Embed Size (px)
Citation preview

Page 1 of 22
1
2 Draft for comment purposes (Not for implementation) 3
4
5
6
7
8
9
10
PROJECT OF GUIDANCE ON REQUIREMENTS FOR 11
CLINICAL INVESTIGATIONS OF MEDICAL DEVICES 12
13
14 15
16
17
18 This document is being distributed for comment purposes only. Comments and 19 suggestions regarding this draft document should be submitted within 30 days of 20 publication on the SFDA’s website. Kindly, submit comments to the 21 [email protected] by using the Commenting Template at 22 <http://www.sfda.gov.sa/en/medicaldevices/resources/Documents/Commenting-23 Template.docx > 24
25 26
27
28
29
30
31

Page 2 of 22
TABLE OF CONTENT 32
33 DEFINITIONS & ABBREVIATIONS ..................................................................................... 3 34
Definitions .............................................................................................................................. 3 35
Abbreviations .......................................................................................................................... 6 36
37
INTRODUCTION ..................................................................................................................... 7 38
Purpose ................................................................................................................................... 7 39
Scope……… ........................................................................................................................... 7 40
Background ............................................................................................................................. 7 41
42
REQUIREMENTS .................................................................................................................... 8 43
REQUIRED DOCUMENTS ................................................................................................... 10 44
FLOWCHART ........................................................................................................................ 13 45
46
ANNEXES ............................................................................................................................... 14 47
Application Form for CIMD .................................................................................................. 15 48
Change Form for CIMD ........................................................................................................ 22 49
50 51

Page 3 of 22
DEFINITIONS & ABBREVIATIONS 52
53
Definitions 54
Medical Device means any instrument, apparatus, implement, machine, appliance,
implant, in vitro reagent or calibrator, software, material or other
similar or related article:
A. Intended by the manufacturer to be used, alone or in
combination, for human beings for one or more of the
specific purpose(s) of:
o Diagnosis, prevention, monitoring, treatment or alleviation of disease,
o Diagnosis, monitoring, treatment, alleviation of or
compensation for an injury or handicap, o Investigation, replacement, modification, or support of
the anatomy or of a physiological process,
o Supporting or sustaining life,
o Control of conception, o Disinfection of medical devices,
o Providing information for medical or diagnostic purposes
by means of in vitro examination of specimens derived from the human body;
and
B. Which does not achieve its primary intended action in or on the human body by pharmacological, immunological or
metabolic means, but which may be assisted in its intended
function by such means.
Investigational Medical Device
is a medical device being assessed for safety or performance in a clinical investigation. The definition cover other product including
prescription eyeglasses, contact lenses and their solutions. This
includes medical devices already on the market, that are being evaluated for new intended uses, new populations, new materials or
design changes.
Medical Devices
National Registry (MDNR)
is the database of registered establishments and the medical devices
they manufacture or import or distribute.
establishment
National Registry
Number
means the number issued to a person by the SFDA under the
establishment registration provisions of the Medical Devices Interim
Regulation.
Medical Device
National Listing
Number
means the code assigned by the SFDA to a single medical device,
that has been included in a marketing authorization application, to
indicate the device is authorized to be placed on the KSA market and facilitate traceability.
National Centre for
Medical Device
Reporting (NCMDR)
means an organization managing a database of information on safety
and/or performance related aspects of medical devices and
employing staff capable of taking appropriate action on any confirmed problems.
Adverse Event means any malfunction or deterioration in the characteristics and/or
performances of a medical device, including any inadequacy in its

