Embed Size (px)
Citation preview

Phylogeny and Genome Biology
Phylogeny and Genome BiologyPhylogeny and Genome BiologyAndrew JacksonAndrew Jackson
Wellcome Trust Sanger InstituteWellcome Trust Sanger Institute
Changes:• Type program name to start• Always Cd to phyml directory before starting program• Beware phyml overwrites tree file with every search• Ctrl C quits phyml and restores terminal• P6 figure refers to all searches generally

Phylogeny and Genome Biology
Introduction
• Phylogeny refers to the ancestry of a biological lineage, but is also synonymous with phylogenetic tree.
• Phylogeny is tree-like, or dichotomous
• Phylogeny provides the historical basis to the comparative method.
• Genomes are historical entities; their structure and function reflect the past.
• There is a need for genomic systematics to establish the identities of genomic phenomena.

Phylogeny and Genome Biology
Principle of phylogenetics
• Inferring relationships is about similarity.
• Homology describes similarity due to common inheritance from an ancestor. Homologous characters are useful similarity.
• Homoplasy describes similarity due to independent acquisitions of the same or superficially similar character state. Homoplasious characters provide a mis-leading picture of phylogeny.
• Distance in a phylogenetic tree reflects a decreasing number of shared, homologous characters (assuming that evolution maximises homology).

