50.0 ml withthe solvent mixture.
Reference solution (a). Dilute 1.0 ml of the test solutionto 100.0 ml with the solvent mixture. Dilute 1.0 ml of thissolution to 10.0 ml with the solvent mixture.
Reference solution (b). Dissolve 5 mg of chlortalidone for peak identification CRS (containing impurities A, B, I, J, Kand L) in the solvent mixture and dilute to 50.0 ml with thesolvent mixture.
Reference solution (c). Dissolve 50.0 mg of chlortalidone CRS in the solvent mixture and diluteto 50.0 ml with the solvent mixture.
Column :
— size : l = 0.25 m, Ø = 4.6 mm;
— stationary phase : octylsilyl silica gel for chromatography R (5 μm)(6).
Temperature : 40 °C.
Mobile phase :
— mobile phase A : dissolve 1.32 g of ammonium phosphate R in about 900 ml of water R and adjust topH 5.5 with dilute phosphoric acid R ; dilute to 1000 mlwith water R ;
— mobile phase B : methanol R2 ;
Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 - 16 65 35
16 - 21 65 → 50 35 → 50
21 - 35 50 50
35 - 40 50 → 65 50 → 35
Flow rate : 1.4 ml/min.
Detection : spectrophotometer at 220 nm.
Injection : 20 μl of the test solution and referencesolutions (a) and (b).
Relative retention with reference to chlortalidone(retention time = about 7 min): impurity A = about 0.4;impurity B = about 0.7; impurity J = about 0.9;impurity K = about 2.7; impurity L = about 3.0;impurity I = about 4.2.
System suitability : reference solution (b):
— resolution : minimum 1.5 between the peaks due toimpurity J and chlortalidone.
Limits :— correction factors : for the calculation of content,multiply the peak areas of the following impurities bythe corresponding correction factor: impurity A = 1.4;impurity I = 1.5;
— impurity B : not more than 6 times the area of theprincipal peak in the chromatogram obtained withreference solution (a) (0.6 per cent) ;
— impurities J, K : for each impurity, not more than 3 timesthe area of the principal peak in the chromatogramobtained with reference solution (a) (0.3 per cent) ;
— impurity L : not more than the area of the principal peakin the chromatogram obtained with reference solution (a)(0.1 per cent);
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity A 4. chlortalidone 7. impurity Fb 10. impurity D 13. impurity I
2. impurity B 5. impurity E 8. impurity K 11. impurity C 14. impurity G
3. impurity J 6. impurity Fa 9. impurity L 12. impurity H
Figure 0546.-1 – Chromatogram of a sample of chlortalidone spiked with impurities
(6) ZORBAX SB-C8 is suitable.
56 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 57/176
Cimetidine
— unspecified impurities : for each impurity, not morethan the area of the principal peak in the chromatogramobtained with reference solution (a) (0.10 per cent);
— total : not more than 12 times the area of the principalpeak in the chromatogram obtained with referencesolution (a) (1.2 per cent);
— disregard limit : 0.5 times the area of the principal peak
in the chromatogram obtained with reference solution (a)(0.05 per cent).
Chlorides ( 2.4.4): maximum 350 ppm.
Triturate 0.3 g, add 30 ml of water R, shake for 5 min andfilter. 15 ml of the filtrate complies with the test. Preparethe standard using 10 ml of chloride standard solution(5 ppm Cl) R.
Loss on drying ( 2.2.32 ): maximum 0.5 per cent, determinedon 1.000 g by drying in an oven at 100-105 °C.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAY
Dissolve 0.200 g in 50 ml of ac etone R . In an atmosphere of nitrogen, titrat e with 0.1 M t etr abutyl ammonium hy dr oxi d e,determining the end-point potentiometrically ( 2.2.20). Carryout a blank titration.
1 ml of 0.1 M t etr abutylammonium hyd r oxid e is equivalent to 33.88 mg of C14H11ClN2O4S.
Liquid chromatography ( 2.2.29) as described in the test forrelated substances with the following modifications.
Injection : 20 μl of the test solutionand reference solution (c).
System suitability :
— repeatability : maximum relative standard deviation of 2.0 per cent after 6 injections of reference solution (c).
Calculate the percentage content of C14H11ClN2O4S from thedeclared content of chlortalidone CRS .
IMPURITIES
Specified impurities: A, B, C, D, E, F, G, H, I, J, K, L.
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : A, C, D, E, F, G, H, I.
A. R = H, R′ = OH: 2-(4-chloro-3-sulphobenzoyl)benzoic acid,
B. R = H, R′ = NH2 : 2-(4-chloro-3-sulphamoylbenzoyl)benzoicacid,
C. R = C2H5, R′ = NH2 : ethyl 2-(4-chloro-3-sulphamoylbenzoyl)benzoate,
I. R = CH(CH3)2, R′ = NH2 : 1-methylethyl2-(4-chloro-3-sulphamoylbenzoyl)benzoate,
D. R = OC2H5, R′ = SO2-NH2 : 2-chloro-5-[(1 RS )-1-ethoxy-3-
oxo-2,3-dihydro-1 H -isoindol-1-yl]benzenesulphonamide,E. R = H, R′ = SO2-NH2 : 2-chloro-5-[(1 RS )-3-oxo-2,3-dihydro-
1 H -isoindol-1-yl]benzenesulphonamide,
G. R = OH, R′ = Cl: (3 RS )-3-(3,4-dichlorophenyl)-3-hydroxy-2,3-dihydro-1 H -isoindol-1-one,
H. R = OCH(CH3)2, R ′ = SO2-NH2 : 2-chloro-5-[(1 RS )-1-(1-methylethoxy)-3-oxo-2,3-dihydro-1 H -isoindol-1-yl]benzenesulphonamide,
F. bis[2-chloro-5-(1-hydroxy-3-oxo-2,3-dihydro-1 H -isoindol-1-yl)benzenesulphonyl]amine,
J. impurity of unknown structure with a relative retentionof about 0.9,
K. impurity of unknown structure with a relative retentionof about 2.7,
L. impurity of unknown structure with a relative retentionof about 3.0.
Reference: PA/PH/Exp. 10C/T (05) 26 ANP
NOTE ON THE MONOGRAPH
The revision includes the replacement of the TLC for related substances by 2 LC methods which allow the control of substances synthesized by different routes and also cover the degradation products. It is also proposed to delete the second identification series as the substance is probably not used in pharmacies.
XXXX:0756
CIMETIDINE
Cimetidinum
C10H16N6S M r 252.3
DEFINITION
2-Cyano-1-methyl-3-[2-[[(5-methyl-1 H -imidazol-4-yl)methyl]sulphanyl]ethyl]guanidine.
Content : 98.5 per cent to 101.5 per cent (dried substance).CHARACTERS
Appearance : white or almost white powder.
© PHARMEUROPA Vol. 18, No. 1, January 2006 57

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 58/176
Cimetidine
Solubility : slightly soluble in water, soluble in ethanol(96 per cent), practically insoluble in methylene chloride. It dissolves in dilute mineral acids.
It shows polymorphism.
IDENTIFICATION
First i d entifi cati on: B.
Second id entific ation: A, C, D.A. Melting point ( 2.2.14): 139 °C to 144 °C. If necessary,
dissolve the substance to be examined in 2 -pr opanol R,evaporate t o dryness and determine the melting point again.
B. Infrared absorption spectrophotometry ( 2.2.24).
Comparison : cimetidine CRS .
If the spectra obtained in the solid state show differences,dissolve the substance to be examined and the referencesubstance separately in 2-propanol R, evaporate todryness and record new spectra using the residues.
C. Examine the chromat ograms obtained in the t est
f or related substances. The principal spot in thechromat ogram obtained with test solution (b) is similarin position, colour and size t o the principal spot in thechromat ogram obtained with ref erence solution (d).
D. Dissolve about 1 mg in a mixture of 1 ml of ethanol R and5 ml of a freshly prepared 20 g/l solution of ci tri c aci d Rin acetic anhydri d e R . Heat in a water-bath f or 10 min to15 min. A reddish-violet colour develops.
TESTS
Appearance of solution. The solution is clear ( 2.2.1) and not more intensely coloured than reference solution Y5 ( 2.2.2, Method II ).
Dissolve 3.0 g in 12 ml of 1 M hydrochloric acid and dilute
to 20 ml with water R.Impurity F. Liquid chromatography ( 2.2.29).
Test solution. Dissolve 20 mg of the substance to beexamined in the mobile phase and dilute to 50 ml with themobile phase.
Reference solution . Dissolve 4.0 mg of cimetidine CRS and4.0 mg of cimetidine impurity F CRS in methanol R anddilute to 50 ml with the same solvent. Dilute 1.0 ml of thissolution to 100.0 ml with the mobile phase.
Column :
— size : l = 0.25 m, Ø = 4.6 mm;
— stationary phase : octadecylsilyl silica gel for
chromatography R (5 μm)(7)
. Mobile phase : dissolve 661 mg of sodiumhexanesulphonate R in 760 ml of water R. Add240 ml of methanol R and mix. Adjust to pH 2.5 with phosphoric acid R.
Flow rate : 2.0 ml/min.
Detection : spectrophotometer at 220 nm.
Injection : 20 μl.
Run time : 25 min.
System suitability : reference solution:
— resolution : minimum 1.0 between the peaks due tocimetidine and impurity F;
— signal-to-noise ratio : minimum 10 for the peak due toimpurity F.
Limit :
— impurity F : not more than the area of the correspondingpeak in the chromatogram obtained with the referencesolution (0.2 per cent).
Related substances. Examine by thin-layer chromatography( 2.2.27 ), using silic a g el GF 254 R as the coating substance.
T est soluti on (a). Dissolve 0.50 g of the substance to beexamined in methanol R and dilut e to 10 ml with the samesolvent.
T est soluti on (b). Dilute 1 ml of t est solution (a) t o 10 mlwith methanol R.
Ref er ence soluti on ( a). Dilut e 1 ml of test solution (a) to100 ml with methanol R. Dilut e 20 ml of this solution t o100 ml with methanol R.
Ref er ence soluti on (b). Dilute 5 ml of ref erence solution (a)to 10 ml with methanol R.
Ref er ence soluti on (c). Dilut e 5 ml of ref erence solution (b)to 10 ml with methanol R.
Ref er ence soluti on ( d). Dissolve 10 mg of cimeti dine CRS in 2 ml of methanol R.
A. Apply separately t o the plat e 4 μl of each solution. Allowthe plate to stand f or 15 min in the chromatographictank saturat ed with vapour from the mobile phase whichconsist s of a mixture of 15 volumes of c onc entr at ed ammoni a R, 20 volumes of methanol R and 65 volumesof ethyl acet ate R and develop immediat ely over a pathof 15 cm using the same mixture of solvents. Dry theplat e in a stream of cold air, expose to iodine vapouruntil maximum contrast of the spots has been obtainedand examine in ultraviolet light at 254 nm. Any spot in the chromat ogram obtained with t est solution (a),apart from the principal spot, is not more int ense thanthe principal spot in the chromat ogram obtained withref erence solution (a) (0.2 per cent) and not more than
two such spots are more int ense than the principalspot in the chromat ogram obtained with ref erencesolution (b) (0.1 per cent). The t est is not valid unless thechromat ogram obtained with ref erence solution (c) showsa clearly visible spot.
B. Apply separately t o the plat e 4 μl of each solution.Develop over a path of 15 cm using a mixture of 8 volumesof conc entr ated ammoni a R, 8 volumes of methanol Rand 84 volumes of ethyl acetate R. Dry the plat e in astream of cold air, expose to iodine vapour until maximumcontrast of the spots has been obtained and examine inultraviolet light at 254 nm. Any spot in the chromat ogramobt ained with the t est solution (a), apart from theprincipal spot, is not more intense than the principal spot in the chromatogram obtained with ref erence solution (a)(0.2 per cent) and not more than two such spots are moreintense than the principal spot in the chromat ogramobtained with reference solution (b) (0.1 per cent). Thet est is not valid unless the chromatogram obtained withref erence solution (c) shows a clearly visible spot.
Liquid chromatography ( 2.2.29).
Test solution. Dissolve 20 mg of the substance to beexamined in the mobile phase and dilute to 50 ml with themobile phase.
Reference solution (a). Dilute 1.0 ml of the test solutionto 100.0 ml with the mobile phase. Dilute 2.0 ml of thissolution to 10.0 ml with the mobile phase.
Reference solution (b). Dissolve 5 mg of cimetidine for peak
identification CRS in the mobile phase and dilute to 10 mlwit h the mobile phase.
(7) Bondapack C18 is suitable.
58 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 59/176
Cimetidine
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity G 5. cimetidine 9. impurity A
2. impurity I 6. impurity D 10. impurity H
3. impurity E 7. impurity C
4. impurity J 8. impurity B
Figure 0756.-1. – Chromatogram for the test for related substances of cimetidine
Column :
— size : l = 0.25 m, Ø = 4.6 mm;
— stationary phase : octadecylsilyl silica gel for chromatography R (5 μm)(8).
Mobile phase : mix 250 volumes of methanol R with amixture of 0.4 volumes of diethylamine R and 780 volumesof a 1.1 g/l solution of sodium hexanesulphonate R,previously adjusted to pH 2.8 with phosphoric acid R.
Flow rate : 1.1 ml/min.
Detection : spectrophotometer at 220 nm.
Injection : 50 μl. Run time : 3 times the retention time of cimetidine.
Identification of impurities : use the chromatogramsupplied with cimetidine for peak identification CRS andthe chromatogram obtained with reference solution (b) toidentify the peaks due to the impurities.
Relative retention with reference to cimetidine (retentiontime = about 17 min): impurity G = about 0.26;impurity E = about 0.38; impurity D = about 1.27;impurity C = about 1.37; impurity B = about 1.86;impurity A = about 2.28; impurity H = about 2.56.
System suitability : reference solution (b):
— resolution : minimum 1.0 between the peaks due toimpurities D and C.
Limits :
— correction factor : for the calculation of content, multiplythe peak area of impurity G by 0.6;
— impurities A, B, C, D, E, G, H : for each impurity, not morethan the area of the principal peak in the chromatogramobtained with reference solution (a) (0.2 per cent) ;
— unspecified impurities : for each impurity, not morethan 0.5 times the area of the principal peak in thechromatogram obtained with reference solution (a)(0.10 per cent);
— total : not more than 5 times the area of the principal peakin the chromatogram obtained with reference solution (a)(1.0 per cent);
— disregard limit : 0.25 times the area of the principal peakin the chromatogram obtained with reference solution (a)(0.05 per cent).
Heavy metals ( 2.4.8 ): maximum 20 ppm.
1.0 g complies with test C. Prepare the reference solutionusing 2 ml of lead standard solution (10 ppm Pb) R.
Loss on drying ( 2.2.32 ) : maximum 0.5 per cent, determinedon 1.000 g by drying in an oven at 100-105 °C.
Sulphated ash ( 2.4.14): maximum 0.2 per cent, determinedon 1.0 g.
ASSAY
Dissolve 0.200 g in 60 ml of anhydrous acetic acid R.Titrate with 0.1 M perchloric acid determining the end-point potentiometrically ( 2.2.20).
1 ml of 0.1 M perchloric acid is equivalent to 25.23 mgof C10H16N6S.
STORAGE
In an airtight container, protected from light.
IMPURITIES
S pecified impurities : A, B, C, D, E, F, G, H.
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : I, J .
(8) Nucleosil 100 C18 is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 59

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 60/176
Cimetidine hydrochloride
A. R1 = CN, R2 = SCH3 : 3-cyano-2-methyl-1-[2-[[(5-methyl-
1 H -imidazol-4-yl)methyl]sulphanyl]ethyl]isothiourea,B. R1 = CN, R2 = OCH3 : 3-cyano-2-methyl-1-[2-[[(5-methyl-
1 H -imidazol-4-yl)methyl]sulphanyl]ethyl]isourea,
C. R1 = CO-NH2, R2 = NH-CH3 : 1-[(methylami-no)[[2-[[(5-methyl-1 H -imidazol-4-yl)methyl]sulphanyl]eth-yl]amino]methylene]urea,
D. R1 = H, R2 = NH-CH3 : 1-methyl-3-[2-[[(5-methyl-1 H -imidazol-4-yl)methyl]sulphanyl]ethyl]guanidine,
E. 2-cyano-1-methyl-3-[2-[[(5-methyl-1 H -imidazol-4-yl)methyl]sulphinyl]ethyl]guanidine,
F. 2-cyano-1,3-bis[2-[[(5-methyl-1 H -imidazol-4-yl)methyl]sulphanyl]ethyl]guanidine,
G. 2-cyano-1,3-dimethylguanidine,
H. 2-cyano-3-[2-[[2-[[(1 Z )-(cyanoamino)(methylamino)methyl-ene]amino]ethyl]disulphanyl]ethyl]-1-methylguanidine,
I. (4-ethyl-1 H -imidazol-5-yl)methanol,
J. 2-[[(5-methyl-1 H -imidazol-4-yl)methyl]sulphanyl]ethan-amine.
Reference: PA/PH/Exp. 10C/T (05) 39 ANP
NOTE ON THE MONOGRAPH
The revision includes the replacement of the TLC for related substances by 2 LC methods which allow the control of substances synthesized by different routes and also cover the degradation products. It is also proposed to delete the
second identification series as the substance is probably not used in pharmacies.XXXX:1500
CIMETIDINE HYDROCHLORIDE
Cimetidini hydrochloridum
C10H17ClN6S M r 288.8
DEFINITION
2-Cyano-1-methyl-3-[2-[[(5-methyl-1 H -imidazol-4-yl)methyl]sulphanyl]ethyl]guanidine hydrochloride.
Content : 98.5 per cent to 101.5 per cent (dried substance).
CHARACTERS
Appearance : white or almost white, crystalline powder.
Solubility : freely soluble in water, sparingly soluble inanhydrous ethanol.
IDENTIFICATION
First i dentifi cati on: B, E.Second i d entificati on: A, C, D, E.
A. Dissolve 70 mg in 0.2 M sulphuric aci d and dilutet o 100.0 ml with the same acid. Dilut e 2.0 ml of thesolution to 100.0 ml with 0.2 M sulphuric aci d . Measurethe absorbance ( 2.2.2 5 ) at the absorption maximum at 218 nm. The specific absorbance at the maximum is 650t o 705.
B. A. Infrared absorption spectrophotometry ( 2.2.24).
Comparison : cimetidine hydrochloride CRS .
C. Examine the chromatograms obtained in the test f or related substances. The principal spot in thechromat ogram obtained with test solution (b) is similar
in position, colour and size t o the principal spot in thechromat ogram obtained with ref erence solution (d).
D. Dissolve about 1 mg in a mixture of 1 ml of ethanol R and5 ml of a freshly prepared 20 g/l solution of citri c aci d Rin ac eti c anhy drid e R . Heat on a water-bath f or 10 min t o15 min. A reddish-violet colour develops.
E. B. It gives reaction (a) of chlorides ( 2.3.1).
TESTS
Appearance of solution. The solution is clear ( 2.2.1) and not more intensely coloured than reference solution Y5 ( 2.2.2, Method II ).
Dissolve 3.0 g in 12 ml of 1 M hydrochloric acid and diluteto 20 ml with water R.
pH ( 2.2.3) : 4.0 to 5.0.Dissolve 100 mg in carbon dioxide-free water R and diluteto 10.0 ml with the same solvent.
60 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 61/176
Cimetidine hydrochloride
Impurity F. Liquid chromatography ( 2.2.29).Test solution. Dissolve 20 mg of the substance to beexamined in the mobile phase and dilute to 50 ml with themobile phase. Reference solution . Dissolve 4.0 mg of cimetidine CRS and4.0 mg of cimetidine impurity F CRS in methanol R anddilute to 50.0 ml with the same solvent. Dilute 1.0 ml of this
solution to 100.0 ml with the mobile phase.Column :— size : l = 0.25 m, Ø = 4.6 mm;— stationary phase : octadecysilyl silica gel for
chromatography R (5 μm)(9). Mobile phase : dissolve 661 mg of sodiumhexanesulphonate R in 760 ml of water R. Add240 ml of methanol R and mix. Adjust to pH 2.5 with phosphoric acid R. Flow rate : 2.0 ml/min. Detection : spectrophotometer at 220 nm. Injection : 20 μl. Run time : 25 min.
System suitability : reference solution:— resolution : minimum 1.0 between the peaks due to
cimetidine and impurity F;— signal-to-noise ratio : minimum 10 for the peak due to
impurity F. Limit :— impurity F : not more than the area of the corresponding
peak in the chromatogram obtained with the referencesolution (0.2 per cent).
Related substances. Examine by thin-layer chromatography( 2.2.27 ), using a TLC silic a g el GF 2 54 pl ate R.T est soluti on ( a). Dissolve 0.50 g of the substance t o beexamined in methanol R and dilut e t o 10 ml with the same
solvent.T est soluti on ( b). Dilut e 1 ml of test solution (a) t o 10 mlwith methanol R. Ref er enc e soluti on ( a). Dilut e 2 ml of t est solution (b) to100 ml with methanol R. Ref er enc e soluti on (b). Dilut e 5 ml of ref erence solution (a)to 10 ml with methanol R.
Ref er ence soluti on (c). Dilut e 5 ml of ref erence solution (b)to 10 ml with methanol R.
Ref er ence soluti on ( d). Dissolve 10 mg of cimetidinehy dr ochl ori de CRS in 2 ml of methanol R.
A. Apply to the plate 4 μl of each solution. Allow the plat et o stand f or 15 min in a chromatographic tank saturatedwith vapour from the mobile phase, which consists of
a mixture of 15 volumes of c onc entr at ed ammoni a R,20 volumes of methanol R and 65 volumes of ethyl acet ate R, and develop immediately over a path of 15 cmusing the same mixture of solvents. Dry the plate in astream of cold air, expose to iodine vapour until maximumcontrast of the spots has been obtained and examine inultraviolet light at 254 nm. Any spot in the chromat ogramobt ained with test solution (a), apart from the principalspot, is not more int ense than the principal spot in thechromat ogram obtained with ref erence solution (a)(0.2 per cent) and at most two such spots are more int ensethan the principal spot in the chromatogram obt ainedwith ref erence solution (b) (0.1 per cent). The test is not valid unless the chromatogram obtained with ref erence
solution (c) shows a clearly visible spot.B. Apply t o the plat e 4 μl of each solution. Developover a path of 15 cm using a mixture of 8 volumes of c onc entr ated ammoni a R, 8 volumes of methanol R and84 volumes of ethyl ac etate R. Dry the plate in a stream of cold air, expose t o iodine vapour until maximum contrast of the spots has been obtained and examine in ultraviolet light at 254 nm. Any spot in the chromat ogram obtainedwith test solution (a), apart from the principal spot, is not more intense than the principal spot in the chromat ogramobtained with ref erence solution (a) (0.2 per cent) and at most two such spots are more int ense than the principalspot in the chromat ogram obtained with ref erencesolution (b) (0.1 per cent). The t est is not valid unless thechromat ogram obtained with ref erence solution (c) showsa clearly visible spot.
Liquid chromatography ( 2.2.29).
Test solution. Dissolve 20 mg of the substance to beexamined in the mobile phase and dilute to 50 ml with themobile phase.
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity G 5. cimetidine 9. impurity A
2. impurity I 6. impurity D 10. impurity H
3. impurity E 7. impurity C
4. impurity J 8. impurity B
Figure 1500.-1. – Chromatogram for the test for related substances of cimetidine hydrochloride
(9) Bondapack C18 is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 61

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 62/176
Cimetidine hydrochloride
Reference solution (a). Dilute 1.0 ml of the test solutionto 100.0 ml with the mobile phase. Dilute 2.0 ml of thissolution to 10.0 ml with the mobile phase.
Reference solution (b). Dissolve 5 mg of cimetidine for peak identification CRS in the mobile phase and dilute to 10 mlwith the mobile phase.
Column :
— size : l = 0.25 m, Ø = 4.6 mm;— stationary phase : octadecylsilyl silica gel for
chromatography R (5 μm)(10).
Mobile phase : mix 250 volumes of methanol R with amixture of 0.4 volumes of diethylamine R and 780 volumesof a 1.1 g/l solution of sodium hexanesulphonate R,previously adjusted to pH 2.8 with phosphoric acid R.
Flow rate : 1.1 ml/min.
Detection : spectrophotometer at 220 nm.
Injection : 50 μl.
Run time : 3 times the retention time of cimetidine.
Identification of impurities : use the chromatogram
supplied with cimetidine for peak identification CRS andthe chromatogram obtained with reference solution (b) toidentify the peaks due to the impurities.
Relative retention with reference to cimetidine (retentiontime = about 17 min): impurity G = about 0.26;impurity E = about 0.38; impurity D = about 1.27;impurity C = about 1.37; impurity B = about 1.86;impurity A = about 2.28; impurity H = about 2.56.
System suitability : reference solution (b):
— resolution : minimum 1.0 between the peaks due toimpurities D and C.
Limits :
— correction factor : for the calculation of content, multiply
the peak area of impurity G by 0.6;— impurities A, B, C, D, E, G, H : for each impurity, not more
than the area of the principal peak in the chromatogramobtained with reference solution (a) (0.2 per cent) ;
— unspecified impurities : for each impurity, not morethan 0.5 times the area of the principal peak in thechromatogram obtained with reference solution (a)(0.10 per cent);
— total : not more than 5 times the area of the principal peakin the chromatogram obtained with reference solution (a)(1.0 per cent);
— disregard limit : 0.25 times the area of the principal peakin the chromatogram obtained with reference solution (a)
(0.05 per cent).Heavy metals ( 2.4.8 ): maximum 20 ppm.
1.0 g complies with test C. Prepare the reference solutionusing 2 ml of lead standard solution (10 ppm Pb) R.
Loss on drying ( 2.2.32 ) : maximum 1.0 per cent, determinedon 1.000 g by drying in an oven at 100-105 °C.
Sulphated ash ( 2.4.14): maximum 0.2 per cent, determinedon 1.0 g.
ASSAY
Dissolve 0.200 g in a mixture of 5 ml of 0.01 M hydrochloric acid and 50 ml of ethanol (96 per cent) R. Carry out a potentiometric titration ( 2.2.20), using 0.1 M sodium
hydroxide. Read the volume added between the 2 pointsof inflexion.
1 ml of 0.1 M sodium hydroxide is equivalent to 28.88 mgof C10H17ClN6S.
STORAGE
In an airtight container, protected from light.
IMPURITIES
Specified impurities : A, B, C, D, E, F, G, H.Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : I, J.
A. R1 = CN, R2 = SCH3 : 3-cyano-2-methyl-1-[2-[[(5-methyl-1 H -imidazol-4-yl)methyl]sulphanyl]ethyl]isothiourea,
B. R1 = CN, R2 = OCH3 : 3-cyano-2-methyl-1-[2-[[(5-methyl-1 H -imidazol-4-yl)methyl]sulphanyl]ethyl]isourea,
C. R1 = CO-NH2,R2=NH-CH3 : 1-[(methylamino)[[2-[[(5-meth-yl-1 H -imidazol-4-yl)methyl]sulphanyl]ethyl]amino]methyl-ene]urea,
D. R1 = H, R2 = NH-CH3 : 1-methyl-3-[2-[[(5-methyl-1 H -imidazol-4-yl)methyl]sulphanyl]ethyl]guanidine,
E. 2-cyano-1-methyl-3-[2-[[(5-methyl-1 H -imidazol-4-yl)methyl]sulphinyl]ethyl]guanidine,
F. 2-cyano-1,3-bis[2-[[(5-methyl-1 H -imidazol-4-yl)methyl]-sulphanyl]ethyl]guanidine,
G. 2-cyano-1,3-dimethylguanidine,
(10) Nucleosil 100 C18 is suitable.
62 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 63/176
Cisplatin
H. 2-cyano-3-[2-[[2-[[(1 Z )-(cyanoamino)(methylamino)methyl-ene]amino]ethyl]disulphanyl]ethyl]-1-methylguanidine,
I. (4-ethyl-1 H -imidazol-5-yl)methanol,
J. 2-[[(5-methyl-1 H -imidazol-4-yl)methyl]sulphanyl]ethan-amine.
Reference: PA/PH/Exp. 10D/T (04) 36 ANP 1R
NOTE ON THE MONOGRAPH
Following the publication of the previous draft revisionin Pharmeuropa 17.1, a new LC method has beendeveloped to quantify transplatin (impurity A) and trichloroamineplatinate (impurity B) in the same run.
XXXX:0599
CISPLATIN
Cisplatinum
[PtCl2(NH3)2] M r 300.0
DEFINITIONcis-Diaminedichloroplatinum (II).Content : 97.0 per cent to 102.0 per cent.
CHARACTERS Appearance : yellow powder or yellow or orange-yellowcrystals.Solubility : slightly soluble in water, sparingly soluble indimethylformamide, practically insoluble in ethanol (96 percent).It decomposes with blackening at about 270 °C.Carry out identification test B, the tests (except that for silver) and the assay protected from light .
IDENTIFICATION First identification : A, B.Second identification: B, C .
A. Infrared absorption spectrophotometry ( 2.2.24). Pr epar ati on : discs of pot assium br omi de R.Comparison : cisplatin CRS .
B. Thin-layer chromatography ( 2.2.27 ).
Test solution. Dilute 1 ml of solution S2 (see Tests) to10 ml with dimethylformamide R.
Reference solution . Dissolve 10 mg of cisplatin CRS in5 ml of dimethylformamide R.
Plate : cellulose for chromatography R1 as the coating
substance. Pretreatment : activate the plate by heating at 150 °C for1 h.
Mobile phase : acetone R, dimethylformamide R(10:90 V/V ).
Application : 2 μl.
Development : over 2/3 of the plate.
Drying : in air.
Detection : spray with a 50 g/l solution of stannouschloride R in a mixture of equal volumes of dilutehydrochloric acid R and water R. Examine after 1 h.
Results : the principal spot in the chromatogram obtained
with the test solution is similar in position, colour andsize to the principal spot in the chromatogram obtainedwith the reference solution.
C. Add 50 mg to 2 ml of dilute sodium hydroxide solution Rin a glass dish. Evaporate to dryness. Dissolve the residuein a mixture of 0.5 ml of nitric acid R and 1.5 ml of hydrochloric acid R. Evaporate to dryness. The residueis orange. Dissolve the residue in 0.5 ml of water R andadd 0.5 ml of ammonium chloride solution R. A yellow,crystalline precipitate is formed.
TESTS
Solution S1. Dissolve 25 mg in a 9 g/l solution of sodiumchloride R prepared with carbon dioxide-free water R and
dilute to 25 ml with the same solvent.Solution S2. Dissolve 0.20 g in dimethylformamide R anddilute to 10 ml with the same solvent.
Appearance of solution S1. Solution S1 is clear ( 2.2.1) andnot more intensely coloured than reference solution GY5
( 2.2.2, Method II ).
Appearance of solution S2. Solution S2 is clear ( 2.2.1).
pH ( 2.2.3) : 4.5 to 6.0 for solution S1, measured immediatelyafter preparation.
Related substances. Examine by thin-layer chromatography( 2.2.27 ), using cellulose f or chr omatogr aphy R1 as thecoating substance. Activat e the plat e by heating at 150 °Cf or 1 h.
T est soluti on (a). Dilute 1 ml of solution S2 to 10 ml withdimethylformami d e R.
T est soluti on (b). Use solution S2.
Ref er ence soluti on (a). Dissolve 10 mg of cisplatin CRS in5 ml of dimethylf ormamid e R.
Ref er ence soluti on (b). Dilut e 1 ml of solution S2 to 50 mlwith dimethylf ormami de R.
Apply separately to the plat e 2.5 μl of t est solution (a), 2.5 μlof ref erence solution (a), 5 μl of t est solution (b) and 5 μl of ref erence solution (b). Develop over a path of 15 cm usinga mixture of 10 volumes of ac et one R and 90 volumes of dimethylformami d e R. Allow the plate t o dry in air and
spray with a 50 g/l solution of st annous chlori d e R in amixture of equal volumes of dilute hydr ochloric acid R andwat er R. Aft er 1 h, the chromatogram obtained with test solution (b) shows no spot with an R f value less than that of
© PHARMEUROPA Vol. 18, No. 1, January 2006 63

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 64/176
Cisplatin
the principal spot and any spot with an Rf value greater thanthat of the principal spot is not more int ense than the spot in the chromat ogram obtained with ref erence solution (b).Liquid chromatography ( 2.2.29). Carry out the test protected from light. Do not heat or sonicate any platinum-containing solution. All solutions are to be used within 4 h.
Test solution. Dissolve 25.0 mg of the substance to be
examined in a 9.0 g/l solution of sodium chloride R anddilute to 25.0 ml with the same solution. Reference solution (a). Dissolve 25.0 mg of cisplatin CRS ina 9.0 g/l solution of sodium chloride R and dilute to 25.0 mlwith the same solution. Reference solution (b). Dissolve 5.0 mg of cisplatinimpurity A CRS in a 9.0 g/l solution of sodium chloride Rand dilute to 50.0 ml with the same solution. Reference solution (c). Dissolve 5.6 mg of cisplatinimpurity B CRS in a 9.0 g/l solution of sodium chloride Rand dilute to 100.0 ml with the same solution. Reference solution (d). Mix 0.050 ml of the test solutionwith 5.0 ml of reference solution (b) and 5.0 ml of referencesolution (c) and dilute to 25.0 ml with a 9.0 g/l solution of
sodium chloride R. Reference solution (e). Dilute 5.0 ml of reference solution (d)to 20.0 ml with a 9.0 g/l solution of sodium chloride R. Blank solution: 9.0 g/l solution of sodium chloride R.Column :— size : l = 0.25 m, Ø = 4.0 mm;— stationary phase : octylsilyl silica gel for
chromatography, base-deactivated R (4 μm)(11);— temperature : 30 °C. Mobile phase: dissolve 1.08 g of sodium octanesulphonate R,1.70 g of tetrabutylammonium hydrogen sulphate R and2.72 g of potassium dihydrogen phosphate R in water for chromatography R and dilute to 950 ml with the same
solvent. Adjust to pH 5.9 with 1 M sodium hydroxide anddilute to 1000 ml with water for chromatography R.
Flow rate : 1.0 ml/min. Detection : spectrophotometer at 210 nm.
Injection : 20 μl of the test solution, reference solutions (d)and (e), and the blank solution. Run time : 3 times the retention time of cisplatin. Relative retention with reference to cisplatin (retentiontime = about 3.8 min): displacement peak = about 0.5;impurity A = about 0.6 ; impurity B = about 0.7.System suitability : reference solution (d):— resolution : minimum 2.5 between the displacement
peak and the peak due to impurity A, and minimum 3.5between the peaks due to impurities A and B.
Limits :— impurity A : not more than the area of the corresponding
peak in the chromatogram obtained with referencesolution (d) (2.0 per cent);
— impurity B : not more than the area of the correspondingpeak in the chromatogram obtained with referencesolution (d) (1.0 per cent);
— unspecified impurities : for each impurity, not more
than 0.5 times the area of the peak due to cisplatin inthe chromatogram obtained with reference solution (d)(0.10 per cent);
— sum of impurities other than A and B : not more than2.5 times the area of the peak due to cisplatin in thechromatogram obtained with reference solution (d)(0.5 per cent);
— disregard limit : the area of the peak due to cisplatin inthe chromatogram obtained with reference solution (e)(0.05 per cent).
Silver : maximum 250 2.50 × 102 ppm.Atomic absorption spectrometry ( 2.2.23, Method I ).Test solution. Dissolve 0.100 g of the substance to be
examined in 15 ml of nitric acid R, heating to 80 °C. Cooland dilute to 25.0 ml with water R.
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. displacement peak 2. impurity A 3. impurity B 4. cisplatin
Figure 0599.-1. – Chromatogram for the test for related substances of cisplatin: cisplatin spiked with 2 per cent of impurity A and 1 per cent of impurity B
(11) Superspher 60 RP select B is suitable.
64 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 65/176
Clonidine hydrochloride
Reference solutions . To suitable volumes (10 ml to 30 ml) of silver standard solution (5 ppm Ag) R add 50 ml of nitric acid R and dilute to 100.0 ml with water R.
Source : silver hollow-cathode lamp, preferably with aspectral slit width of 0.5 nm.
Wavelenth : 328 nm.
Atomisation device : fuel-lean air-acetylene flame.
Carry out a blank determination.
ASSAY
Examine by liquid chromat ography ( 2.2.29).
T est soluti on. Dissolve 50.0 mg of the substance to beexamined in a 9 g/l solution of sodium chl ori de R and dilut eto 100.0 ml with the same solvent.
Ref er enc e soluti on. Dissolve 50.0 mg of cisplatin CRS in a9 g/l solution of sodium chl ori de R and dilut e to 100.0 mlwith the same solvent.
The chromat ographic procedure may be carried out using :
— a column 0.25 m long and 4.6 mm in int ernal diamet erpacked with str ong -ani on - ex change sili ca g el for
chromatogr aphy R (10 μm),— as mobile phase at a flow rat e of 1.2 ml/min a mixture of
10 volumes of a 9 g/l solution of sodium chl orid e R and90 volumes of methanol R,
— as det ect or a spectrophot omet er set at 220 nm.
Use a sample loop. Inject separat ely 20 μl of the t est solutionand 20 μl of the reference solution.
Liquid chromatography ( 2.2.29) as described in the test forrelated substances with the following modification.
Injection : 10 μl of the test solution and reference solution (a).
Calculate the percentage content of [PtCl2(NH3)2] from thedeclared content of cisplatin CRS .
STORAGEIn an airtight container, protected from light.
IMPURITIES
Specified impurities: A, B.
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : C.
A. trans-diaminedichloroplatinum (II) (transplatin),
B. aminetrichloroplatinate(–),
C. tetrachloroplatinate(2–).
Reference: PA/PH/Exp. 11/T (05) 82 ANP
NOTE ON THE MONOGRAPH
It is proposed to revise the monograph to replace the TLC by an LC in the test for related substances.
The second identification series is not needed, so it isdeleted.
XXXX:0477
CLONIDINE HYDROCHLORIDE
Clonidini hydrochloridum
C9H10Cl3N3 M r 266.6
DEFINITION
2,6-Dichloro- N -(imidazolidin-2-ylidene)aniline.Content : 98.5 per cent to 101.0 per cent (dried substance).
CHARACTERS Appearance : white or almost white, crystalline powder.Solubility : soluble in water and in anhydrous ethanol.
IDENTIFICATION First i dentifi cati on: B, D.Second i d entificati on: A, C, D.A. Ultraviolet and visible absorption spectrophotometry
( 2.2.25 ).T est solution. Dissolve 30.0 mg in 0.01 M hydr ochloric
acid and dilut e to 100.0 ml with the same acid.Spectr al r ange : 245-350 nm. Absorpti on maxima : at 272 nm and 279 nm. P oint of infl exi on : at 265 nm.Specific absor bances at the absorption maxima :— at 272 nm: about 18,— at 279 nm: about 16.
B. A. Infrared absorption spectrophotometry ( 2.2.24).Comparison : clonidine hydrochloride CRS .
C. Examine the chromatograms obtained in the test f orrelat ed substances. Results : the principal spot in the chromatogram obtained
with t est solution (b) is similar in position, colour and sizet o the principal spot in the chromat ogram obtained withref erence solution (a).
D. B. It gives reaction (a) of chlorides ( 2.3.1).
TESTS
Solution S. Dissolve 1.25 g in carbon dioxide-free water Rand dilute to 25 ml with the same solvent.
Appearance of solution. Solution S is clear ( 2.2.1) and not more intensely coloured than reference solution Y7 ( 2.2.2, Method II ).
pH ( 2.2.3): 4.0 to 5.0 for solution S.
Related substances. Examine by thin-layer chromatography( 2.2.27 ), using sili c a g el G R as the coating substance.T est soluti on ( a). Dissolve 0.1 g of the substance t o beexamined in methanol R and dilut e to 10 ml with the samesolvent.
© PHARMEUROPA Vol. 18, No. 1, January 2006 65

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 66/176
Clonidine hydrochloride
T est soluti on ( b). Dilute 1 ml of t est solution (a) t o 10 mlwith methanol R.
Ref er enc e solution (a). Dissolve 10 mg of c loni dinehy dr ochl ori de CRS in methanol R and dilute t o 10 ml withthe same solvent.
Ref er enc e soluti on ( b). Dilut e 1 ml of test solution (a) to10 ml with methanol R . Dilut e 5 ml of this solution to 100 ml
with methanol R.Apply t o the plate 10 μl of each solution. Shake a mixture of 10 volumes of gl aci al ac etic aci d R , 40 volumes of but anol Rand 50 volumes of water R. Allow t o separate. Filt er theupper layer and use the filtrat e as the mobile phase. Developover a path of 15 cm. Allow the plat e to dry in air andspray with potassium iodobismuthat e soluti on R2 . Allowthe plat e t o dry in air for 1 h, spray again with potassiumiod obismuthat e soluti on R2 and then immediat ely spraywith a 50 g/l solution of sodium nitrite R. Any spot in thechromatogram obtained with t est solution (a), apart fromthe principal spot, is not more int ense than the spot in thechromatogram obtained with ref erence solution (b) (0.5 percent).
Liquid chromatography ( 2.2.29).
Test solution. Dissolve 50 mg of the substance to beexamined in mobile phase A and dilute to 50.0 ml withmobile phase A.
Reference solution (a). Dilute 1.0 ml of the test solution to100.0 ml with mobile phase A.
Reference solution (b). Dissolve 10 mg of clonidinehydrochloride impurity B CRS in mobile phase A and diluteto 10.0 ml with mobile phase A. To 1 ml of the solution, add1 ml of the test solution and dilute to 10.0 ml with mobilephase A.
Column :
— size : l = 0.15 m, Ø = 3.0 mm;— stationary phase : propylsilyl silica gel for
chromatography R (5 μm)(12) ;
— temperature : 40 °C.
Mobile phase :
— mobile phase A : dissolve 4 g of potassium dihydrogen phosphate R in 1000 ml of water R, and adjust to pH 4.0with phosphoric acid R ;
— mobile phase B : mobile phase A, acetonitrile R(25:75 V/V ) ;
Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 90 10
0 - 15 90 → 30 10 → 70
15 - 15.1 30 → 90 70 → 10
15.1 - 20 90 10
Flow rate : 1.5 ml/min.
Detection : spectrophotometer at 210 nm.
Injection : 5 μl.
System suitability : reference solution (b):
— resolution : minimum 5 between the peaks due toclonidine and impurity B;
— symmetry factor : maximum 2.5 for the peak due toclonidine.
Limits :
— unspecified impurities : for each impurity, not morethan 0.1 times the area of the principal peak in thechromatogram obtained with reference solution (a)(0.10 per cent);
— total : not more than 0.2 times the area of the principalpeak in the chromatogram obtained with reference
solution (a) (0.2 per cent);— disregard limit : 0.05 times the area of the principal peak
obtained with reference solution (a) (0.05 per cent).
Loss on drying ( 2.2.32 ) : maximum 0.5 per cent, determinedon 1.000 g by drying in an oven at 100-105 °C.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAY
Dissolve 0.200 g in 70 ml of ethanol (96 per cent) R. Titratewith 0.1 M ethanolic sodium hydroxide determining theend-point potentiometrically ( 2.2.20).
1 ml of 0.1 M sodium hydroxide is equivalent to 26.66 mgof C9H10Cl3N3.
IMPURITIES
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : A, B, C .
A. 1-acetylimidazolidin-2-one,
B. 1-acetyl- N -(2,6-dichlorophenyl)-4,5-dihydro-1 H -imidazole-2-amine,
C. 2,6-dichloroaniline.
Reagents
Silica gel for chromatography, propylsilyl. XXXXXXX.
A very finely divided silica gel (3-10 μm), chemically modifiedat the surface by the bonding of propylsilyl groups. The
particle size is indicated after the name of the reagent in thetest where it is used.
(12) Zorbax SB-C3 is suitable.
66 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 67/176
Clotrimazole
Reference: PA/PH/Exp. 10A/T (98) 68 ANP 2R
NOTE ON THE MONOGRAPH
A revision of the monograph is proposed to introducean LC for the test for related substances in replacement of the 2 current TLCs. Another LC method was proposed previously in Pharmeuropa, the one now
proposed allows the control of one additional impurity (deschloroclotrimazole). It is also proposed to revise the section Identification and to delete the test for appearanceof solution because the substance is not used in parenteral preparations.
XXXX:0757
CLOTRIMAZOLE
Clotrimazolum
C22H17ClN2 M r 344.8
DEFINITION
1-[(2-Chlorophenyl)diphenylmethyl]-1 H -imidazole.
Content : 98.5 per cent to 100.5 per cent (dried substance).
CHARACTERS
Appearance : white or pale yellow, crystalline powder.
Solubility : practically insoluble in water, soluble in ethanol(96 per cent) and in methylene chloride.
IDENTIFICATION
First identification : B.
Second identification: A, C, D.
A. Melting point ( 2.2.14): 141 °C to 145 °C.
B. Infrared absorption spectrophotometry ( 2.2.24).
Comparison : clotrimazole CRS .
C. Examine bef ore spraying in ultraviolet light at 254 nm,the chromat ograms obt ained in the test f or relat edsubstances. The principal spot in the chromatogramobt ained with test solution (b) is similar in position and
size to the principal spot in the chromatogram obtainedwith ref erence solution (a).
C. Thin-layer chromatography ( 2.2.27 ).
Test solution. Dissolve 50 mg of the substance to beexamined in ethanol (96 per cent) R and dilute to 5 mlwith the same solvent.
Reference solution. Dissolve 50 mg of clotrimazole CRS in ethanol (96 per cent) R and dilute to 5 ml with thesame solvent.
Plate : TLC silica gel GF 254 plate R.
Mobile phase : concentrated ammonia R1, propanol R,toluene R (0.5:10:90 V/V/V ).
Application : 10 μl.
Development : over 2/3 of the plate. Drying : in air.
Detection : examine in ultraviolet light at 254 nm.
Results : the principal spot in the chromatogram obtainedwith the test solution is similar in position and size tothe principal spot in the chromatogram obtained withthe reference solution.
D. Dissolve about 10 mg in 3 ml of sulphuric acid R. Thesolution is pale yellow. Add 10 mg of mer curi c oxi de Rand 20 mg of sodium nitrit e R. Allow t o stand with
occasional shaking. An orange colour develops, becomingorange-brown.
TESTS
Appearance of solution. The solution is clear ( 2.2.1) andnot more intensely coloured than ref erence solution BY6( 2.2.2, M ethod II ).
Dissolve 1.25 g in alcohol R and dilut e t o 25 ml with thesame solvent.
(2-Chlorophenyl)diphenylmethanol. Examine by thin-layerchromat ography ( 2.2.27 ), using a TLC silica gel GF254 plate R.
T est soluti on (a). Dissolve 0.50 g of the substance to beexamined in al c ohol R and dilut e to 5 ml with the samesolvent.
T est soluti on (b). Dilute 1 ml of t est solution (a) t o 10 mlwith alc ohol R.
Ref er ence soluti on (a). Dissolve 50 mg of clotrimazole CRS in al cohol R and dilute t o 5 ml with the same solvent.
Ref er ence solution ( b). Dissolve 10 mg of (2-chloro-phenyl)diphenylmethanol CRS in alc ohol R anddilut e t o 5 ml with the same solvent. Dilut e 1 ml of thesolution t o 10 ml with alc ohol R.
Apply t o the plat e 10 μl of each solution. Develop over a pathof 15 cm using a mixture of 0.5 volumes of c onc entr ated ammoni a R1, 10 volumes of pr opanol R and 90 volumesof toluene R. Allow the plat e to dry in air. Spray the plateswith a 10 per cent V/V solution of sulphuri c aci d R in
al cohol R and heat at 100 °C to 105 °C f or 30 min. Any spot corresponding to (2-chlorophenyl)diphenylmethanol in thechromat ogram obtained with t est solution (a) is not moreint ense than the spot in the chromatogram obtained withref erence solution (b) (0.2 per cent).
Imidazole. Examine by thin-layer chromat ography ( 2.2.27 ),using a TLC silic a g el G plat e R.
T est soluti on. Dissolve 0.50 g of the substance t o beexamined in al c ohol R and dilute to 10 ml with the samesolvent.
Ref er ence soluti on. Dissolve 10 mg of imi d azol e R inal cohol R and dilute to 10 ml with the same solvent. Dilut e1 ml of the solution t o 10 ml with al c ohol R.
Apply t o the plat e 10 μl of each solution. Develop over a path
of 15 cm using a mixture of 0.5 volumes of c onc entr at ed ammoni a R1, 10 volumes of propanol R and 90 volumes of t oluene R . Allow the plat e to dry in air. At the bottom of achromat ography tank, place an evaporating dish cont aininga mixture of 1 volume of hy dr ochl ori c aci d R1, 1 volume of water R and 2 volumes of a 15 g/l solution of potassium permang anat e R, close the tank and allow to stand for15 min. Place the dried plat e in the tank and close thetank. Leave the plat e in contact with the chlorine vapourf or 5 min. Withdraw the plat e and place it in a current of cold air until the excess of chlorine is removed and anarea of coating below the points of application does not give a blue colour with a drop of pot assium i odi de and st ar ch soluti on R. Spray with pot assium iodid e and st ar ch
soluti on R. Any spot corresponding to imidazole in thechromat ogram obtained with the test solution is not moreint ense than the spot in the chromatogram obtained withthe ref erence solution (0.2 per cent).
© PHARMEUROPA Vol. 18, No. 1, January 2006 67

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 68/176
Clotrimazole
Related substances. Liquid chromatography ( 2.2.29).Test solution. Dissolve 50.0 mg of the substance to beexamined in acetonitrile R1 and dilute to 50.0 ml with thesame solvent. Reference solution (a). Dilute 1.0 ml of the test solution to100.0 ml with acetonitrile R1. Dilute 1.0 ml of this solutionto 10.0 ml with acetonitrile R1.
Reference solution (b). Dissolve 5 mg of clotrimazole for peak identification CRS (containing impurities A, B and F)in acetonitrile R1 and dilute to 5 ml with the same solvent. Reference solution (c). Dissolve 5.0 mg of clotrimazoleimpurity D CRS and 5.0 mg of clotrimazole impurity E CRS in acetonitrile R1 and dilute to 100.0 ml with the samesolvent. Dilute 1.0 ml of this solution to 25.0 ml withacetonitrile R1.Column :— size : l = 0.15 m, Ø = 4.6 mm;— stationary phase : spherical end-capped octylsilyl silica
gel for chromatography R (5 μm)(13) ;— temperature : 40 °C.
Mobile phase :— mobile phase A : dissolve 1.0 g of potassium dihydrogen phosphate R and 0.5 g of tetrabutylammonium hydrogen sulphate R1 in water R and dilute to 1000 ml with thesame solvent;
— mobile phase B : acetonitrile R1 ;
Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 - 3 75 25
3 - 25 75 → 20 25 → 80
25 - 30 20 80
30 - 30.1 20 → 75 80 → 25
30.1 - 40 75 25
Flow rate : 1.0 ml/min. Detection : spectrophotometer at 210 nm. Injection : 10 μl. Relative retention with reference to clotrimazole(retention time = about 12 min): impurity D = about 0.1;impurity F = about 0.9; impurity B = about 1.1;impurity E = about 1.5 ; impurity A = about 1.8.
System suitability : reference solution (b):— resolution : minimum 1.5 between the peaks due to
impurity F and clotrimazole;— the chromatogram obtained is similar to the
chromatogram supplied with clotrimazole for peak identification CRS .
Limits :
— impurities A, B : for each impurity, not more than twicethe area of the principal peak in the chromatogramobtained with reference solution (a) (0.2 per cent) ;
— impurities D, E : for each impurity, not more than thearea of the corresponding peak in the chromatogramobtained with reference solution (c) (0.2 per cent) ;
— impurity F : not more than the area of the principal peakin the chromatogram obtained with reference solution (a)(0.1 per cent);
— unspecified impurities : for each impurity, not morethan the area of the principal peak in the chromatogramobtained with reference solution (a) (0.10 per cent);
— total : not more than 5 times the area of the principal peak
in the chromatogram obtained with reference solution (a)(0.5 per cent);— disregard limit : 0.5 times the area of the principal peak
in the chromatogram obtained with reference solution (a)(0.05 per cent).
Loss on drying ( 2.2.32 ) : maximum 0.5 per cent, determinedon 1.000 g by drying in an oven at 100-105 °C.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAYDissolve 0.300 g in 80 ml of anhydrous acetic acid R. Titratewith 0.1 M perchloric acid using 0.3 ml of naphtholbenzein solution R as indicator until the colour changes from
brownish-yellow to green.1 ml of 0.1 M perchloric acid is equivalent to 34.48 mgof C22H17ClN2.
STORAGEProtected from light.
IMPURITIESSpecified impurities: A, B, D, E, F.
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity D 3. clotrimazole 5. impurity E
2. impurity F 4. impurity B 6. impurity A
Figure 0757.-1. – Chromatogram for the test for related substances of clotrimazole
(13) Zorbax Eclipse XDB- C8 is suitable.
68 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 69/176
2.7.28. CFC assay for human haematopoietic progenitor cells
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : C.
A. R = OH : (2-chlorophenyl)diphenylmethanol,
C. R = Cl : 1-chloro-2-(chlorodiphenylmethyl)benzene,
B. 1-[(4-chlorophenyl)diphenylmethyl]-1 H -imidazole,
D. imidazole,
E. (2-chlorophenyl)phenylmethanone,
F. 1-triphenylmethyl-1 H -imidazole (deschloroclotrimazole).
Reference: PA/PH/Exp. CTP/T (05) 27 ANP
XXXX:20728
2.7.28. COLONY-FORMINGCELL ASSAY FOR HUMANHAEMATOPOIETIC PROGENITORCELLS
The haematopoietic system represents a continuum of cellswhose phenotype and properties change as they progressfrom stem cells to differentiated cells. One could therefore
expect that, in a steady-state situation, variations detectedin the more mature compartments will faithfully reflect those occurring in stem cells. Thus, in a unperturbedhaematopoietic system, the presence of colony-forming cellsis an indirect indicator of a functional haematopoietic stemcell compartment. However, all of the compartments arecontrolled by different factors, some of them geneticallydetermined, and their ratio may vary independently fromeach other, especially in the presence of disease or medicaltreatments. This warrants the need for assays that evaluateeach compartment independently. All functional assaysmeasure 2 principal parameters : cell proliferation (measuredby the number of cells produced) and differentiationpotential (estimated by the number of different lineagesrepresented in its progeny).
A hierarchy of haematopoietic progenitor cells (HPCs) hasbeen established, based on the time required in vitro toobtain a colony of mature cells. HPCs with a progressivelyincreasing proliferation capacity and a multilineage naturewill give rise to bigger colonies composed of mature cellslater in the assay. Due to physical constraints in matrix-basedassays, the HPCs with the highest proliferation capacity
can only be functionally tested in a limiting dilution of purified cell populations in liquid cultures. When evaluatinghaematopoiesis, 3 parameters have to be taken into account:
— the lack of reliable correlation between the phenotype of a given cell and its function;
— the possibility that a single assay might score togetherfunctionally heterogeneous progenitor cells ;
— the ontogeny-related changes in haematopoietic cellproliferation and self-renewal.
CELL-SURFACE MARKERS
The capacity of colony-forming cells to give rise tohaematopoietic colonies in vitro and/or to reconstitutethe haematopoietic system has been correlated with theexpression of specific cell-surface antigens. The expressionof the membrane antigen CD34 is an accepted marker formost of the haematopoietic progenitors and stem cells.The expression of other antigens, such as CD33 or CD38,can identify specific subpopulations with a restrictedlineage-differentiation capacity. Percentage contents of colony-forming cells among human CD34+ bone marrowcells average 15-30 per cent for CD34/CD38+ and 5 per cent for the more immature CD34+/CD38neg population.
The total number of transplanted CD34+ cells is used as apredictive factor of engraftment and of the expected lengthof post-transplant aplasia. However, large scale sampling onneonatal blood collections has shown a less-than-perfect correlation between the percentage of CD34+ cells and the
functional capacity to give rise to colonies in vitro and toengraftment in vivo. Similar results have been obtained withthe adult bone marrow, especially in individuals treated withirradiation or chemotherapy.
COLONY ASSAY SPECIFICITY
Colony-forming cells are identified with a nomenclaturebased on the lineages of mature cells present in the colony(for example, CFU-Mix, CFU-GEMM, CFU-GM, CFU-G,CFU-M, BFU-E, CFU-E, CFU-Meg) and are a population of progenitors able to give rise to colonies containing oneor more lineages of haematopoietic cells. No or very lowcapacity for self-renewal has been ascribed to this populationof human HPCs compared to less-differentiated cellular
elements. However, alternative pathways modulate theextent to which a single colony-forming cell will proliferatebefore terminally differentiating, especially in response tostress stimuli, such as anaemia or infection.
© PHARMEUROPA Vol. 18, No. 1, January 2006 69

