Embed Size (px)
Citation preview

THE JOURNAL OPBIOLOGKZAL CHEMISTRY Vol. 238, No.11, November1963
Printed in U.S.A.
Prostaglandins and Related Factors*
15. THE STRUCTURES OF PROSTAGLANDIN Ei, Firr, AND FiB
SUNE BERGSTROM, R. RYHAGE, BENGT SAMUELSSON, AND JAN SJ~VALL
From the Department of Chemistry, Karolinska Institutet, Stockholm, Sweden
(Received for publication, December 21, 1962)
The pharmacodynamic effects of extracts obtained from seminal fluid and male accessory glands were discovered by Goldblatt (1) and von Euler (2). They independently found that this material contains a blood pressure-lowering factor and that it stimulates different smooth muscle organs. von Euler later demonstrated that these effects were due to a lipid-soluble substance with acidic properties, which was called prostaglandin (3). Further work on the biological action of similar extracts has been done by Eliasson (4). Two crystalline compounds (prostaglandin El and F1,) were isolated from vesicular glands of sheep by Bergstrom and Sjijvall (5, 6). The presence of PGEQ and one other factor has been demonstrated in seminal plasma from sheep and man (7, 8). Both PGEi and PGR, have smooth muscle-stimulating actions and PGE, also a blood pressure-reducing activity. Studies on some biological effects of these pure compounds have been reported (9, 10).
In a previous investigation, it was shown that PGEl is a carboxylic acid (C&Ha,Os) containing two hydroxyl groups, one keto group, one trans double bond, and a cyclopentanone ring (11). The formation of two epimeric trihydroxy acids, PGF1, and PGFlp, on sodium borohydride reduction of PGEl was also observed, the former of which proved identical with the com- pound isolated from sheep vesicular glands earlier.
Further work involving degradation by different means showed that the keto group and one of the two hydroxyl groups are present in the cyclopentane ring, which contains two side chains. One of these side chains (-(CH2)rCOOH) carries the carboxyl group and the other (-CH=CH-CH(OH)-(CH&CHs) the trans double bond and one hydroxyl group (12).
This report is concerned with further work, which has led to elucidation of the structures of PGEi, PGFr,, and PGFIp. Pre- liminary reports of the present work have appeared (13, 14).
EXPERIMENTAL PROCEDURES AND RESULTS
Material and Methods
Prostaglandin E1 was isolated in crystalline form (m.p. 114- 115’) from vesicular glands of sheep as described previously (6). The methyl ester was prepared with diazomethane.
Reversed phase partition chromatography was performed with Hyflo Supercel (Johns-Manville) treated with dimethyldichloro-
* Prostaglandin El (PGE1) (earlier PGE) gives on reduction two epimeric compounds prostaglandin F,,(PGFI,) (earlier PGFl or PGF,.,) and prostaglandin F,g (PGFI~) (earlier PGFz or PGF2-I).
1 The abbreviations used are: PGE, prostaglandin E; PGF, prostaglandin F.
silane as support and the solvent systems shown in Table I as moving and stationary phases (15, 16).
We used 3 ml of the stationary phase per 4.5 g of Super-Cel. The chromatograms were run at a constant temperature of +23”.
Gas-Liquid Chromatography-A Pye gas chromatograph (W. G. Pye and Company Ltd., England) with an argon ionization detector was used. Samples were collected for mass spectrom- etry in a glass tube (inner diameter, 2 mm; length, 200 mm), which was attached to the outlet with a glass joint. The proxi- mal end of the tube was kept at a temperature slightly above that of the column, whereas the rest of it was cooled with Dry- Ice. The collected sample was usually located in a narrow zone which was cut out and introduced into the mass spectrometer. This collection device was constructed by Dr. S. Stlillberg- Stenhagen and kindly made available to us.
Silicone grease (15%) (Dow Corning Corporation) or ethylene glycol adipate polyester (17%) was supported on lOO- to 120- mesh Celite (17). The argon pressure was kept constant at 1.0 kg per cm2. Mixtures of methyl esters of fatty acids were used as standards and diagrams were constructed for the different columns by plotting the observed retention times on a logarithmic scale against the number of carbon atoms in the acids on a normal scale (12, 18).
The effect on the retention time of a certain functional group was relatively constant when introduced into normal mono- or dicarboxylic acids. Thus, introduction of a hydroxyl group or a keto group was equivalent to increasing the number of carbon atoms in the acid approximately 0.8. These values were ex- pressed as “M values” which were obtained from the plot of the logarithm of retention time against the number of carbons in the main chain of the fatty acid; e.g. the methyl ester of a Ca mono- carboxylic acid was assigned an M value of 9.0 and the corre- sponding hydroxy acid ester was found to have a value of 9.8.
The introduction of an acetoxy group gave an increment of ill = 2.5 and conversion of the terminal methyl group into a carbomethoxy group raised the M value 3 units compared to the corresponding unsubstituted monocarboxylic acid methyl ester.
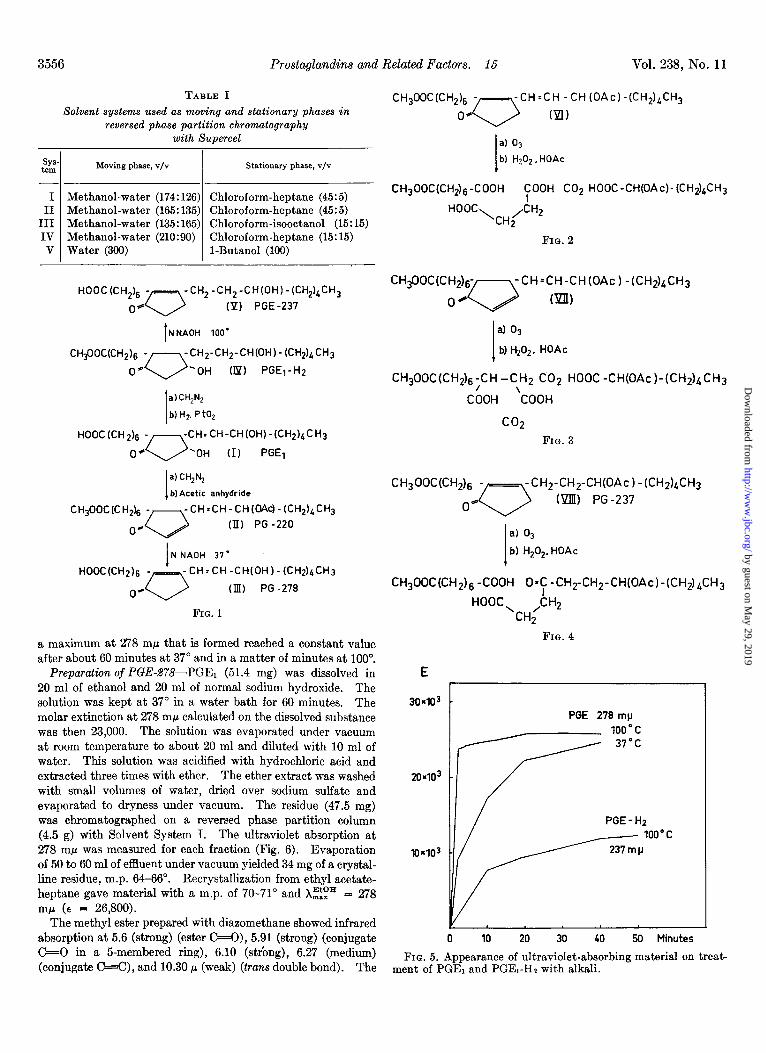
Preparation of Derivatives of Prostaglandin El-In the follow- ing section is described the preparation of derivatives of prosta- glandin E1, i.e. PGE-278, PGE-237, and PGE-220 (Fig. 1) which have been subjected to degradation by oxidative ozonolysis (Figs. 2, 3, and 4).
