Embed Size (px)
Citation preview

Coordination Chemistry Reviews 325 (2016) 102–115
Contents lists available at ScienceDirect
Coordination Chemistry Reviews
journal homepage: www.elsevier .com/ locate /ccr
Review
The concept of photochemical enzyme models – State of the art
http://dx.doi.org/10.1016/j.ccr.2016.06.0060010-8545/� 2016 Elsevier B.V. All rights reserved.
E-mail address: [email protected]
Günther KnörInstitute of Inorganic Chemistry, Johannes Kepler University Linz (JKU), Linz, Austria
a r t i c l e i n f o a b s t r a c t
Article history:Received 4 May 2016Received in revised form 3 June 2016Accepted 3 June 2016Available online 2 July 2016
Keywords:PhotochemistryBiomimetic catalysisGreen chemistryEnzyme modelsPhoto-biocatalytic systems
Synthetic low-molecular-weight catalyst systems with an enzyme-like reactivity can be successfully cre-ated from suitable light-responsive building blocks with rationally designed excited-state properties. Thisunique approach of mimicking natural processes with bio-inspired catalysts based on coordination com-pounds and photoreactive materials offers several important benefits compared to conventional biomi-metic strategies. Such advantages include the convenient triggering and regulation of enzyme-likeactivity by light-intensity variations, efficient substrate conversion even under very mild reaction condi-tions, and the intrinsic possibility of powering energetically uphill processes. Due to these promising fea-tures, the novel field of photochemical enzyme models (artificial photoenzymes) has matured over thelast decade.Several illustrative examples of photocatalytic processes mimicking the functional properties of natural
systems are provided in this short review including some typical applications of artificial photoenzymesin the fields of green chemistry, solar energy conversion, photomedicine and life-sciences. Combiningenzyme-like reactivity to enable stepwise synthetic cascade reactions has meanwhile also been demon-strated, which now opens new avenues for the design of artificial metabolic pathways controlled and dri-ven by light. The creation of directly coupled photo-biocatalytic hybrid systems is also briefly discussedas a straightforward method to trigger more complex reaction sequences at the interface of light-mediated chemistry and natural processes in both cell-free systems and living organisms.
� 2016 Elsevier B.V. All rights reserved.
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1032. Bio-inspired catalysis mediated by light. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 104
2.1. Design principles, benefits and assesment criteria . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1042.2. Proof-of-concept: The first photochemical enzyme models . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 105
3. Artificial photoenzymes in green chemistry and synthesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 105
3.1. Visible-light triggered radical chemistry . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1053.2. Activation and reduction of dioxygen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1063.3. Oxygenase catalysis driven by light . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1074. Artificial photoenzymes for energy conversion and storage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 107
4.1. Catalytic accumulation of reducing equivalents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1074.2. Light-driven hydrogenase enzyme models . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1084.3. Photocatalytic enzyme models for CO2 reduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1085. Artificial photoenzymes in medicine and life sciences . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 109
5.1. Light-controlled induction of apoptosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1095.2. Mimicking cellular response to oxidative stress . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1105.3. Modelling the biosynthesis of hormones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1115.4. Bioanalytical tools based on artificial photoenzymes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1116. Coupled processes involving photocatalytic enzyme models . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 112
6.1. Reaction cascades: Towards metabolic pathways . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1126.2. Photo-biocatalytic hybrid systems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 112
Fig. 1.actingsupero
G. Knör / Coordination Chemistry Reviews 325 (2016) 102–115 103
6.3. Artificial photosynthetic reaction centers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 112
7. Outlook and conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 112Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 113References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 113
1. Introduction
Without enzymes, life as we know it would certainly be impos-sible. Only a few specific reactions are occurring non-enzymatically inside the cell [1]. On the other hand, a remarkablysmall number of different catalysts is responsible for acceleratingthe complex chemistry of all living organisms including humansin a perfect way [2,3]. Already in the pioneering years of enzymol-ogy, researchers were therefore fascinated by the smooth and effi-cient performance of biocatalytic substrate conversions and startedto compare them with other chemical processes [4–7]. In a vision-ary lecture held more than hundred years ago, Nobel prize laureateEmil Fischer encouraged scientists to start creating man-madeenzyme counterparts [8]:
‘‘The chemical exploration of ferments is still at the very beginning[. . .] even today I consider attempts to prepare them [. . . ]in an arti-ficial way as a not too daring enterprise” (E. Fischer, 1907).
Since then, many different approaches have been followed toexploit the biological principles of enzymatic catalysis for abioticand biomimetic chemical transformations. New methods havebeen developed which allowed to obtain the first purely syntheticenzymes with full biological activity [9]. The native protein scaf-folds of biocatalysts have soon been modified and re-designed bygenetic engineering [10]. De-novo design of polypeptide structuresnot directly related to any sequence found in nature also emergedas a powerful tool to control the function of the protein matrix[11]. One of the fascinating more recent goals is to incorporateamino acids not included in the standard genetic code into syn-thetic polypeptides for improving enzymatic catalysis [12,13].Another possibility to create non-natural variants of biocatalystsis the reconstitution of native or engineered apo-proteins withmodified organic coenzymes or synthetic metal-based cofactors[14–18]. Progress and trends in artificial protein design and syn-thetic biology [19] have recently been summarized elsewhere inmore detail [20–22].
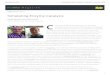
Example of a bioinorganic model compound with competitive enzyme-like reaas a functional superoxide dismutase mimic [26,27] is more than 100-times smaxide dismutase (EC 1.15.1.1, PDB-code: 1N0J).
Low-molecular-weight catalysts which display an enzyme-likereactivity in the absence of a protein matrix in contrast are muchharder to devise. If successful, this alternative approach may signif-icantly broaden the range of technological applications of enzy-matic catalysis by eliminating certain practical drawbacks ofprotein-based systems such as stability problems under non-physiological conditions or the sometimes quite narrow substratescope. Therefore, tremendous efforts have been made in organic,supramolecular and bioinorganic chemistry to develop artificialenzymes or synthetic model enzymes from scratch based on robustmolecular components [23–25]. A typical case of such a stable low-molecular weight non-peptidyl enzyme mimic is illustrated inFig. 1.
Despite of the increasing interest in small biomimetic and bio-inspired catalysts, however, only very few examples of artificialenzyme substitutes with efficiencies and selectivities rivalingthose of natural systems have been reported up to now [25,28].This is due to the fact that in the majority of cases the structureand dynamics of the protein matrix surrounding a catalytic centerplay a crucial role for the correct function of an enzymatic reactionmechanism and thus cannot be neglected. Therefore, many activesite analogs both structurally and spectroscopically representinga perfect copy of a specific substrate binding site may still remaincatalytically inactive. On the other hand, synthetic counterpartsreplacing the highly reactive key-intermediates temporarilyformed in the catalytic cycles of enzymatic processes will of coursehardly be obtained and isolated as robust and stable modelcompounds.
To overcome these serious problems, more flexible designstrategies for the creation of artificial enzymes have to be consid-ered. In this context, the novel concept of photochemical enzymemodels (artificial photoenzymes) was introduced [29–32]. Byexploiting the excited-state properties and dynamics of robustlow-molecular weight catalyst precursors to impose light-controlled electronic, energetic and structural changes at a sub-strate binding site, the selective and efficient chemistry of biocat-
ctivity: The synthetic pentaaza-cyclopentadecan-derived Mn-complex (right side)ller than the macromolecular protein complex of human mitochondrial manganese

104 G. Knör / Coordination Chemistry Reviews 325 (2016) 102–115
alysts can be readily replaced. This new strategy allows to createcompetitive functional enzyme models based on small molecularsystems in the absence of a dynamic protein environment. Somerecent progress in this currently emerging field [28,33–35] of bio-mimetic and bio-inspired photocatalysis with light-driven enzymemodels is summarized in the present review.
2. Bio-inspired catalysis mediated by light
2.1. Design principles, benefits and assesment criteria
Photons at the same time transport a quantum of energy and abit of information [33]. Both aspects can be systematicallyexploited to develop functional model systems mimicking the per-formance of natural systems. Excited-state compounds are selec-tively activated species with their own characteristic physicaland chemical properties thus providing a completely new dimen-sion for the design of molecular devices and functional materials[36]. Many useful reaction intermediates not accessible throughthermal activation may be photochemically generated under verymild operation conditions [37]. Furthermore, thermodynamicallyuphill processes can be powered by light without an additionalinput of other forms of energy [38]. Photons may also be utilizedas an external stimulus to trigger and control highly specificmolecular processes, which offers a straightforward method tosimulate the complex functions of biological regulation mecha-nisms and beyond [33,39].
Among the catalytically relevant features of small molecularsystems that will be directly influenced by the population of elec-tronically excited states are structural changes such as bond distor-tions, enhancement of redox reactivity, acidity and nucleophilicityvariations, modifications of binding constants, changes in spin-multiplicity and the selective generation of open-shell intermedi-ates such as radicals or radical ions. With this versatile toolbox itis possible to rationally construct bespoke photocatalytic systemswith an excited-state electronic structure and reactivity replacingthe otherwise hard to mimic function of short-lived active siteintermediates involved in biocatalytic substrate conversion cycles[28,32,33]. In this context, a special focus should be given to thedesign of photocatalytic reaction cycles that require at least onephoton to be absorbed per product molecule formed (photoas-sisted systems), because they can accelerate endergonic processesand additionally benefit from the possibility of completely switch-ing off their catalytic activity in the dark [33].
To be considered as a properly qualified enzyme mimic or ‘‘ar-tificial enzyme”, synthetic biocatalyst model systems according toKirby [25] should in addition fulfill at least the requirement of sub-strate binding in the course of the catalytic cycle. Evidence for thisimportant criterium of artificial (photo)enzyme function can beeither provided by a direct characterization of the catalyst-substrate complex [32] or by observation of Michaelis–Mententype saturation kinetics with a limiting upper rate of product for-mation reached under optimized conditions (Fig. 2).
Fig. 2. Reaction scheme illustrating the photoassisted accumulation of products (P)from a substrate (S) and an inactive artificial photoenzyme precursor (E). Catalyticactivity requires the formation of an excited-state enzyme substrate complex (ES)⁄
which can be provided by dynamic (hm) or static light-dependent pathways (hm0).Note that the product formation rate of the artificial photoenzyme system dependson the concentration of (ES)⁄ which can be controlled by variations of theirradiance.
Although remarkable analogies can be drawn between biocat-alytic and artificial photoenzyme reactivity, there are also someimportant differences. Under typical irradiation conditions appliedin photochemical experiments, the substrate concentration ismuch higher than the stationary concentration of the excited-state species involved. Artificial photoenzymes can therefore read-ily be characterized while operating in the substrate saturationregime of a Michaelis–Menten type process where product forma-tion simply follows zero-order kinetics with a certain reaction rate(apparent Vmax value). On the other hand, in contrast to conven-tional enzyme kinetics experiments [25], the maximum catalyticconcentration of the active enzyme substrate complex involvedin product formation (Fig. 2) may not be considered as constantfor a sample with given starting concentrations. In homogeneoussolution, the available amount of the excited-state species (ES)⁄
is directly coupled to the incident photon flux and will always besmaller than that of the enzyme precursor (E) initially added tosuch photoassisted model systems. Only under optimal lightabsorption conditions [40], the highest possible steady-state con-centration of the active form of an artificial photoenzyme will bereached, which then defines an upper limit for the reaction rateunder light-saturation (an absolute Vmax value) that can beobtained with a specific catalyst sample (Fig. 3).
Characterization of native enzymes for the most simple case ofsaturation kinetics uses the Michaelis constant (KM) as a quantita-tive measure for substrate affinity [25]. KM is the concentration ofsubstrate, where half of Vmax is reached for biocatalytic productformation, since half of the enzyme is present as enzyme-substrate complex. A corresponding ‘‘Michaelis constant‘‘ KM canalso be defined for the characterization of the excited-state sub-strate affinity of an artificial photoenzyme based on a photoas-sisted reaction cycle. Assuming that no significant ground stateinteraction with the substrate (S) occurs and the artificial photoen-zyme precursor (E) is forming the active enzyme-substrate com-plex (ES)⁄ in a dynamic quenching process (Fig. 2), it can bederived that (Eq. (1)):
KM ¼ 1=KSV with KSV ¼ kq � so ð1ÞKSV is the Stern–Volmer constant [36] obtained for dynamic
bimolecular lifetime quenching of light-activated enzyme modelcompound (E⁄) by a substrate (S). It is given by the product ofthe bimolecular quenching constant (kq) for the specific substrateand the excited-state lifetime (so) of the unquenched active formof the photoenzyme in the absence of this substrate. If static sub-
Fig. 3. Schematic representation of the different activity regimes of an artificialphotoenzyme based on photoassisted substrate conversion. The observed productformation rate Vmax = kcat�[(ES)⁄] varies as a function of actinic light exposure: (a)inactive resting state without excitation; (b) intermediate range; (c) limitingactivity maximum reached with highest possible steady-state concentration of (ES)⁄
at a certain excitation wavelength, temperature and pressure.