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 70/176
2.7.28. CFC assay for human haematopoietic progenitor cells
The amount and type of growth factors supplied duringthe culture partially direct the type of colonies that will beformed. Thus, depending on the amounts and combinationof added or contaminant growth factors, different types of progenitors may give rise to colonies that will be scored inthe same group on the basis of their morphology and cellcontent.
Greater specificity on the general class of HPCs and ontheir relative proliferative potential is provided by the timerequired to differentiate in vitro into mature cells. The timerequired by colony-forming cells to give rise to a colonyformed of mature cells in vitro differs between foetal andadult progenitors (9-10 days and 12-14 days respectively).Neonatal cells progenitor mature according to more adult-than foetal-like kinetics. However, neonatal progenitor cellsare characterised by a higher proliferation, and possiblyself-renewal capacity compared to the adult progenitor cells.Differences in the sensitivity to growth factors have alsobeen reported between neonatal and adult progenitor cells.
Overall, the in vitro assay to assess the presence of colony-forming cells, their number and their proliferationcapacity is a useful, although indirect, evaluation of theoverall HPC health status, and hence of their capacity toengraft and reconstitute within a host.
QUALITY ASSURANCE FOR A CFC ASSAY
It is paramount for the overall quality of the colony-formingcell assay (CFC assay) to apply a strictly standardisedapproach. The source of the materials, including reagents,growth factors and disposables, is codified and referencestandards are identified. The apparatus and its conditionof use are clearly stated and validated over time against acellular standard. Fresh bone marrow cells from a specificstrain of laboratory animals, usually mice, are the most suitable reference standard, as healthy animals of the samestrain, sex and age have a consistent colony-forming cell
content in their bone marrow. Human donors are toovariable to be useful as a standard, especially over a timespan of years or across several laboratories.
The sources of the highest variability in the CFC assay arethe number of cells plated and the scoring procedure. Eventrained individuals in the same laboratory may generatea 5 per cent intra-laboratory variability for the same test.However, if it is necessary to evaluate the colony-forming cellcontent in a purified cell population, it is possible to use alimiting dilution approach where the number of wells positivefor cell proliferation is measured with an automated system.
The other main source of variability stems from the use of undefined materials (for example, foetal bovine serum (FBS)or bovine serum albumin (BSA)) in the CFC assay. These
products derive from the processing of large pools of sourcematerials and provide an undefined stimulation of cellularproliferation. However, it is not uncommon to have batcheswith particular characteristics that selectively stimulate theproliferation of specific haematopoietic lineages.
Finally, a low level of endotoxins (less than 0.01 IU/mlor less than 0.01 IU/mg) in all the materials used for theclonogenic assay is advisable, as higher levels result first in a progressive skewing of the haematopoietic lineagesexpression in the cultures and afterwards in a more generalinhibition of cell proliferation and clonogenesis.
CFC CLONOGENIC ASSAY
The CFC assay is based on the capacity of progenitor cells
to form a colony when plated in a semi-solid medium or in agel in the presence of specific growth factors. Different typesof matrices may be used (for example, agar, plasma-clot andmethylcellulose) depending on the desired readout.
MATERIALS
Matrix. Using methylcellulose as a semi-solid phase insteadof agar improves the efficiency of growth of the coloniesin this assay. This gives tighter colonies that are easier toevaluate and count. The possibility of finely adjusting theviscosity of the medium allows the plated cells to sediment at the bottom of the dish and the colonies to develop
on a single plane. A methylcellulose with a viscosity of 4000 mPa·s is suitable. Prepare a 20 g/l solution in thesame culture medium as that used for the assay. The powderis not easily dissolved in water and care must be taken toobtain a product of homogeneous viscosity. The final invitro concentration ranges around 8-9 g/l.
Culture medium. Various media have been developed forthe clonogenic assay, the latest being the Iscove’s ModifiedDulbecco Medium (IMDM), which includes the addition of sodium selenite and HEPES to improve pH stability duringthe 13-14 days required for the assay. Commercially availablemedia usually give more reproducible results.
Serum. Although serum-deprived culture conditions for theCFC clonogenic assay have often been described, the absence
of detailed information on commercially available mediasupports the continuing use of the serum-supplementedversion. The most common serum used as a source of unspecified nutrients and growth factors is foetal calf serum(FCS). As discussed before it is still advisable to screen thismaterial to avoid skewing the results towards a specifichaematopoietic lineage. The use of FCS at concentrationsranging from 20 to 40 per cent V/V has been described in theliterature. The exact concentration needs to be determinedbased on the characteristics of the particular lot.
Albumin. Some formulae include the use of albumintogether with serum in the CFC assay. The albumin moleculeacts as an aspecific carrier for many small molecules witha cellular-proliferation regulatory activity, such as lipids
and vitamins, and as a generic buffer. A deionised solutionof BSA obtained by the cold precipitation technique(Cohn’s Fraction V) will usually be added to obtain a finalconcentration of 10 g/l.
Growth factors. Due to commercial availability of haematopoietic growth factors, it is no longer necessaryto stimulate progenitor cell proliferation using aconditioned medium from accessory cells or cell lines. Bothmultilineage (Kit-ligand or SCF, interleukin-3, GM-CSF) andlineage-specific (erythropoietin, G-CSF) growth factors arerequired to obtain the highest number of colonies from apopulation containing a mixed population of HPCs. Theamount of each growth factor must be high enough to givethe maximal stimulation of colony formation when added to
the assay cells alone. Concentrations of 10-100 ng/ml areusually sufficient to reach the plateau.
Cells. Single cell suspensions are required for this assay.In the case of bone marrow aspirates, such suspensionscan be obtained by forcing the bone marrow through asieve or through progressively smaller calibre needles.Repeated passages through a 21-gauge needle are usuallysufficient to separate the bone marrow pieces into a singlecell suspension.
Most CFC assays are performed with the mononuclearcell fraction of the sample as the presence of granulocytesand red cells may interfere in the readout. Sterile mediawith a relative density higher than 1.077 intended for theseparation of the mononuclear cells from the other mature
elements are commercially available.
70 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 71/176
Dacarbazine
PLATING
1 ml of the solution containing the liquid medium,the methylcellulose and the cells is usually plated in abacteriological sterile dish (Ø 35 mm).Because of the viscosity of the medium, the solution cannot be plated with air displacement pipettes and the use of syringes equipped with large bore ( 18-gauge) blunt-end
needles is required.The number of cells to be plated depends on the HPCconcentration in the sample to be tested. So that no colonyis derived from 2 different HPCs, the number of cells platedis kept low enough to avoid an excessive number of colonies( 100) per plate (Ø 35 mm). However, too low a numberof colonies ( 10) increases the measurement error. Anumber of colonies per plate between 40 and 80 and a test performed in triplicate usually allow a robust measure of thecolony-forming cell content in the sample.The plates are incubated in aerobic conditions with a carbonedioxide concentration of 5 per cent, at 37 °C for 13-14 days,and the number of colonies is then scored under an invertedmicroscope. Care must be taken when manipulating the
dishes containing the colonies as the methylcellulose-basedmedium is viscous but not jellified. An inclined plate willresult in mixed and ‘comet’-shaped colonies making thescoring likely to be incorrect.
IDENTIFICATION OF THE COLONIES
The size and structure of the colonies depend on the typeof mature cells that are their constituents. 50 cells percolony is usually considered a minimum. The presence of haemoglobinised cells identifies progenitors of the erythroidlineage. As the amount of mature cells for each lineagelargely depends on the growth factors added to the cultures,performing differentiated counts is not recommended unlessotherwise prescribed.
Reference: PA/PH/Exp. 10A/T (01) 2 ANP
XXXX:1691
DACARBAZINE
Dacarbazinum
C6H10N6O M r 182.2
DEFINITION5-(3,3-Dimethyltriazeno)imidazole-4-carboxamide.Content : 98.5 per cent to 101.0 per cent (anhydroussubstance).
CHARACTERS Appearance : white or slightly yellowish crystalline powder.Solubility : slightly soluble in water and in anhydrousethanol.
IDENTIFICATION First identification : B.
Second identification: A, C.
A. Ultraviolet and visible absorption spectrophotometry( 2.2.25 ).Test solution. Dissolve 6.0 mg in 100.0 ml of 0.1 M hydrochloric acid . Dilute 10.0 ml of this solution to100.0 ml with 0.1 M hydrochloric acid .Spectral range : 200-400 nm.
Absorption maximum : at 323 nm.Shoulder : at 275 nm.Specific absorbance at the absorption maximum : 1067to 1088.
B. Infrared absorption spectrophotometry ( 2.2.24).Comparison : dacarbazine CRS .
C. Thin-layer chromatography ( 2.2.27 ).Test solution. Dissolve 2.0 mg of the substance to beexamined in methanol R and dilute to 5.0 ml with thesame solvent. Reference solution . Dissolve 2.0 mg of dacarbazine CRS in methanol R and dilute to 5.0 ml with the same solvent. Plate : TLC silica gel F 254 plate R. Mobile phase : glacial acetic acid R, water R, butanol R(1:2:5 V/V/V ). Application : 10 μl. Development : over a path of 15 cm. Drying : in air. Detection : examine in ultraviolet light at 254 nm. Results : the principal spot in the chromatogram obtainedwith the test solution is similar in position and size tothe principal spot in the chromatogram obtained withthe reference solution.
TESTS
Appearance of solution. The solution is clear ( 2.2.1) and
not more intensely coloured than reference solution BY6( 2.2.2, Method II ).Dissolve 0.5 g in a 210 g/l solution of citric acid R and diluteto 50.0 ml with the same solution.
Impurity A. Liquid chromatography ( 2.2.29). Use freshly prepared solutions and protect them from light.
Test solution. Dissolve 50.0 mg of the substance to beexamined and 75.0 mg of citric acid R in distilled water Rand dilute to 5.0 ml with the same solvent. Reference solution (a). Dissolve 5.0 mg of dacarbazineimpurity A CRS in distilled water R and dilute to 50.0 mlwith the same solvent. Dilute 5.0 ml of this solution to25.0 ml with distilled water R. Reference solution (b). Dissolve 5.0 mg of dacarbazineimpurity B CRS in distilled water R and dilute to 10.0 mlwith the same solvent. Dilute 1.0 ml of this solution to50.0 ml with distilled water R.Column :— size : l = 0.25 m, Ø = 4.5 mm;— stationary phase : octadecylsilyl silica gel for
chromatography R (5 μm)(14). Mobile phase : a 15.63 g/l solution of glacial acetic acid Rcontaining 2.33 g/l of sodium dioctyl sulfosuccinate R. Asthe mobile phase contains sodium dioctyl sulfosuccinate, it must be freshly prepared every day, and the column must beflushed with a mixture of equal volumes of methanol R andwater R after all tests have been completed or at the end of
the day, for at least 2 h. Flow rate : 1.2 ml/min.
(14) Nucleosil or Symmetry C18 are suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 71

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 72/176
Dacarbazine
Detection : spectrophotometer at 254 nm.
Injection : 25 μl of the test solutionand referencesolution (a).
Run time : 3 times the retention time of impurity A.
System suitability : reference solution (a):
— repeatability : maximum relative standard deviation of 2.0 per cent after 5 injections.
Limit :
— impurity A : not more than the area of the principal peakin the chromatogram obtained with reference solution (a)(0.2 per cent).
Impurity D. Head-space gas chromatography ( 2.2.28 ).
Test solution. Introduce 200.0 mg of the substance to beexamined into a 20 ml vial and firmly fix the septum and cap.Using a 10 μl syringe, inject 5 μl of water R into the vial.
Reference solution (a). Dilute 556.2 mg of dimethylamine solution R (impurity D) to 25.0 ml with water R (solution A).Firmly affix the septum and cap to a 20 ml vial. Using a 10 μlsyringe, inject 10 μl of solution A into the vial.
Reference solution (b). Firmly affix the septum and cap to a20 ml vial. Using a 10 μl syringe, inject 10 μl of solution Aand 10 μl of a 10 g/l solution of triethylamine R into the vial.
Column :
— material : fused silica;
— size : l = 30.0 m, Ø = 0.53 mm;
— stationary phase : base-deactivated polyethylene glycol R(film thickness 1.0 μm)(15).
Carrier gas : helium for chromatography R.
Flow rate : 13.2 ml/min.
Split flow : 5.1 ml/min.
Split ratio : 1:1.
Static head-space conditions that may be used :
— equilibration temperature : 60 °C;
— equilibration time : 10 min;
— transfer-line temperature : 90 °C;
— pressurisation time : 30 s.
Temperature :
Time
(min)
Temperature
(°C)
Column 0 - 3 35
3 - 11 35 → 65
Injection port 180
Detector 220
Detection : flame ionisation.
Injection : 1 μl.
System suitability : reference solution (b):
— resolution : minimum 2.5 between the peaks due toimpurity D and triethylamine.
Limit :
— impurity D : not more than the area of the correspondingpeak in the chromatogram obtained with referencesolution (a) (0.05 per cent).
Related substances. Liquid chromatography ( 2.2.29) asdescribed in the test for impurity A with the following
modifications. Mobile phase : mix 45 volumes of a 15.63 g/l solution of glacial acetic acid R containing 2.33 g/l of sodium dioctyl sulfosuccinate R with 55 volumes of methanol R.
Injection : 10 μl of the test solution and reference solution (b).
System suitability : reference solution (b):
— repeatability : maximum relative standard deviation of 2.5 per cent after 6 injections.
Limits :
— impurity B : not more than the area of the principal peakin the chromatogram obtained with reference solution (b)(0.1 per cent);
— unspecified impurities : for each impurity, not morethan the area of the principal peak in the chromatogramobtained with reference solution (b) (0.10 per cent);
— total : not more than 5 times the area of the principal peakin the chromatogram obtained with reference solution (b)(0.5 per cent);
— disregard limit : 0.5 times the area of the principal peakin the chromatogram obtained with reference solution (b)(0.05 per cent).
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity A 2. impurity C
Figure 1691.-1. – Chromatogram for the test for impurity A of dacarbazine
(15) Stabilwax-DB (Restek, N°10855) is suitable.
72 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 73/176
Devil’s claw dry extract
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity B 2. dacarbazine
Figure 1691.-2. – Chromatogram for the test for related substances of dacarbazine
Heavy metals ( 2.4.8 ): maximum 10 ppm.2.0 g complies with test A. Prepare the reference solutionusing 2 ml of lead standard solution (10 ppm Pb) R.
Water ( 2.5.12 ): maximum 0.5 per cent, determined on0.500 g.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAYDissolve 0.150 g in 30 ml of anhydrous acetic acid R. Titratewith 0.1 M perchloric acid , determining the end-point potentiometrically ( 2.2.20).1 ml of 0.1 M perchloric acid is equivalent to 18.22 mgof C6H10N6O.
IMPURITIESSpecified impurities : A, B, D.Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : C .
A. 2-azahypoxanthine,
B. 5-amino-1 H -imidazole-carboxamide,
C. 5-diazoimidazol-4-carboxamide,
D. dimethylamine.
Reagents
Sodium dioctyl sulfosuccinate. C20H37NaO7S. ( M r 444.6). XXXXXXX. [577-11-7].White or almost white, waxy solid.
Dimethylamine solution. XXXXXXX.A 400 g/l solution.Clear colourless solution.Density: about 0.89.bp: about 54 °C.
mp: about
37 °C.Base-deactivated polyethylene glycol. XXXXXXX.
Stationary phase for gas chromatography.Cross-linked base-deactivated polyethylene glycol speciallydesigned for amine analysis.
Reference: PA/PH/Exp. 13B/T (02) 65 ANP 1R
NOTE ON THE MONOGRAPH
The monograph has already been published in Pharmeuropa 16.1; it is republished to introduce a newmethod for the assay. Only comments on this updated assay are requested.
XXXX:1871
DEVIL’S CLAW DRY EXTRACT
Harpagophyti extractum siccumDEFINITIONDry extract obtained from Devil’s claw root (1095).Content : minimum 1.5 per cent of harpagoside (C24H30O11 ; M r 494.5) (dried extract).
PRODUCTION
The extract is produced from the herbal drug by anappropriate procedure using either water or a hydroalcoholicsolvent equivalent in strength to a maximum of 95 percent V/V ethanol.
© PHARMEUROPA Vol. 18, No. 1, January 2006 73

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 74/176
Devil’s claw dry extract
CHARACTERS
Appearance : light brown powder.
IDENTIFICATION
Thin-layer chromatography ( 2.2.27 ).Test solution. To 1.0 g of the extract to be examined add10 ml of methanol R and heat at 60 °C in a water-bath for10 min. Cool and filter.
Reference solution . Dissolve 1.0 mg of harpagoside R and2.5 mg of fructose R in 1.0 ml of methanol R.
Plate : TLC silica gel plate R.
Mobile phase : water R, methanol R, ethyl acetate R(8:15:77 V/V/V ).
Application : 20 μl, as bands.
Development : over a path of 10 cm.
Drying : in a current of warm air.
Detection : spray with a 10 g/l solution of phloroglucinol Rin ethanol (96 per cent) R and then with hydrochloric acid R ; heat at 80 °C for 5-10 min and examine in daylight.
Results : see below the sequence of the zones present in thechromatograms obtained with the reference solution and thetest solution. Furthermore, other zones may be present inthe chromatogram obtained with the test solution.
Top of the plate
_______ _______
Harpagoside: a green zone A green zone (harpagoside)
_______ _______
A yellow zone
A light green zone
Fructose: a yellowish-grey zone A yellowish-grey zone may bepresent (fructose)
A brown zone
Reference solution Test solution
TESTS
Loss on drying ( 2.8.17 ): maximum 5.0 per cent.
ASSAY
Liquid chromatography ( 2.2.29).
Test solution. Transfer 0.350 g of the extract to be examinedto a 100 ml volumetric flask, add 90 ml of methanol R andsonicate for 20 min. After cooling down to room temperaturedilute to 100.0 ml with methanol R and filter through amembrane filter (0.2 μm).
Reference solution . Dissolve 0.015 g of harpagoside R in100.0 ml of methanol R. Dilute 5.0 ml of this solution to10.0 ml with methanol R.
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. harpagoside
Figure 1871.-1. – Chromatogram for the harpagoside assay of Devil’s claw dry extract
Column :
— size : l = 0.10 m, Ø = 4.0 mm;
— stationary phase : octadecylsilyl silica gel for
chromatography R (5 μm).
Mobile phase : methanol R, water R (50:50 V/V ).
Flow rate : 1.5 ml/min.
Detection : spectrophotometer at 278 nm.
Injection : 10 μl.
Run time : 3 times the retention time of harpagoside.
Retention time : harpagoside = about 7 min.
Calculate the percentage content of harpagoside using the
following expression:
F 1 = area of the peak due to harpagoside in thechromatogram obtained with the test solution ;
F 2 = area of the peak due to harpagoside in thechromatogram obtained with the referencesolution;
m1 = mass of the extract to be examined, in grams;m2 = mass of harpagoside R in the reference solution,
in grams.
74 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 75/176
Diethylcarbamazine citrate
Reference: PA/PH/Exp. 10A/T (04) 12 ANP 1R
NOTE ON THE MONOGRAPH
A revision proposal was published in Pharmeuropa 16.2. It is now proposed to use the LC method for the related substances test and the assay. The TLC for impurities A and B is maintained as these impurities are not UV absorbent.
XXXX:0271
DIETHYLCARBAMAZINE CITRATE
Diethylcarbamazini citras
C16H29N3O8 M r 391.4
DEFINITION N , N -Diethyl-4-methylpiperazine-1-carboxamide dihydrogen2-hydroxypropane-1,2,3-tricarboxylate.
Content : 98.0 per cent to 101.0 per cent 102.0 per cent (dried substance).
CHARACTERS
Appearance : white or almost white, crystalline powder,slightly hygroscopic.
Solubility : very soluble in water, soluble in ethanol (96 percent), practically insoluble in acetone.
mp: about 138 °C, with decomposition.
IDENTIFICATION First identification : A, C .
Second identification: B, C .
A. Infrared absorption spectrophotometry ( 2.2.24).
Comparison : diethylcarbamazine citrate CRS .
B. Examine the chromatograms obtained in the test forimpurities A and B.
Results : the principal spot in the chromatogram obtainedwith the test solution is similar in position, colour andsize to the principal spot in the chromatogram obtainedwith reference solution (a).
C. Dissolve 0.1 g in 5 ml of water R. The solution gives the
reaction of citrates ( 2.3.1).TESTS
Solution S. Shake 2.5 g with water R until dissolved anddilute to 25 ml with the same solvent.
Appearance of solution. Solution S is not more opalescent than reference suspension II ( 2.2.1) and not more intenselycoloured than reference solution BY6 ( 2.2.2, Method II ).
Dimethylpiperazine and methylpiperazine Impurities Aand B. Thin-layer chromatography ( 2.2.27 ).
Test solution. Dissolve 0.5 g of the substance to be examinedin methanol R and dilute to 10 ml with the same solvent.
Reference solution (a). Dissolve 0.1 g of diethylcarbamazine
citrate CRS in methanol R and dilute to 2.0 ml with thesame solvent.
Reference solution (b). Dissolve 10 mg of methylpiperazine R in methanol R and dilute to100 ml with the same solvent. Reference solution (c). Dissolve 10 mg of dimethylpiperazine R in methanol R and dilute to100 ml with the same solvent. Plate : TLC silica gel G plate R .
Mobile phase : concentrated ammonia R, methyl ethyl ketone R, methanol R (5:30:65 V/V/V ). Application : 10 μl. Development : over 2/3 of the plate. Drying : at 100-105 °C. Detection : expose to iodine vapour for 30 min. Limits :— impurity A : any spot due to impurity A is not more
intense than the spot in the chromatogram obtained withreference solution (b) (0.2 per cent) ;
— impurity B : any spot due to impurity B is not moreintense than the spot in the chromatogram obtained withreference solution (c) (0.2 per cent).
Related substances. Liquid chromatography ( 2.2.29).Solution A. Dissolve 31.24 g of potassium dihydrogen phosphate R in water R and dilute to 1000 ml with the samesolvent.Test solution (a). Suspend 0.300 g of the substance tobe examined in solution A and dilute to 100.0 ml withsolution A. Filter or centrifuge and use the clear filtrate orsupernatant.Test solution (b). Dissolve 5.0 mg of the substance to beexamined in solution A and dilute to 50.0 ml with solution A. Reference solution (a). Dilute 1.0 ml of test solution (a) to100.0 ml with solution A. Dilute 1.0 ml of this solution to10.0 ml with solution A.
Reference solution (b). Dissolve 10 mg of citric acid R insolution A and dilute to 10 ml with solution A. Reference solution (c). To 3 ml of test solution (a) add 0.5 mlof strong hydrogen peroxide solution R and heat at 80 °Cfor 3 h. Dilute to 100.0 ml with solution A. Reference solution (d). Dissolve 5.0 mg of diethylcarbamazine citrate CRS in solution A anddilute to 50.0 ml with solution A.Column :— size : l = 0.15 m, Ø = 3.9 mm;— stationary phase : end-capped octadecylsilyl silica gel
for chromatography R (5 μm)(16). Mobile phase : mix 100 volumes of methanol R and
900 volumes of a 10 g/l solution of potassium dihydrogen phosphate R. Flow rate : 0.8 ml/min. Detection : spectrophotometer at 220 nm. Injection : 20 μl of test solution (a) and referencesolutions (a), (b) and (c). Run time : twice the retention time of diethylcarbamazine. Identification of impurities : use the chromatogramobtained with reference solution (b) to identify the peak dueto the citrate. Relative retention with reference to diethylcarbamazine(retention time = about 7 min): citrate = about 0.2.System suitability : reference solution (c):
— resolution : minimum 5 between the peaks due todiethylcarbamazine and the degradation product.
(16) Kromasil C18 is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 75

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 76/176
Dimenhydrinate
Limits :— unspecified impurities : for each impurity, not more
than the area of the principal peak in the chromatogramobtained with reference solution (a) (0.10 per cent);
— total : not more than 5 times the area of the principal peakin the chromatogram obtained with reference solution (a)(0.5 per cent);
— disregard limit : 0.5 times the area of the principal peakin the chromatogram obtained with reference solution (a)(0.05 per cent); disregard the peak due to the citrate.
Heavy metals ( 2.4.8 ): maximum 20 ppm.12 ml of solution S complies with test A. Prepare thereference solution using 10 ml of lead standard solution(2 ppm Pb) R.
Loss on drying ( 2.2.32 ): maximum 0.5 per cent, determinedon 1.000 g by drying in vacuo at 60 °C for 4 h.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAY
Dissolve 0.350 g in 25 ml of anhy dr ous ac eti c aci d R andadd 25 ml of ac eti c anhydrid e R. Using 0.2 ml of crystal vi ol et solution R as indicat or, titrate with 0.1 M per chlori c aci d until a greenish-blue colour is obtained.1 ml of 0.1 M per chl ori c acid is equivalent to 39.14 mgof C16H29N3O8.Liquid chromatography ( 2.2.29) as described in the test forrelated substances with the following modification. Injection : test solution (b) and reference solution (d).Calculate the percentage content of C16H29N3O8 from thedeclared content of diethylcarbamazine citrate CRS .
STORAGEIn an airtight container.
IMPURITIESSpecified impurities: A, B.
A. R = H: 1-methylpiperazine,
B. R = CH3 : 1,4-dimethylpiperazine.
Reference: PA/PH/Exp. 11/T (05) 74 ANP
NOTE ON THE MONOGRAPH It is proposed to revise the monograph to replace the TLC by an LC in the test for related substances.
XXXX:0601
DIMENHYDRINATE
Dimenhydrinatum
C24H28ClN5O3 M r 470.0
DEFINITION
Content :— diphenhydramine [2-(diphenylmethoxy)- N,N -
dimethylethylamine] (C17H21NO, M r 255.4): 53.0 per cent to 55.5 per cent (dried substance);
— 8-chlorotheophylline [8-chloro-3,7-dihydro-1,3-dimethyl-1 H -purine-2,6-dione] (C7H7ClN4O2, M r 214.6): 44.0 per
cent to 46.5 per cent (dried substance).
CHARACTERS Appearance : white or almost white, crystalline powder orcolourless crystals.Solubility : slightly soluble in water, freely soluble in ethanol(96 per cent).
IDENTIFICATION
First identification : C .Second identification: A, B, D.A. Melting point ( 2.2.14): 102 °C to 106 °C.B. Dissolve 0.1 g in a mixture of 3 ml of water R and 3 ml of
ethanol (96 per cent) R, add 6 ml of water R and 1 mlof dilute hydrochloric acid R and cool in iced water for30 min, scratching the wall of the tube with a glass rod if necessary to initiate crystallisation. Dissolve about 10 mgof the precipitate obtained in 1 ml of hydrochloric acid R,add 0.1 g of potassium chlorate R and evaporate todryness in a porcelain dish. A reddish residue is obtainedwhich becomes violet-red when exposed to ammoniavapour.
C. Infrared absorption spectrophotometry ( 2.2.24).
Comparison : dimenhydrinate CRS .D. Dissolve 0.2 g in 10 ml of ethanol (96 per cent) R. Add
10 ml of picric acid solution R and initiate cr ystallisationby scratching the wall of the tube with a glass rod. The
precipitate, washed with water R and dried at 100-105 °C,melts ( 2.2.14) at 130 °C to 134 °C.
TESTS
Appearance of solution. The solution is clear ( 2.2.1) andcolourless ( 2.2.2, Method II ).Dissolve 1.0 g in ethanol (96 per cent) R and dilute to 20 mlwith the same solvent.
pH ( 2.2.3): 7.1 to 7.6 for the filtrate.To 0.4 g add 20 ml of carbon dioxide-free water R, shake for2 min and filter.
Theophylline and substances related to diphenhydramine.Examine by thin-layer chromat ography ( 2.2.27 ), using sili ca g el GF 25 4 R as the coating substance.T est soluti on. Dissolve 0.40 g of the substance t o beexamined in methyl ene chl orid e R and dilut e t o 10 ml withthe same solvent. Ref er ence soluti on (a). Dissolve 20 mg of theophylline R inmethyl ene chl ori de R and dilut e t o 100 ml with the samesolvent. Ref er ence soluti on ( b). Dilute 5 ml of the test solution t o100 ml with methyl ene chlorid e R. Dilut e 10 ml of thissolution t o 100 ml with methylene chlorid e R.Apply separately to the plat e 5 μl of each solution. Developover a path of 15 cm using a mixture of 1 volume of conc entr ated ammonia R, 9 volumes of methanol R and90 volumes of methyl ene chl orid e R. Dry the plate in
a current of cold air and examine in ultraviolet light at 254 nm. Any spot corresponding t o theophylline in thechromat ogram obtained with the test solution is not moreint ense than the spot in the chromatogram obtained with
76 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 77/176
Dimenhydrinate
ref erence solution (a) (0.5 per cent). Spray with potassiumiod obismuthat e soluti on R. Allow the plate t o dry in air andspray with dilut e hy dr og en per oxid e solution R. Any spot in the chromat ogram obtained with the t est solution, apart from the principal spot, is not more intense than the spot in the chromat ogram obtained with ref erence solution (b)(0.5 per cent). Disregard any spot ext ending from thestarting point t o an R
f
of about 0.1.
Related substances. Liquid chromatography ( 2.2.29).
Test solution. Dissolve 35 mg of the substance to beexamined in the mobile phase and dilute to 100.0 ml witht he mobile phase.
Reference solution. Dilute 1.0 ml of the test solution to100.0 ml with the mobile phase. Dilute 1.0 ml of this solutionto 10.0 ml with the mobile phase.
Column :
— size : l = 0.25 m, Ø = 4.6 mm;
— stationary phase : diisopropyl cyanopropylsilyl silica gel for chromatography R (5 μm)(17).
Mobile phase. Mix 37 volumes of acetonitrile R and63 volumes of a solution prepared as follows: mix0.5 volumes of triethylamine R and 99.5 volumes of afreshly prepared 1.4 g/l solution of potassium dihydrogen phosphate R and adjust to pH 3.0 with phosphoric acid R.
Flow rate : 1.0 ml/min.
Detection : spectrophotometer at 220 nm.
Injection : 10 μl.
Run time : 4 times the retention time of diphenhydramine. Retention time : diphenhydramine = about 6 min;8-chlorotheophylline = about 5 min.
System suitability : test solution:
— resolution : minimum 3.0 between the peaks due todiphenhydramine and 8-chlorotheophylline.
Limits :
— unspecified impurities : not more than the sum of theareas of the 2 principal peaks in the chromatogramobtained with the reference solution (0.10 per cent);
— total : not more than 5 times the sum of the areas of the2 principal peaks in the chromatogram obtained with thereference solution (0.5 per cent) ;
— disregard limit : 0.5 times the sum of the areas of the2 principal peaks in the chromatogram obtained with thereference solution (0.05 per cent).
Heavy metals ( 2.4.8 ): maximum 20 ppm.
Dissolve 2.5 g in a mixture of 15 volumes of water R and85 volumes of acetone R and dilute to 25 ml with the samemixture of solvents. The solution complies with test B.Prepare the reference solution using lead standard solution(2 ppm Pb) prepared by diluting lead standard solution
(100 ppm Pb) R with a mixture of 15 volumes of water Rand 85 volumes of acetone R.
Loss on drying ( 2.2.32 ) : maximum 0.5 per cent, determinedon 1.000 g by drying in vacuo.
Sulphated ash ( 2.4.14): maximum 0.2 per cent, determinedon 1.0 g.
ASSAY
Diphenhydramine. Dissolve 0.200 g in 60 ml of anhydrousacetic acid R. Titrate with 0.1 M perchloric acid , determiningthe end-point potentiometrically ( 2.2.20).
1 ml of 0.1 M perchloric acid is equivalent to 25.54 mgof C17H21NO.
8-Chlorotheophylline . To 0.800 g add 50 ml of water R, 3 mlof dilute ammonia R1 and 0.6 g of ammonium nitrate Rand heat on a water-bath for 5 min. Add 25.0 ml of 0.1 M silver nitrate and continue heating on a water-bath for15 min with frequent swirling. Cool, add 25 ml of dilutenitric acid R and dilute to 250.0 ml with water R. Filterand discard the first 25 ml of the filtrate. Using 5 ml of ferric ammonium sulphate solution R2 as indicator, titrate
100.0 ml of the filtrate with 0.1 M ammonium thiocyanateuntil a yellowish-brown colour is obtained.
1 ml of 0.1 M silver nitrate is equivalent to 21.46 mgof C7H7ClN4O2.
IMPURITIES
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : A, B.
A. theophylline,
B. 8-[(2-(diphenylmethoxy)ethyl)(methyl)amino]-1,3-dimethyl-3,7-dihydro-1 H -purine-2,6-dione.
Reagents
Silica gel for chromatography, diisopropyl cyanopropylsilyl. XXXXXXX.A very finely divided silica gel chemically modified at thesurface by the bonding of diisopropyl cyanopropylsilyl
groups. The particle size is indicated after the name of thereagent in the tests where it is used.
(17) Nucleosil-CN or Zorbax SB-CN is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 77

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 78/176
Dipyridamole
Reference: PA/PH/Exp. 10C/T (05) 38 ANP
NOTE ON THE MONOGRAPH
A revision of the test for related substances is proposed toreplace the isocratic LC by a gradient LC that allows thecontrol of additional impurities.
XXXX:1199
DIPYRIDAMOLE
Dipyridamolum
C24H40N8O4 M r 504.6
DEFINITION2,2′,2″ ,2′′′-[[4,8-Di(piperidin-1-yl)pyrimido[5,4-d ]pyrimidine-2,6-diyl]dinitrilo]tetraethanol.Content : 98.5 per cent to 101.5 per cent (dried substance).
CHARACTERS Appearance : bright yellow, crystalline powder.Solubility : practically insoluble in water, freely soluble inacetone, soluble in anhydrous ethanol. It dissolves in dilutemineral acids.
IDENTIFICATION First identification : C.
Second identification: A, B, D.
A. Melting point ( 2.2.14): 162 °C to 168 °C.B. Ultraviolet and visible absorption spectrophotometry
( 2.2.25 ).Test solution. Dissolve 10 mg in a mixture of 1 volume of 0.1 M hydrochloric acid and 9 volumes of methanol Rand dilute to 50.0 ml with the same mixture of solvents.Dilute 5.0 ml of this solution to 100.0 ml with a mixtureof 1 volume of 0.1 M hydrochloric acid and 9 volumesof methanol R.
Spectral range : 220-350 nm. Absorption maxima : at 232 nm and 284 nm. Absorbance ratio : A284 / A232 = 1.25 to 1.45.
C. Infrared absorption spectrophotometry ( 2.2.24). Preparation : discs of potassium bromide R.Comparison : dipyridamole CRS .
D. Dissolve about 5 mg in a mixture of 0.1 ml of nitric acid Rand 2 ml of sulphuric acid R. An intense violet colouris produced.
TESTS
Related substances. Examine by liquid chromat ography( 2.2.29).
T est soluti on. Dissolve 10.0 mg in the mobile phase anddilut e t o 20.0 ml with the mobile phase.
Ref er ence soluti on (a). Dilut e 1.0 ml of the test solution to20.0 ml with the mobile phase. Dilut e 5.0 ml of this solutionto 50.0 ml with the mobile phase.
Ref er ence soluti on ( b). Dissolve 10.0 mg of dilti azemhy dr ochlori de CRS in the mobile phase and dilut e t o 10.0 mlwith the mobile phase. Dilute 1.0 ml of this solution to20.0 ml with reference solution (a).
The chromat ographic procedure may be carried out using :— a stainless st eel column 0.25 m long and 4.6 mm in
internal diameter packed with oc tylsi ly l silic a g el for chromatogr aphy R (5 μm),
— as mobile phase at a flow rat e of 1.3 ml/min a mixtureprepared as f ollows: dissolve 0.504 g of pot assiumdihy dr og en phosphat e R in 370 ml of wat er R andadjust to pH 3.0 with phosphori c aci d R ; add 80 ml of acetoni tri le R and 550 ml of methanol R,
— as det ect or a spectrophot omet er set at 290 nm,
maintaining the t emperature of the column at 30 °C.
Inject 20 μl of each solution and continue the
chromat ography of the test solution f or nine times theret ention time of dipyridamole. The t est is not valid unless,in the chromat ogram obtained with ref erence solution (b),the resolution between the peaks corresponding respectivelyto diltiazem and dipyridamole is at least 2.0. In thechromat ogram obtained with the test solution : the area of any peak, apart from the principal peak is not greater thanthe area of the peak in the chromat ogram obtained withref erence solution (a) (0.5 per cent); and the sum of theareas of all the peaks, apart from the principal peak, is not great er than twice the area of the peak in the chromat ogramobtained with ref erence solution (a) (1 per cent). Disregardany peak with an area less than 0.1 times that of the peak inthe chromat ogram obt ained with ref erence solution (a).
Related substances. Liquid chromatography ( 2.2.29).Test solution. Dissolve 0.200 g of the substance to beexamined in methanol R and dilute to 100 ml with the samesolvent.
Reference solution (a). Dilute 1.0 ml of the test solutionto 100.0 ml with methanol R. Dilute 1.0 ml of this solutionto 10.0 ml with methanol R.
Reference solution (b). Dissolve 4 mg of dipyridamoleimpurity D CRS in methanol R and dilute to 10 ml withthe same solvent. To 1 ml of this solution, add 200 mg of the substance to be examined and dilute to 100 ml withmethanol R.
Reference solution (c). Dissolve 5 mg of dipyridamole for
peak identification CRS (containing impurities A, B, C, Dand E) in methanol R and dilute to 10 ml with the samesolvent.
Column :
— size : l = 0.10 m, Ø = 4.0 mm;
— stationary phase : spherical end-capped octadecylsilyl silica gel for chromatography R (5 μm)(18) ;
— temperature : 45 °C.
Mobile phase :
— mobile phase A : dissolve 1.0 g of potassium dihydrogen phosphate R in 900 ml of water R, adjust to pH 7.0 with0.5 M sodium hydroxide and dilute to 1000 ml withwater R ;
— mobile phase B : methanol R ;
(18) Nucleosil C18 is suitable.
78 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 79/176
Dipyridamole
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity B 3. impurity D 5. impurity E 7. impurity A
2. impurity F 4. dipyridamole 6. impurity C
Figure 1199.-1. – Chromatogram for the test for related substances of dipyridamole
Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 - 1 40 60
1 - 15 40 → 5 60 → 95
15 - 20 5 → 40 95 → 60
20 - 25 40 60
Flow rate : 1.2 ml/min.
Detection : spectrophotometer at 295 nm.
Injection : 5 μl.
Identification of impurities : use the chromatogramsupplied with dipyridamole for peak identification CRS and the chromatogram obtained with reference solution (c)to identify the peaks due to impurities A, B, C, D and E.
Relative retention with reference to dipyridamole(retention time = about 6 min): impurity B = about 0.3;impurity D = about 0.9; impurity E = about 1.3;impurity C = about 1.5 ; impurity A = about 2.2.
System suitability : reference solution (b):
— resolution : minimum 1.5 between the peaks due to
impurity D and dipyridamole;— symmetry factor : maximum 2.5 for the peak due to
dipyridamole.
Limits :
— correction factor : for the calculation of content, multiplythe peak area of impurity B by 1.7;
— impurities A, B, C : for each impurity, not more than5 times the area of the principal peak in the chromatogramobtained with reference solution (a) (0.5 per cent) ;
— impurities D, E : for each impurity, not more than twicethe area of the principal peak in the chromatogramobtained with reference solution (a) (0.2 per cent) ;
— unspecified impurities : for each impurity, not morethan the area of the principal peak in the chromatogramobtained with reference solution (a) (0.10 per cent);
— total : not more than 10 times the area of the principalpeak in the chromatogram obtained with referencesolution (a) (1.0 per cent);
— disregard limit : 0.5 times the area of the principal peakin the chromatogram obtained with reference solution (a)(0.05 per cent).
Chlorides ( 2.4.4): maximum 200 ppm.
To 0.250 g add 10 ml of water R and shake vigorously. Filter,rinse the filter with 5 ml of water R and dilute to 15 ml with
water R.Loss on drying ( 2.2.32 ) : maximum 0.5 per cent, determinedon 1.000 g by drying in an oven at 100-105 °C.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAY
Dissolve 0.400 g in 70 ml of methanol R. Titrate with0.1 M perchloric acid , determining the end-point potentiometrically ( 2.2.20).
1 ml of 0.1 M perchloric acid is equivalent to 50.46 mgof C24H40N8O4.
STORAGE
Protected from light.
IMPURITIES
Specified impurities: A, B, C, D, E.
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for
pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : F, G.
© PHARMEUROPA Vol. 18, No. 1, January 2006 79

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 80/176
Dopamine hydrochloride
A. R1 = R2 = R3 = NC5H10, R4 = N(CH2-CH2-OH)2 :
2,2′-[[4,6,8-tri(piperidin-1-yl)pyrimido[5,4-d ]pyrimidin-2-yl]nitrilo]diethanol,
B. R1 = R2 = R4 = N(CH2-CH2-OH)2, R3 = NC5H10 :2,2′,2″ ,2′′′,2′′′′,2′′′′′-[[8-(piperidin-1-yl)pyrimido[5,4-d ]pyrimidine-2,4,6-triyl]trinitrilo]hexaethanol,
C. R1 = R3 = NC5H10, R2 = Cl, R4 = N(CH2-CH2-OH)2 :2,2′-[[2-chloro-4,8-di(piperidin-1-yl)pyrimido[5,4-d ]pyrimidin-6-yl]nitrilo]diethanol,
D. R1 = R3 = NC5H10, R2 = N(CH2-CH2-OH)2, R4 = NH-CH2-CH2-OH: 2,2′-[[6-[(2-hydroxyethyl)amino]-4,8-dipiperidin-1-ylpyrimido[5,4-d ]pyrimidin-2-yl]imino]diethanol,
E. R1 = R4 = N(CH2-CH2-OH)2, R2 = R3 = NC5H10 :2,2′,2″ ,2′′′-[(6,8-dipiperidin-1-ylpyrimido[5,4-d ]pyrimidine-2,4-diyl)dinitrilo]tetraethanol,
F. R1 = NC5H10, R2 = R4 = N(CH2-CH2-OH)2, R3 = NH-CH2-CH2-OH: 2,2′,2″ ,2′′′-[[4-[(2-hydroxyethyl)amino]-8-piperidin-1-ylpyrimido[5,4-d ]pyrimidine-2,6-diyl]dinitrilo]tetraethanol,
G. R1 = R3 = NC5H10, R2 = R4 = Cl : 2,6-dichloro-4,8-dipiperidin-1-ylpyrimido[5,4-d ]pyrimidine.
Reference: PA/PH/Exp. 10A/T (05) 67 ANP NOTE ON THE MONOGRAPH
It is proposed to replace the current TLC test for related substances by an LC test. In addition, the limits of content have been tightened based on batch data, and therecommendations for storage have been supplemented.
XXXX:0664
DOPAMINE HYDROCHLORIDE
Dopamini hydrochloridum
C8H12ClNO2 M r 189.6
DEFINITION
4-(2-Aminoethyl)benzene-1,2-diol hydrochloride.
Content : 98.0 99.0 per cent to 102.0 101.0 per cent (driedsubstance).
CHARACTERS
Appearance : white or almost white, crystalline powder
Solubility : freely soluble in water, soluble in ethanol (96 per
cent), sparingly soluble in acetone and in methylene chloride.IDENTIFICATION
First identification : B, E.
Second identification: A, C, D, E.
A. Ultraviolet and visible absorption spectrophotometry( 2.2.25 ).
Test solution. Dissolve 40.0 mg in 0.1 M hydrochloric acid and dilute to 100.0 ml with the same acid.Dilute 10.0 ml of this solution to 100.0 ml with 0.1 M hydrochloric acid .
Spectral range : 230-350 nm.
Absorption maximum : at 280 nm.
Specific absorbance at the absorption maximum : 136to 150.
B. Infrared absorption spectrophotometry ( 2.2.24).
Preparation : discs of potassium chloride R.
Comparison : dopamine hydrochloride CRS .
C. Dissolve about 5 mg in a mixture of 5 ml of 1 M hydrochloric acid and 5 ml of water R. Add 0.1 mlof sodium nitrite solution R containing 100 g/l of ammonium molybdate R. A yellow colour develops whichbecomes red on the addition of strong sodium hydroxide solution R.
D. Dissolve about 2 mg in 2 ml of water R and add 0.2 mlof ferric chloride solution R2 . A green colour developswhich changes to bluish-violet on the addition of 0.1 g of hexamethylenetetramine R.
E. It gives reaction (a) of chlorides ( 2.3.1).
TESTS
Appearance of solution. The solution is clear ( 2.2.1) andnot more intensely coloured than reference solution B6 orY6 ( 2.2.2, Method II ).
Dissolve 0.4 g in water R and dilute to 10 ml with the samesolvent.
Acidity or alkalinity. Dissolve 0.5 g in carbon dioxide-freewater R and dilute to 10 ml with the same solvent. Add0.1 ml of methyl red solution R and 0.75 ml of 0.01 M sodium hydroxide. The solution is yellow. Add 1.5 ml of 0.01 M hydrochloric acid . The solution is red.
Related substances. Examine by thin-layer chromatography( 2.2.27 ), using sili c a g el G R as the coating substance.
T est soluti on. Dissolve 0.15 g of the substance t o beexamined in methanol R and dilut e t o 5 ml with the samesolvent.
Ref er ence solution ( a). Dissolve 7.5 mg of 4-O-methyl dopamine hy dr ochlori de R in methanol R anddilut e t o 100 ml with the same solvent.
Ref er ence solution ( b). Dissolve 7.5 mg eachof 3-O -methyl dopamine hy dr ochl ori d e R and4-O-methyl dopamine hy dr ochl ori de R in methanol R anddilut e t o 100 ml with the same solvent.
Apply t o the plat e 10 μl of each solution. Develop over a pathof 15 cm using a mixture of 2 volumes of anhy dr ous f ormi c aci d R, 7 volumes of wat er R, 36 volumes of methanol Rand 52 volumes of chl or of orm R. Allow the plate t o dry inair f or 15 min. Spray evenly and abundantly with a mixtureof equal volumes of potassium f erri cyanid e soluti on R andf erri c chl orid e soluti on R1, prepared immediately bef oreuse. Any spot in the chromatogram obtained with the t est solution with an Rf value higher than that of the principal
spot is not more int ense than the spot in the chromat ogramobtained with ref erence solution (a) (0.25 per cent). Thetest is not valid unless the chromatogram obtained withref erence solution (b) shows two clearly separated spots.
80 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 81/176
Dopamine hydrochloride
Related substances. Liquid chromatography ( 2.2.29). Protect the solutions from light.
Buffer solution. Dissolve 21 g of citric acid R in 200 ml of 1 M sodium hydroxide and dilute to 1000 ml with water R.To 600 ml of this solution add 400 ml of 0.1 M hydrochloric acid .Test solution. Dissolve 50.0 mg of the substance to be
examined in mobile phase A and dilute to 25.0 ml withmobile phase A. Reference solution (a). Dilute 1.0 ml of the test solution to100.0 ml with mobile phase A. Dilute 1.0 ml of this solutionto 10.0 ml with mobile phase A. Reference solution (b). Dissolve 10 mg of 3-O-methyldopamine hydrochloride R (impurity B)and 10 mg of 4-O-methyldopamine hydrochloride R(impurity A) in mobile phase A and dilute to 100.0 ml withmobile phase A. Dilute 6.0 ml of this solution to 25.0 ml withmobile phase A.Column :— size : l = 0.15 m, Ø = 3.9 mm;— stationary phase : spherical end-capped octadecylsilyl
silica gel for chromatography R (4 μm)(19). Mobile phase :— mobile phase A : dissolve 1.08 g of sodium
octanesulphonate R in 880 ml of the buffer solution andadd 50 ml of methanol R and 70 ml of acetonitrile R ;
— mobile phase B : dissolve 1.08 g of sodiumoctanesulphonate R in 700 ml of the buffer solution andadd 100 ml of methanol R and 200 ml of acetonitrile R ;
Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 - 5.0 90 10
5.0 - 20.0 90 → 40 10 → 60
20.0 - 25.0 40 60
Flow rate : 1.0 ml/min. Detection : spectrophotometer at 280 nm. Injection : 10 μl.
Retention time : dopamine = about 5 min.System suitability : reference solution (b):— resolution : minimum 5.0 between the peaks due to
impurities B and A. Limits :— unspecificied impurities : for each impurity, not more
than the area of the principal peak in the chromatogram
obtained with reference solution (a) (0.10 per cent);— total : not more than twice the area of the principal peak
in the chromatogram obtained with reference solution (a)(0.2 per cent);
— disregard limit : 0.5 times the area of the principal peakin the chromatogram obtained with reference solution (a)(0.05 per cent).
Heavy metals ( 2.4.8 ): maximum 20 ppm.1.0 g complies with test C. Prepare the reference solutionusing 2 ml of lead standard solution (10 ppm Pb) R.
Loss on drying ( 2.2.32 ) : maximum 0.5 per cent, determinedon 1.000 g by drying in an oven at 100-105 °C for 2 h.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAY In order to avoid overheating in the reaction medium, mix thoroughly throughout the titration and stop the titrationimmediately after the end-point has been reached.
Dissolve 0.1500 g in 10 ml of anhydrous formic acid R. Add50 ml of acetic anhydride R. Titrate with 0.1 M perchloric acid , determining the end-point potentiometrically ( 2.2.20).1 ml of 0.1 M perchloric acid is equivalent to 18.96 mgof C8H12ClNO2.
STORAGEIn an airtight container, under nitrogen, protected from light.
IMPURITIESOther detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limited
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. dopamine 2. impurity B 3. impurity A 4. impurity C
Figure 0664.-1. – Chromatogram for the test for related substances of dopamine hydrochloride
(19) Novapak C18 is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 81

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 82/176
Dorzolamide hydrochloride
by the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : A, B, C.
A. 5-(2-aminoethyl)-2-methoxyphenol (4-O-methyldopamine),
B. 4-(2-aminoethyl)-2-methoxyphenol (3-O-methyldopamine),
C. 2-(3,4-dimethoxyphenyl)ethanamine.
Reference: PA/PH/Exp. 10B/T (04) 104 ANP
XXXX:2359
DORZOLAMIDE HYDROCHLORIDE
Dorzolamidi hydrochloridum
C10H17N2O4S3Cl M r 360.9
DEFINITION
Hydrochloride of (4S ,6S )-4-(ethylamino)-5,6-dihydro-6-methyl-4 H -thieno[2,3-β]thiopyrane-2-sulphonamide7,7-dioxide.
Content : 99.0 per cent to 101.0 per cent (dried substance).
CHARACTERS
Appearance : white or almost white, crystalline powder.
Solubility : soluble in water, slightly soluble in methanol,very slightly soluble in anhydrous ethanol.
It shows polymorphism.
IDENTIFICATION
A. Infrared absorption spectrophotometry ( 2.2.24).
Comparison : dorzolamide hydrochloride CRS .
If the spectra obtained in the solid state show differences,dissolve the substance to be examined and the referencesubstance separately in methanol R, evaporate to dryness
and record new spectra using the residues.B. It gives reaction (a) of chlorides ( 2.3.1).
TESTS
Impurity A. Liquid chromatography ( 2.2.29).Solvent mixture : acetonitrile R, glacial acetic acid R,1,1-dimethylethyl methyl ether R (3:10:87 V/V/V ).
Test solution. In a centrifuge tube, dissolve 20.0 mg of thesubstance to be examined in 4 ml of dilute ammonia R4,add 4 ml of ethyl acetate R, and mix. Separate the organic
layer and transfer it to a separate centrifuge tube. Add4 ml of ethyl acetate R to the aqueous layer, mix, separatethe organic layer, and combine it with the 1st extract.Evaporate the combined organic layers to dryness in a waterbath at 50 °C under a stream of nitrogen R. Dissolve theresidue in 3 ml of acetonitrile R, add 0.06 ml (3 drops) of (S)-(-)- α -methylbenzyl isocyanate R, and heat in a waterbath at 50 °C for 5 min. Evaporate to dryness in a waterbath at 50 °C under a stream of nitrogen R. Dissolve theresidue in 10 ml of the solvent mixture.
Reference solution. In a centrifuge tube, dissolve 18.0 mg of dorzolamide hydrochloride CRS and 2.0 mg of dorzolamideimpurity A CRS in 4 ml of dilute ammonia R4, and proceedas indicated for the test solution beginning with “add 4 ml
of ethyl acetate R, and mix”.Column :— size : l = 0.25 m, Ø = 4.6 mm;— stationary phase : silica gel for chromatography R
(5 μm), with a pore size of 8 nm, a specific area of 180 m2 /g and a porosity of 60 per cent (20).
Mobile phase : water R, acetonitrile R, heptane R,1,1-dimethylethyl methyl ether R (0.2:2:35:63 V/V/V/V ).
Flow rate : 2 ml/min.
Detection : spectrophotometer at 254 nm.
Injection : 10 μl. Run time : twice the retention time of dorzolamide.
Relative retention with reference to dorzolamide (retention
time = about 10 min): impurity A = about 1.4.System suitability : reference solution:— resolution : minimum 4.0 between the peaks due to
dorzolamide and impurity A.Calculate the percentage content of impurity A using thefollowing expression:
A = area of the peak due to impurity A in thechromatogram obtained with the test solution,
B = area of the peak due to dorzolamide in thechromatogram obtained with the test solution.
Limit :— impurity A : maximum 0.5 per cent.
Related substances. Liquid chromatography ( 2.2.29).Test solution. Dissolve 50.0 mg of the substance to beexamined in mobile phase A and dilute to 50.0 ml withmobile phase A.
Reference solution (a). Dissolve 1.0 ml of the test solution to100.0 ml with mobile phase A. Dilute 1.0 ml of this solutionto 10.0 ml with mobile phase A.
Reference solution (b). Dissolve 2 mg of dorzolamidehydrochloride for system suitability CRS (containing
impurities B, C and D) in mobile phase A and dilute to 2 mlwith mobile phase A.
(20) Zorbax Rx-SIL is suitable.
82 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 83/176
Dorzolamide hydrochloride
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity D 2. impurity C 3. dorzolamide 4. impurity B
Figure 2359.-1. – Chromatogram for the test for related substances of dorzolamide hydrochloride: test solution spiked with impurities B, C and D
Column :— size : l = 0.25 m, Ø = 4.6 mm;— stationary phase : end-capped octadecylsilyl silica gel
for chromatography R (5 μm)(21) ;— temperature : 35 °C. Mobile phase :— mobile phase A : mix 65 ml of acetonitrile R and 935 ml of
a 3.7 g/l solution of potassium dihydrogen phosphate R ;— mobile phase B : acetonitrile R ;
Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 - 15 100 0
15 - 30 100 → 50 0 → 50
30 - 37 50 → 100 50 → 0
37 - 44 100 0
Flow rate : 1.5 ml/min. Detection : spectrophotometer at 254 nm. Injection : 10 μl. Relative retention with reference to dorzolamide(retention time = about 11 min): impurity C = about 0.9;impurity B = about 1.1.System suitability : reference solution (b):— resolution : minimum 2.0 between the peaks due to
impurity C and dorzolamide;— peak-to-valley ratio : minimum 2.0, where H p = height
above the baseline of the peak due to impurity B and H v = height above the baseline of the lowest point of
the curve separating this peak from the peak due todorzolamide. Limits :— impurity C : not more than the area of the principal peak
in the chromatogram obtained with reference solution (a)(0.1 per cent);
— unspecified impurities : for each impurity, not morethan the area of the principal peak in the chromatogramobtained with reference solution (a) (0.10 per cent);
— total : not more than twice the area of the principal peakin the chromatogram obtained with reference solution (a)(0.2 per cent);
— disregard limit : 0.5 times the area of the principal peak
in the chromatogram obtained with reference solution (a)(0.05 per cent).
Loss on drying ( 2.2.32 ) : maximum 0.5 per cent, determinedon 1.000 g by drying in an oven at 105 °C.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAY
Dissolve 0.250 g in a mixture of 5.0 ml of 0.01 M hydrochloric acid and 50 ml of ethanol (96 per cent) R, using sonicationif necessary. Carry out a potentiometric titration ( 2.2.20),using 0.1 M sodium hydroxide. Read the volume addedbetween the 1st and the 3rd point of inflexion.
1 ml of 0.1 M sodium hydroxide is equivalent to 18.05 mgof C10H17N2O4S3Cl.
IMPURITIES
Specified impurities: A, C.
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : B, D.
A. (4 R,6 R)-4-(ethylamino)-5,6-dihydro-6-methyl-4 H -thieno[2,3-b]thiopyran-2-sulphonamide 7,7-dioxide,
B. (4 RS ,6S R)-4-(ethylamino)-5,6-dihydro-6-methyl-4 H -thieno[2,3-b]thiopyran-2-sulphonamide 7,7-dioxide,
(21) Inertsil ODS-2 and Kromasil C18 are suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 83

