Embed Size (px)
Citation preview

CLINICAL STUDY PROTOCOL
Study Title.
WORKING INSTRUCTIONSInstructions given in hidden text.
To show hidden text: Office button > Word options > Display > check hidden text.Delete on finalisation.
Short Title:
Version: 1
Date: 06-Sep-2018
CHDR number: CHDR9999
Sponsor number: Z12Y34-32X6
Ethics Committee number: PYY.123
Toetsing Online number: NL3423456634-54
EudraCT number: YYYY-123456-78
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 1 of 48

CHDR9999
SUMMARY OF CHANGES
Study TitleStudy Title.
The following revisions were made to the protocol, which are also reflected in the synopsis, and other documents:
PROTOCOL VERSION 2, AMENDMENT 1, 10-DEC-2012Change Rationale Justification &
ClassificationChanged
Document(s), Section
Upper limit of inclusion criterion age was changed from 35 years to 85 years.
No rationale for previous limit.
Change of age range and inclusion criterion: substantial
- Protocol, Section 4.1.2
- SIS & ICF, Introduction
Monitor details New study monitor Change of name: non-substantial
- Protocol, Contact Details
The font was changed from Times New Roman to Arial.
Consistency with house style.
Stylistic change: non-substantial
- Protocol, Whole Document
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 2 of 48
CHDR9999
CONTACT DETAILS
SPONSOR
(EU legal representative)Full Sponsor NameStreetPostcode, CityCountry
Clinical Trial Leader Name / Qualification(s)Telephone: + inserte-mail: [email protected]
Head of Clinical Pharmacology Name / Qualification(s)Telephone: + inserte-mail: [email protected]
Clinical Pharmacologist Name / Qualification(s)Telephone: + inserte-mail: [email protected]
Monitor Name / Qualification(s)Telephone: + inserte-mail: [email protected]
Medical Expert or Drug Safety Physician
Name / Qualification(s)Telephone: + inserte-mail: [email protected]
Trial Site(visit & delivery address)
Centre for Human Drug ResearchZernikedreef 82333 CL LeidenThe NetherlandsTelephone: + 31 71 5246 400Fax: + 31 71 5246 499Emergency: + 31 71 5246 444
Principal investigator J. (Koos) Burggraaf, MD, PhDTelephone: + inserte-mail: [email protected]
Co-investigator Name / Qualification(s)Telephone: + inserte-mail: [email protected]
Co-investigator Name / Qualification(s)Telephone: + inserte-mail: [email protected]
Manager Operations Unit J. M. (Ria) Kroon, BScTelephone: + 31 71 5246 498e-mail: [email protected]
Manager Clinical Unit C. E. (Emilie) Jonxis, MANPTelephone +31 71 5246433e-mail: [email protected]
Statistician M. L. (Marieke) de Kam, MScTelephone: + 31 71 5246 458e-mail: [email protected]
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 3 of 48

CHDR9999
INDEPENDENT PHYSICIAN Prof. Dr G.J. Blauw, MD, PhDDepartment of Gerontology and Geriatrics LUMCPostbus 96002300 RC LeidenTelephone: + 31 71 5266 640
MONITOR(visit & delivery address)
Full Company NameStreetPostcode, CityCountry
Contact person Name / Qualification(s)Telephone: + inserte-mail: [email protected]
LABORATORY – CHEMISTRY AND HAEMATOLOGY
C.M. Cobbaert, PhDAKCL LUMC, E2-PAlbinusdreef 22333 ZA LeidenThe Netherlands
Contact person R.N. Malhoe Mishre-Lalai
LABORATORY – MICROBIOLOGY A.C.M. Kroes, MD, PhDKML LUMC, E4-PAlbinusdreef 22333 ZA LeidenThe Netherlands
Contact person A.C.M. Kroes
LABORATORY – PK BIOANALYTICAL
Full Laboratory Name
Street
Postcode, City
Country
Contact person Name / Qualification(s)
Telephone: + insert
e-mail: [email protected]
PHARMACY(visit & delivery address)
Apotheek LUMC, L0-P30Goederenontvangst apotheek attn. trialsEinthovenweg 62333 ZC LeidenThe NetherlandsTelephone: + 31 71 5269111Fax + 31 71 5262611
Pharmacist / Trial Manager Marieke Tio, PharmD / Linda van der Hulst
CHDR template v2018.9
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 4 of 48

CHDR9999
SIGNATURE PAGE - PRINCIPAL INVESTIGATOR
Study TitleStudy Title.
I acknowledge accountability for this protocol in accordance with CHDR’s current procedures.
J. (Koos) Burggraaf, MD, PhDPrincipal investigator
Signature Date (dd Mmm yyyy)
[For CHDR sponsored studies, add the following text]This is an investigator-initiated trial, CHDR acts as both the investigator as well as the sponsor of the study, with all applicable responsibilities.
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 5 of 48

CHDR9999
SIGNATURE PAGE - TRIAL SITE STAFFCentre for Human Drug Research
Study TitleStudy Title.
I acknowledge responsibility for this protocol in accordance with CHDR’s current procedures.Name / Qualification(s)Co-investigator
Signature Date (dd Mmm yyyy)
Name / Qualification(s)Co-investigator
Signature Date (dd Mmm yyyy)
J. M. (Ria) Kroon, BScManager Operations Unit
Signature Date (dd Mmm yyyy)
C.E. (Emilie) Jonxis, MANPManager Clinical Unit
Signature Date (dd Mmm yyyy)
M. L. (Marieke) de Kam, MScStatistician
Signature Date (dd Mmm yyyy)
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 6 of 48

CHDR9999
SIGNATURE PAGE - SPONSORFull Sponsor Name
Study TitleStudy Title.
I approve this protocol on behalf of the sponsor.
Name / Qualification(s)Sponsor’s representative / Title
Signature Date (dd Mmm yyyy)
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 7 of 48

CHDR9999
TABLE OF CONTENTSSUMMARY OF CHANGES................................................................................................................................ 2
CONTACT DETAILS.......................................................................................................................................... 3
SIGNATURE PAGE - PRINCIPAL INVESTIGATOR.........................................................................................5
SIGNATURE PAGE - TRIAL SITE STAFF........................................................................................................6
SIGNATURE PAGE - SPONSOR...................................................................................................................... 7
TABLE OF CONTENTS..................................................................................................................................... 7
LIST OF ABBREVIATIONS............................................................................................................................... 7
PROTOCOL SYNOPSIS.................................................................................................................................... 7
1 BACKGROUND AND RATIONALE...........................................................................................................7
1.1 Context............................................................................................................................................... 7
1.2 Non-clinical information.......................................................................................................................7
1.2.1 Non-clinical pharmacology..........................................................................................................7
1.2.2 Non-clinical pharmacokinetics and metabolism..........................................................................7
1.2.3 Non-clinical toxicology and safety pharmacology........................................................................7
1.3 Clinical information.............................................................................................................................. 7
1.3.1 Clinical pharmacology................................................................................................................. 7
1.3.2 Clinical pharmacokinetics and metabolism.................................................................................7
1.3.3 Clinical toxicology and safety pharmacology...............................................................................7
1.4 Study rationale.................................................................................................................................... 7
1.4.1 Benefit and risk assessment.......................................................................................................7
1.4.2 Medical and regulatory background............................................................................................7
1.4.3 Study population.........................................................................................................................7
1.4.4 Study design............................................................................................................................... 7
1.4.5 Comparative drug(s) and/or placebo...........................................................................................7
1.4.6 Safety margin calculations, dose selection, dose escalation, and stopping criteria....................7
1.4.7 Treatment duration......................................................................................................................7
1.4.8 Primary endpoint......................................................................................................................... 7
1.4.9 Statistical hypotheses and sample size.......................................................................................7
2 STUDY OBJECTIVES................................................................................................................................ 7
2.1 Primary objective................................................................................................................................ 7
2.2 Secondary objectives..........................................................................................................................7
3 STUDY DESIGN......................................................................................................................................... 7
3.1 Overall study design and plan.............................................................................................................7
3.1.1 Screening.................................................................................................................................... 7
3.1.2 Treatment and observation period..............................................................................................7
3.1.3 Follow-up.................................................................................................................................... 7
4 STUDY POPULATION............................................................................................................................... 7
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 8 of 48

CHDR9999
4.1 Subject population.............................................................................................................................. 7
4.2 Inclusion criteria.................................................................................................................................. 7
4.3 Exclusion criteria................................................................................................................................. 7
4.4 Concomitant medications....................................................................................................................7
4.4.1 Allowed concomitant medications...............................................................................................7
4.4.2 Prohibited concomitant medications...........................................................................................7
4.4.3 Escape/rescue medications........................................................................................................7
4.5 Lifestyle restrictions............................................................................................................................ 7
4.5.1 Contraception requirements........................................................................................................7
4.6 Study drug discontinuation and withdrawal.........................................................................................7
4.6.1 Study drug interruption or discontinuation...................................................................................7
4.6.2 Subject withdrawal......................................................................................................................7
4.6.3 Replacement policy.....................................................................................................................7
5 INVESTIGATIONAL MEDICINAL PRODUCT............................................................................................7
5.1 Investigational drug and matching placebo.........................................................................................7
5.2 Comparative drug............................................................................................................................... 7
5.3 Study drug dosing scheme.................................................................................................................7
5.4 Study drug up- and down-titration.......................................................................................................7
5.5 Study drug packaging and labelling....................................................................................................7
5.6 Study drug preparation....................................................................................................................... 7
5.7 Drug accountability............................................................................................................................. 7
5.8 Treatment assignment and blinding....................................................................................................7
5.8.1 Treatment assignment................................................................................................................7
5.8.2 Blinding....................................................................................................................................... 7
6 STUDY ENDPOINTS.................................................................................................................................. 7
6.1 Efficacy endpoints............................................................................................................................... 7
6.2 Safety and tolerability endpoints.........................................................................................................7
6.3 Pharmacokinetic endpoints.................................................................................................................7
6.4 Pharmacodynamic endpoints..............................................................................................................7
7 STUDY ASSESSMENTS........................................................................................................................... 7
7.1 Efficacy assessments......................................................................................................................... 7
7.2 Safety and tolerability assessments....................................................................................................7
7.2.1 Specific safety and tolerability assessments...............................................................................7
7.2.2 Vital signs.................................................................................................................................... 7
7.2.3 Weight and height....................................................................................................................... 7
7.2.4 Physical examination.................................................................................................................. 7
7.2.5 Electrocardiography.................................................................................................................... 7
7.2.6 Laboratory assessments.............................................................................................................7
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 9 of 48

