Embed Size (px)
Citation preview

Physiology and productivity of serum-free Spodoptera
frugiperda Sf9 insect cell cultures
Eva Lindskog
M. Sc.
Royal Institute of Technology
Stockholm 2006

Eva Lindskog Stockholm, 2006 ISBN 91-7178-383-0 Royal Institute of Technology School of Biotechnology Department of Bioprocess Technology SE-106 91 Stockholm Sweden Printed at Universitetsservice US-AB Box 700 14 SE-100 44 Stockholm Sweden

Eva Lindskog (2006), Physiology and productivity of serum-free Spodoptera frugiperda Sf9 insect cell cultures. School of Biotechnology, Department of Bioprocess Technology, Royal Institute of Technology, SE-106 91, Stockholm, Sweden.
Abstract The objective of this study was to investigate the mechanisms and factors controlling
growth and proliferation in serum-free Spodoptera frugiperda Sf9 cultures as well as the implications of these factors for protein production in the baculovirus expression system.
The physiology of recently thawed, low passage (Lp) Sf9 cultures, were compared to high passage (Hp) cultures at p>100. Lp cells passed a switch in proliferation kinetics after 30-40 passages, characterized by a shorter lag-phase and an increased maximum specific proliferation rate, µN,max, from 0.03/h to 0.04/h. Conditioned medium (CM), 10 kDa CM filtrate and 10 kDa CM concentrate promoted proliferation of Lp Sf9 cells, but had no effect after the switch. Sf9 cell cycle dynamics were characterized by an initial G2/M arrest, which synchronized the cells and this feature was more pronounced for Hp than for Lp cells. CM addition decreased the initial arrest for Lp cultures, but did not affect Hp cells. Late in the culture, a final G2/M accumulation occurred. An octaploid population emerged during G2/M arrests. Further, a 49 kDa proform and a 39 kDa active form of Sf9 cathepsin L was identified from gelatine zymography of Sf9 CM, on basis of inhibitor profile and substrate range. Removal of procathepsin L during the course of an Sf9 culture had a negative effect on Sf9 proliferation. Procathepsin L was also identified in Trichoplusia ni High five CM. High five CM promoted growth of Sf9 cells, but when procathepsin L was removed no effect was observed. It is suggested that these observations are due to an autocrine system controlling proliferation. One Sf9 mitogen might be a <10 kDa peptide, while the effect of 10 kDa CM concentrate may originate from procathepsin L. A hypothesis is therefore that procathepsin L acts as a mitogen in Sf9 cultures, perhaps in concert with the <10 kDa peptide.
The volumetric product yield (P) in baculovirus infected Sf9 cells increased linearly up to 68-75 h of culture. Beyond this point almost no product was detected. Medium renewal at infection prolonged the productivity phase until 117 h, but generated only a 10% increase in P. The specific product formation rate (YP/N) was highest at µN,max. YP/N of Lp cells decreased by 30-50% when 20% CM or 10 kDa CM filtrate was added, whereas addition of CM to cells having passed the switch on growth kinetics did not affect productivity. Further, Hp cells exhibited a two-fold higher YP/N than Lp cells, when infected during the initial 48 h of culture. This coincided with a high degree of synchronization. Yeastolate limitation was used to achieve artificial synchronization of an Lp culture, and YP/N could thereby be maintained high during a prolonged time, resulting in a 69% increased P. This suggests that a decreasing degree of synchronization during the course of a culture partly explains the cell-density dependent drop in productivity in Sf9 cells.
Finally, ~10 kDa gel filtration fractions from Sf9 and High five CM were found to be bactericidal. Exposure of a Bacillus megaterium culture for eight min to an Sf9 CM fraction killed 99% of the population, and 60 min exposure killed 35% of an Escherichia coli population. In both cases cell lysis was observed. B. megaterium incubated in an High five CM fraction lost 97% viability in 40 min. The effect of the High five CM fraction most probably originated from a lysozyme precursor protein, whereas the Sf9 executor remains unknown.
Keywords: Sf9, High five, Hi5, T. ni, CM, cell cycle, proteolysis, cathepsin L, procathepsin L, synchronization, productivity, antibacterial, autocrine, baculovirus, proliferation.


To my late grandfather Henning Sköld; a natural born engineer who never had the
opportunity of getting a higher education.


Main references
This thesis is based on the following publications, which will be referred to in the text with
their roman numerals.
I. Doverskog, M., Bertram, E., Ljunggren, J., Öhman, L., Sennerstam, R., Häggström, L.
(2000) Cell cycle progression in serum-free cultures of Sf9 insect cells: Modulation by
conditioned medium factors and implications for proliferation and productivity.
Biotechnology Progress 16: 837-846.
II. Svensson, I., Calles, K., Lindskog, E., Henriksson, H., Eriksson, U., Häggström, L.
(2005) Antimicrobial activity of conditioned medium fractions from Spodoptera frugiperda
Sf9 and Trichoplusia ni Hi5 insect cells. Applied Microbiology and Biotechnology 69(1): 47-
53.
III. Lindskog, E., Svensson, I., Häggström, L. (2005) A homologue of cathepsin L
identified in conditioned medium from Sf9 insect cells. Applied Microbiology and
Biotechnology [Epub ahead of print] PMID: 16283300.
IV. Calles, K., Svensson, I., Lindskog, E., Häggström, L. (2006) Effects of conditioned
medium factors and passage numbers on Sf9 physiology and productivity. Biotechnology
Progress 22(2): 394-400.
V. Lindskog, E., Eriksson, U., Häggström, L. (2006) Extracellular proteolytic activity in
non-infected Sf9 and Trichoplusia ni Hi5 insect cell cultures: Implications for proliferation.
Manuscript.
In addition, previously unpublished results are included.



1
Table of contents I Introduction............................................................................................................................................................... 1
1 Background to animal cell cultivation technology ........................................................................................... 1 1.1 History......................................................................................................................................................... 1 1.2 Present situation.......................................................................................................................................... 2
2 Cell growth in vitro............................................................................................................................................. 4 2.1 Cultivation techniques................................................................................................................................ 4 2.2 Growth factors ............................................................................................................................................ 5 2.3 Autocrine regulation................................................................................................................................... 6 2.4 Cell death .................................................................................................................................................... 6
3 Insect cell cultivation.......................................................................................................................................... 7 3.1 History......................................................................................................................................................... 7 3.2 Industrial cell lines ..................................................................................................................................... 7 3.3 Cultivation media ....................................................................................................................................... 8
4 The baculovirus expression vector system (BEVS) technology.................................................................... 10 4.1 Background............................................................................................................................................... 10
4.1.1 The BEVS technology...................................................................................................................... 10 4.1.2 Applications ...................................................................................................................................... 12
4.2 Advantages with BEVS ........................................................................................................................... 12 4.3 Limitations in BEVS ................................................................................................................................ 13
4.3.1 Glycosylation .................................................................................................................................... 14 4.3.2 Proteolysis......................................................................................................................................... 15 4.3.3 The cell density effect ...................................................................................................................... 16
II Objectives .............................................................................................................................................................. 19 III Present investigation............................................................................................................................................ 21
5 Proliferation of Sf9 cells .................................................................................................................................. 21 5.1 Cell cycle dynamics ................................................................................................................................. 23 5.2 Autocrine regulation of proliferation ...................................................................................................... 25 5.3 The conditioned medium effect............................................................................................................... 26 5.4 The culture age effect............................................................................................................................... 28
6 Conditioned medium factors ............................................................................................................................ 30 6.1 Antimicrobial activity .............................................................................................................................. 30 6.2 Peptidases.................................................................................................................................................. 32
6.2.1 Identification of Sf9 and High five cathepsin L ............................................................................. 33 6.2.2 Role of procathepsin L in Sf9 proliferation .................................................................................... 35
7 Protein production ............................................................................................................................................ 37 IV Conclusions.......................................................................................................................................................... 41 V References ............................................................................................................................................................. 43 VI Acknowledgements ............................................................................................................................................. 51 VII Publications I-V ................................................................................................................................................. 53

2

1
I Introduction
1 Background to animal cell cultivation technology
1.1 History
In 1907, Ross Harrison recorded long-term growth of nerve cells in a hanging drop of lymph.
This is referred to as the start of in vitro animal cell cultivation technology, and it showed that
normal cell functions could persist outside the organism. Strict aseptic conditions were
necessary for a successful experiment, but this was difficult to achieve before the
breakthrough of the antibiotics in the 1940s. During the following decade, great progress was
made in animal cell cultivation. One example is the isolation of the first human cell line HeLa
in 1951, which originated from Henrietta Lacks, an American woman who died from a virally
induced cervical cancer. A piece of her tumour was successfully kept alive in a nutrient
solution consisting of cattle embryo extract, fresh chicken plasma and human placental blood.
The HeLa line still persists and is widely used for research purposes. Several other of today’s
cell lines also originate back to the 50s or early 60s; CHO 1, BHK-21 2, and 3T3 3. However,
the varying quality and supply of the biological fluids used in the media so far hampered
animal cell cultivation on a larger scale. An important step forward was therefore Eagle’s
description in 1955 of a chemically defined animal cell cultivation medium known as EMEM,
Eagle’s minimum essential medium 4. Eagle’s medium was developed after extensive analysis
of the requirements of cells in vitro, but serum still had to be added to achieve optimal cell
growth. Not much later the Salk vaccine process, which comprised virus propagation in
primary monkey kidney cells, was the first animal cell process to be set up in industry. A
number of other vaccine processes followed shortly after, for example production of vaccines
against diseases such as measles, rabies, mumps and rubella.
During the late 70s/early 80s two important scientific breakthroughs occurred, which together
laid the ground for the biotech explosion during the last decades of the 20th century. The
hybridoma technology was invented in 1975 when Köhler and Millstein succeeded in fusing
antibody-producing mouse B-lymphocytes with immortal myeloma blood cancer cells. The
resulting immortal hybridoma cell line constitutively produced monoclonal antibodies with a
high quality. Previously, antibodies were produced in processes involving immunized
animals, a feature which implied both a varying quality and a risk of contamination. The
recombinant DNA technology was developed by Stanley Cohen and Herbert Boyer. In 1973

2
they combined their knowledge about plasmids and restriction enzymes and created the
concept genetic engineering. Foreign protein-coding DNA was transfected into host cells,
which expressed the heterologous protein during normal cell growth. The protein products
were used for both diagnostics and research purposes, but also as therapeutic drugs. In 1982,
the first recombinant biopharmaceutical with market approval was launched: Humulin
(human insulin), produced in E. coli by Eli Lilly 5. In 1987 the first recombinant therapeutic
produced in animal cells followed: Activase (human tissue plasminogen activator, tPA),
produced in CHO cells by Genentech.
Historically, the majority of all drugs has consisted of natural-derived compounds such as
morphine and quinine, or small organochemical entities. The few protein biopharmaceuticals
available were purified from their natural sources (e.g. insulin from pancreas). This meant
expensive processes, a non-consistent product quality and risk for contamination from co-
purified pathogenic animal viruses. However, the success of the early recombinant
biopharmaceuticals paved the way for a significant amount of other recombinant products.
1.2 Present situation
At the present date, animal cell processes are not only used in development and production of
protein therapeutics. Perhaps the most widely used application for animal cell processes are
production of human and veterinary vaccines, which constitute wild-type viruses rendered
harmless, or alternatively genetically modified viruses or virus-like particles (VLPs). Animal
cells are further used for production of altered viruses for gene therapy trials and viruses
trophic for noxious larvae, which cause agricultural problems. The animal cells themselves
can also be used, for example in stem cell replacements, for in vitro production of tissues like
skin and cartilage, and possibly in the future as replenishment of whole organs or organ parts
(e.g. liver). Another major product category is proteins used for diagnostics.
Over the past few years, the number of and demand for recombinant drugs from animal cell
culture processes has increased rapidly. In the early eighties 86% of all biopharmaceuticals
were produced in E. coli, but today 60-70% of the novel processes utilise mammalian cells 6.
In the end of 2005, there were 106 FDA approved drugs from processes involving cell
cultures, and approximately 50 of them were produced with animal cell technology (table 1).
In addition, estimates speak of several hundreds of animal cell derived candidate drugs
currently in company pipelines 6, 7. Experts foresee a continuation of this trend, and a veritable
surge for biopharmaceuticals in the next 10-20 years 8.
3
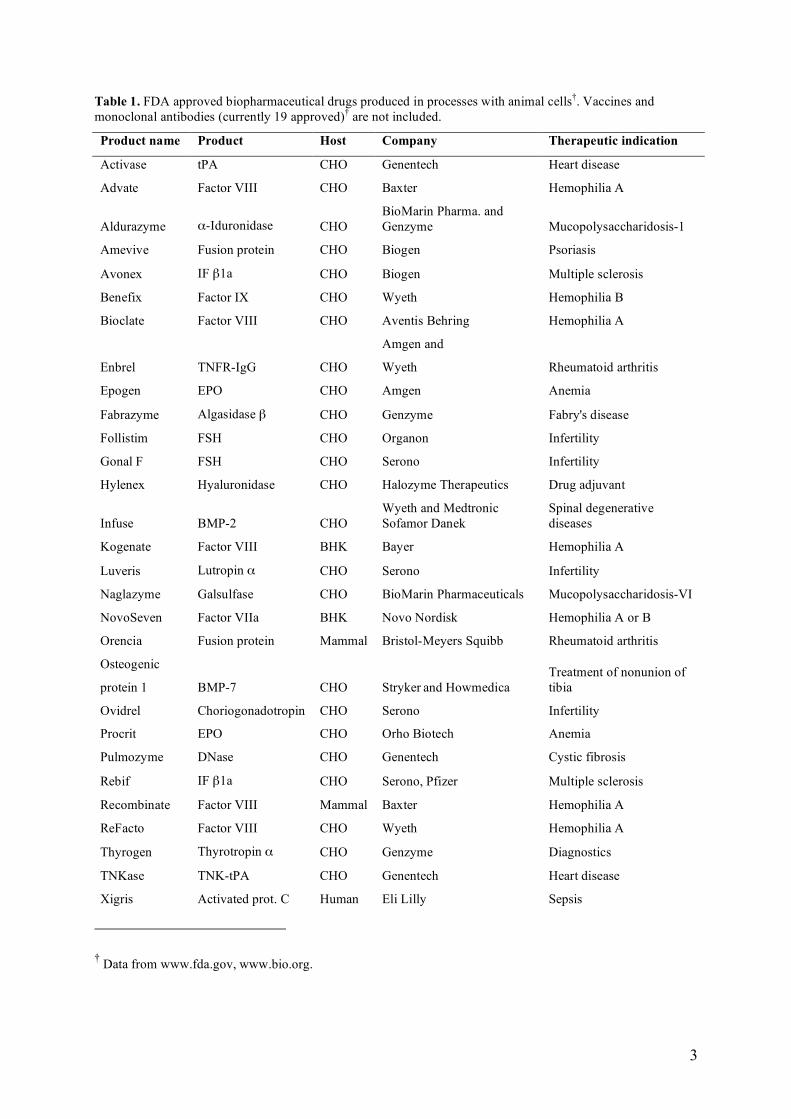
Table 1. FDA approved biopharmaceutical drugs produced in processes with animal cells†. Vaccines and monoclonal antibodies (currently 19 approved)† are not included.
Product name Product Host Company Therapeutic indication
Activase tPA CHO Genentech Heart disease
Advate Factor VIII CHO Baxter Hemophilia A
Aldurazyme α-Iduronidase CHO BioMarin Pharma. and Genzyme Mucopolysaccharidosis-1
Amevive Fusion protein CHO Biogen Psoriasis
Avonex IF β1a CHO Biogen Multiple sclerosis
Benefix Factor IX CHO Wyeth Hemophilia B
Bioclate Factor VIII CHO Aventis Behring Hemophilia A
Enbrel TNFR-IgG CHO
Amgen and
Wyeth Rheumatoid arthritis
Epogen EPO CHO Amgen Anemia
Fabrazyme Algasidase β CHO Genzyme Fabry's disease
Follistim FSH CHO Organon Infertility
Gonal F FSH CHO Serono Infertility
Hylenex Hyaluronidase CHO Halozyme Therapeutics Drug adjuvant
Infuse BMP-2 CHO Wyeth and Medtronic Sofamor Danek
Spinal degenerative diseases
Kogenate Factor VIII BHK Bayer Hemophilia A
Luveris Lutropin α CHO Serono Infertility
Naglazyme Galsulfase CHO BioMarin Pharmaceuticals Mucopolysaccharidosis-VI
NovoSeven Factor VIIa BHK Novo Nordisk Hemophilia A or B
Orencia Fusion protein Mammal Bristol-Meyers Squibb Rheumatoid arthritis
Osteogenic
protein 1 BMP-7 CHO Stryker and Howmedica Treatment of nonunion of tibia
Ovidrel Choriogonadotropin CHO Serono Infertility
Procrit EPO CHO Orho Biotech Anemia
Pulmozyme DNase CHO Genentech Cystic fibrosis
Rebif IF β1a CHO Serono, Pfizer Multiple sclerosis
Recombinate Factor VIII Mammal Baxter Hemophilia A
ReFacto Factor VIII CHO Wyeth Hemophilia A
Thyrogen Thyrotropin α CHO Genzyme Diagnostics
TNKase TNK-tPA CHO Genentech Heart disease
Xigris Activated prot. C Human Eli Lilly Sepsis
† Data from www.fda.gov, www.bio.org.

4
2 Cell growth in vitro
Eukaryotic cells from multicellular organisms are adjusted to a life in vivo, i.e. in a body of an
animal organism. The environment therein is fairly constant regarding, for example, pH and
temperature, but the cells are continuously flushed with body fluids that supply them with
nutrients, energy sources and growth factors, and remove harmful waste products. Explanting
the cells from their natural environment for in vitro cultivation leads therefore to many
challenges.
Several types of in vitro cultures exist. A primary culture is explanted directly from an animal
tissue and has only a limited life span. Primary cultures may express unique characteristics,
and are considered as the best model system for in vitro studies of cell functions. Upon
subculturing (passaging), the culture becomes known as a cell line, which either has a finite or
an indefinite life span. A finite cell line enters a quiescent state called replicative senescence
after a certain number of cell divisions, usually between 20 and 80 population doublings 9.
This state is characterized by several physiologic and metabolic changes, and it may be
induced by e.g. too short telomeres, lack of external growth signals or DNA damage. A
continuous cell line has via transformation escaped from senescence control and thereby
achieved an indefinite life span. Three major phenotypic changes may occur during
transformation: immortalization, non-regulated proliferation and malignancy. Cells can either
transform spontaneously, for example via mutations in the systems that regulate proliferation,
or be transfected with genes (e.g. human telomerase reverse transcriptase (TERT)) or viruses
(e.g. polyoma) known to induce transformation. Examples of transformed cell lines are CHO
DUK XB11, HEK 293 EBNA and COS-1. The ovarian insect cell lines Sf9 and Sf21 can be
regarded as naturally continuous cell lines. They express certain features of a transformed cell
line, such as indefinite life span and capability of suspension growth, but still retain some
normal characteristics, for example contact inhibition when growing adherently.
2.1 Cultivation techniques
Some cell lines grow naturally in single-cell suspension. They include ovarian insect cells,
immortalised cell lines such as HEK 293 EBNA and CHO DUK X B11 cells, and NS0 cells,
which are derived from mouse myeloma (blood cancer) and grow in suspension in their
natural habitat. Suspension cells grow in any vessel, but are commonly cultured in shake
flasks, spinners and bioreactors (fig. 1). In industrial processes the most frequently used

5
bioreactor operational mode is batch 10, but feeding strategies, hollow-fibre bioreactors and
perfusion technologies may be used to enhance product yield.
Primary cell lines and anchorage-dependent cell lines, e.g. HeLa, Vero, and HEK-293, do not
survive without a surface for growth. In small scale they are kept in culture dishes, T-flasks,
and roller bottles (fig. 1). However, scale-up can be a problem. One solution is microcarriers;
spheres specifically designed for optimal cell growth. They provide a surface for cell growth,
and can at the same time be maintained in suspension culture.
Figure 1. Common vessels for animal cell cultivation. From top left: microplate, T-flask, Ehrlenmeyer shake flask. From bottom left: spinner flask, stack with roller bottles and bioreactor. †
2.2 Growth factors
The proliferation of many animal cells in vitro is dependent on growth factors, which may be
added via serum or as purified substances if the requirements of the cells are known.
Examples of common growth factors used for media supplementation are fibroblast growth
factors (FGFs), insulin and insulin growth factors IGF-I and IGF-II 9. Growth factors may act
synergistically or additively with each other, or with other hormones. Further, some are
dependent on the activity of a second growth factor before they can act 11. One example is the
frog neuropeptide bombesin, which is not mitogenic alone, but needs co- or preactivation with
insulin or one of the IGFs 12.
† Pictures from www.costar.com, www.vwr.com, www.biotech.kth.se

6
2.3 Autocrine regulation
Certain cell lines do not need external growth factor stimulation for proliferation in vitro, but
grow by themselves due to the presence of autocrine loops; a mode of signalling in which
cells both express a growth factor ligand and the corresponding receptor for that ligand 13, 14.
Autocrine growth factors are produced in extremely low amounts; some even operate at
submicron levels. Moreover, ligands are commonly bound to specific carrier proteins 15.This
makes discovery of autocrine loops and identification of their compartments exceedingly
difficult 16, 17, but autocrine regulatory cascades have still been identified from various
contexts such as Drosophila development 18, bone remodelling 19 and wound healing 20.
Some autocrine ligands are synthesized as membrane-bound inactive precursors, which are
processed and released into the environment through regulated proteolysis 21. One example of
such a system is found in human mammary epithelial cells (HMEC), which express ligands to
the epidermal growth factor receptor (EGFR) that are cleaved off by a metalloproteinase from
the ADAM family 22. Another example is during Drosophila embryogenesis, where a
peptidase (Rhomboid) in conjunction with a chaperone cleaves off a membrane-bound ligand
precursor (Spitz), which can act on the Drosophila EGFR DER 23.
2.4 Cell death
Cell death may occur by two different mechanisms: necrosis and apoptosis. Necrosis is not
regulated, and takes place due to sudden environmental changes that are impossible for the
cell to counteract, for example too low osmolarity. Apoptosis, or programmed cell death 24, is
a strictly regulated event by which the cell is deconstructed, part-by-part. The onset is caused
by incidents such as DNA damage or extracellular signals, and it progresses via an
intracellular signal cascade with cysteine proteinases, the caspases, as the final executors. The
later stages of apoptosis are characterised by DNA fragmentation, nuclear blebbing and cell
shrinkage. The end products, the apoptotic bodies, are phagocytosed by macrophages to not
cause inflammation of the surrounding tissues in the organism.

