Embed Size (px)
Citation preview

P
Sa
b
a
ARRA
KMCEX
1
cnDitrtlssttbfcs
trp
0d
Materials Chemistry and Physics 118 (2009) 118–124
Contents lists available at ScienceDirect
Materials Chemistry and Physics
journa l homepage: www.e lsev ier .com/ locate /matchemphys
hase-transfer and size-dependent film formation of gold nanoparticles
hweta Hegde a, Ridhima Chadha b, Satyawati Joshi a, Tulsi Mukherjee b, Sudhir Kapoor b,∗
Department of Chemistry, Pune University, Pune 411007, IndiaRadiation & Photochemistry Division, Bhabha Atomic Research Centre, Trombay, Mumbai 400085, India
r t i c l e i n f o
rticle history:eceived 27 January 2009eceived in revised form 2 July 2009ccepted 5 July 2009
eywords:etals
hemical synthesis
a b s t r a c t
In this article, prefunctionalized polyoxometalate, �-[SiW10O36(HSC3H6Si)2O]4−, {[POM(SH)2]4−} cappedAu nanoparticles were synthesized successfully in N,N′-dimethylformamide (DMF). The thiol group of[POM(SH)2]4− provides a capping shell around Au nanoparticles. Zeta potential measurements showedthat [POM(SH)2]4− coated nanoparticles were negatively charged in DMF. A novel method for transfer-ring Au nanoparticles from DMF solution into non-polar organic solvent cyclohexane was developed.Charge neutralization using CTAB enables concentrating nanoparticle dispersion in cyclohexane whichis a prerequisite for transfer and for successful three-dimensional self-assembly. The dispersed particles
lectron microscopy-ray diffraction
were stable for at least 45 days. After the phase-transfer, Au nanoparticles could be separated out fromthe cyclohexane phase in the form of a powder. The hydrophobic particles thus prepared are stable andare readily dispersed in solvent like toluene without further surface treatment. We also report extensionof this wet-chemical method to make films by spontaneous assembly of passivated Au nanoparticles atthe DMF/H2O–cyclohexane liquid interface. The particle size information and collective self-assembling
)2]4−
properties of the [POM(SHFTIR spectroscopy.. Introduction
The synthesis and surface modification of metallic nanoparti-les are significant for their utilization as building blocks in futureanodevices from both fundamental and applied perspectives [1].uring the last few decades, scarcely any field of research especially
n physical chemistry has developed as rapidly as that on nanostruc-ured materials. With decreasing particle size the surface to volumeatio increases to such an extent that finally, the material proper-ies are determined by the surface atoms rather than by the classicattice atoms. Apart from such surface effects, nanoparticles ofemiconductors and metals show a drastic change in the electronictructure. This is called sized-quantization effect and it results fromhe three-dimensional confinement of electrons and holes withinhe particles. The optical properties of metal nanoparticles haveeen exploited in the recent past because of their applications as
unctional materials in optical devices [2], near field scanning opti-al microscopy (NSOM) [3–6], surface enhanced Raman scatteringpectroscopy [7–12] and as sensors [13–17].
Phase transfer of metallic particles into other solvents facilitateshe dispersion of particles into different physicochemical envi-onments. The ability to transfer the nanoparticles into differenthysicochemical environment will aid the development of vari-
∗ Corresponding author. Tel.: +91 22 25590298; fax: +91 22 25505151.E-mail address: [email protected] (S. Kapoor).
254-0584/$ – see front matter © 2009 Elsevier B.V. All rights reserved.oi:10.1016/j.matchemphys.2009.07.014
/CTAB capped Au nanoparticles and film were evaluated by TEM, XRD and
© 2009 Elsevier B.V. All rights reserved.
ous applications [1,18]. In particular, the low interfacial energiesobserved in non-polar solvents may enable better control duringsurface and solution processing. Aqueous to organic phase transferof nanoparticles via alkylthiol stabilization was first developed byBrust et al. [19]. Since that time, numerous stabilizing monolayershave been investigated including tetraalkyl ammonium and den-drimer ligands [20,21]. Synthesis methods have also been expandedto direct synthesis of Au nanoparticles in organic solvents [22].
Nanoparticles anchored to surfaces in the form of film may havepotential use in nanodevices [23–28]. In this regard, films of pre-formed nanoparticles can be more advantageous since the oxidizedproduct of the reducing agent may not get transferred along withthe nanoparticles. Generally, for film formation at the interface, thestrategy being adopted is to use two immiscible liquids with metalprecursor in the organic layer and reducing agent in the aqueouslayer [26,28,29]. In another strategy, thiol is used in the CHCl3 layer[30] or ex situ synthesized metal nanoparticles are obtained in theform of films at the air–liquid or liquid–liquid interface [31–33].
In this context, heteropolyanions (polyoxometalates, POMs)have recently received increasing attention as their metal-oxo clus-ters play a role in many areas such as catalysis, biology, medicineand material science [34–36]. POMs are typical inorganic metal
oxide clusters with a wealth of electrical, magnetic, and opti-cal properties. POM of Keggin structure have the general formula(XM12O40)(8−n)−, where, M = W or Mo and X is a heteroatom suchas P, Si, or Ge with n being the valency of X [37]. It is well knownthat Keggin ions undergo stepwise multielectron processes with-
stry a
ocKtccastAOPfnptp
itia�naaismta
2
2
wC(wc
2�
[[S0res
2r
daf
2
abttrfAs
carried out for [POM(SH)2]4− capped gold nanoparticles. The chargewas determined to be −27.8 mV. Transmission electron micrograph(Fig. 1b) of the gold particles shows that the particles are nearlyspherical with a mean diameter of 5 ± 0.2 nm.