CHDR9999
7.3 Pharmacokinetic and pharmacodynamic assessments......................................................................7
7.3.1 Labelling...................................................................................................................................... 7
7.3.2 Shipping Procedures................................................................................................................... 7
7.3.3 Bioanalysis.................................................................................................................................. 7
7.4 Pharmacodynamic assessments and questionnaires.........................................................................7
7.4.1 Concomitant medications............................................................................................................7
7.5 Sequence of assessments and time windows....................................................................................7
7.6 Total blood volume.............................................................................................................................. 7
8 SAFETY REPORTING............................................................................................................................... 7
8.1 Definitions of adverse events..............................................................................................................7
8.1.1 Intensity of adverse events.........................................................................................................7
8.1.2 Relationship to study drug...........................................................................................................7
8.1.3 Chronicity of adverse events.......................................................................................................7
8.1.4 Action.......................................................................................................................................... 7
8.1.5 Serious adverse events...............................................................................................................7
8.1.6 Suspected unexpected serious adverse reactions......................................................................7
8.1.7 Reporting of serious adverse events...........................................................................................7
8.1.8 Follow-up of adverse events.......................................................................................................7
8.2 Temporary halt for reasons of subject safety......................................................................................7
8.3 Annual safety report or development safety update report.................................................................7
8.4 Pregnancy........................................................................................................................................... 7
8.4.1 Teratogenicity............................................................................................................................. 7
8.4.2 Reporting of pregnancy...............................................................................................................7
8.5 Data safety monitoring board..............................................................................................................7
9 STATISTICAL METHODOLOGY AND ANALYSES..................................................................................7
9.1 Statistical analysis plan.......................................................................................................................7
9.2 Protocol violations/deviations..............................................................................................................7
9.3 Power calculation................................................................................................................................ 7
9.4 Missing, unused and spurious data.....................................................................................................7
9.5 Analysis sets....................................................................................................................................... 7
9.5.1 Safety set.................................................................................................................................... 7
9.5.2 Pharmacokinetic analysis set......................................................................................................7
9.5.3 Pharmacodynamic analysis set...................................................................................................7
9.6 Subject disposition.............................................................................................................................. 7
9.7 Baseline parameters and concomitant medications............................................................................7
9.7.1 Demographics and baseline variables........................................................................................7
9.7.2 Medical history............................................................................................................................ 7
9.7.3 Concomitant Medications............................................................................................................7
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 10 of 48

CHDR9999
9.7.4 Treatment compliance/exposure.................................................................................................7
9.8 Safety and tolerability endpoints.........................................................................................................7
9.8.1 Adverse events........................................................................................................................... 7
9.8.2 Vital signs.................................................................................................................................... 7
9.8.3 ECG............................................................................................................................................ 7
9.8.4 Clinical laboratory tests...............................................................................................................7
9.9 Pharmacokinetic and pharmacodynamic endpoints............................................................................7
9.9.1 Pharmacokinetics........................................................................................................................ 7
9.9.2 Pharmacodynamics.................................................................................................................... 7
9.9.3 Inferential methods..................................................................................................................... 7
9.9.4 PK/PD modelling......................................................................................................................... 7
9.10 Exploratory analyses and deviations...................................................................................................7
9.11 Interim analyses.................................................................................................................................. 7
10 GOOD CLINICAL PRACTICE, ETHICS AND ADMINISTRATIVE PROCEDURES..............................7
10.1 Good clinical practice..........................................................................................................................7
10.1.1 Ethics and good clinical practice.................................................................................................7
10.1.2 Ethics Committee / institutional review board..............................................................................7
10.1.3 Informed consent........................................................................................................................7
10.1.4 Insurance.................................................................................................................................... 7
10.2 Study funding...................................................................................................................................... 7
10.3 Data handling and record keeping......................................................................................................7
10.3.1 Data collection............................................................................................................................ 7
10.3.2 Database management and quality control.................................................................................7
10.4 Access to source data and documents...............................................................................................7
10.5 Quality control and quality assurance.................................................................................................7
10.5.1 Monitoring................................................................................................................................... 7
10.6 Protocol amendments.........................................................................................................................7
10.6.1 Non-substantial amendment.......................................................................................................7
10.6.2 Substantial amendment......................................................Fout! Bladwijzer niet gedefinieerd.
10.7 End of study report.............................................................................................................................. 7
10.8 Public disclosure and publication policy..............................................................................................7
11 STRUCTURED RISK ANALYSIS..........................................................................................................7
11.1 Potential issues of concern.................................................................................................................7
11.1.1 Level of knowledge about mechanism of action..........................................................................7
11.1.2 Previous exposure of human beings with the test product(s) and/or products with a similar biological mechanism................................................................................................................................. 7
11.1.3 Can the primary or secondary mechanism be induced in animals and/or in ex-vivo human cell material? 7
11.1.4 Selectivity of the mechanism to target tissue in animals and/or human beings...........................7
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 11 of 48

CHDR9999
11.1.5 Analysis of potential effect..........................................................................................................7
11.1.6 Pharmacokinetic considerations.................................................................................................7
11.1.7 Study population.........................................................................................................................7
11.1.8 Interaction with other products....................................................................................................7
11.1.9 Predictability of effect.................................................................................................................. 7
11.1.10 Can effects be managed?.......................................................................................................7
11.2 Synthesis............................................................................................................................................ 7
12 REFERENCES....................................................................................................................................... 7
APPENDIX......................................................................................................................................................... 7
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 12 of 48

CHDR9999
LIST OF ABBREVIATIONSABR ABR form, General Assessment and Registration form, is the application form that is
required for submission to the accredited Ethics Committee; in Dutch, ABR = Algemene Beoordeling en Registratie
AE Adverse Event
AeinfCumulative amount of administered dose excreted into the urine from time zero to infinity after single dose
Ae%infPercentage of administered dose excreted into the urine from time zero to infinity after single dose
AelastCumulative amount of administered dose excreted into the urine from time zero to time of last measurable concentration
Ae%lastPercentage of administered dose excreted into the urine from time zero to time of last measurable concentration
AetauCumulative amount of administered dose excreted into the urine within the dosing interval
Ae%tau Percentage of administered dose excreted into the urine within the dosing interval
ALT alanine aminotransferase/serum glutamic pyruvic transaminase (SGPT)ANCOVA Analysis of CovarianceANOVA Analysis of VarianceaPTT activated partial thromboplastin time AST aspartate aminotransferase/serum glutamic oxaloacetic transaminase (SGOT)ATC Anatomic Therapeutic ChemicalAUC Area under the concentration – time curveAUCinf Area under the concentration – time curve from time zero to infinity
AUClastArea under the concentration – time curve from time zero to time of last measurable concentration
AUCtau Area under the concentration – time curve between consecutive dosingb.i.d. bis in diem / twice a dayBLQ Below the Limit of QuantificationBMI Body Mass IndexBP Blood Pressurebpm beats per minuteCA Competent authority (also CCMO)CCMO Central Committee on Research Involving Human Subjects; in Dutch: Centrale
Commissie Mensgebonden OnderzoekCHDR Centre for Human Drug ResearchCI Confidence IntervalCK creatine kinaseCL/F Apparent total clearance following extravascular administrationCLR Renal clearance Cmax Maximum concentrationCtrough Concentration immediately prior to dosing at multiple dosingCRF Case Report FormCV Coefficient of variationDSMB Data Safety and Monitoring BoardEC Ethics Committee (also Medical Research Ethics Committee (MREC); in Dutch:
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 13 of 48

CHDR9999
Medisch Ethische Toetsing Commissie (METC).ECG ElectrocardiogramEDTA Ethylene diamine tetra-acetic acidELISA Enzyme-linked immunosorbant assayEMA European Medicines AgencyEU European UnionFDA Food and Drug AdministrationGCP Good Clinical PracticeICH International Conference on HarmonizationIB investigator’s BrochureIDMC Independent Data Monitoring CommitteeIMPD Investigational Medicinal Product DossierINR International Normalized Ratioi.v. Intravenous(ly)IRB Institutional Review BoardLDH Lactate dehydrogenaseMax MaximumMedDRA Medical Dictionary for Regulatory Activitiesmin MinutesMin Minimumo.d. omnia die/once a dayp.o. per os / orallySAE Serious Adverse EventSAP Statistical Analysis PlanSD Standard DeviationSEM Standard Error of the MeanSOC System Organ ClassSOP Standard Operating ProcedureSPC Summary of Product CharacteristicsSST Serum Separator TubeSUSAR Suspected Unexpected Serious Adverse Reactiont.i.d. ter in diem / three times a dayt½ Terminal Elimination Half-lifetlag Absorption lag timetmax Time to attain Cmax
ULN Upper Limit of NormalVz/F Apparent volume of distribution during the terminal elimination phase after
extravascular administrationWHO World Health OrganizationWBP Personal Data Protection Act; in Dutch: Wet Bescherming PersoonsgevensWMO Medical Research Involving Human Subjects Act; in Dutch: Wet Medisch-
wetenschappelijk Onderzoek met Mensen.
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 14 of 48

