Embed Size (px)
Citation preview

American Journal of Medical Genetics 45297-299 (1993)
Phocomelia, Oligodactyly, and Acrania: The Schinzel-Phocomelia Syndrome
David Chitayat, Heater J. Stalker, Michel Vekemans, Danielle Delneste, and E. Michel Azouz Department of Pediatrics, Division of Medical Genetics (D.C., H.J.S.) , Department of Cytogenetics, Division of Pathology (M.V., D.D.), and Department of Radiology ( E . M A .), Montreal Children’s Hospital, and Centre for Human Genetics, McGill University, Montreal, Quebec, Canada
We report on a girl born with phocomelia of both lower limbs, with 3-toed feet and partial sacral agenesis. She had normal growth of the upper limbs and trunk, and normal intel- ligence. Ultrasound study performed during the subsequent pregnancy documented a large skull defect with an intact brain. Fetal autopsy following the termination of that pregnancy was not done. We think this is a further report of the phocomelia syndrome with additional anomalies as reported by Schinzel [Hum Genet 84.539-541, 19901. 0 1993 Wiley-Lisa, Inc.
KEY WORDS: thalidomide embryopathy, Robert-SC syndrome, auto- soma1 recessive
~ ~~~ ~ ~ ~
INTRODUCTION Phocomelia is a rare limb defect reported mainly in
thalidomide embryopathy and in the pseudothalido- mide (Robert-SC) syndrome. In 1990, Schinzel reported on 2 sisters with phocomelia and additional malforma- tions. One of the sisters also had a skull defect. We report on 2 sibs with findings consistent with this newly re- ported syndrome.
CASE REPORTS Patient 1
The proposita, a 4-year-old girl, was born by vaginal delivery at 40 weeks of gestation to a 33-year-old G2P1 mother and a 34-year-old father. Parents were of Pan- amanian origin and were nonconsanguineous. Both were well and their limb and skull films were normal. The mother had a son from a previous marriage who is well. A paternal aunt has cleft palate. The son of this aunt was deaf and had a kidney transplant (Alport syn-
Received for publication November 6, 1991; revision received August 24, 1992.
Address reprint requests to Dr. D. Chitayat, The Hospital for Sick Children, Departments of Pediatrics and Genetics, 555 Uni- versity Avenue, Toronto, Ontario, Canada M5G 1x8.
drome?). In the paternal family there was a maternal uncle who was married to a woman with lower limb contractures. That couple had 2 sons and a daughter with limb deficiencies (not phocomelia); further infor- mation is unavailable. The mother was treated with metoclopramide (antiemetic medication) during the first trimester of pregnancy. Fetal movements first felt at 4-5 months were feeble. Delivery was by a repeat cesarean section. Post delivery the patient was noted to have bilateral lower limb phocomelia with 3-toed feet (Fig. 1). Psychomotor development has been normal and there were no medical complications noted apart from chronic constipation. On physical examination at age 4, her height was 57 cm (<5th centile), weight 11.4 kg ( 4 t h centile), OFC 50.5 cm (mean), and her sitting height 54 cm ( - 2SD). She was hirsute and had a 3 x 2 cm caf6-au-lait spot on the left abdominal wall and a 1 x 1 cm hypopigmented macule on the right abdominal wall. The left lower limb consisted of an extremely short leg and a foot with 3 toes. The right lower limb consisted of a foot with 3 toes.
Skeletal survey documented some scalloping of the posterior wall of the lumbar vertebral bodies. The sac- rum was hypoplastic (Fig. 2). In the left lower limb was only one spherical ossification center measuring 2 cm in diameter, a 3 x 0.8 cm tubular bone possibly represent- ing a rudimentary tibia or a calcaneum, and 3 metatar- sal bones with corresponding phalanges (Fig. 3A). On the right, a 3.5 x 0.9 cm bone was noted proximal to the foot possibly representing a fused, rudimentary tibia and a tarsal bone. The right foot consists of 2 metatar- sals and their corresponding dysplastic phalanges. The third toe contains a faintly ossified phalanx (Fig. 3B). Films of the skull, chest, and upper limbs were normal. Ultrasonography demonstrated normal kidneys, with the bladder deviated to the right and a normal right ovary. The left ovary and the uterus could not be identi- fied, but there was no pelvic mass or cyst. Chromosomes were normal. Centromere separation and/or chromo- some puffing were not observed.
Patient 2 The pregnancy was uncomplicated and the mother
denied any exposure to medications or teratogens. Fetal ultrasound study at 4 months showed fetal acrania
0 1993 Wiley-Liss, Inc.
