Embed Size (px)
Citation preview

ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2019
Digital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Science and Technology 1787
Probing Catalytic ReactionMechanisms of Biomimetic DiironComplexes through Time-resolvedAbsorption Spectroscopy
SHIHUAI WANG
ISSN 1651-6214ISBN 978-91-513-0610-0urn:nbn:se:uu:diva-380279

Dissertation presented at Uppsala University to be publicly examined in Häggsalen, ÅngströmLaboratory, Lägerhyddsvägen 1, Uppsala, Friday, 10 May 2019 at 10:15 for the degree ofDoctor of Philosophy. The examination will be conducted in English. Faculty examiner:Associate Professor Jillian Dempsey (University of North Carolina at Chapel Hill, USA).
AbstractWang, S. 2019. Probing Catalytic Reaction Mechanisms of Biomimetic Diiron Complexesthrough Time-resolved Absorption Spectroscopy. Digital Comprehensive Summaries ofUppsala Dissertations from the Faculty of Science and Technology 1787. 78 pp. Uppsala:Acta Universitatis Upsaliensis. ISBN 978-91-513-0610-0.
Directed design of improved molecular catalysts for hydrogen evolution reactions relies onrational benchmarking based on a detailed understanding about the mechanism of catalysis.Specifically, investigation of multi-electron redox catalysis, with structural characterization ofcatalytic intermediates, combined with the kinetics of their transformations, can reveal the rate-limiting step of the overall reaction, possible degradation pathways and the function of structuralmotives. However, direct spectroscopic observation of catalytic intermediates is in most casesnot available due to the rapid turnover of efficient catalysts.
In this thesis, time-resolved absorption spectroscopy with UV-Vis and mid-IR detection wasused to identify catalytic reaction intermediates and account for kinetics relevant to elementaryreactions steps of H2 formation on a nanosecond to second time scale. For a class of FeIFeI (S-R-S)(CO)6-n(PMe3)n complexes (R = propyl, benzyl or azapropyl), inspired by the active siteof FeFe-hydrogenase, the key intermediates formed in different catalytic pathways have beencharacterized. These complexes typically feature very similar coordination geometry, but showdifferent structural rearrangements upon reduction. This could be applied to rationalize theirdifferences in protonation dynamics. Protonation kinetics of singly reduced species, forminga bridging hydride, indicate a direct proton transfer step in the FeIFe0 state, in contrast tothat of the neutral complex (FeIFeI state) with phosphine ligands (PMe3) in which the hydrideformation is likely mediated by one of the CO-ligands, as had been proposed. In catalysisof FeFe-hydrogenase, the amine function of the bridgehead is known to assist enzymatic H2
formation by proton shuttling. The same role in catalysis by the synthetic diiron complex withthe azapropyl bridgehead had been proposed. However, our results show that for the syntheticcomplex, the aza-group has no role as a proton shuttle in the hydride formation in the FeIFe0
state. Instead, the effect of nitrogen protonation is to lower the catalyst overpotential, withoutsubstantially slowing down the hydride formation with external protons. The amine acting as aproton shuttle in the hydride formation could be expected in the Fe0Fe0 level. However, slowersecond reduction of FeIFeI (S-azapropyl-S)(CO)6 complex impedes observation of the doublyreduced species under the catalytic conditions. For the benzyldithiolate complex, on the otherhand, the rigid and unsaturated bridging ligand generally leads to less negative potentials andprevent the reduced forms from rapid degradation. This allows characterization of the laterintermediates of the catalytic processes, and to obtain direct kinetic information on the turnoverstep.
Keywords: Artificial photosynthesis, Biomimetic catalysts, H2 formation, Catalyticintermediates
Shihuai Wang, Department of Chemistry - Ångström, Physical Chemistry, Box 523, UppsalaUniversity, SE-75120 Uppsala, Sweden.
© Shihuai Wang 2019
ISSN 1651-6214ISBN 978-91-513-0610-0urn:nbn:se:uu:diva-380279 (http://urn.kb.se/resolve?urn=urn:nbn:se:uu:diva-380279)

3
To my family 致我的家人

4

5
List of Papers
This thesis is based on the following papers, which are referred to in the text by their Roman numerals.
I Structural and Kinetic Studies of Intermediates of a Bio-
mimetic Diiron Proton-Reduction Catalyst Shihuai Wang, Alexander Aster, Mohammad Mirmohades, Re-iner Lomoth and Leif Hammarström Inorg. Chem., 2018, 57, 768-776
II Metal vs. ligand protonation and the alleged proton-shuttling role of the azadithiolate ligand in catalytic H2 for-mation with FeFe hydrogenase model complexes Alexander Aster, Shihuai Wang, Mohammad Mirmohades, Charlene Esmiou, Gustav Berggren, Leif Hammarström and Reiner Lomoth Under minor revisions in Chem. Sci.
III Spectroscopic Observation of Two-electron Reduced Diiron
Azadithiolate Catalyst related to the active site of FeFe-hydrogenase Shihuai Wang, Charlene Esmieu, Gustav Berggren, Matthias Stein, Leif Hammarström and Reiner Lomoth Manuscript
IV Direct Spectroscopic Detection of Key Intermediates and
Turnover Process in Catalytic H2 Formation by a Biomimet-ic Diiron Catalyst Shihuai Wang, Sonja Pullen, Valentin Weippert, Tianfei Liu, Sascha Ott, Reiner Lomoth and Leif Hammarström Submitted

6
Unpublished results
Section 3.2.3 in Chapter 3 Spectroscopic observation of potential tautomerization and catalytic turnover processes for the [FeFe(CO)4(PMe3)2] complex
I planed the study and performed the experimental work.

7
Papers not included in this thesis
I am also a co-author of the following papers, which are not include in the thesis.
IV Eu3+ doped monetite and its use as fluorescent agent for dental res-torations Song Chen#, Shihuai Wang#, Hu Li, Kerstin Forsberg Ceramics International. 2018, 44, 10510 V Efficient Dye-Sensitized Solar Cells with Voltages Exceeding 1 V through Exploring Tris(4-alkoxyphenyl)amine Mediators in Combina-tion with the Tris(bipyridine) Cobalt Redox System Yan Hao, Wenxing Yang, Martin Karlsson, Jiayan Cong, Shihuai Wang, Xing Li, Bo Xu, Jianli Hua, Lars Kloo, and Gerrit Boschloo ACS Energy Lett., 2018, 3, 1929-1937 # The authors contributed equally

8
Contribution to papers
I Planned and performed all the measurements, analyzed and interpreted the data. Wrote the first draft of the manuscript. II Planned and performed the stopped-flow rapid-mixing experiments, con-tributed to laser flash IR experiments, analysis and discussions of data, and inputs of manuscript. III. Conceived the study and performed all the measurements, analyzed and interpreted the data. Wrote the first draft of the manuscript. IV. Performed all the measurements, contributed to the analysis and interpre-tation of data. Wrote the first draft of the manuscript.

9
Contents
1. Introduction ............................................................................................... 13 1.1 Hydrogen Economy ............................................................................ 13 1.2 From Natural to Artificial Photosynthesis .......................................... 14
1.2.1 Proton reduction catalysts ........................................................... 17 1.3 [FeFe] hydrogenases .......................................................................... 17 1.4 Synthetic Molecular Catalysts Inspired by [FeFe]-hydrogenase ........ 18
1.4.1 Structural mimics ........................................................................ 19 1.4.2 Functional mimics ...................................................................... 19
1.5 Proton Reduction Mechanisms ........................................................... 20 1.6 Aim of the Thesis ............................................................................... 22
2. Fundamentals and Methods ...................................................................... 24 2.1 Studied Systems ................................................................................. 24
2.1.1 Photochemical Reduction Systems ............................................. 24 2.1.3 Chemical Reduction Systems ..................................................... 27
2.2 Techniques ......................................................................................... 27 2.2.1 Steady State Absorption Spectroscopy ....................................... 27 2.2.2 Nanosecond Transient Absorption Spectroscopy ....................... 29 2.2.3 Rapid-mixing FT-IR Spectroscopy ............................................. 32 2.2.4 Electrochemical Methods ........................................................... 33
3. Results and Discussion ............................................................................. 34 3.1 Photochemical Reduction of FeFe(pdt)(CO)6 .................................... 34
3.3.1 General Considerations ............................................................... 34 3.1.2 One-electron Reduced Intermediates .......................................... 35 3.1.2 Kinetic Behaviors of Protonation and Detection of Reduced-protonated Species ............................................................................... 37
3.2 Proton-shuttling Role of Azadithiolate Ligand .................................. 40 3.2.1 General ........................................................................................ 40 3.2.2 FeFe(adt)(CO)6 Complex ............................................................ 40 3.2.3 FeFe(adt)(CO)4(PMe3)2 Complex ............................................... 44
3.3 The Factors Influencing the Catalytic Activity of Diiron Complexes with Alkyl Bridging Ligands ................................................. 48
3.3.1 General ........................................................................................ 48 3.3.2 Two-electron Reduced Intermediate of Complex 3 .................... 49 3.3.3 Degradation pathways ................................................................ 50

10
3.4 Photochemical and Chemical Reduction of FeFe(Cl2bdt)(CO)6 Complex ................................................................................................... 53
3.4.1 General ........................................................................................ 53 3.4.2 Key Catalytic Intermediates of FeFe(Cl2bdt)(CO)6 .................... 54 3.4.3 Catalytic Turnover Processes with Stronger Acids .................... 57 3.4.4 Mechanistic Aspects for Catalytic H2 Formation ....................... 58 3.4.5 Hydricity of [5H]- ....................................................................... 59
4. Summary and outlook .............................................................................. 61
5. Sammanfattning på Svenska ..................................................................... 64
6. Acknowledgments ..................................................................................... 67
6. References ................................................................................................ 69

11
Abbreviations
bpy 2, 2’-bipyridine MLCT metal-to-ligand charge transfer UV ultraviolet Vis Visible TTF Tetrathiafuvalene CoCp cobaltocene FTIR frourier transform infrared TOF turnover frequency CCD charge-coupled-device PMT photomultiplier tube QC quantum cascade CV cyclic voltammetry SEC S´spectroelectrochemical pdt propanedithiolate adt azadithiolate bdt benzenedithiolate DCA dichloroacetic acid TsOH p-toluenebenzenesulfonic acid TCA trichloroacetic acid PMe3 trimethylphosphine ET electron transfer PT proton transfer PSI photosystem I PSII photosystem II Cl2BSA 2,5-dichlorobenzenesulfonic acid dmb 4,4’-dimethyl-2,2’-bipyridine PEC photoelectrochemical OEC oxygen evolving complex NADP+ oxidized nicotinamide adenine dinucleotide phosphate NADH reduced nicotinamide adenine dinucleotide phosphate ATP adenosine triphosphate Pt platinum Pd palladium WOC water oxidation catalyst ISC intersystem crossing

12

13
1. Introduction
1.1 Hydrogen Economy Coming into the 21st century, the population growth together with rapid in-dustrialization has led to a surge in the global demand for energy. It has been agreed that the traditional and non-renewable energy sources, such as coal, oil, natural gas, etc. cannot fulfill the need for maintaining the long-term development of modern civilization. At the same time, combustion of the traditional fossil fuels releases a substantial amount of carbon dioxide (CO2), in which the rising of its concentration is the ‘Chief Culprit’ resulting in the greenhouse effect.[1,2] Thus, in response to the energy demand of the world and supporting economic development in the future, scientists are going to great lengths to search for and develop new types of renewable technologies to replace fossil fuels by environmentally friendly and inexpensive alterna-tive fuels.[3-6]
Hydrogen is considered as one of the most promising alternative energy carriers as:
Its combustion generates only water, which can achieve zero emission of greenhouse gases.
It has the highest energy storage density (143 MJ/kg) relative to all compounds that are regarded as energy storage materials, and can easily be applied in fuel cells for electricity generation.[7-11]
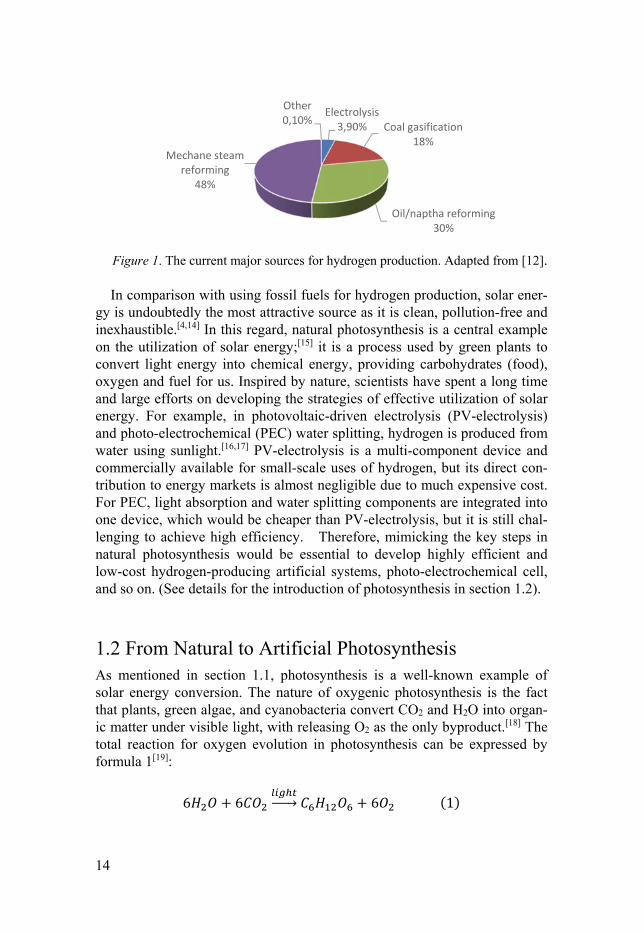
Regarding the production of hydrogen, the traditional strategies are well-known. Figure 1 clearly shows the principal production routes, suggesting that 96% is produced from steam reforming of fossil fuels, and 3.9% from electrolysis of water, a procedure which is rather inefficient and that is very expensive.[12,13] This is obviously not suited for which we achieve the goal of creating a green sustainable society.
14
Electrolysis3,90% Coal gasification
18%
Oil/naptha reforming30%
Mechane steam reforming
48%
Other0,10%
Figure 1. The current major sources for hydrogen production. Adapted from [12]. In comparison with using fossil fuels for hydrogen production, solar ener-
gy is undoubtedly the most attractive source as it is clean, pollution-free and inexhaustible.[4,14] In this regard, natural photosynthesis is a central example on the utilization of solar energy;[15] it is a process used by green plants to convert light energy into chemical energy, providing carbohydrates (food), oxygen and fuel for us. Inspired by nature, scientists have spent a long time and large efforts on developing the strategies of effective utilization of solar energy. For example, in photovoltaic-driven electrolysis (PV-electrolysis) and photo-electrochemical (PEC) water splitting, hydrogen is produced from water using sunlight.[16,17] PV-electrolysis is a multi-component device and commercially available for small-scale uses of hydrogen, but its direct con-tribution to energy markets is almost negligible due to much expensive cost. For PEC, light absorption and water splitting components are integrated into one device, which would be cheaper than PV-electrolysis, but it is still chal-lenging to achieve high efficiency. Therefore, mimicking the key steps in natural photosynthesis would be essential to develop highly efficient and low-cost hydrogen-producing artificial systems, photo-electrochemical cell, and so on. (See details for the introduction of photosynthesis in section 1.2).
1.2 From Natural to Artificial Photosynthesis As mentioned in section 1.1, photosynthesis is a well-known example of solar energy conversion. The nature of oxygenic photosynthesis is the fact that plants, green algae, and cyanobacteria convert CO2 and H2O into organ-ic matter under visible light, with releasing O2 as the only byproduct.[18] The total reaction for oxygen evolution in photosynthesis can be expressed by formula 1[19]:
6 6 6 1

15
For green plants, photosynthesis can be divided into two categories: light reactions and dark reactions. The specific processes of light reactions are as follows: 1) Absorption of photons and excited state energy transfer: Chlorophylls are the most important photosynthetic pigments in green plants. They absorb the visible light ranging from 400 nm to 700 nm and transfer the excited energy to adjacent or pigment molecules in the ground state by means of ‘exciton transfer’ or ‘resonance transfer’,[20] and themselves return back to the ground state; 2) Charge separation: The excited pigment molecules (*P680) initiate a redox reaction with electron acceptors presented in the reaction center in photosys-tem II (PS II), generating charge separation (or electron-hole pairs);[21] 3) Water oxidation: In PS II, the oxidative holes are able to activate the oxy-gen-evolving complex (OEC), a Mn4O5Ca cluster, which in turn oxidizes the water into oxygen (O2) and protons; 4) Electron transport: The generated electrons are transferred to photosystem I (PS I) along with an electron-transport chain. As a result, it causes the re-duction of NADP+ to produce a reducing electron carrier such as NADPH or ATP, as soon as absorbing the second photon in PS I. Such processes can be described by a so-called ‘Z-scheme’ (see Figure 2).[22]
Figure 2. ‘Z-scheme’ of light reactions. Adapted from ref [22].
The dark reactions are actually the processes that carry out the fixation of CO2, where NADPH produced by light reactions is used for reducing CO2 to hydrocarbon such as sugar.[23]
Based on the mechanistic understanding of natural photosystem, artificial photosynthesis aims at mimicking the key steps in photosynthesis. There is no need for replicating all processes like in green leaves, but a successful artificial system for water splitting must be made up of three components: (1) Antenna/reaction center complexes (photosensitizers) that are able to harvest sunlight and generate electrochemical potential; (2) catalysts that

16
oxidize water to produce molecular oxygen; (3) catalysts that reduce protons into hydrogen. The first example of such a system was reported in 1972 by Fujishima et al.[24] They created a photoelectrochemical cell using a platinum (Pt) as the cathode and a TiO2 based photoanode to carry on splitting of wa-ter under ultraviolet (UV) irradiation. However, as already mentioned be-fore, such a system suffers from a low efficient energy conversion mainly because of poor absorption of visible light and low quantum yields. In addi-tion, it generally uses the noble metals, Pt and palladium (Pd), or noble met-als-based complexes as catalysts for hydrogen production. This is obviously not feasible for future sustainable energy demand. Therefore, a new artificial system that contains an earth abundant-element-based molecular catalyst and is able to do direct fuel production will be needed. For this reason, a supra-molecular system that consists of D-P-A units and molecular catalysts has been proposed to perform water splitting into O2 and H2,[25] as shown in Fig-ure 3. In this system, the photo-excited sensitizer (P*) could deliver an elec-tron to the acceptor (A) and the donor (D) should be able to reduce the oxi-dized state of photosensitizer P+, generating a charge separated state, D+-P-A-. Then, it oxidizes water into molecular oxygen on the donor side and reduces protons into hydrogen on the acceptor side, with the help of catalysts. Yet, since the entire reaction in this model is a quite complex process, few examples based on such model have been known to generate molecular hy-drogen and oxygen simultaneously so far.[26-28] A comprehensive helpful approach is to divide this kind of artificial system into two half-reactions, i.e. water oxidation reaction and proton (or CO2) reduction reaction. Water oxi-dation is considered as one of the bottlenecks in the artificial photosystem as it typically involves multiple electron and proton transfers, generating high energetic intermediates. Thus, designing the water oxidation catalyst (WOC) towards high efficiency is crucial to lowering reaction barrier. Several groups have been focusing on developing molecular catalysts for water oxi-dation based on transition metals,[29-32] and the best one is to date Ru-based complex (TOF = 8000 s-1 at pH 7.0) presented by Llobet et al.[33]
Figure 3. An artificial photosystem for water splitting. Adapted from ref. [25].

17
1.2.1 Proton reduction catalysts The work presented in this thesis only focusses on the proton reduction reac-tion, and therefore the water oxidation in the half-reactions will not be fur-ther discussed. Again, the key point of proton reduction in the half-reactions is the design of an efficient catalyst. In nature, photosynthesis for hydrogen evolving reaction employs hydrogenase enzymes capable of catalyzing pro-ton reduction or hydrogen oxidation (Formula 2).[34-36] Many of hydrogenas-es are found in non-photosynthetic organisms such as algae and cyanobacte-ria.[37-40] In terms of the constitution of active sites, hydrogenases can be classified into Fe-only hydrogenase, [Ni-Fe] hydrogenase, and [FeFe] hy-drogenase. In comparison, [FeFe] hydrogenase is a most efficient catalyst for H2 production with the rate reaching to 9000 turnovers per second at close to thermodynamic potential,[41-43] 10~100 times higher than that of other two hydrogenases.
2 2 2
Considering that a molecular catalyst must be efficient, robust and cheap enough to be cost-effective, an inspiration for such catalysts has been taken from the active site structure of the [FeFe] hydrogenase for proton reduction reaction. Today, large efforts have been directed toward the synthetic model-ing of the active site.[44-46] More details about the [FeFe] hydrogenase and the corresponding model complexes will be discussed in the following sections.
1.3 [FeFe] hydrogenases In 1998, Peters and co-workers extracted the [FeFe]-hydrogenase from Clos-tridium pasteurianum (CpI).[47] X-ray crystallography studies showed that structure of this enzyme is so-called ‘H-cluster’, which is made up of a binu-clear Fe cluster [2Fe2S] connected to a [4Fe4S] cubic cluster through a cys-teinate thiol, as shown in Figure 4. The iron connected to [4Fe4S] cubic clus-ter is named as ‘proximal’, Fep, another one ‘distal’, Fed. In the binuclear Fe cluster, the two Fe atoms are bridged by a dithiolate ligand, –S-CH2-X-CH2-S–. It has been well proven that the atoms in the X position are NH.[48,49] Moreover, infrared spectroscopic studies reveal that each Fe in the [2Fe2S] unit is coordinated to one CN- and one CO, as well as one more carbonyl dwells in a bridging position between the two irons. The bridging CO group provides the possibility for an excessively negative Fe atom to transfer part of its electron density to a CO group on a less negatively charged Fe atom.[42] Also, these CN- and CO ligands play a key role in stabilizing the low oxida-tion state of the Fe2 core. The Fe atoms in the binuclear Fe cluster both exist

18
with an octahedral coordination geometry but with reverse direction, which leads to an open coordination position at distal iron Fed.
Figure 4. FeFe-hydrogenase from Clostridium pasteurianum (CpI) and the structure of the active site.
In catalysis of [FeFe]-hydrogenase, two electrons and two protons will be required to produce hydrogen. The mechanism has not been fully revealed so far, especially for catalytic intermediates. However, in general, the [2Fe2S] unit is the only active site, and the [4Fe4S] cubic cluster could serve as an electron reservoir that affords a pathway for electron transfer from and to the active site during catalysis.[50,51] Also, the pendant amine in the azadithiolate ligand acting as proton relay for protonation in the metal center, has been frequently proposed,[49,52-55] and subsequent facilitates the dihydrogen bond formation such as faster intramolecular hydride/proton coupling. In addition, it seemed that involving a PCET reaction in catalysis by FeFe-hydrogenase plays a significant role for lowering overpotential (the extra energy needed to carry on the reaction over the thermodynamic potential),[56,57] because transferring electrons and protons together to the active site is energetically more favorable than stepwise mechanisms.
1.4 Synthetic Molecular Catalysts Inspired by [FeFe]-hydrogenase In practice, [FeFe]-hydrogenase is not available in large quantities and lim-ited to the commercialization for hydrogen production. Therefore, mimick-ing its active site is necessary, and the crystal structure obtained provides the basis for synthetic chemistry. The desirable properties of the synthetic mo-lecular catalysts should resemble those of FeFe-hydrogenase, such as high enough TOF, low overpotential and capable of operating in water at pH 7, etc.
Today, large research has been focusing on the structural and functional mimics of the active site to elucidate the structure-function relationship of the enzyme.