S. Hegde et al. / Materials Chemi
ut a structural change [33,38–40]. Recently, Papaconstantinou ando-workers have shown that photochemically reduced POM of theeggin structure, phosphotungstic acid, [(PW12O40)3−] [41] lead tohe formation of the corresponding metal nanoparticles. Sastry ando-workers have used (PW12O40)3− ions for making phase-pureore shell nanoparticles [42]. They have also used (PW12O40)3− astemplate for the in situ growth of metal nanoparticles [43–46],
tar shaped calcium carbonate crystals [47], and CdS [48] nanopar-icles. Recently, Xie and co-workers [49] have also synthesizedg nanoparticles using tungstosilicate acid (H4SiW12O40) solution.wing to their elegant structural and physicochemical properties,OMs are also regarded as one of the best building blocks for theabrication of functional hybrid materials [50]. As most POMs areegatively charged and soluble in water, many hybrid materials,articularly layered film materials, have been fabricated throughhe electrostatic interactions of POMs with positively charged com-ounds [51,52].
In this report, we have attempted to exploit liquid–liquidnterface for phase transfer as well as to cast metal nanopar-icles into a film in situ. For this purpose, we have used twommiscible liquids cyclohexane and N,N′-dimethylformamide. Toccomplish this, we have used prefunctionalized polyoxometalate,-[SiW10O36(HSC3H6Si)2O]4−, {[POM(SH)2]4−} to prepare metalanoparticles. Although, ingenious ways have been developed toffect the phase transfer at the interface, most of the methodsre laden with their own disadvantages. In this regard, a glar-ng lacuna has been the development of one simple nanoparticleurface capping procedure that leads to the stabilization of theetal nanoparticles in both polar and non-polar environment, and
he film formation for the same particles. In this paper, we havettempted to address this issue.
. Materials and methods
.1. Reagents
Glassware was cleaned using chromic acid before being rinsed with distilledater and Millipore purified water, and dried in an oven at 110 ◦C. HAuCl4 (Aldrich),TAB (Aldrich), N, N′-dimethylformamide, cyclohexane, chlorobenzene and tolueneUV spectroscopy grade, Spectrochem, India) were used as received. All solutionsere prepared just before the experiments and kept in dark to avoid any photo-
hemical reactions.
.2. Synthesis of prefunctionalized polyoxometalate,-[SiW10O36(HSC3H6Si)2O]4− , {[POM(SH)2]4−}
The synthesis of the [POM(SH)2]4− was carried out as per the reported method36]. In brief, 0.28 mL (1.33 × 10−3 mol dm−3) of (3-mercaptopropyl)trimethoxysilaneHSC3H6-Si(Ome)]3 is added to a solution of 2 g (6.66 × 10−4 mol dm−3) of K8(�-iW10O36)·12H2O and 0.64 g of Bu4NBr in 40 mL of acetonitrile and 10 mL of water at◦C. Then the mixture is acidified by 1.2 mL of a 12.0 mol dm−3 solution of hydrochlo-
ic acid and the solution is stirred overnight. The compound is obtained aftervaporation of the organic solvent. The material was further characterized usingpectroscopic technique and results were similar to the one reported.
.3. Synthesis of [POM(SH)2]4− capped Au nanoparticles in DMF by chemicaleduction
Briefly, 1.0 × 10−3 mol dm−3 HAuCl4 and 5.0 × 10−4 mol dm−3 [POM(SH)2]4− wasissolved in 50 mL DMF. Sodium borohydride (NaBH4, 5 mg) was then added to thebove solution and shaken vigorously. The reaction flask was kept aside for 90 minor the reaction to complete.
.4. Characterization
Samples for transmission electron microscopy (TEM) were prepared by puttingdrop of the colloidal solution on a copper grid coated with a thin amorphous car-on film. Samples were dried and kept under vacuum in a desiccator before putting
hem in a specimen holder. Characterization of the film was done by gently liftinghem onto a holey carbon grid from the interface. TEM characterization was car-ied out using a Phillips CM 200 electron microscope. Particle sizes were measuredrom the TEM micrographs and calculated by taking average of at least 100 particles.bsorption measurements were carried out on a JascoV-530 spectrophotometer. Thepectra were recorded at room temperature using 1 cm quartz cuvette. Zeta potentialnd Physics 118 (2009) 118–124 119
measurements for determining the charge were carried out using Malvern Instru-ment, zeta sizer (nanosizer). X-ray diffraction patterns were recorded on a PhillipsX-ray diffractometer (PW 1710) with Ni filtered Cu K� radiation. The FTIR mea-surements were performed using a Nicolet Nexus 870 Fourier-transform infraredspectrometer.
3. Results and discussion
3.1. Synthesis and evolution of gold nanoparticles in DMF
[POM(SH)2]4− capped Au nanoparticles were prepared byNaBH4 reduction. The DMF dispersion of [POM(SH)2]4− shows nocharacteristic absorption maximum in the range of 300–800 nm.The AuCl4
− ions in DMF exhibit an absorption peak at 323 nm thatcan be attributed to the metal-to-ligand charge transfer (MLCT)band of AuCl4
− complexes. Upon addition of NaBH4 to the DMFsolution containing [POM(SH)2]4− and HAuCl4, the yellow color ofthe solution gradually disappeared and after a certain time (1 min),the wine red color developed and persisted. The reaction wasallowed to proceed for 90 min during which most of the nucleationhad occurred. Absorption measurement of this solution showed anabsorption band with a maximum at 514 nm, which corresponds toa typical surface plasmon band of gold nanoparticles. Fig. 1a showsthe absorption spectrum for the formation of gold nanoparticlesobtained in DMF.