G. Knör / Coordination Chemistry Reviews 325 (2016) 102–115 105
strate binding with E already occurs in the absence of light, the sta-bility constant K of the catalytically inactive ground state enzymeprecursor complex (ES) also has to be considered (Fig. 2), whichthen results in a more complex expression for KM.
Besides substrate affinity criteria, the first-order rate constants(kcat) for the reaction of the bound substrates are usually reportedfor comparing the activities of biocatalysts at a certain pH, pressureand temperature [25]. The overall catalytic efficiency of a nativeenzyme is then given by the ratio kcat/KM. Catalytic perfection isreached, when this ratio approaches values above 108–109 M�1 s�1,where more or less every collision with the substrate leads to pro-duct formation (diffusion control) and the classical Michaelis–Menten kinetic model has to be expanded appropriately [41]. Inthe most simple case of a photoassisted process with negligibleground state interaction between the substrate (S) and the artificialphotoenzyme precursor E, a lower limit of the catalytic efficiency(kcat/KM) can be estimated for light-driven biocatalyst mimics asfollows (Eq. (2)):
kcat=KM � /P � kq ð2ÞThe value /P corresponds to the experimentally observed pro-
duct formation quantum yield at a certain irradiation wavelengthand kq is the bimolecular rate constant for dynamic collisionalquenching of E⁄ by a specific substrate as already discussed above(Eq. (1)). With such quantitative criteria for reporting KM and kcatvalues requiring only photochemical standard parameters suchas lifetime data (so), quenching constants (kq) and reaction quan-tum yields (/P) it becomes possible to directly compare the overallperformance of artificial photoenzymes with their native biocata-lyst counterparts.
2.2. Proof-of-concept: The first photochemical enzyme models
The feasibility of designing robust low-molecular weight photo-catalyst systems with an efficient enzyme-like reactivity has ini-tially been demonstrated for oxidoreductase model compounds[30–32]. Different biocatalysts accelerating a specific substrateconversion process in aerobic aqueous solution under otherwiseidentical reaction conditions such as temperature, pressure andpH have been directly compared to artificial photoenzymes operat-ing under visible light exposure [32]. As an example, in Fig. 4 theactive site properties of a copper redox enzyme (galactose oxidase[42]) and its functional photocatalytic counterpart are shown.Alcohols binding to these reactive species are selectively trans-
Fig. 4. Comparison of the key-intermediates accelerating the rate-determiningoxidation step in the active site of a native metalloenzyme and in the correspondingartificial photoenzyme reaction [32]. The substrate binding copper(II)-phenoxyl-radical moiety of galactose oxidase (EC 1.1.3.9.) shown on top provides a reactiveopen-shell electronic structure [42], which is functionally replaced by the excited-state properties of a photocatalytically active coordination compound.
formed into aldehyde products. The reduction equivalents and pro-tons (2e�, 2H+) temporarily stored at the catalytic site (Fig. 4) arefinally coupled to molecular oxygen activation [43] and hydrogenperoxide production, thus closing a two-electron substrate conver-sion cycle.
A unifying mechanistic picture has been proposed with an elec-trophilic oxyl-radical intermediate in direct proximity to thebound substrate. At the same time, such a reactive subunit has tobe efficiently coupled to a reversible multielectron transfer reagent[29] to avoid back reactions and to promote the decisive bond for-mation steps of both natural and artificial enzyme catalyzed trans-formations [32,33]. It could be shown that average turnovernumbers of several thousand productive photoassisted cycles foreach catalyst molecule can be readily achieved that way basedon remarkably simple but carefully selected main-group metallo-porphyrin complexes. These first well-documented examples oflight-driven oxidoreductase model compounds schematicallyshown at the bottom of Fig. 4 are meanwhile considered as the pro-totypes of artificial photoenzymes in the literature [34].
The most significant finding of the pioneering studies describedabove, however, was the fact that a very promising catalytic per-formance of the bio-inspired systems with reaction rates up tothree times higher than that of the natural metalloenzyme coun-terparts could be achieved under otherwise identical reaction con-ditions [32]. This clearly demonstrated that the function of certainbiocatalysts can be fully replaced in a competitive way by syntheticlow-molecular-weight photocatalysts. In the following sections,several examples of such light-driven enzyme model systems arepresented together with some of their potential applications andfuture capabilities.
3. Artificial photoenzymes in green chemistry and synthesis
Following the principles of green chemistry for manufacturingvaluable compounds in an economical, resource-preserving andenvironmentally benign way has been identified as a crucial road-map for the survival of mankind [44,45]. Especially in combinationwith long-wavelength solar radiation as a source of renewableenergy such a strategy could offer interesting new perspectivesfor a sustainable chemistry of the future. Light is a clean and highlyselective reagent that can be readily controlled and dosed. Photo-chemical and photocatalytic key-steps are therefore more andmore recognized as a powerful extension of conventional methodsin organic synthesis [46–52]. This current trend of green and solarphotochemistry will certainly be further strengthened in the nextdecades by the advent of continuous flow (micro)reactors as anew tool for more efficient photochemical synthesis [53–55], andby the gradual progress achieved in light-induced enantioselectivecatalysis, asymmetric photochemistry and photochirogenesis [56–58]. While the reported substrate spectrum of stereoselective pho-toreactions can currently not yet fully compete with the versatilityof biotechnology-based asymmetric catalysis [59], the concept ofartificial photoenzymes is very well suited for the controlled gen-eration of reactive intermediates involved in C–H activation, C–Cbond formation and selective substrate oxidation processes undermild reaction conditions as will be shown below.
3.1. Visible-light triggered radical chemistry
Many enzymes rely on reactive species with unpaired valenceelectrons (radicals) to promote the smooth and rapid conversionof chemically inert compounds. Depending on the kind of interme-diates formed and biocatalytic systems involved, the generation ofprotein- and substrate-based radicals may occur under strictlyanaerobic, oxygen depleted or even aerobic conditions [60–63].

106 G. Knör / Coordination Chemistry Reviews 325 (2016) 102–115
Prominent examples of radical enzymes operating under anoxicconditions are glycyl radical dependent systems such as the classIII ribonucleotide reductases [60] and certain representatives ofthe so-called radical SAM enzyme superfamily [64,65]. The elec-tronic structure properties of several open-shell organic chro-mophores representing potentially photoreactive groups inpeptide and protein radical chemistry have recently been compiled[66].
An important catalytic function of such protein-based radicalintermediates in general is their ability to promote proton-coupled electron transfer processes (PCET) by homolytic hydrogenatom abstraction (HAT) from bound or nearby substrate molecules[25,28,67]. H-atom abstraction mechanisms also play a crucial rolein various metalloenzyme-mediated substrate oxidations, andtherefore triggering such a reactivity in a precise and controlledfashion with biomimetic model systems is a highly desirableresearch goal [32,68–70]. Moreover, especially under anaerobicconditions the short-lived carbon-centered radicals R� generatedfrom R–H substrates in the primary steps of natural or artificialenzyme catalyzed HAT reactions (Fig. 5) are synthetically very use-ful reagents for building up new chemical bonds in coupling andcross-coupling reactions [71,72]. Exploiting visible-light activationto trigger direct HAT and controlled radical chemistry is a promis-ing concept for the design of powerful catalysts in this context.First preliminary studies exploring the synthetic potential of artifi-cial photoenzymes designed for selective C–H functionalizationand carbon–carbon bond formation are currently underway [73].
Another potential source of carbon-based radicals for catalytictransformations is provided by the homolytic cleavage ofbioorganometallic systems such as cobalamins (vitamin B12derivatives). This class of compounds displays a variety of naturallight-responsive functions and a rich photoreactivity followingboth radical and non-radical pathways [74–78]. Based on the pho-tosensitization of redox-active B12 derivatives, Hisaeda andcoworkers have recently developed various bio-inspired reactionsfor the degradation of organohalide pollutants such as DDT andrelated compounds according to Eq. (3) under visible light expo-sure [79,80].
R � CCl3 þ 2e� þ 2Hþ ! R � CHCl2 þHCl ð3ÞThese homogeneous photocatalytic systems can be considered
as synthetic biocatalyst mimics replacing the net function of reduc-tive dehalogenase enzymes (EC 1.97.1.8.) occurring inorganohalide-respiring microorganisms, which under anaerobicconditions can remove halogen atoms from their substrates[81,82].
3.2. Activation and reduction of dioxygen
All higher organisms are designed to capture and activate atmo-spheric oxygen for cellular respiration [83]. The membrane protein
Fig. 5. Photocatalytic substrate conversion cycle producing permanent products (P)based on hydrogen atom transfer (HAT) and subsequent radical chemistry [32,72].Because of the direct involvement of an excited-state species (E⁄) forming anintermediate E–H bond, the light-controlled functionalization of chemically inertR–H substrates such as alkanes with substantial bond dissociation energies even inthe range of 100 kcal/mol can be achieved.
cytochrome c oxidase (EC 1.9.3.1.) acts as the terminal electronacceptor in the mitochondrial respiratory chain driving a protonpump coupled to the four-electron reduction of molecular oxygen[84]. In general, most oxidases are able to catalyze either the for-mation of hydrogen peroxide or water under aerobic conditionsaccording to Eqs. (4) and (5).
O2 þ 2e� þ 2Hþ ! H2O2 ð4Þ
H2O2 þ 2e� þ 2Hþ ! 2H2O ð5ÞThe reduction equivalents required for the transformation of
dioxygen can be provided by a broad variety of different substratesacting as electron donors. Although oxidoreductases capable ofdirect interaction with O2 in its triplet ground state have beenidentified [85], the majority of native oxidase enzymes requiresadditional organic cofactors or redox-active transition metal cen-ters for catalysis. Typical examples are flavoprotein oxidases [86]like glucose oxidase (EC 1.1.3.4) [87], NAD(P)H-oxidase (EC1.6.3.1.) [88], and peroxide generating metalloproteins such asgalacose oxidase oxidase (EC 1.1.3.9.), plant lipoxygenases (EC1.13.11.12), aldehyde oxidase (EC 1.2.3.1) or sulfite oxidase (EC1.8.3.1) containing copper, iron or molybdenum ions in their activesites [42,89–91].
Molecular oxygen from air is a clean, abundant and environ-mentally benign reagent ideally suited for substrate oxidation pro-cesses including selective C–H bond functionalization. Thereforethe utilization of oxidase-like reactivity for green synthetic pro-cesses based on O2 or H2O2 is a highly desirable research goal[92–94].
Versatile photochemical strategies for replacing the activationand two-electron transformation of dioxygen catalyzed by oxyge-nase enzymes (Eq. (4)) have already been identified [43,95]. Akey-process required for triggering an efficient visible-light medi-ated activation of molecular oxygen with high turnover numbersis the photosensitization of metal-centered triplet excited statesof redox-active main group element compounds (Fig. 6). With thisclass of bio-inspired enzyme model systems a stepwise four-electron transfer sequence resembling the respiratory O2 transfor-mation of cytochrome c oxidase has also been demonstrated [43].Among the sensitizer chromophores well-suitable for the construc-tion of photoenzymes with artificial oxidase reactivity aretetrapyrrole-based macrocyclic ligands such as porphyrins,phthalocyanines and corroles [43,96–98].