Treatment of PGEl with Alkali-Solutions of 1.0 mg of PGEr in 5 ml of 0.5 N NaOH in 50% aqueous ethanol were heated at 37” and 100”. The ultraviolet absorption was measured after different periods of time (Fig. 5). The absorption band with
3555
by guest on May 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from
3556 Prostaglandins and Related Factors. 15 Vol. 238, No. 11
-
SYS tern
-
I II
III IV V
-
TABLE I Solvent systems used as moving and stationary phases in
reversed phase partition chromatography
with Supercel
Moving phase, v/v Stationary phase, v/v
Methanol-water (174:126) Chloroform-heptane (45:5) Methanol-water (165: 135) Chloroform-heptane (45:5) Methanol-water (135: 165) Chloroform-isooctanol (15: 15) Methanol-water (210:90) Chloroform-heptane (15:15) Water (390) 1-Butanol (190)
HOOC(CH& -- -CH,-CH,-CH(OH)-(CH,),CH,
0* 0 (P) PGE-237
I NNAOH 100’
CH$OC(CH& - -CH2-CH2-CH(OH)-(CH2)bCH3
0’ 0 ‘OH (Ip) PGEj-H2
1
a)CH,N,
b) HZ, P to,
HOOC (CH 2),j - -CH:CH-CH(OH)-(CH2)bCH3
0. 0 ‘OH (I) PGEl
I
a) CH,N,
b)Acetic anhydride
CH,OOC(C H2)6 - -CH=CH-CH(OAc)-(CH2)~CH3
0. 0 (II) PG -220
1 N NAOH 37’
-CH=CH-CH(OH)-(CH2)&CH3
(Ill) PG -278
FIG. 1
a maximum at 278 rnp that is formed reached a constant value after about 60 minutes at 37” and in a matter of minutes at 100”.
Preparation of PGE-278-PGE1 (51.4 mg) was dissolved in 20 ml of ethanol and 20 ml of normal sodium hydroxide. The solution was kept at 37” in a water bath for 60 minutes. The molar extinction at 278 rnp calculated on the dissolved substance was then 23,000. The solution was evaporated under vacuum at room temperature to about 20 ml and diluted with 10 ml of water. This solution was acidified with hydrochloric acid and extracted three times with ether. The ether extract was washed with small volumes of water, dried over sodium sulfate and evaporated to dryness under vacuum. The residue (47.5 mg) was chromatographed on a reversed phase partition column (4.5 g) with Solvent System I. The ultraviolet absorption at 278 rn@ was measured for each fraction (Fig. 6). Evaporation of 50 to 60 ml of effluent under vacuum yielded 34 mg of a crystal- line residue, m.p. 64-66”. Recrystallization from ethyl acetate- heptane gave material with a m.p. of 70-71” and Xzip = 278 rnp (E = 26,800).
The methyl ester prepared with diazomethane showed infrared absorption at 5.6 (strong) (ester C=O), 5.91 (strong) (conjugate C+O in a 5-membered ring), 6.10 (strbng), 6.27 (medium) (conjugate C=C), and 10.30 p (weak) (tans double bond). The
CH300C(CH,l, - -CH=CH -CH(OAc)-(CH,),CH,
04 0 Nl)
I
a) 03 b) H202. HOAc
CH,OOC(CH$,-COOH ~00” CO2 HOOC-CH(OAC)-(CH~)~CH~
HOOC\CH2/CH2
FIG. 2
CH$0C(CH2)6- -CHsCH-CH(OAc) -(CH$LCH~
OH 0 ml)
I
a) 03
b) Hz02. HOAc
CH300C(CH2)+H-CH2 CO;, HOOC -CH(OAc)-(CH211,CH3
COOH ‘COOH
co2
FIG. 3
CH,OOC(cH,),j - - -CH2-CH2-CH(OAc)-(CH$hCH3
0* 0 (Ylll) PG-237
I
al 03
b) H,O,. HOAc
CH,OOC(CH,)~ -COOH 0’; -CH2-CH2-CH(OAc)-W2)&H3
HOOC CH2 ‘CH;
FIG. 4
30x103
20403
10403
PGE 270 rnp
1oo”c 37°C
PGE - Hz
loo”c
237mp
0 10 20 30 40 50 Minutes
FIG. 5. Appearance of ultraviolet-absorbing material on treat- ment of PGEl and PGEI-H2 with alkali.
by guest on May 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from
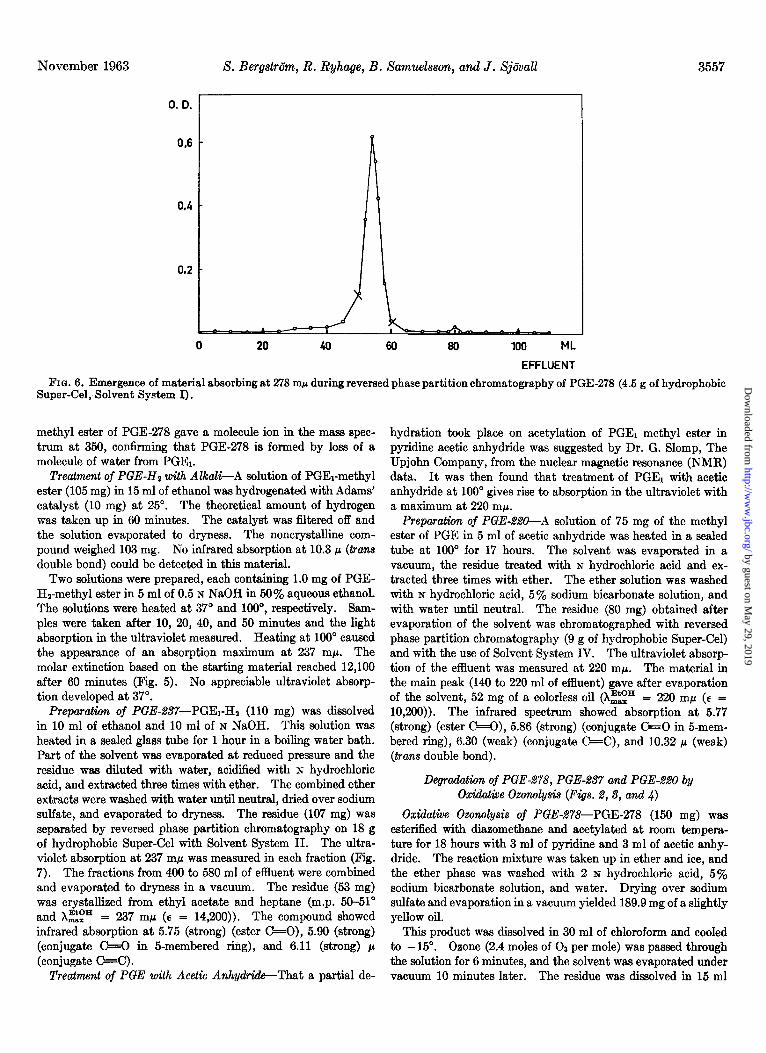
November 1963 S. Berg&&n, R. Ryhage, B. Samu.elsson, and J. Sj6vall 3557
0. D. 1
0 20 40 60 80 100 ML
EFFLUENT
FIG. 6. Emergence of material absorbing at 278 rnp during reversed phase partition chromatography of PGE-278 (4.5 g of hydrophobic Super-Cel, Solvent System I).
methyl ester of PGE-278 gave a molecule ion in the mass spec- trum at 350, confirming that PGE-278 is formed by loss of a molecule of water from PGE1.