The as synthesized nanoparticles were stable in solution for atleast 1 month. It is known that [POM(SH)2]4− get adsorbed on Aunanoparticle via thiol group. As [POM(SH)2]4− consists of a neg-atively charged divacant �-[SiW10O36]8− polyanion, hence afteradsorption onto the Au nanoparticles, the surface would bear anegative charge. To confirm this, zeta potential measurements were
Fig. 1. (a) Optical absorption spectrum for the evolution of gold nanoparticles inDMF and (b) transmission electron micrograph of gold nanoparticles in DMF.

120 S. Hegde et al. / Materials Chemistry and Physics 118 (2009) 118–124
Fn3
3
np[cotfwuaatioantmdrnac
(o1wtcb5aa
tpgmcwaoan
Fig. 3. Photographs showing the color changes of the DMF phase and the phase-transfer process. (A) Au nanoparticles in DMF before phase transfer, (B) Au
ig. 2. The change in the absorption spectrum of [POM(SH)2]4− stabilized goldanoparticles at various concentrations of CTAB: (a) 1.6 × 10−2 mol dm−3 and (b).2 × 10−2 mol dm−3.
.2. Phase-transfer experiments
It was observed that DMF is miscible with most of the organicon-polar solvents except hydrocarbons like cyclohexane. Hence,hase-transfer experiments were attempted using DMF containingPOM(SH)2]4− capped Au nanoparticles (1 × 10−3 mol dm−3) andyclohexane only. The mixture was emulsified by stirring vigor-usly at room temperature. After this, the reaction was allowedo proceed. On standing for one hour, the mixture was transformedrom an emulsion into two immiscible liquid layers. The lower layer
as a colloidal dispersion of gold in DMF colored wine red and thepper layer of cyclohexane remained as transparent. The mixingnd prolonged stirring of the metal nanoparticle solution in DMFnd cyclohexane solution did not result in particle transfer acrosshe interface. The metal nanoparticle solution (in DMF) retainedts original color and the cyclohexane layer was completely col-rless. As the exchange between the two liquids could only occurt the interface between DMF and cyclohexane, the unsuccessfulanoparticle transfer was the result of poor contact between thewo phases because of the lack of mutual solubility. It is important to
ention here that no free [POM(SH)2]4− was present in the solutionuring synthesis of Au nanoparticles (vide infra). The other possibleeason could be that the capped Au nanoparticles are hydrophilic inature, which could have prevented the phase transfer. Therefore,ttempt was made to make their surface hydrophobic by adding aationic stabilizer.
For phase-transfer experiments, aliquots of 5 mL of the sol1 × 10−3 mol dm−3) were taken followed by addition of vari-us amounts (concentration) of CTAB. On addition of (0.03 g).6 × 10−2 mol dm−3 CTAB, minor shift in the absorption spectrumas observed. Further increase in the concentration by a factor of
wo led to change in the color of the Au nanoparticles. The wine redolor of [POM(SH)2]4− stabilized gold nanoparticles solution turnedlue. The further increase in the concentration of CTAB (0.1 g i.e..5 × 10−2 mol dm−3) made the solution turbid. The change in thebsorption spectrum of [POM(SH)2]4− stabilized gold nanoparticlest various concentrations of CTAB is shown in Fig. 2.
The above observations can be due to the fact that on addi-ion of CTAB to [POM(SH)2]4− stabilized gold nanoparticles in DMFartially neutralize the large negative charge on the surface of theold nanoparticles and thus making the surface hydrophobic. Thisight be leading to the aggregation of the particles at high con-
entrations of CTAB. Therefore, optimum concentration of CTAB
as selected after determining the zeta potential of the particlest various concentrations of CTAB. This was followed by carryingut phase-transfer experiments. A typical observation was that onddition of 0.06 g of CTAB to 5 mL the [POM(SH)2]4− stabilized goldanoparticles (1 × 10−3 mol dm−3) in DMF, the charge comes down
nanoparticles after addition of CTAB, (C) biphasic mixture of (B) and cyclohexaneand (D) Au nanoparticles in cyclohexane after phase transfer. (For interpretation ofthe references to color in this figure legend, the reader is referred to the web versionof the article.)
from −27.8 to −13.9 mV. To 5 mL of [POM(SH)2]4− stabilized goldnanoparticles in DMF, 0.06 g of CTAB was added first and the solu-tion was shaken vigorously. After this, 5 mL cyclohexane was addedto the solution. The mixture was transformed into two immisci-ble liquid layers. Spontaneous phase transfer of gold nanoparticlesfrom the DMF layer to the cyclohexane layer occurs on little shak-ing of the mixture. A visible transfer of color from the DMF phaseto cyclohexane indicated the extraction of Au metal nanoparticlesfrom DMF to cyclohexane layer. The lower layer of DMF becomescompletely colorless and cyclohexane layer becomes wine red incolor indicating the transfer of Au nanoparticles. The photographicpresentation of this phase-transfer process is depicted in Fig. 3.
The appearance of Au nanoparticles in cyclohexane shows thatthe surface of Au nanoparticles becomes hydrophobic after inter-action with CTAB. Therefore, zeta potential measurements werecarried out for the particles separated in cyclohexane. It was foundthat the zeta potential changes from −13.9 to −0.1 mV. The presenceof Au nanoparticles transferred to cyclohexane layer was confirmedby the UV–vis spectra. The absorption spectrum of Au nanoparticlesafter phase transfer in cyclohexane is shown in Fig. 4a.
