Embed Size (px)
Citation preview

Protein Structure Prediction With Evolutionary Algorithms
Natalio Krasnogor, U of the West of England
William Hart, Sandia National Laboratories
Jim Smith, U of the West of England
David Pelta, Universidad de Granada
Presenter: Elena Zheleva

Introduction
Problem Description Biology Background
– Protein Folding– HP Protein Folding Model
Genetic Algorithm (GA) Design Factors– Encodings for Internal Coordinates– Potential Energy Formulation– Constraint Management
Methods and Results Conclusion

Problem Description
Computational Biology open problem: protein structure prediction
Genetic algorithms have been used in the research literature
Authors analyze 3 algorithm parameters that impact performance and behavior of GAs
Goal: make suggestions for future algorithm design

Outline
Problem Description Biology Background
– Protein Folding– HP Protein Folding Model
GA Design Factors– Encodings for Internal Coordinates– Potential Energy Formulation– Constraint Management
Methods and Results Conclusion

Protein Folding
Proteins: driving force behind all of the biochemical reactions which make biology work
Protein is an amino acid chain! Amino acid chain -> Structure of a protein Structure of a protein -> Function of a protein
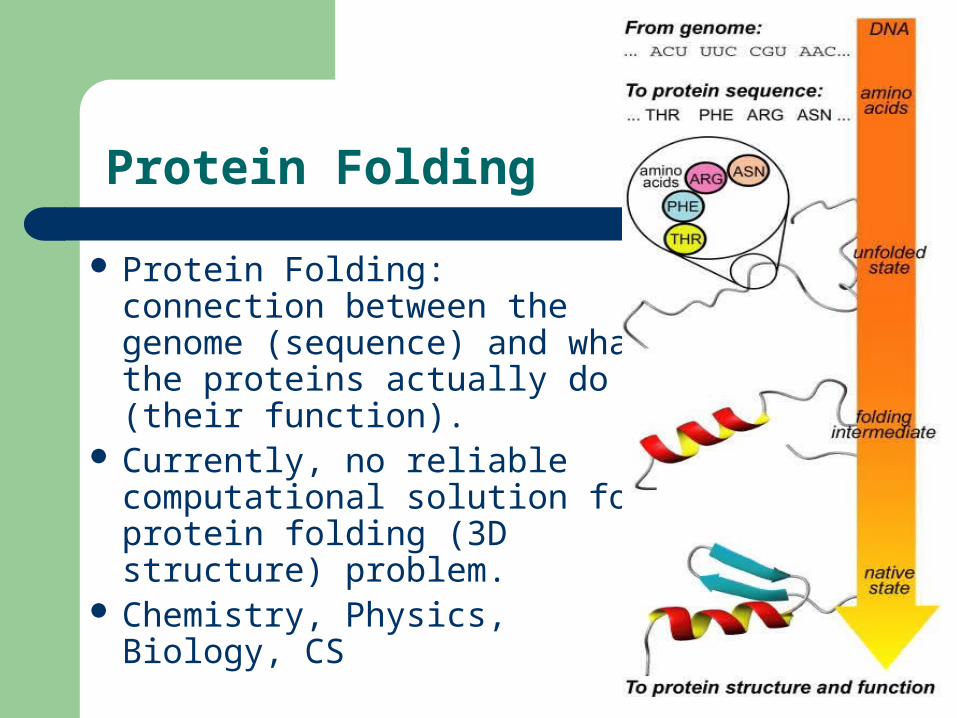
Protein Folding
Protein Folding: connection between the genome (sequence) and what the proteins actually do (their function).
Currently, no reliable computational solution for protein folding (3D structure) problem.
Chemistry, Physics, Biology, CS

Outline
Problem Description Biology Background
– Protein Folding– HP Protein Folding Model
GA Design Factors– Encodings for Internal Coordinates– Potential Energy Formulation– Constraint Management
Methods and Results Conclusion

HP Protein Folding Model
Amino acid chains (proteins) are represented as connected beads on a 2D or 3D lattice
HP: hydrophobic – hydrophilic property
Hydrophobic amino acids can form a hydrophobic core w/ energy potential

HP Protein Folding Model
Model adds energy value e to each pair of hydrophobics that are adjacent on lattice AND not consecutive in the sequence
Goal of GA: find low energy configurations!