Treatment of PGE-Hz with A&al&A solution of PGEI-methyl ester (105 mg) in 15 ml of ethanol was hydrogenated with Adams’ catalyst (10 mg) at 25”. The theoretical amount of hydrogen was taken up in 60 minutes. The catalyst was filtered off and the solution evaporated to dryness. The noncrystalline com- pound weighed 103 mg. No infrared absorption at 10.3 p (trans double bond) could be detected in this material.
Two solutions were prepared, each containing 1.0 mg of PGE- Hz-methyl ester in 5 ml of 0.5 N NaOH in 50% aqueous ethanol. The solutions were heated at 37” and loo”, respectively. Sam- ples were taken after 10, 20, 40, and 50 minutes and the light absorption in the ultraviolet measured. Heating at 100” caused the appearance of an absorption maximum at 237 rnp. The molar extinction based on the starting material reached 12,100 after 60 minutes (Pig. 5). No appreciable ultraviolet absorp- tion developed at 37”.
Preparation of PGE-B7-PGEI-H2 (110 mg) was dissolved in 10 ml of ethanol and 10 ml of N NaOH. This solution was heated in a sealed glass tube for 1 hour in a boiling water bath. Part of the solvent was evaporated at reduced pressure and the residue was diluted with water, acidiied with N hydrochloric acid, and extracted three times with ether. The combined ether extracts were washed with water until neutral, dried over sodium sulfate, and evaporated to dryness. The residue (107 mg) was separated by reversed phase partition chromatography on 18 g of hydrophobic Super-Cel with Solvent System II. The ultra- violet absorption at 237 rnp was measured in each fraction (Pig. 7). The fractions from 400 to 580 ml of effluent were combined and evaporated to dryness in a vacuum. The residue (53 mg) was crystallized from ethyl acetate and heptane (m.p. 50-51” and X~$” = 237 rnp (E = 14,200)). The compound showed infrared absorption at 5.75 (strong) (ester *O), 5.90 (strong) (conjugate C=O in 5-membered ring), and 6.11 (strong) ~1 (conjugate C=C).
Treatment of PGE with Acetic Anhydride-That a partial de-
hydration took place on acetylation of PGEl methyl ester in pyridine acetic anhydride was suggested by Dr. G. Slomp, The Upjohn Company, from the nuclear magnetic resonance (NMR) data. It was then found that treatment of PGEl with acetic anhydride at 100” gives rise to absorption in the ultraviolet with a maximum at 220 rnp.
Preparation of PGE-.%?&A solution of 75 mg of the methyl ester of PGE in 5 ml of acetic anhydride was heated in a sealed tube at 100” for 17 hours. The solvent was evaporated in a vacuum, the residue treated with N hydrochloric acid and ex- tracted three times with ether. The ether solution was washed with N hydrochloric acid, 5% sodium bicarbonate solution, and with water until neutral. The residue (80 mg) obtained after evaporation of the solvent was chromatographed with reversed phase partition chromatography (9 g of hydrophobic Super-Cel) and with the use of Solvent System IV. The ultraviolet absorp- tion of the effluent was measured at 220 mp. The material in the main peak (140 to 220 ml of effluent) gave after evaporation of the solvent, 52 mg of a colorless oil (Xztz” = 220 rnp (E = 10,200)). The infrared spectrum showed absorption at 5.77 (strong) (ester C+O), 5.86 (strong) (conjugate -0 in 5-mem- bered ring), 6.30 (weak) (conjugate C=C), and 10.32 p (weak) (trans double bond).
Degradation of PGE-278, PGE-237 and PGE-220 by Oxidative Ozo~~lysis (Figs. 2, 3, and 4)
Oxiddive Ozorwlysis of PGEd78-PGE-278 (150 mg) was esterified with diazomethane and acetylated at room tempera- ture for 18 hours with 3 ml of pyridine and 3 ml of acetic anhy- dride. The reaction mixture was taken up in ether and ice, and the ether phase was washed with 2 N hydrochloric acid, 5% sodium bicarbonate solution, and water. Drying over sodium sulfate and evaporation in a vacuum yielded 189.9 mg of a slightly yellow oil.
This product was dissolved in 30 ml of chloroform and cooled to -15”. Ozone (2.4 moles of 03 per mole) was passed through the solution for 6 minutes, and the solvent was evaporated under vacuum 10 minutes later. The residue was dissolved in 15 ml
by guest on May 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from
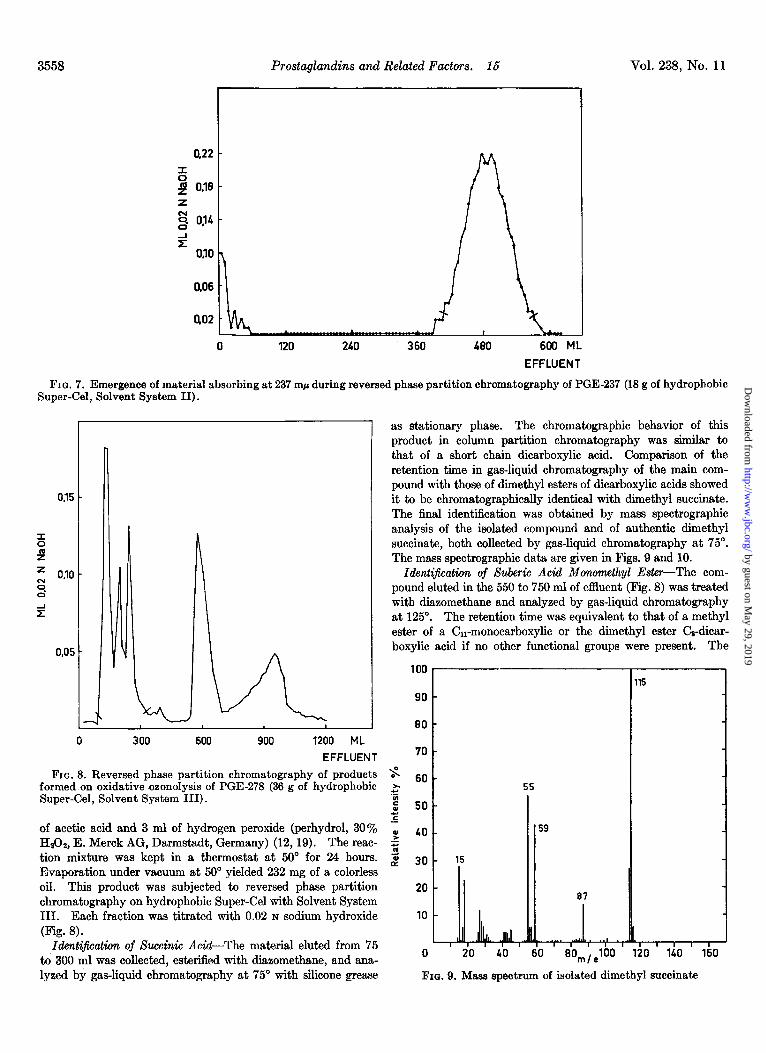
3558 Prostaglandins and Related Factors. 16 Vol. 238, No. 11
0.06
QO2
0 120 240 360 480 600 ML
EFFLUENT
FIG. 7. Emergence of material absorbing at 237 mp during reversed phase partition chromatography of PGE-237 (18 g of hydrophobic Super-Gel, Solvent System II).