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 84/176
Ethambutol hydrochloride
C. [2-[[(4S ,6S )-2-(aminosulphonyl)-6-methyl-7,7-dihydro-4 H -thieno[2,3-b]thiopyran-4-yl]amino]ethyl]boronic acid,
D. (4S ,6S )-4-(amino)-5,6-dihydro-6-methyl-4 H -thieno[2,3-b]thiopyran-2-sulphonamide 7,7-dioxide.
ReagentsAmmonia, dilute R4. XXXXXXX.Content : 8.4 g/l to 8.6 g/l of NH3 ( M r 17.03).Dilute 3.5 g of concentrated ammonia R to 100 ml withwater R.
(S)-(-)- -Methylbenzyl isocyanate. C9H9NO. ( M r 147.2). XXXXXXX. [14649-03-7]. ((1S )-1-Nitrosoethyl)benzene.A colourless liquid.
: about 1.044.: about 1.514.
bp: 55 °C to 56 °C.
NOTE: do not use the reagent if it is coloured.
Reference: PA/PH/Exp. 10B/T (05) 26 ANP
NOTE ON THE MONOGRAPH
The control of impurities has been improved by theintroduction of an LC method which uses a derivatisationwith a chiral reagent. Only the first identification serieshas been kept as the substance is not used in pharmacies.The assay by optical rotation has been replaced by atitration method.
XXXX:0553
ETHAMBUTOL HYDROCHLORIDEEthambutoli hydrochloridum
C10H26Cl2N2O2 M r 277.2
DEFINITION
2,2′-(Ethylenediimino)bis[(2S )-butan-1-ol] dihydrochloride(2S ,2′S )-2,2′-(Ethane-1,2-diyldiimino)dibutan-1-oldihydrochloride.
Content : 99.0 per cent to 101.0 per cent (dried substance).
CHARACTERS
Appearance : white or almost white, crystalline powder,hygroscopic.
Solubility : freely soluble in water, soluble in ethanol (96 percent).
It melts at about 202 °C.
IDENTIFICATION First i dentifi cati on: A, D.
Second i d entificati on: B, C, D.
A. Infrared absorption spectrophotometry ( 2.2.24).
Comparison : ethambutol hydrochloride CRS .
Examine the substances prepared as discs.
B. Examine the chromatograms obtained in the test f or2-aminobutanol impurity A.
Results : the principal spot in the chromatogram obtainedwith t est solution (b) is similar in position, colour and sizet o the principal spot in the chromat ogram obtained withref erence solution (b).
C. Dissolve 0.1 g in 10 ml of wat er R and add 0.2 ml of c opper sulphate soluti on R. Add 0.5 ml of dilut e sodiumhyd r oxid e solution R. A blue colour is produced.
D. B. It gives reaction (a) of chlorides ( 2.3.1).
TESTS
pH ( 2.2.3) : 3.7 to 4.0.
Dissolve 0.2 g in 10 ml of carbon dioxide-free water R.
2-Aminobutanol Impurity A. Thin-layer chromatography( 2.2.27 ).
Test solution (a). Dissolve 0.50 g of the substance to beexamined in methanol R and dilute to 10 ml with the same
solvent.T est soluti on (b). Dilute 1 ml of t est solution (a) t o 10 mlwith methanol R.
Reference solution (a). Dissolve 50.0 mg of 2-aminobutanol R (impurity A) in methanol R anddilute to 10 ml with the same solvent. Dilute 1 ml of thissolution to 10 ml with methanol R.
Reference solution (b). Dissolve 50 mg of ethambutol hydrochloride CRS and 5 mg of 2-aminobutanol R inmethanol R and dilute to 10 ml with the same solvent.
Plate : TLC silica gel G plate R .
Mobile phase : concentrated ammonia R, water R,methanol R (10:15:75 V/V/V ).
Application : 2 μl. Development : over a path of 15 cm.
Drying : in air ; heat at 110 °C for 10 min.
Detection : cool and spray with ninhydrin solution R1 ; heat the plate at 110 °C for 5 min.
System suitability : reference solution (b):
— the chromatogram shows 2 clearly separated spots.
Limit :
— impurity A : any spot corresponding to 2-aminobutanolimpurity A in the chromat ogram obtained with t est solution (a) is not more intense than the spot in thechromatogram obtained with reference solution (a)(1.0 per cent).
Related susbstances. Liquid chromatography ( 2.2.29). Prepare the solutions immediately before use.
84 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 85/176
Ethambutol hydrochloride
Test solution. Suspend 4.0 mg of the substance to beexamined in 4.0 ml of acetonitrile R1 and add 100 μl of triethylamine R. Sonicate the mixture for 5 min. Add 15 μlof R-(+)-phenylethylisocyanate R and heat the mixture for20 min at 70 °C.
Reference solution (a). Dilute 0.50 ml of the test solution to100.0 ml with acetonitrile R1.
Reference solution (b). Treat 4.0 mg of ethambutol for system suitability CRS (containing impurity B) as describedunder test solution.
Column :
— size : l = 0.10 m, Ø = 4.6 mm;
— stationary phase : end-capped octadecylsilyl silica gel for chromatography R (3 μm)(22) ;
— temperature : 40 °C.
Mobile phase :
— mobile phase A : methanol R, water R (50:50 V/V ) ;
— mobile phase B : methanol R ;
Time
(min) Mobile phase A(per cent V/V )
Mobile phase B(per cent V/V )
0 - 30 71 29
30 - 35 71 → 0 29 → 100
35 - 37 0 100
37 - 38 0 → 71 100 → 29
Flow rate : 1.0 ml/min.
Detection : spectrophotometer at 215 nm.
Injection : 10 μl.
Relative retention with reference to ethambutol (retentiontime = about 14 min): impurity B = 1.3.System suitability : reference solution (b):— resolution : minimum 4.0 between the peaks due to
ethambutol and impurity B. Limits :— impurity B : not more than twice the area of the principal
peak in the chromatogram obtained with referencesolution (a) (1.0 per cent);
— unspecified impurities with a relative retention withreference to ethambutol of 0.75 to 1.5 : for each impurity,not more than 0.20 times the area of the peak due toethambutol in the chromatogram obtained with referencesolution (a) (0.10 per cent) ;
— total : not more than twice the area of the principal peakin the chromatogram obtained with reference solution (a)(1.0 per cent);
— disregard limit : 0.1 times the area of the peak due toethambutol in the chromatogram obtained with referencesolution (a) (0.05 per cent).
Heavy metals ( 2.4.8 ): maximum 10 ppm.2.0 g complies with test C. Prepare the ref erence solutionusing 2 ml of l ead standar d soluti on (10 ppm P b) R.Dissolve 2.0 g in water R and dilute to 20 ml with the samesolvent. 12 ml of the solution complies with test A. Preparethe reference solution using 2 ml of lead standard solution(1 ppm Pb) R.
Loss on drying ( 2.2.32 ) : maximum 0.5 per cent, determinedon 0.500 g by drying in an oven at 100-105 °C for 3 h.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity C 2. ethambutol 3. impurity B
Figure 0553.-1. – Chromatogram for the test for related substances of ethambutol hydrochloride: test solution spiked with impurities B and C
(22) LUNA C18(2) is suitable
© PHARMEUROPA Vol. 18, No. 1, January 2006 85

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 86/176
Fenoterol hydrobromide
ASSAY
To 70 ml of dilut e ammonia R2 add 4.0 ml of c opper sulphate soluti on R, mix, add 5.0 ml of dilute sodiumhy dr oxi de soluti on R and dilute to 100 ml with wat er R.Dissolve 0.100 g of the subst ance t o be examined in 20 ml of this solution and dilut e t o 25.0 ml with the same solution.Prepare a ref erence solution in the same manner using
0.100 g of ethambutol hy dr ochl orid e CRS . Measure theangle of optical rotation ( 2.2.7 ) of the solutions at 436 nm.
Calculat e the content of C10H26Cl2N2O2 from the angles of optical rotation measured and the concentrations of thesolutions.
Dissolve 0.150 g in 50 ml of water R and add 1.0 ml of 0.1 M hydrochloric acid . Carry out a potentiometric titration( 2.2.20), using 0.1 M sodium hydroxide. Read the volumeadded between the 2 points of inflection.
1 ml of 0.1 M sodium hydroxide is equivalent to 27.72 mgof C10H26Cl2N2O2.
STORAGE
In an airtight container.
IMPURITIES
Specified impurities: A, B.
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : C.
A. 2-aminobutan-1-ol,
B. (2 R,2′S )-2,2′-(ethane-1,2-diyldiimino)dibutan-1-ol(meso-ethambutol),
C. (2 R,2′ R)-2,2′-(ethane-1,2-diyldiimino)dibutan-1-ol(( R, R)-ethambutol).
Reagents
R -(+)-1-Phenylethyl isocyanate. C9H9NO. ( M r 147.18). XXXXXXX. [33375-06-3]. R-(+)-α-Methylbenzyl isocyanate.Content : minimum 98.0 per cent.A colourless liquid.bp: about 74 °C to 76 °C.
Reference: PA/PH/Exp. 10A/T (05) 57 ANP
NOTE ON THE MONOGRAPH
Currently, the control of impurities of fenoterol hydrobromide is performed using a UV-photometric test for impurity B and an LC test for impurity A (diastereoisomers). In the framework of the special revision programme it is proposed to modify slightly the existing LC test conditionsto cover impurity A and other related substances. A newdetection wavelength has been chosen. Only commentson the tests for impurity A and related substances arerequested.
XXXX:0901
FENOTEROL HYDROBROMIDE
Fenoteroli hydrobromidum
C17H22BrNO4 M r 384.3
DEFINITION(1 RS )-1-(3,5-Dihydoxyphenyl)-2-[[(1 RS )-2-(4-hydroxyphenyl)-1-methylethyl]amino]ethanol hydrobromide.Content : 99.0 per cent to 101.0 per cent (dried substance).
CHARACTERS Appearance : white or almost white, crystalline powder.Solubility : soluble in water and in ethanol (96 per cent).
IDENTIFICATION First identification : B, E .Second identification: A, C, D, E .A. Ultraviolet and visible absorption spectrophotometry
( 2.2.25 ).Test solution. Dissolve 50.0 mg in dilute hydrochloric acid R1 and dilute to 50.0 ml with the same acid. Dilute5.0 ml to 50.0 ml with dilute hydrochloric acid R1.Spectral range : 230-350 nm. Absorption maximum : at 275 nm.Shoulder : at about 280 nm.Specific absorbance at the absorption maximum : 80to 86.
B. Infrared absorption spectrophotometry ( 2.2.24). Pr epar ati on : discs.Comparison : Ph. Eur. reference spectrum of fenoterol hydrobromide.
C. Thin-layer chromatography ( 2.2.27 ).Test solution. Dissolve 10 mg of the substance to beexamined in ethanol (96 per cent) R and dilute to 10 mlwith the same solvent. Reference solution. Dissolve 10 mg of fenoterol hydrobromide CRS in ethanol (96 per cent) R and diluteto 10 ml with the same solvent. Plate : TLC silica gel G plate R. Mobile phase : concentrated ammonia R, water R,aldehyde-free methanol R (1.5:10:90 V/V/V ). Application : 2 μl. Development : over a path of 15 cm.
86 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 87/176
Fenoterol hydrobromide
Drying : in air.
Detection : spray with a 10 g/l solution of potassium permanganate R.
Results : the principal spot in the chromatogram obtainedwith the test solution is similar in position, colour andsize to the principal spot in the chromatogram obtainedwith the reference solution.
D. Dissolve about 10 mg in a 20 g/l solution of disodiumtetraborate R and dilute to 50 ml with the same solution.Add 1 ml of a 10 g/l solution of aminopyrazolone R,10 ml of a 2 g/l solution of potassium ferricyanide Rand 10 ml of methylene chloride R. Shake and allow toseparate. A reddish-brown colour develops in the lowerlayer.
E. It gives reaction (a) of bromides ( 2.3.1).
TESTS
Solution S. Dissolve 2.00 g in carbon dioxide-free water Rand dilute to 50.0 ml with the same solvent.
Appearance of solution. Solution S is clear ( 2.2.1) and not
more intensely coloured than reference solution Y7 ( 2.2.2, Method II ).
pH ( 2.2.3): 4.2 to 5.2 for solution S.
Phenone : maximum 0.2 per cent. The absorbance ( 2.2.25 )of solution S at 330 nm has a maximum of 0.42.
Impurity A Diastereoisomers. Liquid chromatography( 2.2.29). Prepare the solutions immediately before use.
Test solution. Dissolve 25.0 mg 24.0 mg of the substance tobe examined in water R and dilute to 10.0 ml 20.0 ml withthe same solvent.
Reference solution (a). Dissolve 25.0 mg 24.0 mg of fenoterol hydrobromide CRS (containing impurity A) inwater R and dilute to 10.0 ml 20.0 ml with the same solvent.
Reference solution (b). Dissolve 6 mg of fenoterol hydrobromide for peak identification CRS (containingimpurities B and C) in water R and dilute to 5 ml with thesame solvent.
Reference solution (c). Dilute 10.0 ml of the test solutionto 50.0 ml with water R. Dilute 1.0 ml of this solution to100.0 ml wit h water R.
Column :— size : l = 0.25 m 0.15 m, Ø = 4.6 mm;— stationary phase : octadecylsilyl silica gel for
chromatography R (5 μm t o 10 μm)(23). Mobile phase. Dissolve 24 g of disodium hydrogen phosphate R in 1000 ml of water R and adjust to pH 8.5with phosphoric acid R. Mix 69 volumes of this solution
with 1 volume of a 9 g/l solution of potassium of hydrogen phosphate R and 35 volumes of methanol R2 . t o a mixtureof 1 volume of a 9 g/l solution of potassium dihy dr og en phosphat e R and 69 volumes of a 24 g/l solution of disodium hy dr ogen phosphat e R, adjust ed t o pH 8.5 using phosphori c aci d R, add 30 volumes of methanol R. Flow rate : 1 ml/min. Detection : spectrophotometer at 280 nm 215 nm. Injection : 20 μl loop injector of the test solution andreference solution (a). Run time : 3 times the retention time of fenoterol. Relative retention with reference to fenoterol (retentiontime = about 7 min): impurity A = about 1.3.
Sensitivity : the height of the peak due t o thediast ereoisomers eluting immediately after the principal peakis not less than 10 per cent of the full scale of the recorder.System suitability : reference solution (a):— the height of the trough separating the peak due t o the
diastereoisomers from the principal peak is less than4 per cent of the full scale of the recorder,
— the retention time of the principal peak is less than20 min.
— resolution : minimum 3 between the peaks due tofenoterol and impurity A.
Calculat e the content of diastereoisomers by determiningthe height of a perpendicular dropped from the apex of thepeak t o a line drawn from the trough between the 2 peaks t o
the baseline, and taking int o account the declared cont ent of diast ereoisomers in fenot er ol hy dr obr omid e CRS .Calculate the content of impurity A from the areas of thepeaks and the declared content of impurity A in fenoterol hydrobromide CRS . Limit :— impurity A : maximum 4.0 per cent.
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. fenoterol 3. impurity B
2. impurity A 4. impurity C
Figure 0901.-1. – Chromatogram for the test for impurity A and for the test for related substances of fenoterol hydrobromide
(23) Kromasil C18 is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 87

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 88/176
Fexofenadine hydrochloride
Related substances. Liquid chromatography ( 2.2.29) asdescribed in the test for impurity A with the followingmodifications.
Injection : 20 μ l of the test solution and referencesolutions (b) and (c).
Relative retention with reference to fenoterol (retentiontime = about 7 min): impurity B = about 2.0;
impurity C = about 2.2.System suitability : reference solution (b):
— resolution : minimum 1.5 between the peaks due toimpurities B and C.
Limits :
— correction factor : for the calculation of content, multiplythe peak area of impurity B by 0.6;
— impurity B : not more than the area of the principal peakin the chromatogram obtained with reference solution (c)(0.2 per cent);
— impurity C : not more than 1.5 times the area of theprincipal peak in the chromatogram obtained with
reference solution (c) (0.3 per cent) ;— unspecified impurities : for each impurity, not more
than 0.5 times the area of the principal peak in thechromatogram obtained with reference solution (c)(0.10 per cent);
— total : maximum 0.3 per cent;
— disregard limit : 0.25 times the area of the principal peakin the chromatogram obtained with reference solution (c)(0.05 per cent); disregard the peak due to impurity A.
Iron ( 2.4.9): maximum 5 ppm.
Dissolve the residue obtained in the test for sulphated ashin 2.5 ml of dilute hydrochloric acid R and dilute to 10 mlwith water R.
Loss on drying ( 2.2.32 ): maximum 0.5 per cent, determinedon 1.000 g by drying in an oven at 100-105 °C.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAY
Dissolve 0.600 g in 50 ml of water R and add 5 ml of dilutenitric acid R, 25.0 ml of 0.1 M silver nitrate and 2 ml of ferric ammonium sulphate solution R2 . Shake and titratewith 0.1 M ammonium thiocyanate until an orange colouris obtained. Carry out a blank titration.
1 ml of 0.1 M silver nitrate is equivalent to 38.43 mg
of C17H22BrNO4.STORAGE
Protected from light.
IMPURITIES
Specified impurities: A, B, C.
A. (1 RS )-1-(3,5-dihydoxyphenyl)-2-[[(1SR)-2-(4-hydroxyphenyl)-1-methylethyl]amino]ethanol,
B. 1-(3,5-dihydoxyphenyl)-2-[[(1 RS )-2-(4-hydroxyphenyl)-1-methylethyl]amino]ethanone (phenone),
C. (1 RS )-1-(3,5-dihydoxyphenyl)-2-[[(1 RS )-2-(4-hydroxy-3-methylphenyl)-1-methylethyl]amino]ethanol.
Reference: PA/PH/Exp. P4/T (04) 21 ANP
XXXX:2280
FEXOFENADINE HYDROCHLORIDE
Fexofenadini hydrochloridum
C32H40ClNO4 M r 538.1
DEFINITION
2-[4-[(1 RS )-1-Hydroxy-4-(4-hydroxydiphenylmethyl-1-piperidinyl)butyl]phenyl]-2-methylpropanoic acid.
Content : 98.0 per cent to 102.0 per cent (anhydroussubstance).
CHARACTERS
Appearance : white or almost white powder.Solubility : slightly soluble in water, freely soluble inmethanol, very slightly soluble in acetone.
IDENTIFICATION
A. Infrared absorption spectrophotometry ( 2.2.24).
Comparison : fexofenadine hydrochloride CRS .
B. Dissolve 30 mg in a mixture of equal volumes of methanol R and water R. Sonicate if necessary and diluteto 2 ml with the same mixture of solvents. The solutiongives reaction (a) of chlorides ( 2.3.1).
TESTS
Impurity B. Liquid chromatography ( 2.2.29).
Test solution. Dissolve 50.0 mg of the substance to beexamined in the mobile phase and dilute to 100.0 ml withthe mobile phase.
88 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 89/176
Fexofenadine hydrochloride
Reference solution (a). Dissolve the contents of a vial of fexofenadine hydrochloride impurity B CRS in the test solution and dilute to 2.0 ml with the test solution. Reference solution (b). Dilute 1.0 ml of the test solutionto 100.0 ml with the mobile phase. Dilute 1.0 ml of thissolution to 10.0 ml with the mobile phase.Column :
— size : l = 0.25 m, Ø = 4.6 mm;— stationary phase : silica gel BC for chiral
chromatography R1 (5 μm)(24). Mobile phase : mix 20 volumes of acetonitrile for chromatography R and 80 volumes of a buffer solutionprepared as follows: dilute 1.15 ml of glacial acetic acid Rto 1000 ml with water for chromatography R and adjust topH 4.0 ± 0.1 with dilute ammonia R1. Flow rate : 0.5 ml/min. Detection : spectrophotometer at 220 nm. Injection : 20 μl. Run time : 1.2 times the retention time of fexofenadine. Relative retention with reference to fexofenadine (retention
time = 15-23 min): impurity B = about 0.7.System suitability : reference solution (a):— resolution : minimum 3.0 between the peaks due to
fexofenadine and impurity B. Limits :— correction factor : for the calculation of content, multiply
the peak area of impurity B by 1.3;— impurity B : not more than the area of the principal peak
in the chromatogram obtained with reference solution (b)(0.1 per cent).
Related substances. Liquid chromatography ( 2.2.29). Buffer solution. Dissolve 6.64 g of sodium dihydrogen phosphate monohydrate R and 0.84 g of sodium
perchlorate R in water for chromatography R and diluteto 1000.0 ml with the same solvent; adjust to pH 2.0 ± 0.1with phosphoric acid R.Solvent mixture. Mix equal volumes of acetonitrile for chromatography R and the buffer solution.Test solution (a). Dissolve 25.0 mg of the substance to beexamined in 25.0 ml of the solvent mixture.Test solution (b). Dilute 3.0 ml of test solution (a) to 50.0 mlwith the mobile phase. Reference solution (a). Dissolve 25.0 mg of fexofenadinehydrochloride CRS in the solvent mixture and dilute to25.0 ml with the solvent mixture. Dilute 3.0 ml of thissolution to 50.0 ml with the mobile phase.
Reference solution (b). Dilute 1.0 ml of test solution (a) to100.0 ml with the mobile phase. Dilute 1.0 ml of this solutionto 10.0 ml with the mobile phase. Reference solution (c). Dissolve 1 mg each of fexofenadinehydrochloride impurity A CRS and fexofenadinehydrochloride impurity C CRS in 20 ml of referencesolution (a) and dilute to 200.0 ml with the mobile phase.Column :— size : l = 0.25 m, Ø = 4.6 mm;— stationary phase : phenylsilyl silica gel for
chromatography R (5 μm)(25). Mobile phase : mix 350 volumes of acetonitrile for chromatography R and 650 volumes of the buffer solution;add 3 volumes of triethylamine R and mix.
Flow rate : 1.5 ml/min.
Detection : spectrophotometer at 220 nm. Injection : 20 μl of test solution (a) and reference solutions (b)and (c). Relative retention with reference to fexofenadine(retention time = about 9 min): impurity A = about 1.7;impurity D = about 2.3 ; impurity C = about 3.2. Run time : 6 times the retention time of fexofenadine for test
solution (a) and reference solution (c), twice the retentiontime of fexofenadine for reference solution (b).System suitability : reference solution (c):— resolution : minimum 10 between the peaks due to
fexofenadine and impurity A. Limits :— correction factor : for the calculation of content, multiply
the peak area of impurity A by 1.4;— impurity A : not more than 1.8 times the area of the
principal peak in the chromatogram obtained withreference solution (b) (0.18 per cent) ;
— impurity C : not more than 1.5 times the area of theprincipal peak in the chromatogram obtained with
reference solution (b) (0.15 per cent) ;— impurity D : not more than the area of the principal peak
in the chromatogram obtained with reference solution (b)(0.1 per cent);
— unspecified impurities : for each impurity, not morethan the area of the principal peak in the chromatogramobtained with reference solution (b) (0.10 per cent);
— total : not more than 3 times the area of the principal peakin the chromatogram obtained with reference solution (b)(0.3 per cent);
— disregard limit : 0.5 times the area of the principal peakin the chromatogram obtained with reference solution (b)(0.05 per cent).
Heavy metals ( 2.4.8 ): maximum 10 ppm.Dissolve 1.0 g in a mixture of 15 volumes of water R and85 volumes of methanol R, and dilute to 20 ml with thesame mixture of solvents. 12 ml of this solution complieswith test B. Prepare the reference solution using 1 ml of lead standard solution (10 ppm Pb) R.
Water ( 2.5.32 ): maximum 0.5 per cent.Dissolve 1.000 g in 5.0 ml of anhydrous methanol R. Use1.0 ml of this solution.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAYLiquid chromatography ( 2.2.29) as described in the test for
related substances with the following modifications. Injection : test solution (b) and reference solution (a). Run time : twice the retention time of fexofenadine.Calculate the percentage content of fexofenadinehydrochloride from the declared content of fexofenadinehydrochloride CRS .
STORAGE
At a temperature not exceeding 30 °C.
IMPURITIESSpecified impurities: A, B, C, D.
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by one
or other of the tests in the monograph. They are limited
(24) Astec Cyclobond I 2000 is suitable.(25) Agilent Technologies Zorbax SB phenyl is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 89

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 90/176
Fluorescein
by the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : E, F, G.
A. 2-[4-[4-(4-hydroxydiphenylmethyl-1-piperidinyl)-butanoyl]phenyl]-2-methylpropanoic acid,
B. 2-[3-[(1 RS )-1-hydroxy-4-(4-hydroxydiphenylmethyl-1-piperidinyl)butyl]phenyl]-2-methylpropanoic acid,
C. (1 RS )-4-[4-(hydroxydiphenylmethyl)piperidin-1-yl]-1-[4-(1-methylethyl)phenylbutan-1-ol,
D. R = CH3, R′ = CO-OCH3 : methyl 2-[4-[(1 RS )-1-hydroxy-4-(4-hydroxydiphenylmethyl-1-piperidinyl)butyl]phenyl]-2-methylpropanoate,
F. R = H, R′ = CO2H: 2-[4-[(1 RS )-1-hydroxy-4-(4-hydroxydi-phenylmethyl-1-piperidinyl)butyl]phenyl]propanoic acid,
E. 4-hydroxydiphenylmethylpiperidine,
G. 2-[4-[1-hydroxy-4-(4-diphenylmethylidene-1-
piperidinyl)butyl]phenyl]-2-methylpropanoic acid.
Reagents
Silica gel BC for chiral chromatography R1. XXXXXXX.A very finely divided silica gel for chromatography (5 μm)coated with β-cyclodextrin.
Reference: PA/PH/Exp. 10D/T (05) 19 ANP
XXXX:2348
FLUORESCEIN
Fluoresceinum
C20H12O5 M r 332.3
DEFINITION
3′,6′-Dihydroxy-3 H -spiro[2-benzofuran-1,9′-xanthen]-3-one.
Content : 97.0 per cent to 102.0 per cent (dried substance).
CHARACTERS
Appearance : orange-red, fine powder.
Solubility : practically insoluble in water, soluble in hot ethanol (96 per cent). It dissolves in dilute solutions of alkalihydroxides.
IDENTIFICATION
A. Infrared absorption spectrophotometry ( 2.2.24). Preparation : discs.
Comparison : fluorescein CRS .
90 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 91/176
Fluorescein
If the spectra obtained in the solid state show differences,dissolve the substance to be examined and the referencesubstance separately in the minimum volume of ethanol (96 per cent) R, evaporate to dryness and record newspectra using the residues.
B. Dilute 0.1 ml of solution S (see Tests) to 10 mlwith water R. The solution shows a yellowish-green
fluorescence. The fluorescence disappears on addition of 0.1 ml of dilute hydrochloric acid R and reappears onaddition of 0.2 ml of dilute sodium hydroxide solution R.
C. The absorption by a piece of filter paper of 0.05 ml of solution prepared for identification B (before the additionof dilute hydrochloric acid R) colours the paper yellow.On exposing the moist paper to bromine vapour for 1 minand then to ammonia vapour, the colour becomes deeppink.
TESTS
Solution S. Suspend 1.0 g in 35.0 ml of water R and adddropwise with shaking 4.5 ml of 1 M sodium hydroxide.
Adjust to pH 8.5-9.0 with 1 M sodium hydroxide and diluteto 50.0 ml with water R to obtain a clear solution.
Appearance of solution. Solution S is clear ( 2.2.1) andorange-yellow with yellowish-green fluorescence.
Related substances. Liquid chromatography ( 2.2.29).
Solvent mixture : acetonitrile for chromatography R, mobilephase A (30:70 V/V ).
Test solution (a). Dissolve 50.0 mg of the substance to beexamined in 15.0 ml of ethanol (96 per cent) R. Sonicateand dilute to 50.0 ml with the solvent mixture.
Test solution (b). Dilute 5.0 ml of test solution (a) to 250.0 mlwith the solvent mixture.
Reference solution (a). Dissolve 50.0 mg of fluorescein CRS in 15.0 ml of ethanol (96 per cent) R. Sonicate and diluteto 50.0 ml with water R. Dilute 5.0 ml of this solution to250.0 ml with the solvent mixture.
Reference solution (b). Dissolve 10.0 mg phthalic acid CRS (impurity B) and 10.0 mg resorcinol CRS (impurity A) inthe solvent mixture and dilute to 100.0 ml with the solvent mixture. Dilute 1.0 ml of this solution to 100.0 ml with thesolvent mixture. Reference solution (c). Dilute 5.0 ml of test solution (b) to20.0 ml with the solvent mixture.
Reference solution (d). Dilute 10.0 ml of referencesolution (c) to 100.0 ml with the solvent mixture.Column :— size : l = 0.25 m, Ø = 4.6 mm;— stationary phase : octylsilyl silica gel for
chromatography R3 (5 μm)(26) ;— temperature : 35 °C. Mobile phase :— mobile phase A : dissolve 0.610 g of potassium dihydrogen
phosphate R in water for chromatography R, adjust topH 2.0 with phosphoric acid R and dilute to 1000.0 mlwith water for chromatography R ;
— mobile phase B : acetonitrile for chromatography R ;
Time(min)
Mobile phase A(per cent V/V )
Mobile phase B(per cent V/V )
0 - 20 85 → 20 15 → 80
20 - 29 20 80
29 - 30 20 → 85 80 → 15
30 - 40 85 15
Flow rate : 1.0 ml/min. Detection : spectrophotometer at 220 nm. Injection : 20 μl of test solution (a) and referencesolutions (b), (c) and (d). Relative retention with reference to fluorescein (retentiontime = about 15 min): impurity A = about 0.42;
impurity B = about 0.48; impurity C = about 0.86.System suitability : reference solution (b):— resolution : minimum 1.5 between the peaks due to
impurities A and B.
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity A 2. impurity B 3. impurity C 4. fluorescein
Figure 2348.-1. — Chromatogram for the test for related substances of fluorescein spiked with impurities A, B and C
(26) Zorbax SBC8 is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 91

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 92/176
Fluorodopa (18F) injection
Limits :
— correction factor : for the calculation of content, multiplythe peak area of impurity C by 1.9;
— impurities A, B : for each impurity, not more than the areaof the corresponding peak in the chromatogram obtainedwith reference solution (b) (0.1 per cent) ;
— impurity C : not more than 1.2 times the area of theprincipal peak in the chromatogram obtained withreference solution (c) (0.6 per cent) ;
— unspecified impurities : for each impurity, not morethan 0.2 times the area of the principal peak in thechromatogram obtained with reference solution (c)(0.10 per cent);
— sum of impurities other than A, B and C : not morethan 0.4 times the area of the principal peak in thechromatogram obtained with reference solution (c)(0.2 per cent);
— disregard limit : the area of the principal peak in thechromatogram obtained with reference solution (d)
(0.05 per cent).Chlorides ( 2.4.4): maximum 0.25 per cent.
To 10.0 ml of solution S add 90.0 ml of water R and 1.0 ml of dilute nitric acid R, wait for at least 10 min and filter. Dilute10.0 ml of the filtrate to 15.0 ml with water R.
Loss on drying ( 2.2.32 ): maximum 1.0 per cent, determinedon 1.000 g by drying in an oven at 100-105 °C.
ASSAY
Liquid chromatography ( 2.2.29) as described in the test forrelated substances with the following modification.
Injection : test solution (b) and reference solution (a).
Calculate the percentage content of C20H12O5 from thedeclared content of f luorescein CRS .
STORAGE
Protected from light.
IMPURITIES
Specified impurities : A, B, C .
A. resorcinol,
B. benzene-1,2-dicarboxylic acid (phthalic acid),
C. 2-(2,4-dihydroxybenzoyl)benzoic acid.
Reference: PA/PH/Exp. 14/T (03) 01 ANP
XXXX:1918
FLUORODOPA (18F) INJECTION
Fluorodopae (18F) solutio iniectabilis
DEFINITION
Sterile solution of (2S )-2-amino-3-[2-([18F]fluoro)-4,5-dihydroxyphenyl]propanoic acid (6-[18F]fluorolevodopa). It may contain stabilisers such as ascorbic acid and edetic acid.
This monograph applies to an injection containing
6-[18
F]fluorolevodopa produced by electrophilic substitution.Content : 90 per cent to 110 per cent of the declaredfluorine-18 radioactivity at the date and time stated on thelabel.
Content of levodopa : maximum 1 mg per maximumrecommended dose in milliliters.
Content of 6-fluorolevodopa: maximum 15 mg per maximumrecommended dose in milliliters.
PRODUCTION
RADIONUCLIDE PRODUCTION
Fluorine-18 is a radioactive isotope of fluorine that may beproduced by various nuclear reactions, induced by protonirradiation of oxygen-18, deuteron irradiation of neon-20, orhelium-3 or helium-4 irradiation of oxygen-16.In order to obtain fluorine-18 in a chemical form suitable forelectrophilic substitution reactions as fluorine gas or gaseousacetylhypofluorite, a small amount of non-radioactivefluorine gas (0.3-0.8 per cent of the target gas volume) must be added as a carrier at some step in the production process.
RADIOCHEMICAL SYNTHESIS
6-[18F]Fluorolevodopa may be prepared by variousradiochemical synthetic pathways, which lead to different products in terms of yields, specific radioactivity, by-productsand possible impurities. Electrophilic pathways forproduction of 6-[18F]fluorolevodopa may proceed byfluorodemetalation of a stannylated derivative of levodopa,with molecular [18F]fluorine or [18F]acetylhypofluorite,followed by hydrolysis of protecting groups and finalpurification by semipreparative liquid chromatography.Pathways using demercuration and dethallation must not be used.
CHARACTERS
Appearance : clear, colourless solution.
Half-life and nature of radiation of fluorine-18 : see Table of physical characteristics of radionuclides (5.7 ).
IDENTIFICATION
A. Test A for radionuclidic purity (see Tests).
B. Half-life: 105 min to 115 min, determined by 3measurements of the activity of a sample in the samegeometrical conditions at intervals of about 15 min.
C. Examine the chromatograms obtained in the test forradiochemical purity (see Tests).
92 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 93/176
Fluorodopa (18F) injection
Results : the principal peak in the radiochromatogramobtained with the test solution is similar in retention timeto the peak due to 6-fluorolevodopa in the chromatogramobtained with reference solution (a).
TESTS
pH ( 2.2.3): 4.0 to 5.5.
Sterility. It complies with the test for sterility prescribedin the monograph on Radiopharmaceutical preparations(0125). The injection may be released for use beforecompletion of the test.
Bacterial endotoxins ( 2.6.14): less than 175/V IU/ml,V being the maximum recommended dose in millilitres. Theinjection may be released for use before completion of thetest.
Impurities A, B, C and D. Liquid chromatography ( 2.2.29). Prepare the reference solutions immediately before use.Test solution. The preparation to be examined. Reference solution (a). Dissolve 15 mg of fluorodopaimpurity A CRS in 5 ml of the mobile phase and diluteto V ml with the mobile phase, V being the maximumrecommended dose in millilitres. Reference solution (b). Dissolve 1.0 mg of levodopa R in 5 mlof the mobile phase and dilute to V ml with the mobile phase,V being the maximum recommended dose in millilitres. Reference solution (c). Dissolve 1 mg of 6-hydroxydopahydrochloride R(27) in 5 ml of the mobile phase and dilute0.25 ml of this solution to V ml with the mobile phase,V being the maximum recommended dose in millilitres. Reference solution (d). Mix equal volumes of referencesolutions (b) and (c). Reference solution (e). Dissolve 1 mg of trimethyltinchloride R in 2 ml of the mobile phase and dilute 1.0 ml of this solution to V ml with the mobile phase, V being the
maximum recommended dose in millilitres.Column :— size : l = 0.25 m, Ø = 4.0 mm;— stationary phase : spherical end-capped octadecylsilyl
silica gel for chromatography R(28) ;— temperature : maintain at a constant temperature between
20 °C and 30 °C. Mobile phase : a 6.9 g/l solution of sodium dihydrogen phosphate R adjusted to pH 2.4 with a 4.8 g/l solution of phosphoric acid R.
Flow rate : 1 ml/min. Detection : spectrophotometer at 200 nm and radioactivitydetector connected in series.
Injection : 20 μl. Run time : 15 min. Relative retention with reference to impurity A(retention time = about 7 min): impurity C = about 0.7;impurity B = about 0.8.System suitability : reference solution (d):— resolution : minimum 1.5 between the peaks due to
impurities B and C.
Limits : examine the chromatograms obtained with thespectrophotometer:— impurity A : not more than the area of the corresponding
peak in the chromatogram obtained with referencesolution (a) (15 mg/V ) ;
— impurity B : not more than the area of the peak due tolevodopa in the chromatogram obtained with referencesolution (b) (1.0 mg/V ) ;
— impurity C : not more than the area of the correspondingpeak in the chromatogram obtained with referencesolution (c) (0.05 mg/V ) ;
— impurity D : not more than the area of the corresponding
peak in the chromatogram obtained with referencesolution (e) (0.5 mg/V ).
Residual solvents are limited according to the principlesdefined in the general chapter (5.4), using the generalmethod ( 2.4.24). The preparation may be released for usebefore completion of the test.
RADIONUCLIDIC PURITY
Fluorine-18 : minimum 99.9 per cent of the totalradioactivity.
The preparation may be released for use before completionof test B.A. Gamma-ray spectrometry.
Results : the only gamma photons have an energy of
0.511 MeV and, depending on the measurement geometry,a sum peak of 1.022 MeV may be observed.
B. Gamma-ray spectrometry.
Determine the amount of fluorine-18 and radionuclidicimpurities with a half-life longer than 2 h. For thedetection and quantification of impurities, retain thepreparation to be examined for a sufficient time toallow the fluorine-18 to decay to a level that permits thedetection of impurities.
Results : the spectrum obtained with the preparation tobe examined does not differ significantly from that of abackground spectrum.
RADIOCHEMICAL PURITY
Liquid chromatography ( 2.2.29) as described in the test forimpurities A, B, C and D.Examine the chromatogram recorded using the radioactivitydetector and locate the peak due to 6-[18F]fluorolevodopa bycomparison with the chromatogram obtained with referencesolution (a).
Limit :
— 6-[ 18 F]fluorolevodopa : minimum 95 per cent of the totalradioactivity due to fluorine-18.
Enantiomeric purity, impurities E and F. Thin-layerchromatography ( 2.2.27 ).
Test solution. The preparation to be examined.
Reference solution (a). Dissolve 2 mg of DL -fluorodopa R(29)
in water R and dilute to 10 ml with the same solvent. Reference solution (b). Dissolve 2 mg of f luorolevodopa Rin water R and dilute to 10 ml with the same solvent.
Plate : TLC octadecylsily l silica gel plate for chiral separations R(30).
Mobile phase : methanol R, water R (50:50 V/V ).
Application : 2 μl.
Development : over a path of 8 cm.
Drying : in air for 5 min.
Detection : spray with a 2 g/l solution of ninhydrin Rin anhydrous ethanol R and heat at 60 °C for 10 min.Determine the distribution of radioactivity using a suitabledetector.
(27) Available from RBI and Sigma-Aldrich.(28) Merck Rp-18e is suitable.(29) The substance is available from ABX Germany.(30) Chiralplate from Macherey-Nagel is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 93

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 94/176
Fluorouracil
Retention factors : impurity E = about 0;[18F]fluorodopa = about 0.34; impurity F = about 0.56.
System suitability : reference solution (a):
— the chromatogram shows two clearly separated spots.
Limits :
— [ 18 F]fluorolevodopa : minimum 92 per cent of the total
radioactivity due to fluorine-18 ;— impurity E : maximum 4 per cent of the total radioactivity
due to fluorine-18;
— impurity F : maximum 4 per cent of the total radioactivitydue to fluorine-18.
RADIOACTIVITY
Measure the radioactivity using a calibrated instrument.
LABELLING
The label states the maximum recommended dose inmillilitres.
IMPURITIES
A. (2S )-2-amino-3-[2-fluoro)-4,5-dihydroxyphenyl]propanoicacid (6-fluorolevodopa),
B. ( R,S )-2-amino-3-[2-fluoro-4,5-dihydroxyphenyl]propanoic
acid (DL-dopa),
C. (2S )-2-amino-3-(2,4,5-trihydroxyphenyl)propanoic acid(6-hydroxydopa),
D. trimethyltin,
E. (6-[18F]fluorodextrodopa),
F. [18F]fluoride.
Reagents
6-Hydroxydopa hydrochloride. C8H11NO3.( M r 205.6). XXXXXXX. [28094-15-7]. 2-(2,4,5-trihydroxyphenyl)ethylamine hydrochloride.mp: about 233 °C.
Trimethyltin chloride. C3H9ClSn. ( M r 199.3). XXXXXXX.[1066-45-1].
DL-Fluorodopa. XXXXXXX.
Fluorolevodopa. XXXXXXX.
Reference: PA/PH/Exp. 11/T (05) 84 ANP
NOTE ON THE MONOGRAPH
This monograph has already been published in Pharmeuropa 16.1 to revise the test for related substances(replacement of TLC by LC). It has been revised a second time to reintroduce the TLC to cover 2 additional impurities
F and G, not detected by LC. Only comments on this test are requested.XXXX:0611
FLUOROURACIL
Fluorouracilum
C4H3FN2O2 M r 130.1
DEFINITION
5-Fluoropyrimidine-2,4(1 H ,3 H )-dione.
Content : 98.5 per cent to 101.0 per cent (dried substance).
CHARACTERS
Appearance : white or almost white, crystalline powder.
Solubility : sparingly soluble in water, slightly soluble inethanol (96 per cent).
IDENTIFICATION
First i dentifi cati on: A.
Second id entificati on: B, C .
A. Infrared absorption spectrophotometry ( 2.2.24).
Comparison : fluorouracil CRS .
B. Examine the chromatograms obtained in the test f orrelat ed substances in ultraviolet light at 254 nm. Theprincipal spot in the chromatogram obtained with t est solution (b) is similar in position and size to the principalspot in the chromat ogram obtained with ref erencesolution (a).
C. In a t est-tube, heat 0.5 ml of chr omi c aci d cl eansing mixtur e R in a naked flame until white fumes appear inthe upper part of the tube. The solution wets the side of the tube and there is no appearance of greasiness. Addabout 2 mg of the substance to be examined and heat again in a naked flame until white fumes appear. Thesolution does not wet the sides of the tube.
TESTS
Solution S. Dissolve 0.5 g in carbon dioxide-free water Rand dilute to 50 ml with the same solvent.
Appearance of solution. Solution S is clear ( 2.2.1) and not more intensely coloured than reference solution BY7 or Y7
( 2.2.2, Method II ).
pH ( 2.2.3): 4.5 to 5.0 for solution S.
Impurities F and G. Thin-layer chromatography ( 2.2.27 ).
Test solution. Dissolve 0.10 g of the substance to beexamined in a mixture of equal volumes of methanol R andwater R and dilute to 10.0 ml with the same mixture of solvents.
94 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 95/176
Fluorouracil
Reference solution (a). Dissolve 5.0 mg of 2-ethoxy-5-fluorouracil CRS (impurity F) in a mixture of equal volumes of methanol R and water R and dilute to200.0 ml with the same mixture of solvents. Reference solution (b). Dissolve 200.0 mg of urea CRS (impurity G) in methanol R and dilute to 100.0 ml with thesame solvent. Dilute 1.0 ml of the solution to 100.0 ml with
methanol R. Plate : TLC silica gel F 254 plate R. Mobile phase : methanol R, water R, ethyl acetate R(15:15:70 V/V/V ). Application : 10 μl. Development : over a path of 12 cm. Drying : in air. Detection A : examine in ultraviolet light at 254 nm. Limit A :— impurity F : any spot due to impurity F is not more
intense than the spot in the chromatogram obtained withreference solution (a) (0.25 per cent).
Detection B : spray with a mixture of 200 ml of a 10 g/l
solution of dimethylaminobenzaldehyde R in anhydrousethanol R and 20 ml of hydrochloric acid R ; dry in an ovenat 80 °C for 3-4 min, then examine in daylight (impurity Gproduces a yellow spot and f luorouracil is not detected bythe spray). Limit B :— impurity G : any spot due to impurity G is not more
intense than the spot in the chromatogram obtained withreference solution (b) (0.2 per cent).
Related substances. Liquid chromatography ( 2.2.29). Carry out the test protected from light .Test solution. Dissolve 50.0 mg of the substance to beexamined in the mobile phase and dilute to 50.0 ml with themobile phase. Dilute 5.0 ml of this solution to 50.0 ml withthe mobile phase. Reference solution (a). Dissolve 5.0 mg of fluorouracil impurity C CRS in the mobile phase and dilute to 50.0 mlwith the mobile phase.
Reference solution (b) . Dilute 1.0 ml of reference solution (a)to 100.0 ml with the mobile phase. Dilute 1.0 ml of thissolution to 10.0 ml with the mobile phase.
Reference solution (c). Dissolve 5.0 mg of fluorouracil impurity A CRS in the mobile phase and dilute to 50.0 mlwith the mobile phase. Dilute 1.0 ml of the solution to100.0 ml with the mobile phase. Dilute 1.0 ml of this solution
to 10.0 ml with the mobile phase. Reference solution (d). Dissolve 5.0 mg of fluorouracil impurity B CRS in the mobile phase and dilute to 50.0 mlwith the mobile phase. Dilute 1.0 ml of the solution to100.0 ml with the mobile phase. Dilute 1.0 ml of this solutionto 10.0 ml with the mobile phase.
Reference solution (e). Dilute 1.0 ml of the test solutionto 100.0 ml with the mobile phase. Dilute 1.0 ml of thissolution to 10.0 ml with the mobile phase.
Reference solution (f). To 1 ml of reference solution (a) add1 ml of the test solution and dilute to 10 ml with the mobilephase.
Column :
— size : l = 0.25 m, Ø = 4.6 mm;— stationary phase : octadecylsilyl silica gel for chromatography R (5 μm)(31).
Mobile phase : a 6.805 g/l solution of potassium dihydrogen phosphate R adjusted to pH 5.7 ± 0.1 with 5 M potassiumhydroxide.
Flow rate : 1.0 ml/min.
Detection : spectrophotometer at 266 nm.
Injection : 20 μl.
Run time : 3 times the retention time of fluorouracil.
Relative retention with reference to fluorouracil(retention time = about 6 min): impurity A = about 0.5;impurity B = about 0.7; impurity C = about 0.9;
impurity D = about 1.6; impurity E = about 1.9.System suitability : reference solution (f):
— resolution : minimum 2 between the peaks due tofluorouracil and impurity C.
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity A 3. impurity C 5. impurity D
2. impurity B 4. fluorouracil 6. impurity E
Figure 0611.-1. – Chromatogram for the test for related substances of f luorouracil spiked with impurities A, B, C, D, E
(31) Spherisorb ODS 2 is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 95

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 96/176
Glucagon, human
Limits :
— correction factors : for the calculation of content,multiply the peak areas of the following impurities bythe corresponding correction factor: impurity D = 1.5;impurity E = 1.3;
— impurity A : not more than the area of the principal peakin the chromatogram obtained with reference solution (c)
(0.1 per cent);— impurity B : not more than the area of the principal peak
in the chromatogram obtained with reference solution (d)(0.1 per cent);
— impurity C : not more than the area of the principal peakin the chromatogram obtained with reference solution (b)(0.1 per cent);
— impurities D, E : for each impurity, not more than thearea of the principal peak in the chromatogram obtainedwith reference solution (e) (0.1 per cent) ;
— unspecified impurities : for each impurity, not morethan the area of the principal peak in the chromatogramobtained with reference solution (e) (0.10 per cent);
— total : not more than 5 times the area of the principal peakin the chromatogram obtained with reference solution (e)(0.5 per cent);
— disregard limit : 0.5 times the area of the principal peakin the chromatogram obtained with reference solution (e)(0.05 per cent).
Heavy metals ( 2.4.8 ): maximum 20 ppm.
1.0 g complies with test C. Use a platinum crucible. Preparethe reference solution using 2 ml of lead standard solution(10 ppm Pb) R.
Loss on drying ( 2.2.32 ): maximum 0.5 per cent, determinedon 1.000 g by drying in vacuo at 80 °C for 4 h.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g in a platinum crucible.
ASSAY
Dissolve 0.100 g in 80 ml of dimethylformamide R, warminggently. Cool and titrate with 0.1 M tetrabutylammoniumhydroxide, using 0.25 ml of a 10 g/l solution of thymol blue R in dimethylformamide R as indicator. Carry out ablank titration.
1 ml of 0.1 M tetrabutylammonium hydroxide is equivalent to 13.01 mg of C4H3FN2O2.
STORAGE
Protected from light.
IMPURITIES
Specified impurities: A, B, C, D, E, F, G.
A. X1 = H2, X2 = O: pyrimidine-2,4,6(1 H ,3 H ,5 H )-trione(barbituric acid),
B. X1 = O, X2 = H2 : dihydropyrimidine-2,4,5(3 H )-trione(isobarbituric acid or 5-hydroxyuracil),
C. R = H: pyrimidine-2,4(1 H ,3 H )-dione (uracil),
D. R = OCH3 : 5-methoxypyrimidine-2,4(1 H ,3 H )-dione(5-methoxyuracil),
E. R = Cl : 5-chloropyrimidine-2,4(1 H ,3 H )-dione(5-chlorouracil),
F. 2-ethoxy-5-fluoropyrimidin-4(1 H )-one,
G. urea.
Reference: PA/PH/Exp. 6/T (05) 42 ANP
NOTE ON THE MONOGRAPH
A general revision of the monograph is proposed to simplify the test procedures. A change in compositionof mobile phase A in the test for related proteins is also proposed to improve the resolution between glucagon and carbamoylglucagon.
XXXX:1635
GLUCAGON, HUMAN
Glucagonum humanum
C153H225N43O49S M r 3483
DEFINITION
Polypeptide having the same structure (29 amino acids)
as the hormone produced by the α-cells of the humanpancreas, which increases the blood-glucose concentrationby promoting rapid breakdown of liver glycogen.
Content : 92.5 per cent to 105.0 per cent (anhydroussubstance).
PRODUCTION
Human glucagon is produced by a method based onrecombinant DNA (rDNA) technology. During the courseof product development it must be demonstrated that the manufacturing process produces a product having abiological activity of not less than 1 IU/mg using a suitablevalidated bioassay, based on hyperglycaemia measurement.
Host-cell-derived proteins : the limit is approved by thecompetent authority.Host-cell- and vector-derived DNA : the limit is approvedby the competent authority.
96 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 97/176
Glucagon, human
CHARACTERS
Appearance : white or almost white powder.
Solubility : practically insoluble in water and in most organic solvents, soluble in dilute mineral acids and in dilutesolutions of alkali hydroxides.
IDENTIFICATIONA. Peptide mapping. Liquid chromatography ( 2.2.29).
Test solution. Prepare a 10 5 mg/ml solution of thesubstance to be examined in 0.01 M hydrochloric acid . Mix 200 μl of the solution with 800 μl of 0.1 M ammonium carbonate buffer solution pH 10.3 R (dilutedstock solution). Freshly pPrepare a 2.0 mg/ml solutionof α -chymotrypsin for peptide mapping R in 0.1 M ammonium carbonate buffer solution pH 10.3 R and add25 μl of this solution to the diluted stock solution of thesubstance to be examined. Place the test solution in aclosed vial at 37 °C for 2 h. Remove the vial and stop thereaction immediately by the addition of 120 μl of glacial acetic acid R. The α-chymotrypsin solution is stable for
12 months at 70 °C or below. Reference solution . Prepare at the same time and inthe same manner as f or the t est solution but usinghuman glucag on CRS inst ead of the substance t o beexamined. a 1 mg/ml solution of human glucagon CRS in 0.1 M ammonium carbonate buffer solution pH 10.3 R(diluted stock solution) and continue as described for thetest solution.
Column :
— size : l = 0.05 m, Ø = 4 mm;
— stationary phase : octadecylsilyl silica gel for chromatography R (5 μm ) (32).
Mobile phase :— mobile phase A : mix 500 μl of trifluoroacetic acid R
and 1000 ml of water R ;
— mobile phase B : mix 500 μl of trifluoroacetic acid Rwith 600 ml of anhydrous ethanol R and add 400 mlof water R ;
Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 - 35 100 → 53 0 → 47
35 - 45 53 → 0 47 → 100
45 - 46 0 → 100 100 → 0
46 - 75 100 0
Flow rate : 1.0 ml/min.
Detection : spectrophotometer at 215 nm.
Equilibration : mobile phase A for at least 15 min.
Injection : 10 20 μl.
Results : the profile of the chromatogram obtained withthe test solution corresponds to that of the chromatogramobtained with the reference solution.
B. Examine the chromatograms obtained in the assay.
Results : the principal peak in the chromatogram obtainedwith the test solution is similar in retention time to the
principal peak in the chromatogram obtained with thereference solution.
TESTS
Specific absorbance ( 2.2.25 ): 21 t o 25, det ermined at themaximum at 276 nm (anhydrous substance).
Dissolve 2.5 mg in 0.01 M hydr ochloric acid and dilut e t o10.0 ml with the same solvent.
Deamidated glucagon. Liquid chromatography ( 2.2.29): use
the normalisation procedure.Test solution. Dissolve the substance to be examined in0.01 M hydrochloric acid to obtain a concentration of 1.0 mg/ml.
Resolution solution. Dissolve the substance to be examinedin 0.1 M hydrochloric acid to obtain a concentrationof 1.0 mg/ml. Incubate in an oven at 60 °C for 2 h.Immediately after degradation, adjust to pH 2.5 with 1 M sodium hydroxide.
Column :
— material : glass;
— size : l = 0.05 m, Ø = 5 mm;
— stationary phase : anion exchange resin R2 .
Mobile phase :
— mobile phase A : mix 1000 ml of tris-hydrochloride buffer solution pH 8.3 R and 1000 ml of anhydrous ethanol R ;
— mobile phase B : dissolve 29.2 g of sodium chloride R in1000 ml of tris-hydrochloride buffer solution pH 8.3 R ;add 1000 ml of anhydrous ethanol R ;
Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 - 4 100 0
4 - 30 100 → 78 0 → 22
30 - 34 78 → 45 22 → 55
34 - 38 45 → 20 55 → 80
38 - 40 20 → 100 80 → 0
40 - 60 100 0
Flow rate : 0.6 ml/min.
Detection : spectrophotometer at 230 nm.
Equilibration : mobile phase A for at least 15 min.
Injection : 60 μl.
System suitability : resolution solution:
— retention time : glucagon = about 10 min; 4 deamidatedforms: between 15 min and 40 min;
— resolution
: baseline separation of the 4 deamidatedforms and glucagon.
Limit :
— total of the 4 deamidated forms : maximum 0.5 per cent,calculated from the peaks eluting between 10 15 min and40 min.
Related proteins. Liquid chromatography ( 2.2.29): use thenormalisation procedure.
2.8 M urea solution. Dissolve 16.8 g of urea R in 0.01 M hy dr ochl ori c acid and dilute t o 100 ml with the same solvent 100 ml of water R.
Test solution. Dissolve the substance to be examined in0.01 M hydrochloric acid to obtain a concentration of 1.0
0.5 mg/ml. Maintain the solution at 2-8 °C and use within 24 h.
(32) Lichrosphere C-18 is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 97

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 98/176
Glycerol monocaprylate
Reference solution. Dissolve the contents of a vial of human glucagon CRS in 0.01 M hydrochloric acid to obtain aconcentration of 1.0 0.5 mg/ml. Maintain the solution at 2-8 °C and use within 2 4 h.
Resolution solution. Dissolve 10 mg of the substance tobe examined in 10 20 ml of 2.8 M urea solution. Adjust topH 7 with 1 M sodium hydr oxid e and place the sealed vial
at about 50 °C f or about 2 h. Cool and adjust to pH 2.5with 1 M hy dr ochl ori c aci d . Heat at 50 °C for 2 h. Cooland adjust to pH 2.2 with 1 M hydrochloric acid . Maintainthe solution at 2-8 °C and use within 2 h or maintain the solution below 15 °C, then thaw and filter through a0.22 μm filter before use.
Column :
— size : l = 0.25 m, Ø = 4.6 mm;
— stationary phase : octadecylsilyl silica gel for chromatography R (5 μm) with a pore size of 30 nm(33) ;
— temperature : 45 °C.
Mobile phase :— mobile phase A : dissolve 14.2 13.6 g of anhyd r ous
sodium sulphate R potassium dihydrogen phosphate Rin 400 ml of water R, add 1.35 ml of phosphoric acid Rand adjust to pH 2.5 ( 2.2.3) with ethanolamine R phosphoric acid R, and add 100 ml of acetonitrile for chromatography R ;
— mobile phase B : acetonitrile for chromatography R,water R (40:60 V/V ) ;
Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 - 23 57 43
23 - 29 57 → 10 43 → 90
29 - 30 10 90
30 - 31 10 → 57 90 → 43
31 - 75 57 43
Flow rate : 1.0 ml/min.
Detection : spectrophotometer at 214 nm.
Injection : 25 50 μl of the test solution and the resolutionsolution.
Relative retention with reference to glucagon (retentiontime = about 20 min) : carbamoylglucagon = about 1.1.
System suitability : resolution solution:
— r et enti on time : glucagon = about 20 min;carbamoylglucagon = about 22 min,
— resolution : minimum 1.3 1.5 between the peaks due toglucagon and carbamoylglucagon.
— symmetry fact or : 0.6 to 1 f or the peak due to glucagon.
Limit :
— total of all impurities: maximum 2.5 per cent.
Water ( 2.5.12 ): maximum 10 per cent, determined on 20.050 mg.
Bacterial endotoxins ( 2.6.14): less than 10 IU/mg.
ASSAY
Liquid chromatography ( 2.2.29) as described in the test forrelated proteins with the following modifications.
Injection : test solution and reference solution.
Calculate the content of human glucagon (C153H225N43O49S)from the declared content of C153H225N43O49S in human glucagon CRS .
STORAGE
In an airtight container, protected from light, at atemperature lower than 15 °C.
Reference: PA/PH/Exp. 13H/T (03) 60 ANP
XXXX:2213
GLYCEROL MONOCAPRYLATE
Glyceroli monocaprylas
DEFINITION
Mixture of monoacylglycerols, mainly monocaproylglycerol,containing variable quantities of di- and triacylglycerols,obtained by direct esterification of glycerol with caprylic acid.
Content :
— glycerol monocaprylate (type I) :
— monoacylglycerols : 45.0 per cent to 75.0 per cent;
— diacylglycerols : 20.0 per cent to 50.0 per cent;
— triacylglycerols : maximum 10.0 per cent;
— glycerol monocaprylate (type II) :— monoacylglycerols : minimum 80.0 per cent;
— diacylglycerols : maximum 20.0 per cent;
— triacylglycerols : maximum 5.0 per cent.
CHARACTERS
Appearance : colourless or slightly yellow, oily liquid or soft mass.
Solubility : practically insoluble in water, very solublein ethanol (96 per cent) and freely soluble in methylenechloride.
IDENTIFICATION
A. Composition of fatty acids (see Tests).
B. Itcomplies with the limits of the assay (monoacylglycerols).
TESTS
Acid value ( 2.5.1): maximum 4.0.
Iodine value ( 2.5.4, Method A): maximum 1.0.
Composition of fatty acids ( 2.4.22, Method C ). Use themixture of calibrating substances in Table 2.4.22.-2.
Composition of the fatty acid fraction of the substance:
— caproic acid : maximum 1.0 per cent;
— caprylic acid : minimum 80.0 per cent;
— capric acid : maximum 20.0 per cent;
— lauric acid : maximum 1.0 per cent;— myristic acid : maximum 0.5 per cent.
(33) Waters Symmetry C18 and Phenomenex Jupiter C18 are suitable.
98 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 99/176
Glycerol monocaprylate
Free glycerol : maximum 3.0 per cent.
Dissolve 1.20 g in 25.0 ml of methylene chloride R. Heat to about 50 °C then allow to cool. Add 100 ml of water R.Shake and add 25.0 ml of periodic acetic acid solution R.Shake and allow to stand for 30 min. Add 40 ml of a 75 g/lsolution of potassium iodide R. Allow to stand for 1 min.Add 1 ml of starch solution R. Titrate with 0.1 M sodium
thiosulfate. Carry out a blank titration.1 ml of 0.1 M sodium thiosulfate is equivalent to 2.3 mgof glycerol.
Water ( 2.5.12 ): maximum 1.0 per cent, determined on 1.00 g.
Total ash ( 2.4.16 ): maximum 0.5 per cent.
ASSAY
Gas chromatography ( 2.2.28 ): use the normalisationprocedure.
Test solution. To 0.25 g of the substance to be examined,add 5.0 ml of tetrahydrofurane R and shake to dissolve.
Reference solution (a). To 0.25 g of glycerol monocaprylate CRS , add 5.0 ml of tetrahydrofurane R andshake to dissolve.
Reference solution (b). To 50 mg of glycerol mono-octanoate R and 50 mg of glycerol monodecanoate R,add 2.5 ml of tetrahydrofurane R and shake to dissolve.
Column :
— size : l = 10 m, Ø = 0.32 mm;
— stationary phase : poly(dimethyl)(diphenyl)siloxane R(34)
(film thickness 0.1 μm).
Carrier gas : helium for chromatography R.
Flow rate : 2.3 ml/min.
Split ratio : 1:50.
Temperature :
Time
(min)
Temperature
(°C)
Column 0 - 3 60
3 - 38 60 → 340
38 - 50 340
Injection port 350
Detector 370
Detection : flame ionisation.
Injection : 1 μl.
Identification of peaks : use the chromatogram suppliedwith glycerol monocaprylate CRS and the chromatogramobtained with reference solution (a) to identify the peaksdue to mono-, di- and triacylglycerols.
System suitability : reference solution (b):
— resolution : minimum 5 between the peaks due to glycerol
mono-octanoate and glycerol monodecanoate.
For the calculation of content of mono-, di- andtriacylglycerols, disregard the peaks with a retention timeless than that of the monoacylglycerols, which are due toimpurities of the solvent and to the free fatty acids.
Calculate the content of free fatty acids ( B) using thefollowing expression:
I A = acid value of the substance to be examined.
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. monoacylg lycerols C8 2. diacylglycerols C8 3. triacylg lycerols C8
Figure 2213.-1. – Chromatogram for the assay of glycerol monocaprylate
(34) OPTIMA 5 or equivalent: HP5 or CP Sil 8.
© PHARMEUROPA Vol. 18, No. 1, January 2006 99