Page 4 of 22
labeling or the instructions for use which may lead to compromise
the health or safety of patients, users or third parties.
Reportable Adverse
Event
means any adverse event or any technical or medical reason leading
to a Field Safety Corrective Action, which, directly or indirectly, might lead to or may have led (a) to the death of a patient, a user or
another person or (b) to a serious deterioration in their state of
health.
Global Harmonization
Task Force (GHTF)
Countries working to achieve harmonization in medical device
regulation among themselves. These countries are Australia, Canada, Japan, the USA and the EU/EFTA.
Authorized
Representative (AR)
means any natural or legal person established within the KSA who
has received a written mandate from the manufacturer to act on his behalf for specified tasks, including the obligation to represent the
manufacturer in its dealings with the SFDA.
Sponsor means an individual or organization taking responsibility and
liability for the initiation or implementation of a clinical investigation.
Investigator is an individual member of the investigation site team designated and
supervised by the principal investigator at an investigation site to
perform critical clinical-investigation-related procedures or to make important clinical investigation - related decisions.
An individual member of the investigation site team can also be called “sub-investigator” or “co-investigator”.
Clinical Data means safety and/or performance information that are generated
from the clinical use of an investigational device.
Clinical Investigation is a systematic investigation in one or more human subjects, undertaken to assess the safety or performance of a medical device.
Clinical Investigation
Plan (CIP)
document that state(s) the rationale, objectives, design and proposed
analysis, methodology, monitoring, conduct and record-keeping of
the clinical investigation. The term “protocol” sometimes used as a synonymous with “Plan”.
Clinical Investigation
Report
is a written form document describing the identification of the
device(s), the methodology, the design, any deviations from the CIP, execution, data analysis together with statistical analysis, a critical
appraisal of the aims of the clinical investigation, and results of a
clinical investigation.
Ethics Committee (EC)/ Institutional
Review Board (IRB)
an independent body whose responsibility is to review clinical investigations in order to protect the rights, safety and well-being of
human subjects participating in a clinical investigation.
Comparator is medical device, therapy (e.g. active control), placebo or no
treatment, used in the reference group in a clinical investigation.
Completion Date means the date that the final subject was examined or received an
intervention for the purposes of final collection of data for the
primary outcome, whether the clinical investigation concluded according to the prespecified protocol or was terminated.
Contract Research
Organization (CRO)
means a person or organization contracted by the sponsor to perform
one or more of the sponsor's clinical investigation-related duties and
functions.
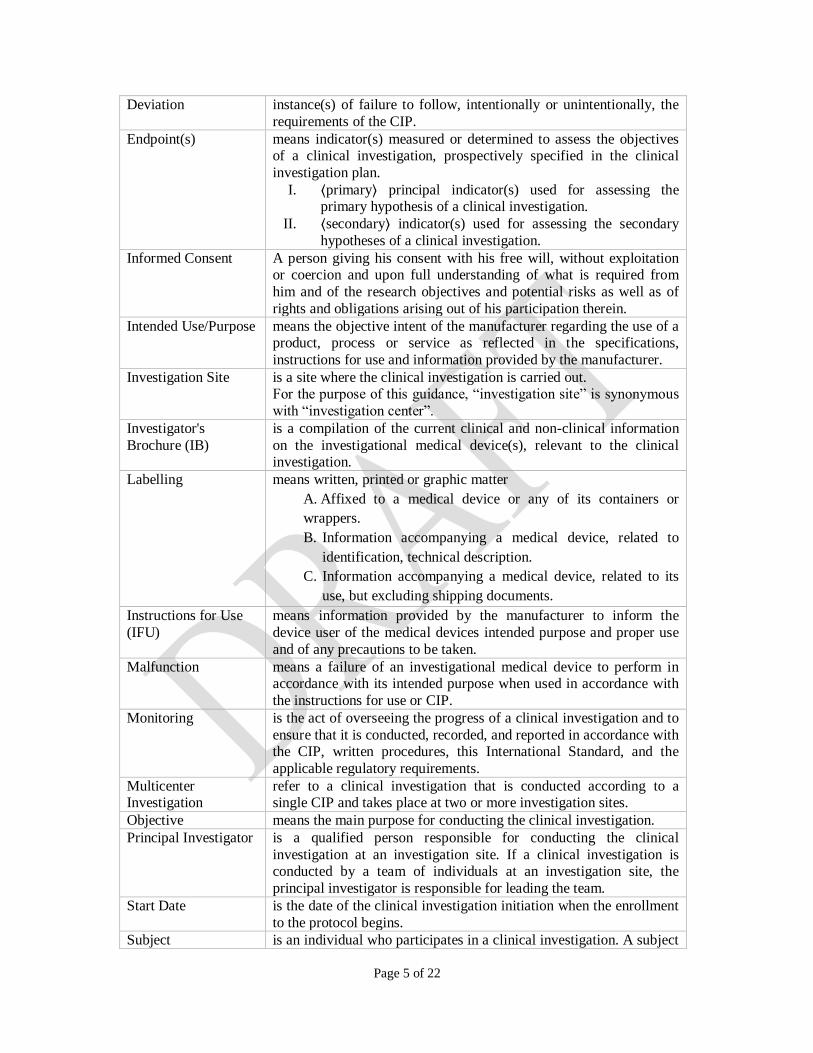
Page 5 of 22
Deviation instance(s) of failure to follow, intentionally or unintentionally, the
requirements of the CIP.
Endpoint(s) means indicator(s) measured or determined to assess the objectives of a clinical investigation, prospectively specified in the clinical
investigation plan.
I. ⟨primary⟩ principal indicator(s) used for assessing the primary hypothesis of a clinical investigation.
II. ⟨secondary⟩ indicator(s) used for assessing the secondary
hypotheses of a clinical investigation.
Informed Consent A person giving his consent with his free will, without exploitation or coercion and upon full understanding of what is required from
him and of the research objectives and potential risks as well as of
rights and obligations arising out of his participation therein.
Intended Use/Purpose means the objective intent of the manufacturer regarding the use of a product, process or service as reflected in the specifications,
instructions for use and information provided by the manufacturer.
Investigation Site is a site where the clinical investigation is carried out. For the purpose of this guidance, “investigation site” is synonymous
with “investigation center”.
Investigator's
Brochure (IB)
is a compilation of the current clinical and non-clinical information
on the investigational medical device(s), relevant to the clinical investigation.
Labelling means written, printed or graphic matter
A. Affixed to a medical device or any of its containers or
wrappers.
B. Information accompanying a medical device, related to
identification, technical description.
C. Information accompanying a medical device, related to its
use, but excluding shipping documents.
Instructions for Use
(IFU)
means information provided by the manufacturer to inform the
device user of the medical devices intended purpose and proper use
and of any precautions to be taken.
Malfunction means a failure of an investigational medical device to perform in accordance with its intended purpose when used in accordance with
the instructions for use or CIP.
Monitoring is the act of overseeing the progress of a clinical investigation and to
ensure that it is conducted, recorded, and reported in accordance with the CIP, written procedures, this International Standard, and the
applicable regulatory requirements.
Multicenter Investigation
refer to a clinical investigation that is conducted according to a single CIP and takes place at two or more investigation sites.
Objective means the main purpose for conducting the clinical investigation.
Principal Investigator is a qualified person responsible for conducting the clinical
investigation at an investigation site. If a clinical investigation is conducted by a team of individuals at an investigation site, the
principal investigator is responsible for leading the team.
Start Date is the date of the clinical investigation initiation when the enrollment
to the protocol begins.
Subject is an individual who participates in a clinical investigation. A subject

