Embed Size (px)
Citation preview

PROTEÍNAS
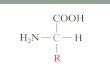
As proteínas são polímeros de aminoácidos ligados covalentemente por pontes peptídicas estabelecidas entre os grupos amino e carboxilo dos aminoácidos
Existindo no corpo humano aos milhares, umas são elementos estruturantes de células e tecidos e outras são solúveis e encontram-se nos espaços intra e extra-celulares
São na sua maioria de síntese hepática de onde partem para a circulação (proteínas séricas) e onde acabam sendo catabolizadas quando o seu teor em ácido siálico diminui ou desaparece de todo
Laboratorialmente podem estudar-se as proteínas do sangue, da urina, do LCR, dos líquidos pleural, perito-neal e amniótico e também as da saliva e fezes

PROTEÍNAS
As proteínas séricas desempenham diversas funções biológicas como:
• Proteínas de fase aguda, respondendo à inflamação ou à destruição celular
• Factores de coagulação que controlam a hemostase• Anticorpos e factores do complemento• Enzimas catalizadoras de reacções• Hormonas reguladoras do metabolismo• Precursoras de diversas substâncias• Proteínas de transporte de diferentes componentes• Elementos fundamentais à manutenção da pressão
oncótica plasmática

PROTEÍNAS
• Proteínas de transporte
– Haptoglobina hemoglobina
– Hemopexina heme
– Transferrina ferro
– Ceruloplasmina cobre
– Proteína ligante do retinol vitamina A
– Transtiretina T3 e T4

PROTEÍNAS
• Proteínas de fase aguda• Surgem como resposta às citocinas libertadas no local
da lesão• As que respondem aumentando (de fase aguda+):
– Proteína C reactiva (PCR)– α1- antitripsina (AAT)– α1- glicoproteína ácida (AAG)– Haptoglobina (HAP)– Ceruloplasmina (Cp)– C3 e C4
• As que respondem diminuindo (de fase aguda-): – Pré-albumina– Albumina– Transferrina (TRF ou siderofilina)
Utilizadas na monitorização da evolução da situação clínica
24h
6-8h
48h

PROTEÍNAS
• Intervenientes em infecção aguda:
• Hipotálamo – febre
• Hipófise – ACTH / Cortisol
• Fígado – síntese de proteínas de infecção
• MO – GBs
• Sistema imunitário – LinfócitosComplementoIgGs

PROTEÍNAS
Intervalos de referência, em adultos dos 20 aos 60 anos, para proteínas séricas:
Pré-albumina 20 a 40 mg/dLAlbumina 3,5 a 5,2 g/dLAAT 50 a 120 mg/dLAAG 25 a 135Cp 20 a 60Hp 30 a 200AMG 130 a 300TRF 200 a 360C3 99 a 180C4 10 a 40IgA 70 a 400IgM 40 a 230IgG 700 a 1600PCR < 0,5 (US <> 0.09 mg/dL)
Relação Albumina / Globulinas = 1,5 a 2,7

PROTEÍNAS
Técnicas electroforéticas
Compreendem:• E. zona
– E. em acetato de celulose– E. em gel (agarose, amido, poliacrilamida)
• E. capilar• Isofocalização • Blotting
– Southern (ADN e fragmentos ADN)– Northern (RNA e fragmentos RNA)– Western (proteínas)
• Imunofixação • E. bidimensional

C
Imunofixação de componentes monoclonais

PROTEÍNAS
Electroforese de alta resoluçãoCorado por Azul Brilhante de Coomassie

PROTEÍNAS
Proteinograma sérico
Acetato de celulose e coloração com Ponceau S

PROTEÍNAS
Electroforese de zonaDesigna-se assim porque a técnica produz zonas de
proteínas que ficam fisicamente separadas entre si
Em meio alcalino, as proteínas carregadas electricamente e sob acção de um campo eléctrico vão-se deslocar num suporte poroso em direcção ao ânodo (+)
A migração depende:– Carga eléctrica das moléculas– Tamanho e forma das moléculas– Intensidade do campo eléctrico– Propriedades do meio de suporte– Temperatura da operação– Tempo de migração– Electroendosmose

