Embed Size (px)
Citation preview

Photophysics of trioxatriangulenium ion. Electrophilic reactivityin the ground state and excited singlet state†
Jóhannes Reynisson,*‡a Robert Wilbrandt,§a Vibeke Brinck,b Bo W. Laursen,b
Kasper Nørgaard,b Niels Harrit¶b and Albert M. Brouwer c
a Condensed Matter Physics and Chemistry Department, Risø National Laboratory,DK-4000 Roskilde, Denmark
b Nano-Science Center, University of Copenhagen, H.C. Ørsted Institute, Universitetsparken 5,DK-2100, Copenhagen, Denmark
c Laboratory of Organic Chemistry, University of Amsterdam, Nieuwe Achtergracht 129,NL-1018 WS Amsterdam, The Netherlands
Received 22nd May 2002, Accepted 13th August 2002First published as an Advance Article on the web 17th September 2002
Trioxatriangulenium (TOTA�, 4,8,12-trioxa-4,8,12,12c-tetrahydro-dibenzo[cd,mn]-pyrenylium) is a closed shellcarbenium ion, which is stable in non-nucleophilic polar solvents at ambient temperatures. In alcohols, smallquantities of the leuco ether are formed in a reversible reaction. The physical and chemical properties of the excitedsinglet state of the trioxatriangulenium (TOTA�) carbenium ion are investigated by experimental and computationalmeans. The degeneracy of the lowest excited states is counteracted by Jahn–Teller-type distortion, which leads tovibronic broadening of the long wavelength absorption band. A strong fluorescence is observed at 520 nm (τfl = 14.6ns, �fl = 0.12 in deaerated acetonitrile). The fluorescence is quenched by 10 aromatic electron donors predominantlyvia a dynamic charge transfer mechanism, but ground state complexation is shown to contribute in varying degrees.Quenching is also observed in the presence of halide ions. Quenching rate constants are derived from lifetimemeasurements while charge transfer (CT) complex formation constants follow from the steady-state Stern–Volmerplots. CT-complex formation with three discogenic triphenylenes is studied separately. Phosphorescence spectra,triplet lifetimes, and triplet–triplet absorption spectra are provided. In the discussion, TOTA� is compared tothe unsubstituted xanthenium ion and its 9-phenyl derivative with respect to the excited state properties.
1 IntroductionPhotolytic generation of carbenium ions has proved a very suc-cessful means for obtaining rate constants and thermodynamicparameters for their ground state reactions with nucleophilesand solvent molecules.1 Others have used spectroscopic tech-niques to investigate the structures of carbenium ions generatedas stable species in strongly acidic media, often at low temper-ature. In view of the fact that ground state reactivity is generallyaugmented in the excited state as a result of enhanced exo-thermicity upon electronic excitation, it is of interest to explorethe properties and interactions of singlet and triplet states ofcarbenium ions. Much work has been done in this respect. Inparticular, the triphenylmethane dyes (TPMs) have been exten-sively studied.2 The role of carbenium ions as electron acceptorsin fluorescence quenching 3–6 and ground-state charge transfercomplexation 7 is well established.
Generally, the lifetime of the excited state of di- and tri-phenylmethane dyes is in the ps range in non-viscous solutiondue to efficient internal conversion coupled to torsionalmovements of the phenyl groups. When this movement ishindered either by a rigid medium or by binding to natural orsynthetic polymers, the fluorescence efficiency increases.2
† Electronic supplementary information (ESI) available: experimentaldetails and supplementary figures. See: http://www.rsc.org/suppdata/pp/b2/b204954f/‡ Present address: Universität Leipzig, Permosrestrasse 15, D-04318Leipzig, Germany.§ Present address: Bornholms Amtsgymnasium, DK-3700 Roenne,Denmark.¶ Author to whom correspondence should be addressed.
Strong fluorescence is also observed when the rotation ishindered by oxygen bridging as in the closed shell ions xanth-ylium (X�) and 9-phenylxanthylium (PX�) (Fig. 1).8
In the trioxatriangulenium ion (4,8,12-trioxa-4,8,12,12c-tetrahydrodibenzo[cd,mn]-pyrenylium, TOTA�, Fig. 1) themolecular framework is further stiffened by two oxygen bridges.The planar, rigid structure of TOTA� 9 permits extensivedelocalisation of the lone pairs on the three oxygen atoms in theground state.10 Consequently, TOTA� is far less susceptible tonucleophilic attack than X� and PX�, as reflected in their pKR�
values, being 9.05,11 �0.84,12 and 1.0,3 respectively. TOTA� isnot responsive to nucleophilic attack by water at neutral pH.However, it is accessible to attack from more nucleophilicsolvents like methanol, as will be shown in this paper.
Since publication of the synthesis 11 of TOTA� only littleattention has been given to its one-electron reduction.14 Proper-ties of the excited states of TOTA� have not been reportedexcept for a conference proceeding,15 a PhD thesis,16 and arecent short communication by Dileesh and Gopidas.17 Here,
Fig. 1 Trioxatriangulenium (TOTA�), 9-phenylxanthylium (PX�) andxanthylium (X�) ions.
DOI: 10.1039/b204954f Photochem. Photobiol. Sci., 2002, 1, 763–773 763
This journal is © The Royal Society of Chemistry and Owner Societies 2002
Publ
ishe
d on
17
Sept
embe
r 20
02. D
ownl
oade
d by
Lom
onos
ov M
osco
w S
tate
Uni
vers
ity o
n 16
/12/
2013
07:
33:3
6.
View Article Online / Journal Homepage / Table of Contents for this issue

our initial investigations 15,16 of the physical and chemicalproperties of the S0, S1, and T1 states of TOTA� are presented.
2 ExperimentalMost experiments were carried out according to standardphotochemical procedures. Details are reported in thesupplementary material.
In order to demonstrate formation of ground state donor–acceptor complexes and observe their spectra, a double com-partmentalized optical quartz cell—or so-called tandem cell(Helma 238-QS)—was used. In this, the light beam passesthrough both chambers, which have identical path lengths.Equal volumes of concentrated solutions of TOTA� and the π-donor, respectively, were placed separately in the compartmentsof the tandem cell and the absorption spectrum was run.Thereafter, the solutions were mixed through a small windowpositioned in the glass wall separating the compartments at alevel above the liquid surfaces. The absorption spectrum wasrecorded anew. By the mixing, all concentrations are cut in half,but since the path length is doubled for each component,absorbancies of the donor and the acceptor remain constantprovided only a negligible amount of complex is formed. Onthis condition, the changes observed when going from the firstspectrum to the second will reflect the absorption of any chargetransfer complexes formed.
3 Results
3.1 Reaction with methanol
The UV-vis absorption spectrum of TOTA� in alcoholsdepends on concentration. Traces a, b, and c in Fig. 2 shows the
long-wavelength absorption band of TOTA� (see next sectionfor a more in-depth treatment of the optical transitions) inMeOH at concentrations spanning two orders of magnitude.The optical path length is varied reversely. Obviously, Lambert–Beers law is not obeyed, indicating a concentration dependentchemical equilibrium. When the solution is made acidic byadding either aqueous HBF4 or CF3COOH, the absorbanceincreases (trace d). The same phenomena were observed inEtOH. No concentration dependence was observed in H2O,MeCN and CH2Cl2. In each of the latter solvents, an absorp-tion intensity and spectral profile superimposable on trace dwere obtained (spectra not shown).
Fig. 2 Long-wavelength absorption band of TOTA� in MeOH; (a)5.48 × 10�6 M, 10 cm path length; (b) 5.48 × 10�5 M, 1 cm path length;(c) 5.48 × 10�4 M, 1 mm path length; (d) 5.48 × 10�6 M added 1 dropaqueous HBF4 (ca. 50%), 10 cm path length; (e) spectrum of the leucomethyl ether (TOTA-OMe) at 5.48 × 10�5 M obtained by reactingTOTA� with t-BuOK in MeOH.
While the absorption coefficients above 300 nm decrease(Fig. 2, traces c, b, and a) as the concentration of TOTA� isdiminished, the short wavelength peak (ε282 =2,73 × 104
M�1cm�1) undergoes a bathochromic shift and displays aremanent absorption. When a catalytic amount of t-BuOKwas added to a solution of TOTA� in methanol, all the bandsof TOTA� disappeared and were replaced with a spectrumdisplaying a peak at λmax = 288 nm (Fig. 2, trace e). No absorp-tions were found in the near-UV/visible range (part of spectrumnot shown). This spectrum is believed to represent the leucomethyl ether TOTA-OMe (ε288 = 8.9 × 103 M�1cm�1). Additionof a single drop of concentrated HCl regenerated the spectrumof TOTA� in a completely reversible reaction.
While many cationic dyes fail to obey Lambert–Beers law dueto aggregational phenomena, the concentration dependenceillustrated in Fig. 2 is attributable to a reaction with the solvent.This was confirmed by 1H-NMR spectra of TOTA� tetrafluoro-borate in CD3OD, C2D5OD, D2O and CD3CN. In D2O andCD3CN only two absorptions with triplet and doubletmultiplicities were observed with δ = 8.4 (J = 9 Hz, 1H) and δ =7.8 ppm (J = 9 Hz, 2H), respectively. The triplet absorption isassigned to the hydrogen atoms in the meta positions relative tothe carbon–oxygen bond and the doublet to the neighbouringortho-hydrogens. In case of the deuterated alcohol solvents,however, an additional set of absorptions was observed (seesupplementary material for spectra†). They are shifted upfieldrelative to the TOTA� absorptions by 1.1 ppm (triplet) and0.8 ppm (doublet), respectively. The coupling constant isunchanged. When the solution was diluted, the intensity of theupfield signals increased relative to the TOTA� absorptions. Inanother experiment, addition of a small volume of CD3ONadissolved in CD3OD to TOTA� dissolved in CD3CN causeddisappearance of the proton absorptions assigned to TOTA�
and emergence of the upfield absorptions observed in CD3OD.The upfield signals are due to the leuco ether TOTA-OMe 18
formed as a consequence of nucleophilic attack by the solvent(Scheme 1).