600 1200 ML
EFFLUENT 70
FIG. 8. Reversed phase partition chromatography of products s formed on oxidative ozonolysis of PGE-278 (36 g of hydrophobic
69 ,X
7 as stationary phase. The chromatographic behavior of this product in column partition chromatography was similar to that of a short chain dicarboxylic acid. Comparison of the retention time in gas-liquid chromatography of the main com- pound with those of dimethyl esters of dicarboxylic acids showed it to be chromatographically identical with dimethyl succinate. The final identification was obtained by mass spectrographic analysis of the isolated compound and of authentic dimethyl succinate, both collected by gas-liquid chromatography at 75“. The mass spectrographic data are given in Figs. 9 and 10.
Ident$cation of Suberic Acid Monomethyl Ester-The com- pound eluted in the 550 to 750 ml of eflluent (Fig. S) was treated with diazomethane and analyzed by gas-liquid chromatography at 125”. The retention time was equivalent to that of a methyl ester of a Cii-monocarboxylic or the dimethyl ester Ce-dicar- boxylic acid if no other functional groups were present. The
Super-Cel, Solvent System HI).
of acetic acid and 3 ml of hydrogen peroxide (perhydrol, 30% f 49 HrOz, E. Merck AG, Darmstadt, Germany) (12,19). The reac- 2 tion mixture was kept in a thermostat at 50” for 24 hours. 2 39 Evaporation under vacuum at 50” yielded 232 mg of a colorless oil. This product was subjected to reversed phase partition 20 chromatography on hydrophobic Super-Cel with Solvent System III. Each fraction was titrated with 0.02 N sodium hydroxide 10
- 15
(Fig. 8). Identiificattin of Succinic Acid-The material eluted from 75
to 300 ml was collected, esterified with diizomethane, and ana- 0 20 40 60 80,,,100 120 140 160
15
lyzed by gas-liquid chromatography at 75” with silicone grease FIG. 9. Mass spectrum of isolated dimethyl succinate
by guest on May 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from
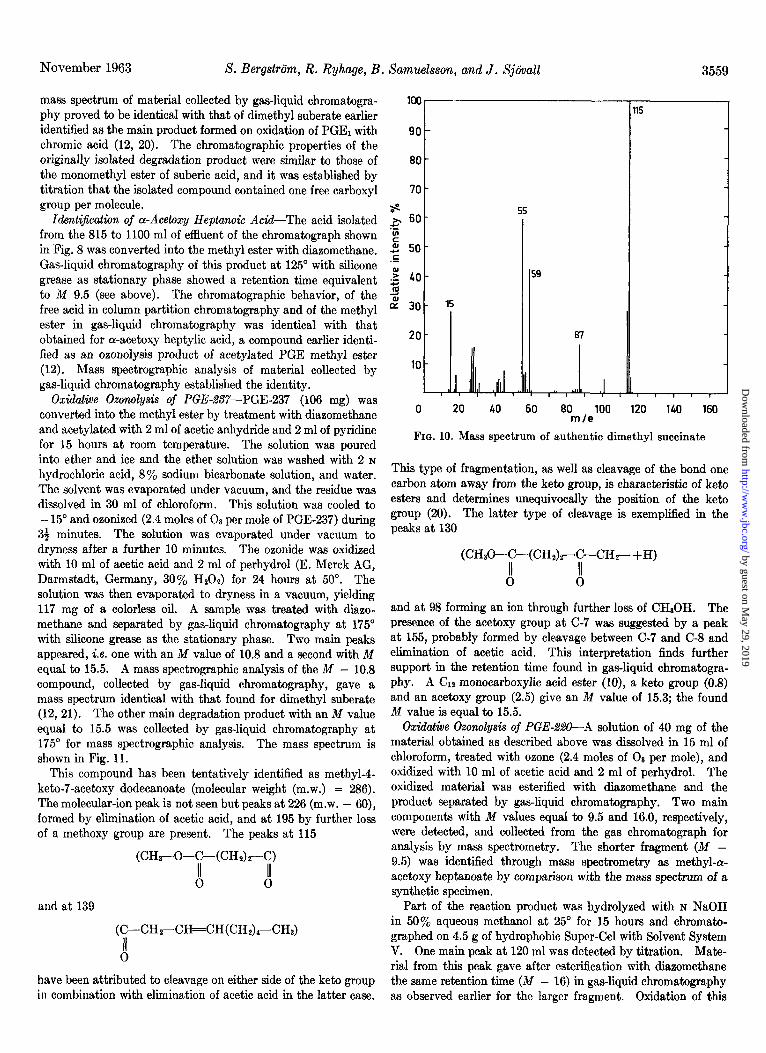
November 1963 S. Bergstrtim, R. Ryhage, B
mass spectrum of material collected by gas-liquid chromatogra- phy proved to be identical with that of dimethyl suberate earlier identified as the main product formed on oxidation of PGE, with chromic acid (12, 20). The chromatographic properties of the originally isolated degradation product were similar to those of the monomethyl ester of suberic acid, and it was established by titration that the isolated compound contained one free carboxyl group per molecule.
IdentQication of a-Acetoxy Heptanok Acid-The acid isolated from the 815 to 1100 ml of effluent of the chromatograph shown in Fig. 8 was converted into the methyl ester with diazomethane. Gas-liquid chromatography of this product at 125” with silicone grease as stationary phase showed a retention time equivalent to M 9.5 (see above). The chromatographic behavior, of the free acid in column partition chromatography and of the methyl ester in gas-liquid chromatography was identical with that obtained for a-acetoxy heptylic acid, a compound earlier identi- fied as an ozonolysis product of acetylated PGE methyl ester (12). Mass spectrographic analysis of material collected by gas-liquid chromatography established the identity.
Oxidatioe Ozonolysis of PGE-,%?7-PGE-237 (106 mg) was converted into the methyl ester by treatment with diazomethane and acetylated with 2 ml of acetic anhydride and 2 ml of pyridine for 15 hours at room temperature. The solution was poured into ether and ice and the ether solution was washed with 2 N
hydrochloric acid, 8% sodium bicarbonate solution, and water. The solvent was evaporated under vacuum, and the residue was dissolved in 30 ml of chloroform. This solution was cooled to - 15” and ozonized (2.4 moles of 03 per mole of PGE-237) during
33 minutes. The solution was evaporated under vacuum to dryness after a further 10 minutes. The ozonide was oxidized with 10 ml of acetic acid and 2 ml of perhydrol (E. Merck AG, Darmstadt, Germany, 30% HzO.J for 24 hours at 50”. The solution was then evaporated to dryness in a vacuum, yielding 117 mg of a colorless oil. A sample was treated with diazo- methane and separated by gas-liquid chromatography at 175” with silicone grease as the stationary phase. Two main peaks appeared, i.e. one with an M value of 10.8 and a second with M equal to 15.5. A mass spectrographic analysis of the M - 10.8 compound, collected by gas-liquid chromatography, gave a mass spectrum identical with that found for dimethyl suberate (12,21). The other main degradation product with an M value equal to 15.5 was collected by gas-liquid chromatography at 175’ for mass spectrographic analysis. The mass spectrum is shown in Fig. 11.