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 100/176
Glycerol monocaprylocaprate
Calculate the content of mono-, di- and triacylglycerols usingthe following expressions :
A = percentage content of free glycerol ;
X = monoacylglycerols content obtained bynormalisation;
Y = diacylglycerols content obtained by normalisation;
Z = triacylglycerols content obtained bynormalisation.
LABELLING
The label states the type of glycerol monocaprylate (type Ior II).
Reagents
Glycerol mono-octanoate. C11H22O4. ( M r 218.3). XXXXXXX.[19670-49-6]. 1-Octanoyl-rac -glycerol(35).Content : about 99 per cent.
Glycerol monodecanoate. C13H26O4. ( M r 246.3). XXXXXXX.
[26402-22-2]. 1-Decanoyl-rac -glycerol(36).Content : about 99 per cent.
Reference: PA/PH/Exp. 13H/T (05) 67 ANP
XXXX:2392
GLYCEROL MONOCAPRYLOCAPRATE
Glyceroli monocaprylocapras
DEFINITION
Mixture of monoacylglycerols, mainly monocaproylglyceroland monocaprilylglycerol, containing variable quantities of di- and triacylglycerols, obtained by direct esterification of glycerol with caprylic and capric acids.
Content :
— glycerol monocaprylocaprate (type I):
— monoacylglycerols : 45.0 per cent to 75.0 per cent;
— diacylglycerols : 20.0 per cent to 50.0 per cent;
— triacylglycerols : maximum 10.0 per cent;
— glycerol monocaprylocaprate (type II) :
— monoacylglycerols : minimum 80.0 per cent;
— diacylglycerols : maximum 20.0 per cent;
— triacylglycerols : maximum 5.0 per cent.
CHARACTERS
Appearance : colourless or slightly yellow, oily liquid or soft mass.
Solubility : practically insoluble in water, very solublein ethanol (96 per cent) and freely soluble in methylenechloride.
IDENTIFICATION
A. Composition of fatty acids (see Tests).
B. Itcomplies with the limits of the assay (monoacylglycerols).
TESTS
Acid value ( 2.5.1): maximum 4.0.
Iodine value ( 2.5.4, Method A): maximum 1.0.
Composition of fatty acids ( 2.4.22, Method C ). Use themixture of calibrating substances in Table 2.4.22.-2.
Composition of the fatty acid fraction of the substance:— caproic acid : maximum 3.0 per cent;
— caprylic acid : 50.0 per cent to 90.0 per cent;
— capric acid : 10.0 per cent to 50.0 per cent;
— lauric acid : maximum 3.0 per cent;
— myristic acid : maximum 1.0 per cent.
Free glycerol : maximum 3.0 per cent.
Dissolve 1.20 g in 25.0 ml of methylene chloride R. Heat to about 50 °C then allow to cool. Add 100 ml of water R.Shake and add 25.0 ml of periodic acetic acid solution R.Shake and allow to stand for 30 min. Add 40 ml of a 75 g/lsolution of potassium iodide R. Allow to stand for 1 min.
Add 1 ml of starch solution R. Titrate with 0.1 M sodiumthiosulphate. Carry out a blank titration.
1 ml of 0.1 M sodium thiosulphate is equivalent to 2.3 mgof glycerol.
Water ( 2.5.12 ): maximum 0.5 per cent, determined on 1.00 g.
Total ash ( 2.4.16 ): maximum 0.5 per cent.
ASSAY
Gas chromatography ( 2.2.28 ): use the normalisationprocedure.
Test solution. To 0.25 g of the substance to be examined,add 5.0 ml of tetrahydrofuran R and shake to dissolve.
Reference solution (a). To 0.25 g of glycerol monocaprylocaprate CRS , add 5.0 ml of tetrahydrofuran Rand shake to dissolve.
Reference solution (b). To 50 mg of glycerol mono-octanoate R and 50 mg of glycerol monodecanoate R,add 2.5 ml of tetrahydrofuran R and shake to dissolve.
Column :
— size : l = 10 m, Ø = 0.32 mm;
— stationary phase : poly(dimethyl)(diphenyl)siloxane R(37)
(film thickness 0.1 μm).
Carrier gas : helium for chromatography R.
Flow rate : 2.3 ml/min.
Split ratio : 1:50.(35) Sigma Ref. M-2265.(36) Sigma Ref. M-2140.(37) OPTIMA 5 or equivalent: HP 5 or CP Sil 8.
100 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 101/176
Glycerol monocaprylocaprate
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. monoacylglycerols C8 3. diacylglycerols C8 5. triacylglycerols
2. monoacylglycerols C10 4. diacylglycerols C10
Figure 2392.-1. – Chromatogram for the assay of glycerol monocaprylocaprate
Temperature :
Time
(min)
Temperature
(°C)
Column 0 - 3 60
3 - 38 60 →
340
38 - 50 340
Injection port 350
Detector 370
Detection : flame ionisation.
Injection : 1 μl.
Identification of peaks : use the chromatogram supplied with glycerol monocaprylocaprate CRS and the chromatogramobtained with reference solution (a) to identify the peaksdue to mono-, di- and triacylglycerols.
System suitability : reference solution (b):
— resolution : minimum 5 between the peaks due to glycerolmono-octanoate and glycerol monodecanoate.
For the calculation of content of mono-, di- andtriacylglycerols, disregard the peaks with a retention timeless than that of the monoacylglycerols, which are due toimpurities of the solvent and to the free fatty acids.
Calculate the content of free fatty acids (B) using thefollowing expression:
I A = acid value of the substance to be examined.
Calculate the content of mono-, di- and triacylglycerols usingthe following expressions :
A = percentage content of free glycerol;
X = monoacylglycerols content obtained bynormalisation;
Y = diacylglycerols content obtainedby normalisation;
Z = triacylglycerols content obtained by
normalisation.
LABELLINGThe labelling states the type of glycerol monocaprylocaprate(type I or II).
Reagents
Glycerol mono-octanoate. C11H22O4. ( M r 218.3). XXXXXXX.[19670-49-6]. 1-Octanoyl-rac -glycerol(38).Content : about 99 per cent.
Glycerol monodecanoate. C13H26O4. ( M r 246.3). XXXXXXX.[26402-22-2]. 1-Decanoyl-rac -glycerol(39).
Content : about 99 per cent.
(38) Sigma Ref. M-2265.(39) Sigma Ref. M-2140.
© PHARMEUROPA Vol. 18, No. 1, January 2006 101

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 102/176
Human anti-D immunoglobulin
Reference: PA/PH/Exp. 6B/T (05) 49 ANP
NOTE ON THE MONOGRAPH
It is proposed in the revised draft below, in order to increasethe level of safety with regard to B19 virus contamination,to require to carry out the test for B19 virus DNA onalbumin when it is used as stabiliser.
XXXX:0557
HUMAN ANTI-D IMMUNOGLOBULIN
Immunoglobulinum humanum anti-D
DEFINITION
Human anti-D immunoglobulin is a liquid or freeze-driedpreparation containing immunoglobulins, mainlyimmunoglobulin G. The preparation is intended forintramuscular administration. It contains specific antibodiesagainst erythrocyte D-antigen and may also contain smallquantities of other blood-group antibodies. Human normal
immunoglobulin (0338) may be added.It complies with the monograph on Human normal immunoglobulin (0338), except for the minimum number of donors and the minimum total protein content. For productsprepared by a method that eliminates immunoglobulins withspecificities other than anti-D, where authorised, the test forantibodies to hepatitis B surface antigen is not required.
PRODUCTION
Human anti-D immunoglobulin is preferably obtained fromthe plasma of donors with a sufficient titre of previouslyacquired anti-D antibodies. Where necessary, in order toensure an adequate supply of human anti-D immunoglobulin,it is obtained from plasma derived from donors immunised
with D-positive erythrocytes that are compatible in relevant blood group systems in order to avoid formation of undesirable antibodies.
ERYTHROCYTE DONORS
Erythrocyte donors comply with the requirements fordonors prescribed in the monograph Human plasma for fractionation (0853).
IMMUNISATION
Immunisation of the plasma donor is carried out underproper medical supervision. Recommendations concerningdonor immunisation, including testing of erythrocyte donors,have been formulated by the World Health Organisation( Requirements for the collection, processing and quality
control of blood, blood components and plasma derivatives,WHO Technical Report Series, No. 840, 1994 or subsequent revision).
POOLED PLASMA
To limit the potential B19 virus burden in plasma pools usedfor the manufacture of anti-D immunoglobulin, the plasmapool is tested for B19 virus using validated nucleic acidamplification techniques ( 2.6.21).
B19 virus DNA : maximum 10.0 IU/μl.
A positive control with 10.0 IU of B19 virus DNA permicrolitre and, to test for inhibitors, an internal controlprepared by addition of a suitable marker to a sample of theplasma pool are included in the test. The test is invalid if thepositive control is non-reactive or if the result obtained withthe internal control indicates the presence of inhibitors. B19 virus DNA for NAT testing BRP is suitable for use as apositive control.
If Human normal immunoglobulin (0338) is added tothe preparation, the plasma pool from which it is derivedcomplies with the above requirement for B19 virus DNA.If Human albumin solution (0255) is added to thepreparation, it complies with the above requirement forB19 virus DNA.
POTENCYCarry out the assay of human anti-D immunoglobulin( 2.7.13, Method A). The estimated potency is not less than90 per cent of the stated potency. The confidence limits( P = 0.95) are not less than 80 per cent and not more than120 per cent of the estimated potency.Method B or C ( 2.7.13) may be used for potencydetermination if a satisfactory correlation with the resultsobtained by Method A has been established for the particularproduct.
STORAGESee Human normal immunoglobulin (0338).
LABELLING
See Human normal immunoglobulin (0338).The label states the number of International Units percontainer.
Reference: PA/PH/Exp. 6B/T (05) 50 ANP
NOTE ON THE MONOGRAPH
It is proposed in the revised draft below, in order to increasethe level of safety with regard to B19 virus contamination,to require to carry out the test for B19 virus DNA onalbumin when it is used as stabiliser.
XXXX:1527
HUMAN ANTI-D IMMUNOGLOBULINFOR INTRAVENOUS
ADMINISTRATION
Immunoglobulinum humanum anti-Dad usum intravenosum
DEFINITIONHuman anti-D immunoglobulin for intravenousadministration is a liquid or freeze-dried preparationcontaining immunoglobulins, mainly immunoglobulin G.It contains specific antibodies against erythrocyte
D-antigen and may also contain small quantities of otherblood-group antibodies. Human normal immunoglobulinfor intravenous administration (0918) may be added.It complies with the monograph on Human normal immunoglobulin for intravenous administration (0918),except for the minimum number of donors, the minimumtotal protein content, the limit for osmolality and the limit for prekallikrein activator. For products prepared by amethod that eliminates immunoglobulins with specificitiesother than anti-D: where authorised, the test for antibodiesto hepatitis B surface antigen is not required ; a suitable test for Fc function is carried out instead of that described inchapter 2.7.9, which is not applicable to such a product.
PRODUCTION
Human anti-D immunoglobulin is preferably obtained fromthe plasma of donors with a sufficient titre of previouslyacquired anti-D antibodies. Where necessary, in order to
102 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 103/176
Human plasma for fractionation
ensure an adequate supply of human anti-D immunoglobulin,it is obtained from plasma derived from donors immunisedwith D-positive erythrocytes that are compatible in relevant blood group systems in order to avoid formation of undesirable antibodies.
ERYTHROCYTE DONORS
Erythrocyte donors comply with the requirements for
donors prescribed in the monograph Human plasma for fractionation (0853).
IMMUNISATION
Immunisation of the plasma donor is carried out underproper medical supervision. Recommendations concerningdonor immunisation, including testing of erythrocyte donors,have been formulated by the World Health Organisation( Requirements for the collection, processing and quality control of blood, blood components and plasma derivatives,WHO Technical Report Series, No. 840, 1994 or subsequent revision).
POOLED PLASMA
To limit the potential B19 virus burden in plasma pools used
for the manufacture of anti-D immunoglobulin, the plasmapool is tested for B19 virus using validated nucleic acidamplification techniques ( 2.6.21).
B19 virus DNA : maximum 10.0 IU/μl.
A positive control with 10.0 IU of B19 virus DNA permicrolitre and, to test for inhibitors, an internal controlprepared by addition of a suitable marker to a sample of theplasma pool are included in the test. The test is invalid if thepositive control is non-reactive or if the result obtained withthe internal control indicates the presence of inhibitors.
B19 virus DNA for NAT testing BRP is suitable for use as apositive control.
If Human normal immunoglobulin for intravenousadministration (0918) is added to the preparation, theplasma pool from which it is derived complies with the aboverequirement for B19 virus DNA.
If Human albumin solution (0255) is added to thepreparation, it complies with the above requirement forB19 virus DNA.
POTENCY
Carry out the assay of human anti-D immunoglobulin( 2.7.13, Method A). The estimated potency is not less than90 per cent of the stated potency. The confidence limits( P = 0.95) are not less than 80 per cent and not more than
120 per cent of the estimated potency.Method B or C ( 2.7.13) may be used for potencydetermination if a satisfactory correlation with the resultsobtained by Method A has been established for the particularproduct.
STORAGE
See Human normal immunoglobulin for intravenousadministration (0918).
LABELLING
See Human normal immunoglobulin for intravenous
administration (0918).The label states the number of International Units percontainer.
Reference: PA/PH/Exp. 6B/T (05) 44 ANP
NOTE ON THE MONOGRAPH
In order to clarify the requirements for freezing plasmaintended for the recovery of labile proteins, whether it isobtained by plasmapheresis or from whole blood, and inorder to ensure homogeneous quality, it is proposed toreplace the chamber temperature indication ( 30 °C or below) by the following conditions: freezing conditionshave to be validated to ensure that a temperature of 25 °C or lower is attained at the core of each plasma unit within12 h of placing in the freezing chamber. These data arebased on scientific studies on the freezing conditions for plasma. One of these studies is published in the Scientific Notes section of this edition of Pharmeuropa, and will alsobe published in the next Pharmeuropa Scientific Notes.
XXXX:0853
HUMAN PLASMA FORFRACTIONATION
Plasma humanum ad separationemDEFINITIONHuman plasma for fractionation is the liquid part of humanblood remaining after separation of the cellular elements fromblood collected in a receptacle containing an anticoagulant,or separated by continuous f iltration or centrifugationof anticoagulated blood in an apheresis procedure; it isintended for the manufacture of plasma-derived products.
PRODUCTION
DONORS
Only a carefully selected, healthy donor who, as far as canbe ascertained after medical examination, laboratory bloodtests and a study of the donor’s medical history, is free fromdetectable agents of infection transmissible by plasma-derivedproducts may be used. Recommendations in this fieldare made by the Council of Europe [ Recommendation No. R (95) 15 on the preparation, use and quality assuranceof blood components, or subsequent revision]; a directive of the European Union also deals with the matter : Commission Directive 2004/33/EC of 22 March 2004 implementing Directive 2002/98/EC of the European Parliament and of the Council as regards certain technical requirements for blood and blood components.
Immunisation of donors. Immunisation of donors to obtainimmunoglobulins with specific activities may be carriedout when sufficient supplies of material of suitable qualitycannot be obtained from naturally immunised donors.
Recommendations for such immunisations are formulatedby the World Health Organisation ( Requirements for thecollection, processing and quality control of blood, blood components and plasma derivatives, WHO Technical Report Series, No. 840, 1994 or subsequent revision).
Records. Records of donors and donations made are kept insuch a way that, while maintaining the required degree of confidentiality concerning the donor’s identity, the originof each donation in a plasma pool and the results of thecorresponding acceptance procedures and laboratory testscan be traced.
Laboratory tests. Laboratory tests are carried out for eachdonation to detect the following viral markers :1. antibodies against human immunodeficiency virus 1
(anti-HIV-1);2. antibodies against human immunodeficiency virus 2
(anti-HIV-2);
© PHARMEUROPA Vol. 18, No. 1, January 2006 103

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 104/176
Human plasma for fractionation
3. hepatitis B surface antigen (HBsAg);
4. antibodies against hepatitis C virus (anti-HCV).
Pending complete harmonisation of the laboratory tests tobe carried out, the competent authority may require that atest for alanine aminotransferase (ALT) also be carried out.
The test methods used are of suitable sensitivity and
specificity and comply with the regulations in force. If arepeat-reactive result is found in any of these tests, thedonation is not accepted.
INDIVIDUAL PLASMA UNITS
The plasma is prepared by a method that removes cells andcell debris as completely as possible. Whether preparedfrom whole blood or by plasmapheresis, the plasma isseparated from the cells by a method designed to prevent the introduction of micro-organisms. No antibacterial orantifungal agent is added to the plasma. The containerscomply with the requirements for glass containers ( 3.2.1)or for plastic containers for blood and blood components( 3.2.3). The containers are closed so as to prevent anypossibility of contamination.
If 2 or more units are pooled prior to freezing, the operationsare carried out using sterile connecting devices or underaseptic conditions and using containers that have not previously been used.
When obtained by plasmapheresis, plasma intended for therecovery of proteins that are labile in plasma is frozen bycooling rapidly in a chamber at 30 °C or below as soon aspossible in conditions validated to ensure that a temperatureof 25 °C or below is attained at the core of each plasmaunit within 12 h of placing in the freezing apparatus andat t he latest within 24 h of collection.
When obtained by plasmapheresis, plasma intended solelyfor the recovery of proteins that are not labile in plasma isfrozen by cooling rapidly in a chamber at 20 °C or below
as soon as possible and at the latest within 24 h of collection.When obtained from whole blood, plasma intended for therecovery of proteins that are labile in plasma is separatedfrom cellular elements and is frozen by cooling rapidlyin a chamber at 30 °C or below as soon as possible inconditions validated to ensure that a temperature of 25 °Cor below is attained at the core of each plasma unit within12 h of placing in the freezing apparatus and at the lat est within 24 h of collection.
When obtained from whole blood, plasma intended solelyfor the recovery of proteins that are not labile in plasma isseparated from cellular elements and frozen in a chamber at 20 °C or below as soon as possible and at the latest within72 h of collection.
It is not intended that the determination of total proteinand factor VIII shown below be carried out on each unit of plasma. They are rather given as guidelines for good manufacturing practice, the test for factor VIII being relevant for plasma intended for use in the preparation of concentrates of labile proteins.
The total protein content of a unit of plasma depends onthe serum protein content of the donor and the degree of dilution inherent in the donation procedure. When plasmais obtained from a suitable donor and using the intended proportion of anticoagulant solution, a total protein content complying with the limit of 50 g/l is obtained. If a volumeof blood or plasma smaller than intended is collected into
the anticoagulant solution, the resulting plasma is not necessarily unsuitable for pooling for fractionation. Theaim of good manufacturing practice must be to achieve the prescribed limit for all normal donations.
Preservation of factor VIII in the donation depends onthe collection procedure and the subsequent handling of the blood and plasma. With good practice, 0.7 IU/ml can usually be achieved, but units of plasma with a lower activity may still be suitable for use in the productionof coagulation factor concentrates. The aim of good manufacturing practice is to conserve labile proteins asmuch as possible.
Total protein. Carry out the test using a pool of not fewerthan 10 units. Dilute the pool with a 9 g/l solution of sodium chloride R to obtain a solution containing about 15 mg of protein in 2 ml. To 2.0 ml of this solution in around-bottomed centrifuge tube add 2 ml of a 75 g/l solutionof sodium molybdate R and 2 ml of a mixture of 1 volumeof nitrogen-free sulphuric acid R and 30 volumes of water R. Shake, centrifuge for 5 min, decant the supernatant liquid and allow the inverted tube to drain on filter paper.Determine the nitrogen in the residue by the method of sulphuric acid digestion ( 2.5.9) and calculate the proteincontent by multiplying the quantity of nitrogen by 6.25. Thetotal protein content is not less than 50 g/l.
Factor VIII. Carry out the test using a pool of not fewer than10 units. Thaw the samples to be examined, if necessary,at 37 °C. Carry out the assay of factor VIII ( 2.7.4), usinga reference plasma calibrated against the InternationalStandard for human coagulation factor VIII in plasma. Theactivity is not less than 0.7 IU/ml.
STORAGE AND TRANSPORT
Frozen plasma is stored and transported in conditionsdesigned to maintain the temperature at or below 20 °C;for accidental reasons, the storage temperature may riseabove 20 °C on one or more occasions during storage andtransport but the plasma is nevertheless considered suitablefor fractionation if all the following conditions are fulfilled :
— the total period of time during which the temperature
exceeds 20 °C does not exceed 72 h;— the temperature does not exceed 15 °C on more than
one occasion;
— the temperature at no time exceeds 5 °C.
POOLED PLASMA
During the manufacture of plasma products, the first homogeneous pool of plasma (for example, after removal of cryoprecipitate) is tested for HBsAg and for HIV antibodiesusing test methods of suitable sensitivity and specificity; thepool must give negative results in these tests.
The plasma pool is also tested for hepatitis C virus RNAusing a validated nucleic acid amplification technique
( 2.6.21). A positive control with 100 IU/ml of hepatitis Cvirus RNA and, to test for inhibitors, an internal controlprepared by addition of a suitable marker to a sample of theplasma pool are included in the test. The test is invalid if thepositive control is non-reactive or if the result obtained withthe internal control indicates the presence of inhibitors. Theplasma pool complies with the test if it is found non-reactivefor hepatitis C virus RNA.
CHARACTERS
Before freezing, a clear to slightly turbid liquid without visible signs of haemolysis ; it may vary in colour from light yellow to green.
LABELLINGThe label enables each individual unit to be traced to aspecific donor.
104 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 105/176
Human prothrombin complex
Reference: PA/PH/Exp. 6B/T (05) 48 ANP
NOTE ON THE MONOGRAPH
The main indication of the human prothrombin complex has changed: it is now more linked to the factor II content than to the factor IX content. Furthermore, a factor II (prothrombin) overload could lead to complications like
thrombosis. For both reasons, this revised draft includes aratio limit between factor II and factor IX, with factor II content between 0.7 and 1.5 times factor IX content. Thismakes it possible to guarantee a better level of safety for the product.
Moreover, in order to clarify the assays’ requirements, it is specifically stated under Definition that limits of theconfidence interval apply only when the coagulation factor content is stated by a single value. When the content is stated as a range, the estimated potency is not less thanthe lower limit and not greater than the upper limit of therange stated.
XXXX:0554
HUMAN PROTHROMBIN COMPLEX
Prothrombinum multiplex humanum
DEFINITION
Human prothrombin complex is a plasma protein fractioncontaining blood coagulation factor IX together with variableamounts of coagulation factors II, VII and X; the presenceand proportion of these additional factors depends on themethod of fractionation. It is obtained from human plasmathat complies with the monograph on Human plasma for fractionation (0853).
The potency of the preparation, reconstituted as stated onthe label, is not less than 20 IU of factor IX per millilitre.
If a factor content is stated as a single value, the estimatedpotency is not less than 80 per cent and not more than125 per cent of the stated potency; if a factor content isstated as a range, the estimated potency is not less than thelower limit and not greater than the upper limit of the range.
PRODUCTION
The method of preparation is designed to minimiseactivation of any coagulation factor (to minimise potentialthrombogenicity) and includes a step or steps that havebeen shown to remove or to inactivate known agents of
infection; if substances are used for inactivation of virusesduring production, the subsequent purification proceduremust be validated to demonstrate that the concentration of these substances is reduced to a suitable level and that anyresidues are such as not to compromise the safety of thepreparation for patients.
The specific activity is not less than 0.6 IU of factor IX permilligram of total protein, before the addition of any proteinstabiliser.
The prothrombin complex fraction is dissolved in a suitableliquid. Heparin, antithrombin and other auxiliary substancessuch as a stabiliser may be added. No antimicrobialpreservative is added. The solution is passed through a
bacteria-retentive f ilter, distributed aseptically into thefinal containers and immediately frozen. It is subsequentlyfreeze-dried and the containers are closed under vacuumor under an inert gas.
CHARACTERS
A white or slightly coloured powder or friable solid, veryhygroscopic.
Reconstitute the preparation to be examined as stated onthe label immediately before carrying out the identification,tests (except those for solubility and water) and assay.
IDENTIFICATIONIt complies with the limits of the assay for coagulationfactor IX activity and, where applicable, those for factors II,VII and X.
TESTS
Solubility. To a container of the preparation to be examinedadd the volume of the liquid stated on the label at therecommended temperature. The preparation dissolvescompletely with gentle swirling within 10 min, giving a clearsolution that may be coloured.
pH ( 2.2.3): 6.5 to 7.5.
Osmolality ( 2.2.35 ): minimum 240 mosmol/kg.
Total protein. If necessary, dilute an accurately measuredvolume of the reconstituted preparation with a 9 g/1solution of sodium chloride R to obtain a solution expectedto contain about 15 mg of protein in 2 ml. To 2.0 ml of thesolution in a round-bottomed centrifuge tube add 2 ml of a 75 g/l solution of sodium molybdate R and 2 ml of amixture of 1 volume of nitrogen-free sulphuric acid R and30 volumes of water R. Shake, centrifuge for 5 min, decant the supernatant liquid and allow the inverted tube to drainon filter paper. Determine the nitrogen in the residue by themethod of sulphuric acid digestion ( 2.5.9) and calculate theamount of protein by multiplying the result by 6.25.
Activated coagulation factors ( 2.6.22 ). If necessary, dilutethe preparation to be examined to contain 20 IU of factor IX
per millilitre. For each of the dilutions, the coagulation timeis not less than 150 s.
Heparin. If heparin has been added during preparation,determine the amount present by the assay of heparin incoagulation factor concentrates ( 2.7.12 ). The preparation tobe examined contains not more than the amount of heparinstated on the label and in any case not more than 0.5 IU of heparin per International Unit of factor IX.
Thrombin. If the preparation to be examined containsheparin, determine the amount present as described in thetest for heparin and neutralise it by addition of protamine sulphate R (10 μg of protamine sulphate neutralises 1 IUof heparin). In each of 2 test tubes, mix equal volumesof the reconstituted preparation and a 3 g/l solution of
fibrinogen R. Keep one of the tubes at 37 °C for 6 h and theother at room temperature for 24 h. In a third tube, mix avolume of the fibrinogen solution with an equal volume of asolution of human thrombin R (1 IU/ml) and place the tubein a water-bath at 37 °C. No coagulation occurs in the tubescontaining the preparation to be examined. Coagulationoccurs within 30 s in the tube containing thrombin.
Water. Determined by a suitable method, such as thesemi-micro determination of water ( 2.5.12 ), loss on drying( 2.2.32 ) or near-infrared spectrometry ( 2.2.40), the watercontent is within the limits approved by the competent authority.
Sterility ( 2.6.1). It complies with the test for sterility.
Pyrogens ( 2.6.8 ). It complies with the test for pyrogens.Inject per kilogram of the rabbit’s mass a volume of thereconstituted preparation equivalent to not less than 30 IUof factor IX.
© PHARMEUROPA Vol. 18, No. 1, January 2006 105

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 106/176
Influenza vaccine (surface antigen, inactivated, virosome)
ASSAY
Factor IX. Carry out the assay of human coagulationfactor IX ( 2.7.11).
The estimated potency is not less than 80 per cent andnot more than 125 per cent of the stated potency. Theconfidence interval ( P = 0.95) of the estimated potency is not
greater than 80 per cent to 125 per cent.Factor II. Carry out the assay of human coagulation factor II( 2.7.18 ).
The estimated potency is not less than 80 per cent andnot more than 125 per cent of the stated potency. Theconfidence interval ( P = 0.95) of the estimated potency is not greater than 90 per cent to 111 per cent.
The measured factor II potency is not less than 70 per cent and not more than 150 per cent of the measured factor IXpotency.
Factor VII. If the label states that the preparation contains
factor VII, carry out the assay of human coagulationfactor VII ( 2.7.10).
The estimated potency is not less than 80 per cent andnot more than 125 per cent of the stated potency. Theconfidence interval ( P = 0.95) of the estimated potency is not greater than 80 per cent to 125 per cent.
Factor X. Carry out the assay of human coagulation factor X( 2.7.19).
The estimated potency is not less than 80 per cent andnot more than 125 per cent of the stated potency. Theconfidence interval ( P = 0.95) of the estimated potency is not greater than 90 per cent to 111 per cent.
STORAGE
In an airtight container, protected from light.
LABELLING
The label states:
— the number of International Units of factor IX, factor IIand factor X per container;
— where applicable, the number of International Units of factor VII per container;
— where applicable, that the preparation contains protein Cand/or protein S;
— the amount of protein per container;
— the name and quantity of any added substances, includingwhere applicable, heparin ;
— the name and quantity of the liquid to be used forreconstitution;
— that the transmission of infectious agents cannot betotally excluded when medicinal products prepared fromhuman blood or plasma are administered.
Reference: PA/PH/Exp. 15/T (05) 15 ANP
NOTE ON THE MONOGRAPH
It is proposed to revise this monograph so that it covers 2 slightly different manufacturing processes. Furthermore,the virosome size test has been revised in order to comply with the ISO guide 13321, and an upper limit on the polydispersity index has been included.
XXXX:2053
INFLUENZA VACCINE (SURFACEANTIGEN, INACTIVATED, VIROSOME)
Vaccinum influenzae inactivatum ex corticisantigeniis praeparatum virosomale
DEFINITION
Influenza vaccine (surface antigen, inactivated, virosome) is asterile, aqueous suspension of a strain or strains of influenzavirus, type A or B, or a mixture of strains of the 2 typesgrown individually in fertilised hens’ eggs, inactivated andtreated so that the preparation consists predominantly of haemagglutinin and neuraminidase antigens reconstitutedto virosomes with phospholipids and without diminishingthe antigenic properties of the antigens. The stated amount of haemagglutinin antigen for each strain present in thevaccine is 15 μg per dose, unless clinical evidence supportsthe use of a different amount.The vaccine is a slightly opalescent liquid.
PRODUCTION
GENERAL PROVISIONS
The production method shall have been shown t o yieldconsistently vaccines comparable with the vaccine of provenclinical efficacy and saf ety in man.The production method is validated to demonstrate that theproduct, if tested, would comply with the test for abnormaltoxicity for immunosera and vaccines for human use ( 2.6.9).
CHOICE OF VACCINE STRAIN
The World Health Organisation reviews the worldepidemiological situation annually and if necessaryrecommends the strains that correspond to thisepidemiological evidence.Such strains are used in accordance with the regulationsin force in the signatory states of the Convention on theElaboration of a European Pharmacopoeia. It is nowcommon practice to use reassorted strains giving high yieldsof the appropriate surface antigens. The origin and passagehistory of virus strains shall be approved by the competent authority.SUBSTRATE FOR VIRUS PROPAGATION
Influenza virus seed to be used in the production of vaccineis propagated in fertilised eggs from chicken flocks freefrom specified pathogens (SPF) (5.2.2 ) or in suitable cellcultures (5.2.4), such as chick-embryo fibroblasts or chickkidney cells obtained from SPF chicken flocks (5.2.2 ). Forproduction, the virus of each strain is grown in the allantoiccavity of fertilised hens’ eggs from healthy f locks.
VIRUS SEED LOT
The production of vaccine is based on a seed lot system.Working seed lots represent not more than 15 passages fromthe approved reassorted virus or the approved virus isolate.
The final vaccine represents 1 passage from the workingseed lot. The haemagglutinin and neuraminidase antigens of each seed lot are identified as originating from the correct strain of influenza virus by suitable methods.
106 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 107/176
Influenza vaccine (surface antigen, inactivated, virosome)
Only a working virus seed lot that complies with thefollowing requirements may be used in the preparation of the monovalent pooled harvest.
Bacterial and fungal contamination. Carry out the test forsterility ( 2.6.1), using 10 ml for each medium.
Mycoplasmas ( 2.6.7 ). Carry out the test for mycoplasmas,using 10 ml.
VIRUS PROPAGATION AND HARVEST
An antimicrobial agent may be added to the inoculum. Afterincubation at a controlled temperature, the allantoic f luidsare harvested and combined to form a monovalent pooledharvest. An antimicrobial agent may be added at the timeof harvest.
MONOVALENT POOLED HARVEST
To limit the possibility of contamination, inactivation isinitiated as soon as possible after preparation. The virusis inactivated by a method that has been demonstrated on3 consecutive batches to be consistently effective for themanufacturer. The inactivation process shall have beenshown to be capable of inactivating the influenza virus
without destroying its antigenicity; the process is designedso as to cause minimum alteration of the haemagglutininand neuraminidase antigens. The inactivation processshall also have been shown to be capable of inactivatingavian leucosis viruses and mycoplasmas. If the monovalent pooled harvest is stored after inactivation, it is held at atemperature of 5 ± 3 °C. If formaldehyde solution is used,the concentration does not exceed 0.2 g/l of CH2O at anytime during inactivation; if betapropiolactone is used, theconcentration does not exceed 0.1 per cent V/V at any timeduring inactivation.Before or after the inactivation process, the monovalent pooled harvest is concentrated and purified by high-speedcentrifugation or other suitable method.
Only a monovalent pooled harvest that complies with thefollowing requirements may be used for the preparation of virosomes.Provided the tests for haemagglutinin antigen, neuraminidaseantigen and residual infectious virus have been carriedout with satisfactory results on the monovalent virosomalpreparation, they may be omitted on the monovalent pooledharvest when the manufacturing process is continuousbetween the monovalent pooled harvest and the monovalent virosomal preparation.
Haemagglutinin antigen. Determine the content of haemagglutinin antigen by an immunodiffusion test ( 2.7.1),by comparison with a haemagglutinin antigen referencepreparation or with an antigen preparation calibrated against it (40). Carry out the test at 20-25 °C.
Neuraminidase antigen. The presence and type of neuraminidase antigen are confirmed by suitable enzymaticor immunological methods on the first 3 monovalent pooledharvests from each working seed lot.
Residual infectious virus. Carry out the test describedunder Tests.
PREPARATION OF MONOVALENT VIROSOMES
Virus particles are disrupted into component subunitsby a suitable det ergent approved procedures and furtherpurified so that the monovalent bulk consists mainly of haemagglutinin and neuraminidase antigens. After additionof suitable phospholipids, solubilisation by ultrasonicationand st erile filtration, virosomal preparations Additional
phospholipids may be added and virosomes may be
formed by removal of the detergent either by adsorptionchromatography or another suitable technique. Severalmonovalent virosomal preparations may be pooled.Only a monovalent virosomal preparation that complies withthe following requirements may be used in the preparationof the final bulk vaccine.
Haemagglutinin antigen. Determine the content of
haemagglutinin antigen by an immunodiffusion test ( 2.7.1),by comparison with a haemagglutinin antigen referencepreparation or with an antigen preparation calibrated against it (40). Carry out the test at 20-25 °C.
Neuraminidase antigen. The presence and type of neuraminidase antigen are confirmed by suitable enzymaticor immunological methods on the first 3 virosomalpreparations from each working seed lot.
Residual infectious virus. Carry out the test describedunder Tests. Provided this test has been carried out withsatisfactory results on the monovalent pooled harvest, it maybe omitted on the preparation of monovalent virosomes.
Sterility ( 2.6.1). Carry out the test for sterility, using 10 mlfor each medium.
Purity. The purity of the monovalent virosomal preparationis examined by polyacrylamide gel electrophoresis ( 2.2.31)or by other approved techniques. Mainly haemagglutininand neuraminidase antigens are present.
Residual chemicals Chemicals used for disruption andpurification. Tests for the chemicals used for disruption andpurification are carried out on the monovalent virosomalpreparation, the limits being approved by the competent authority.
Phospholipid. The content and identity of thephospholipids is determined by suitable immunochemical orphysico-chemical methods.
Ratio of haemagglutinin to phospholipid. The ratio of
haemagglutinin content to phospholipid content is withinthe limits approved for the particular product.
Virosome size distribution. The size of the virosomesaverage virosome diameter, determined by a suitablemethod such as laser light scatt ering photon correlationspectroscopy, is not less than 100 nm and not greater than500 300 nm. The polydispersity index is not greater than 0.4.
FINAL BULK VACCINE
Appropriate quantities of the monovalent virosomalpreparations are blended to make the final bulk vaccine.Only a f inal bulk vaccine that complies with the followingrequirements may be used in the preparation of the final lot.
Antimicrobial preservative. Where applicable, determine
the amount of antimicrobial preservative by a suitablechemical method. The content is not less than 85 per cent and not greater than 115 per cent of the intended amount.
Sterility ( 2.6.1). Carry out the test for sterility, using 10 mlfor each medium.
FINAL LOT
The final bulk vaccine is distributed aseptically into sterile,tamper-proof containers. The containers are closed so as toprevent contamination.Only a final lot that is satisfactory with respect to eachof the requirements given under Tests and Assay may bereleased for use. Provided that the test for residual infectiousvirus has been performed with satisfactory results on eachmonovalent pooled harvest or, where appropriate, on the
monovalent virosomal preparations, and that the tests for
(40) Reference haemagglutinin antigens are available from the National Institute for Biological Standards and Control (NIBSC), Blanche Lane, South Mimms, Potters Bar, HertfordshireEN6 3QC, United Kingdom.
© PHARMEUROPA Vol. 18, No. 1, January 2006 107

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 108/176
Isotretinoin
phospholipid, ratio of haemagglutinin to phospholipid,free formaldehyde, ovalbumin and total protein have beenperformed with satisfactory results on the final bulk vaccine,they may be omitted on the final lot.
IDENTIFICATION
The assay serves to confirm the antigenic specificity of the
vaccine.TESTS
Residual infectious virus. Inoculate 0.2 ml of the vaccineinto the allantoic cavity of each of 10 fertilised eggs andincubate at 33-37 °C for 3 days. The test is not valid unlessat least 8 of the 10 embryos survive. Harvest 0.5 ml of theallantoic fluid from each surviving embryo and pool thefluids. Inoculate 0.2 ml of the pooled fluid into a further10 fertilised eggs and incubate at 33-37 °C for 3 days.The test is not valid unless at least 8 of the 10 embryossurvive. Harvest about 0.1 ml of the allantoic fluid from eachsurviving embryo and examine each individual harvest forlive virus by a haemagglutination test. If haemagglutinationis found for any of the fluids, carry out for that fluid afurther passage in eggs and test for haemagglutination; nohaemagglutination occurs.
pH ( 2.2.3): 6.5 to 7.8
Phospholipid. The content and identity of the phospholipidsis determined by a suitable immunochemical orphysico-chemical method.
Ratio of haemagglutinin to phospholipid. The ratio of haemagglutinin content to phospholipid content is withinthe limits approved for the particular product.
Antimicrobial preservative. Where applicable, determinethe amount of antimicrobial preservative by a suitablechemical method. The content is not less than the minimumamount shown to be effective and is not greater than 115 per
cent of the quantity stated on the label.Free formaldehyde ( 2.4.18 ): maximum 0.2 g/l, whereapplicable.
Ovalbumin. Not more than the quantity stated on thelabel and in any case not more than 1 μg per human dose,determined by a suitable immunochemical method ( 2.7.1)using a suitable reference preparation of ovalbumin.
Total protein. Not more than 40 μg of protein other thanhaemagglutinin per virus strain per human dose andnot more than a total of 120 μg of protein other thanhemagglutinin per human dose.
Sterility ( 2.6.1). It complies with the test for sterility.
Virosome size distribution. The size of the virosomesaverage virosome diameter, determined by a suitablemethod such as laser light scatt ering photon correlationspectroscopy, is not less than 100 nm and not greater than500 300 nm. The polydispersity index is not greater than 0.4.
Bacterial endotoxins ( 2.6.14): maximum less than 100 IUper human dose.
ASSAY
Determine the content of haemagglutinin antigen byan immunodiffusion test ( 2.7.1), by comparison with ahaemagglutinin antigen reference preparation or with anantigen preparation calibrated against it (40). Carry out thetest at 20-25 °C. The confidence limits ( P = 0.95) are not
less than 80 per cent and not more than 125 per cent of the estimated haemagglutinin antigen content. The lowerconfidence limit ( P = 0.95) is not less than 80 per cent of theamount stated on the label for each strain.
LABELLING
The label states:— that the vaccine has been prepared on eggs ;
— the strain or strains of influenza virus used to preparethe vaccine;
— the method of inactivation;
— the haemagglutinin content, in micrograms per virusstrain per dose;— the maximum amount of ovalbumin;
— the season during which the vaccine is intended toprotect.
Reference: PA/PH/Exp. VIT/T (05) 3 ANP 1R
NOTE ON THE MONOGRAPH
It is proposed to revise the test for related substances inorder to obtain a better separation between impurities B and C and the principal peak. In order to reduce the
amount of toxic solvents/reagents used, it is proposed todelete the 2 nd identification series, and to modify the assay slightly. As IR alone is sufficient for identification, test Ais also deleted. It is also proposed that the determinationof water replaces the test for loss on drying, which isunreliable due to the potential of the oxidation process tocause a weight increase; in addition, the drying time isvery long (16 h).
XXXX:1019
ISOTRETINOIN
Isotretinoinum
C20H28O2 M r 300.4
DEFINITION
(2 Z ,4 E ,6 E ,8 E )-3,7-Dimethyl-9-(2,6,6-trimethylcyclohex-1-enyl)nona-2,4,6,8-tetraenoic acid.
Content : 98.0 per cent to 102.0 per cent (dried anhydroussubstance).
CHARACTERS
Appearance : yellow or light orange, crystalline powder.
Solubility : practically insoluble in water, soluble inmethylene chloride, slightly soluble in ethanol (96 per cent).It is sensitive to air, heat and light, especially in solution.
Carry out all operations as rapidly as possible and avoid exposure to actinic light; use freshly prepared solutions.
IDENTIFICATION
First i dentifi cati on: A, B.
Second i d entificati on: A, C, D.
A. Ultraviolet and visible absorption spectrophotometry( 2.2.25 ).
T est solution. Dissolve 75.0 mg in 5 ml of methylenechl ori de R and dilute immediat ely t o 100.0 ml withacidified 2-propanol (prepared by diluting 1 ml of 0.01 M hydr ochlori c acid t o 1000 ml with 2 -pr opanol R). Dilute
108 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 109/176
Isotretinoin
5.0 ml of this solution t o 100.0 ml with the acidified2-propanol (solution A). Dilut e 5.0 ml of solution A t o50.0 ml with the acidified 2-propanol.Spectr al r ange : 300-400 nm. Absorpti on maximum : at 354 nm.Specific absor banc e at the absorpti on maximum : 1290to 1420.
B. Infrared absorption spectrophotometry ( 2.2.24).Comparison : isotretinoin CRS .Examine the substances prepared as discs.
C. Examine by thin-layer chromat ography ( 2.2.27 ), using aTLC si li ca gel GF 2 54 pl ate R.T est solution. Dissolve 10 mg of the substance t o beexamined in methyl ene chl orid e R and dilute to 10 mlwith the same solvent. Refer ence solution ( a). Dissolve 10 mg of isotr etinoin CRS in methy lene chl orid e R and dilute t o10 ml with the same solvent. Refer ence soluti on (b). Dissolve10mg of isotr etinoinCRS and 10 mg of tr etinoin CRS in methylene chlori d e R and
dilut e to 10 ml with the same solvent.Apply separat ely t o the plat e 5 μl of each solution.Develop over a path of 15 cm using a mixture of 2 volumesof gl aci al ac eti c aci d R, 4 volumes of acet one R,40 volumes of per oxid e-fr ee ether R and 54 volumes of cyclohex ane R. Allow the plat e to dry in air and examinein ultraviolet light at 254 nm. The principal spot inthe chromat ogram obtained with the t est solution issimilar in position and size t o the principal spot in thechromat ogram obtained with ref erence solution (a).The test is not valid unless the chromatogram obtainedwith ref erence solution (b) shows two clearly separat edprincipal spot s.
D. Dissolve about 5 mg in 2 ml of antimony trichlorid e
soluti on R. An intense red colour develops and lat erbecomes violet.
TESTS
Related substances. Examine by liquid chromat ography( 2.2.29).T est soluti on. Dissolve 0.100 g of the subst ance to beexamined in methanol R and dilut e to 50.0 ml with the samesolvent. Ref er enc e soluti on ( a). Dissolve 10.0 mg of tr etinoin CRS inmethanol R and dilute t o 10.0 ml with the same solvent. Ref er enc e solution (b). Dilut e 1.0 ml of reference solution (a)to 25.0 ml with methanol R. Ref er enc e soluti on ( c). Mix 1.0 ml of ref erence solution (a)
with 0.5 ml of the t est solution and dilut e t o 25.0 ml withmethanol R. Ref er enc e soluti on ( d). Dilute 0.5 ml of the t est solution t o100.0 ml with methanol R.The chromat ographic procedure may be carried out using :— a stainless st eel column 0.15 m long and 4.6 mm in
internal diameter packed with oct adecylsilyl silica gel for chromatogr aphy R (3 μm),
— as mobile phase at a flow rat e of 1.0 ml/min a mixtureof 5 volumes of g laci al ac etic aci d R, 225 volumes of wat er R and 770 volumes of methanol R.
— as det ect or a spectrophot omet er set at 355 nm.Inject separately 10 μl of each of ref erence solutions (b),
(c) and (d) and of the test solution. Adjust the sensitivityof the detector so that the height of the principal peak in
the chromat ogram obtained with ref erence solution (b) isnot less than 70 per cent of the full scale of the recorder.The test is not valid unless the resolution between thepeaks due t o isotretinoin and tretinoin in the chromatogramobtained with ref erence solution (c) is at least 2.0. In thechromat ogram obtained with the test solution : the area of any peak due to tretinoin is not greater than the area of theprincipal peak in the chromat ogram obtained with referencesolution (b) (2.0 per cent); the sum of the areas of any peaks,apart from the principal peak and any peak due t o tretinoin,is not greater than the area of the principal peak in thechromat ogram obtained with ref erence solution (d) (0.5 percent).
Related substances. Liquid chromatography ( 2.2.29).
Test solution. Dissolve 0.30g of thesubstance to be examinedin 10 ml of a 1.0 g/l solution of butylhydroxytoluene R intetrahydrofuran R and dilute to 100.0 ml with acetonitrile R.
Reference solution (a). Dilute 10.0 ml of the test solution to100.0 ml with acetonitrile R. Dilute 1.0 ml of this solution to100.0 ml with acetonitrile R.
Reference solution (b). Dilute 10.0 ml of the test solution
to 50 ml with methanol R. Heat to 50 °C in daylight for2 h. Allow to cool, then dilute to 100.0 ml with methanol R(in situ degradation to obtain impurities A, B and C).
Column :
— size : l = 0.25 m, Ø = 3.0 mm;
— stationary phase : octadecylsilyl silica gel for chromatography R (5 μm)(41) ;
— temperature : 35 °C.
Mobile phase : glacial acetic acid R, water R, methanol R(2.5:200:1800 V/V/V ).
Flow rate : 1.0 ml/min.
Detection : spectrophotometer at 355 nm.
Injection : 10 μl. Run time : twice the retention time of isotretinoin.
Identification of impurities : use the chromatogramobtained with reference solution (b) to identify the peaksdue to impurities A and B + C.
Relative retention with reference to isotretinoin (retentiontime = about 16 min): impurity B + C = about 1.2 ;impurity A = about 1.6.
System suitability : reference solution (b):
— resolution : minimum 3.0 between the peaks due toisotretinoin and impurities B + C.
Limits :
— impurity A : not more than 5 times the area of theprincipal peak in the chromatogram obtained withreference solution (a) (0.5 per cent) ;
— sum of impurities B and C : not more than twice the areaof the principal peak in the chromatogram obtained withreference solution (a) (0.2 per cent) ;
— unspecified impurities : for each impurity, not morethan the area of the principal peak in the chromatogramobtained with reference solution (a) (0.10 per cent);
— total : not more than 7 times the area of the principal peakin the chromatogram obtained with reference solution (a)(0.7 per cent);
— disregard limit : 0.5 times the area of the principal peak
in the chromatogram obtained with reference solution (a)(0.05 per cent).
(41) MERCK Purospher RP18 non-endcapped is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 109

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 110/176
Liquorice dry extract, quantified
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. isotretinoin 2. impurities B + C 3. impurity A
Figure 1019.-1. – Chromatogram for the test for related substances of isotretinoin: test solution
Heavy metals ( 2.4.8 ): maximum 20 ppm.
0.5 g complies with test D. Prepare the reference solution
using 1 ml of lead standard solution (10 ppm Pb) R.Loss on drying ( 2.2.32 ): maximum 0.5 per cent, det erminedon 1.000 g by drying in vacuo f or 16 h.
Water ( 2.5.12 ): maximum 0.5 per cent, determined on1.000 g.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAY
Dissolve 0.200 g in 70 ml of acetone R. Titrate with 0.1 M tetrabutylammonium hydroxide in 2-propanol determiningthe end-point potentiometrically ( 2.2.20).
1 ml of 0.1 M tetrabutylammonium hydroxide in 2-propanol
is equivalent to 30.04 mg of C20H28O2.
STORAGE
In an airtight container, protected from light, at atemperature not exceeding 25 °C.
It is recommended that the contents of an opened containerbe used as soon as possible and any unused part be protectedby an atmosphere of an inert gas.
IMPURITIES
Specified impurities : A, B, C .
A. tretinoin,
B. R = CO2H, R ′ = H: (2 Z ,4 E ,6 Z ,8 E )-3,7-dimethyl-9-(2,6,6-trimethylcyclohex-1-enyl)nona-2,4,6,8-tetraenoic acid(9,13-di-cis-retinoic acid),
D. R = H, R′ = CO2H : (2 E ,4 E ,6 Z ,8 E )-3,7-dimethyl-9-(2,6,6-trimethylcyclohex-1-enyl)nona-2,4,6,8-tetraenoic acid(9-cis-retinoic acid), deleted,
C. (2 Z ,4 Z,6 E ,8 E )-3,7-dimethyl-9-(2,6,6-trimethylcyclohex-1-enyl)nona-2,4,6,8-tetraenoic acid (11,13-di-cis-retinoicacid).
E. oxidation products of isotretinoin. deleted.
Reference: PA/PH/Exp. 13A/T (05) 28 ANP
NOTE ON THE MONOGRAPH
The monograph presented below is a quality high in glycyrrhizic acids suitable for flavouring purposes. Thereaders are requested to inform us, if there is a necessity for a quality low in glycyrrhizic acids suitable for other purposes.
XXXX:2378
LIQUORICE DRY EXTRACT,QUANTIFIED
Liquiritiae extractum siccum quantificatumDEFINITIONDried extract produced from Liquorice root (0277).
Content : 5.0 per cent to 7.0 per cent of glycyrrhizic acid(C42H62O16 ; M r 823) (dried extract).
PRODUCTION
The extract is prepared from the cut herbal drug by waterextraction according to the monograph Extracts (0765).
CHARACTERS Appearance : yellowish-brown or brown powder.Very sweet taste.
IDENTIFICATIONThin layer chromatography ( 2.2.27 ).Test solution. Add 30 ml of hydrochloric acid R1 to 0.30 g
of the extract t o be examined and heat on a water-bath undera reflux condenser for 60 min. After cooling, extract themixture with 2 quantities, each of 20 ml, of ethyl acetate R.Combine the organic layers and filter through a filter covered
110 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 111/176
Liquorice dry extract, quantified
with anhydrous sodium sulphate R. Evaporate the filtrateto dryness in vacuo and dissolve the residue in 2.0 ml of amixture of equal volumes of ethyl acetate R and methanol R.
Reference solution . Dissolve 5.0 mg of glycyrrhetic acid Rand 5.0 mg of thymol R in 5.0 ml of ether R.
Plate : TLC silica gel F 254 plate R (5-40 μm).
Mobile phase : concentrated ammonia R, water R, ethanol (96 per cent) R, ethyl acetate R (1:9:25:65 V/V/V/V ).
Application : 20 μl, as bands.
Development : over a path of 15 cm.
Drying : in air for 5 min.
Detection A : examine in ultraviolet light at 254 nm.
Results A : see below the sequence of the zones present inthe chromatograms obtained with the reference solutionand the test solution. The zone due to glycyrrhetic acid inthe chromatogram obtained with the test solution is similarin intensity to the zone due to glycyrrhetic acid in the
chromatogram obtained with the reference solution.
Top of the plate
Thymol: a quenching zone
_______ _______
_______ _______
Glycyrrhetic acid: a quenchingzone
A quenching zone (glycyrrheticacid)
Reference solution Test solution
Detection B : spray with anisaldehyde solution R, and heat at 100-105 °C for 5-10 min; examine in daylight.
Results B : see below the sequence of the zones present inthe chromatograms obtained with the reference solution andthe test solution. Furthermore, other zones may be present in the chromatogram obtained with the test solution.
Top of the plate
Thymol: a red zone
A yellow zone
_______ _______
_______ _______
Glycyrrhetic acid: a violet zone A violet zone (glycyrrhetic acid)
Reference solution Test solution
TESTS
Water-insoluble substances : maximum 1.0 per cent.
Shake 1.00 g with 50 ml of water R for 15 min. Filter themixture through a tared sintered glass f ilter crucible (100).Wash the residue with 2 quantities, each of 20 ml, of water Rand with 1 quantity of 10 ml of water R. Dry the residue
in an oven at 100-105 °C and determine the weight aftercooling. The residue weighs a maximum of 10 mg.
Loss on drying ( 2.8.17 ): maximum 7.0 per cent.
ASSAY
Liquid chromatography ( 2.2.29).
Test solution. Place 0.250 g of the extract to be examined ina 150 ml ground glass conical flask. Add 100.0 ml of an 8 g/lsolution of ammonia R and treat in an ultrasonic bath for30 min. Centrifuge a part of the supernatant layer and dilute
1.0 ml of the supernatant to 5.0 ml with an 8 g/l solution of ammonia R. Filter through a 0.45 μm filter.
Reference solution (a). Dissolve 0.130 g of monoammonium glycyrrhizate CRS in an 8 g/l solution of ammonia R anddilute to 100.0 ml with the same solvent (solution A). Dilute5.0 ml of solution A to 100.0 ml with an 8 g/l solution of ammonia R (solution A).
Reference solution (b). Dilute 10.0 ml of solution A to100.0 ml with an 8 g/l solution of ammonia R.
Reference solution (c). Dilute 15.0 ml of solution A to100.0 ml with an 8 g/l solution of ammonia R.
Column :
— size : l = 0.10 m, Ø = 4.0 mm;
— stationary phase : octadecylsilyl silica gel for chromatography R (5 μm).
Mobile phase : glacial acetic acid R, acetonitrile R, water R(6:30:64 V/V/V ).
Flow rate : 1.5 ml/min.
Detection : spectrophotometer at 254 nm.
Injection : 10 μl.
Run time : 3 times the retention time of monoammoniumglycyrrhizate.
Retention time : monoammonium glycyrrhizate = about 9 min.
Establish a calibration curve with the concentration of thereference solutions as the abscissa and the correspondingareas as the ordinate. Calculate the percentage content of glycyrrhizic acid using the following expression:
A = concentration of monoammoniumglycyrrhizate in the test solution determinedfrom the calibration curve, in g/100 ml ;
B = declared percentage content of monoammonium glycyrrhizate CRS ;
m = mass of the extract to be examined, in grams;
822 = molecular weight of glycyrrhizic acid;840 = molecular weight of the monoammonium
glycyrrhizate (anhydrous substance).
© PHARMEUROPA Vol. 18, No. 1, January 2006 111