In dilute solutions, the reaction towards equilibrium is slowon a time scale of hours. Thus, attempts to obtain an equi-librium constant K = [TOTA-OR][D�]/[TOTA�][ROD] fromUV-vis data produced scattered values. Instead, these werederived from the integrated 1H-NMR signals. The concentra-tions of the neat solvents ([CD3OD] = 24.7 M and [CD3CD2-OD] = 17.0 M) were calculated from their densities at 20 �C.Values of K = 1.06 × 10�5 for C2D5OD and K = 1.38 × 10�6 forCD3OD were obtained. The difference may be due to the highernucleophilicity of ethanol as reflected in the ion pair associationconstant (KA), being 18 for methanol and 133 for ethanol.19
Methanolic solutions of TOTA� were found to be sensitive todaylight and actinic light. Since TOTA� is completely photo-stable when dissolved in non-alcoholic solvents—includingwater—the light induced degradation observed in alcohol isprobably due to photoreactions of the leuco ether.20
3.2 Absorption and emission
The absorption and fluorescence spectra of TOTA� tetrafluoro-borate in acetonitrile are shown in Fig. 3 as they were recordedon a wavelength scale. A broad absorption band is observed
Scheme 1
764 Photochem. Photobiol. Sci., 2002, 1, 763–773
Publ
ishe
d on
17
Sept
embe
r 20
02. D
ownl
oade
d by
Lom
onos
ov M
osco
w S
tate
Uni
vers
ity o
n 16
/12/
2013
07:
33:3
6.
View Article Online
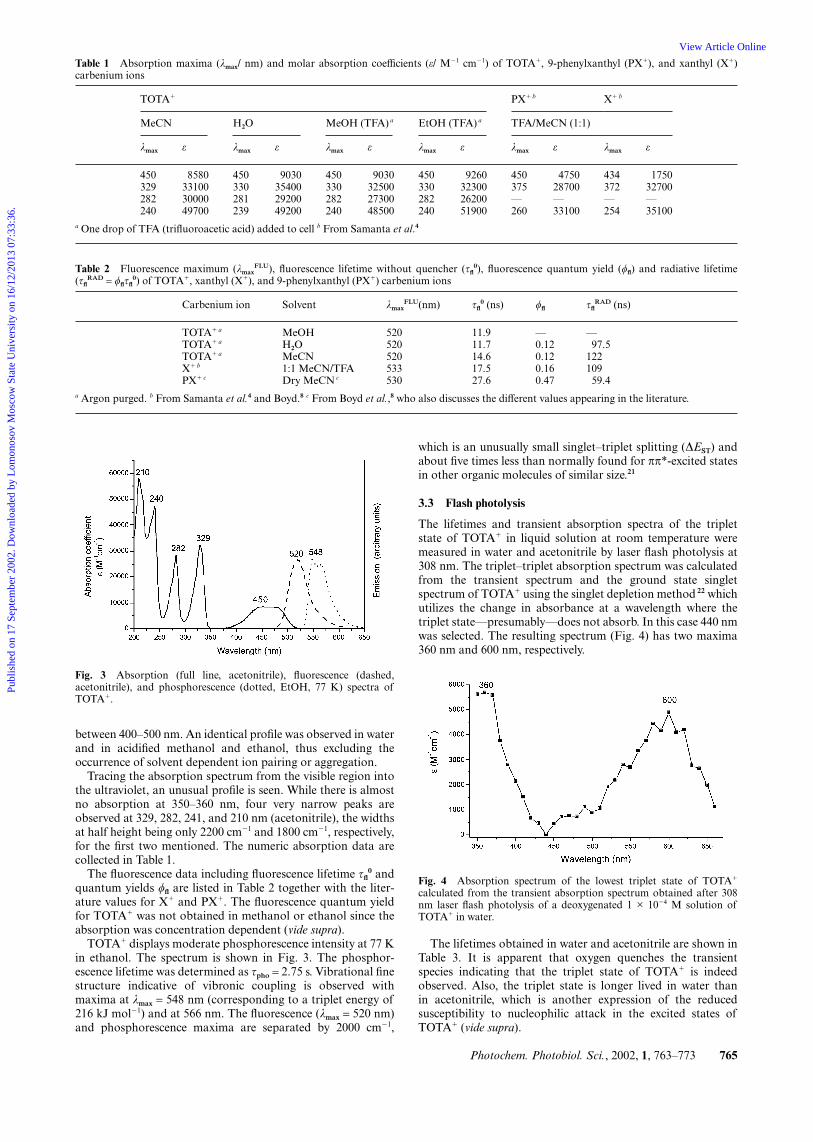
Table 1 Absorption maxima (λmax/ nm) and molar absorption coefficients (ε/ M�1 cm�1) of TOTA�, 9-phenylxanthyl (PX�), and xanthyl (X�)carbenium ions
TOTA� PX� b X� b
MeCN H2O MeOH (TFA) a EtOH (TFA) a TFA/MeCN (1:1)
λmax ε λmax ε λmax ε λmax ε λmax ε λmax ε
450 8580 450 9030 450 9030 450 9260 450 4750 434 1750329 33100 330 35400 330 32500 330 32300 375 28700 372 32700282 30000 281 29200 282 27300 282 26200 — — — —240 49700 239 49200 240 48500 240 51900 260 33100 254 35100
a One drop of TFA (trifluoroacetic acid) added to cell b From Samanta et al.4
Table 2 Fluorescence maximum (λmaxFLU), fluorescence lifetime without quencher (τfl
0), fluorescence quantum yield (�fl) and radiative lifetime(τfl
RAD = �flτfl0) of TOTA�, xanthyl (X�), and 9-phenylxanthyl (PX�) carbenium ions
Carbenium ion Solvent λmaxFLU(nm) τfl
0 (ns) �fl τflRAD (ns)
TOTA� a MeOH 520 11.9 — —TOTA� a H2O 520 11.7 0.12 97.5TOTA� a MeCN 520 14.6 0.12 122X� b 1:1 MeCN/TFA 533 17.5 0.16 109PX� c Dry MeCN c 530 27.6 0.47 59.4
a Argon purged. b From Samanta et al.4 and Boyd.8 c From Boyd et al.,8 who also discusses the different values appearing in the literature.
between 400–500 nm. An identical profile was observed in waterand in acidified methanol and ethanol, thus excluding theoccurrence of solvent dependent ion pairing or aggregation.
Tracing the absorption spectrum from the visible region intothe ultraviolet, an unusual profile is seen. While there is almostno absorption at 350–360 nm, four very narrow peaks areobserved at 329, 282, 241, and 210 nm (acetonitrile), the widthsat half height being only 2200 cm�1 and 1800 cm�1, respectively,for the first two mentioned. The numeric absorption data arecollected in Table 1.
The fluorescence data including fluorescence lifetime τfl0 and
quantum yields �fl are listed in Table 2 together with the liter-ature values for X� and PX�. The fluorescence quantum yieldfor TOTA� was not obtained in methanol or ethanol since theabsorption was concentration dependent (vide supra).
TOTA� displays moderate phosphorescence intensity at 77 Kin ethanol. The spectrum is shown in Fig. 3. The phosphor-escence lifetime was determined as τpho = 2.75 s. Vibrational finestructure indicative of vibronic coupling is observed withmaxima at λmax = 548 nm (corresponding to a triplet energy of216 kJ mol�1) and at 566 nm. The fluorescence (λmax = 520 nm)and phosphorescence maxima are separated by 2000 cm�1,
Fig. 3 Absorption (full line, acetonitrile), fluorescence (dashed,acetonitrile), and phosphorescence (dotted, EtOH, 77 K) spectra ofTOTA�.