7
3 Insect cell cultivation
3.1 History
Insect cell cultivation dates back to 1915 when Goldstein cultured moth tissue in a mixture of
salts and insect hemolymph. The first longer cultivation of insect cells took place in 1935,
when a primary culture from silkworm ovary was kept alive for 3 weeks in a medium
composed of maltose, salts, egg albumin extract and serum 25. In 1962 Grace isolated the first
four insect cell lines 26, and the baculovirus expression vector system (BEVS) technology was
introduced 1983 27. During the 1990s the versatility of the insect cell/BEVS became
widespread, and it is today one of the most common platforms for recombinant protein
expression.
3.2 Industrial cell lines
The most frequently used industrial insect cell lines originate from Trichoplusia ni (cabbage
looper; niflyfjäril) (fig. 2), Spodoptera frugiperda (fall army worm; nattflyfjäril) (fig. 2), and
Drosophila melanogaster (fruit fly; bananfluga). Cell lines from T. ni and S. frugiperda are
primarily used for transient expression, whilst D. melanogaster is also used for generation of
stable cell lines. However, many cell lines from other insect species also exist. Examples
include Bombyx mori Bm-5, Lymantria dispar LD652Y, Estigmene acrea Ea4 and Mamestra
brassicae MB0503. In addition, larvae have been used successfully for production of foreign
proteins, but larval production systems is out of the scope of this thesis and will not be further
discussed.
The first Trichoplusia ni cell line TN-368, was established 1970 from minced adult ovaries 28.
TN-368 grows as a monolayer and tends to clump in suspension culture 29. Another T. ni cell
line, BTI-TN-5B1-4, or High five, was established 1994 at Boyce Thompson Institute (New
York, USA)† as a clone from the T. ni egg cell line BTI-TN-5B1-28 30. High five cells grow
well in suspension, are readily infected with baculoviruses and have been reported to produce
some recombinant proteins in higher levels than cell lines from S. frugiperda 31-34. However,
baculovirus propagation in T. ni cells is not as effective as in S. frugiperda cell lines 35.
In 1977 the Spodoptera frugiperda cell line IPLB-Sf21 was isolated by Vaughn and co-
workers 36. This cell line was derived from a pupal ovary tissue culture at the USDA Insect
† U.S. patent no. 5,300,435 http://www.nal.usda.gov/bic/Biotech_Patents/1994patents/05300435.html

8
Pathology Laboratory (IPLB) in Maryland, USA. Sf21 AE is a subclone of Sf21, adapted to
serum-free media in England (AE = Adapted in England), and Sf9 is an isolate of this line,
cloned by Smith and Cherry 1983. Sf9 is perhaps the most widely used of all insect cell lines
today. Sf9 and Sf21 display similar characteristics, but Sf21 are reported to have a wider size
distribution and to form more irregular monolayers and plaques than Sf9 does 29, 37.
Genetically modified S. frugiperda cell lines are commercially available. The Mimic cells
from Invitrogen are genetically engineered to generate a human-like glycosylation pattern 38
(section 4.3.1). Another recently developed S. frugiperda cell line is expresSF+ from Protein
Sciences, USA. It grows in serum-free media and is marketed as high cell density, high
yielding, stable and easy scalable.
Figure 2. a) Spodoptera frugiperda (fall army worm), b) Trichoplusia ni (cabbage looper).††
One drawback with using S. frugiperda and T. ni cell lines is the lytic nature of the systems. If
a non-lytic expression system is desired, Drosophila melanogaster S2 Schneider cells may
serve as an alternative host. The S2 cells originate from dissociated embryos near hatching 39.
The commonly used baculoviral very late promoters (polyhedrin and p10) are not effective in
S2 cells, and therefore earlier promoters are used 40.
3.3 Cultivation media
In 1956 Wyatt was the first to develop a defined medium for insect cells in culture, after
extensive analysis of the composition of insect hemolymph. However, a small part of
hemolymph addition was still necessary for cell proliferation. Grace modified the Wyatt
medium 1962, and Grace’s medium is one of the most common insect cell media today. Other
examples of early developed and widely used insect cell media are TNM-FH, which consists
of Grace’s medium supplemented with lactalbumin hydrolysates and yeastolate 28, TC-100 41
and IPL-41 42. These media need in most cases supplementation of serum or insect
†† Pictures from www.cbif.gc.ca
a b

9
hemolymph to ensure proliferation, however, IPL-41 was in 1988 successfully used for large-
scale serum-free production after addition of yeastolate and lipids 43.
When media for insect cells and mammalian cells are compared, several major differences are
apparent, since they mimic the compositions of insect hemolymph and mammalian blood
serum, respectively. The Na+/K+ ratio in insect media is ∼0.3, and in mammalian media ∼20 44, because K+ is the major monovalent cation in insect hemolymph and Na+ in mammalian
blood. Moreover, insect media have higher osmotic pressure than mammalian media (340-390
and 280-320 mOsm/kg, respectively) 45, 46, and lower pH (~6.3 and ~7.2, respectively). A
lower pH enables the application of phosphate buffers, instead of the CO2-bicarbonate system
commonly used in mammalian cultivation. Consequently CO2 addition is not needed in insect
cell cultures. Finally, a striking difference is the colour of the media. Mammalian cell media
often contains the bright red pH indicator phenol red, whilst insect cell media are clear, or
golden yellow due to addition of yeastolate hydrolysates.
Recently, large efforts have been made to develop serum-free media (SFM) for bioproduction,
for reasons such as easier down-stream processing of products, lower cost and a reduced risk
for contamination with for example mammalian viruses. The first insect SFM was developed
in 1980 47 and recent examples of commercial SFM include Sf900II SFM (Invitrogen) 48,
Express five SFM (Invitrogen) 49, Insect-XPRESS (Cambrex), HyQ SFX Insect (HyClone)
and EX-CELL 400, 405 and 420 (JRH Biosciences). All of these media supply the cells with
sugars, vitamins, trace elements, amino acids and organic acids, but the relative amounts of
the inherent compounds differ.
One drawback with most commercially available media is that their composition remains
confidential, and it is not possible, or very expensive, to alter the original recipe. Therefore
much of the work in this thesis is performed with KBM10 50, which is a serum-free,
chemically defined Sf9 medium developed by Lena Häggström’s animal cell group at KTH in
collaboration with Karobio AB 51. The only not thoroughly defined components in KBM10
are cod liver oil, which contains omega-3 oils, and yeastolate ultrafiltrate. Yeastolate is a
water-soluble fraction of autolysed yeast cells, which has been widely used in insect cell
cultures and is regarded as a key growth-promoting component 52-58. Yeastolate provides the
culture with nucleotides, lipids and vitamins 59, amino acids, peptides and carbon compounds.
It is not necessary for cell survival, but for proliferation (IV). Cholesterol, α-
tocopherolacetate, and Tween 80 are also added to KBM10, to compensate for the lack of
serum.

10
4 The baculovirus expression vector system (BEVS) technology
4.1 Background
Baculoviruses are a diverse group of double stranded DNA-viruses trophic for a variety of
arthropods, and Baculo refers to the rod-shaped capsids of the virus particles. Traditionally,
baculoviruses have been used as biopesticides, since they are capable of infecting noxious
agricultural larvae whilst being non-pathogenic to plants and vertebrates. The most widely
used baculovirus in biotechnological respects is Autographa californica nuclear polyhedrosis
virus (AcNPV). The AcNPV genome is ~130 kbp and it has been completely sequenced 60. It
contains a polyhedrin (polh) gene that is strongly expressed late in infection, but not essential
for viral replication in cell culture 61. Based on this knowledge, the first baculovirus
expression vector was designed. In the vector polh was simply exchanged for a heterologous
gene under the polh promoter, and the modified vector was used to transfect insect cells 29.
4.1.1 The BEVS technology
Protein production in the BEVS begins with construction of the recombinant expression
vector. Direct insert of a product gene into the baculoviral DNA is difficult due to the large
AcPNV genome size, and instead, transfer vectors are commonly used. A transfer vector
contains the sequences adjacent to the polh gene in the baculovirus genome, whilst polh is
exchanged for the product gene. Upon co-transfection of the transfer vector and the viral
genome into insect cells, homologous recombination between the product gene and the
polyhedrin gene takes place, and recombinant viruses can subsequently be harvested.
Examples of homologous recombination systems are BacVector (Merck Biosciences),
Baculo-Gold (BD Biosciences), pBacPAK (BD Biosciences), and Bac-N-Blue (Invitrogen).
A drawback with homologous recombination is that the efficiency is low; recombination
frequency is typically only about 0.1% 62. Consequently, the first viral generation will be a
mixture between parental and recombinant virus, thus making time-consuming purification
steps necessary. To circumvent this, new methods utilising bacterial artificial chromosomes
(bacmids) have been developed. A bacmid contains viral elements for infection and
replication within the insect cell, but is propagated in E. coli. After bacmid transfection into
insect cells, baculoviruses are generated, which can be used for further infections 63. Bacmids
generate a homogenous viral stock and decrease the time for identification and purification of

11
recombinant viruses from 4-6 weeks to 7-19 days 64. One example of a bacmid system is Bac-
to-Bac from Invitrogen. It is compatible with the Gateway cloning system, which includes
commercial vectors for a number of different expression systems. Another recently evolved
bacmid technology is flashBAC (Oxford expression technologies). It is based on homologous
recombination, but in the parental baculoviruses, a part of a sequence necessary for viral
replication (ORF 1629) has been deleted. Homologous recombination with a bacmid
containing the recombinant product gene and a complete ORF 1629 is therefore the only
possibility of creating a functional virus, and the result is a homogenous virus stock.
After generation of the first passage of recombinant viruses, two or three virus multiplication
steps follow for creation of a high-titre virus stock. The virus titre of the final stock has
traditionally been determined with end-point dilution and plaque assays, where recombinant
non-occluded virus particles are visually distinguished from occluded parental viruses 29.
Nowadays several faster and more reliable methods are commercially available, for example
antibody-based assays 65-67. Methods utilising flow cytometry 68 and real-time PCR 69 have
also been reported.
The protein production phase consists of cultivating the host insect cells up to a specific cell
density, where the recombinant baculoviruses are added in a previously determined amount.
The ratio between virus particles and cells is defined as the multiplicity of infection (MOI). A
low MOI generates subsequent infections of secondary order or higher whereas a high MOI
(>5 pfu/cell) results in simultaneous infection of the whole culture 29. However, a high MOI is
unpractical in larger scale, due to difficulties in scaling up the virus stock. The time of
infection, is crucially important, and this will be further discussed in sections 4.3.3 and 7. The
viruses bind to receptors at the cell surface and enter cells by receptor-mediated endocytosis 70. Subsequently, expression of viral and recombinant genes follows. The protein production
phase continues for up to 96 hours, since the most commonly used promoters, polh and p10,
are inactive until a very late stage in the viral life cycle. Finally, cell lysis will occur, whereby
the product and cell constituents become mixed in the medium. The maximum product
synthesis rate usually occurs at 48-72 hours 29, but the optimal time for harvest varies with the
application. Intracellular products are often harvested before the cells lyse, whereas harvest of
extracellular products depend on for example degradation susceptibility. Down-stream
processing commonly involves addition of peptidase inhibitors, centrifugation and filtration,
followed by several chromatographic steps.

12
4.1.2 Applications
The most important applications of BEVS are in research, drug and vaccine development. The
combination of eukaryotic host cell characteristics and a short time frame between cloning
and production, makes the system ideal for quick screening purposes. No insect-made
therapeutic has yet made it to the market, but several drug candidates are currently in phase III
trials. Examples are the flu vaccine FluBlØk™ (Protein Sciences Corporation) and the
prostate cancer vaccine Provenge (Dendreon), which are produced in S. frugiperda cell lines.
Moreover, recombinant BEVS-made vaccines for AIDS, malaria, cancer, and influenza has
been tested in clinical phase I and II trials 71. In 1998 one of the greatest successes for the
BEVS so far was accomplished when an insect-made vaccine against the highly pathogenic
avian influenza strain H5N1 had FDA approval for compassionate use1, and the BEVS is still
today utilised in avian influenza virus research 72, 73.
The BEVS has also been subject for commercial interest in areas beyond recombinant protein
production. Recombinant viruses have been utilised in agriculture to kill noxious larvae 74.
The surface of the baculovirus particles can be used for displaying foreign peptides and
proteins 62, 75, and such altered viruses can be used as effective immunogens 75. Moreover,
research is ongoing about using baculoviruses as vectors in mammalian systems. In contrast
to insect cells, the cytopathic effect of baculoviral infection of mammalian cells is small or
even not observable 62. No replication of viral genes occurs, and subsequently no host cell
lysis follows. These features, amongst others, make baculoviruses attractive as a gene
delivery tool in human gene therapy 76. However, a mammalian promoter has to be inserted in
the baculovirus genome to achieve protein expression.
4.2 Advantages with BEVS
Each expression system available for recombinant protein production has its own unique
advantages as well as drawbacks, and no one is suitable for all applications. Prokaryotic
systems such as E. coli are simple and may give high yields in a short time. However, the lack
of post-translational modifications and the risk for inclusion body formation are two serious
disadvantages. Yeasts, for example Pichia pastoris, express certain proteins in very high
yields, but suffer severely from a non-mammalian glycosylation pattern, including
immunogenic epitopes 77. Mammalian systems are today the only alternative if the product
1 www.proteinsciences.com

13
has to be glycosylated, but they generate commonly low yields and the media cost is high.
Moreover, a stable mammalian system takes a long time to generate due to time-consuming
selection procedures, subcloning and amplification strategies. Transient systems are less time-
consuming, but the mammalian vectors available for gene-delivery are far from optimal 76, 78.
The BEVS has found its niche in between the two extremes of E. coli and mammalian
systems, and it has successfully been used for production of a large variety of proteins. Insect
cells are considered to be more stress-resistant, easier to handle and more productive as
compared with mammalian systems 79. They are easily adapted to suspension growth and
serum-free conditions, and they tolerate a wide osmolarity range. In contrast to prokaryotic
systems, a range of post-translational modifications that confer correct folding, biological
activity and antigenicity on expressed proteins can be performed. These include
phosphorylation, glycosylation, myristylation and palmitylation, signal peptide processing,
post-translational cleavage and disulphide bond formation 80, 81. However, the glycosylation
pattern exhibits certain non-human anomalies, and these will be further discussed in section
4.3.1.
Moreover, the baculoviral vectors are not trophic for mammals and therefore safe for humans.
In contrast, the utilisation of mammalian viruses for gene transfer requires several precautions
to avoid unwanted human infections, for example deletions of viral genes and virus
propagation in specific helper cell lines. In addition, the genetically modified non-occluded
baculoviruses used in laboratories are harmless for their natural hosts 62, and the laboratory
waste is therefore environmentally safe, in that respect. Further, baculoviral vectors may
harbour large inserts 76, 82; possibly 100 kb or even more 29, and be propagated to very high
titres, which allows for generation of a high-titre virus stock with a minimum number of virus
passages in the host cells. Long-term passaging implies a risk for genetic changes, resulting in
a heterogeneous virus stock. Most mammalian-based viral vectors are hampered by the small
size of the maximum DNA inserts, and do not generate high titres 76, 78. Finally, the time from
cloning to ready-made product in BEVS is very fast as compared to other systems, see further
section 4.1.1.
4.3 Limitations in BEVS
The most serious limitations of BEVS can be summarized in three points: 1) a non-human
glycosylation pattern, 2) a high proteolytic activity and 3) the so-called "cell density effect",
which leads to a drop in productivity late in cultivation. These features will be further

14
discussed in the following sections. For commercial applications, scale-up related issues such
as oxygen supply 83, carbon dioxide accumulation 84 and bioreactor-type to be employed 85
have to be considered.
4.3.1 Glycosylation
There are fundamental differences between glycosylation in insects and higher eukaryotes 86,
87 and the processing of N-linked glycans is one of the most serious limitations of the insect
cell/BEVS. Mammalian N-glycans are predominantly complex with terminal sialic acid
residues, whilst insect N-glycan end products tend to be paucimannosidic (Man3-GlcNAc2-N-
Asn) or of high-mannose type (Man5-9-GlcNAc2-N-Asn) 86. This is a severe problem since
complex N-glycans, and specifically terminal sialic acid residues contribute to mammalian
glycoprotein functions in many different ways. Besides, some insects α(1-3)fucosylate the
core Asn-linked GlcNAc residue in the glycan 88. This α(1-3) linked fucose is a potential
human allergenic epitope and a protein with such glycans will never be accepted as a
candidate drug.
Still, inherent N-glycosylation characteristics similar to that of mammalian cells exist in
certain insect cultures. Tn-4h, a clonal isolate from High five, was shown to produce
sialylated glycans when cultured in T-flasks supplemented with the sialic acid precursor N-
acetylmannosamine, or in serum-containing media in a HARV (High Aspect Ratio Vessel) 89
Moreover, the cell line Ea4 from E. acrea, is capable of synthesizing N-glycans containing
terminal GalNAc residues 90, 91. However, in the majority of the industrially used insect cell
lines, the inherent N-glycosylation capabilities are not sufficient.
To address the problem with inadequate N-glycosylation, insect cells have been stably
transformed with mammalian enzymes involved in generation of glycans 92-95. This has
resulted in the SFSWT-3 cell line, which is capable of producing biantennary, complex N-
glycans in serum-free media supplemented with N-acetylmannosamine. However, terminal
sialic acid residues are only present at one glycan branch 95. A parental line, SFSWT-1, is
commercially available as Mimic cells (section 3.2). It has the same glycosylation capabilities
as SFSWT-3, but has to be cultivated in serum-containing medium 75, 94, 95. In summary, the
humanization of the insect cell glycan pathways is well in progress, but many additional
developments are still needed 62.