19
1.4.1 Structural mimics
Figure 5. Three parent complexes for a structural mimic of the active site of [FeFe]-hydrogenase. When research interest about mimicking the active site of FeFe-hydrogenase germinated from 1999, synthetic chemists only sought to focus on the [2Fe2S] unit and make the mimics resemble the structure of the active site as close as possible. Then, the synthetic pdt-functionalized (pdt = propanedithi-olate) complex as biomimetic catalysts for H2 production, the first structural mimic of the complete [2Fe2S] cluster, has been reported by Darensbourg.[58] The next generation of mimics carried out along different routes, with the group of Rauchfuss primarily focusing on structural mimics containing the azadithiolate (μ-adt) bridge,[59-61] and Lichtenberger et al. as well as other groups focusing on mimics containing aromatic bridges such as the ben-zenedithiolate (μ-bdt) bridge.[62-64] Later on, many research efforts extend to taking care of the ligands substituent based on the three parent complexes (see Figure 5). More than 300 compounds have been synthesized so far. Ex-periments in the electrochemical and visible-light-driven reduction of pro-tons show that most of them are active,[44,65,66] but unfortunately they all are much less efficient than the enzyme. Thus, mechanistic understanding for catalysts evaluation (heart of the thesis) will be needed.
1.4.2 Functional mimics Earlier DFT calculations[67] showed that the reaction site for proton reduction catalysis by the FeFe-hydrogenase mainly focused on the open coordination position at distal iron Fed. From the crystal structure of FeFe-hydrogenase, a pendant amine is placed in the second coordination sphere, which brings proton and hydride close enough to react and release as molecular hydrogen, or to help binding H2 for heterolytic cleavage. This suggests that one could make not only the structural mimics, but also directly mimic the reaction site at open coordination position, i.e. functional mimics of FeFe-hydrogenase. Significant progress has been achieved by incorporating the pendant amine into the second coordination sphere of a mononuclear nickel complex, [Ni(PR
2NR'2)2] (see Figure 6b), to mimicking the function of the proton shut-
tle in the active site of FeFe-hydrogenase.[68-72] Indeed, such catalyst shows
20
impressive enhancements in rate for H2 production,[72] which in turn provide insight into the function of the enzyme. Later on, such a functional approach is extended to introduce additional enzyme-like functionality such as pep-tides into the outer coordination sphere to create the proton channels and pursue more efficient and faster catalysis.[70,73-76]
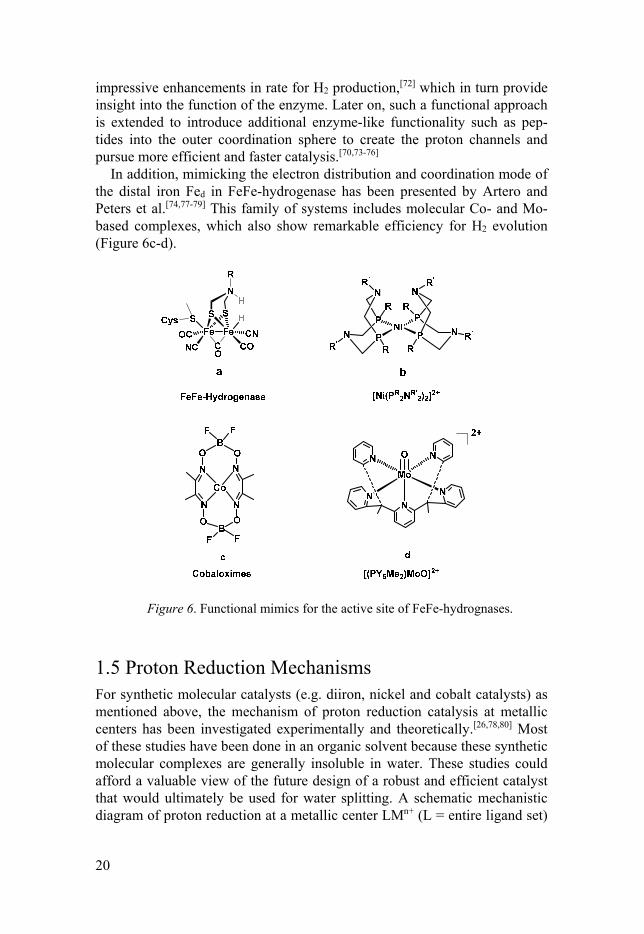
In addition, mimicking the electron distribution and coordination mode of the distal iron Fed in FeFe-hydrogenase has been presented by Artero and Peters et al.[74,77-79] This family of systems includes molecular Co- and Mo-based complexes, which also show remarkable efficiency for H2 evolution (Figure 6c-d).
Figure 6. Functional mimics for the active site of FeFe-hydrognases.
1.5 Proton Reduction Mechanisms For synthetic molecular catalysts (e.g. diiron, nickel and cobalt catalysts) as mentioned above, the mechanism of proton reduction catalysis at metallic centers has been investigated experimentally and theoretically.[26,78,80] Most of these studies have been done in an organic solvent because these synthetic molecular complexes are generally insoluble in water. These studies could afford a valuable view of the future design of a robust and efficient catalyst that would ultimately be used for water splitting. A schematic mechanistic diagram of proton reduction at a metallic center LMn+ (L = entire ligand set)

21
is presented in Figure 7, which basically summarizes the all possibilities of the catalytic cycle for H2 production. In spite of the apparent simplicity of 2 2 ⇌ reaction, the coupling of two electrons and two protons at the metallic center LMn+ would follow a number of different pathways.[81] It depends on many things such as catalyst itself and reaction conditions in-cluding solvent, the strength of acid added, temperature, reducing agent and concentrations. A common route is that reduction of LMn+ is followed by protonation to form the key intermediate hydride, [LM-H](n-1)+. [LM-H](n-1)+
can react in different ways: 1) it could involve the protonation of hydride to form dihydrogen bond, accompanied by release of H2 and regeneration of the starting material (pathway I) ; 2) further reduction of hydride [LM-H]n+ re-sults in a stronger base, which directly undergoes electrophilic attack from external proton to eliminate H2 (pathway II).[26,82] The protonation of the metal center can occur prior to reduction, depending on the strength of pro-ton source and the basicity of the metal center. In other words, whether the first step of these processes consists of the addition of an electron to LMn+ or that of a proton depends on the conjugated acid pKa of LMn+ and of acid used. When the catalyst LMn+ contains a basic ligand site, an additional lig-and protonation can occur before or after protonation of metal center, giving rise to a product that bears a proton and hydride. It can also be regarded as a potential proton relay to facilitate protonation at metal center.[82-84] Protona-tion at ligand site and metal center both shifts the reduction potential to be positive relative to LMn+, leading to decrease of thermodynamic cost. In addition, it is also critical to investigate the redox behavior of the metal complex LMn+ in the absence of acid when the catalytic reaction is initiated by an electron transfer step, because electron transfers may result in cleavage of bonds and bring about new protonation sites. Regardless of which step occurs first, the catalytic reaction of LMn+ follows either ET/PT sequence or PT/ET sequence. Few examples for possible coupled proton and electron transfers have been identified in the literature.[72]
While the general principles of catalytic H2 formation can easily be known, spectroscopic characterization on reduced and/or protonated intermediates associated with hydrogen evolution reaction is very rare. To gain a deeper understanding of proton reduction catalysis of metal complexes, relevant spectroscopic need to be developed to identify catalytic reaction intermedi-ates and account for potential reaction kinetics.

22
Figure 7. A general mechanistic scheme of proton reduction at metallic center LMn+.
1.6 Aim of the Thesis Mechanistic insight and kinetic characterization of catalytic intermediates for molecular systems of H2 formation is a prerequisite for the rational de-sign of molecular catalysts.
The thesis focusses on mimics of the active site of FeFe-hydrogenase. Most of the synthetic diiron complexes generally yield short-lived interme-diates upon reduction and protonation. Even under particular conditions (i.e. low temperature, CO atmosphere), the lifetimes of reduced states just extend into the limit of the spectroelectrochemistry (SEC) time scale. As a result, mechanistic information, specifically spectroscopic characterization, on those reduced and/or protonated species formed under turnover conditions has escaped observation by conventional techniques. For this reason, the thesis aims to employ time-resolved spectroscopies with better time resolu-tion addressing remaining questions of biomimetic diiron complexes. Specif-ic aims and/or questions are:
To build up what conditions for laser flash photolysis and stopped-flow experiments are suitable to initiate electron transfer and redox

23
catalysis so that the key catalytic intermediates of biomimetic diiron complexes can be identified and time-resolved.
To elucidate how chemical bonds in diiron complexes (e.g. FeFe bond, FeS bond,) detach and rebind upon reduction and protonation by investigating the structure of catalytic intermediates, and in what states the terminal carbonyl (CO) can be shifted to a bridging posi-tion between two irons.
To understand how protonation kinetics depend on driving force by studying the reactivity of reduced catalysts.
To study how and to what extent reduction does affect the prefer-ence for ligand versus metal protonation for those catalysts that con-tain a basic ligand site, and whether tautomerization/proton shuttling between a nitrogen base and the catalytic metal center can be ob-served on time scales relevant for catalysis.
The overall aim of the thesis is to obtain the understanding that is neces-sary to make the rational design of molecular photocatalytic systems for solar fuels formation like H2.

24
2. Fundamentals and Methods
2.1 Studied Systems 2.1.1 Photochemical Reduction Systems
Photosensitizer As mentioned in the introduction, a photocatalytic system for hydrogen for-mation is typically made up of a catalyst, an electron donor, and possibly photosensitizer, as well as an acid to afford the proton source. Such systems are used in chapter 3 and in Papers to obtain the one-electron reduced inter-mediates of biomimetic catalysts, and subsequent their reactivity towards protons. The catalysts investigated in the thesis are listed in Figure 9 in the following section, which are the structural mimics of the active site of FeFe-hydrogenase.
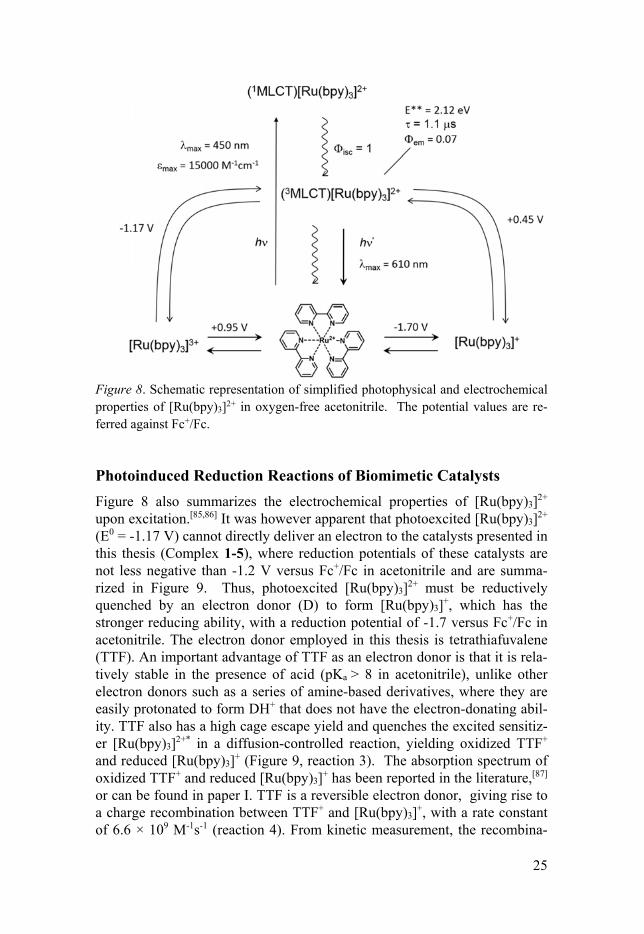
The photosensitizer is particularly important in photocatalytic systems, because the catalysts presented in this thesis are not intrinsically photoactive, mainly the reduced states and protonated states relevant to catalysis (see below). The photosensitizer used in this thesis is Ru(bpy)3(PF6)2 (bpy = 2, 2’-bipyridine), or its derivatives to tune the redox properties. [Ru(bpy)3]2+ is a quite photostable compound and shows an absorption maximum of 459 nm in acetonitrile that corresponds to a metal-to-ligand charge transfer (MLCT), with an extinction coefficient of about 14700 M-1cm-1 (see Figure 8).[85] Up-on excitation with visible light, [Ru(bpy)3]2+ can be initially excited to a singlet MLCT state, which then rapidly converts into its lowest triplet MLCT state via intersystem crossing (ISC). The nature of the MLCT excita-tion is to show a metal-centered oxidation process and six distinct ligand-centered reduction processes. Thus, [Ru(bpy)3]2+, in its 3MLCT excited-state, is both a good oxidant and a good reductant. In addition, the 3MLCT state has a lifetime of about 1s in oxygen-free acetonitrile, which is long-lived enough for electron transfer reactions occurring in homogenous solution if the donor-acceptor pair has a high cage yield, or gives a high yield of charge separation. When exciting the [Ru(bpy)3]2+ sample, the generating transient species generated is limited to the optical path length. It typically generates an excited concentration of about 2 M in UV-Vis (1 cm optical path length) and 20 M in IR (1 mm) experiments; details about experimental setup will be discussed in section 2.3.
25
Figure 8. Schematic representation of simplified photophysical and electrochemical properties of [Ru(bpy)3]2+ in oxygen-free acetonitrile. The potential values are re-ferred against Fc+/Fc. Photoinduced Reduction Reactions of Biomimetic Catalysts
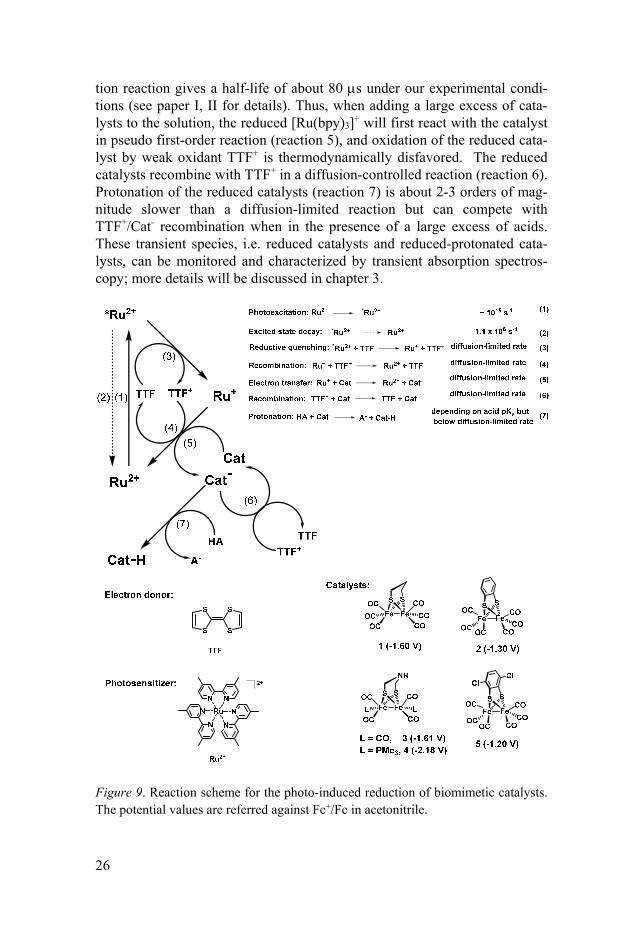
Figure 8 also summarizes the electrochemical properties of [Ru(bpy)3]2+ upon excitation.[85,86] It was however apparent that photoexcited [Ru(bpy)3]2+ (E0 = -1.17 V) cannot directly deliver an electron to the catalysts presented in this thesis (Complex 1-5), where reduction potentials of these catalysts are not less negative than -1.2 V versus Fc+/Fc in acetonitrile and are summa-rized in Figure 9. Thus, photoexcited [Ru(bpy)3]2+ must be reductively quenched by an electron donor (D) to form [Ru(bpy)3]+, which has the stronger reducing ability, with a reduction potential of -1.7 versus Fc+/Fc in acetonitrile. The electron donor employed in this thesis is tetrathiafuvalene (TTF). An important advantage of TTF as an electron donor is that it is rela-tively stable in the presence of acid (pKa > 8 in acetonitrile), unlike other electron donors such as a series of amine-based derivatives, where they are easily protonated to form DH+ that does not have the electron-donating abil-ity. TTF also has a high cage escape yield and quenches the excited sensitiz-er [Ru(bpy)3]2+* in a diffusion-controlled reaction, yielding oxidized TTF+ and reduced [Ru(bpy)3]+ (Figure 9, reaction 3). The absorption spectrum of oxidized TTF+ and reduced [Ru(bpy)3]+ has been reported in the literature,[87] or can be found in paper I. TTF is a reversible electron donor, giving rise to a charge recombination between TTF+ and [Ru(bpy)3]+, with a rate constant of 6.6 × 109 M-1s-1 (reaction 4). From kinetic measurement, the recombina-
26
tion reaction gives a half-life of about 80 s under our experimental condi-tions (see paper I, II for details). Thus, when adding a large excess of cata-lysts to the solution, the reduced [Ru(bpy)3]+ will first react with the catalyst in pseudo first-order reaction (reaction 5), and oxidation of the reduced cata-lyst by weak oxidant TTF+ is thermodynamically disfavored. The reduced catalysts recombine with TTF+ in a diffusion-controlled reaction (reaction 6). Protonation of the reduced catalysts (reaction 7) is about 2-3 orders of mag-nitude slower than a diffusion-limited reaction but can compete with TTF+/Cat- recombination when in the presence of a large excess of acids. These transient species, i.e. reduced catalysts and reduced-protonated cata-lysts, can be monitored and characterized by transient absorption spectros-copy; more details will be discussed in chapter 3.
Figure 9. Reaction scheme for the photo-induced reduction of biomimetic catalysts. The potential values are referred against Fc+/Fc in acetonitrile.

27
2.1.3 Chemical Reduction Systems In the photoinduced reduction of biomimetic catalysts as described above, the photogenerated reducing equivalent (about 2 M) reacts with a large excess of catalyst to reach pseudo first-order reaction, which favorably com-petes with charge recombination of TTF+ with [Ru(bpy)3]+. This leads to the fact that only one-electron reduced product can be obtained, even if further reduction of the one-electron reduced intermediate is thermodynamically favorable. A chemical reduction could, by contrast, achieve the goal of mul-ti-electron reduction of a catalyst if thermodynamically allowed. The reduct-ant employed in the chemical reduction experiments of biomimetic catalysts is cobaltocene (CoCp, E0 = -1.33 V vs Fc+/Fc) or decamethylcobaltocene (CoCp2*, E0 = -1.95 V).[88,89] The reactivity of CoCp2 and/or CoCp2* with a catalyst is addressed by stopped-flow IR spectroscopy, where the precursors of the chemical reaction are rapidly mixed; more details will be discussed in the following sections or seen in the Paper II, III and IV.
2.2 Techniques For the above systems, electron transfer and proton transfer reactions could result in changes in the electronic configuration, and structural geometry of catalysts. This, in general, leads to spectral differences in the absorption measurements. In this thesis, most relevant to the studies of the above sys-tems is absorption changes in the UV-Vis and mid-IR region. These con-cepts and techniques, including stopped-flow rapid mixing, steady state and transient absorption spectroscopy, will be described in the following sec-tions. In addition, electrochemistry is also a powerful tool to investigate re-dox catalysts. This is because the power of electrochemistry can provide the thermodynamic and kinetic parameter relevant for above systems involving redox processes of chemical species. It is a straightforward way to estimate and control the driving force of a chemical reaction. The corresponding techniques, i.e. cyclic voltammetry and bulk electrolysis, will be briefly ex-plained below.
2.2.1 Steady State Absorption Spectroscopy Spectroscopy is a tool that is used to describe the interaction between light with the material. Components of molecular systems mentioned in section 2.1 have well-defined energy levels and therefore point to different absorp-tion properties, which take the possibility to identify and distinguish molecu-lar species. The absorption spectrum of a sample is commonly recorded us-ing a steady state spectrophotometer, where a typical setup is presented in Figure 10a. It describes that a lamp passes through a monochromator, which

28
then allows one to do measurements at a single wavelength. The light is split into two beams: one passes through the cuvette containing samples to meas-ure the intensity of light after being absorbed by molecules, defined as I; the other beam is directed into a reference (in general only solvent) to record the intensity of light that is not absorbed by sample, defined as I0. The collected spectrum shows the fraction of incident light absorbed by the sample over a range of frequencies. The absorption extent by the samples is reported as absorbance (Abs) or optical density (OD), which can be described by the Beer-Lambert law (equation 2.1),
∙ ∙ (2.1)
where is the extinction coefficient of a molecule and the wavelength () dependent, c stands for the concentration of the sample, and l represents the path length. The shape of the spectrum can also give insights about the prop-erties of the electronic transition. For example, the absorption spectrum of [Ru(bpy)3]2+ features π → ∗ transition of ligands around 360 nm and MLCT absorption around 450 nm.
FT-IR (Fourier Transform Infrared) Spectroscopy is used to probe vibra-tional transitions of a molecule. Figure 10b presents a common setup for FTIR spectrometer, which is composed of an IR lamp, interferometer, sam-ple compartment, detector, processor, and a computer. The IR light passes the sample through the interferometer and reaches the detector. Then the signal is processed and converted to an interferogram. Finally, the result of the interferogram is transferred to a computer in which the Fourier transform is carried out. This results in a frequency domain trace, commonly intensity vs. wavenumber (cm-1). The basic measurement obtained in the IR spectros-copy is an IR spectrum, which provides more insights about the structure of a molecule; more details will be continuously discussed in the following sections (transient IR absorption spectroscopy).