which is an unusually small singlet–triplet splitting (∆EST) andabout five times less than normally found for ππ*-excited statesin other organic molecules of similar size.21
3.3 Flash photolysis
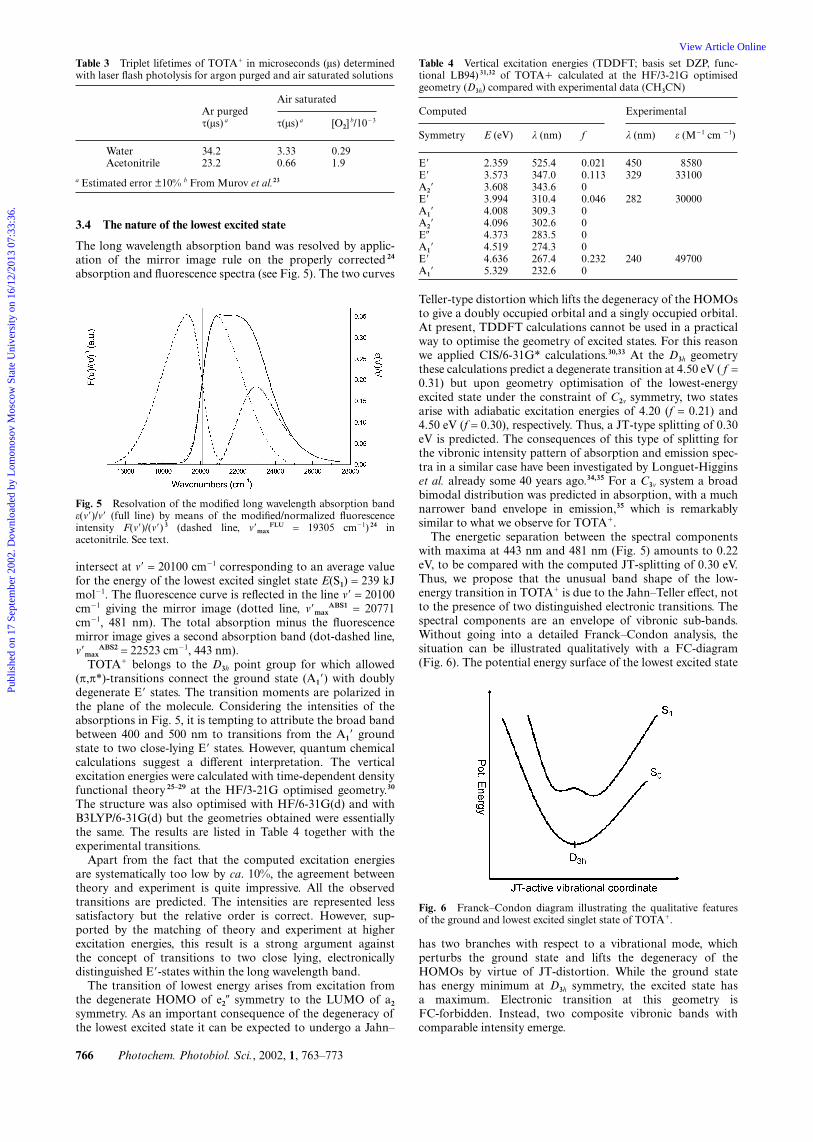
The lifetimes and transient absorption spectra of the tripletstate of TOTA� in liquid solution at room temperature weremeasured in water and acetonitrile by laser flash photolysis at308 nm. The triplet–triplet absorption spectrum was calculatedfrom the transient spectrum and the ground state singletspectrum of TOTA� using the singlet depletion method 22 whichutilizes the change in absorbance at a wavelength where thetriplet state—presumably—does not absorb. In this case 440 nmwas selected. The resulting spectrum (Fig. 4) has two maxima360 nm and 600 nm, respectively.
The lifetimes obtained in water and acetonitrile are shown inTable 3. It is apparent that oxygen quenches the transientspecies indicating that the triplet state of TOTA� is indeedobserved. Also, the triplet state is longer lived in water thanin acetonitrile, which is another expression of the reducedsusceptibility to nucleophilic attack in the excited states ofTOTA� (vide supra).
Fig. 4 Absorption spectrum of the lowest triplet state of TOTA�
calculated from the transient absorption spectrum obtained after 308nm laser flash photolysis of a deoxygenated 1 × 10�4 M solution ofTOTA� in water.
Photochem. Photobiol. Sci., 2002, 1, 763–773 765
Publ
ishe
d on
17
Sept
embe
r 20
02. D
ownl
oade
d by
Lom
onos
ov M
osco
w S
tate
Uni
vers
ity o
n 16
/12/
2013
07:
33:3
6.
View Article Online
3.4 The nature of the lowest excited state
The long wavelength absorption band was resolved by applic-ation of the mirror image rule on the properly corrected 24
absorption and fluorescence spectra (see Fig. 5). The two curves
intersect at ν� = 20100 cm�1 corresponding to an average valuefor the energy of the lowest excited singlet state E(S1) = 239 kJmol�1. The fluorescence curve is reflected in the line ν� = 20100cm�1 giving the mirror image (dotted line, ν�max
ABS1 = 20771cm�1, 481 nm). The total absorption minus the fluorescencemirror image gives a second absorption band (dot-dashed line,ν�max
ABS2 = 22523 cm�1, 443 nm).TOTA� belongs to the D3h point group for which allowed
(π,π*)-transitions connect the ground state (A1�) with doublydegenerate E� states. The transition moments are polarized inthe plane of the molecule. Considering the intensities of theabsorptions in Fig. 5, it is tempting to attribute the broad bandbetween 400 and 500 nm to transitions from the A1� groundstate to two close-lying E� states. However, quantum chemicalcalculations suggest a different interpretation. The verticalexcitation energies were calculated with time-dependent densityfunctional theory 25–29 at the HF/3-21G optimised geometry.30
The structure was also optimised with HF/6-31G(d) and withB3LYP/6-31G(d) but the geometries obtained were essentiallythe same. The results are listed in Table 4 together with theexperimental transitions.
Apart from the fact that the computed excitation energiesare systematically too low by ca. 10%, the agreement betweentheory and experiment is quite impressive. All the observedtransitions are predicted. The intensities are represented lesssatisfactory but the relative order is correct. However, sup-ported by the matching of theory and experiment at higherexcitation energies, this result is a strong argument againstthe concept of transitions to two close lying, electronicallydistinguished E�-states within the long wavelength band.
The transition of lowest energy arises from excitation fromthe degenerate HOMO of e2� symmetry to the LUMO of a2
symmetry. As an important consequence of the degeneracy ofthe lowest excited state it can be expected to undergo a Jahn–
Fig. 5 Resolvation of the modified long wavelength absorption bandε(ν�)/ν� (full line) by means of the modified/normalized fluorescenceintensity F(ν�)/(ν�) 3 (dashed line, ν�max
FLU = 19305 cm�1) 24 inacetonitrile. See text.
Table 3 Triplet lifetimes of TOTA� in microseconds (µs) determinedwith laser flash photolysis for argon purged and air saturated solutions
Ar purgedAir saturated
τ(µs) a τ(µs) a [O2]
b/10�3
Water 34.2 3.33 0.29 Acetonitrile 23.2 0.66 1.9
a Estimated error ±10% b From Murov et al.23
Teller-type distortion which lifts the degeneracy of the HOMOsto give a doubly occupied orbital and a singly occupied orbital.At present, TDDFT calculations cannot be used in a practicalway to optimise the geometry of excited states. For this reasonwe applied CIS/6-31G* calculations.30,33 At the D3h geometrythese calculations predict a degenerate transition at 4.50 eV ( f =0.31) but upon geometry optimisation of the lowest-energyexcited state under the constraint of C2v symmetry, two statesarise with adiabatic excitation energies of 4.20 (f = 0.21) and4.50 eV (f = 0.30), respectively. Thus, a JT-type splitting of 0.30eV is predicted. The consequences of this type of splitting forthe vibronic intensity pattern of absorption and emission spec-tra in a similar case have been investigated by Longuet-Higginset al. already some 40 years ago.34,35 For a C3v system a broadbimodal distribution was predicted in absorption, with a muchnarrower band envelope in emission,35 which is remarkablysimilar to what we observe for TOTA�.
The energetic separation between the spectral componentswith maxima at 443 nm and 481 nm (Fig. 5) amounts to 0.22eV, to be compared with the computed JT-splitting of 0.30 eV.Thus, we propose that the unusual band shape of the low-energy transition in TOTA� is due to the Jahn–Teller effect, notto the presence of two distinguished electronic transitions. Thespectral components are an envelope of vibronic sub-bands.Without going into a detailed Franck–Condon analysis, thesituation can be illustrated qualitatively with a FC-diagram(Fig. 6). The potential energy surface of the lowest excited state
has two branches with respect to a vibrational mode, whichperturbs the ground state and lifts the degeneracy of theHOMOs by virtue of JT-distortion. While the ground statehas energy minimum at D3h symmetry, the excited state hasa maximum. Electronic transition at this geometry isFC-forbidden. Instead, two composite vibronic bands withcomparable intensity emerge.
Fig. 6 Franck–Condon diagram illustrating the qualitative featuresof the ground and lowest excited singlet state of TOTA�.
Table 4 Vertical excitation energies (TDDFT; basis set DZP, func-tional LB94) 31,32 of TOTA� calculated at the HF/3-21G optimisedgeometry (D3h) compared with experimental data (CH3CN)
Computed Experimental
Symmetry E (eV) λ (nm) f λ (nm) ε (M�1 cm �1)
E� 2.359 525.4 0.021 450 8580E� 3.573 347.0 0.113 329 33100A2� 3.608 343.6 0 E� 3.994 310.4 0.046 282 30000A1� 4.008 309.3 0 A2� 4.096 302.6 0 E� 4.373 283.5 0 A1� 4.519 274.3 0 E� 4.636 267.4 0.232 240 49700A1� 5.329 232.6 0
766 Photochem. Photobiol. Sci., 2002, 1, 763–773
Publ
ishe
d on
17
Sept
embe
r 20
02. D
ownl
oade
d by
Lom
onos
ov M
osco
w S
tate
Uni
vers
ity o
n 16
/12/
2013
07:
33:3
6.
View Article Online

Fig. 7 Atomic coefficients of two degenerate HOMO and non-degenerate LUMO orbitals.
By analogy, the same effect should be important in othertriarylmethyl carbocations with threefold symmetry, such asthe triphenylmethyl cation, which has an absorption spectrumstrikingly similar to that of TOTA�.36 Likewise, a JT-splittingshould be observed in the corresponding triplet states, whichhave the same occupation of spatial orbitals. A geometry opti-misation of the T1 state of TOTA� under C2v symmetry usingthe B3LYP hybrid functional 37 (6-31G(d) basis set) indeed ledto an energy lowering. A subsequent TDDFT calculation withthe same DFT method showed an energy splitting of 0.084 eVbetween the two triplet JT-states, which is almost four timessmaller than was calculated for the corresponding singletJT-states. Experimentally, triplet state EPR spectra of TOTA�
did not reveal Jahn–Teller splitting, in contrast to some otherthreefold symmetric carbocations,38 but this may be due torapid interconversion of the three equivalent relaxed species,leading to an averaged symmetric structure, even in thelow-temperature glass.
