Embed Size (px)
Citation preview

Acta Neurol. Scand., 1988:77:461-467
Key words: lysosomes; muscles; muscular dystrophy; neuromuscular diseases.
Phosphomannosyl receptors of lysosomal enzymes of skeletal muscle in neuromuscular diseases
A. Salminen', V. Marjornbki', U. Tolonenz, V. V. MyllylBz Departments of 'Cell Biology, University of Jyvaskylb, and 2Neurology, University of Oulu, Finland
ABSTRACT - The phosphomannosyl receptor system is responsible for both the receptor-mediated endocytosis and the intracellular transport of lysosomal enzymes. In the present study this receptor system was examined in affected muscles of patients with various neuromuscular diseses. The total activity of B-N-acetyl- glucosaminidase, a marker enzyme of lysosomal hydrolases, was significantly ele- vated in the patients with myopathies (polymyositis and muscular dystrophies) but only slightly increased in those with neurogenic muscle atrophies (amyotrophic lateral sclerosis, polyneuropathy or other neurogenic muscle disease). The increase was most prominent in the group of polymyositis. The content of phosphomannosyl receptors was increased in the patients with myogenic muscle disease but not in those with neurogenic disease. The receptor binding of lysosomal enzymes was saturable and inhibited with mannose 6-phosphate showing the typical characteristics of phosphomannosyl receptors. The characteristics of the receptors were very similar both to control and to diseased muscle samples. When surveying all the material, the content of phosphomannosyl receptors correlated highly significantly with the mus- cular activity of R-N-acetylglucosaminidase, muscle atrophy index, and serum creatine kinase activity.
Accepted for publication December 8, 1987
The increase of lysosomal enzyme activities (1-3) and the decrease in the membrane-linked latency of these enzyme activities (4) are typical observations in dystrophic muscles. A great part of these changes is due to invaded inflam- matory phagocytes which are a rich source of lysosomal enzymes. However, the activities of lysosomal enzymes and the number of auto- phagic vacuoles are also increased in surviving muscle fibers ( 5 , 6 ) as well as abnormal charac- teristics, such as decreased membrane-linked latencies of lysosomal enzymes and appearance of lysosomal storage, are also described in mutant fibroblasts (7, 8).
Lysosomal enzymes are transported in intra-
cellular membranes by the means of phos- phomannosyl receptors (9). Phosphomannosyl receptors also mediate the endocytic uptake of lysosomal enzymes. Recent studies (10-12) have shown the presence of phosphomannosyl recep- tors in skeletal muscle membranes. For instance, Reuser et al. (11) have shown that the lysosomal enzyme uptake by cultured human myotubes from culture medium is mediated by mannose- 6-phosphate-dependent receptors and that the enzyme uptake can completely correct the lysosomal storage disease. However, Thesleff et al. (13) have shown that the micropinocytic uptake of extracellular macromolecules, mainly by the transverse tubular membranes, is promi-

462 SALMINEN ET AL
nently increased in dytrophic muscle fibers in association with the activation of lysosomal enzymes and the degeneration of muscle fibers. Christie and Stoward (14) have shown that in dystrophic muscle fibers of hamster there are tubuli which contain a strong activity of acid phosphatase. Thus, it was interesting to study the contents and some characteristics of phos- phomannosyl receptors in neuromuscular dis- eases which in our earlier studies (15, 3) have shown increased lysosomal enzyme acitivities in skeletal muscles.