PROTEÍNAS
Perfil normal

PROTEÍNAS
Separação electroforética das Proteínas• Zona de migração da albumina:
– Proteína fixadora do retinol (RBP)– Pré-albumina – Albumina
(α1- lipoproteína/HDL)
• Zona de migração da alfa-1 globulina :– AAT– AAG (Orosomucóide)– α1- Fetoproteína (AFP)
• Zona de migração da alfa-2 globulina :– HAP– α2- Macroglobulina (AMG)– Cp
(Plasminogénio, pré-βlipoproteína)

PROTEÍNAS
• Zona de migração da beta-1 globulina :– TRF– Hemopexina – C4
• Zona de migração da beta-2 globulina :– C3– β2- Microglobulina (BMG)
(Fibinogénio; β-lipoproteínas)
• Zona de migração da gama globulina :– IgA– IgM– IgG– PCR

PROTEÍNAS

PROTEÍNAS
PCR (Proteína C Reactiva)
Sintetizada no fígado e regularmente estável em circulação, é a primeira proteína de inflamação a manifestar-se e fá-lo com aumentos drásticos
Clinicamente útil em:
Despiste de uma doença orgânica (em EAM, a subida faz-se em 6h e atinge valores 2.000 vezes o normal)
Perceber da actividade de uma doença inflamatória (placas de ateroma)
Detectar uma infecção intercorrente em LED, leucémia e após cirurgia (colocação de stent)
Avaliar da possibilidade de uma septicémia neonatal ou meningi-te, quando as amostras para exame bacteriológico são difíceis de obter
De uma forma ainda mal esclarecida, a PCR parece ser, só por si, um factor de risco para a doença cardiovascular

PROTEÍNAS
AAT (α1- antitripsina)
Inactiva serino proteases (tripsina) no que é semelhante a:
α1- antiquimotripsina α2-plasminaATIII C1 inibidor
Uma deficiência congénita associa-se a um alto risco de desenvolver enfisema e doença hepática, como a colestase neonatal, cirrose e carcinoma
• Aumenta em:
– Inflamações– Por acção dos estrogéneos

PROTEÍNAS
Diminuição de alfa 1 antitripsina

PROTEÍNAS
Heterozigotia Alfa 1 antitripsina

PROTEÍNAS
Proteínas do Complemento
São sintetizadas no fígado e em pequenas quantidades em células como os monócitos
As que mais interessam ao clínico são:
• C1 e C1 inibidor• C3 e C3 proactivador • C4
C3 e C4 são proteínas de fase aguda com resposta fraca e tardia. Também aumentam na obstrução biliar
Deficiências e variantes genéticas destes componentes podem acontecer e têm expressão clínica

PROTEÍNAS
Proteínas do ComplementoDeficiência genética de C2 e C4 associa-se a doenças
autoimunes (LED, polimiosite e glomerulonefrite)Deficiência em C3, ou seu inibidor, associa-se a infecções,
especialmente de bactérias capsuladas Deficiência nos elementos de ataque à membrana (C5 a C9),
pode relacionar-se com infecção persistente e recorrente a Neisseria
Deficiência em C1 inibidor provoca angioedema hereditário, com actividade continuada de C1, C2 e C4, que se cara-cteriza por ataques recorrentes de edema subcutâneo, laríngeo, brônquico e GI que pode pôr o doente em risco de vida
Para destrinçar entre situações genéticas e adquiridas é necessário estudos familiares, fenotipagem e estudos de ADN