Page 6 of 22
can be either a healthy volunteer or a patient.
Use Error means an act, or omission of an act relating to the use of a product
within the scope of this Regulation, whereby the resulting outcome is different from that intended by the manufacturer or expected by the
user.
Vulnerable Subject refer to an individual whose willingness to volunteer in a clinical investigation could be unduly influenced by the expectation, whether
justified or not, of benefits associated with participation or of
retaliatory response from senior members of a hierarchy in case of
refusal to participate. Example Individuals with lack of or loss of autonomy due to
immaturity or through mental disability, persons in nursing homes,
children, impoverished persons, subjects in emergency situations, ethnic minority groups, homeless persons, nomads, refugees, and
those incapable of giving informed consent. Other vulnerable
subjects include, for example, members of a group with a
hierarchical structure such as university students, subordinate hospital and laboratory personnel, employees of the sponsor,
members of the armed forces, and persons kept in detention.
55
Abbreviations 56
KSA Kingdom of Saudi Arabia
SFDA Saudi Food and Drug Authority
MDS Medical Devices Sector
GHTF Global Harmonization Task Force
MDNR Medical Device National Registry
MDMA Medical Devices Marketing Authorization
AR Authorized Representative
CIMD Clinical Investigations of Medical Devices
IFU Instructions for Use
CRO Contract Research Organization
CIP Clinical Investigation Plan
EC Ethics Committee
IRB Institutional Review Board
NCMDR National Centre for Medical Device Reporting
IB Investigator's Brochure
NCBE National Committee of Bio Ethics
57

Page 7 of 22
INTRODUCTION 58
59
Purpose 60
The purpose of this document is to clarify requirements of conducting CIMD within KSA. 61
62
63
Scope……… 64
This document is applicable to any party wishes to conduct CIMD or its accessories within KSA. 65
66
Background 67
Medical devices may be placed on the market and/or put into service only if they comply with the 68
applicable provisions of the Medical Devices Interim Regulation, as signified by the SFDA 69
issuing the manufacturer with a written marketing authorization (MDMA). 70
71
Devices that may access KSA for purposes rather than marketing, such as for clinical 72
investigations, are exempt from marketing authorization requirements. However, SFDA requires 73
approval for conducting CIMD or its accessories within KSA, in order to protect the rights, safety 74
and well-being of participants during the clinical investigation, and to ensure the scientific 75
conduct of the clinical investigation and the credibility of the clinical investigation results. For 76
achieving that, SFDA issued this document. 77
78
79 80
81

