Embed Size (px)
Citation preview

Preparation and characterization of thermally expandable microcapsules
by in situ polymerization
Xuebao Wang
Department of Fire Protection Engineering, The Armed Police Forces Academy
Langfang 065000, P. R. China
Keywords: thermally expandable microcapsules, in situ polymerization, polyacrylonitrile, polyethyl
acrylate
Abstract. In the present work, we synthesized microcapsules with copolymer of acrylonitrile and
ethyl acrylate as shell and isooctane as core by in situ polymerization, and then optimized the
preparative method. Synthesized microcapsules were characterized by Fourier transform infrared
spectroscopy (FTIR), 13
C nuclear magnetic resonance (13
C-NMR), scanning electron microscopy
(SEM), particle diameter analysis and thermogravimetry (TG).
Introduction
Microencapsulation is a process of enveloping micro-sized materials of solids, liquids or gasses
in a thin hollow microsphere, for the purpose of protecting encapsulated materials from degrading
reagents [1-9]. It has been widely used in the fields of pharmaceutical, biotechnology, agricultural,
intumescent fire retarding, flavor preservation, etc [10-12].
Thermally expandable microcapsules (TEMs) are particles made up of a thermoplastic shell filled
with liquid hydrocarbon. The shell softens and the liquid hydrocarbon boils upon heating. The
hydrocarbon gas works as a blowing agent because the shell expands as the inner pressure increases.
The volume of TEMs expands rapidly to even 100 times [13, 14].
Acrylonitrile is used primarily as a co-monomer in the production of acrylic material. It is a
chemical intermediate in the synthesis of various antioxidants, pharmaceuticals, dyes, and
surface-active agents. In the present work, microcapsules with copolymer of acrylonitrile and ethyl
acrylate as shell and isooctane as core were synthesized by in situ polymerization for the first time
and characterized by Fourier transform infrared spectroscopy (FTIR), 13
C nuclear magnetic
resonance (13
C-NMR), particle diameter analysis, scanning electron microscopy (SEM) and
thermogravimetry (TG).
Experimental
Preparation of thermally expandable microcapsules
Placed distilled water, MgCl2, NaOH, carboxymethyl cellulose (CMC) and NaNO2 into an
oven-dried three-necked round-bottomed flask equipped with a machine stirrer and a condenser,
then added prepared acrylonitrile (ACN), ethyl acrylate (EA), isooctane (ISO) and AIBN mixture.
After stirred for 10 hr at 65 ℃, we adjusted the mixture to proper pH. The mixture was then cooled
to room temperature, filtered, washed with distilled water, and dried. The TEMs powder was finally
obtained.
Fourier Transform Infrared Spectra
The Fourier transform infrared (FTIR) spectra were recorded using a Nicolet 8700
spectrophotometer to identify the chemical structure of the specimens. Samples were mixed with
KBr powders, and the mixture was pressed into a tablet.
Advanced Materials Research Vol. 496 (2012) pp 130-133Online available since 2012/Mar/27 at www.scientific.net© (2012) Trans Tech Publications, Switzerlanddoi:10.4028/www.scientific.net/AMR.496.130
All rights reserved. No part of contents of this paper may be reproduced or transmitted in any form or by any means without the written permission of TTP,www.ttp.net. (ID: 128.104.46.196, University of Wisconsin-Madison, Madison, USA-04/09/14,17:15:22)
13C nuclear magnetic resonance
The 13
C nuclear magnetic resonance (13
C-NMR) measurement was carried out by using a
JNM-AL300 NMR spectrometer with DMSO-d6 as the solvent and TMS the internal standard.
Scanning electron microscopy
The morphology of the TEMs and after gold-sputtered were studies by S-4300FEG scanning
electron microscope (SEM).
Particle Size Distributions Measurement
The mean particle size and distribution of samples were measured by a LA-920 optical particle
sizing instrument.
Thermogravimetric analysis
Thermogravimetric analysis (TGA) was carried out using a TA 2950 thermogravimetric analyzer
at a linear heating rate of 20 ℃ min-1
in air. The weight of sample was kept within 5–10 mg and the
airflow rate was 40ml per minute.
