Embed Size (px)
Citation preview

Photooxidation of Anhydride-Cured Epoxies: FTIR Studyof the Modifications of the Chemical Structure
V. OLLIER-DUREAULT,1 B. GOSSE2
1 Schneider Electric, Materials Research Division, Site A3, 38050 Grenoble Cedex 9, France
2 CNRS–LEMD, 25 rue des Martyrs, 38040 Grenoble Cedex 9, France
Received 5 August 1997; accepted 6 April 1998
ABSTRACT: Anhydride-cured epoxies are used in electrical engineering to manufactureinsulators. The purpose of this study is to determine the modifications undergone bythese resins when exposed to solar-type radiation (radiation with a wavelength of morethan 300 nm). Two epoxy–anhydride systems have been studied. Using transmissionFourier transform infrared (FTIR) and attenuated total reflection (ATR), we havedetermined the structural modifications and the groups formed during photooxidation.The degradation is very heterogeneous, more significant on the surface than in the bulkof the material, and a considerable decrease in almost all the initial functional groupsis observed. The flexibilized system, richer in ester groups but containing fewer phenylgroups, degrades more rapidly during the first hours of exposure than the nonflexibil-ized system. Two explanations accounting for this faster degradation in the first hoursof exposure are drawn from ultraviolet absorbance analysis and degradation mecha-nisms considerations. After 40 h of irradiation, the disappearance rate of the aromaticgroups depends only on their initial concentration, whereas the formation kinetics forthe hydroxyl groups is limited by the diffusion of oxygen in the material. Chemicaltreatments using SF4, NH3, and NaCl have revealed the formation of hydrophilicproducts, acids, and alcohols. Their presence and particularly high concentration at thesurface could very well be responsible for the degradation of the insulating propertiesof the electrical insulators. © 1998 John Wiley & Sons, Inc. J Appl Polym Sci 70: 1221–1237, 1998
Key words: epoxy–anhydride resins; photodegradation; FTIR spectroscopy; aging ofinsulators; hydrophilic surface
INTRODUCTION
Cast epoxy resins based on di-glycidyl ether ofBisphenol A (DGEBA) are widely used in electri-cal engineering to manufacture indoor insulators.Such insulators can undergo an oxidative surfaceattack by ultraviolet radiation, for instance, whenexposed to sunlight during transport or whenelectrical discharges occur.
The purpose of this study is to analyze struc-tural modifications in 2 DGEBA-based epoxy–an-hydride systems during photooxidation by ultra-violet (UV) radiation in the solar wavelengthrange and to analyze the degradation products. Athird epoxy–anhydride system based on a cy-cloaliphatic epoxide will be studied for compari-son purpose.
As far as we know, there has been no previouswork on the photodegradation of DGEBA basedepoxy–anhydride systems in air. On the otherhand, a number of studies have dealt with thethermal or photochemical degradation of phenoxyresins,1,2 noncrosslinked DGEBA resins,3 and, es-
Correspondence to: B. Gosse.Journal of Applied Polymer Science, Vol. 70, 1221–1237 (1998)© 1998 John Wiley & Sons, Inc. CCC 0021-8995/98/061221-17
1221

pecially, epoxy resins crosslinked by amines. Forthe latter, refer to the article by Lin et al.,4 whichcontains numerous references, or the more recentwork of Bellenger and Verdu5 on the influence ofthe chemical structure on the photooxidation of ep-oxy–amine systems. The work of Le Huy et al.6,7
concerns the thermooxidative degradation of epoxy-–anhydride systems, while that of Lin et al.4 con-cerns an Fourier transform infrared (FTIR) study ofthe thermal and photochemical degradation of ep-oxy systems crosslinked by trimethoxyboroxine. Fi-nally, relatively recent studies8,9 on the pyrolysis ofepoxy resins crosslinked by different hardeners, in-cluding anhydrides, constitute further references onthe relative thermal stability of the different struc-tural groups of these systems.
In the present work, we propose a structuralrepresentation of the 2 DGEBA-based epoxy sys-tems that accounts for the major differences be-tween the 2 formulations, as observed by Fouriertransform infrared (FTIR) spectroscopy. Next, wepresent the FTIR analysis of the structural mod-ifications produced and the products formed whenthese materials are exposed to UV radiation. Weparticularly investigate the quantities and thedistribution of the hydrophilic products, acids,and alcohols, their presence and concentrationson the surface being largely responsible for thedegradation of the insulating properties of elec-trical insulators.
EXPERIMENTAL
Materials
The 2 studied formulations, referred to as the EA(epoxy–anhydride) and EAF (flexibilized epoxy–anhydride) systems, respectively, are commercialformulations composed of a DGEBA-type epoxyresin, an anhydride hardener based on methyltetrahydrophtalic anhydride (MTHPA), and a cat-alyst of the tertiary amine-type. The EAF systemfurther contains a diester–dicarboxylic acid flexi-bilizer. The cycloaliphatic system studied for com-parison, referred to as the EAC system, is also acommercial formulation composed of a cyclo-aliphatic epoxy resin and an anhydride hardenerbased on HHPA (hexahydrophtalic anhydride). Itis thus aromatic free.
Analysis by gel permeation chromatography(GPC) and by FTIR spectroscopy of the basic con-stituents and subsequently of the crosslinked sys-tems (Fig. 1) have led us to the following conclu-sions for the aromatic systems.
The EA system is made up of a mixture ofDGEBA oligomers that can be assimilated withDGEBA n 5 1.5 (long chains), crosslinked byMTHPA. The constitutional repeating half-unit(CRU) is represented in Figure 2(A).
The EAF system is made up of a mixture ofDGEBA oligomers with n 5 0 and 2, crosslinked
Figure 1 FTIR spectra of the EA (——) and EAF (. . .) systems.
1222 OLLIER-DUREAULT AND GOSSE

