Embed Size (px)
DESCRIPTION
biologia
Citation preview

Proyecto RLA/1/010
Mejora de la Gestión de la Contaminación de Masas de Aguas
Superficiales Contaminadas con Metales, ARCAL
PROPUESTA de un
MA%UAL DE PROTOCOLOS
armonizados y evaluados PARA LA
TOMA DE MUESTRA Y EL A%ALISIS
DE AGUAS Y SEDIME%TOS para la Región de Latinoamérica y del Caribe
REPORTE FI%AL
Mayo de 2009

1
INDICE 1. EL PROYECTO 2
1.1. Objetivo General 3
1.2. Objetivos Específicos 3
2. TALLER DE ELABORACION DE UN MANUAL DE PROTOCOLOS
ARMONIZADOS Y EVALUADOS 6
2.1. Objetivo 6
2.2. Agenda 7
2.3. Conclusiones del Taller 9
2.3.1. Protocolos Consensuados y Evaluados 9
2.3.1.1. Plan Muestreo General (PT-Mu-01) 10
2.3.1.2. Muestreo Agua Superficial (PT-MuA-01) 34
2.3.1.3. Demanda Bioquímica de Oxígeno (DBO5) en aguas superficiales (PT-FQ-01) 50
2.3.1.4. Amonio (NH4+) - Método titulométrico previa destilación (PT-FQ-02) 65
2.3.1.5. Determinación de amonio (NH4+) - Método fenato (PT-FQ-03) 69
2.3.1.6. Determinación de coliformes fecales - Método de Filtración por
Membrana (PT-FQ-04) 73
2.3.1.7. Determinación de coniformes fecales - Método del número más probable
(PT-FQ-05) 85
2.3.1.8. Análisis de Turbiedad o Turbidez (PT-FQ-06) 90
2.3.1.9. Determinación de Nitratos (NO3-) previa reducción con cadmio o hidracina
(PT-FQ-07) 94
2.3.1.10. Determinación de nitratos (NO3-). Método del electrodo de nitrato (PT-FQ-08) 104
2.3.1.11. Determinación de oxígeno disuelto por el método iodométrico - modificación
azida (PT-FQ-09) 109
2.3.1.12. Determinación de fósforo total - Método del ácido Ascórbico (PT-FQ-10) 115
2.3.1.13. Determinación de fósforo total - Método de molibdato de amonio y cloruro de
estaño II (PT-FQ-11) 122
2.3.1.14. Determinación de fósforo total - Método del molibdovanadato de amonio
(PT-FQ-12) 129
2.3.1.15. Determinación de sólidos totales disueltos secados a 180 °C (PT-FQ-13) 136
2.3.1.16. Determinación de sólidos totales suspendidos (PT-FQ-14) 141
2.3.1.17. Nitratos por cromatografía iónica (PT-FQ-15) 146
2.3.1.18. Determinación de pH por medición potenciométrica (PT-FQ-16) 150
2.3.1.19. Determinación de oxígeno disuelto por el método electrométrico (PT-FQ-17) 155
2.3.1.20. Determinación de solutos ionizables (PT-FQ-18) 160
2.3.1.21 Digestión ácida de aguas para análisis de metales recuperables totales o

2
metales disueltos por GFAAS (PT-DA-02) 166
2.3.1.22. Determinación de cadmio, cromo, plomo, arsénico y cobre en aguas
superficiales por ICP-OES (PT-MA-01) 169
2.3.1.23. Determinación de cadmio, cobre, cromo y plomo total en agua superficial
mediante GFAAS (PT-MA-02) 179
2.3.1.24. Determinación de As, Co, Cr, Cs, Fe, Rb, Sb, Sr, U, Zn en muestras de agua
mediante el análisis por activación neutrónica, método del k subcero (PT-MA-04) 187
2.3.1.25. Determinación de Cadmio por el Método de Espectrometría de Absorción
Atómica con Aspiración Directa (PT-MA-05) 194
2.3.1.26. Determinación de Cromo por el Método de Espectrometría de Absorción
Atómica con Aspiración Directa (PT-MA-06) 199
2.3.1.27. Determinación de Cobre por el Método de Espectrometría de Absorción
Atómica con Aspiración Directa (PT-MA-07) 204
2.3.1.28. Determinación de Plomo por el Método de Espectrometría de Absorción
Atómica con Aspiración Directa (PT-MA-08) 208
2.3.1.29. Determinación de Arsénico Total por el Método de Espectrometría de
Absorción Atómica con Generación de Hidruros (PT-MA-09) 213
2.3.1.30. Determinación de Mercurio total por el método de Espectrometría de
Absorción Atómica con Generación de Vapor Atómico de Mercurio -
Vapor Frío (PT-MA-10) 223
2.3.1.31. Pretratamiento y Preparación de Muestras para análisis de Sedimentos
(PT-PS-01) 229
2.3.1.32. Digestión ácida total de sedimentos utilizando microondas (PT-DS-01) 233
2.3.1.33. Digestión ácida total de sedimentos en sistema abierto (PT-DS-02) 236
2.3.1.34. Digestión ácida total de sólidos en suspensión (PT-DS-03) 239
2.3.1.35. Determinación de cadmio, cromo, plomo, arsénico y cobre en sedimentos
por ICP-OES (PT-MS-01) 242
2.3.1.36. Determinación de cadmio, cobre y plomo total en sedimentos mediante
GFAAS (PT-MS-02) 251
2.3.1.37. Determinación de As, Co, Cr, Cs, Fe, Rb, Sb, Sr, U, Zn en muestras de
sedimentos mediante el análisis por activación neutrónica, método del
k subcero (PT-MS-04) 259
2.3.1.38. Determinación de Cadmio total en muestras de sedimentos por el Método
de Espectrometría de Absorción Atómica (PT-MS-06) 265
2.3.1.39. Determinación de Cromo en sedimentos por el Método de Espectrometría de
Absorción Atómica (PT-MS-07) 269
2.3.1.40. Determinación de Cobre en sedimentos por el Método de Espectrometría de

3
Absorción Atómica (PT-MS-08) 274
2.3.1.41. Determinación de Plomo en sedimentos por el Método de Espectrometría de
Absorción Atómica (PT-MS-09) 280
2.3.1.42. Determinación de Arsénico Total en sedimentos por el Método de
Espectrometría de Absorción Atómica con Generación de Hidruros (PT-MS-10) 285
2.3.1.43. Determinación de Mercurio en sedimentos por el método de Espectrometría
de Absorción Atómica con Generación de Vapor Atómico de Mercurio - Vapor
Frío (PT-MS-11) 293

4
PROPUESTA DE UN MANUAL DE PROTOCOLOS
ARMONIZADOS Y EVALUADOS PARA LA TOMA DE AGUA Y
SEDIMENTOS
1. EL PROYECTO
El Proyecto ARCAL RLA/1/010 “Mejora de la gestión d e las masas de agua
que están contaminadas con metales (ARCAL)”
El proyecto propone armonizar protocolos y capacitar los recursos humanos
necesarios para la evaluación de la calidad del agua y el transporte de metales
en cuerpos de agua superficiales en países de la región de Latinoamérica con
problemas de contaminación con metales (natural o antropogénica) aplicando
técnicas analíticas nucleares y complementarias, incluyendo el empleo de
trazadores.
Se trata de un proyecto ARCAL (Acuerdo Regional de Cooperación para la
Promoción de la Ciencia y Tecnología Nucleares en América Latina y El Caribe)
que financia el OIEA (Organismo Internacional de Energía Atómica) y que dió
comienzo el 1 de enero de 2007, con una duración de 2 años.
Participan del mismo, Argentina, Bolivia, Brasil, Chile, Costa Rica, Cuba, El
Salvador, México, Perú, República Dominicana, Uruguay y Venezuela. Cada uno
de estos países ha elegido un ecosistema relevante donde poder desarrollar el
proyecto.
Existen instituciones gubernamentales y no gubernamentales ocupadas de la
gestión del recurso agua en la región. Los usuarios directos de los productos de
este proyecto serán aquellas instituciones que velan por la calidad y uso
sustentable del recurso hídrico y las dedicadas a la formulación de leyes,
normas y criterios regulatorios de la calidad del agua. Estos usuarios se

5
beneficiarán con la disponibilidad de protocolos armonizados y con recursos
humanos formados para la evaluación integral de la calidad del agua y el
transporte de contaminantes en cuerpos de aguas superficiales. Así mismo
coadyuvará al desarrollo de normas y criterios en materia de agua de los países
participantes.
1.1. Objetivo General
Armonizar protocolos y capacitar los recursos humanos necesarios para la
evaluación de la calidad del agua y el transporte de metales en cuerpos de agua
superficiales en países de la región de Latinoamérica y el Caribe con problemas
de contaminación con metales (natural o antropogénica) aplicando técnicas
analíticas nucleares y complementarias, incluyendo el empleo de trazadores.
1.2. Objetivos Específicos
• Proponer índices de calidad del agua (ICA) que puedan ser aplicados en los
países de la región.
• Desarrollar criterios para el diseño y establecimiento de bases de datos que
permitan soportar modelos de dispersión de contaminantes en aguas
superficiales, sedimentos y biota.
• Armonizar y evaluar protocolos; en particular, de diseño muestral, toma de
muestra, medición, análisis de resultados y reporte para la evaluación de la
calidad de los cuerpos de aguas superficiales con elementos ecotóxicos,
utilizando técnicas analíticas nucleares, complementarias y trazadores.
• Capacitar recursos humanos en la aplicación de estrategias y técnicas
quimiométricas y de modelado de dispersión de contaminantes.
Como resultado de la ejecución del proyecto se espera:
• Mejorar la gestión del recurso agua superficial a nivel regional por el
empleo de Índices de Calidad del Agua armonizados y evaluados.

6
• Mejorar la capacidad de predicción de los modelos utilizados en
evaluación de la calidad del agua y el transporte de elementos ecotóxicos
en cuerpos de agua superficiales.
• Mejorar la confiabilidad, reproducibilidad y aplicabilidad de los resultados
para su comparación, de manera de poder afrontar problemáticas
comunes en la región.
• Mejorar las capacidades de los grupos de la región para asumir nuevos
desafíos y sus efectos multiplicadores en la formación de recursos
humanos, contribuyendo a la sustentabilidad del proyecto.
2. TALLER DE ELABORACION DE UN MANUAL DE PROTOCOLOS
ARMONIZADOS Y EVALUADOS PARA LA TOMA DE MUESTRAS Y
ANÁLISIS DE AGUA Y SEDIMENTO (SAN SALVADOR, 5 AL 9 DE MAYO
DE 2008)
2.1. Objetivo
En concordancia con el tercer objetivo específico, se desarrolló un Taller
Regional de elaboración de un Manual de Protocolos Armonizados y Evaluados
para la Toma de Muestras y Análisis de Agua y Sedimento del 5 al 9 de Mayo de
2008 en San Salvador, El Salvador. En esta reunión se acordó evaluar y
consensuar protocolos de técnicas de muestreo y análisis de aguas superficiales
y sedimentos; que incluyeron los correspondientes a los parámetros
(indicadores) utilizados por el índice de calidad de aguas (ICA) consensuado en
el Taller de Río de Janeiro, para los países de la región.
Este taller es producto de la necesidad de proponer protocolos armonizados,
con la finalidad de minimizar los errores de muestreo y análisis de agua y
sedimento que inciden significativamente sobre la calidad de los datos
ambientales (en particular los correspondientes a los parámetros del índice de
calidad de agua), para todos los países de la región de Latinoamérica y del
Caribe.