15
4.3.2 Proteolysis
The BEVS is known for its high proteolytic activity. Since the system is lytic by nature, the
infection process will eventually result in disruption of the cells 3-5 days after infection. This
releases intracellular peptidases of insect origin, as well as peptidases encoded by the
baculoviruses, introduced via the infection. In addition, truly extracellular insect peptidases
may already be present in the medium. In a number of cases, researchers have reported
decreased product yields due to proteolytic activity 96-100. The utilisation of serum-free culture
media may aggravate the problem 101, since serum contains natural peptidase inhibitors 9.
The best-characterized proteolytic enzyme within the BEVS is v-cath, a cysteine proteinase
encoded by AcPNV 102. V-cath is a homologue of the mammalian lysosomal peptidase
cathepsin L, and has an active form of 27.5 kDa and two precursor forms of 35.5 and 32 kDa 103. It plays an essential role in liquefaction of insect larvae in baculovirus infection, thereby
releasing virus particles for infection of more hosts 104. Moreover, AcNPV also encodes a 58
kDa chitinase (chiA) 105. During the protein production phase in BEVS, chiA accumulates in
ER and forms a para-chrystalline array, which hamper trafficking of extracellular and
membrane recombinant products 106, 107. To avoid unwanted effects from these AcNPV-
encoded peptidases, novel vectors have been created with deletions of the chiA and v-cath
genes. In flashBAC from Oxford technologies, chiA has been removed, and BacVector-3000 108 from Merck Biosciences has deletions of both chiA and v-cath. The utilisation of
BacVector-3000 was shown to greatly reduce proteolysis of two model proteins (GUS and β-
galactosidase) 108.
A number of less well characterized peptidases have been identified from insect cell cultures.
In lysates of infected Sf9 cells, a caseinolytic peptidase with an unusually high molecular
weight of 120 kDa was detected 109. Moreover, several intracellular Sf9 peptidases (36-52
kDa) have been visualized on gelatine zymography gels 110-112. All but one originated from
baculovirus infected cells, a feature that could make their true nature (viral- or insect) difficult
to interpret. However, a 49 kDa papain-like cysteine peptidase was present also in non-
infected cells 110. Further, BTI-Tn-5B1-4 High five cultures have been found to secrete
metalloproteinases. Two enzymes at 32-33 and 41-42 kDa, respectively, have been identified 113, as well as precursor and degradation forms of a metalloproteinase with an active form at
48 kDa 114.
A popular strategy for controlling the proteolytic activity in the culture medium is to add
protease inhibitor cocktails. However, this strategy has drawbacks. Many inhibitors are

16
expensive and toxic, and some are reported to hamper cell growth 115 and recombinant protein
production 116, when added to cultures. Further, the half-life of inhibitors in water solutions
might be short, which leads to the necessity of repeated additions for optimal peptidase
inhibition. Inhibitors might also co-purify together with the product. To avoid problems
arising with overuse of inhibitors, and at the same time achieve optimal inhibition within the
production system in question, it is necessary to obtain a good knowledge of the peptidase
background.
Another strategy for avoiding product degradation is to separate the recombinant protein
production phase from the cell lysis stage late in infection, by using earlier promoters than the
most common very late ones (p10 and polyhedrin). One example is the basic protein promoter
(bpp), which has been used to produce a variety of recombinant proteins 117-119. The bpp
production phase begins as early as 6 h p.i. 118, and in one report a vector including the bpp
promoter generated the highest product yield, in a comparison with vectors with the p10 and
polh promoters 117. Yet another strategy against proteolysis is to utilise a viral vector with a
reduced capability for cell lysis 120.
4.3.3 The cell density effect
Insect cells can easily be grown to high densities. In our laboratory, the maximum batch cell
density for Sf9 is about 107 /mL and for High five about 6·106/mL. However, the problem is
not the cell density, but the fact that a drastic decrease in product yield occurs at an early time
point in culture 43, 52, 55, 121-129. This is referred to as “the cell density effect”, and it takes place
at a batch cell density of 2-3·106/mL for Sf9, and immediately after inoculation for High five 130. In practice, this leads to the consequence that infection is commonly done no more than
one or two days after inoculation, well before the maximum cell density is reached. If the
inherent potential of the cells could be fully used, i.e. if productivity could be sustained at the
maximum cell density, the total product yield would increase significantly.
A tremendous amount of research has been done to counteract the cell density effect. Early
results indicated that the product drop was due to nutrient depletion 131, and large efforts have
been made to study the influence of nutrients and medium exchange on cell proliferation and
productivity in S. frugiperda and T. ni cell lines, as reviewed recently 84. However, the
productivity drop was shown to occur prior to nutrient depletion 132, and no nutrient has been
shown to be responsible for the lower yield at high cell densities 128, 133. Moreover, nutrient
limitation was in one case favourable, resulting in an increased yield of active proteins 134.

17
Nevertheless, several researchers have achieved beneficial effects of nutrient additions and
feeding strategies 54, 55, 58, 135-137. No specific medium component has been critical for
enhancing growth and product yield, but different combinations of glucose, glutamine, amino
acids, yeastolate ultrafiltrate, lipids, vitamins, iron and trace metals have been utilised for
medium supplementation. The Sf9 batch culture with the highest maximum cell density
reported so far (5.2·107/mL) was given semi-continuous additions of concentrated nutrient
solutions, and a similar culture could be infected at a cell density as high as 1.7·107/mL 137.
Still, the maximum volumetric product yield was low in comparison with yields from other
experiments, despite additions of nutrient concentrates both pre- and post infection. One
reason for this could be a too high osmolarity due to the addition of nutrient concentrates.
A different strategy for improving productivity has been to replenish the medium at the time
of infection 52, 55, 58, 124, 127, 129, 138, but such a procedure is both expensive and impractical on a
large scale. Perfusion cultures have also been used successfully, and resulted in high cell
densities and likewise high productivities 139 128, 140, 141. However, a perfusion process may be
complicated to set up, and is not optimal for transient systems such as the BEVS.
The drop in productivity has also been suggested to originate from toxic by-product
accumulation. The major metabolic by-products in S. frugiperda cultures is alanine during
glucose excess, and ammonia under glucose limitation 51, 56, 129, 142, whilst High five cells
accumulate lactate, alanine and ammonia during normal growth conditions 133, 143, 144 145.
However, insect cells are not as sensitive as mammalian cells to ammonia 56, 128, 142, and high
alanine concentrations were shown not to affect growth 56, 142 or recombinant protein
production 55. Lactate may be inhibitory at high levels 146, but this effect seems to be cell line
specific. Irrespective of what effects these by-products may have at high levels, they will not
have accumulated to a large extent at the early time when the drop in productivity occurs.
Toxic by-product formation is therefore not a plausible explanation for the cell density effect.
In summary, total medium replenishment at infection and nutrient additions have enhanced
the productivity in many cases, but the mechanisms behind the increased product yields are
not fully understood. The final conclusion is therefore that the relation between nutrients and
product yield in the BEVS is far from straightforward, and the intriguing cell density effect
remains unexplained.

18

19
II Objectives
The ultimate goal for this work has been to increase productivity in the Sf9/BEVS. The
strategy was to investigate the mechanisms controlling growth and proliferation in normal
cultures, and to use that knowledge for increasing product yields. The specific aims were
therefore:
• To investigate how nutrients affect productivity and proliferation.
• To investigate whether the proliferation is under autocrine control.
• To search Sf9 conditioned medium (CM) for the presence of autocrine mitogens. If an
enhancing mitogen could be identified, supply the culture with surplus amounts. If an
inhibitory component was found, render it harmless.
• To investigate the underlying causes to the cell density dependent drop in
productivity.

20
21
III Present investigation
5 Proliferation of Sf9 cells
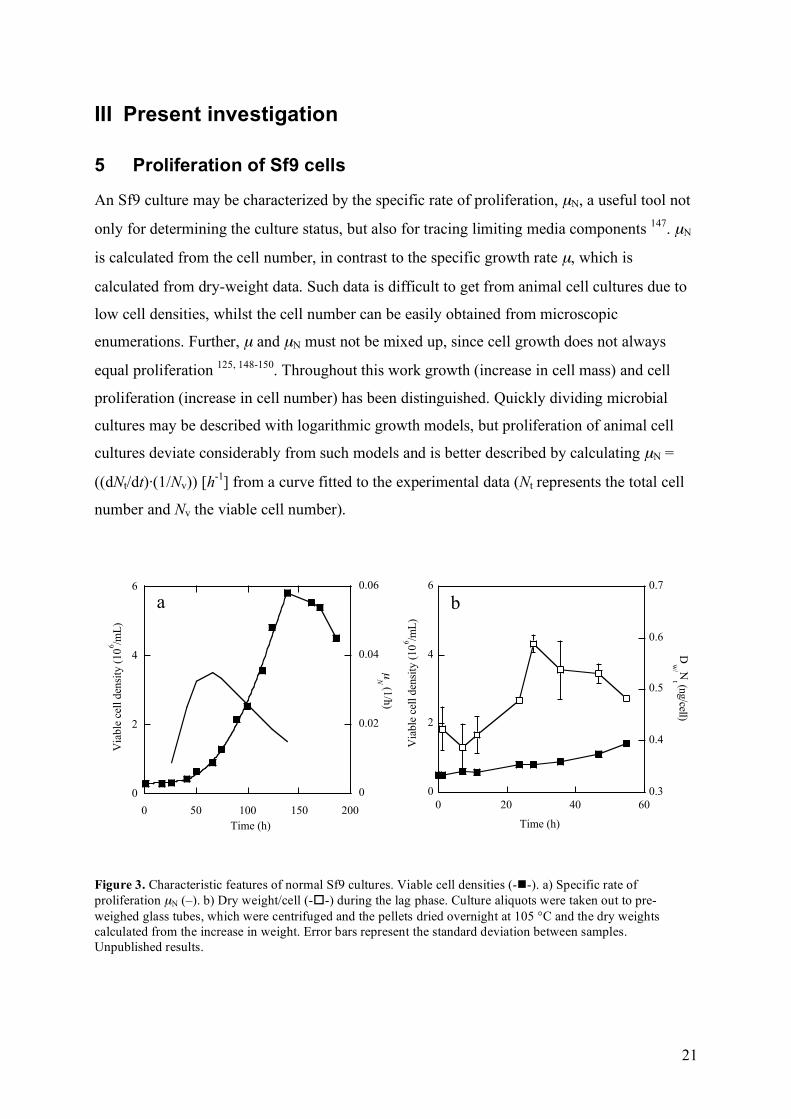
An Sf9 culture may be characterized by the specific rate of proliferation, µN, a useful tool not
only for determining the culture status, but also for tracing limiting media components 147. µN
is calculated from the cell number, in contrast to the specific growth rate µ, which is
calculated from dry-weight data. Such data is difficult to get from animal cell cultures due to
low cell densities, whilst the cell number can be easily obtained from microscopic
enumerations. Further, µ and µN must not be mixed up, since cell growth does not always
equal proliferation 125, 148-150. Throughout this work growth (increase in cell mass) and cell
proliferation (increase in cell number) has been distinguished. Quickly dividing microbial
cultures may be described with logarithmic growth models, but proliferation of animal cell
cultures deviate considerably from such models and is better described by calculating µN =
((dNt/dt)·(1/Nv)) [h-1] from a curve fitted to the experimental data (Nt represents the total cell
number and Nv the viable cell number).
0
2
4
6
0.3
0.4
0.5
0.6
0.7
0 20 40 60
Via
ble
cel
l den
sity
(10
6 /mL
)
Dw
/ Nt (n
g/cell)
Time (h)
0
2
4
6
0 50 100 150 200
0
0.02
0.04
0.06
Time (h)
Via
ble
cel
l den
sity
(10
6 /mL
)
µN
(1/h
)
a b
Figure 3. Characteristic features of normal Sf9 cultures. Viable cell densities (--). a) Specific rate of proliferation µN (–). b) Dry weight/cell (--) during the lag phase. Culture aliquots were taken out to pre-weighed glass tubes, which were centrifuged and the pellets dried overnight at 105 °C and the dry weights calculated from the increase in weight. Error bars represent the standard deviation between samples. Unpublished results.
22
Upon inoculation, the Sf9 cultures exhibit a lag phase, where the cells adjust to their new
environment and µN is low. When proliferation starts, µN increases for a short period, but
µN,max is only transiently reached before the proliferation rate declines again, well before the
onset of the stationary phase (fig. 3a, I) 50, 51, 53, 56. Moreover, dry weight (fig. 3b) and cell
volume measurements (III) showed that biomass growth continued during the initial lag
phase, although cell division was stalled.
The Sf9 cultures were routinely passaged every third day to a start density of 0.3·106
cells/mL, and during such conditions the lag phase lasted for approximately 24 hours. The
length of the lag phase depended on the inoculum concentration, a typical feature for the
presence of an autocrine growth controlling system 151, as will be discussed in section 5.2.
The minimum inoculum density used in this work was 105 cells/mL, due to difficulties in
accurately determining lower viable cell densities. Further, cell growth has found to be
hampered at a seeding density of <5·104 /mL 152, 153.
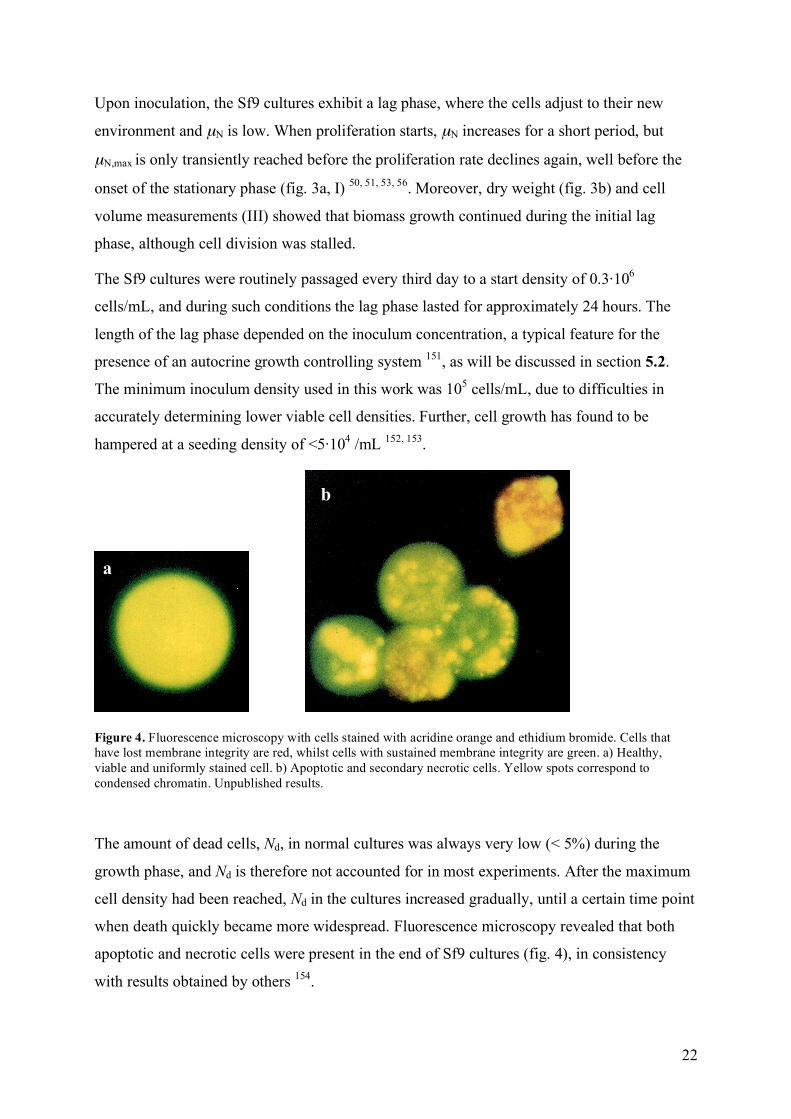
Figure 4. Fluorescence microscopy with cells stained with acridine orange and ethidium bromide. Cells that have lost membrane integrity are red, whilst cells with sustained membrane integrity are green. a) Healthy, viable and uniformly stained cell. b) Apoptotic and secondary necrotic cells. Yellow spots correspond to condensed chromatin. Unpublished results.
The amount of dead cells, Nd, in normal cultures was always very low (< 5%) during the
growth phase, and Nd is therefore not accounted for in most experiments. After the maximum
cell density had been reached, Nd in the cultures increased gradually, until a certain time point
when death quickly became more widespread. Fluorescence microscopy revealed that both
apoptotic and necrotic cells were present in the end of Sf9 cultures (fig. 4), in consistency
with results obtained by others 154.
a
b
23
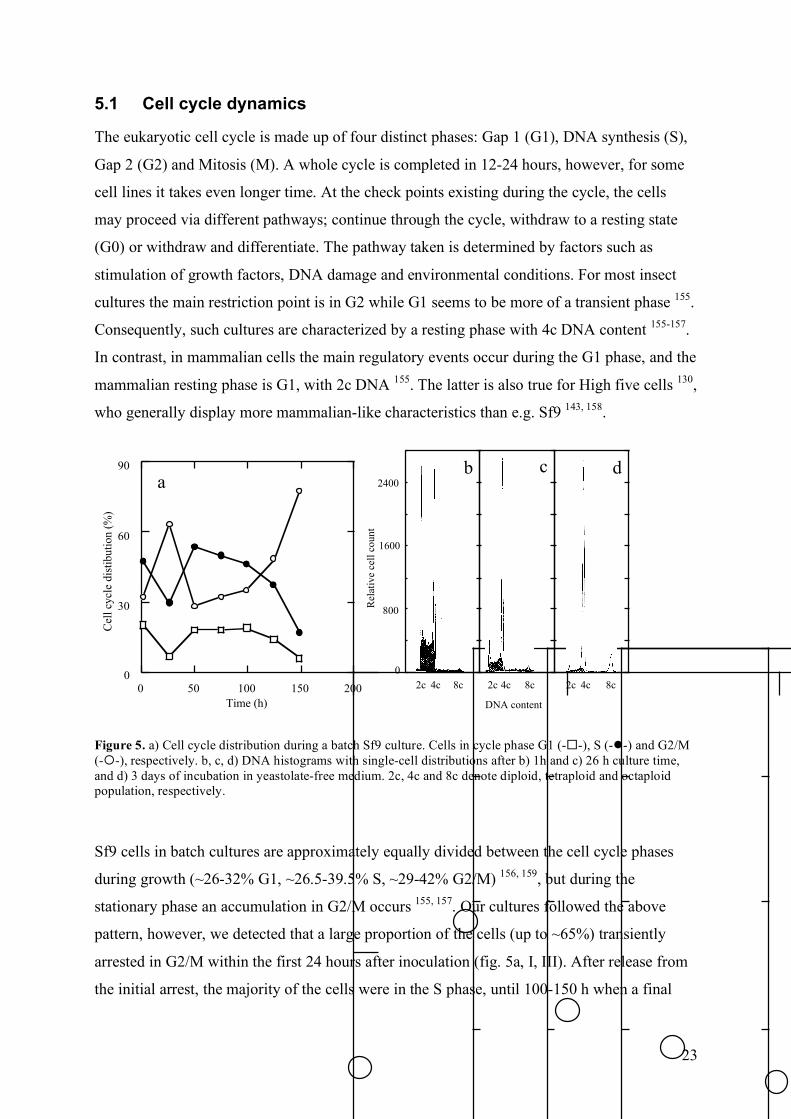
5.1 Cell cycle dynamics
The eukaryotic cell cycle is made up of four distinct phases: Gap 1 (G1), DNA synthesis (S),
Gap 2 (G2) and Mitosis (M). A whole cycle is completed in 12-24 hours, however, for some
cell lines it takes even longer time. At the check points existing during the cycle, the cells
may proceed via different pathways; continue through the cycle, withdraw to a resting state
(G0) or withdraw and differentiate. The pathway taken is determined by factors such as
stimulation of growth factors, DNA damage and environmental conditions. For most insect
cultures the main restriction point is in G2 while G1 seems to be more of a transient phase 155.
Consequently, such cultures are characterized by a resting phase with 4c DNA content 155-157.
In contrast, in mammalian cells the main regulatory events occur during the G1 phase, and the
mammalian resting phase is G1, with 2c DNA 155. The latter is also true for High five cells 130,
who generally display more mammalian-like characteristics than e.g. Sf9 143, 158.
0
30
60
90
0 50 100 150 200
Cel
l cyc
le d
istib
utio
n (%
)
Time (h)2c 4c 8c 2c 4c 8c 2c 4c 8c
a
DNA content
Rel
ativ
e ce
ll co
unt
800
2400
1600
0
b c d
Figure 5. a) Cell cycle distribution during a batch Sf9 culture. Cells in cycle phase G1 (--), S (--) and G2/M (--), respectively. b, c, d) DNA histograms with single-cell distributions after b) 1h and c) 26 h culture time, and d) 3 days of incubation in yeastolate-free medium. 2c, 4c and 8c denote diploid, tetraploid and octaploid population, respectively.
Sf9 cells in batch cultures are approximately equally divided between the cell cycle phases
during growth (~26-32% G1, ~26.5-39.5% S, ~29-42% G2/M) 156, 159, but during the
stationary phase an accumulation in G2/M occurs 155, 157. Our cultures followed the above
pattern, however, we detected that a large proportion of the cells (up to ~65%) transiently
arrested in G2/M within the first 24 hours after inoculation (fig. 5a, I, III). After release from
the initial arrest, the majority of the cells were in the S phase, until 100-150 h when a final
24
G2/M arrest occurred. Further, a polyploid population emerged during G2/M arrests, both
during the lag-phase and during the stationary phase (fig. 5b, I). Polyploidy has also been
observed by others 155, 157, 160-162, but it seems not to be a general feature 156. Jarman-Smith and
co-workers (2002) characterised Sf9 clones obtained from different laboratories, and found a
polyploid fraction in all clones, but the relative amount of polyploid cells varied between the
clones.
The main cell cycle characteristics (initial and final G2/M arrest, fig. 5a) were present in most
cultures, but addition of 20% conditioned medium (CM) decreased the initial arrest, resulting
in a less synchronous growth pattern during the remaining culture (I). Implications of this
result and other effects of CM additions are discussed in section 5.3.
25
50
75
100
0 50 100 150 200
Time (h)
G2
/M (%
)
0
2
4
6
0 50 100 150
Time (h)
Via
ble
cel
l den
tiy (
10
6/m
L)
a b
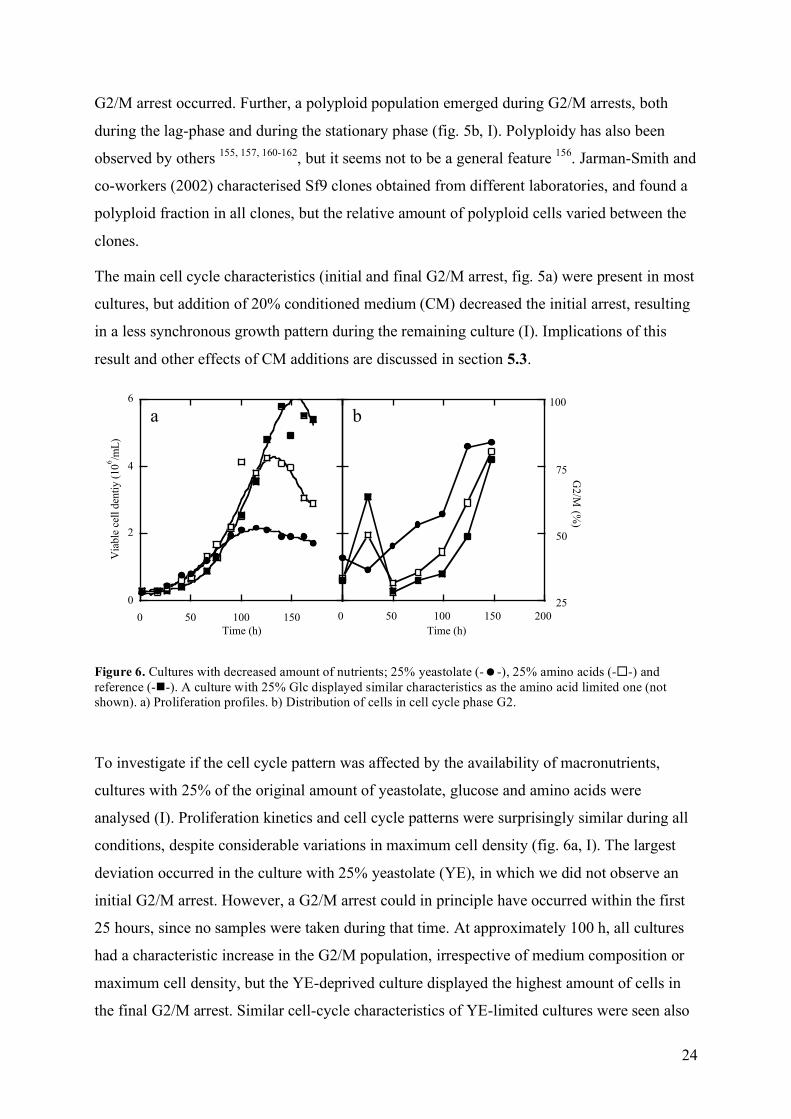
Figure 6. Cultures with decreased amount of nutrients; 25% yeastolate (--), 25% amino acids (--) and reference (--). A culture with 25% Glc displayed similar characteristics as the amino acid limited one (not shown). a) Proliferation profiles. b) Distribution of cells in cell cycle phase G2.
To investigate if the cell cycle pattern was affected by the availability of macronutrients,
cultures with 25% of the original amount of yeastolate, glucose and amino acids were
analysed (I). Proliferation kinetics and cell cycle patterns were surprisingly similar during all
conditions, despite considerable variations in maximum cell density (fig. 6a, I). The largest
deviation occurred in the culture with 25% yeastolate (YE), in which we did not observe an
initial G2/M arrest. However, a G2/M arrest could in principle have occurred within the first
25 hours, since no samples were taken during that time. At approximately 100 h, all cultures
had a characteristic increase in the G2/M population, irrespective of medium composition or
maximum cell density, but the YE-deprived culture displayed the highest amount of cells in
the final G2/M arrest. Similar cell-cycle characteristics of YE-limited cultures were seen also