In contrast to many biological systems, where the presence ofantioxidants, catalases and dismutases usually keeps the con-trolled levels of activated oxygen species low enough to protect tis-sues from serious damage by oxidative stress [99], a catalyticaccumulation of reactive compounds such as peroxides in largeramounts can be readily achieved with robust artificial photoen-zymes [100,101]. Besides its prominent role as a clean oxidantfor synthetic applications, H2O2 obtained from air, water and sun-light therefore can also be exploited as an attractive energy-richproduct for reversibly storing chemical bond energy [33,100–102]. The most important role of dioxygen activation by artificialphotoenzymes, however, is the controlled generation of catalyti-cally competent metal-peroxo and metal-oxo species [28] driving
Fig. 6. Initial steps in photocatalytic dioxygen reduction and peroxide productionmediated by low-valent main group metal complexes irradiated under aerobicconditions [43].

G. Knör / Coordination Chemistry Reviews 325 (2016) 102–115 107
remarkable mono- and dioxygenase-type substrate conversionprocesses, which will be briefly discussed in the next section.
3.3. Oxygenase catalysis driven by light
The unique chemistry of high-valent metal oxo-species such asferryl FeIV@O intermediates and MnIV-oxyl radicals plays a promi-nent role in some of themost difficult to achieve bioinorganic redoxprocesses like selective alkane functionalization and photosyn-thetic water oxidation [92,103–107]. With the help of reactiveoxo and peroxo species, oxygenase enzymes can catalyze the trans-fer of one or two O-atoms originating from dioxygen to their sub-strates. Many oxygenases are therefore also investigated asattractive candidates for applications in biotechnology and organicsynthesis [108]. Typical examples of oxidase metalloproteins arethe soluble and particulate methane monooxygenases (EC1.14.13.25, EC 1.14.18.3) [92,109], chloroperoxidase (EC 1.11.1.10)[110], nitric oxide synthase (EC 1.14.1.13.39) [111] and aromaticperoxygenase (EC 1.11.2.1) [112]. Among the most widely studiedrepresentatives of this class of redox enzymes with catalyticallyactive metal-oxo intermediates are the heme-thiolate monooxyge-nases of the cytochrome P450 (EC.1.14.14.1) superfamily [113].These enzymes are able to catalyze a broad range of different O-atom transfer reactions including substrate hydroxylations andepoxidations under mild reaction conditions (Fig. 7).
Artificial photoenzymes with visible-light mediated oxygenasereactivity have been successfully constructed. Already about20 years ago, very close functional similarities between the reac-tive oxoferryl intermediates of cytochrome P450-type monooxyge-nase enzymes and the low-lying charge transfer excited states ofcertain photocatalytic systems both competent for substrate con-versions such as alkene-epoxidations have been discovered [43].These analogies between photochemical activation of certain metalcomplexes and metalloenzyme catalysis have been further system-atically studied in the authors own group investigating main groupmetal derivatives [28,30], but also several transition metal com-plexes seem to show a similar photocatalytic reactivity [72,114].In this context, it should be pointed out that currently emergingtheories meanwhile clearly confirm the critical role of low-lyingexcited-state population, spin-state control and specific transitionstate electronic structures on the chemical reactivity and selectiv-ity of many metal-oxo enzymes and their model compounds [115–
Fig. 7. Schematic representation of a net two-electron substrate oxidation process(alkene epoxidation) catalyzed at the high-valent metal-oxo site of an enzyme withmonooxygenase reactivity.
Table 1Selected examples of artificial oxidoreductase enzyme reactions driven by visible-light.
Photocatalyzed substrate oxidation Enzymatic function rep
RCH2OH? RCHO + 2e� + 2H+ Monooxygenase activiRH + H2O? ROH + 2e� + 2H+ Monooxygenase activiRCHO + H2O? RCOOH + 2e� + 2H+ Dioxygenase activityRH + HX? RX + 2e� + 2H+ Haloperoxidase activitRH + R0H? R-R0 + 2e� + 2H+ Reversed lyase activityRCH2OH + H2O? RCOOH + 4e� + 4H+ Coupled dehydrogenasR2S + H2O? R2S = O + 2e� + 2H+ Peroxygenase activity
a Irradiation wavelength.b Polychromatic irradiation with simulated sunlight.
117]. Such a theoretical framework will certainly turn out to bevery helpful for a rational design of new artificial photoenzymescapable of accelerating mechanistically demanding substrate oxi-dations. Detailed knowledge about the catalytic role of high-valent M@O excited-state species with an electrophilic oxyl-radical character will also be of prime interest for replacing theenzymatic function of the light-driven water:plastoquinone oxi-doreductase (EC 1.10.3.9.) of natural photosystem II, which is crit-ically required for artificial photosynthetic water splitting and O–Obond formation [35]. The development of bio-inspired photocata-lysts with controlled oxyl-radical reactivity capable of selectivepartial oxidation of methane to methanol is another fundamentalresearch goal in this field [28]. In Table 1, a collection of somelight-driven oxygenase model reactions already available today isprovided.
4. Artificial photoenzymes for energy conversion and storage
While the synthetic applications of biomimetic photocatalysisdescribed above are typically restricted to the acceleration of ener-getically downhill reactions, the concept of artificial photoenzymesalso offers a straightforward route to achieve direct chemical stor-age of light energy. Many different energy-rich products (solarfuels) may be targeted in artificial photosynthetic devices basedon molecular catalyst systems with an enzyme-like reactivity.The state of the art of this important branch of bio-inspiredresearch has recently been discussed in more detail [35]. Possibleapplications of artificial photoenzymes for the design of chemicalenergy conversion and storage systems are therefore only brieflysummarized here.
4.1. Catalytic accumulation of reducing equivalents
Enzymatic reductions in biological systems can handle redoxpotentials slightly more negative than �0.62 V vs. NHE underanoxic conditions [122]. The involvement of short-lived excited-state intermediates in natural photoenzymes allows to expandthe available redox scale to more negative values with larger driv-ing force for electron transfer processes. For example, by absorp-tion of red light corresponding to a photon energy of 1.77 eV, thechlorophyll-based primary electron donor P700 present in photo-system I (PS I) of many photosynthetic organisms can reach a redoxpotential limit of about �1.37 V vs. NHE [123,124]. These photo-systems can be considered as the strongest reductants availablein the biosphere. One of the most challenging tasks of enzymaticand artificial photoenzyme catalysis is to rapidly accumulate thesingle electron reduction equivalents released from such excited-state species and to store them reversibly as electron pairs in anew chemical bond [35]. This essential proton-coupled multielec-tron transfer reactivity can lead to a variety of energy-rich productmolecules available for many other secondary energy conversionprocesses.
laced kirr (nm) a Refs.
ty 546 [32,98]ty AM 1.5 b [118]
AM 1.5 b [119]y <620 [101,120]
410 [73]e activity AM 1.5 b [119]
>455 [121]

108 G. Knör / Coordination Chemistry Reviews 325 (2016) 102–115
In terms of energy density, the most efficient way to stabilizeand store a photochemically released pair of two electrons froma donor substrate is to connect it to a single proton, forming ahydride anion (Eq. (6)).
2e� þHþ ! H� ð6Þ
H� þHþ ! H2 ð7ÞFurther protonation of such a species will lead to an exergonic
release of molecular H2 (Eq. (7)) or a transfer of the correspondingtwo-electron reduction equivalents (2e�, 2H+) available for bio-chemical or biomimetic reactions. In living systems, the reversibleformation of nicotinamide adenine dinucleotide cofactors NAD(P)Hhas evolved to provide a universal reagent for such reductivebiosynthetic substrate transformations [28,125]. A hydride equiva-lent can be efficiently stored and transported by the reduced formof this class of organic cofactors (Eq. (8)).
NADðPÞþ þ 2e� þHþ ! NADðPÞH ð8ÞRepresenting the biochemical version of solar hydrogen as a
fuel, NAD(P)H is also generated as the energy-storing final productterminating the light-reactions of natural photosynthesis. Thisimportant last step in the photosynthetic electron transfer chainis catalyzed by the flavin-dependent enzyme ferredoxin-NADP+
reductase (EC 1.18.1.2) capable of accelerating proton-coupledmultielectron transfer reactions [35,126]. Successful replacementof this important catalytic function with an artificial photoenzymein a solar energy storing reaction sequence rivalling nature in herown game has recently been demonstrated [35,125].
Reversible recycling of redox cofactors such as NAD(P)H andrelated compounds powered by renewable energy or direct expo-sure to sunlight is a topic of considerable interest [127,128]. Thehydride equivalents intermediately stored in these cofactors andthe catalyst systems involved in their generation can serve as ver-satile reagents for various chemoenzymatic processes [129], forstereoselective reductions of organic substrates and for the pro-duction of solar fuels such as alcohols and other energy-rich stor-age molecules using CO2 as the carbon source [35,130–133].
4.2. Light-driven hydrogenase enzyme models
Hydrogenases catalyze the reversible interconversion betweenprotons, electrons and molecular hydrogen involving a heterolyticH–H bond cleavage mechanism (Eq. (9)).
2e� þ 2Hþ � ðH� þHþÞ�H2 ð9ÞAs a function of the electron acceptor involved in the hydrogen
activation process, this family of metalloenzymes can be systemati-cally divided into various subgroups including hydrogen:NAD+
oxidoreductase (EC 1.12.1.2), hydrogen:ferricytochrome-c3 oxi-doreductase (EC 1.12.2.1), ferredoxin hydrogenase (EC 1.12.7.2), orcoenzyme F420 hydrogenase (EC 1.12.98.1). Frequently, an alterna-tive classification into [NiFe]-, [FeFe]- and [Fe]-hydrogenases is cho-
Fig. 8. Advanced organometallic hydrogenase model compound combining aferrocene-based donor subunit and a robust catalytically active iron-sulfur clustercore for proton reduction [140].
sen according to the nature of the catalytically active site [134,135].With a focus on the required structural features includingmultistepelectron transfer chains andhydride attachment sites (Fig. 8), a largenumber of different sulfur ligated low-valent iron containing com-plexes has been synthesized and investigated as biomimetic hydro-genase model systems [136–140].
Visible-light mediated proton reduction with bioinspired modelcomplexes has already been extensively studied by several groups[141–149]. In most cases, a combination of different photosensitiz-ers with electron-rich mono- and polynuclear Fe and Ni, but alsoCo, Mo and W complexes has been tested. Although significantrecent progress has been made in the field, a competitive artificialphotoenzyme with long-lasting hydrogenase-like activity in aque-ous solution has not yet been identified so far [142,150]. Neverthe-less, homogeneous photocatalytic generation of hydrogen withrobust and deeply-coloured enzyme model complexes as thoseshown in Fig. 8 might become feasible in the near future.
4.3. Photocatalytic enzyme models for CO2 reduction
Assimilation of CO2 to generate carbon-based fuels is an impor-tant pathway for the production of renewable energy carriers[151,152]. Binding of the substrate molecule itself under mild reac-tion conditions represents the most critical initial step for convert-ing carbon dioxide into energy-rich products. In order to activateCO2 for chemical reactions, it is advantageous to destabilize thisrather inert molecule. An important strategy to control and influ-ence the reactivity of carbon dioxide is its coordination to amines,metal-bound imido moieties, or low-valent metal centers [28],which leads to a decrease of the C–O bond order, while the mole-cule in most cases becomes considerably bent. In nature this diffi-cult task is achieved by metalloenzymes such as CO-dehydrogenases (CODHs), which are able to catalyze the reversiblemultielectron transfer interconversion between carbon dioxideand carbon monoxide according to Eq. (10) [152–154].
CO2 þ 2e� þ 2Hþ �COþH2O ð10ÞThe crystal structure of substrate loaded Ni-CODH (EC 1.2.99.2)
at atomic resolution has recently been reported [155]. From thesedata it becomes clear that carbon dioxide attached to the active sitecan form a two-electron reduced intermediate CO2
2� in the enzy-matic reaction mechanism (Fig. 9).
Acceleration of such a two-electron substrate reduction isessential for the overall efficiency of the enzymatic process. Whilea moderately low reduction potential of E� = �0.53 V is required for
Fig. 9. Substrate binding at the multinuclear nickel–iron active site (C-cluster) ofthe bacterial enzyme Ni-CODH [155]. The carbon atom of the CO2 molecule iscoordinated to a nucleophilic Ni center. Carbon dioxide reaches a carboxylate-likebent structure with a O–C–O angle of 117� facilitating the uptake of two electrons.The highly activated carbon dioxide adduct is further stabilized by interaction witha neighbouring electrophilic Fe site present in the [NiFe4S4]-cluster of the enzyme.