In order to obtain a qualitative picture of redistribution ofcharge in the excited state, the atomic coefficients of theHOMO/LUMO orbitals were calculated by means of HF/6-31G* calculations. The results are depicted in Fig. 7. TheLUMO has a large coefficient on the central carbon atom, whilethe electron density of the HOMOs are mostly distributed atthe periphery of the π-system and have a node on the centralcarbon. Thus, a transfer of negative charge accompanies excit-ation to the central carbon atom. This explains why the excitedstate has a much-reduced electrophilic character (vide supra).
3.5 Quenching of fluorescence lifetime by aromatic compounds
The fluorescence of TOTA� was partly quenched in thepresence of the following aromatic compounds: pyrene,anthracene, phenanthrene, trans-stilbene, triphenylene, naphth-alene, biphenyl, durene, mesitylene and p-xylene. This quench-ing was evident from the gradual shortening of the observedlifetime (Fig. 8) and from the progressive decrease in steady-state emission intensity (see next section) with increasingquencher concentration.
In every case, the lifetime data gave linear Stern–Volmer plots(e.g. inset Fig. 8) in agreement with eqn. (1), where KTR is theSV-constant, τfl is the lifetime measured without quencher, andkq
TR the dynamic quenching rate constant.
None of the aromatic quenchers have excited singlet statesbelow S1 of TOTA�. Therefore, singlet–singlet energy transferis excluded and the quenching mechanism is most probablyelectron transfer. Together with the dynamic quenchingconstants (kq
TR), Table 5 displays the ionisation (IP) and halfwave oxidation (E½
ox) potentials of the donors. The free energy
τfl0/τfl = 1 � KTR[Q] = 1 � kq
TRτfl0[Q] (1)
change of photoinduced electron transfer (∆GET) was derivedby application of eqn. (2).43 A coulombic energy work-term isnot included since no new charges are created in the electrontransfer process.44
The excited singlet energy (Es) was obtained from the inter-section between the corrected fluorescence and absorptionspectra (Fig. 5). The reduction (half-wave) potential of TOTA�
(E½red = �0.385 V (versus. SCE)) has been reported by Nem-
cová and Nemec.14,45
The resulting values for ∆GET are compiled in Table 5. Theleast exergonic electron transfers have ∆G = ca. �5 kcal mol�1,i.e. they are close to the onset value (∆G = ca. �10 kcal mol�1)for diffusionally controlled bimolecular electron transfer as out-lined in the classical Rehm–Weller theory.43 Accordingly, therate constants are all close to the diffusionally controlled limitin acetonitrile (2 × 1010 M�1 s�1). Still, a weak, qualitativecorrelation to the driving force can be seen.
3.6 Quenching of fluorescence intensity by aromaticcompounds
Shortening of the fluorescence lifetime of TOTA� was para-lleled by progressive decrease in steady-state emission intensityupon addition of one of the 10 aromatic quenchers as illus-trated in Fig. 9.
The Stern–Volmer plots derived from the steady statefluorescence measurements all showed upward curvature, asillustrated by the inset of Fig. 9. This feature is indicative ofstatic quenching, in this case probably due to charge transfercomplex formation in the ground state according to eqn. (3):
with association constant
When both static and dynamic quenching occur, the modifiedStern–Volmer expression applies: 46
where KTR = kqTRτf is the Stern–Volmer constant obtained
from the time resolved measurements [eqn. (1)]. The groundstate association constants were estimated by fitting the data toeqn. (5). The values obtained are listed in Table 6. They spanthe range of very weak (mesitylene) to moderately strong(pyrene) complexes. Formation of a donor–acceptor complexresults from an intricate combination of forces.47 In this case,
∆GET = E½ox � E½
red � Es (2)
TOTA� � Q TOTA-Q� (3)
KA = [TOTA-Q�]/[TOTA�][Q] (4)
l0/I = 1 � (KTR � KA)[Q] � KTR KA [Q]2 (5)
Photochem. Photobiol. Sci., 2002, 1, 763–773 767
Publ
ishe
d on
17
Sept
embe
r 20
02. D
ownl
oade
d by
Lom
onos
ov M
osco
w S
tate
Uni
vers
ity o
n 16
/12/
2013
07:
33:3
6.
View Article Online

Table 5 Bimolecular rate constants (kqTR) for quenching of fluorescence lifetime of TOTA� in argon purged acetonitrile. Steady state quenching
rate constants (kqSS) of 9-phenylxanthylium (PX�) fluorescence in acetonitrile and xanthylium (X�) fluorescence in 1:1 (v/v) MeCN/TFA at room
temperature. The half-wave oxidation (E½ox) and ionisation (Ip) potentials of the aromatic donors are also given
TOTA�
X� PX�
QuencherIp
a
(eV)E½
ox b
(V)kq
TR c
(109 M�1 s�1)∆GET
(eV)∆GET
(kcal mol�1)kq
SS d
(109 M�1 s�1)kq
SS d
(109 M�1 s�1)
Mesitylene 8.41 1.90 e 9.04 �0.20 �4.6 i) 13p-Xylene 8.44 1.85 f 9.28 �0.24 �5.5 20 14Durene 8.04 1.62 e 11.6 �0.48 �11.1 i) 18Naphthalene 8.14 1.72 e 12.3 �0.38 �8.8 23 18Biphenyl 7.95 1.91 e 12.5 �0.19 �4.4 19 16Triphenylene 7.842 1.55 g 13.5 �0.55 �12.7 i) i)Anthracene 7.45 1.31 g 14.8 �0.79 �18.2 28 26Phenanthrene 7.86 1.52 f 15.5 �0.58 �13.3 i) i)Pyrene 7.41 1.15 f 15.7 �0.95 �21.9 i) i)trans-Stilbene 7.7 1.51 h 16.9 �0.59 �13.6 i) i)
a Taken from Siegerman.39 b Versus SCE c Estimated error ±5 % d From Samanta et al.4,6 e From O. Hammerich.40 f Converted41 from value measuredversus Ag/0.1 N Ag� in acetonitrile.39 g From Jones and Fox42 h In AcOH.39 i Not determined.
the ranking is probably determined by ionisation potentials ofthe donors (Table 5), sterical factors (p-xylene, mesitylene,durene, and biphenyl), and extended π–π-interaction (pyrene,anthracene, triphenylene).
In the solvents used in this study, no emission, which couldbe attributed to exciplex formation between TOTA� and thearomatic donors, was observed at longer wavelengths.
The absorption spectra of the ground state complexes wereinvestigated by means of tandem cells (see experimental
Fig. 8 Quenching of the fluorescence lifetime of TOTA� (1.0 × 10�5
M in acetonitrile) by trans-stilbene in concentrations graduallyincreasing from 0.0 mM (a) to 2.59 mM (b). Inset: The accompanyingStern–Volmer plot.
Fig. 9 Quenching of TOTA� (1.0 × 10�5 M in acetonitrile) steady statefluorescence (λex = 450 nm) by pyrene in concentrations increasing from0.0 mM (a) to 9.53 mM (b) and the accompanying Stern–Volmer plot.
section). It was necessary to use highly concentrated solutionsof TOTA� and the π-donor in order to observe absorptionchanges induced by mixing, a fact indicating weak absorptionsof the complexes and/or small constants of formation. In gen-eral, an apparent red shift of the long wavelength absorptionband of TOTA� was observed meaning that new transition(s)of the CT complex occur around 500 nm. Furthermore, adistinctive increase in absorption in the 350–400 nm region wasa common observation for all the π-donors. In these experi-ments, pyrene and anthracene differed qualitatively from theothers due to a colour change from yellow to brownish redand the appearance of a distinctive charge transfer band atlonger wavelength upon mixing of the highly concentratedsolutions of the components in acetonitrile (see supplementarymaterial†).
3.7 CT-complexes with alkoxy- and alkylthio-substitutedtriphenylenes
In Table 6, triphenylene ranks among the better donors forCT-complex formation with TOTA�. Hexakis(alkyloxy)- andhexakis(alkylsulfanyl)-substituted triphenylenes are expected tobe even better electron donors due to the π-donating hetero-atoms.48 Furthermore, some of these derivatives can exist indiscotic liquid crystalline phases 49,50 provided the alkyl chainsare long enough (>C5) and unbranched. Aggregationalbehaviour in solution has been shown to relate to this ability.48,51
On this background, complex formation between TOTA�
and (2,3,6,7,10,11)-hexakis-(n-hexyloxy)triphenylene (HHOTP,discogenic 50), (2,3,6,7,10,11)-hexakis-(n-hexylsulfanyl)triphen-ylene(HHSTP, discogenic 49), and (2,3,6,7,10,11)-hexakis(4-tert-butylphenylsulfanyl)triphenylene (HTBPSTP, not discogenic)(Fig. 10) was investigated.
Table 6 Association constants (KA) for CT-complexes betweenTOTA� and 10 aromatic fluorescence quenchers in acetonitrile
π-Donor KAa (M�1)
p-Xylene 15Mesitylene 5Durene 10Biphenyl 15Naphthalene 40Phenanthrene 90Pyrene 560Anthracene 470Triphenylene 325trans-Stilbene 250
a Estimated error, ±15%.
768 Photochem. Photobiol. Sci., 2002, 1, 763–773
Publ
ishe
d on
17
Sept
embe
r 20
02. D
ownl
oade
d by
Lom
onos
ov M
osco
w S
tate
Uni
vers
ity o
n 16
/12/
2013
07:
33:3
6.
View Article Online

Table 7 Long wavelength absorption maxima of CT-complexes between TOTA� and substituted triphenylenes
Triphenylene CT Abs max (nm) εmax (M�1 cm �1) (±10%) K (M�1) (±10%)
HHOTP in CH2Cl2 661 152 a 2290 a
HHOTP in CH3CN 632 185 b 1630 b
HHOTP in CHCl3 636 — —HHSTP in CH2Cl2 620 — —HTBPSTP in CH2Cl2 580 — —
a From Benesi–Hildebrand plots with HHOTP in excess. b From Benesi–Hildebrand plots with TOTA� in excess.
