Embed Size (px)
Citation preview
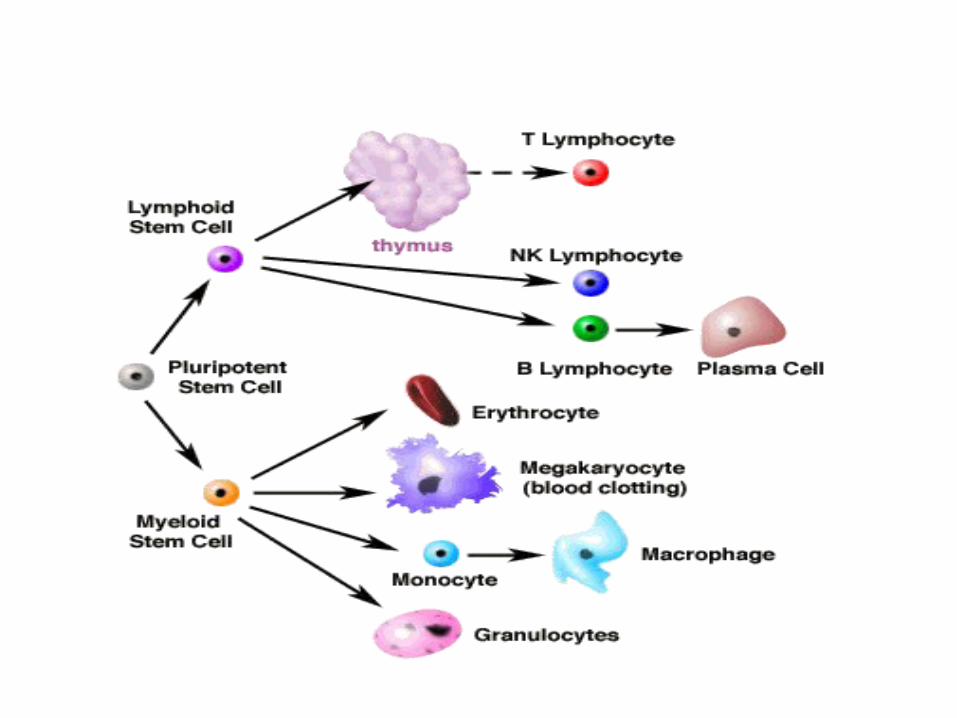
Products of haemopoiesis

ABNORMALITIES IN THE HEMOPOIETIC SYSTEM
• CAN LEAD TO
• HEMOGLOBINOPATHIES
• HEMOPHILIA
• DEFECTS IN HEMOSTASIS/THROMBOSIS
• HEMATOLOGICAL MALIGNANCY

MUTATIONS AND DNA
• VARIOUS TYPES OF MUTATIONS CAN OCCUR LEADING TO DISEASE PHENOTYPE
• POINT MUTATIONS• INSERTIONS OR DELETIONS• TRANSLOCATIONS• COMPLEX CHROMOSOMAL
REARRANGEMENTS
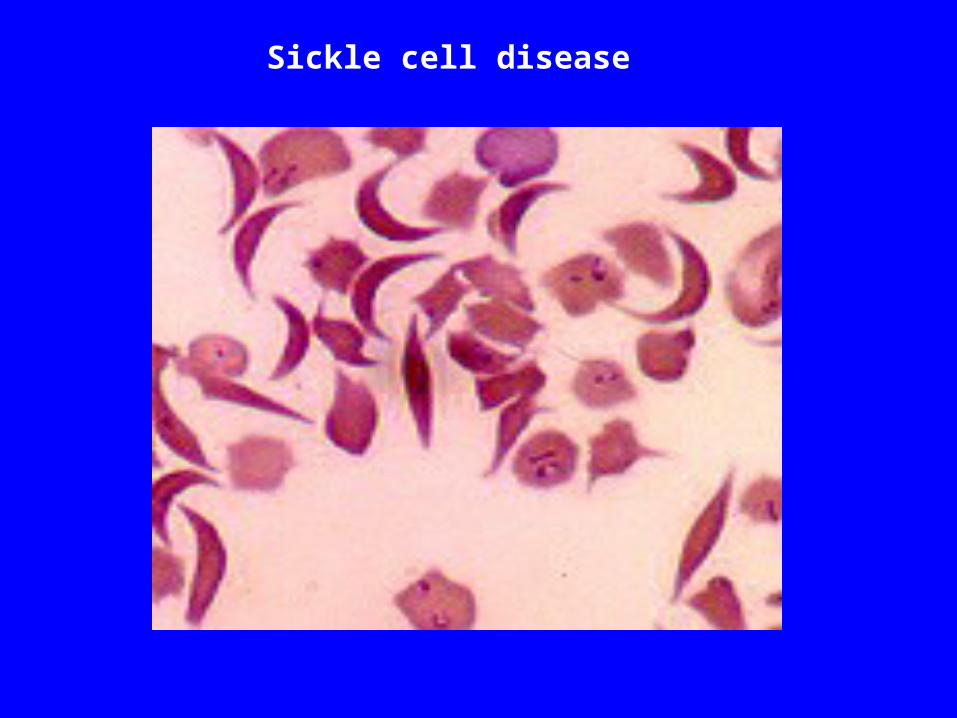
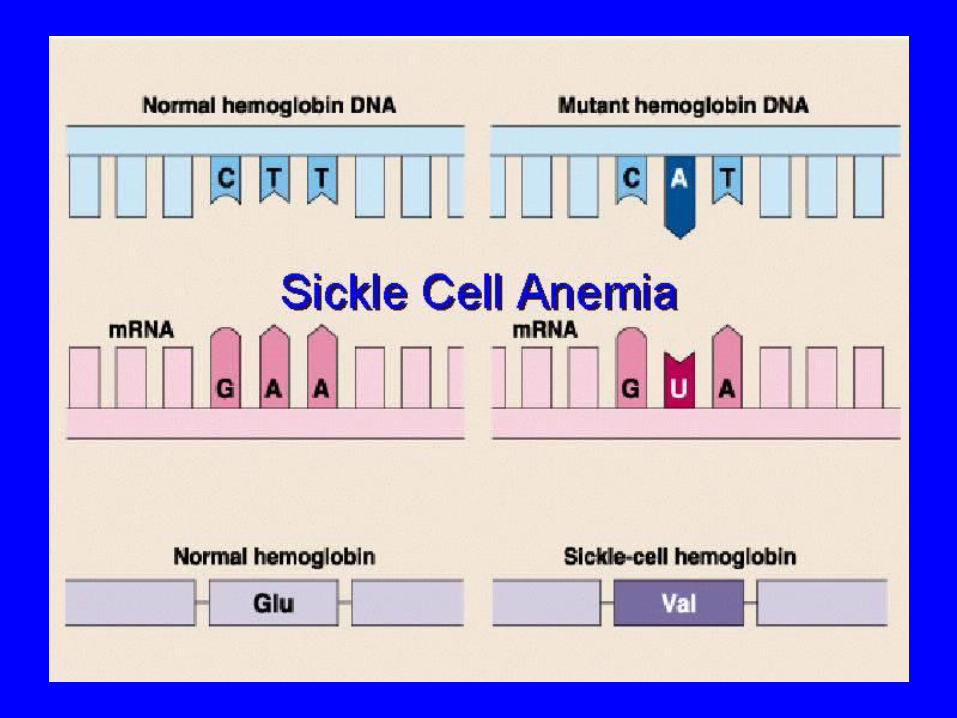
Sickle cell disease

