Embed Size (px)
Citation preview
Phylogenetic reconstruction

Phylogeny
The evolutionary relationships among organisms; the patterns of lineage branching produced by the true evolutionary history of the organisms being considered

What is Molecular Phylogeny?
is the inference of lines of ancestry for organisms based on DNA, RNA, or protein sequences for those organisms obtained in the laboratory
We can infer relation between
Organism to Organism
Genes of different organisms
Or genes within one organism

Gene function prediction
Speciation
Origin of genes & gene transfer

The tree of life
A phylogenetic tree, also known as a tree of life or simply a phylogeny, describes branching relationships among species, showing which species shares its most recent common ancestor with which other species.

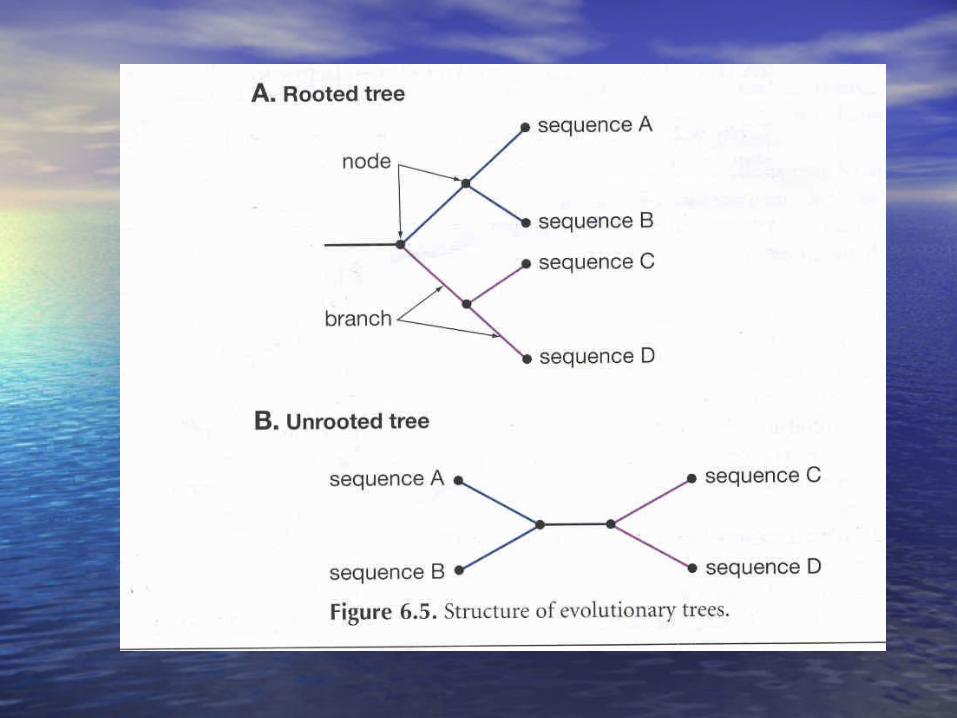
Rooted trees
infers the existence of an actual common ancestor and defines the evolutionary paths leading to the development of each organism.
It provides an indication of the direction of the evolutionary process, defining ancestral and derived characters or species
Unrooted trees
shows only the evolutionary relationships between the organisms in the tree, and does not actually infer the placement of a common ancestor in the structure or the evolutionary path used to obtain the current relationships

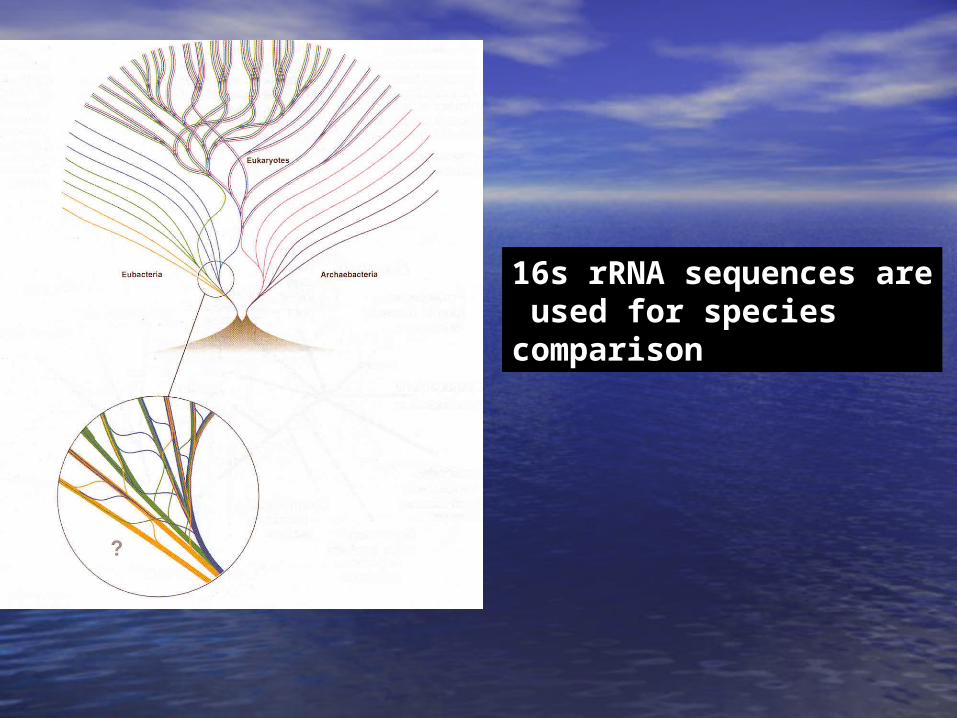
16s rRNA sequences are used for species comparison

Molecular Clocks
concept based on the assumption that mutations occur at some regular, more or less predictable rate.