25
in a later experiment (IV). Implications of this result for productivity are discussed in section
7.
The cell cycle phase at the time of infection was already 30 years ago suggested to play a role
for the progression of the viral infection 163, and the majority of the prevailing literature
indicates that infection in cell cycle phase S is most effective 156, 164-166, possibly due to
enhanced viral replication 165. In concordance, no expression of late viral genes was detected
in Sf9 cells which did not proceed through the S phase 156, and the product yield was 1.5- to
1.8-fold higher in Sf9 cells infected in G1 or S, as compared to G2/M 166. However, viral
early genes were replicated irrespective of cell cycle phase at infection, and budded virus
progeny was detected also in cultures arrested at the G1/S transition point 156. The importance
of the cell cycle phase on recombinant product yield thus seems to be dependent of the viral
promoter. If very late promoters are used, infection should preferably take place to allow
replication of the recombinant product genes during S, whereas the function of viral early
promoters seems to be less dependent of the cell cycle phase. In addition, AcNPV infection
prevents normal cell cycle progression. Cells infected in G1 or S are arrested in S 159, 166,
whereas cells infected in G2/M do not proceed through mitosis, but arrest at an early mitosis
stage 156, 159, 166. Braunagel, (1998) found that 84% of the Sf9 cells arrested in G2/M after
infection.
5.2 Autocrine regulation of proliferation
Sf9 insect cells grow and proliferate in serum-free media, without any growth factor addition.
The reason is either a totally deregulated cell cycle, similar to the situation in cancer cells, or
production of autocrine factors (see further section 2.3). In fact, Sf9 cultures display several
features characteristic for the presence of an autocrine regulatory system. First, an increase of
the start cell density resulted in an augmented µN,max in several different media and a
shortened lag-phase, whilst a decreased inoculation density prolonged the lag phase (I). This
has also been observed by others 132, 138, 153, 165, and indicates that autocrine mitogens are
secreted during proliferation, as suggested by Kioukia et al. (1995). A high inoculation cell
density would thereby result in a fast build-up of autocrine factors, and consequently a short
lag-phase. Second, the final G2/M arrest occurs at approximately the same time in different
cultures, irrespective of medium composition (section 5.1). Further, changes in the uptake
rates of several amino acids have also been shown to take place, shortly before the onset of
the stationary phase 50, 132, 167. Amino acid transport is generally tightly linked to cell growth
26
and proliferation 167, 168. The synchronized changes in transport rates and cell cycle
progression may indicate that their onsets are timed by the cells’ own autocrine clock.
5.3 The conditioned medium effect
Proliferation of certain cells is stimulated by conditioned medium (CM), i.e. medium in which
cells of the same kind have been growing previously 169. This phenomenon has been reported
for cell lines of both vertebrate and invertebrate origin. Insect cell lines that are stimulated by
addition of CM include NIH-Sape-4 cells from the flesh fly Sarcophaga peregrina 170, 171,
Trichoplusia ni BTI-Tn-5B1-4 (High five) 58, 114 and the Drosophila wing-disc cell line C1.8+ 172. The stimulatory effect in NIH-Sape-4 CM originated from a novel 52 kDa cytokine
designated IDGF (insect derived growth factor) 170, shown to have adenosine deamidase
activity indispensable for the growth factor activity 171. In High five cultures, CM taken from
the growth phase stimulated proliferation, and the enhancing factor in this case was identified
as a metalloproteinase 114. Further, C1.8+, a Drosophila wing-disc cell line, was shown to
secrete three members from a novel family designated IDGFs (imaginal disc growth factors),
and two related genes were also identified by sequence homology. These imaginal disc
cytokines were structurally related to chitinases, but had no chitinase activity. They
cooperated with insulin to stimulate the proliferation, polarization and motility of Drosophila
imaginal discs 172.
0
2
4
6
8
10
0 50 100 150 200 250
Via
ble
cel
l den
sity
(10
6/m
L)
Time (h)
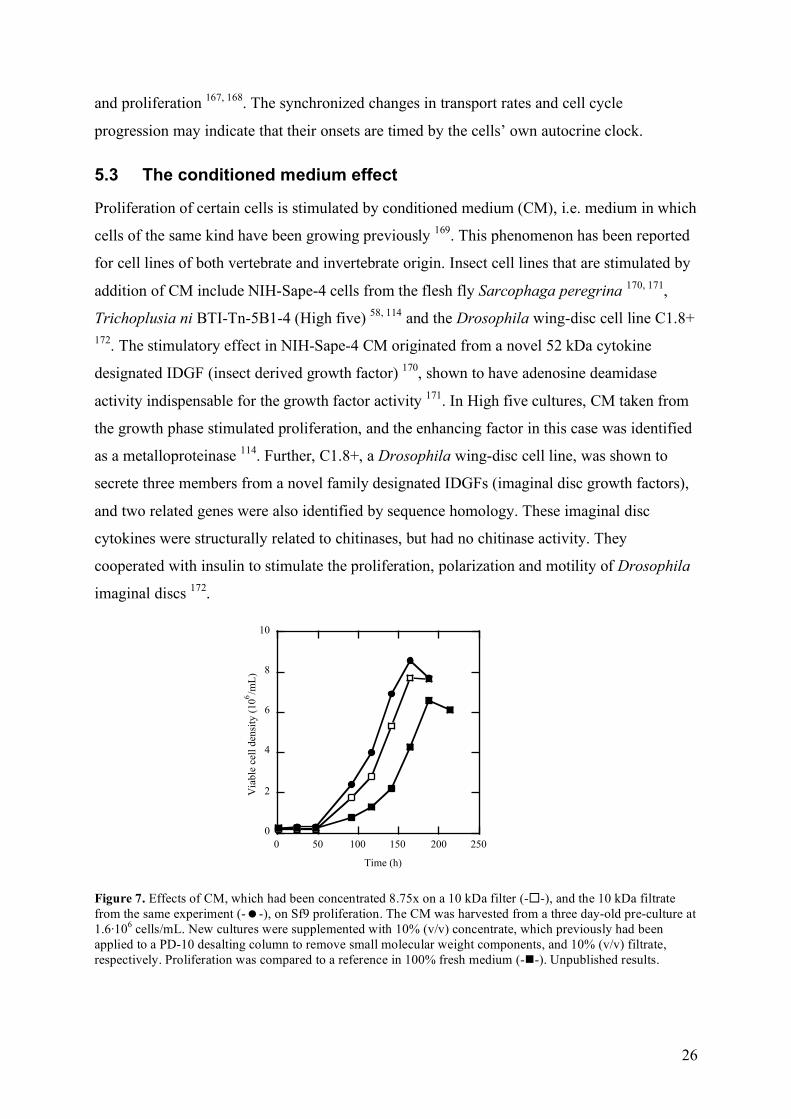
Figure 7. Effects of CM, which had been concentrated 8.75x on a 10 kDa filter (--), and the 10 kDa filtrate from the same experiment (--), on Sf9 proliferation. The CM was harvested from a three day-old pre-culture at 1.6·106 cells/mL. New cultures were supplemented with 10% (v/v) concentrate, which previously had been applied to a PD-10 desalting column to remove small molecular weight components, and 10% (v/v) filtrate, respectively. Proliferation was compared to a reference in 100% fresh medium (--). Unpublished results.

27
CM additions have also been reported to stimulate proliferation of Sf9 cultures 58, 173, and it
has been suggested to contain endogenous, secreted growth-promoting factors 127, 165. Wu et
al. (1998) noted higher growth rates and maximum cell densities in spent medium fortified
with glucose, glutamine and yeastolate, than in purely fresh medium. However, the factors
responsible for the observed effect are not yet identified. On the other hand, an inhibitory
effect of spent medium carry over has been observed for Sf21 cells 158. This might be
explained by the fact that the CM had been taken from the stationary phase of the previous
culture, and a previous enhancing effect could thereby possibly be lost, as observed for High
five cells 114.
Our results showed that Sf9 CM taken from a previous culture at 28 h, 39 h or three days
stimulated proliferation (I), as well as CM taken at even earlier time points 173. To further
characterize this effect, CM was subjected to low molecular (10 kDa) cut-off filtration.
Surprisingly both the filtrate and the retentate affected Sf9 proliferation in a positive manner
(fig. 7), and the conclusion was that the stimulation originated from at least two different
compounds: a low molecular weight (<10 kDa) and a higher molecular weight (>10 kDa).
Addition of CM also had direct physiological effects on the cells; CM or 10 kDa filtrated CM
decreased the average cell size (IV). A smaller average cell size indicates that the number of
cells in G2/M was less than in a standard culture, since cells in G2/M are larger than cells in
G1/S. This might be explained by the fact that addition of CM decreased the initial G2/M lag-
phase arrest (I), a feature that signifies the presence of mitosis-promoting autocrine factors,
and thus earlier onset of proliferation.
To facilitate purification of the unknown stimulatory factors, KBM10 medium without
yeastolate (YE) was used for production of CM, since YE largely contributes to the peptide
background in complete medium. During such YE-free conditions the culture remains viable,
but the cells cannot proceed through mitosis and arrest in cell cycle phase G2/M (I, IV). CM
and 10 kDa CM retentate from such arrested cultures exerted a positive effect on cell
proliferation, similarly to CM from complete medium, but 10 kDa CM filtrate inhibited
proliferation (not shown). Thus, the larger (>10 kDa) autocrine mitogen was produced also
during a resting cell cycle state, but YE was essential for production of the smaller (<10 kDa)
factor.
28
0
1
2
3
4
50 150 250
Via
ble
cel
l den
sity
(1
06 /m
L)
Time (h)
a b
0
0,1
0,2
0,3
0,4
0,5
10 20 30 40
Ab
s 2
80
nm
Fraction no
12
3
4
5
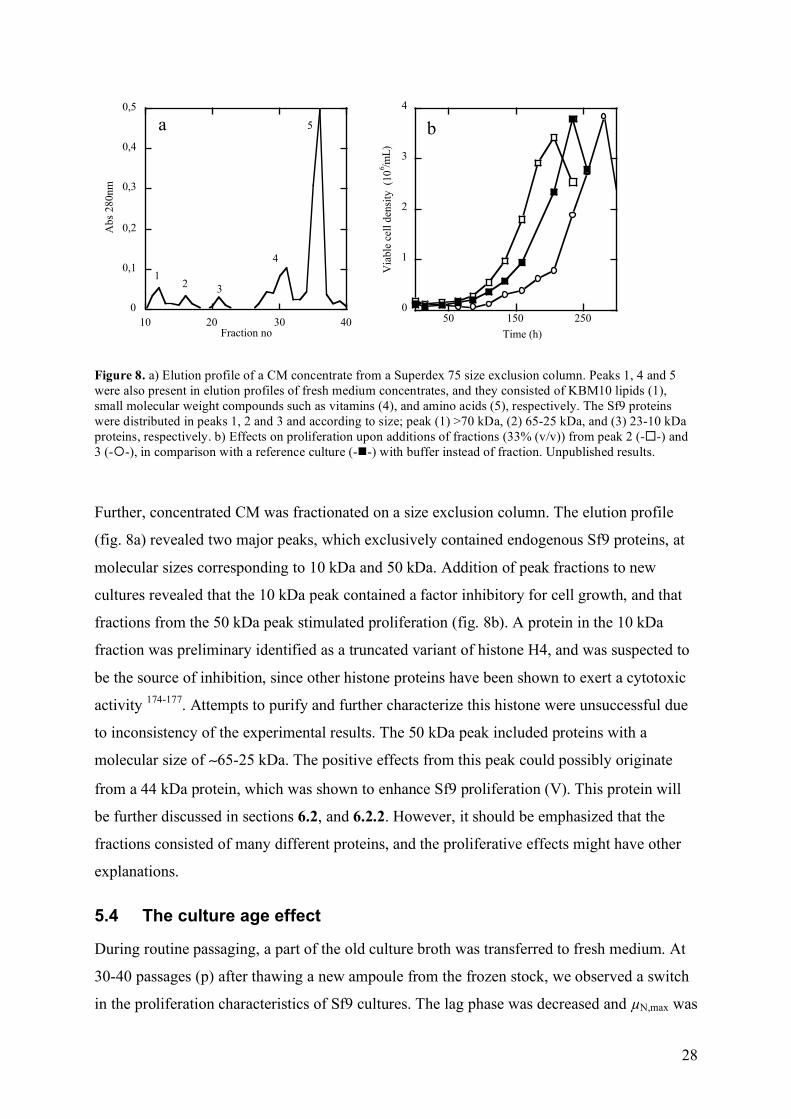
Figure 8. a) Elution profile of a CM concentrate from a Superdex 75 size exclusion column. Peaks 1, 4 and 5 were also present in elution profiles of fresh medium concentrates, and they consisted of KBM10 lipids (1), small molecular weight compounds such as vitamins (4), and amino acids (5), respectively. The Sf9 proteins were distributed in peaks 1, 2 and 3 and according to size; peak (1) >70 kDa, (2) 65-25 kDa, and (3) 23-10 kDa proteins, respectively. b) Effects on proliferation upon additions of fractions (33% (v/v)) from peak 2 (--) and 3 (--), in comparison with a reference culture (--) with buffer instead of fraction. Unpublished results.
Further, concentrated CM was fractionated on a size exclusion column. The elution profile
(fig. 8a) revealed two major peaks, which exclusively contained endogenous Sf9 proteins, at
molecular sizes corresponding to 10 kDa and 50 kDa. Addition of peak fractions to new
cultures revealed that the 10 kDa peak contained a factor inhibitory for cell growth, and that
fractions from the 50 kDa peak stimulated proliferation (fig. 8b). A protein in the 10 kDa
fraction was preliminary identified as a truncated variant of histone H4, and was suspected to
be the source of inhibition, since other histone proteins have been shown to exert a cytotoxic
activity 174-177. Attempts to purify and further characterize this histone were unsuccessful due
to inconsistency of the experimental results. The 50 kDa peak included proteins with a
molecular size of ∼65-25 kDa. The positive effects from this peak could possibly originate
from a 44 kDa protein, which was shown to enhance Sf9 proliferation (V). This protein will
be further discussed in sections 6.2, and 6.2.2. However, it should be emphasized that the
fractions consisted of many different proteins, and the proliferative effects might have other
explanations.
5.4 The culture age effect
During routine passaging, a part of the old culture broth was transferred to fresh medium. At
30-40 passages (p) after thawing a new ampoule from the frozen stock, we observed a switch
in the proliferation characteristics of Sf9 cultures. The lag phase was decreased and µN,max was
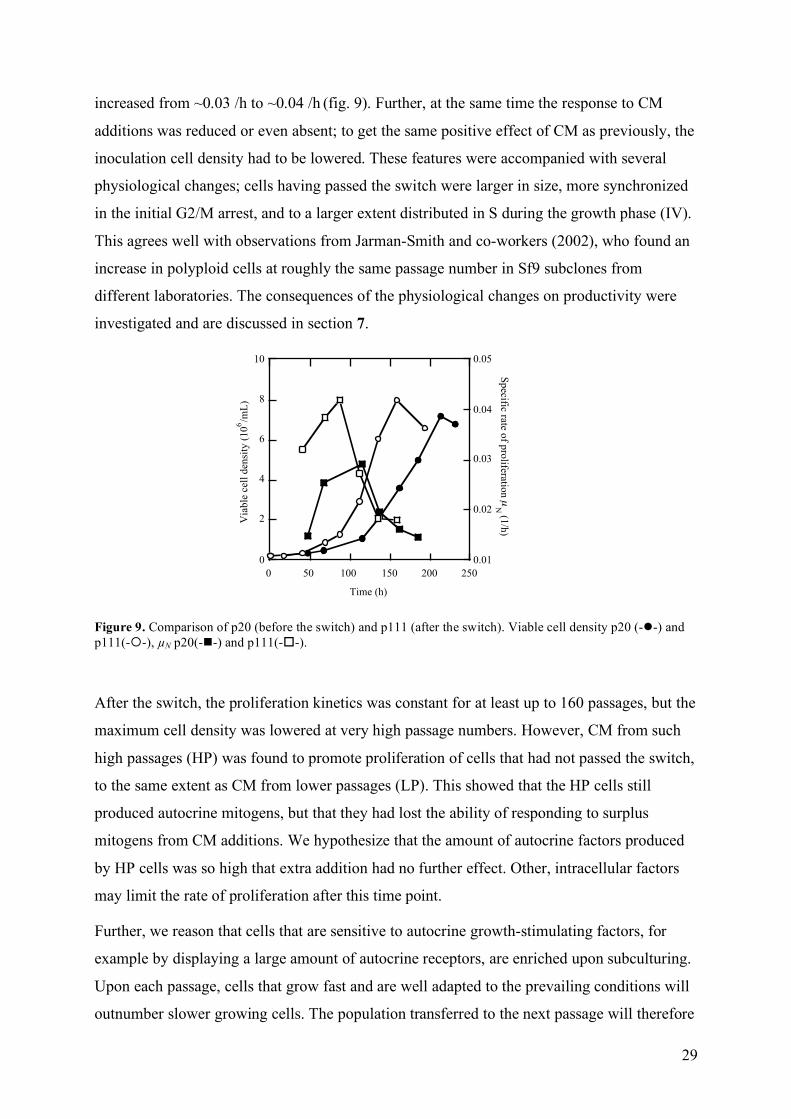
29
increased from ~0.03 /h to ~0.04 /h (fig. 9). Further, at the same time the response to CM
additions was reduced or even absent; to get the same positive effect of CM as previously, the
inoculation cell density had to be lowered. These features were accompanied with several
physiological changes; cells having passed the switch were larger in size, more synchronized
in the initial G2/M arrest, and to a larger extent distributed in S during the growth phase (IV).
This agrees well with observations from Jarman-Smith and co-workers (2002), who found an
increase in polyploid cells at roughly the same passage number in Sf9 subclones from
different laboratories. The consequences of the physiological changes on productivity were
investigated and are discussed in section 7.
0
2
4
6
8
10
0.01
0.02
0.03
0.04
0.05
0 50 100 150 200 250
Time (h)
Via
ble
cel
l den
sity
(10
6/m
L)
Specific rate o
f pro
liferation µ
N (1
/h)
Figure 9. Comparison of p20 (before the switch) and p111 (after the switch). Viable cell density p20 (--) and p111(--), µN p20(--) and p111(--).
After the switch, the proliferation kinetics was constant for at least up to 160 passages, but the
maximum cell density was lowered at very high passage numbers. However, CM from such
high passages (HP) was found to promote proliferation of cells that had not passed the switch,
to the same extent as CM from lower passages (LP). This showed that the HP cells still
produced autocrine mitogens, but that they had lost the ability of responding to surplus
mitogens from CM additions. We hypothesize that the amount of autocrine factors produced
by HP cells was so high that extra addition had no further effect. Other, intracellular factors
may limit the rate of proliferation after this time point.
Further, we reason that cells that are sensitive to autocrine growth-stimulating factors, for
example by displaying a large amount of autocrine receptors, are enriched upon subculturing.
Upon each passage, cells that grow fast and are well adapted to the prevailing conditions will
outnumber slower growing cells. The population transferred to the next passage will therefore