CHDR9999
PROTOCOL SYNOPSISTitleShort TitlePrincipal investigator & Trial SiteName / Qualification(s), Centre for Human Drug Research, Zernikedreef 8, 2333 CL Leiden, The Netherlands
Background & RationaleObjective(s)Primary ObjectiveSecondary ObjectivesDesignInvestigational drug
Comparative drug
Subjects / GroupsInclusion criteriaExclusion criteriaConcomitant medicationsTolerability / safety endpoints
Pharmacokinetic endpoints
Pharmacodynamic endpoints
Efficacy endpoints
Sample Size Justification
Statistical methodologyStudy committee(s)
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 15 of 48
CHDR9999
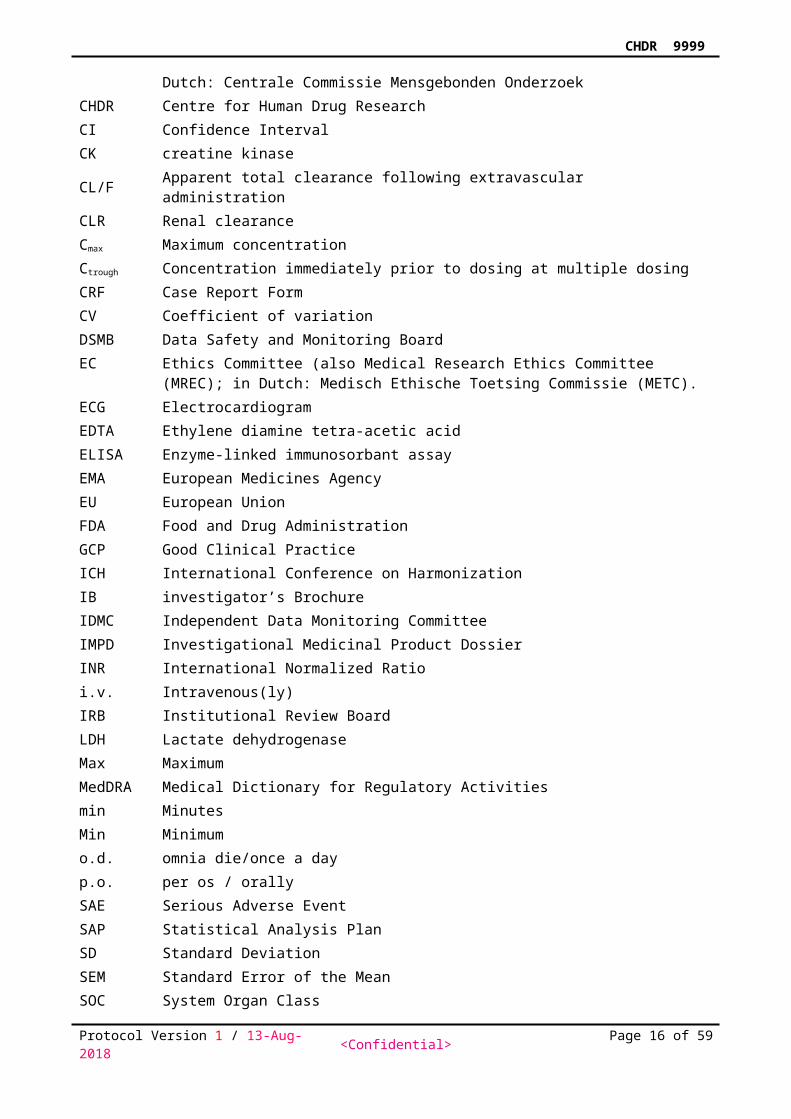
Table 1 Visit and Assessment ScheduleSCR Treatment days 1, 8, 22, 30, 44, 52, 66, 75 FU1
Time point Assessment
Up to-42 d
-2h
-1h
-30min
-15min
0h
1h
2h
3h
4h
5h
6h
8h
12h
+14 d± 2 d
Informed consent XDemography XInclusion and exclusion criteria XMedical history XPhysical examination X XConcomitant medication XVirology XBsHaem, BsChem2, Urinalysis X XUrDrug, BrAlc X XTemperature X X XECG X X X XGeneral symptoms3 X X X X X X X X X X XVital Signs (Pulse Rate, BP, RR)4
Meals / snack5 X XDrug (-placebo) administration X X XPK sample X X X X X X X X XPD tests X6 X7 X X XDischarge X X(S)AE/Con-meds <----- continuous ----->
BP = Blood Pressure, HR = Heart Rate, RR = Respiratory Rate, SCR = Screening, EEG = Electroencephalogram, PK = Pharmacokinetics, AE = Adverse Event, UrDrug = Urine Drug Screen, BsHaem =
Blood Sample Haematology, BsChem = Blood Sample Chemistry, BrAlc = Breath Alcohol Test.
1 Follow up (FU) visit after last treatment. 2 Samples are taken after at least …hours of fasting3 Including nausea scale on inpatient study days.4 Supine/Standing5 Fasting will be required from midnight the night before the inpatient study day. A light snack will be offered after the last nociceptive tests is performed. 6 PD tests training session. 7 PD tests will be done twice for baseline measurements.
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 16 of 48

CHDR9999
1 BACKGROUND AND RATIONALE
1.1 Context1.2 Non-clinical information
1.2.1 Non-clinical pharmacology
1.2.2 Non-clinical pharmacokinetics and metabolism
1.2.3 Non-clinical toxicology and safety pharmacology
1.3 Clinical information1.3.1 Clinical pharmacology
1.3.2 Clinical pharmacokinetics and metabolism
1.3.3 Clinical toxicology and safety pharmacology
1.4 Study rationale
1.4.1 Benefit and risk assessmentFor a structured risk assessment see Section 11.
1.4.2 Medical and regulatory background1.4.3 Study population1.4.4 Study design1.4.5 Comparative drug(s) and/or placebo1.4.6 Safety margin calculations, dose selection, dose escalation, and stopping criteria
Safety margin calculationsSafety margins for <drug> were calculated for the planned starting dose of 5 mg and a high dose of 1000 mg based on NOAEL/MABEL human equivalent doses including a safety factor of X as well as modelled exposures (AUC0–24h) in humans.
Dose escalationDoses will be administered in an escalating manner following thorough review of the safety, tolerability, and PD data for at least 24 hours post dose after all subjects have completed the scheduled safety and PD assessments in the preceding dose group. The review of the data and the decision on the next dose level will be made jointly by the investigator and the sponsor.
Stopping criteriaDose escalation will be stopped in case of an unacceptable tolerability profile based on the nature, frequency, and intensity of observed AEs judged jointly by the investigator and the sponsor.
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 17 of 48

CHDR9999
1.4.7 Treatment duration1.4.8 Primary endpoint1.4.9 Statistical hypotheses and sample size
This is an exploratory study; therefore, the sample size is not based on statistical considerations.
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 18 of 48
CHDR9999
2 STUDY OBJECTIVES2.1 Primary objective
2.2 Secondary objectives
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 19 of 48

CHDR9999
3 STUDY DESIGN
3.1 Overall study design and planThe total duration of the study for each subject will be up to xx days divided as follows:
Screening: Up to 42 days before dosing;
Treatment and study assessments: Days -1 to 99
In Clinic period: Days -2 to 3;
Follow-up visit: 7 to 10 days after last dose.
Subjects will be admitted to the study unit on Day -2 and will be discharged approximately 48 hours after study drug administration.
3.1.1 Screening
3.1.2 Treatment and observation period3.1.3 Follow-up
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 20 of 48

CHDR9999
4 STUDY POPULATION
4.1 Subject populationSubjects will be recruited via media advertisement or from the subjects’ database of the Centre for Human Drug Research, Leiden, the Netherlands.
4.2 Inclusion criteria4.3 Exclusion criteria4.4 Concomitant medicationsAll medications (prescription and over-the-counter [OTC]) taken within 30 days of study screening will be recorded.
No prescription medications and OTC medications will be permitted within 14 days prior to study drug administrations, or less than 5 half-lives (whichever is longer, and during the course of the study. In addition, no vitamin, mineral, herbal, and dietary supplements will be permitted within 7 days prior to study drug administrations, or less than 5 half-lives (whichever is longer, and during the course of the study.
Exceptions are paracetamol (up to 4 g/day) and ibuprofen 1 g/day). Other exceptions will only be made if the rationale is clearly documented by the investigator.
4.4.1 Allowed concomitant medications4.4.2 Prohibited concomitant medications4.4.3 Escape/rescue medications
4.5 Lifestyle restrictions
Approximate meal times will be according to the study schedule. In-clinic meals will be comparable in composition and time of administration across all cohorts in each study part.
For the screening and the follow up visit, subjects will be required to fast for at least 4 hours. Subjects will be required to fast minimally 8 hours overnight (no food and no fluid including water) before study dose administration. Water is allowed as required.
Any nutrients known to modulate CYP enzymes activity (e.g., grapefruit or Seville orange containing products or quinine containing drinks (tonic water or bitter lemon)) will not be permitted from 3 days before dosing until <EOS/ collection of the final pharmacokinetic blood sample/ discharge from the study unit >.
The use of (illicit) drugs including cannabis can influence the measurements. Therefore, using 'drugs' is not permitted from 3 days before dosing and until <EOS/ collection of the final pharmacokinetic blood sample/ discharge from the study unit >. Since poppy seeds can cause a positive 'drugs' result; these should be avoided. If a positive result occurs without an explanation, the subject cannot participate in the study. However, positive urine drug screen for prescribed medication is allowed at the discretion of the PI.
Alcohol will not be allowed from at least 24 hours before screening, dosing, at each scheduled visit, and whilst in the study unit until <EOS/ collection of the final pharmacokinetic blood sample/ discharge from the study unit>. At other times throughout the study, subjects should not consume more than 2 units of alcohol daily on average (one unit is 10 grams of alcohol). Subjects may undergo an alcohol breath test at the discretion of the investigator.
Subjects will not be allowed to have excessive caffeine consumption, defined as >800 mg per day from 7 days prior to the first dose of the study drug until 24 hours prior to dosing. Subjects will abstain from caffeine-containing products for 24 hours prior to the start of dosing until <EOS/ collection of the final pharmacokinetic blood sample/ discharge from the
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 21 of 48

CHDR9999
study unit>. Caffeine quantities defined as: one cup of coffee contains 100 mg of caffeine; one cup of tea, or one glass of cola, or potion of chocolate (dark:100 g, milk 200 g) contains approximately 40 mg of caffeine; one bottle of Red Bull contains approximately 80 mg of caffeine.
Subjects will abstain from the use of tobacco-or nicotine-containing products (including e-cigarettes and patches) for 24 hours prior to dosing until <EOS/ collection of the final pharmacokinetic blood sample/ discharge from the study unit>.
Strenuous physical activity (e.g., heavy lifting, weight or fitness training) is not allowed from 48 hours prior to each study day until <EOS/ collection of the final pharmacokinetic blood sample/ discharge from the study unit>. Light ambulatory activities (e.g. walking at normal pace) will be permitted, with the level of activities kept as similar as possible on all days in the study unit.
Subjects will be confined to the procedure room for the first <x> hours after dosing during continuous ECG monitoring, except to use the toilet. After this, if the equipment setup allows, subjects may be ambulatory during the ECG monitoring period, but should not engage in strenuous activities.
4.5.1 Contraception requirementsAll women of child bearing potential <and all males> must practice effective contraception during the study and be willing and able to continue contraception for at least 90 days after their last dose of study treatment.Women of child bearing potential are defined as all women physiologically capable of becoming pregnant, unless they meet one of the following conditions:
Postmenopausal: 12 months of natural (spontaneous) amenorrhea or 6 weeks after surgical bilateral oophorectomy with or without hysterectomy;
Posthysterectomy.
For the purposes of the study, effective contraception is defined as follows:
Females: Using 1 or more of the following acceptable methods of contraception: surgical sterilization (e.g., bilateral tubal ligation), intrauterine contraception/device, hormonal contraception, or any 2 barrier methods (a combination of male or female condom with diaphragm, sponge or cervical cap).
Males: Effective male contraception includes a vasectomy with negative semen analysis at follow up, or the use of condoms.
Abstinence can be considered an acceptable method of contraception at the discretion of the investigator. Periodic abstinence (e.g., calendar, ovulation, symptothermal, post ovulation methods) and withdrawal are not considered acceptable methods of contraception.
4.6 Study drug discontinuation and withdrawal4.6.1 Study drug interruption or discontinuation
Before trial medication is administered, changes in the subject health status including laboratory results if applicable, since the previous visit or previous dose in case of a multiple dose regimen, have to be checked. The investigator must temporally interrupt or permanently discontinue the study drug if continued administration of the study drug is believed to be contrary to the best interests of the subject. The interruption or premature discontinuation of study drug might be triggered by an Adverse Event (AE), a diagnostic or therapeutic procedure, an abnormal assessment (e.g., ECG or laboratory abnormalities), or for administrative reasons in particular withdrawal of the subject’s consent. The reason for study drug interruption or premature discontinuation must be documented.
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 22 of 48