Indeed, long wavelengths absorption maxima were observed(Table 7) upon mixing of concentrated solutions of the tri-phenylenes and TOTA� by means of absorption tandem-cells(see experimental section). Since CT-complexes with betterelectron donors are expected to show lower excitationenergies,47 the maximum wavelength values imply, that theelectron donor capability increases in the series HTBPSTP,HHSTP, and HHTOP. This result is in agreement with theoxidation potentials being E½ = 0.820 V for HHTOP andE½ = 1.03 V for HHSTP (measured 52 versus SCE in CH2Cl2/CF3COOH (9:1)), respectively.
Assuming 1:1 stoichiometry for the complex formationbetween TOTA� and the triphenylenes, attempts were made todetermine the equilibrium constants by application of thestandard Benesi–Hildebrand procedure.53 These titrations werecarried out by varying an excess concentration of donor(HHOTP in CH2Cl2) or by varying an excess concentration ofacceptor (TOTA� in CH3CN). As can be seen from Table 7, thetwo procedures led to different results, which may reflectaggregational behaviour of HHOTP in the highly concentratedsolution in CH2Cl2. In a model study, Markovitsi et al.54 foundno indication of hexalkoxytriphenylenes forming aggregates inCH2Cl2. However, the HHOTP concentrations used in theirstudy were in the range 10�6 M, i.e. orders of magnitude lessthan applied here with HHOTP in excess. In n-heptane, wherediscogenic triphenylenes show aggregational behaviour atlow concentrations, Markovitsi et al.51 found that formation ofCT complexes between (2,3,6,7,10,11)-hexakis-(n-pentyloxy)-triphenylene and 2,4,7-trinitrofluoren-9-one is greatly favoured.Despite this uncertainty, the CT-complex formation betweenHHOTP and TOTA� must be characterized as relatively strong.
3.8 Quenching of fluorescence by inorganic ions and oxygen
No ground state complex formation between TOTA� andchloride, bromide or iodide ions were detectable in water bymeans of absorption spectroscopy in tandem cells, employingsaturated solutions of the halide ions. However, the fluor-escence of TOTA� is quenched efficiently by bromide andiodide ions in millimolar concentrations in water while muchhigher concentrations (0.5 M) of chloride are required for thesame effect. The steady-state Stern–Volmer plots (see figures insupplementary material†) are linear for bromide and iodide inthe range studied while some downward curvature is discernible
Fig. 10 Alkoxy- and alkylthio-substituted triphenylenes.
for chloride. Since this feature is observed in steady-state as wellas in the lifetime SV-plot it must be ascribed to the increasingionic strength. Being a charge annihilation, the quenchingprocess is expected to slow down when the polarity increases.
The following rate constants (kqSS (M�1s�1)) were obtained
from the slopes of the steady-state SV-plots: 2.0 × 108 forchloride, 1.4 × 1010 for bromide, and 2.1 × 1010 for iodide. Fromthe chloride/lifetime plot (initial slope), kq
TR = 1.8 × 108 M�1s�1.Quenching experiments were also performed with acetate,
nitrate, phosphate, and tetrafluoroborate anions. The two firstmentioned were selected since their ionisation potentials areintermediate between chloride and bromide.47 However,none of the four anions caused any noticeable reduction offluorescence intensity even at very high quencher concen-trations. For phosphate this result is important for inter-pretation of the photophysical interactions between TOTA�
and DNA.17,55
The fluorescence of TOTA� is only moderately quenched bymolecular oxygen. The quenching rate constants in methanoland acetonitrile are the same, kq
oxygen = 4 (± 1) × 109 M�1 s�1.This value was derived both from steady-state and lifetimemeasurements in oxygen saturated, aerated, and argon purgedsolutions.
4 Discussion
4.1 Reaction with alcohols
Carbenium ions with localized charge are not stable inalcoholic solvents due to nucleophilic attack. In contrast, manyTPMs are considered completely stable—or appear as such ifthey are only partially converted to the leuco ether. The xanth-ylium ions represent intermediate cases, since they can bestudied in water-free solvents with low nucleophilicity liketrifluoroethanol.8 Formation of a leuco ether by attack of analcohol molecule is conceptually very simple. Still, it seems tobe a largely unnoticed phenomenon among TPMs in spite ofthe extensive studies of their absorption spectra in differentsolvents.2,56 As an exception, Jones and Goswami 20 studied thephotobleaching of crystal violet (CV�) in 2-propanol in thepresence of halide ions. The dye photobleaches upon excitationin the UV but not in the visible. The photoactive species turnedout to be the leuco ether, the existence of which was establishedby independent experiments (ε263 = 5.4 × 104 M�1cm�1). Jonesand Goswami also reported an equilibrium constant for thedye/leuco ether reaction but it was incorrectly defined. Theynoticed that it takes more than 8 h for a dilute solution of CV�
in propanol to equilibrate. Likewise, Goldacre and Phillips 57
found the hydrolysis reactions of a series of TPMs to be surpris-ingly slow. Thus, in the reaction between crystal violet and H2O/OH� at pH = 4.7 it takes more than 7 h to reach 50% of theequilibrium carbinol concentration.
Thermodynamically TOTA� (pKR� = 9.05 11) and crystalviolet (pKR� = 9.4 57) show very comparable reactivity towardsnucleophilic reagents while the xanthyl carbenium ions havemuch greater affinity. Calculations based on density functionaltheory (DFT) on the ground state of TOTA� have indicatedthat the charge on the central carbon atom follows the TPMs inbeing extensively delocalized.10 Beyond the significance of
Photochem. Photobiol. Sci., 2002, 1, 763–773 769
Publ
ishe
d on
17
Sept
embe
r 20
02. D
ownl
oade
d by
Lom
onos
ov M
osco
w S
tate
Uni
vers
ity o
n 16
/12/
2013
07:
33:3
6.
View Article Online

delocalisation, attention should also be drawn to the stereo-chemical aspects of ether formation, which may be determiningfor the rates by which equilibrium is reached in each case. Whilethe central carbon atom of TPMs is screened from nucleophilicattack by the ortho hydrogen atoms, it is more accessible in thexanthyl carbenium and TOTA� systems. On the other hand,rehybridisation means disrupture of the planar structure of thewhole molecule (Scheme 1).
The scarcity of published work on the equilibrium reactionbetween ground state TPMs and alcohols is in contrast to theuniversal character of the reaction. Eventually, many absorp-tion coefficients reported in the literature await correction forthis reason and – since the TPM leuco ether absorb in the UV-region only—spectral profiles of TPMs in alcohols might beinaccurate.
4.2 Absorption and fluorescence spectra
In general, a good mirror-image relationship is observedbetween the fluorescence and the low-energy absorption bandof xanthyl carbenium ions.8 Thus, Samanta et al.4 derivedthe radiative lifetime for X� and PX� by means of the Strickler–Berg formula and found excellent agreement with the experi-mental values obtained from the fluorescence lifetimes andquantum yields. This result implies that only one electronictransition is contributing to the low energy absorption band ofthe xanthyl carbenium ions. In contrast, Fig. 5 shows that thelong wavelength absorption band of TOTA� does not connectto the fluorescence in a mirror-image relationship. As describedabove, this feature is believed to originate in a Jahn–Teller likedistortion of the excited singlet state. It is noteworthy, thatthe long wavelength absorption band of the unsubstitutedtriphenylmethyl carbenium ion shows a striking resemblanceto that of TOTA�.36 Neither in this case nor among othersymmetrically substituted TPMs of D3 symmetry does JT-distortion of excited states seem to have been investigatedcomputationally.2 Considering the (perturbed) degeneracy ofthe TOTA� transitions, it is natural that its long wavelengthabsorption coefficient is almost twice as big as that of PX�
(Table 1).PX� and X� also display intense bands at 375 and 372 nm,
respectively, (Table 1). They correspond to the peak at 330 nmin the spectrum of TOTA�. The absorption coefficients areroughly the same for all three, but the TOTA� band is muchnarrower (FWHM = 2200 cm�1) probably as a consequence ofthe rigidity of the TOTA� molecular framework. The samecomparable remark can be made on the absorption banddisplayed by each of the three compounds in the 240–260 nmrange.
The Stokes shift in acetonitrile, obtained as the energeticdistance between the first vibronic absorption band at 481 nm(Fig. 5) and fluorescence maximum amounts to 1460 cm�1. Thismodest value is ascribed to internal reorganization, sinceneither absorption nor the fluorescence spectra of TOTA�
show any noticeable solvatochromy in the solvents investigated(Table 2). Neither do TPMs show significant solvatochromy.2
Apparently, the change in coulombic solvation energyaccompanying redistribution of charge upon excitation is out-balanced by a reverse change in dispersion solvation energy. Incontrast to TOTA�, X� and PX�show larger Stokes shifts of4227 cm�1 and 3720 cm�1, respectively, in the same solvent,which indicate major differences in conformational andsolvation energies between the ground states and excited states.
4.3 Fluorescence quenching by nucleophilic attack and electrontransfer
Dynamic electron transfer quenching of excited states caneither occur as a single electron transfer or as a nucleophilicattack involving an electron pair. The last process leads to an
adduct which eventually may dissociate homolytically thuscreating the same products as the single electron transfer.