29
Figure 10. Schematic representations of the working principles of a steady-state UV-Vis absorption (a) and (b) FTIR spectroscopy.
2.2.2 Nanosecond Transient Absorption Spectroscopy UV-Vis Spectroscopy. As discussed before, most of the studies in this thesis are based on the fact that the light absorbed by the sample should be able to trigger a chemical reaction, followed by absorption changes of the sample. This is not the case for steady-state techniques since the signal obtained in the steady-states measurements is averaged over the time scale of seconds, which is much longer than the rate of a chemical reaction. To investigate the reaction dynamics or mechanism for molecular systems as presented in sec-tion 2.1.1, nanosecond (ns) transient absorption spectroscopy, or called laser flash photolysis in the thesis, was used. Specifically, a nanosecond laser (pump light) is used to excite the sample, and probe light is then to measure the transmission of the sample. A chemical reaction can be monitored as a function of time by probing the absorption changes after excitation has oc-curred with pump light, expressed by equation 2.2,
∆ ∆
(2.2)
Figure 11 shows a sketch for ns transient absorption experiments. Specifical-ly, a laser pulse (about 8-12 ns duration) is generated from a Q-switched Nd:YAG laser (Quanta-Ray Pro-230), which incorporates the dual-rod oscil-lators with two amplifier stages. Consequently, each pulse that undergoes the amplification produces output energies up to about 1250 mJ and emits a photon at 1064 nm, which then interacts with a nonlinear crystal to produce

30
a secondary wave with a wavelength of 532 nm, called ‘double harmonic generation’. The resulting 532 nm wave can be mixed in the nonlinear crys-tal with the residual 1064 nm fundamental to produce a light at 355 nm, called ‘triple harmonic generation’. The 355 nm light then passes through an optical parametric oscillator (OPO), which converts it into a beam of a tuna-ble wavelength in the visible region (410-700 nm). The resulting visible light is then directed into the sample chamber and used to excite the sample (10 × 10 mm cuvette) at right-angle with respect to the probe light, which is pro-duced by a Xe arc lamp that has broad and strong output energy ranging from ultraviolet to near-infrared light. The probe light hits the sample and passes through a detector in which the data is analyzed by either a charge-coupled-device (CCD) camera or a photomultiplier tube (PMT) detector coupled with a monochromator in the UV-Vis region (200-850 nm). The CCD camera (in our case an iStar CCD 320) is an array of 1024 × 256 pix-els, so it can monitor multiple wavelengths and analyze the whole spectrum of probe light at a specific delay time. The PMT instead is to monitor one single wavelength, which is selected with the monochromator, as a function of time. This measurement generates a kinetic trace, which is collected with a digital oscilloscope, ultimately sending it to the computer.
Figure 11. Schematic representation of nanosecond transient UV-Vis spectroscopy. OPO: optical parametric oscillator; PMT: photomultiplier tube; CCD: charge-coupled-device camera. IR Spectroscopy. When involving the structural characterization of a sam-ple, UV-Vis absorption measurements typically cannot give more infor-mation on the structure of the molecule. The IR spectrum of a molecule, by contrast, provides valuable information to analyze the electron density and structural changes, since electronic and structural changes often alter the vibrational frequencies in a predictable way. After absorption of light in the mid-IR region, vibrations involving chemical bonds in a molecule typically give distinct signals and narrow peaks at higher wavenumbers than the elec-

31
tronic spectrum. This could be more easily assigned than UV/VIS-absorptions. In this thesis, the IR studies focused on the characteristic bands of carbonyl groups coordinated to the Fe centers in the synthetic diiron com-plexes. We know that carbon monoxide (CO) has a characteristic absorption band at 2143 cm-1. When it is coordinated to a metal, the IR absorption shifts to a lower wavenumber. This is caused by the π*-backbonding interaction, where the electrons are transferred from a d-orbital of the metal to the anti-bonding π* orbital of the CO.[90,91] This leads to the strengthening of the met-al-C bond and weakening of the C-O bond, which is reflected in decreases of the stretching frequencies for the C-O bond. It indicates that the stretching frequency of the CO ligand is very sensitive to the electron density of the metal center, and therefore measuring the vibrational spectrum of CO is an excellent strategy to gain insights into the structures of a biomimetic diiron catalyst upon oxidation, reduction, and protonation.
By combining IR spectroscopy with flash photolysis technique, we could achieve better time resolution down to a few nanoseconds for IR absorption measurements,[87] compared to a modern rapid-scan FTIR spectrometer where one can measure on time scales of tens of milliseconds, see details below. In the setup of transient IR spectroscopy, we use the same pump light as mentioned in the UV-Vis setup to excite the sample. Two continuous-wave quantum cascade (QC) mid-IR QC lasers were equipped to provide the IR light for probing, instead of Xe arc lamp. The IR probe light is over-lapped with the pump light in a suitable angle (see Figure 12). After the IR probe light passes through the sample, it is directed into a liquid-nitrogen-cooled mercury-cadmium-telluride (MCT) detector. Transient IR measure-ments do not provide an entire spectrum since the two mid-IR QC lasers are tunable with output wavelength between 1300-1965 cm-1 and 1960-2150 cm-
1, respectively. In other words, the IR probe light itself is monochromatic. Thus, the MCT detector monitors the transient event at a single wavelength as a function of time and generates the kinetic trace that is collected with the same oscilloscope as described in the UV-Vis detection.
Figure 12. Simplified scheme of nanosecond transient infrared experiments, probing in the mid-IR region.
32
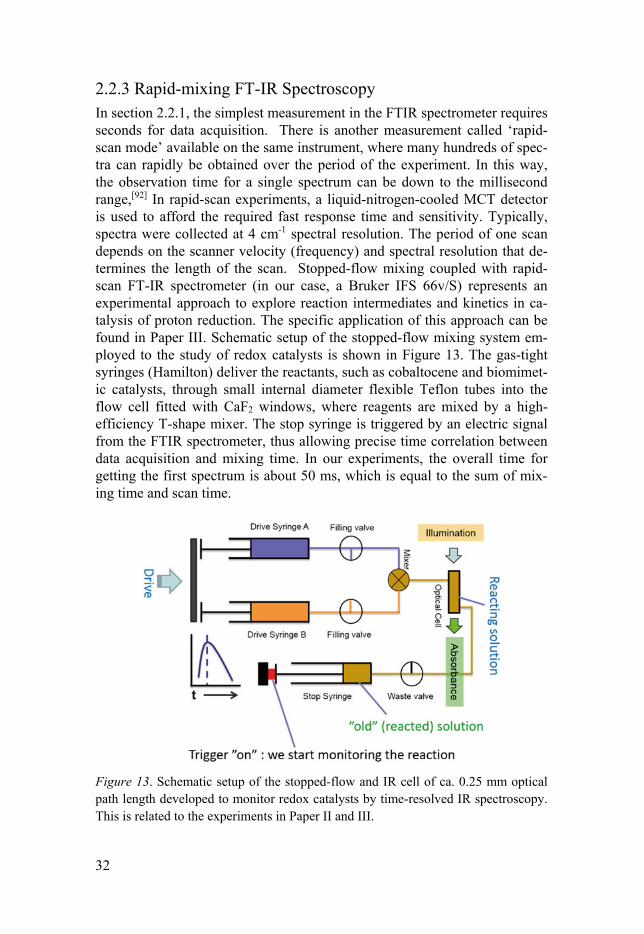
2.2.3 Rapid-mixing FT-IR Spectroscopy In section 2.2.1, the simplest measurement in the FTIR spectrometer requires seconds for data acquisition. There is another measurement called ‘rapid-scan mode’ available on the same instrument, where many hundreds of spec-tra can rapidly be obtained over the period of the experiment. In this way, the observation time for a single spectrum can be down to the millisecond range,[92] In rapid-scan experiments, a liquid-nitrogen-cooled MCT detector is used to afford the required fast response time and sensitivity. Typically, spectra were collected at 4 cm-1 spectral resolution. The period of one scan depends on the scanner velocity (frequency) and spectral resolution that de-termines the length of the scan. Stopped-flow mixing coupled with rapid-scan FT-IR spectrometer (in our case, a Bruker IFS 66v/S) represents an experimental approach to explore reaction intermediates and kinetics in ca-talysis of proton reduction. The specific application of this approach can be found in Paper III. Schematic setup of the stopped-flow mixing system em-ployed to the study of redox catalysts is shown in Figure 13. The gas-tight syringes (Hamilton) deliver the reactants, such as cobaltocene and biomimet-ic catalysts, through small internal diameter flexible Teflon tubes into the flow cell fitted with CaF2 windows, where reagents are mixed by a high-efficiency T-shape mixer. The stop syringe is triggered by an electric signal from the FTIR spectrometer, thus allowing precise time correlation between data acquisition and mixing time. In our experiments, the overall time for getting the first spectrum is about 50 ms, which is equal to the sum of mix-ing time and scan time.
Figure 13. Schematic setup of the stopped-flow and IR cell of ca. 0.25 mm optical path length developed to monitor redox catalysts by time-resolved IR spectroscopy. This is related to the experiments in Paper II and III.

33
2.2.4 Electrochemical Methods Electrochemistry is a powerful tool for the study of redox catalysts. In the electrochemical experiments, an electrode can be regarded as an electron donor. Thus, a catalytic system can be reduced to fewer components than discussed above. For example, only a catalyst and an acid are in practice needed in electrocatalytic H2 formation. Electrochemical behavior of a cata-lyst can be studied by cyclic voltammetry (CV), bulk electrolysis, and so on.
CV describes an experiment where the current is measured as a function of applied potential. This allows us to determine the redox potential of a couple if both oxidation states are stable over the time scales of measure-ment that is determined by the scan rate.
Bulk electrolysis describes an experiment where the working electrode is held at a constant potential (E) and current (A) is monitored over time (sec-onds). In this thesis, bulk electrolysis is used for the quantitatively conver-sion of the catalyst from its original oxidation state to a new oxidation state, either reduced or oxidized. The current is decreasing as the starting material is consumed, approaching zero when the conversion of the original oxidation state into a new oxidation state gets close to completion.
When combined electrochemistry with spectroscopic techniques, i.e. spec-troelectrochemical methods, it also affords deeper insights into the reactivity of a catalyst. Yet, it, in general, has a time resolution less than stopped-flow techniques.

34
3. Results and Discussion
In this chapter, the mechanistic proposals for proton reduction catalyzed by structural mimics of FeFe-hydrogenase are presented. The results will be summarized into three parts in terms of the differences amongst their bridg-ing ligands:
1) Photochemical reduction of FeFe(pdt)(CO)6 2) Proton-shuttling role of the azadithiolate ligand in the second coor-
dination sphere 3) The factors influencing the catalytic activity of diiron complexes
with alkyl bridging ligands 4) Photochemical and chemical reduction on FeFe(Cl2-bdt)(CO)6
This chapter will only summarize the significance of the results and implica-tions; details of what has been done can be found in the enclosed papers.
3.1 Photochemical Reduction of FeFe(pdt)(CO)6 3.3.1 General Considerations As already mentioned before, FeFe(pdt)(CO)6 (1) is the first member of a well-defined set of [(-SR)2Fe2L] complexes inspired by FeFe-hydrogenase. To make the structure resemble the enzyme as closely as possible, two CO ligands of hexacarbonyl can be replaced by electron donating ligands, such as cyanide (CN-), trimethylphosphine (PMe3) etc. The first example that was used to catalyze proton reduction to molecular hydrogen was the [FeFe(pdt)(CO)4(PMe3)(CN)]- compound,[58] including one CN- ligand and one PMe3 ligand. Later on, proton reduction catalyzed by the parent complex 1 was also investigated extensively.[93] Despite various strategies that have been employed to achieve better efficiency for hydrogen production,[84,94-97] there still is a large discrepancy in comparison with the natural enzyme both at turnover frequency and overpotential so far.[98] Understanding the mecha-nism, especially the electron transfer chemistry in catalysis, is fundamental for the design of better artificial systems. For compound 1, earlier spectro-/electrochemical studies showed that the major electron transfer step was a reversible one-electron step to give a singly reduced species 1-. However, the singly reduced state is unstable typically on the time scale of electrochemical experiments.[99] It has hence escaped characterization by direct spectroscopic

35
observation and information regarding structure and reactivity of the one-electron reduced intermediate has remained elusive. Understanding of this intermediate has been limited to results from computational studies and ki-netic data indirectly inferred by electrochemistry from catalytic currents under turnover conditions. In contrast, photochemical reduction using laser flash-quench methods is an excellent solution to characterize these short-lived catalyst intermediates, because in general it has a faster time resolution compared to those of electrochemical techniques.
In this section, I will primarily address the structures of the one-electron reduced intermediate of 1 triggered by the flash-quench-generated [Ru(dmb)3]+ reductant, and subsequent reactivity with acids. More details on the reaction scheme and explanation of flash-quench techniques can be found in chapter 2.
3.1.2 One-electron Reduced Intermediates In our methods, the reduction of complex 1 (E0 = 1.60 V vs. Fc+/Fc in ace-tonitrile)[100] by flash-quench-generated [Ru(dmb)3]+ (E0 = 1.81 V)[85] typi-cally takes place on the sub-microsecond time scales. Meanwhile, only 2 M or 20 M of a transient concentration of reduced intermediate is generated depending on the optical path length. This excludes any kind of bimolecular reaction, such as disproportionation or dimerization of reduced catalyst 1- as the half-life of these reactions will not be faster than several tens of micro-seconds under the conditions of the flash-quench experiment. In addition, further reduction of 1- will not be expected to occur. This is because the pho-togenerated [Ru(dmb)3]+ (assuming 2 M transient concentration) reacts with a large excess of 1, minimizing the possibility of double reduction caused by two subsequent encounters with [Ru(dmb)3]+. This allows for the detection of the one-electron reduced state during a long-time window. The reduction of complex 1 forming singly reduced intermediate gives three new absorption peaks (390 nm, 580 nm and 700 nm) in the UV-Vis region (see Figure 14a), which match previously reported spectrum after electrochemical reduction of 1.[93]
The reduction reaction of complex 1 was also followed by transient mid-IR spectroscopy, where one uses the frequencies of carbonyl ligands as a sensitive probe for differences in electron density at the metal center, as well as geometry changes. Of major importance is that the shift of the CO fre-quencies allows for the structural identification of singly reduced intermedi-ate. Reduction of complex 1 with flash-quench-generated [Ru(dmb)3]+ leads to the bleaches of all three absorption bands of 1 in the carbonyl region, and accompanying rise of 1- bands shifted by 75-90 cm-1 to lower frequencies, Figure 14c. Such a shift and similar spectral envelopes for 1 and 1- indicate that the geometry does not change significantly upon reduction. Supported by earlier DFT calculations,[101,102] the primary structural change on the one-
36
electron reduction of 1 is the lengthening of the distance of Fe-Fe bond by 0.28 Å but retention of an intact coordination sphere.
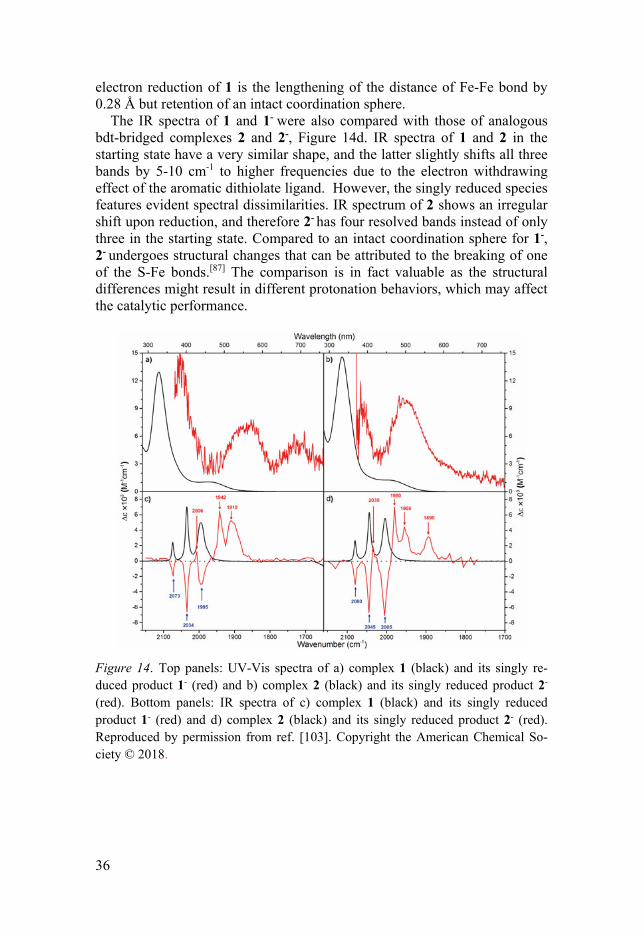
The IR spectra of 1 and 1- were also compared with those of analogous bdt-bridged complexes 2 and 2-, Figure 14d. IR spectra of 1 and 2 in the starting state have a very similar shape, and the latter slightly shifts all three bands by 5-10 cm-1 to higher frequencies due to the electron withdrawing effect of the aromatic dithiolate ligand. However, the singly reduced species features evident spectral dissimilarities. IR spectrum of 2 shows an irregular shift upon reduction, and therefore 2- has four resolved bands instead of only three in the starting state. Compared to an intact coordination sphere for 1-, 2- undergoes structural changes that can be attributed to the breaking of one of the S-Fe bonds.[87] The comparison is in fact valuable as the structural differences might result in different protonation behaviors, which may affect the catalytic performance.
Figure 14. Top panels: UV-Vis spectra of a) complex 1 (black) and its singly re-duced product 1- (red) and b) complex 2 (black) and its singly reduced product 2- (red). Bottom panels: IR spectra of c) complex 1 (black) and its singly reduced product 1- (red) and d) complex 2 (black) and its singly reduced product 2- (red). Reproduced by permission from ref. [103]. Copyright the American Chemical So-ciety © 2018.

37
3.1.2 Kinetic Behaviors of Protonation and Detection of Reduced-protonated Species The reactivity of singly reduced species of complex 1 towards different strength of acids was investigated by transient nanosecond UV-Vis- and mid-IR- spectroscopy. The acids examined in this investigation include p-toluenesolfonic acid (TsOH, pKa ~ 8.6), trichloroacetic acid (TCA, pKa ~ 10.7) and dichloroacetic acid (DCA, pKa ~ 13.3). Catalysis of proton reduc-tion with complex 1 has been proposed by electrochemical techniques.[63,93] One-electron reduced species was followed by the protonation reaction in the overall catalytic cycle. This means that the reaction rate for this step should be faster than dimerization or disproportionation of 1- under electro-chemical experiment conditions. However, it is typically difficult to measure the exact rate constant for protonation of the reduced catalyst using electro-chemical techniques.[93,100] With flash-quench methods, one can provide direct measurements for protonation reaction by following kinetic traces of singly reduced species signals. In Figure 15, in the presence of three differ-ent acids, the accelerated decays of kinetic traces at 700 nm, which is one of the absorption bands of 1-, were obtained. The observation of the accelerated decay suggests that 1- was protonated by acids. Fitting the decay with expo-nential function gives the pseudo-first-order rate constants kobs for protona-tion reaction, and plots of kobs as a function of acid concentration result in the second order rate constant of protonation. The rate constants with three acids examined in this investigation can be found in Paper I.[103]
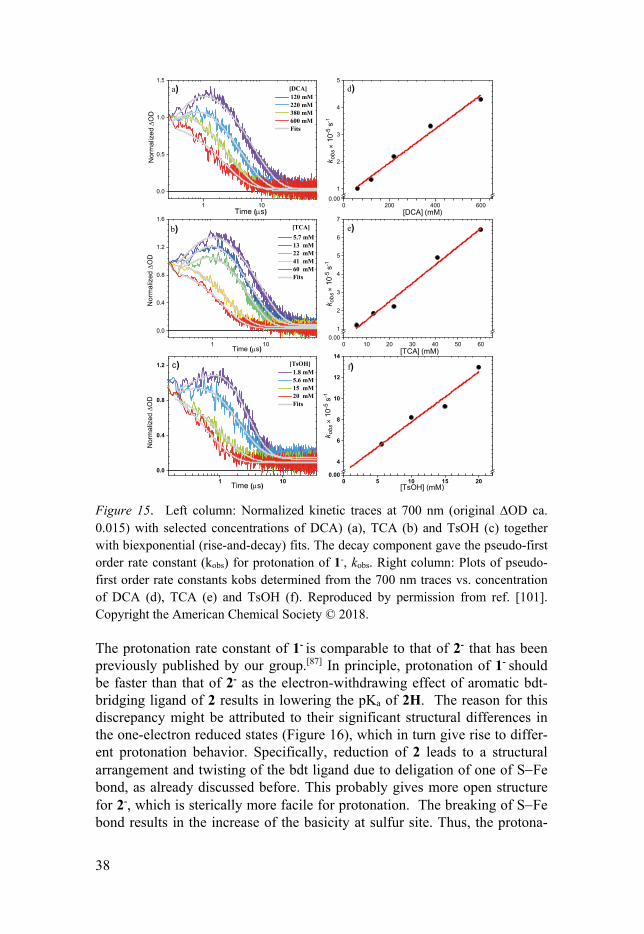
38
Figure 15. Left column: Normalized kinetic traces at 700 nm (original OD ca. 0.015) with selected concentrations of DCA) (a), TCA (b) and TsOH (c) together with biexponential (rise-and-decay) fits. The decay component gave the pseudo-first order rate constant (kobs) for protonation of 1-, kobs. Right column: Plots of pseudo-first order rate constants kobs determined from the 700 nm traces vs. concentration of DCA (d), TCA (e) and TsOH (f). Reproduced by permission from ref. [101]. Copyright the American Chemical Society © 2018. The protonation rate constant of 1- is comparable to that of 2- that has been previously published by our group.[87] In principle, protonation of 1- should be faster than that of 2- as the electron-withdrawing effect of aromatic bdt-bridging ligand of 2 results in lowering the pKa of 2H. The reason for this discrepancy might be attributed to their significant structural differences in the one-electron reduced states (Figure 16), which in turn give rise to differ-ent protonation behavior. Specifically, reduction of 2 leads to a structural arrangement and twisting of the bdt ligand due to deligation of one of SFe bond, as already discussed before. This probably gives more open structure for 2-, which is sterically more facile for protonation. The breaking of SFe bond results in the increase of the basicity at sulfur site. Thus, the protona-
1 10
0.0
0.5
1.0
1.5
1 10
0.0
0.4
0.8
1.2
1.6
0 200 400 6000.00
1
2
3
4
5
0 10 20 30 40 50 600.00
1
2
3
4
5
6
7
0 5 10 15 200.00
4
6
8
10
12
14
1 10
0.0
0.4
0.8
1.2
a)
No
rma
lize
d O
D
Time (s)
120 mM 220 mM 380 mM 600 mM Fits
[DCA]
b)
Time (s)
Nor
ma
lize
d
OD
5.7 mM 13 mM 22 mM 41 mM 60 mM Fits
[TCA]
d)
[DCA] (mM)
k obs
1
0-5 s
-1
e)
[TCA] (mM)
k ob
s
10- 5
s-1
f)
[TsOH] (mM)
k obs
1
0-5
s-1
c)
Time (s)
No
rmal
ize
d O
D
[TsOH] 1.8 mM 5.6 mM 15 mM 20 mM Fits