Outline
Problem Description Biology Background
– Protein Folding– HP Protein Folding Model
GA Design Factors– Encodings for Internal Coordinates– Potential Energy Formulation– Constraint Management
Methods and Results Conclusion

Encodings for Internal Coordinates
Proteins are represented using internal coordinates (vs. Cartesian)
Absolute vs. Relative encoding Absolute Encoding: specifies an absolute
direction cubic lattice: {U,D,L,R,F,B} Relative Encoding: specifies direction relative
to the previous amino acid cubic lattice: {U,D,L,R,F}
n-1
n-1

Encodings for Internal Coordinates
Encoding impacts global search behavior of GA Example: One-point Mutations Relative Encoding:
FLLFRRLRLLR->
FLLFRFLRLLR Absolute Encoding:
RULLURURULU->
RULLUULULDL

Outline
Problem Description Biology Background
– Protein Folding– HP Protein Folding Model
GA Design Factors– Encodings for Internal Coordinates– Potential Energy Formulation– Constraint Management
Methods and Results Conclusion
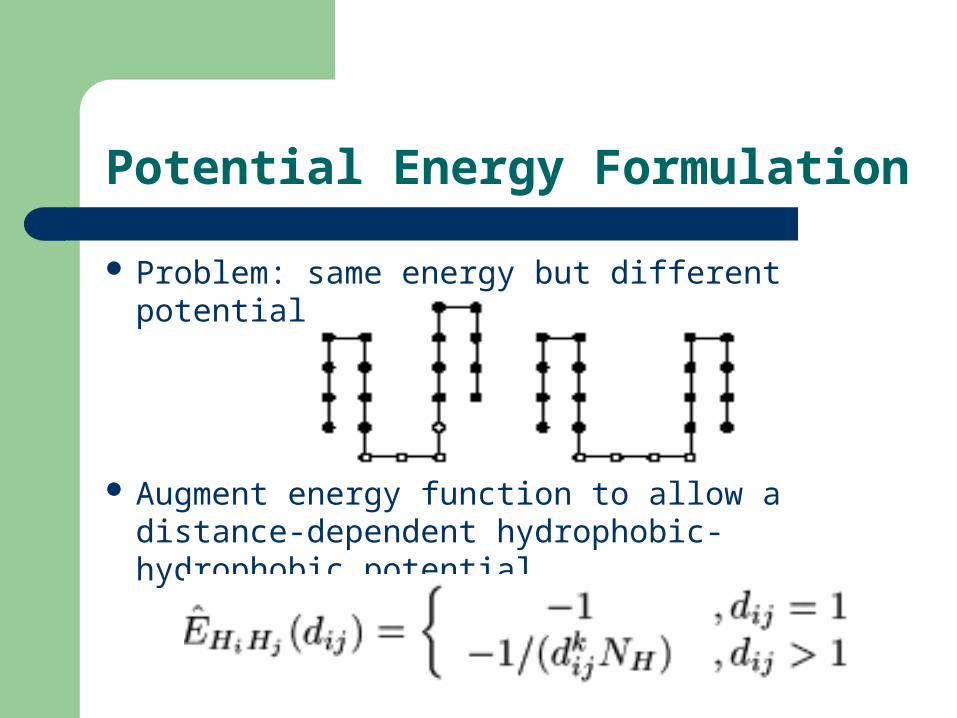
Potential Energy Formulation
Problem: same energy but different potential
(Picture )
Augment energy function to allow a distance-dependent hydrophobic-hydrophobic potential
(Formula)

Outline
Problem Description Biology Background
– Protein Folding– HP Protein Folding Model
GA Design Factors– Encodings for Internal Coordinates– Potential Energy Formulation– Constraint Management
Methods and Results Conclusion

Constraint Management
Methods for penalizing infeasible conformations Method 1: Consider only feasible conformations
– Weakness: shortest path from one feasible conformation to another may be very long
Method 2: Fixed Penalty Approach– Violations:
2 amino acids lying on the same lattice point Lattice point at which there are 2 or more amino acids
– Penalty per violation = 2*number of hydrophobics + 2
(any infeasible conformation has positive energy)

Outline
Problem Description Biology Background
– Protein Folding– HP Protein Folding Model
GA Design Factors– Encodings for Internal Coordinates– Potential Energy Formulation– Constraint Management
Methods and Results Conclusion