Thalassemia
• The thalassemias are a diverse group of genetic blood diseases characterized by absent or decreased production of normal hemoglobin, resulting in a microcytic anemia of varying degree
• The alpha () thalassemias are concentrated in Southeast Asia, Malaysia, and southern China.
• The beta () thalassemias are seen primarily in the areas surrounding Mediterranean Sea, Africa and Southeast Asia.
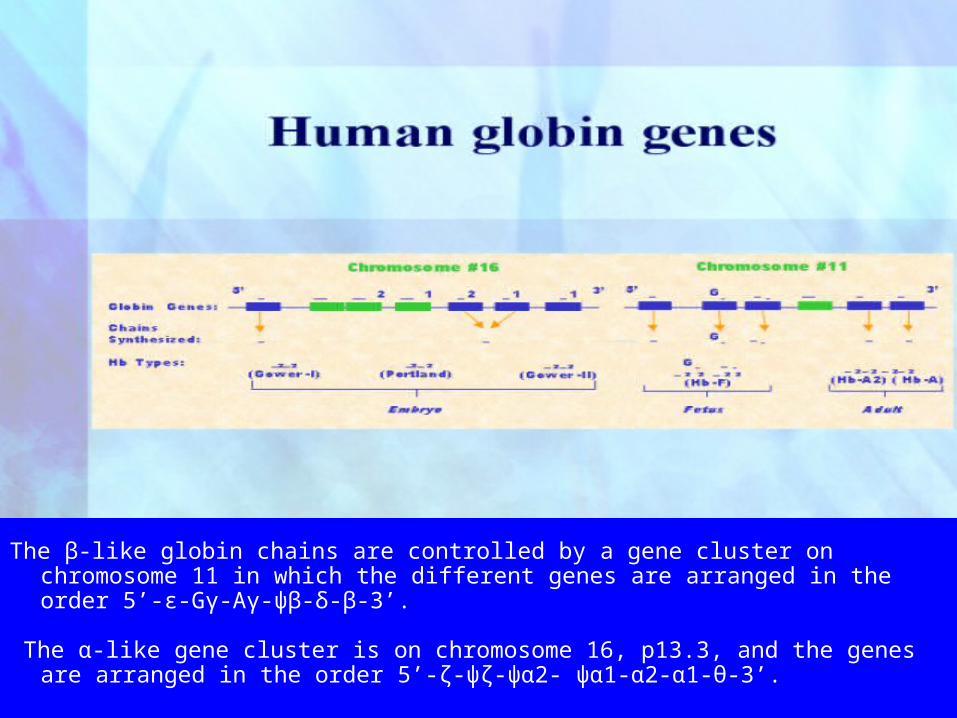
The β-like globin chains are controlled by a gene cluster on chromosome 11 in which the different genes are arranged in the order 5’-ε-Gγ-Aγ-ψβ-δ-β-3’.
The α-like gene cluster is on chromosome 16, p13.3, and the genes are arranged in the order 5’-ζ-ψζ-ψα2- ψα1-α2-α1-θ-3’.
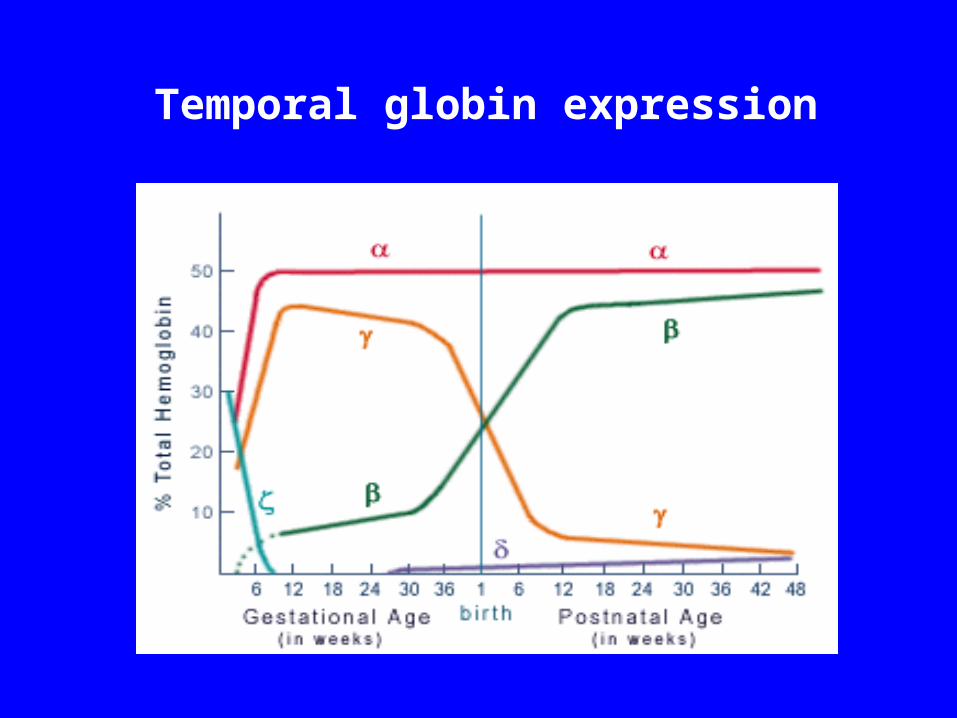
Temporal globin expression

Temporal Globin expression globin expression is rather stable in fetal and adult life, because it is
needed for both fetal and adult hemoglobin production
globin appears early in fetal life at low levels and rapidly increases after 30 weeks gestational age, reaching a maximum about 30 weeks postnatally
globin molecule is expressed at a high level in fetal life ( 6 weeks) and begins to decline about 30 weeks gestational age, reaching a low level about 48 weeks postgestational age.
globin appears at a low level at about 30 weeks gestational age and maintains a low profile throughout life.
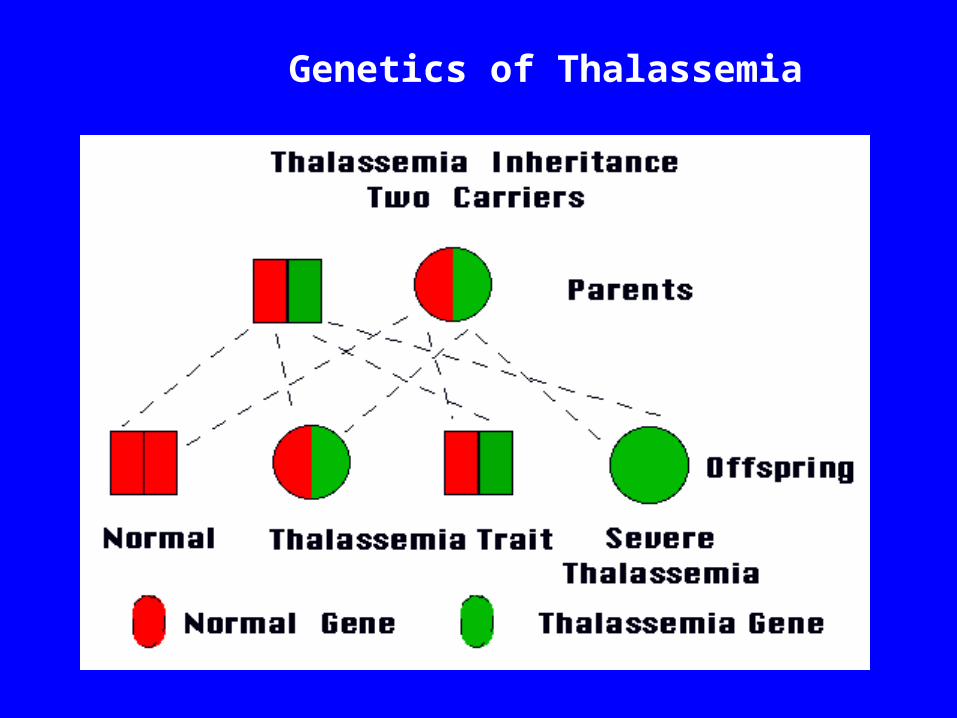
Genetics of Thalassemia

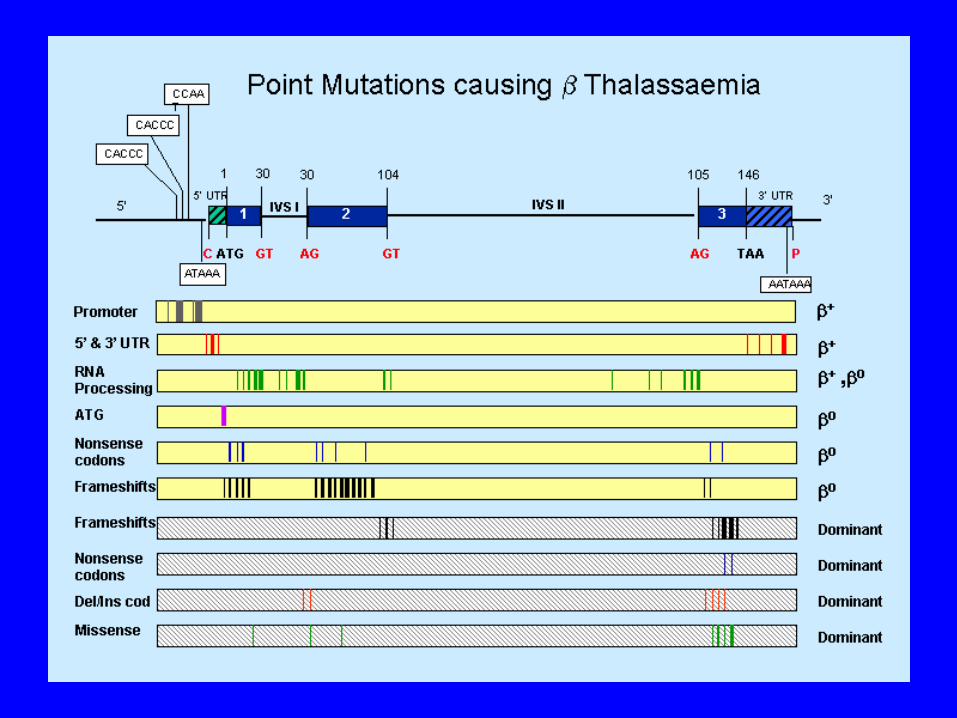
Types of Thalassemia thal: excess of globins, leading to formation of globin
tetramers (4) that accumulate in the erythroblast , leading to ineffective erythropoiesis. Two types of mutations, the β0 in which no β globin chains are produced and β+, in which some β chains are produced but at a reduced rate.
thal : excess of globins, leading to the formation of globin tetramers (4) called hemoglobin H. Results in hemolysis, generally shortening the life span of the red cell. Hemoglobin H-Constant Spring disease is a more severe form of this hemolytic disorder. Most severe form is thalassemia major, in which fetus produces no globins, which is generally incompatible with life.