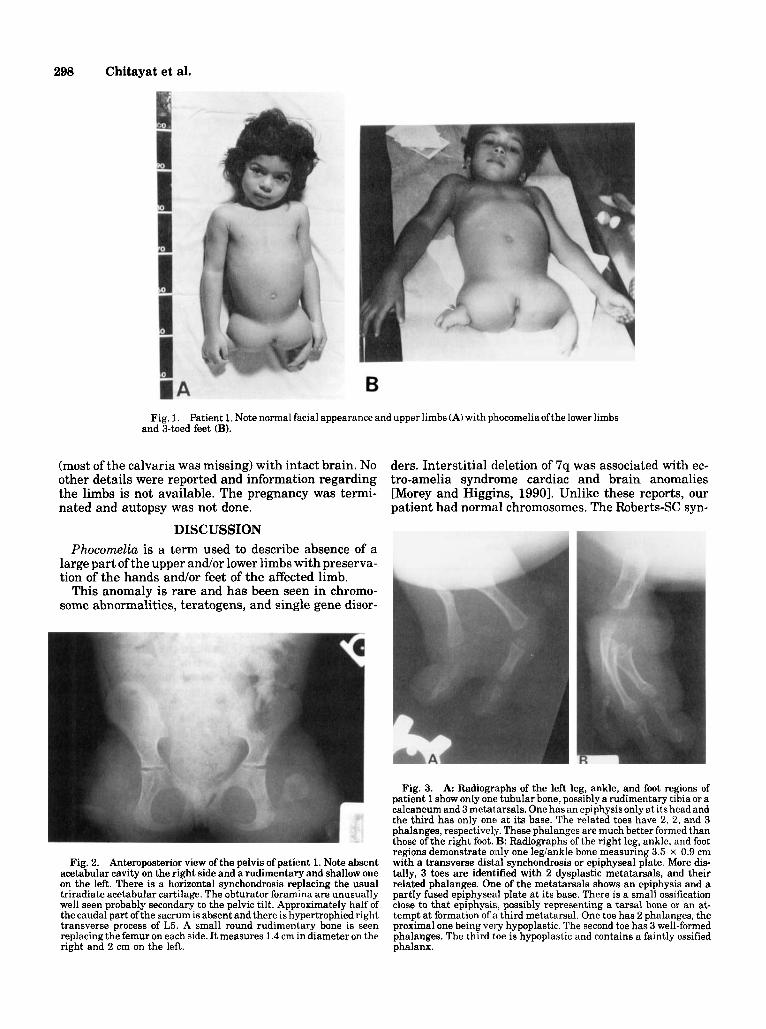
298 Chitayat et al.
Fig. 1. Patient 1. Note normal facial appearance and upper limbs (A) with phocomeliaofthe lower limbs and %toed feet (B).
(most of the calvaria was missing) with intact brain. No ders. Interstitial deletion of 7q was associated with ec- other details were reported and information regarding tro-amelia syndrome cardiac and brain anomalies the limbs is not available. The pregnancy was termi- [Morey and Higgins, 19901. Unlike these reports, our nated and autopsy was not done. patient had normal chromosomes. The Roberts-SC syn-
DISCUSSION Phocomelia is a term used to describe absence of a
large part of the upper and/or lower limbs with preserva- tion of the hands andlor feet of the affected limb.
This anomaly is rare and has been seen in chromo- some abnormalities, teratogens, and single gene disor-
Fig. 2. Anteroposterior view of the pelvis of patient 1. Note absent acetabular cavity on the right side and a rudimentary and shallow one on the left. There is a horizontal synchondrosis replacing the usual triradiate acetabular cartilage. The obturator foramina are unusually well seen probably secondary to the pelvic tilt. Approximately half of the caudal part ofthe sacrum is absent and there is hypertrophied right transverse process of L5. A small round rudimentary bone is seen replacing the femur on each side. It measures 1.4 cm in diameter on the right and 2 cm on the left.
Fig. 3. A: Radiographs of the left leg, ankle, and foot regions of patient 1 show only one tubular bone, possibly a rudimentary tibia or a calcaneum and 3 metatarsals. One has an epiphysis only at its head and the third has only one at its base. The related toes have 2, 2, and 3 phalanges, respectively. These phalanges are much better formed than those of the right foot. B: Radiographs of the right leg, ankle, and foot regions demonstrate only one legiankle bone measuring 3.5 x 0.9 cm with a transverse distal synchondrosis or epiphyseal plate. More dis- tally, 3 toes are identified with 2 dysplastic metatarsals, and their related phalanges. One of the metatarsals shows an epiphysis and a partly fused epiphysed plate at its base. There is a small ossification close to that epiphysis, possibly representing a tarsal bone or an at- tempt at formation of a third metatarsal. One toe has 2 phalanges, the proximal one being very hypoplastic. The second toe has 3 well-formed phalanges. The third toe is hypoplastic and contains a faintly ossified phalanx.

Phocomelia 299
drome is an autosomal recessive disorder associated with phocomelia [Qazi et al., 19791. However, unlike the child reported here, patients with this syndrome also have abnormalities of the upper limbs, cleft lip and palate, and mental retardation [Riimke et al., 19871. Premature separation of the centromere is a cytogenetic marker for this syndrome. However, Zimmer et al. [19851 reported 6 male fetuses born to consanguineous Arab Moslem parents with phocomelia and craniofacial abnormalities consistent with the Roberts-SC syndrome without the characteristic chromosome abnormality.