Results and discussion
Characterization of TEMs
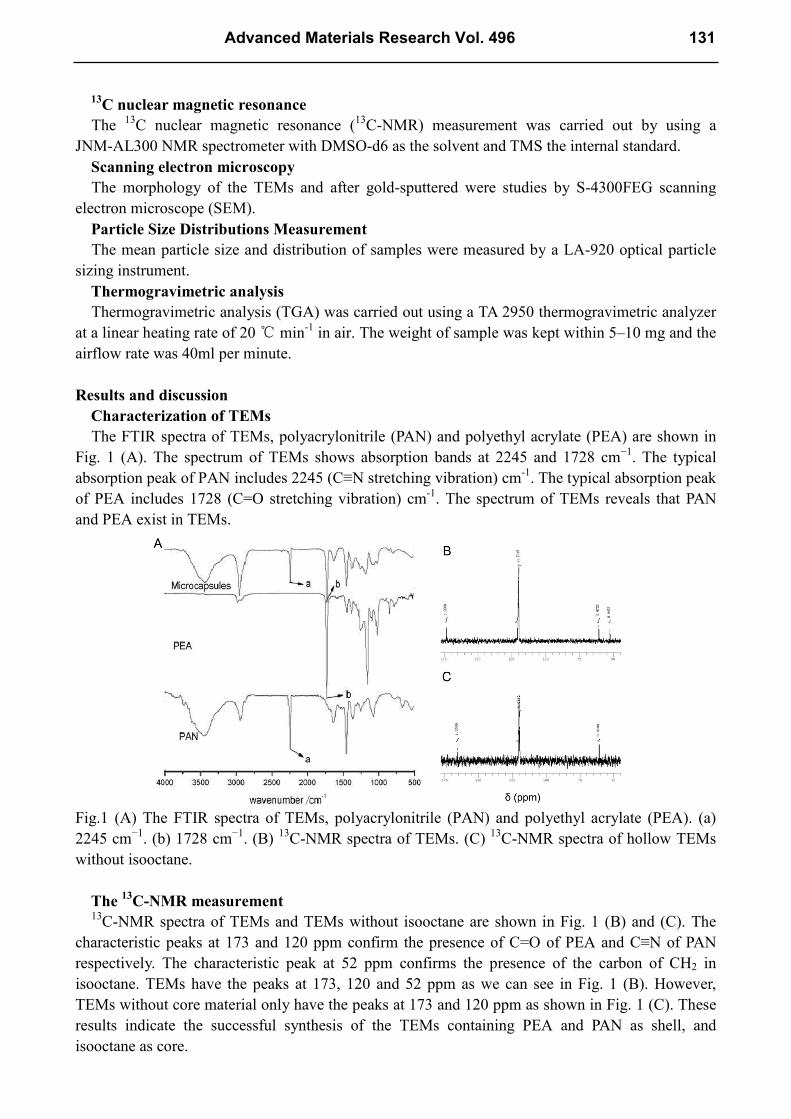
The FTIR spectra of TEMs, polyacrylonitrile (PAN) and polyethyl acrylate (PEA) are shown in
Fig. 1 (A). The spectrum of TEMs shows absorption bands at 2245 and 1728 cm−1
. The typical
absorption peak of PAN includes 2245 (C≡N stretching vibration) cm-1
. The typical absorption peak
of PEA includes 1728 (C=O stretching vibration) cm-1
. The spectrum of TEMs reveals that PAN
and PEA exist in TEMs.
Fig.1 (A) The FTIR spectra of TEMs, polyacrylonitrile (PAN) and polyethyl acrylate (PEA). (a)
2245 cm−1
. (b) 1728 cm−1
. (B) 13
C-NMR spectra of TEMs. (C) 13
C-NMR spectra of hollow TEMs
without isooctane.