by the MTHPA anhydride after an initial stepinvolving the elongation of the DGEBA n 5 0chains (short chains) by a tetrahydrophtalic di-acid–diester type flexibilizer. Thus, 3 CRUs arenecessary to describe the EAF system. The first isidentical to that of the EA system [Fig. 2(A)] andcorresponds to the reaction of the MTHPA anhy-dride with the DGEBA n 5 2 di-epoxides. Thesecond, shown in Figure 2(B), corresponds to thereaction of MTHPA with the di-epoxide molecules
that have been elongated by the flexibilizer. Fi-nally, the third CRU corresponds to the reactionof MTHPA with the DGEBA n 5 0 di-epoxidemolecules that have not reacted with the flexi-bilizer. The proportions of these 3 CRUs are 20,40, and 40% by mass, respectively.
The ratio of identical groups per mass unit inthe EA system [or functional index IFEA(X)] ver-sus that of the EAF system [IFEAF(X)] is given inTable I. It appears that the EA system contains
Figure 2 (A) Constitutional repeating half-unit of the EA network. (B) Constitutionalrepeating half-unit of the EAF network characteristic of this system and accounting for40% (w/w) of the material.
Table I Attribution of the IR Bands of the 2 Crosslinked Systemsand the Calculated Ratios of Functional Indices in the 2 Systems
Functional Groups Wavelength Number (cm21)
Ratios of Functional IndicesIFEA(X)/IFEAF(X)
Calculated Ratios(from CRU)
AbsorbanceRatios
Hydroxyle (OOH) 3520 1,54 1,43Arylene (COH) 3052, 3033 1,34 1,31Methyl (CH3) 2966, 2872 1,07 1,04Methylene (CH2) 2931 0,97 0,93Dimethyl OC(CH3)2 1381, 1363,807Phenylene (CAC) 1610, 1580, 1510, 1,43 1,4Phenyl ortho di-substituted 1458PhenylOC(CH3)2OPhenyl 1184p-phenylene 830Ester (OCAO) 1739, 1152 0,68 0,68aromatic Ether FOOOC 1240 1,38 1,48aliphatic Ether FOOOC 1042
PHOTOOXIDATION OF ANHYDRIDE-CURED EPOXIES 1223

1.5 times more aromatic groups than the EAFsystem, whereas the percentage for ester groupsis approximately 1.5 times lower.
The cycloaliphatic epoxy–anhydride (CEA) is madeup of di-glycidyl ether of cyclohexane crosslinked byhexa-hydrophtalic-anhydride (HHPA).
For the entire study, except for the determina-tion of oxydation profiles, the samples are filmsabout 10 m thick, cut using a microtome (univer-sal motorized JUNG Autocut 2055, LEICA) fromthe center of test pieces cast under atmosphericpressure in air. For the determination of oxyda-tion profiles, those test pieces, previously agedunder UV irradiation, are cut from surface tocenter to get a 10-m thick film allowing micro-scopic FTIR mapping (by transmission) 20 by 20 mstarting from the surface edge. Table II gives theprocedure used for the preparation of these cylin-drical test pieces (6 mm height and 35 mm diam-eter). The proportions of resin and hardener used
are those recommended by the supplier and cor-respond to the stoichiometric mixture. The quan-tity of accelerator added in the case of the EAsystem is 0.2% by weight with respect to the ep-oxy resin. For the EAF system, the accelerator isincluded in the resin and the flexibilizer in thehardener.
The difference in preparation temperatures isdue to the fact that the resin in the EA system isa solid at room temperature and must be heatedbefore mixing, whereas the resin in the EAF sys-tem is already a liquid at room temperature. Inaddition, the difference in curing temperaturesmakes it possible to compensate for the differencein reactivities and to ensure an identical jellifica-tion time, about 18 min, for the 2 systems.
Aging Apparatus
The apparatus used for the aging process is aclosed 40-L chamber with 3 overhead high-pres-sure mercury arc lamps (OSRAM Ultravitalux300 W). The samples are placed at a distance of 25cm from the lamps on a revolving plate, thusensuring identical irradiation of all the samples.In order to filter the IR radiation and to limitthermodegradation as compared to photodegrada-tion, an IR filter is placed between the lamps andthe samples. With the lamps turned on, the tem-
Table III Measurements of Energy Densities Received at the Surface of theSamples
Spectral Region Centered on Energy Densities 254 nm 310 nm 365 nm
Lamps placed at a distance of 25 cm 0.7 W/m2 5.5 W/m2 20 W/m2
In Grenoble at summertime in the midday sun 0.8 W/m2 6.4 W/m2 23,6 W/m2
Table II Procedure for the Preparation of Cast Test PiecesFrom Which 10-m Films Are Cut Using a Microtome
Operations System EASystem
EAF
Preparation of the mixture 40°C 130°CDegassing under vacuum Few minutes
CastingIn the preheated mold at the curing
temperatureCuring (duration : 3 3 gel-time) 140°C 130°CDemolding Before coolingPostcuring 140°C, 8-h duration
1224 OLLIER-DUREAULT AND GOSSE