G. Knör / Coordination Chemistry Reviews 325 (2016) 102–115 109
this multielectron redox process [155], the uncatalyzed one-electron reduction of CO2 would require a much more negativepotential (E� = �1.90 V vs. NHE at pH = 7), by far exceeding the lim-iting constraints of biological systems (see Section 4.1). Protona-tion of the two-electron reduced intermediate CO2
2� could eitherlead to the production of formate (HCO2
�) as a permanent productor, as is the case with Ni-CODH, to a fragmentation into OH� andCO. This latter step is facilitated by the presence of a suitably ori-entated FeII center acting as a Lewis acid able to form a Fe–OH bond(Fig. 9). Release of the Ni-coordinated CO-ligand and a second pro-tonation step at the Fe-bound hydroxo ligand to form water closesthe reversible catalytic cycle according to Eq. (10).
Carbon dioxide fixation mimicking these proton-coupled multi-electron transfer reaction pathways with molecular catalysts andphotochemical key-steps is a vibrant research field [156–160].Similar to the enzymatic substrate activation process describedabove, in many of the investigated systems the formation of aninner-sphere CO2 adduct is part of the proposed reaction mecha-nisms [161–167]. Only a few photocatalysts are able to reach a cer-tain degree of selectivity for the type of CO2 reduction products.Sometimes also the substrate specificity of the excited-state spe-cies is low, leading to simultaneous proton reduction in competi-tion with the formation of carbon-based reaction products suchas CO. In the context of solar energy conversion and storage, how-ever, the combination of carbon monoxide with photogeneratedhydrogen offers a very attractive route to provide synthesis gas(H2/CO-mixtures) suitable for the production of hydrocarbons asa renewable substitute for fossil fuels [133,168]. Alternatively,the conversion of carbon dioxide can be coupled to the photogen-eration of hydride reducing equivalents (Section 4.1) to generateprotonated C1-reduction products such as formic acid (2e�, 2H+)and formaldehyde (4e�, 4H+) as valuable chemical feedstocks ormethanol (6e�, 6H+) as a liquid solar fuel [35,127].
Among the plethora of systems proposed for the photoreduc-tion of CO2 some remarkable examples combine controlled sub-strate binding and activation with highly selective productformation in a single light-responsive molecule acting both as asensitizer and a catalyst [169–171]. These systems are promisingcandidates for the development of artificial carbon dioxide con-verting photoenzymes. For example, an interesting Fe-based pho-tocatalyst for the selective two-electron reduction of CO2 into COhas been reported recently [171], which can be considered as asimple light-driven CO-dehydrogenase model. The receptor prop-erties around the low-valent metal center are further assisted byadditional hydrogen bonding motifs to stabilize the CO2 substrateadduct formed. Unfortunately, triggering of the observedenzyme-like redox activity still requires short-wavelenght UV-light leading to catalyst stability problems, and the activity of thecatalytic Fe-center may also be hampered by self-inhibition withthe reaction product carbon monoxide [172]. In general, selectiveCO2 reduction systems utilizing a significant share of visible lightin the absence of any additional photosensitizer are currently stillvery rare [173–175] and usually contain expensive transition met-als with limited availability such as rhenium or iridium instead ofusing earth-abundant raw materials. Therefore, more researchefforts in this direction will be necessary to provide competitiveCO2 converting enzyme models powered by photons.
5. Artificial photoenzymes in medicine and life sciences
Beneficial and destructive effects of light exposure on livingorganisms have been observed since ancient times. Systematicapplications of sensitizing compounds in photodynamic therapy(PDT) are dating back to the nineteenth century [176] and continueto play a prominent role in the field of molecular photomedicine
[28,34,177]. Today, in medicine and life sciences light is alsoexploited as a valuable tool to precisely manipulate cells and tis-sues [178] or to release physiologically relevant substances ondemand from biocompatible precursors by irreversible cleavageof photolabile protecting (caging) groups [179]. Such optochemicaltools can also be successfully employed for indirectly influencingmore complex intracellular processes. For example gene expres-sion controlled by riboswitches [180] can be manipulated usinglight as an external trigger [181] and sequential gene silencingbecomes possible by introducing caged oligonucleotides [182]which has possible implications for the development of futuretherapeutic agents such as antisense drugs [183]. In addition,new techniques for bio-orthogonal ligation based on the photo-cleavage of protecting groups are now available to carry out selec-tive and spatially resolved labelling of biomolecules in their nativesetting even within living organisms [184].
The reversible photo-control of biological processes in life cellsrequires the design of more sophisticated light-responsive compo-nents which can be selectively addressed multiple times. Thisimportant functionality can be achieved with photochromic sys-tems or on–off switchable photoassisted catalysts[30,33,185,186]. Repeated contactless activation with spatial andtemporal precision is for instance possible in vivo by using geneti-cally encodable photoactuators which arose from the field of opto-genetics [186–189]. Control of intracellular signalling cascades andcell behaviour with engineered blue-light responsive (LOV-domaincarrying) receptor tyrosine kinases has also been demonstrated[190]. Reversible activation and inhibition of chemically modifiednative enzymes by photochromic switches already is a well-established optochemical technique [191,192]. In contrast, theexploitation of artificial photoenzyme catalysis as a versatile newtool for providing innovative solutions in medicine and lifesciences is still in its infancy. Only a few selected examples ofpotential applications can be presented in this review with theintention to stimulate further research in this new transdisci-plinary field, which together with optogenetic strategies has thepotential to revolutionize molecular biology, genetics, neuro-sciences and molecular photomedicine.
5.1. Light-controlled induction of apoptosis
Programmed cell death (apoptosis) is a fundamental biologicalprocess of paramount importance for the unobstructed mainte-nance and development of all animal and plant tissues. This cellsuicide program can be triggered on demand and plays a crucialrole for the control of regular morphogenesis [193] and for awell-functioning immune defense against viral and bacterial inva-sion [194]. Disrupted or altered apoptotic pathways on the otherhand are closely related to pathological mechanisms of many dis-eases including neurodegenerative disorders or cancer [195,196],where tumor cell resistance against apoptosis is observed.
The hydrolytic enzymes taking part in the reaction cascade ofapoptosis are present in a deactivated resting stage in all healthytissues waiting for an apoptotic death signal to trigger the suicideprogram. A tightly regulated network of coupled processes involv-ing proteolytic enzymes such as caspases [197] finally leads to anendonuclease catalyzed degradation of DNA into nucleosomal frag-ments which is regarded as a classical hallmark for the operation ofprogrammed cell death. It has been shown that apoptosis can alsobe photochemically triggered in animal or plant tissues underharsh experimental conditions such as UV-C light exposure[198,199]. Selective and precisely controlled induction of apoptoticresponse at the molecular level, however, requires to mimic indi-vidual key-steps of the amplifying proteolytic cascade leading toprogrammed cell death.

Fig. 11. Structure of a water-soluble gold porphyrin complex able to act as anartificial photonuclease enzyme for catalytic double-strand cleavage of DNAsubstrates under visible-light exposure [202].
110 G. Knör / Coordination Chemistry Reviews 325 (2016) 102–115
A possible target for achieving photoinduced apoptosis with anartificial enzyme is to create a light-sensitive counterpart of thenative hydrolytic enzyme CAD (caspase activated DNAse, EC3.1.30.2), which is one of the major apoptotic nucleases responsi-ble for catalytic double strand cleavage leading to DNA fragmenta-tion in the course of programmed cell death [200]. It has recentlybeen confirmed that initializing the CAD nuclease activity is suffi-cient to drive caspase-dependent apoptosis [201]. In the naturalsystem, an up-regulation of the nuclease enzyme activity requiresproteolytic cleavage of the chaperone ICAD acting as an inhibitor ofCAD in the inactive pronuclease state or CAD/ICAD [200,201]. Thiskey-step of switching on the enzymatic activity can be substitutedwith an artificial photoenzyme requiring light instead of caspaseactivation as a mediator for catalytic DNA double strand cleavage(Fig. 10).
Photochromic switches have been successfully employed forinfluencing the rate of RNA or DNA single strand damage withmodified nucleases or synthetic metal complexes [203–206]. Inearly attempts to create artificial endonuclease-like enzyme coun-terparts (photonucleases) the possibility of UV-light induced DNA-damage has also been explored [207]. However, in such systems nosignificant double-strand cleavage activity could be achieved[208]. Control of apoptosis-like DNA fragmentation with a light-driven molecular scissors (E⁄) as schematically described inFig. 10, in contrast, essentially requires a compound with catalyticphotonuclease activity in the excited state and an inactive, non-toxic and highly biocompatible dark-state (E). In Fig. 11 the struc-ture of such an artificial endonuclease photoenzyme is displayed,which has been rationally designed and optimized in our owngroup [30,202,209,210].
In the electronic ground state this cationic metalloporphyrincomplex can be regarded as an inhibited pronuclease form of aDNAse, which upon visible-light exposure transforms into an acti-vated artificial nuclease enzyme (Fig. 10). The catalytic perfor-mance of this class of gold pyrenyl-porphyrin systems is basedon visible-light induced electron transfer steps reaching turnovernumbers of several hundred cycles [210], which is an importantrequirement to accelerate a larger series of nucleic acid doublestrand cleavage steps in a row leading to DNA fragmentation. Arelated red-light sensitive photonuclease system has also beendescribed, carrying a high-valent metal-oxo (M@O) core suitable
Fig. 10. Possible strategies for controlling the selective destruction of individualcells by DNA cleavage into nucleosomal fragments. Left side: The hydrolytic enzymeCAD (caspase-activated DNAse) is complexed with an inhibitory protein (ICAD) inhealthy tissues. Apoptotic stimuli leading to caspase activation launch programmedcell death by releasing the active form of CAD [200]. Right side: A similar apoptosis-like DNA fragmentation process can also be triggered and sustained by switching onartificial nuclease enzyme catalysis photochemically [28,202].
for photocatalytic hydrogen atom transfer (HAT) from the sugar-phosphate backbone of the deoxyribose subunits in the minorgroove of the DNA double helix [209]. The design of this latterhydrolytic photoenzyme was inspired by the cytostatic mode ofaction of the naturally occurring antitumor antibiotic compoundbleomycin [211].
Besides the ability of switching on and off apoptosis-like reac-tivity and cytostatic effects as described above, selective artificialnuclease catalysis is also an attractive target for many other appli-cations in molecular biology and genetics [212]. A prominentexample is the search for future therapeutic methods based onantisense drugs for silencing disease genes [213–215]. Moreover,programmable designer nucleases such as zinc finger nucleases(ZFNs), transcription activator-like effector nucleases (TALENs) orthe so-called CRISPR-Cas9 system encoded for catalyzing highlysequence specific double-strand breaks play a central role for mod-ern genome modification tools [216–219]. Combining the precisetargeting strategies of such systems with the light-controlled andswitchable DNA-cleavage activity of an artificial nuclease photoen-zyme could open a novel route for safely controllable genomeediting.
5.2. Mimicking cellular response to oxidative stress
Living organisms rely on a set of antioxidant species and pro-tecting mechanisms to save their cells from undesired damage.Besides easily oxidizable compounds simply acting as sacrificialtraps for capturing reactive oxygen species (ROS), the protectionagainst oxidative stress also includes specialized enzymes withan antioxidant function such as catalase or superoxide dismutase(Fig. 1). In addition to these protective redox enzymes transform-ing potentially toxic oxidants into a harmless molecules for cells,nature in the course of evolution also established more complexcellular defense systems with a different mode of action that canbe precisely up-regulated and controlled. Mimicking the functionof such enzymes could open new approaches for the cure of disor-ders that are characterized by persistent oxidative stress andinflammation.