It was noticed that the surface plasmon absorption band is slightred shifted to 532 nm after phase transfer. The two-dimensionalassembly of the particles (Fig. 4b) after the phase transfer showsthat the particles are of average size 5 ± 0.2 nm. They are spheri-cal, highly monodisperse and are equally spaced throughout. Oncomparing the results obtained in Fig. 4b with TEM picture of Aunanoparticles before phase transfer (Fig. 1b), it can be seen thatthe transferred particles were highly dense and fully packed. Thiscould be due to the presence of optimum charge at the surface ofthe particles. However, after phase transfer, no significant changein the size of the particles was observed. There exists a possibil-ity that the addition of CTAB displaces the [POM(SH)2]4− from thesurface of the gold particles. It is known that reduction of POM onheating of the DMF solution leads to the development of blue color.To delineate this possibility, DMF solution after the phase trans-fer of gold nanoparticles in cyclohexane was heated. No changein the color of the DMF solution was observed. As no blue colorwas observed on heating the DMF solution, it indicates that allthe [POM(SH)2]4− got transferred with the Au nanoparticles tocyclohexane phase. To further substantiate the above arguments,FTIR experiments were carried out. The presence of a covalent linkbetween the [POM(SH)2]4− and gold nanoparticles can be con-
firmed by studying the vibrational band of the thiol group. Theresults are summarized in Table 1. It can be noted that for boththe samples, that is, before (in DMF) and after phase transfer (i.e. incyclohexane), the IR spectrum showed the same frequency bands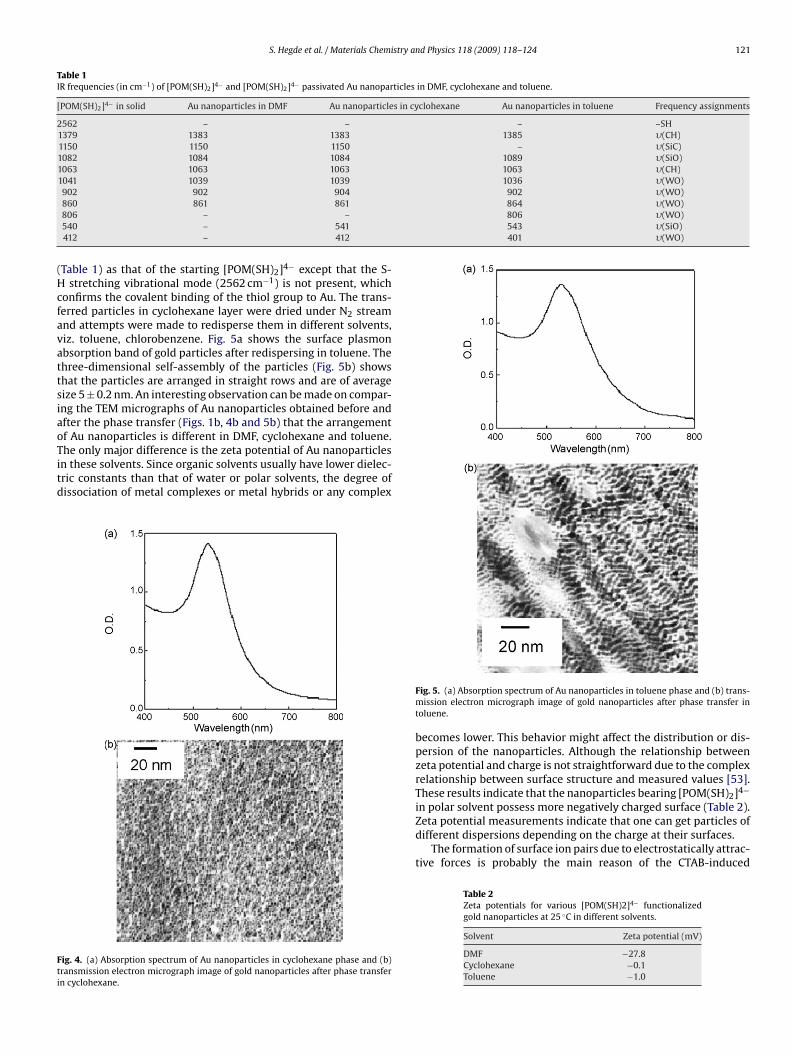
S. Hegde et al. / Materials Chemistry and Physics 118 (2009) 118–124 121
Table 1IR frequencies (in cm−1) of [POM(SH)2]4− and [POM(SH)2]4− passivated Au nanoparticles in DMF, cyclohexane and toluene.
[POM(SH)2]4− in solid Au nanoparticles in DMF Au nanoparticles in cyclohexane Au nanoparticles in toluene Frequency assignments
2562 – – – –SH1379 1383 1383 1385 �(CH)1150 1150 1150 – �(SiC)1082 1084 1084 1089 �(SiO)1063 1063 1063 1063 �(CH)1041 1039 1039 1036 �(WO)902 902 904 902 �(WO)
864 �(WO)806 �(WO)543 �(SiO)401 �(WO)
(HcfavattsiaoTitd
Fti
860 861 861806 – –540 – 541412 – 412
Table 1) as that of the starting [POM(SH)2]4− except that the S-stretching vibrational mode (2562 cm−1) is not present, which
onfirms the covalent binding of the thiol group to Au. The trans-erred particles in cyclohexane layer were dried under N2 streamnd attempts were made to redisperse them in different solvents,iz. toluene, chlorobenzene. Fig. 5a shows the surface plasmonbsorption band of gold particles after redispersing in toluene. Thehree-dimensional self-assembly of the particles (Fig. 5b) showshat the particles are arranged in straight rows and are of averageize 5 ± 0.2 nm. An interesting observation can be made on compar-ng the TEM micrographs of Au nanoparticles obtained before andfter the phase transfer (Figs. 1b, 4b and 5b) that the arrangementf Au nanoparticles is different in DMF, cyclohexane and toluene.
he only major difference is the zeta potential of Au nanoparticlesn these solvents. Since organic solvents usually have lower dielec-ric constants than that of water or polar solvents, the degree ofissociation of metal complexes or metal hybrids or any complex
ig. 4. (a) Absorption spectrum of Au nanoparticles in cyclohexane phase and (b)ransmission electron micrograph image of gold nanoparticles after phase transfern cyclohexane.