7
2.2. Agenda
Lunes, 5 de Mayo
08:00 - 08.30 Inscripción de participantes 08:30 - 09:00 Apertura de la reunión y bienvenida
� Bienvenida, a cargo de Lic. Maribel Quintanilla, Directora de Cooperación Multilateral del Ministerio de Relaciones Exteriores de El Salvador, Enlace Oficial de El Salvador con el OIEA.
� Descripción del proyecto y del eveno, a cargo de Sra. E. Zeiller. Oficial Técnico, OIEA
� Inauguración del Evento, a cargo del Ing. Carlos José Guerrero Contreras, Ministro de Medio Ambiente y Recursos Naturales de El Salvador.
09:00 - 09:15 Presentación de las actividades durante el Taller (Sra Deisy
Lopez) 09:15 - 09:30 Presentación de los participantes 09:30 - 10:00 Aseguramiento de la Calidad de Muestreo. (Lisa Zeiller) 10:00 - 10:30 Café 10:30 - 12:00 Resumen de las metodologías utilizadas para muestreo
(Representante del grupo de trabajo) 12:00 - 13:30 Almuerzo 13:30 - 15:00 Discusión metodologías muestreo parámetros índice de calidad
de agua 15:00 - 15:30 Café 15:30 - 17:00 Continúa discusión
Martes, 6 de Mayo 08:30 - 10:00 Presentación of compilación medición de elementos traza
(Representante del grupo de trabajo) 10:00 - 10:30 Café 10:30 - 12:00 Presentación resultados ensayo de aptitud de laboratorio
regional (Sr. Luis Muñoz) 12:00 - 13:30 Almuerzo 13:30 - 15:00 Discusión resultados ensayo de aptitud de laboratorio regional 15:00 - 15:30 Café 15:30 - 17:00 Discusión de los métodos de medición de elementos traza del
ICA

8
Miércoles, 7 de Mayo 08:30 - 10:00 Presentación métodos compilados para la medición de los 9
parámetros de calidad general del agua (pH, turbidez, etc.). (Representante del grupo de trabajo)
10:00 - 10:30 Café 10:30– 12:00 Discusión sobre métodos compilados para la medición de los 9
parámetros de calidad del agua. 12:00 - 13:30 Almuerzo 13:30 - 15:00 Continúa discusión 15:00 - 15:30 Café 15:30 - 17:00 Continúa discusión
Jueves, 8 de Mayo
08:30 - 10:00 Trabajo por grupos para redacción propuestas metodologías armonizadas. Escribir los protocolos para el muestreo y el análisis de los parámetros y elementos traza (1 o 2 métodos), incluyendo referencias y estándares internacionales
10:00 - 10:30 Café 10:30 - 12:00 Continúa trabajo por grupos 12:00 - 13:30 Almuerzo 13:30 - 15:00 Presentación de los resultados del trabajo en grupos y discusión 15:00 -15:30 Café 15:30 - 17:00 Continúa Discusión 19:00 Cena oficial
Viernes, 9 de Mayo
08:30 - 10:00 Redacción informe Final 10:00 - 10:30 Café 10:30 - 12:30 Redacción informe Final 12:30 - 13:30 Almuerzo 13:30 - 15:00 Última revisión del Manual de protocolos armonizados 15:00 - 15:30 Café 15:30 - 17:00 Aprobación del informe final de la reunión. Discusión sobre
futuras acciones en el proyecto. Clausura
A la reunión asistieron profesionales de las instituciones participantes
relacionados con la elaboración e implementación de planes de monitoreo, de
análisis de las muestras y evaluación de la calidad de los resultados analíticos
de los 12 países participantes del proyecto.

9
2.3. Conclusiones del Taller
La Tabla 1 presenta los procedimientos, formularios o registros
consensuados en este taller.
Tabla 1
Procedimiento Técnicos
/ Formularios, PT & F
Grupos de procedimientos y
formularios
Número (01, 02..)
PT o F DA (Digestión Agua)
PT o F DS (Digestión Sedimentos)
PT o F MA (Metal Agua)
PT o F MS (Metal Sedimentos)
PT o F MuA (Muestreo Agua)
PT o F MuS (Muestreo Sedimentos)
PT o F Mu (Muestreo general)
PT o F FQA (Fisicoquímico Agua)
Ejemplo: Código Procedimiento PT.FQA.04
2.3.1. Protocolos Consensuados y Evaluados
A continuación se presentan los protocolos que han sido consensuados y
evaluados.

10
2.3.1.1. Plan Muestreo General
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales”
Código del procedimiento: PT-Mu-01
Nombre del procedimiento: Plan Muestreo General
Fecha de aprobación: 2008-05-08
Fecha de revisión: 2008-05-08
1. OBJETIVOS
1.1. Establecer los lineamientos generales para planificar una campaña de
muestreo en aguas superficiales (ríos, lagos, embalses, arroyos, canales,
estanques y ambientes costeros) y sedimentos.
1.2. Recolectar una parte representativa de una población, que sea lo
suficientemente pequeña para ser transportada y lo suficientemente
grande para propósitos analíticos
1.3. Garantizar que no se produzcan alteraciones significativas en su
composición física, química o biológica que pudieran alterar su
representatividad.
1.4. Armonizar las actividades de colección de muestras de agua y
sedimentos para todos los Grupos de trabajo en América Latina que se
interesen en calcular el ICA (Taller Río de Janeiro 2007) en aguas y que
quieren mejorar su métodos de muestreo.
2. ALCANCE
Este procedimiento es aplicable a todos los parámetros seleccionados para
medir el índice de calidad de agua del proyecto ARCAL RLA 1010 (Mejora
de la Gestión de la Contaminación de Masas de Agua que están
contaminadas con Metales).
2.1. PARÁMETROS DE MEDICIÓN EN CAMPO

11
pH, Temperatura, Conductividad, Oxígeno Disuelto (OD)
2.2. PARÁMETROS GENERALES Y ELEMENTALES DE MEDICIÓN EN
LABORATORIO
Turbiedad y/o Sólidos totales suspendidos, Nitratos, Ión Amonio, Fósforo
total, Demanda Bioquímica de Oxígeno después de 5 días (DBO5)
Elementos químicos: Cd, Cr, Pb, Cu, Hg y As
2.3. PARÁMETROS BIOLÓGICOS
Coliformes fecales
Nota: temperatura no es un parámetro necesario para calcular el ICA pero es
necesario para establecer factores de corrección en la medición de otros
parámetros.
3. ACCIONES PREVIAS:
Todos los grupos de trabajo pueden seguir los procedimientos relacionados a
tópicos de muestreo.
4. COLECCIÓN DE INFORMACIÓN
Para preparar un PLAN DE MUESTREO se debe coleccionar al menos la
siguiente información:
4.1. Objetivo del muestreo
• Razón de muestreo y el uso de los resultados que se obtendrán de las
muestras colectadas
• Parámetros que van ha ser medidos
• Limites de cuantificación necesarios para decisiones
Nota: La razón del muestreo esta definida por el proyecto RLA1010

12
Nota: Considerar la posibilidad de medir los 15 parámetros de ICA
4.2. Descripción del cuerpo de agua que va ha ser evaluado
4.2.1. Extensión y volumen de agua: profundidad, caudal y batimetría
4.2.2. Topografía (Cartografía de la región)
4.2.3. Afluentes y tributarios (ríos, canales, acequias etc.)
4.2.4. Fuentes de contaminación fijas y difusas naturales y
antropogénicas (actividades reflejadas en el uso del suelo de la
cuenca: industriales, agrícolas, turísticas, extractivas como la
minería, centros urbanos, escorrentía, etc.)
4.2.5. Información sobre la zona de muestreo
4.2.6. Recopilación de información de campañas de muestreo previas y
sus resultados para evaluar:
4.2.7. Variabilidad espacial y temporal
4.2.8. Heterogeneidad debido a: contaminación de fuentes puntuales y
difusas, topografía, batimetría, caudal, etc.
4.2.9. Accesibilidad de los puntos de muestreo
4.2.10. Tiempo necesario para el traslado y muestreo desde cada
zona de interés
4.3. Métodos que están disponibles para las mediciones y el muestreo
4.3.1. Cantidad de agua o sedimento necesarios para medir los
parámetros con una incertidumbre aceptable
4.4. Métodos no disponibles en bibliografía
4.4.1. Prever desarrollo de métodos de muestreo y medida

13
5. Consideraciones y decisiones previas
5.1. Selección de puntos para muestrear
Para seleccionar los puntos de muestreo se pueden utilizar todas las
informaciones compiladas acordes a la razón del muestreo. Se
recomiendan las siguientes zonas de muestreo:
5.1.1. El origen de la fuente (para obtener información de base y la
composición natural del agua)
5.1.2. Para cada tributario y afluentes importantes, seleccionar dos sitios
de muestreo – uno arriba y uno abajo del punto de inserción.
5.1.3. Puntos de fuentes de contaminación o contaminación previsible.
5.1.4. El sitio extremo
5.1.5. Cuando el muestreo se realize en cuerpos lénticos tales como
lagos, presas, embalses y esteros, se colectarán las muestras de
afluentes y efluentes
Nota: Si existe estratificación horizontal o vertical en lagunas o estuarios, se
deben considerar muestras de aguas adicionales.
Hay factores adicionales que influyen en la decisión de la selección de los
sitios de muestreo. Estos son factores relacionados con los recursos
económicos de un proyecto y la logística de una campaña de muestreo:
5.1.6. Accesibilidad del sitio para humanos y/o vehículos
5.1.7. Disponibilidad de vehículos terrestres y fluviales apropiados
5.1.8. Existencia y accesibilidad de las rutas para transportes (tiempo
necesario entre las zonas seleccionadas)