Page 8 of 22
REQUIREMENTS 82
General 1 Any CIMD or its accessories within KSA shall be approved by SFDA
before commence.
Regulations and
Standards
2 CIMD shall be complied with the Law of Ethics of Research on
Living Creatures.
3 CIMD should be complied with:
o Declaration of Helsinki and
o GCP/ISO 14155.
Labeling
Requirements
4 The labeling of device shall be complied with the requirements that
described in SFDA’s guidance document entitled MDS – G10
Guidance on Labelling Requirements for Medical Devices.
Adverse Events
Reporting
5 Sponsor shall report to the SFDA’s NCMDR, IRBs, and investigators
any reportable adverse event of which it becomes aware, that involves
the medical device. This shall be reported without delay but not later
than 10 working days of occurrence.
For more details see the SFDA’s guidance document entitled MDS –
G6 Guidance on Post-Marketing Surveillance.
Pre-requisite
6 Before applying for CIMD:
- the sponsor located within the KSA are required to have an
Establishment National Registry Number that is issued
through SFDA’s MDNR. Independent individuals are
exempt.
- the sponsor located outside the KSA are required to assign an
AR, the AR is required to have:
- an establishment National Registry Number that is
issued through SFDA’s MDNR, and
- an AR license. Information on this procedure see
SFDA’s guidance document entitled MDS – G3
Guidance on for Authorized Representatives.
Submitting
Documents to
SFDA and the
approval
7 Sponsor (either is located within KSA, or outside the KSA, by its
AR) shall submit the required documents by email to
1. prior to CIMD, the required documents are specified in
section (Required Documents-A). SFDA responds to the
applicant within a week with the missing required documents
and once satisfied, SFDA will send a “no objection letter”
together with an importation license, if needed, to the