CHDR9999
4.6.2 Subject withdrawalSubjects have the right to withdraw from the study at any time for any reason. Should a subject decide to withdraw from the study, all efforts should be made to complete and report the observations, particularly the follow-up examinations, as thoroughly as possible.
4.6.3 Replacement policySubjects withdrawing for reasons other than adverse events or any other tolerability issues with the treatment will be replaced.
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 23 of 48

CHDR9999
5 INVESTIGATIONAL MEDICINAL PRODUCT
5.1 Investigational drug and matching placeboStudy drug or placebo will be administered to the subjects as detailed in Table 1. x strengths of active, xx mg and xx mg <IMP name>, and matching placebo formulations will be manufactured.
Doses in the study can be adapted with ongoing assessment of safety, tolerability and systemic exposure prior to initiation of the next dose. At each dose level x subjects will be randomized to receive <IMP name> and x to receive placebo. <IMP name>/placebo will be administered following a 4-hour period of fasting with xxx mL of still water. The following dose groups are planned:
Cohort 1: 50 mg <IMP name> / matching placebo
Cohort 2: 200 mg <IMP name> / matching placebo
Cohort 3: 400 mg <IMP name> / matching placebo
Cohort 4: 800 mg <IMP name> / matching placebo
Cohort 5: 1600 mg <IMP name> / matching placebo
Cohort 6: 3200 mg <IMP name> / matching placebo
5.2 Comparative drug 5.3 Study drug dosing scheme5.4 Study drug up- and down-titrationNot applicable.5.5 Study drug packaging and labelling Bulk study drug will be supplied in bottles of XX tablets each, packaged and labelled by <manufacturer> in accordance with local regulations. Upon arrival at the pharmacy, the investigational products should check them for damage and verify proper identity, quantity, integrity of seals and temperature conditions, and report any deviations or product complaints upon discovery. The dispensing of the study drug will be performed by the pharmacy. Study drug will be dispensed for each subject according to the randomization list. Study drug packaging will be overseen by the <Leiden University Medical Centre Pharmacy> and bearing a label with the identification required by local law, the protocol number, drug identification, and dosage.
<IMP name> tablet storage should not exceed 25 ºC in the containers provided. All drug supplies must be stored in a secure, temperature-controlled area with limited access. For batch-specific storage instructions, see the packaging.
<For internal studies insert the label(s) provided by the pharmacy. For sponsored studies provide as separate document D3 in the trial dossier >
5.6 Study drug preparation5.7 Drug accountabilityDrug accountability will be maintained by the Leiden University Medical Centre Pharmacy and assessed by maintaining adequate study drug dispensing records. The investigator is responsible for ensuring that dosing is administered in compliance with the protocol. Delegation of this task must be clearly documented and approved by the investigator. All study drug administration will occur under medical supervision.
5.8 Treatment assignment and blinding
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 24 of 48

CHDR9999
5.8.1 Randomization and treatment assignmentSubjects must be randomized in a consecutive order starting with the lowest number.
Subject numbers consist of 1 to 3 digits, as depicted in the table below
Digit 1 Status: 0 for planned subjects, 1 for first set replacement subjects, 2 for second set replacement numbers and 9 for reserve subjects
Digit 2-3 Consecutive subject number
E.g for a study with 24 subjects and 2 replacement sets and 2 reserve subjects: planned set 001-024, replacement set 1 101-124, replacement set 2 201-224 and reserves 925, 926
Subject numbers consist of 4 or 5 digits, as depicted in the table below
Digit 1-2 Cohort number
Digit 3 Status: 0 for planned subjects, 1 for first set replacement subjects, 2 for second set replacement numbers and 9 for reserve subjects
Digit 4-5 Consecutive subject number within the cohort
E.g. for a study with 3 cohorts, 8 subjects per cohort, 1 replacement set and 2 reserves per cohort: planned set 01001-01008, 02001-02008, 03001-03008, replacement set 01101-01108, 02101-02108, 03101-03108, and reserves 01909, 01910, 02909, 02910, 03909, 03910
Stratification for gender takes place in Promasys, by adding key value “F” to subject numbers for females and “M” to subject numbers for males.
The randomization code will be generated using specify the name of the software and its version by a study-independent, CHDR statistician. The randomization code will be unblinded/broken and made available for data analysis only after study closure, i.e., when the study has been completed, the protocol deviations determined, and the clinical database declared complete, accurate and locked. The randomization code will be kept strictly confidential. Sealed individual randomization codes, per subject and per treatment, will be placed in a sealed envelope containing the and labelled 'emergency decoding envelopes' will be kept in a safe cabinet at CHDR.
5.8.2 BlindingNot applicable.This study will be performed in a double-blind fashion. The investigator, study staff, subjects, sponsor, and monitor will remain blinded to the treatment until study closure. The investigational drug and its matching placebo and/or active comparative drug are indistinguishable and will be packaged in the same way.This study will be performed in a double-blind fashion. With the exceptions described in this section, the randomization list will not be available to the investigator, study staff, subjects, sponsor, or monitors. The investigational drug and its matching placebo and/or active comparative drug are indistinguishable and will be packaged in the same way.The randomization list will be made available to the pharmacist preparing the study drug, to the individual responsible for PK sample bioanalysis and to statisticians or programmers involved in preparing blinded summaries, graphs and listings to support the dose decisions.
The summaries, graphs and listings provided by the statisticians or programmers will be produced in an area to which other team members do not have access. The reports will be made available to project leader and the DSMB for the dose decision. The DSMB may share the reports with other
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 25 of 48

CHDR9999
individuals, experts or decision bodies if it is necessary that they participate in the dose decision process.
This study will performed in a double-blind fashion. With the exceptions described in thissection, the randomization list will not be available to the investigator, study staff, subjects, sponsor, or monitor. The investigational drug and its matching placebo and/or active comparative drug are indistinguishable and will be packaged in the same way.In order for the sponsor to make recommendations or decisions regarding further development of the drug, the study management team may unblind the individual cohort after the data for all subjects on these cohorts have been entered into the database, cleaned and locked (except possibly for data to be received directly from third parties e.g. laboratories in electronic form).
The investigator will receive a set of sealed emergency codes to be broken in case of emergency situations and duplicates will be kept by the <sponsor>. If the identity of the study drug administered needs to be known in order to manage the subject’s condition i.e., in case of a medical emergency or in the case a SUSAR occurs, the treatment emergency code for that subject may be broken and the study drug identified. All such occurrences should be documented in the study file. Treatment emergency codes should not be broken except in emergency situations and, if possible, the investigator/sponsor should be contacted before the emergency code is opened. <At the final monitoring visit/Just prior to database lock> the unused emergency code labels will be checked and a statement to the effect that all are intact (or not as the case may be) will be made on the database lock form.
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 26 of 48

CHDR9999
6 STUDY ENDPOINTS
6.1 Efficacy endpoints6.2 Safety and tolerability endpoints
Treatment-emergent (serious) adverse events ((S)AEs). Concomitant medication
Clinical laboratory tests
o Haematology
o Chemistry
o Urinalysis
Vital signs
o Pulse Rate (bpm)
o Systolic blood presuure (mmHg)
o Diastolic blood pressure (mmHg)
ECG
o Heart Rate (HR) (bpm), PR, QRS, QT, QTcB, QTcF
6.3 Pharmacokinetic endpointsThe following endpoints will be determined for <compound> for each treatment <and day/period>. They will be derived by non-compartmental analysis of the <plasma/serum> concentration-time data <and urine data>:
Single ascending dose
<Plasma/Serum> PK parameters of <compound>: AUCinf, AUClast, CL/F, Cmax, t1/2, tlag, tmax, Vz/F
Dose-normalized <plasma/serum> PK parameters of <compound>: AUCinf, AUClast, Cmax
<Urine PK parameters of compound: Aelast, Aelast%, CLR> Multiple ascending dose
<Plasma/Serum> PK parameters of <compound>: AUCtau, CL/F, Cmax, Ctrough, t1/2, tmax, Vz/F
Dose-normalized <plasma/serum> PK parameters of <compound>: AUCtau, Cmax, Ctrough
<Urine PK parameters of compound: Aetau, Aetau%, CLR>
6.4 Pharmacodynamic endpoints Saccadic eye movements:
o saccadic reaction time (second), o saccadic peak velocity (degrees/second), and o saccadic inaccuracy (%);
Smooth pursuit eye movements:o percentage of time the eyes of the subjects are in smooth pursuit of the target (%);
Body sway:o antero-posterior sway (mm);
Adaptive tracking:o average performance (%);
Visual Analog Scales (VAS) according to Bond and Lader to assess: o mood (mm),
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 27 of 48

CHDR9999
o alertness (mm), and o calmness (mm).
During each treatment period:
Visual Verbal Learning Test (VVLT) memory testing o Immediate recall trial 3 (number correct), o Delayed recall (number correct), ando Delayed recognition (number correct)o Delayed recognition (reaction time correct) (msec)
Baseline is defined as the < last/average> value prior to dosing.
Total and free cholesterol, triglycerides, phospholipids and levels of apoproteins, LCAT activity and ex vivo cholesterol efflux capacity.
Cytokines induced by ex vivo LPS challenge,
Circulating cytokines induced by in vivo LPS challenge,
C-reactive protein (CRP) levels and vital signs (temperature, heart rate, blood pressure) after in vivo LPS challenge can be regarded as PD parameters.
Thrombin generation assay output (at variable assay conditions), including lag time, time topeak, peak, and endogenous thrombin potential (ETP); activated partial thromboplastin time(aPTT); prothrombin time (PT). Besides its use in the PK analyses, FVIIa activity is alsoregarded a PD parameter.
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 28 of 48