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 112/176
Magnesium citrate, anhydrous
Reference: PA/PH/Exp. INC/T (04) 26 ANP 1R
NOTE ON THE MONOGRAPH
In order to allow a better identification of the substance,it is proposed to add a cross reference to the tests for pH and for loss on drying. The testing conditions for the losson drying have been revised in order to dry the substancecompletely. Limits of content have consequently beenmodified.
XXXX:2339
MAGNESIUM CITRATE, ANHYDROUS
Magnesii citras anhydricus
Mg3(C6H5O7)2 M r 451.1
DEFINITIONTrimagnesium bis(2-hydroxypropane-1,2,3-tricarboxylate).Content : 15.0 per cent to 17.0 16.5 per cent of Mg (driedsubstance).
CHARACTERS Appearance : white or almost white, fine powder.Solubility : soluble in water, practically insoluble in ethanol(96 per cent). It dissolves in dilute hydrochloric acid.
IDENTIFICATIONA. It gives the reaction of citrates ( 2.3.1).B. It gives the reaction of magnesium ( 2.3.1).
C. pH (see Tests).D. Loss on drying (see Tests).
TESTS
Solution S. Dissolve 5.0 g in carbon dioxide-free water Rprepared from distilled water R by heating at 60 °C, cooland dilute to 100 ml with the same solvent.
Appearance of solution. Solution S is not more opalescent than ref erence suspension III clear ( 2.2.1) and not moreintensely coloured than reference solutions Y7 or BY6
( 2.2.2, Method II ).
pH ( 2.2.3): 6.0 to 8.5 for solution S.
Oxalates : maximum 280 ppm.
Dissolve 0.50 g in 4 ml of water R. Add 3 ml of hydrochloric acid R and 1 g of activated zinc R. Allow to stand for 5 min.Transfer the liquid to a tube containing 0.25 ml of a 10 g/lsolution of phenylhydrazine hydrochloride R. Heat toboiling. Cool rapidly, transfer to a graduated cylinder andadd an equal volume of hydrochloric acid R and 0.25 mlof potassium ferricyanide solution R. Shake and allow tostand for 30 min. Any pink colour in the solution is not moreintense than that in a standard prepared at the same timeand in the same manner using 4 ml of a 50 mg/l solutionof oxalic acid R.
Sulphates ( 2.4.13): maximum 0.2 per cent.Dilute 1.5 ml of solution S to 15 ml with distilled water R.
Calcium ( 2.4.3): maximum 0.2 per cent.
Dilute 1.0 ml of solution S to 15 ml with distilled water R.Iron ( 2.4.9): maximum 100 ppm.Dilute 2.0 ml of solution S to 10 ml with distilled water R.
Heavy metals ( 2.4.8 ): maximum 10 ppm.Dissolve 2.0 5.0 g in 7 15 ml of dilute hydrochloric acid Rand heat with heating. Adjust to pH 3.5 with ammonia R.Dilute to 20 50 ml with distilled water R. 12 ml of thissolution complies with test A. Prepare the reference solutionusing lead standard solution (1 ppm Pb) R .
Loss on drying ( 2.2.32 ): maximum 2.5 3.5 per cent,
determined on 1.000 g by drying in an oven at 130 180 °Cfor 16 5 h.
ASSAYDissolve 0.150 g in 50 ml of water R. Carry out thecomplexometric titration of magnesium ( 2.5.11).1 ml of 0.1 M sodium edetate is equivalent to 2.431 mg of Mg.
Reference: PA/PH/Exp. 10C/T (05) 22 ANP
NOTE ON THE MONOGRAPH
The TLC for related substances has been replaced by an
LC. The corresponding transparency statement has beenintroduced. Based on the low contents found, the impuritieshave been classified under “other detectable impurities”. However, the TLC is maintained for identification purposes. The 2 nd identification series is maintained as the substance may be used in pharmacies. It is also proposed to tighten the limits of content based on batch results.
XXXX:0622
MECLOZINE HYDROCHLORIDE
Meclozini hydrochloridum
C25H29Cl3N2 M r 463.9
DEFINITION1-[( RS -(4-Chlorophenyl)phenylmethyl]-4-(3-methylbenzyl)piperazine dihydrochloride.Content : 98.0 99.0 per cent to 102.0 101.0 per cent (anhydrous substance).
CHARACTERS Appearance : yellow white or yellowish-white, crystallinepowder.Solubility : slightly soluble in water, soluble in ethanol(96 per cent) and in methylene chloride.
IDENTIFICATION First identification : B, D.Second identification: A, C, D.A. Ultraviolet and visible absorption spectrophotometry
( 2.2.25 ).Test solution. Dissolve 15.0 mg in 0.1 M hydrochloric acid and dilute to 100.0 ml with the same acid.Dilute 10.0 ml of this solution to 100.0 ml with 0.1 M
hydrochloric acid .Spectral range : 220-350 nm. Absorption maximum : at 232 nm.
112 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 113/176
Meclozine hydrochloride
Specific absorbance at the absorption maximum : 345 to380 (anhydrous substance).
The test solution shows a weak absorbance without adefined maximum between 260 nm and 300 nm.
B. Infrared absorption spectrophotometry ( 2.2.24).
Preparation : discs of potassium chloride R.
Comparison: meclozine hydrochloride CRS
.
C. Examine the chromat ograms obtained in the t est f or related substances. The principal spot in thechromat ogram obtained with test solution (b) is similarin position, colour and size t o the principal spot in thechromat ogram obtained with ref erence solution (a).
C. Thin-layer chromatography ( 2.2.27 ).
Solvent mixture : methanol R, methylene chloride R(50:50 V/V ).
Test solution. Dissolve 50 mg of the substance to beexamined in the solvent mixture and dilute to 10 ml withthe solvent mixture.
Reference solution. Dissolve 50 mg of meclozinehydrochloride CRS in the solvent mixture and dilute to10 ml with the solvent mixture. Plate : TLC silica gel G plate R.
Mobile phase : concentrated ammonia R, methanol R,toluene R, methylene chloride R (0.5:5:30:60 V/V/V/V ).
Application : 10 μl.
Development : over a path of 15 cm.
Drying : in air.
Detection : spray with dilute potassium iodobismuthate solution R.
Results : the principal spot in the chromatogram obtainedwith the test solution is similar in position, colour andsize to the principal spot in the chromatogram obtained
with the reference solution.D. Dissolve about 15 mg in 2 ml of ethanol (96 per cent) R .The solution gives reaction (a) of chlorides ( 2.3.1).
TESTS
Appearance of solution. The solution is clear ( 2.2.1) and not more intensely coloured than reference solution Y6 ( 2.2.2, Method II ).
Dissolve 0.50 g in ethanol (96 per cent) R and dilute to25 ml with the same solvent.
Acidity or alkalinity. Calculate the acidity or alkalinityfrom the titration volumes obtained in the assay using thefollowing expression:
V 1 = volume of 0.1 M sodium hydroxide added at the1st point of inflexion;
V 2 = volume of 0.1 M sodium hydroxide added at the2nd point of inflexion.
A is not less than 0.3 ml and not more than 0.3 ml for0.3500 g of the substance to be examined.
Related substances. Examine by thin-layer chromatography( 2.2.27 ), using si li c a g el G R as the coating substance.
T est soluti on ( a). Dissolve 0.50 g of the substance t o beexamined in a mixture of equal volumes of methanol R and
methyl ene chl ori d e R and dilute to 10 ml with the samemixture of solvents.
T est soluti on ( b). Dilute 1 ml of test solution (a) t o 10 ml witha mixture of equal volumes of methanol R and methyl enechl ori de R.
Ref er ence soluti on ( a). Dissolve 50 mg of mecl ozinehy d r ochl ori de CRS in a mixture of equal volumes of methanol R and methylene chlorid e R and dilute to 10 mlwith the same mixture of solvent s.
Ref er ence soluti on ( b). Dilut e 0.5 ml of test solution (b) t o10 ml with a mixture of equal volumes of methanol R andmethyl ene chl ori de R.
Apply separately to the plat e 10 μl of each solution. Developover a path of 15 cm using a mixture of 0.5 volumes of conc entr ated ammonia R, 5 volumes of methanol R,30 volumes of t oluene R and 60 volumes of methyl enechlori de R. Allow the plat e t o dry in air and spray withdilute potassium i od obismuthate solution R. Any spot in thechromat ogram obtained with t est solution (a), apart fromthe principal spot, is not more int ense than the spot in thechromat ogram obtained with ref erence solution (b) (0.5 percent). Disregard any yellowish-whit e spot at the startingpoint.
Liquid chromatography ( 2.2.29).Solvent mixture : water R, acetonitrile R (30:70 V/V ).
Test solution. Dissolve 50 mg of the substance to beexamined in the solvent mixture and dilute to 100 ml withthe solvent mixture.
Reference solution (a). Dilute 1.0 ml of the test solutionto 100.0 ml with the solvent mixture. Dilute 10.0 ml of thissolution to 50.0 ml with the solvent mixture.
Reference solution (b). Dissolve 10 mg of the substanceto be examined and 10 mg of 4-chlorobenzophenone R(impurity C) in the solvent mixture and dilute to 100.0 mlwith the solvent mixture. Dilute 5.0 ml of this solution to50.0 ml with the solvent mixture.
Column :— size : l = 0.30 m, Ø = 3.9 mm;
— stationary phase : octadecylsilyl silica gel for chromatography R (10 μm)(42).
Mobile phase : dissolve 1.5 g of sodium heptanesulphonate Rin 300 ml of water R and adjust to pH 4 with dilute sulphuric acid R ; mix 30 volumes of this solution with 70 volumes of acetonitrile R.
Flow rate : 1.3 ml/min.
Detection : spectrophotometer at 230 nm.
Injection : 20 μl.
Run time : 3 times the retention time of meclozine.
Retention time : meclozine = about 4 min.System suitability : reference solution (b):
— resolution : minimum 2.0 between the peaks due tomeclozine and impurity C.
Limits :
— unspecified impurities : for each impurity, not morethan 0.5 times the area of the principal peak in thechromatogram obtained with reference solution (a)(0.10 per cent);
— total : not more than the area of the principal peak inthe chromatogram obtained with reference solution (a)(0.2 per cent);
— disregard limit : 0.25 times the area of the principal peak
in the chromatogram obtained with reference solution (a)(0.05 per cent).
(42) μ-Bondapack C18 is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 113

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 114/176
Meclozine hydrochloride
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity A 2. impurity B 3. meclozine 4. impurity C
Figure 0622.-1. – Chromatogram for the test for related substances of meclozine hydrochloride
Water ( 2.5.12 ): maximum 5.0 per cent, determined on0.200 g.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAY
Dissolve 0.3500 g in 50 ml of ethanol (96 per cent) R. Carryout a potentiometric titration ( 2.2.20), using 0.1 M sodiumhydroxide. Read the volume added between the 2 pointsof inflexion.
1 ml of 0.1 M sodium hydroxide is equivalent to 46.39 mgof C25H29Cl3N2.
STORAGE
In an airtight container.
IMPURITIES
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for
pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : A, B, C.
A. 3-methylbenzaldehyde,
B. ( RS )-(4-chlorophenyl)(phenyl)methanol,
C. (4-chlorophenyl)(phenyl)methanone.
Reagents
4-Chlorobenzophenone. C13H9ClO. ( M r 216.7). XXXXXXX.[134-85-0]. (4-Chlorophenyl)(phenyl)methanone.
114 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 115/176
Molsidomine
Reference: PA/PH/Exp. 10A/T (01) 78 ANP 2R
NOTE ON THE MONOGRAPH
A proposal was published in Pharmeuropa 14.1. Following the comments received, additional experimental work was performed and the draft monograph has been completely revised. The lower content limit was tightened based on batch data, the pH range was enlarged based on theregistered limits, and the limit for sulphated ash wasreduced based on batch data. The gradient LC method proposed allows the control of impurity B at 3 ppm. A specific test for the control of impurity E, which is a potential degradation product, has also been introduced.
XXXX:1701
MOLSIDOMINE
Molsidominum
C9H14N4O4 M r 242.2
DEFINITION
N -(Ethoxycarbonyl)-3-(morpholin-4-yl)sydnonimine.Content : 99.0 per cent to 101.0 per cent (dried substance).
CHARACTERS Appearance : white or almost white, crystalline powder.Solubility : sparingly soluble in water, soluble in anhydrousethanol, methylene chloride and ethyl acetate.
mp: about 142 °C.IDENTIFICATIONInfrared absorption spectrophotometry ( 2.2.24).
Comparison : molsidomine CRS .
TESTS
Appearance of solution. The solution is clear ( 2.2.1) and not more intensely coloured than reference solution B7 ( 2.2.2, Method II ).Dissolve 1.0 g in anhydrous ethanol R and dilute to 20.0 mlwith the same solvent.
pH ( 2.2.3): 5.5 to 7.5.Dissolve 0.50 g in carbon dioxide-free water R and dilute to50.0 ml with the same solvent.
Impurity B. Liquid chromatography ( 2.2.29) as describedin the test for related substances with the followingmodifications.
Detection : spectrophotometer at 240 nm. Injection : 20 μl of test solution (a) and reference solution (c). Relative retention with reference to molsidomine (retentiontime = about 9 min): impurity B = about 0.43.System suitability : reference solution (c):— signal-to-noise ratio : minimum 20 for the principal peak.
Limit :— impurity B : not more than the area of the corresponding
peak in the chromatogram obtained with referencesolution (c) (3 ppm).
Impurity E. Liquid chromatography ( 2.2.29).
Test solution. Dissolve 200.0 mg of the substance to beexamined in 100.0 ml of the mobile phase. Reference solution (a). Dissolve 1.000 g of molsidomineimpurity E CRS in 1000 ml of water for chromatography R.Dilute 25.0 ml of this solution to 500.0 ml with water for chromatography R. Dilute 20.0 ml of this solution to
500.0 ml with water for chromatography R. Dilute 10.0 mlof this solution to 100.0 ml with the mobile phase.
Reference solution (b). Mix 10.0 ml of the test solution with10.0 ml of reference solution (a).
Column :— size : l = 0.25 m, Ø = 4.0 mm;— stationary phase : resin for reversed-phase ion
chromatography R(43) ;
— temperature : 25 °C. Mobile phase : dilute 3.0 ml of methanesulphonic acid Rand 75 ml of acetonitrile R in water for chromatography Rand dilute to 1000 ml with the same solvent.
Suppressor regenerant : water for chromatography R.
Flow rate : 1.0 ml/min. Expected background conductivity : less than 0.5 μS.
Detection : conductivity detector with a sensitivity of 10 μS.
Injection : 50 μl.
Run time : 20 min.
Retention time : impurity E = about 7 min.
System suitability :— signal-to-noise ratio : minimum 6 for the peak due to
impurity E in the chromatogram obtained with referencesolution (b);
— recovery : the area of the peak due to impurity E in thechromatogram obtained with reference solution (b) is not
less than 80 per cent of the peak area of the principal peakin the chromatogram obtained with reference solution (a).
Limit :
— impurity E : not more than the area of the principal peakin the chromatogram obtained with reference solution (a)(0.01 per cent).
Related substances. Liquid chromatography ( 2.2.29). Protect the solutions from light.
Solvent mixture : methanol R, mobile phase B (1:9 V/V ).
Test solution (a). Dissolve 0.200 g of the substance to beexamined in 2.5 ml of methanol R using an ultrasonic bathand keeping at 20 °C. Dilute to 5.0 ml with mobile phase B.
Test solution (b). Dilute 1.0 ml of test solution (a) to 20.0 ml
with the solvent mixture. Reference solution (a). Dilute 1.0 ml of test solution (b)to 100.0 ml with the solvent mixture. Dilute 1.0 ml of thissolution to 10.0 ml with the solvent mixture.
Reference solution (b). Dissolve 10 mg of the substance tobe examined in 2.0 ml of 2 M hydrochloric acid and boilfor 30 min to produce impurity C. Allow to cool. Neutralisewith 2.0 ml of 2 M sodium hydroxide and dilute to 10.0 mlwith methanol R. Dilute 1.0 ml of this solution to 10.0 mlwith the solvent mixture.
Reference solution (c). Dissolve 2.4 mg of molsidomineimpurity B CRS in 80 ml of methanol R using an ultrasonicbath and keeping at 20 °C. Dilute to 100.0 ml withmethanol R. Dilute 2.0 ml of the solution to 100.0 ml with
the solvent mixture. Dilute 5.0 ml of this solution to 20.0 mlwith the solvent mixture.
(43) Dionex IonPac CS14 is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 115

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 116/176
Molsidomine
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity A 2. impurity D 3. impurity C 4. molsidomine
Figure 1701.-1. – Chromatogram for the test for related substances of molsidomine : test solution spiked with 0.1 per cent of impurities A, C and D
Reference solution (d). Dissolve 10 mg of linsidominehydrochloride R (impurity A) and 5 mg of molsidomineimpurity D CRS in 10 ml of methanol R and dilute to 50.0 ml
with the solvent mixture. Dilute 5.0 ml of this solution to50.0 ml with the solvent mixture.
Column :
— size : l = 0.15 m, Ø = 4.6 mm;
— stationary phase : end-capped octadecylsilyl silica gel for chromatography R (5 μm) with a specific surface areaof 430 m2 /g and a pore size of 9.5 nm(44) ;
— temperature : 30 °C.
Mobile phase :
— mobile phase A : methanol R ;
— mobile phase B: dissolve 4.0 g of potassium dihydrogen phosphate R in water for chromatography R and dilute
to 1000 ml with the same solvent;Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 - 3 10 90
3 - 10 10 → 80 90 → 20
10 - 13 80 20
13 - 13.1 80 → 10 20 → 90
13.1 - 15 10 90
Flow rate : 1.3 ml/min.
Detection : spectrophotometer at 210 nm.
Injection : 20 μl of test solution (b) and reference
solutions (a), (b) and (d).
Relative retention with reference to molsidomine(retention time = about 9 min): impurity A = about 0.23;impurity D = about 0.32; impurity C = about 0.86.
System suitability : reference solution (d):
— resolution : minimum 6.0 between the peaks due toimpurities A and D.
Limits :
— correction factor : for the calculation of content, multiplythe peak area of impurity C by 1.5;
— impurity C : not more than the area of the peak dueto molsidomine in the chromatogram obtained withreference solution (a) (0.1 per cent) ;
— unspecified impurities : for each impurity, not morethan the area of the peak due to molsidomine in thechromatogram obtained with reference solution (a)
(0.10 per cent);— total : not more than 3 times the area of the peak dueto molsidomine in the chromatogram obtained withreference solution (a) (0.3 per cent) ;
— disregard limit : 0.5 times the area of the peak dueto molsidomine in the chromatogram obtained withreference solution (a) (0.05 per cent).
Heavy metals ( 2.4.8 ): maximum 20 ppm.
1.0 g complies with test C. Prepare the reference solutionusing 2 ml of lead standard solution (10 ppm Pb) R.
Loss on drying ( 2.2.32 ) : maximum 0.5 per cent, determinedon 1.000 g by drying in an oven at 105 °C.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determined
on 1.0 g.
(44) Phenomenex Luna 5μ C18 100Å is suitable.
116 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 117/176
Moxidectin for veterinary use
ASSAY
Dissolve 0.200 g in a mixture of 5 ml of acetic anhydride Rand 50 ml of acetic acid R. Titrate with 0.1 M perchloric acid , determining the end-point potentiometrically ( 2.2.20).
1 ml of 0.1 M perchloric acid is equivalent to 24.22 mgof C9H14N4O4.
STORAGE
Protected from light.
IMPURITIES
Specified impurities: B, C, E.
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to
identify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : A, D.
A. 3-(morpholin-4-yl)sydnonimine (linsidomine),
B. R = NO: 4-nitrosomorpholine,
D. R = CHO : morpholine-4-carbaldehyde,
E. R = H : morpholine,
C. (2 E )-(morpholin-4-ylimino)acetonitrile.
Reagents
Linsidomine hydrochloride. C6H11ClN4O2. ( M r 206.6). XXXXXXX. [33876-97-0]. 3-(Morpholin-4-yl)sydnoniminehydrochloride.White or almost white powder.
Reference: PA/PH/Exp. 7/T (04) 60 ANP
XXXX:1656
MOXIDECTIN FOR VETERINARY USE
Moxidectinum ad usum veterinarium
C37H53NO8 M r 640
DEFINITION
(2a E ,4 E ,4′ E ,5′S ,6 R,6′S ,8 E ,11 R,13 R,15S ,17a R,20 R,20a R,20bS )-6′-[(1 E )-1,3-Dimethyl-1-buten-1-yl]-20,20b-dihydroxy-5′,6,8,19-tetramethyl-5′,6′,10,11,14,15,17a,20,20a,20b-decahydro-2 H ,7 H -spiro[11,15-methanofuro[4,3,2- pq ][2,6]benzodioxacyclooctadecine-13,2 ′-pyran]-4′,17(3′ H ,6 H )-dione 4′-(O-methyloxime). Semi-synthetic product derivedfrom a fermentation product. It may contain suitablestabilisers such as antioxidants.
Content : 92.0 per cent to 102.0 per cent (anhydroussubstance).
CHARACTERS
Appearance : white or almost white, amorphous powder.
Solubility : practically insoluble in water, very soluble inethanol (96 per cent), slightly soluble in hexane.
IDENTIFICATION
Infrared absorption spectrophotometry ( 2.2.24).
Comparison : moxidectin CRS .
TESTS
Appearance of solution. The solution is clear ( 2.2.1) andnot more intensely coloured than reference solution GY6
( 2.2.2, Method II ).
Dissolve 0.40 g in benzyl alcohol R and dilute to 20 ml withthe same solvent.
Related substances. Liquid chromatography ( 2.2.29).
A. Test solution. Dissolve 25.0 mg of the substance to beexamined in acetonitrile R and dilute to 25.0 ml withwith the same solvent.
Reference solution (a). Dilute 1.0 ml of the test solutionto 100.0 ml acetonitrile R.
Reference solution (b). Dissolve 5 mg of moxidectin for system suitability CRS in 5 ml of acetonitrile R.
Reference solution (c). Dissolve 25.0 mg of moxidectin CRS in acetonitrile R and dilute to 25.0 mlwith acetonitrile R.
© PHARMEUROPA Vol. 18, No. 1, January 2006 117

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 118/176
Moxidectin for veterinary use
Column :
— size : l = 0.15 m, Ø = 3.9 mm;
— stationary phase: end-capped octadecylsilyl silica gel for chromatography R (4 μm)(45) ;
— temperature : 50 °C.
Mobile phase : dissolve 7.7 g of ammonium acetate R in
400 ml of water R, adjust to pH 4.8 with glacial acetic acid R, and add 600 ml of acetonitrile R.
Flow rate : 2.5 ml/min.
Detection : spectrophotometer at 242 nm.
Injection : 10 μl of the test solution and referencesolutions (a) and (b).
Run time : 3 times the retention time of moxidectin.
Identification of impurities : use the chromatogramsupplied with moxidectin for system suitability CRS andthe chromatogram obtained with reference solution (b)to identify the peaks due to impurities A, B, C, D, E + Fand G.
Relative retention with reference to moxidectin
(retention time = about 12 min): impurity A = about 0.5;impurity B = about 0.7; impurity C = about 0.75;impurity D = about 0.94; impurities E + F = about 1.3-1.5;impurity G = about 1.6.
System suitability : reference solution (b):
— peak-to-valley ratio : minimum 3, where H p = height above the baseline of the peak due to impurity D and H v = height above the baseline of the lowest point of the curve separating this peak from the peak due tomoxidectin.
Limits :
— impurity D : not more than 2.5 times the area of theprincipal peak in the chromatogram obtained withreference solution (a) (2.5 per cent) ;
— sum of impurities E and F : not more than 1.5 timesthe area of the principal peak in the chromatogramobtained with reference solution (a) (1.5 per cent) ;
— impurities A, C, G: for each impurity, not morethan 1.5 times the area of the principal peak in thechromatogram obtained with reference solution (a)(1.5 per cent);
— impurity B : not more than 0.5 times the area of theprincipal peak in the chromatogram obtained withreference solution (a) (0.5 per cent) ;
— any other impurity : for each impurity, not morethan 0.5 times the area of the principal peak in thechromatogram obtained with reference solution (a)
(0.5 per cent);— disregard limit : 0.1 times the area of the principal
peak in the chromatogram obtained with referencesolution (a) (0.1 per cent); disregard the peak due tothe stabiliser.
B. Test solution. Dissolve 75.0 mg of the substance to beexamined in acetonitrile R and dilute to 25.0 ml with thesame solvent.
Reference solution (a). Dilute 1.0 ml of the test solutionto 100.0 ml with acetonitrile R.
Reference solution (b). Dissolve 5 mg of moxidectin for system suitability CRS in 5 ml of acetonitrile R.
Column :
— size : l = 0.15 m, Ø = 3.9 mm;
— stationary phase : end-capped octadecylsilyl silica gel for chromatography R (4 μm)(46) ;
— temperature : 35 °C. Mobile phase : dissolve 3.8 g of ammonium acetate R in250 ml of water R, adjust to pH 4.2 with acetic acid R,and add 750 ml of acetonitrile R. Flow rate : 2.0 ml/min.
Detection : spectrophotometer at 242 nm. Injection : 10 μl. Run time : 10 times the retention time of moxidectin. Identification of impurities : use the chromatogramsupplied with moxidectin for system suitability CRS andthe chromatogram obtained with reference solution (b) toidentify the peaks due to impurities H + I, J and K. Relative retention with reference to moxidectin (retentiontime = about 4 min): impurities H + I = about 2;impurity J = about 2.2 ; impurity K = about 3.4.System suitability : reference solution (b):— resolution : minimum 2 between the peaks due to
impurities H + I and J.
Limits :— sum of impurities H and I : not more than the area of
the principal peak in the chromatogram obtained withreference solution (a) (1.0 per cent);
— impurities J, K : for each impurity, not more than0.5 times the area of the principal peak in thechromatogram obtained with reference solution (a)(0.5 per cent);
— any other impurity : for each impurity, not morethan 0.5 times the area of the principal peak in thechromatogram obtained with reference solution (a)(0.5 per cent);
— disregard limit : 0.1 times the area of the principalpeak in the chromatogram obtained with referencesolution (a) (0.1 per cent); disregard the peak due tothe stabiliser.
Total of all impurities. Calculate the total of the impuritieseluting from the start of the run to impurity G in test A, andfrom impurities H + I to the end of the run in test B. The sumof all impurities is not more than 7.0 per cent.
Heavy metals ( 2.4.8 ): maximum 20 ppm.1 g complies with test C. Prepare the reference solutionusing 2 ml of lead standard solution (10 ppm Pb) R.
Water ( 2.5.12 ) : maximum 1.3 per cent, determined on 0.5 g.
Sulphated ash ( 2.4.14): maximum 0.2 per cent, determinedon 1.0 g.
ASSAYLiquid chromatography ( 2.2.29) as described in test A forrelated substances with the following modification. Injection : test solution and reference solution (c).Calculate the percentage content of C37H53NO3 using thedeclared content of moxidectin CRS .
IMPURITIESSpecified impurities: A, B, C, D, E, F, G, H, I, J, K.
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for
pharmaceutical use (2034). It is therefore not necessary to
(45) Waters Nova-Pak C18 and Waters Pico-Tag C18 are suitable.(46) Waters Nova-Pak C18 and Waters Pico-Tag C18 are suitable.
118 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 119/176
Moxidectin for veterinary use
identify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : L.
A. (2a E ,4 E ,4′ E ,5′S ,6 R,6′S ,8 E ,11 R,13 R,15S ,17a R,20 R,20a R,20bS )-6′-[(1 E )-1,3-dimethyl-1-buten-1-yl]-20,20b-dihydroxy-5′,6,8,19-tetramethyl-5′,6′,10,11,14,15,17a,20,20a,20b-
decahydro-2 H ,7 H -spiro[11,15-methanofuro[4,3,2- pq ][2,6]benzodioxacyclooctadecine-13,2 ′-pyran]-4′,17(3′ H ,6 H )-dione 4′-(O-methyloxime),
B. (2a E ,4 E ,4′ Z ,6 R,6′S ,8 E ,11 R,13 R,15S ,17a R,20 R,20a R,20bS )-6′-[(1 E )-1,3-dimethyl-1-buten-1-yl]-20,20b-dihydroxy-6,8,19-trimethyl-5′,6′,10,11,14,15,17a,20,20a,20b-decahydro-2 H ,7 H -spiro[11,15-methanofuro[4,3,2- pq ][2,6]benzodioxacyclooctadecine-13,2 ′-pyran]-4′,17(3′ H ,6 H )-dione 4′-(O-methyloxime),
C. (2a E ,4 E ,4′ E ,5′S ,6 R,6′S ,8 E ,11 R,13 R,15S ,17a R,20 R,20a R,20bS )-20,20b-dihydroxy-5′,6,8,19-tetramethyl-6′-[(1 E )-
1-methyl-1-buten-1-yl]-5′,6′,10,11,14,15,17a,20,20a,20b-decahydro-2 H ,7 H -spiro[11,15-methanofuro[4,3,2- pq ][2,6]benzodioxacyclooctadecine-13,2 ′-pyran]-4′,17(3′ H ,6 H )-dione 4′-(O-methyloxime),
D. (2a E ,4 E ,4′ E ,5′S ,6 R,6′S ,8 E ,11 R,13 R,15S ,17aS ,20 R,20a R,20bS )-6′-[(1 E )-1,3-dimethyl-1-buten-1-yl]-20,20b-dihydroxy-5′,6,8,19-tetramethyl-5′,6′,10,11,14,15,17a,20,20a,20b-decahydro-2 H ,7 H -spiro[11,15-methanofuro[4,3,2- pq ][2,6]benzodioxacyclooctadecine-13,2 ′-pyran]-4′,17(3′ H ,6 H )-dione 4′-(O-methyloxime),
E. (2a E ,4 E ,4′ E ,5′S ,6 R,6′S ,8 E ,11 R,13 R,15S ,19S ,20 R,20a R,20b R)-6′-[(1 E )-1,3-dimethyl-1-buten-1-yl]-20,20b-dihydroxy-5′,6,8,19-tetramethyl-5′,6′,10,11,14,15,19,20,20a,20b-decahydro-2 H ,7H-spiro[11,15-methanofuro[4,3,2- pq ][2,6]benzodioxacyclooctadecine-13,2 ′-pyran]-4′,17(3′ H ,6 H )-dione 4′-(O-methyloxime),
F. one of R1 to R5 is ethyl, the others are methyl,
© PHARMEUROPA Vol. 18, No. 1, January 2006 119

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 120/176
Moxidectin for veterinary use
G. (3S ,4′ E ,5 R,5′S ,6′S ,7 R,9 E ,12 R,13 E ,15 E ,16aS ,18S ,20a R)-6′-[(1 E )-1,3-dimethyl-1-buten-1-yl]-16a,18-dihydroxy-5′,10,12,16,19-pentamethyl-3,4,5′,6′,7,8,11,12,16a,17,18,20a-dodecahydro-1 H -spiro[3,7-methano[2,6]benzodioxacyclooctadecine-5,2 ′-pyran]-1,4′(3′ H )-dione4′-(O-methyloxime),
H. (2a E ,4 E ,4′ E ,5′S ,6 R,6′S ,8 E ,11 R,13 R,15S ,20a R,
20b R)-6′-[(1 E )-1,3-dimethyl-1-buten-1-yl]-20b-hydroxy-5′,6,8,19-tetramethyl-5′,6′,10,11,14,15,20a,20b-octahydro-2 H ,7 H -spiro[11,15-methanofuro[4,3,2- pq ][2,6]benzodioxacyclooctadecine-13,2 ′-pyran]-4′,17(3′ H ,6 H )-dione 4′-(O-methyloxime),
I. (2a E ,4 E ,4′S ,5′S ,6 R,6′S ,8 E ,11 R,13S ,15S ,17a R,20 R,20a R,20bS )-6′-[(1 E )-1,3-dimethyl-1-buten-1-yl]-20,20b-dihydroxy-5′,6,8,19-tetramethyl-4′-[(methylthio)methoxy]-3′,4′,5′,6,6′,7,10,11,14,15,17a,20,20a,20b-tetradecahydro-2 H ,17 H -spiro[11,15-methanofuro[4,3,2- pq ][2,6]benzodioxacyclooctadecine-13,2′-pyran]-17-one,
J. R = CH2-S-CH3, R′ = H: (2a E ,4 E ,4′ E ,5′S ,6 R,6′S ,8 E ,11 R,13 R,15S ,17a R,20 R,20a R,20bS )-6′-[(1 E )-1,3-dimethyl-1-buten-1-yl]-20-hydroxy-5′,6,8,19-tetramethyl-20b-[(methylthio)methoxy]-5 ′,6′,10,11,14,15,17a,20,20a,20b-decahydro-2 H ,7 H -spiro[11,15-methanofuro[4,3,2- pq ][2,6]benzodioxacyclooctadecine-13,2 ′-pyran]-4′,17(3′ H ,6 H )-
dione 4′-(O-methyloxime),
K. R = H, R′ = CO-C6H4- pNO2 : (2a E ,4 E ,4′ E ,5′S ,6 R,6′S ,8 E ,11 R,13 R,15S ,17a R,20 R,20a R,20bS )-6′-[(1 E )-1,3-dimethyl-1-buten-1-yl]-20b-hydroxy-4′-(methoxyimino)-5′,6,8,19-tetramethyl-17-oxo-3′,4′,5′,6,6′,10,11,14,15,17,17a,20,20a,20b-tetradecahydro-2 H ,7 H -spiro[11,15-methanofuro[4,3,2- pq ][2,6]benzodioxacyclooctadecine-13,2 ′-pyran]-20-yl4-nitrobenzoate,
L. (2a E ,4 E ,4′ Z ,5′S ,6 R,6′S ,8 E ,11 R,13 R,15S ,17a R,20 R,20a R,20bS )-6′-[(1 E )-1,3-dimethyl-1-buten-1-yl]-20,20b-dihydroxy-5′,6,8,19-tetramethyl-5′,6′,10,11,14,15,17a,20,20a,20b-decahydro-2 H ,7 H -spiro[11,15-methanofuro[4,3,2- pq ][2,6]benzodioxacyclooctade cine-13,2′-pyran]-4′,17(3′ H ,6 H )-dione 4′-(O-methyloxime).
120 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 121/176
Norgestimate
Reference: PA/PH/Exp. 10B/T (05) 74 ANP
NOTE ON THE MONOGRAPH
Norgestimate is a steroid with progestative action. The substance has been known for more than 25 years and isused in combination with estrogenic compounds as an oral contraceptive. A typical daily dose is 250 μ g. Norgestimateis already described in the USP. This Ph. Eur. proposal isbased on the USP monograph, however, now only 1 related substance test is proposed. It has been shown that theimpurities detected with the 2 nd USP related substance test are far below the current identification threshold.
XXXX:1732
NORGESTIMATE
Norgestimatum
C23H31NO3 M r 369.5
DEFINITION( EZ )-13β-Ethyl-3-hydroxyimino-18,19-dinor-17α-pregn-4-en-20-yn-17β-yl acetate.Content : 99.0 per cent to 101.0 per cent (dried substance).
CHARACTERS Appearance : white or almost white powder.
Solubility : practically insoluble in water, freely soluble inmethylene chloride, soluble in acetone.
IDENTIFICATIONInfrared absorption spectrophotometry ( 2.2.24).Comparison : norgestimate CRS .
TESTS
Specific optical rotation ( 2.2.7 ): + 42.0 to + 50.0 (driedsubstance).Dissolve 0.200 g in methylene chloride R and dilute to20.0 ml with the same solvent.
Related substances. Liquid chromatography ( 2.2.29).
Solvent mixture : water R, methanol R (1:4 V/V ).Test solution. Dissolve 25.0 mg of the substance to beexamined in the solvent mixture and dilute to 50.0 ml withthe solvent mixture.
Reference solution (a). Dilute 2.0 ml of the test solutionto 100.0 ml with the solvent mixture. Dilute 1.0 ml of thissolution to 20.0 ml with the solvent mixture. Reference solution (b). Dissolve 2 mg of norgestimate for system suitability CRS (containing impurity A) in 4 ml of thesolvent mixture.Column :— size : l = 0.10 m, Ø = 4.6 mm;— stationary phase : spherical end-capped octadecylsilyl
silica gel for chromatography R (5 μm)(47) ;— temperature : 40 °C. Mobile phase : acetonitrile R, tetrahydrofuran for chromatography R, water R (18:22:60 V/V/V ). Flow rate : 1.0 ml/min.
Detection : spectrophotometer at 244 nm. Injection : 25 μl. Run time : 3 times the retention time of the ( E )-isomer of norgestimate.
Identification of impurities : use the chromatogramsupplied with norgestimate for system suitability CRS andthe chromatogram obtained with reference solution (b) toidentify the peak due to impurity A.
Relative retention with reference to the ( E )-isomerof norgestimate (retention time = about 14 min):impurityA = about 0.7; ( Z )-isomer of norgestimate = about 0.9.System suitability : reference solution (b):— resolution : minimum 1.5 between the peaks due to the
( E )- and ( Z )-isomers of norgestimate.
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity B 3. impurity D 5. ( Z )-isomer of norgestimate
2. impurity C 4. impurity A 6. ( E )-isomer of norgestimate
Figure 1732.-1. – Chromatogram for the test for related substances of norgestimate: test solution spiked with impurities A, B, C and D
(47) Luna C18 100A is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 121

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 122/176
2.7.29. Nucleated cell count and viability
Limits :
— correction factor : for the calculation of content, multiplythe peak area of the ( Z )-isomer of norgestimate by 1.33;
— impurity A : not more than twice the sum of the areas of the peaks due to the ( Z )- and ( E )-isomers of norgestimatein the chromatogram obtained with reference solution (a)(0.2 per cent);
— unspecified impurities : for each impurity, not morethan the sum of the areas of the peaks due to the ( Z )-and ( E )-isomers of norgestimate in the chromatogramobtained with reference solution (a) (0.10 per cent);
— total : not more than 3 times the sum of the areas of thepeaks due to the ( Z )- and ( E )-isomers of norgestimate inthe chromatogram obtained with reference solution (a)(0.3 per cent);
— disregard limit : 0.5 times the sum of the areas of thepeaks due to the ( Z )- and ( E )-isomers of norgestimate inthe chromatogram obtained with reference solution (a)(0.05 per cent).
Ratio of ( E )- to ( Z )-isomers. Liquid chromatography ( 2.2.29)
as described in the test for related substances with thefollowing modification.
Injection : test solution (a).
Calculate the ( E )- to ( Z )-isomer ratio by dividing the area of the peak due to the ( E )-isomer by 1.33 times the area of thepeak due to the ( Z )-isomer. The ratio is 1.27 to 1.78.
Loss on drying ( 2.2.32 ): maximum 0.5 per cent, determinedon 0.500 g by drying in an oven at 100-105 °C for 3 h.
ASSAY
Dissolve 0.300 g in 40 ml of tetrahydrofuran R. Add10 ml of a 100 g/l solution of silver nitrate R and titratewith 0.1 M sodium hydroxide, determine the end-point
potentiometrically ( 2.2.20). Rinse the electrode withacetone R after each titration.
If necessary, after multiple titrations re-equilibrate theelectrode in water R for 15 min to obtain sharper titrationcurves.
1 ml of 0.1 M sodium hydroxide is equivalent to 36.95 mgof C23H31NO3.
IMPURITIES
Specified impurities: A.
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limited
by the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : B, C, D.
A. 13β-ethyl-3-oxo-18,19-dinor-17α-pregn-4-en-20-yn-17β-ylacetate (levonorgestrel acetate),
B. levonorgestrel,
C. ( E )-13β-ethyl-3-hydroxyimino-18,19-dinor-17α-pregn-4-en-20-yn-17β-ol (( E )-norelgestromin),
D. ( Z )-13β-ethyl-3-hydroxyimino-18,19-dinor-17α-pregn-4-en-20-yn-17β-ol (( Z )-norelgestromin).
Reference: PA/PH/Exp. CTP/T (05) 28 ANP
XXXX:20729
2.7.29. NUCLEATED CELL COUNT ANDVIABILITY
The determination of the overall health of cell culturesrequires accurate measurements of both cell concentrationand percentage of viable cells. These data are essential to thedecision-making process for basic cell-culture passage and formaintaining optimum culture conditions. The cell count maybe expressed as the number of cells per volume of mediumor per area of attached surface (for anchorage-dependent cells). The cell-count procedure may be performed manually(haemocytometer) or with an apparatus based on light measurement (for example, particle counter, flow cytometer,fluorimeter).
CELL NUMBER
HAEMOCYTOMETER
Test principle and description of the apparatus. Ahaemocytometer is a specialised microscope slide that hasbeen etched with a grid over a defined area and is available indifferent designs. The most commonly used haemocytometermodel (see example shown in Figure 2.7.29.-1) is a modifiedthick slide with 2 counting chambers separated by deepgrooves to avoid cross-filling. The counting chamberis etched in the glass and contains 9 large squares of 1 mm × 1 mm. A large square is bounded by a triple line
and almost fills the field of view when the microscope lensand the oculars both have a 10× magnification. Different large-square grid patterns are used for different purposesin haematology laboratories.
122 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 123/176
2.7.29. Nucleated cell count and viability
A. covership B. ruled grid area
.Figure 2.7.29.-1. – Counting chamber of an improved
Neubauer haemocytometer
Dimensions in millimetres
The other special feature of a haemocytometer is thethickened coverslip. When placed on the haemocytometer,the typical rainbow sheen of Newton’s rings may be seenon the side of the chamber. The haemocytometer is sealedso that each chamber is maintained at a defined distancefrom the grid (0.1 mm). The volume of the chamber istherefore 0.1 mm3. The haemocytometer can therefore beused to quantify the number of cells in a given solution bycalculation of the cell concentration per millilitre (C ) usingthe following expression:
a = number of cells in the large square;
d = dilution factor (where applicable).
Some cells are found on the triple lines delimiting the
large squares: cells crossing the middle line on the topand left sides are usually counted while those on the right
and bottom sides are ignored. It is possible to distinguishbetween mixed cell populations provided they differ in sizeor pigmentation (for example, leukocytes and erythrocytes).However, haemocytometers have a significant intrinsic error.Different operators analysing the same cell populationobtain a spread of results due to the subjective nature of the determination.
Materials. The following materials are required:— a haemocytometer with coverslip ;
— a hand-held counter;
— a light microscope - low power 40× to 100× magnification;
— hand-held pipettes of a 10-100 μl volume range andcorresponding tips.
Test preparation and analysis. Mount the haemocytometerand the coverslip, cleaned beforehand with deionised waterand ethanol (70 per cent V/V ) or a 70 per cent V/V solutionof isopropyl alcohol. Move the coverslip back and forth overthe chamber, pressing slightly until the rainbow sheen isseen on the sides of the etched counting chambers.
Add 5-8 μl of the cell suspension to the counting chamber.
The liquid is added to the border of the coverslip andis drained inside the chamber by capillarity. For most haemocytometers, this is sufficient to fill the chamber. Theuse of a variable-volume micropipette is suggested to avoidspilling. Carefully place the haemocytometer under themicroscope and focus on a large-square chamber. Count thecells for each large square. Calculate the cell concentrationper millilitre in the diluted and original samples.
To increase the accuracy of the measurement, it is important to respect the following basic precautions:
— use only suitably thickened coverslips ;
— ensure that the rainbow sheen is present on the sides of the counting chambers;
— wherever possible, count more than 200 cells (i.e. count more large squares) ;
— where cell clustering is detected (i.e. the cell suspensionis not monocellular), resuspend the cells before samplingand repeat the procedure;
— avoid underfilling or overflowing the chamber, otherwisethe volume will no longer be accurate;
— count the cells twice.
FLOW CYTOMETER
A flow cytometer ( 2.7.24)(48) is an instrument in which cellsare made to flow in 1 single file through an orifice (usually70-100 μm in diameter) where they intersect a laser beam.The cells cause the light to be scattered and may f luoresce
if stained with a f luorescent dye. A set of photomultiplierspositioned around the point of intersection collect thescattered light and measure the number and intensity of events. A computer analyses the events and expresses theresults in terms of a ratio between any 2 sets of parametersmeasured.
Measurements taken include light scatter, fluorescent emission (wavelength and intensity) and optical density. Thissystem has the following advantages:
— it allows the counting of different groups of cells in thesame sample at the same time;
— it may count up to 10 000 events per second, allowing theenumeration of rare populations ;
— it may give structural information such as the complexityof the intracellular organelles or the cell’s volume.
(48) See Pharmeuropa 16.4 (October 2004).
© PHARMEUROPA Vol. 18, No. 1, January 2006 123

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 124/176
2.7.29. Nucleated cell count and viability
It needs to be calibrated with reference beads of knownconcentration to give an absolute cell number per volume.However, a calibrating solution is no longer necessary insome instruments using 2 electrodes inserted in the samplingchamber. The fixed size of the sampling chamber anddistance between the 2 electrodes allow the measurement of the content of a fixed volume. This type of instrument rarelyneeds to be calibrated after the initial setting.
PARTICLE COUNTERS
Particle counters based on conductivity variation.Electronic particle counting devices measure the size andnumber of particles in a solution. The oldest approach isbased on the change in conductivity with respect to that of the medium alone when a particle crosses an orifice of a roughly similar size. The cell suspension is drawn byvacuum through a fine orifice 70-100 μm in diameter and theimpulses are recorded by 2 electrodes as an analogue signal,which is measured and counted. The intensity of the pulseis used to measure the relative size of the cell, as a fractionof the orifice is occupied by a non-conductive material (i.e.the cell). The volume of liquid analysed is measured usingan internal calibrated tube. As cell debris may also generatepulses that may cause errors, counters are also fitted witha threshold control allowing only larger particles to becounted.
Particle counters are calibrated before use with a solutionof synthetic latex beads of known concentration. To allowthe counting of larger particles, tubes f itted with differentlycalibrated orifices are available. These apparatuses do not allow the discrimination between dead and live cells.
VIABILITY
This section applies to cell staining by viability dyes andanalysis, under direct microscope light or by flow cytometry,of a cell suspension in order to determine the percentage
of viable cells.Viability may be determined by staining with dyes suchas trypan blue (manual or automated method) or by flowcytometry using nucleic acid intercalating agents such aspropidium iodide (PI), 7-aminoactinomycin D (7-AAD) orother suitable dyes.
Depending on the type of cells, results obtained with the2 methods may differ.
MANUAL DYE-EXCLUSION METHOD
Test principle. This test is based on the exclusion of thedye from viable cells whereas dead or damaged cells absorbthe dye and are coloured. It provides information on thecytoplasmic membrane integrity but its results do not necessarily reflect cell functionality. Recently trypsinisedliving cells or cells thawed after cryopreservation in amedium containing dimethylsulfoxide may have leakymembranes, causing them to absorb the dye.
Dye. Trypan blue is the stain most commonly used todistinguish between viable and non-viable cells, but othersuitable dyes such as erythrosin B or nigrosin may also beused. It is an acid dye ( M r 961), an anion with 4 sulphonategroups that can easily bind to proteins. Binding to celluloseand proteins may also result from van der Waals forces orfrom the formation of hydrogen bonds.
Test conditions. Dye fixation is strongly influenced by pH,within a range of 6.6 to 7.6. Fixation is optimal at pH 7.5.The other conditions, such as the dye concentration, the
temperature and the staining time are standardised. Becausetrypan blue is easily fixed on soluble proteins, such as serumproteins, the protein concentration of the preparation to betested must be as low as possible.
Because old staining solutions are prone to aggregationand polymerisation causes bad staining, freshly prepareddye solutions are used.Storage conditions of dye solution: Generally a 0.4 or 0.5 percent trypan blue solution in sterile phosphate-bufferedsaline is used. Ready-made solutions are also commerciallyavailable. Store protected from light and air, at room
temperature.Test preparation and analysis. Stain the cell suspension at the required dilution (usually in phosphate-buffered saline)with a solution of, for example, 0.1 to 0.2 per cent trypanblue. Mix gently. Incubate for not more than 2-4 min at room temperature. Mix gently and place a suitable volume ina counting chamber. Count without delay. The number of squares or lines that have to be read for a correct estimationis validated previously.Determine the percentage of viable cells from the ratio of thenumber of unstained cells to the total number of cells undera direct light microscope, considering all blue cells as deadcells. Viability (V ) is calculated as a percentage using thefollowing expression:
n = number of unstained (viable) cells;
N = total number of cells (blue and unstained).
It is essential that the incubation time be less than 5 minas the number of stained cells may increase significantlyafterwards. For a new determination, it may therefore benecessary to prepare a new test.
AUTOMATED METHODS
Flow cytometry
Test principle. The test is based on the capacity of certain
dyes to cross damaged membranes and bind DNA byintercalating between bases so that dead cells may f luoresceand be detected by flow cytometry ( 2.7.24)(48). Non-viablecells are evaluated and discriminated by focusing on positivestaining whereas viable cells remain unstained. This analysisis generally performed with 7-AAD or PI but other suitabledyes may also be used. Dye. 7-AAD and PI are given as examples of membrane-impermeants that may be used as viability dyes.7-AAD is an analogue of actinomycin D that contains asubstituted amino group at position 7 of the chromophore.It intercalates between cytosine and guanine DNA bases.The spectral properties of 7-AAD make this moleculeparticularly suitable for f low-cytometry analysis. The
maximum absorption of the 7-AAD/DNA complex is situatedin the green spectral region and is thus suitable for anargon laser-equipped cytometer (excitation wavelength of 488 nm). The deep red fluorescence emission of the 7-AADviability dye (635 nm to 675 nm) eases the use of the probein combination with fluorescein isothiocyanate (FITC)and phycoerythrin (PE)-conjugated antibodies, because incontrast to PI, the 7-AAD/DNA complex shows minimaloverlap with FITC and PE.PI binds to double-stranded DNA by intercalating betweenbases with little or no sequence preference and with astoichiometry of 1 dye molecule per 4-5 DNA base pairs.Once the dye is bound to nucleic acids, its fluorescenceis enhanced 20- to 30-fold, the fluorescence excitation
maximum is shifted around 30-40 nm towards the red andthe f luorescence emission maximum (615 nm) is shiftedaround 15 nm towards the blue. Although its absorptivityis quite low, PI exhibits a sufficiently large Stokes shift
124 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 125/176
Paraffin, white soft
to allow simultaneous detection of nucleic acids andfluorescein-labelled antibodies, provided that the properoptical filters are used.Storage conditions of nucleic acid dye solution: Store at 5 ± 3 °C.Test preparation and analysis. In the case of haematopoieticcells, the dye may be used after CD45 labelling to obtain
a better separation of cells from debris and platelets witha side scatter (SS)/CD45 gating region. The incubationconditions (time and concentration) of the cell suspensionwith the dye are validated previously.Incubation is performed at room temperature in the dark.Where necessary, lysis of red blood cells is performed using,for example, ammonium chloride. If not, add buffer alone.Acquisition of sample tubes is performed according to thestandardised operating procedure.Percentages of viable cells are directly given by the f lowcytometer and deduced from the analysis of positive cells(dead cells) in the SS/7-AAD or SS/PI cytogram (dot plots).Positive controls may consist of stabilised cells (dead cells)mixed with fresh viable cells at a target value.
Other automated dye-exclusion methods. The development of digital imaging has allowed the automation of dye-exclusion methods. The cell suspension and viability-dyesolution are directly mixed by a machine. The system, whichallows sample aspiration, reagent handling, and subsequent instrument cleaning is fully automated. Once the cellularsuspension has been aspirated and mixed with the dyesolution, it is pumped to the flow cell for imaging. Thestained cell suspension is aspirated through a chamberwhere stroboscopic light allows a camera to photograph theflowing cells. The images are digitalised and the number of dark (dead) or bright (live) cells counted by the software.The first step is to digitise the collected video image andtransform this from a continuous smooth image into an
array of distinct elements or pixels. Each element is assigneda grey level or brightness value from 0 (black) to 255 (white).Thresholds within the software then determine which cellshave absorbed the dye (dead cells) and which have not (viable cells).
Reference: PA/PH/Exp. 11/T (05) 61 ANP
NOTE ON THE MONOGRAPH
The revision proposal for this monograph is published below as part of the framework of harmonisation with the JP and the USP. The stage 4 draft provided by the USP is published in the International harmonisation section. This
draft will not lead to complete harmonisation for reasonsdescribed in the following briefing note:
— Definition. The term ‘antioxidants’ is replaced by ‘stabilisers’. The statement concerning unsuitability for oral use will be maintained since this reflects regulatory requirements in Europe.
— Identification. The second identification will bemaintained as a local provision.
The determination of drop point is replaced by adetermination of melting point by a specific method.
For the identification by IR, the reference spectrum isto be replaced. The test cites a number of bands typical of paraffin.
— Appearance. The reference solution is slightly modified.
— Consistency. This functionality-related characteristic (FRC) is placed in the special section according tocurrent policy. The limits in the existing monograph
correspond to a range of products, rather than atolerance for a given product. They are to be deleted in accordance with the general policy on FRCs, theacceptance criterion being a matter for agreement between supplier and user.
— Polycyclic aromatic hydrocarbons. A tighter limit isapplied for oral use. This test is to be maintained as
a local European requirement, since it is required by regulatory authorities.XXXX:1799
PARAFFIN, WHITE SOFT
Vaselinum albumDEFINITION
Purified and wholly or nearly decolorised mixture of semi-solid hydrocarbons, obtained from petroleum. It maycontain a suitable antioxidant stabiliser. White soft paraffindescribed in this monograph is not suitable for oral use.
CHARACTERS Appearance : white or almost white, translucent, soft unctuous mass, slightly fluorescent in daylight when melted.
Solubility : practically insoluble in water, soluble inmethylene chloride, practically insoluble in ethanol (96 percent) and in glycerol.
IDENTIFICATION
First identification : A, B, D.
Second identification: A, C, D.
A. The drop point is between 35 °C and 70 °C and does not diff er by more than 5 °C from the value stat ed on thelabel, according t o method ( 2.2.17 ) with the f ollowingmodification t o fill the cup: heat the substance t o be
examined at a t emperature not exceeding 80 °C, withstirring t o ensure unif ormity. Warm the metal cup at at emperature not exceeding 80 °C in an oven, remove it from the oven, place on a clean plat e or ceramic tile andpour a sufficient quantity of the melted sample int o thecup to fill it completely. Allow the filled cup to cool f or30 min on the plat e or the ceramic tile and place it in awat er bath at 24-26 °C f or 30-40 min. Level the surface of the sample with a single stroke of a knif e or razor blade,avoiding compression of the sample.
A. Melting range: 38 °C to 60 °C.
Melt a quantity of the substance to be examined slowly,while stirring, until it reaches a temperature of 90-92 °C.Remove the source of the heat and allow the molten
substance to cool to a temperature of 8-10 °C abovet he expected melting point. Chill the bulb of a suitablethermometer to 5 °C, wipe it dry, and while it is still colddip it into the molten substance so that approximatelythe lower half of the bulb is submerged. Withdraw it immediately, and hold it vertically away from the heat until the wax surface dulls, then dip it for 5 min into awater-bath at a temperature not higher than 16 °C.
Fix the thermometer securely in a test tube so that thelower point is 15 mm above the bottom of the test tube.Suspend the test tube in a water-bath adjusted to about 16 °C, and raise the temperature of the bath at the rateof 2 °C/min to 30 °C, then change to a rate of 1 °C/min,and note the temperature at which the first drop of
melted substance leaves the thermometer. Repeat thedetermination twice on a freshly melted portion of thetest substance. If the variation of 3 determinations is lessthan 1 °C, take the average of the 3 determinations as the
© PHARMEUROPA Vol. 18, No. 1, January 2006 125