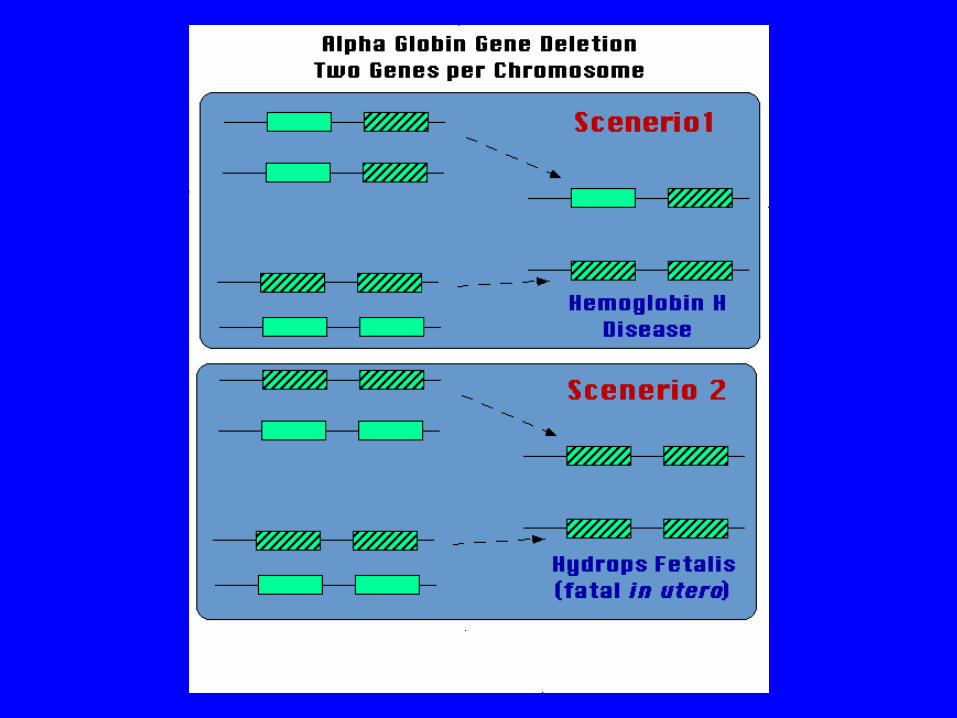

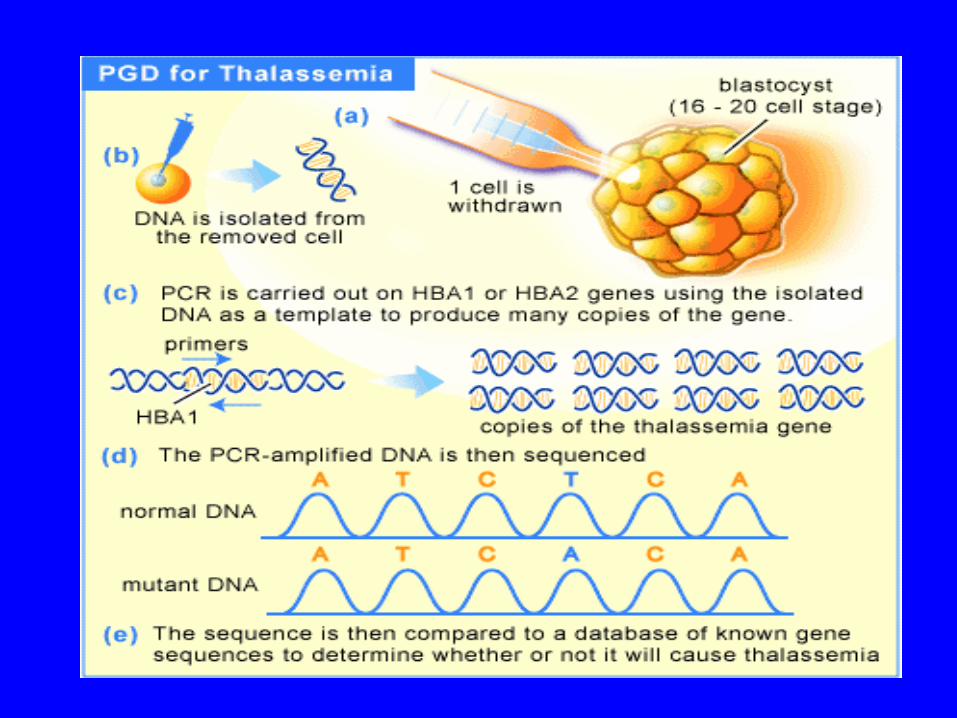
Thalassemia Prevention
• Preventive programs in (i) public education, (ii) population screening, genetic counseling and prenatal diagnosis have been very effective in reducing the birth rate of β-thalassemia major.
• Combination of hematological and molecular techniques offers the most reliable and accurate strategy for β-thalassemia prenatal diagnosis
• Development of molecular techniques not only made it possible to offer prenatal diagnosis at an early stage of the pregnancy but they can help to resolve diagnostic problems.

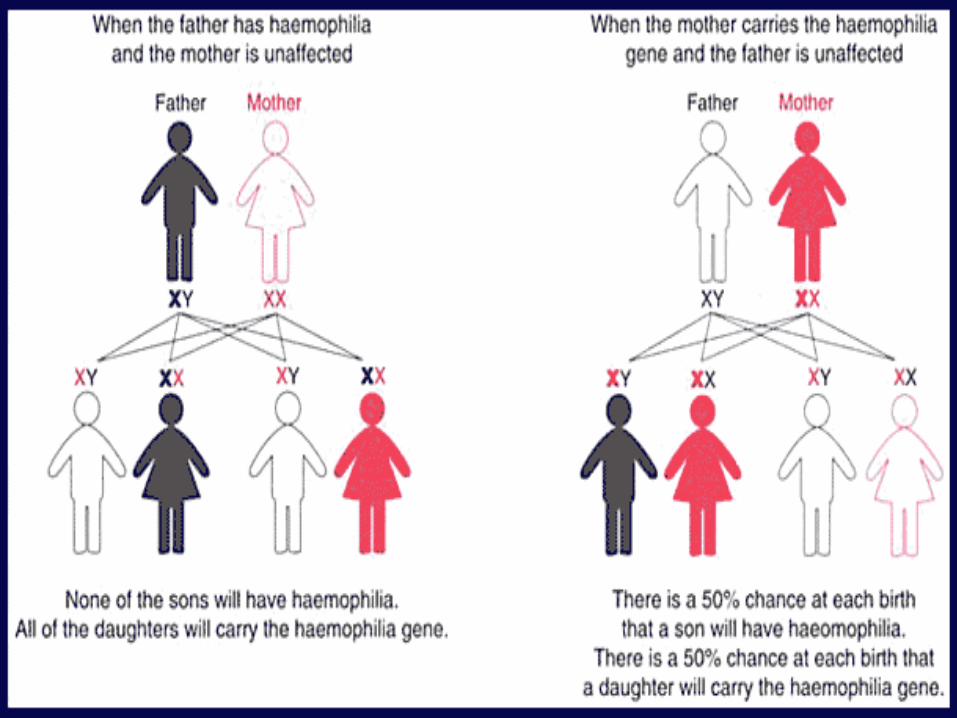
HAEMOPHILIA
X LINKED RECESSIVE DISORDER
HAEMOPHILIA A – MUTATIONS IN FACTOR VIII GENE
HAEMOPHILIA B – MUTATIONS IN FACTOR IX GENE
SIMPLE AND COMPLICATED MUTATIONS
THE FLIP TIP MUTATION