The 13
C-NMR measurement
13
C-NMR spectra of TEMs and TEMs without isooctane are shown in Fig. 1 (B) and (C). The
characteristic peaks at 173 and 120 ppm confirm the presence of C=O of PEA and C≡N of PAN
respectively. The characteristic peak at 52 ppm confirms the presence of the carbon of CH2 in
isooctane. TEMs have the peaks at 173, 120 and 52 ppm as we can see in Fig. 1 (B). However,
TEMs without core material only have the peaks at 173 and 120 ppm as shown in Fig. 1 (C). These
results indicate the successful synthesis of the TEMs containing PEA and PAN as shell, and
isooctane as core.
Advanced Materials Research Vol. 496 131
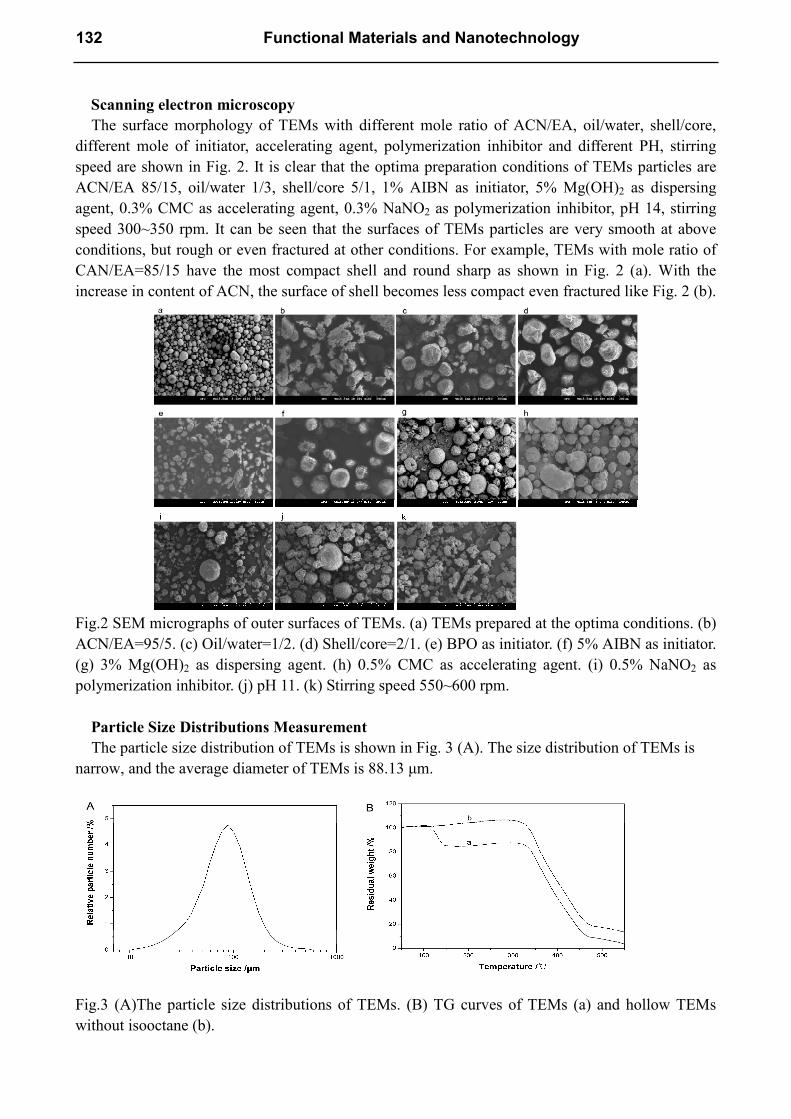
Scanning electron microscopy
The surface morphology of TEMs with different mole ratio of ACN/EA, oil/water, shell/core,
different mole of initiator, accelerating agent, polymerization inhibitor and different PH, stirring
speed are shown in Fig. 2. It is clear that the optima preparation conditions of TEMs particles are
ACN/EA 85/15, oil/water 1/3, shell/core 5/1, 1% AIBN as initiator, 5% Mg(OH)2 as dispersing
agent, 0.3% CMC as accelerating agent, 0.3% NaNO2 as polymerization inhibitor, pH 14, stirring
speed 300~350 rpm. It can be seen that the surfaces of TEMs particles are very smooth at above
conditions, but rough or even fractured at other conditions. For example, TEMs with mole ratio of
CAN/EA=85/15 have the most compact shell and round sharp as shown in Fig. 2 (a). With the
increase in content of ACN, the surface of shell becomes less compact even fractured like Fig. 2 (b).