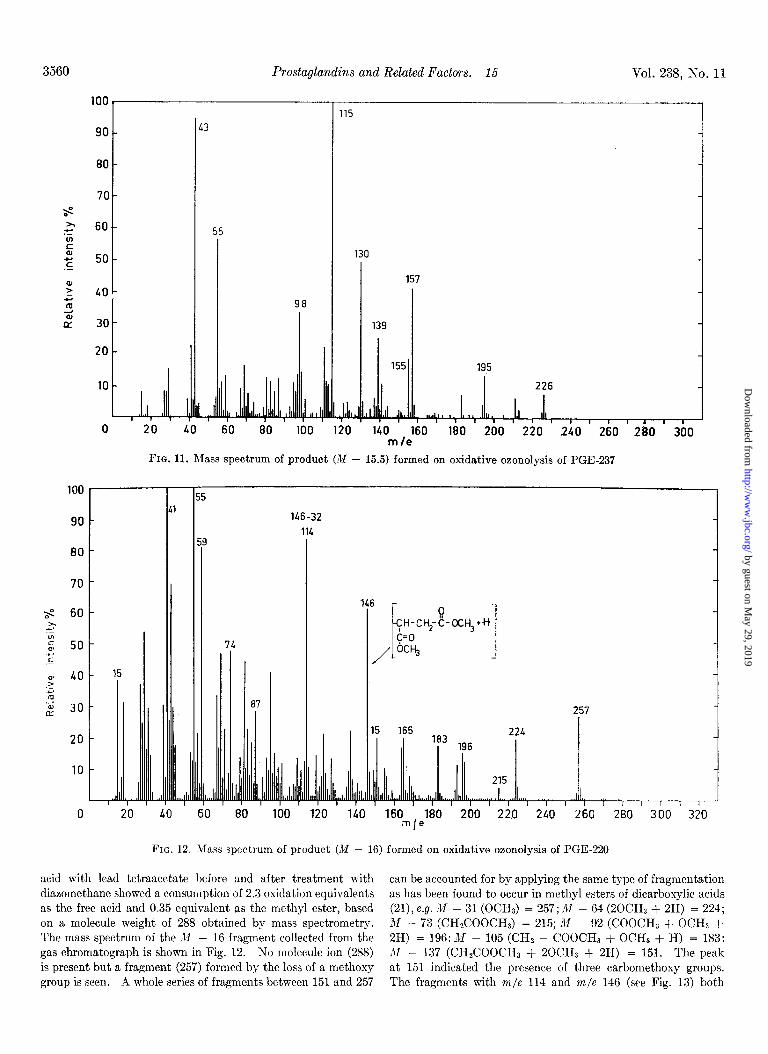
This compound has been tentatively identified as methyl-4- keto-7-acetoxy dodecanoate (molecular weight (m.w.) = 286). The molecular-ion peak is not seen but peaks at 226 (m.w. - SO), formed by elimination of acetic acid, and at 195 by further loss of a methoxy group are present. The peaks at 115
(CH3-0-C-(CH&--C)
!i d
and at 139
(C-CHrCH=CH(CH&-CHa)
6
have been attributed to cleavage on either side of the keto group in combination with elimination of acetic acid in the latter case.
Samuelssm, and J. Sjiivall
100
90
80
70
s x 60 Y z 2 50 .s
2 40
J
B 30
20
10
l-
/-
55
59
1 97
0 20 40 60 80 100 120 140 160 m/e
FIG. 10. Mass spectrum of authentic dimethyl succinate
This type of fragmentation, as well as cleavage of the bond one carbon atom away from the keto group, is characteristic of keto esters and determines unequivocally the position of the keto group (20). The latter type of cleavage is exemplified in the peaksat
(CH&-C-(CH&-C-CHz-+H)
8 d
and at 98 forming an ion through further loss of CHaOH. The presence of the acetoxy group at C-7 was suggested by a peak at 155, probably formed by cleavage between C-7 and C-8 and elimination of acetic acid. This interpretation finds further support in the retention time found in gas-liquid chromatogra- phy. A Cl2 monocarboxylic acid ester (lo), a keto group (0.8) and an acetoxy group (2.5) give an M value of 15.3; the found M value is equal to 15.5.
Oxidative Ozonolysis of PGE-!&%W---A solution of 40 mg of the material obtained as described above was dissolved in 15 ml of chloroform, treated with ozone (2.4 moles of 03 per mole), and oxidized with 10 ml of acetic acid and 2 ml of perhydrol. The oxidized material was esterified with diazomethane and the product separated by gas-liquid chromatography. Two main components with M vaIues equal to 9.5 and 16.0, respectively, were detected, and collected from the gas chromatograph for analysis by mass spectrometry. The shorter fragment (M - 9.5) was identified through mass spectrometry as methyl-a- acetoxy heptanoate by comparison with the mass spectrum of a synthetic specimen.
Part of the reaction product was hydrolyzed with N NaOH in 50% aqueous methanol at 25” for 15 hours and chromato- graphed on 4.5 g of hydrophobic Super-Cel with Solvent System V. One main peak at 120 ml was detected by titration. Mate- rial from this peak gave after esterification with diazomethane the same retention time (M - 16) in gas-liquid chromatography as observed earlier for the larger fragment. Oxidation of this
by guest on May 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from
3560 Prostaglandins and Related Factors. 15 Vol. 238, n-o. 11
loo1 90 1 13
80
70 s t x .z 60
P 2 .E
50
: 40 I z m G CY 30
115
130
I 157
139
155 195
226
I II I I , I I , I I I I , 120 140 160 180 200 220 240 260
m/e 2&l 300 20 40 60 80 la0
FIG. 11. RIass spectrum of product (M - 15.5) formed on oxidative oaonolysis of PGE-237
80
$ 60 s .- g 50 7 .- F 40
.-
2 30
r
4 I 8
146-32
114
' 100 ' 110 ' 160 180 200 220 240 260 280 300 320 mle
15
'I 20
87 257 1
FIG. 12. RIass spectrum of product (M - 16) formed on oxidative ozonolysis of PGE-220
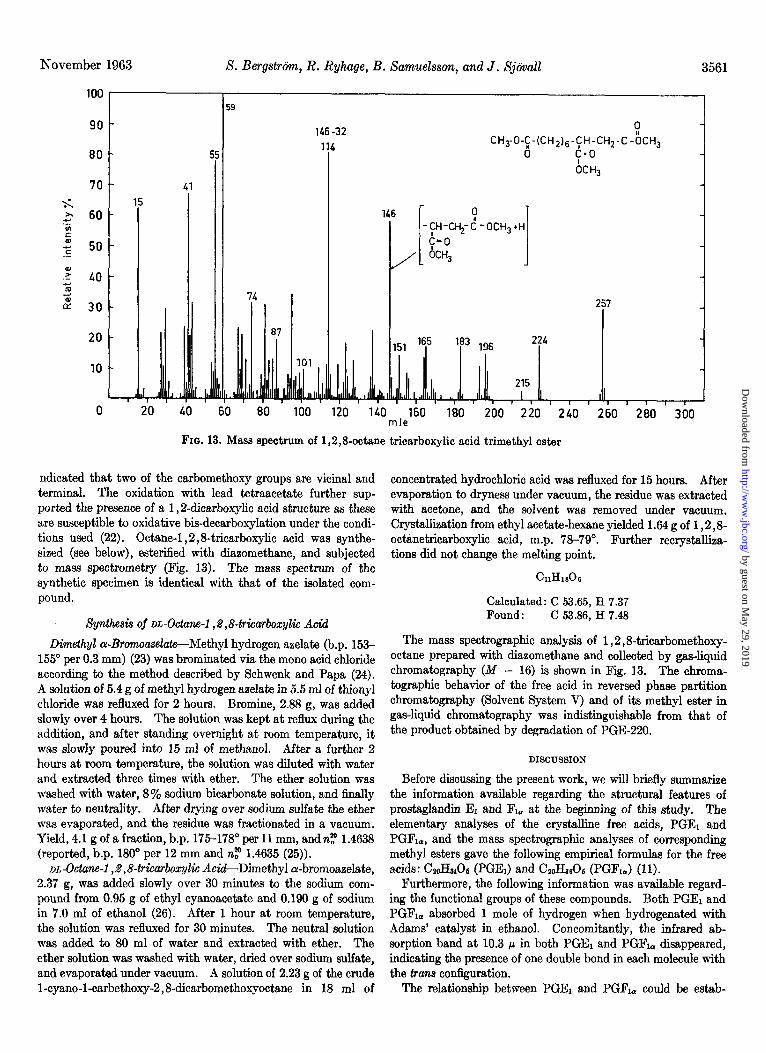
acid with lead tetraacetate before and after treatment with can be accounted for by applying the same type of fragmentation diazomethane showed a consumption of 2.3 oxidation equivalents as has been found to occur in methyl esters of dicarboxylic acids as the free acid and 0.35 equivalent as the methyl ester, based (21), e.g. X - 31 (OCHJ = 257; M - 64 (20CHa + 2H) = 224; on a molecule weight of 288 obtained by mass spectrometry. M - 73 (CH#ZOOCH3 = 215; M - 92 (COOCH, + OCHS + The mass spectrum of the M - 16 fragment collected from the 2H) = 196: M - 105 (CHZ - COOCH3 + OCHB + H) = 183: gas chromatograph is shown in Fig. 12. No molecule ion (288) X - 137 (CH&OOCH, + 20CH3 + 2H) = 151. The peak is present but a fragment (257) formed by the loss of a methoxy at 151 indicated the presence of three carbomethoxy groups. group is seen. A whole series of fragments between 151 and 257 The fragments with m/e 114 and m/e 146 (see Fig. 13) both
by guest on May 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from
November 1963 S. Bergstriim, R. Ryhage, B. Samuelsson, and J. Sjiivall 3561
90 -
80 -
70 - .