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 126/176
Paraffin, yellow soft
melting point. If the variation of 3 determinations is 1 °Cor greater than 1 °C, rate 2 additional determinationsand take the average of the 5 determinations.
B. Infrared absorption spectrophotometry ( 2.2.24).
Preparation : spread a thin film of the melted substanceto be examined between sodium chloride plates.
Comparison : Ph. Eur. r ef er enc e spectrum of white soft par affin white soft paraffin CRS .High intensity bands are obtained between 3000 cm -1 and2800 cm-1, medium intensity bands between 1500 cm -1
and 1300 cm-1, and low intensity bands between 750 cm-1
and 700 cm-1.
C. Melt 2 g and when a homogeneous phase is obtained,add 2 ml of water R and 0.2 ml of 0.05 M iodine. Shake.Allow to cool. The solid upper layer is violet-pink.
D. Appearance Colour (see Tests).
TESTS
Appearance. The substance is whit e. Melt 12 g on awater-bath. The melt ed mass is not more intensely coloured
than a mixture of 1 volume of yellow primary solution and9 volumes of a 1 per cent m/V solution of hy d r ochl ori c aci d R ( 2.2.2, Method II ).
Colour. Melt about 10 g on a steam bath, and pour 5 ml of the resulting liquid into a clear-glass, 16 mm × 150 mm test tube: the warm, melted liquid is not more intensely colouredthan a solution prepared by mixing 1.6 ml of yellow primarysolution ( 2.2.2 ) and 3.4 ml of water R. The comparison of the 2 preparations being made in reflected light against awhite background, the tubes being held directly against thebackground at such an angle that there is no fluorescence.
Acidity or alkalinity. To 10 g add 20 ml of boiling water Rand shake vigorously for 1 min. Allow to cool and decant. To10 ml of the aqueous layer add 0.1 ml of phenolphthalein solution R. The solution is colourless. Not more than 0.5 mlof 0.01 M sodium hydroxide is required to change the colourof the indicator to red.
Consistency ( 2.9.9): 60 to 300.
Polycyclic aromatic hydrocarbons : maximum 300 ppm.
Use reagents for ultraviolet absorption spectrophotometry.Dissolve 1.0 g in 50 ml of hexane R which has beenpreviously shaken with 2 quantities, each of 10 ml, of dimethyl sulphoxide R. Transfer the solution to a 125 mlseparating funnel with unlubricated ground-glass parts(stopper, stopcock). Add 20 ml of dimethyl sulphoxide R.Shake vigorously for 1 min and allow to stand until 2 clearlayers are formed. Transfer the lower layer to a second
separating funnel. Repeat the extraction with a further 20 mlof dimethyl sulphoxide R. Shake vigorously the combinedlower layers with 20 ml of hexane R for 1 min. Allow to standuntil 2 clear layers are formed. Separate the lower layer anddilute to 50.0 ml with dimethyl sulphoxide R. Measure theabsorbance ( 2.2.25 ) between 260 nm and 420 nm using apath length of 4 cm and using as the compensation liquidthe clear lower layer obtained by vigorously shaking 10 mlof dimethyl sulphoxide R with 25 ml of hexane R for 1 min.Prepare a 6.0 mg/l reference solution of naphthalene Rin dimethyl sulphoxide R and measure the absorbance of this solution at the absorption maximum at 278 nm using apath length of 4 cm and using dimethyl sulphoxide R asthe compensation liquid. At no wavelength in the range260-420 nm does the absorbance of the test solution exceedthat of the reference solution at 278 nm.Sulphated ash ( 2.4.14) : maximum 0.05 per cent, determinedon 2.0 10.0 g.
STORAGE
Protected from light.
LABELLING
The label states:
— the nominal drop point,
— where applicable, the name and concentration of anyadded antioxidant stabiliser.
FUNCTIONALITY-RELATED CHARACTERISTICS
The following test is not a mandatory requirement but inview of its known importance for achieving consistency in manufacture, quality and performance of medicinal products, it is recommended that suppliers should verify this characteristic and provide information on the result and the analytical method applied to users. The method indicated below has been found suitable but other methodsmay be used.
The following characteristic is relevant for white soft paraffinused as an emollient in semi-solid dosage forms.
Consistency ( 2.9.9). Carry out a minimum of 3 tests, eachspaced such that there is no overlapping of the areas of penetration. Where the penetration exceeds 20 mm, use aseparate container of the test substance for each test. Readthe penetration to the nearest 0.1 mm. Calculate the averageof the 3 or more readings, and conduct further tests to atotal of 10 if the individual results differ from the averageby more than ± 3 per cent: each millimetre of penetrationcorresponds to a consistency value of 10.
Reference: PA/PH/Exp. 11/T (05) 60 ANP
NOTE ON THE MONOGRAPH
The revision proposal for this monograph is published below as part of the framework of harmonisation with the JP and the USP. The stage 4 draft provided by the USP is published in the International harmonisation section. Thisdraft will not lead to complete harmonisation for reasonsdescribed in the following briefing note:
— Definition. The term ‘antioxidants’ is replaced by ‘stabilisers’.
— Identification. The second identification will bemaintained as a local provision.
The determination of drop point is replaced by adetermination of melting point by a specific method.
For the identification by IR, the reference spectrum isto be replaced. The test cites a number of bands typical of paraffin.
— Appearance. The reference solution is slightly modified.
— Consistency. This functionality-related characteristic (FRC) is placed in the special section according to current policy. The limits in theexisting monograph correspond to a range of products,rather than a tolerance for a given product. They areto be deleted in accordance with the general policy on FRCs, the acceptance criterion being a matter for agreement between supplier and user.
— Polycyclic aromatic hydrocarbons. This test is to bemaintained as a local European requirement, since it isrequired by regulatory authorities.
126 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 127/176
Paraffin, yellow soft
XXXX:1554
PARAFFIN, YELLOW SOFT
Vaselinum flavum
DEFINITIONPurified mixture of semi-solid hydrocarbons, obtained frompetroleum. It may contain a suitable antioxidant stabiliser.
CHARACTERS
Appearance : yellow, translucent, unctuous mass, slightlyfluorescent in daylight when melted.
Solubility : practically insoluble in water, soluble inmethylene chloride, practically insoluble in ethanol (96 percent) and in glycerol.
IDENTIFICATION
First identification : A, B, D.
Second identification: A, C, D.A. The drop point ( 2.2.17 ) is 40 °C t o 60 °C and does not
diff er by more than 5 °C from the value stated on thelabel, with the f ollowing modification to fill the cup: heat the substance t o be examined at 118 °C to 122 °C, withstirring t o ensure unif ormity, then cool t o 100 °C t o107 °C. Warm the metal cup at 103 °C to 107 °C in anoven, remove it from the oven, place on a clean plat e orceramic tile and pour a sufficient quantity of the melt edsample int o the cup t o fill it complet ely. Allow the filledcup to cool f or 30 min on the ceramic tile and place it in a wat er-bath at 24 °C to 26 °C f or a further 30 minto 40 min. Level the surface of the sample with a singlestroke of a knif e or razor blade, avoiding compression of
the sample.A. Melting range: 38 °C to 60 °C.
Melt a quantity of the substance to be examined slowly,while stirring, until it reaches a temperature of 90-92 °C.Remove the source of the heat and allow the moltensubstance to cool to a temperature of 8-10 °C abovethe expected melting point. Chill the bulb of a suitablethermometer to 5 °C, wipe it dry, and while it is still colddip it into the molten substance so that approximatelythe lower half of the bulb is submerged. Withdraw it immediately, and hold it vertically away from the heat until the wax surface dulls, then dip it for 5 min into awater-bath at a temperature not higher than 16 °C.
Fix the thermometer securely in a test tube so that the
lower point is 15 mm above the bottom of the test tube.Suspend the test tube in a water-bath adjusted to about 16 °C, and raise the temperature of the bath at the rateof 2 °C/min to 30 °C, then change to a rate of 1 °C/min,and note the temperature at which the first drop of melted substance leaves the thermometer. Repeat thedetermination twice on a freshly melted portion of thetest substance. If the variation of 3 determinations is lessthan 1 °C, take the average of the 3 determinations as themelting point. If the variation of 3 determinations is 1 °Cor greater than 1 °C, rate 2 additional determinationsand take the average of the 5 determinations.
B. Infrared absorption spectrophotometry ( 2.2.24).
Preparation : spread a thin film of the related substanceto be examined between sodium chloride plates.Comparison : Ph. Eur. ref er enc e spectrum of y ellow soft par affin CRS yellow soft paraffin CRS .
High intensity bands are obtained between 3000 cm1 and2800 cm,1 medium intensity bands between 1500 cm1
and 1300 cm1, and low intensity bands between 750 cm1
and 700 cm1.
C. Melt 2 g and when a homogenous phase is obtained, add2 ml of water R and 0.2 ml of 0.05 M iodine. Shake.Allow to cool. The solid upper layer is violet-pink.
D. Appearance Colour (see Tests).
TESTS
Appearance. The substance is yellow. Melt 12 g on awater-bath. The melt ed mass is not more intensely colouredthan a mixture of 7.6 volumes of yellow primary solution and2.4 volumes of red primary solution ( 2.2.2, M ethod II ).
Colour. Melt about 10 g on a steam bath, and pour 5 mlof the resulting liquid into a clear-glass, 16 mm × 150 mmtest tube: the warm, melted liquid is not more intenselycoloured than a solution made by mixing 3.8 ml of yellowprimary solution and 1.2 ml of red primary solution ( 2.2.2 )in a similar tube, the comparison of the 2 being made inreflected light against a white background, the tubes being
held directly against the background at such an angle that there is no fluorescence.
Acidity or alkalinity. To 10 g add 20 ml of boiling water Rand shake vigorously for 1 min. Allow to cool and decant. To10 ml of the aqueous layer add 0.1 ml of phenolphthalein solution R. The solution is colourless. Not more than 0.5 mlof 0.01 M sodium hydroxide is required to change the colourof the indicator to red.
Consistency ( 2.9.9). The consistency is 100 to 300.
Polycyclic aromatic hydrocarbons. Use reagents for ultraviolet absorption spectrophotometry. Dissolve 1.0 gin 50 ml of hexane R which has been previously shakenwith 2 quantities, each of 10 ml, of dimethyl sulphoxide R.
Transfer the solution to a 125 ml separating funnel withunlubricated ground-glass parts (stopper, stopcock). Add20 ml of dimethyl sulphoxide R. Shake vigorously for 1 minand allow to stand until 2 clear layers are formed. Transferthe lower layer to a second separating funnel. Repeat theextraction with a further 20 ml of dimethyl sulphoxide R.Shake vigorously the combined lower layers with 20 mlof hexane R for 1 min. Allow to stand until 2 clear layersare formed. Separate the lower layer and dilute to 50.0 mlwith dimethyl sulphoxide R. Measure the absorbance( 2.2.25 ) between 260 nm and 420 nm using a path length of 4 cm and using as the compensation liquid the clear lowerlayer obtained by vigorously shaking 10 ml of dimethyl sulphoxide R with 25 ml of hexane R for 1 min. Prepare a9.0 mg/l reference solution of naphthalene R in dimethyl
sulphoxide R and measure the absorbance of this solution at the absorption maximum at 278 nm using a path length of 4 cm and using dimethyl sulphoxide R as the compensationliquid. At no wavelength in the range 260-420 nm does theabsorbance of the test solution exceed that of the referencesolution at 278 nm.
Sulphated ash ( 2.4.14) : maximum 0.05 per cent, determinedon 2.0 10.0 g.
STORAGE
Protected from light.
LABELLING
The label states:
— the nominal drop point,— where applicable, the name and concentration of any
added antioxidant stabiliser.
© PHARMEUROPA Vol. 18, No. 1, January 2006 127

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 128/176
Pentaerythrityl tetranitrate, diluted
FUNCTIONALITY-RELATED CHARACTERISTICS
The following test is not a mandatory requirement but inview of its known importance for achieving consistency in manufacture, quality and performance of medicinal products, it is recommended that suppliers should verify this characteristic and provide information on the result and the analytical method applied to users. The method
indicated below has been found suitable but other methodsmay be used.
The following characteristic is relevant for yellow soft paraffin used as an emollient in semi-solid dosage forms.
Consistency ( 2.9.9). Carry out a minimum of 3 tests, eachspaced such that there is no overlapping of the areas of penetration. Where the penetration exceeds 20 mm, use aseparate container of the test substance for each test. Readthe penetration to the nearest 0.1 mm. Calculate the averageof the 3 or more readings, and conduct further tests to atotal of 10 if the individual results differ from the averageby more than ± 3 per cent: each millimetre of penetrationcorresponds to a consistency value of 10.
Reference: PA/PH/Exp. 10D/T (05) 34 ANP
NOTE ON THE MONOGRAPH
The current method in the test for related substancesdoes not adequately control impurity C. A new method is proposed in replacement.
XXXX:1355
PENTAERYTHRITYL TETRANITRATE,DILUTED
Pentaerythrityli tetranitras dilutus
C5H8N4O12 M r 316.1
DEFINITIONDry mixture of 2,2-bis(hydroxymethyl)propane-1,3-dioltetranitrate (pentaerythrityl tetranitrate) and Lactosemonohydrate (0187) or Mannitol (0559).
Content : 95.0 per cent m/m to 105.0 per cent m/m of thedeclared content of pentaerythrityl tetranitrate.
CHARACTERS
Appearance of pentaerythrityl tetranitrate : white or slightlyyellowish powder.
Solubility of pentaerythrityl tetranitrate : practicallyinsoluble in water, soluble in acetone, slightly soluble inethanol (96 per cent V/V ).The solubility of diluted pentaerythrityl tetranitrate dependson the diluent and its concentration.
IDENTIFICATION
First identification : A, B, D.
Second identification: A, C, D.
A. Melting point ( 2.2.14): 138 °C to 142 °C, for theresidue obtained with the substance to be examined inidentification test B.
B. Infrared absorption spectrophotometry ( 2.2.24).
Preparation : separately shake a quantity of the substanceto be examined and a quantity of the reference substance,each corresponding to 25 mg of pentaerythrityltetranitrate, with 10 ml of acetone R for 5 min. Filter,evaporate to dryness at a temperature below 40 °Cand dry the residue, over diphosphorus pentoxide R at a pressure of 0.7 kPa for 16 h. Examine the residuesprepared as discs.Comparison : diluted pentaerythrityl tetranitrate CRS .
C. Thin-layer chromatography ( 2.2.27 ).Test solution. Shake a quantity of the substance to beexamined corresponding to 10 mg of pentaerythrityltetranitrate with 10 ml of ethanol (96 per cent) R for5 min and filter. Reference solution. Shake a quantity of diluted pentaerythrityl tetranitrate CRS corresponding to 10 mgof pentaerythrityl tetranitrate with 10 ml of ethanol (96 per cent) R for 5 min and filter. Plate : TLC silica gel G plate R. Mobile phase : ethyl acetate R, toluene R (20:80 V/V ). Application : 10 μl. Development : over a path of 15 cm. Drying : in air. Detection : spray with freshly prepared potassium iodideand starch solution R. Expose to ultraviolet light at 254 nm for 15 min. Examine in daylight. Results : the principal spot in the chromatogram obtainedwith the test solution is similar in position, colour andsize to the principal spot in the chromatogram obtainedwith the reference solution.
D. Thin-layer chromatography ( 2.2.27 ).Test solution. Shake a quantity of the substance to beexamined corresponding to 0.10 g of lactose or mannitolwith 10 ml of water R. Filter if necessary.
Reference solution (a) Dissolve 0.10 g of lactose R inwater R and dilute to 10 ml with the same solvent. Reference solution (b). Dissolve 0.10 g of mannitol R inwater R and dilute to 10 ml with the same solvent. Reference solution (c). Mix equal volumes of referencesolutions (a) and (b). Plate : TLC silica gel G plate R. Mobile phase : water R, methanol R, anhydrous acetic acid R, ethylene chloride R (10:15:25:50 V/V/V/V ).Measure the volumes accurately since a slight excess of water produces cloudiness. Application : 1 μl; thoroughly dry the starting points. Development A : over a path of 15 cm.
Drying A : in a current of warm air. Development B : immediately, over a path of 15 cm, afterrenewing the mobile phase. Drying B : in a current of warm air. Detection : spray with 4-aminobenzoic acid solution R.Dry in a current of cold air until the acetone is removed.Heat at 100 °C for 15 min. Allow to cool and spray with a2 g/l solution of sodium periodate R. Dry in a current of cold air. Heat at 100 °C for 15 min.System suitability : reference solution (c):— the chromatogram shows 2 clearly separated spots. Results : the principal spot in the chromatogram obtainedwith the test solution is similar in position, colour and size
to the principal spot in the chromatogram obtained withreference solution (a) for lactose or to the principal spot in the chromatogram obtained with reference solution (b)for mannitol.
128 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 129/176
Pentaerythrityl tetranitrate, diluted
TESTS
Inorganic nitrates. Thin-layer chromatography ( 2.2.27 ).
Test solution. Shake a quantity of the substance to beexamined corresponding to 0.10 g of pentaerythrityltetranitrate with 5 ml of ethanol (96 per cent) R and filter.
Reference solution . Dissolve 10 mg of potassium nitrate R
in 1 ml of water R and dilute to 100 ml with ethanol (96 per cent) R.
Plate : TLC silica gel plate R.
Mobile phase : glacial acetic acid R, acetone R, toluene R(15:30:60 V/V/V ).
Application : 10 μl.
Development : over a path of 15 cm.
Drying : in a current of air until the acetic acid is completelyremoved.
Detection : spray copiously with freshly prepared potassiumiodide and starch solution R. Expose the plate to ultraviolet light at 254 nm for 15 min. Examine in daylight.
Limit :— nitrate : any spot due to nitrate is not more intense than
the spot in the chromatogram obtained with the referencesolution (0.5 per cent, calculated as potassium nitrate).
Related substances. Examine by liquid chromat ography( 2.2.29) as described under Assay.
Adjust the sensitivity of the system so that the height of theprincipal peak in the chromatogram obtained with ref erencesolution (c) is at least 20 per cent of the full scale of therecorder.
The test is not valid unless in the chromat ogram obtainedwith ref erence solution (e), the resolution between the peakscorresponding to glyceryl trinitrate and t o pent aerythrityl
tetranitrate is at least 2.0.Inject 20 μl of t est solution (a) and 20 μl of referencesolution (c) and record the chromat ogram of t est solution (a)f or at least five times the ret ention time of pentaerythrityltetranitrate. In the chromat ogram obtained with test solution (a); the area of any peak, apart from the principalpeak is not great er than the area of the principal peak inthe chromat ogram obtained with ref erence solution (c)(0.3 per cent); the sum of the areas of all the peaks, apart from the principal peak, is not great er than twice the areaof the principal peak in the chromat ogram obtained with
ref erence solution (c) (0.6 per cent). Disregard any peak withan area less than 0.2 times that of the principal peak in thechromat ogram obtained with ref erence solution (c).Liquid chromatography ( 2.2.29).Test solution (a). Sonicate for 15 min a quantity of thesubstance to be examined corresponding to 25.0 mg of pentaerythrityl tetranitrate in 20 ml of the mobile phase and
dilute to 25.0 ml with the mobile phase. Filter through asuitable membrane filter.Test solution (b). Dilute 1.0 ml of test solution (a) to 10.0 mlwith the mobile phase. Reference solution (a). Sonicate for 15 min a quantity of diluted pentaerythrityl tetranitrate CRS corresponding to25.0 mg of pentaerythrityl tetranitrate in 20 ml of the mobilephase and dilute to 25.0 ml with the mobile phase. Filterthrough a suitable membrane filter. Reference solution (b) . Dilute 1.0 ml of reference solution (a)to 10.0 ml with the mobile phase. Reference solution (c) . Dilute 0.3 ml of reference solution (b)to 10.0 ml with the mobile phase. Reference solution (d). Dilute 200 μl of glyceryl trinitrate
solution CRS to 25.0 ml with the mobile phase. Reference solution (e). To 1 ml of reference solution (b)add 1 ml of reference solution (d) and dilute to 10 ml withthe mobile phase. Reference solution (f). Dilute 1.0 ml of reference solution (a)to 20.0 ml with the mobile phase. Dilute 0.5 ml of thissolution to 50.0 ml with the mobile phase.Column :— size : l = 0.15 m, Ø = 3.9 mm;— stationary phase : octylsilyl silica gel for
chromatography R (5 μm)(49). Mobile phase : water R, acetonitrile R (35:65 V/V ). Flow rate : 1.4 ml/min.
Detection : spectrophotometer at 220 nm. Injection : 20 μl of test solution (a) and referencesolutions (c), (e) and (f). Run time : 5 times the retention time of pentaerythrityltetranitrate. Retention time : pentaerythrityl tetranitrate = about 2.4 min;impurity C = about 7.1 min.System suitability : reference solution (e):— resolution : minimum 3.0 between the peaks due to
glyceryl trinitrate and pantaerythrityl tetranitrate.
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity B 3. impurity D
2. pentaerythrityl tetranitrate 4. impurity C
Figure 1355.-1. – Chromatogram for the test for related substances of diluted pentaerythrityl tetranitrate: substance spiked with impurities B, C and D
(49) Symmetry C8 is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 129

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 130/176
Pentaerythrityl tetranitrate, diluted
Limits :
— impurity C : not more than the area of the principal peakin the chromatogram obtained with reference solution (c)(0.3 per cent);
— unspecified impurities : for each impurity, not more than
twice the area of the principal peak in the chromatogramobtained with reference solution (f) (0.10 per cent);
— total : not more than twice the area of the principal peakin the chromatogram obtained with reference solution (c)(0.6 per cent);
— disregard limit : the area of the principal peak in thechromatogram obtained with reference solution (f)(0.05 per cent).
ASSAY
Liquid chromatography ( 2.2.29) as described in the test forrelated substances with the following modification.
Injection : test solution (b) and reference solution (b).
Calculate the percentage content of C5H8N4O12 from thedeclared content of pentaerythrityl tetranitrate CRS .
When the chromat ograms are recorded in the prescribedconditions the retention time of pentaerythrityl t etranitrat eis about 8 min. Inject 20 μl of ref erence solution (b).Adjust the sensitivity of the system so that the height of the principal peak in the chromat ogram obtained is at least 50 per cent of the full scale of the recorder. Inject ref erence
solution (b) six times. The assay is not valid unless therelative standard deviation f or the area of the principal peakis at most 2.0 per cent. Inject test solution (b) and ref erencesolution (b) alt ernately.
T est soluti on ( a). Sonicate f or 15 min a quantity of thesubstance to be examined corresponding to 25.0 mg of pentaerythrityl t etranitrat e in 20 ml of methanol R anddilut e t o 25.0 ml with the mobile phase. Filt er through asuitable membrane filter.
T est soluti on (b). Dilut e 1.0 ml of test solution (a) t o 10.0 mlwith the mobile phase.
Ref er enc e soluti on ( a). Sonicat e f or 15 min a quantity of di lut ed pentaery thrity l tetr ani tr ate CRS correspondingto 25.0 mg of pentaerythrityl tetranitrate in 20 ml of methanol R and dilut e t o 25.0 ml with the mobile phase.Filt er through a suitable membrane filter.
Ref er enc e solution (b). Dilut e 1.0 ml of ref erence solution (a)to 10.0 ml with the mobile phase.
Ref er enc e solution (c). Dilute 0.3 ml of ref erence solution (b)to 10.0 ml with the mobile phase.
Ref er enc e soluti on ( d). Sonicat e f or 15 min a quantity of gly ceryl trinitr ate soluti on CRS corresponding t o 20.0 mg
of glyceryl trinitrate in 20 ml of methanol R and dilute t o25.0 ml with the mobile phase. Filt er through a suitablemembrane filter. Dilute 1.0 ml of the filtrate t o 10.0 ml withthe mobile phase.
Ref er ence soluti on ( e). To 1 ml of ref erence solution (b),add 1 ml of ref erence solution (d) and dilute t o 10 ml withthe mobile phase.
The chromat ographic procedure may be carried out using :
— a stainless st eel column 0.25 m long and 4.6 mm ininternal diamet er packed with oct adecylsilyl sili ca gel for chromatog r aphy R (10 μm),
— as mobile phase at a flow rate of 2 ml/min a mixture of 40 volumes of wat er R and 60 volumes of methanol R,
— as det ect or a spectrophot omet er set at 230 nm.
STORAGE
Store protected from light and heat.
LABELLING
The label states:
— the content of pentaerythrityl tetranitrate as a percentage ;
— the diluent used.
IMPURITIES
Specified impurities : A, C.
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : B, D.
A. inorganic nitrates,
B. pentaerythrityl trinitrate,
C. tripentaerythrityl octanitrate,
D. dipentaerythrityl hexanitrate.
130 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 131/176
Perindopril tert -butylamine
Reference: PA/PH/Exp. 10C/T (05) 33 ANP
NOTE ON THE MONOGRAPH
The current LC method for related substances has beencriticised for lack of ease of laboratory transfer, the separation of impurity D and perindopril seeming tobe especially critical. Also, new degradation productshave been detected under severe conditions of storage(temperature and humidity). These degradation productsmay be co-eluted with impurity B in the present method. Amore robust and easy-to-use chromatographic system that also allows separation and detection of these new impuritieswas developed and is now proposed. A peak-to-valley ratiocriterion for the separation between impurities B and K is proposed. The limits for impurities are the same as inthe present monograph. The new degradation productsare listed as impurities J and K under “other detectableimpurities”, limited by the standard acceptance criterionof not more than 0.10 per cent. Impurities L, M, N and O,which are potential synthesis impurities not normally found in the batches, have also been included in the list as“other detectable impurities” for information. In routine
quality control it was also observed that the solubility in methylene chloride is slightly better that at present described, therefore an adjustment is proposed.
XXXX:2019
PERINDOPRIL tert -BUTYLAMINE
tert -Butylamini perindoprilum
C23H43N3O5 M r 441.6
DEFINITION2-Methylpropan-2-amine (2S ,3aS ,7aS )-1-[(2S )-2-[[(1S )-1-(ethoxycarbonyl)butyl]amino]propanoyl]octahydro-1 H -indole-2-carboxylate.Content : 99.0 per cent to 101.0 per cent (anhydroussubstance).
CHARACTERS Appearance : white or almost white, slightly hygroscopic,crystalline powder.Solubility : freely soluble in water and in ethanol (96 percent), soluble or sparingly soluble in methylene chloride.It shows polymorphism.
IDENTIFICATIONA. Specific optical rotation ( 2.2.7 ) : 66 to 69 (anhydrous
substance).Dissolve 0.250 g in ethanol (96 per cent) R and dilute to25.0 ml with the same solvent.
B. Infrared absorption spectrophotometry ( 2.2.24). Preparation : discs.Comparison : perindopril tert-butylamine CRS .If the spectra obtained show differences, dissolve thesubstance to be examined and the reference substance
separately in methylene chloride R, evaporate to drynessand record new spectra using the residues.
C. Examine the chromatograms obtained in the test forimpurity A. Results : in the chromatogram obtained with the test solution a spot is observed with the same Rf as the spot with the higher R f in the chromatogram obtained withreference solution (c) (tert -butylamine).
TESTS
Impurity A. Thin-layer chromatography ( 2.2.27 ).Test solution. Dissolve 0.20 g of the substance to beexamined in methanol R and dilute to 10 ml with the samesolvent. Reference solution (a). Dissolve 5 mg of perindopril impurity A CRS in methanol R and dilute to 25.0 ml withthe same solvent. Reference solution (b). Dilute 5 ml of reference solution (a)to 20 ml with methanol R. Reference solution (c). To 5 ml of reference solution (a) add5 ml of a 20 g/l solution of 1,1-dimethylethylamine R inmethanol R. Plate : TLC silica gel plate R. Mobile phase : glacial acetic acid R, toluene R, methanol R(1:40:60 V/V/V ). Application : 10 μl of the test solution and referencesolutions (b) and (c). Development : in a saturated tank, over 2/3 of the plate. Drying : in a current of warm air. Detection : expose to iodine vapour for at least 20 h.System suitability : reference solution (c):— the chromatogram shows 2 clearly separated spots. Limit :— impurity A : any spot due to impurity A is not more
intense than the spot in the chromatogram obtained with
reference solution (b) (0.25 per cent).Stereochemical purity. Liquid chromatography ( 2.2.29).Test solution. Dissolve 20 mg of the substance to beexamined in ethanol (96 per cent) R and dilute to 10.0 mlwith the same solvent. Reference solution (a). Dilute 1.0 ml of the test solution to200.0 ml with ethanol (96 per cent) R. Reference solution (b). Dissolve 10 mg of perindopril for stereochemical purity CRS in ethanol (96 per cent) R anddilute to 5.0 ml with the same solvent. Reference solution (c). Dilute 10.0 ml of referencesolution (a) to 50.0 ml with ethanol (96 per cent) R.Column :
— size : l = 0.25 m, Ø = 4.6 mm;— stationary phase : spherical octadecylsilyl silica gel for
chromatography R (5 μm)(50) with a specific surface areaof 450 m2 /g and a pore size of 10 nm ;
— temperature : 50 °C for the column and at least 30 cm of the tubing preceding the column.
Mobile phase : mix, in the following order, 21.7 volumes of acetonitrile R, 0.3 volumes of pentanol R and 78 volumesof a 1.50 g/l solution of sodium heptanesulphonate R,previously adjusted to pH 2.0 with a mixture of equalvolumes of perchloric acid R and water R. Flow rate : 0.8 ml/min. Detection : spectrophotometer at 215 nm.
Equilibration : minimum 4 h. Injection : 10 μl.
(50) Inertsil ODS3 is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 131

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 132/176
Perindopril tert -butylamine
Run time : 1.5 times the retention time of perindopril.
Retention time : perindopril = about 100 min.
System suitability :
— signal-to-noise ratio : minimum 3 for the principal peak inthe chromatogram obtained with reference solution (c) ;
— peak-to-valley ratio : minimum 3, where H p = height above
the baseline of the peak due to impurity I and H v = height above the baseline of the lowest point of the curveseparating this peak from the peak due to perindopril inthe chromatogram obtained with reference solution (b) ;
— the chromatogram obtained with reference solution (b) issimilar to the chromatogram provided with perindopril for stereochemical purity CRS .
Limits :
— any impurity : not more than 0.2 times the area of the principal peak in the chromatogram obtained withreference solution (a) (0.1 per cent); disregard any peakwith a retention time less than 0.6 times the retentiontime of perindopril and any peak with a retention timegreater than 1.4 times the retention time of perindopril.
Related substances. Liquid chromatography ( 2.2.29). Prepare the solutions immediately before use or maintainthem at a temperature lower than 10 °C .
Test solution. Dissolve 60 mg of the substance to beexamined in mobile phase A and dilute to 20.0 ml withmobile phase A.
Reference solution (a). Dissolve 15 mg of perindopril for system suitability peak identification CRS (containingimpurities B, E, F, H and K) in mobile phase A and dilute to5.0 ml with mobile phase A.
Reference solution (b). Dilute 1.0 ml of the test solution to200.0 ml with mobile phase A.
Reference solution (c). Dilute 1.0 ml of reference solution (b)
to 10.0 ml with mobile phase A.
Column :— size : l = 0.25 0.15 m, Ø = 4.6 4 mm;— stationary phase : spherical octylsilyl silica gel for
chromatography R (5 μm)(51) with a pore size of 15 nm;— temperature : 70 60 °C. Mobile phase :— mobile phase A : dissolve 0.92 g of sodium
hept anesulphonate R in 1000 ml of wat er R, add 1 ml of tri ethyl amine R and adjust to pH 2.0 with a mixture of equal volumes of per chloric acid R and wat er R,
— mobile phase B : ac etonitril e R1,
Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 - 1 70 30
1 - 20 70 → 40 30 → 60
20 - 25 40 60
25 - 35 40 → 20 60 → 80
35 - 40 20 → 0 80 → 100
40 - 45 0 → 70 100 → 30
Mobile phase :— mobile phase A : water R adjusted to pH 2.5 with a
mixture of equal volumes of perchloric acid R andwater R ;
— mobile phase B : a 0.03 per cent (V /V ) solution of perchloric acid R in acetonitrile R1 ;
Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 - (5 t ) 95 5
(5 t ) - (60 t ) 95 → 40 5 → 60
(60
t ) - (65
t ) 40 → 95 60 → 5
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity J 4 . impurity L 7. impurity E 10. impurity N 13. impurity H
2. impurity B 5. impurity M 8. impurity C 11. impurity O 14 - 15. impurity G
3. impurity K 6. perindopril 9. impurity D 12. impurity F
Figure 2019.-1. – Chromatogram for the test for related substances of perindopril tert-butylamine: substance withabout 0.5 per cent of the impurities
(51) Lichrospher RP8 ec Inertsil C8 is suitable.
132 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 133/176
Perindopril tert -butylamine
The isocratic step is described for a dwell volume of 2 mlof the chromatographic system. If the dwell volume ( D) isdifferent from 2 ml, correct each time of the gradient withthe value (t ), calculated using the following expression:
Flow rate : 1.5 1.0 ml/min.
Detection : spectrophotometer at 215 nm.
Injection : 20 μl.
Relative retention with reference to perindopril (retentiontime = about 8 25 min): impurity B = about 0.4;impurity C = about 0.8; impurity D = about 0.9;impurity E = about 1.4; impurity F = about 1.7;impurity G = about 2.2 and 2.3; impurity H = about 3.6and 3.7 impurity B = about 0.68; impurity K = about 0.72;impurity E = about 1.16; impurity F = about 1.57;impurity H = about 1.80 (impurity H may be eluted as 1 or2 peaks).
System suitability : reference solution (a):
— peak-to-valley ratio : minimum 10, where H p = height above the baseline of the peak due to impurity D and H v = height above the baseline of the lowest point of the curve separating this peak from the peak due t operindopril. If necessary, adjust the concentration of tri ethy lamine R in mobile phase A minimum 3, where H p = height above the baseline of the peak due toimpurity B and H v = height above the baseline of thelowest point of the curve separating this peak from thepeak due to impurity K.
Limits :
— impurity B : not more than 0.6 times the area of theprincipal peak in the chromatogram obtained withreference solution (b) (0.3 per cent) ;
— impurity E : not more than 0.8 times the area of theprincipal peak in the chromatogram obtained withreference solution (b) (0.4 per cent) ;
— impurities F, H : for each impurity, notmore than 0.4 timesthe area of the principal peak in the chromatogramobtained with reference solution (b) (0.2 per cent) ;
— unspecified impurities : for each impurity, not morethan 0.2 times the area of the principal peak in thechromatogram obtained with reference solution (b)(0.10 per cent);
— total : not more than twice the area of the principal peakin the chromatogram obtained with reference solution (b)(1 per cent);
— disregard limit : 0.1 times the area of the principal peakin the chromatogram obtained with reference solution (b)(c) (0.05 per cent).
Water ( 2.5.12 ): maximum 1.0 per cent, determined on 0.50 g.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAY
Dissolve 0.160 g in 50 ml of anhydrous acetic acid R. Titratewith 0.1 M perchloric acid , determining the end-point
potentiometrically ( 2.2.20).1 ml of 0.1 M perchloric acid is equivalent to 22.08 mgof C23H43N3O5.
STORAGE
In an airtight container.
IMPURITIES
Specified impurities: A, B, E, F, H, I.
Other detectable impurities (the following substances would,if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the generalacceptance criterion for other/unspecified impurities and/orby the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify theseimpurities for demonstration of compliance. See also 5.10.Control of impurities in substances for pharmaceutical use) : C, D, G, J, K, L, M, N, O.
A. (2S ,3aS ,7aS )-octahydro-1 H -indole-2-carboxylic acid,
B. R = H: (2S ,3aS ,7aS )-1-[(2S )-2-[[(1S )-1-carboxybutyl]ami-no]propanoyl]octahydro-1 H -indole-2-carboxylic acid,
E. R = CH(CH3)2 : (2S ,3aS ,7aS )-1-[(2S )-2-[[(1S )-1-[(1-methyl-ethoxy)carbonyl]butyl]amino]propanoyl]octahydro-1 H -in-dole-2-carboxylic acid,
C. R = H: (2S )-2-[(3S ,5aS ,9aS ,10aS )-3-methyl-1,4-dioxodecahydropyrazino[1,2-a]indol-2(1 H )-yl]pentanoicacid,
F. R = C2H5 : ethyl (2S )-2-[(3S ,5aS ,9aS ,10aS )-3-methyl-1,4-dioxodecahydropyrazino[1,2-a]indol-2(1 H )-yl]pentanoate,
D. (2S )-2-[(3S ,5aS,9aS ,10a R)-3-methyl-1,4-dioxodecahydropy-razino[1,2-a]indol-2(1 H )-yl]pentanoic acid,
© PHARMEUROPA Vol. 18, No. 1, January 2006 133

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 134/176
Potassium clavulanate
G. (2S ,3aS ,7aS )-1-[(2S )-2-[(5 RS )-3-cyclohexyl-2,4-dioxo-5-propylimidazolidin-1-yl]propanoyl]octahydro-1 H -indole-2-carboxylic acid,
H. (2S ,3aS ,7aS )-1-[(2S )-2-[(5 RS )-3-cyclohexyl-2-(cyclohexylimino)-4-oxo-5-propylimidazolidin-1-yl]propanoyl]octahydro-1 H -indole-2-carboxylic acid,
I . (2S ,3aS ,7aS )-1-[(2S )-2-[[(1 R)-1-(ethoxycarbonyl)butyl]ami-no]propanoyl]octahydro-1 H -indole-2-carboxylic acid,
J. (2S ,3aS ,7aS )-1-((2S )-2-aminopropanoyl)octahydro-1 H -indole-2-carboxylic acid,
K. (3S ,5aS ,9aS ,10aS )-3-methyldecahydropyrazino[1,2-a]indole-1,4-dione,
L. (2S ,3aS ,7aS )-1-acetyloctahydro-1 H -indole-2-carboxylicacid,
M. (2S ,3aS ,7aS )-1-((2S )-2-(((1S )-1-((methyloxy)carbonyl)but-yl)amino)propanoyl)octahydro-1 H -indole-2-carboxylicacid,
N. (2S )-3-cyclohexyl-2-(((2S )-2-(((1S )-1-((ethyloxy)carbonyl)-butyl)amino)propanoyl)amino)propanoic acid,
O. (2S ,3aS ,7aS )-1-(((2S ,3aS ,7aS )-1-((2S )-2-(((1S )-1-((ethyloxy)carbonyl)butyl)amino)propanoyl)octahydro-1 H -indol-2-yl)carbonyl)octahydro-1 H -indole-2-carboxylic acid.
Reference: PA/PH/Exp. 7/T (05) 78 ANP
NOTE ON THE MONOGRAPH The present transparency statement and impurity limits donot accurately reflect the quality of potassium clavulanatein approved products. The list of specified impuritiesdetected by LC has therefore been reduced to C, E and G; the limit for ‘any other impurity’ has been tightened to 0.2 per cent; and a system suitability CRS has beenintroduced.
XXXX:1140
POTASSIUM CLAVULANATE
Kalii clavulanas
C8H8KNO5 M r 237.3
DEFINITIONPotassium (2 R,3 Z ,5 R)-3-(2-hydroxyethylidene)-7-oxo-4-oxa-1-azabicyclo[3.2.0]heptane-2-carboxylate, the potassium salt of a substance produced by the growth of certain strains of Streptomyces clavuligerus or obtained by any other means.Content : 96.5 per cent to 102.0 per cent (anhydroussubstance).
CHARACTERS Appearance : white or almost white, crystalline powder,hygroscopic.
134 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 135/176
Potassium clavulanate
Solubility : freely soluble in water, slightly soluble in ethanol(96 per cent), very slightly soluble in acetone.
PRODUCTION
The methods of production, extraction and purification aresuch that clavam-2-carboxylate is eliminated or present at alevel not exceeding 0.01 per cent.
IDENTIFICATIONA. Infrared absorption spectrophotometry ( 2.2.24).
Comparison : Ph. Eur. reference spectrum of potassiumclavulanate.
B. It gives reaction (b) of potassium ( 2.3.1).
TESTS
Solution S. Dissolve 0.400 g in carbon dioxide-free water Rand dilute to 20.0 ml with the same solvent.
pH ( 2.2.3): 5.5 to 8.0.
Dilute 5 ml of solution S to 10 ml with carbon dioxide-freewater R.
Specific optical rotation ( 2.2.7 ): + 53 to + 63 (anhydroussubstance), determined on solution S.
Absorbance ( 2.2.25 ): maximum 0.40 at 278 nm.
Dissolve 50.0 mg in 0.1 M phosphate buffer solution pH 7.0 R and dilute to 50.0 ml with the same buffer solution.Measure the absorbance immediately.
Related substances. Liquid chromatography ( 2.2.29). Prepare the solutions immediately before use .
Test solution. Dissolve 0.250 g of the substance to beexamined in mobile phase A and dilute to 25.0 ml withmobile phase A.
Reference solution (a). Dilute 1.0 ml of the test solution to100.0 ml with mobile phase A.
Reference solution (b). Dissolve 10 mg of lithiumclavulanate CRS and 10 mg of amoxicillin trihydrate CRS in mobile phase A and dilute to 100 ml with mobile phase A.
Reference solution (c). Dissolve 5 mg of potassiumclavulanate for system suitability CRS in 1 ml of mobilephase A.
Column :
— size : l = 0.10 m, Ø = 4.6 mm;
— stationary phase : octadecylsilyl silica gel for chromatography R (5 μm);
— temperature : 40 °C.
Mobile phase :
— mobile phase A: a 7.8 g/l solution of sodium dihydrogen phosphate R adjusted to pH 4.0 with phosphoric acid Rand filtered through a 0.5 μm filter;
— mobile phase B : a mixture of equal volumes of mobilephase A and methanol R ;
Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 - 4 100 0
4 - 15 100 → 50 0 → 50
15 - 18 50 50
18 - 24 50 → 100 50 → 0
24 - 39 100 0
Flow rate : 1 ml/min.
Detection : spectrophotometer at 230 nm.
Injection : 20 μl.
Identification of impurities : use the chromatogram suppliedwith potassium clavulanate for system suitability CRS andthe chromatogram obtained with reference solution (c) toidentify the peaks due to impurities C, E and G.
Relative retention with reference to clavulanate(retention time = about 3 min): impurity E = about 2.3;impurity G = about 3.6; impurity C = about 7.4.
System suitability : reference solution (b):
— resolution : minimum 13 between the peaks due toclavulanate (1st peak) and amoxicillin (2nd peak).
Limits :
— any impurity impurities C, E, G : for each impurity,not more than t he area of the principal peak in thechromatogram obtained with reference solution (a)(1.0 per cent);
— any other impurity : for each impurity, not morethan 0.2 times the area of the principal peak in thechromatogram obtained with reference solution (a)
(0.2 per cent);— total : not more than twice the area of the principal peak
in the chromatogram obtained with reference solution (a)(2.0 per cent);
— disregard limit : 0.05 times the area of the principal peakin the chromatogram obtained with reference solution (a)(0.05 per cent).
Aliphatic amines. Gas chromatography ( 2.2.28 ).
The method shown below can be used to determinethe following aliphatic amines : 1,1-dimethylethylamine;diethylamine; N , N , N ′ , N ′ -tetramethylethylenediamine;1,1,3,3-tetramethylbutylamine; N , N ′ -diisopropylethylenedi-amine; 2,2′-oxydi( N , N )dimethylethylamine.
Internal standard solution : dissolve 50 μ l of 3-methylpentan-2-one R in water R and dilute to 100.0 mlwith the same solvent.
Test solution. Weigh 1.00 g of the substance to be examinedinto a centrifuge tube. Add 5.0 ml of the internal standardsolution, 5.0 ml of dilute sodium hydroxide solution R,10.0 ml of water R, 5.0 ml of 2-methylpropanol R and 5 g of sodium chloride R. Shake vigorously for 1 min. Centrifugeto separate the layers.
Reference solution. Dissolve 80.0 mg of each of the followingamines: 1,1-dimethylethylamine R ; diethylamine R ;tetramethylethylenediamine R ; 1,1,3,3-tetramethyl- butylamine R ; N,N ′ -diisopropylethylenediamine Rand 2,2 ′ -oxybis(N,N-dimethylethylamine) R in dilutehydrochloric acid R and dilute to 200.0 ml with the sameacid. Introduce 5.0 ml of this solution into a centrifuge tube.Add 5.0 ml of the internal standard solution, 10.0 ml of dilute sodium hydroxide solution R, 5.0 ml of 2-methylpropanol Rand 5 g of sodium chloride R. Shake vigorously for 1 min.Centrifuge to separate the layers.
Column :
— material : fused silica;
— size : l = 50 m, Ø = 0.53 mm;
— stationary phase : poly(dimethyl)(diphenyl)siloxane R(film thickness 5 μm)(52).
Carrier gas : helium for chromatography R.
Flow rate : 8 ml/min.Split ratio : 1:10.
(52) Chrompack CPSil 8CB is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 135

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 136/176
Potassium clavulanate
Temperature :
Time
(min)
Temperature
(°C)
Column 0 - 7 35
7 - 10.8 35 → 150
10.8 - 25.8 150
Injection port 200
Detector 250
Detection : flame ionisation.
Injection : 1 μl of the upper layers obtained from the test solution and the reference solution.
Relative retention with reference to 3-methylpentan-2-one(retention time = about 11.4 min): impurity H = about 0.55;impurity I = about 0.76; impurity J = about 1.07;impurity K = about 1.13; impurity L = about 1.33;impurity M = about 1.57.
Limit :
— aliphatic amines : maximum 0.2 per cent.
2-Ethylhexanoic acid ( 2.4.28 ): maximum 0.8 per cent.
Water ( 2.5.12 ): maximum 0.5 per cent, determined on 1.00 g.
Bacterial endotoxins ( 2.6.14): less than 0.03 IU/mg if intended for use in the manufacture of parenteral dosageforms without a further appropriate procedure for theremoval of bacterial endotoxins.
ASSAY
Liquid chromatography ( 2.2.29). Prepare the solutionsimmediately before use.
Test solution. Dissolve 50.0 mg of the substance tobe examined in a 4.1 g/l solution of sodium acetate Rpreviously adjusted to pH 6.0 with glacial acetic acid R, anddilute to 50.0 ml with the same solution.
Reference solution (a). Dissolve 50.0 mg of lithiumclavulanate CRS in a 4.1 g/l solution of sodium acetate Rpreviously adjusted to pH 6.0 with glacial acetic acid R anddilute to 50.0 ml with the same solution.
Reference solution (b). Dissolve 50.0 mg of lithiumclavulanate CRS and 50.0 mg of amoxicillin trihydrate CRS in a 4.1 g/l solution of sodium acetate R previously adjustedto pH 6.0 with glacial acetic acid R and dilute to 50.0 mlwith the same solution.
Column :— size : l = 0.3 m, Ø = 4.6 mm;
— stationary phase : octadecylsilyl silica gel for chromatography R (5 μm).
Mobile phase : mix 5 volumes of methanol R1 and95 volumes of a 15 g/l solution of sodium dihydrogen phosphate R previously adjusted to pH 4.0 with dilute phosphoric acid R.
Flow rate : 1 ml/min.
Detection : spectrophotometer at 230 nm.
Injection : 10 μl.
System suitability : reference solution (b):— resolution : minimum 3.5 between the peaks due to
clavulanate (1st peak) and amoxicillin (2nd peak).
1 mg of clavulanate (C8H9NO5) is equivalent to 1.191 mgof C8H8KNO5.
STORAGE
In an airtight container, at a temperature of 2 °C to8 °C. If the substance is sterile, store in a sterile, airtight,tamper-proof container.
LABELLING
The label states, where applicable, that the substance is freefrom bacterial endotoxins.
IMPURITIES
Specified impurities: A, B, C, D, E, G, H, I, J, K, L, M.
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.
See also 5.10. Control of impurities in substances for pharmaceutical use) : A, B, D, E, F.
By liquid chromatography: A, B, C, D, E, F, G.
By gas chromatography: H, I, J, K, L, M.
A. R = H: 2,2′-(pyrazine-2,5-diyl)diethanol,
B. R = CH2-CH2-CO2H: 3-[3,6-bis(2-hydroxyethyl)pyrazin-2-yl]propanoic acid,
C. R = CH2-CH3 : 2,2′-(3-ethylpyrazine-2,5-diyl)diethanol,
D. 4-(2-hydroxyethyl)pyrrole-3-carboxylic acid,
E. (2 R,4 R,5 Z )-2-(carboxymethyl)-5-(2-hydroxyethylidene)-3-[[(2 R,3 Z ,5 R)-3-(2-hydroxyethylidene)-7-oxo-4-oxa-1-azabibyclo[3.2.0]hept-2-yl]carbonyl]oxazolidine-4-carboxylic acid,
F. 4-[[[[4-(2-hydroxyethyl)-1 H -pyrrol-3-yl]carbonyl]oxy]meth-yl]-1 H -pyrrole-3-carboxylic acid,
136 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 137/176
Potassium clavulanate, diluted
G. 4-[[(1S )-1-carboxy-2-(4-hydroxyphenyl)ethyl]amino]-4-oxobutanoic acid ( N -succinyltyrosine),
H. 2-amino-2-methylpropane (1,1-dimethylethylamine),
I. diethylamine,
J. 1,2-bis(dimethylamino)ethane ( N , N , N ′, N ′-
tetramethylethylenediamine),
K. 2-amino-2,4,4-trimethylpentane (1,1,3,3-tetramethylbutylamine),
L. N , N ′-bis(1-methylethyl)-1,2-ethanediamine( N , N ′-diisopropylethylenediamine),
M. bis(2-dimethylamino)ethyl ether [2,2′-oxybis( N , N -dimethylethylamine)].
Reference: PA/PH/Exp. 7/T (05) 79 ANP
NOTE ON THE MONOGRAPH
The present transparency statement and impurity limits donot accurately reflect the quality of potassium clavulanatein approved products. The list of specified impuritiesdetected by LC has therefore been reduced to C, E and G; the limit for ‘any other impurity’ has been tightened to 0.2 per cent; and a system suitability CRS has beenintroduced.
XXXX:1653
POTASSIUM CLAVULANATE, DILUTED
Kalii clavulanas dilutus
C8H8KNO5 M r 237.3
DEFINITIONDry mixture of Potassium clavulanate (1140) andCellulose, microcrystalline (0316) or Silica, colloidal anhydrous (0434) or Silica, colloidal hydrated (0738).
Content : 91.2 per cent to 107.1 per cent of the content of potassium clavulanate stated on the label.
CHARACTERS
Appearance of diluted potassium clavulanate : white oralmost white powder, hygroscopic.
Solubility of potassium clavulanate : freely soluble in water,slightly soluble in ethanol (96 per cent), very slightly solublein acetone.The solubility of the diluted product depends on the diluent and its concentration.
IDENTIFICATIONA. Examine the chromatograms obtained in the assay.
Results : the principal peak in the chromatogram obtainedwith the test solution is similar in retention time tothe principal peak in the chromatogram obtained withreference solution (a).
B. It gives reaction (b) of potassium ( 2.3.1).
C. Depending on the diluent used, carry out thecorresponding identification test (a) or (b).
(a) A quantity of the substance to be examined,corresponding to 20 mg of cellulose, when placed ona watch-glass and dispersed in 4 ml of iodinated zinc chloride solution R, becomes violet-blue.(b) It gives the reaction of silicates ( 2.3.1).
TESTS
pH ( 2.2.3): 4.8 to 8.0.Suspend a quantity of the substance to be examinedcorresponding to 0.200 g of potassium clavulanate in 20 mlof carbon dioxide-free water R.
Absorbance ( 2.2.25 ): maximum 0.40 at 278 nm.
Disperse a quantity of the substance to be examined
corresponding to 50.0 mg of potassium clavulanate in 10 mlof 0.1 M phosphate buffer solution pH 7.0 R, dilute to50.0 ml with the same buffer solution and filter. Measurethe absorbance immediately.
© PHARMEUROPA Vol. 18, No. 1, January 2006 137

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 138/176
Potassium clavulanate, diluted
Related substances. Liquid chromatography ( 2.2.29). Prepare the solutions immediately before use .Test solution. Disperse a quantity of the substance to beexamined corresponding to 0.250 g of potassium clavulanatein 5 ml of mobile phase A, dilute to 25.0 ml with mobilephase A and filter. Reference solution (a). Dilute 1.0 ml of the test solution to
100.0 ml with mobile phase A. Reference solution (b). Dissolve 10 mg of amoxicillintrihydrate CRS in 1 ml of the test solution and dilute to100 ml with mobile phase A. Reference solution (c). Dissolve 5 mg of potassiumclavulanate for system suitability CRS in 1 ml of mobilephase A.Column :— size : l = 0.10 m, Ø = 4.6 mm;— stationary phase : octadecylsilyl silica gel for
chromatography R (5 μm);— temperature : 40 °C. Mobile phase :
— mobile phase A: a 7.8 g/l solution of sodium dihydrogen phosphate R adjusted to pH 4.0 with dilute phosphoric acid R ;
— mobile phase B : a mixture of equal volumes of mobilephase A and methanol R ;
Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 - 4 100 0
4 - 15 100 → 50 0 → 50
15 - 18 50 50
18 - 24 50 → 100 50 → 0
24 - 39 100 0
Flow rate : 1 ml/min. Detection : spectrophotometer at 230 nm. Injection : 20 μl. Identification of impurities : use the chromatogram suppliedwith potassium clavulanate for system suitability CRS andthe chromatogram obtained with reference solution (c) toidentify the peaks due to impurities C, E and G. Relative retention with reference to clavulanate(retention time = about 3 min): impurity E = about 2.3;impurity G = about 3.6 ; impurity C = about 7.4.System suitability : reference solution (b):— resolution : minimum 13 between the peaks due to
clavulanate (1st peak) and amoxicillin (2nd peak).
Limits :— any impurity impurities C, E, G : for each impurity,
not more than the area of the principal peak in thechromatogram obtained with reference solution (a)(1.0 per cent);
— any other impurity : for each impurity, not morethan 0.2 times the area of the principal peak in thechromatogram obtained with reference solution (a)(0.2 per cent);
— total : not more than twice the area of the principal peakin the chromatogram obtained with reference solution (a)(2.0 per cent);
— disregard limit : 0.05 times the area of the principal peakin the chromatogram obtained with reference solution (a)(0.05 per cent).
Water ( 2.5.12 ): maximum 2.5 per cent, determined on1.000 g.
ASSAY
Liquid chromatography ( 2.2.29). Prepare the solutionsimmediately before use.
Test solution. Disperse a quantity of the substance to beexamined corresponding to 50.0 mg of potassium clavulanatein a 4.1 g/l solution of sodium acetate R previously adjustedto pH 6.0 with glacial acetic acid R, dilute to 50.0 ml with
the same solution and f ilter. Reference solution (a). Dissolve 50.0 mg of lithiumclavulanate CRS in a 4.1 g/l solution of sodium acetate Rpreviously adjusted to pH 6.0 with glacial acetic acid R anddilute to 50.0 ml with the same solution.
Reference solution (b). Dissolve 10 mg of amoxicillintrihydrate CRS in 10 ml of reference solution (a).
Column :
— size : l = 0.3 m, Ø = 4.6 mm;
— stationary phase : octadecylsilyl silica gel for chromatography R (10 μm).
Mobile phase : mix 5 volumes of methanol R1 and95 volumes of a 15 g/l solution of sodium dihydrogen
phosphate R previously adjusted to pH 4.0 with dilute phosphoric acid R.
Flow rate : 1 ml/min.
Detection : spectrophotometer at 230 nm.
Injection : 10 μl.
System suitability : reference solution (b):
— resolution : minimum 3.5 between the peaks due toclavulanate (1st peak) and amoxicillin (2nd peak).
1 mg of C8H9NO5 is equivalent to 1.191 mg of C8H8KNO5.
STORAGE
In an airtight container.
LABELLINGThe label states the m/m percentage content of potassiumclavulanate and the diluent used to prepare the mixture.
IMPURITIES
Specified impurities: A, B, C, D, E, G.
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for
pharmaceutical use) : A, B, D, E, F.
A. R = H: 2,2′-(pyrazine-2,5-diyl)diethanol,
B. R = CH2-CH2-CO2H: 3-[3,6-bis(2-hydroxyethyl)pyrazin-2-yl]propanoic acid,
C. R = CH2-CH3 : 2,2′-(3-ethylpyrazine-2,5-diyl)diethanol,
D. 4-(2-hydroxyethyl)-1 H -pyrrole-3-carboxylic acid,
138 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 139/176
Rectal preparations
E. (2 R,4 R,5 Z )-2-(carboxymethyl)-5-(2-hydroxyethylidene)-3-[[(2 R,3 Z ,5 R)-3-(2-hydroxyethylidene)-7-oxo-4-oxa-1-azabicyclo[3.2.0]hept-2-yl]carbonyl]oxazolidine-4-carboxylic acid,
F. 4-[[[[4-(2-hydroxyethyl)-1 H -pyrrol-3-yl]carbonyl]oxy]meth-yl]-1 H -pyrrole-3-carboxylic acid,
G. 4-[[(1S )-1-carboxy-2-(4-hydroxyphenyl)ethyl]amino]-4-oxobutanoic acid ( N -succinyltyrosine).
Reference: PA/PH/Exp. 12/T (05) 49 ANP
NOTE ON THE MONOGRAPH
In the current monograph, the test for uniformity of dosage units applies only to solid single-dose preparations. However, certain preparations are presented as liquid or semi-solid preparations in single-dose containers, for which the total content corresponds to a precise dose of themedicine (expressed as unit amount of active substance). It is proposed to modify the corresponding paragraph to takethese preparations into account.
XXXX:1145
RECTAL PREPARATIONS
Rectalia
DEFINITION
Rectal preparations are intended for rectal use in order toobtain a systemic or local effect, or they may be intendedfor diagnostic purposes.
Where applicable, containers for rectal preparationscomply with the requirements for Materials used for themanufacture of containers ( 3.1 and subsections) andContainers ( 3.2 and subsections).
Several categories of rectal preparations may bedistinguished:
— suppositories;
— rectal capsules;
— rectal solutions, emulsions and suspensions;
— powders and tablets for rectal solutions and suspensions ;
— semi-solid rectal preparations;
— rectal foams;
— rectal tampons.
PRODUCTION
During the development of a rectal preparation whoseformulation contains an antimicrobial preservative, theneed for and the efficacy of the chosen preservative shall bedemonstrated to the satisfaction of the competent authority.A suitable test method together with criteria for judging thepreservative properties of the formulation are provided inthe text on Efficacy of antimicrobial preservation (5.1.3).
During development, it must be demonstrated that thenominal content can be withdrawn from the containerof liquid and semi-solid rectal preparations presented insingle-dose containers.
In the manufacture, packaging, storage and distribution of rectal preparations, suitable measures are taken to ensuretheir microbial quality ; recommendations on this aspect are provided in the text on Microbiological quality of pharmaceutical preparations (5.1.4).
In the manufacture of semi-solid and liquid rectalpreparations containing dispersed particles, measures aretaken to ensure a suitable and controlled particle size withregard to the intended use.
TESTS
Uniformity of dosage units. Liquid and semi-solidsingle-dose rectal preparations comply with the test foruniformity of dosage units ( 2.9.40). Solid single-doserectal preparations comply with the test for uniformity of dosage units ( 2.9.40) or, where justified and authorised, withthe tests for uniformity of content and/or uniformity of massshown below. Herbal drugs and herbal drug preparationspresent in the dosage form are not subject to the provisionsof this paragraph.
Uniformity of content ( 2.9.6 ). Unless otherwise prescribedor justified and authorised, solid single-dose rectalpreparations with a content of active substance less than2 mg or less than 2 per cent of the total mass comply withtest A (tablets) or test B (suppositories, rectal capsules)for uniformity of content of single-dose preparations. If the preparation contains more than one active substance,
this requirement applies only to those substances that correspond to the above conditions.
Uniformity of mass ( 2.9.5 ). Solid single-dose rectalpreparations comply with the test for uniformity of mass. If the test for uniformity of content is prescribed for all activesubstances, the test for uniformity of mass is not required.
Dissolution. A suitable test may be required to demonstratethe appropriate release of the active substance(s) from solidsingle-dose rectal preparations, for example the Dissolutiontest for lipophilic solid dosage forms ( 2.9.42 ).
Where a dissolution test is prescribed, a disintegration test may not be required.
LABELLINGThe label states the name of any added antimicrobialpreservative.
© PHARMEUROPA Vol. 18, No. 1, January 2006 139