Page 9 of 22
applicant’s email within 60 calendar days.
2. during the CIMD, the required documents are specified in
section (Required Documents-B).
3. at the end of the CIMD, the required documents are specified
in section (Required Documents-C).
Reviewing Fees 8 No fees are required for reviewing CIMD applications.
83
84
85
86
87
88
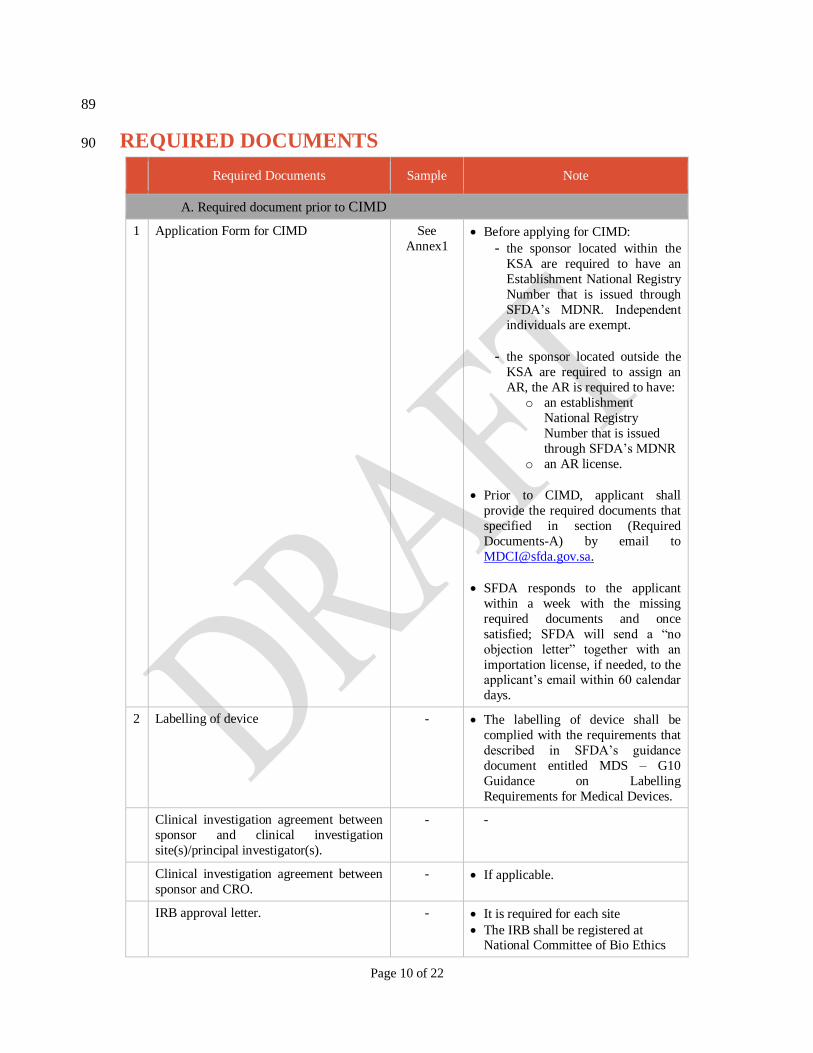
Page 10 of 22
89
REQUIRED DOCUMENTS 90
Required Documents Sample Note
A. Required document prior to CIMD
1 Application Form for CIMD See
Annex1 Before applying for CIMD:
- the sponsor located within the
KSA are required to have an
Establishment National Registry
Number that is issued through
SFDA’s MDNR. Independent
individuals are exempt.
- the sponsor located outside the
KSA are required to assign an
AR, the AR is required to have:
o an establishment
National Registry
Number that is issued
through SFDA’s MDNR
o an AR license.
Prior to CIMD, applicant shall
provide the required documents that
specified in section (Required
Documents-A) by email to
SFDA responds to the applicant
within a week with the missing
required documents and once
satisfied; SFDA will send a “no
objection letter” together with an
importation license, if needed, to the applicant’s email within 60 calendar
days.
2 Labelling of device
- The labelling of device shall be
complied with the requirements that
described in SFDA’s guidance
document entitled MDS – G10
Guidance on Labelling
Requirements for Medical Devices.
Clinical investigation agreement between
sponsor and clinical investigation
site(s)/principal investigator(s).
- -
Clinical investigation agreement between
sponsor and CRO.
- If applicable.
IRB approval letter. - It is required for each site
The IRB shall be registered at National Committee of Bio Ethics
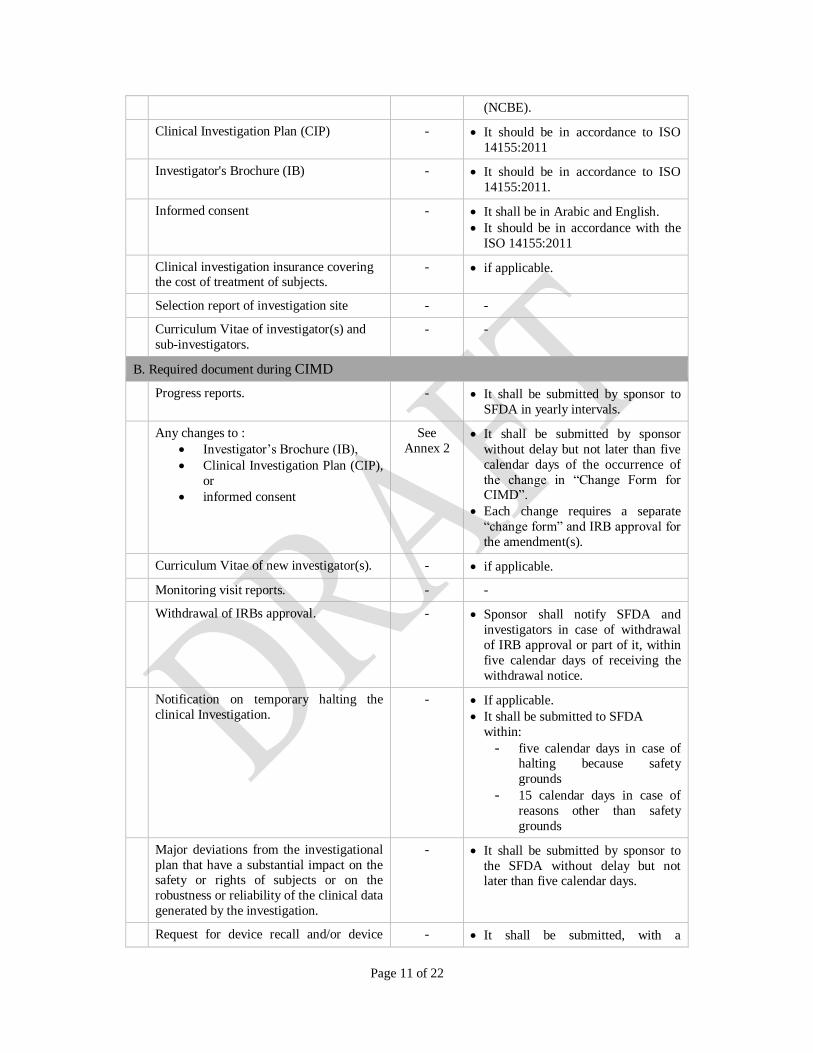
Page 11 of 22
(NCBE).
Clinical Investigation Plan (CIP) - It should be in accordance to ISO
14155:2011
Investigator's Brochure (IB) - It should be in accordance to ISO
14155:2011.
Informed consent - It shall be in Arabic and English.
It should be in accordance with the
ISO 14155:2011
Clinical investigation insurance covering the cost of treatment of subjects.
- if applicable.
Selection report of investigation site - -
Curriculum Vitae of investigator(s) and
sub-investigators.
- -
B. Required document during CIMD
Progress reports. - It shall be submitted by sponsor to
SFDA in yearly intervals.
Any changes to :
Investigator’s Brochure (IB),
Clinical Investigation Plan (CIP),
or
informed consent
See
Annex 2 It shall be submitted by sponsor
without delay but not later than five
calendar days of the occurrence of
the change in “Change Form for CIMD”.
Each change requires a separate
“change form” and IRB approval for
the amendment(s).
Curriculum Vitae of new investigator(s). - if applicable.
Monitoring visit reports. - -
Withdrawal of IRBs approval. - Sponsor shall notify SFDA and
investigators in case of withdrawal
of IRB approval or part of it, within
five calendar days of receiving the
withdrawal notice.
Notification on temporary halting the
clinical Investigation.
- If applicable.
It shall be submitted to SFDA
within:
- five calendar days in case of halting because safety
grounds
- 15 calendar days in case of
reasons other than safety
grounds
Major deviations from the investigational
plan that have a substantial impact on the safety or rights of subjects or on the
robustness or reliability of the clinical data
generated by the investigation.
- It shall be submitted by sponsor to
the SFDA without delay but not later than five calendar days.
Request for device recall and/or device - It shall be submitted, with a