14
5.1.9. Recursos humanos disponibles con conocimiento y entrenamiento
necesario para las actividades previstas
5.1.10. Calidad y cantidad de recipientes (envases), materiales,
reactivos y equipos indispensables
Nota: Puede ser recomendable seleccionar menos puntos, pero no reducir la
calidad del muestreo.
6. LINEAMIENTOS GENERALES PARA UN MUESTREO REPRESENTATIVO
6.1. Las muestras deben ser representativas de las condiciones que existan
en el sitio a la hora de muestreo y tener el volumen suficiente, para
efectuar con él las determinaciones correspondientes.
6.2. Evitar zonas de embalse o turbulencias no características del cuerpo de
agua, a menos que sean el objeto de la evaluación.
6.3. Elegir un punto donde el río esté lo mas regular, accesible y uniforme en
profundidad.
6.4. Restricciones al muestreo
6.4.1. Después de un período de lluvia las muestras se tomarán cuando
el cuerpo de agua haya recuperado sus condiciones hidrológicas
normales (caudal y cota).
6.4.2. Si el cuerpo de agua no es caudaloso y voluminoso no se debe
muestrear al medio día o con temperatura ambiente elevada (a
menos que este sea el objeto del estudio).
6.5. Considerar varios tipos de cuerpos de agua según su profundidad

15
Tabla 1
Profundidad del punto de muestreo (m)
Profundidad de muestreo por debajo de la superficie (cm)
≤ 1,5 entre 20 -50
> 1,5 entre 20 -50 y profundidad media
6.6. Cuando se muestrea un punto aguas abajo de un afluente debe hacerse
a una distancia tal que se haya producido el mezclado completo de las
aguas. Esta distancia depende del caudal de rio, de la temperatura de las
dos corrientes de agua, de la densidad y del flujo de ambos.
6.7. El punto de mezclado se determina en forma precisa utilizando
trazadores conservativos, por ejemplo: conductividad, cloruros, etc.
En caso de no disponer de esta tecnología, puede estimarse como:
El flujo mayor de las dos corrientes x tiempo estimado de mezcla (entre 3
y 5 minutos).
Ejemplo para 5 minutos y flujo de 1,25 m/s.
Distancia=1,25 (m/s) x 300 s=375 m
Nota: La zona de mezcla completa puede a veces apreciarse directamente
(por ejemplo apreciación de color, conductividad o temperatura uniformes)
6.8. Elegir la distancia de muestreo desde la orilla del cuerpo de agua: Si es
posible colectar una muestra en el centro del cuerpo de agua y entre 1 y
2.5 m de distancia a ambos lados de la orilla.
6.9. Seleccionar una distancia apropiada para la toma de muestra, corriente
arriba del afluente. si es posible al menos 100m.
6.10. Cuando se trate de cuerpos de bajo flujo que desembocan en el
mar, o en estuarios, considerar la influencia de la contaminación marina
de la zona costera en ese sector del río. Esta influencia debe

16
determinarse a partir del seguimiento de trazadores conservativos tales
como: CE, concentración de cloruros o trazadores radiactivos naturales.
6.11. En caso de no poder seguirse las consideraciones anteriores, se
recomienda tomar los valores presentados en la Tabla 2
Tabla 2
TIPO DE CORRIENTE SEGMENTO A CONSIDERAR
Rio grande (más de 20 m de ancho) 10 km arriba del sitio de muestreo
Rio mediano (10 a 20 m de ancho) 5 km arriba del sitio de muestreo
Rio pequeño (3 a 9 m de ancho) 3 km arriba del sito de muestreo
Arroyo (menos de 3 m) 1 km arriba del sitio de muestreo
7. Número y tipo de muestra
El número de muestras a tomar depende de la variabilidad espacial, temporal
(estacional), cotas máxima y mínima del cuerpo de agua en estudio, la clase
de muestreo seleccionado (ver tabla 3), numero de afluentes y fuentes de
contaminación existentes. El número de muestras puede ser el balance
realista y practicable entre un número ideal de muestras (estadístico),
usualmente numeroso y un número elegido por juicio profesional. El muestreo
simple contiene información detallada del punto muestreado y momento de
muestreo. Por el contrario el muestreo compuesto reúne información
promedio de los sitios muestreados disminuyendo el número de muestras
pero perdiéndose la información de los puntos y momentos críticos de
contaminación.
Tabla 3
Tipos de muestreo
Definición
Muestra Simple Muestra individual tomada en un punto, a una profundidad y en un tiempo determinado.
Muestra Compuesta
Muestra obtenida mezclando en un recipiente varias muestras discretas o simples de volúmenes iguales o

17
ponderados
Azaroso Muestreo realizado sobre el tendido de una malla imaginaria en la cual se seleccionan nodos al azar
Sistemático Muestreo realizado sobre el tendido de una malla imaginaria en la cual se seleccionan nodos en forma regular
Juicio profesional
Muestreo realizado de acuerdo al criterio sostenido por el profesional responsable
8. CONSIDERACIONES ESTADÍSTICAS
Hay más de un modo de calcular el número apropiado de muestras. El mas
simple depende de un aceptable nivel de incertidumbre (U), de la desviación
estándar relativa del sistema (s) y de la desviante de la distribución de
Student (t), que a su vez depende del número de muestras (N) y de la
confiabilidad requerida en la medida (p).
Nota: Observe que el cálculo de N es implícito porque depende de t, que a su
vez depende de N.
2
),( .
≥
U
st�
p� ec1
El valor de s puede estar referido a diferentes consideraciones de la
variabilidad, por ejemplo la variabilidad espacial, temporal o del método de
medida.
Ejemplo para un sitio:
Cálculo usando la variabilidad espacial: U=5% , s=5% , p=0.95
N≥5
Cálculo usando la variabilidad del método de medida ICP-MS: U=5%, s=0.1%,

18
p=0.95
N≥1
En este ejemplo se muestra que se deberían tomar 5 muestras y hacer una
medida para cada una de ellas obteniéndose una confiabilidad final del 0.90
(0.95x0.95) dentro de una incertidumbre del 5%.
Si el valor estimado se encuentra entre cerca del límite de detección del
método se debe utilizar un algoritmo de cálculo más complejo, ver
www.acs-envchem.duq.edu/dqopro.htm
9. CONSIDERACIONES PRÁCTICAS
El número de muestras esta dictado por consideraciones prácticas tales
como:
9.1. Recursos disponibles (humanos y económicos)
9.2. Materiales y reactivos disponibles para tomar, conservar y garantizar la
calidad de muestreo.
9.3. Tiempo necesario para muestrear y transportar
9.4. Capacidad de análisis del laboratorio
9.5. Tiempo límite para su procesamiento (maximum holding time MHT)
10. REDUCCIÓN DEL NÚMERO DE MUESTRAS
Para reducir el número de muestras estimadas sin perder información
importante se puede aplicar el juicio profesional. Algunas posibilidades son:
10.1. Reducción del número de sitios y/o puntos de muestreo
o Similar topografía y geología
o Similares resultados para muchos parámetros

19
10.2. Reducción del número de muestras simples utilizando la técnica de
“muestras compuestas”
o Pequeña variabilidad en un sitio
Ambas decisiones pueden depender de los resultados de las visitas al campo,
campaña exploratoria o como consecuencia de los resultados de la primera
campaña de muestreo.
11. FRECUENCIA
La frecuencia de muestreo esta relacionada con el período de la variabilidad
estudiada. Esta puede ser debida a estaciones del año, periodos de sequía o
lluvias, cotas máxima y mínima del sistema, frecuencia de volcado de
contaminantes, etc.
El ICA seleccionado en el proyecto RLA 1010 requiere un mínimo de 4
campañas por año.
12. EQUIPOS de MUESTREO
12.1. Manuales
Si se dispone de una embarcación sea a motor o a remos, se debe
orientar la proa en contra de la corriente y muestrear desde ella con un
recipiente de acero inoxidable o plástico.
Desde la costa se puede muestrear empleando una vara que suspende
al recipiente.
12.2. Automaticos
Bombas centrífugas o peristáltica conectadas a mangueras de silicona de
calidad médica.

20
SEDIMENTOS
El muestreo y análisis del sedimento permite muchas veces observar
contaminación adsorbida en el material particulado en suspensión (MPS) que no
es posible detectar en el agua, debido a sus bajas concentraciones. En este
proyecto se decidió medir en MPS, solamente los elementos trazas.
Los sedimentos pertenecientes al lecho del cuerpo de agua superficial no
requieren una frecuencia estacional de muestreo ya que registran cambios en
períodos de años o décadas. Se debe prestar particular atención a la fracción de
tamaño de partícula arcilla.
13. SELECCIÓN DE PUNTOS DE MUESTREO
13.1. Para muestreo de MPS los puntos de muestreo deben coincidir
con los de muestreo de aguas.
13.2. Se debe considerar que los puntos elegidos sean accesibles al
muestreo en todas las campañas previstas.
13.3. Para muestrear el lecho se deben elegir zonas de acumulación de
sedimentos.
14. SELECCIÓN DEL TIPO DE MUESTREADOR
Para MPS se recomienda filtrar la muestra de agua en el sitio de muestreo
con filtro de 0.45 µm de tamaño de poro y dispositivo de vacío.
Hay dos tipos de muestreadores utilizados comúnmente para tomar muestras
de sedimentos de fondo: draga (grab) y cilíndricos (core). Los muestreadores
tipo draga no permiten controlar adecuadamente la ubicación y profundidad
de la muestra.
Para tomar muestras de sedimentos del lecho se utilizan los colectores
manuales o cilíndricos que se cierran al levantarlos.

21
14.1. Muestreadores Dragas
• Son recomendados para flujos muy lentos (lagunas, embalses,
etc.).
• Son utilizados para tomar muestras de la porción superficial del
sedimento de fondo y para comparaciones espaciales y temporales. Son
susceptibles a perder las fracciones más finas del sedimento y dispersar
material frente a la onda de presión generada por el muestreador.
14.2. Colectores Manuales
Se trata de recipientes o cuencos de plástico para colectar
muestras de sedimentos del lecho en su porción más superficial.
Son recomendados para cursos pequeños que pueden
muestrearse por vadeo.
14.3. Muestreadores Cilíndricos
• Se utilizan para tomar muestras de sedimentos de fondo para
comparaciones temporales y espaciales y para estudios estratigráficos.
• Pueden o no ser susceptibles a perder las fracciones más finas del
sedimento y dispersar material frente a la onda de presión generada por
el muestreador y compactar el material.
Nota: en cuerpos de agua con poca profundidad pueden utilizarse también
muestreadotes cilíndricos para suelos.
15. DETERMINACIÓN DEL NÚMERO DE MUESTRAS
El análisis de MPS debe realizarse para cada muestra de agua tomada.
El análisis del sedimento en el lecho es una información adicional para el
proyecto. Por razones prácticas (demanda de tiempo de muestreo, de
pretratamiento de muestras y análisis) un número elevado de muestras tales
como lo exigiría un muestreo estadístico no es económicamente justificable.