39
tion of 2- might initially occur at the sulfur site. Reduction of 1 significantly lengthens the distance of Fe-Fe bond and weakens it a lot, whereas the pro-tonation of reduced catalyst corresponds to the two metal centers resulting in a bridging hydride.[104] Instead, the breaking of one SFe bond in 2- mini-mizes the weakening of FeFe bond, which could also lead to protonation in 2- more easily.[64]
Figure 16. Structural indication of all three states of [Fe2(pdt)(CO)6] (1) and [Fe2(bdt)(CO)6] (2). The protonation states are also transient intermediates in catalysis of proton reduction. Detecting the reduced and protonated species could be an im-portant step to understand the kinetic bottlenecks limiting the catalytic effi-ciency. The singly reduced intermediate 1- can be easily detected using flash-quench methods coupled with mid-IR probing. However, it’s much more challenging to detect the reduced-protonated species, as protonation of 1-, forming 1H, leads to a blue shift in the IR spectrum back to higher frequen-cies where the neutral complex 1 is bleaching.
Protonation of 1- was in contrast to those of neutral complex bearing elec-tron-donating ligands PMe3, FeFe(pdt)(CO)4(PMe3)2.[105] Two PMe3 ligands resulted in the enhanced electron density at the metal center, FeFe(pdt)(CO)4(PMe3)2 complex can be therefore protonated in the Fe2(0,0) state. In comparison, protonation of 1- (~107 M-1s-1 with TsOH) is much fast-er even with weaker acid than those of neutral complex (ca. 103 M-1s-1 with HBF4). This could be attributed to the fact that larger PMe3 groups in those neutral complexes significantly lower the proton accessibility. Alternatively, the protonation mechanism in 1- is considered as direct proton transfer from the acid to FeFe bond, in contrast to the proposed indirect proton transfer via the CO ligands rotation of the metal centers in those neutral complexes; see more details in Paper I.[103]

40
3.2 Proton-shuttling Role of Azadithiolate Ligand 3.2.1 General The pendant amine in the azadithiolate (adt) bridge has been regarded as potential proton relay for facilitating either protonation of the iron center of model compounds or reaction with terminal hydride in catalysis of [FeFe]-hydrogenase. Therefore, mimicking the function of proton relays in the ac-tive site of this enzyme has been applied to the development of synthetic molecular electrocatalysts for H2 evolution. In this context, DuBois and co-workers perhaps provide the most comprehensive studies of the proton relay functions of amines with Nickel (Ni)-containing catalysts.[72] Upon reduction, one of their catalysts could convert to a transition state that contains a proto-nated nitrogen at a pendant amine of the second coordination sphere and a hydride on the Ni center.[68] Regarding structural mimics of FeFe-hydrogenase with adt ligand, the same role has been more recently proposed in an electrochemical and computational study by Gloaguen and co-workers.[106] Their studies pointed out that the superior catalytic performance of adt-based complexes relative to their pdt- and bdt-based analogues was attributed to the formation of hydride intermediate by proton shuttling from the nitrogen site of adt ligand. The work presented in this section was there-fore motivated by these proposed hypotheses regarding the role of the adt ligand as a proton relay, and if the formation of terminal hydride intermedi-ates in these model complexes could be inferred from direct spectroscopic investigation.
In this section, I will address these electron and proton transfer reactions of diiron complexes, FeFe(adt)(CO)6 (3) and [FeFe(adt)(CO)4(PMe3)2] (4), with the adt ligand investigated by time-resolved IR and UV-Vis spectrosco-py to elucidate whether the bridged-head nitrogen atom acts as proton relay to the metal center in catalytic H2 formation.
3.2.2 FeFe(adt)(CO)6 Complex Laser flash photolysis. The redox potential of complex 3 is very similar to that of complex 1, and therefore we could use the same experimental proce-dure as that presented in section 3.1 to initiate one-electron reduction of complex 3 (see reaction equations below); more details about the reaction scheme were explained in chapter 2.
Ru dmb ∗ TTF → Ru dmb TTF 1
Ru dmb Ru dmb 2
TTF.
TTF 3
The resulting electronic absorption bands of 3- (390 nm, 580 nm and 700 nm) and its vibrational spectrum in the carbonyl region (2006 cm-1, 1945 cm-1 and

41
1910 cm-1, Fig. 18a) resemble closely those of 1-.[103] Yet, unlike complexes 1 and 2, where protonation must be initiated by a reduction step, complex 3 contains a protonable NH group that allows one to investigate either an ET/PT or a PT/ET mechanism depending on the strength of the acid.
Figure 17. Structural indications of complex 3 in different oxidation and protonated states. According to the results reported by Gloaguen and co-workers,[106] with weaker acid such as ClCH2COOH (pKa = 15.3 in acetonitrile),[107] the H2 production is catalyzed via an ET/PT mechanism. In contrast, a PT/ET se-quence was followed when a stronger acid such as 2, 5-dichlorobenzenesulfonic acid (Cl2BSA, pKa = 6.7) was used as proton source. Our experimental results suggest that no matter what mechanism is involved, the initial protonation occurs at adt-N site, Figure 18b, c. This ex-cludes the possibility of a ET/PT route that results in direct formation of a hydride complex, 3Hy, without involvement of nitrogen site, as has been presented by Gloaguen et al. based on DFT calculated pKa values of 18.8 (bridging hydride), 17.1 (terminal hydride) and 13.8 (adt-N protonation).[106] The protonation of the adt-N site results in a shift of about 20 cm-1 to the higher wavenumber of the C-O bands, characteristic shift from the protona-tion of the bridged-head nitrogen. Both ET/PT and PT/ET mechanisms pro-duce the same initial product 3H (2025, 1965 and 1935 cm-1). 3H formed by ET/PT sequence decays with second-order kinetics, which is attributed to charge recombination of 3H with TTF+ in a diffusion-controlled reaction. No tautomerization from 3H to 3Hy was observed on the time scales of charge recombination. The decay of 3H generated by a PT/ET route with Cl2BSA is on the other hand much faster than the former. The accelerated decay cannot be assigned to a tautomerization process (Figure 18c). This is because 3Hy
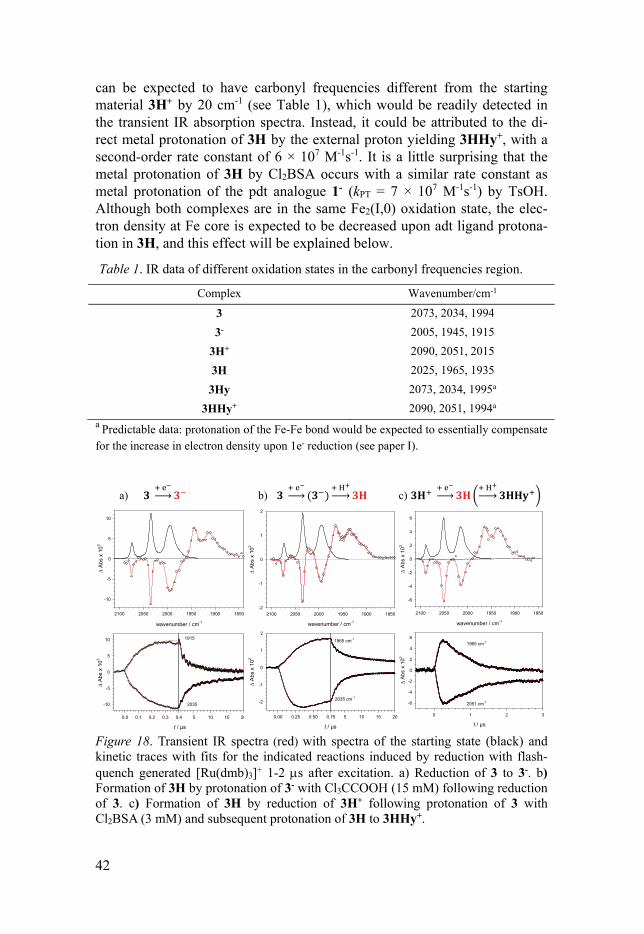
42
can be expected to have carbonyl frequencies different from the starting material 3H+ by 20 cm-1 (see Table 1), which would be readily detected in the transient IR absorption spectra. Instead, it could be attributed to the di-rect metal protonation of 3H by the external proton yielding 3HHy+, with a second-order rate constant of 6 × 107 M-1s-1. It is a little surprising that the metal protonation of 3H by Cl2BSA occurs with a similar rate constant as metal protonation of the pdt analogue 1- (kPT = 7 × 107 M-1s-1) by TsOH. Although both complexes are in the same Fe2(I,0) oxidation state, the elec-tron density at Fe core is expected to be decreased upon adt ligand protona-tion in 3H, and this effect will be explained below.
Table 1. IR data of different oxidation states in the carbonyl frequencies region.
Complex Wavenumber/cm-1
3 2073, 2034, 1994
3- 2005, 1945, 1915
3H+ 2090, 2051, 2015
3H 2025, 1965, 1935
3Hy 2073, 2034, 1995a
3HHy+ 2090, 2051, 1994a
a Predictable data: protonation of the Fe-Fe bond would be expected to essentially compensate for the increase in electron density upon 1e- reduction (see paper I).
a)
b)
c)
Figure 18. Transient IR spectra (red) with spectra of the starting state (black) and kinetic traces with fits for the indicated reactions induced by reduction with flash-quench generated [Ru(dmb)3]+ 1-2 s after excitation. a) Reduction of 3 to 3-. b) Formation of 3H by protonation of 3- with Cl3CCOOH (15 mM) following reduction of 3. c) Formation of 3H by reduction of 3H+ following protonation of 3 with Cl2BSA (3 mM) and subsequent protonation of 3H to 3HHy+.
wavenumber / cm-1
185019001950200020502100
A
bs x
10
3
-10
-5
0
5
10
t / µs
0.0 0.1 0.2 0.3 0.4
A
bs x
103
-10
-5
0
5
10
5 10 15 20
1915
2035
wavenumber / cm-1
185019001950200020502100
A
bs
x 10
2
-2
-1
0
1
2
t / µs
0.00 0.25 0.50 0.75
A
bs x
102
-2
-1
0
1
2
5 10 15 20
1965 cm-1
2035 cm-1
wavenumber / cm-1
185019001950200020502100
A
bs
x 10
3
-6
-4
-2
0
2
4
6
t / µs
0 1 2 3
A
bs
x 1
03
-6
-4
-2
0
2
4
61965 cm-1
2051 cm-1
43
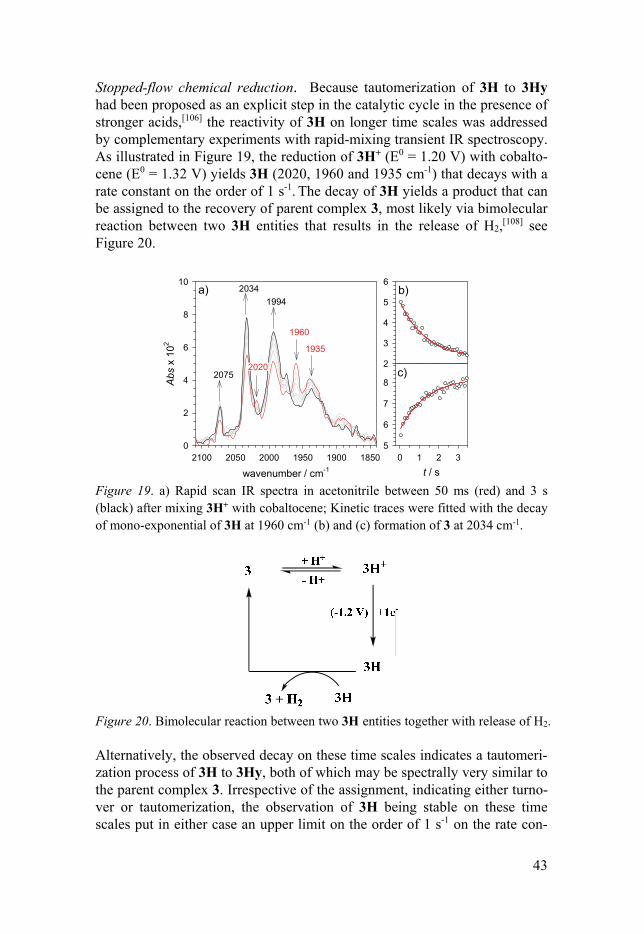
Stopped-flow chemical reduction. Because tautomerization of 3H to 3Hy had been proposed as an explicit step in the catalytic cycle in the presence of stronger acids,[106] the reactivity of 3H on longer time scales was addressed by complementary experiments with rapid-mixing transient IR spectroscopy. As illustrated in Figure 19, the reduction of 3H+ (E0 = 1.20 V) with cobalto-cene (E0 = 1.32 V) yields 3H (2020, 1960 and 1935 cm-1) that decays with a rate constant on the order of 1 s-1. The decay of 3H yields a product that can be assigned to the recovery of parent complex 3, most likely via bimolecular reaction between two 3H entities that results in the release of H2,[108] see Figure 20.
Figure 19. a) Rapid scan IR spectra in acetonitrile between 50 ms (red) and 3 s (black) after mixing 3H+ with cobaltocene; Kinetic traces were fitted with the decay of mono-exponential of 3H at 1960 cm-1 (b) and (c) formation of 3 at 2034 cm-1.
Figure 20. Bimolecular reaction between two 3H entities together with release of H2. Alternatively, the observed decay on these time scales indicates a tautomeri-zation process of 3H to 3Hy, both of which may be spectrally very similar to the parent complex 3. Irrespective of the assignment, indicating either turno-ver or tautomerization, the observation of 3H being stable on these time scales put in either case an upper limit on the order of 1 s-1 on the rate con-
a)
wavenumber / cm-1
185019001950200020502100
Abs
x 1
02
0
2
4
6
8
10
1935
1960
2075
2034
1994b)
X Data
0 1 2 32
3
4
5
6
c)
t / s
0 1 2 35
6
7
8
2020

44
stant of tautomerization. For the electrochemical H2 formation with 3, pseu-do-first-order catalytic rate constant on the order of 103 to 104 s-1 have been determined and the proposed mechanism involves intramolecular isomeriza-tion of 3H to 3Hy according to computational results.[106] To account for the observed turnover frequencies from electrochemical experiments, the tau-tomerization rate constant would have to exceed the upper limit determined from the direct observation of 3H by several orders of magnitude. Our re-sults, however, exclude that the catalytic cycle passes through a tautomeriza-tion step. Instead, the electrochemical reaction most likely involves the for-mation of 3HHy+ by direct protonation of 3H from the external acid. This is due to the fact that the acid concentration is much larger in these experi-ments, the same manner as in the flash photolysis experiments.
Regarding the superior catalytic performance of complex 3 in direct com-parison with its pdt (1) and bdt (2) counterparts, protonation at nitrogen site shifts the reduction potential by about 300 mV to a less negative position, and therefore lowers the overpotential in the catalytic cycle.[106] In principle, shift of about 300 mV in reduction potential for the 3H+/3H compared to 1/1- should drop in pKa by about 5 upon adt protonation. However, the low-ered driving force for protonation of adt-protonated catalyst does not slow down the catalytic rate constant (see above), which is faster by one to two orders of magnitude than that expected. This could be hence attributed to the fact that the barrier for metal protonation might be lowered by structural changes at Fe core induced by the adt ligand protonation.[83,109] Also, the higher catalytic rates of 3 could possibly be attributed to the fact that bridg-ing hydride undergoes a structural rearrangement forming a transient inter-mediate with a terminally bound hydride ligand that is more active for catal-ysis,[110-113] as shown in Figure 21. The follow-up reaction of intramolecular hydride/proton coupling could precede the release of H2 more rapidly rela-tive to its pdt (1) and bdt (2) analogues.
Figure 21. The proposed route for H2 elimination from 3HHy+ through a transient intermediate with terminally bound hydride ligand.
3.2.3 FeFe(adt)(CO)4(PMe3)2 Complex In case of complex 3, the reason why the amine of the adt ligand does not work as a proton shuttle to metal center could be the poor basicity of the hexacarbonyl metal core, giving a small driving force for tautomerization of

45
3H to 3Hy. In this text, trimethylphosphine (PMe3) ligands have been intro-duced at the diiron center to prepare complex 4 (Figure 22), and also mimic the electron donating CN- ligand in the active site of [FeFe]-hydrogenase enzyme[114]. The introduction of PMe3 ligands significantly increases the electron density of the Fe core, which allows for the investigation of the possibility of the amine functionality working as proton relay to the metal core. Indeed, previous experimental studies for complex 4 showed that tau-tomerization of 4H+ to 4Hy+ can be catalyzed by chloride ions (Cl-), but the rate is remarkably slow (ktauto. = 2 M-1s-1).[83]
Figure 22. Structural indications of complex 4 in different oxidation states
The focal interest is in the tautomerization upon reduction of 4H+ induced by a laser flash-quench-generated photoreductant, without the help of chloride ions as catalysts. The protonation of complex 4 (E0 = -2.19 V vs. Fc+/Fc) in the bridged-head nitrogen atom shifts the reduction potential by about 610 mV to less negative potentials for 4H+ (E0 = -1.58 V).[114] Thus, the flash-quench-generated [Ru(dmb)3]+ (E0 = 1.81 V) is thermodynamically able to reduce 4H+. A larger driving force for tautomerization of 4H to 4Hy would be expected upon reduction. This allows for the study of a potential PCET process if the tautomerization of 4H to 4Hy is rapid enough. The complex 4 is protonated by Cl-anilinium (pKa = 9.7)[115] to prepare the 4H+, which combined with sensitizer [Ru(dmb)3]2+ and electron donor TTF constitutes
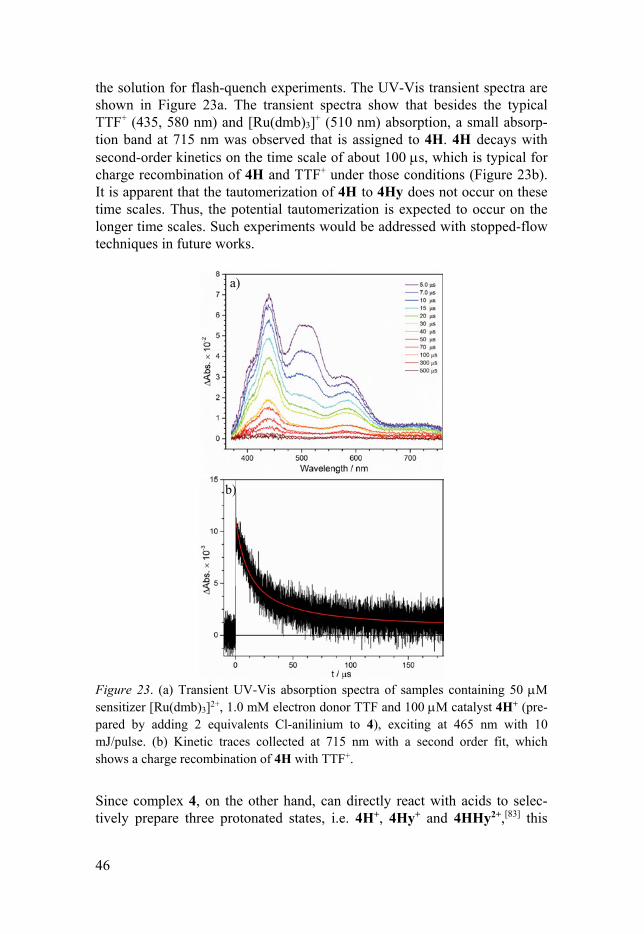
46
the solution for flash-quench experiments. The UV-Vis transient spectra are shown in Figure 23a. The transient spectra show that besides the typical TTF+ (435, 580 nm) and [Ru(dmb)3]+ (510 nm) absorption, a small absorp-tion band at 715 nm was observed that is assigned to 4H. 4H decays with second-order kinetics on the time scale of about 100 s, which is typical for charge recombination of 4H and TTF+ under those conditions (Figure 23b). It is apparent that the tautomerization of 4H to 4Hy does not occur on these time scales. Thus, the potential tautomerization is expected to occur on the longer time scales. Such experiments would be addressed with stopped-flow techniques in future works.
Figure 23. (a) Transient UV-Vis absorption spectra of samples containing 50 M sensitizer [Ru(dmb)3]2+, 1.0 mM electron donor TTF and 100 M catalyst 4H+ (pre-pared by adding 2 equivalents Cl-anilinium to 4), exciting at 465 nm with 10 mJ/pulse. (b) Kinetic traces collected at 715 nm with a second order fit, which shows a charge recombination of 4H with TTF+.
Since complex 4, on the other hand, can directly react with acids to selec-tively prepare three protonated states, i.e. 4H+, 4Hy+ and 4HHy2+,[83] this
a)
b)
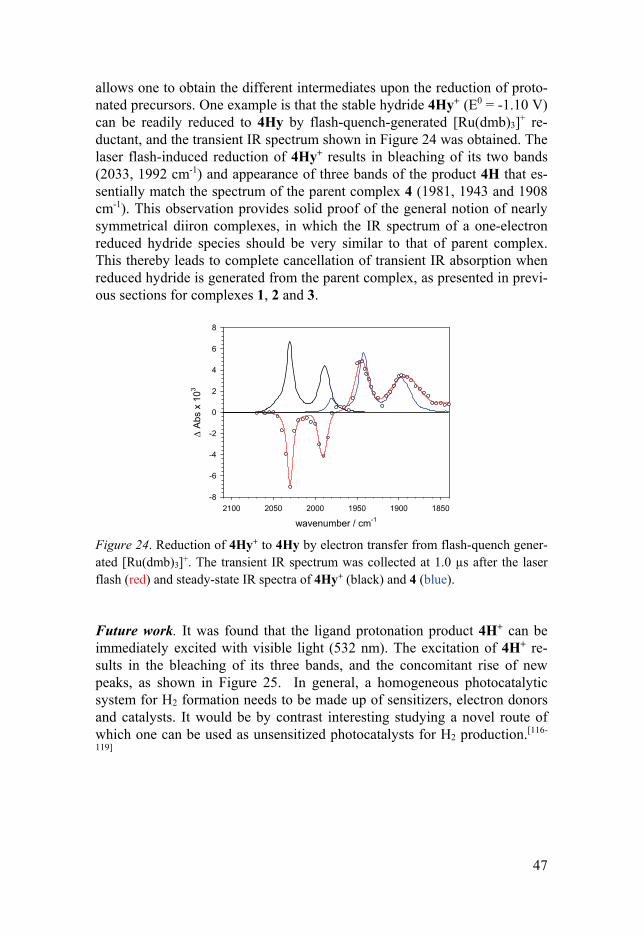
47
allows one to obtain the different intermediates upon the reduction of proto-nated precursors. One example is that the stable hydride 4Hy+ (E0 = -1.10 V) can be readily reduced to 4Hy by flash-quench-generated [Ru(dmb)3]+ re-ductant, and the transient IR spectrum shown in Figure 24 was obtained. The laser flash-induced reduction of 4Hy+ results in bleaching of its two bands (2033, 1992 cm-1) and appearance of three bands of the product 4H that es-sentially match the spectrum of the parent complex 4 (1981, 1943 and 1908 cm-1). This observation provides solid proof of the general notion of nearly symmetrical diiron complexes, in which the IR spectrum of a one-electron reduced hydride species should be very similar to that of parent complex. This thereby leads to complete cancellation of transient IR absorption when reduced hydride is generated from the parent complex, as presented in previ-ous sections for complexes 1, 2 and 3.
Figure 24. Reduction of 4Hy+ to 4Hy by electron transfer from flash-quench gener-ated [Ru(dmb)3]+. The transient IR spectrum was collected at 1.0 µs after the laser flash (red) and steady-state IR spectra of 4Hy+ (black) and 4 (blue).
Future work. It was found that the ligand protonation product 4H+ can be immediately excited with visible light (532 nm). The excitation of 4H+ re-sults in the bleaching of its three bands, and the concomitant rise of new peaks, as shown in Figure 25. In general, a homogeneous photocatalytic system for H2 formation needs to be made up of sensitizers, electron donors and catalysts. It would be by contrast interesting studying a novel route of which one can be used as unsensitized photocatalysts for H2 production.[116-
119]
wavenumber / cm-1
185019001950200020502100
A
bs x
10
3
-8
-6
-4
-2
0
2
4
6
8
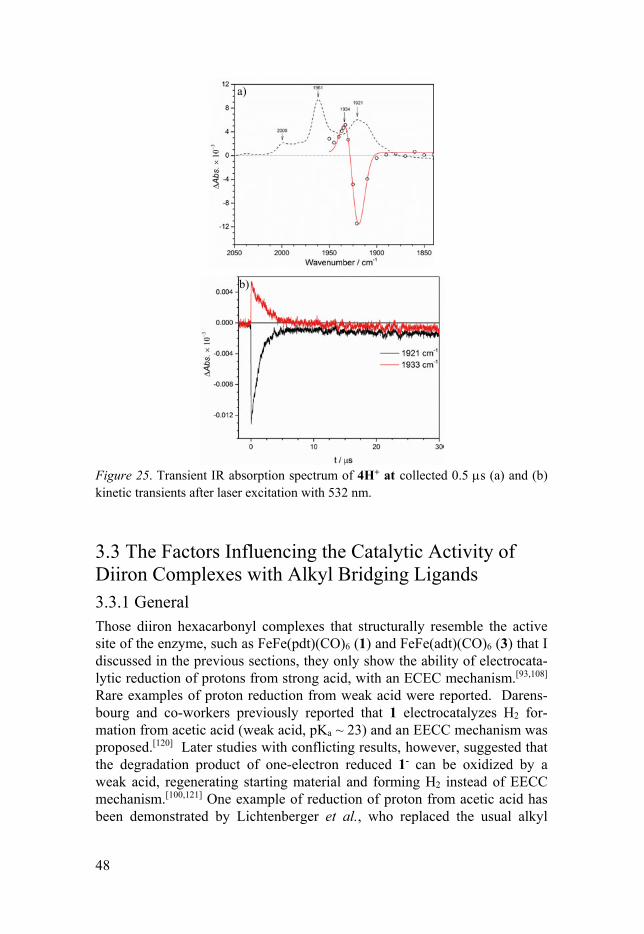
48
Figure 25. Transient IR absorption spectrum of 4H+ at collected 0.5 s (a) and (b) kinetic transients after laser excitation with 532 nm.
3.3 The Factors Influencing the Catalytic Activity of Diiron Complexes with Alkyl Bridging Ligands 3.3.1 General Those diiron hexacarbonyl complexes that structurally resemble the active site of the enzyme, such as FeFe(pdt)(CO)6 (1) and FeFe(adt)(CO)6 (3) that I discussed in the previous sections, they only show the ability of electrocata-lytic reduction of protons from strong acid, with an ECEC mechanism.[93,108] Rare examples of proton reduction from weak acid were reported. Darens-bourg and co-workers previously reported that 1 electrocatalyzes H2 for-mation from acetic acid (weak acid, pKa ~ 23) and an EECC mechanism was proposed.[120] Later studies with conflicting results, however, suggested that the degradation product of one-electron reduced 1- can be oxidized by a weak acid, regenerating starting material and forming H2 instead of EECC mechanism.[100,121] One example of reduction of proton from acetic acid has been demonstrated by Lichtenberger et al., who replaced the usual alkyl
a)
b)