Material and methods
Patients Muscle biopsies and serum samples were obtain- ed from patients for diagnostic purposes. All muscle samples were obtained with the permis- sion of the patient in accord with the principles of the Declaration on Helsinki. The patients, aged 18-77 years (27 M, 1 1 F), comprised the following diagnostic groups: 4 with polymyositis (mean age & S.D., 64 & 22 years); 4 with muscu- lar dystrophies (age 32 ? 10 years); 1 with Duch- enne dystrophy, 7 with amyotrophic lateral scle- rosis (ALS) (age 57 & 11 years); 7 with po- lyneuropathy (age 57 f 18 years), 5 with other neurogenic muscle diseases (age 43 f 17 years), and 1 1 as controls (age 41 & 8 years). The con- trols had no muscle disease that could be verified by laboratory tests for serum enzymes, by clini- cal neurophysiologic examination, or by the his- tologic investigation of muscle biopsy specimens. The diagnosis of the neuromuscular disease in each patient was based on clinical examination, laboratory tests for enzymes characteristic of neuromuscular and systemic disease (i.e. creatine kinase, lactic dehydrogenase and aldolase), elec- troneuromyography, and histological examina- tion of the muscle biopsy specimen. The magni- tude of atrophy in the biopsied muscle (scale of 0-3; 0 indicates no atrophy; 1 mild atrophy; 2 moderate atrophy; and 3 severe atrophy) was evaluated by a clinical neurophysiologist. In all the cases muscle atrophies were verified in more than one limb.
Biopsies The muscle biopsies from the patients with neu- romuscular disorders were taken from affected muscles i.e., 25: tibialis anterior, 2: vastus later- a h or 1 deltoideus, and the biopsies from con- trols: 10 tibialis anterior and 1 vastus lateralis. The values of vastus lateralis and deltoideus did not differ from those of tibialis anterior in any group. The muscle samples were taken with a Weil-Blakeslay conchotome under local 1 % lido- caine-epinephrine anesthesia. The specimen was washed with cold 0.9% sodium chloride solu- tion, and stored at -80°C until assayed.
Assays The assays of the content of phosphomannosyl- enzyme receptors were adapted from our earlier studies (12). The frozen muscle biopsy specimens were washed to remove blood and homogenized in a Potter-Elvehjem homogenizer in ice-cold buffer (0.25 M sucrose, 5 mM EDTA, and 25 mM Tris-HC1, pH 7.0). Homogenates were made to a concentration of 3% (w/v). The homoge- nate was centrifuged at 35,000 g-av ( 5 T ) for 25 min (Sorvall, RC-25, Du Pont). The pellet was resuspended in 0.5% saponin and incubated for 30 min in an ice-bath to open the membrane vesicles (10). After incubation the fraction was centrifuged at 35,000 g-av for 25 min and the pellet was washed once with homogenization buffer. The pellet was resuspended in the homog- enization buffer and stored at -20°C for a few days.
High-uptake lysosomal enzymes with a recog- nition marker were produced by cultured fibro- blasts separated from rat embryos with trypsin digestion. Cultured fibroblasts were stimulated to secrete lysosomal enzymes by maintaining them overnight in serum-free Dulbecco medium (KC Biologicals) containing 1 mg/ml albumin and 50 mM NH,Cl (10). For partial purification (10, 12), the pooled media were first filtered by vacuum on a Buchner funnel (Whatman No 40 paper) and then through a membrane filter (Gel- man, 0.45 pm) and concentrated by ultrafiltra- tion using Diaflo PM 30 membranes (Amicon). The concentrated sample was applied to a

LYSOSOMAL RECEPTORS IN MUSCULAR DISEASES 463
DEAE-Sephadex A-25 column. After that the enzyme preparation was dialyzed overnight against 10 mM sodium phosphate and 10 mM Tris-HC1, pH 7.2, and stored at -20°C.
The binding assays of phosphomannosyl- enzyme receptors were carried out with respect to R-N-acetylglucosaminidase (EC 3.2.1.30). The binding assays were adapted from the methods of Fischer et al. (10) and Salminen (12) after testing that enzyme binding was linear in relation to the amount of fraction added. The activity of R-N-acetylglucosaminidase in the enzyme prepa- ration used in the binding assays was 140 pkat/ 100 pl.