22
16. PRETRATAMIENTO DE MUESTRAS DE SEDIMENTO (MPS O LECHO)
16.1. Los pretratamientos de muestras deben evitar la incorporación de
contaminantes provenientes de los contenedores y equipos utilizados y
evitar pérdidas por evaporación de los componentes de la muestra o
calentamiento durante las operaciones de molienda y secado.
16.2. El secado del sedimento puede realizarse en estufa a ≤40ºC ó a
temperatura ambiente. En ambos casos debe evitarse la contaminación
por el aire ambiente y la contaminación cruzada.
16.3. Para el tamizado de la muestra se deben utilizar tamices de
plástico.
16.4. Para la molienda se deben utilizar morteros de ágata o de un
material ausente en el objetivo del estudio (por ejemplo wolframio, titanio,
hierro, óxido de circonio, etc)
16.5. Para los ataques químicos se deben utilizar utensilios de Teflon® y
métodos que eviten contaminación y pérdidas de elementos.
Nota: considerar la posibilidad de incluir blancos de molienda, tamizado y
ataque químico o en cualquier proceso que pudiese contaminar la muestra
17. CONSIDERACIONES PARA EL MUESTREO DE MEDICIONES EN EL
SITIO
17.1. Seleccionar los elementos de infraestructura, mecánicos y
eléctricos necesarios para las mediciones (provisión de energía,
transporte de instrumental, baterías, bombas, agitadores, muestreadores,
recipientes para residuos, etc.)
17.2. Seleccionar los materiales de limpieza adecuados para los
instrumentos de medición (solventes para lavar los electrodos, materiales
de secado, paños, papeles, etc.).

23
17.3. Seleccionar los estándares de calibración de los instrumentos de
medición.
17.4. Decidir si la medida se efctuará por inmersión directa de los
electrodos o con toma de muestras
17.5. Determinar el número de mediciones en cada punto
17.6. Considerar los elementos para realizar los blancos de equipo antes
y después de las mediciones
17.7. Considerar los elementos para el acondicionamiento posterior a las
mediciones del instrumental utilizado (preservación, embalaje y
transporte)
18. SELECCIÓN DE PROCEDIMIENTOS
Los procedimientos son dependientes del parámetro a medir, del método de
análisis y su límite de detección, de la cantidad de muestra necesaria para
alcanzar este límite, del método y equipo de muestreo.
Tomar conocimiento de los protocolos de muestreo y analisis para cada
parámetro.
19. SELECCIÓN DE RECIPIENTES
Los recipientes deben ser adecuados para el muestreo de cada parámetro,
método de análisis y muestreo. Los siguientes puntos pueden ser
considerados:
19.1. Material adecuado al parámetro de medición, por ejemplo: vidrio
borosilicato, polietileno de alta densidad, teflón.
19.2. Limpiar con un procedimiento apropiado para el método de
muestreo y preservación
19.3. Tomar en cuenta los problemas de contaminación y evaporación

24
19.4. Tamaño adecuado para la cantidad de muestra
19.5. Resistencia a la rotura o bien protegido para el transporte
19.6. Resistencia a la temperatura de almacenamiento
La persona responsable para preparar los recipientes debe estar entrenada
con todos los métodos para garantizar su acondicionamiento.
20. PRESERVACION Y ALMACENAMIENTO
Consideraciones necesarias para la preservación y almacenamiento son:
20.1. Las muestras requieren almacenamiento a temperatura de 4ºC y/o
preservación con químicos para mantener su integridad durante el
transporte y antes del análisis en el laboratorio.
20.2. Los preservantes químicos más comunes son ácido clorhídrico,
nítrico, sulfúrico e hidróxido de sodio del grado de pureza analítica
requerida por el método de análisis. Cumplir los protocolos de
manipulación adecuados.
20.3. Las preservaciones deben ser adecuadas a los parámetros a
determinar y deben tomar en cuenta los métodos de análisis
20.4. Las cajas conservdoras, cooler o coleman usadas para el
transporte de las muestras deberán ser apropiadas y de suficiente
tamaño para almacenar las muestras tomadas, materiales de empaque y
de enfriamiento (hielo, “ice pack”, etc).
El personal responsable de la preparación de los reactivos debe estar
capacitado para todos los procedimientos de preservación y
almacenamiento.
21. CADENA DE CUSTODIA
La cadena de custodia es un requisito de ISO 17025. Se deben seguir los
procedimientos establecidos en el laboratorio. Es importante que el código de

25
identificación sea adecuado para las distintas muestras de cada lugar, para
diferentes parámetros, diferentes puntos de muestreo (muestras simples),
diferentes métodos de preservación, tipo de muestreo, etc.
Se recomienda que la lista del tipo de muestras a tomar y sus códigos estén
definidos antes del muestreo.
22. PLANIFICACIÓN DE LA SECUENCIA Y DEL TIEMPO
La lista de muestras puede ser también la base para decidir la secuencia y el
tiempo para el muestreo. En general es importante que
22.1. El tiempo sea suficiente para:
o trasladarse entre los sitios de muestreo
o la operación de muestreo
o registrar las muestras
22.2. La secuencia sea planificada para evitar
o recorridos innecesarios o repetidos
o problemas de contaminación
Nota: Si se trata de muestrear un río se debe avanzar siempre en
contracorriente.
Para la planificación de la preservación de las muestras, se debe tener en
cuenta prioritariamente:
22.3. estabilidad de la muestra
22.4. tiempo límite para su procesamiento (maximum holding time MHT)
22.5. capacidad de almacenamiento y refrigeración de las muestras en el
laboratorio
22.6. capacidad de análisis del laboratorio

26
23. Contenido de protocolo de muestreo
El formato de contenido de protocolo de muestreo debe ser fijado antes de la
campaña de muestreo. Informaciones que son esenciales:
• Fecha y hora del muestreo
• Código del punto de muestreo
• Código del muestro (siguiendo la cadena de custodia)
• Descripción clara y definida del punto de muestreo (coordenadas de
ubicación del punto de muestreo (GPS), fotos, observaciones, etc)
• Localidad, distrito, provincia y departamento
• Fuentes de contaminación (si son visibles)
• Las condiciones climáticas
• Responsable del muestreo: datos personales de quien realizó la toma de
muestra
• Tipo de muestras (agua, aguas compuestas, sedimentos)
• Tipo de equipo o método utilizado para el muestreo
• Tipo de preservación
• Resultados de todas las mediciones realizadas en campo
• Informaciones en muestras compuestas
• Vigilancia y custodia de las muestras (por ejemplo numero de caja
transporte)
• Otras observaciones pertinentes en el punto de muestreo
• Firmas del personal participante en el proceso de control
Nota: además completar un formato de campo se recomienda etiquetar las
muestras de modo que permitan la trazabilidad de toda la información del
muestreo.
24. ASEGURAMIENTO Y CONTROL DE CALIDAD

27
Aseguramiento y control de calidad (AC y CC) son parte esencial de todo
sistema de monitoreo. Comprende un programa de actividades (capacitación,
calibración de equipos y registro de datos) que garantizan que la medición
cumple normas definidas y apropiadas de calidad con un determinado nivel de
confianza, o puede ser visto como una cadena de actividades diseñadas para
obtener datos fiables y precisos.
Las funciones de control de calidad influyen directamente en las actividades
relacionadas con la medición en campo, la calibración de los equipos de
campo, registro de datos y la capacitación. Para garantizar el éxito del
programa, es necesario que cada componente del esquema del
aseguramiento y control de calidad se implemente de manera adecuada, para
lo cual debe tenerse en cuenta lo siguiente:
24.1. Asegurarse que los frascos de muestreos cumplan con los
requisitos técnicos establecidos en el protocolo.
24.2. Enviar toda la documentación (formatos, cadena de custodia,
etiqueta, oficios, etc) de las muestras asegurando que los datos de
campo no varíen en su descripción.
24.3. Es esencial que el personal de campo este entrenado para aplicar
las metodologías estandarizadas y aprobadas.
Para realizar el control de calidad aplicado al muestreo se requiere considerar
los siguientes blancos y duplicados de acuerdo a las determinaciones analíticas:
24.4. Físico Químicos
24.4.1. Los blancos de equipo:
Son envases que contienen el agua del enjuague final de la
descontaminación de los equipos. Una vez analizados, muestran la
efectividad de la limpieza de los equipos de campo. Colecte aquellos
blancos de equipo, después del muestreo de agua subterránea o
superficial, que posean la contaminación más alta. Uno por día de
muestreo es suficiente.

28
24.4.2. Los blancos de campo
Son envases que contienen agua desionizada llenados, etiquetados,
empaquetados, sellados en la estación de muestreo y se envian al
laboratorio con las otras muestras. Este tipo de blancos de campo se
utilizan para investigar la contaminación del laboratorio y durante la
colecta y envío de las muestras. Se requiere un blanco de campo por
cada día de muestreo.
24.4.3. Los blancos viajeros
Son envases que contienen agua desionizada preparados en el
laboratorio y se trasladan junto a los envases que se utilizaran en el
muestreo. Se mantienen en la misma hielera que las otras muestras
en cada fase del proceso de colecta, manejo y envío. Si se encuentran
contaminados, podría ser que la contaminación ocurriera durante el
transporte de muestra o en el almacenaje en el laboratorio. Se
requiere por lo menos uno para cada envío de muestra.
24.4.4. Las muestras duplicadas
Se usan para verificar la precisión de la colecta de campo o el análisis
de laboratorio. Cada diez muestras se debe preparar una muestra
duplicada de muestreo, que consiste en llenar dos envases con una
misma muestra de agua extraída del mismo lugar y en el mismo
tiempo. De esta forma se verifica la variabilidad en los resultados
debido al manipuleo, conservación o contaminación de las muestras
corrientes.
También colecte una muestra duplicada de una estación en dónde se
cree que hay niveles altos de un compuesto en particular.
24.5. Microbiológico
24.5.1. Blanco Viajero:

29
Se coloca agua destilada estéril en un envase de muestreo, se realiza
un análisis de recuento de bacterias heterótrofas, para determinar que
el agua no contiene ningún microorganismo presente.
El blanco viajero se coloca en la misma caja de muestreo con el resto
de frascos, este se mantendrá cerrado durante todo el tiempo de
muestreo, para luego ser analizado conjuntamente con las muestras.
Este blanco permite comprobar una posible contaminación por el
transporte y procedimientos de almacenamiento en campo.
24.5.2. Duplicados de Muestreo:
Cada diez muestras se debe preparar una muestra duplicada de
muestreo, que consiste en llenar dos frascos con una misma muestra
de agua extraída del mismo lugar y en el mismo tiempo. De esta forma
se verifica la variabilidad en los resultados debido al manipuleo,
conservación o contaminación de las muestras corrientes.
25. LISTA DE VERIFICACIÓN DE ACTIVIDADES PARA ELABORAR EL PLAN
DE MUESTREO
Después de haber considerado todos los puntos del plan de muestreo es
necesario hacer un inventario de las acciones que se necesitan llevar a cabo
a fin de evitar que la posible falta de previsión u olvido provoque el fracaso
parcial o total de la campaña. Para ello es conveniente contar con una lista
de items para tener en cuenta.
26. LISTA DE VERIFICACIÓN DE ACTIVIDADES
• Razón del muestreo
• Parámetros a muestrear
• Procedimientos de muestreo específicos o especiales de cada parámetro
• Mapa de los sitios que se van a muestrear

30
• Lugar dónde se efectuarán las mediciones (a que distancia del grupo de
muestreo, en que tipo de terreno: llanura, desierto, selva, etc.)
• Mapa de las rutas de acceso a los sitios de muestreo
• Época del año en que se realizarán las campañas
• Condiciones meteorológicas de la época y de la zona (lluvias, nieve,
temperatura ambiente, etc.)
• Número de muestras que se van a tomar en cada sitio
• Lista de codificación y documentación de las muestras
• Equipos existentes o que es necesario proveer
• Periodicidad con que se realizarán las campañas
• Horarios en que se efectuarán los muestreos
• Infraestructura necesaria (energía, espacios de almacenamiento y
refrigeración, etc.)
• Comodidades para el personal: habitacional (hoteles, refugios, carpas,
etc.), facilidades para la alimentación higiene y descanso.
• Materiales y equipos que se necesitan transportar
• Tipos y número de vehículos necesarios (terrestres y fluviales)
• Tipo de personal que se necesita, cantidad y previsión de entrenamiento.
• Elementos especiales de vestimenta para el personal
• Medidas de seguridad que deben tomarse
27. REFERENCIAS:
[1] Mudroch A.M. y Azcue J.M., Manual of aquatic Sediment Sampling, Lewis
Publishers, (1995).
[2] Salomons W., Sediments and Water Quality. Sci. Technol. Lett., 6:315-
326, (1985).
[3] Secretaría de Comercio y Fomento Industrial (SCFI) Proyecto de Norma
Mexicana PROY-MNX-AA-121-SCFI-2005: Muestreo de aguas naturales
epicontinentales, costeras y marinas, (2005).

31
[4] Protocolo de monitoreo de la calidad sanitaria de los recursos hídricos
superficiales, Dirección de Ecología y Protección del Ambiente, Area de
Protección de los Recursos Hídricos, MINISTERIO DE SALUD, Dirección
General de Salud Ambiental “DIGESA”, Perú (2007)
[5] Standard Methods For The Examination Of Water And Wastewater. APHA.
AWWA. WEF. 21th Edition, (2005)
[6] Normas Chilenas:
a) Norma Chilena NCh 411/1. Of.95 Calidad de Agua – Muestreo - Parte 1:
Guía para el diseño de programas de muestreo.
b) Norma Chilena NCh 411/2. Of.96 Calidad de Agua – Muestreo - Parte 2:
Guía sobre técnicas de muestreo.
c) Norma Chilena NCh 411/3. Of.96 Calidad de Agua – Muestreo - Parte 3:
Guía sobre la preservación y manejo de las muestras.
Protocolo Cadena de Custodia

32
*Debe registrarse para DBO5 y coliformes
RLA/1/010
Cadena de Custodia Muestreo de Agua Superficial No._______
PI- Fecha Aprobación 30-06-2008
Fecha de Revisión 30-06-2008
GENERALES Fecha: Ubicación de la zona (UTM)
Descripción de la zona: Identificación de la(s) muestra (s):
Hora de muestreo
Tª del agua (ºC)
MUESTREO
Preservantes: Si / No Para análisis de:
Preservante:
Responsable:
Observaciones:
TRANSPORTE Fecha: No. de Horas: Tº de Transporte (ªC) No. de Coleman o conservadora Responsable:
Observaciones:
ANÁLISIS IN SITU Fecha Hora T emp. (ºC) pH C (µS/cm) OD (mg/L) Responsable
RECEPCIÓN EN EL LABORATORIO Envases en condiciones adecuadas? Si / No Fecha: Hora:
Etiquetas en condiciones adecuadas? Si / No
*Tº (ºC) del blanco de transporte
Observaciones: Identificación de la muestra:
Código dado por el Laboratorio
Observaciones:
Responsable: Firma:

33
Fecha: Ubicación de la zona (UTM)
Descripción de la zona: Identificación de la(s) muestra (s):
Hora de muestreo
MUESTREO Tipo de muestreador:
Tipo de muestreo:
Responsable:
Observaciones:
TRANSPORTE Fecha: No. de Horas: Tº de Transporte (ªC) No. de Coleman o
conservadora Responsable:
Observaciones:
RLA/1/010
Cadena de Custodia Muestreo de Sedimento No._______
PI- Fecha Aprobación 30-06-2008
Fecha de Revisión 30-06-2008
GENERALES
RECEPCIÓN EN EL LABORATORIO Envases en condiciones adecuadas? Si / No Fecha: Hora:
Etiquetas en condiciones adecuadas? Si / No
*Tº (ºC) del blanco de transporte
Observaciones: Identificación de la muestra:
Código dado por el Laboratorio
Observaciones: Responsable:
Firma:

34
2.3.1.2. Muestreo de Agua Superficial
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales
PT-MuA-01 MUESTREO DE AGUA SUPERFICIAL
Fecha Aprobación 30-06-2008
Fecha de Revisión 30-06-2008
1. OBJETIVO
Establecer los lineamientos generales para el muestreo de aguas
superficiales que garanticen la representatividad de la muestra (en su
composición física, química o biológica) para su posterior análisis.
2. ALCANCE
Este procedimiento será aplicado dentro del marco del proyecto ARCAL
RLA/1/010 de la OIEA, el cual contempla la mejora en la gestión de aguas
contaminadas con elementos ecotóxicos Además podrá ser aplicado por el
personal encargado de muestreo de agua, dentro de las actividades de
servicio, investigación y monitoreo de aguas superficiales en los países
participantes.
3. PRINCIPIO DEL METODO
La etapa de recolección de muestras es de suma importancia. Los resultados
de los mejores procedimientos analíticos serán inútiles si no se recolectan y
manipulan adecuadamente las muestras – American Public Heal Association,
American Waer Works, Association Water Pollution Control Federation 20th
Edition, 1998.
4. EQUIPOS
4.1. EQUIPOS PARA MEDICIÓN EN CAMPO
Analizadores multipruebas o sondas multiparámetros. (Conductímetro,

35
Oxímetro, Potenciómetro, termómetros, etc) para la medición de pH, T,
OD, CE.
4.2. EQUIPOS DE MUESTREO
4.2.1. Botella de tipo Van-Dorn, Niskin o botellas de laboratorio
adecuados a los parámetros a analizar.
4.2.2. Cubeta de plástico para mezclado de muestras compuestas con
capacidad aproximada de 20 L.
4.2.3. Disco de Secchi.
4.2.4. Sistema de posicionamiento global (GPS).
4.3. PRESERVANTES
Los preservantes químicos más comunmente utilizados en análisis de
agua son:
4.3.1. Acido Nítrico (HNO3)
4.3.2. Acido Sulfúrico (H2SO4)
4.3.3. Acido Clorhídrico (HCl)
4.3.4. Alcali (NaOH)
4.3.5. Agentes declorantes (tiosulfato de sodio y otros)
4.3.6. Agente quelante (EDTA)
4.3.7. Refrigeración a 4±2 ºC
4.4. MATERIALES
4.4.1. Bitácora o carpeta de campo.
4.4.2. Lista de chequeo.
4.4.3. Cadenas de custodia.
4.4.4. Tarjetas de consulta rápida para la operación y calibración de
equipos de medición.
4.4.5. Cinta adhesiva.
4.4.6. Cinta métrica (Wincha).