CHDR9999
7 STUDY ASSESSMENTSSee Table 1 for the time points of the assessments.
7.1 Efficacy assessments7.2 Safety and tolerability assessmentsThe definitions, reporting and follow-up of AEs, SAEs and potential pregnancies are described in section 8.
7.2.1 Specific safety and tolerability assessments7.2.2 Vital signs
Evaluations of systolic and diastolic blood pressure, pulse rate, respiratory rate, and temperature will be performed throughout the study. Pulse and blood pressure will be taken after <5> minutes in the supine position. Automated oscillometric blood pressures and pulse rate will be measured using a Dash 3000, Dash 4000, Dynamap 400 or Dynamap ProCare 400.
7.2.3 Weight and heightWeight (kg) will be recorded at screening and the follow-up visit or upon early termination. Height (cm) will be recorded and body mass index (BMI) calculated at screening.
7.2.4 Physical examinationPhysical examination (i.e., inspection, percussion, palpation and auscultation) is performed during the course of the study. Clinically relevant findings that are present prior to study drug initiation must be recorded with the subject’s Medical History. Clinically relevant findings found after study drug initiation and meeting the definition of an AE (new AE or worsening of previously existing condition) must be recorded.
7.2.5 ElectrocardiographyECGs will be obtained during the course of the study using Marquette 800/2000/5500 or Dash 3000 and stored using the MUSE Cardiology Information System. ECGs will be taken after <5> minutes in the supine position. The investigator will assess the ECG recording as 'normal', 'abnormal - not clinically significant', or 'abnormal - clinically significant' and include a description of the abnormality as required. The ECG parameters assessed will include heart rate, PR, QRS, QT, and QTc<B/F> (calculated using <Bazett’s/Fredericia's> method).
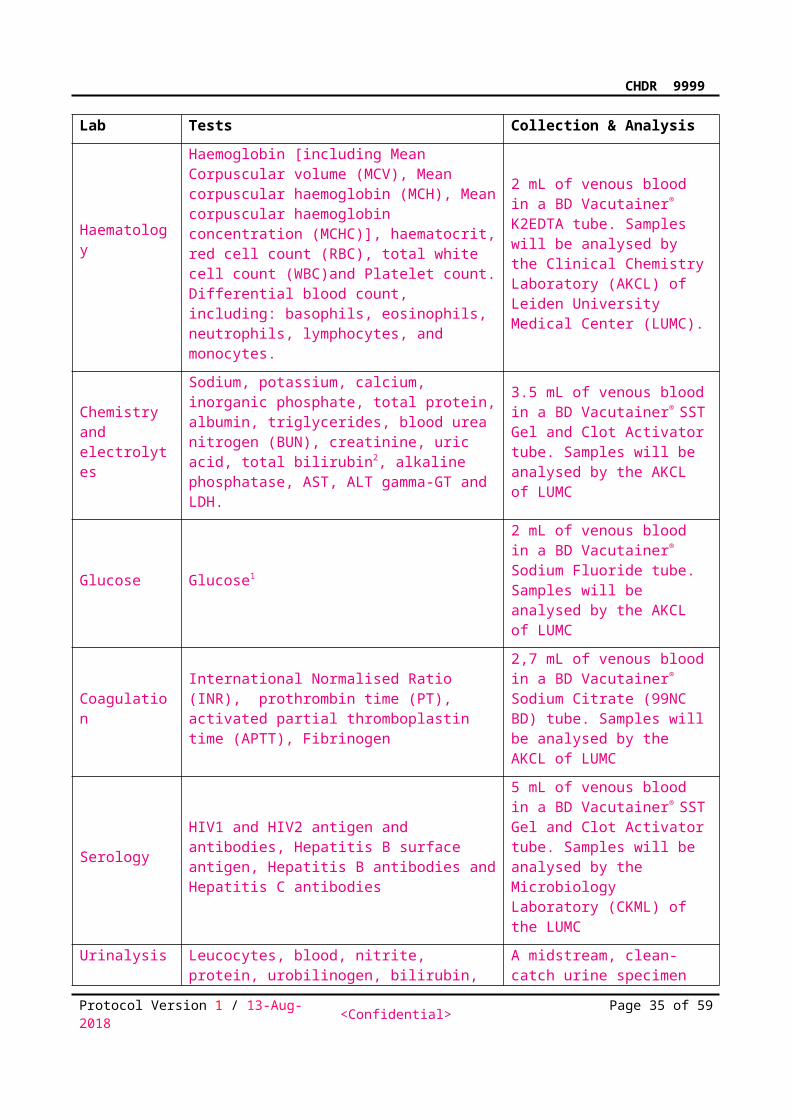
7.2.6 Laboratory assessmentsLaboratory parametersBlood and other biological samples will be collected for the following clinical laboratory tests:Lab Tests Collection & Analysis
Haematology
Haemoglobin [including Mean Corpuscular volume (MCV), Mean corpuscular haemoglobin (MCH), Mean corpuscular haemoglobin concentration (MCHC)], haematocrit, red cell count (RBC), total white cell count (WBC)and Platelet count. Differential blood count, including: basophils, eosinophils, neutrophils, lymphocytes, and monocytes.
2 mL of venous blood in a BD Vacutainer® K2EDTA tube. Samples will be analysed by the Clinical Chemistry Laboratory (AKCL) of Leiden University Medical Center (LUMC).
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 29 of 48

CHDR9999
Chemistry and electrolytes
Sodium, potassium, calcium, inorganic phosphate, total protein, albumin, triglycerides, blood urea nitrogen (BUN), creatinine, uric acid, total bilirubin2, alkaline phosphatase, AST, ALT gamma-GT and LDH.
3.5 mL of venous blood in a BD Vacutainer® SST Gel and Clot Activator tube. Samples will be analysed by the AKCL of LUMC
Glucose Glucose1
2 mL of venous blood in a BD Vacutainer® Sodium Fluoride tube. Samples will be analysed by the AKCL of LUMC
CoagulationInternational Normalised Ratio (INR), prothrombin time (PT), activated partial thromboplastin time (APTT), Fibrinogen
2,7 mL of venous blood in a BD Vacutainer® Sodium Citrate (99NC BD) tube. Samples will be analysed by the AKCL of LUMC
SerologyHIV1 and HIV2 antigen and antibodies, Hepatitis B surface antigen, Hepatitis B antibodies and Hepatitis C antibodies
5 mL of venous blood in a BD Vacutainer® SST Gel and Clot Activator tube. Samples will be analysed by the Microbiology Laboratory (CKML) of the LUMC
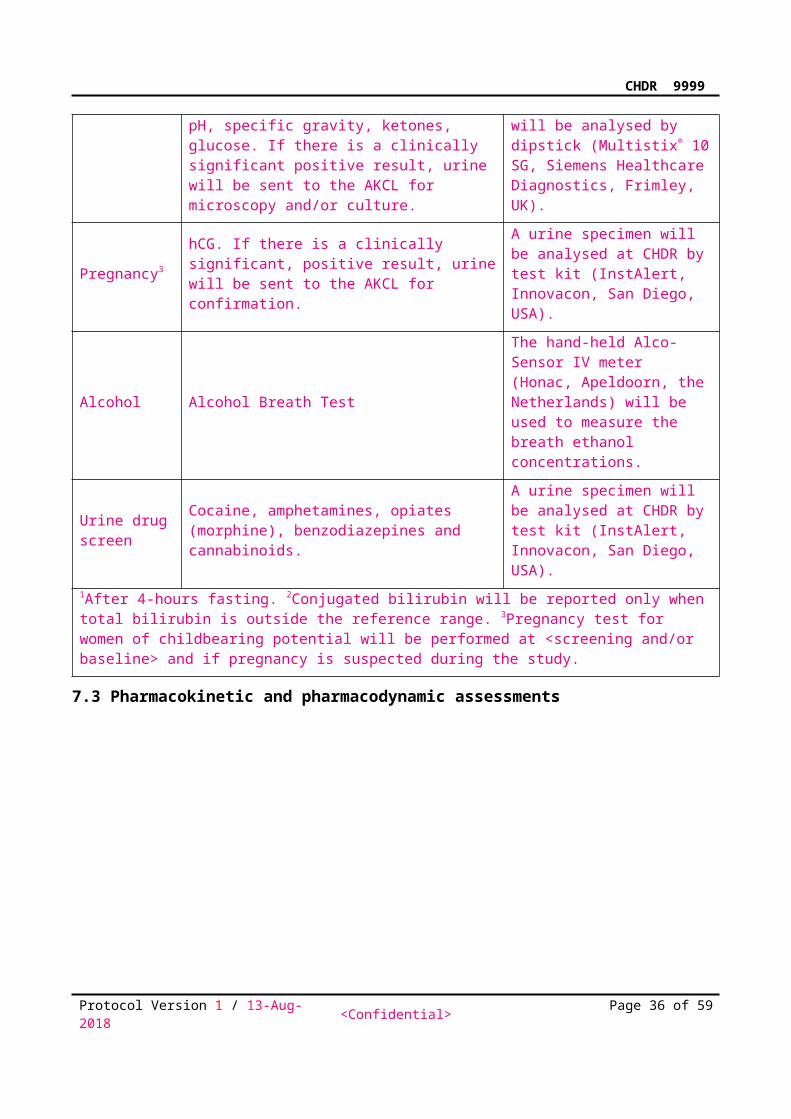
Urinalysis
Leucocytes, blood, nitrite, protein, urobilinogen, bilirubin, pH, specific gravity, ketones, glucose. If there is a clinically significant positive result, urine will be sent to the AKCL for microscopy and/or culture.
A midstream, clean-catch urine specimen will be analysed by dipstick (Multistix® 10 SG, Siemens Healthcare Diagnostics, Frimley, UK).
Pregnancy3hCG. If there is a clinically significant, positive result, urine will be sent to the AKCL for confirmation.
A urine specimen will be analysed at CHDR by test kit (InstAlert, Innovacon, San Diego, USA).
Alcohol Alcohol Breath Test
The hand-held Alco-Sensor IV meter (Honac, Apeldoorn, the Netherlands) will be used to measure the breath ethanol concentrations.
Urine drug screen
Cocaine, amphetamines, opiates (morphine), benzodiazepines and cannabinoids.
A urine specimen will be analysed at CHDR by test kit (InstAlert, Innovacon, San Diego, USA).
1After 4-hours fasting. 2Conjugated bilirubin will be reported only when total bilirubin is outside the reference range. 3Pregnancy test for women of childbearing potential will be performed at <screening and/or baseline> and if pregnancy is suspected during the study.
7.3 Pharmacokinetic and pharmacodynamic assessments
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 30 of 48