The photophysical properties of the xanthyl carbenium ionshave been investigated in a number of polar solvents.4,6,44,58–61
Since the excited singlet states of these carbenium ions are verysensitive to nucleophilic attack, e.g. by traces of water, severalvalues have been reported for the fluorescence lifetimes. Thenumbers quoted in Table 2 for compounds X� and PX� havebeen obtained in 1:1 trifluoroacetic acid/MeCN and dryMeCN, respectively.8
Boyd and coworkers 8,44,58,60,62 and others 63 have deliberatelystudied quenching by water, alcohols and ethers of thefluorescence from a range of substituted 9-phenylxanthyl and9-phenylthioxanthyl carbenium ions. The quenching rates couldnot be correlated with the reduction potential of the cationsaccording to Rehm–Weller theory, as otherwise wouldhave been expected if single electron transfer was operating.Consequently, it was concluded that the quenching action wasinitiated by a nucleophilic attack on the excited state followedby ground state dissociation and regeneration of the carbeniumion, eventually via a homolytic pathway.60,62 Minto and Das 59
measured the electrophilicity of the lowest excited singletstate of 9-phenylxanthylium in terms of rate constants of bi-molecular quenching by anions and lone-pair containingmolecules. They find it considerably more pronounced thanthat of the ground state, that is, kq’s being higher by severalorder of magnitudes.
In contrast, the excited singlet state of TOTA� seems to beonly marginally affected by nucleophiles. The fluorescencelifetime (Table 2) is the same in water and methanol in spite ofthe increased nucleophilicity of the latter. Further, τfl
0 is onlyreduced from 14.6 ns to 11.7 ns when going from acetonitrile towater (Table 2). If this difference had its origin in a nucleophilicattack by water the rate constant for this hypothetical reaction(approximating the activity of water to 55.5 M) would be only3.4 × 105 M�1s�1.
Thus, the relative reactivity towards nucleophiles amongground state molecules of TPMs (like crystal violet), PX�, X�,and TOTA� is retained in the excited state. The reactions mayspeed up by many orders of magnitude due to increased exo-thermicity 59 but due to even faster, competing processes, thesolvolysis does not manifest itself in the excited state lifetimesexcept in case of the xanthylium carbenium ions.
On the other side, single electron transfer has been demon-strated to occur as a general phenomenon between excitedcarbenium ions and aromatics or alkenes.8 In particular,Samanta et al.,4,6 Wan et al.,13 and Azarani et al.5 have deter-mined the quenching rates for a series of aromatic donors ofthe fluorescence from X� and PX� by means of steady stateStern–Volmer kinetics. The first mentioned authors applied nsflash photolysis and established the quenching process aselectron transfer by observing the radical cations of thearomatic quenchers.4,6 They also specifically looked for—butdid not observe—any ground state complex formation.
From the 18 aromatic donors investigated by Samanta et al.relevant values for the quenching rate constants kq
SS areselected and listed in Table 5. Even though a levelling effect isexpected owing to the diffusional limit, corresponding rateconstants for fluorescence quenching of X�(kq
SS), PX�(kqSS)
TOTA�(kqTR) consistently decrease in this order. This trend
seems reasonable considering the decrease in electrophilicity asindicated by the pKR� values of the three carbenium ions, but aquantitative estimate is difficult. The half-wave reductionpotential of TOTA� has been reported as E½
red = �0.385 Vversus SCE in acetonitrile.14 For PX�, E½
red = �0.25 V, and forX�, E½
red = �0.25 V versus SCE (converted from ferrocenium/ferrocene reference; the numeric identity is accidental) 64
However, these values refer to solutions in sulfolane/3-methylsulfolane(5%). The driving force of the reaction alsodepends on the excitation energy E(S1) of the acceptor (Rehm–
770 Photochem. Photobiol. Sci., 2002, 1, 763–773
Publ
ishe
d on
17
Sept
embe
r 20
02. D
ownl
oade
d by
Lom
onos
ov M
osco
w S
tate
Uni
vers
ity o
n 16
/12/
2013
07:
33:3
6.
View Article Online

Weller, eqn. (2)). Thus, E(S1) increases by 16 kJ mol�1 whengoing from TOTA� to PX� and further by 10 kJ mol�1 whengoing to X� (Table 1). This feature adds to the increasingexothermicity for electron transfer quenching in the series. Eventhough the rate constants for fluorescence quenching ofX�(kq
SS), PX�(kqSS) are in the diffusional limit, the marginal
correlation with the donor oxidation potential has been takenas credit in support of an electron transfer mechanism.5,6
However, Samanta et al.6 erroneously quote the electrochemicaldata of Arnett et al.64
In a very recent communication, Dileesh and Gopidas 17
report quenching of TOTA� fluorescence by 13 aromaticquenchers in acetonitrile. Two of these, naphthalene andbiphenyl, are also used in the work presented here, and for thesethe quenching rate constants given by Dileesh and Gopidas aregreater than ours by ca. 30%. Since their values are derivedfrom steady state measurements, we suggest that the differencerelies on a contribution from static quenching beingunrecognised for their part.
4.4 Quenching by inorganic anions and oxygen
The relative quenching efficiency of the three halide ions inwater clearly reflects their increasing reductive power Cl� < Br�
< I�. In contrast to Jones and Goswami 20 who observed form-ation of a CT-complex between CV� and iodide (but notchloride or bromide), there was no indication for formation ofCT-complexes between TOTA� and the halides. In addition, allsteady-state CV-plots were linear. Shukla and Wan 65 observedquenching of the fluorescence from thioxanthylium ion by N3
�
and CN�. They found kq = 1.5 × 1010 and 1.3 × 109 M�1s�1,respectively, but found no experimental evidence to distinguishbetween single electron transfer versus nucleophilic attack by anelectron pair for the quenching mechanism.
An interesting general feature of carbocationic fluorescenceis its resistance to quenching by oxygen, kq
oxygen ∼ 109 M�1s�1 forxanthylium and 9-phenylxanthylium ions,4–6,44,59 which is slowerby an order of magnitude compared to the rates normallyobserved for quenching of singlet excited (π,π*)-states ofaromatic hydrocarbons. This behaviour illustrates that the rateconstant for molecular oxygen singlet quenching falls markedlywith increase in ionisation potential of the substrate.66 Theexcited singlet state of TOTA� is a little more reactive (kq
oxygen =4 (± 1) × 109 M�1s�1) than xanthylium ions in accordance withan electron transfer mechanism (and the trend in Table 5) sincethe positive charge is more efficiently delocalised in TOTA�. Inaddition to electron transfer, molecular oxygen can also exercisea quenching effect by virtue of paramagnetically induced inter-system crossing.
4.5 Formation of ground state CT-complexes
Dynamic quenching results from collisional encountersbetween the fluorophore and quencher. Non-collisional orstatic quenching can occur when a non-fluorescent complex isformed between the fluorophore and quencher in their groundstates. In case of TOTA�, the steady-state Stern–Volmer plotsshowed upward curvature (Fig. 9) as a clear indication ofpartial static quenching within ground state complexes formedbetween TOTA� and the electron donors.46 Among those,pyrene and anthracene stand out with the largest Ks values(Table 6) and distinct long-wavelength absorption bands of theCT-complexes. The other donors form weaker ground statecomplexes as seen from their lower KA values (Table 6). Asidefrom reductive power, structural properties of the donors alsoplay a role in their ability to form ground state complexes.47
The individual nodal structures of the HOMOs and LUMOsinvolved determine the orbital overlap in each case. Further-more, the polymethylated benzenes are hindered by the sub-stituents. For biphenyl, a planar configuration and unhindered
interaction with the TOTA� π-system is hampered by repulsionamong its ortho-hydrogens.
Samanta et al.6 were unable to detect any ground state com-plexation between a range of aromatic donors and X� or PX�
even at the highest concentrations obtainable. Other carbeniumions like the tropylium system 7,67 follow TOTA� in its ability toform CT-complexes with aromatic electron donors.
Formation of CT-complexes between TOTA� and the substi-tuted triphenylenes was evidenced by appearance of distinctnew absorption bands (Table 7). According to common theoryfor charge transfer complexation, the energy of the CT-excitation decreases with the oxidation potential of thedonor.7,47 The CT-complexes of HHOTP and HHSTP complywith this rule, since the former, being a better electron donor(E½
ox = 0.82 V (SCE)), absorbs at higher wavelength (661 nm)than the latter (E½
ox = 1.03 V (SCE), 620 nm)).Non-conformational behaviour is indicated by the batho-
chromic shift undergone by the CT-band of HHOTP/TOTA�
when going from the less polar CHCl3 (ε = 4.8) to the morepolar CH2Cl2 (ε = 9.1). In contrast to CT-complexes betweenneutral partners, the optical transition of these TOTA� com-plexes represent charge delocalisation instead of chargeseparation. Therefore, one should expect the smallest excitationenergy in the least polar solvent, contrary to observation.However, this picture may be too simple. Feldman and Graves 68
measured absorption spectra of CT-complexes between thetropylium ion and donors pyrene and phenothiazine in eightdifferent solvents. In their data, there were no correlations tobe found between excitation energy and solvent parameterslike dielectrical constant or refraction index. Taken togetherwith the unsuccessful attempts to determine unambiguousassociation constants by a conventional Benesi–Hildebrandapproach, it must also be questioned whether the premiseof one-to-one stoichiometry still holds. Eventually, the sub-stituted triphenylenes form aggregates in the concentratedsolutions and contributions from polaron states may becomeimportant.