Methods and Results
1-point and 2-point Mutation operators 1-point, 2-point and Uniform Crossover
operators 5 polymer sequences (< 50 amino acids) Each run of GA: 200 generations
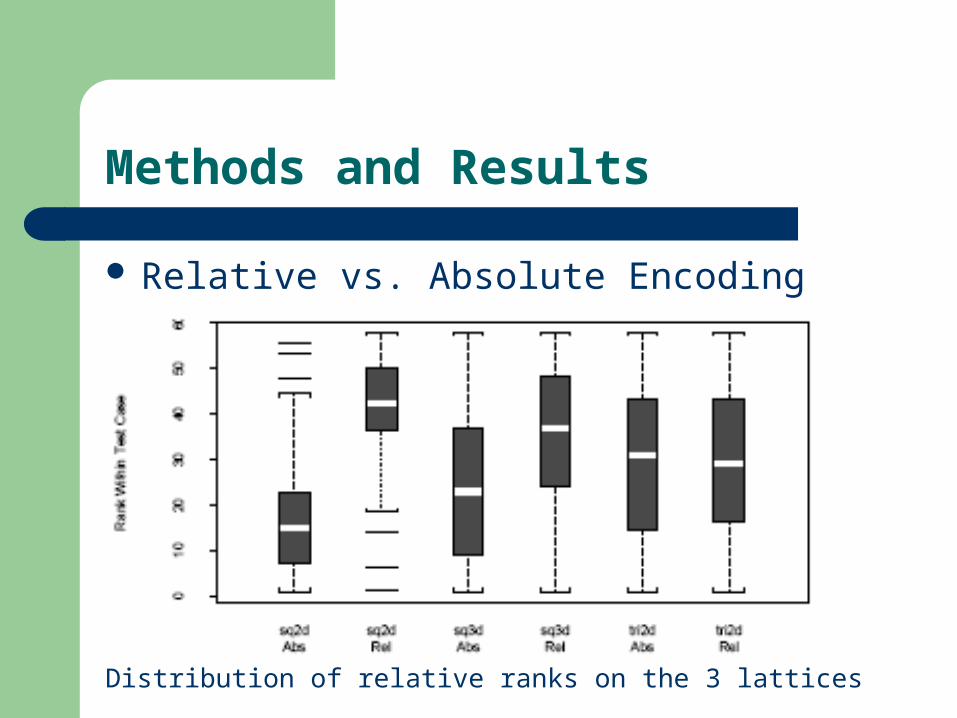
Methods and Results
Relative vs. Absolute Encoding
(Diagram )
Distribution of relative ranks on the 3 lattices

Methods and Results
Standard vs. Distant Energy Does the modified energy potential improve the
search capabilities of the GA? No significant difference on test sequences A guess: there might be on longer sequences

Conclusion
GAs applied to Protein Structure Prediction problem have 3 important factors to consider
Relative encoding is at least as good as absolute encoding, in some cases much better
Modified energy potential does not improve search capabilities of GA
The proposed constraint/penalty method ensures feasibility of the optimal solution
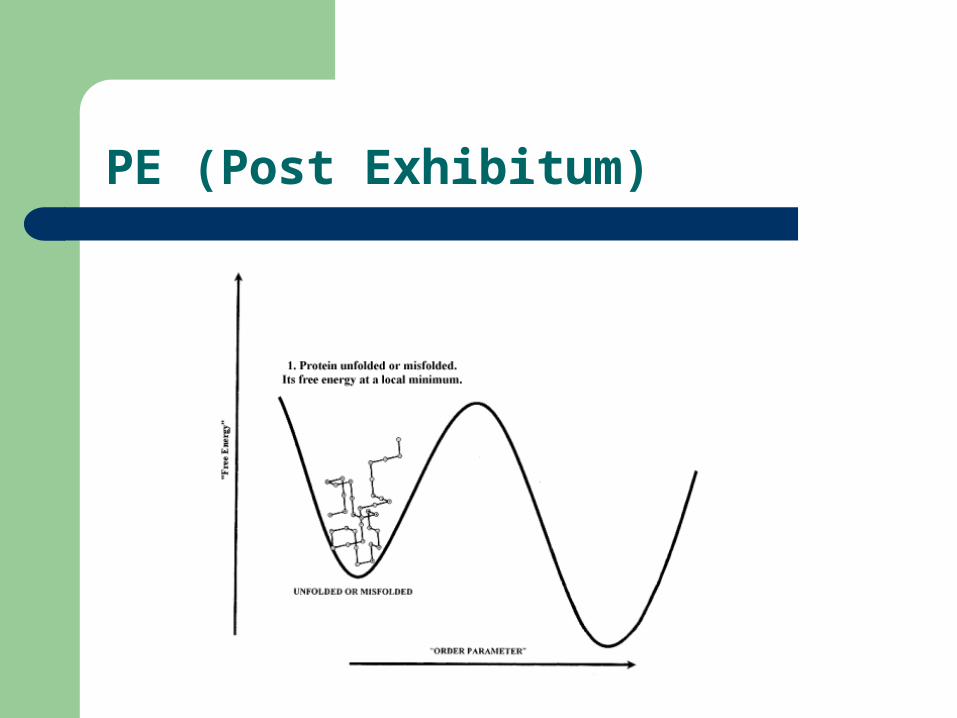
PE (Post Exhibitum)