Hemophilia Mutations
• Deletions
• Point mutations
• Flip tip mutations
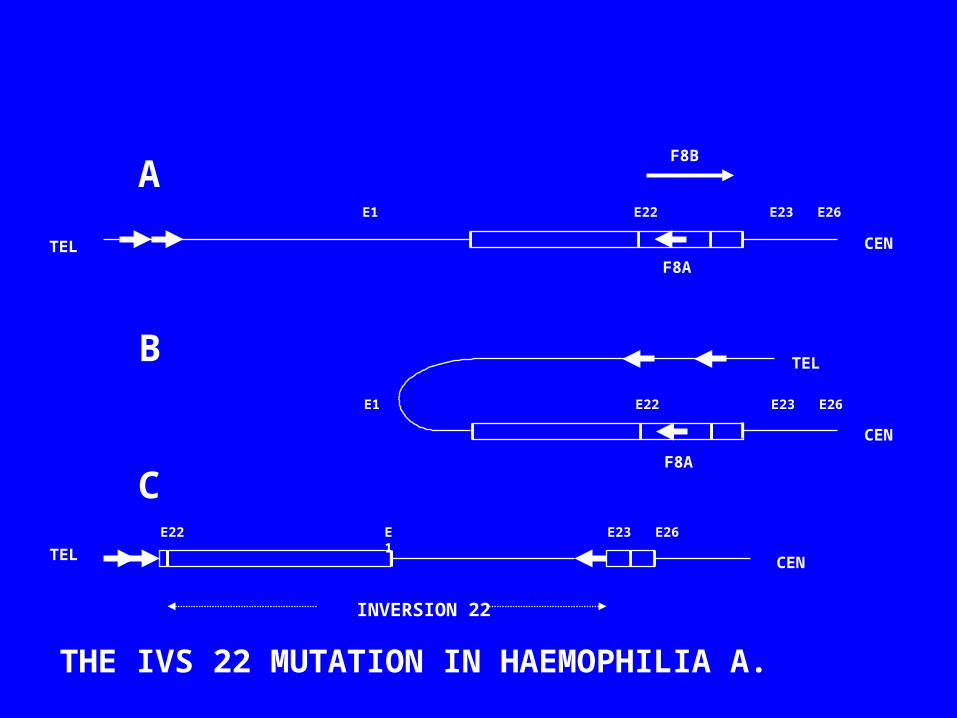
F8A
E1 E22 E23 E26
F8A
E1 E22 E23 E26
TEL CEN
CEN
CENTEL
TEL
INVERSION 22
E1E22 E23 E26
A
B
C
THE IVS 22 MUTATION IN HAEMOPHILIA A.
F8B
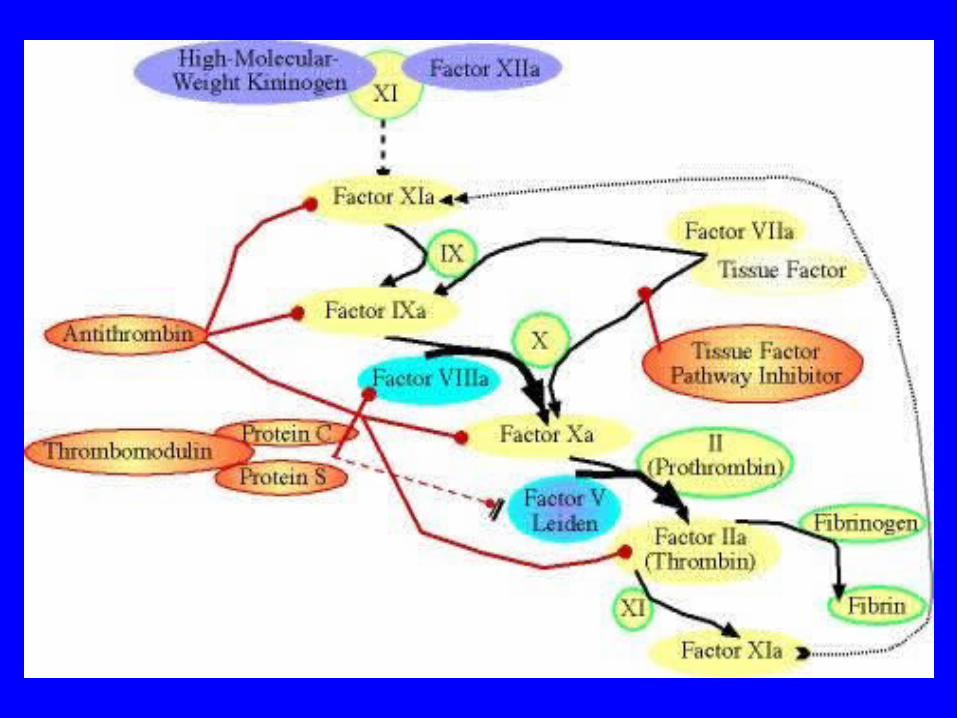
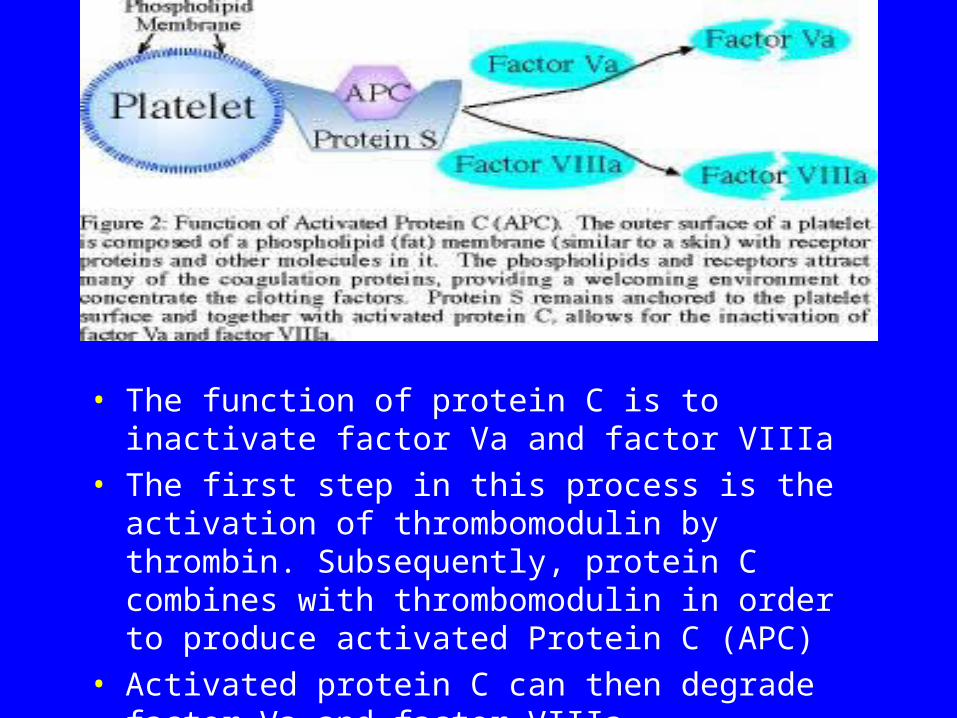
Activated Protein C and Factor V
• The function of protein C is to inactivate factor Va and factor VIIIa
• The first step in this process is the activation of thrombomodulin by thrombin. Subsequently, protein C combines with thrombomodulin in order to produce activated Protein C (APC)
• Activated protein C can then degrade factor Va and factor VIIIa

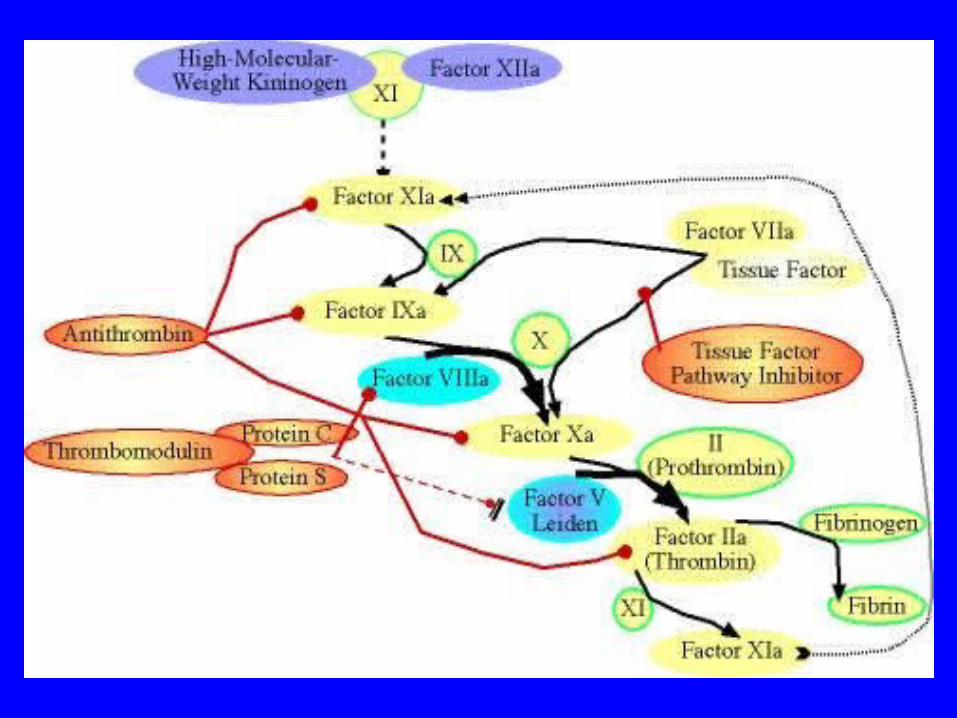
Factor V Leiden
• Factor V Leiden is a genetically acquired trait that can result in a thrombophilic (hypercoaguable) state resulting in the phenomenon of activated protein C resistance (APCR)
• Over 95% of patients with APCR have factor V Leiden.

Activated Protein C and Factor V Leiden
• When one has factor V Leiden, the factor Va is resistant to the normal effects of activated protein C, thus the term activated protein C resistance
• The result is that factor V Leiden is inactivated by activated protein C at a much slower rate (see Figure 3), thus leading to a thrombophilic (propensity to clot) state by having increased activity of factor V in the blood

Prevalence of FVL
• Factor V Leiden is seen more commonly in the northern European populations
• About 4-7% of the general population is heterozygous for factor V Leiden. About 0.06 to 0.25% of the population is homozygous for factor V Leiden.
• The factor V Leiden mutation is relatively uncommon in the native populations of Asia, Africa and North America. In contrast, in Greece and southern Sweden, rates above 10% have been reported.

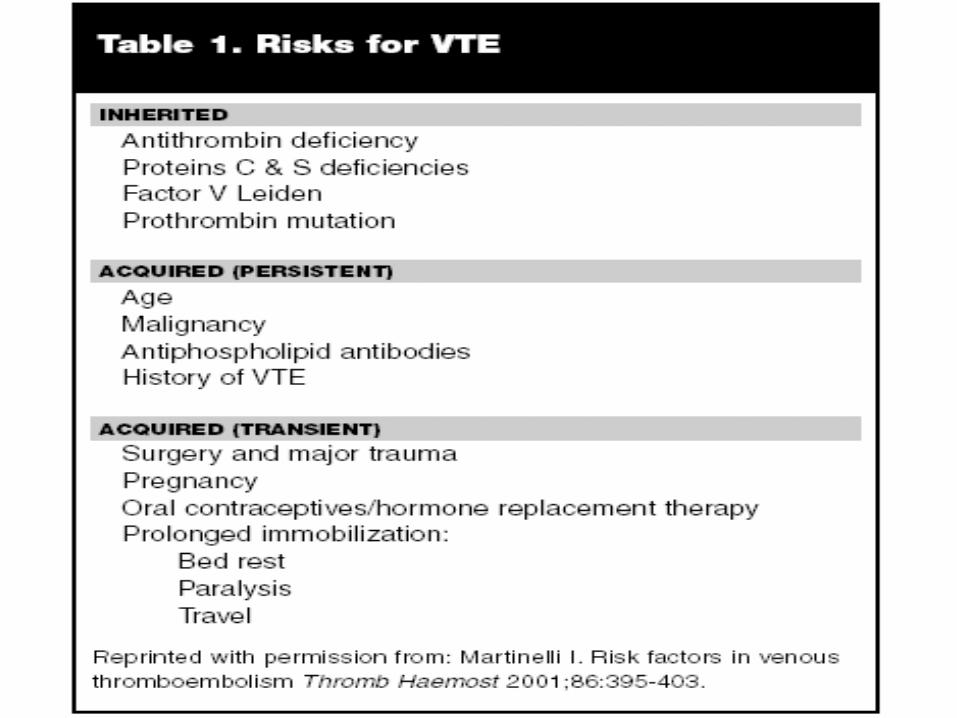
Prothrombin and Deep Vein Thrombosis
• Prothrombin is the precursor to thrombin in the coagulation cascade
• Thrombin is required in order to convert fibrinogen into fibrin, which is the primary goal of the coagulation cascade
• The gene has a mutation at position 20210, hence the disorder being referred to as prothrombin mutation 20210
• The prothrombin gene mutation is seen more commonly in the Caucasian population. About 1-2% of the general population is heterozygous for the prothrombin gene mutation

Relative Risk of Venous Thrombosis
• Normal Risk 1• Use of OCP 4• FVL heterozygous 5-7• + OCP 30-35• Homozygous 80• + OCP >100• Prothrombin heterozygous 3• + OCP 16