4.6 Triplet state properties
Experimentally, the singlet–triplet splitting for TOTA� wasfound to be ∆EST = 2000 cm�1, while the TDDFT calculationdescribed above gave ∆EST(TOTA�) = 3100 cm�1. Thisunusually small value is due to the relatively small spatialoverlap of HOMO and LUMO (Fig. 7).69
Noteworthy, small singlet–triplet splittings are typicallyfound among TPMs as well,2 as was recognized by Kasha morethan 50 years ago.27
Dileesh and Gopidas 17 have also obtained the triplet–tripletabsorption spectrum of TOTA� in acetonitrile by means of nsflash photolysis. The two measurements agree on the mainfeatures, i.e. both spectra display dominant maxima at 360 nmand 600 nm (Fig. 4). However, the spectrum of Dileesh andGopidas also display less pronounced maxima at 450 nm and550 nm, respectively. This is rather unfortunate. Both groupsapply the singlet depletion method 22 for determination of theT–T-absorption coefficients. To do this, one must decide a wave-length at which the ε(T–T) is set at zero. We have used 450 nm,while Dileesh and Gopidas fail to report this assignment. Thus,we obtain ε600(T–T) = 5000 M�1cm�1, while they report 6400M�1cm�1. Considering the circumstances, the discrepancyseems minor.
5 ConclusionsThe lowest excited state of TOTA� is doubly degenerate if D3h
symmetry is maintained. Calculations, however, indicate thatthe state is split by a vibrational mode due to the Jahn–Tellereffect, giving rise to a broad, long wavelength absorption bandcomposed of two vibronic sub-bands.
Photochem. Photobiol. Sci., 2002, 1, 763–773 771
Publ
ishe
d on
17
Sept
embe
r 20
02. D
ownl
oade
d by
Lom
onos
ov M
osco
w S
tate
Uni
vers
ity o
n 16
/12/
2013
07:
33:3
6.
View Article Online

The electron deficiency of carbenium ions can be met bysingle electron transfer or by nucleophilic attack, i.e. transfer ofan electron pair. As such, the term nucleophilicity is confined tospecies with lone-pairs. In this work, both types of electrontransfer are involved. Together with the basic photophysicalproperties of TOTA�, electrophilic reactivity of the groundstate and electron transfer to the excited singlet state arereported and discussed.
While triphenylmethane dyes owe their stability partly toconfigurational shielding of the centre carbon atom, the groundstate of TOTA� escapes nucleophilic attack by e.g. water—andinteracts only weakly with methanol—because the positivecharge is effectively delocalized.10 This feature is further inten-sified upon excitation. The excited state lifetimes are barely(singlet) or not at all (triplet) influenced by the nucleophilicityof the solvent. This divergent behaviour is rationalized by theorbital pictures of the HOMOs and LUMO orbitals (Fig. 7).The LUMO has a large coefficient on the central carbon atomwhile the HOMOs have a node there and are mostly located atthe periphery of the π-system. Consequently, excitation isaccompanied by a transfer of negative charge to the centralcarbon atom.
The structurally related xanthylium and 9-phenylxanthyliumcarbenium ions have a less favourable electron distribution.They are much more sensitive to nucleophilic solvents and aremore easily reduced. The relative reactivities of these systemswith respect to electron transfer and nucleophilic attack areretained in the excited singlet states with necessary modifica-tions due to different excitation energies.
In sum, TOTA� distinguishes itself by combining severalmolecular properties: 1) It is stable under normal circumstancesin solvents with modest nucleophilic power, 2) it is planar andpossesses high symmetry in the ground state (D3h), 3) it is anelectron acceptor by virtue of its cationic nature, 4) it absorbs inthe visible region but has a region of very low absorbance in thenear-UV, and 5) it fluoresces with a fair quantum yield. Takentogether, these properties make TOTA� and its derivatives 70,71
worthy of further investigations with reference to applicationsas fluorescence probes and as building blocks in supramolec-ular structures.72
Acknowledgements
Professor Klaus Bechgaard (Research Center Risø, Denmark)measured the oxidation potentials of HHTOP and HHSTP,while Professor Steen Steenken (MPI, Mülheim) and ProfessorKurt Mikkelsen (University of Copenhagen) provided somevaluable comments.
References and notes1 R. A. McClelland, N. Mathivanan and S. Steenken, Laser flash
photolysis of 9-fluorenol. Production and reactivities of the 9-fluorenol radical cation and the 9-fluorenyl cation, J. Am. Chem.Soc., 1990, 112(12), 4857.
2 D. F. Duxbury, The photochemistry and photophysics oftriphenylmethane dyes in solid and liquid media, Chem. Rev., 1993,93(1), 381.
3 L. J. Johnston and D. F. Wong, Electron Transfer Reactionsof Triplet 9-Arylxanthenium and 9-Arylthioxanthenium Cations,J. Phys. Chem., 1993, 97, 1589.
4 A. Samanta, K. R. Gopidas and P. K. Das, Carbocationicfluorescence and its efficient electron-transfer quenching, J. Phys.Chem., 1993, 97, 1583.
5 A. Azarani, A. B. Berinstain, L. J. Johnston and S. Kazanis, Electrontransfer reactions between excited diarylmethyl and triarylmethylcarbocations and aromatic donors, J. Photochem. Photobiol., 1991,57, 175.
6 A. Samanta, K. R. Gopidas and P. K. Das, Electron acceptorbehavior of 9-phenylxanthenium carbocation singlet, Chem. Phys.Lett., 1990, 167, 165.
7 Y. Takahashi, S. Sankararaman and J. K. Kochi, Carbocations aselectron acceptors. Photoexcitation of the charge-transfer complexes
of tropylium salts and aromatic hydrocarbons, J. Am. Chem. Soc.,1989, 111(8), 2954.
8 M. K. Boyd, Photochemistry and Photophysics of Carbocations, inOrganic Photochemistry, eds. V. Ramamurthy and K. S. Schanze,Marcel Dekker, New York, 1997, p. 147.
9 F. C. Krebs, B. W. Laursen, I. Johannsen, A. Faldt, K. Bechgaard,C. S. Jacobsen, N. Thorup and K. Boubekeur, The geometryand structural properties of the 4 8,12-trioxa-4,8,12,12c-tetra-hydrobenzo[cd,mn]pyrene system in the cationic state. Structures ofa planar organic cation with various monovalent and divalentanions, Acts Cryst., 1999, B55, 410.
10 J. Reynisson, G. Balakrishnan, R. Wilbrandt and N. Harrit,Vibrational spectroscopic and quantum chemical studies of thetrioxatriangulenium carbocation, J. Mol. Struct., 2000, 520, 63.
11 J. C. Martin and R. G. Smith, Factors Influencing the Basicities ofTriarylcarbinols. The Synthesis of Sesquixanthydrol, J. Am. Chem.Soc., 1964, 86, 2252.
12 D. Bethelland and V. Gold, Carbonium Ions, An Introduction,Academic Press, London, 1967, 77.
13 D. Shukla and P. Wan, Product studies of electron transfer fromdimethoxybenzene and trimethoxybenzene to photoexcitedxanthenium cations in S1 in aqueous acid solution, J. Photochem.Photobiol. A: Chem, 1993, 76, 47.
14 I. Nemcová and I. Nemec, The voltammetry of triarylmethane dyesin acetonitrile, J. Electroanal. Chem., 1971, 30, 506.
15 N. Harrit, J. Reynisson, B. Laursen and R. Wilbrandt, Photophysicsof trioxatriangulenes, Book of Abstracts, 216th ACS NationalMeeting, Boston C.A., 1998 : 526577, 1998.
16 J. Reynisson, Spectroscopic and Photophysical Properties of theTrioxatriangulenium Carbocation and its Interactions withSupramolecular Systems, PhD thesis, 2000, http://www.risoe.dk/rispubl/fys/ris%2Dr%2D1191.htm.
17 S. Dileesh and K. R. Gopidas, Photophysical and electron transferstudies of a stable carbocation, Chem. Phys. Lett, 2000, 330(3,4),397.
18 H. Kessler, A. Moosmayer and A. Rieker, Das ausmass sterischerhinderung in substituirten triarylmethanen und triarylmethyl-radikalen, Tetrahedron, 1969, 25, 287.
19 N. Isaacs, Physical Organic Chemistry, 2. ed., Longman Scientific &Technical, Essex, 1994, 272.
20 G. Jones II and K. Goswami, Photoreduction of crystal violet inisopropyl alcohol. Mechanisms involving a leuco ether derivativeand dye ion pairs, J. Phys. Chem. A., 1986, 90, 5414.
21 N. J. Turro, Modern Molecular Photochemistry, Benjamin/Cummings, Menlo Park, 1978, 183.
22 I. Carmichael and G. L. Hug, Triplet-triplet absorption spectra oforganic molecules in condensed phases, J. Phys. Chem. Ref. Data,1986, 15(1), 1.
23 S. L. Murov, I. Carmichael and G. L. Hug, Handbook ofPhotochemistry, 2. ed., Marcel Dekker, Inc., New York, 1993,p. 264.
24 J. B. Birks, Photophysics of Aromatic Molecules, Wiley-Interscience,London, 1970, p. 84.
25 S. J. A. Van Gisbergen, J. G. Snijders and E. J. Baerends,Implementation of time-dependent density functional responseequations, Comput. Phys. Commun., 1999, 118(2–3), 119.
26 R. E. Stratmann, G. E. Scuseria and M. J. Frisch, An efficientimplementation of time-dependent density-functional theory for thecalculation of excitation energies of large molecules, J. Chem. Phys.,1998, 109(19), 8218.
27 M. Kasha, Phosphorescence and the role of the triplet state in theelectronic excitation of complex molecules, Chem. Rev., 1947, 41,401.
28 C. Jamorski, J. B. Foresman, C. Thilgen and H. P. Luthi, Assessmentof time-dependent density-functional theory for the calculation ofcritical features in the absorption spectra of a series of aromaticdonor-acceptor systems, J. Chem. Phys., 2002, 116, 8761.
29 J. M. Zwier, J. W. Hoeth and A. M. Brouwer, Computational studyof radical cations of saturated compounds with sigma-type andpi-type N-N bonds, J. Org. Chem., 2001, 66, 466.