Fig. 5. (a) Absorption spectrum of Au nanoparticles in toluene phase and (b) trans-mission electron micrograph image of gold nanoparticles after phase transfer intoluene.
becomes lower. This behavior might affect the distribution or dis-persion of the nanoparticles. Although the relationship betweenzeta potential and charge is not straightforward due to the complexrelationship between surface structure and measured values [53].These results indicate that the nanoparticles bearing [POM(SH)2]4−
in polar solvent possess more negatively charged surface (Table 2).Zeta potential measurements indicate that one can get particles ofdifferent dispersions depending on the charge at their surfaces.
The formation of surface ion pairs due to electrostatically attrac-tive forces is probably the main reason of the CTAB-induced
Table 2Zeta potentials for various [POM(SH)2]4− functionalizedgold nanoparticles at 25 ◦C in different solvents.
Solvent Zeta potential (mV)
DMF −27.8Cyclohexane −0.1Toluene −1.0

122 S. Hegde et al. / Materials Chemistry and Physics 118 (2009) 118–124
F ce bouo had –m partic
pstattttoistfropawhi[uodbis
rtmcaoteoi
3
Df
TAi
S
NCT
ig. 6. Schematic showing the formation of surface ion pair between CTAB and surfaf charges at the surface by one end of POM, which has CTAB group. As the other endeasurement. Addition of CTAB in non-polar solvents makes the shell of gold nano
hase transfer of gold nanoparticles. The mechanism is illustratedchematically in Fig. 6. Zeta potential measurements have shownhat the gold nanoparticle surface is negatively charged. These neg-tively charged surfaces strongly attract CTA+ group. This results inhe formation of ion pairs due to electrostatically attractive interac-ions. Due to the formation of surface ion pairs, the protecting layerurns from hydrophilic to hydrophobic. Addition of CTAB results inhe formation of surface ion pairs at larger distance from the surfacef the gold nanoparticles. This is similar to the changes occurring
n the bilayer (double layer) around the particles. The formation ofurface ion pairs at the other end of the gold nanoparticles makeshem complete hydrophobic from hydrophilic. This drives the trans-er of gold nanoparticles from DMF to cyclohexane phase and theiredispersibility in other non-polar solvents. Interestingly, the sizef the gold nanoparticles is not changing significantly in the ionair formations. That is, surface area to volume ratio is remaininglmost constant. Although the gold nanoparticles are solvophobicith respect to cyclohexane, they can be transferred into cyclo-
exane by complexation with CTAB cations. This process is calledon-pair extraction and can be interpreted by solvophobic theory54]. The observation corroborates the results reported recentlysing TOABr [55]. It is important to mention here that passivationf Ag nanoparticles by thiol-derivative makes them able to be han-led as a simple organic compound, and the “nanocompound” cane precipitated and isolated in solid forms [19,56]. Further stud-
es have shown that they tend to form spontaneously 2D and 3Duperlattices upon solvent removal [57–65].
However, in the present case, the dried particles could not beedispersed in DMF or any other polar solvents. This could be dueo the reasons mentioned above that addition of CTAB molecules
akes the surface of the [POM(SH)2]4− stabilized gold nanoparti-les hydrophobic rendering them insoluble in polar solvents as wells in DMF. It seems that CTAB molecules after charge neutralizationf [POM(SH)2]4− stabilized Au nanoparticles orient their tail groupsowards solution making them ideal for non-polar solvents. Murrayt al. [56] have also suggested that alkane-thiolated coated dry filmf Au, Ag and Pd nanoparticles have highly charged surfaces that
nhibit the inter-digitations of monolayer chains.
.3. Solvent effects on Au surface plasmon absorption
Table 3 gives the refractive index and dielectric constant ofMF, cyclohexane and toluene as well as the position of the sur-
ace plasmon band of [POM(SH)2]4− capped Au nanoparticles in
able 3bsorption maximum of [POM(SH)2]4− capped Au nanoparticles in solvents of vary-
ng refractive indices.
olvent Refractive index (n) Dielectric constant �max (nm)
,N′-dimethylformamide 1.4305 36.7 513yclohexane 1.4266 2.023 531oluene 1.4961 2.379 531
nd anions. The surface of gold nanoparticle is hydrophobic due to the neutralizationSH group that makes the overall surface of gold ionic as observed by zeta potential
les hydrophobic resulting in phase transfer.
these solvents, respectively. It is known that the position of thesurface plasmon band of metal nanoparticles is greatly influencedby the surrounding media [20]. Recently, Malinsky et al. [66] haveinvestigated the structural changes in unmodified Ag nanoparticles.The unmodified nanoparticles were reported to undergo structuralchanges on exposing to various solvents. It can be seen from thetable that [POM(SH)2]4− capped Au nanoparticles shows similarspectrum in cyclohexane and toluene, whereas a shift of 18 nm wasobserved in DMF. There is no significant shift in the absorption max-imum of gold nanoparticles in cyclohexane and toluene which arenon-polar solvents. It is known that chemisorption of anions orinjection of electrons on the surface of Ag nanoparticles leads toa blue shift in the surface plasmon absorption band. Thus, the blueshift in DMF solution could be due to very large negative charge onthe surface of the Au nanoparticles (Table 2). In general, the posi-tion of the plasmon band of particles will change according to therefractive indices if particles can sense changes in the surroundingenvironment, that is, if the solvent infiltrates through the grainsaround the particles [67]. We propose that [POM(SH)2]4− being abulky ligand prevents the solvent from wetting the particles sur-face. This was evident from the FTIR results obtained in differentsolvents (Table 1). This could be the reason for not observing anychange in the absorption maximum with the change in the polarityof the matrix.