The DK phocomelia syndrome [Cherstovy et al., 19801 is another autosomal recessive disorder that is associ- ated with phocomelia and oligodactyly. However, unlike our patient this syndrome is associated with thrombocy- topenia, renal, and brain malformations. The CHILD syndrome may also present with unilateral phocomelia; however, our patient did not have other findings charac- teristic of this syndrome, including brain and cardiac malformations [Happle et al., 19801. Hecht and Scott [19811 reported on half-sibs with limb deficiencies. The first sib had a severe cardiac abnormality, absence of the hands, the right foot, and the distal part of the left leg. The other child had absence of the right hand and fore- arm, total syndactyly of 2 digits with a common nail, shortness and bowing of both tibiae, and absent left fibula. The feet contained only 4 metatarsals and, un- like the first sib, cardiac anomalies were absent. The mother was unaffected. The limb defects in our patient are different from those described in this syndrome. Moreover, neither of these reported patients had acrania or skull defects.
McKusick reported on 2 males from an inbred Amish pedigree with left focal femoral deficiency, congenital cataracts, and scoliosis [McKusick et al., 19681. How- ever, unlike our patient, both sibs in this report had radiological findings of spondyloepiphysial dysplasia.
Ohdo et al. [19871 reported a brother and a sister, the offspring of consanguineous parents, with tetra-amelia, minor dysmorphic facial anomalies with a large mouth, and complete absence of the scalp hair and eyebrows. The facial changes are inconsistent with those of our patient.
Rosenak et al. [1991] reported on 3 sibs born to a nonconsanguineous Moslem couple with tetra-amelia, pulmonary hypoplasia, and multiple congenital malfor- mations. Our patient did not have pulmonary hypo- plasia and only her lower limbs were affected.
In 1990, Schinzel reported on 2 sisters with lower limb phocomelia with 4-toed feet born to nonconsanguineous normal parents. The older sister also had phocomelia of the left upper limb with 5 finger rays and multiple congenital abnormalities including: diaphragmatic her- nia and agenesis of the gallbladder with normal female genitalia. The younger sister had agenesis ofthe uterus, vaginal atresia, sacral hypoplasia, and dysplasia of the pelvic bones. She also had an occipital skull defect. The radiological findings in our patient 1 resemble the find- ings in the second patient reported by Schinzel [19901, including the normal facial structure and mental devel- opment. Moreover, patient 2 in our report had acrania which was found in the other patient in Schinzel’s report [19901. Unfortunately, fetal autopsy was not done and further details on patient 2 are missing.
REFERENCES Cherstovy E, Lazjuk G, Lurie I, Ostrovskaya T, Shved I (1980): Syn-
drome of multiple congenital malformations including phocomelia, thrombocytopenia, encephalocele, and urogenital abnormalities. Lancet 2:485 only.
Happle R, Koch H, Lenz W (1980): The CHILD syndrome: Congenital hemidysplasia with ichthyosiform erythroderma and limb defects. Eur J Pediatr 13427-33.
Hecht JT, Scott C Jr (1981): Limb deficiency syndrome in half sibs. Clin Genet 20:432-437.
McKusick VA, Weilbaecher RG, Gragg GW (1968): Recessive inheri- tance of a congenital malformation syndrome: Unilateral absence deformity of leg and congenital cataracts. JAMA 204:lll-118.
Morey MA, Higgins RR (1990): Ectro-amelia syndrome associated with an interstitial deletion of 7q. Am J Med Genet 35:95-99.
Ohdo S, Madokoro H, Sonoda T, Takei M, Yasuda H, Mori N (1987): Association of tetra-amelia, ectodermal dysplasia, hypoplastic lac- rimal ducts and sacs opening towards the exterior, peculiar face and developmental retardation. J Med Genet 24:609-612.
Qazi QH, Kassner EG, Masakawa A, Madahar C, Choi SJ (1979): The Sc phocomelia syndrome: Report of two cases with cytogenetic ab- normality. Am J Med Genet 4:231-238.
RGmke C, F’roster-Iskenius U, Heyne K, Hohn W, Hof M, Gnejszczyk G, Rauskolb R, Rehder H, Schwinger E (1987): Robert syndrome and SC phocomelia. A single genetic entity. Clin Genet 31:170-177.
Rosenak D, Ariel I, Arnon J , Diamant YZ, Chetrit AB, Nadjari M, Zilberman R, Yaffe H, Cohen T, Ornoy A (1991): Recurrent tetra- amelia and pulmonary hypoplasia with multiple malformations in sibs. Am J Med Genet 38:25-28.
Schinzel A (1990): Phocomelia and additional anomalies in two sisters. Hum Genet 84:539-541.
Zimmer EZ, Taub E, Sova Y, Divon MY, Pery M, Peretz BA (1985): Tetra-amelia with multiple malformations in six male fetuses of one kindred. Eur J Paediatr 144412-414.





