perature in the chamber during degradation is60°C. Also owing to the IR filter, the differences insurface temperatures between samples with dif-ferent IR absorptions may be disregarded. In ad-dition, the variation of the energy density withthe wavelength was checked using an UV radiom-eter (ORIEL UVX) equipped with filters centeredon 3 wavelengths characteristic of mercury arclamps: 254, 310, and 365 nm (Table III). Thevalues measured in Grenoble in summer, atground level, at noon, in the sun and centered onthe same wavelength are very similar (Table III).We have checked that the structural modifica-tions caused by irradiation in this laboratorychamber are representative of those caused by annatural outdoor exposure.
Methods of Characterizing the Aging
FTIR transmission microscopy was used to ob-serve the structural evolution of the 10-m thick
films during degradation. The employed appara-tus was a Nicolet 510 equipped with a Spectra-tech IR-plan microscope.
The IR bands (Table I) were attributed usingreference works,10–12 the results of Lin et al.,4
and especially those of Stevens,13,14 and by com-paring the spectra of the constituents with thoseof the 2 crosslinked systems (Fig. 1).
Methods of Analyzing the Photoproducts
The degradation products, and, more specifically,the carboxylic acids and alcohols, were identifiedby combining the FTIR spectroscopy with specificchemical reactions, allowing certain IR bands tobe shifted towards less crowded regions of thespectrum.
Thus, gaseous SF4 reacts with acids, alcohols,and hydroperoxides according to the followingscheme15,16:
After treatment, the bands characteristic of ac-ids [mainly nOH around 3300 cm21, overtonesand combinations of carboxylic acid dimersaround 2600 cm21,12 nCAO intense band around1710 cm21, and nCOO strong band between 1315and 1280 cm21] should disappear, leaving roomfor the absorption bands characteristic of the cor-responding fluorinated derivatives. In particular,the nCAO band of acyl fluorides (I) should appearbetween 1850 and 1800 cm21, and its gradualtransformation into RCF3.17 Likewise, the bandscharacteristic of alcohols or hydroperoxides (nOHat 3450 cm21) should disappear, leaving room forthe absorption bands characteristic of the corre-sponding monofluorinated compounds (around1080 cm21). Pure aldehydes or ketones can reactwith SF4; however, inside a polymeric matrix,they do not generally react when the treatmenttakes place at room temperature.16 The absenceof reaction with esters was confirmed.
Gaseous NH3 reacts with carboxylic acids accord-ing to a classical acid–base reaction to yield thecorresponding ammonium and carboxylate ions:
This reaction should therefore lead to the dis-appearance of the bands characteristic of acids(3300, 2600, and 1700 cm21) and the appearanceof bands characteristic of ammonium ions (nCHaround 3200 cm21) and carboxylate ions (asym-metrical nCAO between 1550 and 1600 cm21 andsymmetrical nCAO between 1300 and 1400cm21).16 NH3 may also react with esters or anhy-drides to yield amides, the absorption of whichappears around 1665 cm21.17 Finally, by carryingout an acid return with HCl (vapors), the carbox-ylate ions immediately reform the carboxylicacid.17,18
PHOTOOXIDATION OF ANHYDRIDE-CURED EPOXIES 1225