3.4. Size dependent film formation of Au nanoparticles at theinterface
It is known that MELLFs (metal liquid like films) are systems ofhighly concentrated metal colloid films separating two immiscibleliquid phases [68]. MELLFs are special cases of interfacial colloids,that is, colloids confined to the interface between two liquids orto the surface of a single liquid phase. Surfactants are known tobe of importance in MELLFs. Surfactants with an anion and cationheadgroups promote the production of MELLFs. It is reported that[POM(SH)2]4− is soluble in organic solvent [36]. Thus, the presenceof water in DMF might affect the stability and/or the size of the Aunanoparticles. Indeed, we had observed the similar phenomenon.Fig. 7 shows the UV–vis absorption spectra of Au nanoparticles pre-pared by borohydride reduction in various ratios of DMF/water. Itwas observed that a red shift of the surface plasmon band occurredwith increasing amounts of water in the solution. Typical TEMimages of Au nanoparticles at 4:1 and 1:1 DMF/water ratios areshown in Fig. 8. It can be noticed that at the ratio of 4:1 DMF/waterratio, the nanoparticles were spherical in shape and approximatelyof the size of 6–8 nm, whereas, particles of about 10–12 nm wereobtained at 1:1 ratio of DMF/water. At 1:1 ratio, the particles were
found to be more aggregated and less monodisperse than at 4:1ratio.As mentioned above, MELLFs formation at the interfacerequires the presence of surfactants of opposite charge. There-fore, to neutralize the charge over the Au nanoparticles, 0.06 g

S. Hegde et al. / Materials Chemistry and Physics 118 (2009) 118–124 123
F3
(sstlattcttwa
Fo
ig. 7. Absorption spectrum of Au nanoparticles obtained in (a) 4:1 DMF/water, (b):1 DMF/water, (c) 2:1 DMF/water and (d) 1:1 DMF/water ratio.
3.2 × 10−2 mol dm−3) of CTAB was added and the solution washaken vigorously. After this, 5 mL cyclohexane was added to theolution. After vigorous stirring and on standing for 6 h, the mix-ure was transformed from a wine red colored emulsion into twoiquid layers. The upper and the lower layer turned transparentnd Au particles gradually accumulated at the interface. On fur-her standing for 3–4 h, the film at the interface turns into a sturdyhree-dimensional membrane-like film and acquires a red opales-
ence. No film formation was observed when CTAB was not addedo the DMF/water solution. Also, it appears that the concentra-ion of CTAB plays a significant role as no film formation was seenhen CTAB concentration was lower than 3.2 × 10−2 mol dm−3. Inddition, it was observed that without Au nanoparticles in the
ig. 8. (a) TEM image of Au nanoparticles at 4:1 DMF/water ratio and (b) TEM imagef Au nanoparticles at 1:1 DMF/water ratio.
Fig. 9. (a) TEM image of an as-prepared film of Au nanoparticles that was obtainedin solution of 4:1 DMF/water containing Au nanoparticles and (b) TEM image of anas-prepared film of Au nanoparticles that was obtained in solution of 1:1 DMF/watercontaining Au nanoparticles.
DMF/water layer, but with CTAB and [POM(SH)2]4− present inDMF/water there is no opalescence observed at the DMF/water-cyclohexane interface. These experiments indicate that the filmformation is entirely dependent on the presence of Au nanopar-ticles, CTAB, [POM(SH)2]4− and concentration of water in DMF.
Formation of the film at the interface can be explained as below.It appears that due to the low solubility of [POM(SH)2]4− in waterand the “push–pull” mechanism particles get accumulated at theinterface. The as-prepared film was quite sturdy and stable. Wenoted that the as-prepared film retain their initial appearance for aminimum of 2 days at the experimental conditions used.
The above reaction mixture was kept for 8 h with the reactantsbefore lifting the film over the Cu grid. The size of the particlesin the film was determined using TEM. In Fig. 9, we show a rep-resentative TEM image of an as-prepared film of Au nanoparticlesthat was obtained in solution of 4:1 and 1:1 DMF/water containingAu nanoparticles. It can be seen that as-prepared film consists ofnanoparticles with diameters in the range of 5–20 nm with a meanof ∼12 nm. Fig. 10 shows the XRD patterns of the films that werelifted over a glass plate. The films show moderate crystalline natureand the 2� values were found to match the ASTM data obtained for
Au metal.The recent work of Reincke et al. [69] discussed the assembly ofcharged Au nanoparticles at the oil–water interface. Reincke et al.infer from their data that the particles assemble at the interface toform a monolayer. It is suggested that changes in interfacial energy,

124 S. Hegde et al. / Materials Chemistry a
F4
etogeAcwtdifaaiatid
4
viifiniwr
R
[
[[
[
[
[[[
[[
[[
[[
[[
[[[[
[
[[
[[[
[
[
[[[[
[
[[[[[[[[[[[[[[[
[
[[[[[
[
[[
[
ig. 10. XRD patterns of interfacial Au films obtained in solutions containing (a):1DMF/water and (b) 1:1: DMF/water.