An important representative for this latter case is the catalyticactivity of heme oxygenase (EC 1.14.99.3) playing an essential pro-tective and regulatory role in both plants and animals [220]. Theparticular antioxidant function of this type of redox enzymes istriggered by degradation of the natural substrate heme, whilesimultaneously releasing the bile pigment biliverdin and carbonmonoxide which display unique cell protective characteristics.Therefore especially the activity of the inducible isoform hemeoxygenase-1 (HO-1) currently is an emerging target for pharmacol-ogy and drug discovery [221]. In this context, many researchgroups have focussed on the prominent role of endogeneous car-bon monoxide (CO) released as one of the main HO-1 metabolites

G. Knör / Coordination Chemistry Reviews 325 (2016) 102–115 111
and acting as a small gaseous cell-signalling molecule. Similar tonitric oxide (NO), carbon monoxide has been shown to activate sol-uble guanylyl cyclase (EC 4.6.1.2), which catalyses the conversionof guanosine triphosphate to cyclic guanosine monophosphate(cGMP). Cyclic GMP has been reported to ameliorate medical con-ditions such as pulmonary hypertension, thrombosis and arthe-riosclerosis among others. In addition to this, also non-cGMPdependent intracellular pathways and several other potential sitesof action of CO are discussed in the literature [222]. Besides itsimportant bio-regulatory and signaling properties especially inthe cardiovascular system, the physiological effects of smallamounts of endogeneous carbon monoxide are mainly anti-inflammatory and cell-protective including the promotion ofwound healing processes and the suppression of organ rejectionafter transplantation [221].
Similar to an oxidative stress-induced up-regulation of HO-1,the controlled release of carbon monoxide can also be achievedby using light as the external signal (Fig. 12). This strategy basedon photo-activated carbon monoxide releasing moieties (photo-CORMs) allows one to exactly define the location and timing ofthe CO delivery step [223], and therefore can serve to developlow-molecular weight enzyme model systems mimicking someof the beneficial cell-protective, anti-inflammatory and anti-hypertensive functions of inducible heme oxygenase [224].
The development and characterization of biocompatible com-pounds able to act as photo-CORMs is an active research topic.CO-loss from metal carbonyl complexes belongs to the earliestobservations made in biological inorganic photochemistry andphysiology [28,225], and in the last decade many different newmaterials have been tested for their photochemically triggereddecarbonylation reactivity under biomedically relevant conditions.Decomposition of CO-carrying compounds and carbon monoxidedelivery to tissues can be readily achieved with high efficienciesor quantum yields.
A major problem for the future therapeutic application of suchpotential drug precursor systems with CO-releasing moieties(CORMs) in general, however, is the fact that an exposure to carbonmonoxide exceeding the naturally regulated concentration levelsbecomes highly toxic to humans, due to the fatal function of COas a respiratory inhibitor [221,224]. In contrast, the activity of nat-ural heme oxygenase enzymes responsible for the controlledrelease of carbon monoxide in cells and tissues is typically onlyleading to very low local concentrations in the range of around1 nmol CO/h/mg [226,227]. In order to trigger the desired benefi-cial instead of toxic effects by light as an external stimulus, thedesign of any artificial photoenzyme replacing the physiologicalfunctions of the heme oxygenase system therefore should closelymatch the catalytic CO-delivery rate of the natural enzymecounterpart.
Fig. 12. Possible approaches for a pharmacological exploitation of the inducibleheme oxygenase-1 (HO-1) system are coupled to the controlled release of smallamounts of CO inside cells and tissues [221]. With a light-responsive CO-releasingmoiety (photo-CORM function), the physiological effects related to carbon monox-ide delivery can also be triggered by irradiation of an artificial heme-oxygenasephotoenzyme precursor (E), which can be up-regulated on demand [224].
We have recently presented a biocompatible system withphoto-CORM properties fulfilling these important requirements[224]. The water-soluble carbonyl complex investigated showsselective cellular uptake, no toxicity and an excellent dark-stability. Moreover, the observed quantum yields for light-triggered CO release are in an advantageous range, which shouldallow to photochemically mimick the desired effects of an up-regulated inducible heme oxygenase (HO-1) function on demandwithout risking to reach otherwise toxic levels of carbon monoxideunder physiological conditions. At the present stage, however, fur-ther ongoing tests will be necessary to confirm beneficial cardio-vascular, anti-inflammatory or cell-protective carbon monoxideeffects upon light exposure.
5.3. Modelling the biosynthesis of hormones
Hormones and their receptor binding analogues regulate a vari-ety of essential functions in all living systems [228]. Photocleav-able protecting groups can be attached to synthetic precursorcompounds, which allows to trigger physiological effects in bothanimal and plant systems based on caged hormones and phero-mones [229–231]. Mimicking the biocatalytic synthesis and thereversible dosage of the bioactive forms of hormones andhormone-like substances, in contrast, has not yet been systemati-cally targeted with photochemical methods.
Several of the artificial photoenzymes described in this revieware covering a substrate spectrum that could be exploited for alight-controlled catalytic production of hormones and phero-mones. For example, the amino acid halogenation steps catalyzedby thyroid peroxidase (TPO, EC 1.11.1.8.) in the presence of hydro-gen peroxide are important steps in the biosynthesis of thyroidhormones triiodo- and tetraiodothyronine [232,233]. In the syn-thesis of steroid hormones (steroidogenesis) the type of reactionscatalyzed by monooxygenases of the cytochrome P450 family(EC.1.14.14.1) plays an important role when bioactive steroidsare generated from cholesterol [234]. Artificial photoenzymemodel compounds replacing the redox function of these hemopro-teins as already described in the previous sections (Table 1) couldtherefore also be applied for a light-controlled generation of hor-mones and hormone-like compounds in future studies.
5.4. Bioanalytical tools based on artificial photoenzymes
Many other currently still unexplored applications of light-driven enzyme models in the field of medicine and life-sciencescould also be envisioned. For example, bioanalytics is one of themajor branches that might benefit from employing artificialenzymes as robust functional components. To inspire further workin this direction, the potential development of artificial photoen-zyme linked immunoassay systems will be suggested here.Enzyme-linked immunosorbent assays (ELISA) belong to the mostwidely used analytical tools for measuring trace biomarkers. Forthe selective detection of target analytes, a specific antibody-enzyme conjugate is formed in the ELISA-method. The role of theattached redox enzymes such as peroxidases in this type ofimmunoassay is to greatly amplify the detected signals in a cat-alytic substrate conversion step, which typically involves the pro-duction of intensely coloured, fluorescent or chemiluminescentproducts for convenient signal read-out (Fig. 13).
Attempts to use synthetic catalysts labeled with antibodies inorder to replace expensive and instable native enzymes requiredfor immunosorbent assays is currently gaining increasing attention[235]. It could already be shown in preliminary studies [236], thatbioconjugates of artificial oxidoreductase photoenzymes of thetype described in Section 2.2 can be covalently attached to anti-bodies without any loss of their photocatalytic activity. These early

Fig. 13. Functional components required for an artificial photoenzyme basedimmunoassay system related to the well-established ELISA technique. A light-driven oxidoreductase model compound is covalently conjugated to an antibodymolecule for specific recognition of an analyte. After formation of an antibody-target-complex, the artificial photoenzyme can be activated by light-exposure toachieve catalytic signal amplification strongly increasing the sensitivity limit of theassay [236].
112 G. Knör / Coordination Chemistry Reviews 325 (2016) 102–115
findings could serve as a successful starting point for developingthe first examples of an artificial photoenzyme-linked immunosor-bent assay (photo-ELISA) as schematically displayed in Fig. 13.
Fig. 14. Recycling of reduced ferredoxin by photocatalytic processes and artificialphotoenzyme reactions has the potential to open attractive light-powered multi-electron and proton transfer pathways in chemical-biological hybrid systems withimportant implications for the conversion and chemical storage of solar energy[247].
6. Coupled processes involving photocatalytic enzyme models
6.1. Reaction cascades: Towards metabolic pathways
In living organisms, biocatalytic reaction networks are used toproduce the vast variety of molecules found in nature. The combi-nation of several enzymatic transformations in concurrent one-potprocesses (multi-enzymatic cascade reactions) also plays animportant role in biotechnology and modern synthetic chemistry[237,238]. In this context, attempts are also made to establish sim-ple model systems for mimicking metabolism-like substrate trans-formations using multiple enzymatic cascade reactions in chemicalenvironments resembling artificial cell structures [239].
Substrate transformation in a sequence of consecutive photo-chemical steps has already been suggested in the initial studies onlight-driven enzyme model systems [29,30]. As a first succesfulexample into this important direction of controlling artificial meta-bolic pathways by visible-light, a coupled and highly selective two-step substrate oxidation cascade based on artificial oxidoreductasephotoenzymes has been reported recently [119]. Such a generalstrategy of combining individual enzyme model compounds asdescribed in this review inone-pot systemswill have tobemuch fur-ther elaborated to fully exploit the versatile potential of this novelapproach. Moreover, the combination of different wavelength-selectively addressable artificial photoenzymes will be able to adda new dimension on the control of such reaction networks.
6.2. Photo-biocatalytic hybrid systems
Despite of the increasing success of the concepts presented inthis review, the development of a new artificial photoenzyme sys-tem completely replacing the performance of its native biocatalystcounterpart is not an easy task. Therefore, especially in the case ofmore complex enzymatic reaction cascades, an alternativeapproach is to construct artificial light-controlled systems com-posed from both artificial photoenzyme and native biocatalystcomponents [33,127,240]. This very powerful shortcut pioneeredby Willner and others [241] allows much faster progress for theoptimization and practical application of light-mediated substrateconversion processes involving enzymatic catalysis steps. Researchon such chemical-biochemical hybrid systems combining syntheticbiology and photochemistry is therefore a rapidly emerging fieldwhich has been attracting growing interest within the last decade[242–246].
One of the possible strategies investigated is to regenerate andshuttle cofactors that will be accepted by natural enzymes to drivesubsequent reaction steps of an enzymatic cascade. Typical exam-ples are the recycling of the redox intermediates such as NAD(P)H[35] or reduced ferredoxin [247,248], which can also serve forpowering some of the most important biocatalytic processes suchas CO2 assimilation or nitrogen fixation (Fig. 14).
It is also possible to re-direct the naturally occuring flow ofelectron transfer chains and to build up enzyme combinationsnot available in nature for this purpose. These approaches are alsoopen for applications involving living systems and to couple themto artificial photoenzyme reactions, which is an attractive route forthe photochemical control of physiological and bioenergetic pro-cesses from outside the cell. A comprehensive treatment of thesefascinating possibilities, however, is not possible within the scopeof the present review.
6.3. Artificial photosynthetic reaction centers
The concept of light-driven enzyme model compounds recentlyalso led to a fully operating and competitive substitution of thebiochemical function of one of the most complex enzymes presentin nature. The light-driven plastocyanin: ferredoxin oxidoreduc-tase (EC 1.97.1.12) better known as Photosystem I (PS I) of photo-synthetic organisms could be replaced by a set of small-molecularcomponents and the first successful synthetic model reaction for abiomimetic accumulation of NADH with red-light photons hasbeen described [35,125]. While comparably high quantum yieldsin the range of the naturally evolved photosynthetic energy con-version systems have already been achieved [125], the artificialphotosynthetic reaction sequence described even operates with amore versatile set of electron delivering donors than PS I. Sincedonors with much less reduction power than that of the naturalsubstrate plastocyanine can be coupled to the artificial photosyn-thetic system, the light-reactions catalyzed are able to reach evenbetter maximum energy-storage efficiencies and fractional energyyields than those observed in nature, while providing the identicalchemical storage product as a solar fuel [35]. Such an approachbased on synthetic red-light powered enzyme model systems forcatalyzing artificial photosynthetic fuel production could help tominimize the considerable loss of two-thirds of absorbed photonenergy occurring in natural photosystems [249].
7. Outlook and conclusions
The unique concept of constructing photochemical enzymemodels is continuously expanding. Many succesful applicationsin the fields of chemistry, biology and medicine have already been

G. Knör / Coordination Chemistry Reviews 325 (2016) 102–115 113
demonstrated, which otherwise could hardly have been achieved.Compared to non-photochemical approaches for the design of arti-ficial enzymes, the strategy of employing catalytic reaction cyclesdriven by light offers several important advantages. In their inac-tive resting state such catalytic systems can be readily synthesized,characterized and stored. Upon light exposure they are activatedfor catalytic turnover on demand, which allows to reversiblyswitch on and fully regulate their performance in a reversible man-ner and also to directly power and accelerate endergonic processesfor chemical energy conversion. Light-driven enzyme-like com-pounds may also be designed for catalyzing abiotic substrate trans-formations or technologically relevant processes under reactionconditions for which no native biocatalysts are accessible. Due tothese very advantageous features, a bright future can be expectedfor the further development of new artificial photoenzymes as key-components of more complex assemblies such as photo-biochemical hybrid systems or artificial metabolic reaction net-works, as well as many other creative applications in chemistry,biology and medicine including the control of cellular functionsusing light and the exploitation of photocatalytic processes insideliving organisms.