$ 60-
: z 50- .-
z ;;; 40-
i 30-
20 -
10 -
7
146 -32
114
1 46
FI CH~-O-$-(CH~),-~H-CH~-C-OCH~
0 c=o
bCH3
151 ‘65 le3 196 224
215 ll LI8 I 1. 11 1 I I I I I,,,,,,,
I 80 100 120 140 160 180 200 220 240 mle
260 280 300
FIG. 13. Mass spectrum of 1,2,8octane tricarboxylic acid trimethyl ester
ndicated that two of the carbomethoxy groups are vicinal and terminal. The oxidation with lead tetraacetate further sup- ported the presence of a 1,2dicarboxylic acid structure as these are susceptible to oxidative bis-decarboxylation under the condi- tions used (22). Octane-l ,2,8-tricarboxylic acid was synthe- sized (see below), esterified with diazomethane, and subjected to mass spectrometry (Fig. 13). The mass spectrum of the synthetic specimen is identical with that of the isolated com- pound.
Synthesis of DL-Octane-l ,%,8-triearboxylic Acid
Dimethyl cY-Bronwase2ate--Methyl hydrogen azelate (b.p. 153- 155” per 0.3 mm) (23) was brominated via the mono acid chloride according to the method described by Schwenk and Papa (24). A solution of 5.4 g of methyl hydrogen aaelate in 5.5 ml of thionyl chloride was refluxed for 2 hours. Bromine, 2.88 g, was added slowly over 4 hours. The solution was kept at reflux during the addition, and after standing overnight at room temperature, it was slowly poured into 15 ml of methanol. After a further 2 hours at room temperature, the solution was diluted with water and extracted three times with ether. The ether solution was washed with water, 8% sodium bicarbonate solution, and 6nally water to neutrality. After drying over sodium sulfate the ether was evaporated, and the residue was fractionated in a vacuum. Yield, 4.1 g of a fraction, b.p. 175-178” per 11 mm, and n$’ 1.4638 (reported, b.p. 180” per 12 mm and n? 1.4635 (25)).
DL-Octane-l , 2 ,84&arboxylic A&--Dimethyl cr-bromoazelate, 2.37 g, was added slowly over 30 minutes to the sodium com- pound from 0.95 g of ethyl cyanoacetate and 0.190 g of sodium in 7.0 ml of ethanol (26). After 1 hour at room temperature, the solution was refluxed for 30 minutes. The neutral solution was added to 80 ml of water and extracted with ether. The ether solution was washed with water, dried over sodium sulfate, and evaporated under vacuum. A solution of 2.23 g of the crude l-cyano-1-carbethoxy-2,8-dicarbomethoxyoctane in 18 ml of
concentrated hydrochloric acid was refluxed for 15 hours. After evaporation to dryness under vacuum, the residue was extracted with acetone, and the solvent was removed under vacuum. Crystallization from ethyl acetate-hexane yielded 1.64 g of 1,2,8- octanetricarboxylic acid, m.p. 78-79”. Further recrystalliza- tions did not change the melting point.
Calculated: C 53.65, H 7.37 Found : C 53.86, H 7.48
The mass spectrographic analysis of 1,2,8tricarbomethoxy- octane prepared with diazomethane and collected by gas-liquid chromatography (Af - 16) is shown in Fig. 13. The chroma- tographic behavior of the free acid in reversed phase partition chromatography (Solvent System V) and of its methyl ester in gas-liquid chromatography was indistinguishable from that of the product obtained by degradation of PGE-220.
DISCUSSION
Before discussing the present work, we will briefly summarize the information available regarding the structural features of prostaglandin El and Fr, at the beginning of this study. The elementary analyses of the crystalline free acids, PGEr and PGR,, and the mass spectrographic analyses of corresponding methyl esters gave the following empirical formulas for the free acids: C&Ha06 (PGEJ and CaoHssOa (PGFr,) (11).
Furthermore, the following information was available regard- ing the functional groups of these compounds. Both PGEI and PGFr, absorbed 1 mole of hydrogen when hydrogenated with Adams’ catalyst in ethanol. Concomitantly, the infrared ab- sorption band at 10.3 p in both PGEr and PGR, disappeared, indicating the presence of one double bond in each molecule with the trans configuration.
The relationship between PGEl and PGFr, could be estab-
by guest on May 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from

3562 Prostaglandins and Related Factors. 15 Vol. 238, No. 11
lished by the formation of the latter on reduction of PGEi with sodium borohydride. This reaction apparently involved reduc- tion of a keto group in PGEi and also gave rise to a second reac- tion product, PGF,B, epimeric with PGF,,. Further proof for this view was afforded by the following experiments. i4C- Labeled methyl esters of PGE1 and PGF1, were prepared with W-labeled diazomethane and acylated with p-nitrobenzoyl- chloride in pyridine. The ratio between the ultraviolet absorp- tion at 257 rnp (due to the p-nitrobenzoyl group) and the radio- activity demonstrated that PGEl contained two (1.4 to 1.8) and PGFi, three acylable groups per molecule (11). The low value obtained for PGEi was later shown to be due to partial dehydra- tion during the acylation.