Leukaemia, the current hypothesis
• Defect in maturation of white blood cells• May involve a block in differentiation and/or a
block in apoptosis• Acquired genetic defect• Initiating events unclear• Transformation events involve acquired genetic
changes• Chromosomal translocation implicated in many
forms of leukaemia

Chronic Myeloid Leukaemia• Malignancy of the haemopoietic system• Transformation of the pluripotent stem cell• 9;22 translocation giving rise to the
Philadelphia (Ph’) chromosome• Creation of a leukaemia specific mRNA
(BCR-ABL)• Resistance to apoptosis, abnormal
signalling and adhesion

Clinical Course: Phases of CML
Chronic phase
Median 4–6 yearsstabilization
Accelerated phase
Median durationup to 1 year
Blastic phase (blast crisis)
Median survival3–6 months
Terminal phase
Advanced phases
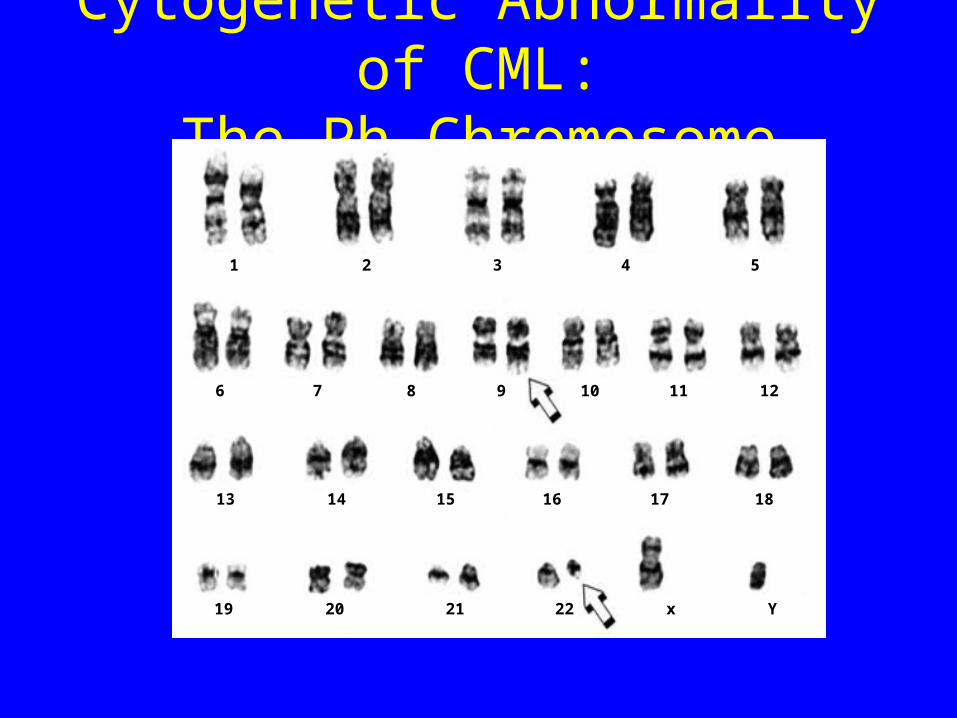
Cytogenetic Abnormality of CML:The Ph Chromosome
1 2 3 4 5
6 7 8 10 119 12
13 14 15 16 17 18
19 20 21 22 x Y
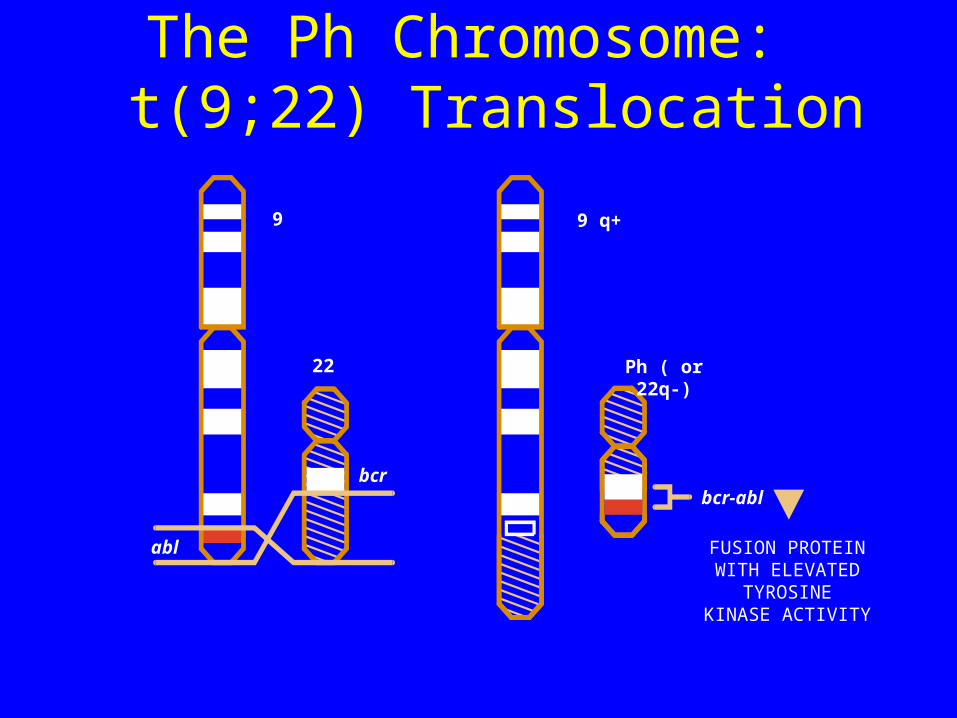
The Ph Chromosome: t(9;22) Translocation
22
bcr
abl
Ph ( or 22q-)
bcr-abl
FUSION PROTEINWITH ELEVATED
TYROSINEKINASE ACTIVITY
9 9 q+
abl
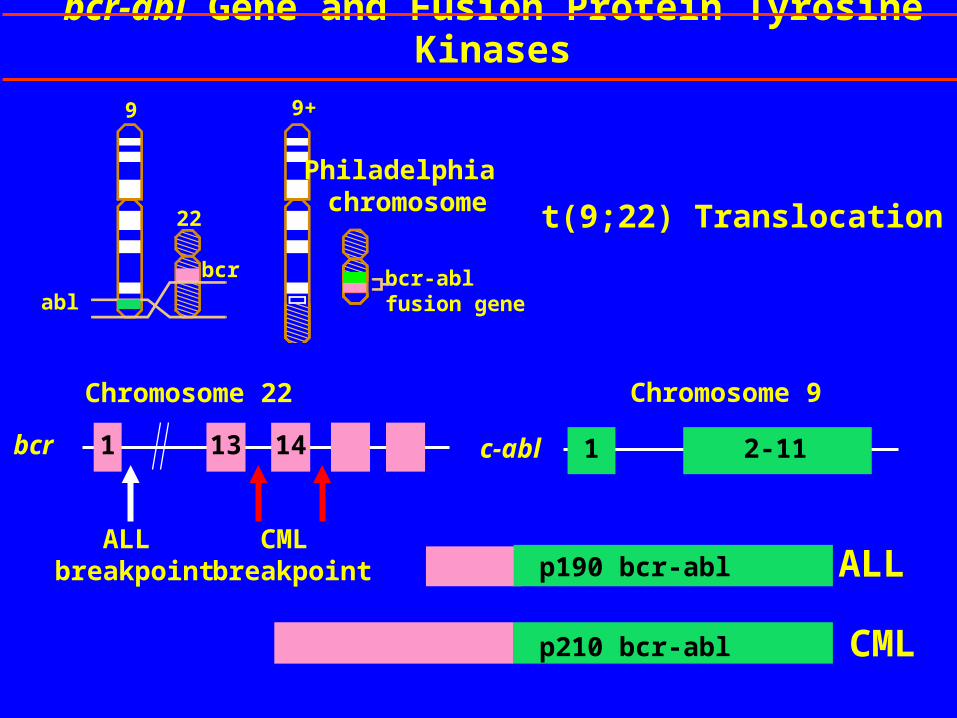
t(9;22) Translocation
bcr-abl fusion gene
Philadelphia chromosome
9
22
bcr
p210 bcr-abl
p190 bcr-abl ALL
CML
Chromosome 22
1 13 14bcr
Chromosome 9
2-111c-abl
ALL breakpoint
CML breakpoint
bcr-abl Gene and Fusion Protein Tyrosine Kinases
9+