30
be progressively more enriched in fast-growing cells, and after a certain amount of passages
this results in altered culture characteristics. It is well known that long-term passaging affects
both mammalian and insect cells with respect to size, shape, DNA content, proliferation
kinetics and ability to express recombinant protein 125, 153, 161. Hence, a number of
contradictory results in the literature with respect to e.g. oxygen uptake, MOI, product yields
and nutrient consumption are likely due to the physiological heterogeneity of subclones of the
Sf9 cell line, with different origin and culture age. For example, polyploid Sf9 cells were
shown to have a higher oxygen consumption rate than diploid Sf9 cells, and to consume twice
as much glucose as diploid cells upon infection 162. Comparisons and generalizations of data
should therefore be done with care.
6 Conditioned medium factors
6.1 Antimicrobial activity
During our attempts to identify factors that affected proliferation, we observed by coincidence
that CM from Sf9 cultures exhibited antimicrobial activity. The first observation was made
after fractionating CM with a size exclusion column. It appeared that all CM fractions but one
were heavily infected when left during non-sterile conditions at room temperature. The non-
infected fraction contained proteins at ~10 kDa. The second observation was that Sf9 cultures
were capable of recovering from bacterial infections. At one occasion, the presence of
bacillus-shaped bacteria was unquestionable at one culture day, but the next morning no
infectious organism could be detected. It appeared as the cells had fought off the bacteria
themselves.
Results from experiments performed under more controlled forms confirmed the preliminary
observations. A size exclusion fraction containing proteins at 9-25 kDa exhibited strong
antimicrobial activity against the Gram-negative Escherichia coli and the Gram-positive
Bacillus megaterium (II). The toxic effect on B. megaterium was extremely potent; after 8
min incubation in the specific CM fraction, close to 100% of the B. megaterium cells were
dead (fig. 10). E. coli were more resistant; after 60 min incubation together with the CM
fraction 65% of the original population had died. Microscopic analyses revealed odd-shaped
and ghost-like bacteria upon exposure to the fraction, and the presence of protein bands with
bacterial origin within the incubation media showed that cell lysis occurred. Many known
antimicrobial toxins act by forming holes in the cell membrane, and it is likely that the Sf9
toxin works in a similar way. This would also explain the better resistance of the Gram-

31
negative E. coli, since it has an extra, protective outer cell membrane serving as a first defence
barrier.
Insects are well known for their ability to fight off microbial infections. Often, this involves
expression of small, broad-spectrum antimicrobial peptides, such as cecropins 178, attacins 179
and defensins 180. Similar peptides have also been identified from other species, such as frogs
or fish 181, 182. The interest for using such small-molecule compounds as, for example, human
antibiotics and anticancer drugs, has increased lately 183.
0
5 107
1 108
1.5 108
2 108
0 20 40 60 80 100
Time (min)
Via
ble
count
(cfu
/mL
)
0
2
4
6
8
10
0 20 40 60 80 100
Via
ble
count
(log c
fu/m
L)
Time (min)
a b
Figure 10. Viability of B. megaterium incubated with (--) and without (--) the CM fraction from an Sf9 culture. a) Linear scale, b) log scale. Cfu denotes colony-forming units.
Due to sequence homology with known proteins from other species, four proteins in the
antibacterial 10 kDa fraction were identified by N-terminal amino acid sequencing and mass
spectrometry (MS); ubiquitin, a thioredoxin-like protein and two cyclophilin-like proteins
(table 2). The only one of these four proteins with known antimicrobial activity is ubiquitin 184, but ubiquitin in the range 0.05-80 µm had no effect on bacterial survival during our assay
conditions. A fifth protein was also present in the antimicrobial fraction, but amino acid
sequences from this protein did not generate any matches in database searches. Previously, a
truncated variant of histone H4 (11.3 kDa) was identified in 10 kDa size exclusion fractions
(section 5.3), but no similarity between the fifth protein and histone H4 was detected. Since
none of the four identified proteins in the antimicrobial fraction could be linked to the
cytotoxic effect, our conclusion was that it possibly originated from the fifth, unknown
protein.

32
Table 2. Identification of protein bands in the antimicrobial Sf9 10 kDa CM size exclusion fraction.
MW protein band (kDa)a
Sequences obtained by mass spectrometry
Database matches
9.6 TITLEVEPSDTIENVK
ESTLHLVLR
FAGKQLEDGR
Ubiquitinb (many species)
11.9 IHIKDSDDLKNR
KLEEFSGANVDKLR
Thioredoxin-like protein (Manduca Sexta)
18.4 VFFDVSADGSALGR Cyclophilin like protein (many species)
26.5 ILEGMDVVR
FEDENFKLR
Cyclophilin like proteinb (many species)
a Molecular masses estimated from SDS-PAGE b Also confirmed by N-terminal amino acid sequencing
Size exclusion fractions from High five cultures eluting at about 10 kDa also exerted
antimicrobial activity: 97% of a B. megaterium population died after 40 min exposure to the
fraction. This effect most probably originated from a 16.3 kDa lysozyme precursor protein,
identified by sequence homology. Lysozyme exerts hydrolytic activity against β(1–4)
glycosidic linkages in the bacterial peptidoglycan, and has been reported to have an
antibacterial activity independent of the enzymatic activity. However, the activity spectrum of
lysozyme seems to be limited to specific gram-positive bacteria 185.
6.2 Peptidases
Peptidases are important components of many autocrine systems, where their most common
task is to activate and/or liberate hormone precursors. Previously, a metalloproteinase was
identified in High five CM 114. Inhibition of this metalloproteinase totally hampered the
positive effect of CM on cell proliferation, and it was therefore concluded to be the main
cause of the High five CM effect.
Sf9 peptidase activity has previously been found intracellularly 110-112, 186, and in cell lysates
after infection with baculoviruses 101, 109, 112, 115, 116, 121, 186-188. Gotoh et al. (2001a) found
endogenous peptidase activities in the medium during the Sf9 death phase, but reports about
true extracellular Sf9 peptidases were scarce. We therefore wanted to investigate Sf9 CM for
the presence of endogenous peptidases, and in that case study if they were involved in
controlling proliferation.

33
6.2.1 Identification of Sf9 and High five cathepsin L
Initial analysis of proteolysis in Sf9 CM was performed with zymography. No caseinolytic
activity was found, but two gelatinolytic bands, a strong 39 kDa and a fainter, approximately
49 kDa, were identified (fig 11, III). The molecular weight of the 49 kDa band was not
constant; during sample treatment it migrated to lower molecular weights, and in some
samples it was not visible at all. We therefore suspected that the two bands were precursor
form and active form of the same peptidase.
0
2
4
6
8
10
0 50 100 150
Via
ble
cel
l den
sity
(10
6/m
L)
Time (h)
1 23
4
5
6
7
Figure 11. Peptidase activity during the course of an Sf9 culture. a) Batch of Sf9 cells. b) Gelatine zymogram with samples from a. Arrows indicate the 49 kDa band.
Inhibition profiling revealed that the gelatinolytic peptidase was inhibited by the cysteine
proteinase inhibitor E-64 and the serine proteinase inhibitor PMSF, and therefore belonged to
the papain family of cysteine proteinases. To facilitate identification and quantification,
further enzymatic analyses were performed with fluorometry instead of zymography.
Peptidase activity was recorded on the flurophore Z-Phe-Arg-AMC, a substrate for kallikrein,
cathepsins B and L, papain and soybean trypsin-like enzyme, but not on Bz-Arg-AMC, a
substrate for papain, soybean trypsin-like enzyme and trypsin. In summary this showed that
the Sf9 peptidase was a homologue of either cathepsin B or L. The activity was completely
repressed by the cathepsin L-specific inhibitor Z-Phe-Tyr-t(Bu)-DMK 189, and the 39 kDa
peptidase was therefore finally identified as an Sf9 homologue of cathepsin L. Cathepsin L
(cL) is a lysosomal peptidase, which is synthesised as a preproenzyme 190. The proform can
either be transported out of the cell or be stored intracellularly until it is activated, by for
example the low pH in the lysosomes. Most probably the 49 kDa protein band in the
zymogram represented the proform of Sf9 cL. Procathepsin L (pro-cL) from other species was
a
1 2 3 4 5 6 7
116 80
52
35
30 22
MW
kDa
b

34
shown to have one-tenth or lower activity than mature cL 191, 192, but the activity might be
restricted to low-molecular weight compounds 193. Therefore autoactivation of the proform
may have occurred during sample preparation and electrophoresis, as observed elsewhere 187,
194.
Further, an additional, 22 kDa form of Sf9 cL could be evoked by incubation of CM samples
at pH 3.5 (III). Substrate specificity, inhibitor susceptibility and pH optimum (5.0) was the
same for the 22 kDa form as for the other active form. Therefore, it appears that Sf9 cL
possesses two active forms: one at 22 kDa and one at 39 kDa. The activity of Sf9 cL was
measured during an Sf9 cultivation and found to be low in the beginning, but increase
throughout the culture. Inhibition of the enzyme with the non membrane-permeable E-64
analogue E-64c added directly to the culture had no effect on cell proliferation (III).
A homologue of cL/pro-cL was likewise discovered in CM from High five cells (V). The
molecular weight of this enzyme was 44 kDa, as estimated from bands in zymography gels,
and it was occasionally accompanied by another band at 37.5-42 kDa. The two bands could
either be precursor form and active form, similar to the situation in Sf9 cultures, or different
glycoforms of the same protein. However, no active cL was found in High five CM. In
addition, the N-terminal sequence from a 44 kDa High five protein (VSFFDLVRE) was found
to be 88% similar to a sequence from the propeptide of Tenebrio molitor cL (VSFFDLVQE) 195, and also similar to the N-terminal of pro-cL from Sf21 (VSLLDLVRE) 192. The two
positions that differ between Sf21 pro-cL and the High five sequence are occupied by neutral
amino acids: leucin in Sf21 and phenylalanin in High five. The conclusion was therefore that
the 44 kDa High five band most likely represented High five pro-cL, and that the band with
lower molecular weight may represent an ongoing transformation to the fully active form.
Pro-cL is autocatalytically converted to mature cL during incubation at low pH 190, 196, 197, a
feature that was used to indirectly measure the amount of pro-cL during Sf9 and High five
cultures (fig. 12, V). This revealed a low quantity of pro-cL during the growth phase in High
five cultures, but a drastic augmentation in the very end during cell lysis. In Sf9 cultures the
situation was different; an increase in activity from pro-cL occurred at approximately 100 h,
but when cell proliferation had ceased, the amount of pro-cL decreased again. These results
may indicate that the extracellular pro-cL in Sf9 cultures is actively transported to the medium
from viable cells. The presence of pro-cL in High five cultures remains unexplained, but at
least in the end it is seemingly caused by cell lysis.
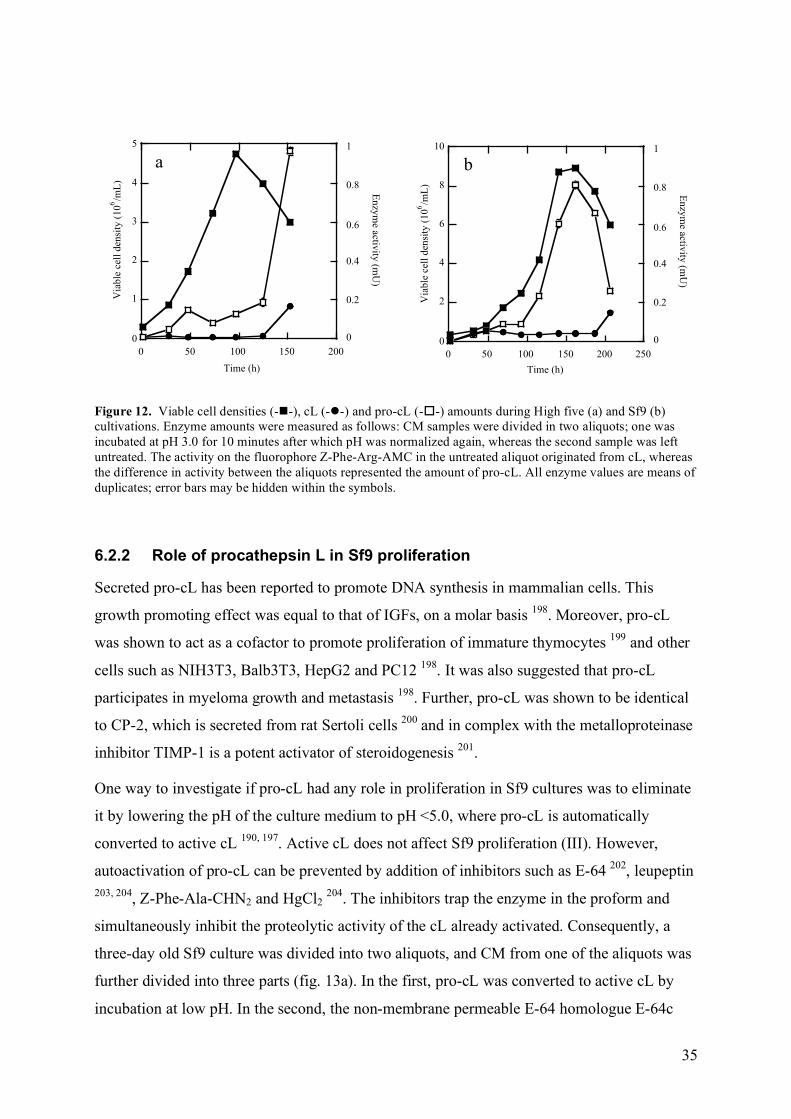
35
0
2
4
6
8
10
0 50 100 150 200 250
Via
ble
cel
l d
ensi
ty (
10
6/m
L)
En
zym
e activity
(mU
)
Time (h)
0.8
0.6
0.4
0.2
0
1
0
1
2
3
4
5
0 50 100 150 200
En
zym
e activity
(mU
)
Time (h)
0.8
0.6
0.4
0.2
0
1
Via
ble
cel
l d
ensi
ty (
10
6/m
L)
a b
Figure 12. Viable cell densities (--), cL (--) and pro-cL (--) amounts during High five (a) and Sf9 (b) cultivations. Enzyme amounts were measured as follows: CM samples were divided in two aliquots; one was incubated at pH 3.0 for 10 minutes after which pH was normalized again, whereas the second sample was left untreated. The activity on the fluorophore Z-Phe-Arg-AMC in the untreated aliquot originated from cL, whereas the difference in activity between the aliquots represented the amount of pro-cL. All enzyme values are means of duplicates; error bars may be hidden within the symbols.
6.2.2 Role of procathepsin L in Sf9 proliferation
Secreted pro-cL has been reported to promote DNA synthesis in mammalian cells. This
growth promoting effect was equal to that of IGFs, on a molar basis 198. Moreover, pro-cL
was shown to act as a cofactor to promote proliferation of immature thymocytes 199 and other
cells such as NIH3T3, Balb3T3, HepG2 and PC12 198. It was also suggested that pro-cL
participates in myeloma growth and metastasis 198. Further, pro-cL was shown to be identical
to CP-2, which is secreted from rat Sertoli cells 200 and in complex with the metalloproteinase
inhibitor TIMP-1 is a potent activator of steroidogenesis 201.
One way to investigate if pro-cL had any role in proliferation in Sf9 cultures was to eliminate
it by lowering the pH of the culture medium to pH <5.0, where pro-cL is automatically
converted to active cL 190, 197. Active cL does not affect Sf9 proliferation (III). However,
autoactivation of pro-cL can be prevented by addition of inhibitors such as E-64 202, leupeptin 203, 204, Z-Phe-Ala-CHN2 and HgCl2 204. The inhibitors trap the enzyme in the proform and
simultaneously inhibit the proteolytic activity of the cL already activated. Consequently, a
three-day old Sf9 culture was divided into two aliquots, and CM from one of the aliquots was
further divided into three parts (fig. 13a). In the first, pro-cL was converted to active cL by
incubation at low pH. In the second, the non-membrane permeable E-64 homologue E-64c
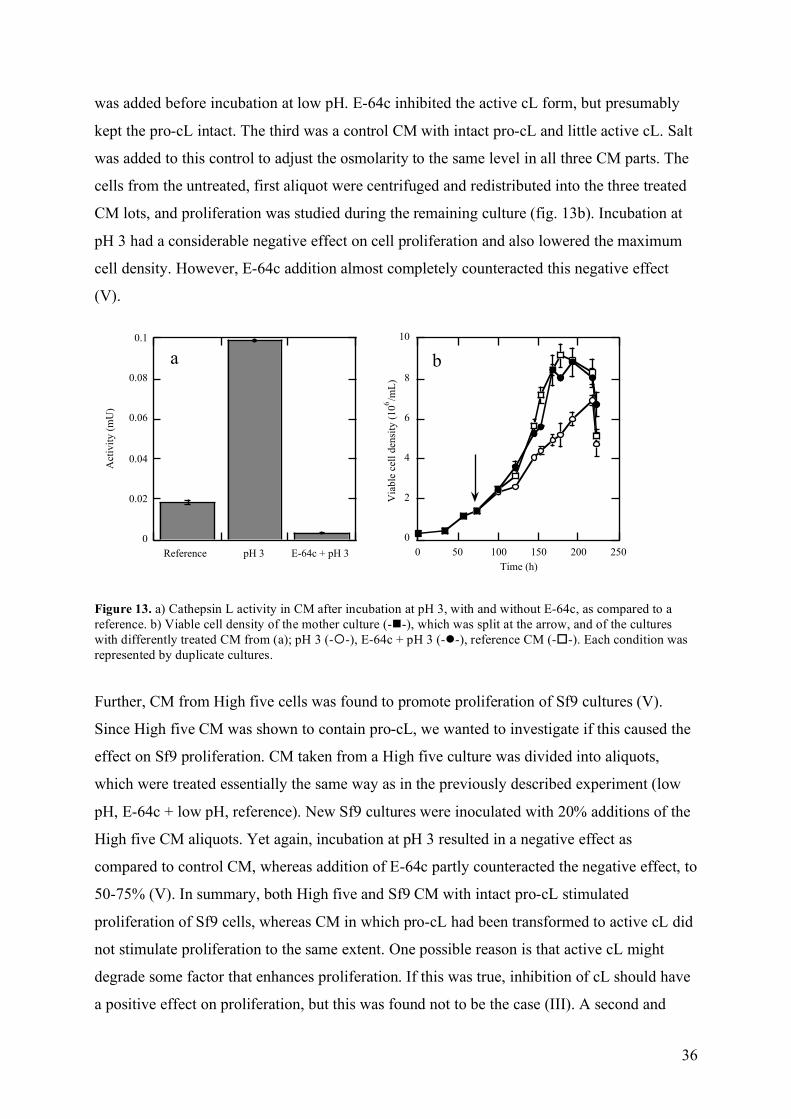
36
was added before incubation at low pH. E-64c inhibited the active cL form, but presumably
kept the pro-cL intact. The third was a control CM with intact pro-cL and little active cL. Salt
was added to this control to adjust the osmolarity to the same level in all three CM parts. The
cells from the untreated, first aliquot were centrifuged and redistributed into the three treated
CM lots, and proliferation was studied during the remaining culture (fig. 13b). Incubation at
pH 3 had a considerable negative effect on cell proliferation and also lowered the maximum
cell density. However, E-64c addition almost completely counteracted this negative effect
(V).
0
2
4
6
8
10
0 50 100 150 200 250
Via
ble
cel
l den
sity
(10
6/m
L)
Time (h)
0
0.02
0.04
0.06
0.08
0.1
Reference pH 3 E-64c + pH 3
Act
ivit
y (
mU
)
a b
Figure 13. a) Cathepsin L activity in CM after incubation at pH 3, with and without E-64c, as compared to a reference. b) Viable cell density of the mother culture (--), which was split at the arrow, and of the cultures with differently treated CM from (a); pH 3 (--), E-64c + pH 3 (--), reference CM (--). Each condition was represented by duplicate cultures.
Further, CM from High five cells was found to promote proliferation of Sf9 cultures (V).
Since High five CM was shown to contain pro-cL, we wanted to investigate if this caused the
effect on Sf9 proliferation. CM taken from a High five culture was divided into aliquots,
which were treated essentially the same way as in the previously described experiment (low
pH, E-64c + low pH, reference). New Sf9 cultures were inoculated with 20% additions of the
High five CM aliquots. Yet again, incubation at pH 3 resulted in a negative effect as
compared to control CM, whereas addition of E-64c partly counteracted the negative effect, to
50-75% (V). In summary, both High five and Sf9 CM with intact pro-cL stimulated
proliferation of Sf9 cells, whereas CM in which pro-cL had been transformed to active cL did
not stimulate proliferation to the same extent. One possible reason is that active cL might
degrade some factor that enhances proliferation. If this was true, inhibition of cL should have
a positive effect on proliferation, but this was found not to be the case (III). A second and