Phylogeny and Genome Biology
Methods
• Stages in phylogenetic analysis:
1. Data preparation; multiple alignment of DNA or protein sequences. Similar principle applies to character states.
2. Data scoring; producing genetic distances or character states (‘distance’ or ‘discrete’ data).
3. Tree sorting; processes for searching ‘tree-space’, e.g., hill-climbing or MCMC.4. Estimation; identifying the most acceptable tree topology and model parameters using
a variety of methods (‘clustering’ or ‘optimising’ methods).
Clustering Optimising
Distance Neighbour-joining
UPGMA
Minimum evolution
Discrete Maximum parsimony
Maximum likelihood
Bayesian inference
• Phylogenetic methods:
• A phylogenetic method is judged according to its ‘accuracy’ (i.e., obtains the true tree) and ‘precision’ (i.e., always produces the same tree).
Phylogeny and Genome Biology
Applications to genome biology
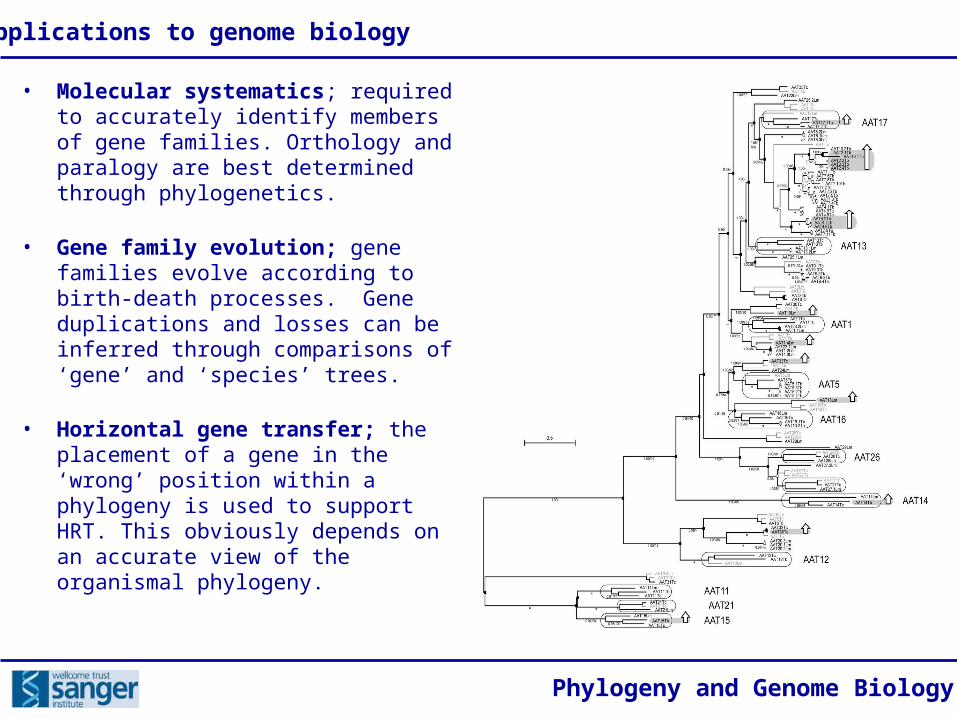
• Molecular systematics; required to accurately identify members of gene families. Orthology and paralogy are best determined through phylogenetics.
• Gene family evolution; gene families evolve according to birth-death processes. Gene duplications and losses can be inferred through comparisons of ‘gene’ and ‘species’ trees.
• Horizontal gene transfer; the placement of a gene in the ‘wrong’ position within a phylogeny is used to support HRT. This obviously depends on an accurate view of the organismal phylogeny.
Phylogeny and Genome Biology
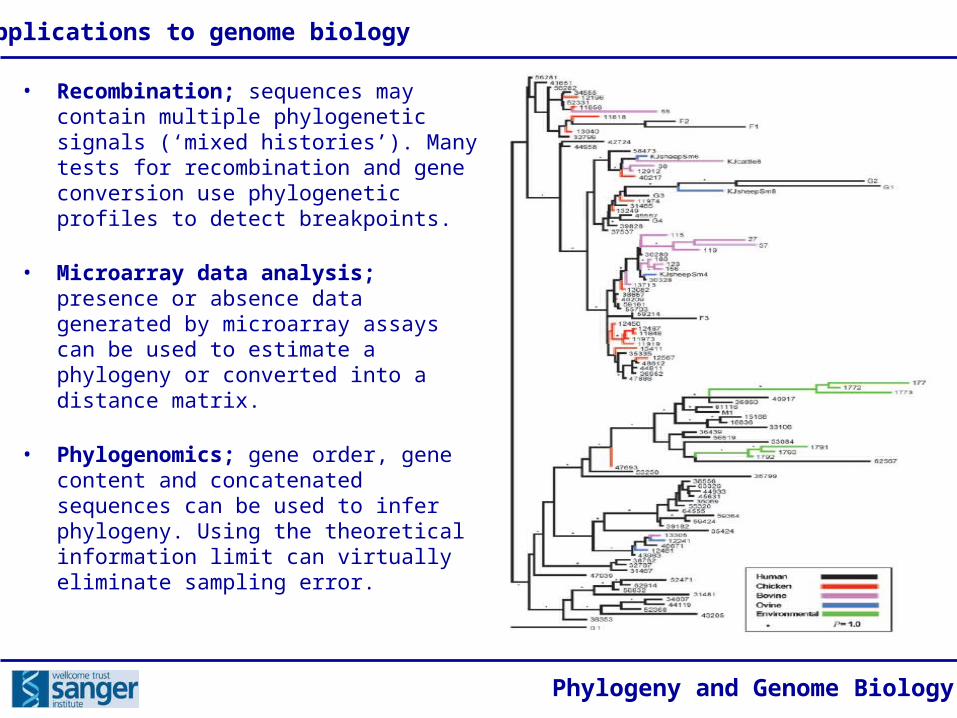
• Recombination; sequences may contain multiple phylogenetic signals (‘mixed histories’). Many tests for recombination and gene conversion use phylogenetic profiles to detect breakpoints.
• Microarray data analysis; presence or absence data generated by microarray assays can be used to estimate a phylogeny or converted into a distance matrix.
• Phylogenomics; gene order, gene content and concatenated sequences can be used to infer phylogeny. Using the theoretical information limit can virtually eliminate sampling error.
Applications to genome biology
Phylogeny and Genome Biology
Clustering methods (e.g., Neighbour-joining)
A B C D E F G H I
A ·
B 0.001 ·
C 0.025 0.024 ·
D 0.003 0.002 0.019 ·
E 0.336 0.331 0.219 0.231 ·
F 0.021 0.019 0.001 0.018 0.233 ·
G 0.001 0.001 0.025 0.002 0.256 0.023 ·
H 0.056 0.044 0.005 0.042 0.132 0.051 0.043 ·
I 0.325 0.300 0.116 0.195 0.005 0.122 0.366 0.213 ·
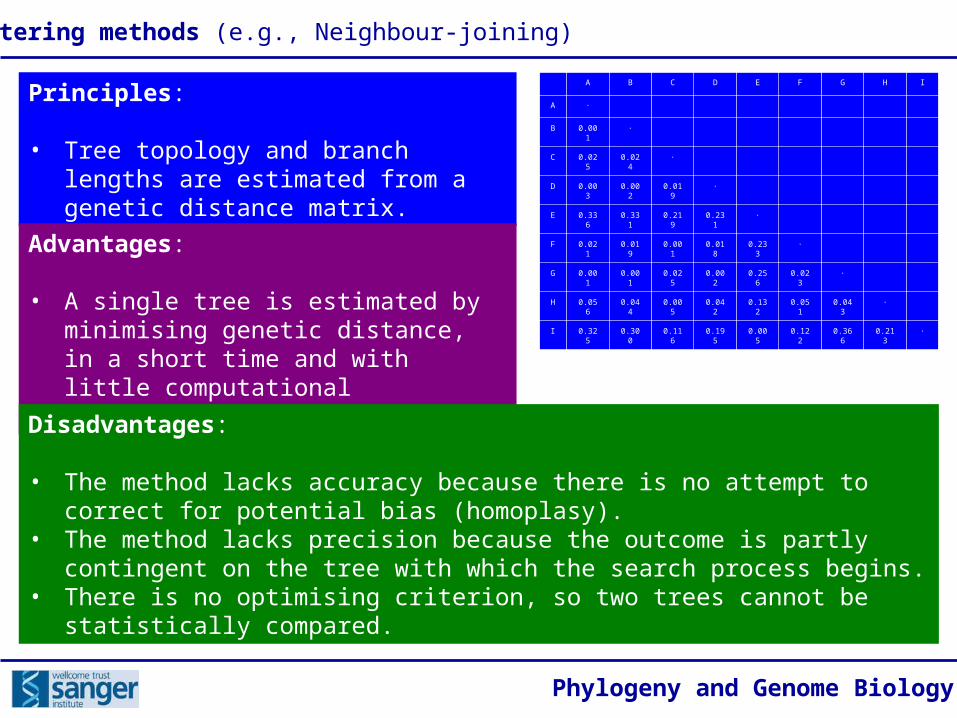
Principles:
• Tree topology and branch lengths are estimated from a genetic distance matrix.
Advantages:
• A single tree is estimated by minimising genetic distance, in a short time and with little computational expenditure.
Disadvantages:
• The method lacks accuracy because there is no attempt to correct for potential bias (homoplasy).
• The method lacks precision because the outcome is partly contingent on the tree with which the search process begins.
• There is no optimising criterion, so two trees cannot be statistically compared.