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 140/176
Rectal preparations
Suppositories
DEFINITION
Suppositories are solid, single-dose preparations. Theshape, volume and consistency of suppositories are suitablefor rectal administration.
They contain 1 or more active substances dispersed ordissolved in a suitable basis that may be soluble or dispersiblein water or may melt at body temperature. Excipients suchas diluents, adsorbents, surface-active agents, lubricants,antimicrobial preservatives and colouring matter, authorisedby the competent authority, may be added if necessary.
PRODUCTION
Suppositories are prepared by compression or moulding. If necessary, the active substance(s) are previously ground andsieved through a suitable sieve. When prepared by moulding,the medicated mass, sufficiently liquefied by heating, ispoured into suitable moulds. The suppository solidifies oncooling. Various excipients are available for this process,such as hard fat, macrogols, cocoa butter, and variousgelatinous mixtures consisting of, for example, gelatin, waterand glycerol. Where applicable, the determination of thesoftening time of lipophilic suppositories ( 2.9.22 ) and/or thedetermination of the resistance to rupture of suppositories( 2.9.24) are carried out.
A suitable test is carried out to demonstrate the appropriaterelease of the active substance(s) from suppositoriesintended for modified release or for prolonged local action.
In the manufacture of suppositories containing dispersedactive substances, measures are taken to ensure a suitableand controlled particle size.
TESTS
Disintegration. Unless intended for modified release orfor prolonged local action, they comply with the test fordisintegration of suppositories and pessaries ( 2.9.2 ). Forsuppositories with a fatty base, examine after 30 min, andfor suppositories with a water-soluble base, examine after60 min, unless otherwise justified and authorised.
Rectal capsules
DEFINITION
Rectal capsules (shell suppositories) are solid, single-dosepreparations generally similar to soft capsules as defined inthe monograph on Capsules (0016) except that they mayhave lubricating coatings. They are of elongated shape, aresmooth and have a uniform external appearance.
PRODUCTION
A suitable test is carried out to demonstrate the appropriaterelease of the active substance(s) from rectal capsulesintended for modified release or for prolonged local action.
TESTS
Disintegration. Unless intended for modified release
or for prolonged local action, they comply with the test for disintegration of suppositories and pessaries ( 2.9.2 ).Examine the state of the capsules after 30 min, unlessotherwise justified and authorised.
Rectal solutions, emulsionsand suspensions
DEFINITION
Rectal solutions, emulsions and suspensions are liquidpreparations intended for rectal use in order to obtaina systemic or local effect, or they may be intended for
diagnostic purposes.Rectal solutions, emulsions and suspensions are suppliedin single-dose containers and contain 1 or more activesubstances dissolved or dispersed in water, glycerol ormacrogols or other suitable solvents. Emulsions may showevidence of phase separation but are readily redispersed onshaking. Suspensions may show a sediment that is readilydispersible on shaking to give a suspension that remainssufficiently stable to enable the correct dose to be delivered.
Rectal solutions, emulsions and suspensions may containexcipients, for example to adjust the viscosity of thepreparation, to adjust or stabilise the pH, to increase thesolubility of the active substance(s) or to stabilise thepreparation. These substances do not adversely affect the
intended medical action or, at the concentrations used,cause undue local irritation.
Rectal solutions, emulsions and suspensions are suppliedin containers containing a volume in the range of 2.5 ml to2000 ml. The container is adapted to deliver the preparationto the rectum or is accompanied by a suitable applicator.
Powders and tablets for rectal solutionsand suspensions
DEFINITION
Powders and tablets intended for the preparation of rectalsolutions or suspensions are single-dose preparations
that are dissolved or dispersed in water at the time of administration. They may contain excipients to facilitatedissolution or dispersion or to prevent aggregation of theparticles.
After dissolution or suspension, they comply with therequirements for rectal solutions or rectal suspensions, asappropriate.
TESTS
Disintegration. Tablets for rectal solutions or suspensionscomply with the test for disintegration of tablets and capsules( 2.9.1) but using water R at 15-25 °C. Carry out the test on6 tablets. Examine the state of the tablets after 3 min. Thetablets comply with the test if all 6 have disintegrated.
LABELLINGThe label states:
— the method of preparation of the rectal solution orsuspension;
— the conditions and duration of storage of the solution orsuspension after constitution.
Semi-solid rectal preparationsDEFINITION
Semi-solid rectal preparations are ointments, creams or gels.
They are often supplied as single-dose preparations incontainers provided with a suitable applicator.
Semi-solid rectal preparations comply with the requirementsof the monograph on Semi-solid preparations for cutaneousapplication (0132).
140 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 141/176
Ropivacaine hydrochloride monohydrate
Rectal foamsDEFINITIONRectal foams comply with the requirements of themonograph on Medicated foams (1105).
Rectal tampons
DEFINITIONRectal tampons are solid, single-dose preparations intendedto be inserted into the lower part of the rectum for a limitedtime.They comply with the requirements of the monograph on Medicated tampons (1155).
Reference: PA/PH/Exp. P4/T (04) 29 ANP
XXXX:2335
ROPIVACAINE HYDROCHLORIDE
MONOHYDRATE
Ropivacaini hydrochloridummonohydricum
C17H27ClN2O,H2O M r 328.9
DEFINITION
S ()-1-Propyl- N -(2,6-dimethylphenyl)piperidine-2-carboxamide hydrochloride monohydrate.Content : 99.0 per cent to 101.0 per cent (anhydroussubstance).
CHARACTERS Appearance : white or almost white, crystalline powder.Solubility : soluble in water and in ethanol (96 per cent),slightly soluble in methylene chloride.
IDENTIFICATIONA. Infrared absorption spectrophotometry ( 2.2.24).
Comparison : ropivacaine hydrochloridemonohydrate CRS .
B. Specific optical rotation ( 2.2.7 ) : 64.0 to 74.0(anhydrous substance).Mix 2 ml of a 200 g/l solution of sodium hydroxide Rand 30 ml of water R and dilute to 100.0 ml with ethanol (96 per cent) R (solution A). Dissolve 0.500 g of the
substance to be examined in solution A and dilute to50.0 ml with solution A.C. It gives reaction (a) of chlorides ( 2.3.1).
TESTS
Solution S. Dissolve 0.50 g of the substance to be examinedin water R and dilute to 25.0 ml with the same solvent.
Appearance of solution. Solution S is clear ( 2.2.1).
pH ( 2.2.3): 4.5 to 6.0.Dissolve 0.250 g in carbon dioxide-free water R and diluteto 25 ml with the same solvent.
Absorbance ( 2.2.25 ): maximum 0.030 at 405 nm andmaximum 0.025 at 436 nm, determined on solution S,
prepared immediately before use, with a path length of 5 cmand using water R as compensation liquid.
Related substances. Liquid chromatography ( 2.2.29).Test solution. Dissolve 55.0 mg of the substance to beexamined in the mobile phase and dilute to 20.0 ml with themobile phase. Reference solution (a). Dilute 1.0 ml of the test solutionto 100.0 ml with the mobile phase. Dilute 1.0 ml of thissolution to 10.0 ml with the mobile phase. Reference solution (b). Dissolve 5 mg of the substance to beexamined and 5 mg of bupivacaine hydrochloride CRS inthe mobile phase and dilute to 5 ml with the mobile phase.Dilute 1.0 ml of this solution to 100.0 ml with the mobilephase.
Column :— size : l = 0.15 m, Ø = 3.9 mm;— stationary phase : end-capped octadecylsilyl silica gel
for chromatography R (4 μm)(53). Mobile phase : mix 1.3 ml of a 156 g/l solution of sodiumdihydrogen phosphate R and 32.5 ml of an 89 g/l solutionof disodium hydrogen phosphate R and dilute to 1000 mlwith water R ; mix equal volumes of this solution andacetonitrile R. Flow rate : 1.0 ml/min. Injection : 20 μl.
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity B 3. impurity D 5. ropivacaine
2. impurity C 4. impurity E 6. impurity A
Figure 2335.-1. – Chromatogram for the test for related substances of ropivacaine hydrochloride monohydrate: solutionof ropivacaine spiked with impurities A, B, C, D and E
(53) Nova-Pak C18 is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 141

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 142/176
Ropivacaine hydrochloride monohydrate
Detection : spectrophotometer at 240 nm. Run time : 2.5 times the retention time of ropivacaine. Relative retention with reference to ropivacaine (retentiontime = about 6 min): impurity A = about 1.6.System suitability : reference solution (b):— resolution : minimum 6.0 between the peaks due to
ropivacaine and impurity A. Limits :— impurity A : not more than twice the area of the principal
peak in the chromatogram obtained with referencesolution (a) (0.2 per cent);
— unspecified impurities : for each impurity, not morethan the area of the principal peak in the chromatogramobtained with reference solution (a) (0.10 per cent);
— total : not more than 5 times the area of the principal peakin the chromatogram obtained with reference solution (a)(0.5 per cent);
— disregard limit : 0.5 times the area of the principal peakin the chromatogram obtained with reference solution (a)(0.05 per cent).
Enantiomeric purity. Capillary electrophoresis ( 2.2.47 ): usethe normalisation procedure.Test solution. Dissolve 50.0 mg of the substance to beexamined in water R and dilute to 25.0 ml with the samesolvent. Reference solution (a). Dilute 1.0 ml of the test solution to200.0 ml with water R. Reference solution (b). Dissolve 1.5 mg of the substance to beexamined and 1.5 mg of (R)-ropivacaine hydrochloride CRS in water R and dilute to 100.0 ml with the same solvent.Capillary :— material : fused silica;— size : effective length = about 72 cm, Ø = 50 μm.
Temperature : 30 °C.CZE buffer : prepare a 133 g/l solution of dimethyl- β -cyclodextrin R in a 9.8 g/l solution of phosphoric acid R previously adjusted to pH 3.0 withtriethanolamine R. The CZE buffer is prepared and f ilteredthrough a 0.45 μm membrane filter immediately before use.
Detection : spectrophotometer at 206 nm.
Preconditioning of the capillary : rinse the capillary withwater R for 1 min, with 0.1 M sodium hydroxide for 10 minand with water R for 3 min. If the capillary is new or dryincrease the sodium hydroxide rinse to 30 min.
Between-run-rinsing : rinse the capillary, at 100 kPa withwater R for 1 min, with 0.1 M sodium hydroxide for 4 min,with water R for 1 min and with CZE buffer for 4 min.
Injection : under pressure (5 kPa) for 5 s.
Migration : apply a field strength of 375 V/cm, initialramping 500 V/s, positive polarity and resulting current 40-45 μA.
Run time : 30 min.
System suitability :
— resolution : minimum 3.7 between the peaks due to( R)-ropivacaine (impurity G) (1st peak) and (S )-ropivacaine.If necessary increase the dimethyl-β-cyclodextrinconcentration in the CZE buffer or lower the temperature;
— signal-to-noise ratio : minimum 10 for referencesolution (a).
Limit :
— impurity G : maximum 0.5 per cent.
Heavy metals ( 2.4.8 ): maximum 10 ppm.
Dissolve 2.0 g in a mixture of 15 volumes of water R and85 volumes of methanol R and dilute to 20 ml with the samemixture of solvents. 12 ml of the solution complies withtest B. Prepare the reference solution using lead standardsolution (1 ppm Pb) obtained by diluting lead standard solution R (100 ppm Pb) with a mixture of 15 volumes of water R and 85 volumes of methanol R.
2,6-Dimethylaniline . Liquid chromatography ( 2.2.29)as described in the test for related substances with thefollowing modifications.
Test solution. Dissolve 0.100 g of the substance to beexamined in the mobile phase and dilute to 10.0 ml with themobile phase.
The following electropherogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity G 2. ropivacaine
Figure 2335.-2. – Electropherogram for the test for enantiomeric purity of ropivacaine hydrochloride monohydrate:reference solution (b)
142 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 143/176
Sertraline hydrochloride
Reference solution. Dissolve 13.0 mg of 2,6-dimethylanilinehydrochloride R(54) in the mobile phase and dilute to100.0 ml with the mobile phase. Dilute 1.0 ml of the solutionto 100.0 ml with the mobile phase. Dilute 1.0 ml of thissolution to 10.0 ml with the mobile phase.
Retention time : 2,6-dimethylaniline = about 2-3 min.
Limit :
— 2,6-dimethylaniline : not more than the area of theprincipal peak in the chromatogram obtained with thereference solution (10 ppm).
Water ( 2.5.12 ): 5.0 per cent to 6.0 per cent, determined on0.100 g.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAY
Dissolve 0.250 g in a mixture of 10 ml of water R and 40 ml of ethanol (96 per cent) R. Add 5.0 ml of 0.01 M hydrochloric acid . Carry out a potentiometric titration ( 2.2.20) using0.1 M sodium hydroxide. Read the volume added between
the 2 points of inflexion.1 ml of 0.1 M sodium hydroxide is equivalent to 31.09 mgof C17H27ClN2O.
IMPURITIES
Specified impurities: A.
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for
pharmaceutical use) : B, C, D, E, F.
A. R = [CH2]3-CH3 : bupivacaine,
B. R = H: ()-1- H - N -(2,6-dimethylphenyl)piperidine-2-carboxamide,
C. R = CH3 : ()-1-methyl- N -(2,6-dimethylphenyl)piperidine-2-carboxamide (mepivacaine),
D. R = CH2-CH3 : ()-1-ethyl- N -(2,6-dimethylphenyl)piperidine-2-carboxamide
E. R = CH(CH3)-CH3 : ()-1-isopropyl- N -(2,6-dimethylphenyl)piperidine-2-carboxamide,
F. acetone adduct,
G. ( R)-ropivacaine.
Reagents
Dimethyl- -cyclodextrin. C56H98O35. ( M r 1331). XXXXXXX.[51166-71-3]. 2,6-Di-O-methyl-β-cyclodextrin.White or almost white powder.
2,6-Dimethylaniline hydrochloride. C8H12ClN. ( M r 157.6). XXXXXXX. [21436-98-6]. 2,6-Xylidine hydrochloride.
Reference: PA/PH/Exp. 10A/T (00) 72 ANP
XXXX:1705
SERTRALINE HYDROCHLORIDE
Sertralini hydrochloridum
C17H18Cl3N M r 342.7
DEFINITION
(1S ,4S )-4-(3,4-Dichlorophenyl)-1,2,3,4-tetrahydro- N -methyl-1-naphthalenamine hydrochloride.
Content : 97.5 per cent to 102.0 per cent (anhydroussubstance).
CHARACTERS
Appearance : white or almost white, crystalline powder.
Solubility : slightly soluble in water, freely soluble inanhydrous ethanol, slightly soluble in acetone and in
isopropanol.It shows polymorphism.
IDENTIFICATION
A. Specific optical rotation ( 2.2.7 ): + 39.0 to + 43.0.
Dissolve 0.250 g in a 103 g/l solution of hydrochloric acid R diluted to 20 volumes with methanol R and diluteto 25 ml with the same solution. Measure the specificoptical rotation at 25 °C.
B. Infrared absorption spectrophotometry ( 2.2.24).
Preparation : 10 g/l solutions in methylene chloride R.
Comparison : sertraline hydrochloride CRS .
C. Dissolve 10 mg in 5 ml of anhydrous ethanol R and
add 5 ml of water R. The solution gives reaction (a) of chlorides ( 2.3.1).
(54) Available from TCI Europe product N°X0029, purity > 98 per cent.
© PHARMEUROPA Vol. 18, No. 1, January 2006 143

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 144/176
Sertraline hydrochloride
TESTS
Enantiomeric purity. Liquid chromatography ( 2.2.29).
Solvent mixture : diethylamine R, hexane R, 2-propanol R(1:40:60 V/V/V ).
Test solution. Dissolve 60.0 mg of the substance to beexamined in the solvent mixture and dilute to 10.0 ml withthe solvent mixture.
Reference solution (a). Dissolve 30.0 mg of sertralinehydrochloride CRS in the solvent mixture and dilute to10.0 ml with the solvent mixture.
Reference solution (b). Dissolve 3 mg of sertralineimpurity G CRS in the solvent mixture, add 1 ml of referencesolution (a) and dilute to 2 ml with the solvent mixture.
Reference solution (c). Dilute 1.0 ml of reference solution (a)to 100.0 ml with the solvent mixture.
Column :
— size : l = 0.25 m, Ø = 4.6 mm;
— stationary phase : silica gel for chiral separation R(5 μm)(55).
Mobile phase : mix 30 volumes of hexane R and 70 volumesof a mixture of 1 volume of diethylamine R, 25 volumes of 2-propanol R and 975 volumes of hexane R.
Flow rate : 0.4 ml/min.
Detection : spectrophotometer at 275 nm.
Injection : 20 μl.
Run time : 30 min.
Elution order : sertraline, impurity G.
System suitability :
— resolution : minimum 1.5 between the peaks due tosertraline and impurity G in the chromatogram obtainedwith reference solution (b) ;
— signal-to-noise ratio : minimum 10 for the peak due tosertraline in the chromatogram obtained with referencesolution (c).
Limit :
— impurity G : not more than the area of the principal peakin the chromatogram obtained with reference solution (c)(0.5 per cent).
Impurity E. Thin-layer chromatography ( 2.2.27 ).
Test solution. Dissolve 0.556 g of the substance to beexamined in a mixture of equal volumes of methanol R andmethylene chloride R and dilute to 10.0 ml with the samemixture of solvents.
Reference solution. Dissolve 5.0 mg of sertraline
impurity E CRS in a mixture of equal volumes of
methanol Rand methylene chloride R and dilute to 50.0 ml with thesame mixture of solvents.
Plate : TLC silica gel F 254 plate R.
Mobile phase : ammonium hydroxide R, methanol R,methylene chloride R (15:50:120 V/V/V ).
Application : 100 μl as bands of about 8 cm. Allow to dry.
Development : over 2/3 of the plate.
Drying : in air.
Detection : examine in ultraviolet light at 254 nm.
Limit :
— impurity E : any zone due to impurity E is not moreintense than the zone in the chromatogram obtained with
the reference solution (0.2 per cent).
Related substances. Gas chromatography ( 2.2.28 ): use thenormalisation procedure.Test solution. Introduce 0.250 g of the substance to beexamined into a 15 ml stoppered centrifuge tube, add 2.0 mlof methanol R and 0.20 ml of a 25 per cent solution of potassium carbonate R and mix in a vortex mixer for 30 s.Add 8 ml of methylene chloride R, stopper the tube and mix
in a vortex mixer for 60 s. Add 1 g of anhydrous sodium sulphate R, mix well and then centrifuge for about 5 min.Use the methylene chloride layer. Reference solution. Introduce 50 mg of sertraline for peak identification CRS into a 5 ml stoppered centrifuge tube,add 0.5 ml of methanol R and 0.05 ml of a 25 per cent solution of potassium carbonate R and mix in a vortex mixerfor 30 s. Add 2 ml of methylene chloride R, stopper the tubeand mix in a vortex mixer for 60 s. Add 0.25 g of anhydrous sodium sulphate R, mix well and then centrifuge for about 5 min. Use the methylene chloride layer.Column :— material : fused silica;— size : l = 30 m, Ø = 0.53 mm;
— stationary phase : poly[phenyl(50)methyl(50)]- siloxane R(56) (film thickness 1.0 μm).
Carrier gas : helium for chromatography R. Flow rate : 9 ml/min.Split ratio : 1:10.Temperature :
Time
(min)
Temperature
(°C)
Column 0 - 1 200
1 - 31 200 → 260
31 - 39 260
Injection port 250
Detector 280
Detection : flame ionisation. Injection : 1 μl. Identification of impurities : use the chromatogramsupplied with sertraline for peak identification CRS andthe chromatogram obtained with the reference solution toidentify the peaks due to the impurities. Relative retention with reference to sertraline (retentiontime = about 24 min): impurity B = about 0.5;impurities C and D = about 0.7; impurity A = about 1.05;impurity F = about 1.1.System suitability : reference solution:
— the chromatogram obtained is similar to thechromatogram supplied with sertraline for peak identification CRS .
Limits :— impurities A, B, F : for each impurity, maximum 0.2 per
cent;— sum of impurities C and D: maximum 0.8 per cent;— unspecified impurities : for each impurity, maximum
0.10 per cent;— total : maximum 1.5 per cent;— disregard limit : 0.05 per cent.
Heavy metals ( 2.4.8 ): maximum 20 ppm.1.0 g complies with test C. Prepare the reference solution
using 2 ml of lead standard solution (10 ppm Pb) R.
(55) Chiralpak Daicel AD is suitable.(56) OV 17 is suitable.
144 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 145/176
Sertraline hydrochloride
Water ( 2.5.12 ) : maximum 0.5 per cent, determined on 2.0 g.
Sulphated ash ( 2.4.14): maximum 0.2 per cent, determinedon 1.0 g.
ASSAY
Liquid chromatography ( 2.2.29).
Buffer solution. To 28.6 ml of glacial acetic acid R slowly
add, while stirring and cooling, 34.8 ml of triethylamine R,and dilute to 100 ml with water R. Dilute 10 ml of thissolution to 1000 ml with water R.
Test solution. Dissolve 55.0 mg of the substance to beexamined in the mobile phase and dilute to 50.0 ml with themobile phase. Dilute 5.0 ml of this solution to 100.0 ml withthe mobile phase.
Reference solution. Dissolve 55.0 mg of sertralinehydrochloride CRS in the mobile phase and dilute to 50.0 mlwith the mobile phase. Dilute 5.0 ml of this solution to100.0 ml with the mobile phase.
Column :
— size : l = 0.15 m, Ø = 3.9 mm;
— stationary phase : octadecylsilyl silica gel for chromatography R (4 μm)(57) ;
— temperature : 30 °C.
Mobile phase : methanol R, buffer solution, acetonitrile R(15:40:45 V/V/V ).
Flow rate : 1.8 ml/min.
Detection : spectrophotometer at 254 nm.
Injection : 20 μl.
Run time : twice the retention time of sertraline.
Calculate the percentage content of C17H18Cl3N from thedeclared content of sertraline hydrochloride CRS .
STORAGEProtected from light.
IMPURITIES
Specified impurities: A, B, C, D, E, F, G.
A. (±)-trans-4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydro- N -methyl-1-naphthalenamine,
B. (±)-cis-4-phenyl-1,2,3,4-tetrahydro- N -methyl-1-naphthalenamine,
C. (±)-cis-4-(4-chlorophenyl)-1,2,3,4-tetrahydro- N -methyl-1-naphthalenamine,
D. (±)-cis-4-(3-chlorophenyl)-1,2,3,4-tetrahydro- N -methyl-1-naphthalenamine,
E. mandelic acid,
F. 4-(3,4-dichlorophenyl)-3,4-dihydro-1(2 H )-naphthalenone(tetralone),
G. 4-(3,4-dichlorophenyl)-1,2,3,4-tetrahydro- N -methyl-1-naphthalenamine ((1 R,4 R) enantiomer).
Reagents
Poly[phenyl(50)methyl(50)]siloxane. XXXXXXX.Stationary phase for gas chromatography.
Contains 50 per cent of methyl groups and 50 per cent of phenyl groups.
(57) Nova-Pak Waters Cat. No. 086355 is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 145

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 146/176
Sodium acetate trihydrate
Reference: PA/PH/Exp. INC/T (05) 35 ANP
NOTE ON THE MONOGRAPH
As the test for reducing substances does not give satisfactory results, it is proposed to revise it.
XXXX:0411
SODIUM ACETATE TRIHYDRATE
Natrii acetas trihydricus
C2H3NaO2,3H2O M r 136.1
DEFINITION
Sodium ethanoate trihydrate.
Content : 99.0 per cent to 101.0 per cent (dried substance).
CHARACTERS Appearance : colourless crystals.
Solubility : very soluble in water, soluble in ethanol (96 percent).
IDENTIFICATION
A. 1 ml of solution S (see Tests) gives reaction (b) of acetates( 2.3.1).
B. 1 ml of solution S gives reaction (a) of sodium ( 2.3.1).
C. Loss on drying (see Tests).
TESTS
Solution S. Dissolve 10.0 g in carbon dioxide-free water R
prepared from distilled water R and dilute to 100 ml withthe same solvent.
Appearance of solution. Solution S is clear ( 2.2.1) andcolourless ( 2.2.2, Method II ).
pH ( 2.2.3): 7.5 to 9.0.
Dilute 5 ml of solution S to 10 ml with carbon dioxide-freewater R.
Reducing substances. Dissolve 1.0 5.0 g in 100 50 ml of boiling water R, then add 5 ml of dilute sulphuric acid Rand 0.5 ml of 0.002 M potassium permanganate, mix andboil gently for 5 min. The pink colour is not complet elydischarged persists for at least 1 h.
Chlorides ( 2.4.4): maximum 200 ppm.Dilute 2.5 ml of solution S to 15 ml with water R.
Sulphates ( 2.4.13): maximum 200 ppm.
Dilute 7.5 ml of solution S to 15 ml with distilled water R.
Aluminium ( 2.4.17 ): maximum 0.2 ppm, if intended for usein the manufacture of dialysis solutions.
Prescribed solution. Dissolve 20 g in 100 ml of water R andadjust to pH 6.0 by the addition of 1 M hydrochloric acid (about 10 ml).
Reference solution. Mix 2 ml of aluminium standard solution (2 ppm Al) R, 10 ml of acetate buffer solution pH 6.0 R and 98 ml of water R.
Blank solution. Mix 10 ml of acetate buffer solution pH 6.0 R and 100 ml of water R.Arsenic ( 2.4.2, Method A): maximum 2 ppm, determinedon 0.5 g.
Calcium and magnesium : maximum 50 ppm, calculatedas Ca.
To 200 ml of water R add 10 ml of ammonium chloridebuffer solution pH 10.0 R, 0.1 g of mordant black 11triturate R, 2.0 ml of 0.05 M zinc chloride and, dropwise,0.02 M sodium edetate until the colour changes from violet to blue. Add to the solution 10.0 g of the substance to be
examined and shake to dissolve. Titrate with 0.02 M sodiumedetate until the blue colour is restored. Not more than0.65 ml of 0.02 M sodium edetate is required.
Heavy metals ( 2.4.8 ): maximum 10 ppm.
12 ml of solution S complies with test A. Prepare thereference solution using lead standard solution (1 ppm Pb) R.
Iron ( 2.4.9): maximum 10 ppm, determined on 10 ml of solution S.
Loss on drying ( 2.2.32 ): 39.0 per cent to 40.5 per cent,determined on 1.000 g by drying in an oven at 130 °C.Introduce the substance to be examined into the oven whilethe latter is cold.
ASSAY
Dissolve 0.250 g in 50 ml of anhydrous acetic acid R, add5 ml of acetic anhydride R, mix and allow to stand for30 min. Using 0.3 ml of naphtholbenzein solution R asindicator, titrate with 0.1 M perchloric acid until a greencolour is obtained.
1 ml of 0.1 M perchloric acid is equivalent to 8.20 mgof C2H3NaO2.
STORAGE
In an airtight container.
LABELLING
The label states, where applicable, that the substance issuitable for use in the manufacture of dialysis solutions.
Reference: PA/PH/Exp. INC/T (05) 36 ANP
NOTE ON THE MONOGRAPH
The current assay method often causes problems due to theunsatisfactory solubility of the substance in the titrationmedium. Another method is therefore proposed as areplacement.
XXXX:0514
SODIUM FLUORIDE
Natrii fluoridum
NaF M r 41.99
DEFINITION
Content : 98.5 per cent to 100.5 per cent (dried substance).
CHARACTERS
Appearance : white or almost white powder or colourlesscrystals.Solubility : soluble in water, practically insoluble in ethanol(96 per cent).
146 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 147/176
Vaginal preparations
IDENTIFICATION
A. To 2 ml of solution S (see Tests) add 0.5 ml of calciumchloride solution R. A gelatinous white precipitate isformed that dissolves on adding 5 ml of ferric chloride solution R1.
B. To about 4 mg add a mixture of 0.1 ml of alizarin S solution R and 0.1 ml of zirconyl nitrate solution R andmix. The colour changes from red to yellow.
C. Solution S gives reaction (a) of sodium ( 2.3.1).
TESTS
Solution S. Dissolve 2.5 g in carbon dioxide-free water Rwithout heating and dilute to 100 ml with the same solvent.
Appearance of solution. Solution S is clear ( 2.2.1) andcolourless ( 2.2.2, Method II ).
Acidity or alkalinity. Dissolve 2.5 g of potassium nitrate Rin 40 ml of solution S and dilute to 50 ml with carbondioxide-free water R. Cool to 0 °C and add 0.2 ml of phenolphthalein solution R. If the solution is colourless,
not more than 1.0 ml of 0.1 M sodium hydroxide is requiredto produce a red colour that persists for at least 15 s. If thesolution is red, not more than 0.25 ml of 0.1 M hydrochloric acid is required to change the colour of the indicator.
Chlorides ( 2.4.4): maximum 200 ppm.
Dilute 10 ml of solution S to 15 ml with water R.
Fluorosilicates. Heat to boiling the neutralised solutionobtained in the test for acidity or alkalinity and titrate whilst hot. Not more than 0.75 ml of 0.1 M sodium hydroxide isrequired to change the colour of the indicator to red.
Sulphates ( 2.4.13): maximum 200 ppm.
Dissolve 0.25 g in 10 ml of a saturated solution of boric acid R in distilled water R. Add 5 ml of distilled water R
and 0.6 ml of hydrochloric acid R1. Prepare the standardby mixing 0.6 ml of hydrochloric acid R1, 5 ml of sulphate standard solution (10 ppm SO4 ) R and 10 ml of a saturatedsolution of boric acid R in distilled water R.
Loss on drying ( 2.2.32 ): maximum 0.5 per cent, determinedon 1.000 g by drying in an oven at 130 °C for 3 h.
ASSAY
To 80.0 mg add a mixture of 5 ml of ac eti c anhy drid e Rand 20 ml of anhydrous acetic acid R and heat t o dissolve.Allow t o cool and add 20 ml of di ox an R. Using 0.1 ml of cryst al vi ol et solution R as indicator, titrat e with 0.1 M per chlori c aci d until a green colour is obtained. Carry out
a blank titration.1 ml of 0.1 M per chl ori c acid is equivalent to 4.199 mgof NaF.
Dissolve 0.100 g in water R and dilute to 60 ml with the samesolvent. Titrate with 0.1 M lanthanum nitrate determiningthe end-point potentiometrically using a fluoride-selectiveindicator electrode and a silver-silver chloride referenceelectrode ( 2.2.20).
1 ml of 0.1 M lanthanum nitrate is equivalent to 12.60 mgg of NaF.
Reagents
0.1 M Lanthanum nitrate. XXXXXXX.
Dissolve 43.30 g of lanthanum nitrate R in water R anddilute to 1000.0 ml with the same solvent.
Standardisation. To 20 ml of the lanthanum nitrate solution,add 15 ml of water R and 25 ml of 0.1 M sodium edetate.Add about 50 mg of xylenol orange triturate R and about 2 g of hexamethylenetetramine R. Titrate with 0.1 M zinc sulphate until the colour changes from yellow to violet-pink.
Reference: PA/PH/Exp.12/T (05) 48 ANP
NOTE ON THE MONOGRAPH
In the current monograph, the test for uniformity of dosage units applies only to solid single-dose preparations. However, certain preparations are presented as liquid or semi-solid preparations in single-dose containers, for which the total content corresponds to a precise dose of themedicine (expressed as unit amount of active substance). It is proposed to modify the corresponding paragraph to takethese preparations into account.
XXXX:1164
VAGINAL PREPARATIONS
Vaginalia
DEFINITION
Vaginal preparations are liquid, semi-solid or solidpreparations intended for administration to the vaginausually in order to obtain a local effect. They contain 1 ormore active substances in a suitable basis.
Where appropriate, containers for vaginal preparationscomply with the requirements for Materials used for themanufacture of containers ( 3.1 and subsections) andContainers ( 3.2 and subsections).
Several categories of vaginal preparations may bedistinguished:
— pessaries;
— vaginal tablets;
— vaginal capsules;
— vaginal solutions, emulsions and suspensions;
— tablets for vaginal solutions and suspensions ;
— semi-solid vaginal preparations;
— vaginal foams;
— medicated vaginal tampons.
PRODUCTION
During development, it must be demonstrated that thenominal content can be withdrawn from the container of liquid and semi-solid vaginal preparations presented insingle-dose containers.
In the manufacturing, packaging, storage and distributionof vaginal preparations, suitable measures are taken toensure their microbial quality; recommendations on thisaspect are provided in the text on Microbiological quality of pharmaceutical preparations (5.1.4).
TESTS
Uniformity of dosage units. Liquid and semi-solidsingle-dose vaginal preparations comply with the test for
uniformity of dosage units ( 2.9.40). Solid single-dosevaginal preparations comply with the test for uniformity of dosage units ( 2.9.40) or, where justified and authorised, withthe tests for uniformity of content and/or uniformity of mass
© PHARMEUROPA Vol. 18, No. 1, January 2006 147

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 148/176
Vaginal preparations
shown below. Herbal drugs and herbal drug preparationspresent in the dosage form are not subject to the provisionsof this paragraph.
Uniformity of content ( 2.9.6 ). Unless otherwise prescribedor justified and authorised, solid single-dose vaginalpreparations with a content of active substance less than2 mg or less than 2 per cent of the total mass comply with
test A (vaginal tablets) or test B (pessaries, vaginal capsules)for uniformity of content of single-dose preparations.If the preparation has more than one active substance,the requirement applies only to those substances whichcorrespond to the above conditions.
Uniformity of mass ( 2.9.5 ). Solid single-dose vaginalpreparations comply with the test for uniformity of massof single-dose preparations. If the test for uniformity of content is prescribed for all the active substances, the test for uniformity of mass is not required.
Dissolution. A suitable test may be carried out todemonstrate the appropriate release of the activesubstance(s) from solid single-dose vaginal preparations,for example one of the tests described in Dissolution test
for solid dosage forms ( 2.9.3) or in Dissolution test for lipophilic solid dosage forms ( 2.9.42 ).
When a dissolution test is prescribed, a disintegration test may not be required.
Pessaries
DEFINITION
Pessaries are solid, single-dose preparations. They havevarious shapes, usually ovoid, with a volume and consistencysuitable for insertion into the vagina. They contain 1 or moreactive substances dispersed or dissolved in a suitable basis
that may be soluble or dispersible in water or may melt at body temperature. Excipients such as diluents, adsorbents,surface-active agents, lubricants, antimicrobial preservativesand colouring matter authorised by the competent authoritymay be added, if necessary.
PRODUCTION
Pessaries are usually prepared by moulding. Whereappropriate in the manufacture of pessaries, measures aretaken to ensure a suitable and controlled particle size of theactive substance(s). If necessary, the active substance(s) arepreviously ground and sieved through a suitable sieve.
When prepared by moulding, the medicated mass, sufficiently
liquefied by heating, is poured into suitable moulds. Thepessary solidifies on cooling. Various excipients are availablefor this process, such as hard fat, macrogols, cocoa butter,and various gelatinous mixtures consisting, for example, of gelatin, water and glycerol.
A suitable test is carried out to demonstrate the appropriaterelease of the active substance(s) from pessaries intended forprolonged local action.
Where appropriate, the determination of the resistance torupture of pessaries ( 2.9.24) is carried out.
TESTS
Disintegration. Unless intended for prolonged local action,they comply with the test for disintegration of suppositoriesand pessaries ( 2.9.2 ). Examine the state of the pessariesafter 60 min, unless otherwise justified and authorised.
Vaginal tablets
DEFINITION
Vaginal tablets are solid, single-dose preparations.They generally conform to the definitions of uncoated orfilm-coated tablets given in the monograph on Tablets (0478).
PRODUCTIONA suitable test is carried out to demonstrate the appropriaterelease of the active substance(s) from vaginal tabletsintended for prolonged local action.
TESTS
Disintegration. Unless intended for prolonged local action,they comply with the test for disintegration of suppositoriesand pessaries (special method for vaginal tablets, 2.9.2 ).Examine the state of the tablets after 30 min, unlessotherwise justified and authorised.
Vaginal capsules
DEFINITIONVaginal capsules (shell pessaries) are solid, single-dosepreparations. They are generally similar to soft capsulesas defined in the monograph on Capsules (0016), differingonly in their shape and size. Vaginal capsules have variousshapes, usually ovoid. They are smooth and have a uniformexternal appearance.
PRODUCTION
A suitable test is carried out to demonstrate the appropriaterelease of the active substance(s) from vaginal capsulesintended for prolonged local action.
TESTS
Disintegration. Unless intended for prolonged local action,they comply with the test for disintegration of suppositoriesand pessaries ( 2.9.2 ). Examine the state of the capsules after30 min, unless otherwise justified and authorised.
Vaginal solutions, emulsionsand suspensions
DEFINITION
Vaginal solutions, emulsions and suspensions are liquidpreparations intended for a local effect, for irrigation orfor diagnostic purposes. They may contain excipients, forexample to adjust the viscosity of the preparation, to adjust or stabilise the pH, to increase the solubility of the activesubstance(s) or to stabilise the preparation. The excipientsdo not adversely affect the intended medical action or, at theconcentrations used, cause undue local irritation.
Vaginal emulsions may show evidence of phase separationbut are readily redispersed on shaking. Vaginal suspensionsmay show a sediment that is readily dispersed on shaking togive a suspension that remains sufficiently stable to enable ahomogeneous preparation to be delivered.
They are supplied in single-dose containers. The containeris adapted to deliver the preparation to the vagina or it isaccompanied by a suitable applicator.
PRODUCTION
In the manufacture of vaginal suspensions measures aretaken to ensure a suitable and controlled particle size withregard to the intended use.
148 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 149/176
Vinpocetine
Tablets for vaginal solutionsand suspensions
DEFINITION
Tablets intended for the preparation of vaginal solutions andsuspensions are single-dose preparations which are dissolved
or dispersed in water at the time of administration. Theymay contain excipients to facilitate dissolution or dispersionor to prevent caking.
Apart from the test for disintegration, tablets for vaginalsolutions or suspensions conform with the definition forTablets (0478).
After dissolution or dispersion, they comply with therequirements for vaginal solutions or vaginal suspensions, asappropriate.
TESTS
Disintegration. Tablets for vaginal solutions or suspensions
comply with the test for disintegration of tablets and capsules( 2.9.1), but using water R at 15-25 °C. Carry out the test on6 tablets. Examine the state of the tablets after 3 min. Thetablets comply with the test if all 6 have disintegrated.
LABELLING
The label states:
— the method of preparation of the vaginal solution orsuspension;
— the conditions and duration of storage of the solution orsuspension after constitution.
Semi-solid vaginal preparations
DEFINITION
Semi-solid vaginal preparations are ointments, creams orgels.
They are often supplied in single-dose containers. Thecontainer is provided with a suitable applicator.
Semi-solid vaginal preparations comply with the requirementsof the monograph on Semi-solid preparations for cutaneousapplication (0132).
Vaginal foams
DEFINITION
Vaginal foams comply with the requirements of themonograph on Medicated foams (1105).
Medicated vaginal tampons
DEFINITION
Medicated vaginal tampons are solid, single-dosepreparations intended to be inserted in the vagina for a
limited time.They comply with the requirements of the monograph on Medicated tampons (1155).
Reference: PA/PH/Exp. 11/T (05) 81 ANP
NOTE ON THE MONOGRAPH
The test for related substances has been modified to cover all the specified impurities.
XXXX:2139
VINPOCETINE
Vinpocetinum
C22H26N2O2 M r 350.5
DEFINITIONEthyl (13aS ,13bS )-13a-ethyl-2,3,5,6,13a,13b-hexahydro-1 H -indolo[3,2,1-de]pyrido[3,2,1-ij ][1,5]naphthyridine-12-carboxylate.
Content : 98.5 per cent to 101.5 per cent (dried substance).
CHARACTERS
Appearance : white or slightly yellow, crystalline powder.
Solubility : practically insoluble in water, soluble inmethylene chloride, slightly soluble in anhydrous ethanol.
IDENTIFICATION
A. It complies with the test for specific optical rotation (see
Tests).B. Infrared absorption spectrophotometry ( 2.2.24).
Comparison : vinpocetine CRS .
TESTS
Specific optical rotation ( 2.2.7 ): + 127 to + 134 (driedsubstance).
Dissolve 0.25 g in dimethylformamide R and dilute to25.0 ml with the same solvent.
Related substances. Liquid chromatography ( 2.2.29).
Test solution. Dissolve 50.0 mg of the substance to beexamined in ac et onitrile R the mobile phase and dilute to50.0 ml with the mobile phase.
Reference solution (a). Dilute 1.0 ml of the test solution to20.0 ml 50.0 ml with acet onitrile R the mobile phase. Dilut e1.0 ml of this solution t o 50.0 ml with ac etonitril e R.
Reference solution (b). Dissolve 5.0 mg of vinpocetineimpurity B CRS , 6.0 mg of vinpocetine impurity A CRS ,5.0 mg of vinpocetine impurity C CRS and 5.0 mg of vinpocetine impurity C D CRS in acet onitrile R the mobilephase and dilute to 50.0 ml with the mobile phase. Dilut e1.0 ml of the solution t o 20.0 ml with the test solution.
Reference solution (c). Dissolve 5.0 mg of vinpoc etineimpurity A CRS in ac et onitril e R and dilute to 50.0 ml withthe same solvent. Dilut e 1.0 ml of the solution t o 20.0 mlwith ac et onitril e R. Dilute 1.0 ml of reference solution (a)and 1.0 ml of reference solution (b) to 20.0 ml with the
mobile phase.Column :
— size : l = 0.25 m, Ø = 4.6 mm;
© PHARMEUROPA Vol. 18, No. 1, January 2006 149

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 150/176
Vinpocetine
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity A 3. impurity B 5. vinpocetine
2. impurity D 4. impurity C
Figure 2139.-1. – Chromatogram for the test for related substances of vinpocetine
— stationary phase : octadecylsilyl silica gel for chromatography R (5 μm)(58).
— t emper atur e : 40 °C.
Mobile phase : 0.1 M ammonium c ar bonat e a 15.4 g/lsolution of ammonium acetate R, acetonitrile R (32:68 V/V 45:55 V/V ).
Flow rate : 1.0 ml/min.
Detection : spectrophotometer at 280 nm.
Injection : 15 μl.
Run time : twice 3 times the retention time of vinpocetine.
Relative retention with reference to vinpocetine (retentiontime = about 17 min 16 min): impurity A = about 0.4;impurity D = about 0.68; impurity B = about 0.8 0.75;impurity C = about 0.85 0.83.
System suitability : reference solution (b) (c):
— resolution : minimum 1.5 2.0 between the peaks due to
impurities B and C D. Limits :
— impurity A : not more than the area of the principalcorresponding peak in the chromatogram obtained withreference solution (c) (0.5 per cent 0.6 per cent);
— impurity B : not more than the area of the correspondingpeak in the chromatogram obtained with referencesolution (b) (c) (0.5 per cent);
— impurity C : not more than 0.4 times 0.6 times the areaof the corresponding peak in the chromatogram obtainedwith reference solution (b) (c) (0.2 per cent 0.3 percent per cent);
— impurity D : not more than the area of the correspondingpeak in the chromatogram obtained with referencesolution (c) (0.5 per cent);
— any other impuri ty unspecified impurities : for each
impurity, not more than the area of the principal peakdue to vinpocetine in the chromatogram obtained withreference solution (a) (c) (0.10 per cent);
— total : maximum 1.0 per cent not more than 10 times thearea of the peak due to vinpocetine in the chromatogramobtained with reference solution (c) (1.0 per cent) ;
— disregard limit : 0.5 times the area of the principal peakdue to vinpocetine in the chromatogram obtained withreference solution (a) (c) (0.05 per cent).
Loss on drying ( 2.2.32 ) : maximum 0.5 per cent, determinedon 1.000 g by drying in vacuo in an oven at 100 °C for 3 h.
Sulphated ash ( 2.4.14): maximum 0.1 per cent, determinedon 1.0 g.
ASSAY
Dissolve 0.300 g in 50 ml of a mixture of equal volumes of acetic anhydride R and anhydrous acetic acid R. Titratewith 0.1 M perchloric acid , determining the end-point potentiometrically ( 2.2.20).
1 ml of 0.1 M perchloric acid is equivalent to 35.05 mgof C22H26N2O2.
IMPURITIES
Specified impurities : A, B, C, D.
(58) Waters spherisorb® S5 ODS2 is suitable. Supelco discovery C18 is suitable.
150 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 151/176
Warfarin sodium
A. ethyl (12 RS ,13aSR,13bSR)-13a-ethyl-12-hydroxy-2,3,5,6,12,13,13a,13b-octahydro-1 H -indolo[3,2,1-de]pyrido[3,2,1-ij ][1,5]naphthyridine-12-carboxylate (ethyl vincaminate),
B. R1 = CH3, R2 = H: methyl (13aS ,13bS )-13a-ethyl-2,3,5,6,13a,13b-hexahydro-1 H -indolo[3,2,1-de]pyrido[3,2,1-ij ][1,5]naphthyridine-12-carboxylate (apovincamine),
C. R1 = C2H5, R2 = OCH3 : ethyl (13aS ,13bS )-13a-ethyl-9-methoxy-2,3,5,6,13a,13b-hexahydro-1 H -indolo[3,2,1-de]pyrido[3,2,1-ij ][1,5]naphthyridine-12-carboxylate(methoxyvinpocetine),
D. dihydrovinpocetine.
Reference: PA/PH/Exp. 10B/T (05) 5 ANP
NOTE ON THE MONOGRAPH
It is proposed to replace the TLC test for related substancesby a LC test and to delete the 2 nd identification series.
XXXX:0698
WARFARIN SODIUM
Warfarinum natricum
Cl9H15NaO4 M r 330.3
DEFINITION
Sodium 2-oxo-3-[(1 RS )-3-oxo-1-phenylbutyl]-2 H -1-benzopyran-4-olate.Content : 98.0 per cent to 102.0 per cent (anhydrous
substance).CHARACTERS
Appearance : white or almost white, hygroscopic powder.
Solubility : very soluble in water and in ethanol (96 percent), soluble in acetone, very slightly soluble in methylenechloride.
IDENTIFICATION First i dentifi cati on: B, D, E .Second i d entificati on: A, C, D, E .
A. Dissolve 1 g in 25 ml of water R, add 2 ml of dilut ehydr ochlori c acid R and filter. Reserve the filtrat e f oridentification test E. The precipitate, washed with water Rand dried at 100 °C to 105 °C, melts ( 2.2.14) at 159 °Ct o 163 °C.
B. A. Infrared absorption spectrophotometry ( 2.2.24).Dissolve 1 g in 25 ml of water R, add 2 ml of dilut ehyd r ochlori c acid R and filter. Reserve the filtrat e f oridentification t est E. Examine the precipitate.Comparison : warfarin sodium CRS .
C. Examine the chromatograms obtained in the test f or related substances. The principal spot in thechromat ogram obtained with test solution (b) issimilar in position and size to the principal spot in the
chromat ogram obtained with ref erence solution (b).D. B. Dissolve 1 g in 10 ml of water R, add 5 ml of nitric
acid R and filter. To the filtrate add 2 ml of potassiumdichromate solution R1 and shake for 5 min. Allow tostand for 20 min. The solution is not greenish-blue whencompared with a blank.
E. C. The filtrat e obtained in identification test A It givesreaction (b) of sodium ( 2.3.1).
TESTS
Appearance of solution. The solution is clear ( 2.2.1) andcolourless ( 2.2.2, Method II ).Dissolve 1.0 g in water R and dilute to 20 ml with the samesolvent.
pH ( 2.2.3): 7.6 to 8.6.Dissolve 1.0 g in carbon dioxide-free water R and dilute to100 ml with the same solvent.
Related substances. Examine by thin-layer chromatography( 2.2.27 ), using silic a g el GF 2 54 R as the coating subst ance.
T est soluti on (a). Dissolve 0.20 g of the substance to beexamined in ac et one R and dilut e t o 10 ml with the samesolvent.T est soluti on (b). Dilute 2 ml of t est solution (a) t o 10 mlwith ac et one R. Ref er ence soluti on ( a). Dilut e 1 ml of test solution (b) t o200 ml with acet one R.
Ref er ence soluti on ( b). Dissolve 40 mg of warf arin sodium CRS in acet one R and dilut e t o 10 ml with the samesolvent. Ref er ence soluti on ( c). Dissolve 10 mg of acenoc oumar ol CRS in acet one R, add 1 ml of t est solution (a) and dilut e t o 10 ml with ac et one R.Apply separately to the plat e 20 μl of each solution. Developover a path of 15 cm using a mixture of 20 volumes of g laci al acetic acid R, 50 volumes of methyl ene chl ori de R and50 volumes of cy cl ohexane R. Allow the plat e to dry in airand examine in ultraviolet light at 254 nm. Any spot in thechromat ogram obtained with t est solution (a), apart fromthe principal spot, is not more int ense than the spot in thechromat ogram obtained with ref erence solution (a) (0.1 per
cent). The test is not valid unless the chromat ogram obtainedwith ref erence solution (c) shows two clearly separat ed spotsand the chromat ogram obtained with ref erence solution (a)shows a clearly visible spot.
© PHARMEUROPA Vol. 18, No. 1, January 2006 151

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 152/176
Warfarin sodium
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity B 2. impurity C 3. warfarin 4. impurity A
Figure 0698.-1. – Chromatogram for the test for related substances of warfarin sodium
Related substances. Liquid chromatography ( 2.2.29).Solvent mixture : methanol R, water R (25:75 V/V ).
Test solution. Dissolve 40.0 mg of the substance to beexamined in the solvent mixture and dilute to 50.0 ml withthe solvent mixture.
Reference solution (a). Dissolve 2.0 mg of warfarinimpurity B CRS and 2.0 mg of warfarin impurity C CRS in25 ml of methanol R and dilute to 100.0 ml with water R.
Reference solution (b). Dilute 1.0 ml of the test solutionto 100.0 ml with the solvent mixture. Dilute 1.0 ml of thissolution to 10.0 ml with the solvent mixture.
Column :
— size : l = 0.25 m, Ø = 4.0 mm;— stationary phase : spherical nitrile silica gel for
chromatography R (5 μm)(59) ;
— temperature : 30 °C.
Mobile phase : glacial acetic acid R, acetonitrile R, water R(1:25:75 V/V/V ).
Flow rate : 1.5 ml/min.
Detection : spectrophotometer at 260 nm.
Injection : 20 μl.
Run time : twice the retention time of warfarin.
Relative retention with reference to warfarin (retention
time = about 9 min): impurity B = about 0.4;impurity C = about 0.6.
System suitability : reference solution (a):
— resolution : minimum 2.0 between the peaks due toimpurities B and C.
Limits :
— correction factors : for the calculation of content,multiply the peak areas of the following impurities bythe corresponding correction factor: impurity B = 0.5;impurity C = 0.4;
— impurities B, C : for each impurity, not more than thearea of the principal peak in the chromatogram obtainedwith reference solution (b) (0.1 per cent) ;
— unspecified impurities : for each impurity, not morethan the area of the principal peak in the chromatogramobtained with reference solution (b) (0.10 per cent);
— total : not more than 3 times the area of the principal peakin the chromatogram obtained with reference solution (b)(0.3 per cent);
— disregard limit : 0.5 times the area of the principal peakin the chromatogram obtained with reference solution (b)(0.05 per cent).
Phenolic ketones : the absorbance ( 2.2.25 ) is maximum 0.20measured at 385 nm within 15 min of preparing the solution.
Dissolve 1.25 g in a 20 g/l solution of sodium hydroxide Rand dilute to 10.0 ml with the same solvent.
Water ( 2.5.12 ): maximum 4.0 per cent, determined on0.750 g.
ASSAY
Dissolve 0.1000 g in 0.01 M sodium hydroxide and dilute to100.0 ml with the same solvent. Dilute 10.0 ml of the solutionto 100.0 ml with 0.01 M sodium hydroxide. Dilute 10.0 mlof this solution to 100.0 ml with 0.01 M sodium hydroxide.Measure the absorbance ( 2.2.25 ) at the absorption maximumat 308 nm.
Calculate the content of C19H15NaO4 taking the specificabsorbance to be 431.
STORAGEIn an airtight container, protected from light.
IMPURITIES
Specified impurities: B, C.
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary toidentify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : A.
(59) LiChrospher 100 CN is suitable.
152 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 153/176
Warfarin sodium clathrate
A. 3-(2-hydroxyphenyl)-5-phenylcyclohex-2-en-1-one,
B. 4-hydroxycoumarin,
C. benzalacetone.
Reference: PA/PH/Exp. 10B/T (05) 6 ANP
NOTE ON THE MONOGRAPH
It is proposed to replace the TLC test for related substancesby a LC test and to delete the 2 nd identification series. Based on batch results, the limit in the test for water hasbeen increased to 0.3 per cent, which is also in line withthe USP monograph.
XXXX:0699
WARFARIN SODIUM CLATHRATE
Warfarinum natricum clathratum
DEFINITION
Mixture, in the form of a clathrate, of warfarin sodium(sodium2-oxo-3-[(1 RS )-3-oxo-1-phenylbutyl]-2 H -1-benzopyran-4-olate)and propan-2-ol in molecular proportions 2:1 (equivalent toabout 92 per cent of warfarin sodium).
Content :
— warfarin sodium : 98.0 per cent to 102.0 per cent (anhydrous and propan-2-ol-free substance);
— propan-2-ol : 8.0 per cent to 8.5 per cent.
CHARACTERS
Appearance : white or almost white powder.
Solubility : very soluble in water, freely soluble in ethanol(96 per cent), soluble in acetone, very slightly soluble inmethylene chloride.
IDENTIFICATION
First i dentifi cati on: B, D, E .Second i d entificati on: A, C, D, E .A. Dissolve 1 g in 25 ml of water R, add 2 ml of dilut e
hydr ochlori c acid R and filter. Reserve the filtrat e f oridentification test E. The precipitate, washed with water Rand dried at 100 °C to 105 °C, melts ( 2.2.14) at 159 °C
t o 163 °C.B. A. Infrared absorption spectrophotometry ( 2.2.24).
Dissolve 1 g in 25 ml of water R, add 2 ml of dilut ehyd r ochlori c acid R and filter. Reserve the filtrat e f oridentification t est E. Examine the precipitate.
Comparison : the precipitat e prepared in the samemanner from warfarin sodium clathrate CRS .
C. Examine the chromatograms obtained in the test f or related substances. The principal spot in thechromat ogram obtained with test solution (b) issimilar in position and size to the principal spot in thechromat ogram obtained with ref erence solution (b).
D. B. Dissolve 1 g in 10 ml of water R, add 5 ml of nitric
acid R and filter. To the filtrate add 2 ml of potassiumdichromate solution R1 and shake for 5 min. Allow tostand for 20 min. The solution is greenish-blue whencompared with a blank.
E. C. The filtrat e obtained in identification test A It givesreaction (b) of sodium ( 2.3.1).
TESTS
Appearance of solution. The solution is clear ( 2.2.1) andcolourless ( 2.2.2, Method II ).
Dissolve 1.0 g in water R and dilute to 20 ml with the samesolvent.
pH ( 2.2.3): 7.6 to 8.6.Dissolve 1.0 g in carbon dioxide-free water R and dilute to100 ml with the same solvent.Related substances. Examine by thin-layer chromatography( 2.2.27 ), using silic a g el GF 2 54 R as the coating substance.T est soluti on (a). Dissolve 0.20 g of the substance to beexamined in ac et one R and dilut e t o 10 ml with the samesolvent.
T est soluti on (b). Dilute 2 ml of t est solution (a) t o 10 mlwith ac et one R.
Ref er ence soluti on ( a). Dilut e 1 ml of test solution (b) t o200 ml with acet one R.
Ref er ence soluti on ( b). Dissolve 40 mg of warf arin sodium CRS in acet one R and dilut e t o 10 ml with the samesolvent.
Ref er ence soluti on ( c). Dissolve 10 mg of acenoc oumar ol CRS in acet one R, add 1 ml of t est solution (a) and dilut e t o 10 ml with ac et one R.Apply separately to the plat e 20 μl of each solution. Developover a path of 15 cm using a mixture of 20 volumes of glaci al acetic acid R, 50 volumes of methyl ene chl ori de R and50 volumes of cy cl ohexane R. Allow the plat e to dry in airand examine in ultraviolet light at 254 nm. Any spot in thechromat ogram obtained with t est solution (a), apart fromthe principal spot, is not more int ense than the spot in thechromat ogram obtained with ref erence solution (a) (0.1 percent). The test is not valid unless the chromat ogram obtainedwith ref erence solution (c) shows two clearly separat ed spotsand the chromat ogram obtained with ref erence solution (a)
shows a clearly visible spot.Related substances. Liquid chromatography ( 2.2.29).
Solvent mixture : methanol R, water R (25:75 V/V ).
© PHARMEUROPA Vol. 18, No. 1, January 2006 153

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 154/176
Warfarin sodium clathrate
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. impurity B 2. impurity C 3. warfarin 4. impurity A
Figure 0699.-1. – Chromatogram for the test for related substances of warfarin sodium clathrate
Test solution. Dissolve 40.0 mg of the substance to beexamined in the solvent mixture and dilute to 50.0 ml withthe solvent mixture. Reference solution (a). Dissolve 2.0 mg of warfarinimpurity B CRS and 2.0 mg of warfarin impurity C CRS in25 ml of methanol R and dilute to 100.0 ml with water R. Reference solution (b). Dilute 1.0 ml of the test solutionto 100.0 ml with the solvent mixture. Dilute 1.0 ml of thissolution to 10.0 ml with the solvent mixture.Column :— size : l = 0.25 m, Ø = 4.0 mm;— stationary phase : spherical nitrile silica gel for
chromatography R (5 μm)(60) ;— temperature : 30 °C. Mobile phase : glacial acetic acid R, acetonitrile R, water R(1:25:75 V/V/V ). Flow rate : 1.5 ml/min. Detection : spectrophotometer at 260 nm. Injection : 20 μl. Run time : twice the retention time of warfarin. Relative retention with reference to warfarin (retentiontime = about 9 min): impurity B = about 0.4;impurity C = about 0.6.System suitability : reference solution (a):— resolution : minimum 2.0 between the peaks due to
impurities B and C. Limits :— correction factors : for the calculation of content,
multiply the peak areas of the following impurities bythe corresponding correction factor: impurity B = 0.5;impurity C = 0.4;
— impurities B, C : for each impurity, not more than thearea of the principal peak in the chromatogram obtainedwith reference solution (b) (0.1 per cent) ;
— unspecified impurities : for each impurity, not morethan the area of the principal peak in the chromatogramobtained with reference solution (b) (0.10 per cent);
— total : not more than 3 times the area of the principal peakin the chromatogram obtained with reference solution (b)(0.3 per cent);
— disregard limit : 0.5 times the area of the principal peakin the chromatogram obtained with reference solution (b)(0.05 per cent).
Phenolic ketones : the absorbance ( 2.2.25 ) is maximum 0.20measured at 385 nm within 15 min of preparing the solution.
Dissolve 1.25 g in a 20 g/l solution of sodium hydroxide Rand dilute to 10.0 ml with the same solvent.
Propan-2-ol. Gas chromatography ( 2.2.28 ).
Internal standard solution. Dilute 1.0 ml of propanol R to200.0 ml with water R.
Test solution (a). Dissolve 0.250 g of the substance to beexamined in water R and dilute to 5.0 ml with the same
solvent.Test solution (b). Dissolve 0.50 g of the substance to beexamined in the internal standard solution and dilute to10.0 ml with the internal standard solution.
Reference solution . Dilute 0.50 ml of 2-propanol R to100.0 ml with the internal standard solution.
Column :
— size : l = 1.5 m, Ø = 4 mm ;
— stationary phase : ethylvinylbenzene-divinylbenzenecopolymer R (125-150 μm).
Carrier gas : nitrogen for chromatography R.
Flow rate : 40 ml/min.
Temperature :— column : 150 °C;
— injection port : 180 °C;
— detector : 200 °C.
Detection : flame ionisation.
Injection : the chosen volume of the test solutions and thereference solution.
Calculate the content of propan-2-ol taking its density at 20 °C to be 0.785 g/ml.
Limit :
— propan-2-ol : 8.0 per cent to 8.5 per cent.
Water ( 2.5.12 ): maximum 0.1 0.3 per cent, determined on2.500 g.
(60) LiChrospher 100 CN is suitable.
154 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 155/176
2.9.45. Wettability of porous solids including powders
ASSAY
Dissolve 0.1000 g in 0.01 M sodium hydroxide and dilute to100.0 ml with the same solvent. Dilute 10.0 ml of the solutionto 100.0 ml with 0.01 M sodium hydroxide. Dilute 10.0 mlof this solution to 100.0 ml with 0.01 M sodium hydroxide.Measure the absorbance ( 2.2.25 ) at the absorption maximumat 308 nm.
Calculate the content of warfarin sodium (C19H15NaO4)taking the specific absorbance to be 431.
STORAGE
In an airtight container, protected from light.
IMPURITIES
Specified impurities: B, C.
Other detectable impurities (the following substanceswould, if present at a sufficient level, be detected by oneor other of the tests in the monograph. They are limitedby the general acceptance criterion for other/unspecifiedimpurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to
identify these impurities for demonstration of compliance.See also 5.10. Control of impurities in substances for pharmaceutical use) : A.
A. 3-(2-hydroxyphenyl)-5-phenylcyclohex-2-en-1-one,
B. 4-hydroxycoumarin,
C. benzalacetone.
Reference: PA/PH/Exp. POW/T (03) 12 ANP
XXXX:20945
2.9.45. WETTABILITY OF POROUSSOLIDS INCLUDING POWDERS
INTRODUCTION
Three methods for the determination of wettability are
described below. The methods are capable of measuringthe wettability of porous solids like powders or granules.All 3 methods express the wettability by a contact anglemeasurement between the porous solid and a given liquid.
With the Washburn method and the Wilhelmy plate methodthe contact angles are indirectly measured. The methodsare based on the capillary effect of the powder pores. Theeffect (mass gain) is recorded instantly by special electronicbalances starting the moment that the powder sampletouches the surface of the liquid. The measurement hasvery little or no effect on the state of the powder. However,the indirect measurements do not yield the true values of the contact angle. Only apparent values that significantlydepend on the true contact angle are obtained. TheWilhelmy plate method needs the lowest sample mass andis therefore important in pharmaceutical development. TheWashburn method and the Wilhelmy plate method can becarried out on the same type of electronic balance; only thesample holders differ.
The sessile drop method is a simple method, which makesit reasonably easy to measure directly the contact angle of a sessile drop on a compacted powder disc. However, thedrop technique and the analysis of the drop shape are beingfurther developed.
Any pre-treatment of the sample to be examined is
disadvantageous, since the properties may be significantlyaltered. For example, the compression of a powder as a discmay decrease the surface free energy when the crystallisationbehaviour of the powder is changed (metastable forms), ormay increase surface free energy by creating crystal defects.
The wettability of solid surfaces is commonly characterisedby direct or indirect contact angle measurements. Thecontact angle (θ) between a liquid and a solid is the anglenaturally formed when a drop of a liquid is placed on a solidsurface. This is depicted in Figure 2.9.45.-1. Wettable solidsshow a contact angle of less than 90°, non-wettable solidsshow a contact angle of 90° or more.
Figure 2.9.45.-1. – Contact angle ( θ ) of a sessile dropobserved on a non-porous surface
WASHBURN METHOD
INTRODUCTION
The Washburn method is able to measure the contact angle of porous solids with a contact angle in the rangeof 0° to 90°. Usually the method is applied to examine thefollowing parameters :
— batch-to-batch consistency of substances or formulationsreferring to wettability ;
— effect of liquid viscosity on wettability (e.g. reconstitutionof a powder);
— effect of liquid surface tension on wettability;
— alteration of surface properties of formulations (e.g. toprovide more complete and/or rapid wetting; testing of different formulations of a non-wettable active substancein a blend with wettable excipients).
The tested material is the combination of the sample, theholder and the f ilter system. Therefore, an estimation or
determination of the true value is not possible and apparent values of the contact angle are determined instead. However,the contact angle of the sample is the functional property onwhich the result is significantly dependent. The outcome of
© PHARMEUROPA Vol. 18, No. 1, January 2006 155