Correspondingly, the amount of fraction pro- tein used in the assay was 63.2 +- 14.2 pg (+ SD). The standard binding assay contained 200 p1 muscle fraction, 100 pl enzyme preparation, and 100 pl homogenization buffer or mannose 6-phosphate (50 mM in the final volume). Man- nose 6-phosphate is the specific inhibitor of phosphomannosyl receptor binding (10). The enzyme binding was carried out in ice-bath for 60 min. After incubation 3 ml ice-cold homogeniza- tion buffer was added and the tubes were cen- trifuged at 35,000 g-av for 25 min. The pellet was washed with homogenization buffer and cen- trifuged at 35,000 g-av. The activity of R-N- acetylglucosaminidase (EC 3.2.1.30) and the pro- tein content were assayed from the suspended pellets as described earlier (16).
Standard procedures were used to calculate means, standard deviations and errors. Statistical significances between groups were tested by one way analysis of variance and by the modified Student’s t-test where the confidence limit was determined by the Bonferroni method. The cor- relations between biochemical variables were cal- culated with Pearson’s least-square method. Atrophy index and biochemical findings were correlated with the non-parametric Spearman’s rank correlation.
skeletal muscles of patients with myogenic mus- cle disease but only a slight increake ( P < 0.05) was found in those with neurogenic muscle disea- se (Table 1). The increase was most prominent in the groups of polymyositis (P < 0.001) and muscular dystrophies (P < 0.01) (Fig. 1).
The specific binding capacity of phosphoman- nosy1 receptors was significantly (P < 0.05) ele- vated in the myogenic muscle disease but not in neurogenic diseases (Table 1). The increase was most prominent in the group of muscular dystro- phies (P < 0.05) (Fig. 1).
No differences occurred between males and females in any of the diagnostic groups, further- more, there were no relationships between the biochemical parameters and age within the con- trol group or in any other group (data not shown).
Correlations between variables The total activity of B-N-acetylglucosaminidase showed a highly significant positive correlation (r = 0.649) with the specific binding capacity of phosphomannosyl receptors in skeletal muscle samples (Fig. 2). There also occurred highly sig- nificant correlations between the creatine kinase activity in serum and phosphomannosyl receptor binding capacities (r = 0.547) and R-N-acety- lglucosaminidase activities (r = 0.830) in muscle samples, respectively. The atrophy index of skel- etal muscle samples correlated highly signifi- cantly (P < 0.001) both with the activities of creatine kinase in serum and R-N-acetylglu-
Table 1 Total activity of R-N-acetylglucosaminidase and specific bin- ding capacity of phosphomannosyl receptors in myogenic and neurogenic muscle diseases.
Group (n) R-N-acetyl- Phosphomannosyl glucosaminidase receptors
Control (11) 11.4 f 0.8 42.6 -C 2.7 Myogenic disease (8) 36.6 f 6.4.’ 59.6 f 6.7* Neurogenic disease (19) 16.0 f 1.1; 47.5 f 2.8
Biochemical observations The total activity of O-N-acetylglucosaminidase was significantly (P <: 0.001) elevated in the
___
Enzyme activities (p kat X kg-1 prot.) and specific enzyme binding of phosphomannosyl receptors (p kat x kg-1 prot.) are means f S.E. Significances (compared to controls): P c 0.05
** P c 0.001

464 SALMINEN ET AL
Total activity (pkat
0 :: 68.2
6 0 .
50-
- 40- 0
0
30-
0 20-
--k
0,
C PM MD ALS
kg’ prot.)
PN PNP
Receptors (pkat . k g ’ prot.)
PM MD ALS PN PNP
Fig. I . The activities of R-N-acetylglucosaminidase and the contents of phosphomannosyl receptors in various diagnostic groups: (C) control group, (PM) polymyositis, (MD) muscular dystrophies, (ALS) amyotrophic lateral sclerosis, (PN) polyneuropathy, and (PNP) other neurogenic muscle diseases. Horizontal solid and dashed lines indicate the mean and the limit of 5 SD for controls. Short solid lines indicate the means for the patient groups.
cosaminidase in muscle samples and with the binding capacity of phosphomannosyl receptors.