NH3 in the gaseous state does not react withalcohols, nor with aldehydes or ketones. We haveverified that it does not react with esters underour operating conditions by treating unirradiatedfilms with NH3.
However, assuming the possible faculty of NH3to favor the formation of carboxylic acid by hydro-lysis of esters on oxidized and, thus, hydratedsurfaces, a second chemical treatment character-istic of acids was employed, using sodium chlorideNaCl. Indeed, NaCl may react by ion exchangewith ions in the matrix according to the reaction:
Na1Cl2 1 RCOO2H13 RCOO2Na1 1 H1Cl2
The hydrochloric acid HCl, in turn, is then ableto react, but its presence requires a previous ex-change between the Na1 and the H1 stemmingfrom the oxidized polymer.
In addition, FTIR microscopy by attenuatedtotal reflectance (ATR), with which the acquisi-tion of a spectrum is achieved with a single reflec-tion on the surface, was used to compare thesurface (at a depth of about a micron) and bulkcompositions of the aged films. Finally, the UVabsorbance of the photoproducts was determinedby UV absorption spectroscopy.
RESULTS AND DISCUSSION
Modification of the Chemical Structure
Analysis by Transmission FTIR
The FTIR analysis as a function of the irradiationtime (1 spectrum every 10 h from 0 to 100 h, thenevery 24 h until 180 h, and, finally, a spectrum at240 h) shows a rapid spectral evolution of the 2systems under UV radiation, which can be ob-served from the very first hours of exposure, asillustrated in Figure 3, as follows.
● A major decrease of the bands characteristicof aromatics is shown (para-disubstituted,aromatic CAC bands at 1610 and 1510 cm21,aromatic CH bands at 3032 and 830 cm21)[Fig. 3(A) and (C)].
● A decrease of the ether groups (band at 1042cm21) associated with a decrease of the aro-matic ether band (band at 1245 cm21), [Fig.3(C)] is shown.
● A decrease of the aliphatics [C(CH3)2 band at1184 cm21, (CH3)2) band at 807 cm21, CH3
bands at 2966 and 2874 cm21, and CH2 bandat 2933 cm21] is shown [Fig. 3(A) and (C)].
● A strong increase of the hydroxyls (shoulder at3600 cm21 and maximum at 3450 cm21) grad-ually yielding more tightly bound hydroxyls(shoulder at about 3250 cm21 and a broad bandaround 2600 cm21) is shown [Figures 3(A) and10]. The increase in the OH band varies lin-early with the irradiation time until approxi-mately 30 to 40 h. After 40 h, the increasevaries as a function of t0.5 (Fig. 4). It is gener-ally accepted that such a variation indicates anoxidation process limited by the diffusion ofoxygen in the polymer.7 It results in a hetero-geneous volume distribution of the material’soxidation products in thick samples, as isshown in Figure 5.
● The appearance of several carbonyl and car-boxyl groups absorbing in the range of 1800to 1680 cm21 is shown, which may be attrib-uted to lactones or peroxyesters (1800–1760cm21) and ketones or carboxylic acids (1720–1680 cm21) [Figure 3(B)].
For the EAF system, which is rich in estergroups, we note a particularly fast and significantdisappearance of the CAO and COO bands of es-ters between 0 and 50 h (Fig. 6). An identical de-crease of all the other bands is associated with thesemodifications, such as, for instance, that of the ar-omatic groups at 830 cm21 (Fig. 7). After 50 h, theEA and EAF systems degrade according to an ap-parent first-order reaction and with an identicalrate. After 240 h of irradiation, the intensity of thearomatic bands has decreased by over 40%.
Note that it was not possible to take into ac-count the possible loss of matter due to the for-mation of volatile products. The decrease of allbands, including that of the aromatic band at1610 cm21, which was chosen as a reference forthermodegradation,7 forced us to work with filmsof a given thickness, which we had to assumeconstant throughout the aging process.
The modification or disappearance of numer-ous absorption bands, associated with the strongincrease of the hydroxyl band and the broadeningof the carbonyl band, is evidence of an importantoxidation phenomenon in both systems. As for theattacks that are common to both formulations,the obtained results show that UV irradiationleads to a specific attack upon (1) the aromaticfunctions [disappearance of aromatic CAC andCOH bands], and (2) the aliphatic functions lo-cated near aromatic centers in the chain [disap-
1226 OLLIER-DUREAULT AND GOSSE
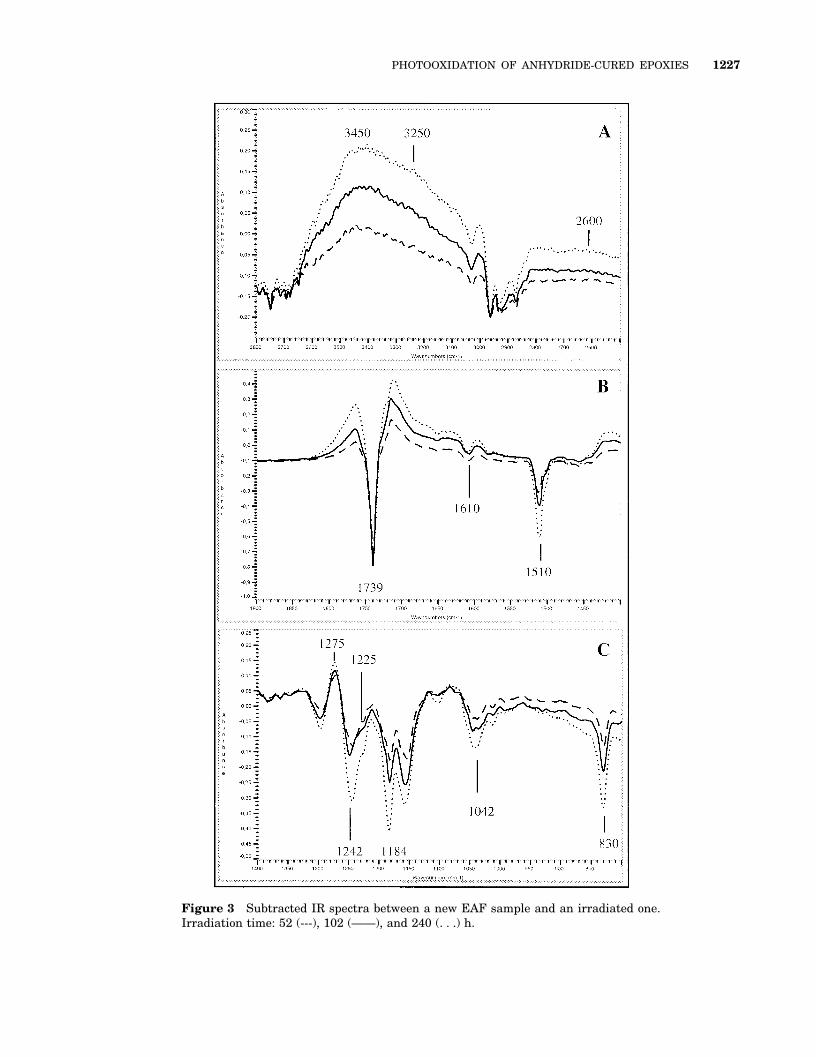
Figure 3 Subtracted IR spectra between a new EAF sample and an irradiated one.Irradiation time: 52 (---), 102 (——), and 240 (. . .) h.
PHOTOOXIDATION OF ANHYDRIDE-CURED EPOXIES 1227