PROTEÍNAS
AAG (α1-glicoproteína ácida / orosomucóide)
Função pouco conhecidaDiminui a biodisponibilidade de alguns fármacos• Aumenta em:
– Inflamações, especialmente GI– Neoplasias– Por acção de corticosteróides e alguns anti-inflama-
tórios não esteróides
• Diminui em:– Sindroma nefrótico – Por acção de estrogénios (gravidez, contracepção oral)

PROTEÍNAS
Hp (Haptoglobina)
A Hb livre que for libertada na circulação liga-se de forma irreversível à Hp. Este complexo é fagocitado pelo sistema mononuclear fagocítico e retirado da circulação. Isto promove a ↓Hp em α2
• Diminui em:– Ausência genética (a-haptoglobulinémia)– Hemólise intravascular– Corticosteróides e estrogéneos– Doença hepatocelular (hepatite viral, cirrose)
• Aumenta em:
– Infecção / Inflamação (tardia como a Cp)– Sindroma nefrótico– Presença de alguns fenótipos– Obstrução biliar

PROTEÍNAS
Hemólise intravascular

PROTEÍNAS
Hemólise intravascular
A hemopexina transportadora do heme também se encarrega de transportar a Hb livre até às células de Kupffer onde a liberta, deixando o Ferro para ser incorporado na ferritina e o heme para seguir o metabolismo das bilirrubinas
Por causa da hemólise intravascular a hemopexina, que sempre volta à circulação, não altera o padrão electro-forético

PROTEÍNAS
Cp (Ceruloplasmina)
É uma α2-globulina que contém cerca de 95% do cobre (S)
Cp no soro, envolve-se principalmente em reacções redoxe neste contexto oxida Fe2+ em Fe3+, o que é necessá-rio à incorporação do ferro na transferrina
A ligação do cobre é feita por uma ATPase intracelular que não existe no doença de Wilson
A clínica da doença de Wilson ou degenerescência hepato-lenticular apresenta depósitos de cobre em diversos tecidos como o hepático, cerebral e periferia da íris onde se forma o característico o anel de Kayser-Fleischer
A sintomatologia surge em média, por volta da 2ª ou 3ª década de vida

PROTEÍNAS
Cp (Ceruloplasmina)
• Aumentada em:
Resposta a inflamações, mas com um atraso de uma semana em relação a outras proteínas de fase aguda
Influência de estrogéneos

PROTEÍNAS
Cp (Ceruloplasmina)
• Diminuída em:
- Deficiência primária
Apresenta um quadro clínico de hemocromatose com aumento de ferro em depósito
O cobre é normal nos tecidos
- Deficiência secundária a:
Insuficiência na ingestão Impossibilidade de libertar Cu2+ das células do epitélio GIImpossibilidade de inserir Cu2+ na Cp (D. de Wilson)Perdas sanguíneas, sindroma nefrótico e GI

PROTEÍNAS
Pré-albumina (Transtiretina / TTR)
Circula em complexo de 1:1 com RBP
Transporta T3 e T4 (10%)
Tem muitas variantes genéticas e algumas associam-se ao depósito tissular de substância amilóide
• Aumenta em:– Tumores – Doença de Hodgkin– Glucocorticóides
• Diminui em:– Doença hepática– Má nutrição proteica

PROTEÍNAS
Albumina
É a mais pequena e a mais abundante proteína
60% encontra-se no espaço extravascular
Os seus aumentos séricos decorrem da diminuição do volume plasmático
A sua síntese hepática aumenta de acordo com a pressão oncótica do plasma e a ingestão proteica e diminui por acção das citocinas inflamatórias
Há mais de 80 variantes genéticas que se transmitem de forma autossómica dominante
Nos heterozigóticos, muitos isotipos podem alterar o padrão electroforético exibindo bisalbuminémia