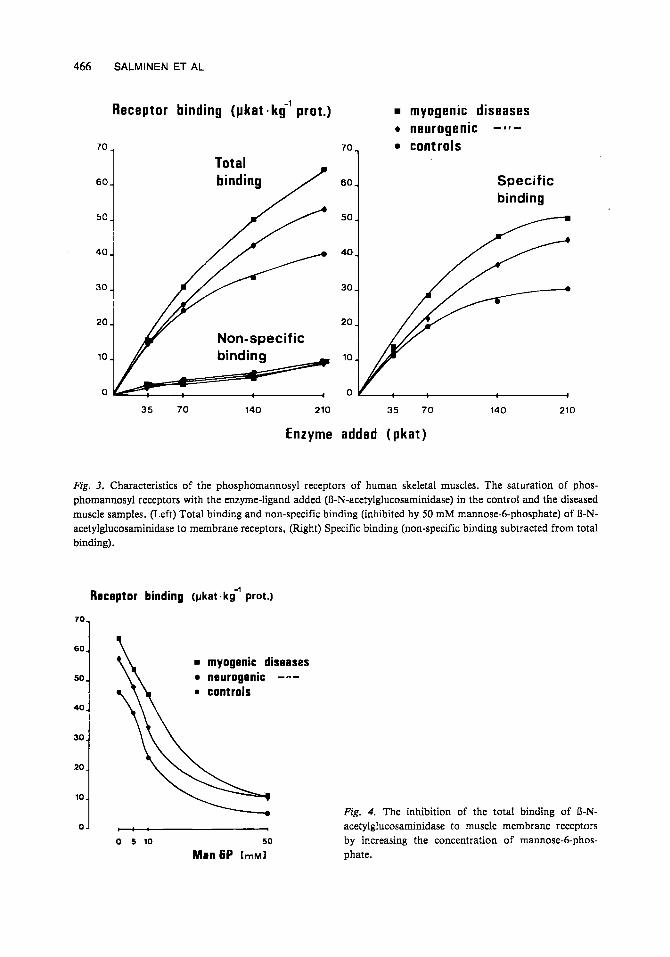
Characterization of p hosp homan nosy1 receptors Four to five muscle samples processed from the groups of controls and myogenic and neu- rogenic muscle diseases were pooled and some characteristics of phosphomannosyl receptors of control and diseased muscles were analyzed. The receptor binding of R-N-acetylglucosa- minidase was inhibited by mannose 6-phosphate (Figs. 3, 4). A 50% inhibition occurred with mannose 6-phosphate a t the concentration of less than 20 mM (Fig. 4). The specific receptor binding of R-N-acetylglucosaminidase was sat-
urable with increasing the enzyme-ligand in the binding assay (Fig. 3). A 50% saturation occurred with the enzyme activity of 55 pkat in the binding assay. All these characteristics are typical for phosphomannosyl receptors (17, 9, 10). The characteristics of receptors were very similar both to control and to diseased muscle samples (Figs. 3, 4).