CHDR9999
Approximately XX mL blood will be collected via an i.v. catheter placed in an antecubital vein in the arm in <appropriate/K2EDTA/K3EDTA/Lithium Heparin/Plain/etc> tubes. The indwelling catheter will be kept patent by saline flush after each blood sampling. Immediately following collection of the required blood volume, the tubes will be slowly tilted backwards and forwards (no shaking) to bring the anti-coagulant into solution <delete for plain tubes>, <protected from light> and <immediately cooled on ice>. Within 30 minutes of collection, the tubes will be centrifuged at approximately 2000 g for 10 minutes at 2 to 8 °C. The <plasma/serum> will be [transferred into one]/[split into two] labelled polypropylene tube, avoiding carryover of erythrocytes. All samples will be stored in an upright position at - 40 °C or lower. The exact actual clock time of withdrawal of the blood sample will be recorded.
7.3.1 LabellingPre-printed, waterproof labels will be used to identify the tubes used during sample collection and for storage of separated plasma. Each label will contain the following information:
CHDR Protocol number
Subject Number & Initials
Occasion number (date)
Protocol (delta) time
Activity: Sample type (blood) & purpose (PK)
Last line optional: Sponsor ID or free text [~16 characters]
7.3.2 Shipping ProceduresCHDR will arrange shipment of the samples. The samples must be packed securely together with completed shipment forms in polystyrene insulated shipping containers together with enough dry ice to last for 48 hours. Samples must be shipped to <sponsor or central laboratory name> at time intervals agreed with the sponsor.
7.3.3 Bioanalysis<Plasma/Serum> <drug> concentrations will be measured by a validated <LC/MS/MS> method. <Plasma/Serum> samples may also be used for exploratory assessment of the <pro-drug> and/or metabolites, if applicable.
7.4 Pharmacodynamic assessments and questionnaires7.4.1 Concomitant medications
Concomitant medications initiated, stopped, up-titrated or down-titrated for an AE will be recorded.
7.5 Sequence of assessments and time windowsWhen the following assessments are scheduled to be performed at the same time-point, the order (of priority) will be as follows: ECG, vital signs, physical examination, blood sampling for safety, activity 1, activity 2, etc. When a PK assessment is scheduled for the same nominal time as another scheduled assessment, the PK sample will take precedence.
The deviations of actual time points from the expected time points will be within ten percent, calculated from the zero point (time of drug administration) or the last relevant activity. The expected timepoints are defined as the timepoints in Promasys. Deviations of more than 10% will be explained in a note. These time deviations only apply for ECG, vital signs, blood sampling for PK and PD, activity 1, activity 2, etc.
Pre-dose assessments are given in indicative expected times.
In case of multiple dosing the time window for the second dosing at CHDR is plus or minus ten minutes. In case of further dosing this will take place at the initial dosing time with a window of plus
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 31 of 48

CHDR9999
or minus 10 minutes. If dosing takes place twice or thrice per 24 hours, subsequent time of dosing must be related to the first dosing time of the day.
When subjects are dosed on return visits a time window of plus or minus one hour is acceptable. This must always be related to the initial dosing time.
7.6 Total blood volumeSample Samples taken Sample Volume* VolumeHaematology 6 x 2 mL = 12 mLChemisty 6 x 3.5 mL = 21 mLGlucose 6 x 2 mL = 12 mLCoagulation 1 x 2.7 mL = 2.7 mLSerology 1 x 5 mL = 5 mLPharmacokinetics 36 x 6 mL = 216 mLWet PD 36 x 2 mL = 72 mL* exclusive discarded volume Total blood volume/subject 340.7 mL
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 32 of 48

CHDR9999
8 SAFETY REPORTING
8.1 Definitions of adverse eventsAn Adverse Event (AE) is any untoward medical occurrence in a subject who is participating in a clinical study performed. The AE does not necessarily have to follow the administration of a study drug, or to have a causal relationship with the study drug. An AE can therefore be any unfavourable and unintended sign (including an abnormal laboratory or vital sign finding), symptom, or disease temporally associated with the study participation, whether or not it is related to the study drug.
8.1.1 Intensity of adverse events The intensity of clinical AEs is graded three-point scale as defined below:
Mild: discomfort noticed but no disruption of normal daily activity;
Moderate: discomfort sufficient to reduce or affect normal daily activity;
Severe: inability to work or perform daily activity.
8.1.2 Relationship to study drug For each AE the relationship to drug as judged by the investigator:
Probable;
Possible;
Unlikely;
Unrelated.
8.1.3 Chronicity of adverse eventsThe chronicity of the AE will be classified by the investigator on a three-item scale as defined below:
Single occasion: single event with limited duration;
Intermittent: several episodes of an event, each of limited duration;
Persistent: event which remained indefinitely.
8.1.4 Action
Eventual actions taken will be recorded.
8.1.5 Serious adverse eventsA Serious Adverse Event (SAE) is defined by the International Conference on Harmonization (ICH) guidelines as any AE fulfilling at least one of the following criteria:
- results in death;
- is life threatening (at the time of the event);
- requires hospitalisation or prolongation of existing inpatients’ hospitalisation;
- results in persistent or significant disability or incapacity;
- is a congenital anomaly or birth defect; or
- any other important medical event that did not result in any of the outcomes listed above due to medical or surgical intervention but could have been based upon appropriate judgement by the investigator.
An elective hospital admission will not be considered as a SAE.
8.1.6 Suspected unexpected serious adverse reactions
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 33 of 48

CHDR9999
A SUSAR (Suspected Unexpected Serious Adverse Reaction) is a SAE that is unexpected, (nature or severity of which is not consistent with the applicable product information (e.g., investigator's brochure for an unauthorised investigational product or summary of product characteristics for an authorised product)) and suspected (a reasonable possibility of causal relationship with investigational drug, regardless of the administered dose).
8.1.7 Reporting of serious adverse eventsSAEs and SUSAR's will be reported according to the following procedure.
The sponsor/investigator will report the SAEs through the web portal ToetsingOnline (see https://toetsingonline.nl/) to the accredited EC that approved the protocol, within 24 hours of first knowledge of the SAE. SUSARS must be reported to the EC that approved the study, the CA and the Dutch Medicines Evaluation Board (College ter Beoordeling van Geneesmiddelen).
SUSARS must be reported to the EC that approved the study, the CA and the Dutch Medicines Evaluation Board (College ter Beoordeling van Geneesmiddelen).
The sponsor/investigator will report the SAEs through the web portal ToetsingOnline (see https://toetsingonline.nl/) to the accredited EC that approved the protocol, within 7 days of first knowledge for SAEs that result in death or are life threatening followed by a period of maximum of 8 days to complete the initial preliminary report. All other SAEs will be reported within a period of maximum 15 days after the sponsor has first knowledge of the SAE.
SUSARS must be reported to the EC that approved the study, the CA and the Dutch Medicines Evaluation Board (College ter Beoordeling van Geneesmiddelen).
The sponsor will report expedited the following SUSARs to the EC:
SUSARs that have arisen in the clinical trial that was assessed by the EC;
SUSARs that have arisen in other clinical trials of the same sponsor and with the same medicinal product, and that could have consequences for the safety of the subjects involved in the clinical trial that was assessed by the EC.
The remaining SUSARs are recorded in an overview list (line-listing) that will be submitted once every half year to the EC. This line-listing provides an overview of all SUSARs from the study medicine, accompanied by a brief report highlighting the main points of concern.
The expedited reporting of SUSARs through the web portal EMA Eudravigilance is sufficient as notification to the CA and the Dutch Medicines Evaluation Board, a separate notification is not necessary.
The sponsor can delegate reporting SUSARS to the EC to CHDR, but CHDR cannot take over the sponsor responsibility of reporting a SUSAR to the EMA EudraVigilance database.
The sponsor will report expedited all SUSARs to the competent authorities in other Member States, according to the requirements of the Member States.
The expedited reporting will occur not later than 15 days after the sponsor has first knowledge of the adverse reactions. For fatal or life threatening cases the term will be maximal 7 days for a preliminary report with another 8 days for completion of the report.
The sponsor can prepare additional reports for other authorities (e.g. FDA).
The investigator will report expedited the following SUSARs through the web portal ToetsingOnline to the EC:
• SUSARs that have arisen in the clinical trial that was assessed by the EC;
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 34 of 48
CHDR9999
• SUSARs that have arisen in other clinical trials of the same sponsor and with the same medicinal product, and that could have consequences for the safety of the subjects involved in the clinical trial that was assessed by the EC.
The expedited reporting of SUSARs through the web portal ToetsingOnline is sufficient as notification to the EC, CA and the Dutch Medicines Evaluation Board, a separate notification is not necessary. To prevent a double notification, it must be indicated in ToetsingOnline if the SUSAR is reported in the EMA EudraVigilance database, this will prevent the notification of the CA and the Dutch Medicines Evaluation Board through the web portal ToetsingOnline.
For international investigator initiated studies: The investigator will report expedited all SUSARs to the competent authorities in other Member States, according to the requirements of the Member States.
The expedited reporting will occur not later than 15 days after the first knowledge of the adverse reactions. For fatal or life threatening cases the term will be maximal 7 days for a preliminary report with another 8 days for completion of the report.
8.1.8 Follow-up of adverse eventsAll AEs will be followed until they have abated, returned to baseline status or until a stable situation has been reached. Depending on the event, follow up may require additional tests or medical procedures as indicated, and/or referral to the general physician or a medical specialist.
8.2 Temporary halt for reasons of subject safetyIn accordance to section 10, subsection 4, of the WMO, the investigator will inform the subjects and the EC if anything occurs, on the basis of which it appears that the disadvantages of participation may be significantly greater than was foreseen in the research proposal. The study will be suspended pending further review by the EC, except insofar as suspension would jeopardise the subjects’ health. The investigator will ensure that all subjects are kept informed.
8.3 Annual safety report or development safety update reportIn addition to the expedited reporting of SUSARs, the sponsor/investigator will submit, once a year throughout the clinical trial, a safety report to the EC, CA and CAs of the concerned Member States.This safety report consists of:
a list of all suspected (unexpected or expected) serious adverse reactions, along with an aggregated summary table of all reported serious adverse reactions, ordered by organ system, per study;
a report concerning the safety of the subjects, consisting of a complete safety analysis and an evaluation of the balance between the efficacy and the harmfulness of the medicine under investigation.
8.4 Pregnancy
8.4.1 TeratogenicityIf a woman becomes pregnant when on study drug, permanent discontinuation of study drug should be considered as appropriate. The investigator must counsel the subject and discuss the risks of continuing with the pregnancy and the possible effects on the foetus. Monitoring of the subject should continue until the outcome of the pregnancy is known.
8.4.2 Reporting of pregnancyIrrespective of the treatment received by the subject, any pregnancy occurring during study drug administration or <during the x months following study drug discontinuation/until follow-up>, must be reported within 24 hours of the investigator's knowledge of the event to the sponsor.
8.5 Data safety monitoring board
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 35 of 48
CHDR9999
Screening (enrolment) for the next cohort (the next planned dose level) will not begin until the DSMB has approved dosing for that cohort. The DSMB will comprise the principal investigator, a clinical pharmacologist from CHDR (non-voting member), a medical director from the sponsor, a statistician (non-voting member), and ad hoc members (non-voting), as required. The DSMB is empowered to recommend modifications of the protocol (to enhance subject safety) or to recommend early termination of the study if major concerns arise about the subject's safety at any time during the course of this study or during any other study with the same investigational drug. There is no limit to the number and timing of interim analyses aimed at guaranteeing the safety of the subjects.
The data to be reviewed by the DSMB is listed in Section 9.11.
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 36 of 48