49
dithiolate bridging ligand with an aromatic dithiolate ligand such as ben-zenedithiolate (bdt).[64] This stabilized the reduced forms of the catalyst and led to inverted potentials for the first two reduction steps (FeIFeI FeIFe0 Fe0Fe0). The bdt analogue will be discussed in Chapter 3.4. Apparently, the ability of diiron complexes with alkyl dithiolate bridging ligands to catalyze H2 formation is limited by the poor stability in oxidation states below FeIFeI level (starting state) particularly under weakly acidic conditions.
In general, for these types of complexes (i.e. 1 and 3), two-electron reduc-tion to form the dianion is essential to catalyze proton reduction from weak acid. Potential degradation of one-electron reduced intermediates would dominate if second-reduction step slows down in the system. In this chapter (also related to Paper IV), we investigate the reaction of complex 3 with a chemical reductant, decamethylcobaltocene (CoCp2*, E0 = -1.95 V vs. Fc+/Fc), both in the absence and in the presence of weak acids, exploring the formation and degradation pathways of catalytic intermediates. The follow-ing sections will summarize the findings.
3.3.2 Two-electron Reduced Intermediate of Complex 3 In Paper IV, reduction of 3 was addressed by stopped-flow IR spectroscopy, where mixing of complex 3 with CoCp2* is finished in 50-80 ms.[122] In the absence of acid, complex 3 is chemically reduced by CoC2*. This reaction cleanly generates two-electron reduced intermediate 32- because of the shift of IR absorption bands by ca. 125 cm-1 to lower wavenumber (Figure 26).[87] The bridging carbonyl at 1680 cm-1 is in general regarded as the unique fea-ture of the two-electron reduced product. IR absorption bands of 32- in the carbonyl region were given in Table 2. Note that previous electrochemical studies did not lead to the formation of cleanly two-electron reduced spe-cies.[93] This might be attributed to the notion that in electrochemical exper-iments singly reduced species reacts with starting material on the electrode prior to further reduction. In our case, complex 3 could sufficiently react with excess CoCp2* in homogeneous solution as long as two reagents mix.
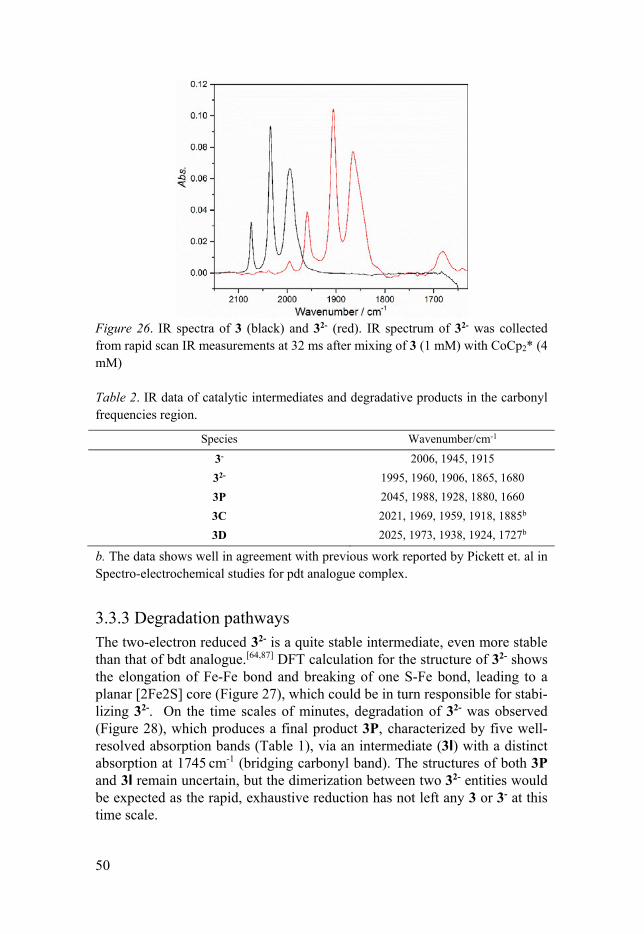
50
Figure 26. IR spectra of 3 (black) and 32- (red). IR spectrum of 32- was collected from rapid scan IR measurements at 32 ms after mixing of 3 (1 mM) with CoCp2* (4 mM) Table 2. IR data of catalytic intermediates and degradative products in the carbonyl frequencies region.
Species Wavenumber/cm-1
3- 2006, 1945, 1915
32- 1995, 1960, 1906, 1865, 1680
3P 2045, 1988, 1928, 1880, 1660
3C 2021, 1969, 1959, 1918, 1885b
3D 2025, 1973, 1938, 1924, 1727b
b. The data shows well in agreement with previous work reported by Pickett et. al in Spectro-electrochemical studies for pdt analogue complex.
3.3.3 Degradation pathways The two-electron reduced 32- is a quite stable intermediate, even more stable than that of bdt analogue.[64,87] DFT calculation for the structure of 32- shows the elongation of Fe-Fe bond and breaking of one S-Fe bond, leading to a planar [2Fe2S] core (Figure 27), which could be in turn responsible for stabi-lizing 32-. On the time scales of minutes, degradation of 32- was observed (Figure 28), which produces a final product 3P, characterized by five well-resolved absorption bands (Table 1), via an intermediate (3I) with a distinct absorption at 1745 cm-1 (bridging carbonyl band). The structures of both 3P and 3I remain uncertain, but the dimerization between two 32- entities would be expected as the rapid, exhaustive reduction has not left any 3 or 3- at this time scale.
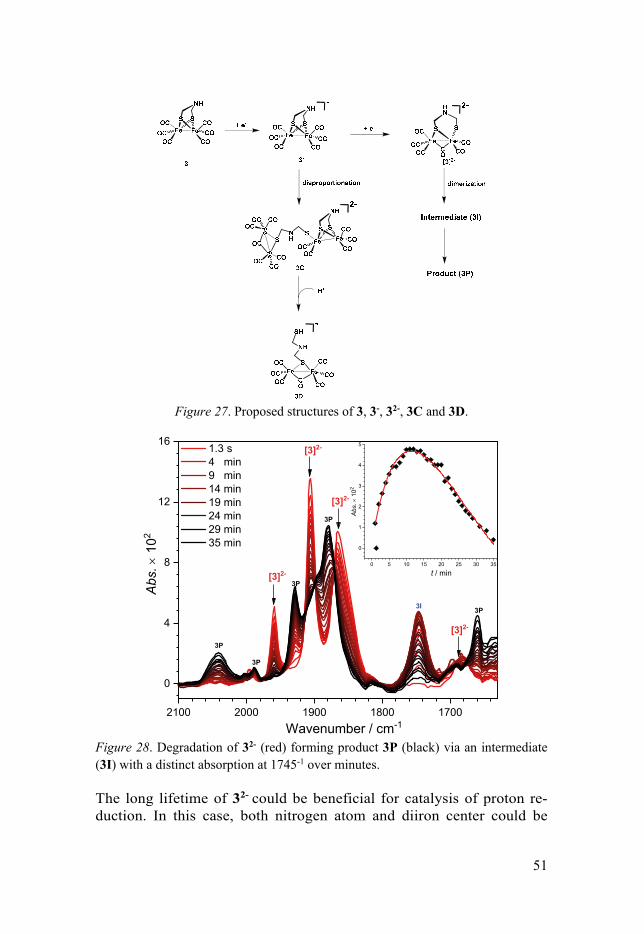
51
Figure 27. Proposed structures of 3, 3-, 32-, 3C and 3D.
Figure 28. Degradation of 32- (red) forming product 3P (black) via an intermediate (3I) with a distinct absorption at 1745-1 over minutes.
The long lifetime of 32- could be beneficial for catalysis of proton re-duction. In this case, both nitrogen atom and diiron center could be
2100 2000 1900 1800 1700
0
4
8
12
16
0 5 10 15 20 25 30 35
0
1
2
3
4
5
3P
3P
3P
3P
3P
[3]2-
[3]2-
[3]2-
3I
Abs
. 1
02
Wavenumber / cm-1
1.3 s 4 min 9 min 14 min 19 min 24 min 29 min 35 min
[3]2-
Abs
. 1
02
t / min
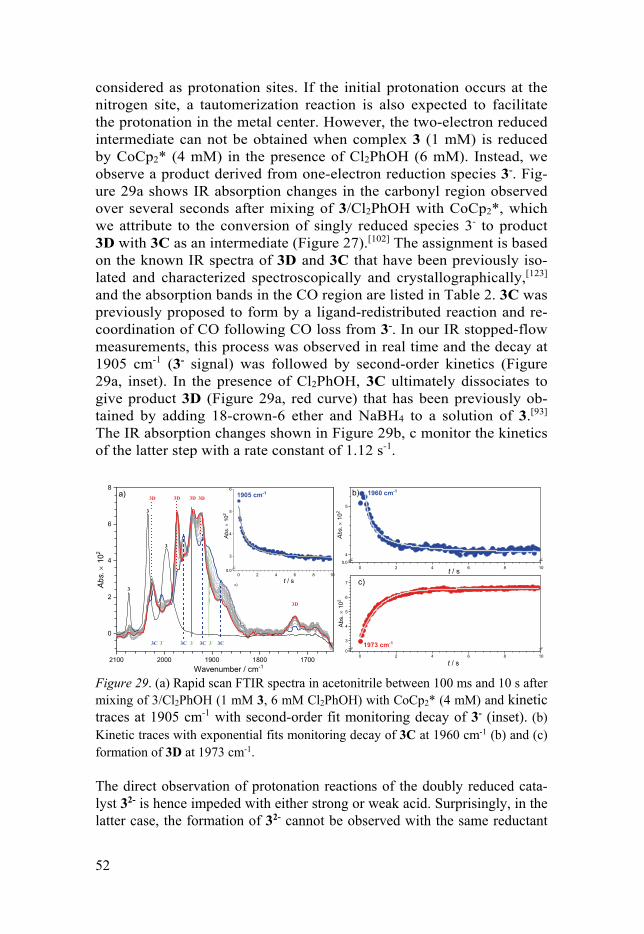
52
considered as protonation sites. If the initial protonation occurs at the nitrogen site, a tautomerization reaction is also expected to facilitate the protonation in the metal center. However, the two-electron reduced intermediate can not be obtained when complex 3 (1 mM) is reduced by CoCp2* (4 mM) in the presence of Cl2PhOH (6 mM). Instead, we observe a product derived from one-electron reduction species 3-. Fig-ure 29a shows IR absorption changes in the carbonyl region observed over several seconds after mixing of 3/Cl2PhOH with CoCp2*, which we attribute to the conversion of singly reduced species 3- to product 3D with 3C as an intermediate (Figure 27).[102] The assignment is based on the known IR spectra of 3D and 3C that have been previously iso-lated and characterized spectroscopically and crystallographically,[123] and the absorption bands in the CO region are listed in Table 2. 3C was previously proposed to form by a ligand-redistributed reaction and re-coordination of CO following CO loss from 3-. In our IR stopped-flow measurements, this process was observed in real time and the decay at 1905 cm-1 (3- signal) was followed by second-order kinetics (Figure 29a, inset). In the presence of Cl2PhOH, 3C ultimately dissociates to give product 3D (Figure 29a, red curve) that has been previously ob-tained by adding 18-crown-6 ether and NaBH4 to a solution of 3.[93] The IR absorption changes shown in Figure 29b, c monitor the kinetics of the latter step with a rate constant of 1.12 s-1.
Figure 29. (a) Rapid scan FTIR spectra in acetonitrile between 100 ms and 10 s after mixing of 3/Cl2PhOH (1 mM 3, 6 mM Cl2PhOH) with CoCp2* (4 mM) and kinetic traces at 1905 cm-1 with second-order fit monitoring decay of 3- (inset). (b) Kinetic traces with exponential fits monitoring decay of 3C at 1960 cm-1 (b) and (c) formation of 3D at 1973 cm-1. The direct observation of protonation reactions of the doubly reduced cata-lyst 32- is hence impeded with either strong or weak acid. Surprisingly, in the latter case, the formation of 32- cannot be observed with the same reductant
0 2 4 6 8 100.0
4
5
0 2 4 6 8 100
3
4
5
6
7
2100 2000 1900 1800 1700
0
2
4
6
8
0 2 4 6 8 100.0
3
4
5
6
1905 cm-1
1973 cm-1
Abs
. 1
02
t / s
1960 cm-1
Ab
s.
102
t / s
b)
c)3
3
3D
3C 3- 3- 3-3C
3
3D 3D
3D
3C 3C
Abs
. 1
02
Wavenumber / cm-1
3D a)
c)
Abs
. 1
02
t / s

53
that generates 32- in the absence of acid. This could be rationalized by an equilibrium between 3- and 3C. While this would not prevent exhaustive reduction of 3 to 32- in absence of acid, protonation-driven dissociation of 3C to 3D could compete with the second reduction even if the acid is too weak to protonate 3-. An alternative explanation is the direct reaction of the reductant with the acid that might simply deplete the reductant before the second reduction of the catalyst.
Future Perspectives. While initial attempts to directly observe protona-tion reactions of 32- were unsuccessful so far, 32-, which can be suspected to be an important catalytic intermediate under sufficiently reducing conditions like electrolysis at E< -1.95 V. This is based on the consideration that in the Fe2(0,0) state amine function could act as a proton relay to facilitate the pro-tonation at the metal center. Catalytic H2 formation from very weak acids at these potentials might benefit from the very stable Fe2(0,0) state of the cata-lyst, in contrast to the Fe2(0,I) intermediate that much more rapidly converts into non-reactive products.
3.4 Photochemical and Chemical Reduction of FeFe(Cl2bdt)(CO)6 Complex 3.4.1 General In section 3.1, we compared the IR spectra of one-electron reduced interme-diates for FeFe(pdt)(CO)6 (1) and FeFe(bdt)(CO)6 (2) to identify their struc-tural differences, which were then applied to elucidate their different proto-nation behavior. For electrochemical studies that were not discussed in the previous sections, complex 1 undergoes an initial quasi-reversible one-electron reduction at about -1.6 V versus Fc+/Fc in acetonitrile, and a second irreversible reduction of 1 occurs at about 0.6 V more negative than the ini-tial reduction.[100]
The complex 3, adt-analogue, has a similar electrochemical behaviour with that of 1. In comparison, complex 2 having the benzenedithiolate (bdt) ligand is directly reduced to its dianion in a reversible two-electron process with a mild reduction potential (E0 = -1.32 V).[62,64,124] The mild reduction potential has been explained by Lichtenberger and co-workers, who studied the electronic effect of bdt bound to transition metals. They demonstrated that the bdt ligand has an ability to buffer the change in electron density at diiron core upon reduction, and therefore minimizes the changes in electron energies. The buffering is attributed to the ability of the benzenedithiolate to adapt its geometry relative to the diiron center to the change in electron con-figuration. The geometry change upon reduction is confirmed by experi-mental data and DFT calculations in which the IR spectrum is not only shift-ed to lower wavenumber, but also shows a different shape compared to the

54
spectrum for the oxidized form.[87] As a result, the reduction potentials of the two one-electron processes are inverted, which means that the second reduc-tion is thermodynamically more favorable than first one, leading to an over-all two-electron reduction observed by cyclic voltammetry (CV). Thus, the coupling of a bdt ligand with the diiron core in molecular analogues to the active site of the enzyme should have benefits for saving thermodynamic cost and stabilizing the reduced states in the overall catalytic cycle. The re-duction potential of the two-electron process is further shifted to more posi-tive values by the installation of electron-withdrawing chloro-groups in or-tho-position of bdt ligand. The resulting FeFe(-Cl2bdt)(CO)6 complex (5, E0 = -1.2 V vs. Fc+/Fc)[125,126] essentially maintains an intact structure geome-try and has the same electrochemical properties as the parent complex 2 . It has been demonstrated that complex 5 is capable of catalyzing the hydrogen production both in electrochemical and visible-light-driven photochemical experiments.[126] However, the introduction of the electron-withdrawing chloro groups is expected to slow down those protonation steps in the cata-lytic cycle, because it becomes less basic than complex 2 in each oxidized and protonated states. This provides a possibility to characterize more cata-lytic key intermediates, and therefore allows for the understanding of the fundamental steps in proton reduction catalysis of complex 5 via spectro-scopic techniques with resolution ranging from nanosecond to seconds time scales.
3.4.2 Key Catalytic Intermediates of FeFe(Cl2bdt)(CO)6
Since the properties of complex 5 are very similar to that of parent complex 2, photochemically one-electron reduction can be readily achieved using flash-quench techniques. Complexes 2 and 5 both in the neutral and reduced states feature very similar IR spectra with a minor shift of all bands in the complex 5 by about 5 cm-1 to higher wavenumbers due to the electron with-drawing effect of chloro groups, as shown in Figure 30.
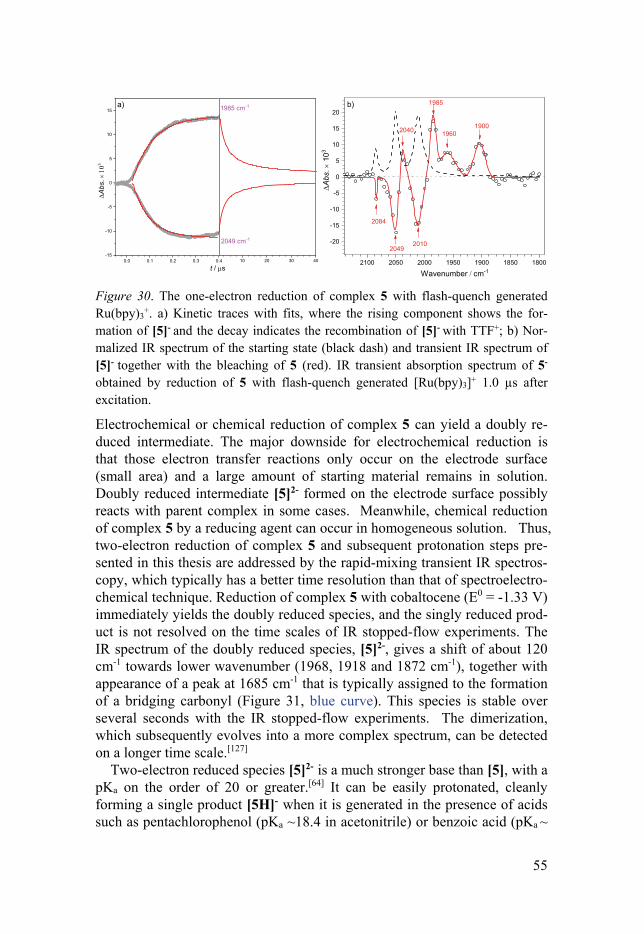
55
Figure 30. The one-electron reduction of complex 5 with flash-quench generated Ru(bpy)3
+. a) Kinetic traces with fits, where the rising component shows the for-mation of [5]- and the decay indicates the recombination of [5]- with TTF+; b) Nor-malized IR spectrum of the starting state (black dash) and transient IR spectrum of [5]- together with the bleaching of 5 (red). IR transient absorption spectrum of 5- obtained by reduction of 5 with flash-quench generated [Ru(bpy)3]+ 1.0 µs after excitation.
Electrochemical or chemical reduction of complex 5 can yield a doubly re-duced intermediate. The major downside for electrochemical reduction is that those electron transfer reactions only occur on the electrode surface (small area) and a large amount of starting material remains in solution. Doubly reduced intermediate [5]2- formed on the electrode surface possibly reacts with parent complex in some cases. Meanwhile, chemical reduction of complex 5 by a reducing agent can occur in homogeneous solution. Thus, two-electron reduction of complex 5 and subsequent protonation steps pre-sented in this thesis are addressed by the rapid-mixing transient IR spectros-copy, which typically has a better time resolution than that of spectroelectro-chemical technique. Reduction of complex 5 with cobaltocene (E0 = -1.33 V) immediately yields the doubly reduced species, and the singly reduced prod-uct is not resolved on the time scales of IR stopped-flow experiments. The IR spectrum of the doubly reduced species, [5]2-, gives a shift of about 120 cm-1 towards lower wavenumber (1968, 1918 and 1872 cm-1), together with appearance of a peak at 1685 cm-1 that is typically assigned to the formation of a bridging carbonyl (Figure 31, blue curve). This species is stable over several seconds with the IR stopped-flow experiments. The dimerization, which subsequently evolves into a more complex spectrum, can be detected on a longer time scale.[127]
Two-electron reduced species [5]2- is a much stronger base than [5], with a pKa on the order of 20 or greater.[64] It can be easily protonated, cleanly forming a single product [5H]- when it is generated in the presence of acids such as pentachlorophenol (pKa ~18.4 in acetonitrile) or benzoic acid (pKa ~
0.0 0.1 0.2 0.3 0.4-15
-10
-5
0
5
10
15
10 20 30 40 2100 2050 2000 1950 1900 1850 1800
-20
-15
-10
-5
0
5
10
15
20
Ab
s.
t / s
1985 cm-1
2049 cm-1
Ab
s.
10
3
Wavenumbercm-1
2084
20492010
2040
1985
19601900
a) b)
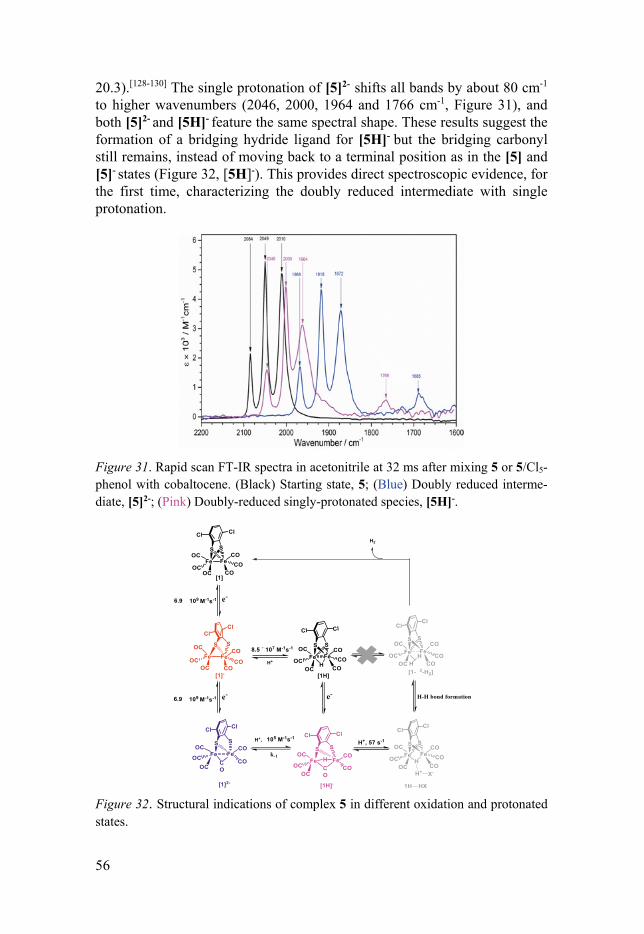
56
20.3).[128-130] The single protonation of [5]2- shifts all bands by about 80 cm-1 to higher wavenumbers (2046, 2000, 1964 and 1766 cm-1, Figure 31), and both [5]2- and [5H]- feature the same spectral shape. These results suggest the formation of a bridging hydride ligand for [5H]- but the bridging carbonyl still remains, instead of moving back to a terminal position as in the [5] and [5]- states (Figure 32, [5H]-). This provides direct spectroscopic evidence, for the first time, characterizing the doubly reduced intermediate with single protonation.
Figure 31. Rapid scan FT-IR spectra in acetonitrile at 32 ms after mixing 5 or 5/Cl5-phenol with cobaltocene. (Black) Starting state, 5; (Blue) Doubly reduced interme-diate, [5]2-; (Pink) Doubly-reduced singly-protonated species, [5H]-.
Figure 32. Structural indications of complex 5 in different oxidation and protonated states.