lectrostatic energy, or van der Waals interactions among the par-icles, water and oil determine if spontaneous film formation willccur at the interface. Though, Reincke et al. work appears to be aeneral method for film synthesis using metal nanoparticles, how-ver, we observe some differences that make our system unique.s mentioned above, no film formation was observed when DMFontaining CTAB/water/POM without Au nanoparticles were mixedith cyclohexane. If POM used as a stabilizing agent is physisorbed
o the Au nanoparticles, it may serve as a transient ligand thatesorbs from the nanoparticles at the DMF–cyclohexane liquid
nterface due to the low solubility. This leads to the reduction in sur-ace charge and allows the film formation. It appears that POM playrole in altering the interfacial energy among passivated particles
nd thus allowing the particle to assemble at the DMF–cyclohexanenterface. It is pertinent to mention here that very recently Feng etl. [70] have proposed a simple method for aqueous–organic phaseransfer of Au, Ag and Pt nanoparticles by decreasing the solubil-ty of PVP. Thus, our results corroborate their findings, albeit in aifferent way.
. Conclusion
The present study shows how the liquid–liquid interface pro-ides a unique way to produce crystalline ultrathin film of gold. Thenterface method described hence is simple and the uniqueness oft is by changing the ingredient one can get either phase transfer orlm formation. In conclusion, a simple protocol for the preparationear-monodisperse gold organosol in the small size regime approx-
mately 6 nm has been developed. We believe that this new protocolill be useful whenever precise size control and monodispersity are
equired.
eferences
[1] G. Schmid, Chem. Rev. 92 (1992) 1709.[2] Y. Dixrix, C. Bastiaansen, W. Caseri, P. Smith, Adv. Mater. 11 (1999) 223.[3] E.J. Sanchez, L. Novotny, X.S. Xie, Phys. Rev. Lett. 82 (1999) 4014.[4] B. Knoll, F. Kellmann, Nature 391 (1999) 134.[5] H.F. Hamann, A. Gallagher, D.J. Nesbitt, Appl. Phys. Lett. 73 (1998) 1469.[6] M.R. Pufalt, A. Berger, S.J. Schultz, Appl. Phys. 81 (1997) 5689.[7] P.J. Tarcha, J. Desaja-Gonzalez, S. Rodriguez-Llorente, R. Aroca, Appl. Spectrosc.
53 (1993) 43.
[8] S. Mie, S.R. Emory, Science 275 (1997) 1102.[9] W.-H. Yang, J. Hulteen, G.C. Schatz, R.P. Van Duyne, J. Chem. Phys. 104 (1996)4313.10] R.G. Freeman, K.C. Grabar, K.J. Allison, R.M. Bright, J.A. Davis, A.P. Guthrie, M.B.
Hommer, M.A. Jackson, P.C. Smith, D.G. Walter, M.J. Natan, Science 267 (1995)1629.
[[
[
nd Physics 118 (2009) 118–124
11] A.C.R. Pipino, R.P. Van Duyne, G.C. Schatz, Phys. Rev. B 53 (1996) 4162.12] N. Biswas, S. Thomas, S. Kapoor, A. Mishra, S. Wategaonkar, S. Venkateswaran,
T. Mukherjee, J. Phys. Chem. A 110 (2006) 1805.13] J.J. Storhoff, R. Elghanian, R.C. Mucic, C.A. Mirkin, R.L. Letsinger, J. Am. Chem.
Soc. 120 (1998) 1959.14] R. Elghanian, J.J. Storhoff, R.C. Mucic, R.L. Letsinger, C.A. Mirkin, Science 277
(1997) 1078.15] U. Kriebig, M. Gartz, A. Hilger, Ber. Bunsenges. Phys. Chem. 101 (1997) 1593.16] R.P. Van Duyne, J.C. Hulteen, D.A. Treichel, J. Chem. Phys. 99 (1993) 2101.17] M.D. Malinsky, K.L. Kelly, G.C. Schatz, R.P. Van Duyne, J. Phys. Chem. B 105 (2001)
2343.18] M.J. Hostetler, J.J. Stokes, M.W. Murray, Langmuir 12 (1996) 3604.19] M. Brust, M. Walker, D. Bethell, D.J. Schiffrin, R. Whyman, J. Chem. Soc. Chem.