CHDR9999
9 STATISTICAL METHODOLOGY AND ANALYSES
9.1 Statistical analysis planAll safety and statistical programming is conducted with SAS 9.4 for Windows or newer (SAS Institute Inc., Cary, NC, USA). PK variable programming is conducted with R 3.5.3 for Windows or newer (R Foundation for Statistical Computing/R Development Core Team, Vienna, Austria, 2010.A Statistical Analysis Plan (SAP) will be written and finalized before the study closure, i.e., database closure and unblinding of the randomization code (delete if not applicable). The SAP will provide full details of the analyses, the data displays and the algorithms to be used for data derivations. The SAP will include the definition of major and minor protocol deviations and the link of major protocol deviations to the analysis sets.
9.2 Protocol violations/deviationsProtocol deviations will be identified based on conditions related to the categories below:
Protocol entry criteria Forbidden concomitant medications Missing evaluations for relevant endpoints Other protocol deviations occurring during study conduct.
Major protocol deviations will be identified before the study closure, and listed where appropriate.
9.3 Power calculationNot applicable.Power calculations were performed for the parameter <parameter> using the data available at CHDR/based on literature. For <parameter>, a sample size of xx will have 80% power to detect a difference in means of xx.xxx, assuming a standard deviation of differences of xx.xxx, using a paired t-test with a 0.050 two-sided significance level. The estimated average score under placebo is xxx.xx, so a true improvement of circa x% will be significant in x out of x studies with xx subjects.
9.4 Missing, unused and spurious dataAll missing or incomplete safety and PD data, including dates and times, are treated as such. Missing test results or assessments will not be imputed. Missing PD data, indicated as ‘M’ in the data listing, will be estimated within the statistical mixed model using SAS PROC MIXED.
For graphical and summary purposes PD and safety values below the limit of quantification will be set to half (½) of the limit of quantification. For analysis no undetermined values will be replaced.
If single data points for <plasma/serum> <drug> concentrations are missing, the AUC parameters will be derived by interpolating with regard to the two neighboring non-missing concentrations.
For calculation of PK parameters, all <plasma/serum> <drug>concentration values below the quantification limit (BLQ) occurring prior to Cmax will be replaced by 0, except for embedded BLQ values (between two measurable time points) which will be treated as “missing”. All BLQ values after Cmax will be treated as “missing”.
The handling of missing, unused and spurious data will be documented in the study report.9.5 Analysis setsData of all subjects participating in the study will be included in the analyses if the data can meaningfully contribute to the objectives of the study.
9.5.1 Safety setThe safety population will be defined as all subjects who were validated (randomised) and received at least 1 dose of study treatment.
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 37 of 48

CHDR9999
9.5.2 Pharmacokinetic analysis setThe PK analysis population is defined as all subjects who were validated (randomised), received at least one dose of study treatment, and have at least one measurable drug concentration in samples collected.
9.5.3 Pharmacodynamic analysis setThe analysis population for <pharmacodynamics/efficacy> is defined as all subjects who were validated (randomised), received at least one dose of study treatment, and have at least one post-baseline assessment of the parameter being analyzed.
9.6 Subject dispositionSubject disposition will be listed by subject.
The following subject data will be summarized:
number and percentage of subjects screened,
number and percentage of subjects enrolled,
number and percentage of subjects completed,
number and percentage of subjects included in safety population and
number and percentage of subjects included in the PD analysis population.
A subject who completed the study is defined as a subject where the last (72 hour) PK blood sample was assessed.
9.7 Baseline parameters and concomitant medications9.7.1 Demographics and baseline variables
Continuous demographic variables (e.g., age, height, weight, BMI) will be summarized by descriptive statistics (n, mean, SD, median, Min, Max).
Qualitative demographic characteristics (sex, race/ethnicity) will be summarized by counts and percentages.
The results of the <name> questionnaire at screening will only be listed.
9.7.2 Medical history Medical history will only be listed.
9.7.3 Concomitant MedicationsPrevious and concomitant medications will be listed by International Nonproprietary Names, dose, regimen, route and for which indication it was prescribed.
9.7.4 Treatment compliance/exposureExposure to study treatment is described in terms of duration of treatment and average infusion rate. The average infusion rate (mL/hr) is summarized by mean, SD, median, Q1, Q3, Min, Max.
9.8 Safety and tolerability endpoints The safety set is used to perform all safety analyses. Baseline is defined as the <last/average> value prior to dosing. Change from baseline will be calculated for all continuous safety parameters.
9.8.1 Adverse events
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 38 of 48

CHDR9999
The AE coding dictionary for this study will be Medical Dictionary for Regulatory Activities (MedDRA). It will be used to summarize AEs by primary system organ class (SOC) and preferred term (PT).All adverse events will be displayed in listings.
A treatment-emergent adverse event (TEAE) is defined as an adverse event observed after starting administration of the specific treatment, and <prior to the start of another treatment, if any OR up to 5 days (120 hours) after study drug administration>. If a subject experiences an event both prior to and after starting administration of a treatment, the event will be considered a TEAE (of the treatment) only if it has worsened in severity (i.e., it is reported with a new start date) after starting administration of the specific treatment, and prior to the start of another treatment, if any. All TEAEs collected during the investigational period will be summarized.
<The number of and/or the number of subjects with> treatment emergent AEs will be summarized by:
1. treatment, MedDRA SOC and PT;2. treatment, MedDRA SOC, PT and severity;3. treatment, MedDRA SOC, PT and drug relatedness.
9.8.2 Vital signsAt each time point, absolute values and change from baseline of supine blood pressure and pulse rate will be summarized with n, mean, SD, SEM, median, Min, and Max values. The number of available observations and out-of-range values (absolute and in percentage) will be presented. Values outside the reference range will be flagged in the listing. ‘H’ and ‘L’, denoting values above or below the investigator reference range (when present), will flag out-of-range results.
9.8.3 ECGAt each time point, absolute values and change from baseline of ECG numeric variables will be summarized with n, mean, SD, SEM, median, Min, and Max values. The number of available observations and out-of-range values (absolute and in percentage) will be presented. Values outside the investigator’s normal range will be flagged in the listing. ‘H’ and ‘L’, denoting values above or below the investigator reference range (when present), will flag out-of-range results.
9.8.4 Clinical laboratory testsAt each time point, absolute values and change from baseline of clinical laboratory variables will be summarized with n, mean, SD, SEM, median, Min, and Max values. The number of available observations and out-of-range values (absolute and in percentage) will be presented. All laboratory data (including re-check values if present) will be listed chronologically. ‘H’ and ‘L’, denoting values above or below the investigator reference range (when present), will flag out-of-range results.
9.9 Pharmacokinetic and pharmacodynamic endpoints9.9.1 Pharmacokinetics
The individual <plasma/serum> <drug> concentrations will be listed by treatment, subject, visit and time. Individual <plasma/serum> <drug> concentrations versus time will be plotted in panel plots for each treatment using both a linear and log y-axis. <The amount excreted of <compound> at each collection interval will be listed by treatment, subject, and visit.>
The individual <plasma/serum> <drug> concentrations will be summarised (n, mean, SD, %CV, median, Min and Max values) by treatment and time, and will also be presented graphically as mean over time, with standard deviation as error bars. If more than one third of the concentrations are BLQ then Mean and SD will not be provided. If one concentration is BLQ then %CV will not be provided. <The amount of <compound> in urine will be summarized (n, mean, SD, %CV, median, Min and Max values) by treatment, visit and collection interval.>
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 39 of 48

CHDR9999
When an actual sampling time of a drug concentration sample differs from the protocol time by more than <10% and at least 5 minutes>, the concentration will be excluded from calculation of descriptive statistics and a note will be added to the sample in the listing.
The individual PK parameters (except tmax and tlag) will be summarized (n, mean, SD, %CV, geometric mean, geometric %CV, median, Min and Max) per treatment group and will be presented graphically as boxplots. For tmax and tlag,the n, median, Min and Max statistics will be reported. .
9.9.2 PharmacodynamicsThe final analysis will be preceded by a <blind > data review which consists of individual graphs per visit by time of all pharmacodynamic measurements by time. The graphs will be used to detect outliers and measurements unsuitable for analysis.The PD parameters will be listed by treatment, subject, visit and time. Individual graphs by time will be generated.
All <repeatedly measured> PD endpoints will be summarised (n, mean, SD, SEM, median, Min and Max values) by treatment and time, and will also be presented graphically as mean over time, with standard deviation as error bars. All <single measured> PD endpoints will be summarised (mean, SD, SEM, median, Min and Max values) by treatment, and will also be presented graphically as mean in a bargraph, with standard deviation as error bars.
Parameters will initially be analyzed without transformation, but if the data suggest otherwise, log-transformation may be applied. Log-transformed parameters will be back-transformed after analysis where the results may be interpreted as percentage change.
To establish whether significant treatment effects can be detected on the <repeatedly measured> PD parameters, each parameter will be analyzed with a mixed model analysis of covariance (ANCOVA) with treatment, time, <period> and treatment by time as fixed factors and subject, subject by treatment and subject by time as random factors and the (average) baseline measurement as covariate.
Single measured PD parameters will be analyzed with a mixed model analysis of variance (ANOVA) with treatment <and period> as fixed factor<s> and subject as random factor.
The Kenward-Roger approximation will be used to estimate denominator degrees of freedom and model parameters will be estimated using the restricted maximum likelihood method.
The general treatment effect and specific contrasts will be reported with the estimated difference and the 95% confidence interval, the least square mean estimates and the p-value. Graphs of the Least Squares Means(LSM) estimates over time by treatment will be presented with 95% confidence intervals as error bars, as well as change from baseline LSM estimates.
The following contrasts will be calculated within the model:
Treatment 1 - Placebo
Treatment 2 - Placebo
9.9.3 Inferential methodsThe study is exploratory and no formal null hypothesis is set. No adjustments for multiple comparisons will be applied.
9.9.4 PK/PD modellingPopulation PK and PK/PD models may be developed to address objectives that require an integrative interpretation of the study results. These include assessment of the dose proportionality, investigation of the nature of the PK/PD relationship, and the use of study results as part of a larger model-based data analysis. If population PK/PD models are developed, a separate Pharmacometric Analysis Plan will be written.
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 40 of 48