Page 12 of 22
disposition regarding return, repair, or
dispose the device or a part of it.
justification, by:
- sponsor to IRB within 30
calendar days after receiving
the request from the principal investigator
- sponsor to SFDA within 30
calendar days after receiving
the request.
C. Required document at the end of the CIMD
Notification of clinical investigation
termination.
- If applicable.
Premature termination of a clinical
investigation.
- If applicable
It shall be submitted by sponsor
within 15 calendar days of the
termination with justifications of the
reason for terminating the
investigation.
Close-out notification of the investigation. - It shall be submitted by sponsor to
SFDA without delay but not later
than 15 calendar days of the
termination.
Close-out report of the investigation. - It shall be submitted by sponsor to
the SFDA and IRBs without delay
but not later than two months after
the termination.
Investigational device accountability. - -
Clinical investigation report. - It shall be submitted by sponsor to
the SFDA and IRBs without delay
but not later than six months after
the termination.
91 92
93

Page 13 of 22
94
FLOWCHART 95
At end of the clinical investigation,
Submit the required documents
specified in section (Required
Documents-C).
At end of the clinical investigation,
Submit the required documents
specified in section (Required
Documents-C).
Submit the required documents
specified in section (Required
Documents-A) by email to
Submit the required documents
specified in section (Required
Documents-A) by email to
During the clinical investigation,
Submit the required documents
specified in section (Required
Documents-B).
During the clinical investigation,
Submit the required documents
specified in section (Required
Documents-B).
SFDA responds to the applicant
within a week with the missing
required documents and once
satisfied; SFDA will send a “no
objection letter” together with an
importation license, if needed, to the
applicant’s email within 60 calendar
days.
SFDA responds to the applicant
within a week with the missing
required documents and once
satisfied; SFDA will send a “no
objection letter” together with an
importation license, if needed, to the
applicant’s email within 60 calendar
days.
Start
End
Sponsor located
outside the KSA
Sponsor located
within the KSA
Organization or
independent individual
Organization or
independent individual
It shall appoint an AR to
to act on his behalf within the KSA
It shall appoint an AR to
to act on his behalf within the KSA
Determine
the
applicant
Determine
the
applicant
The assigned AR The assigned AR
Before applying for approval of a
clinical investigation,
It is required to have:
- an establishment National
Registry Number that is issued
through SFDA’s MDNR, and
- an AR license.
Before applying for approval of a
clinical investigation,
It is required to have:
- an establishment National
Registry Number that is issued
through SFDA’s MDNR, and
- an AR license.
Before applying for approval of a
clinical investigation, It is required
to have an Establishment National
Registry Number that is issued
through SFDA’s MDNR.
Independent individuals are
excepted.
Before applying for approval of a
clinical investigation, It is required
to have an Establishment National
Registry Number that is issued
through SFDA’s MDNR.
Independent individuals are
excepted.
96