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 156/176
2.9.45. Wettability of porous solids including powders
the test is a ranking order listing the wettability of different substances or formulations characterised by an apparent contact angle.
PRINCIPLE
If a porous solid is brought into contact with a liquid, suchthat the solid is not submerged in the liquid, but rather is just touching the liquid surface, then the rise of liquid into
the pores of the solid due to capillary action will be governedby the following equations:
(1)
m = mass of liquid sucked into the solid;
t = time elapsed since the solid and the liquid werebrought into contact;
A = constant, dependent on the properties of theliquid and the solid to be examined, calculatedusing the following equation:
(2)
η = viscosity of the liquid;
ρ = density of the liquid;
σ = surface tension of the liquid;
θ = contact angle between the solid and the liquid ;
c = material constant, dependent on the porousarchitecture of the solid.
Combining equations (1) and (2), followed by rearrangement,
leads to equation (3), which is the useful form of Washburn’sequation.
(3)
In setting up a Washburn determination, a liquid with knowndensity (ρ), viscosity (η), and surface tension (σ) is used. Aninspection of equation (3) leads to the conclusion that, if thisis the case, and the mass of liquid that rises into the poroussolid can be monitored as a function of time (such that capillary velocity ( ) is the raw data), then 2 unknownsremain: the contact angle (θ) of the liquid on the solid, and
the solid material constant (c ).Once the material constant (c ) has been determined for thesolid to be examined (see below), a sample of the solid canbe tested for wettability by another liquid. The materialconstant determined by the n-heptane test is used in theWashburn equation, in combination with the capillaryvelocity ( ) data obtained while testing the substanceto be examined in the prescribed liquid. This allows thecalculation of the contact angle.
Determination of the material constant (c ). The materialconstant for a porous solid is theoretically given by thefollowing formula :
(4)
r = average capillary radius within the porous solid;
n = number of capillaries per volumetric unit.
The Washburn equation contains 2 unknowns, the materialconstant (c ) and the contact angle (θ).
However, if a Washburn determination is performedwith a liquid that is known to have a contact angle of 0°
(cos 0° = 1) on the solid, then the solid material constant (c )is the only remaining unknown in equation (3) and canthus be determined. n-Heptane is the liquid of choice fordetermining material constants because of its low surfacetension (20.3 mN·m1 at room temperature). n-Hexane mayalso be used (18.4 mN·m1 at room temperature) but is morevolatile. If the powder dissolves too quickly in these liquids,hexamethyldisiloxane may be used instead (15.9 mN·m1 at 25 °C).
If a series of liquids (at least 2 liquids in addition to the liquidused to determine the material constant) is tested against agiven solid then the resultant contact-angle data can be usedto calculate the surface energy of the porous solid.
APPARATUS
A. electronic balance D. f ilter
B. computer E. immersion liquid
C. sample holder F. lift
Figure 2.9.45.-2. – Apparatus for the contact anglemeasurement by the Washburn method
Figure 2.9.45.-2 shows the principal components of theapparatus. The main device is an electronic balance with asuitable processor ensuring a suitable resolution in forcemeasurement and a suitable resolution in lifting up theimmersion liquid towards the sample.
Table 2.9.45.-1 indicates parameters of the electronic balancethat are generally considered suitable.
Table 2.9.45.-1. – Technical parameters of the electronic balance
Lift Mass measurement
Range > 110 mm 0 - 210 g
Resolution 0.1 μm 10 μg
Speed 0.099-500 mm/min -
156 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 157/176
2.9.45. Wettability of porous solids including powders
Sample holders. The sample holder may be a small glasscylinder with a sintered-glass filter at one end.
Other powder material holders (see Figure 2.9.45-3) aremade of aluminium, are less fragile than glass, with smallholes in the bottom that render them easier to clean than asintered-glass filter. The cover for the cell is equipped with
2 screw threads. One connects it with the sample chamberwhile the other allows the user to guide a piston down ontothe sample itself and compress it. The apparatus is similar toan automatic tensiometer, except for the sample holder.
A. f ixing C. thread E. capillary holes
B. cover D. plunger F. capillary holes
Figure 2.9.45.-3. – Example of sample holder with plunger for compaction of a powder
PROCEDURE
Place the porous solid in an appropriate sample holderand suspend it just above the surface of a test liquid (seeFigure 2.9.45.-2), using the lift.
The liquid is further raised until it just touches the bottom of the porous sample. Mass versus time data is then collectedas liquid penetrates into the solid. At the end of theexperiment data can be output in either graphical or tabularformat. The apparatus may perform the whole determinationautomatically.
Filling of the sample holder. Place a disc of filter paper inthe bottom of the aluminium or glass sample holder. Thisprevents powder from leaking out of the bottom of the cell.
The filter does not have to be paper, but it must be a materialthat is easily wetted by the liquid to be tested. A black-bandfilter (used for reverse osmosis) is recommended because of its high porosity and minimum f low resistance.
Place a known amount of powder into the cell. This amount is big enough so that the compressed powder obtainedduring the last step of this procedure is sufficient. If thisis the case, then reproducibility of material constants andcontact angles will depend almost solely on the ability toweigh out the same amount of powder for each test.
For most powders, a correct amount is in the range of 1-2 g,which normally equals 2/3 of the capacity of the holder.Place a second piece of filter paper on top of the powder inthe cell. This will prevent powder from rising through theholes in the piston during the compression process and/orduring the determination.
Tapping/compaction of the powder. A bulk powder bed isvery porous and thus very sensitive to small influences that can easily alter the porosity. Therefore a tapped powder maybe advantageous and will show more reproducible results.The appropriate number of taps must first be evaluated:50 to 100 taps are usually appropriate.
If the aluminium sample holder is used then it may bemounted in the cylinder of a stamp volumeter, which canrun the evaluated number of taps.
If tapping is not appropriate, the powder bed is compactedby screwing the piston of the aluminium sample holder or bya piston weighted down by a specific mass such as 1 kg.
Where applicable, a compacted disc of the powder sampleis mounted on the electronic balance. A sample holder isomitted in this case.
CRITICAL PARAMETERS The following points must be considered :
— the compaction state of the different powder samplesmust be uniform;
— it may be advisable to sieve the sample (e.g. using a250 μm sieve) before testing;
— the optimal compaction parameters (amount of sample,number of taps or piston mass) must be determined;
— uniformity of the results is improved by using a sampleholder made of aluminium;
— the sample holder or, if used, the glass frit must becarefully cleaned ;
— specifications of the immersion liquid must be indicated.
SESSILE DROP METHOD
This method may be used to directly characterise thewettability of coatings and compacted formulations suchas tablets. Moreover, it is sometimes possible to use thesessile drop instrument in a dynamic measurement (dynamiccontact angle measurement, Figure 2.9.45.-4) of porous
solid/liquid systems where the contact angle becomes lessthan 90°. By taking several contact angle measurements asa function of time, the rate of penetration of a liquid droplet into a slightly porous solid may be studied.
© PHARMEUROPA Vol. 18, No. 1, January 2006 157

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 158/176
Willow bark dry extract
Figure 2.9.45.-4. – Sessile drop determination with visual inspection of the droplet: evolution of the contact angle
over time
PROCEDURE
Since powders are unable to form a completely flat surface,the powder is usually compressed as a disc in an attempt to make the surface smoother. A drop with a given size isplaced on the disc (see Figure 2.9.45.-4.) allowing a direct measurement of the contact angle using a goniometer fittedwith an eyepiece protractor, or by geometric constructionon a photomicrograph. Other physical and mathematicalprocedures of data analysis may also be appropriate. Thedrop size may influence the result.
WILHELMY PLATE METHOD
PRINCIPLE
This method is a dynamic contact angle (DCA) measurement,where an apparent contact angle between a given liquidand a system consisting of plate, adhesive and sample isdetermined according to the following details.
By immersing the plate in a liquid, it is possible to measurethe immersion load as the force ( F 0), using the followingequation:
σ = surface tension of the immersion liquid;
p = effective wet perimeter of the powder-coveredplate;
θ = contact angle;
P = resistance factor for the roughness of the plate,representing the elastic energy released duringwetting as a result of surface defects.
Both the perimeter and the resistance factor are regardedto be independent of the liquid and have to be determinedinitially. A preliminary determination is carried out with aliquid of low surface tension, resulting in perfect wetting,and the contact angle can be assumed to be 0°.
Other measurements in liquids exhibiting finite contact angles (determined using another method such as sessiledrop) on a particular coating material with which the roughsurface is covered must also be carried out.
This coating is used afterwards to measure the contact angleon a smooth plate:
p = perimeter of the smooth plate, equal to thegeometric perimeter.
With this information, the effective wet perimeter and theresistance factor can be calculated. Testing can start.
APPARATUS
The apparatus used for the Wilhelmy plate methodis similar to that used for the Washburn method (seeFigure 2.9.45.-2) except that the sample holder is replacedby a powder-covered glass plate or powder-covered adhesivetape (see Figure 2.9.45.-5).
Figure 2.9.45.-5. – Wilhelmy plate just touching the liquid
CRITICAL PARAMETERS
The Wilhelmy plate method is advantageous since lesspowder is needed and there is no risk of altering the surfaceby compression. Considering the rapid measurement, thesolubility will not disturb the measurement as much as inother techniques. The measurements are dependent on theatmospheric humidity at the time of measuring.
Therefore, the reproducibility can be improved bypre-equilibration of the environment with vapour. By thismethod, it is possible to attain a correct ordering of thesamples at all angles irrespective of whether the contact angle is greater than or less than 90°.
Reference: PA/PH/Exp. 13B/T (04) 21 ANP
XXXX:2312
WILLOW BARK DRY EXTRACT
Salicis corticis extractum siccumDEFINITION
Dry extract produced from Willow bark (1583).
Content : minimum 5.0 per cent of total salicylic derivatives,expressed as salicin (C13H18O7 ; M r 286.3) (dried extract).
PRODUCTION
The extract is produced from the herbal drug using ethanol(60-80 per cent V/V ) by an appropriate procedure.
CHARACTERS
Appearance : yellowish-brown amorphous powder.IDENTIFICATION
Thin-layer chromatography ( 2.2.27 ).
158 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 159/176
Willow bark dry extract
Test solution (a). To 0.200 g of the extract to be examinedadd 5 ml of methanol R. Sonicate for 5 min, filter and diluteto 10 ml with methanol R.
Test solution (b). To 5.0 ml of test solution (a) add 1.0 ml of a 50 g/l solution of anhydrous sodium carbonate R andheat in a water-bath at about 60 °C for 10 min. Cool andfilter if necessary.
Reference solution . Dissolve 2.0 mg of salicin R and 2.0 mgof chlorogenic acid R in 1.0 ml of methanol R.
Plate : TLC silica gel plate R (5-40 μm) [or TLC silica gel plate R (2-10 μm)].
Mobile phase : water R, methanol R, ethyl acetate R(8:15:77 V/V/V ).
Application : 10 μl [or 2 μl] as bands.
Development : over a path of 15 cm [or 6 cm].
Drying : in a current of warm air.
Detection : spray with a mixture of 5 volumes of sulphuric acid R and 95 volumes of methanol R. Heat at 100-105 °Cfor 5 min and examine in daylight.
Results : see below the sequence of the zones present inthe chromatograms obtained with the reference solutionand the test solutions. Furthermore, several reddish-violet zones may be present in the chromatogram obtained withtest solution (a).
Top of the plate
_______ _______
Salicin: a reddish-violet zone
A weak reddish-violet zone (salicin)
A reddish-violet zone(salicin)
_______ _______
Chlorogenic acid: abrown zone
Reference solution Test solution (a) Test solution (b)
TESTS
Loss on drying ( 2.8.17 ): maximum 5.0 per cent.
ASSAY
Liquid chromatography ( 2.2.29).
The following chromatogram is shown for information but will not be published in the European Pharmacopoeia.
1. salicin 2. picein
Figure 2312.-1. – Chromatogram for the assay of willowbark dry extract: test solution
Test solution. To 0.300 g of the extract to be examinedadd 40 ml of methanol R and 40.0 ml of 0.1 M sodiumhydroxide. Heat in a water-bath at about 60 °C under areflux condenser, with frequent shaking, for about 1 h. Aftercooling, add 4.0 ml of 1 M hydrochloric acid . Filter thesuspension into a 100 ml volumetric f lask, then wash anddilute to 100.0 ml with a mixture of equal volumes of water Rand methanol R. Filter through a 0.45 μm membrane filter.
Reference solution . Dissolve 5.0 mg of picein R in 25.0 mlof a mixture of 20 volumes of water R and 80 volumes of methanol R (solution A). Dissolve 15.0 mg of salicin R in25 ml of a mixture of 20 volumes of water R and 80 volumesof methanol R. Add 5.0 ml of solution A and dilute to 50.0 mlwith a mixture of 20 volumes of water R and 80 volumes of methanol R.
Column:
— size : l = 0.10 m, Ø = 4.6 mm;
— stationary phase : octadecylsilyl silica gel for chromatography R (3 μm)(61).
Mobile phase:— mobile phase A : tetrahydrofuran R, a 0.5 per cent V/V
solution of phosphoric acid R (1.8:98.2 V/V ) ;
— mobile phase B : tetrahydrofuran R ;
Time
(min)
Mobile phase A
(per cent V/V )
Mobile phase B
(per cent V/V )
0 - 15 100 0
15 - 17 100 → 90 0 → 10
17 - 23 90 10
23 - 25 90 → 100 10 → 0
25 - 40 100 0
Flow rate : 1.0 ml/min.
Detection : spectrophotometer at 270 nm.
Injection : 10 μl.
System suitability : reference solution:
— retention time : salicin = about 6.4 min;picein = about 7.7 min;
— resolution : minimum 1.5 between the peaks due tosalicin and picein.
Calculate the percentage content of total salicylic derivatives,expressed as salicin, from the following expression:
A1 = area of the peak due to salicin in the
chromatogram obtained with the test solution ; A2
= areaofthepeakduetosalicininthechromatogramobtained with the reference solution;
m1 = mass of the extract to be examined used in thepreparation of the test solution, in milligrams;
m2 = mass of salicin R in the reference solution, inmilligrams;
p = percentage content of salicin in salicin R.
(61) Waters Spherisorb 3 μm ODS2 is suitable.
© PHARMEUROPA Vol. 18, No. 1, January 2006 159

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 160/176
160 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 161/176
Ribwort plantain
Illustrations of Powdered Drugsin Herbal Monographs
It has been decided to progressively introduce illustrations of powdered drugs into herbal monographs in order tocomplement the corresponding identification section (usually Identification B). These drawings will be published
in Pharmeuropa as they become available.
Reference: PA/PH/Exp. 3/T (97) 79 DEF
NOTE ON THE MONOGRAPH
This monograph is currently published in the 5 th Edition.01/2005:1297
CALENDULA FLOWER
Calendulae flos
A. Epidermis of the corolla D. Stigma fragment
B. Epidermis of the corolla at theapex, with anomocytic stomata
E. Covering trichomes
C. Pollen grains F. Glandular trichomes
Figure 1297.-1. — Illustration of powdered herbal drug of calendula f lower (see Identification B)
Reference: PA/PH/Exp. 13B/T (03) 19 DEF
NOTE ON THE MONOGRAPH
This monograph is currently published in the 5 th Edition.01/2005:1884
RIBWORT PLANTAIN
Plantaginis lanceolatae folium
A. Covering trichome of the leaf D. Vein fragment with vessels
B. Lower epidermis of lamina, withglandular trichomes
E. Epidermis of scape
C. Upper epidermis of lamina F. Palisade parenchyma
Figure 1884.-1. — Illustration of powdered herbal drug of ribwort plantain (see Identification B)
© PHARMEUROPA Vol. 18, No. 1, January 2006 161

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 162/176
162 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 163/176
Carmellose
International HarmonisationThis section contains proposals for monographs and general texts, new or revised, elaborated under the international harmonisation procedure (see chapter 5.8 of the European Pharmacopoeia). After these texts have undergone theharmonisation procedure and have been adopted, they will be included in the European Pharmacopoeia and the
pharmacopoeias of the United States and Japan.The draft harmonised texts are published for comment (stage 4: official public enquiry in the forum of each of the three pharmacopoeias). You may send your comments through the appropriate national pharmacopoeia authority at theaddress listed on the back cover page of this issue. Readers whose country is not a signatory state of the European Pharmacopoeia Convention can send their comments directly to the European Directorate for the Quality of Medicines(see address of the EDQM on the cover of this issue). To facilitate the processing of comments received by the Secretariatsof the national authorities and the EDQM please mention in any correspondence the PA/PH reference number at the beginning of each text. If you are requesting a change in the limits or are proposing other methods of analysis, please support your proposal by providing appropriate analytical data obtained on a significant number of samplesand the results of a comparative study between the official method and the proposed method. Comments sent before31 March 2006 will be considered for the preparation of the final version of the harmonised texts.
We wish to emphasise that these draft texts have not yet been adopted by the European Pharmacopoeia Commissionand therefore cannot be considered to be official texts.
It should be noted that for monographs, a version drafted in the European Pharmacopoeia style is usually published at
the same time in the section on Draft monographs and general texts for comments.
Reference: PA/PH/Exp. CEL/T (05) 9 ANP
NOTE ON THE MONOGRAPH
Stage 4
The JP is the coordinating Pharmacopoeia.
Although there is no production of carmellose actually in Europe, it seems that the substance could be of interest for the Pharmaceutical industry in the near future.
XXXX:2360
CARMELLOSE
Carmellosum
DEFINITION
Polycarboxymethylether of cellulose.
CHARACTERS
Appearance : white or almost white powder, hygroscopic.
Solubility : practically insoluble in anhydrous ethanol. It swells with water to form suspension and becomes viscid in1 M sodium hydroxide.
IDENTIFICATION
A. pH ( 2.2.3): 3.5 to 5.0.
Suspend 1.0 g in 100 ml of water R.
B. Infrared absorption spectrophotometry ( 2.2.24).
Preparation : discs.
Comparison : carmellose CRS .
C. Shake 0.1 g with 10 ml of water R, add 2 ml of 1 M sodiumhydroxide and allow to stand for 10 min (solution A).Dilute 1 ml of solution A to 5 ml with water R. To 0.05 mlof this solution add 0.5 ml of chromotropic acid-sulphuric acid concentrated solution - acid solution R and heat ona water bath for 10 min. A red-purple colour develops.
D. Shake 5 ml of solution A (obtained in identificationtest C) with 1 ml of a 90 g/l solution of ferric chloride R.A brown f locculent precipitate is produced.
TESTS
Chlorides : maximum 0.36 per cent.Shake 0.8 g with 50 ml of water R, dissolve in 10 ml of 1 M sodium hydroxide and dilute to 100 ml with water R. Heat on a water-bath a mixture of 10 ml of dilute nitric acid Rand 20 ml of this solution until a flocculent precipitate isproduced. Cool, centrifuge and take out the supernatant liquid. Wash the precipitate with 3 quantities, each of 10 ml, of water R, by centrifuging each time. Combine thesupernatant liquid and the washings and dilute to 100 ml
with water R. To 25 ml of this solution add 6 ml of dilutenitric acid R and dilute to 50 ml with water R (test solution).Prepare the reference solution with 0.40 ml of 0.01 M hydrochloric acid . Add 1 ml of silver nitrate solution R2 tothe test solution and the reference solution. After standingprotected from light for 5 min, any opalescence in the test solution is not more intense than that in the referencesolution.
Sulphates : maximum 0.75 per cent.Shake 0.40 g with 25 ml of water R, dissolve in 5 ml of 1 M sodium hydroxide and add 20 ml of water R. Heat this solution with 2.5 ml of hydrochloric acid R in awater-bath until a flocculent precipitate is produced. Cool,centrifuge, and take out the supernatant liquid. Wash the
precipitate with 3 quantities, each of 10 ml, of water R, bycentrifuging each time. Combine the supernatant liquid andthe washings, and dilute to 100 ml with water R. Filter, anddiscard the first 5 ml of the filtrate. To 25 ml of the filtrateadd 1 ml of dilute hydrochloric acid R and dilute to 50 mlwith water R (test solution). Prepare the reference solutionwith 1.5 ml of 0.005 M sulphuric acid . Add 2 ml of a 120 g/lsolution of barium chloride R to the test solution and thereference solutions. Mix and allow to stand for 10 min. Thewhite turbidy produced in the test solution is not thickerthan that in the reference solution.
Heavy metals : maximum 20 ppm.Place 1.0 g in a quartz or porcelain crucible. Cover looselywith a lid and carbonise by gentle ignition. Cool and add
2 ml of nitric acid R and 5 drops of sulphuric acid R. Heat cautiously until white fumes are no longer evolved andincinerate by ignition at 500-600 °C. Cool and add 2 ml of hydrochloric acid R. Evaporate to dryness on a water-bath.
© PHARMEUROPA Vol. 18, No. 1, January 2006 163

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 164/176
White Petrolatum Jelly
Moisten the residue with 3 drops of hydrochloric acid R,add 10 ml of hot water R and heat for 2 min. Add 1 dropof phenolphthalein solution R1, add dilute ammonia R1dropwise until the solution develops a pale red color. Add2 ml of dilute acetic acid R, filter if necessary, and washwith 10 ml of water R. Transfer the f iltrate and washings toa test tube, and dilute to 50 ml with water R (test solution).Prepare the reference solution as follows: evaporate amixture of 2 ml of nitric acid R, 5 drops of sulphuric acid Rand 2 ml of hydrochloric acid R on a water bath, thenevaporate to dryness on a sand bath. Moisten the residuewith 3 drops of hydrochloric acid R. Proceed as describedfor the test solution, then add 2.0 ml of lead standard solution (10 ppm Pb) R and dilute to 50 ml with water R.
Loss on drying ( 2.2.32 ): maximum 8.0 per cent, determinedon 1.000 g by drying in an oven at 105 °C for 4 h.
Residue on ignition : maximum 1.5 per cent, determinedon 1.000 g.
STORAGE
In an airtight container.
Reagents
Chromotropic acid-sulphuric acid concentrated solution. XXXXXXX.Suspend 0.5 g of chromotropic acid, sodium salt R in 50 mlof sulphuric acid R, centrifuge and use the supernatant liquid. Prepare immediately before use.
Reference: PA/PH/Exp. 11/T (05) 59 ANP
NOTE ON THE MONOGRAPH
Stage 4
The USP is the coordinating pharmacopoeia for thismonograph.
The draft for harmonisation presented below is essentially for information. In the section on monographs for comment of this issue of Pharmeuropa, a revision proposal for the European Pharmacopoeia monograph on white soft paraffin is published. This shows how the stage 4 draft would affect the European monograph. Comments areinvited on the revision proposal (rather than on the USP draft).
XXXX:1799
WHITE PETROLATUM JELLY
White Petrolatum is a purified and wholly or nearlydecolorized mixture of semisolid hydrocarbons obtainedfrom petroleum. It may contain a suitable stabilizer.
Packaging and storage- Preserve in well-closed containers,protected from light.
Labeling- The labeling indicates the name and concentrationof any added stabilizer. Where the labeling indicates theconsistency, determine compliance using Consistency .
Color- Melt about 10 g on a steam bath, and pour 5 mL of the liquid into a clear-glass, 16- × 150-mm bacteriological test tube: the warm, melted liquid is not darker than a solution
made by mixing 1.6 mL of ferric chloride CS and 3.4 mL of water in a similar tube, the comparison of the two beingmade in reflected light against a white background, thetubes being held directly against the background at such anangle that there is no fluorescence.
Identification - Its infrared absorption spectrum, obtainedby spreading a thin film of melted test specimen between
sodium chloride plates, exhibits high-intensity bands between3000 cm1 and 2800 cm1, medium intensity bands between1500 cm1 and 1300 cm1, and low intensity bands between750 cm1 and 700 cm1.
Melting range, Class III <741>: between 38° and 60°.
Consistency-
Apparatus- Determine the consistency of White Petrolatum Jelly by means of a penetrometer fitted with a polishedcone-shaped metal plunger weighing 150 g, having adetachable steel tip of the following dimensions: the tip of the cone has an angle of 30°, the point being truncatedto a diameter of 0.381 ± 0.025 mm, the base of the tip is8.38 ± 0.05 mm in diameter, and the length of the tip is14.94 ± 0.05 mm. The remaining portion of the cone has anangle of 90°, is about 28 mm in height, and has a maximumdiameter at the base of about 65 mm. The containers forthe test are f lat-bottom metal cylinders that are 100 ± 6 mmin diameter and not less than 65 mm in height. They areconstructed of at least 1.6-mm (16-gauge) metal, and areprovided with well-fitting, water-tight covers.
Procedure- Place the required number of containers in anoven, and bring them and a quantity of White Petrolatum Jelly to a temperature of 82 ± 2.5°. Pour the WhitePetrolatum Jelly into one or more of the containers, fillingto within 6 mm of the rim. Cool to 25 ± 2.5° over a periodof not less than 16 hours, protected from drafts. Twohours before the test, place the containers in a water bathat 25 ± 0.5°. If the room temperature is below 23.5° or
above 26.5°, adjust the temperature of the cone to 25 ± 0.5°by placing it in the water bath.
Without disturbing the surface of the substance under test,place the container on the penetrometer table, and lower thecone until the tip just touches the top surface of the test substance at a spot 25 mm to 38 mm from the edge of thecontainer. Adjust the zero setting and quickly release theplunger, then hold it free for 5 seconds. Secure the plunger,and read the total penetration from the scale. Make threeor more trials, each so spaced that there is no overlappingof the areas of penetration. Where the penetration exceeds20 mm, use a separate container of the test substance foreach trial. Read the penetration to the nearest 0.1 mm.Calculate the average of the three or more readings, and
conduct further trials to a total of 10 if the individual resultsdiffer from the average by more than ± 3%: each mm of penetration corresponds to a consistency value of 10.
Acidity or alkalinity- To 10 g add 20 mL of boiling water,and shake vigorously for 1 minute. Allow to cool, and decant.To 10 mL of the aqueous layer add 0.1 mL of a 1 in 10solution in alcohol of phenolphthalein TS. The solution iscolorless. Not more than 0.5 mL of 0.01 N sodium hydroxideis required to change the color of the indicator to red.
Residue on ignition <281>- Heat 10 g in an open porcelainor platinum dish over a Bunsen flame: on ignition it yieldsnot more than 0.05% of residue.
164 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 165/176
Yellow Petrolatum Jelly
Reference: PA/PH/Exp. 11/T (05) 58 ANP
NOTE ON THE MONOGRAPH
Stage 4
The USP is the coordinating pharmacopoeia for thismonograph.
The draft for harmonisation presented below is essentially
for information. In the section on monographs for comment of this issue of Pharmeuropa, a revision proposal for the European Pharmacopoeia monograph on yellow soft paraffin is published. This shows how the stage 4 draft would affect the European monograph. Comments areinvited on the revision proposal (rather than on the USP draft).
XXXX:1554
YELLOW PETROLATUM JELLY
Petrolatum is a purified mixture of saturated semisolid andbleached hydrocarbons obtained from petroleum. It maycontain a suitable stabilizer.
Packaging and storage- Preserve in well-closed containers,protected from light.
Labeling- The labeling indicates the name and concentrationof any added stabilizer. Where the labeling indicates theconsistency, determine compliance using Consistency.
Color- Melt about 10 g on a steam bath, and pour 5 mL of the liquid into a clear-glass, 16- × 150-mm bacteriologicaltest tube: the warm, melted liquid is not darker than asolution made by mixing 3.8 mL of ferric chloride CS and1.2 mL of cobaltous chloride CS in a similar tube, thecomparison of the two being made in reflected light against awhite background, the tubes being held directly against thebackground at such an angle that there is no fluorescence.
Identification - Its infrared absorption spectrum, obtainedby spreading a thin film of melted test specimen betweensodium chloride plates, exhibits high-intensity bands between3000 cm1 and 2800 cm1, medium intensity bands between1500 cm1 and 1300 cm1, and low intensity bands between750 cm1 and 700 cm1.
Melting range, Class III <741>: between 38° and 60°.
Consistency-
Apparatus- Determine the consistency of Yellow Petrolatum Jelly by means of a penetrometer fitted with a polished
cone-shaped metal plunger weighing 150 g, having adetachable steel tip of the following dimensions: the tip of the cone has an angle of 30°, the point being truncatedto a diameter of 0.381 ± 0.025 mm, the base of the tip is8.38 ± 0.05 mm in diameter, and the length of the tip is14.94 ± 0.05 mm. The remaining portion of the cone has anangle of 90°, is about 28 mm in height, and has a maximumdiameter at the base of about 65 mm. The containers forthe test are f lat-bottom metal cylinders that are 100 ± 6 mmin diameter and not less than 65 mm in height. They areconstructed of at least 1.6-mm (16-gauge) metal, and areprovided with well-fitting, water-tight covers.
Procedure- Place the required number of containers in anoven, and bring them and a quantity of Yellow Petrolatum Jelly to a temperature of 82 ± 2.5°. Pour the YellowPetrolatum Jelly into one or more of the containers, fillingto within 6 mm of the rim. Cool to 25 ± 2.5° over a periodof not less than 16 hours, protected from drafts. Two hoursbefore the test, place the containers in a water bath at 25 ± 0.5°. If the room temperature is below 23.5° or above26.5°, adjust the temperature of the cone to 25 ± 0.5° byplacing it in the water bath.
Without disturbing the surface of the substance under test,place the container on the penetrometer table, and lower thecone until the tip just touches the top surface of the test substance at a spot 25 mm to 38 mm from the edge of thecontainer. Adjust the zero setting and quickly release theplunger, then hold it free for 5 seconds. Secure the plunger,and read the total penetration from the scale. Make threeor more trials, each so spaced that there is no overlappingof the areas of penetration. Where the penetration exceeds20 mm, use a separate container of the test substance foreach trial. Read the penetration to the nearest 0.1 mm.Calculate the average of the three or more readings, andconduct further trials to a total of 10 if the individual resultsdiffer from the average by more than ± 3%: each mm of
penetration corresponds to a consistency value of 10.Acidity or alkalinity- To 10 g add 20 mL of boiling water,and shake vigorously for 1 minute. Allow to cool, and decant.To 10 mL of the aqueous layer add 0.1 mL of a 1 in 10solution in alcohol of phenolphthalein TS. The solution iscolorless. Not more than 0.5 mL of 0.01 N sodium hydroxideis required to change the color of the indicator to red.
Residue on ignition <281>- Heat 10 g in an open porcelainor platinum dish over a Bunsen flame: on ignition it yieldsnot more than 0.05% of residue.
© PHARMEUROPA Vol. 18, No. 1, January 2006 165

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 166/176
166 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 167/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 167
International Harmonisation
STATE OF WORK
OF INTERNATIONAL HARMONISATION
(Updated November 2005)
Item
CP Stage
General Methods Relevant to Q6A
Dissolution USP 6
Disintegration USP 6
Uniformity of Content/Mass USP 6
Microbial Contamination EP
Tests for specified microorganism EP 6
Microbial enumeration EP 6
Microbial contamination limits for non-sterile products EP 6
Bacterial Endotoxins (rev. 1) JP 2
Color (instrumental method) EP 2Extractable Volume of Parenterals (rev. 1) EP 6
Test for particulate contamination: subvisible particles (rev. 1) EP 6
Residue on Ignition (rev. 2) JP 6
Sterility test EP 6
General Chapters
Analytical sieving USP 6
Bulk Density and Tapped Density EP 4
Conductivity EP 2
Density of Solids EP 4
Flowability (Powder Flow) USP 6
Tablet Friability USP 6
Heavy Metals USP 3
Inhalation EP 4Optical Microscopy USP 6
Powder Fineness USP 4 rev.
Specific Surface Area EP 6
Porosimetry by Mercury Intrusion EP 4
Laser diffraction measurement of particle size EP 4
X-Ray powder diffraction EP 4
Water-solid interactions EP 3
Thermal behaviour of powders EP 2
Methods for Biotechnology Products
Amino acid determination USP 6
Capillary electrophoresis EP 6
Isoelectric focusing EP 6
Protein determination USP 6
Peptide mapping USP 6
Polyacrylamide Gel Electrophoresis EP 6
Excipients
Alcohol (rev. 2) EP 5B
Dehydrated Alcohol (rev. 2) EP 5B
Benzyl Alcohol (rev. 1) EP 6
Calcium Disodium Edetate JP 6
Calcium Phosphate Dibasic (and anhydrous) JP 6
Carmellose Calcium (rev. 1) USP 6
Carmellose Sodium USP 4
Croscarmellose Sodium USP 6
Microcrystalline Cellulose (rev. 1) USP 6Cellulose, Powdered (rev. 1) USP 6
Cellulose Acetate (rev. 1) USP 6

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 168/176
168 © PHARMEUROPA Vol. 18, No. 1, January 2006
International Harmonisation
Cellulose Acetate Phthalate USP 6
Citric Acid, Anhydrous (rev. 1) EP 6
Citric Acid, monohydrate (rev. 1) EP 6
Crospovidone EP 4
Ethylcellulose EP 6
Hydroxyethylcellulose EP 4-2
Hydroxypropylcellulose USP 4
Hydroxypropylcellulose, Low Substituted USP 4
Hydroxypropylmethylcellulose JP 6
Hydroxypropylmethylcellulose Phthalate USP 5B
Lactose, Anhydrous (rev. 2) USP 5A
Lactose, Monohydrate USP 6
Magnesium Stearate USP 4 rev.
Methylcellulose JP 6
Methyl paraben EP 6
Petrolatum USP 4
Petrolatum, White USP 4
Polyethylene Glycol USP 4
Polysorbate 80 EP 4
Povidone JP 5ASaccharin USP 6
Saccharin, Sodium (rev. 1) USP 6
Saccharin, Calcium USP 6
Silicon Dioxide JP 4 rev.
Silicon Dioxide, Colloidal JP 4 rev.
Sodium Chloride (rev. 2) EP 6
Sodium Starch Glycolate (rev. 1) USP 6
Starch, Corn (rev. 1) USP 6
Starch, Potato EP 6
Starch, Rice EP 5A
Starch, Wheat EP 6
Stearic Acid EP 5A
Sucrose EP 4Talc EP 6
Titanium Dioxide JP 5A2
Ethyl Paraben EP 6
Propyl Paraben EP 6
Butyl Paraben EP 6
Glycerin USP 3
Carmellose JP 4
Calcium carbonate USP 3
Copovidone JP 4
Gelatin EP 2
Glucose monohydrate / anhydrous EP 3
Glyceryl monostearate USP 2
Mannitol EP 3Propylene glycol EP 4
Sodium laurylsulfate USP 3
Starch, pregelatinised JP 2
Water for Injection in Containers USP 2
Stage 1: Identification
Stage 2: Investigation
Stage 3: Proposal for Expert Committee Review
Stage 4: Official Inquiry
Stage 5A: Provisional Consensus
Stage 5B: Draft sign-off Consensus
Stage 6: Regional adoption and implementation
Stage 7: Inter-regional implementation

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 169/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 169
International Harmonisation
OTHER GENERAL CHAPTERS (November 2005)
PROJECTED TIMETABLE FOR PUBLICATION ANDIMPLEMENTATION OF TEXTS SIGNED OFF BY
THE PHARMACOPOEIAL DISCUSSION GROUP (PDG) Effective harmonisation of monographs and general chapters requires that texts signed off by the PDG be published and implemented by all three pharmacopoeias. This process of adoption and implementation takes place according
to the particular procedure of each pharmacopoeia. The table below shows the projected timetable for publication and implementation of all signed-off texts, based on information provided by each pharmacopoeia.
Q6A GENERAL CHAPTERS (November 2005)
Item Sign-off Publication
Stage 6B Stage 7
Regional Implementation Inter-regional Implementation
EP JP USP EP JP USP EP JP USP
Amino acid
determination 2002/9 2003/6 2004/12 2001/12
2005/1 2005/1 2002/1 TBD 2006/4 TBD
Capillary
electrophoresis 2002/9 2003/6 2004/12 2003/7
2005/1 2005/1 2003/8 TBD 2006/4 TBD
Isoelectric focusing 2002/9 2003/6 2004/12 2003/7 2004/1 2005/1 2003/8 TBD 2006/4 TBD
Protein determination 2002/9 2003/6 2004/12 2002/12 2004/1 2005/1 2003/1 TBD 2006/4 TBD
Peptide mapping 2002/9 2003/6 2004/12 2001/12 2004/1 2005/1 2002/1 TBD 2006/4 TBD
Polyacrylamide gel
electrophoresis1999/10 2001/9 2002/12 2001/1 2002/1 2003/1 2001/3 TBD 2006/4 TBD
Tablet Friability 2004/2 2004/12 2006/3 2006/8 2005/7 2006/4 2006/8 2007/1 2006/4 2006/8
Specific Surface Area 2003/11 2004/9 2006/3 2004/7 2005/4 2006/4 2005/4 2007/1 2006/4 2006/5
Analytical Sieving 2004/6 2005/6 2006/3 2004/11 2006/1 2006/4 2005/8 TBD 2006/4 TBD
Powder Flow 2004/6 2005/6 2006/3 2004/11 2006/1 2006/4 2005/8 TBD 2006/4 TBD
Optical Microscopy 2004/6 2005/6 2006/3 2004/11 2006/1 2006/4 2005/8 TBD 2006/4 TBD
Item Sign-off Publication
Stage 6B Stage 7
Regional Implementation Inter-regional Implementation
EP JP USP EP JP USP EP JP USP
Dissolution 2004/6 2005/6 2006/3 2005/3 2006/1 2006/4 2006/4 2007/1 2006/4 2006/4
Disintegration 2004/6 2005/6 2006/3 2005/3 2006/1 2006/4 2006/4 2007/1 2006/4 2006/4
Content
uniformity
2004/2 2004/12 2006/3 2005/1 2005/6 2006/4 2007/1 2007/1 2006/4 2007/1
Extractable
volume of
parenterals
2000/7 2001/6 - 2000/7 2002/1 - 2002/1
Rev. 1 2004/6 2005/6 2005/7` 2005/11 2006/1 2005/7 2006/8 2007/1 2005/7 2006/8
Particulate
matter in
injectables
2001/5 2002/6 - 2001/3 2003/1 - 2002/1
Rev. 1 2004/6 2005/6 2006/3 2005/2 2006/1 2006/4 2005/4 2006/7 2006/4 2006/4
Sterility 2002/10 2003/6 2004/12 2003/5 2004/1 2005/1 2004/1 2006/1 2006/4 2004/1
Microbiological
quality
Bacterial
endotoxins
2000/1 2000/6 2001/3 2000/7 2001/1 2001/4 2001/1 2003/8
Sulphated ash/
Residue on
ignition
2000/2 2001/6 2002/12 2002/11 2002/1 2003/1 -
Rev. 1 2002/9 (2004/12) (2002/11) (2005/1) (2003/8) 2005/4Rev. 2 2004/10 2005/6 2006/3 2005/5 2006/1 2006/4 2006/4 2006/4 2006/4
Colour and
clarity of
solution

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 170/176
170 © PHARMEUROPA Vol. 18, No. 1, January 2006
International Harmonisation
MONOGRAPHS (November 2005)
Item Sign-off Publication
Stage 6B Stage 7
Regional Implementation Inter-regional Implementation
EP JP USP EP JP USP EP JP USP
Alcohol (Rev. 1) 2002/9 2003/6 2006/3 2004/9 2004/1 2006/4 2005/8 2006/8
Alcohol,dehydrated
2002/9 2003/6 2006/3 2004/9 2004/1 2006/4 2005/8 2006/8
Benzyl alcohol 2000/7 2001/10 2004/12 2004/3 2002/4 2005/1 2005/1 2006/8
Carmellose
calcium (Rev. 1)2003/7 2003/9 2004/12 2004/3 2004/4 2005/1 2005/1 2006/8
Calcium
disodium edetate2005/11 2006/10
Calcium
phosphate,
dibasic,
anhydrous
2005/11 2006/10
Calcium
phosphate,
dibasic, dihydrate
2005/11 2006/10
Croscarmellose
sodium 2001/10 2001/9 2006/3 2004/3 2002/4 2006/4 2005/1 2006/8Cellulose acetate
(Rev. 1)2003/2 2003/9 2004/3 2004/4 2005/1 2006/8
Cellulose acetate
phthalate2001/10 2002/6 2004/12 2004/3 2003/1 2005/1 2005/1 2006/8
Cellulose,
microcrystalline2004/2 2004/7 2005/4
Rev. 1 2005/5 2006/6 2006/3 2006/3 2007/1 2006/4 2007/1 2006/8
Cellulose powder 2004/2 2004/7 2005/4
Rev. 1 2005/5 2006/6 2006/3 2006/3 2007/1 2006/4 2007/1 2006/8
Citric acid
anhydrous2001/5 2003/6 2002/12 2004/3 2004/1 2003/1 2005/1
Rev. 1 2003/11 2006/3 2004/9 2006/4 2005/8 2006/8
Citric acid
monohydrate2001/5 2003/6 2002/12 2004/3 2004/1 2003/1 2005/1
Rev .1 2003/11 2006/3 2004/9 2006/4 2005/8 2006/8
Ethylcellulose 2002/2 2002/11 2007/9 2004/3 2003/6 2007/10 2005/1 2006/8
Hypromellose 2003/11 2006/6 2006/3 2006/4 2007/1 2006/4 2007/1 2006/8
Lactose
anhydrous2003/2 2003/6 2006/3 2006/4 2004/1 2006/4 2007/1 2006/8
Rev. 1
Rev. 2
Lactose
monohydrate2002/9 2003/6 2006/3 2006/4 2004/1 2006/4 2007/1 2006/8
Methyl cellulose 2003/11 2006/6 2006/3 2006/4 2007/1 2006/4 2007/4 2006/8
Methyl Paraben 2004/2 2004/2 2006/3 2004/7 2004/2 2006/4 2005/4 2006/8
Saccharin 2003/2 2005/6 2006/3 2004/7 2006/1 2006/4 2006/4
Saccharin
calcium (Rev. 1)2003/2 2004/7 2006/4 2006/8
Saccharinsodium
2003/2 2005/6 2004/7 2006/1 2006/4
Rev. 1 2004/2 2006/3 2006/4 2006/8
Sodium chloride 2001/5 2003/6 2002/12 2004/3 2004/1 2003/1 2005/1
Rev. 1 2001/10
Rev. 2 2003/11 2006/3 2006/4 2006/8
Sodium starch
glycolate2003/11 2004/12 2004/7 2005/7 2006/4 2005/4
Rev. 1 2005/5 2006/6 2007/9 2005/9 2007/1 2007/10 2006/4 2006/8
Starch, corn 2001/10 2002/6 2004/3 2005/1
Rev. 1 2004/2 2004/12 2005/1 2006/8
Starch, potato 2001/10 2002/6 2004/12 2004/3 2005/1 2005/1 2006/8
Starch, wheat 2001/10 2002/6 2004/12 2004/3 2005/1 2005/1 2006/8
Talc 2003/11 2004/9 2007/9 2004/9 2005/4 2007/10 2005/8 2006/8
Ethyl Paraben 2004/2 2004/2 2006/3 2004/7 2004/2 2006/4 2005/4 2006/8
Propyl Paraben 2004/2 2004/2 2006/3 2004/7 2004/2 2006/4 2005/4 2006/8Butyl Paraben 2004/2 2004/2 2006/3 2004/7 2004/2 2006/4 2006/1 2006/8

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 171/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 171
Contents of the JP Forum
JAPANESE PHARMACOPOEIAL FORUM (JP)
Vol. 14, No. 3 September 2005
CONTENTS
Revision Drafts for the JP 15
1. General Rules for Preparations
1. General Notices for Preparations 2. Aerosols 3. Aromatic Waters
4. Capsules 5. Cataplasms /Gel Patches 6. Elixirs 7. Extracts 8. Fluidextracts 9. Granules 10. Infusions and Decoctions 11. Injections 12. Lemonades 13. Liniments 14. Liquids and Solutions 15. Lotions 16. Ointments 17. Ophthalmic Ointments
18. Ophthalmic Solutions 19. Pills 20. Plasters and Pressure Sensitive Adhesives Tapes 21. Powders 22. Spirits 23. Suppositories 24. Suspensions and Emulsions 25. Syrups 26. Tablets 27. Tinctures 28. Transdermal Systems 29. Troches
2. General Tests, Processes and Apparatus
(1) Revision
1.11. Arsenic Limit Test 2.57. Boiling Point and Distilling Range Test 2.60. Melting Point Determination 3.02. Specific Surface Area by Gas Adsorption 3.04. Particle Size Determination 4.02. Microbial Assay for Antibiotics 5.01. Crude Drugs Test 5.02. Microbial Limit Test for Crude Drugs 9. Reference Standards; Reagents, Test
Solutions; Standard Solutions for Volumetric Analysis; Standard Solutions;Matching Fluids for Color; Optical Filters
for Wavelength and Transmission RateCalibration; and Measuring Instruments,
Appliances
9.41. Reagents,Teat Solutions3. Official Monographs
(1) Addition
L-Aspartic Acid Benincasa Seed Celmoleukin (Genetical Recombination) Croscarmellose Sodium Dolichos Seed Eleutherococcus Senticosus Rhizome Faropenem Sodium for Syrup Human Menopausal Gonadotrophin Nelumbo Seed Polygonatum Rhizome
Pullulan Rifampicin Capsules Teceleukin (Genetical Recombination) Teceleukin for Injection (Genetical
Recombination) Verapamil Hydrochloride Tablets
(2) Revision
Achyranthes Root Alisma Rhizome Powdered Alisma Rhizome L-Arginine Hydrochloride Astragalus Root Atractylodes Lancea Rhizome
Powdered Atractylodes Lancea Rhizome Benidipine Hydrochloride Tablets Bupleurum Root Calcium Gluconate Calcium Gluconate Hydrate L-Carbocisteine Cefalotin Sodium Cefditoren Pivoxil Microcrystalline Cellulose Powdered Cellulose Clindamycin Hydrochloride Dexamethasone Dicloxacillin Sodium Enviomycin Sulfate
10% Ephedrine Hydrochloride Powder Erythromycin Ethanol for Disinfection Famotidine Powder Famotidine Tablets Powdered Fennel Flopropione Capsules Ginger Powdered Ginger Glycyrrhiza Powdered Glycyrrhiza Idarubicin Hydrochloride Imipenem Insulin
Insulin Injection Japanese Angelica Root Powdered Japanese Angelica Root
PUBLICATION OF THE LISTS OF CONTENTS
APPEARING IN THE JP FORUM AND USP FORUM
Partners in International Harmonisation

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 172/176
172 © PHARMEUROPA Vol. 18, No. 1, January 2006
Kitasamycin Tartrate Lincomycin Hydrochloride Moutan Bark Powdered Moutan Bark Ophiopogon Tuber Oxytocin Oxytocin Injection
Powdered Peach Kernel Peony Root Powdered Peony Root Phytonadione Pinellia Tuber Pirarubicin Platycodon Root Powdered Platycodon Root Polymixin B Sulfate Poria Sclerotium Powdered Poria Sclerotium Povidone Powdered Rhubarb Pueraria Root
Rehmannia Root Rhubarb Roxithromycin Scutellaria Root Powdered Scutellaria Root Spectinomycin Hydrochloride Talampicillin Hydrochloride Teicoplanin Tranexamic Acid Capsules Tranexamic Acid Tablets Trichosanthes Root Vasopressin Injection Wine Zinostatin Stimalamer
4. General Information
(1) Revision
14. Tablet Friability Test
Additional Revision Drafts for JP 15
1. General Tests, Processes and Apparatus
(1) Revision
14. Disintegration Test
2. Official Monographs
(1) Addition
Amphotericin B Tablets L-Arginine Betamethasone Tablets Cefditoren Pivoxil Tablets Clindamycin Hydrochloride Capsules Faropenem Sodium Tablets Metronidazole Tablets Trimetazidine Hydrochloride Tablets Voglibose Tablets
(2) Revision
Calcium Para-aminosalicylate Granules
Chlorpheniramine Maleate Tablets Norepinephrine Noradrenaline Norepinephrine Injection
International Harmonization
2. Stage 4 (Official Inquiry Stage Draft)
(1) Characterisation of Crystalline Solids by X-rayPowder Diffraction Polyethylene Glycol (XRPD)(by EP)
Contents Pages of Pharmacopeial Forum and Pharmeuropa
Japanese Pharmacopoeia Reference Standards and OfficialNon-pharmacopoeial Reference Standards available fromthe Society of Japanese Pharmacopoeia
- Ordering Information
Contents of the USP Forum
PHARMACOPOEIAL FORUM (USP)
_______________________________________________________________________________
Vol. 31, No. 6 Nov–Dec 2005
CONTENTS*
STANDARDS DEVELOPMENT
HOW TO USE PF
Section DescriptionsCommittee DesignationsStaff Directory
POLICIES AND ANNOUNCEMENTS
USP Seeks Submission of Proposals for StabilityIndicating Assay Procedures for Steroids
Call for High Priority Monographs for Drug Substancesand Products, and Excipients
Pharmacopeial Education Courses Visit the USP Web Site at http://www.usp.orgInternational CorrespondenceHow to Submit Comments
Publication Schedules
SIXTH INTERIM REVISION ANNOUNCEMENT
NOTICE OF OFFICIAL STATUS
Vinorelbine Injection
NOTICE OF POSTPONEMENT
<905> Uniformity of Dosage Units
GENERAL CHAPTERS
<1> Injections
ERRATA LIST FOR USP 28–NF 23
* The USP–NF (USP 29–NF 24), the Supplement (Supp), or the Interim Revision Announcement (IRA) for which the revision proposal is targetedis shown in parentheses next to each proposed item.

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 173/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 173
Contents of the USP Forum
IN-PROCESS REVISION
USP MONOGRAPHS
Amitriptyline Hydrochloride (USP 30)Calcium Lactate (USP 30)Calcium Lactate Tablets (USP 30)Cladribine [ new] (USP 30)
Diphenoxylate Hydrochloride and Atropine Sulfate OralSolution (USP 30)Diphenoxylate Hydrochloride and Atropine Sulfate
Tablets (USP 30)Diphtheria Toxin for Schick Test (USP 30)Ensulizole (USP 30)Estradiol Vaginal Tablets [ new] (USP 30)Synthetic Conjugated Estrogens [ new] (USP 30)Etidronate Disodium (USP 30)Fentanyl [ new] (USP 30)Flumazenil (USP 30)Gemcitabine for Injection (USP 30)Glipizide and Metformin Hydrochloride Tablets [ new]
(USP 30)
Glucagon (Proposal for 2nd
IRA)Goserelin Acetate [ new] (USP 30)Hepatitis B Virus Vaccine Inactivated (USP 30)Sodium Iodide I 123 Capsules (USP 30)Sodium Iodide I 123 Solution (USP 30)Sodium Iodide I 131 Solution (USP 30)Diluted Isosorbide Mononitrate (Proposal for 2nd
IRA)
Ivermectin (USP 30)Levocabastine Hydrochloride [ new] (USP 30)Lindane (USP 30)Mangafodipir Trisodium(USP 30)Mirtazapine (USP 30)Ondansetron Injection (USP 30)Orphenadrine Citrate Injection (USP 30)
Oxybutynin Chloride Extended-Release Tablets [ new](USP30)Prednicarbate Cream [ new] (USP30)Prednicarbate Ointment [ new] (USP 30)Risperidone [ new] (USP 30)Rubella and Mumps Virus Vaccine Live (USP 30)Schick Test Control (USP 30)Talc (USP 30)
EXCIPIENTS
NF MONOGRAPHS
Canola Oil [ new] (NF 25)Ethylcellulose Aqueous Dispersion (NF 25)
Glyceryl Monostearate (NF 25)Oleyl Oleate [ new] (NF 25)Polacrilin Potassium (NF 25)
Polyoxyl 35 Castor Oil (NF 25) Anhydrized Liquid Sorbitol (NF 25)Tetrafluoroethane [ new] (NF 25)
GENERAL CHAPTERS
<11> USP Reference Standards (USP 30)<621> Chromatography (USP 30)<711> Dissolution (Proposal for 2nd IRA) (2nd Supp to
USP 29)
GENERAL INFORMATION CHAPTERS
<1217> Tablet Breaking Force [ new] (USP 30)
REAGENTS, INDICATORS AND SOLUTIONS
Reagent Specifications
Geneticin [ new] (USP 30)Hydroxypropyl-beta-cyclodextrin [ new] (USP 30)IsopropylIodide (USP 30)Sodium Carbonate, Monohydrate [ new] (USP 30)1-Vinyl-2-pyrrolidinone (USP 30)
REFERENCE T ABLES
Container Specifications for Capsules and Tablets(USP 30)
Description and Solubility (USP 30)
PENDING PROPOSALS
CANCELED PROPOSALS
HARMONIZATION
GENERAL INFORMATION CHAPTERS
<1216> Tablet Friability (Proposal for 2nd IRA)
PHARMACOPEIAL PREVIEWS
STIMULI TO THE REVISION PROCESS
Instructions to AuthorsUSP Advisory Panels on the USP Performance Test,
L. Shargel, and T. FosterCritical Quality and Performance Parameters for
Modified-Release Parenteral Dosage Forms, Diane J.Burgess, Brian C. Clark, Mary Joan Hampson-Carlin,and Pankaj Shah
Compendial Calculations: Improving Calculations inUSP–NF, Philip Travis, Kerrie Heck, Deborah Teitz,Luciano Virgili, and Mark Wiggins
Comments on ‘‘Compendial Calculations: Improving theCalculations in USP–NF’’, USP Staff
NOMENCLATURE
INDEX
CORRESPONDENCE
Individuals who wish to correspond with the JP or the USP concerning any of the monographs/articles mentionedcan do so at the following addresses:
Pharmacopeial Forum
The United States PharmacopeiaDivision of Standards Development, USP-NF12601 Twinbrook Parkway
Rockville, Maryland 20852USA
Japanese Pharmacopoeial Forum
Secretariat of the Japanese PharmacopoeiaEvaluation and Licensing DivisionPharmaceutical and Medical Safety Bureau
Ministry of Health and Welfare1-2-2, Kasumigaseki, Chiyoda-kuTokyo, 100-8045, JAPAN
_______________________________________________________________________________

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 174/176
174 © PHARMEUROPA Vol. 18, No. 1, January 2006

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 175/176
© PHARMEUROPA Vol. 18, No. 1, January 2006 175

8/18/2019 PHARMEUROPA 18.1 January 2006
http://slidepdf.com/reader/full/pharmeuropa-181-january-2006 176/176
ADDRESSES OF THE NATIONAL PHARMACOPOEIAAUTHORITIES
Correspondence relating to the draft monographs submitted for comment inthe present issue are to be sent to the relevant Authority.
AUSTRIABundesministerium für Gesundheit und Frauen,PharmazeutischeAngelegenheitenRadetzkystrasse 2A 1030 [email protected]
BELGIUMService public fédéralSanté publiqueDirection générale MédicamentsPlace Victor Horta 40BP 10
B 1060 [email protected]
BOSNIA ANDHERZEGOVINAInstitute for the MedicinesQuality ControlM. Titova, 9P.O. Box 0413BA 71000 [email protected]
FINLANDLääkelaitos Nat. Agency forMedicinesP.O. Box 55FIN 00301 [email protected]
FRANCEAgence française de SécuritéSanitaire des Produits de SantéDirection des Laboratoires etdes ContrôlesUnité Pharmacopée143-147, bld. Anatole FranceF 93285 SAINT-DENIS CEDEXpharmacopeefrancaise@ afssaps.sante.fr
GERMANYBundesinstitut für Arzneimittelund MedizinprodukteGeschäftsstelle der DeutschenArzneibuch-KommissionenKurt-Georg-Kiesinger Allee 3D 53175 [email protected]
LITHUANIAState Medicines Control AgencyMinistry of HealthTraku str. 14LT 01132 [email protected]
LUXEMBOURGMinistère de la SantéDivision de la Pharmacie etdes MédicamentsVilla Louvigny, Allée MarconiL 2120 LUXEMBOURG
jacqueline.genoux-hames@ ms.etat.lu
MALTAMalta Medicines Authority198, Rue D’Argens,Gzira GZR 03MT [email protected]
NETHERLANDSSecretariat of the DutchPharmacopoeia AuthorityLab. for Quality Control of
SPAINReal Farmacopea EspanolaAgencia Espanola deMedicamentos y ProductosSanitariosC/ Alcala 56Office n° 554E 28014 [email protected]
SWEDENSwedish PharmacopoeiaCommissionMedical Products AgencyP.O. Box 26S 751 03 [email protected]
SWITZERLANDSwissmedic, Swiss Agencyfor Therapeutic Products,Pharmacopoeia UnitHallerstrasse 7P. O. BoxCH 3000 BERN [email protected]