Discussion The results of the present study show the pre- sence of phophomannosyl receptors in the mem- branes of human skeletal muscle. Reuser et al. (1 1) have shown the mannose-6-phosphate-de- pendent uptake of lysosomal enzymes to the
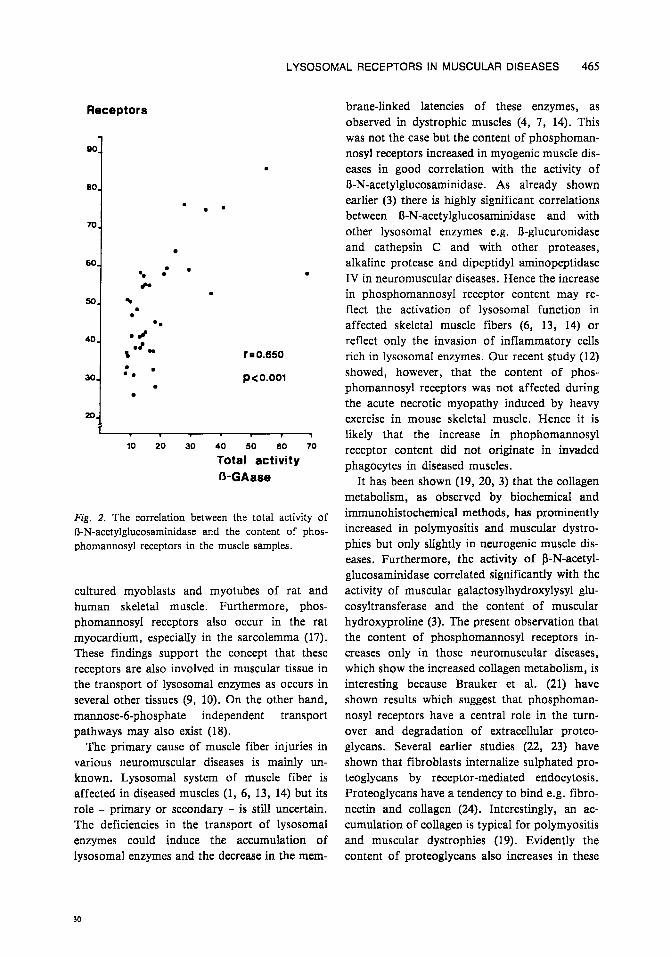
LYSOSOMAL RECEPTORS IN MUSCULAR DISEASES 465
Receptors
90
80
70
60
50
40.
30.
20.
.
- . -. c.
.
? 0.650
p<o.oor
1
10 20 30 40 50 60 70
Total activity fi-GAase
Fig. 2. The correlation between the total activity of R-N-acetylglucosaminidase and the content of phos- phomannosyl receptors in the muscle samples.
cultured myoblasts and myotubes of rat and human skeletal muscle. Furthermore, phos- phomannosyl receptors also occur in the rat myocardium, especially in the sarcolemma (17). These findings support the concept that these receptors are also involved in muscular tissue in the transport of lysosomal enzymes as occurs in several other tissues (9, 10). On the other hand, mannose-6-phosphate independent transport pathways may also exist (18).
The primary cause of muscle fiber injuries in various neuromuscular diseases is mainly un- known. Lysosomal system of muscle fiber is affected in diseased muscles (1, 6, 13, 14) but its role - primary or secondary - is still uncertain. The deficiencies in the transport of lysosomal enzymes could induce the accumulation of lysosomal enzymes and the decrease in the mem-
brane-linked latencies of these enzymes, as observed in dystrophic muscles (4, 7, 14). This was not the case but the content of phosphoman- nosyl receptors increased in myogenic muscle dis- eases in good correlation with the activity of R-N-acetylglucosarninidase. As already shown earlier (3) there is highly significant correlations between R-N-acetylglucosaminidase and with other lysosomal enzymes e.g. 8-glucuronidase and cathepsin C and with other proteases, alkaline protease and dipeptidyl aminopeptidase IV in neuromuscular diseases. Hence the increase in phosphomannosyl receptor content may re- flect the activation of lysosomal function in affected skeletal muscle fibers (6, 13, 14) or reflect only the invasion of inflammatory cells rich in lysosomal enzymes. Our recent study (12) showed, however, that the content of phos- phomannosyl receptors was not affected during the acute necrotic myopathy induced by heavy exercise in mouse skeletal muscle. Hence it is likely that the increase in phophomannosyl receptor content did not originate in invaded phagocytes in diseased muscles.