Acknowledgements
Financial support by the Austrian Science Foundation (FWF pro-jects P25038 ‘‘Functional Light-Responsive Metal Carbonyl Sys-tems” and W1250 ‘‘NanoCell”), the Austrian Climate and EnergyFunds (FFG project 841186 ‘‘Artificial Photosynthesis”), and theEuropean Commission COST Action CM1202 ‘‘Supramolecular Pho-tocatalytic Water Splitting” (PERSPECT-H2O) is gratefullyacknowledged.
References
[1] M.A. Keller, G. Piedrafita, M. Ralser, Curr. Opin. Biotechnol. 34 (2015) 153–161.
[2] J.M. Thornton, C.A. Orengo, A.E. Todd, F.M.G. Pearl, J. Mol. Biol. 293 (1999)333–342.
[3] P. Romero, J. Wagg, M.L. Green, D. Kaiser, M. Krummenacker, P.D. Karp,Genome Biol. 6 (2004) R2.
[4] E. Buchner, Ber. Dt. Chem. Ges. 31 (1898) 568–574.[5] G. Bredig, Anorg. Fermente, Leipzig (1901).[6] W. Langenbeck, Die organischen Katalysatoren und ihre Beziehung zu den
Fermenten, Springer, Berlin, 1935.[7] L. Pauling, Nature 161 (1948) 707–709.[8] Sitzungsberichte d. Königl. Preuß. Akad. d. Wissenschaften zu Berlin (1907),
35. (Reports of Proceedings of the Royal Prussian Academy of Sciences:Translated by the author from a public speech of Emil Fischer on ‘‘Thechemistry of proteins and their relationships to biology‘‘ held in Berlin onJanuary 24th 1907).
[9] B. Gutte, R.B. Merrifield, J. Biol. Chem. 246 (1971) 1922–1941.[10] K.M. Ulmer, Science 219 (1983) 666–671.[11] J.W. Bryson, S.F. Betz, H.S. Lu, D.J. Suich, H.X. Zhou, K.T. O’Neil, W.F. DeGrado,
Science 270 (1995) 935–941.[12] M.G. Hoesl, C.G. Acevedo-Rocha, S. Nehring, M. Royter, C. Wolschner, B.
Wiltschi, N. Budisa, G. Antranikian, ChemCatChem 3 (2011) 213–221.[13] C. Hu, S.I. Chan, E.B. Sawyer, Y. Yu, J. Wang, Chem. Soc. Rev. 43 (2014) 6498–
6510.[14] L. Fruk, C.-H. Kuo, E. Torres, C.M. Niemeyer, Angew. Chem. Int. Ed. 48 (2009)
1550–1574.[15] N. Kawakami, O. Shoji, Y. Watanabe, ChemBioChem 13 (2012) 2045–2047.[16] L. Curatti, J.A. Hernandez, R.Y. Igarashi, B. Soboh, D. Zhao, L.M. Rubio, Proc.
Natl. Acad. Sci. U.S.A. 104 (2007) 17626–17631.[17] G. Berggren, A. Adamska, C. Lambertz, T.R. Simmons, J. Esselborn, M. Atta, S.
Gambarelli, J.-M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero, M.Fontecave, Nature 499 (2013) 66–69.
[18] J.C. Lewis, ACS Catal. 3 (2013) 2954–2975.[19] S.A. Benner, A.M. Sismour, Nat. Rev. Genet. 6 (2005) 533–543.[20] F. Yu, V.M. Cangelosi, M.L. Zastrow, M. Tegoni, J.S. Plegaria, A.G. Tebo, C.S.
Mocny, L. Ruckthong, H. Qayyum, V.L. Pecoraro, Chem. Rev. 114 (2014) 3495–3578.
[21] M. Dürrenberger, T.R. Ward, Curr. Opin. Chem. Biol. 19 (2014) 99–106.[22] M. Singh, A. Vaidya, Syst. Synth. Biol. 9 (2015) 191–195.[23] Z. Dong, Q. Luo, J. Liu, Chem. Soc. Rev. 41 (2012) 7890–7908.[24] R. Breslow (Ed.), Artificial Enzymes, Wiley-VCH, Weinheim, 2005.
[25] A.J. Kirby, F. Hollfelder, From Enzyme Models to Model Enzymes, RSCPublishing, Cambridge, 2009.
[26] D.P. Riley, Chem. Rev. 99 (1999) 2573–2588.[27] J.S. Thompson, Y. Chu, J. Glass, A.A. Tapp, S.A. Brown, Free Rad. Res. 44 (2010)
529–540.[28] G. Knör, U. Monkowius, Adv. Inorg. Chem. 63 (2011) 235–289.[29] G. Knör, Coord. Chem. Rev. 171 (1998) 61–70.[30] G. Knör, Habilitation Thesis, University of Regensburg, 2001.[31] G. Knör, 14th International Symposium on the Photochemistry and
Photophysics of Coordination Compounds, Vesprém, Hungary, 2001, Bookof Abstracts, p. 59.
[32] G. Knör, ChemBioChem 2 (2001) 593–596.[33] G. Knör, Chem. Eur. J. 15 (2009) 568–578.[34] G. Stochel, M. Brindell, W. Macyk, Z. Stasicka, K. Szaciłowski, Bioinorganic
Photochemistry, Wiley, Chichester, 2009.[35] G. Knör, Coord. Chem. Rev. 304–305 (2015) 102–108.[36] V. Balzani, P. Ceroni, A. Juris, Photochemistry and Photophysics: Concepts,
Research, Applications, Wiley, Weinheim.[37] A. Albini, M. Fagnoni, Photochemically Generated Intermediates in Synthesis,
Wiley, Hoboken, 2013.[38] R.H. Crabtree (Ed.), Energy Production and Storage, Wiley, Chichester, 2010.[39] P. Hegemann, S. Sigrist (Eds.), Optogenetics, de Gruyter, Berlin, 2013.[40] For a given photon flux, the observed reaction rates are proportional to the
catalyst concentration in the optically dilute regime, reach an optimum levelat total absoprption of the incident light, and may then even level off again athigher catalyst concentrations as a result of reduced penetration depth of thelight beam and several other side effects. For a critical discussion onnormalizing photocatalytic reaction rates see: H. Kisch, Angew. Chem. 122(2010) 9782–9783.
[41] S. Park, N. Agmon, J. Phys. Chem. B 112 (2008) 5977–5987.[42] F. Himo, L.A. Eriksson, F. Maseras, P.E.M. Siegbahn, J. Am. Chem. Soc. 122
(2000) 8031–8036.[43] G. Knör, A. Vogler, Inorg. Chem. 33 (1994) 314–318.[44] P.T. Anastas, M.M. Kirchhoff, Acc. Chem. Res. 35 (2002) 686–694.[45] R. Noyori, Tetrahedron 66 (2010) 1028.[46] H. Kisch, in: D.C. Neckers, G. von Bünau, W.S. Jencks (Eds.), Adv. in
Photochem., vol. 26, Wiley, Hoboken, 2001.[47] M. Oelgemöller, C. Jung, J. Mattay, Pure Appl. Chem. 79 (2007) 1939–1947.[48] N. Hoffmann, J. Photochem. Photobiol. C: Photochem. Rev 9 (2008) 43–60.[49] S. Protti, M. Fagnoni, Photochem. Photobiol. Sci. 8 (2009) 1499–1516.[50] A. Albini, M. Fagnoni (Eds.), Handbook of Synthetic Photochemistry, Wiley-
VCH, Weinheim, 2010.[51] T.P. Yoon, M.A. Ischay, J. Du, Nat. Chem. 2 (2010) 527–532.[52] C.K. Prier, D.A. Rankic, D.W.C. MacMillan, Chem. Rev. 113 (2013) 5322–5363.[53] M. Oelgemöller, O. Shvydkiv, Molecules 16 (2011) 7522–7550.[54] S. Mumatz, C. Sattler, M. Oelgemöller, in: T. Letcher, J. Scott, D. Patterson
(Eds.), Chemical Processes for a Sustainable Future, RSC, Cambridge, UK, 2015.[55] R. Porta, M. Benaglia, A. Puglisi, Org. Process Res. Dev. 20 (2016) 2–25.[56] C. Jang, Y. Inoue, Chem. Soc. Rev. 43 (2014) 4123–4143.[57] J. Du, K.L. Skubi, D.M. Schultz, T.P. Yoon, Science 344 (2014) 392–396.[58] R. Brimioulle, D. Lehnart, M.M. Maturi, T. Bach, Angew. Chem. Int. Ed. 54
(2015) 3872–3890.[59] A. Liese, Adv. Biochem. Eng. Biotechnol. 92 (2005) 197–224.[60] J.A. Stubbe, W.A. van der Donk, Chem. Rev. 98 (1998) 705–762.[61] P.A. Frey, Ann. Rev. Biochem. 70 (2001) 121–148.[62] W. Buckel, Angew. Chem. Int. Ed. 48 (2009) 6779–6787.[63] J.B. Broderick, B.R. Du, K.S. Duschene, E.M. Shepard, Chem. Rev. 114 (2014)
4229–4317.[64] J.L. Vey, C.L. Drennan, Chem. Rev. 111 (2011) 2487–2506.[65] K.A. Shisler, J.B. Broderick, Arch. Biochem. Biophys. 546 (2014) 64–71.[66] For a recent compilation of peptide radical structural motifs see: F. Turecek, J.
Phys. Chem. A 119 (2015) 10101–10111.[67] J.J. Warren, J.M. Mayer, Biochem 54 (2015) 1863–1878.[68] T. Ramasarma, Ind. J. Biochem. Biophys. 49 (2012) 295–305.[69] X. Wanga, R. Ullrich, M. Hofrichter, J.T. Groves, Proc. Natl. Acad. Sci. U.S.A. 112
(2015) 3686–3691.[70] N. Gagnon, W.B. Tolman, Acc. Chem. Res. 48 (2015) 2126–2131.[71] V. Resch, J.H. Schrittwieser, E. Siirola, W. Kroutil, Curr. Opin. Biotechnol. 22
(2011) 793–799.[72] S. Protti, M. Fagnoni, D. Ravelli, ChemCatChem 7 (2015) 1516–1523.[73] D. Ravelli, G. Knör, unpublished results.[74] B. Kräutler, Coord. Chem. Rev. 111 (1991) 215–220.[75] A. Vogler, H. Kunkely, J. Organomet. Chem. 453 (1993) 269–272.[76] R.J. Kutta, S.J. Hardman, L.O. Johannissen, B. Bellina, H.L. Messiha, J.M. Ortiz-
Guerrero, M. Elias-Arnanz, S. Padmanabhan, P. Barran, N.S. Scrutton, A.R.Jones, Nat. Commun. 6 (2015) 7907, http://dx.doi.org/10.1038/ncomms8907.
[77] M. Giedyk, K. Goliszewska, D. Gryko, Chem. Soc. Rev. 44 (2015) 3391–3404.[78] M. Giedyk, K. Goliszewska, K.Ó. Proinsias, D. Gryko, Chem. Comm. 52 (2016)
1389–1392.[79] Y. Hisaeda, K. Tahara, H. Shimakoshi, T. Masuko, Pure Appl. Chem. 85 (2013)
1415–1426.[80] J. Xu, H. Shimakoshi, Y. Hisaeda, J. Organomet. Chem. 782 (2015) 89–95.[81] M. Bommer, C. Kunze, J. Fesseler, T. Schubert, G. Diekert, H. Dobbek, Science
346 (2014) 455–458.[82] K.A.P. Payne, C.P. Quezada, K. Fisher, M.S. Dunstan, F.A. Collins, H. Sjuts, C.
Levy, S. Hay, S.E.J. Rigby, D. Leys, Nature 517 (2015) 513–516.

114 G. Knör / Coordination Chemistry Reviews 325 (2016) 102–115
[83] G.T. Babcock, M. Wikström, Nature 356 (1992) 301–309.[84] C. Ostermeier, S. Iwata, H. Michl, Curr. Opin. Struct. Biol. 6 (1996) 460–466.[85] A. Hernández-Ortega, M.G. Quesne, S. Bui, D.J. Heyes, R.A. Steiner, N.S.