PROTEÍNAS
Bisalbuminémia

PROTEÍNAS
Variações na concentração da albuminaDiminuições:• Inflamações agudas ou crónicas
– Passagem para o espaço extravascular– Aumento de consumo celular– Diminuição da síntese
• Doença hepática (> 95% de perda da função)– Aumento das imunoglobulinas– Perda para o espaço extravascular– Inibição de síntese por acção directa de álcool e toxinas
• Perda renal• Perda gastrointestinal• Estados de má nutrição Presença de edemas e ascite (secundários a aumento da
permeabilidade vascular)

PROTEÍNAS
Analbuminémia
Corresponde a uma rara doença genética em que o teor de albumina é < 0,5 g/L
Promove um anormal transporte de lípidos (fosfolípidos e ácidos gordos livres)
Outros componentes que transporta:
– Iões metálicos– Aminoácidos– Fármacos– Hormonas– Bilirrubina

PROTEÍNAS
Analbuminémia

PROTEÍNAS
TRF (Transferrina)
É a principal proteína transportadora de ferro e de outros catiões divalentes
Cada molécula fixa dois Fe3+, se a Cp estiver presente para actuar como ferroxidase, e leva-os para serem incorporados nos citocromos, Hb, mioglobina e consti-tuir depósitos
Algumas variantes genéticas podem alterar a mobilidade electroforética e simular a presença de uma para-proteinémia

PROTEÍNAS
TRF (Transferrina)
• Aumentada em:
– Estrogéneos – Anemia ferropénica
• Diminuída em:
– A-transferrinémia– Inflamação – Neoplasia– Sindroma nefrótico e perdas GI

PROTEÍNAS
Hipertransferrinémia em anemia

PROTEÍNAS
Padrão inflamatórioAgudo:
- O aumento da fracção α1 depende mais da AAT uma vez que o elevado teor do orosomucóide em HC (40%) prejudica a sua coloraçãoNas vasculites o aumento de produção da AAT pode não se perceber, dado o seu grande consumo intravascular como antiprotease
- O aumento da fracção α2 depende mais da Hp do que da Cp cujo aumento é discreto
- Aumentos muito elevados de PCR podem simular uma gamapatia monoclonal

PROTEÍNAS
Inflamação aguda

PROTEÍNAS
Inflamação crónica

PROTEÍNAS
AFP (α -Fetoproteína)
É a primeira e a principal α-globulina sérica do período embrionário, sintetizada pelo saco univitelino e fígado
Numa electroforese de soro fetal ou de RN, coloca-se entre a albumina e a AAT
• Aumentos:
No soro materno e líquido amniótico apontam para a possibilidade de uma abertura do tubo neural ou defeitos na parede abdominal do feto
Em carcinomas hepatocelulares e de células germinativas na criança
• Diminuições:
No soro materno de gravidez com trisomíase 18 ou 21

PROTEÍNAS
AMG (α2-Macroglobulina)
É a maior inibidora de proteases e ao contrário das outras não se difunde do plasma para o espaço extravascular de forma significativa
• Aumenta em:– Crianças (2 a 3 vezes o adulto), como elemento de
protecção contra a exposição a proteases bacterianas e leucocitárias durante a infância)
– Sindroma nefrótico– Por acção dos estrogéneos
• Diminui em:– Pancreatite aguda severa

PROTEÍNAS
BMG (β2-Microglobulina)
É uma pequena proteína que se encontra à superfície de todas as células nucleadas
Constitui a cadeia leve (ou β) dos Ags leucocitários humanos (HLA)
• Aumenta em:– Insuficiência renal– Inflamações– Neoplasias, especialmente as que envolvam os
linfócitos B
Útil para testar a função tubular renal, só por si ou após exposição a metais pesados ou transplante
Usada na monitorização de tumores a células B

PROTEÍNAS
Imunoglobulinas
O aumento de imunoglobulinas pode ser:
Policlonal - mistura de diversos Acs
Monoclonal - classe, subclasse ou idiotipo produzido por um só clone de plasmócitos ou linfócitos B
Oligoclonal - algumas proteínas “monoclonais” de diferentes especificidades