Commun. (1994) 801.20] K.S. Mayya, F. Caruso, Langmuir 79 (2003) 6987.21] A.C. Templeton, M.J. Hosteler, E.K. Warmoth, S. Chen, C.M. Hartshorn, V.M. Kris-
namurthy, M.D.E. Forbes, R.W. Murray, J. Am. Chem. Soc. 120 (1998) 4845.22] R.W.J. Scott, O.M. Wilson, R.M. Crooks, J. Phys. Chem. B 109 (2005) 692.23] S. Erokhina, V. Erokhin, C. Nicolini, Langmuir 19 (2003) 766, and references
therein.24] Y. Lin, H. Skaff, A.D. Dinsmore, T.P. Russell, Science 299 (2003) 226.25] P.R. Selvakannan, P.S. Kumar, A.S. More, R.D. Shingte, P. Wadgaonkar, M. Sastry,
Adv. Mater. 76 (2004) 966.26] S.U. Pickering, J. Chem. Soc. 91 (2001) 1907.27] U.K. Gautam, M. Ghosh, C.N.R. Rao, Langmuir 20 (2004) 10776.28] V.V. Agrawal, G.U. Kulkarni, C.N.R. Rao, J. Phys. Chem. B 109 (2005) 7300.29] J.K. Sakata, A.D. Dwoskin, J.L. Vigorita, E.M. Spain, J. Phys. Chem. B 109 (2005)
138.30] A. Kumar, S. Mandal, S.P. Mathew, P.R. Selvakannan, A.B. Mandale, R.V. Chaud-
hari, M. Sastry, Langmuir 18 (2002) 6478.31] J.R. Heath, C.M. Knobler, D.V. Leff, J. Phys. Chem. 107 (1997) 189.32] L.F. Chi, S. Rakers, M. Hartig, H. Fuchs, G. Schmid, Thin Solid Films 327–329
(1998) 520.33] M.T. Pope, A. Muller, Angew. Chem. Int. Ed. 30 (1991) 34.34] T. Yamase, Chem. Rev. 98 (1998) 307.35] T. Yamase, M.T. Pope, Polyoxometalate Chemistry for Nano-composite Design,
Kluwer Academic, Plenum Publishers, New York, 2002.36] C.R. Mayer, S. Neveu, V. Cabuil, Angew. Chem. Int. Ed. 41 (2002) 501, and refer-
ences therein.37] J.E. Huheey, Inorganic Chemistry. Principles of structure and reactivity, 3rd ed.,
Harper and Row, New York, p 698, 1983.38] V. Kogan, Z. Aizenshtat, R. Neumann, Angew. Chem. Int. Ed. 38 (1999) 3331.39] M. Sakamoto, M. Fujistuka, T. Majima, J. Photochem. Photobiol. C 10 (2009) 33.40] B. Keita, T. Liu, L. Nadjo, J. Mater. Chem. 19 (2009) 19.41] A. Troupis, A. Hiskia, E. Papaconstantinou, Angew. Chem. Int. Ed. 41 (2002)
(1911).42] S. Mandal, P.R. Selvakannan, R. Pasricha, M. Sastry, J. Am. Chem. Soc. 125 (2003)
8440.43] S. Mandal, D. Rautaray, M. Sastry, J. Mater. Chem. Soc. 13 (2003) 3002.44] A. Sanyal, S. Mandal, M. Sastry, Adv. Funct. Mater. 15 (2005) 273.45] S. Mandal, A.B. Mandale, M. Sastry, J. Mater. Chem. Soc. 14 (2004) 2868.46] S. Mandal, A. Das, R. Srivastava, M. Sastry, Langmuir 21 (2005) 2408.47] D. Rautaray, S.R. Sainkar, M. Sastry, Langmuir 19 (2003) 10095.48] S. Mandal, D. Rautaray, A. Sanyal, M. Sastry, J. Phys.Chem. B 108 (2004) 7126.49] L. Yang, Y. Shen, A. Xie, B. Zhang, J. Phys. Chem. C 111 (2007) 5300.50] E. Coronado, C. Gomez-Garcoa, J. Chem. Rev. 98 (1998) 273.51] I. Mariguchi, J.H. Fendler, Chem. Mater. 10 (1998) 2205.52] S.Q. Liu, D.G. Kurth, H. Mohwald, D. Volkmer, Adv. Mater. 14 (2002) 225.53] B. Kirby, E.F. Hasselbrink, J. Electrophoresis 25 (2004) 187.54] K. Myabe, S. Taguchi, I. Kasahara, K. Goto, J. Phys. Chem. B 104 (2000) 8481.55] W. Cheng, E. Wang, J. Phys. Chem. B 108 (2004) 24.56] D.E. Cliffel, F.P. Zamborini, S.M. Gross, R.W. Murray, Langmuir 16 (2000) 9699.57] J.E. Martin, J.P. Wilcoxon, J. Odinek, P. Provencio, J. Phys. Chem. B 106 (2002)
971.58] S. He, J. Yao, P. Jiang, D. Shi, H. Zhang, S. Xie, S. Pang, H. Gao, Langmuir 17 (2001)
1571.59] A. Ulman, Chem. Rev. 96 (1996) 1533.60] S.Y. Zhao, S. Wang, K. Kimura, Langmuir 20 (2004) 1977.61] S.A. Harfenist, Z.I. Wang, J. Phys. Chem. B 103 (1999) 4342.62] H. Hiramatsu, F.E. Osterloh, Chem. Mater. 16 (2004) 2509.63] S. Fullam, H. Rensmo, S.N. Rao, D. Fitzmaurice, Chem. Mater. 14 (2002)
3643.64] B.L.V. Prasad, S.I. Stoeva, C.M. Sorensen, K.J. Klabunde, Chem. Mater. 15 (2003)
935.65] M.J. Hosteller, J.J. Stokes, R.W. Murray, Langmuir 12 (1996) 2.66] M. Malinsky, K.L. Kelly, G.C. Schatz, R.P. Van Duyne, J. Am. Chem. Soc. 123 (2001)
1471.67] G. Chumanov, K. Sokolov, B.W. Gregory, T.M. Cotton, J. Phys. Chem. 99 (1995)
9466.68] D. Yogev, S.J. Efrima, J. Phys. Chem. 92 (1988) 5761.69] F. Reincke, S.G. Hickey, W.K. Kegel, D. Vanmaekelbergh, Angew. Chem. Int. Ed.
43 (2004) 458.70] X. Feng, H. Ma, S. Huang, W. Pan, X. Zhang, F. Tian, C. Gao, Y. Cheng, J. Luo, J.
Phys. Chem. B 110 (2006) 12311.





