Scrutton, S.P. de Visser, J. Am. Chem. Soc. 137 (2015) 7474–7487.[86] W.P. Dilkman, G. Gonzalo, A. Mattevi, M.W. Fraaije, Appl. Microbiol.
Biotechnol. 97 (2013) 5177–5188.[87] S.B. Bankar, M.V. Bule, R.S. Singhal, L. Anathanaryan, Biotechnol. Adv. 27
(2009) 489–501.[88] A. Panday, M.K. Sakoo, D. Osorio, S. Batra, Cell. Mol. Immunol. 12 (2015) 5–23.[89] M.E. Newcomer, A.R. Brash, Protein Sci. 24 (2015) 298–309.[90] D.C. Pryde, D. Dalvie, Q. Hu, P. Jones, R.S. Obach, T.-D. Tran, J. Med. Chem. 53
(2010) 8160–8441.[91] U. Kappler, J.H. Enemark, J. Biol. Inorg. Chem. 20 (2015) 253–264.[92] W. Wong, A.D. Liang, S.J. Lippard, Acc. Chem. Res. 48 (2015) 2632–2639.[93] T. Kurahashi, Inorg. Chem. 54 (2015) 8356–8366.[94] S.L. Collom, A.J. Bloomfield, P.T. Anastas, Ind. Eng. Chem. Res. (2015), http://
dx.doi.org/10.1021/acs.iecr.5b03674.[95] R. Lechner, S. Kümmel, B. König, Photochem. Photobiol. Sci. 9 (2010) 1367–
1377.[96] G. Knör, Inorg. Chem. 35 (1996) 7916–7918.[97] W. Schöfberger, F. Lengwin, L.M. Reith, M. List, G. Knör, Inorg. Chem.
Commun. 13 (2010) 1187–1190.[98] L.M. Reith, M. Himmelsbach, W. Schöfberger, G. Knör, J. Photochem.
Photobiol. A: Chem. 218 (2011) 247–253.[99] E.T. Denisov, I.B. Afanasev, Oxidation and Antioxidants in Organic Chemistry
and Biology, CRC, 2005.[100] G. Knör, A. Vogler, S. Roffia, F. Paolucci, V. Balzani, Chem. Comun. (1996)
1643–1644.[101] M. Ertl, E. Wöß, G. Knör, Photochem. Photobiol. Sci. 14 (2015) 1826–1830.[102] Y. Yamada, M. Yoneda, S. Fukuzumi, Energy Environ. Sci. 8 (2015) 1698–1701.[103] K.D. Karlin, Nature 463 (2010) 168–169.[104] J. Hohenberger, K. Ray, K. Meyer, Nat. Commun. 3 (2012), http://dx.doi.org/
10.1038/ncomms1718.[105] E. Tordin, M. List, U. Monkowius, S. Schindler, G. Knör, Inorg. Chim. Acta 402
(2013) 90–96.[106] W. Nam, Acc. Chem. Res. 18 (2015) 2415–2423.[107] N. Cox, M. Retegan, F. Neese, D.A. Pantazis, A. Boussac, W. Lubitz, Science 345
(2014) 804–808.[108] G. Torrelo, U. Hanefeld, F. Hollmann, Catal. Lett. 145 (2015) 309–345.[109] R. Balasubramanian, S.M. Smith, S. Rawat, L.A. Yatsunyk, T.L. Stemmler, A.C.
Rosenzweig, Nature 465 (2010) 115–119.[110] M.T. Green, J.H. Dawson, H.B. Gray, Science 304 (2004) 1653–1656.[111] U. Förstermann, W.L. Sessa, Eur. Heart J. 33 (2012) 829–837.[112] X. Wang, R. Ullrich, M. Hofrichter, J.T. Groves, Proc. Natl. Acad. Sci. U.S.A. 112
(2015) 3686–3691.[113] P.R. Ortiz de Montellano (Ed.), Cytoochrome P450 – Structure, Mechanism
and Biochemistry, Springer, Heidelberg, 2015.[114] D. Vuka, O. Horváth, Z. Marinicc, I. Škorica, J. Mol. Struct. 1107 (2016) 70–76.[115] S. Shaik, H. Chen, D. Janardanan, Nat. Chem. 3 (2011) 19–27.[116] N. Dietl, M. Schlangen, H. Schwarz, Angew. Chem. 124 (2012) 5638–5650.[117] D. Usharani, D. Janardanan, C. Li, S. Shaik, Acc. Chem. Res. 46 (2013) 471–482.[118] M. Schulz, C. Paulik, G. Knör, J. Mol. Catal. A: Chem. 347 (2011) 60–64.[119] M. Hajimohammadi, C. Schwarzinger, G. Knör, RSC Adv. 2 (2012) 3257–3260.[120] G. Knör, Chem. Phys. Lett. 330 (2000) 383–388.[121] L.M. Reith, M. Stiftinger, U. Monkowius, G. Knör, W. Schöfberger, Inorg. Chem.
50 (2011) 6788–6797.[122] T. Weinert, S.G. Huwiler, J.W. Kung, S. Weidenweber, P. Hellwig, H.-J. Stärk, T.
Biskup, S. Weber, J.J.H. Cotelesage, G.N. George, U. Ermler, M. Boll, Nat. Chem.Biol. 11 (2015), http://dx.doi.org/10.1038/nchembio.1849.
[123] J.H. Golbeck (Ed.), Photosystem I: The Light-driven Plastocyanine:FerredoxinOxidoreductase, Advances in Photosynthesis and Respiration,vol. 24, Springer, Dordrecht, 2006.
[124] A. Nakamura, T. Suzawa, Y. Kato, T. Watanabe, Plant Cell Physiol. 52 (2011)815–823.
[125] K.T. Oppelt, E. Wöß, M. Stiftinger, W. Schöfberger, W. Buchberger, G. Knör,Inorg. Chem. 52 (2013) (1922) 11910–11911.
[126] N. Carrillo, E.A. Ceccarelli, Eur. J. Biochem. 270 (2003) 1900–1915.[127] M. Aresta, A. Dibenedetto, W. Macyk, in: E.A. Rozhkova, K. Ariga (Eds.), From
Molecules to Materials: Pathways to Artificial Photosynthesis, Springer,Heidelberg, 2015, pp. 149–169.
[128] A. McSkimming, S.B. Colbran, Chem. Soc. Rev. 42 (2013) 5439–5488.[129] M. de Torres, J. Dimroth, I.W.C.E. Arends, J. Keilitz, F. Hollmann, Molecules 17
(2012) 9835–9841.[130] K.T. Oppelt, J. Gasiorowski, D.A.M. Egbe, J.P. Kollender, M. Himmelsbach, A.W.
Hassel, N.S. Sariciftci, G. Knör, J. Am. Chem. Soc. 136 (2014) 12721–12729.[131] R.H. Fish, J. Organomet. Chem. 782 (2015) 3–16.[132] T. Quinto, D. Häussinger, V. Köhler, T.R. Ward, Org. Biomol. Chem. 13 (2015)
357–360.[133] E. Portenkirchner, K.T. Oppelt, D.A.M. Egbe, G. Knör, N.S. Sariciftci,
Nanomater. Energy 2 (2013) 134–147.[134] J.C. Fontecilla-Camps, A. Volbeda, C. Cavazza, Y. Nicolet, Chem. Rev. 107
(2007) 4273–4303.[135] W. Lubitz, H. Ogata, O. Rüdiger, E. Reijerse, Chem. Rev. 114 (2014) 4081–
4148.[136] I.P. Georgakaki, L.M. Thomson, E.J. Lyon, M.B. Hall, M.Y. Darensbourg, Coord.
Chem. Rev. 238–239 (2003) 255–266.
[137] C. Tard, C.J. Pickett, Chem. Rev. 109 (2009) 2245–2274.[138] B.E. Barton, M.T. Olsen, T.B. Rauchfuss, Curr. Opin. Biotechnol. 21 (2010) 292–
297.[139] J.A. Wright, C.J. Pickett, in: W. Weigand, P. Schollhammer (Eds.), Bioinspired
Catalysis – Metal-Sulfur Complexes, Wiley-VCH, Weinheim, Germany, 2015,pp. 161–198.
[140] M. Kaiser, G. Knör, Eur. J. Inorg. Chem. (2015) 4199–4206.[141] D. Streich, Y. Astuti, M. Orlandi, L. Schwartz, R. Lomoth, L. Hammarström, S.
Ott, Chem. Eur. J. 16 (2010) 60–63.[142] C. Topf, U. Monkowius, G. Knör, Inorg. Chem. Commun. 21 (2012) 147–150.[143] F. Wang, W.-G. Wang, H.-Y. Wang, G. Si, C.-H. Tung, L.-Z. Wu, ACS Catal. 2
(2012) 407–416.[144] W.R. McNamara, Z. Han, C.-J.M. Yin, W.W. Brennessel, P.L. Holland, R.
Eisenberg, Proc. Natl. Acad. Sci. U.S.A. 109 (2012) 15594–15599.[145] Z. Han, W.R. McNamara, M.-S. Eum, P.L. Holland, R. Eisenberg, Angew. Chem.
124 (2012) 1699–1702.[146] W.T. Eckenhoff, W.W. Brennessel, R. Eisenberg, Inorg. Chem. 53 (2014) 9860–
9869.[147] Z. Han, R. Eisenberg, Acc. Chem. Res. 47 (2014) 2537–2544.[148] T.R. Simmons, G. Berggren, M. Bacchi, M. Fontecave, V. Artero, Coord. Chem.
Rev. 270–271 (2014) 127–150.[149] M. Gomez-Mingot, J.-P. Porcher, T.-K. Todorova, T. Fogeron, C. Mellot-
Draznieks, Y. Li, M. Fontecave, J. Phys. Chem. B 119 (2015) 13524–13533.[150] W. Wang, T.B. Rauchfuss, L. Bertini, G. Zampella, J. Am. Chem. Soc. 134 (2012)
4525–4528.[151] M. Aresta, A. Dibenedetto, E. Quaranta, Reaction Mechanisms in Carbon
Dioxide Conversion, Springer, Heidelberg, 2016.[152] C.A. Eckert, C.T. Trinh (Eds.), Biotechnology for Biofuel Production and
Optimization, Elsevier, Amsterdam, 2016. 480.[153] J.H. Jeoung, J. Fesseler, S. Goetzl, H. Dobbek, Met. Ions Life Sci. 14 (2014) 37–
69.[154] M.W. Ribbe, Angew. Chem. 127 (2015) 8455–8457.[155] J. Fesseler, J.H. Jeoung, H. Dobbek, Angew. Chem. 127 (2015) 8680–8684.[156] M.D. Doherty, D.C. Grills, J.T. Muckerman, D.E. Polyansky, E. Fujita, Coord.
Chem. Rev. 254 (2010) 2472–2482.[157] R. Reithmeier, C. Bruckmeier, B. Rieger, Catalysts 2 (2012) 544–571.[158] A.M. Appel, J.E. Bercaw, A.B. Bocarsly, H. Dobbek, D.L. Dubois, M. Dupuis, J.G.
Ferry, E. Fujita, R. Hille, P.A. Kennis, C.A. Kerfeld, R.H. Morris, C.H.F. Peden, A.R.Portis, S.W. Ragsdale, T.B. Rauchfuss, N.H. Reek, L.C. Seefeldt, R.K. Thauer, G.L.Waldrop, Chem. Rev. 113 (2013) 662–6658.
[159] L. Gan, D. Jennings, J. Laureanti, A.K. Jones, Top. Organomet. Chem. (2015),http://dx.doi.org/10.1007/3418_2015146.
[160] Y. Yamazakia, H. Takeda, O. Ishitani, J. Photochem. Photobiol. C: Photochem.Rev. 25 (2015) 106–137.
[161] K. Koike, H. Hori, M. Ishizuka, J.R. Westwell, K. Takeuchi, T. Ibusuki, K. Enjouji,H. Konno, K. Sakamoto, O. Ishitani, Organometallics 16 (1997) 5724–5729.
[162] E. Barton Cole, P.S. Lakkaraju, D.M. Rampulla, A.J. Morris, E. Abelev, A.B.Bocarsly, J. Am. Chem. Soc. 132 (2010) 11539–11551.