pearance of the aliphatic ether bands, decrease ofthe aromatic ether band, theOC(CH3)2, the CH2,and CH3 bands]. In addition, it may be possiblethat the aliphatic groups, which are slightly fur-ther from these aromatic centers, may also beattacked, but the corresponding bands are verydifficult to observe by IR (this is the case fortertiary CH groups, notably those with a very lowmolecular absorption coefficient «, as well as thesecondary alcohols, for which the absorption bandis overlapped by the others).
For the EAF system, a rapid attack upon allgroups, and, particularly the esters, takes place
at the beginning of the degradation (Fig. 6). Atthe same time, we note a rapid disappearance of aband at 665 cm21 corresponding to the cis-dialkylvinyl group of the flexibilizer’s tetrahydrophtalicdiacid–diester. Based on the presented structuralmodels, the quantity of ester groups belonging tothe flexibilizer in the EAF system (21%) is of thesame order of magnitude as the percentage ofdegradation of ester groups in the EAF system, asmeasured at 50 h (approx. 28%). These observa-tions suggest a rapid attack of the flexibilizer,which possesses ester groups of a different config-uration than those common to the 2 systems. Inaddition, they are located near the network nodesand are therefore particularly fragile. Once theflexibilizer has been consumed, the EAF systemscontinues to degrade. The disappearance of thearomatics then follows the same kinetics for bothsystems (Fig. 7).
Identification of the Photoproducts
Polymers that have undergone a thermal or pho-tochemical aging process in the presence of oxy-gen generally give rise to a complex mixture of thefollowing oxidation products: alcohols, hydroper-oxides, ketones, aldehydes, carboxylic acids, es-ters, peracids, peroxyesters, and anhydrides. As amatter of fact, we observe the appearance of newabsorption bands in regions that are characteris-
Figure 4 Absorbance of the OH groups (taken at the maximum) as a function of theirradiation time: EA (——); EAF (---).
Figure 5 Oxidation profile in a thick sample: absor-bance at 3500 cm21 as a function of the distance to theirradiated face (EAF system; 200-h irradiation time).
1228 OLLIER-DUREAULT AND GOSSE

tic of hydroxyls, carbonyls and ethers (Fig. 3).These bands are often very broad and result fromthe overlapping of bands that are characteristic ofseveral photoproducts, as follows:
● The hydroxyl OH region, with several bands(3600, 3465, 3250, and 2600 cm21) that maycorrespond to alcohols, phenols, hydroperox-ides, or carboxylic acids [Fig. 3(A)];
Figure 6 Subtracted IR spectra between a new sample and a sample irradiated for102 h: EA (——); EAF (. . .).
Figure 7 Decrease of the aromatic IR band versus irradiation time for an EA (——)and EAF (---) system.
PHOTOOXIDATION OF ANHYDRIDE-CURED EPOXIES 1229

● The carbonyl CAO region, with several max-ima or shoulders [Fig. 3(B)], 1764 cm21 (lac-tones or peroxyesters), 1715 cm21 (ketones orcarboxylic acids), 1787 cm21 (acid anhy-drides), 1724 cm21 (aldehydes), 1704 cm21
(carboxylic acids), and 1690 cm21 (conju-gated ketones or benzoic acid);
● COO region, with a main band at 1275 cm21,a shoulder around 1225 cm21, and an up-surge in the 1000–1100 cm21 range thatmay correspond to the COO vibrations ofcarboxylic acids, phenols, alcohols, or ethers[Fig. 3(C)].
All of these new bands are common to both theEA and the EAF system but are different fromthose observed by Le Huy et al.7 for thermodeg-radation. In this study, the major products wereanhydrides, which may be identified by a charac-teristic doublet at 1850 and 1785 cm21, whereaswe only observe a small shoulder at 1787 cm21
and a very weak band at 1850 cm21. Thus, it isclear that these are not the major products in thecase of photodegradation. The following was ob-served.
● The reaction with gaseous SF4 (Fig. 8) pro-vokes the disappearance of the broad hy-droxyl band between 3700 and 3000 cm21
[Fig. 8(A)] and the appearance of severalbands corresponding to fluorinated alkyls inthe 1000 to 1100 cm21 range [Fig. 8(C)].These observations lead us to the conclusionthat alcohols are present in the oxidized poly-mer in a rather high proportion. The RFproducts seem rather unstable or volatilesince a notable fluctuation is observed in thecorresponding bands as a function of the du-ration of treatment. The decrease of theCAO band between 1690 and 1725 cm21,associated with the appearance of fluori-nated derivatives absorbing around 1850 and1800 cm21, proves the presence of carboxylicacids in a relatively low concentration. In-deed, the intensity of the decrease of the acidbands as well as that of the increase of thecorresponding fluorinated derivatives re-mains rather low. In addition, we observe thedecrease of the carbonyl CAO band at 1739cm21 [Fig. 8(B)]. This might be explained bya very slow reaction between SF4 and thepolymer’s ester functions. The reaction is notobserved with unaged polymer but would bemade possible by the general degradation of
the polymer. Finally, the bands appearing at1262 cm21 and between 800 and 900 cm21
[Fig. 8(C)] are attributed either to RF3 prod-ucts (ultimate derivatives of the fluorinatedacids), or more probably to fluorinated ben-zene groups, which might result from theaction of SF4 on phenols.
● The evolution observed due to the action ofNH3 (Fig. 9) confirms the presence of carbox-ylic acids in small quantities; in this case, anappearance between 1550 and 1590 cm21 ofshoulders on the aromatic band at 1610 cm21
is observed, which is characteristic of carbox-ylate ions, associated with the disappearanceof the shoulder around 1710 cm21. Mean-while, the treatment leads to a decrease ofalmost all the bands of the initial spectrum ofthe aged film, a phenomenon which we at-tribute to the formation of a strongly absor-bent superficial layer resulting from the re-action of NH3 with the degradation productspresent at a high concentration at the sur-face of the film. After treatment with HCl,followed by a rinsing with distilled water, theresulting spectrum is comparable to the oneobtained before treatment with NH3; onlythe bands corresponding to products havingreacted with NH3 and eliminated by rinsingwith distilled water have disappeared.
● Finally, the treatment with NaCl (assumedto exchange Na1 with H1 of carboxylic acid)has given an IR evolution that is very diffi-cult to interpret as a whole (decrease of thecarbonyls at 1750 and 1730 cm21 and in-crease of the hydroxyls); but the appearanceof two bands characteristic of carboxylateions (1575 cm21 and approximately 1400cm21), associated with the disappearance ofthe carboxylic acid band at 1710 cm21 ob-tained by a simple ion exchange, confirms thepresence of carboxylic acids in the oxidizedpolymer.
These results as a whole show that the UVirradiation of epoxy–anhydride resins must leadto a disappearance of material due to the forma-tion of volatile molecules (CO, CO2, H2O, aro-matic products of low molecular mass, etc.) notdetected in this study. In addition, the followingcompounds are formed in relatively small quan-tities: (1) aliphatic alcohols, to which we attributethe OH band at 3450 cm21, the correspondingCOO band at 1080 cm21 and the different absorp-tions around 1000 cm21; (2) phenols, to which we
1230 OLLIER-DUREAULT AND GOSSE