Fig.2 SEM micrographs of outer surfaces of TEMs. (a) TEMs prepared at the optima conditions. (b)
ACN/EA=95/5. (c) Oil/water=1/2. (d) Shell/core=2/1. (e) BPO as initiator. (f) 5% AIBN as initiator.
(g) 3% Mg(OH)2 as dispersing agent. (h) 0.5% CMC as accelerating agent. (i) 0.5% NaNO2 as
polymerization inhibitor. (j) pH 11. (k) Stirring speed 550~600 rpm.
Particle Size Distributions Measurement
The particle size distribution of TEMs is shown in Fig. 3 (A). The size distribution of TEMs is
narrow, and the average diameter of TEMs is 88.13 µm.
Fig.3 (A)The particle size distributions of TEMs. (B) TG curves of TEMs (a) and hollow TEMs
without isooctane (b).
132 Functional Materials and Nanotechnology

Thermogravimetric analysis
Fig. 3 (B) demonstrates the thermal characteristic of TEMs and TEMs without isooctane. It can
be seen that the there are two steps on the degradation of TEMs. At the first step, TEMs decompose
with about 20% weight lost at 120 ℃. At the second step, TEMs begin to decompose at about
330 ℃, which is equal with the curve of TEMs without core material. This result indicates that the
weight lost at 120 ℃ is because of the volatilization of isooctane, which is the core material of
TEMs. The boiling point and flash point of isooctane is 99.3℃ and -12℃ respectively. It is clear
that microencapsulation of isooctane with PAN and PEA shell possesses better heat and
volatilizable gases resistance at high temperature. This makes TEMs suitable for many materials
because of relatively high initial decomposition temperature.
Conclusions
In the present work, we synthesized microcapsules with copolymer of acrylonitrile and ethyl
acrylate as shell and isooctane as core by in situ polymerization, and then optimized the preparative
method. Synthesized microcapsules were characterized by Fourier transform infrared spectroscopy
(FTIR), 13
C nuclear magnetic resonance (13
C-NMR), particle diameter analysis, scanning electron
microscopy (SEM) and thermogravimetry (TG). These results indicate that the polymer of
acrylonitrile and ethyl acrylate encapsulates isooctane very well in TEMs. The size distribution of
TEMs is narrow, and the average diameter of TEMs is 88.13 µm. TEMs have nice heat and
volatilizable gases resistance at high temperature, which makes it suitable for many materials
because of relatively high initial decomposition temperature.
References
[1] Y. H. Lee, C. A. Kim, W. H. Jang, H. J. Choi and M. S. Jhon: Polymer Vol. 42 (2001), p.
8277–8283
[2] X. D. Liu, T. T. Ataroshi, H. F. Furuta, S. Yoshii, M. O. Ashima and P. Linko: Dry. Technol. Vol.
19 (2001), p. 1361–1374
[3] S. J. Park, Y. S. Shin and J. R. J. Lee: Colloid Interface Sci Vol. 241 (2001), p. 502–508
[4] B. J. Park, J. Y. Lee, J. H. Sung and H. J. Choi: Curr. Appl. Phys. Vol. 6 (2006), p. 632–635
[5] G. Nelson: Int. J. Pharm. Vol. 242 (2002), p. 55–62
[6] G. Orive, R. M. Hernandez, A. Rodriguez Gasco, T. M. S. Chang, P. Vos, G. Hortelano, D. H. I.
Lacyk and J. L. Pedraz: Trends Biotechnol Vol. 22 (2004), p. 87–92
[7] G. Sukhorukov, A. Fery and H. Mohwald: J. Prog. Polym. Sci. Vol. 30 (2005), p. 885–897
[8] H. B. Ji, G. J. Kuang and Y. Qian: Catal. Today Vol. 105 (2005), p. 605–611
[9] J. Eukaszczyk and P. Urba: React Funct. Polym. Vol. 33 (1997), p. 233–239
[10] A.R. Hemsley and P.C. Griffiths: Colloid. Surface. B. Vol. 25 (2002), p. 163-70
[11] R. Arshady: Microspheres, Microcapsules and Liposomes (Citrus Books, London 1999).