37
more plausible explanation is that the pro-cL itself has direct or indirect mitogenic effects in
Sf9 cultures.
Growth factors can be divided into different categories: competence factors such as PDGF,
which make the cells receptive for stimulation, and progression factors including insulin and
IGFs, which stimulate competent cells directly and induce mitogenesis. Pro-cL was suggested
to act on mammalian cells as a progression factor 198, since pro-cL alone had no mitogenic
effect, but needed previous stimulation by priming growth factors such as IL-1 199. This might
also be true for Sf9 cultures, since the small factor, which passes 10 kDa filters, exhibits a
positive effect on Sf9 proliferation (I, III, section 5.3). The small factor and pro-cL may act
together in regulating Sf9 proliferation, either cooperatively or as competence and progression
factors.
7 Protein production
An abrupt drop in productivity in the insect cell/BEVS has been associated with a critical cell
density at the time of infection (section 4.3.3). However, it is not the cell density per se that is
important, but rather the physiological state of the cells at the time of infection 165. Renewal of
medium at infection is, according to the literature, known as the best way of improving the
productivity in laboratory scale, and nutrient feed strategies have also generated positive
effects (see references in 4.3.3). However, despite tremendous efforts to increase growth and
productivity in the insect cell/BEVS, the underlying cause of the cell-density dependent
productivity drop remains unknown.
To characterize the productivity of Sf9 cultures, we investigated changes in recombinant β-
galactosidase production throughout all culture stages (I). The product yield in old medium
was compared to the yield from cells that were resuspended in fresh medium prior to infection
(fig. 14, I). During the initial 75 h, volumetric production increased linearly, but the yield in
renewed medium was approximately 12% lower than the yield in spent medium. The
maximum volumetric yield (~600 U/mL) in old medium occurred at a cell density of 2.6-
3.6·106 /mL, but after this time point the productivity decreased drastically to levels below the
detection limit. The productivity in renewed medium could be sustained until 117 h (6.8·106
cells/mL), when a final drop occurred. However, during this extended production phase (75-
117 h) the volumetric yield did not increase by more than 10%, to 660 U/mL. The maximum
specific yield occurred for both conditions at 43 h culture time (1.6·106 cells/mL), which
coincided with µN, max.
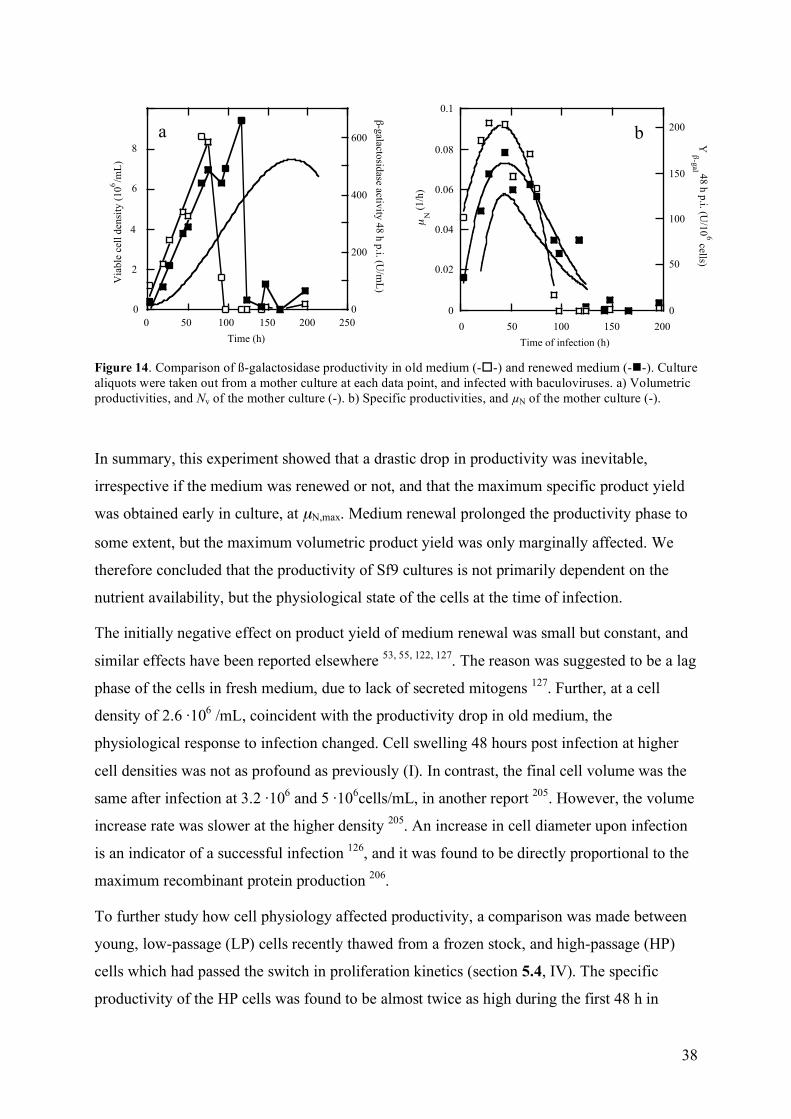
38
0
200
400
600
0 50 100 150 200 250
Via
ble
cel
l d
ensi
ty (
10
6/m
L)
!-g
alactosid
ase activity
48
h p
.i. (U/m
L)
Time (h)
8
6
4
2
0
a b
0
0.02
0.04
0.06
0.08
0.1
0 50 100 150 200
0
50
100
150
200
Y !
-gal 4
8 h
p.i. (U
/10
6 cells)
µN
(1
/h)
Time of infection (h)
b
Figure 14. Comparison of ß-galactosidase productivity in old medium (--) and renewed medium (--). Culture aliquots were taken out from a mother culture at each data point, and infected with baculoviruses. a) Volumetric productivities, and Nv of the mother culture (-). b) Specific productivities, and µN of the mother culture (-).
In summary, this experiment showed that a drastic drop in productivity was inevitable,
irrespective if the medium was renewed or not, and that the maximum specific product yield
was obtained early in culture, at µN,max. Medium renewal prolonged the productivity phase to
some extent, but the maximum volumetric product yield was only marginally affected. We
therefore concluded that the productivity of Sf9 cultures is not primarily dependent on the
nutrient availability, but the physiological state of the cells at the time of infection.
The initially negative effect on product yield of medium renewal was small but constant, and
similar effects have been reported elsewhere 53, 55, 122, 127. The reason was suggested to be a lag
phase of the cells in fresh medium, due to lack of secreted mitogens 127. Further, at a cell
density of 2.6 ·106 /mL, coincident with the productivity drop in old medium, the
physiological response to infection changed. Cell swelling 48 hours post infection at higher
cell densities was not as profound as previously (I). In contrast, the final cell volume was the
same after infection at 3.2 ·106 and 5 ·106cells/mL, in another report 205. However, the volume
increase rate was slower at the higher density 205. An increase in cell diameter upon infection
is an indicator of a successful infection 126, and it was found to be directly proportional to the
maximum recombinant protein production 206.
To further study how cell physiology affected productivity, a comparison was made between
young, low-passage (LP) cells recently thawed from a frozen stock, and high-passage (HP)
cells which had passed the switch in proliferation kinetics (section 5.4, IV). The specific
productivity of the HP cells was found to be almost twice as high during the first 48 h in
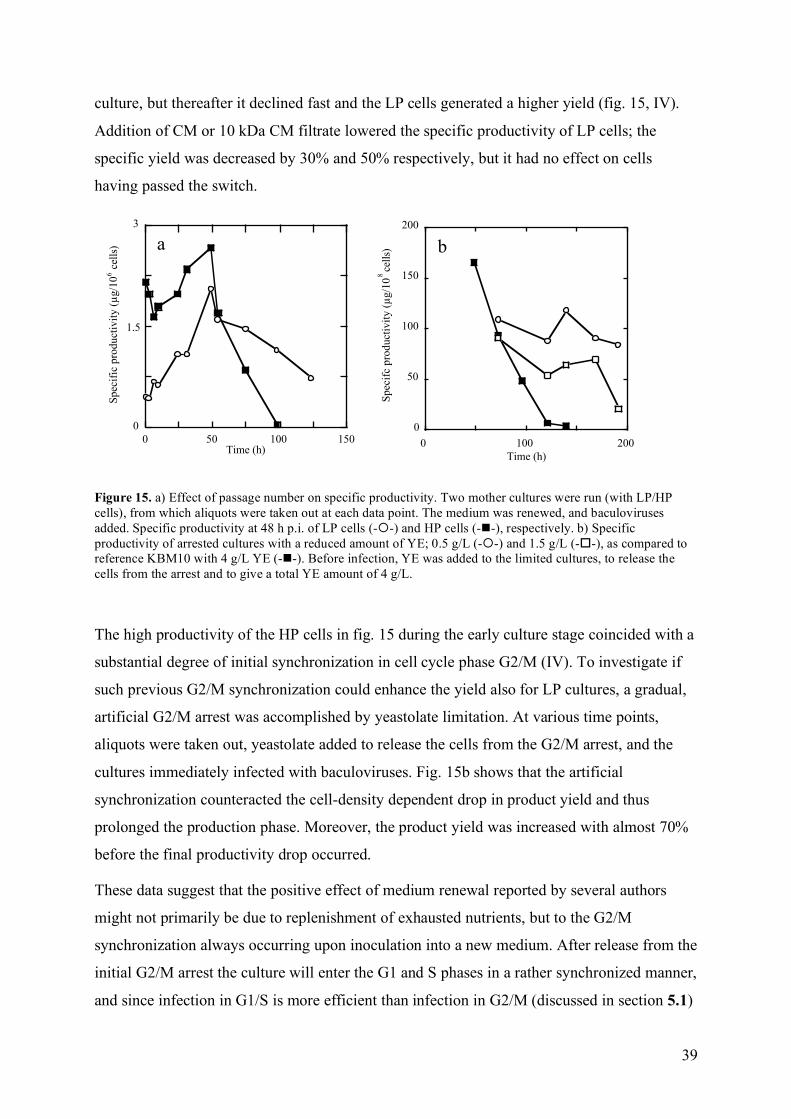
39
culture, but thereafter it declined fast and the LP cells generated a higher yield (fig. 15, IV).
Addition of CM or 10 kDa CM filtrate lowered the specific productivity of LP cells; the
specific yield was decreased by 30% and 50% respectively, but it had no effect on cells
having passed the switch.
0
50
100
150
200
0 100 200
Sp
ecif
c p
rod
uct
ivit
y (!
g/1
08
cell
s)
Time (h)
0
0 50 100 150
Spec
ific
pro
duct
ivit
y (!
g/1
06 c
ells
)
Time (h)
1.5
3
a b
Figure 15. a) Effect of passage number on specific productivity. Two mother cultures were run (with LP/HP cells), from which aliquots were taken out at each data point. The medium was renewed, and baculoviruses added. Specific productivity at 48 h p.i. of LP cells (--) and HP cells (--), respectively. b) Specific productivity of arrested cultures with a reduced amount of YE; 0.5 g/L (--) and 1.5 g/L (--), as compared to reference KBM10 with 4 g/L YE (--). Before infection, YE was added to the limited cultures, to release the cells from the arrest and to give a total YE amount of 4 g/L.
The high productivity of the HP cells in fig. 15 during the early culture stage coincided with a
substantial degree of initial synchronization in cell cycle phase G2/M (IV). To investigate if
such previous G2/M synchronization could enhance the yield also for LP cultures, a gradual,
artificial G2/M arrest was accomplished by yeastolate limitation. At various time points,
aliquots were taken out, yeastolate added to release the cells from the G2/M arrest, and the
cultures immediately infected with baculoviruses. Fig. 15b shows that the artificial
synchronization counteracted the cell-density dependent drop in product yield and thus
prolonged the production phase. Moreover, the product yield was increased with almost 70%
before the final productivity drop occurred.
These data suggest that the positive effect of medium renewal reported by several authors
might not primarily be due to replenishment of exhausted nutrients, but to the G2/M
synchronization always occurring upon inoculation into a new medium. After release from the
initial G2/M arrest the culture will enter the G1 and S phases in a rather synchronized manner,
and since infection in G1/S is more efficient than infection in G2/M (discussed in section 5.1)

40
the above situation should be optimal for recombinant protein production. The
synchronization will gradually be lost after the initial release from the lag-phase, and such a
simultaneous entry into G1 and S will not take place. The mean specific productivity should
therefore be highest directly after the lag-phase and thereafter inevitably decrease, which is
also confirmed in fig. 14b. These results also explain why medium renewal at infection early
in culture does not have any positive effect on protein production; the culture will already be
synchronous, and as such maximally productive. However, it should be emphasized that if a
medium with an unsatisfactory nutritional level is used, addition of depleted components will
of course be beneficial.

41
IV Conclusions
Physiology
• Proliferation and physiology of Sf9 insect cells was controlled by an autocrine system
consisting of several different factors, amongst them one low-molecular mass (<10 kDa)
and one higher molecular mass (>10 kDa). Yeastolate was a prerequisite for the
production of the smaller factor.
• Sf9 cell cycle progression during a batch culture was characterized by a G2/M arrest
during the lag phase, and a final G2/M arrest when proliferation ceased. The initial arrest
was shortened by autocrine conditioned medium (CM) factors, but the final arrest was
inevitable.
• Extensive changes in Sf9 physiology and proliferation kinetics occurred at certain passage
numbers after thawing a new vial. The cells appeared larger in size, displayed a shorter lag
phase, an increased µN,max and an increased initial G2/M arrest. Further, the response for
CM additions was weaker.
• Two novel, endogenous and extracellular insect peptidases were identified in Sf9 and
High five CM; proforms and active forms of cathepsin L. Cathepsin L activity was low at
culture pH, but increased at a decreased pH.
• Removal of procathepsin L from Sf9 CM had a negative effect on Sf9 proliferation. Thus,
procathepsin L may constitute the >10 kDa autocrine CM factor.
• Sf9 and High five cultures expressed antimicrobial factors. The High five antimicrobial
substance was most likely lysozyme, whilst the Sf9 factor remains unidentified.
Continued on next page.

42
Conclusions
Productivity
• Medium renewal at infection with baculoviruses had a limited effect on the maximal
product concentration. The physiological state of the cells was more important.
• The cell-density dependent drop in productivity was, at least partly, caused by a decreased
synchronization of the culture, leading to a decreased amount of cells in a beneficial cell
cycle phase at infection.
• The productivity drop was counteracted by an artificial G2/M synchronization before
infection, which lead to a prolonged production phase and an augmented maximum
volumetric product yield.

43
V References 1. Puck, T.T., Cieciura, S.J. & Robinson, A. Genetics of somatic mammalian cells: III. Long-term
cultivation of euploid cells from human and animal subjects. J. Exp. Med. 108, 945-956 (1958). 2. Macpherson, I. & Stoker, M. Polyoma transformation of hamster cell clones--an investigation of genetic
factors affecting cell competence. Virology 16, 147-151 (1962). 3. Todaro, G.J. & Green, H. Quantitative studies of the growth of mouse embryo cells in culture and their
development into established lines. J. Cell Biol. 17, 299-313 (1963). 4. Eagle, H. Nutrition needs of mammalian cells in tissue culture. Science 122, 501-504 (1955). 5. Johnson, J.S. Human insulin from recombinant DNA technology. Science 219, 632-637 (1983). 6. Wurm, F.M. Production of recombinant protein therapeutics in cultivated mammalian cells. Nature
Biotechnol. 22, 1393-1398 (2004). 7. Walsh, G. Biopharmaceutical benchmarks - 2003. Nature Biotechnol. 21, 865-870 (2003). 8. Henry, C.M. The next pharmaceutical century. Chem. Eng. News, 85-100 (2000). 9. Freshney, R.I. Culture of animal cells: a manual of basic techniques, Edn. 5. (John Wiley & Sons, Inc.,
Hoboken, New Jersey; 2005). 10. Molowa, D.T. & Mazanet, R. The state of biopharmaceutical manufacturing. Biotechnol. Ann. Rev. 9,
285-302 (2003). 11. Phillips, P.D. & Cristofalo, V.J. Classification system based on the functional equivalency of mitogens
that regulate WI-38 cell proliferation. Exp. Cell. Res. 175, 396-403 (1988). 12. Aaronson, S.A., Bottaro, D.P., Miki, T., Ron, D. & Finch, P.W. Keratinocyte growth factor: a fibroblast
growth factor family member with an unusual target cell specificity. Ann. NY Acad. Sci. 638, 62-77 (1991).
13. Sporn, M.B. & Todaro, G.J. Autocrine secretion and malignant transformation of cells. New Eng. J. Med. 303, 878-880 (1980).
14. Sporn, M.B. & Roberts, A.B. Autocrine secretion--10 years later. Ann. Intern. Med. 117, 408-414 (1992).
15. Jones, J.I. & Clemmons, D.R. Insulin-like growth factors and their binding proteins: biological actions. Endocr. Rev. 16, 3-34 (1995).
16. Lauffenburger, D.A., Oehrtman, G.T., Walker, L. & Wiley, H.S. Real-time quantitative measurement of autocrine ligand binding indicates that autocrine loops are spatially localized. PNAS 95, 15368-15373 (1998).
17. DeWitt, A.E., Dong, J.Y., Wiley, H.S. & Lauffenburger, D.A. Quantitative analysis of the EGF receptor autocrine system reveals cryptic regulation of cell response by ligand capture. J. Cell Sci. 114, 2301-2313 (2001).
18. Casci, T. & Freeman, M. Control of EGF receptor signalling: lessons from fruit flies. Can. Metast. Rev. 18, 181-201 (1999).
19. Goldring, S.R. & Goldring, M.B. Cytokines and skeletal physiology. Clin. Orthop. 323, 13-23 (1996). 20. Tokumaru, S. et al. Ectodomain shedding of epidermal growth factor receptor ligands is required for
keratinocyte migration in cutaneous wound healing. J. Cell Biol. 151, 209-220 (2000). 21. Massague, J. & Pandiella, A. Membrane-anchored growth-factors. Ann. Rev. Biochem. 62, 515-554
(1993). 22. Dong, J. et al. Metalloprotease-mediated ligand release regulates autocrine signaling through the
epidermal growth factor receptor. PNAS 96, 6235-6240 (1999). 23. Klambt, C. EGF receptor signalling: roles of star and rhomboid revealed. Curr. Biol. 12, R21-23
(2002). 24. Al-Rubeai, M. & Singh, R.P. Apoptosis in cell culture. Curr. Opin. Biotechnol. 9, 152-156 (1998). 25. Trager, W. Cultivation of the virus of Grasserie in silkworm tissue cultures. J. Exp. Med. 61, 501-514
(1935). 26. Grace, T.D. Establishment of four strains of cells from insect tissues grown in vitro. Nature 195, 788-
789 (1962). 27. Smith, G.E., Summers, M.D. & Fraser, M.J. Production of human beta interferon in insect cells infected
with a baculovirus expression vector. Mol. Cell. Biol. 3, 2156-2165 (1983a). 28. Hink, W.F. Established insect cell line from the cabbage looper, Trichoplusia ni. Nature 226, 466-467
(1970). 29. O'Reilley, D.R., Miller, L.K. & Luckow, V.A. Baculovirus expression vectors: a laboratory manual.
(Oxford University Press, New York; 1994).