57
3.4.3 Catalytic Turnover Processes with Stronger Acids [5H]- cannot be further protonated by weak acid as presented above, and is thereby a significantly weaker base than the doubly reduced species [5]2-. Thus, for proton reduction catalysis in the presence of weak acids, [5H]- needs to be further reduced to [5H]2- that undergoes electrophilic attack from the external proton, followed by the dissociation of molecular hydrogen from the complex without an energy barrier.[64] Upon inspection of cyclic volt-ammetry, however, it is clear that the catalytic current occurs near -1.94 V, giving rise to a large requirement of overpotential, about 0.65 V.
Instead, when the catalyst is reduced by two-electrons in the presence of stronger acids such as TsOH (pKa ~ 8.6), the doubly reduced species [5]2- is rapidly protonated, which is not resolved under experimental conditions. The resulting [5H]- can react with the second proton. [5H]- decays with a rate constant on the time scales of seconds, followed by regeneration of the start-ing complex 5, as shown in Figure 33a. This suggests that the catalysis clos-es the cycle by a facile release of H2 from the two-electron two-proton in-termediate, with the second protonation as the rate-limiting step; more de-tails about the mechanism will be discussed in the following section. The decay of [5H]- and the rise of starting complex 5 were accelerated with the increasing amount of TsOH concentration (Figure 33b, c). The observed pseudo-first-order rate constants are proportional to the concentration of TsOH, leading to a second order rate constant of 57 M-1s-1 (turnover fre-quency, TOF) for catalytic molecular hydrogen formation (Figure 33d). The same measurements were repeated using acids spanning over several pKa units to estimate the hydricity or pKa of the [5H]-. With Cl2BSA (pKa ~ 6.7), the decay of [5H]- is faster as expected, and the process of catalytic turnover is not resolved with acid concentration up to 6 mM under the stopped-flow IR conditions, observing the complete recovery of starting complex 5; the time-resolved IR spectra with different concentrations of Cl2BSA can be found in paper III. The Cl3CCOOH (pKa ~ 10.7), a moderately strong acid, essentially does not catalyze the formation of H2, and more complex spectra were observed with either higher concentration or longer time scales. The detailed mechanism will be discussed in the following section.
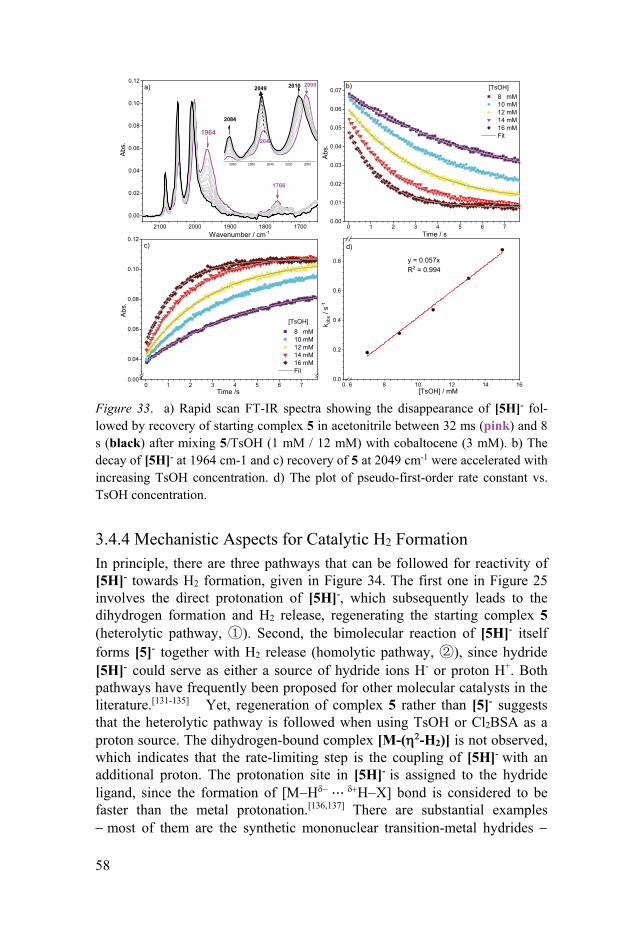
58
Figure 33. a) Rapid scan FT-IR spectra showing the disappearance of [5H]- fol-lowed by recovery of starting complex 5 in acetonitrile between 32 ms (pink) and 8 s (black) after mixing 5/TsOH (1 mM / 12 mM) with cobaltocene (3 mM). b) The decay of [5H]- at 1964 cm-1 and c) recovery of 5 at 2049 cm-1 were accelerated with increasing TsOH concentration. d) The plot of pseudo-first-order rate constant vs. TsOH concentration.
3.4.4 Mechanistic Aspects for Catalytic H2 Formation In principle, there are three pathways that can be followed for reactivity of [5H]- towards H2 formation, given in Figure 34. The first one in Figure 25 involves the direct protonation of [5H]-, which subsequently leads to the dihydrogen formation and H2 release, regenerating the starting complex 5 (heterolytic pathway, ①). Second, the bimolecular reaction of [5H]- itself forms [5]- together with H2 release (homolytic pathway, ②), since hydride [5H]- could serve as either a source of hydride ions H- or proton H+. Both pathways have frequently been proposed for other molecular catalysts in the literature.[131-135] Yet, regeneration of complex 5 rather than [5]- suggests that the heterolytic pathway is followed when using TsOH or Cl2BSA as a proton source. The dihydrogen-bound complex [M-(-H2)] is not observed, which indicates that the rate-limiting step is the coupling of [5H]- with an additional proton. The protonation site in [5H]- is assigned to the hydride ligand, since the formation of [ ⋯ HX] bond is considered to be faster than the metal protonation.[136,137] There are substantial examples most of them are the synthetic mononuclear transition-metal hydrides
0 1 2 3 4 5 6 70.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0 1 2 3 4 5 6 70.00
0.04
0.06
0.08
0.10
0.12
2100 2000 1900 1800 1700
0.00
0.02
0.04
0.06
0.08
0.10
0.12
2080 2060 2040 2020 2000
0 6 8 10 12 14 160.0
0.2
0.4
0.6
0.8
8 mM 10 mM 12 mM 14 mM 16 mM Fit
Abs
.
Time / s
[TsOH]
c)
8 mM 10 mM 12 mM 14 mM 16 mM Fit
Ab
s.
Time /s
[TsOH]
b)
Wavenumber / cm-1
Ab
s.
1964
1766
a)
2084
2046
2049 2010 2000
d)
[TsOH] / mM
k obs
/ s-1
y = 0.057xR2 = 0.994

59
showing that the kinetic protonation site is the hydride ligand and not the metal center,[135,138-141] ultimately leading to dihydrogen bond formation [M-(-H2)] and elimination of H2. No catalytic turnover was observed in the presence of Cl3CCOOH even with larger concentration, indicating that there is low or no driving force for proton transfer from the acid to hydride [5H]-. Instead, more complex IR spectra were detected on longer time scales. This is because the equilibrium rate constant between [5H]- and [5]2- is too small, leading to the deprotonation of [5H]- to again form [5]2- that involves a di-merization process. The dimerization is accelerated with the assistance of acid compared to that without acid. Further reduction of [5H]- (pathway ③) is not thermodynamically favored when using cobaltocene as reductant, but it has been investigated in an electrochemical and computational study by Lichtenberger et.al.[64]
Figure 34. Three possible pathways of reactivity of [5H]- towards ultimately H2 formation.
3.4.5 Hydricity of [5H]-
The diiron hydride [5H]− may be capable of hydride transfer to CO2, result-ing in the reduction of CO2 to Formate (HCO2
-). To estimate the free energy change of this hydride transfer reaction, the hydricity (G0
H-) of [5H]−,
which quantifies the thermodynamic ability of the [5H]− hydride species to transfer a H- ions to a substrate, needs to be compared to that of HCO2
-. G0
H- is derived from the thermodynamic cycle in which the thermodynamic

60
hydricity is expressed by the sum of the bond dissociation free energy (BDFE) of the metal hydride, the free energy for two-electron reduction of the starting complex 5, 2F ⁄ , and the free energy for proton re-duction to a hydride anion,∆ ⁄ , given by equation 1-5, [142]
⇌ H (1) ⇌ 2 2 ⁄ (2)
H 2 ⇌ H ∆ ⁄ (3)
⇌ H ∆ (4) ∆ 1.364 46.1 ⁄ 76.6 (5) Applying the pKa of ca. 21 for [5H]− in equation 5, the hydricity, ∆ , of [5H]− can be calculated to be 45 kcal/mol. While the determined hydricity here is slightly greater than 44 kcal/ mol for the hydricity of formate ΔG°H− (HCO2
−) in acetonitrile,[143,144] 5H]− might still act as a hydride donor to CO2, resulting in formate formation under non-standard conditions. CO2 reduction to format with this compound will be addressed in the future works.

61
4. Summary and outlook
Structural mimics of FeFe-hydrogenase are a class of attractive catalysts, which only contain earth-abundant transition metal iron. Yet, they generally are much less efficient than the enzyme. For this reason, investigating the catalytic cycle of these catalysts is essential to facilitate the rational design of future improved catalysts. Figure 36 summarizes all the relevant struc-tures of the complexes discussed in the thesis.
Figure 35. Overview of all complexes discussed in the thesis
For biomimetic diiron complexes 1-5, I have used time-resolved UV-Vis and mid-IR spectroscopy to elucidate their structures and measure their reactivity in proton- and electron transfer reactions of the catalytic cycle. The key in-termediates formed in different catalytic pathways have been characterized.
For complexes 1-2 (chapter 3.1), I have found fundamentally different structural rearrangements upon reduction between these two complexes. Specifically, complex 1, with the bridging pdt ligand, maintains an intact coordination geometry upon one-electron reduction, accompanied by a lengthening of FeFe bond. This is in contrast to the studies for complex 2 with the aromatic bdt ligand, where one-electron reduction leads to structural changes caused by the breaking of one of FeS bonds and the twisting of the bdt ligand. The significant structural differences between 1- and 2- may be applied to explain differences in the protonation dynamics. For the complex

62
2 with the bdt ligand, the bond cleavage may generate a new basic site (i.e. sulfur site) for proton attack, which may also facilitate the protonation in the metal center mediated by S-protonation. Moreover, the kinetics of protona-tion, forming a bridging hydride, suggest that the mechanism is direct proton transfer from the acid to FeIFe0 state. This is in contrast to that of the neutral complex (FeIFeI state) with phosphine ligands (PMe3) in which the hydride formation is much slower and likely mediated by one of the CO-ligands. Alternatively, for the neutral complex the protonation in the metal center is sterically affected by the larger PMe3 groups, which lower the proton acces-sible to the metal center.
In chapter 3.2 (Paper II), the introduction of proton relays in the secondary coordination sphere of catalysts has been investigated, aiming to facilitate protonation in the metal center. For complex 3, the adt bridge has been taken as an example design for proton shuttling to the metal center. I have shown, by direct spectroscopy measurements, that the nitrogen site in the adt ligand has no role as a proton shuttle in the FeIFe0 state, as had been proposed. In-stead the kinetics of the proton transfer from bulk acid to the adt ligand and, with stronger acids, to the metal center suggest that the superior activity of the adt complex arises from lower reorganization energy for hydride for-mation once the adt ligand is protonated. The finding hence excludes a pro-ton relay function of the adt ligand in the FeIFe0 state of this particular cata-lyst. This points more generally to the importance of alternative functions of basic ligand sites for enhanced activity of molecular catalysts. Chapter 3.2 also contains unpublished results. The PT/ET sequence of complex 4, where introduction of two electron-donating PMe3 groups increases the elec-tron density in the Fe2 core, was studied by nanosecond transient absorption spectroscopy with UV-Vis and mid-IR detection to look at potential proton transfer from the nitrogen site to the metal center. Preliminary data does not show evidence of a tautomerization process after tens of microseconds. Thus, the potential tautomerization is expected to occur on longer time scales that could be explored with stopped-flow techniques in the future.
In chapter 3.3 (Paper III), reduction of complex 3 was further investigated by using stopped-flow IR spectroscopy both in the absence and in the pres-ence of weak acid. The overall two-electron reduction process was spectro-scopically revealed in the absence of acid. Surprisingly, the two-electron reduced intermediate 32- is stable over minutes. This was attributed to a pla-nar 2Fe2S core caused by FeS bond cleavage and formation of a bridging carbonyl, which might contribute to the stability of the dianion. In the pres-ence of acid, the second reduction is however impeded by other competitive reactions like the direct reaction of acid with reductant, ultimately leading to the formation of catalytically unreactive products. While initial attempts to directly observe protonation reactions of 32- were unsuccessful so far, 32- can be suspected to be an important catalytic intermediate under sufficiently reducing conditions.

63
Chapter 3.4 summarizes the investigation of complex 5 with the Cl2bdt ligand (Paper IV). As observed by combining the stopped-flow mixing method with IR spectroscopy, reduction of complex 5 with cobaltocene im-mediately yields the doubly reduced intermediate [5]2- due to the inverted potentials for the 50/1- and 51-/2-. [5]2- can easily be protonated even with weak acids like benzoic acid and Cl5PhOH. The major finding is that the structure of the protonation product of [5]2- can be spectroscopically charac-terized, as well as deprotonation of [5H]- and dimerization of [5]2- can then be followed when using weak acids. In the presence of strong acid, the pro-tonation of [5H]- leads to, for the first time, direct spectroscopic observation of the turnover process, providing a straightforward method for determina-tion of turnover frequency for hydrogen evolution. Estimation of [5H]- hy-dricity allows one to further explore the application of complex 5 in the re-duction of CO2 in the future works.
Overall, the aim of the thesis was to contribute to a better understanding of the structure and reactivity of catalyst intermediates in proton- and elec-tron transfer reactions in the catalytic cycle. Investigating these can allow us to identify if bond cleavage occurs upon reduction. In a catalyst, bond cleavages may lead to the generation of a new protonation site on the ligand, facilitating protonation of the metal site. Moreover, a bridging CO-ligand is generally not observed upon one-electron reduction, but occurs in the two-electron reduced state, Fe0Fe0. When the two-electron reduced species is protonated to form the bridging hydride, the bridging CO is not released back to the terminal position. Thus, such a hydride is relatively stable and may be less active for catalysis. A design for proton shuttling to the metal center is a promising strategy to obtain an efficient molecular catalyst. How-ever, such proton shuttling does sometimes not occur. Important factors that need to be considered are the distance between the basic ligand site and met-al center, as well as the driving force for this proton transfer reaction. Meanwhile, an improved catalyst needs the proton transfer steps to compete sufficiently with potential side reaction of reduced intermediates such as dimerization. These model complexes typically show the poor stability in solution compared to the active site of FeFe-hydrogenase surrounded by the protein matrix, which prevents dimerization of reduced forms. Thus, immo-bilization on model complexes would be a great strategy towards the im-provement of the stability under catalytic conditions, such as incorporating the molecular catalyst into a porous material.

64
5. Sammanfattning på Svenska
Strukturella härmningar av FeFe-hydrogenaser är en klass av attraktiva kata-lysatorer, som endast innehåller den rikligt förekommande övergångsmetal-len järn. Dock är dessa generellt mycket mindre effektiva än enzymet. För denna anledning är det viktigt att undersöka den katalytiska cykeln för dessa katalysatorer, för att underlätta rationell utveckling av framtida förbättrade katalysatorer. Figur 36 sammanfattar de relevanta strukturerna för kom-plexen som diskuteras i denna avhandling.
Figur 36. Översikt av alla komplex som diskuteras i denna avhandling
För biomimetiska dijärnkomplexen 1-5 har jag använt tidsupplöst UV-vis och mid-IR spektroskopi för att klargöra strukturerna och mäta deras aktivi-tet i proton och elektrontransferreaktioner i deras katalytiska cykel. Nyck-elintermediärerna som formas i olika katalytiska mekanismer har karakteri-serats.
För komplexen 1-2 (kapitel 3.1) har jag hittat fundamentalt olika struktu-rella förändringar efter reduktion. Specifikt behåller komplex 1, med den överbryggande pdt-liganden samma koordinationsgeometri efter enelektron-reduktion, med en förlängd Fe-Fe bindning. Detta skiljer sig från komplex 2, med den aromatiska bdt-liganden, där enelektronreduktion leder till struktu-rella förändringar, orsakad av brytningen av en av Fe-S bindningar och vrid-ningen av bdt-liganden. De signifikanta strukturella förändringarna mellan 1-

65
och 2- kan användas för att förklara skillnaderna i protoneringshastigheten. För komplex 2, med bdt-liganden, skulle bindningsbrytningen kunna gene-rera ett nytt basiskt säte (svavelatomen) för proton-attacken, som också skulle underlätta protoneringen av metallcentret genom vidare protonöverfö-ring. Vidare indikerar kinetiken av protoneringen, som resulterar i en över-bryggande hydrid, att mekanismen är protonöverföring från syran direkt till FeIFe0-tillståndet. Detta skiljer sig från det neutrala komplexet (i FeIFeI-tillståndet) med fosfinligander (PMe3), där hydridformation är mycket lång-sammare och antagligen medierat via en av CO-liganderna. Alternativt är protoneringen av metallcentret i det neutrala komplexet steriskt påverkad av de stora PMe3-grupperna, som minskar protontillgängligheten av metallcent-ret.
I kapitel 3.2 (Artikel II) har introduktionen av protonrelä i andra ko-ordineringssfären av katalysatorer undersökts, med målet att underlätta pro-tonering av metallcentret. För komplex 3 har adt-bron tagits som ett exempel för utveckling av ett protonrelä till metallcentret. Jag har visat, med spektro-skopiska mätningar, att kvävesätet i adt-liganden inte har någon roll som protonrelä i FeIFe0-tillståndet, i motsats till vad som hade föreslagits tidigare. Istället indikerar kinetiken av protoneringen av adt-liganden med bulksyra, och till metallcentret, med starkare syra, att den högre aktiviteten av adt-komplexet härstammar från den lägre reorganisationsenergin för hydrid-formation när adt-liganden har protonerats. Dessa upptäckter exkluderar därför en funktion av adt-liganden som protonrelä i FeIFe0-tillståndet av denna specifika katalysator. Detta tyder mer generellt på betydelsen av alter-nativa funktioner av basiska ligandsäten för förbättrad aktivitet av mole-kylära katalysatorer. Kapitel 3.2 innehåller också opublicerade resultat. PT/ET sekvensen för komplex 4, där introduktionen av två elektrongivande PMe3-grupper ökar elektrondensiteten i Fe2-centret, har studerats med nano-sekundsupplöst transient absorptionsspektroskopi med UV-vis och mid-IR detektion, för att observera potentiell protonöverföring från kvävesätet till metallcentret. Preliminära resultat visar inga bevis på tautomerisering efter ett tiotal mikrosekunder. Därför väntas potentiell tautomerisering ske på en längre tidsskala. Ytterligare experiment skulle kunna adresseras med stop-ped-flow tekniker i framtiden.
I kapitel 3.3 (Artikel III) undersöktes reduktionen av komplex 3 vidare, både i när- och frånvaro av svag syra. Tvåelektronreduktionsprocessen ob-serverades spektroskopisk när ingen syra fanns närvarande. Förvånansvärt är den tvåelektron-reducerade mellanprodukten stabil under minuter. Detta hänfördes till en plan 2Fe2S kärna orsakad av FeS bindningsklyvning och bildning av en överbryggande karbonyl, vilket kan bidra till stabiliteten hos dianionen. I närvaro av syra hindras den andra reduktionen av andra konkur-renskraftiga reaktioner, såsom den direkta reaktionen av syra med reduktant, vilket slutligen leder till bildandet av katalytiskt oreaktiva produkter. Medan inledande försök att direkt observera protoneringsreaktioner av hade miss-