Phylogeny and Genome Biology
Non-Parametric Methods - Maximum parsimony
Principles:• Searches through tree topologies in ‘tree-space’ using a ‘hill-climbing’ algorithm.• Applies an optimising criterion: maximum parsimony.• Scores trees on their ‘length’, i.e., the number of character state changes required to
explain the distribution of characters on a given tree topology.• Selects the topology with the fewest character changes overall.
Advantages:• Generally accurate method with few assumptions.• Phylogenetic hypotheses can be statistically tested by comparing the lengths of different
trees.• All available information on sites is used in reconstruction.• Tree estimation is relatively fast and undemanding.
Disadvantages:• There are typically several shortest trees, resulting in a potentially ambiguous consensus
topology.• There is no explicit model of evolution and so the method is prone to error under certain
circumstances, e.g., long-branch attraction (homoplasy), making MP trees positively misleading in many cases.
Phylogeny and Genome Biology
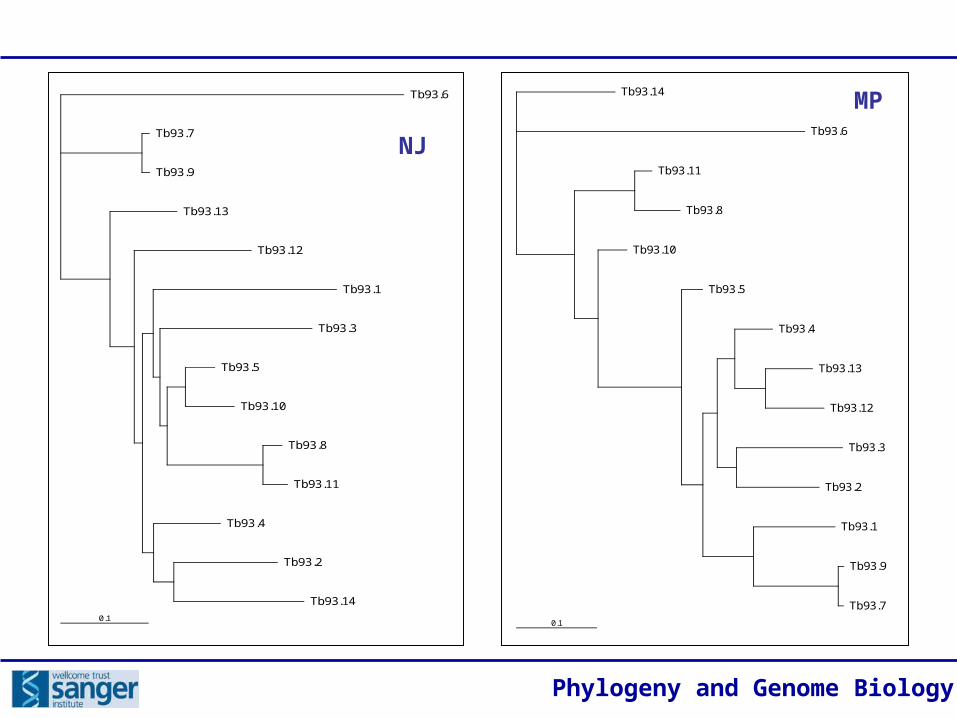
NJ
0.1
Tb93.6
Tb93.7
Tb93.9
Tb93.13
Tb93.12
Tb93.1
Tb93.3
Tb93.5
Tb93.10
Tb93.8
Tb93.11
Tb93.4
Tb93.2
Tb93.14
0.1
Tb93.14
Tb93.6
Tb93.11
Tb93.8
Tb93.10
Tb93.5
Tb93.4
Tb93.13
Tb93.12
Tb93.3
Tb93.2
Tb93.1
Tb93.9
Tb93.7
MP