Page 14 of 22
97
98
99
100
101
102
103
104
105
ANNEXES 106
107

Page 15 of 22
Annex 1 108
Application Form for CIMD 109
Date Received (For SFDA use only)
CIMD Application Number (For SFDA use only)
1. Status
1.1 Type of submission
First submission
Amendments to previous
submission.
1.2 Aim of Study Pre-marketing approval for
new device
Pre-marketing approval for
new claims
Post-Marketing study
Non Marketing study
1.3 Does this clinical
investigation involve
first in human use?
Yes
No
1.4 Importation license for the investigational
device is required?
Yes No
2. Sponsor Details
2.1 Manufacturer Name
establishment National
Registry Number, that is issued
through SFDA’s MDNR (if
applicable)
Address
Phone
Fax
E- mail
Contact person name
Contact person phone
Contact person e-mail
2.1 AR, if applicable Name
establishment National
Registry Number, that is issued
through SFDA’s MDNR (if
applicable)
AR license number
Address
Phone
Fax
E- mail
Contact person name
Contact person phone
Contact person e-mail
2.3 Sponsor, if other than manufacturer
Name
establishment National Registry Number, that is issued

Page 16 of 22
through SFDA’s MDNR (if
applicable)
Address
Phone
Fax
E- mail
Contact person name
Contact person phone
Contact person e-mail
2.5 Person responsible for
completing the
application.
Name
Position
Phone
3. CRO Details
3.1 CRO, if applicable Name
establishment National
Registry Number, that is issued
through SFDA’s MDNR (if
applicable)
Address
Phone
Fax
E- mail
Contact person name
Contact person phone
Contact person e-mail
4. Sponsorship details
4.1 Type of Sponsorship Commercial
Non-commercial
4.2 Type of sponsor
local manufacturer
AR
Hospital
Independent individuals
Foundation
University or Institution
Other, please specify:
……………………………….
4.3 Type of aid Material support Funding support
Other, please specify:
………………………………...
5. Investigational Device Information
5.1 Is the device registered
at SFDA?
Yes, Medical Device
National Listing Number
issued through SFDA’s
MDMA is: ………………
………………………….. No, but registered in:

Page 17 of 22
Australia
Canada
Japan
USA
EU
Other, please specify:
……………………………..…
Not registered anywhere
1.4 Importation license for
the investigational device is required?
Yes
No
5.2 Investigational Device Name
5.3 Device Generic name (if not specified above)
5.4 Alternative name for the device used elsewhere (if
applicable)
5.5 Is the device approved
to be marketed
elsewhere for other use
than intended for this
clinical investigation?
No
Yes, explain:………………
……………………………..…
………………………………..
5.6 Device Category ☐ active implantable devices
☐ anesthetic and respiratory
devices
☐ dental devices
☐ electro mechanical medical
devices
☐ hospital hardware
☐ non-active implantable
devices; ophthalmic and
optical devices
☐ reusable devices
☐ single use devices
☐ assistive products for
persons with disability
☐ diagnostic and therapeutic
radiating devices
☐ complementary therapy
devices
☐ biologically derived devices
☐ healthcare e facility
products and adaptations
☐ Laboratory equipment
☐ Other: ………………..
5.7 Does the device is an
implantable?
No
Yes,
Brief description:……..
……………………
……………………
Is the device intended
to remain permanently in patient:
No
Yes