30 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria,M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. Montgomery,R. E. Stratmann, J. C. Burant, S. Dapprich, A. D. Daniels, K. N.Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi,R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford,J. Ochterski, G. A. Petersson, P. Y. Ayala, K. Morokuma, D. K.Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman,J. Cioslowski, J. V. Ortiz, B. B Stefanov, G. Liu, A. Liashenko,P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox,T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara,C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen,M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon,
772 Photochem. Photobiol. Sci., 2002, 1, 763–773
Publ
ishe
d on
17
Sept
embe
r 20
02. D
ownl
oade
d by
Lom
onos
ov M
osco
w S
tate
Uni
vers
ity o
n 16
/12/
2013
07:
33:3
6.
View Article Online

E. S. Replogle and J. A. Pople, Gaussian 98, Revision A.7, Gaussian,Inc., Pittsburgh, 1998.
31 R. van Leeuwen and E. J. Baerends, Exchange-correlation potentialwith correct asymptotic behavior, Phys. Rev. A: At., Mot., Opt.Phys., 1994, 49(4), 2421.
32 G. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra,S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, Chemistry withADF, J. Comput. Chem., 2001, 22(9), 931.
33 J. B. Foresman, M. Head-Gordon, J. A. Pople and M. J. Frisch,Toward a systematic molecular orbital theory for excited states,J. Phys. Chem., 1992, 96(1), 135.
34 H. C. Longuet-Higgins, U. Opik, M. L. Pryce and R. A. Sack,Studies of the Jahn-Teller effect. II. The dynamical problem, Proc.R. Soc. London, Ser. A, 1958, 244, 1.
35 G. Herzberg, Molecular Spectra and Molecular Structure. III.Electronic Spectra and Electronic Structure of PolyatomicMolecules., D. van Nostrand, Princeton, 1966, 166.
36 H. P. J. M. Dekkers and E. C. M. Kielman-van Luyt, Magneticcircular dichroism of the triphenyl carbenium ion and somesymmetrically para-substituted derivatives, Mol. Phys., 1976, 31(4),1001.
37 A. D. Becke, Density-functional thermochemistry. IV. A newdynamic correlation functional and implications for exact-exchangemixing, J. Chem. Phys., 1996, 104(3), 1040.
38 M. S. De Groot, A. M. Hesselmanm and J. H. v. d. Waals,Paramagnetic resonance in phosphorescent aromatic compounds IV.Ions in orbitally degenerate states, Mol. Phys., 1966, 10, 242.
39 H. Siegerman, Oxidation and Reduction Half-Wave Potentials ofOrganic Compounds, in Technique of Electroorganic Synthesis,Techniques of Chemistry, ed. N. L. Weinberg, John Wiley & sons,New York, 1975, p. 667.
40 O. Hammerich, Anodic Oxidation of Hydrocarbons, in OrganicElectrochemistry, eds. O. Hammerich and H. Lund, Marcel Dekker,New York, 2001, p. 471.
41 G. J. Kavarnos, Fundamentals of Photoinduced Electron Transfer,VCH Publishers, Inc., New York, 1993, 35.
42 W. E. Jones, Jr. and M. A. Fox, Determination of Excited-StateRedox Potentials by Phase-Modulated Voltammetry, J. Phys. Chem.,1994, 98(19), 5095.
43 D. Rehm and A. Weller, Kinetics of fluorescence quenching byelectron and H-atom transfer, Isr. J. Chem., 1970, 8, 259.
44 M. R. Valentino and M. K. Boyd, Quenching behavior of singletexcited 9-arylxanthylium cations, J. Org. Chem., 1993, 58, 5826.
45 To calculate ∆G-values, Dileesh and Gopidas apply the irreversiblepeak potential of 0.106 V versus SCE.
46 J. R. Lakowicz, Principles of Fluorescence Spectroscopy, PlenumPress, New York, 1983, p. 266.
47 G. Jones II, Photochemistry and Photophysics of Organic ChargeTransfer Complexes, in Photoinduced Electron Transfer, part A,eds. M. A. Fox, and M. Chanon, Elsevier, Amsterdam, 1988,p. 245.
48 D. Baunsgaard, M. Larsen, N. Harrit, J. Frederiksen, R. Wilbrandtand H. Stapelfeldt, Photophysical properties of 2,3,6,7,10,11-hexakis(n-hexylsulfanyl)triphenylene and 2 3,6,7,10,11-hexakis-(n-hexylsulfonyl)triphenylene in solution, J. Chem. Soc., FaradayTrans., 1997, 93, 1893.
49 B. Kohne, W. Poules and K. Praefcke, Erste flüssigkristallineHexakis(alkylthio)triphenylene, Chem. Zeit., 1984, 108, 113.
50 C. Destrade, M. C. Mondon and J. Malthête, Hexasubstitutedtriphenylenes A new mesomorphic order, J. Phys. (Paris), 1979, 40,17.
51 D. Markovitsi, H. Bengs and H. Ringsdorf, Charge-transferabsorption in doped columnar liquid crystals, J. Chem. Soc.,Faraday Trans., 1992, 88, 1275.
52 K. Bechgaard, Unpublished work, 2002.53 H. A. Benesi and J. H. Hildebrand, A spectrophotometric
investigation of the interaction of iodine with aromatichydrocarbons, J. Am. Chem. Soc., 1949, 71, 2703.
54 D. Markovitsi, F. Rigaut, M. Mouallem and J. Malthete, One-dimensional energy migration in crystalline and columnar liquid-crystalline phases of 2,3,6,7,10,11-Hexa-n-hexyloxytriphenylene,Chem. Phys. Lett., 1987, 135, 236.
55 J. Reynisson, G. B. Schuster and N. Harrit, The Interaction betweentrioxatriangulenium and DNA, to be published.
56 J. Korppi-Tommola and R. W. Yip, Solvent effects on the visibleabsorption spectrum of crystal violet, Can. J. Chem., 1981, 59, 191.
57 R. J. Goldacre and J. N. Phillips, The ionization of basictriphenylmethane dyes, J. Chem. Soc., 1949, 1724.
58 M. K. Boyd, H. L. Lai and K. Yates, Water quenching behaviorof excited 9-xanthylium cations in aqueous sulfuric acid solutions,J. Am. Chem. Soc., 1991, 113, 7294.
59 R. E. Minto and P. K. Das, A laser flash photolysis study ofphotodehydroxylation phenomena of 9-phenylxanthen-9-ol andphotobehavior of related intermediates Enhanced electrophilicity of9-phenylxanthenium cation singlet, J. Am. Chem. Soc., 1989, 111,8858.
60 M. R. Valentino and M. K. Boyd, Ether Quenching of SingletExcited 9-Arylxanthyl Cations, J. Photochem. Photobiol. A: Chem.,1995, 89, 7.
61 J. M. Bedleck, M. R. Valentino and M. K. Boyd, Substituent effectson carbocation photophysics 9-arylxanthyl and 9-arylthioxanthylcarbocations, J. Photochem. Photobiol. A: Chem., 1996, 94, 7.
62 M. K. Boyd, Photochemistry and photophysics of xanthyl andthioxanthyl carboxcations, Spectrum, 1998, 11(3), 6.
63 R. A. McClelland, N. Banait and S. Steenken, Electrophilicreactions of xanthylium carbocations produced by flash photolysisof 9-xanthenols, J. Am. Chem. Soc., 1989, 111, 2929.
64 E. M. Arnett, K. Amarnath, N. G. Harvey and J. Cheng,Determination and interrelation of bond heterolysis and homolysisenergies in solution, J. Am. Chem. Soc., 1990, 112(1), 344.
65 D. Shukla and P. Wan, Adiabatically photogeneratedthioxanthenium cations: Probes of reactivity of nucleophiles towardexcited state carbocations in aqueous solution, J. Photochem.Photobrol. A: Chem., 1994, 79, 55.
66 R. G. Brown and D. Phillips, Quenching of the first excited singletstate of substituted benzenes by molecular oxygen, Trans. FaradaySoc. 2, 1974, 70, 630.
67 H. J. Dauben and J. D. Wilson, Carbonium ion charge-transfercomplexes, J. Chem. Soc., Chem. Commun., 1968, 1629.
68 M. Feldman and B. G. Graves, Solvent shifts in charge-transferspectra of tropylium complexes, J. Phys. Chem., 1966, 70, 955.
69 J. Michl and E. W. Thulstrup, Why is azulene blue and anthracenewhite? A simple MO picture, Tetrahedron, 1976, 32(2), 205.
70 B. W. Laursen, F. C. Krebs, M. F. Nielsen, K. Bechgaard, J. B.Christensen and N. Harrit, 2,6,10-Tris(dialkylamino)-trianguleniumions: Synthesis, structure, and properties of exceptionally stablecarbenium ions, J. Am. Chem. Soc., 1998, 120, 12255.
71 B. W. Laursen and F. C. Krebs, Synthesis of a triazatrianguleniumsalt, Angew. Chem., Int. Ed., 2000, 39(19), 3432.
72 M. Lofthagen, R. VernonClark, K. K. Baldridge and J. S. Siegel,Synthesis of trioxatricornan and derivatives Useful keystones for theconstruction of rigid molecular cavities, J. Org. Chem., 1992, 57, 61.
Photochem. Photobiol. Sci., 2002, 1, 763–773 773
Publ
ishe
d on
17
Sept
embe
r 20
02. D
ownl
oade
d by
Lom
onos
ov M
osco
w S
tate
Uni
vers
ity o
n 16
/12/
2013
07:
33:3
6.
View Article Online





