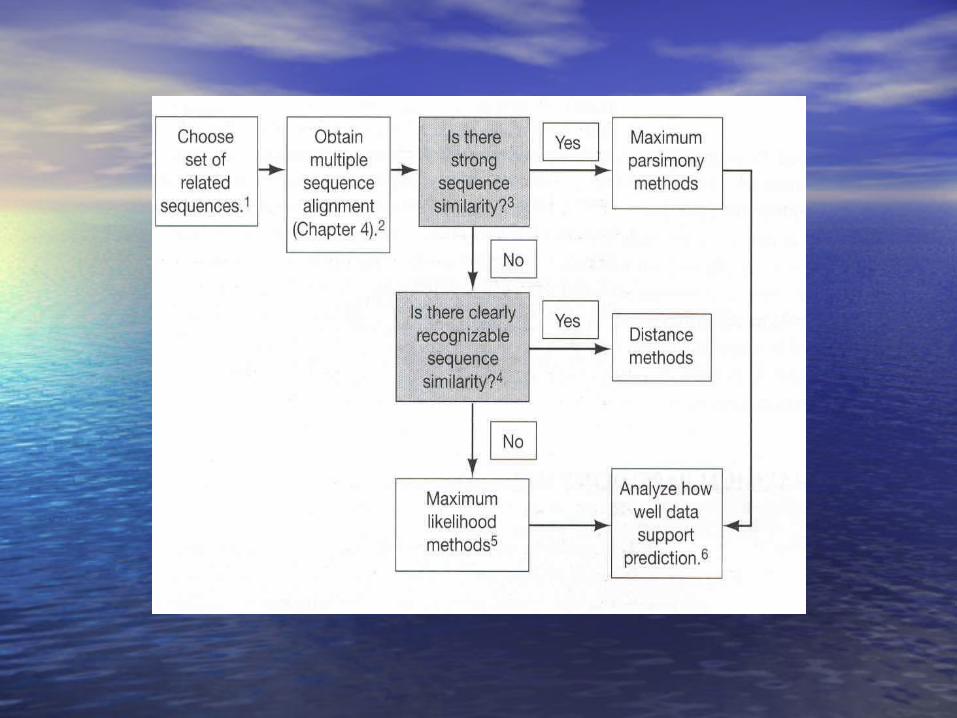
Evolutionary Tree Construction Methods
Maximum Parsimony
Maximum Likelihood
Distances / transformed distances

Maximum Parsimony
Attempt to create an evolutionary tree for the organisms in question by using the minimum number of evolutionary steps

Phylogenetic inference using parsimony proceeds in two stages:
1. Infer the unrooted tree for a set of species. An unrooted tree shows the branching relations between the species but does not show the position of the deepest common ancestor. It is a phylogenetic tree with the time dimension removed.
2. Locate the root. This means calculating character polarity for a group: finding the shared derived homologies which reveal whether A evolved from B or the other way round.

How can we infer the unrooted tree?
Imagine there are four species: human, chimpanzee, magnolia tree and amoeba.
Write out all the possible unrooted trees for the species; that is (a), (b) and (c)

count the minimum number of evolutionary events (that is, changes in character states) implied by each
The best estimate of the true unrooted tree is the one requiring the least evolutionary change.
Suppose we know that 1000 characters are shared in all four species, that each species has 10 characters unique to itself and that 100 characters are shared between humans and chimps, but are absent in amoebas and magnolias

the 1000 common characters could have evolved once each in the common ancestor of all four species and been retained throughoutthe phylogeny; that would require a total of 1000 evolutionary events. Each species' 10 unique characters can have evolved in the lineage leading to that species, making 40 evolutionary events.
The smallest number of events to produce the distribution of characters between humans and chimps is 200: they could have evolved separately in the human lineage and in the chimp lineage (100 events in each, making 200).
1000 + 40 + 200 = 1240 evolutionary events.

In (c), however, the 100 characters shared by humans and chimps only need to have evolved once, in the common ancestor of humans and chimps, and would not have to be lost again; (c) therefore only requires 1140 events.
(c) is therefore the most parsimonious unrooted tree, and (according to the principle of parsimony) the best estimate of the real unrooted tree.

Distance Methods

Distance is a principle of phylogeny referring to the quantitatively measured difference between two groups of organisms, such as two species.
Distance can be measured in two ways:
The difference between the phenetic appearance of two groups (phenetic distance).
The difference in their gene frequencies (genetic difference).
Distance is simply a more precise method of the 'look more similar' approach which classifies human and chimpanzees (opposite) as having a more recent common ancestor than humans and rabbits because they share more phenetic characters.

The distances method creates a matrix of all distances (difference scores) between all organisms for which the tree is to be constructed.
Having calculated the matrix, the pair of organisms which have the smallest distance score are connected, with a root in between them
The average of the distances from each member of the pair to a third node is use for the next iteration of the distances matrix.
The process is repeated, until all organisms have been placed in the tree.
There is an extremely critical assumption when creating a tree like this, and that is that there is a "molecular clock" and that all organisms are mutating at the same rate.

Maximum Likelihood
Maximum likelihood methods create all possible trees containing the set of organisms in question, and then uses statistics to evaluate which tree is most likely.

Steps in Phylogeny construction
BLAST
Convert to FASTA
CLUSTALX
Treeview





