Phylogeny and Genome Biology
Maximum likelihood
Principles:
• Applies a complex model of DNA or protein sequence evolution that estimates parameters for specific substitutions and other qualities of molecular sequences.
• Calculates the ‘likelihood’ of each individual character in each candidate tree and applies an optimising criterion, maximising the likelihood of the data, given the model.
• Locates the most likely tree topology through a hill-climbing algorithm.
• Various models accommodate sources of molecular homoplasy that might result in the wrong tree:
• ‘Multiple hits’ (substitutional saturation)
• Rate convergence• Rate heterogeneity• Base composition bias• Codon usage bias• Secondary structure• Covariance

Phylogeny and Genome Biology
Advantages:
• Highly accurate because considerable biological realism is introduced through the substitutional model. This allows various forms of homoplasy to be corrected for.
• Phylogenetic estimation within the likelihood framework provides a robust statistical context in which to evaluate specific hypotheses.
• A single tree is produced that is generally precise.
Maximum likelihood
Disadvantages:
• The complexity of the estimation process means that it is slow and computationally demanding.
• The hill-climbing algorithm is susceptible to local optima and so does not guarantee to return the most optimal solution.
• NP-hard problem. Similar objection to all methods dependent on heuristic searches.
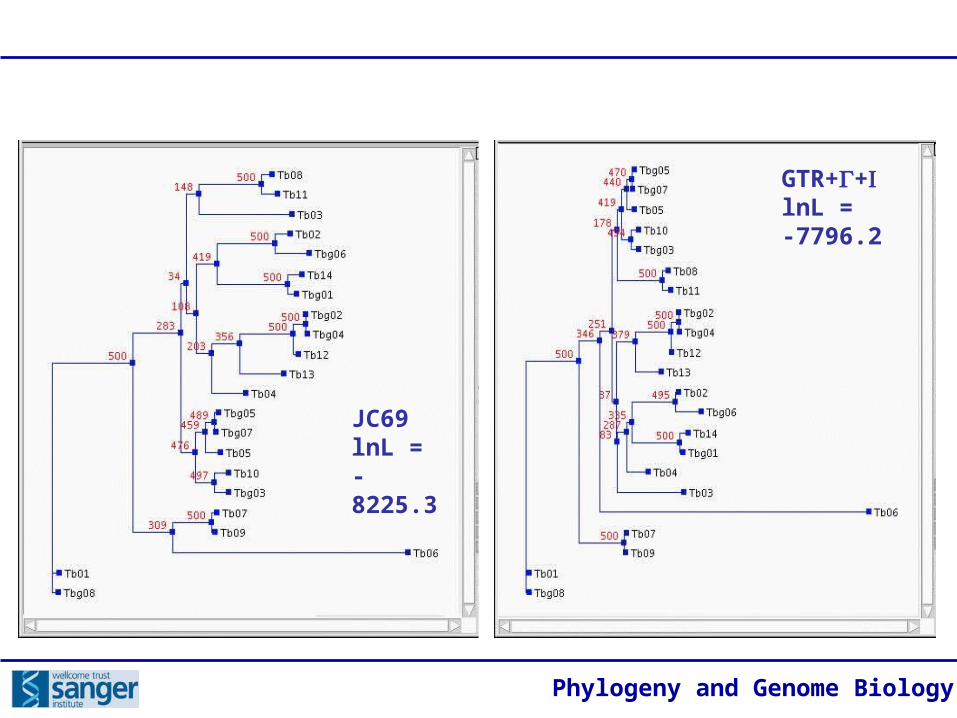
Phylogeny and Genome Biology
GTR++lnL = -7796.2
JC69lnL = -8225.3