The location of the above mentioned reducible keto group was inferred from the infrared data. The infrared spectrum of the methyl ester of PGEi showed only one strong band in the car- bony1 region at 5.77 ~.r, while the free acid absorbed at both 5.77 p and 5.87 p. The absorption band at 5.77 p in the former case is due to the carbonyl group in both the keto group and the carbomethoxy group whereas in the latter case the carbonyl of the carboxyl group absorbs at 5.87 p. The keto carbonyl absorp- tion (5.77 cc) of PGEi appears at approximately the same wave length as reported for cyclopentanones. Since aldehydes, aliphatic ketones as well as cyclohexanones absorb at longer wave lengths, these data strongly indicated that the keto group is present in a B-membered ring (11). The data obtained at this stage thus indicated that prostaglandin E1 contains 1 carboxyl group, 1 keto group (cyclopentanone), 2 hydroxyl groups, 1 double bond (trans), and a 5-membered ring.
This work was continued by different types of degradations in order to identify the carbon skeleton and to localize the func- tional groups (12). From the reaction product obtained on chromic acid oxidation of PGEi, suberic acid (CJ was isolated in high yield, demonstrating that six unsubstituted methylene groups are present in one chain. Oxidative ozonolysis of the methyl ester acetate of PGEl gave one large unidentified frag- ment and cY-acetoxyheptanoic acid, indicating that seven other carbon atoms are located in a side chain with the following structure:
=CH-CH(OH)-(CH&CHs
The possibility that the remaining five carbon atoms were present in the cyclopentane ring and that the Cr and Cs chains were directly attached to this ring appeared as one possibility. If this were the case, the CT chain should be directly bound to the ring by the double bond. A possible alternative would be that the Cr chain was attached via a -CH= group and that one of the carboxyl groups in the suberic acid originated in the ring carbons. Additional alternatives were of course the presence of three side chains attached to the ring.
The question whether the double bond of the Cr side chain is attached directly to the cyclopentane ring or placed one or more carbon atoms away was resolved by periodate-permanganate oxidation of the trimethyl ether of the methyl ester of PGFi, followed by mass spectrographic analyses of the larger fragment isolated by gas chromatography. This contained one new carboxyl group, indicating that the double bond is disubstituted and placed at least one carbon atom away from the ring. The newly formed carboxyl group of the suberic acid must therefore originate in the B-membered ring and the second hydroxyl group in PGEi must also be present in the ring.
In the present work, a number of derivatives of prostaglandin E1 have been prepared and degraded by oxidative ozonolysis. The proposed structures of these compounds are shown in Fig. 1.
Treatment of PGEi with 0.5 N sodium hydroxide solution gave a compound (PGE-278) with X,,, at 278 rnp. The acetylated methyl ester of this derivative (VI) gave on oxidative ozonolysis, suberic acid monomethyl ester, succinic acid, and a-acetoxy- heptanoic acid (Fig. 2), i.e. all but one of the carbon atoms of VI were accounted for. The isolation of suberic acid monomethyl ester and a-acetoxyheptanoic acid was in consonance with in- formation earlier obtained regarding the structures of the side chains. However, the isolation of succinic acid, which must originate in the ring, was of particular importance since it showed that two vicinal methylene groups are present in the 5-membered ring of this derivative and, accordingly, the three carbon atoms carrying the side chains and the keto group must be adjacent. Furthermore, on the basis of these considerations, the isolation of suberic acid monomethyl ester, produced by oxidation of the vicinal diketone originally formed indicated that the carboxyl side chain must be attached to the carbon atom that is a! to the keto group. If the positions of the two side chains were reversed, a Cu y-ketodicarboxylic acid would have been formed instead of suberic acid and succinic acid.
The indicated structure of PGE-278 is also supported by the ultraviolet absorption with X,, at 278 rnl.c (27, 28) and by the infrared absorption band attributable to the ketonic C=O stretching vibrations, which is shifted from 5.77 /J to PGEi to 5.91 p in PGE-278 due to the conjugation. The latter compound also exhibits absorption bands at 6.10 and 6.27 p due to C=C stretching vibrations (29).
The saturated analogue (PGEi-HZ) of PGEi gave on treatment with alkali a less hydrophilic derivative with X,,, at 237 rnp (Fig. 1). The methyl ester acetate of this compound gave on oxidative ozonolysis two products, i.e. the monomethyl ester of suberic acid and 4-keto-7-acetoxydodecanoic acid, which were identified by gas chromatography and mass spectrometry (Fig. 4). In this case all of the 20 carbon atoms of the prostaglandin skeleton were identified. The formation of these products has been visualized as an initial attack on the tetrasubstituted double bond giving two keto groups, one of which is vicinal to the original keto group. Oxidative cleavage of the vicinal keto groups by the peracetic acid gives rise to the two carboxyl groups in suberic acid monomethyl ester and in 4-keto-7-acetoxydodecanoic acid, respectively.
Further proof for the proposed structure of PGE-237 was obtained by various physical methods. Thus, the infrared spectrum showed absorption at 5.90 /.L (conjugated B-membered ring ketone) and 6.11 p (C+C) and the ultraviolet absorption gave a Amax at 237 rnp, which is completely in accordance with the values obtained for a number of trisubstituted cy ,@-un- saturated cyclopentenones (28, 30). Furthermore, the tetra- substitution of the double bond was confirmed by the absence of olefinic protons in the NMR spectrum of PGE-237.
By treatment of PGEi with acetic anhydride or weak alkali, a derivative (PGE-220) was obtained, which absorbed in the ultraviolet with X,,, at 220 rnp. Oxidative ozonolysis of the methyl ester acetate of this compound gave 1,2,8-octanetricar- boxylic acid and ru-acetoxyheptanoic acid (Fig. 3). The isola- tion of these degradation products, formed as illustrated in Fig. 3, gives firm support for the proposed structure of PGE-220. This evidence is further amplified by the ultraviolet absorption of this compound (X,,, at 220 mp) which is in agreement with
by guest on May 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from

November 1963 S. Berg&&n, R. Ryhage, B. Samuelsson, and J. Sjiivall 3563
HOOC(CH2)6 - -CH=CH-CH (OHb(CH2)4CH3
HO’ 0 ‘OH (x)
FIQ. 14
that observed for monosubstituted LY ,&unsaturated cyclo- pentenones (27,28).
Independent proof for the carbon skeleton of PGEl has been obtained by a direct identification of a derivative in the following manner. Catalytic reduction of PGEi in acetic acid followed by treatment of one of the products with alkali gave a derivative, the physical data of which indicated Structure IX. A com- pound with this structure has been synthesized by two different methods and proved to be identical with the derivative obtained from PGEi (31).
The physical data on the derivatives (PGE-278, PGE-237, and PGE-220) of prostaglandin E1 and the identification of the degradation products obtained from these compounds have, as discussed above, rigorously established the structures of these compounds. From this work follows the structures of the side chains in PGE, as well as their attachments to the ring and the position of the keto group. In this way, the structural problem of PGEl has been minimized to establishing the position of the hydroxyl group in the 5-membered ring.
There are formally four diierent carbon atoms in the ring of PGEi to which this hydroxyl group might be attached. Of these carbons, the tertiary ones could be excluded by acylation experi- ments in which this hydroxyl group was easily esterified by different acylating agents. In accordance with these experi- ments, the NMR data indicated the presence of two and three carbinol hydrogens, respectively, in PGEi and PGFi,. Further- more, the placing of the hydroxyl group at the secondary carbon atom that is a: to the keto group was ruled out by the failure of PGE1, PGFb, and PGFle to undergo oxidation with periodate or lead tetraacetate.
Taken together, these experiments leave the secondary carbon atom /? to the keto group as the only possible site for the ring hydroxyl in PGEi (see Structure I). In addition, the facile elimination of water with formation of the monosubstituted ar,&unsaturated ketone (PGE-220) on treatment of PGEi with acetic anhydride or weak alkali serves as a further adjunct in assigning the P-ketol structure of PGEi.