It has been shown (19, 20, 3) that the collagen metabolism, as observed by biochemical and immunohistochemical methods, has prominently increased in polymyositis and muscular dystro- phies but only slightly in neurogenic muscle dis- eases. Furthermore, the activity of 8-N-acetyl- glucosaminidase correlated significantly with the activity of muscular galactosylhydroxylysyl glu- cosyltransferase and the content of muscular hydroxyproline (3). The present observation that the content of phosphomannosyl receptors in- creases only in those neuromuscular diseases, which show the increased collagen metabolism, is interesting because Brauker et al. (21) have shown results which suggest that phosphoman- nosyl receptors have a central role in the turn- over and degradation of extracellular proteo- glycans. Several earlier studies (22, 23) have shown that fibroblasts internalize sulphated pro- teoglycans by receptor-mediated endocytosis. Proteoglycans have a tendency to bind e.g. fibro- nectin and collagen (24). Interestingly, an ac- cumulation of collagen is typical for polymyositis and muscular dystrophies (19). Evidently the content of proteoglycans also increases in these
30
466 SALMINEN ET AL
Receptor binding ( p k a t . k g ’ prot.)
60.
50
40.
30.
20.
10
70
60 -
50 -
40
30
70
Tota l binding / 601
5 4
rnyogenic diseases + neurogenic - 1 1 -
controls
Specific binding
35 70 140 210 35 70 140 210
Enzyme added ( p k a t )
Fig. 3. Characteristics of the phosphomannosyl receptors of human skeletal muscles. The saturation of phos- phomannosyl receptors with the enzyme-ligand added (R-N-acetylglucosaminidase) in the control and the diseased muscle samples. (Left) Total binding and non-specific binding (inhibited by 50 mM mannose-6-phosphate) of R-N- acetylglucosaminidase to membrane receptors, (Right) Specific binding (non-specific binding subtracted from total binding).
Receptor binding (pkat.k<’ prot.)
701
myogenic diseases neurogenic - - a - . controls
01 I : :
0 5 10 50
M a n 6 P [mM]
Fig. 4. The inhibition of the total binding of R-N- acetylglucosaminidase to muscle membrane receptors by increasing the concentration of mannose-6-phos- phate.

LYSOSOMAL RECEPTORS IN MUSCULAR DISEASES 467
diseases, as well as the number of fibroblasts. We suggest that the increased content of phos- phomannosyl receptors in polymyositis and mus- cular dystrophies is associated with increased metabolism of connective tissue.
Acknowledgement This study was supported by the Academy of Finland and the Ministry of Education in Finland.
References 1.
2.
3.
4.
5.
6 .
7.
8.
9.
10.
11.
Kar N C, Pearson C M. Muscular dystrophy and activa- tion of proteinases. Muscles Nerve 1978:1:308-313. Owens K. Biochemical studies of dystrophy in the young chicken: lysosomal and sarcolemmal enzymes. Ann NY Acad Sci 1979:317247-262. Takala T E S , Myllyla V V, Salminen A, Tolonen U, Hassinen I E, Vihko V. Lysosomal and non-lysosomal hydrolases of skeletal muscle in neuromuscular diseases. Arch Neurol 1983:40:541-544. Baxter J H, Suelter C H. Skeletal muscle lysosomes: com- parison of lysosomes from normal and dystrophic avian pectoralis muscle as a function of age. Muscle Nerve 1983 :6: 187-194. Hudgson P, Pearce G W. Ultramicroscopic studies of dis- eased muscle. In: Walton J N, ed. Disorders of voluntary muscle. London: Churchill. 1969:277-317. Weinstock I M. Iodice A A. Acid hydrolase activity in muscular dystrophy and denervation atrophy. In: Din- gle J T, Fell H B, eds. Lysosomes in biology and pathol- ogy. Amsterdam: North-Holland, 1969:1:450468. Davis M H, Gelman B B, Gruenstein E. Decreased struc- turelinked latency of lysosomes dipeptidyl aminopeptidase- I-activity in Duchenne muscular dystrophy fibroblasts. Neurology 1982:32:486-491. Gelman B B, Davis M H, Morris R E, Gruenstein E. Structural changes in lysosomal from cultured human fibroblasts in Duchenne's muscular dystrophy. J Cell Biol 1981 :88:329-337. Von Figura K, Hasilik A. Lysosomal enzymes and their receptors. Ann Rev Biochem 1986:55:167-193. Fischer H D. Gonzalez-Noriega A. Sly W S, Morre D J. Phosphomannosyl-enzyme receptors in rat liver. Subcellu- lar distribution and role in intracellular transport of lysosomal enzymes. J Biol Chem 1980:225:9608-9615. Reuser A J J , Kroos M A, Ponne N J, et al. Uptake and stability of human and bovine acid a-glucosidase in cultured fibroblasts and skeletal muscle cells from gly- cogenosis type I1 patients. Exp Cell Res 1984:155:178-189.