CHDR9999
9.10 Exploratory analyses and deviationsExploratory data-driven analyses can be performed with the caveat that any statistical inference will not have any confirmatory value. Deviations from the original statistical plan will be documented in the clinical study report.
9.11 Interim analysesNo interim analysis is planned.The following blinded safety data will be reviewed by the DSRC for the ongoing cohorts and used to determine if dose escalation can occur:
AEs and SAEs through <xx> days after the last dose of study treatment; Post-dosing physical examination results up to <xx> days after the last dose of study
treatment; and Pharmacodynamic results at up to <xx> days after the last dose of study treatment.
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 41 of 48

CHDR9999
10 GOOD CLINICAL PRACTICE, ETHICS AND ADMINISTRATIVE PROCEDURES
10.1 Good clinical practice
10.1.1 Ethics and good clinical practiceThe investigator will ensure that this study is conducted in full compliance with the protocol, the principles of the Declaration of Helsinki (www.wma.net), ICH GCP guidelines (http://www.ich.org/products/guidelines.html), and with the laws and regulations of the country in which the clinical research is conducted.
10.1.2 Ethics committee / institutional review boardThe investigator will submit this protocol and any related documents to an Ethics Committee (EC) and the Competent Authority (CA). Approval from the EC and the statement of no objection from the CA must be obtained before starting the study, and should be documented in a dated letter/email to the investigator, clearly identifying the trial, the documents reviewed and the date of approval. A list of EC members must be provided, including the functions of these members. If study staff were present, it must be clear that none of these persons voted.
Modifications made to the protocol after receipt of the EC approval must also be submitted as amendments by the investigator to the EC in accordance with local procedures and regulations.
10.1.3 Informed consentIt is the responsibility of the investigator to obtain written informed consent from each individual participating in this study after adequate explanation of the aims, methods, objectives and potential hazards of the study. The investigator must also explain to the subjects that they are completely free to refuse to enter the study or to withdraw from it at any time for any reason.
The Informed Consent and Subject Information will be provided in the local language/Dutch.
10.1.4 InsuranceThe sponsor/investigator has a liability insurance which is in accordance with article 7, subsection 6 of the WMO.The sponsor/investigator (also) has an insurance which is in accordance with the legal requirements in the Netherlands (Article 7 WMO and the Measure regarding Compulsory Insurance for Clinical Research in Humans of 23rd June 2003). This insurance provides cover for damage to research subjects through injury or death caused by the study.
€ 650,000.- (i.e., six hundred and fifty thousand Euro) for death or injury for each subject who participates in the Research;
€ 5,000,000.- (i.e., five million Euro) for death or injury for all subjects who participate in the Research;
€ 7,500,000.- (i.e., seven million and five hundred thousand Euro) for the total damage incurred by the organisation for all damage disclosed by scientific research for the Sponsor as ‘verrichter’ in the meaning of said Act in each year of insurance coverage.
The insurance applies to the damage that becomes apparent during the study or within 4 years after the end of the study.
10.2 Study fundingxxx is the sponsor of the study and is funding the study. All financial details are provided in the separate contract(s) between the CHDR, and the sponsor. CHDR is the sponsor of the study and is funding the study.
10.3 Data handling and record keeping10.3.1 Data collection
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 42 of 48

CHDR9999
Data will be recorded on electronic data collection forms in Promasys and will be entered after quality control in a sponsor eCRF for subsequent tabulation and statistical analysis. The data will be handled confidentially.A Subject Screening and Enrolment Log will be completed for all eligible or non-eligible subjects with the reasons for exclusion.
Data will be recorded on paper data collection forms and will be entered after quality control in a Promasys database and subsequently in a sponsor eCRF for tabulation and statistical analysis. The data will be handled confidentially.A Subject Screening and Enrolment Log will be completed for all eligible or non-eligible subjects with the reasons for exclusion.
10.3.2 Database management and quality controlAll data from paper source will be entered into the Promasys database twice, by two different individuals. A quality control check will be done by CHDR staff on all data entered in the Promasys database, using data entry progress checks and database listings (blind data review). Errors with obvious corrections will be corrected before database lock. Results of computer (NeuroCart/PainCart/ECG) tests and electronically captured questionnaires, clinical laboratory and pharmacokinetic analyses will be sent electronically to CHDR and loaded into the database.After the database has been declared complete and accurate, the database will be locked. Any changes to the database after that time can only be made by joint written agreement between the investigator, sponsor and the statistician.
10.4 Access to source data and documentsAll study data will be handled confidential. The investigator will retain the originals of all source documents generated at CHDR for a period of 2 years after the report of the study has been finalised, after which all study-related documents will be archived (at a minimum) on micro-film which will be kept according to GCP regulations. After 2 years the sponsor will be notified that the source documents can be retained with the sponsor or destroyed.The investigator will permit trial-related monitoring, audits, EC review and regulatory inspections, providing direct access to source data and documents.
10.5 Quality control and quality assuranceThis study will be conducted according to applicable Standard Operating Procedures (SOPs). Quality assurance will be performed under the responsibility of CHDR’s Quality Assurance manager.
10.5.1 MonitoringAn initiation visit will be performed before the first subject is included. Monitoring visits and contacts will occur at regular intervals thereafter, according to a frequency defined in the study-specific monitoring plan. A close-out visit will be performed after study closure.
10.6 Protocol amendmentsAny change to a protocol has to be considered as an amendment.
10.6.1 Substantial amendmentSignificant changes that affect subject safety and/or the scientific value of a trial require a substantial amendment. Examples of significant changes are given in EU guidelines on the request to the competent authorities for authorisation of a clinical trial on a medicinal product for human use, the notification of substantial amendments and the declaration of the end of the trial (CT-1, 2010/C 82/01). The need for submitting a substantial amendment is the responsibility of the sponsor. Substantial amendments are to be approved by the appropriate EC and the CA will need to provide
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 43 of 48

CHDR9999
a ‘no grounds for non-acceptance’ notification prior to the implementation of the substantial amendment.
10.6.2 Non-substantial amendmentNon substantial amendments do not affect subject safety or the scientific integrity of the trial. Non-substantial amendments will be approved (signed) by the investigator(s) and will be recorded and filed by the investigator/sponsor. Non-substantial amendments will be submitted to the EC for information only. The CA will only be notified by changes in Eudract form and ABR form (if applicable) at toetsingonline. The implementation of a non-substantial amendment can be done immediately.
The EU guideline CT-1 2010/C 82/01 stipulates the importance of preventing over-reporting. Therefore the following changes are by definition non-substantial in this study:
- change in amount and timing of the samples (maximum of 2 samples without a > 50 ml increase in the amount of blood taken and not exceed 500 ml of blood in total)
- changes in assay-type and / or institution where an assay will be performed, provided that validated assays will be used;
- editorial changes to documents in the submission dossier including the volunteer information sheets and the protocol. An editorial change is defined as a modification in the documents of typographical errors and other modifications that in no way alter the meaning or content of the document
- determination of additional parameters in already collected materials, which are in agreement with the study objectives and do not provide prognostic or genetic information;
- other statistical analyses than described in the protocol.- A change in clinical staff, including the principal investigator, when this concerns regular staff
members of CHDR who comply with internal regulations for training and authorisation.- A change in dosing schedule in an ascending dose trial, provided the expected exposure of
the subjects does not exceed the preset values indicated in this protocol.
10.6.3 Urgent amendmentAn urgent amendment might become necessary to preserve the safety of the subjects included in the study. The requirements for approval should in no way prevent any immediate action being taken by the investigators or the sponsor in the best interests of the subjects. Therefore, if deemed necessary, an investigator can implement an immediate change to the protocol for safety reasons. This means that, exceptionally, the implementation of urgent amendments will occur before submission to and approval by the EC(s) and CA.
10.7 End of study reportThe sponsor will notify the EC and the CA of the end of the study within a period of 90 days. The end of the study is defined as the last subject’s last visit. In case the study is ended prematurely, the investigator/sponsor will notify the EC and the CA within 15 days, including the reasons for the premature termination.
The sponsor will notify the EC immediately of a temporary halt of the study, including the reason of such an action.
Within one year after the end of the study, the investigator and/or sponsor will submit a final study report with the results of the study, including any publications/abstracts of the study, to the EC and the CA. The principal investigator and the sponsor’s representative will be the signatories for the study report.
The investigator will notify the EC of the end of the study within a period of 8 weeks. The end of the study is defined as the last subject’s last visit. In case the study is ended prematurely, the
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 44 of 48

CHDR9999
investigator/sponsor will notify the EC within 15 days, including the reasons for the premature termination.
The sponsor will notify the EC immediately of a temporary halt of the study, including the reason of such an action.
Within one year after the end of the study, the investigator and/or sponsor will submit a final study report with the results of the study, including any publications/abstracts of the study, to the EC. The principal investigator and the sponsor’s representative will be the signatories for the study report.
10.8 Public disclosure and publication policyIn accordance with standard editorial and ethical practice, the results of the study will be published, if applicable. The authorship guidelines of the Vancouver Protocol8 will be followed regarding co-authorship.
The principal investigator will have the opportunity to review the analysis of the data and to discuss with the sponsor the interpretation of the study results prior to publication.
Any study-related article or abstract written independently by investigators should be submitted to the sponsor for review at least 60 days prior to submission for publication or presentation.
The list of authors of any formal publication or presentation of study results may include, as appropriate, representatives of the sponsor and will be determined by mutual agreement.
In accordance with standard editorial and ethical practice, the results of the study will be published, if applicable. Conditions are subject to a separate contract between CHDR and the sponsor. The authorship guidelines of the Vancouver Protocol9 will be followed regarding co-authorship.
8 http://www.icmje.org/9 http://www.icmje.org/Protocol Version 1 / 13-Aug-2018 <Confidential> Page 45 of 48

CHDR9999
11 STRUCTURED RISK ANALYSIS
11.1 Potential issues of concern11.1.1 Level of knowledge about mechanism of action11.1.2 Previous exposure of human beings with the test product(s) and/or products with a similar biological mechanism11.1.3 Can the primary or secondary mechanism be induced in animals and/or in ex-vivo human cell material?11.1.4 Selectivity of the mechanism to target tissue in animals and/or human beings11.1.5 Analysis of potential effect11.1.6 Pharmacokinetic considerations11.1.7 Study population11.1.8 Interaction with other products11.1.9 Predictability of effect11.1.10 Can effects be managed?
11.2 Synthesis
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 46 of 48

CHDR9999
12 REFERENCES
In the seminal research investigating the CNS effects of lamotrigine … [1].
1. Cohen AF, Ashby L, Crowley D, Land G, Peck AW, Miller AA. Lamotrigine (BW430C), a potential anticonvulsant. Effects on the central nervous system in comparison with phenytoin and diazepam. Br J Clin Pharmacol 1985; 20: 619-629.
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 47 of 48

CHDR9999
APPENDIX 1 TITLE
Protocol Version 1 / 13-Aug-2018 <Confidential> Page 48 of 48





