The data obtained thus support Structure I for PGE1, PGR, and PGFia (X in Fig. 14) differ in the steric position of the hydroxyl group formed by reduction of the keto group in PGEi
(11).
SUMMARY
Prostaglandm E1 isolated from sheep vesicular glands has been shown to have Structure I, i.e. 2-(6-carboxyhexyl)-3-(3-hy- droxyocten - 1 - yl) - 4 - hydroxycyclopentanone. Reduction of prostaglandin Ei yields two epimeric compounds, prostaglandin F1, and prostaglandin F1o, which differ in the steric position of the hydroxyl group formed by reduction of the keto group.
Acknowbdgments-We wish to thank Dr. G. Slomp, The Upjohn Company, Kalamazoo, Michigan, for determining and interpreting the NMR spectra of prostaglandin Ei and deriva- tives referred to in this report.
We are indebted to Dr. I. Fischmeister for some of the infrared spectra.
The technical assistance of Miss C. Carlon, Miss A. Ivarsson, Mr. L. Krabisch, Mrs. G. Pettersson, and Miss M. Zetterbom is gratefully acknowledged.
This work has been supported by the Foundations “Therese och Johan Anderssons Mix-me” and “Gustaf och Tyra Svenssons Minne.” We are also greatly indebted to The Upjohn Company, Kalamazoo, Michigan.
Addendum-Recent isolations of additional prostaglanclms (32) have necessitated introduction of a new nomenclature for this class of compounds. This is being based on the trivial name prostanoic acid for the parent C&o acid, numbered as shown in XI. The stereochemical features of the structures shown below have been provided by recent. x-ray diffraction studies (quoted by Abrahamsson et al. (33)).
XI
Prostaglandin El (llcu,l5-dihydroxy-9-ketoprost-13-enoic acid)
CHa-CHrCH2-CHrCH2-CH(OH)-CH=CH CH~-CH~CHTCH~-CH~-CH~-COOH
XII
Prostaglandin Fla(9a,110,15-trihydroxyprost-13-enoic acid)
CHa-CHz-CHt--CHs-CHz-CH(OH)-CH=CH CH2-CH~--CH2-CH~-CH2-CH2-COOH
XIII
Prostaglandin F&9@,11Lu,l5-trihydroxyprost-13-enoic acid)
by guest on May 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Prostaglandins and Related Factors. 15 Vol. 238, No. 11
1.
2.
3.
4. 5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
RTTFlZR.RNCRR ------ --.--- 16. NORMAN. A.. Acta Chem. Stand.. 7. 141” IlQxQ\ 17. GOLDBLATT, M. W., Chem. & Ind. (London), 62, 1056 (1933);
J. Physiol. (London), 64, 208 (1935). VON EULER, U. S., Arch. exptl. Pathol. Pharmakol., 176, 78
- . - - . ~ - - - - , . ~ . , ~~ L” \~“V” , .
FARQUHAR, J. W., INSULL, W., jR' ROSEN, P., STOFFEL, W., AND AHRENS, E. H., JR., Nutritzon Revs., 17, Suppl. No. 8, Part 2 (1959).
18. 19.
JAMBS, A. T., AND MARTIN, A. J. P., Biochem. J., 63,144 (1956). KLENK, E., AND BONGARD, W., 2. physiol. Chem., 260, 181
(1952).
(1934). ’ ’ - vow EULER, U. S., J. Physiol. (London), 88,213 (1936); Skand.
Arch. Phvsiol.. 81, 65 (1939). ELIASSON, k., Acta Physiol. &and., 46, Suppl. 158 (1959). BERQSTR~M, S., AND SJ~VALL, J., Acta Chem. Stand., 14. 1693
(1960). BERQSTRBM,S., AND SJBVALL,J., Acta Chem.Scand.,14,1701
(1960). BEROSTR~M, S., KRABISCH, L., AND SJBVALL, J., Acta Chem.
Stand., 14, 1706 (1960). BERQSTRBM, S., AND SAMUELSSON, B., J. Biol. Chem., 237,
PC3605 (1962). BERGSTRBM.S.,ELIASSON, R., VONEULER, U.S., ANDSJ~VALL,
J., Acta @hy&ol. &and:, 45, 133 (1959); BERQSTR~M.S..DUN&R. H.. VON EULER. U.S.. PERNOW. B..
AND SJ~V~LL; J., Act; Phisiol. Scand.,‘BS, 145 (1959). ’ ’ BERGSTRBM,S.,KRABISCH, L., SAMUELSSON, B., AND SJ~VALL,
J., Acta Chem. Stand., 16, 969 (1962). BERGSTROM, S., RYHAQE, R., SAMUELSSON, B., AND SJBVALL,
J., Acta Chem. Stand., in press. BERGSTRBM,S.,SAMUELSSON,B., AND SJ~VALL, J., Federation
Proc., 21, 281 (1962). BERQSTR~Y, S., RYHAQE, R., SAMUELSSON, B., AND SJ~VALL,
J., Acta Chem. Stand., 16, 501 (1962). SJBVALL, J., Acta Physiol. Stand., 29, 232 (1953).
20. RYHA~E, R., AND STENHAGEN, E., Arkiv Kemi, 16, 545 (1960). 21. RYHAGE, R., AND STENHAGEN, E., Arkiv Kemi, 14, 497 (1959). 22. GROB, C. A., OHTA, M., RENK, E., AND WEISS, A.,Helv.Chim.
Acta, 41, 1191 (1958). 23. MORGAN, G. T., AND WALTON, E., J. Chem. Sot., 292 (1935). 24. SCHWENK, E., AND PAPA, D., J. Am. Chem. Sot., 70, 3622,
25. 26.
TREIBS, W., AND LEICHSSEURING,G., Chem.Ber.,64.52 (1951). LINSTEAD,R. P., NOBLE, E.G., AND WRIGHT, J.M., J.Chem.
sot., 911 (1937). 27. 28. 29.
WOODWARD, R. B., J. Am. Chem. Sot., K&l123 (1941). GILLAM, A. E., AND WEST, T. F., J. Chem. Sot., 486 (1942). BELLAMY, L. J., The infra-red spectra of complex molecules,
John Wiley and Sons, Inc., New York. 1958. 30.
31.
32.
33.
FRANK, R.L:, ARMSTRO&,R:,KWIATE<J.,~~DPRICE, H.A., J. Am. Chem. Sot., 70, 1379 (1948).
SAMUELSSON, B., AND STXLLBERG, G., Acta Chem. Stand., 17 810 (1963).
BERQSTR~M,S.,DRESSLER, F., RYHAQE, R., SAMUELSSON, B., AND SJ~VALL, J., Arkiv Kemi, 19,563 (1962).
ABRAHAMSSON,S., BEROSTR~M,~., AND S~UELSSON, B., Proc. Chem. Sot., 332 (1962).
by guest on May 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Sune Bergström, R. Ryhage, Bengt Samuelsson and Jan Sjövallβ, AND F1αPROSTAGLANDIN E1, F1
Prostaglandins and Related Factors: 15. THE STRUCTURES OF
1963, 238:3555-3564.J. Biol. Chem.
http://www.jbc.org/content/238/11/3555.citation
Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/238/11/3555.citation.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on May 29, 2019
http://ww
w.jbc.org/
Dow
nloaded from





