Figure 8 Subtracted spectra between a sample irradiated for 100 h and the samesample treated with SF4. Treatment time: 5 (---), 1200 (——), and 3600 (. . .) min.
PHOTOOXIDATION OF ANHYDRIDE-CURED EPOXIES 1231

attribute the intense absorption bands at 3300(OH), 1225 (COCOO), and 1420 cm21 (COOH)12;and (3) carboxylic acids, to which we attribute theCAO bands at 1700 and 1710 cm21, as well as theCOO band at 1275 cm21, the broad hydroxylband between 2600 and 2900 cm21, and the ab-sorption band around 950 cm21 (OHOO interac-tion).
Concerning the other products, we suggest thepresence of saturated ketones (1724 cm21) andconjugated ketones (1690 cm21) or benzoic acid,as well as lactones (1764 cm21). In addition, asmall quantity of anhydrides is observed around1787 and 1820 cm21, but in relatively low concen-trations, indicating that the photooxidation leadsto the formation of different products from thoseof the thermooxidation.
The classical photooxidation processes, such asthe molecular cleavage mechanisms of the Nor-rish I or II type, may be invoked in order toexplain the formation of most of the observedgroups. Part of the radicals that are formed dur-ing cleavage undergo a decarboxylation or a de-carbonylation and successive cleavage, leading tothe formation of a low-molecular-weight and,thus, volatile species.
Analysis of the Surface by ATR Microscopy
The penetration of the degradation inside thebulk of the polymer depends on several factors, asfollows: its absorbance in the considered spectralregion; the possibility of the formation of absorb-ing photoproducts at the surface; and, a last im-portant parameter, its permeability to oxygen.This is the reason why the degradation is oftenconfined to the surface.
In order to determine the chemical compositionat the extreme surface of the films, which maythus be different from the one detected by thetechnique of transmission FTIR, we have carriedout an analysis by ATR microscopy of the side ofthe film that was exposed to the UV radiation.The spectra obtained by ATR for a 50-m film irra-diated for 160 h are shown in Figure 10. Thespectrum obtained for the aged film has beenmultiplied by a factor 1.5.
Compared to the ATR spectrum of the newfilm, the ATR spectrum of the aged film shows ageneral decrease of all the initial bands. Thisdecrease, which had already been observed byFTIR transmission reveals a strong surface deg-radation of the polymer (increase in roughness,
Figure 9 IR spectra of an EAF sample irradiated for 102 h (——), then treated for20 h with NH3 (. . .), and finally exposed to HCl vapors, followed by rinsing withdistilled water (— — —).
1232 OLLIER-DUREAULT AND GOSSE

cracking, etc.), which is responsible for the film’sopacification. In spite of a multiplication of thespectrum obtained with aged film by a factor of1.5, we note the very strong decrease of the aro-matic bands (CAC at 1510 cm21; COH at 830cm21); the strong decrease of the bands charac-teristic of aromatic ethers (FOCOO at 1245cm21) and of aliphatic ethers (1042 cm21), accom-panied by a broadening of these bands; and, also,the strong decrease of the aliphatics [OC(CH3)2at 1184 cm21 and CH3 and CH2 at 2966, 2933,and 2974 cm21]. Concurrently to the general de-crease of all the initial bands of the polymer, wenote the presence of particularly strong oxidationbands: the CAO band is intense and stronglyshifted towards the low wave numbers (maximumat 1727 cm21 with important shoulders at 1710and 1690 cm21), thus indicating a high concentra-tion of acid products. In addition, the shouldersalready observed by transmission at 1820, 1787,1764, and 1724 cm21 are observed once again. Inthe hydroxyl region [Fig. 10(A)], besides a verybroad band with a maximum at 3450 cm21 (at-tributed to aliphatic alcohols), we note the pres-ence of a band of strong intensity with a maxi-mum at 3250 cm21, probably corresponding tophenolic products, which are the classical photo-products of aromatic polymers. In addition, wenote the importance of the intensity of the broadacid OH band at 2600 cm21, which confirms thepresence of high concentrations of acids at thesurface.
Finally, in the COO region [Fig. 10(B)], wenote the emergence of 2 strong shoulders at 1225cm21 (which may be attributed to phenols) and at1180 cm21 (which may be attributed to aliphaticalcohols), as well as the increase of the absor-bance around 1275 cm21 (which may be attrib-uted to carboxylic acids).12
Screening Effect
In order to assess the absorbance of the formedphotoproducts, a UV absorption spectrum was ob-tained for a new film and, subsequently, for a filmaged for 100 h (Fig. 11). The absorbance’s evolu-tion towards wavelengths well above 300 nmshows the formation during the degradation ofstrongly UV absorbing photoproducts. These
Figure 10 ATR spectra of an EA sample, new (. . .), and after 160 h of irradia-tion (——).
Figure 11 UV absorption spectra of an EA film (1)nonirradiated and (2) after an irradiation time of 100 h.
PHOTOOXIDATION OF ANHYDRIDE-CURED EPOXIES 1233