Page 18 of 22
58 Whether the device
intended to be used for
cosmetic rather than
medical purposes
No
Yes, Select:
A non-corrective
contact lens
An implant for
augmentation, fixation,
or sculpting of body
parts
A facial or other skin filler
Equipment for
liposuction
Surgical laser
equipment
5.10 Does the device
incorporate, as an
integral part or
substance, a medicinal
product in achieving its
primary intended
action?
No
Yes
Brand name of
drug:………………
………………….. Active
ingredient:…………
……………………
……………………
Drug
manufacturer:………
………………….. SFDA Drug
Registration Number (if
Applicable):………...
……………………
5.11 Does the device
incorporate a substance
of animal origin?
No
Yes
Type of tissue, cell, or
substance:……………
………………………
……………………….
5.12 Does the device
incorporate human tissue, cell, or
substance?
No
Yes Type of tissue, cell, or
substance:……………
………………………
………………………
5.13 Does the device
incorporate cells or
substance of microbial
origin?
No
Yes
Type of microorganism:
………………………
………………………
5.14 The intended purpose of the device
5.15 Targeted patient
population as intended
All patient
Specific group of patients

Page 19 of 22
by the manufacturer
Clearly
defined:……….……..
5.16 Nomenclature code
number (if any):
GMDN:…………………….
UMDNS:…………...……...
Other:………………………
5.17 Device classification
based on GHTF
guidance “Principles of
Medical Devices
Classification”
A
B
C
D
5.18 Device Classification
in other countries
Country:……………………
Class:………………………
Country:……………………
Class:………………………
Country:……………………
Class:………………………
6. Design of Clinical Investigation
6.1 Clinical Investigational
Plan title
Scientific title
Abbreviated title
6.2 Clinical Investigational
Plan (CIP) information
CIP number
CIP date
CIP version
6.3 Clinical investigation
objective(s)
Primary objective(s)
Secondary objective(s)
6.4 Clinical investigation
endpoint(s)
Primary endpoint(s)
Secondary endpoint(s)
6.5 Type of Design
Open-label non-randomized clinical investigation
Randomization,
Randomized controlled
clinical investigation
o Parallel group:
………………………
o Cross over:
………………………
Blinding
Single blinded
Double blinded
Other Comparator used
Placebo
Comparator device,
identify:
………………………
6.6 Subject health status
Healthy volunteers
Patients
Both
6.7 Subjects Gender
Male
Female
Both
6.8 Does this study No

Page 20 of 22
includes vulnerable
subjects?
Yes
6.9 Size of the sample
population
Planned total number of
subjects involved in the clinical
investigation
Planned number of subjects
involved in the KSA
6.10 Number of study centers in the KSA
6.11 Other countries where this clinical investigation is carried
out
6.12 Inclusion / Exclusion Criteria
Reference page in the CIP for inclusion criteria
Reference page in the CIP
exclusion criteria
6.13 Duration of the study
Planned start date
Planned completion date
6.14 Is there a Data Safety
Monitoring Committee
for this study?
Yes
No
7. Investigation Site(s) in the KSA
7.1 Site 1 Name
Address
Phone
Name of principal investigator
IRBs name
IRBs address
IRBs phone
IRBs e-mail
Protocol number approved by
HREC/IRB
7.2 Site 2 Name
Address
Phone
Name of principal investigator
IRB name
IRB address
IRB phone
IRB e-mail
Protocol number approved by
HREC/IRB
Add
8. Declaration
8.1 I, the sponsor defined in this application, :
undertake that I comply with the Law of Ethics of Research on Living Creatures.
undertake that I will report to the SFDA’s NCMDR, IRBs, and investigators any reportable

Page 21 of 22
adverse event of which I become aware that involves the medical device; without delay but
not later than 10 working days of occurrence. Details on reporting are found in SFDA’s
guidance document entitled MDS – G6 Guidance on Post-Marketing Surveillance.
undertake that I will provide the documents specified in sections (II-B) and (II-C) in
SFDA’s guidance document entitled MDS – G.. Guidance on Clinical Investigations of
Medical Devices.
undertake to notify IRBs and investigators in case of withdrawal of SFDA approval or part
of it, within five calendar days of receiving the withdrawal notice.
undertake, under any request from the IRBs, and SFDA, to response by providing accurate, current, and complete information about any aspects of the study.
declare that all information provided in this application is true and complete.
Name:
Position:
Address:
Date:
Signature:
110
111

Page 22 of 22
Annex 2 112
Change Form for CIMD 113
Date:
CIMD Application
Number:
1. The document type
where the change
occur
2. The original
statement
3. The changed
statement
4. Reason of change
Note: Each change requires a separate change form. 114
115
116





