Prevalence of the Ph Chromosome in Haematological Malignancies
Leukaemia % of Ph+ Patients
CML 95
ALL (Adult) 15–30
ALL (Paediatric) 5
AML 2
Faderl S et al. Oncology (Huntingt). 1999;13:169-184.
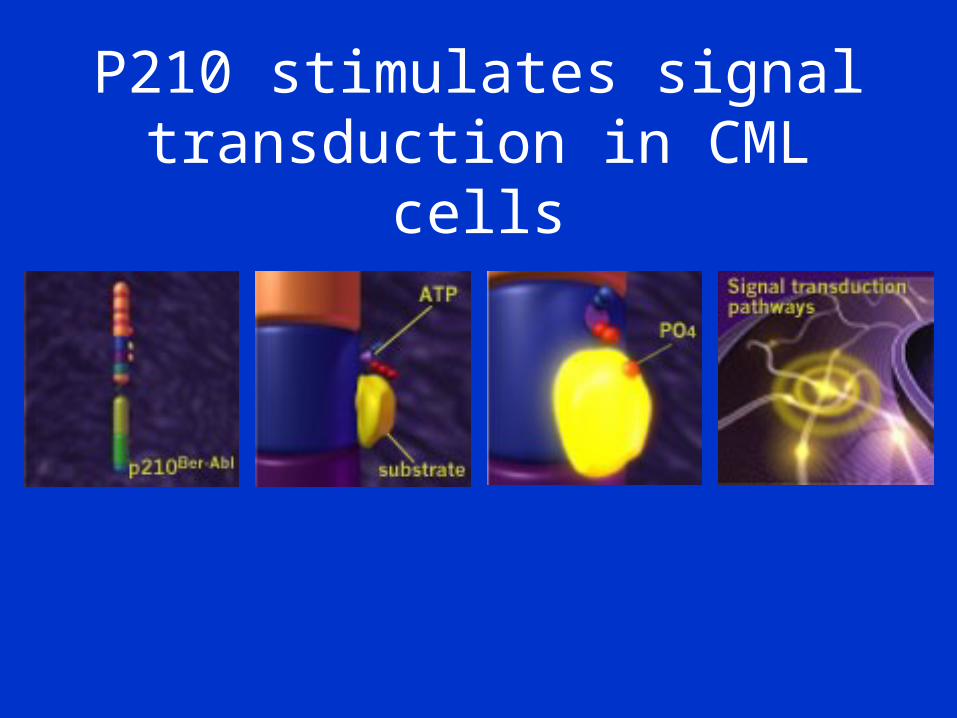
P210 stimulates signal transduction in CML cells
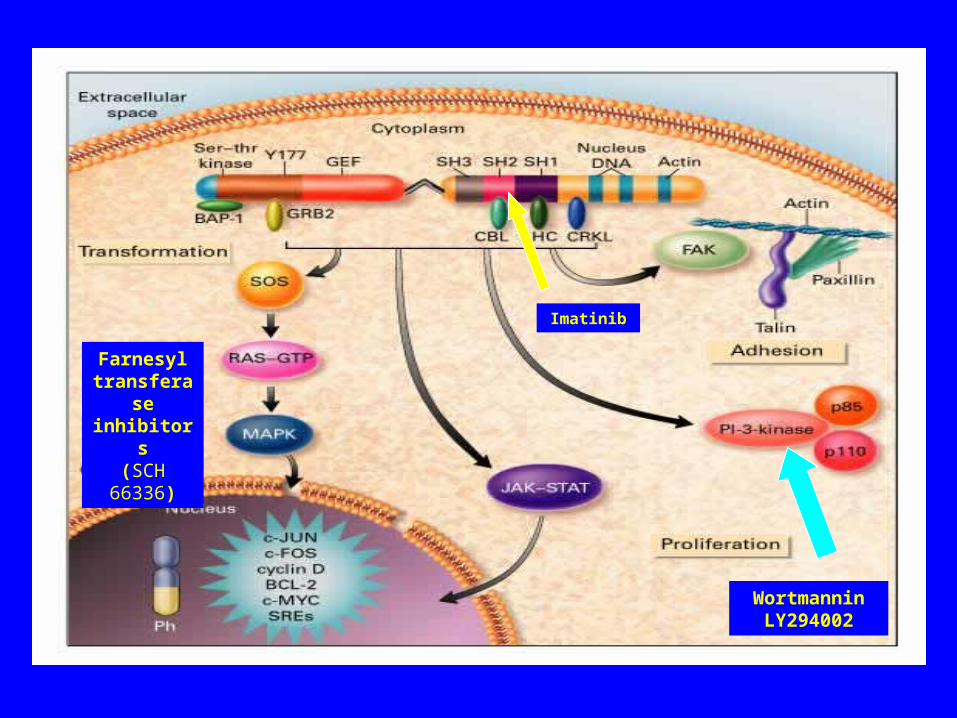
WortmanninLY294002
Farnesyl transferase inhibitors
(SCH 66336)
Imatinib

ACUTE LEUKEMIA
• Translocation is a major mechanism• Involves genes whose normal function is to
control cell division, haematological development etc
• These genes are known as master genes• MLL, AML1• Mutatation of these genes through
translocation leads to leukemia
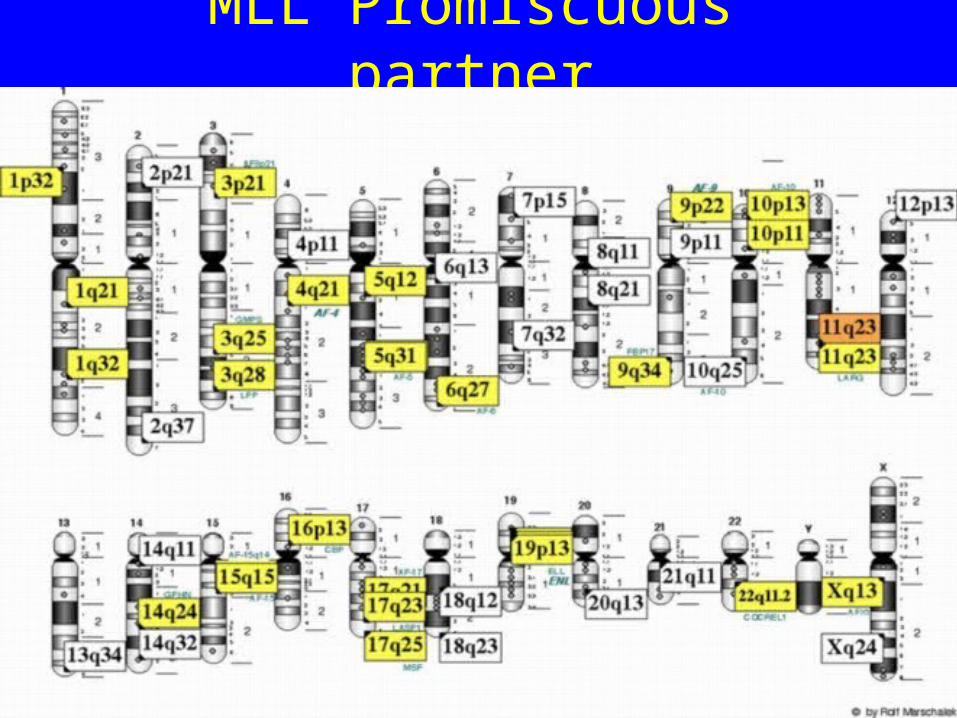
MLL Promiscuous partner


AML1
• 21q
• AML1-ETO t(8;21)
• T(3;21)
• TEL-AML t(12;21)
• Loss of transactivation domain critical to t(8;21) and t(3;21) abnormalities
• Inv (16)
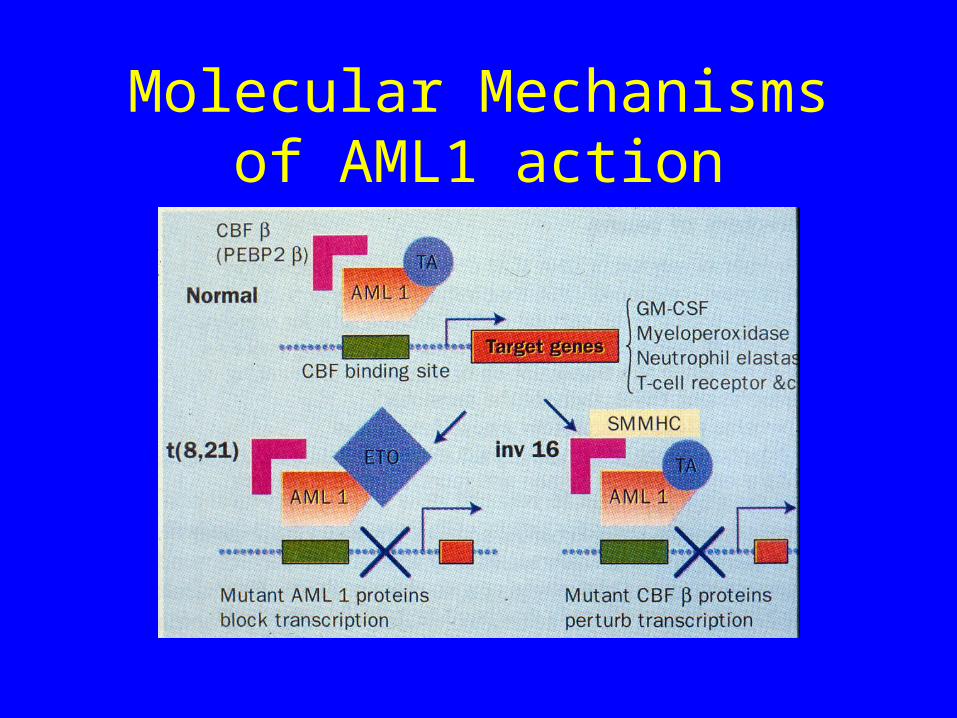
Molecular Mechanisms of AML1 action

AML1
• 21q
• AML1-ETO t(8;21)
• T(3;21)
• TEL-AML t(12;21)
• Loss of transactivation domain critical to t(8;21) and t(3;21) abnormalities
• Inv (16)