36
4.4.7. Cuerdas.
4.4.8. Pizeta o Frasco lavador
4.4.9. Pipetas, goteros y/o jeringas.
4.4.10. Cajas conservadoras, cooler, hieleras o coleman.
4.4.11. Baldes o Tinas.
4.4.12. Hielo, refrigerante o Icepack (gel 3M).
4.4.13. Botiquín.
4.4.14. Equipo de protección personal (Botas de goma, filtro solar,
gorras, etc.)
4.4.15. Cámara fotográfica.
4.4.16. Linternas.
4.4.17. Brazo muestreador.
4.4.18. Cronómetro.
4.4.19. Agua destilada.
4.4.20. Preservantes.
5. REACTIVOS Y MATERIALES
5.1. REQUISITOS DE ENVASES Y PRESERVANTES
La función de la preservación radica fundamentalmente en evitar o
disminuir al máximo posible, las reacciones químicas, físicas y biológicas
que se puedan producir durante el transporte y almacenamiento de las
muestras en el periodo transcurrido entre su recolección y análisis. Entre
estas se mencionan: actividad bacteriana, disolución o precipitación de
metales, adsorción, volatilización, etc.
Todos los productos químicos utilizados como preservantes deben ser de
calidad p.a. y dependiendo del tipo de ensayo y analito a determinar,
estos deben ser agregados a los envases, preferentemente como parte
de su preparación o bien a las muestras inmediatamente después de la
recolección, de manera de comenzar la preservación desde el mismo
momento del muestreo.
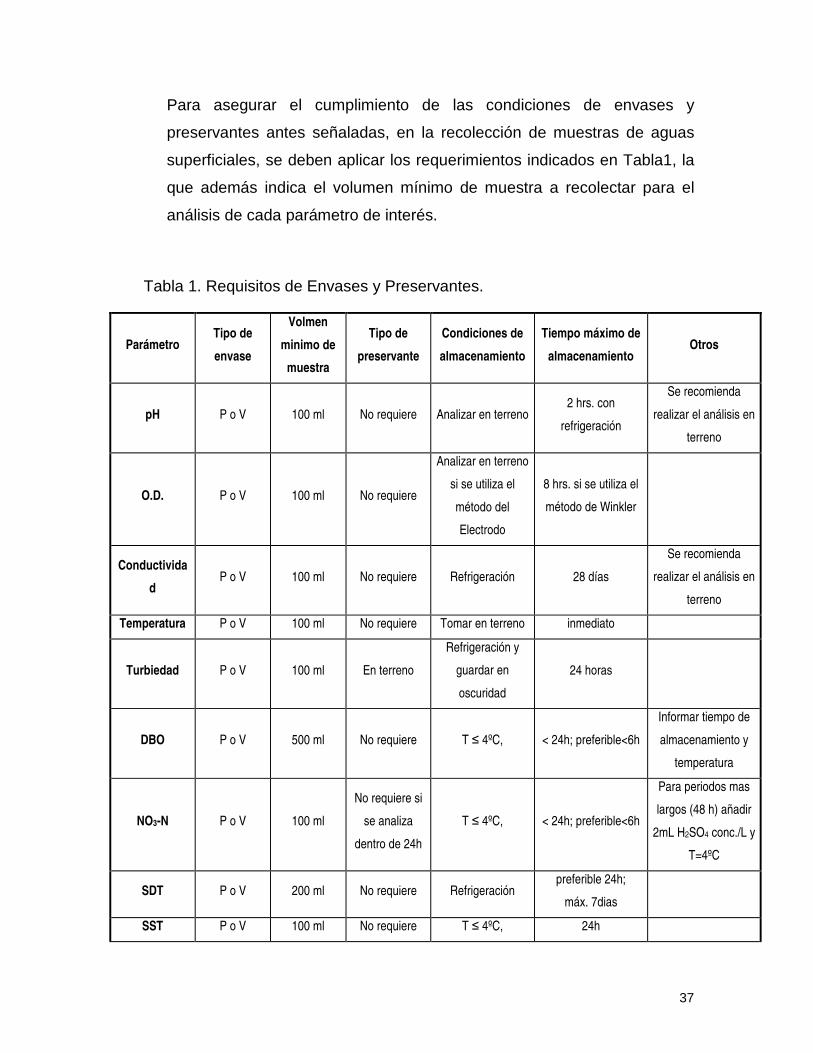
37
Para asegurar el cumplimiento de las condiciones de envases y
preservantes antes señaladas, en la recolección de muestras de aguas
superficiales, se deben aplicar los requerimientos indicados en Tabla1, la
que además indica el volumen mínimo de muestra a recolectar para el
análisis de cada parámetro de interés.
Tabla 1. Requisitos de Envases y Preservantes.
Parámetro Tipo de
envase
Volmen
minimo de
muestra
Tipo de
preservante
Condiciones de
almacenamiento
Tiempo máximo de
almacenamiento Otros
pH P o V 100 ml No requiere Analizar en terreno 2 hrs. con
refrigeración
Se recomienda
realizar el análisis en
terreno
O.D. P o V 100 ml No requiere
Analizar en terreno
si se utiliza el
método del
Electrodo
8 hrs. si se utiliza el
método de Winkler
Conductivida
d P o V 100 ml No requiere Refrigeración 28 días
Se recomienda
realizar el análisis en
terreno
Temperatura P o V 100 ml No requiere Tomar en terreno inmediato
Turbiedad P o V 100 ml En terreno
Refrigeración y
guardar en
oscuridad
24 horas
DBO P o V 500 ml No requiere T ≤ 4ºC, < 24h; preferible<6h
Informar tiempo de
almacenamiento y
temperatura
NO3-N P o V 100 ml
No requiere si
se analiza
dentro de 24h
T ≤ 4ºC, < 24h; preferible<6h
Para periodos mas
largos (48 h) añadir
2mL H2SO4 conc./L y
T=4ºC
SDT P o V 200 ml No requiere Refrigeración preferible 24h;
máx. 7dias
SST P o V 100 ml No requiere T ≤ 4ºC, 24h
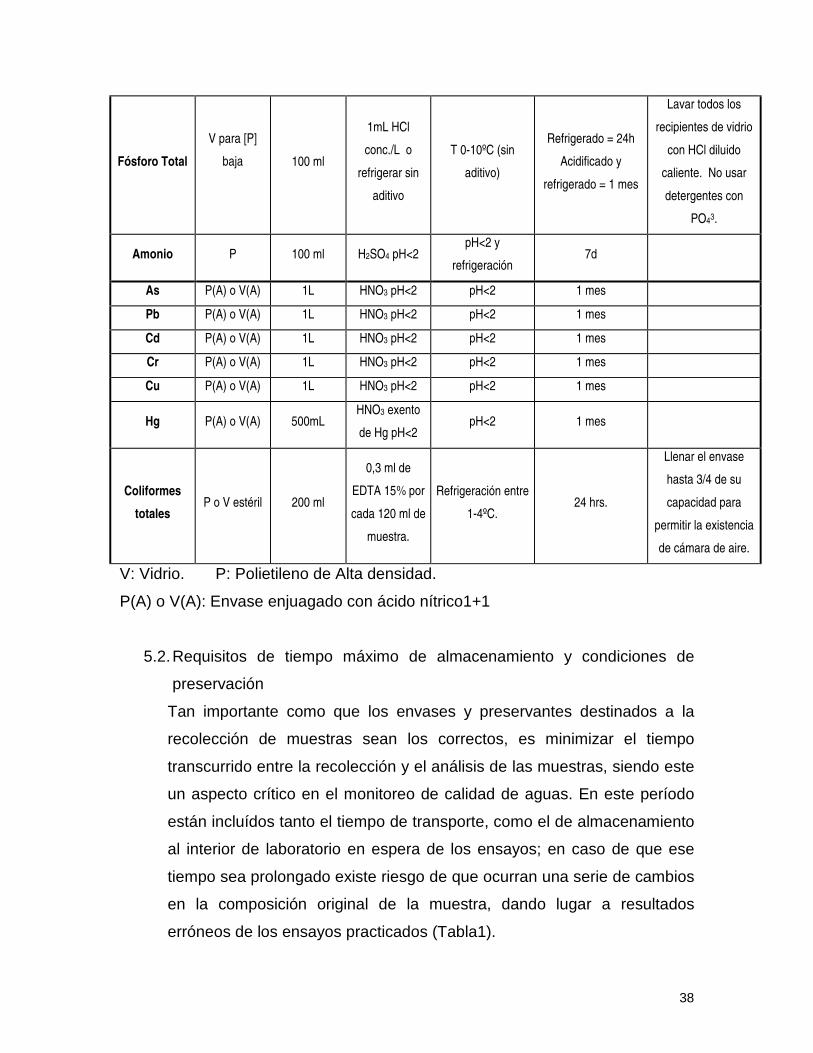
38
Fósforo Total
V para [P]
baja
100 ml
1mL HCl
conc./L o
refrigerar sin
aditivo
T 0-10ºC (sin
aditivo)
Refrigerado = 24h
Acidificado y
refrigerado = 1 mes
Lavar todos los
recipientes de vidrio
con HCl diluido
caliente. No usar
detergentes con
PO43.
Amonio P 100 ml H2SO4 pH<2 pH<2 y
refrigeración 7d
As P(A) o V(A) 1L HNO3 pH<2 pH<2 1 mes
Pb P(A) o V(A) 1L HNO3 pH<2 pH<2 1 mes
Cd P(A) o V(A) 1L HNO3 pH<2 pH<2 1 mes
Cr P(A) o V(A) 1L HNO3 pH<2 pH<2 1 mes
Cu P(A) o V(A) 1L HNO3 pH<2 pH<2 1 mes
Hg P(A) o V(A) 500mL HNO3 exento
de Hg pH<2 pH<2 1 mes
Coliformes
totales P o V estéril 200 ml
0,3 ml de
EDTA 15% por
cada 120 ml de
muestra.
Refrigeración entre
1-4ºC. 24 hrs.
Llenar el envase
hasta 3/4 de su
capacidad para
permitir la existencia
de cámara de aire.
V: Vidrio. P: Polietileno de Alta densidad.
P(A) o V(A): Envase enjuagado con ácido nítrico1+1
5.2. Requisitos de tiempo máximo de almacenamiento y condiciones de
preservación
Tan importante como que los envases y preservantes destinados a la
recolección de muestras sean los correctos, es minimizar el tiempo
transcurrido entre la recolección y el análisis de las muestras, siendo este
un aspecto crítico en el monitoreo de calidad de aguas. En este período
están incluídos tanto el tiempo de transporte, como el de almacenamiento
al interior de laboratorio en espera de los ensayos; en caso de que ese
tiempo sea prolongado existe riesgo de que ocurran una serie de cambios
en la composición original de la muestra, dando lugar a resultados
erróneos de los ensayos practicados (Tabla1).

39
5.3. REACTIVOS DE CALIBRACION PARA MEDICIONES IN SITU.
Deberán ser de acuerdo a los procedimientos XXXXXXX
6. ACCIONES PREVIAS:
6.1. Revisión del itinerario adecuado a la cronología del muestreo.
6.2. Comprobación del buen funcionamiento de los equipos de muestreo.
6.3. Revisión y elaboración de las listas de equipos y materiales.
6.4. Preparación de reactivos químicos (preservadores) y estándares de
calibración en campo.
6.5. Preparación del blanco viajero.
6.6. Revisión, verificación y calibración de equipos de medición en campo.
Esta tarea se llevará a cabo siguiendo las recomendaciones del
fabricante de cada equipo.
6.7. Revisión y preparación de recipientes (lavado y etiquetado) de
recipientes.
6.8. Revisión y verificación de equipos de muestreo.
6.9. Preparación de cadena de custodia y bitácora de campo.
6.10. Etiquetado de muestras
6.11. Nombre de Institución/Laboratorio.
6.12. Codificación de la muestra.
6.13. Georeferenciación del punto de muestreo (GPS).
6.14. Fecha y hora de muestreo.
6.15. Parámetro a determinar.
6.16. Tipo de muestra.
6.17. Identificación del responsable del muestreo.
7. DESCRIPCION DEL PROCEDIMIENTO

40
Para la toma de muestras de agua superficiales, evitar las áreas de
turbulencia excesiva, considerando la profundidad, velocidad de la corriente y
la distancia de separación entre ambas orillas (Homogeneidad del punto de
muestreo).
Para la toma de muestras de agua superficial, se debe considerar el tipo de
cuerpo de agua:
7.1. CUERPOS DE AGUA LÓTICOS
7.1.1. Tomar datos de temperatura ambiental, temperatura del río en el
punto de muestreo, condiciones meteorológicas, hora, y ubicación; y
anotarlo en el formato de cadena de custodia (Anexo 12.2).
7.1.2. Ubicarse en el punto de muestreo a través de una lancha o el
medio adecuado para ello (Puente, brazo muestreador, etc).
7.1.3. De acuerdo a la profundidad, se tomarán muestras de agua
superficial (AS) o de media profundidad, cuando esta sea mayor a
1.5 m. (AMP) (Fig. 1)
7.1.4. Considerando la sección transversal se tomarán 3 muestras
simples de 5lt. cada una, distribuidas a lo largo de la sección
transversal, una en el centro y 2 a las orillas.
7.1.5. Homogeneizar las muestras simples recolectadas en una tina para
obtener una muestra compuesta (aproximadamente 15 lts.)
Determinar en esta, lectura de pH, T, y CE (Procedimientos XXXXX)
y reportar los valores obtenidos en la cadena de custodia.
7.1.6. Medir el OD en los 3 puntos de muestreo sobre el río y reportar los
valores obtenidos en la cadena de custodia.
7.1.7. Se llenarán los frascos (considerando un espacio libre de
seguridad del 1% aproximadamente de la capacidad total) para la
determinación de cada uno de los parámetros considerados según el
plan de muestreo y especificaciones del laboratorio previa
preservación de las muestras (Tabla 1).