PROTEÍNAS
Imunoglobulinas• Aumento policlonal em:
– Infecções crónicas ou recorrentes (parasitárias)– Doenças autoimunes (AR, LED)– Doença hepatocelular
Aumentos de IgA relacionam-se mais com infecções da pele, rim, intestino e brônquio
A IgM aumenta nas infecções primárias a vírus e intra-vasculares, tipo malária
A IgG tende a predominar na resposta autoimune
• Aumento oligoclonal em:Infecções, doenças autoimunes e transplante de Mo
podem fazer proliferar vários clones em simultâneo

PROTEÍNAS
Perfil biclonal

PROTEÍNAS
Perfil oligoclonal

PROTEÍNAS
Hipergamaglobulinémia

PROTEÍNAS
Imunoglobulinas
• Aumento monoclonal em:
60% dos casos - Mieloma múltiplo (proliferação difusa na MO / Paraproteína)
Plasmocitoma (tumor sólido)
15% dos casos - Hiper-produção de linfócitos B:LLC (IgM)Linfomas (IgM)Macroglobulinémia de Waldenström (IgM)Doença de cadeias pesadas (cadeias α)Crioglobulinémia
Os aumentos de IgM causam aumento de viscosidade sanguínea

PROTEÍNAS
Banda monoclonal com diminuição de gama

PROTEÍNAS
Banda monoclonal na região gama com gama policlonal

PROTEÍNAS
Banda monoclonal na região beta

PROTEÍNAS
ImunodeficiênciaCaracteriza-se pela recorrência de infecções que podem
resultar da falência de um dos seguintes sistemas:
– Imunidade humoral (Acs)– Imunidade celular (linfócitos T)– Sistema mononuclear fagocítico– Sistema do Complemento
A imunodeficiência, que é perceptível, por redução ou ausência de IgG na electroforese pode ser:
– Fisiológica (entre os 2 e 5 meses de idade)– Congénita– Secundária a perda de proteína– Falha transitória na síntese da proteína

PROTEÍNAS
Hipogamaglobulinémia

PROTEÍNAS
Síndrome nefrótico
Quadro clínico que cursa com o aumento da permeabili-dade glomerular para proteínas de elevado PM como as IgG e IgA (160 e 180 KDa)
Todas as pequenas proteínas, como a Albumina são expoliadas e no caso desta há um esforço de compen-sação por aumento da sua síntese hepática com vista à manutenção da pressão oncótica do plasma
Em simultâneo há a síntese de outras proteínas como a α2-macroglobulina (PM=800 KDa) e apolipoproteína B (PM=3.000 KDa) que se vão acumulando em circulação

PROTEÍNAS
Síndrome nefrótico

PROTEÍNAS
Padrão de cirrose hepática (Ponte beta-gama)Na cirrose hepática há destruição do parênquima hepático
(↓ síntese proteica) e exclusão do fígado (função de filtro) da circulação portal o que vai impedir a degrada-ção da IgA intestinal e de outro material antigénico da mesma procedência
Disto resulta uma baixa de Albumina e uma subida das Igs com o desenvolvimento da ponte beta-gama
É um padrão que se observa em estadios muito avançados de deterioração hepática
Melhor e mais precoce indicação dão as provas de coagula-ção sobre a síntese dos factores
Padrão idêntico em infecções crónicas respiratórias e GI

PROTEÍNAS
Ponte beta-gama

PROTEÍNAS
Padrão de gravidez
Caracteriza-se por elevados níveis plasmáticos de estrogénios e uma tendência para a baixa de ferro
Electroforeticamente apresenta um aumento de α2 e β1 (α2-macroglobulina e transferrina)
Em relação à primeira o aumento também se observa no aporte exógeno de estrogénios como acontece na terapêutica anticontraceptiva

PROTEÍNAS pt
Presença de fibrinogénio