products are probably responsible for the addi-tional screening effect by UV absorption in theaged film’s superficial layer. This effect and theabsorption by the aromatic chromophores of theEA and EAF systems would partly explain thevery heterogeneous degradation of the film andthe formation of the thin, very strongly degradedsurface layer that we have observed.
UV Absorption Spectraand Degradation Mechanisms
In a first approach, Norrish-type mechanismsmay be invoked to explain the disappearance ofesters and the formation of numerous oxidizedproducts as observed for EA and EAF systems.But for the same conditions of irradiation (UVlight intensity and irradiation time), FTIR anal-ysis didn’t show any modification of the EAC (cy-cloaliphatic) system, which contains, however, alot of ester groups in a similar configuration. Thecomparison of UV absorption spectra of the 3 newsystems (Fig. 12) allows one to understand whythe EAC system doesn’t degrade under UV asboth EA and EAF systems do. Indeed its aro-matic-free structure doesn’t absorb UV of wave-length longer than 300 nm. Thus, we could at-tribute the strong degradation of both aromaticsystems to the absorption of UV light by aromaticstructures followed by an intrinsic and/or induceddegradation. Moreover, the shoulder observed inthe EAF system UV absorption spectra (Fig. 12)may explain the rapid attack of the flexibilizer.Indeed, the comparison of UV absorption spectraof EA and EAF hardeners allowed us to attributethis shoulder to the flexibilizer contained in theEAF hardener. However, further considerationcan be taken into account for the degradation
mechanisms of EA and EAF systems. Some exam-ples in the literature show in the case of aromaticchromophores that bond scissions may occur notonly on the bonds directly linked to the ring butalso on the bonds in the vicinity.19 Figure 13shows possible mechanisms for intrinsic degrada-tion according to experimental observations andconsidering bonds dissociation energies.20 In fact,the strong increase of hydroxyl bands confirmsthe formation of tertiary alcohols [a maximumaround 3450 cm21; Fig. 10(A)] and phenols [amarked shoulder at 3250 cm21 in Figure 10(A)and 1225 cm21 in Fig. 10(B)]. The formation ofconjugated ketones is more hypothetic; theslightly marked shoulder at 1690 cm21 (Fig. 10)might be characteristic for conjugated ketonesbut also for benzoic acid. Further analysis is nec-essary to confirm the formation of these products.
From the proposed mechanism can be drawnanother explanation for the rapid degradation ofthe flexibilizer in the EAF system. Indeed, if rad-icals CH3
• issued from reaction (1) [Fig. 13] willlead to the formation of a formaldehyde and, fur-ther, of a formic acid, radicals OCH2
• from reac-tion 2(b) can give rise to different situations:
On one hand, it can give rise to the rapid prop-agation of oxidation in the EAF system, in thelong linear chain constituted by the flexibilizer,by nibbling; on the other hand, it can give rise toa rapid annihilation of the oxidation processes inthe EA system by formation of relatively stablephenol or quinone groups when reaching the ar-omatic group. Those mechanisms are depicted inFigure 14.
Another mechanism of induced photooxidationcould explain the fast degradation of EA and EAFsystems compared to the EAC. It is the oxidativeattack of the hardener part of the EA and EAFmolecules. Indeed, MTHPA (EA and EAF hard-ener) contains a double bond that is weakeningthe OCH2 bond in its vicinity, although HHPA(EAC hardener) doesn’t. This mechanism leads tocarboxylic acids (1718 cm21) and conjugated ke-tones (1690 cm21) that we observe in FTIR on EAand EAF aged sample surfaces. As an inducedoxidative one, this mechanism alone cannot ex-plain the high degradation rate of EA and EAFsystems compared to EAC, but it can account forthe appearance of several observed degradationproducts. Finally, a last mechanism can be citedhere as highly probable. This is the oxidative at-tack of the tertiary carbon–hydrogen bond ofhardener cycles. This mechanism leads to conju-gated ketones linked to an aliphatic segment that
Figure 12 UV absorption spectra of films of EA, EAF,and CEA systems.
1234 OLLIER-DUREAULT AND GOSSE