12. Salminen A. Latencies and phosphomannosyl-enzyme receptors of lysosomal enzymes during the appearance and
13.
14.
15.
16.
17.
18.
19.
20.
21.
repair of exercise injuries in mouse skeletal muscles. Exp Mol Pathol 1984:41:409-418. Thesleff S, Libelius R, Lundquist I. Endocytosis as induc- er of degenerative changes in skeletal muscle. In: Kid- man A D, Tomkins J K, eds. Muscle, nerve and brain degeneration. Amsterdam: Excerpta Medica, 1979:119- 138. Christie K N, Stoward P J. A cytochemical study of acid phosphatase in dystrophic hamster muscle. J Ultrastruct Res 1977:58:219-234. Myllyla V, Kihlstrom M, Takala T E S, Tolonen U, Salminen A, Vihko V. Activities of some antioxidative and hexose monophosphate shunt enzymes of skeletal muscle in neuromuscular diseases. Acta Neurol Scand 1986:74: 17-24. Salminen A. Lysosomal changes in skeletal muscles during the repair of exercise injuries in muscle fibers. Acta Phys- iol Scand 1985924 (Suppl 539):l-31. Marjomaki V S, Salminen A. Characteristics of lysosomal phosphomannosyl-enzyme receptors in the rat heart. Basic Res Cardiol 1987:82:252-260. Den H, Shanske S, DiMauro S. Targeting of lysosomal enzymes: N-acetylglucosaminy-I-phosphotransferase dur- ing muscle development. Muscle Nerve 1986:9:261-264. Myllyla R, Myllyla V V, Tolonen U, Kivirikko K I. Changes in collagen metabolism in diseased muscle I. Bio- chemical studies. Arch Neurol 1982:39752-755. Peltonen L, Myllyla R, Tolonen U, Myllyla V V. Changes in collagen metabolism in diseased muscle 11. Immu- nohistochemical studies. Arch Neurol 1982:39:756-759. Brauker J H, Roff C F, Wang J L. The effect of mannose 6-phosphate on the turnover of the proteoglycans in the extracellular matrix of human fibroblasts. Exp Cell Res 1986:164:115-126.
22. Kresse H, Truppe W. Studies on the pinocytosis of hyaluronate and proteoglycans. Prog Biochem Pharmacol 1977:1419-25.
23. Prim R, Schwermann J, Buddecke E, von Figura K. Endocytosis of sulphated proteglycans by cultured skin fibroblasts. Biochem J 1978:176671-676.
24. Timpl R, Fujiwara S, Dziadek M, Aumailley M, WeberS, Engel J. Laminin, proteoglycan, nidogen and collagen IV: structural models and molecular interactions. Ciba Foun- dation Symposium 1984:108:2543.
Address
Varpu Marjomaki, University of Jyvaskyla Department of Cell Biology Vapaudenkatu 4 SF-40100 Jyviskyla Finland
30'





