44
30. Granados, R.R., Guoxun, L., Derksen, A.C.G. & McKenna, K.A. A new insect cell line from Trichoplusia ni (BTI-Tn-5B1-4) susceptible to Trichoplusia ni single enveloped nuclear polyhedrosis virus. J. Invertebr. Pathol. 64, 260-266 (1994).
31. Wickham, T.J., Davis, T., Granados, R.R., Shuler, M.L. & Wood, H.A. Screening of insect cell lines for the production of recombinant proteins and infectious virus in the baculovirus expression system. Biotechnol. Prog. 8, 391-396 (1992).
32. Davis, T.R. et al. Comparative recombinant protein production of eight insect cell lines. In Vitro Cell. Dev. Biol. Anim. 29A, 388-390 (1993).
33. Wickham, T.J. & Nemerow, G.R. Optimization of growth methods and recombinant protein production in BTI-Tn-5B1-4 insect cells using the baculovirus expression system. Biotechnol. Prog. 9, 25-30 (1993).
34. Saarinen, M.A., Troutner, K.A., Gladden, S.G., Mitchell-Logean, C.M. & Murhammer, D.W. Recombinant protein synthesis in Trichoplusia ni BTI-Tn-5B1-4 insect cell aggregates. Biotechnol. Bioeng. 63, 612-617 (1999).
35. Wang, P., Granados, R.R. & Shuler, M.L. Studies on serum-free culture of insect cells for virus propagation and recombinant protein production. J. Invertebr. Pathol. 59, 46-53 (1992).
36. Vaughn, J.L., Goodwin, R.H., Tompkins, G.L. & McCawley, P. The establishment of two cell lines derived from the insect Spodoptera frugiperda (Lepidoptera: Noctuidae). In Vitro 13, 213-217 (1977).
37. Hink, W.F., Thomsen, D.R., Davidson, D.J., Meyer, A.L. & Castellino, F.J. Expression of three recombinant proteins using baculovirus vectors in 23 insect cell lines. Biotechnol. Prog. 7, 9-14 (1991).
38. Jarvis, D.L., Kawar, Z.S. & Hollister, J.R. Engineering N-glycosylation pathways in the baculovirus-insect cell system. Curr. Opin. Biotechnol. 9, 528-533 (1998).
39. Schneider, I. Cell lines derived from late embryonic stages of Drosophila melanogaster. J. Embryol. Exp. Morphol. 27, 353-365 (1972).
40. Benting, J., Lecat, S., Zacchetti, D. & Simons, K. Protein expression in Drosophila Schneider cells. Anal. Biochem. 278, 59-68 (2000).
41. Gardiner, G.R. & Stockdale, H. Two tissue culture media for the production of lepidopteran cells and nuclear polyhedrosis viruses. J. Invertebr. Pathol. 25, 363-370 (1975).
42. Weiss, S.A., Smith, G.C., Kalter, S.S. & Vaughn, J.L. Improved method for the production of insect cell cultures in large volumes. In Vitro 17, 495-502 (1981).
43. Maiorella, B., Inlow, D., Shauger, A. & Harano, D. Large-scale insect cell-culture for recombinant protein production. Bio/Technology 6 (1988).
44. English, L.H. & Cantley, L.C. Characterization of monovalent ion transport systems in an insect cell line (Manduca sexta embryonic cell line che). J. Cell. Physiol. 121, 125-132 (1984).
45. Schlaeger, E.-J. Medium design for insect cell culture. Cytotechnol. 20, 57-70 (1996). 46. Schmid, G. Insect cell cultivation: growth and kinetics. Cytotechnol. 20, 43-56 (1996). 47. Wilkie, G.E.I., Stockdale, H. & Pirt, S.V. Chemically-defined media for production of insect cells and
viruses in vitro. Dev. Biol. Stand. 46, 29-37 (1980). 48. Weiss, S.A. et al. Growth of insect cells in a serum-free medium and production of recombinant
proteins using various bioreactors. In Vitro Cell. Dev. Biol. 26, 30A (1990). 49. Godwin, G., Danner, D. & Gorfien, S. Express Five TM SFM: a new serum-free medium for growth of
BTI-TN-5B1-4 cells and expression of recombinant proteins. Focus 17, 58-60 (1995). 50. Doverskog, M., Han, L. & Häggström, L. Cystine/cysteine metabolism in cultured Sf9 cells: influence
of cell physiology on biosynthesis, amino acid uptake and growth. Cytotechnol. 26, 91-102 (1998). 51. Öhman, L., Ljunggren, J. & Häggström, L. Induction of a metabolic switch in insect cells by substrate-
limited fed batch cultures. App. Microbiol. Biotechnol. 43, 1006-1013 (1995). 52. Caron, A.W., Archambault, J. & Massie, B. High-level recombinant protein production in bioreactors
using the baculovirus-insect cell expression system. Biotechnol. Bioeng. 36, 1133-1140 (1990). 53. Reuveny, S., Kim, Y.J., Kemp, C.W. & Schiloach, J. Production of recombinant proteins in high-
density insect cell cultures. Biotechnol. Bioeng. 42, 235-239 (1993b). 54. Nguyen, B., Jarnagin, K., Williams, S., Chan, H. & Barnett, J. Fed-batch culture of insect cells: a
method to increase the yield of recombinant human nerve growth factor (rhNGF) in the baculovirus expression system. J. Biotechnol. 31, 205-217 (1993).
55. Bédard, C., Kamen, A., Tom, R. & Massie, B. Maximization of recombinant protein yield in the insect cell/baculovirus system by one-time additions of nutrients to high-density batch cultures. Cytotechnol. 15, 129-138 (1994).
56. Drews, M., Paalme, T. & Vilu, R. The growth and nutrient utilization of the insect cell line Spodoptera frugiperda Sf9 in batch and continuous culture. J. Biotechnol. 40, 187-198 (1995).
57. Wu, J. & Lee, K.-l.D. Growth promotion by yeastolate and related components on insect cells. Biotechnol. Techn. 12, 67-70 (1998a).

45
58. Wu, J., Ruan, W. & Lam, H.Y.P. Evaluation of spent medium recycle and nutrient feeding strategies for recombinant protein production in the insect-cell baculovirus process. J. Biotechnol. 66, 109-116 (1998b).
59. Ferrance, J.P., Goel, A. & Ataai, M.M. Utilization of glucose and amino acids in insect cell cultures: quantifying the metabolic flows within the primary pathways and medium development. Biotechnol. Bioeng. 42, 697-707 (1993).
60. Ayres, M.D., Howard, S.C., Kuzio, J., Lopez-Ferber, M. & Possee, R.D. The complete DNA sequence of Autographa californica nuclear polyhedrosis virus. Virology 202, 586-605 (1994).
61. Smith, G.E., Fraser, M.J. & Summers, M.D. Molecular engineering of the Autographa californica nuclear polyhedrosis virus genome: deletion mutations within the polyhedrin gene. J. Virol. 46, 548-593 (1983b).
62. Kost, T.A., Condreay, J.P. & Jarvis, D.L. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nature Biotechnol. 23, 567-575 (2005).
63. Luckow, V.A., Lee, S.C., Barry, G.F. & Olins, P.O. Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli. J. Virol. 67, 4566-4579 (1993).
64. Hunt, I. From gene to protein: a review of new and enabling technologies for multi-parallel protein expression. Prot. Exp. Purif. 40, 1-22 (2005).
65. Kitts, P.A. & Green, G. An immunological assay for determination of baculovirus titers in 48 hours. Anal. Biochem. 268, 173-178 (1999).
66. Kwon, M.S., Dojima, T., Toriyama, M. & Park, E.Y. Development of an antibody-based assay for determination of baculovirus titers in 10 hours. Biotechnol. Prog. 18, 647-651 (2002).
67. Mulvania, T., Hayes, B. & Hedin, D. A flow cytometric assay for rapid, accurate determination of baculovirus titers. Bioproc. J. 3, 47-53 (2004).
68. Shen, C.F., Meghrous, J. & Kamen, A. Quantitation of baculovirus particles by flow cytometry. J. Virol. Meth. 105, 321-330 (2002).
69. Lo, H.R. & Chao, Y.C. Rapid titer determination of baculovirus by quantitative real-time polymerase chain reaction. Biotechnol. Prog. 20, 354-360 (2004).
70. Dee, K.U. & Shuler, M.L. Optimization of an assay for baculovirus titer and design of regimens for the synchronous infection of insect cells. Biotechnol. Prog. 13, 14-24 (1997).
71. Cox, M.M. Commercial production in insect cells: one company's perspective. Bioproc. Int., 2-5 (2004).
72. Wang, K. et al. Expression and purification of an influenza hemagglutinin--one step closer to a recombinant protein-based influenza vaccine. Vaccine 24, 2176-2185 (2006).
73. Hu, Y.C. et al. Dual expression of the HA protein of H5N2 avian influenza virus in a baculovirus system. J. Virol. Meth. In Press, Corrected Proof (2006).
74. Shuler, M.L., Wood, H.A., Granados, R.R. & Hammer, D.A. Baculovirus expression systems and biopesticides. (Wiley-Liss, Inc., New York; 1995).
75. Jarvis, D.L. Developing baculovirus-insect cell expression systems for humanized recombinant glycoprotein production. Virology 310, 1-7 (2003).
76. Kost, T.A. & Condreay, J.P. Recombinant baculoviruses as mammalian cell gene-delivery vectors. Trends Biotechnol. 20, 173-180 (2002).
77. Coligan, J.E., Dunn, B.M., Speicher, D.W. & Wingfield, P.T. Current protocols in protein science. (John Wiley & Sons, Inc., New York; 2000).
78. Colosimo, A. et al. Transfer and expression of foreign genes in mammalian cells. Biotechniques 29, 314-331 (2000).
79. Schmidt, F.R. Recombinant expression systems in the pharmaceutical industry. Appl. Microbiol. Biotechnol. 65, 363-372 (2004).
80. Luckow, V.A. & Summers, M.D. Trends in the development of baculovirus expression vectors. Bio/Technology 6, 47-55 (1988).
81. Miller, L.K. Baculoviruses as gene expression vectors. Ann. Rev. Microbiol. 42, 177-199 (1988). 82. Philipps, B., Forstner, M. & Mayr, L.M. A baculovirus expression vector system for simultaneous
protein expression in insect and mammalian cells. Biotechnol. Prog. 21, 708-711 (2005). 83. Reiter, M. & Bluml, G. Large-scale mammalian cell culture. Curr. Op. Biotechnol. 5, 175-179 (1994). 84. Ikonomou, L., Schneider, Y.J. & Agathos, S.N. Insect cell culture for industrial production of
recombinant proteins. App. Microbiol. Biotechnol. 62, 1-20 (2003). 85. Birch, J.R. & Froud, S. Mammalian cell culture systems for recombinant protein production.
Biologicals 22, 127-133 (1994). 86. Altmann, F., Staudacher, E., Wilson, I.B.H. & Marz, L. Insect cells as hosts for the expression of
recombinant glycoproteins. Glycoconj. J. 16, 109-123 (1999).

46
87. Marchal, I., Jarvis, D.L., Cacan, R. & Verbert, A. Glycoproteins from insect cells: sialylated or not? Biol. Chem. 382, 151-159 (2001).
88. Tomiya, N., Narang, S., Lee, Y.C. & Betenbaugh, M.J. Comparing N-glycan processing in mammalian cell lines to native and engineered lepidopteran cell lines. Glycoconj. J. 21, 343-360 (2004).
89. Joshi, L., Shuler, M.L. & Wood, H.A. Production of a sialylated N-Linked glycoprotein in insect cells. Biotechnol. Prog. 17, 822-827 (2001).
90. Ogonah, O.W., Freedman, R.B., Jenkins, N., Patel, K. & Rooney, B. Isolation and characterization of an insect cell line able to perform complex N-linked glycosylation on recombinant proteins. Bio/Technology 14, 197-202 (1996).
91. Wagner, R., Geyer, H., Geyer, R. & Klenk, H.D. N-acetyl-beta-glucosamidase accounts for differences in glycosylation of influenza virus hemagglutinin expressed in insect cells from a baculovirus vector. J. Virol. 70, 4103-4109 (1996).
92. Hollister, J.R., Shaper, J.H. & Jarvis, D.L. Stable expression of mammalian 1,4-galactosyltransferase extends the N-glycosylation pathway in insect cells. Glycobiol. 8, 473-480 (1998).
93. Hollister, J. & Jarvis, D.L. Engineering lepidopteran cell lines for sialoglycoprotein production by genetic transformation with mammalian 1,4-galactosyltransferase and 2,6-sialyltransferase genes. Glycobiol. 11, 1-9 (2001).
94. Hollister, J.R., Grabenhorst, E., Nimtz, M., Conradt, H.O. & Jarvis, D.L. Engineering the protein N-glycosylation pathway in insect cells for production of biantennary, complex glycans. Biochem. Mol. Biol. Internat. 41, 15093-15104 (2002).
95. Aumiller, J.J., Hollister, J.R. & Jarvis, D.L. A transgenic insect cell line engineered to produce CMP-sialic acid and sialylated glycoproteins. Glycobiol. 6, 497-507 (2003).
96. Whitefleet-Smith, J. et al. Expression of human plasminogen cDNA in a baculovirus vector-infected insect cell system. Arch. Biochem. Biophys. 1, 390-399 (1989).
97. Ramabhadran, T.V. et al. Proteolytic processing of human amyloid β protein precursor in insect cells. Major carboxyl-terminal fragment is identical to its human counterpart. J. Biol. Chem. 268, 2009-2012 (1993).
98. Sugiyama, K., Ahorn, H., Maurer-Fogy, I. & Voss, T. Expression of human interferon-beta alpha 2 in Sf9 cells. Characterization of O-linked glycosylation and protein heterogeneities. Eur. J. Biochem. 217, 921-927 (1993).
99. Pyle, L.E., Barton, P., Fujiwara, Y., Mitchell, A. & Fidge, N. Secretion of biologically active human proapolipoprotein A-I in a baculovirus-insect cell system: protection from degradation by protease inhibitors. J. Lip. Res. 36, 2355-2361 (1995).
100. Heitz, F., Nay, C. & Guenet, C. Expression of functional human muscarinic M2 receptors in different insect cell lines. J. Recept. Sign. Transd. Res. 17, 305 (1997).
101. Grosch, H.-W. & Hasilik, A. Protection of proteolysis-prone recombinant proteins in baculovirus expression systems. Biotechniques 24, 930-934 (1998).
102. Rawlings, N.D., Pearl, L.H. & Butte, D.J. The baculovirus Autographa californica nuclear polyhedrosis virus genome includes a papain-like sequence. Biol. Chem. Hoppe Seyler 373, 1211-1215 (1992).
103. Slack, J.M., Kuzio, J. & Faulkner, P. Characterization of v-cath, a cathepsin L-like proteinase expressed by the baculovirus Autographa californica multiple nuclear polyhedrosis virus. J. Gen. Virol. 76, 1091-1098 (1995).
104. Hawtin, R.E. et al. Liquefaction of Autographa californica nucleopolyhedrovirus-infected insects is dependent on the integrity of virus-encoded chitinase and cathepsin genes. Virology 238, 243-253 (1997).
105. Hawtin, R.E. et al. Identification and preliminary characterization of a chitinase gene in the Autographa californica nuclear polyhedrosis virus genome. Virology 212, 673-685 (1995).
106. Thomas, C.J. et al. Localization of a baculovirus-Induced chitinase in the insect cell endoplasmic reticulum. J. Virol. 72, 10207-10212 (1998).
107. Saville, G.P., Patmanidi, A.L., Possee, R.D. & King, L.A. Deletion of the Autographa californica nucleopolyhedrovirus chitinase KDEL motif and in vitro and in vivo analysis of the modified virus. J. Gen. Virol. 85, 821-831 (2004).
108. Monsma, S.A. & Scott, M. Bacvector-3000: an engineered baculovirus for foreign protein expression, lacking viral chitinase and cathepsin protease activities. Innovations, 16-19 (1997).
109. Schmid, G. & Bischoff, A. Proteolytic activities in the baculovirus-insect cell expression system. 303-306 (1998).
110. Naggie, S. & Bentley, W.E. Appearance of protease activities coincides with p10 and polyhedrin-driven protein production in the baculovirus expression system: effects on yield. Biotechnol. Prog. 14, 227-232 (1998).

47
111. Naggie, S., Hu, Y.C., Pulliam-Holoman, T.R. & Bentley, W.E. Substrate (gelatin) gel electrophoretic method for analysis of protease activity in insect (Sf-9) cells. Biotechnol. Techn. 11, 297-300 (1997).
112. Wang, M.Y., Pulliam, T.R., Valle, M., Vakharia, V.N. & Bentley, W.E. Kinetic analysis of alkaline protease activity, recombinant protein production and metabolites for infected insect (Sf9) cells under different DO levels. J. Biotechnol. 46, 243-254 (1996).
113. Ikonomou, L., Peeters-Joris, C., Schneider, Y.J. & Agathos, S.N. Supernatant proteolytic activities of High-Five insect cells grown in serum-free culture. Biotechnol. Lett. 24, 965-969 (2002).
114. Eriksson, U., Hassel, J., Lüllau, E. & Häggström, L. Metalloproteinase activity is the sole factor responsible for the growth-promoting effect of conditioned medium in Trichoplusia ni insect cell cultures. J. Biotechnol. 119, 76-86 (2005a).
115. Hu, Y.C. & Bentley, W.E. Enhancing yield of infectious bursal disease virus structural proteins in baculovirus expression systems: focus on media, protease inhibitors, and dissolved oxygen. Biotechnol. Prog. 15, 1065-1071 (1999).
116. Gotoh, T., Miyazaki, Y., Kikuchi, K.I. & Bentley, W.E. Investigation of sequential behaviour of carboxyl protease and cysteine protease activities in virus-infected Sf-9 insect cell culture by inhibition assay. App. Microbiol. Biotechnol. 56, 742-749 (2001a).
117. Bonning, B.C., Roelvink, P.W., Vlak, J.M., Possee, R.D. & Hammoch, B.D. Superior expression of juvenile hormone esterase and beta-galactosidase from the basic protein promoter of Autographa californica nuclear polyhedrosis virus compared to the p10 protein and polyhedrin promoters. J. Gen. Virol. 75, 1551-1556 (1994).
118. Lawrie, A.M., King, L.A. & Ogden, J.E. High level synthesis and secretion of human urokinase using a late gene promoter of the Autographa californica nuclear polyhedrosis virus. J. Biotechnol. 39, 1-8 (1995).
119. Kost, T.A. et al. Production of a urokinase plasminogen activator-IgG fusion protein (uPA-IgG) in the baculovirus expression system. Gene 190, 139-144 (1997).
120. Ho, Y., Ho, H.-R., Lee, T.-C., Wu, C.P. & Chao, Y.-C. Enhancement of correct protein folding in vivo by a non-lytic baculovirus. Biochem. J. 382, 695-702 (2004).
121. Licari, P. & Bailey, J.E. Factors influencing recombinant protein yields in an insect cell-baculovirus expression system: multiplicity of infection and intracellular protein degradation. Biotechnol. Bioeng. 37, 238-246 (1991).
122. Lindsay, D.A. & Betenbaugh, M.J. Quantification of cell culture factors affecting recombinant protein yields in baculovirus-infected insect cells. Biotechnol. Bioeng. 39, 614-618 (1992).
123. Reuveny, S., Kim, Y.J., Kemp, C.W. & Schiloach, J. Effect of temperature and oxygen on cell growth and recombinant protein production in insect cell cultures. Appl. Microbiol. Int. 38, 619-623 (1993a).
124. Hensler, W.T. & Agathos, S.N. Evaluation of monitoring approaches and effects of culture conditions on recombinant protein production in baculovirus-infected insect cells. Cytotechnol. 15, 177-186 (1994).
125. Wong, K.T., Peter, C.H., Greenfield, P.F., Reid, S. & Nielsen, K. Low multiplicity infection of insect cells with a recombinant baculovirus: the cell yield concept. Biotechnol. Bioeng. 49, 659-666 (1996).
126. Taticek, R.A. & Shuler, M.L. Effect of elevated oxygen and glutamine levels on foreign protein production at high cell densities using the insect cell-baculovirus expression system. Biotechnol. Bioeng. 54, 142-152 (1997).
127. Jesionowski, G.A. & Ataai, M.M. An efficient medium for high protein production in the insect cell/baculovirus expression system. Biotechnol. Prog. 13, 355-360 (1997).
128. Chico, E. & Jäger, V. Perfusion culture of baculovirus-infected BI-Tn-5B4 insect cells: a method to restore cell-specific β-trace glycoprotein productivity at high cell density. Biotechnol. Bioeng. 70, 574-586 (2000).
129. Radford, K., Reid, S. & Greenfield, P.F. Substrate limitation in the baculovirus expression system. Biotechnol. Bioeng. 56, 32-44 (1997).
130. Eriksson, U. in Department of Biotechnology, Vol. Licentiate (Royal Institute of Technology, Stockholm; 2005).
131. Stockdale, H. & Gardiner, G.R. The influence of the condition of cells and medium on production of polyhedra of Autographa californica nuclear polyhedrosis virus in vitro. J. Invertebr. Pathol. 30, 330-336 (1977).
132. Doverskog, M., Ljunggren, J., Öhman, L. & Häggström, L. Physiology of cultured animal cells. J. Biotechnol. 59, 103-115 (1997).
133. Rhiel, M., Mitchell-Logean, C.M. & Murhammer, D.W. Comparison of Trichoplusia ni BTI-Tn-5B1-4 (High Five) and Spodoptera frugiperda Sf-9 insect cell line metabolism in suspension cultures. Biotechnol. Bioeng. 55, 909-920 (1997).