66
lyckats hittills kan misstänks vara en viktig katalytisk mellanprodukt under tillräcklig reducerande betingelser.
Kapitel 3.4 sammanfattar undersökningen av komplex 5 med Cl2bdt-liganden (Artikel IV). Som observerad genom att kombinera stopped-flow blandningsmetoden med IR-spektroskopi, leder reduktion av komplex 5 med cobaltocen direkt till formation av [5]2-, på grund av de inverterade potentia-lerna för 50/1- och 51-/2-. [5]2- kan enkelt protoneras även med svaga syror, såsom bensoesyra och Cl5PhOH. Den största upptäckten är att strukturen för den protonerade produkten av [5]2- kunde karakteriseras, och att deprotone-ring av [5H]- och dimerisering av [5]2- sedan kan följas när svaga syror an-vänds. I närvaron av starka syror leder protoneringen av [5H]-, för första gången, till direkt observation av omsättningsprocessen, vilket medför en enkel metod för bestämningen av omsättningsfrekvensen för vätgasevolut-ion. Uppskattningen av hydriciteten av [5H]- tillåter oss att undersöka til--lämpning av komplex 5 för reduktionen av CO2 i framtiden.
Sammantaget har målet av denna avhandling varit att bidra till en bättre förståelse av strukturen och reaktiviteten av katalysatorintermediärer i proton och elektronöverföringsreaktioner i den katalytiska cykeln. Undersökningar av dessa tillåter oss att identifiera om bindningsbrytningar sker efter redukt-ion. Bindningsbrytningar kan leda till bildning av nya protoneringssäten på liganden, vilket också kan underlätta protonering av metallcentret. Vidare observeras generellt ingen överbryggande CO-ligand efter enelektronredukt-ion, medan den förekommer i det tvåelektronreducerade tillståndet, Fe0Fe0. När det tvåelektronreducerade komplexet protoneras för att bilda den över-bryggande hydriden, släpps inte den överbryggande CO-liganden tillbaka till terminala positionen. Därför är en sådan hydrid någorlunda stabil och möj-ligtvis mindre aktiv i katalys. Utveckling av protonrelä för vidare protonö-verföring till metallcentret är en lovande strategi för att skapa en effektiv molekylär katalysator. Dock förekommer sådan medierad protonöverföring ibland inte. Viktiga faktorer som måste tas hänsyn till är avståndet mellan det basiska ligandsätet och metallcentret, såsom drivkraften för denna proto-növerföringsreaktion. I mellantid måste dessa protonöverföringsreaktioner i en förbättrad katalysator konkurrera tillräckligt med potentiella sidoreaktion-er av de reducerade intermediärerna, såsom dimerisering. Dessa modellkom-plex har vanligtvis dålig stabilitet i ett lösningsmedel i jämförelse med reakt-ionscentret av FeFe-hydrogenas, omgiven av proteinmatrisen som förhindrar dimerisering av reducerade former. Därför skulle immobilisering vara en bra strategi för förbättringen av stabiliteten under katalytiska omständigheter, genom till exempel inkorporation av den molekylära katalysatorn i ett poröst material.

67
6. Acknowledgments
I have received much support and help from supervisors, colleagues, collab-orators, friends and my family during the past four years. This is a great chance to express my gratitude to all of them when I come to this section.
First and foremost, I would like to express my deepest gratitude to my su-pervisors, Leif Hammarström and Reiner Lomoth. Thank you for accept-ing me to work in the Fyskem group, and to pursue a doctoral degree at Uppsala University. You have guided me throughout my whole PhD studies. I really appreciate all the scientific discussions that we had together, and large efforts on manuscripts, experiments and my thesis that you have spent. My PhD studies would not be possible without the help from both of you.
I also want to thank Mohammad Mirmohades, for teaching me how to use the Quanta-Ray and prepare samples with remarkable patience when I started my PhD work. Many thanks go to Edgar Mijangors for the help with the Glove Box, and Luca D’Amario for teaching me to use Brilliant B laser when the Quanta-Ray was out of order.
I would like to thank Robin Tyburski, a great person in the lab, for the kind help with the Swedish Summary of my thesis, but also valuable sugges-tions for parts of my thesis. I am also grateful to Vincent Wang, Mohamed Qenawy, Aijie Liu and Nidhi Kaul for the kind help with proof reading of my thesis, which made some of sentences and languages much better.
Many thanks go to my collaborators: Alexander Aster for all the discus-sions that we had; Sonja Pullen, Charlène Esmieu, Gustav Berggren and Sascha Ott for providing the diiron catalysts so that I can carry out my PhD work.
I am truly thankful to Erik Johansson, Haining Tian, Burkhard Zietz, Ana Moranderia and Jacinto Sá for being such good teachers during courses, and taking more attention about lab safety.
Great thanks go to the administrator staff: Eva Larsson, Susanne Söder-berg, Lina Rosén, Patrik Lindahl, Jessica Stålberg and Sven Johansson for being always available whenever I need a hand.
To all the former and present colleagues in the Fyskem group. I am thank-ful to Lei Zhang, Bo Xu, Jing Huang, Aijie Liu, Lei Tian, Lin Li and Tianfei Liu for the help in the lab, as well as those lunches and talks that we had. Great thanks go to Starla Glover for the help, being always energetic and making funny things in the lab. I am also grateful to Belinda Pettersson Rimgard, Astrid Nilsen-Moe, Jens Föhlinger, Martin Axelsson, Kelly

68
Materna, Melina Gilbert Gatty, Wesley Swords, Nidhi Kaul, Lea Hohmann, Prateek Dongare and Nicolas Queyriaux for being helpful when I had questions to ask; it has been nice to share the office with all of you. Many thanks go to Muhammad Anwar Shameem Yocefu Hattori, Noémie Lalaoui and Mariia Pavliuk for useful discussions in the lab .
I have met so many Chinese friends in Sweden. I very much appreciate their company, which made my PhD life easier in the past four years. Thank you Meiyuan Guo, Wenxing Yang, Yi Ren, Lichuan Wu, Song Chen, Xin Chen, Teng Zhang, Feiyan Liang, Dou Du, Liyang Shi, Junxin Wang, Ruisheng Xiong, Yu Zhang, Changqing Ruan, Xiao Xiao, Jiaojiao Yang, Jun Luo, Rui Sun, Le Fu and Mingzhi Jiao for those won-derful dinners that we had together, for sharing stories, information and hap-piness. Please forgive me if I forgot someone here, I do not have perfect memory.
I gratefully acknowledge the financial support from China Scholarship Council (CSC) for my PhD study. I am also thankful to Liljewalchs Schol-arship, Åforsk Foundation for financing my conferences travels.
Last but not least, I want to thank my parents for always giving me the best you had, unconditional support whatever I wanted to do and wherever I wanted to go, 谢谢爸爸妈妈 . Special thanks go to my dear girlfriend, Kaixiu Luo, for support, understanding, love, and especially encouragement when I felt stressful during my PhD work. I also thank myself because I manage to survive and get positive under the stressful situations.

69
6. References
(1) Hansen, J.; Nazarenko, L.; Ruedy, R.; Sato, M.; Willis, J.; Del Genio, A.; Koch, D.; Lacis, A.; Lo, K.; Menon, S.et al., Earth's Energy Imbalance: Con-firmation and Implications. Science 2005, 308, 1431-1435.
(2) Marshall, J.; John, M.; Plumb, R. A.; Academic Press, 2008. (3) Armaroli, N.; Balzani, V. Energy for a sustainable world: from the oil age to
a sun-powered future; Wiley-VCH: Weinheim, 2011. (4) Lewis, N. S.; Nocera, D. G., Powering the planet: Chemical challenges in
solar energy utilization. Proceedings of the National Academy of Sciences 2006, 103, 15729-15735.
(5) Balzani, V.; Credi, A.; Venturi, M., Photochemical Conversion of Solar En-ergy. ChemSusChem 2008, 1, 26-58.
(6) Faunce, T.; Styring, S.; Wasielewski, M. R.; Brudvig, G. W.; Rutherford, A. W.; Messinger, J.; Lee, A. F.; Hill, C. L.; deGroot, H.; Fontecave, M.et al., Artificial photosynthesis as a frontier technology for energy sustainability. Energy & Environmental Science 2013, 6, 1074-1076.
(7) Orimo, S.-i.; Nakamori, Y.; Eliseo, J. R.; Züttel, A.; Jensen, C. M., Complex Hydrides for Hydrogen Storage. Chemical Reviews 2007, 107, 4111-4132.
(8) Dincă, M.; Long, J. R., Hydrogen Storage in Microporous Metal–Organic Frameworks with Exposed Metal Sites. Angewandte Chemie International Edition 2008, 47, 6766-6779.
(9) Schlapbach, L.; Züttel, A., Hydrogen-storage materials for mobile applica-tions. Nature 2001, 414, 353.
(10) Liu, C.; Li, F.; Ma, L.-P.; Cheng, H.-M., Advanced Materials for Energy Storage. Advanced Materials 2010, 22, E28-E62.
(11) Dresselhaus, M. S.; Thomas, I. L., Alternative energy technologies. Nature 2001, 414, 332.
(12) Ewan, B. C. R.; Allen, R. W. K., A figure of merit assessment of the routes to hydrogen. International Journal of Hydrogen Energy 2005, 30, 809-819.
(13) Jacobsson, T. J.; Fjällström, V.; Edoff, M.; Edvinsson, T., Sustainable solar hydrogen production: from photoelectrochemical cells to PV-electrolyzers and back again. Energy & Environmental Science 2014, 7, 2056-2070.
(14) Esswein, A. J.; Nocera, D. G., Hydrogen Production by Molecular Photoca-talysis. Chemical Reviews 2007, 107, 4022-4047.
(15) Nealson, K. H.; Conrad, P. G., Life: past, present and future. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 1999, 354, 1923-1939.
(16) van de Krol, R.; Parkinson, B. A., Perspectives on the photoelectrochemical storage of solar energy. MRS Energy & Sustainability 2017, 4, E13.
(17) Ardo, S.; Fernandez Rivas, D.; Modestino, M. A.; Schulze Greiving, V.; Abdi, F. F.; Alarcon Llado, E.; Artero, V.; Ayers, K.; Battaglia, C.; Becker, J.-P.et al., Pathways to electrochemical solar-hydrogen technologies. Energy & Environmental Science 2018, 11, 2768-2783.

70
(18) Lambers, H.; Chapin, F. S., III; Pons, T. L. Plant physiological ecology; 2nd;Second; ed.; Springer: New York, 2008.
(19) Blankenship, R. E. Molecular mechanisms of photosynthesis; 1. Aufl. ed.; Blackwell Science: Oxford;Malden, MA;, 2002.
(20) Ferreira, K. N.; Iverson, T. M.; Maghlaoui, K.; Barber, J.; Iwata, S., Archi-tecture of the Photosynthetic Oxygen-Evolving Center. Science 2004, 303, 1831-1838.
(21) Barber, J.; Tran, P. D., From natural to artificial photosynthesis. Journal of The Royal Society Interface 2013, 10.
(22) Bunt, J., Light and photosynthesis in aquatic ecosystems. Aquatic Botany 1995, 50, 111-112.
(23) Prof, H. S. S. F. f. S.; Society; Prof, H. S. S. F. f. S.; Technology; Prof. H.S. Srivastava Foundation for Science and Technology: Lucknow;New Delhi;, 1995.
(24) Fujishima, A.; Honda, K., Electrochemical Photolysis of Water at a Semi-conductor Electrode. Nature 1972, 238, 37.
(25) Sun, L.; Hammarström, L.; Åkermark, B.; Styring, S., Towards artificial photosynthesis: ruthenium–manganese chemistry for energy production. Chemical Society Reviews 2001, 30, 36-49.
(26) Berardi, S.; Drouet, S.; Francàs, L.; Gimbert-Suriñach, C.; Guttentag, M.; Richmond, C.; Stoll, T.; Llobet, A., Molecular artificial photosynthesis. Chemical Society Reviews 2014, 43, 7501-7519.
(27) Tong, L.; Iwase, A.; Nattestad, A.; Bach, U.; Weidelener, M.; Götz, G.; Mishra, A.; Bäuerle, P.; Amal, R.; Wallace, G. G.et al., Sustained solar hy-drogen generation using a dye-sensitised NiO photocathode/BiVO4 tandem photo-electrochemical device. Energy & Environmental Science 2012, 5, 9472-9475.
(28) Arai, T.; Sato, S.; Kajino, T.; Morikawa, T., Solar CO2 reduction using H2O by a semiconductor/metal-complex hybrid photocatalyst: enhanced efficiency and demonstration of a wireless system using SrTiO3 photoanodes. Energy & Environmental Science 2013, 6, 1274-1282.
(29) Duan, L.; Bozoglian, F.; Mandal, S.; Stewart, B.; Privalov, T.; Llobet, A.; Sun, L., A molecular ruthenium catalyst with water-oxidation activity compa-rable to that of photosystem II. Nature Chemistry 2012, 4, 418.
(30) Blakemore, J. D.; Crabtree, R. H.; Brudvig, G. W., Molecular Catalysts for Water Oxidation. Chemical Reviews 2015, 115, 12974-13005.
(31) Garrido-Barros, P.; Gimbert-Suriñach, C.; Moonshiram, D.; Picón, A.; Monge, P.; Batista, V. S.; Llobet, A., Electronic π-Delocalization Boosts Cat-alytic Water Oxidation by Cu(II) Molecular Catalysts Heterogenized on Gra-phene Sheets. Journal of the American Chemical Society 2017, 139, 12907-12910.
(32) Gimbert-Suriñach, C.; Moonshiram, D.; Francàs, L.; Planas, N.; Bernales, V.; Bozoglian, F.; Guda, A.; Mognon, L.; López, I.; Hoque, M. A.et al., Structur-al and Spectroscopic Characterization of Reaction Intermediates Involved in a Dinuclear Co–Hbpp Water Oxidation Catalyst. Journal of the American Chemical Society 2016, 138, 15291-15294.
(33) Matheu, R.; Ertem, M. Z.; Benet-Buchholz, J.; Coronado, E.; Batista, V. S.; Sala, X.; Llobet, A., Intramolecular Proton Transfer Boosts Water Oxidation Catalyzed by a Ru Complex. Journal of the American Chemical Society 2015, 137, 10786-10795.
(34) Kim, D.-H.; Kim, M.-S., Hydrogenases for biological hydrogen production. Bioresource Technology 2011, 102, 8423-8431.

71
(35) Khetkorn, W.; Rastogi, R. P.; Incharoensakdi, A.; Lindblad, P.; Madamwar, D.; Pandey, A.; Larroche, C., Microalgal hydrogen production – A review. Bioresource Technology 2017, 243, 1194-1206.
(36) Verhaart, M. R. A.; Bielen, A. A. M.; Oost, J. v. d.; Stams, A. J. M.; Kengen, S. W. M., Hydrogen production by hyperthermophilic and extremely thermo-philic bacteria and archaea: mechanisms for reductant disposal. Environmen-tal Technology 2010, 31, 993-1003.
(37) Oren, A., A proposal for further integration of the cyanobacteria under the Bacteriological Code. International Journal of Systematic and Evolutionary Microbiology 2004, 54, 1895-1902.
(38) Schaeffer, D. J.; Krylov, V. S., Anti-HIV Activity of Extracts and Com-pounds from Algae and Cyanobacteria. Ecotoxicology and Environmental Safety 2000, 45, 208-227.
(39) Tamagnini, P.; Axelsson, R.; Lindberg, P.; Oxelfelt, F.; Wünschiers, R.; Lindblad, P., Hydrogenases and Hydrogen Metabolism of Cyanobacteria. Microbiology and Molecular Biology Reviews 2002, 66, 1-20.
(40) Vignais, P. M.; Billoud, B., Occurrence, Classification, and Biological Func-tion of Hydrogenases: An Overview. Chemical Reviews 2007, 107, 4206-4272.
(41) Vignais, P. M.; Billoud, B.; Meyer, J., Classification and phylogeny of hy-drogenases1. FEMS Microbiology Reviews 2001, 25, 455-501.
(42) Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E., Hydrogenases. Chemical Reviews 2014, 114, 4081-4148.
(43) Fontecilla-Camps, J. C.; Volbeda, A.; Cavazza, C.; Nicolet, Y., Struc-ture/Function Relationships of [NiFe]- and [FeFe]-Hydrogenases. Chemical Reviews 2007, 107, 4273-4303.
(44) Tard, C.; Pickett, C. J., Structural and Functional Analogues of the Active Sites of the [Fe]-, [NiFe]-, and [FeFe]-Hydrogenases. Chemical Reviews 2009, 109, 2245-2274.
(45) Du, P.; Eisenberg, R., Catalysts made of earth-abundant elements (Co, Ni, Fe) for water splitting: Recent progress and future challenges. Energy & En-vironmental Science 2012, 5, 6012-6021.
(46) Merki, D.; Hu, X., Recent developments of molybdenum and tungsten sul-fides as hydrogen evolution catalysts. Energy & Environmental Science 2011, 4, 3878-3888.
(47) Peters, J. W.; Lanzilotta, W. N.; Lemon, B. J.; Seefeldt, L. C., X-ray Crystal Structure of the Fe-Only Hydrogenase (CpI) from <em>Clostridium pasteuri-anum</em> to 1.8 Angstrom Resolution. Science 1998, 282, 1853-1858.
(48) Silakov, A.; Wenk, B.; Reijerse, E.; Lubitz, W., 14N HYSCORE investiga-tion of the H-cluster of [FeFe] hydrogenase: evidence for a nitrogen in the di-thiol bridge. Physical Chemistry Chemical Physics 2009, 11, 6592-6599.
(49) Berggren, G.; Adamska, A.; Lambertz, C.; Simmons, T. R.; Esselborn, J.; Atta, M.; Gambarelli, S.; Mouesca, J. M.; Reijerse, E.; Lubitz, W.et al., Bio-mimetic assembly and activation of [FeFe]-hydrogenases. Nature 2013, 499, 66.
(50) Adamska-Venkatesh, A.; Krawietz, D.; Siebel, J.; Weber, K.; Happe, T.; Reijerse, E.; Lubitz, W., New Redox States Observed in [FeFe] Hydrogenas-es Reveal Redox Coupling Within the H-Cluster. Journal of the American Chemical Society 2014, 136, 11339-11346.
(51) Adamska, A.; Silakov, A.; Lambertz, C.; Rüdiger, O.; Happe, T.; Reijerse, E.; Lubitz, W., Identification and Characterization of the “Super-Reduced” State of the H-Cluster in [FeFe] Hydrogenase: A New Building Block for the

72
Catalytic Cycle? Angewandte Chemie International Edition 2012, 51, 11458-11462.
(52) Greco, C.; Bruschi, M.; De Gioia, L.; Ryde, U., A QM/MM Investigation of the Activation and Catalytic Mechanism of Fe-Only Hydrogenases. Inorgan-ic Chemistry 2007, 46, 5911-5921.
(53) Knörzer, P.; Silakov, A.; Foster, C. E.; Armstrong, F. A.; Lubitz, W.; Happe, T., Importance of the protein framework for catalytic activity of [FeFe]-hydrogenases. Journal of Biological Chemistry 2012, 287, 1489-1499.
(54) Greco, C.; Bruschi, M.; Fantucci, P.; Ryde, U.; De Gioia, L., Mechanistic and Physiological Implications of the Interplay among Iron–Sulfur Clusters in [FeFe]-Hydrogenases. A QM/MM Perspective. Journal of the American Chemical Society 2011, 133, 18742-18749.
(55) Olsen, M. T.; Rauchfuss, T. B.; Wilson, S. R., Role of the Azadithiolate Co-factor in Models for [FeFe]-Hydrogenase: Novel Structures and Catalytic Implications. Journal of the American Chemical Society 2010, 132, 17733-17740.
(56) Mulder, D. W.; Ratzloff, M. W.; Bruschi, M.; Greco, C.; Koonce, E.; Peters, J. W.; King, P. W., Investigations on the Role of Proton-Coupled Electron Transfer in Hydrogen Activation by [FeFe]-Hydrogenase. Journal of the American Chemical Society 2014, 136, 15394-15402.
(57) Peters, J. W.; Schut, G. J.; Boyd, E. S.; Mulder, D. W.; Shepard, E. M.; Bro-derick, J. B.; King, P. W.; Adams, M. W. W., [FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2015, 1853, 1350-1369.
(58) Lyon, E. J.; Georgakaki, I. P.; Reibenspies, J. H.; Darensbourg, M. Y., Car-bon Monoxide and Cyanide Ligands in a Classical Organometallic Complex Model for Fe-Only Hydrogenase. Angewandte Chemie International Edition 1999, 38, 3178-3180.
(59) Lawrence, J. D.; Li, H.; Rauchfuss, T. B.; Bénard, M.; Rohmer, M.-M., Diiron Azadithiolates as Models for the Iron-Only Hydrogenase Active Site: Synthesis, Structure, and Stereoelectronics. Angewandte Chemie Internation-al Edition 2001, 40, 1768-1771.
(60) Barton, B. E.; Olsen, M. T.; Rauchfuss, T. B., Aza- and Oxadithiolates Are Probable Proton Relays in Functional Models for the [FeFe]-Hydrogenases. Journal of the American Chemical Society 2008, 130, 16834-16835.
(61) Schilter, D.; Rauchfuss, T. B., And the Winner is…Azadithiolate: An Amine Proton Relay in the [FeFe] Hydrogenases. Angewandte Chemie International Edition 2013, 52, 13518-13520.
(62) Capon, J.-F.; Gloaguen, F.; Schollhammer, P.; Talarmin, J., Activation of proton by the two-electron reduction of a di-iron organometallic complex. Journal of Electroanalytical Chemistry 2006, 595, 47-52.
(63) Capon, J.-F.; Gloaguen, F.; Schollhammer, P.; Talarmin, J., Electrochemical proton reduction by thiolate-bridged hexacarbonyldiiron clusters. Journal of Electroanalytical Chemistry 2004, 566, 241-247.
(64) Felton, G. A. N.; Vannucci, A. K.; Chen, J.; Lockett, L. T.; Okumura, N.; Petro, B. J.; Zakai, U. I.; Evans, D. H.; Glass, R. S.; Lichtenberger, D. L., Hydrogen Generation from Weak Acids: Electrochemical and Computational Studies of a Diiron Hydrogenase Mimic. Journal of the American Chemical Society 2007, 129, 12521-12530.
(65) Song, L.-C., Investigations on Butterfly Fe/S Cluster S-Centered Anions (μ-S-)2Fe2(CO)6, (μ-S-)(μ-RS)Fe2(CO)6, and Related Species. Accounts of Chemical Research 2005, 38, 21-28.