PE
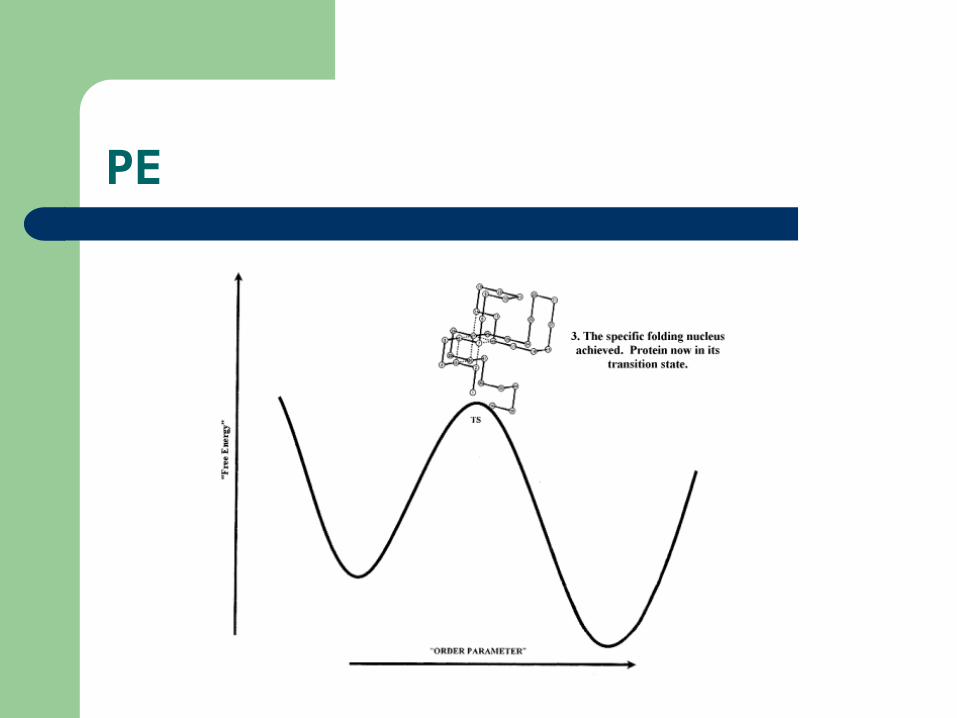
PE
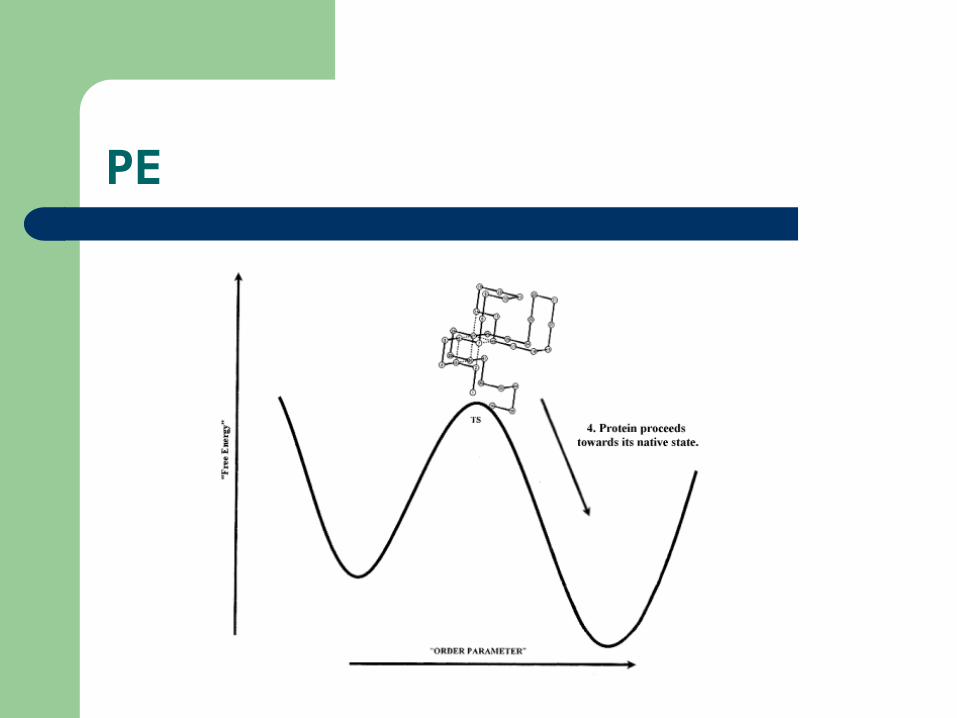
PE

PE