41
7.1.8. Tomar el número de muestras de acuerdo a lo establecido en el
plan de muestreo y el parámetro a determinar.
7.1.9. Etiquetar y codificar cada una de las muestras.
7.1.10. Cada muestra será sellada y colocada en una hielera
acondicionada a una temperatura entre 1 y 4 grados Celcius.
7.1.11. Considerar un espacio de alrededor del 1%
aproximadamente de la capacidad del envase (espacio de cabeza)
para permitir el espacio de la muestra.
7.1.12. La caja térmica y/o cooler deberá ser sellada con cinta de
embalaje.
7.1.13. Las muestras y el formato de Cadena de Custodia con datos
de campo deberán ser transportadas al laboratorio en el menor
tiempo posible.
7.2. CUERPOS DE AGUA LÉNTICOS
7.2.1. Se procederá de acuerdo a los mismos pasos del punto 7.1.2, con
la diferencia de que no se toma en cuenta una sección transversal
para la toma de las 3 muestras simples.
7.2.2. Para la toma de muestras en Lagos y pantanos, se evitara la
presencia de espumas superficiales.
Primera Sección Segunda Sección
SEGMENTACIÓN DE LA TRANSVERSAL DEL RIO
50 CM 50 CM a. AS:20-50 cm
b. AMP.
MS1 MS2 MS
3

42
Figura No. 1 Segmentación de la Transversal del Río
7.3. Registros del muestreo
7.3.1. Nombre y firma del muestreador;
7.3.2. Fecha y hora del muestreo;
7.3.3. Localización del punto de muestreo;
7.3.4. Identificación de la muestra;
7.3.5. Hora de muestreo;
7.3.6. Condiciones meteorológicas;
7.3.7. Número y tipo de recipientes;
7.3.8. Volumen de muestra;
7.3.9. Preservadores;
7.3.10. Analitos a determinar;
7.3.11. Análisis de campo (pH, conductividad y temperatura, OD);
7.3.12. Condiciones de transporte;
7.3.13. Fecha y hora de recepción;
7.3.14. Condiciones de recepción;
7.3.15. Nombre y firma de quien recibe, y
7.3.16. Observaciones
8. MUESTREO PARA ANALISIS BACTERIOLOGICO
Las botellas para este propósito, limpias y esterilizadas, deben estar
protegidas hasta el momento en que se necesite llenarla.
La tapa debe estar recubierta con una cubierta de tela, papel resistente o
papel de aluminio para protegerla en el momento del muestreo.
Inmediatamente antes del muestreo la cubierta de papel y la tapa, deben ser
removidos de la botella, evitando contaminarlos.

43
Se llena la botella sin enjuagar y la tapa se coloca inmediatamente. Las
muestras se toman sosteniendo, la botella por la base y sumergiéndola
hacia abajo, a una profundidad de 0.3 m de la superficie.
La boca de la botella o recipiente se dirige hacia la dirección del flujo de
modo que los puntos del cuello queden hacía arriba aproximadamente 45º.
El agua, que entra a la botella no debe tener contacto con la mano que
sostiene la botella, si esto ocurre la muestra debe rechazarse.
Si este problema es debido a la turbulencia, se busca un punto donde esto
no ocurra o se usa un objeto para amarrar la botella.
Para la toma de muestras a profundidades específicas, se emplean
dispositivos de muestreo esterilizados.
9. ASEGURAMIENTO Y CONTROL DE CALIDAD
Aseguramiento y control de calidad (AC y CC) son parte esencial de todo
sistema de monitoreo. Comprende un programa de actividades (capacitación,
calibración de equipos y registro de datos) que garantizan que la medición
cumple normas definidas y apropiadas de calidad con un determinado nivel de
confianza, o puede ser visto como una cadena de actividades diseñadas para
obtener datos fiables y precisos.
Las funciones de control de calidad influyen directamente en las actividades
relacionadas con la medición en campo, la calibración de los equipos de
campo, registro de datos y la capacitación. Para garantizar el éxito del
programa, es necesario que cada componente del esquema del
aseguramiento y control de calidad se implemente de manera adecuada,
para lo cual debe tenerse en cuenta lo siguiente:
9.1. Asegurarse que los frascos de muestreos cumplan con los requisitos
técnicos establecidos en el presente protocolo.
9.2. Enviar toda la documentación (formatos, cadena de custodia, etiqueta,
oficios, etc) de las muestras asegurando que los datos de campo no
varíen en su descripción.

44
9.3. Es esencial que el personal de campo este capacitado para aplicar las
metodologías estandarizadas y aprobadas.
Para realizar el control de calidad aplicado al muestreo se requiere
considerar los siguientes blancos y duplicados de acuerdo a las
determinaciones analíticas:
9.4. Físico Químicos
9.4.1. Los blancos de equipo: Consisten de envases llenos con el agua
final del enjuague de la descontaminación de los equipos. Una vez
analizados, muestran la efectividad de la limpieza de los equipos de
campo. Colecte los blancos de equipo después del muestreo del
agua subterránea o superficial en la estación con la contaminación
más alta. Uno por día del muestreo es suficiente.
9.4.2. Los blancos de campo: Son envases de agua desionizada que se
llenan en la estación de muestreo, etiquetan, empaquetan, sellan y
se mandan al laboratorio con las otras muestras. Se usan los blancos
de campo para investigar la contaminación en el laboratorio, y
durante la colecta y envío de las muestras. El laboratorio requiere un
blanco de campo por cada día del muestreo.
9.4.3. Los blancos viajeros: Son envases de agua desionizada
preparados en el laboratorio que se envía con los frascos de
muestreo. Se mantienen en la misma hielera que las otras muestras
en cada fase del proceso de colecta, manejo y envío. Si se
encuentran contaminados, podría ser que la contaminación ocurriera
durante el transporte de muestra o en el almacenaje en el
laboratorio. Se requiere por lo menos uno para cada envío de
muestra.
9.4.4. Las muestras duplicadas: Se usan para verificar la precisión de la
colecta de campo o el análisis de laboratorio. Se colectan los
duplicados a la vez que la muestra de la calidad del agua a una
cantidad de una en cada diez o 10% al día, lo que sea más grande.

45
Colecte una muestra duplicada de una estación en dónde se cree
que hay niveles altos de un compuesto en particular.
9.5. Microbiológico
9.5.1. Blanco Viajero: Se coloca agua destilada estéril en un frasco de
muestreo, se realiza un análisis de recuento de bacterias
heterótrofas, para determinar que el agua no contiene ningún
microorganismo presente.
9.5.2. El blanco viajero se coloca en la misma caja de muestreo con el
resto de frascos, este se mantendrá cerrado durante todo el tiempo
de muestreo, para luego ser analizado conjuntamente con las
muestras.
9.5.3. Este blanco permite comprobar una posible contaminación por el
transporte y procedimientos de almacenamiento en campo.
9.5.4. Duplicados de Muestreo: Cada diez muestras se debe preparar
una muestra duplicada de muestreo, que consiste en llenar dos
frascos con una misma muestra de agua extraída del mismo lugar y
en el mismo tiempo. De esta forma se verifica la variabilidad en los
resultados debido al manipuleo, conservación o contaminación de las
muestras corrientes.

46
10. REFERENCIAS
[1] Standard Methods For The Examination Of Water And Wastewater.
APHA. AWWA. WEF. 21th Edition, (2005)
[2] Secretaría de Comercio y Fomento Industrial (SCFI) (2005). Proyecto de
Norma Mexicana PROY-MNX-AA-121-SCFI-2005: Muestreo de aguas naturales
epicontinentales, costeras y marinas
[3] Protocolo de monitoreo de la calidad sanitaria de los recursos hídricos
superficiales, Dirección de Ecología y Protección del Ambiente, Area de
Protección de los Recursos Hídricos, MINISTERIO DE SALUD, Dirección
General de Salud Ambiental “DIGESA”, Perú (2007)
[4] Normas Chilenas:
a) Norma Chilena NCh 411/1. Of.95 Calidad de Agua – Muestreo - Parte 1:
Guía para el diseño de programas de muestreo.
b) Norma Chilena NCh 411/2. Of.96 Calidad de Agua – Muestreo - Parte 2:
Guía sobre técnicas de muestreo.
c) Norma Chilena NCh 411/3. Of.96 Calidad de Agua – Muestreo - Parte 3:
Guía sobre la preservación y manejo de las muestras.
11. GLOSARIO
Aguas naturales: Se define como agua natural el agua cruda, subterránea, de
lluvia, de tormenta, residual y superficial.
Agua de tormenta: Escurrimiento torrencial de agua superficial que fluye
hacia un cauce de agua como resultado de una lluvia intensa.
Bitácora de campo: Cuaderno de campo debidamente foliado e identificado,
en el cual los monitoreadores y/o analistas anotan todos los datos de los
procedimientos que siguen en el análisis de una muestra, así como todas las
informaciones pertinentes y relevantes a su trabajo en el laboratorio. Es a

47
partir de dichas bitácoras que los inspectores pueden reconstruir el proceso
de análisis de una muestra tiempo después de que se llevó a cabo
Calibración: Conjunto de operaciones que establecen, bajo condiciones
específicas, la relación entre los valores de una magnitud indicados por un
instrumento o sistema de medición, y los valores representados por una
medida estándar.
Cadena de Custodia: Documento en el cual se lleva el registro de la
información que permite identificar las muestras, el sitio de muestreo y los
parámetros fisicoquímicos medidos in situ.
Cuerpos de agua superficiales. Son los lagos, lagunas, estuarios, redes
colectoras con excepción de los sistemas de drenaje y alcantarillado urbano
o municipal, ríos y sus efluentes directos o indirectos, permanentes o
intermitentes, presas, cuencas, cauces, canales, embalses, asequias, y
demás corrientes de agua.
Demanda bioquímica de oxígeno (DBO): Concentración del oxígeno disuelto
consumido bajo condiciones especificadas por la oxidación biológica de la
materia orgánica, inorgánica o ambas, contenidas en el agua.
Ecosistema léntico: son cuerpos de agua cerrados que permanecen en un
mismo lugar sin correr ni fluir, como los lagos, las lagunas, los esteros, los
pantanos, etc.
Ecosistema lótico: sistema de agua corriente como en los ríos, arroyos y
manantiales.
Ficha de registro de campo: Es un formato utilizado en la toma de muestras y
contiene la siguiente información: código del punto de muestreo, origen de la
fuente, descripción clara y definida del punto de muestreo, hora y fecha de
muestreo, localidad, distrito, provincia y departamento, coordenadas de
ubicación del punto de muestreo, datos personales de quien realizó la toma
de muestra, las condiciones climáticas y otras observaciones pertinentes en
el punto de muestreo.