73
(66) Lomoth, R.; Ott, S., Introducing a dark reaction to photochemistry: photo-catalytic hydrogen from [FeFe] hydrogenase active site model complexes. Dalton Transactions 2009, 9952-9959.
(67) Siegbahn, P. E. M.; Tye, J. W.; Hall, M. B., Computational Studies of [NiFe] and [FeFe] Hydrogenases. Chemical Reviews 2007, 107, 4414-4435.
(68) Ginovska-Pangovska, B.; Dutta, A.; Reback, M. L.; Linehan, J. C.; Shaw, W. J., Beyond the Active Site: The Impact of the Outer Coordination Sphere on Electrocatalysts for Hydrogen Production and Oxidation. Accounts of Chemi-cal Research 2014, 47, 2621-2630.
(69) Yang, J. Y.; Bullock, R. M.; Shaw, W. J.; Twamley, B.; Fraze, K.; DuBois, M. R.; DuBois, D. L., Mechanistic Insights into Catalytic H2 Oxidation by Ni Complexes Containing a Diphosphine Ligand with a Positioned Amine Base. Journal of the American Chemical Society 2009, 131, 5935-5945.
(70) O’Hagan, M.; Ho, M.-H.; Yang, J. Y.; Appel, A. M.; DuBois, M. R.; Raugei, S.; Shaw, W. J.; DuBois, D. L.; Bullock, R. M., Proton Delivery and Removal in [Ni(PR2NR′2)2]2+ Hydrogen Production and Oxidation Cata-lysts. Journal of the American Chemical Society 2012, 134, 19409-19424.
(71) Dutta, A.; DuBois, D. L.; Roberts, J. A. S.; Shaw, W. J., Amino acid modi-fied Ni catalyst exhibits reversible H<sub>2</sub> oxidation/production over a broad pH range at elevated temperatures. Proceedings of the National Academy of Sciences 2014, 111, 16286-16291.
(72) Helm, M. L.; Stewart, M. P.; Bullock, R. M.; DuBois, M. R.; DuBois, D. L., A Synthetic Nickel Electrocatalyst with a Turnover Frequency Above 100,000 s<sup>−1</sup> for H<sub>2</sub> Production. Science 2011, 333, 863-866.
(73) O’Hagan, M.; Shaw, W. J.; Raugei, S.; Chen, S.; Yang, J. Y.; Kilgore, U. J.; DuBois, D. L.; Bullock, R. M., Moving Protons with Pendant Amines: Proton Mobility in a Nickel Catalyst for Oxidation of Hydrogen. Journal of the American Chemical Society 2011, 133, 14301-14312.
(74) Dutta, A.; Lense, S.; Hou, J.; Engelhard, M. H.; Roberts, J. A. S.; Shaw, W. J., Minimal Proton Channel Enables H2 Oxidation and Production with a Water-Soluble Nickel-Based Catalyst. Journal of the American Chemical So-ciety 2013, 135, 18490-18496.
(75) Wiese, S.; Kilgore, U. J.; Ho, M.-H.; Raugei, S.; DuBois, D. L.; Bullock, R. M.; Helm, M. L., Hydrogen Production Using Nickel Electrocatalysts with Pendant Amines: Ligand Effects on Rates and Overpotentials. ACS Catalysis 2013, 3, 2527-2535.
(76) Hoffert, W. A.; Roberts, J. A. S.; Morris Bullock, R.; Helm, M. L., Produc-tion of H2 at fast rates using a nickel electrocatalyst in water–acetonitrile so-lutions. Chemical Communications 2013, 49, 7767-7769.
(77) Wang, M.; Chen, L.; Sun, L., Recent progress in electrochemical hydrogen production with earth-abundant metal complexes as catalysts. Energy & En-vironmental Science 2012, 5, 6763-6778.
(78) Varma, S.; Castillo, C. E.; Stoll, T.; Fortage, J.; Blackman, A. G.; Molton, F.; Deronzier, A.; Collomb, M.-N., Efficient photocatalytic hydrogen production in water using a cobalt(iii) tetraaza-macrocyclic catalyst: electrochemical generation of the low-valent Co(i) species and its reactivity toward proton re-duction. Physical Chemistry Chemical Physics 2013, 15, 17544-17552.
(79) Thoi, V. S.; Sun, Y.; Long, J. R.; Chang, C. J., Complexes of earth-abundant metals for catalytic electrochemical hydrogen generation under aqueous con-ditions. Chemical Society Reviews 2013, 42, 2388-2400.

74
(80) Andreiadis, E. S.; Jacques, P.-A.; Tran, P. D.; Leyris, A.; Chavarot-Kerlidou, M.; Jousselme, B.; Matheron, M.; Pécaut, J.; Palacin, S.; Fontecave, M.et al., Molecular engineering of a cobalt-based electrocatalytic nanomaterial for H2 evolution under fully aqueous conditions. Nature Chemistry 2012, 5, 48.
(81) Artero, V.; Chavarot-Kerlidou, M.; Fontecave, M., Splitting Water with Co-balt. Angewandte Chemie International Edition 2011, 50, 7238-7266.
(82) Tschierlei, S.; Ott, S.; Lomoth, R., Spectroscopically characterized interme-diates of catalytic H2 formation by [FeFe] hydrogenase models. Energy & Environmental Science 2011, 4, 2340-2352.
(83) Eilers, G.; Schwartz, L.; Stein, M.; Zampella, G.; de Gioia, L.; Ott, S.; Lo-moth, R., Ligand versus Metal Protonation of an Iron Hydrogenase Active Site Mimic. Chemistry – A European Journal 2007, 13, 7075-7084.
(84) Gloaguen, F.; Lawrence, J. D.; Rauchfuss, T. B., Biomimetic Hydrogen Evo-lution Catalyzed by an Iron Carbonyl Thiolate. Journal of the American Chemical Society 2001, 123, 9476-9477.
(85) Arias-Rotondo, D. M.; McCusker, J. K., The photophysics of photoredox catalysis: a roadmap for catalyst design. Chemical Society Reviews 2016, 45, 5803-5820.
(86) Balzani, V.; Bergamini, G.; Marchioni, F.; Ceroni, P., Ru(II)-bipyridine complexes in supramolecular systems, devices and machines. Coordination Chemistry Reviews 2006, 250, 1254-1266.
(87) Mirmohades, M.; Pullen, S.; Stein, M.; Maji, S.; Ott, S.; Hammarström, L.; Lomoth, R., Direct Observation of Key Catalytic Intermediates in a Photoin-duced Proton Reduction Cycle with a Diiron Carbonyl Complex. Journal of the American Chemical Society 2014, 136, 17366-17369.
(88) Connelly, N. G.; Geiger, W. E., Chemical Redox Agents for Organometallic Chemistry. Chemical Reviews 1996, 96, 877-910.
(89) MacInnes, M. M.; Hlynchuk, S.; Acharya, S.; Lehnert, N.; Maldonado, S., Reduction of Graphene Oxide Thin Films by Cobaltocene and Decame-thylcobaltocene. ACS Applied Materials & Interfaces 2018, 10, 2004-2015.
(90) Spessard, G. O.; Miessler, G. L. Organometallic chemistry; 2nd ed.; Oxford University Press: New York, 2010.
(91) Allian, A. D.; Wang, Y.; Saeys, M.; Kuramshina, G. M.; Garland, M., The combination of deconvolution and density functional theory for the mid-infrared vibrational spectra of stable and unstable rhodium carbonyl clusters. Vibrational Spectroscopy 2006, 41, 101-111.
(92) Fabian, H.; Naumann, D., Methods to study protein folding by stopped-flow FT-IR. Methods 2004, 34, 28-40.
(93) Borg, S. J.; Behrsing, T.; Best, S. P.; Razavet, M.; Liu, X.; Pickett, C. J., Electron Transfer at a Dithiolate-Bridged Diiron Assembly: Electrocatalytic Hydrogen Evolution. Journal of the American Chemical Society 2004, 126, 16988-16999.
(94) Nann, T.; Ibrahim, S. K.; Woi, P.-M.; Xu, S.; Ziegler, J.; Pickett, C. J., Water Splitting by Visible Light: A Nanophotocathode for Hydrogen Production. Angewandte Chemie International Edition 2010, 49, 1574-1577.
(95) Samuel, A. P. S.; Co, D. T.; Stern, C. L.; Wasielewski, M. R., Ultrafast Pho-todriven Intramolecular Electron Transfer from a Zinc Porphyrin to a Readily Reduced Diiron Hydrogenase Model Complex. Journal of the American Chemical Society 2010, 132, 8813-8815.
(96) Jian, J.-X.; Ye, C.; Wang, X.-Z.; Wen, M.; Li, Z.-J.; Li, X.-B.; Chen, B.; Tung, C.-H.; Wu, L.-Z., Comparison of H2 photogeneration by [FeFe]-

75
hydrogenase mimics with CdSe QDs and Ru(bpy)3Cl2 in aqueous solution. Energy & Environmental Science 2016, 9, 2083-2089.
(97) Wang, F.; Wang, W.-G.; Wang, X.-J.; Wang, H.-Y.; Tung, C.-H.; Wu, L.-Z., A Highly Efficient Photocatalytic System for Hydrogen Production by a Ro-bust Hydrogenase Mimic in an Aqueous Solution. Angewandte Chemie In-ternational Edition 2011, 50, 3193-3197.
(98) Nicolet, Y.; Piras, C.; Legrand, P.; Hatchikian, C. E.; Fontecilla-Camps, J. C., Desulfovibrio desulfuricans iron hydrogenase: the structure shows unusual coordination to an active site Fe binuclear center. Structure 1999, 7, 13-23.
(99) Hunt, N. T.; Wright, J. A.; Pickett, C., Detection of Transient Intermediates Generated from Subsite Analogues of [FeFe] Hydrogenases. Inorganic Chemistry 2016, 55, 399-410.
(100) Capon, J.-F.; Ezzaher, S.; Gloaguen, F.; Pétillon, F. Y.; Schollhammer, P.; Talarmin, J.; Davin, T. J.; McGrady, J. E.; Muir, K. W., Electrochemical and theoretical investigations of the reduction of [Fe2(CO)5L{μ-SCH2XCH2S}] complexes related to [FeFe] hydrogenase. New Journal of Chemistry 2007, 31, 2052-2064.
(101) Tye, J. W.; Darensbourg, M. Y.; Hall, M. B., Correlation between computed gas-phase and experimentally determined solution-phase infrared spectra: Models of the iron–iron hydrogenase enzyme active site. Journal of Compu-tational Chemistry 2006, 27, 1454-1462.
(102) Eckert, F.; Leito, I.; Kaljurand, I.; Kütt, A.; Klamt, A.; Diedenhofen, M., Prediction of acidity in acetonitrile solution with COSMO-RS. Journal of Computational Chemistry 2009, 30, 799-810.
(103) Wang, S.; Aster, A.; Mirmohades, M.; Lomoth, R.; Hammarström, L., Struc-tural and Kinetic Studies of Intermediates of a Biomimetic Diiron Proton-Reduction Catalyst. Inorganic Chemistry 2018, 57, 768-776.
(104) Greco, C.; Zampella, G.; Bertini, L.; Bruschi, M.; Fantucci, P.; De Gioia, L., Insights into the Mechanism of Electrocatalytic Hydrogen Evolution Mediat-ed by Fe2(S2C3H6)(CO)6: The Simplest Functional Model of the Fe-Hydrogenase Active Site. Inorganic Chemistry 2007, 46, 108-116.
(105) Jablonskytė, A.; Webster, L. R.; Simmons, T. R.; Wright, J. A.; Pickett, C. J., Electronic Control of the Protonation Rates of Fe–Fe Bonds. Journal of the American Chemical Society 2014, 136, 13038-13044.
(106) Bourrez, M.; Steinmetz, R.; Gloaguen, F., Mechanistic Insights into the Ca-talysis of Electrochemical Proton Reduction by a Diiron Azadithiolate Com-plex. Inorganic Chemistry 2014, 53, 10667-10673.
(107) Kolthoff, I. M., Acid-base equilibriums in dipolar aprotic solvents. Analytical Chemistry 1974, 46, 1992-2003.
(108) Capon, J.-F.; Ezzaher, S.; Gloaguen, F.; Pétillon, F. Y.; Schollhammer, P.; Talarmin, J., Electrochemical Insights into the Mechanisms of Proton Reduc-tion by [Fe2(CO)6{μ-SCH2N(R)CH2S}] Complexes Related to the [2Fe]H Subsite of [FeFe]Hydrogenase. Chemistry – A European Journal 2008, 14, 1954-1964.
(109) Löscher, S.; Schwartz, L.; Stein, M.; Ott, S.; Haumann, M., Facilitated Hy-dride Binding in an Fe−Fe Hydrogenase Active−Site Biomimic Revealed by X-ray Absorption Spectroscopy and DFT Calculations. Inorganic Chemistry 2007, 46, 11094-11105.
(110) Ezzaher, S.; Capon, J.-F.; Gloaguen, F.; Pétillon, F. Y.; Schollhammer, P.; Talarmin, J.; Pichon, R.; Kervarec, N., Evidence for the Formation of Termi-nal Hydrides by Protonation of an Asymmetric Iron Hydrogenase Active Site Mimic. Inorganic Chemistry 2007, 46, 3426-3428.

76
(111) Zaffaroni, R.; Rauchfuss, T. B.; Gray, D. L.; De Gioia, L.; Zampella, G., Terminal vs Bridging Hydrides of Diiron Dithiolates: Protonation of Fe2(dithiolate)(CO)2(PMe3)4. Journal of the American Chemical Society 2012, 134, 19260-19269.
(112) Rauchfuss, T. B., Diiron Azadithiolates as Models for the [FeFe]-Hydrogenase Active Site and Paradigm for the Role of the Second Coordina-tion Sphere. Accounts of Chemical Research 2015, 48, 2107-2116.
(113) Filippi, G.; Arrigoni, F.; Bertini, L.; De Gioia, L.; Zampella, G., DFT Dissec-tion of the Reduction Step in H2 Catalytic Production by [FeFe]-Hydrogenase-Inspired Models: Can the Bridging Hydride Become More Re-active Than the Terminal Isomer? Inorganic Chemistry 2015, 54, 9529-9542.
(114) Schwartz, L.; Eilers, G.; Eriksson, L.; Gogoll, A.; Lomoth, R.; Ott, S., Iron hydrogenase active site mimic holding a proton and a hydride. Chemical Communications 2006, 520-522.
(115) Elgrishi, N.; Kurtz, D. A.; Dempsey, J. L., Reaction Parameters Influencing Cobalt Hydride Formation Kinetics: Implications for Benchmarking H2-Evolution Catalysts. Journal of the American Chemical Society 2017, 139, 239-244.
(116) Bertini, L.; Fantucci, P.; De Gioia, L.; Zampella, G., Excited State Properties of Diiron Dithiolate Hydrides: Implications in the Unsensitized Photocataly-sis of H2 Evolution. Inorganic Chemistry 2013, 52, 9826-9841.
(117) Wang, W.; Rauchfuss, T. B.; Bertini, L.; Zampella, G., Unsensitized Photo-chemical Hydrogen Production Catalyzed by Diiron Hydrides. Journal of the American Chemical Society 2012, 134, 4525-4528.
(118) Perutz, R. N.; Procacci, B., Photochemistry of Transition Metal Hydrides. Chemical Reviews 2016, 116, 8506-8544.
(119) Frederix, P. W. J. M.; Adamczyk, K.; Wright, J. A.; Tuttle, T.; Ulijn, R. V.; Pickett, C. J.; Hunt, N. T., Investigation of the Ultrafast Dynamics Occurring during Unsensitized Photocatalytic H2 Evolution by an [FeFe]-Hydrogenase Subsite Analogue. Organometallics 2014, 33, 5888-5896.
(120) Chong, D.; Georgakaki, I. P.; Mejia-Rodriguez, R.; Sanabria-Chinchilla, J.; Soriaga, M. P.; Darensbourg, M. Y., Electrocatalysis of hydrogen production by active site analogues of the iron hydrogenase enzyme: structure/function relationships. Dalton Transactions 2003, 4158-4163.
(121) Borg, S. J.; Tye, J. W.; Hall, M. B.; Best, S. P., Assignment of Molecular Structures to the Electrochemical Reduction Products of Diiron Compounds Related to [Fe−Fe] Hydrogenase: A Combined Experimental and Density Functional Theory Study. Inorganic Chemistry 2007, 46, 384-394.
(122) Jablonskytė, A.; Wright, J. A.; Pickett, C. J., Mechanistic aspects of the pro-tonation of [FeFe]-hydrogenase subsite analogues. Dalton Transactions 2010, 39, 3026-3034.
(123) Aguirre de Carcer, I.; DiPasquale, A.; Rheingold, A. L.; Heinekey, D. M., Active-Site Models for Iron Hydrogenases: Reduction Chemistry of Dinu-clear Iron Complexes. Inorganic Chemistry 2006, 45, 8000-8002.
(124) Quentel, F.; Gloaguen, F., Kinetic and thermodynamic aspects of the electro-catalysis of acid reduction in organic solvent using molecular diiron-dithiolate compounds. Electrochimica Acta 2013, 110, 641-645.
(125) Schwartz, L.; Singh, P. S.; Eriksson, L.; Lomoth, R.; Ott, S., Tuning the elec-tronic properties of Fe2(μ-arenedithiolate)(CO)6−n(PMe3)n (n=0, 2) com-plexes related to the [Fe–Fe]-hydrogenase active site. Comptes Rendus Chimie 2008, 11, 875-889.

77
(126) Streich, D.; Astuti, Y.; Orlandi, M.; Schwartz, L.; Lomoth, R.; Ham-marström, L.; Ott, S., High-Turnover Photochemical Hydrogen Production Catalyzed by a Model Complex of the [FeFe]-Hydrogenase Active Site. Chemistry – A European Journal 2010, 16, 60-63.
(127) Singh, P. S.; Rudbeck, H. C.; Huang, P.; Ezzaher, S.; Eriksson, L.; Stein, M.; Ott, S.; Lomoth, R., (I,0) Mixed-Valence State of a Diiron Complex with Per-tinence to the [FeFe]-Hydrogenase Active Site: An IR, EPR, and Computa-tional Study. Inorganic Chemistry 2009, 48, 10883-10885.
(128) Bae, H. Y.; Höfler, D.; Kaib, P. S. J.; Kasaplar, P.; De, C. K.; Döhring, A.; Lee, S.; Kaupmees, K.; Leito, I.; List, B., Approaching sub-ppm-level asym-metric organocatalysis of a highly challenging and scalable carbon–carbon bond forming reaction. Nature Chemistry 2018, 10, 888-894.
(129) Kaupmees, K.; Tolstoluzhsky, N.; Raja, S.; Rueping, M.; Leito, I., On the Acidity and Reactivity of Highly Effective Chiral Brønsted Acid Catalysts: Establishment of an Acidity Scale. Angewandte Chemie International Edition 2013, 52, 11569-11572.
(130) Vazdar, K.; Kunetskiy, R.; Saame, J.; Kaupmees, K.; Leito, I.; Jahn, U., Very Strong Organosuperbases Formed by Combining Imidazole and Guanidine Bases: Synthesis, Structure, and Basicity. Angewandte Chemie International Edition 2014, 53, 1435-1438.
(131) Costentin, C.; Dridi, H.; Savéant, J.-M., Molecular Catalysis of H2 Evolu-tion: Diagnosing Heterolytic versus Homolytic Pathways. Journal of the American Chemical Society 2014, 136, 13727-13734.
(132) Marinescu, S. C.; Winkler, J. R.; Gray, H. B., Molecular mechanisms of cobalt-catalyzed hydrogen evolution. Proceedings of the National Academy of Sciences 2012.
(133) Elgrishi, N.; McCarthy, B. D.; Rountree, E. S.; Dempsey, J. L., Reaction Pathways of Hydrogen-Evolving Electrocatalysts: Electrochemical and Spec-troscopic Studies of Proton-Coupled Electron Transfer Processes. ACS Ca-talysis 2016, 6, 3644-3659.
(134) Queyriaux, N.; Jane, R. T.; Massin, J.; Artero, V.; Chavarot-Kerlidou, M., Recent developments in hydrogen evolving molecular cobalt(II)–polypyridyl catalysts. Coordination Chemistry Reviews 2015, 304-305, 3-19.
(135) Gloaguen, F., Electrochemistry of Simple Organometallic Models of Iron–Iron Hydrogenases in Organic Solvent and Water. Inorganic Chemistry 2016, 55, 390-398.
(136) Belkova, N. V.; Filippov, O. A.; Shubina, E. S., Z−H Bond Activation in (Di)hydrogen Bonding as a Way to Proton/Hydride Transfer and H2 Evolu-tion. Chemistry – A European Journal 2018, 24, 1464-1470.
(137) Papish, E. T.; Rix, F. C.; Spetseris, N.; Norton, J. R.; Williams, R. D., Proto-nation of CpW(CO)2(PMe3)H: Is the Metal or the Hydride the Kinetic Site? Journal of the American Chemical Society 2000, 122, 12235-12242.
(138) Belkova, N. V.; Epstein, L. M.; Filippov, O. A.; Shubina, E. S., Hydrogen and Dihydrogen Bonds in the Reactions of Metal Hydrides. Chemical Re-views 2016, 116, 8545-8587.
(139) Gordon, J. C.; Kubas, G. J., Perspectives on How Nature Employs the Princi-ples of Organometallic Chemistry in Dihydrogen Activation in Hydrogenas-es. Organometallics 2010, 29, 4682-4701.
(140) Morris, R. H., Brønsted–Lowry Acid Strength of Metal Hydride and Dihy-drogen Complexes. Chemical Reviews 2016, 116, 8588-8654.
(141) Crabtree, R. H., Dihydrogen Complexation. Chemical Reviews 2016, 116, 8750-8769.

78
(142) Schilter, D.; Camara, J. M.; Huynh, M. T.; Hammes-Schiffer, S.; Rauchfuss, T. B., Hydrogenase Enzymes and Their Synthetic Models: The Role of Metal Hydrides. Chemical Reviews 2016, 116, 8693-8749.
(143) Waldie, K. M.; Ostericher, A. L.; Reineke, M. H.; Sasayama, A. F.; Kubiak, C. P., Hydricity of Transition-Metal Hydrides: Thermodynamic Considera-tions for CO2 Reduction. ACS Catalysis 2018, 8, 1313-1324.
(144) Ilic, S.; Pandey Kadel, U.; Basdogan, Y.; Keith, J. A.; Glusac, K. D., Ther-modynamic Hydricities of Biomimetic Organic Hydride Donors. Journal of the American Chemical Society 2018, 140, 4569-4579.


Acta Universitatis UpsaliensisDigital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Science and Technology 1787
Editor: The Dean of the Faculty of Science and Technology
A doctoral dissertation from the Faculty of Science andTechnology, Uppsala University, is usually a summary of anumber of papers. A few copies of the complete dissertationare kept at major Swedish research libraries, while thesummary alone is distributed internationally throughthe series Digital Comprehensive Summaries of UppsalaDissertations from the Faculty of Science and Technology.(Prior to January, 2005, the series was published under thetitle “Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Science and Technology”.)
Distribution: publications.uu.seurn:nbn:se:uu:diva-380279
ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2019





