[12] B. J. Blaiszik, M. M. Caruso, D. A. McIlroy, J. S. Moore, S. R. White and N. R. Sottos:
Polymer Vol. 50 (2009), p. 990-7
[13] P. Griss, H. Andersson and G. Stemme: Lab on a Chip Vol. 2 (2002), p. 117-20
[14] C. J. McDonald and M. J. Devon: Adv. Colloid. Interface. Vol. 99 (2002), p. 181-213
Advanced Materials Research Vol. 496 133

Functional Materials and Nanotechnology 10.4028/www.scientific.net/AMR.496 Preparation and Characterization of Thermally Expandable Microcapsules by In Situ Polymerization 10.4028/www.scientific.net/AMR.496.130
DOI References
[1] Y. H. Lee, C. A. Kim, W. H. Jang, H. J. Choi and M. S. Jhon: Polymer Vol. 42 (2001), p.8277–8283.
http://dx.doi.org/10.1016/S0032-3861(01)00342-1 [2] X. D. Liu, T. T. Ataroshi, H. F. Furuta, S. Yoshii, M. O. Ashima and P. Linko: Dry. Technol. Vol. 19
(2001), p.1361–1374.
http://dx.doi.org/10.1081/DRT-100105293 [3] S. J. Park, Y. S. Shin and J. R. J. Lee: Colloid Interface Sci Vol. 241 (2001), p.502–508.
http://dx.doi.org/10.1006/jcis.2001.7727 [4] B. J. Park, J. Y. Lee, J. H. Sung and H. J. Choi: Curr. Appl. Phys. Vol. 6 (2006), p.632–635.
http://dx.doi.org/10.1016/j.cap.2005.04.009 [5] G. Nelson: Int. J. Pharm. Vol. 242 (2002), p.55–62.
http://dx.doi.org/10.1016/S0378-5173(02)00141-2 [6] G. Orive, R. M. Hernandez, A. Rodriguez Gasco, T. M. S. Chang, P. Vos, G. Hortelano, D. H. I. Lacyk
and J. L. Pedraz: Trends Biotechnol Vol. 22 (2004), p.87–92.
http://dx.doi.org/10.1016/j.tibtech.2003.11.004 [7] G. Sukhorukov, A. Fery and H. Mohwald: J. Prog. Polym. Sci. Vol. 30 (2005), p.885–897.
http://dx.doi.org/10.1016/j.progpolymsci.2005.06.008 [8] H. B. Ji, G. J. Kuang and Y. Qian: Catal. Today Vol. 105 (2005), p.605–611.
http://dx.doi.org/10.1016/j.cattod.2005.06.006 [9] J. Eukaszczyk and P. Urba: React Funct. Polym. Vol. 33 (1997), p.233–239.
http://dx.doi.org/10.1016/S1381-5148(97)00044-8 [10] A.R. Hemsley and P.C. Griffiths: Colloid. Surface. B. Vol. 25 (2002), pp.163-70.
http://dx.doi.org/10.1016/S0927-7765(01)00316-2 [12] B. J. Blaiszik, M. M. Caruso, D. A. McIlroy, J. S. Moore, S. R. White and N. R. Sottos: Polymer Vol. 50
(2009), pp.990-7.
http://dx.doi.org/10.1016/j.polymer.2008.12.040 [13] P. Griss, H. Andersson and G. Stemme: Lab on a Chip Vol. 2 (2002), pp.117-20.
http://dx.doi.org/10.1039/b111040n [14] C. J. McDonald and M. J. Devon: Adv. Colloid. Interface. Vol. 99 (2002), pp.181-213.
http://dx.doi.org/10.1016/S0001-8686(02)00034-9





