will probably further degrade into low-molecular-weight by-products.20
CONCLUSION
1. The 2 studied aromatic epoxy–anhydridesystems are rapidly degraded by UV irra-diation in the solar wavelength range. Thestructural modifications are considerableafter only 50 h of irradiation.
2. The flexibilized system is degraded morerapidly during the first hours of exposure.We suppose a specific attack upon the flexi-
bilizer’s structural groups, the characteris-tic chemical functions of which disappearrapidly. Afterwards, this system continuesto degrade, but only according to first-orderkinetics, identically to the EA system.
3. The formed products are similar for the 2systems. They react in a similar way to thedifferent chemical treatments applied.
4. After 50 h of irradiation, the disappearancerate of aromatic groups does not depend ontheir initial concentration, whereas the in-crease of hydroxyl groups varies as thesquare root of time, which is generally ac-
Figure 13 Mechanisms for intrinsic photodegradation of EA and EAF systems.
PHOTOOXIDATION OF ANHYDRIDE-CURED EPOXIES 1235

cepted for an oxidation process limited bythe diffusion of oxygen in the material.
5. The considerable decrease of practically allthe bands characteristic of the initialchemical functions, associated with the ap-pearance of new, low-intensity bands, sug-gests the formation of a considerable quan-tity of volatile products.
6. The strong evolution of the UV absorptionspectra towards higher wavelengths, indi-cating the formation of strongly UV absor-bent products, suggests a rather markedscreening effect. This effect, associatedwith the limitation of oxygen in the bulk ofthe material, is responsible for the veryheterogeneous degradation of the films.
7. Norrish-type mechanisms may be in-volved to explain the disappearance ofesters and the formation of numerous ox-
idized products, as observed for EA andEAF systems. But from the comparisonwith the cycloaliphatic system (aromaticfree) EAC, we could attribute the strongdegradation of both aromatic systems tothe absorption of UV light by aromaticstructures, followed by an induced pho-tooxidative degradation. The fast degra-dation of the flexibilizer in the EAF sys-tem may be attributed either to a specificabsorbance of UV light by the flexibilizeror to a nibbling phenomenon along theflexibilizer chemical structure (fast prop-agation of the induced photooxidation).Two other mechanisms of photooxidativedegradation by a radical attack of thehardener cycles are cited, as they arehighly probable. They could account forthe formation of several photoproducts,
Figure 14 Mechanisms for induced photooxidation showing the initiation of a nib-bling degradation mechanism of an EAF system.
1236 OLLIER-DUREAULT AND GOSSE

including carboxylic acids that we ob-served in our study by FTIR analysis.
These results as a whole indicate that ultravio-lets should have a marked effect upon the electri-cal properties at the surface. The assessment ofthis impact on the functional properties of theepoxy resins used in electrical engineering will bethe objective of the continuation of this work.
The authors thank the Clermont Ferrand Photochem-istry laboratory for carrying out the SF4 treatmentexperiments and the acquisition of the correspondingspectra. They also thank J. L. Gardette, Research Di-rector at the CNRS, and J. Verdu, Professor at ENSAMParis, for fruitful discussions.
REFERENCES
1. B. D. Gesner and P. G. Kelleher, J. Appl. Polym.Sci., 13, 2183 (1969).
2. P. G. Kelleher and B. D. Gesner, J. Appl. Polym.Sci., 13, 9 (1969).
3. N. Grassie, M. Guy, and N. H. Tennent, Polym.Degrad. Stab., 13, 249 (1985).
4. S. C. Lin, B. J. Bulkin, E. M. Pearce, J. Polym. Sci.,17, 3121 (1979).
5. V. Bellenger and J. Verdu, J. Appl. Polym. Sci., 28,2677 (1983).
6. H. M. Le Huy, V. Bellenger, M. Paris, and J. Verdu,Polym. Degrad. Stab., 35, 77 (1992).
7. H. M. Le Huy, V. Bellenger, M. Paris, and J. Verdu,Polym. Degrad. Stab., 35, 171 (1992).
8. H. Nakagawa, S. Tsuge, and T. Koyama, J. Anal.Appl. Pyrolysis, 12, 97 (1987).
9. B. Plage and H. R. Schulten, Macromolecules, 21,2018 (1988).
10. L. J. Bellamy, The Infra-Red Spectra of ComplexMolecules, Methuen, London, 1958.
11. Nakanishi, IR Absorption Spectroscopy—Practical,1962.
12. D. Lin Vien, N. B. Colthup, W. G. Fateley, and J. G.Grasseli, The Handbook of Infrared and RamanCharacteristic Frequencies of Organic Molecules,Academic Press, New York, 1991.
13. G. C. Stevens, J. Appl. Polym. Sci., 26, 4259 (1981).14. G. C. Stevens, J. Appl. Polym. Sci., 26, 4279 (1981).15. J. F. Heacock, J. Appl. Polym. Sci., 7, 2319 (1963).16. S. Wilheim and J. L. Gardette, J. Appl. Polym. Sci.,
51, 1411 (1994).17. A. Rivaton, Polym. Degrad. Stab., 41, 283 (1993).18. J. Lacoste and D. J. Carlsson, J. Appl. Polym. Sci.,
30, 493 (1992).19. W. Schnabel and J. Kiwi, in Aspects of Degradation
and Stabilization of Polymers, Elsevier, New York,1978.
20. V. Ollier-Dureault, M.S. thesis, Joseph FourierUniversity of Grenoble, 1995.
PHOTOOXIDATION OF ANHYDRIDE-CURED EPOXIES 1237





![Photooxidation of Cinnamaldehyde in Methanol — Prod … Photooxidation of Cinnamaldehyde in Methanol — Prod ... fied by vacuum distillation [4]. Pure cinnamaldehyde was stored](https://img.pdfslide.net/doc/110x75/5aceeb767f8b9ac1478c345b/photooxidation-of-cinnamaldehyde-in-methanol-prod-of-cinnamaldehyde-in-methanol.jpg)