[163] Y.-E. Kim, J. Kim, Y. Lee, Chem. Commun. 50 (2014) 11458–11561.[164] Y. Kou, Y. Nabetani, D. Masui, T. Shimada, S. Takagi, H. Tachibana, H. Inoue, J.
Am. Chem. Soc. 136 (2014) 6021–6030.[165] J. Bonin, M. Robert, M. Routier, J. Am. Chem. Soc. 136 (2014) 16768–16771.[166] T. Morimoto, T. Nakajima, S. Sawa, R. Nakanishi, D. Imori, O. Ishitani, J. Am.
Chem. Soc. 135 (2013) 16825–16828.[167] P.L. Cheung, C.W. Machan, A.Y.S. Malkhasian, J. Agarwal, C.P. Kubiak, Inorg.
Chem. 55 (2016) 3192–3198.[168] P.G. Alsabeh, A. Rosas-Hernández, E. Barsch, H. Junge, R. Ludwig, M. Beller,
Catal. Sci. Technol. (2016), http://dx.doi.org/10.1039/C5CY01129A.[169] J. Hawecker, J.-M. Lehn, R. Ziessel, J. Chem. Soc., Chem. Commun. (1983) 536–
538.[170] A. Morris, G.J. Meyer, E. Fujita, Acc. Chem. Res. 42 (2009) 1983–1994.[171] J. Bonin, M. Chaussemier, M. Robert, M. Routier, ChemCatChem 6 (2014)
3200–3207.[172] B. Mondal, A. Rana, P. Sen, A. Dey, J. Am. Chem. Soc. 137 (2015) 11214–11217.[173] E. Portenkirchner, E. Kianfar, N.S. Sariciftci, G. Knör, ChemSusChem 7 (2014)
1347–1351.[174] R.O. Reithmeier, S. Meister, A. Siebel, B. Rieger, Dalton Trans. 44 (2015) 6466–
6472.[175] A.J. Huckaba, E.A. Sharpe, J.H. Delcamp, Inorg. Chem. 55 (2016) 682–690.[176] H. von Tappeiner, O. Raab, Münch. Med. Wochenschr. 47 (1900) 5.[177] Z. Li, K.B. Grant, RSC Adv. 6 (2016) 24617–24634.[178] M.W. Berns, K.O. Greulich (Eds.), Methods in Cell Biology, Laser Manipulation
of Cells and Tissues, vol. 82, Academic Press, 2007.[179] P. Klán, T. Šolomek, C.G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A.
Kostikov, J. Wirz, Chem. Rev. 113 (2013) 119–191.[180] P. Machtel, K. Bakowska- _Zywicka, M. _Zywicki, J. Appl. Genet. (2016), http://
dx.doi.org/10.1007/s13353-016-0341-x.[181] S. Walsh, L. Gardner, A. Deiters, G.J. Williams, ChemBioChem 15 (2014) 1346–
1351.[182] S. Yamazoe, Q. Liu, L.E. McQuade, A. Deiters, J.K. Chen, Angew. Chem. Int. Ed.
53 (2014) 10114–10118.[183] C.F. Bennett, E.E. Swayze, Annu. Rev. Pharmacol. Toxicol. 50 (2010) 259–
293.[184] L. Shah, S.T. Laughlin, I.S. Carrico, J. Am. Chem. Soc. 138 (2016) 5186–5189.[185] C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer, A. Heckel, Angew. Chem. Int.
Ed. Engl. 51 (2012) 8446–8476.

G. Knör / Coordination Chemistry Reviews 325 (2016) 102–115 115
[186] A. Gautier, C. Gauron, M. Volovitch, D. Bensimon, L. Jullien, S. Vriz, Nature,Chem. Biol. 10 (2014) 533–541.
[187] B.V. Zemelman, N. Nesnan, G.A. Lee, G. Miesenböck, Proc. Natl. Acad. Sci. U.S.A. 100 (2003) 1352–1357.
[188] G. Miesenböck, Science 326 (2009) 395–399.[189] K. Deisseroth, Nat. Methods 8 (2010) 26–29.[190] M. Grusch, K. Schelch, R. Riedler, E. Reichhart, C. Differ, W. Berger, Á.-.. Inglés-
Prieto, H. Janovjak, EMBO J. 33 (2014) 1713–1726.[191] T. Shimoboji, E. Larenas, T. Fowler, S. Kulkarni, A.S. Hoffman, P.S. Stayton,
Proc. Natl. Acad. Sci. U.S.A. 99 (2002) 16592–16596.[192] M. Goeldner, R. Givens (Eds.), Dynamic Studies in Biology: Phototriggers,
Photoswitches and Caged Biomolecules, Wiley-VCH, Weinheim, 2005.[193] M. Suzanne, H. Steller, Nature 20 (2013) 669–675.[194] M. Bots, J.P. Medema, J. Cell Sci. 119 (2006) 5011–5014.[195] B. Favaloro, N. Allocati, V. Graziano, C. Di Ilio, V. De Laurenzi, Aging 4 (2012)
330–349.[196] J.M. Brown, L.D. Attardi, Nat. Rev. 5 (2005) 231–237.[197] J. Li, J. Yuan, Oncogene 27 (2008) 6144–6206.[198] S.J. Martin, T.G. Cotter, Int. J. Radiat. Biol. 59 (1991) 1001–1016.[199] A. Danon, P. Gallois, FEBS Lett. 437 (1998) 131–136.[200] M. Enari, H. Sakahira, H. Yokoyama, K. Okawa, A. Iwamatsu, S. Nagata, Nature
391 (1998) 43–50.[201] K. Samejima, H. Ogawa, A.V. Ageichik, K.L. Peterson, S.H. Kaufmann, M.T.
Kanemaki, W.C. Earnshaw, J. Biol. Chem. 289 (2014) 31617–31623.[202] M. Häubl, L. Reith, B. Gruber, U. Karner, N. Müller, G. Knör, W. Schöfberger, J.
Biol. Inorg. Chem. 14 (2009) 1037–1052.[203] Y. Liu, D. Sen, J. Mol. Biol. 341 (2004) 887–892.[204] S. Keiper, J.S. Vyle, Angew. Chem. Int. Ed. 45 (2006) 3306–3309.[205] H. Prakash, A. Shodai, H. Yasui, H. Sakurai, S. Hirota, Inorg. Chem. 47 (2008)
5045–5047.[206] B. Schierling, A.-J. Noël, W. Wende, L.T. Hien, E. Volkov, E. Kubareva, T.
Oretskaya, M. Kokkinidis, A. Römpp, B. Spengler, A. Pingoud, Proc. Natl. Acad.Sci. U.S.A. 107 (2010) 1361–1366.
[207] B. Armitage, C. Yu, C. Devadoss, G.B. Schuster, J. Am. Chem. Soc. 116 (1994)9847–9859.
[208] B. Armitage, Chem. Rev. 98 (1998) 1171–1200.[209] G. Knör, J. Inf. Recording 25 (2000) 111–117.[210] G. Knör, Inorg. Chem. Commun. 4 (2001) 160–163.[211] C.A. Claussen, E.C. Long, Chem. Rev. 99 (1999) 2797–2816.[212] M. Murtola, M. Wenska, R. Strömberg, J. Am. Chem. Soc. 132 (2010) 8984–
8990.[213] S.M. Hammond, E. Bernstein, D. Beach, G.J. Hannon, Nature 404 (2000) 293–
296.[214] C.R. Dass, P.F.M. Choong, L.M. Khachigian, Mol. Cancer Ther. 7 (2008) 243–
251.[215] A.I. Taylor, V.B. Pinheiro, M.J. Smola, A.S. Morgunov, S. Peak-Chew, C. Cozens,
K.M. Weeks, P. Herdewijn, P. Holliger, Nature 518 (2015) 427–430.[216] S. Chandrasegaran, D. Carroll, J. Mol. Biol. 428 (2016) 963–989.
[217] H.R. Siddique, MOJ Cell Sci. Rep. 3 (2016) 00046, http://dx.doi.org/10.15406/mojcsr.2016.03.00046.
[218] M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J.A. Doudna, E. Charpentier,Science 337 (2014) 816–821.
[219] C. Anders, O. Niewoehner, A. Duerst, M. Jinek, Nature 513 (2014) 569–573.[220] A. Wilks, Antioxid. Redox Signal. 4 (2002) 603–614.[221] R. Motterlini, R. Foresti, Antioxid. Redox Signal. 20 (2014) 1810–1826.[222] B.E. Mann, Roberto Motterlini, Chem. Commun. 4197–4208 (2007).[223] R.D. Rimmer, A.E. Pierri, P.C. Ford, Coord. Chem. Rev. 256 (2012) 1509–1519.[224] E. Kianfar, C. Schäfer, M.R. Lornejad-Schäfer, E. Portenkirchner, G. Knör, Inorg.
Chim. Acta 435 (2015) 174–177.[225] J. Haldane, J. Smith, J. Physiol. 20 (1896) 497–520.[226] H.J. Vreman, D.K. Stevenson, Anal. Biochem. 168 (1988) 31–38.[227] E.F. Chang, R.J. Wong, H.J. Vreman, T. Igarashi, E. Galo, F.R. Sharp, D.K.
Stevenson, L.J. Noble-Haeusslein, J. Neurosci. 23 (2003) 3689–3696.[228] C.S. Westfall, A.M. Muehler, J.M. Jez, J. Biol. Chem. 288 (2013) 19304–19311.[229] J.L. Ward, M.H. Beale, Phytochemistry 38 (1995) 811–816.[230] K. Hayashi, K. Hashimoto, N. Kusaka, A. Yamazoe, H. Fukaki, M. Tasaka, H.
Nozaki, Bioorg. Med. Chem. Lett. 16 (2006) 2470–2474.[231] A. Herrmann, Photochem. Photobiol. Sci. 11 (2012) 446–459.[232] M. Morrison, G.R. Schonbaum, Ann. Rev. Biochem. 45 (1976) 861–888.[233] L. Wang, X. Zhou, M. Fredimoses, S. Liao, Y. Liu, RSC Adv. 4 (2014) 57350–
57376.[234] I. Hanukoglu, J. Steroid Biochem. Mol. Biol. 43 (1992) 779–804.[235] Z. Wang, S. Zheng, J. Cai, P. Wang, J. Feng, X. Yang, L. Zhang, M. Ji, F. Wu, N. He,
N. Wan, Anal. Chem. 85 (2013) 11602–11609.[236] G. Knör, B. Oswald, O. Wolfbeis, unpublished results.[237] S. Panke, A. Kümmel, M. Schümperli, M. Heinemann, Chim. Oggi Chem. Today
22 (2004) 44–47.[238] E. Ricca, B. Brucher, J.H. Schrittwieser, Adv. Synth. Catal. 253 (2011) 2239–
2262.[239] L. Hosta-Rigau, M.J. York-Duran, Y. Zhang, K.N. Goldie, B. Städler, ACS Appl.
Mater. Interfaces 6 (2014) 12771–12779.[240] D.D. Young, A. Deiters, Org. Biomol. Chem. 5 (2007) 999–1005.[241] I. Willner, N. Lapidot, J. Am. Chem. Soc. 113 (1991) 3625–3626.[242] S. Rumpel, J.F. Siebel, C. Farès, J. Duan, E. Reijerse, T. Happe, W. Lubitz, M.
Winkler, Energy Environ. Sci. 7 (2014) 3296–3301.[243] L.M. Lassen, A.Z. Nielsen, B. Ziersen, T. Gnanasekaran, B. Lindberg Møller, P.E.
Jensen, ACS Synth. Biol. 3 (2014) 1–12.[244] L.M. Utschig, S.R. Soltau, D.M. Tiede, Curr. Opin. Chem. Biol. 25 (2015) 1–8.[245] Y. Ni, F. Hollmann, in: Advances in Biochemical Engineering/Biotechnology,
Springer, 2016, pp. 1–22.[246] B. Kandemir, S. Chakraborty, Y. Guo, K.L. Bren, Inorg. Chem. 55 (2016) 467–
477.[247] G. Knör, K. Oppelt, T. Happe, unpublished results.[248] I. Akkerman, M. Janssen, J. Rocha, R.H. Wijffels, Int. J. Hydr. En. 27 (2002)
1195–1208.[249] R.T. Ross, M. Calvin, Biophys. J. 7 (1967) 595–614.