48
134. Carpentier, E. et al. Increased production of active human β-adregenic Gαs fusion receptor in Sf-9 cells using nutrient limiting conditions. Protein Expr. Purif. 23, 66-74 (2001).
135. Chan, L.C.L., Greenfield, P.F. & Reid, S. Optimizing fed-batch of recombinant proteins using the baculovirus expression vector system. Biotechnol. Bioeng. 59, 178-188 (1998).
136. Bédard, C., Perret, S. & Kamen, A.A. Fed-batch culture of Sf-9 cells supports 3x107 cells per ml and improves baculovirus-expressed recombinant protein yields. Biotechnol. Lett. 19, 629-632 (1997).
137. Elias, C.B., Zeiser, A., Bédard, C. & Kamen, A.A. Enhanced growth of Sf-9 cells to a maximum density of 5.2x107 cells per mL and production of β-galactosidase at high-cell density by fed-batch culture. Biotechnol. Bioeng. 68, 381-388 (2000).
138. Lazarte, J.E., Tosi, P.-F. & Nicolau, C. Optimization of the production of full-length rCD4 in baculovirus-infected Sf9 cells. Biotechnol. Bioeng. 40, 214-217 (1992).
139. Jäger, V. Perfusion bioreactors for the production of recombinant proteins in insect cells. Cytotechnol. 20, 191-198 (1996).
140. Caron, A.W., Tom, R.L., Kamen, A.A. & Massie, B. Baculovirus expression system scaleup by perfusion of High-Density Sf-9 cell cultures. Biotechnol. Bioeng. 43, 881-891 (1994).
141. Zhang, J., Collins, A., Chen, M., Knyazev, I. & Gentz, R. High-density perfusion culture of insect cells with a biosep ultrasonic filter. Biotechnol. Bioeng. 59, 351-359 (1998).
142. Bédard, C., Tom, R. & Kamen, A.A. Growth, nutrient consumption, and end-product accumulation in Sf-9 and BTI-EAA insect cell cultures: insights into growth limitation and metabolism. Biotechnol. Prog. 9, 615-624 (1993).
143. Sugiura, T. & Amann, E. Properties of two insect cell lines useful for the baculovirus expression system in serum-free culture. Biotechnol. Bioeng. 51, 494-499 (1996).
144. Yang, J.D. et al. Rational scale-up of a baculovirus-insect cell batch process based on medium nutritional depth. Biotechnol. Bioeng. 52, 696-706 (1996).
145. Benslimane, C., Elias, C.B., Hawari, J. & Kamen, A. Insights into the central metabolism of Spodoptera frugiperda (Sf-9) and Trichoplusia ni BTI-Tn-5B1-4 (Tn5) insect cells by radiolabeling studies. Biotechnol. Prog. 21, 78-86 (2005).
146. Stavroulakis, D.A., Kalogerakis, N., Behie, L.A. & Latrou, K. Kinetic data for the BM-5 insect cell line in repeated-batch suspension cultures. Biotechnol. Bioeng. 38, 116-126 (1991).
147. Ljunggren, J. & Häggström, L. Specific growth rate as a parameter for tracing growth-limiting substances in animal cell cultures. J. Biotechnol. 42, 163-175 (1995).
148. Sennerstam, R. & Strömberg, J.-O. Cell cycle progression: computer simulation of uncoupled subcycles of DNA replication and cell growth. J. Theor. Biol. 175, 177-189 (1995).
149. Su, T.T. & O'Farrell, P.H. Size control: cell proliferation does not equal growth. Curr. Biol. 8, R687-689 (1998).
150. Kim, J.H., Kim, E.J. & Park, T.H. Fed-batch culture of insect cells with exponential feeding of amino acid and yeastolate solution. Bioproc. Eng. 23, 367-370 (2000).
151. Lauffenburger, D.A. & Cozens, C. Regulation of mammalian cell growth by autocrine growth factors: analysis of consequences for inoculum cell density effects. Biotechnol. Bioeng. 33, 1365-1378 (1989).
152. Murhammer, D.W. & Goochee, C.F. Scaleup of insect cell cultures: protective effects of pluronic F-68. Bio/Technology 6, 1411-1415 (1988).
153. Neutra, R., Levi, B.-Z. & Shoham, Y. Optimization of protein-production by the baculovirus expression vector system in shake flasks. App. Microbiol. Biotechnol. 37, 74-78 (1992).
154. Meneses-Acosta, A., Mendonca, R.Z., Merchant, H., Covarrubias, L. & Ramirez, O.T. Comparative characterization of cell death between Sf9 insect cells and hybridoma cultures. Biotechnol. Bioeng. 72, 441-457 (2000).
155. Fertig, G., Klöppinger, M. & Miltenburger, H.G. Cell cycle kinetics of insect cell cultures compared to mammalian cell cultures. Exp. Cell Res. 189, 208-212 (1990).
156. Braunagel, S.C., Parr, R., Belyavskyi, M. & Summers, M.D. Autographa californica nucleopolyhedrovirus infection results in Sf9 cell cycle arrest at G2/M phase. Virology 244, 195-211 (1998).
157. Meneses-Acosta, A., Mendonca, R.Z., Merchant, H., Covarrubias, L. & Ramirez, O.T. Comparative characterization of cell death between Sf9 insect cells and hybridoma cultures. Biotechnol. Bioeng. 72, 441-457 (2001).
158. Zhang, Y.H., Enden, G. & Merchuk, J.C. Insect cells-baculovirus system: factors affecting growth and low MOI infection. Biochem. Eng. J. 27, 8-16 (2005).
159. Ikeda, M. & Kobayashi, M. Cell-cycle perturbation in Sf9 cells infected with Autographa californica nucleopolyhedrovirus. Virology 258, 176-188 (1999).
160. Léry, X., Charpentier, G. & Belloncik, S. DNA content analysis of insect cell lines by flow cytometry. Cytotechnol. 29, 103-113 (1999).

49
161. Jarman-Smith, R.F., Armstrong, S.J., Mannix, C.J. & Al-Rubeai, M. Chromosome instability in Spodoptera frugiperda Sf-9 cell line. Biotechnol. Prog. 18, 623-628 (2002).
162. Jarman-Smith, R.F., Mannix, C. & Al-Rubeai, M. Characterisation of tetraploid and diploid clones of Spodoptera frugiperda cell line. Cytotechnol. 44, 15-25 (2004).
163. Brown, M. & Faulkner, P. Factors affecting the yield of virus in cloned cell line of Trichoplusia ni infected with nuclear polyhedrosis virus. J. Invertebr. Pathol. 26, 251-257 (1975).
164. Lynn, D.E. & Hink, W.F. Infection of synchronized TN-368 cell cultures with alfalfa looper nuclear polyhedrosis virus. J. Inv. Pathol. 32, 1-5 (1978).
165. Kioukia, N., Nienow, A.W., Emery, A.N. & Al-Rubeai, M. Physiological and environmental factors affecting the growth of insect cells and infection with baculovirus. J. Biotechnol. 38, 243-251 (1995).
166. Saito, T., Dojima, T., Toriyama, M. & Park, E.Y. The effect of cell cycle on GFPuv gene expression in the baculovirus expression system. J. Biotechnol. 93, 121-129 (2002).
167. Collarini, E.J. & Oxender, D.L. Mechanisms of transport of amino acids across cell membranes. Ann. Rev. Nutr. 7, 75-90 (1987).
168. Wonderlin, W.F. & Strobl, J.L. Potassium channels, proliferation and G1 progression. J. Membr. Biol. 154, 91-107 (1996).
169. Rubin, H.A. A small substance in conditioned medium which enhances the growth of chick embryo cells. Exp. Cell. Res. 41, 138-148 (1966).
170. Homma, K.-i., Matsushita, T. & Natori, S. Purification, characterization, and cDNA cloning of a novel growth factor from the conditioned medium of NIH-Sape-4, an embryonic cell line of Sarcophaga peregrina (flesh fly). J. Biol. Chem. 271, 13770-13775 (1996).
171. Homma, K.J. et al. Adenosine deaminase activity of insect-derived growth factor Is essential for Its growth factor activity. J. Biol. Chem. 276, 43761-43766 (2001).
172. Kawamura, K., Shibata, T., Saget, O., Peel, D. & Bryant, P.J. A new family of growth factors produced by the fat body and active on Drosophila imaginal disc cells. Development 126, 211-219 (1999).
173. Doverskog, M. in Department of Biotechnology, Vol. Doctoral (Royal Insititute of Technology, Stockholm; 2000).
174. Class, R. et al. Histone H1 suppresses tumour growth of leukemia cells in vivo, ex vivo and in animal model suggesting extracellular functions of histones. Am. J. Clin. Oncol. 19, 522-531 (1996).
175. Currie, J.R. et al. Reduction of histone cytotoxicity by the alzheimer β-amyloid peptide precursor. Biochim. Biophys. Acta 1355, 248-258 (1997).
176. Abakushin, D.N., Zamulaeva, I.A. & Poverenny, A.M. Histones evoke thymocyte death in vitro: Histone-binding immunoglobulins decrease their cytotoxicity. Biochem. (Moscow) 64, 693-698 (1999).
177. Pohlmeyer, K. et al. The recombinant human histones H1° and H1.2 cause different toxicity profiles on the human leukemia cell line K562. Anticancer Res. 20, 2499-2504 (2000).
178. Steiner, H., Hultmark, D., Engström, A., Bennich, H. & Boman, H.G. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 292, 246-248 (1981).
179. Hultmark, D. et al. Insect immunity. Attacins, a family of antibacterial proteins from Hyalophora cecropia. EMBO J. 4, 571-576 (1983).
180. Hoffman, J.A. & Hetru, C. Insect defensins: inducible antibacterial peptides. Immunol. Today 10, 411-415 (1992).
181. Xu, X. et al. Two antimicrobial peptides from skin secretions of Rana grahami. Toxicon. 47, 459-464 (2006).
182. Fernandes, J.M.O., Kemp, G.D., Molle, M.G. & Smith, V.J. Anti-microbial properties of histone 2A from skin secretions of rainbow trout, Onchorhynchus mykiss. Biochem. J. 368, 611-620 (2002).
183. Papo, N. & Shai, Y. Host defence peptides as new weapons in cancer treatment. Cellular and molecular life sciences (CMLS) 62, 784 (2005).
184. Kieffer, A.-E. et al. The N- and C-terminal fragments of ubiquitin are important for the antimicrobial activities. FASEB J. 17, 776-778 (2003).
185. McKenzie, H.A. & White, F.H.J. Lysozyme and alpha-lactalbumin: structure, function, and interrelationships. Adv. Protein. Chem. 41, 173-315 (1991).
186. Cruz, P.E. et al. Proteolytic activity in infected and noninfected insect cells: degradation of HIV-1 Pr55gag particles. Biotechnol. Bioeng. 65, 133-143 (1999).
187. Hom, L.G. & Volkman, L.E. Preventing proteolytic artefacts in the baculovirus expression system. Biotechniques 25, 18-20 (1998).
188. Gotoh, T., Miyazaki, Y., Sato, W., Kikuchi, K.-I. & Bentley, W.E. Proteolytic activity and recombinant protein production in virus-infected Sf-9 insect cell cultures supplemented with carboxyl and cysteine protease inhibitors. J. Biosci. Bioeng. 92, 248-255 (2001b).

50
189. Shaw, E., Mohanty, S., Colic, A., Stoka, V. & Turk, V. The affinity-labelling of cathepsin S with peptidyl diazomethyl ketones: comparison with the inhibition of cathepsin L and calpain. FEBS Lett. 334, 340-342 (1993).
190. Turk, B., Turk, D. & Turk, V. Lysosomal cysteine proteases: more than scavengers. Biochim. Biophys. Acta 1477, 98-111 (2000).
191. Mason, R.W., Gal, S. & Gottesman, M.M. The identification of the major excreted protein (MEP) from a transformed mouse fibroblast cell line as a catalytically active precursor form of cathepsin L. Biochem. J. 248, 449-454 (1987).
192. Johnson, G.D. & Jiang, W. Characterization of cathepsin L secreted by Sf21 insect cells. Arch. Biochem. Biophys. 444, 7-14 (2005).
193. Dickinson, D.P. Cysteine peptidases of mammals: their biological roles and potential effects in the oral cavity and other tissues in health and disease. Crit. Rev. Oral Biol. Med. 13, 238-275 (2002).
194. Kleiner, D.E. & Stetler-Stevenson, W.G. Quantitative zymography: detection of picogram quantities of gelatinases. Anal. Biochem. 218, 325-329 (1994).
195. Cristofoletti, P.T., Ribeiro, A.F., Terra, W.R. & The cathepsin L-like proteinases from the midgut of Tenebrio molitor larvae: sequence, properties, immunocytochemical localization and function. Insect Biochem. Mol. Biol. 35, 883-901 (2005).
196. Takahashi, S.Y., Yamamoto, Y., Watabe, S. & Kageyama, T. Autolytic activation mechanism of Bombyx acid cysteine protease (BCP). Biochem. Mol. Biol. Int. 42, 591-600 (1997).
197. Turk, V., Turk, B. & Turk, D. Lysosomal cysteine proteases: facts and opportunities. EMBO J. 20, 4629-4633 (2001).
198. Ishidoh, K. & Kominami, E. Gene regulation and extracellular functions of procathepsin L. Biol. Chem. 379, 131-135 (1998).
199. Kasai, M. et al. Proenzyme form of cathepsin L produced by thymic epithelial cells promotes proliferation of immature thymocytes in the presence of IL-1, IL-7, and anti-CD3 antibody. Cell. Immunol. 150, 124-136 (1993).
200. Erickson-Lawrence, M., Zabludoff, S.D. & Wright, W.W. Cyclic protein-2, a secretory product of rat Sertoli cells, is the proenzyme form of cathepsin L. Mol. Endocrinol. 5, 1789-1798 (1991).
201. Boujrad, N. et al. Identification of a stimulator of steroid hormone synthesis isolated from testis. Science 268, 1609-1612 (1995).
202. Takahashi, S.Y., Yamamoto, Y., Shionoya, Y. & Kageyama, T. Cysteine proteinase from the eggs of the silkmoth, Bombyx mori: identification of a latent enzyme and characterization of activation and proteolytic processing in vivo and in vitro. J. Biochem. (Tokyo) 114, 267-272 (1993).
203. Nishimura, Y., Kato, K., Furono, K. & Himeno, M. Inhibitory effect of leupeptin on the intracellular maturation of lysosomal cathepsin L in primary cultures of rat hepatocytes. Biol. Pharm. Bull. 7, 945-950 (1995).
204. Nomura, T. & Fujisawa, Y. Processing properties of recombinant human procathepsin L. Biochem. Biophys. Res. Comm. 230, 143-146 (1997).
205. Rosinski, M., Reid, S. & Nielsen, L.K. Osmolarity effects on observed insect cell size after baculovirus infection are avoided using growth medium for sample dilution. Biotechnol. Prog. 16, 782-785 (2000).
206. Palomares, L., Pedroza, J. & Ramirez, O. Cell size as a tool to predict the production of recombinant protein by the insect-cell baculovirus expression system. Biotechnol. Lett. 23, 359-364 (2001).

51
VI Acknowledgements CBioPT and its partners. For financing and taking part of my project, even though I got pregnant twice along the time.
Lena. For accepting me as a PhD student and never failing to lose engagement, not even when times went from good to bad to worse. For making me always feeling welcome for more or less stupid questions and discussions of the latest lack of results. And finally, for your ability to write beautiful texts. I like!
The bioprocess seniors: Olle, Gen and Andres. For being open and friendly, and for never hesitating to share me a bit of your large treasure of bioprocess knowledge.
The animal cell girls (in order of appearance): Karin, Erika, Ulrika and Ingrid. For laughter and joy during nice dinners and endless hours in the lab, and maybe most importantly for sharing despair and frustration when nothing went right. Karin, for good company, inspiration and advice. The histone is now history, may it rest in peace. Erika, for helping me survive the last year, and for deep-talk behind closed class 2-signed doors. Ulrika, for invaluable assistance during the last months in the laboratory, and for your kindness. Ingrid, for doing some of the most boring work in the project during my maternity leaves, and thereby largely contributing to this thesis. Without all of you, girls, I would not be here today.
The animal cell boy: Magnus. For fruitful discussions, both during the too short time we spent together at the department, and afterwards.
The Bioprocess staff, past and present. Fredrik, for serious conversations about life and girls, Atefeh, my longest shortest room-mate, you showed me that everything is possible, Anna Maria, for the atmosphere of joie de vivre you emit, Marie, a good company in the baby-caused lack-of-sleep delirium, Bato, hvala for bad jokes and nice guitar play, Tommy for fixing everything that needed to be fixed, Ela for polish schwissh-schwissch and help in the lab, Magda, for nice discussions over Chinese noodle soup, Marita, for your organized mind, Helène, Emma, Katrin, Mattias, Maria, Ling, for good lunch company, Eric2 for computer help, and all the rest, not mentioned but not forgotten.
My friends: Syjuntan with families, Vinprovargänget, pH-95 and other KTH leftovers, Ulli, Malla and all the rest. For happy get-togethers, which continuously make me not forgetting that life is so much more than work. The Lindskog clan: For always creating a friendly and warm atmosphere for me, and my family, during nice gatherings in Stockholm, the mountains and Stenskär. For helpful babysitting, reckless off-piste tours and excellent wine servings.
My family: Mum and Dad for spoiling my whole family, and Erik for understanding me inside-and-out, and thereby being the absolutely best Bro in da world. Come home to Stockholm soon, SL is even cheaper than Ryanair! Hugo and Filippa. For bringing sunshine back into my life end helping me believe in the future. You make me so very, very proud, every single day. My true little sweethearts, I love you so much.
Fredrik. I don't think I can ever repay you for the support you have been given me during these years. You have not only coped with my strange working-schedule, my depressions and frustrations over lack of results, and my all-of-a-sudden exercise fanatics, but along the time you have both married me and helped me produce two perfect children. I'm so happy that I met you, happier than I can say. I love you deeply, always and forever.

52

53
VII Publications I-V
The three oddest words
When I pronounce the word Future,
the first syllable already belongs to the past.
When I pronounce the word Silence,
I destroy it.
When I pronounce the word Nothing,
I make something no nonbeing can hold.
Wislawa Szymborska





