What is AML1
• Subunit of a multifactorial transcription factor known as Core Binding Factor
• AML1 is also known as Core Binding FactorA• It has homology to the drosophila developmental
gene “runt” in its DNA binding region• Also has a transactivation domain at its carboxy
terminus
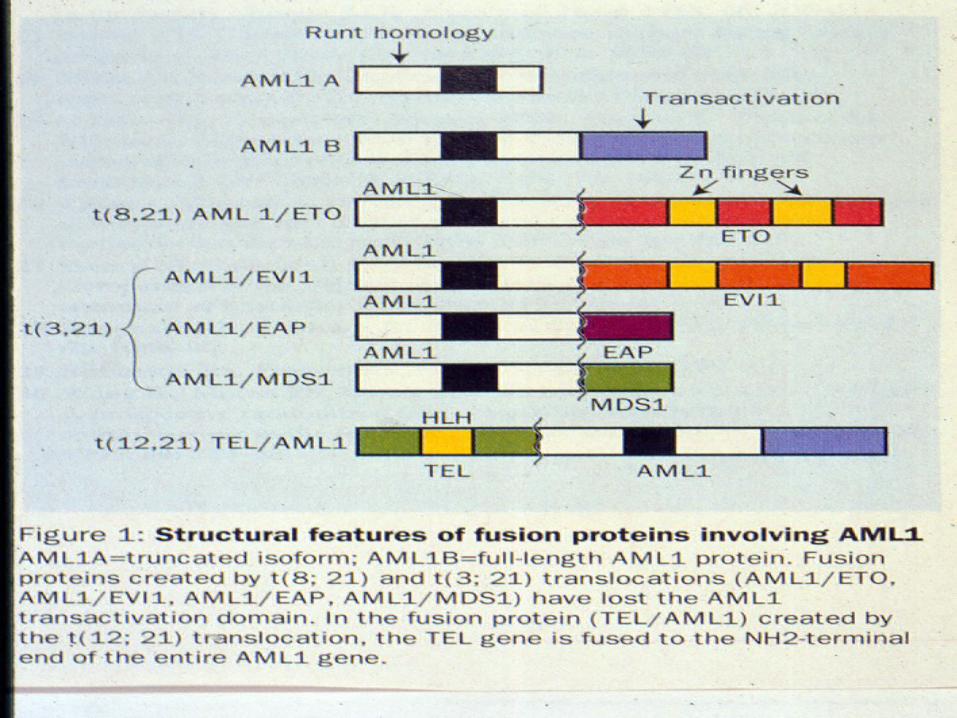

What does AML 1 do?
• Binds DNA• Binding site for AML1 is a core enhancer that is
located at the 5’ control region of genes that are involved in controlling lineage differentiation
• T cell receptor , myeloperoxidase, IL3, GM-CSF, CSF1
• AML1 plays a pivotal role in hemopoietic differentiation by orchestrating expression of appropriate lineage specific genes

What do translocations involving AML1 do?
• T(8;21) Generates AML1-ETO fusion• T(3;21) generates AML1-EVI1, AML1-
EAP1 or AML1-MDS1• All of the above involve replacement of the
transactivation domain• These new fusion proteins can no longer
activate AML1 binding sites in lineage specific genes
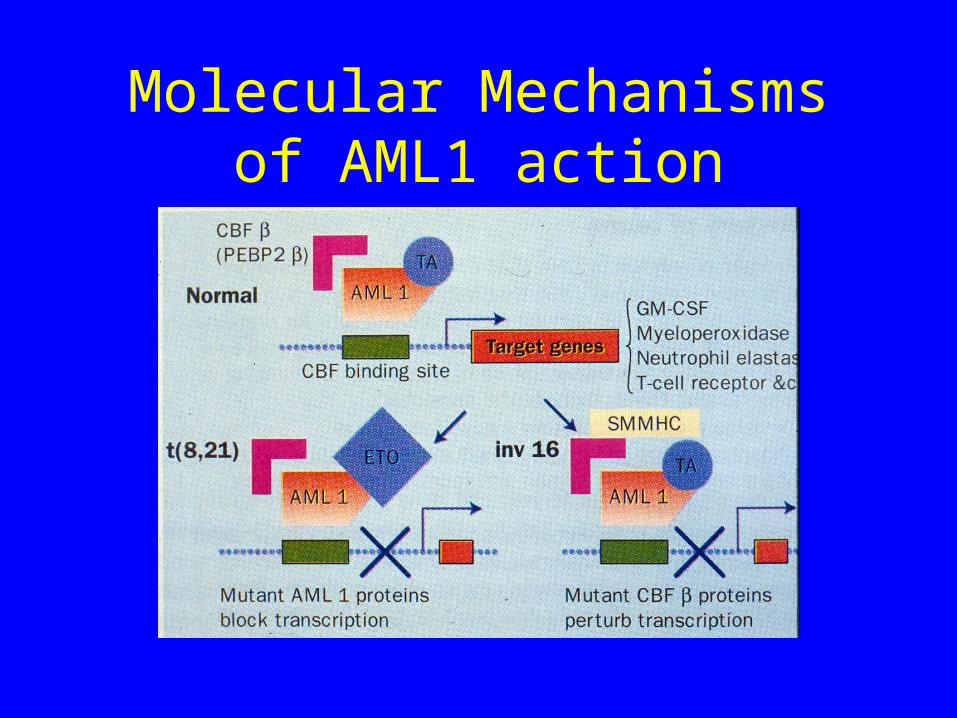
Molecular Mechanisms of AML1 action

Inversion 16
• Here AML1 is not involved
• However the other member of the Core Binding Factor complex (CBFb) is mutated
• Net result is a pertubation of transcription of genes with AML1 binding sites
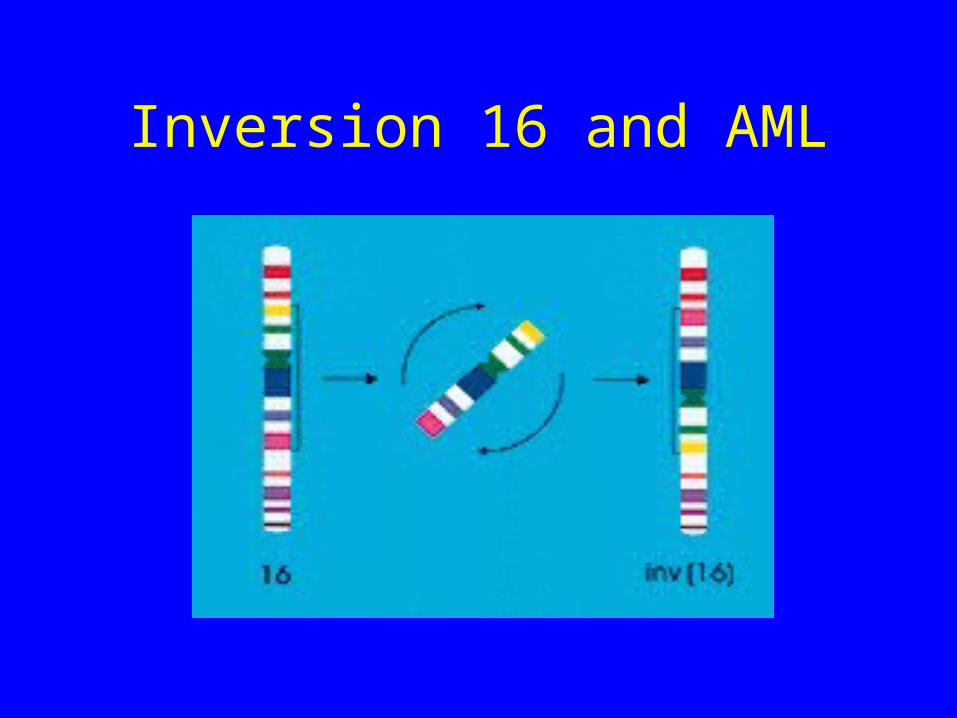
Inversion 16 and AML
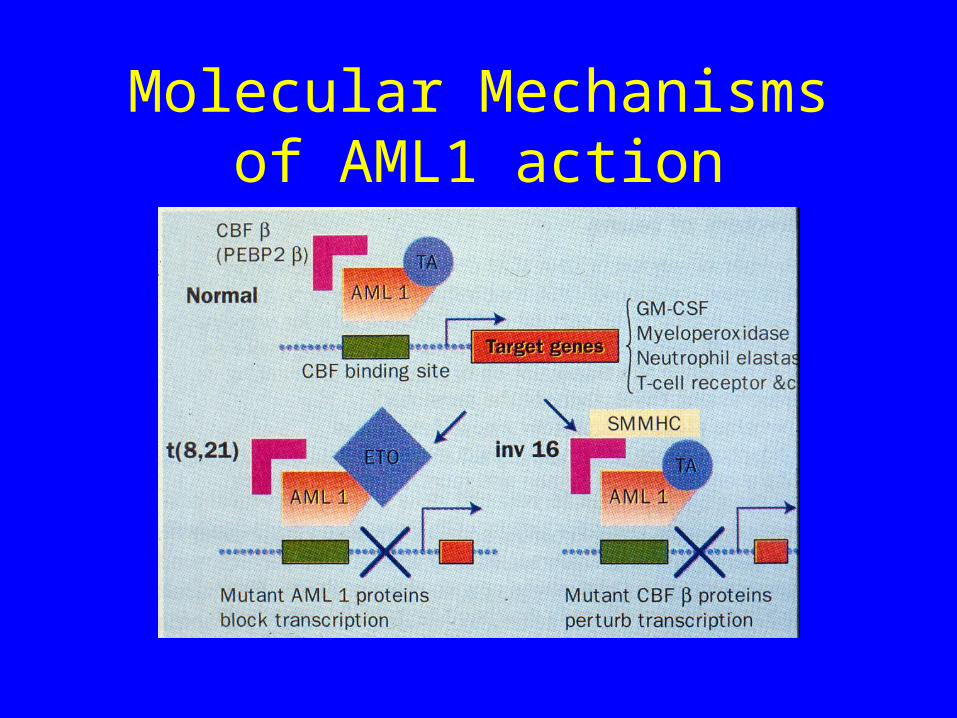
Molecular Mechanisms of AML1 action

Summary
• Master genes such as AML1 and MLL control lineage specific gene expression, thus orchetrating lineage specific development of hemopoiesis
• Mutations in these genes disrupt this control, thus leading to aberrant hemopoiesis and development of leukemia