Phylogeny and Genome Biology
Bayesian inference
Principles:
• Frequentist approaches make long-term frequency statements about random data, conditional on a model. “Given the tree and model, how unlikely are the data?”
• Bayesian inference estimates probability of random parameters, conditional on data. “Given the data, what are the probabilities of the tree and model?”
• Uses an MCMC process to search through tree-space.• Selects the tree-topology with the highest probability, given the data.
Advantages:
• Intuitive• Potential for any complex model.• Provides both parameter estimates (i.e., tree) and their probabilities in a single analysis. • Many different hypotheses can be evaluated in a single analysis.• The MCMC algorithm makes integrating over all parameter values fast and accurate.• Coupled MCMC’s are able to break out of local optima.
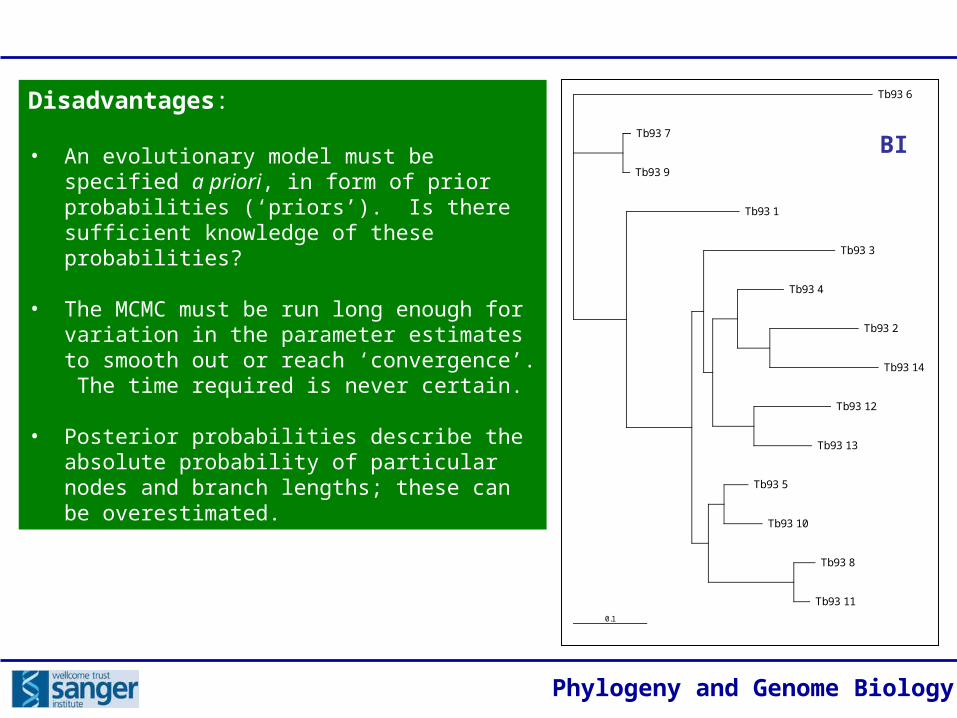
Phylogeny and Genome Biology
Disadvantages:
• An evolutionary model must be specified a priori, in form of prior probabilities (‘priors’). Is there sufficient knowledge of these probabilities?
• The MCMC must be run long enough for variation in the parameter estimates to smooth out or reach ‘convergence’. The time required is never certain.
• Posterior probabilities describe the absolute probability of particular nodes and branch lengths; these can be overestimated.
0.1
Tb93 6
Tb93 7
Tb93 9
Tb93 1
Tb93 3
Tb93 4
Tb93 2
Tb93 14
Tb93 12
Tb93 13
Tb93 5
Tb93 10
Tb93 8
Tb93 11
BI

Phylogeny and Genome Biology
Remember…
All trees are wrong
There are no free lunches

Phylogeny and Genome Biology
Rate Heterogeneity
• All methods are prone to long branch attraction artefacts.
• All but MP can be accounted for in some way.
• Most commonly used - gamma correction.

Phylogeny and Genome Biology
Further details
Textbooks:
Page & Holmes (1999) Molecular Evolution: A Phylogenetic Approach. Blackwell Science.
Felsenstein (2004) Inferring Phylogenies. Sinauer Associates.
Website:
Felsenstein’s Phylogeny program page (links to available software): http://evolution.genetics.washington.edu/phylip/software.html
Software:
PAUP* (NJ, MP, ML): http://paup.csit.fdsu.eduPHYLIP (NJ, MP, ML): http://evolution.genetics.washington.edu/phylip.htmlMrBayes (BI): http://mrbayes.csit.fdsu.eduSplitstree (Networks) http://www.splitstree.org





