APML MOLECULAR GENETICS
• M3 FORM OF AML
• NON RANDOM CHROMOSOMAL ABNORMALITY
• t(15;17) IN 95% OF CASES
• RARa GENE ON CHROMSOME 17
• PML GENE ON CHROMOSOME 15
• t(11;17); t(5;17)
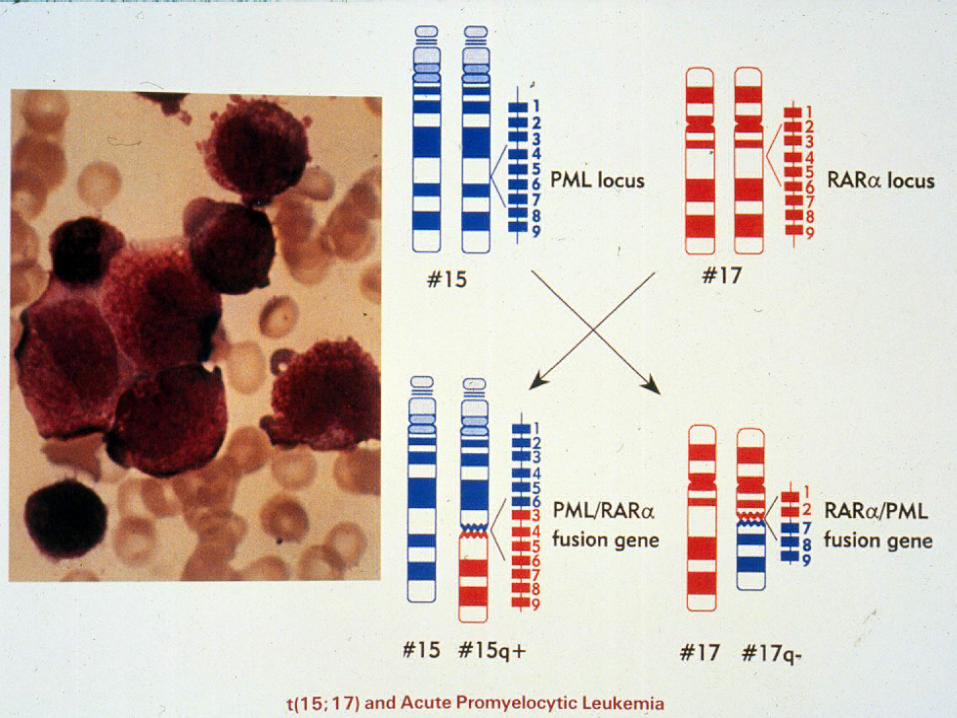

MOLECULAR MEDICINE INTO ACTION
• PRESENCE OF RARa CRITICAL TO THE TREATMENT OF THIS DISEASE
• STANDARD CHEMOTHERAPY ONLY PARTIALLY EFFECTIVE
• TREATMENT WITH RA REMOVES DIFFERENTIATION BLOCKADE
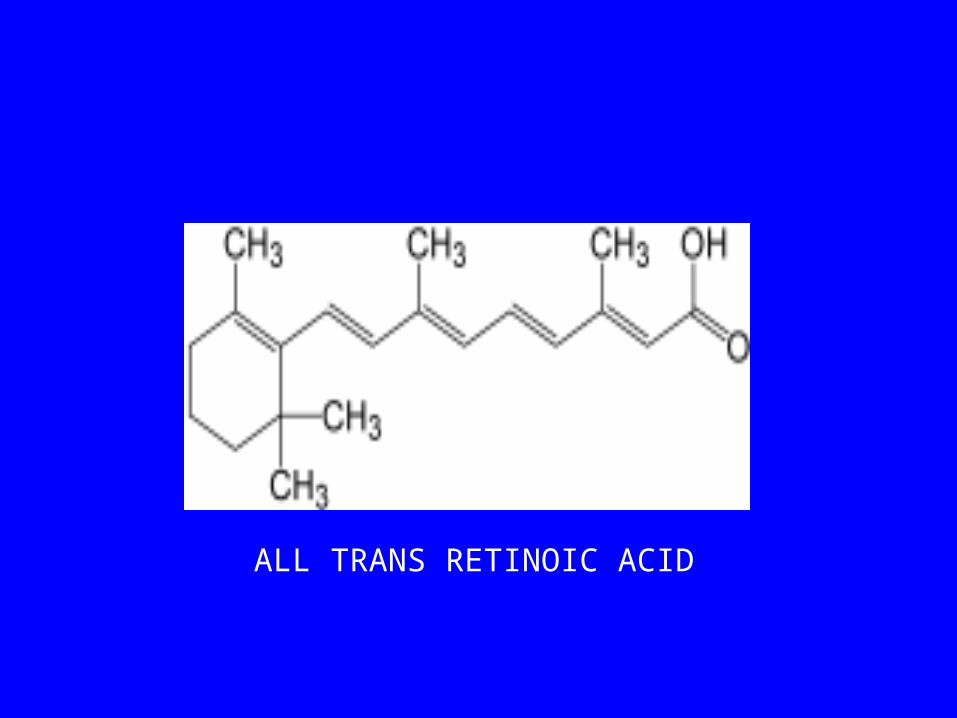
ALL TRANS RETINOIC ACID
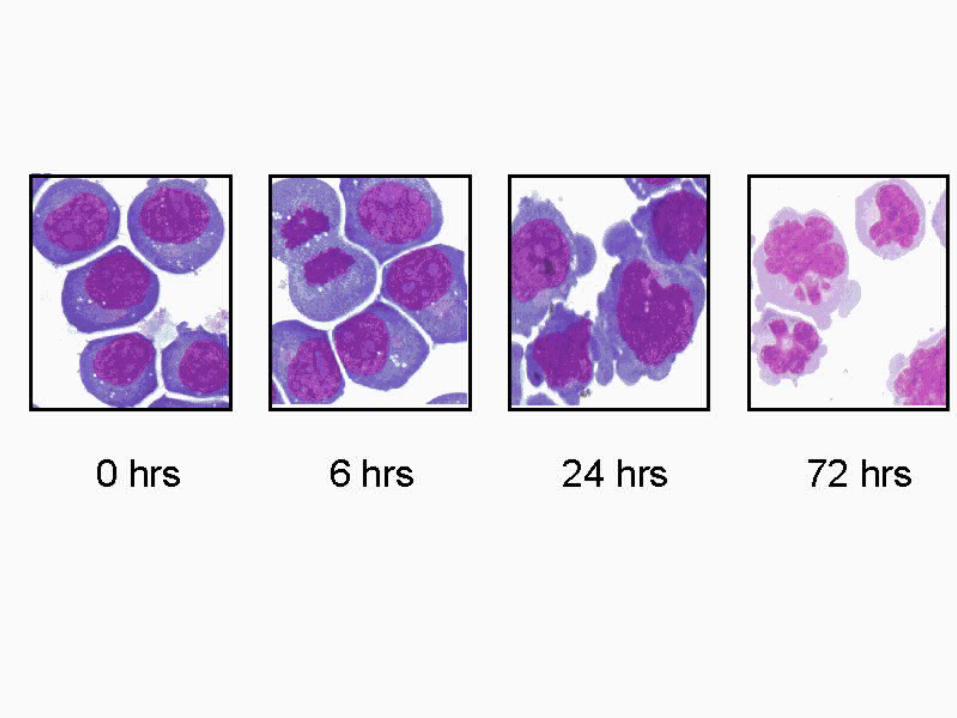

NON HODGKINS LYMPHOMA
• B CELL FOLLICULAR LYMPHOMA
• t(14;18)(q21;q14)
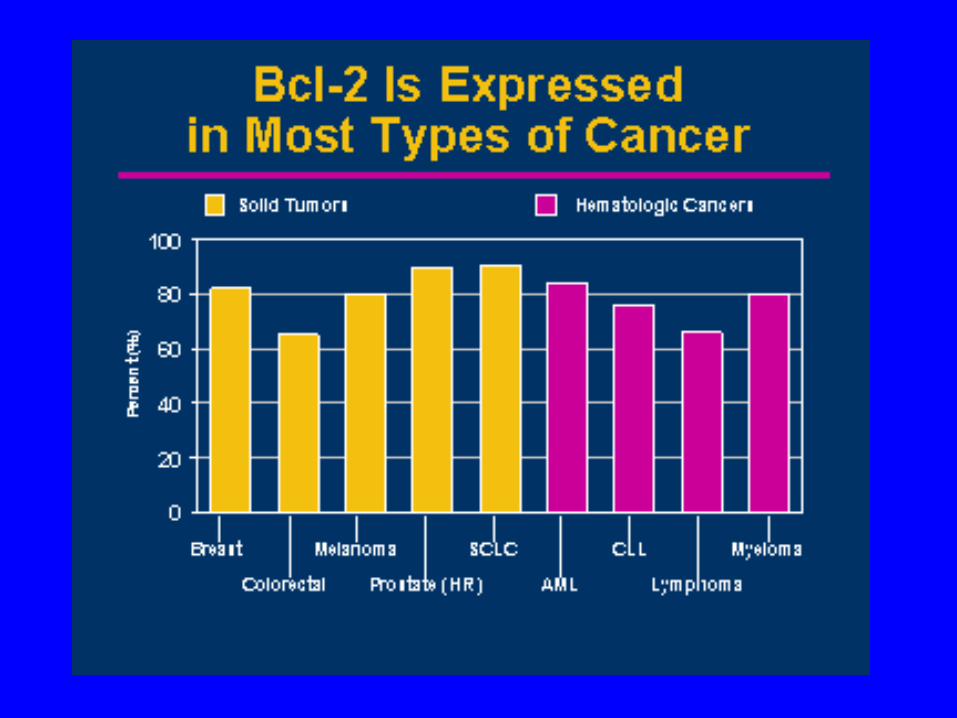
• BCL 2 AND IMMUNOGLOBULIN GENES INVOLVED
• DYSREGULATION OF BCL 2
• FAILURE OF APOPTOSIS


Summary• Molecular changes implicated in haemoglobinopathies• Factor VIII and Factor IX in Haemophilia• Factor V leiden and Prothrombin in Deep vein Thrombus• Molecular abnormalities in Leukemia, particularly
translacations• CML, a paradigm for malignancy• Mutations in master genes disrupt control of hemopoiesis
leading to development of leukemia• Knowledge of molecular changes can influence
diagnosis, prognosis and treatment





