48
Muestra Compuesta: Aquella que se obtiene mezclando varias muestras
discretas o simples de volúmenes iguales.
Muestra representativa: Una porción de agua o material que está
estrechamente identificado, lo más posible en contenido y consistencia con el
gran cuerpo de agua o materia a ser muestreada
Muestra Simple: Muestra individual tomada en una localidad, a una
profundidad y en un tiempo determinado.
Muestreador: Aparato utilizado para tomar una muestra de agua, de manera
intermitente o continua, con el propósito de examinar diversas características
definidas.
Organismos coliformes fecales (termotolerantes): Comprende todos los
bacilos aerobios o anaerobios facultativos , Gram negativos , no esporulados
que fermentan la lactosa con producción de ácido y gas a 44ºC ± 1ºC en un
plazo de 24 h.
Preservación de la muestra: Proceso en el cual, por medio de adición de
productos químicos o la modificación de las condiciones físicas o ambas, se
reducen al mínimo los cambios de las características de la muestra a
terminar durante el tiempo que transcurre entre el muestreo y al análisis.
Punto de muestreo: Lugar preciso donde se tomará la muestra.
Red de muestreo: Sistema de zonas de muestreo preestablecidas a fin de
monitorear uno o más lugares definidos.
Zona de muestreo: Lugar de estudio o sitio de medición.
12. ANEXOS
12.1. MEDICION Y CALCULO DE CANTIDAD DE AGUA EN LOS
SITIOS DE TOMA DE MUESTRAS.
Para la medición de caudales se realiza el aforo y batimetría del sitio de
toma de muestras, para ello se divide en tramos el ancho de la sección
transversal del río y en cada uno de ellos se miden las velocidades a

49
diferentes profundidades a través de los molinetes; además de ello se
reportan los datos de profundidad de cada uno de los tramos del río.
Entonces el caudal obtenido en un aforo es la suma de los productos del
área de cada subsección y la velocidad promedio en la subsección
respectiva.
Para calcular descargas individuales en cada subsección se utiliza la
fórmula general siguiente:
i
ii
ii pdd
Vq *2
)1()1(
−= −+
qi = descarga en la subsección.
Vi= Velocidad promedio de la subsección
d = distancia de la subsección al punto del inicio del aforo
pi= profundidad de cada sub- sección
La sumatoria de cada descarga nos dará el caudal de toda la sección.

50
2.3.1.3. Determinación de la demanda bioquímica de oxígeno
(DBO5) en aguas superficiales
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales” Código del procedimiento: PT-FQ-01
Nombre del procedimiento: Determinación de la demanda bioquímica de oxígeno (DBO5) en aguas superficiales
Fecha de aprobación: 2008-05-02
Fecha de revisión: 2008-05-02
1. OBJETIVO
Establecer el método de análisis para la determinación de la demanda
bioquímica de oxígeno.
2. ALCANCE
Se aplica a muestras de aguas superficiales.
3. PRINCIPIO DEL METODO
El método se basa en medir la cantidad de oxígeno que requieren los
microorganismos para efectuar la oxidación de la materia orgánica presente.
La muestra o una dilución adecuada de la misma es incubada por 5 días a 20
ºC en la oscuridad y la DBO5 se determina por la diferencia entre el oxígeno
disuelto inicial y el oxígeno disuelto al cabo de los 5 días de incubación. Para
la determinación de oxígeno disuelto se puede emplear diferentes métodos:
a) El método iodométrico de la modificación de azida (titulación)
b) El método de electrodo de membrana. 3.1. INTERFERENCIAS
• pH alcalinos o ácidos presentes en las muestras.

51
• La presencia de cloro residual, metales o compuestos orgánicos
tóxicos no permiten el desarrollo de las bacterias que degradan la
materia orgánica.
4. EQUIPOS
Debe utilizarse el equipo normal de laboratorio, además de:
4.1. Balanza analítica.
4.2. Incubadora para DBO controlado por termostato a 20 ± 1°C. Eliminar
toda la luz para evitar la posibilidad de producción fotosintética de
oxígeno disuelto.
4.3. Electrodo de membrana selectiva al oxígeno, con compensación
automática de temperatura y medidor apropiado (Oxímetro [ver 3. (b)])
5. REACTIVOS Y MATERIALES
5.1. REACTIVOS
Utilizar únicamente reactivos de grado analítico reconocido y agua
destilada o de pureza equivalente.
• Sulfito de sodio (Na2SO3)
• Cloruro férrico hexahidratado (FeCl3.6H2O)
• Cloruro de calcio anhidro (CaCl2)
• Sulfato de magnesio heptahidratado (MgSO4.7H2O)
• Fosfato dibásico de potasio (K2HPO4)
• Fosfato monobásico de potasio(KH2PO4)
• Fosfato dibásico de sodio heptahidratado(Na2HPO4.7H2O)
• Cloruro de amonio (NH4Cl)
• Hidróxido de sodio (NaOH)
• 2-cloro-6 (triclorometil) piridina

52
• Glucosa (C6H12O6)
• Ácido glutámico (C5H9NO4)
• Ácido sulfúrico concentrado (H2SO4)
• Ácido nítrico (HNO3)
5.2. SOLUCIONES
• Solución de cloruro férrico: Se disuelven 0,25 g de cloruro férrico
hexahidratado en agua destilada hasta completar un litro.
• Solución de cloruro de calcio: Se disuelven 27,5 g de cloruro de calcio
anhidro o su equivalente de sal hidratada en un litro de agua destilada.
• Solución de sulfato de magnesio: Se disuelven 22,5 g de sulfato de
magnesio heptahidratado en un litro de agua destilada.
• Solución amortiguadora de fosfato: Se disuelven 8,5 g de fosfato
monobásico de potasio, 21,75 g de fosfato dibásico de potasio, 1,7 g
de cloruro de amonio y 33,4 g de fosfato dibásico de sodio
heptahidratado (si no hay se sustituye por 20,0 g de fosfato dibásico de
sodio monohidratado ó 44,6 g de fosfato dibásico de sodio
dodecahidratado). Se ajusta el pH a 7,2 con hidróxido de sodio al 30 %
y se lleva a 1 L.
• Solución ácida y alcalina 1N, para neutralización de muestras de
desecho alcalinas ó ácidas.
• Solución ácida: lentamente y mientras se mezcla, agregar 28 mL de
ácido sulfúrico concentrado en agua destilada. Diluir a 1 litro
• Solución alcalina: disolver 40 g de hidróxido de sodio en agua
destilada. Diluir a 1 litro.
• Inhibidor de nitrógeno, 2-cloro-6(triclorometil) piridina.
• Solución de glucosa-ácido glutámico: Secar la glucosa y el ácido
glutámico a 103 °C por 1 hora. Agregar 150 mg de gl ucosa y 150 mg
de ácido glutámico en agua destilada y diluir a 1 litro. Preparar solución
fresca inmediatamente antes de su uso.

53
• Solución de cloruro de amonio: Disolver 1,15g de cloruro de amonio en
aproximadamente 500 mL de agua destilada, ajustar el pH a 7,2 con
solución de hidróxido de sodio, y diluir a 1 L. La solución contiene 0,3
mg N/mL.
• Solución de sulfito de sodio: Disolver 1,575g de sulfito de sodio en
1000 mL de agua destilada. Esta solución es poco estable, debe
prepararse diariamente.
5.3. MATERIALES
Debe emplearse cristalería normal de laboratorio, así como:
• Frascos Winkler de vidrio para incubación de 300 mL aforo total y con
boca estrecha, reborde y tapón de vidrio esmerilado, de forma cónica.
• Contratapa de politetrafloroetileno u otro material plástico para botella
Winkler.
6. ACCIONES PREVIAS
6.1. LIMPIEZA DEL MATERIAL.
Todo material usado en la determinación debe ser exclusivo para este
tipo de procedimiento. Para el lavado de material remojar durante una
hora en una solución de ácido sulfúrico al 10% y enjuagar con agua. Los
detergentes con base de amoníaco no deben usarse para la limpieza del
material.
En los casos de que el material presente grasas, enjuagar con acetona
y/o hexano.
6.2. MUESTRAS

54
No se debe agregar ningún preservador a las muestras. Se debe
conservar a 4°C previo al análisis. Reportar la dur ación y la temperatura
de almacenamiento con los resultados. Si el análisis es comenzado
dentro de 2 horas de la colecta, el almacenamiento en frío es
innecesario.
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Preparación del agua de dilución:
Colocar un volumen deseado de agua en un frasco apropiado y agregar
1 mL de cada una de las soluciones de buffer fosfatos, sulfato de
magnesio, cloruro de calcio y cloruro férrico por litro de agua.
Antes de utilizar el agua de dilución debe llevarse a 20 °C. Saturar con
oxígeno disuelto por agitación en un frasco parcialmente lleno o por
medio de aireación con aire filtrado y libre de orgánicos.
Alternativamente, almacenar en frascos tapados con algodón y que
sean lo suficientemente grandes como para saturarse con OD. Proteger
la calidad del agua utilizando cristalería limpia, pipetas y botellas.
7.2. Chequeo del agua de dilución:
Si el agotamiento del oxígeno de un agua excede de 0,2 mg/L, se
obtiene un agua satisfactoria por mejoramiento de la purificación a partir
de otra fuente. Alternativamente si la inhibición de la nitrificación es
usada, almacenar el agua de dilución, inoculada y en un lugar oscuro a
temperatura ambiental hasta que el consumo de oxígeno esté lo
suficientemente reducido para cumplir con el criterio de chequeo del
agua de dilución. Chequear la calidad del agua de dilución almacenada
en uso, pero no agregarle el inóculo al agua almacenada para
mejoramiento de la calidad. El almacenamiento no es recomendable

55
cuando las DBO no serán determinadas sin inhibición de la nitrificación
debido a que los organismos nitrificantes pueden desarrollarse durante
el almacenamiento. Chequear el agua de dilución almacenada para
determinar si existen remanentes suficientes de amonio después del
almacenamiento. Si no lo posee agregar solución de cloruro de amonio
para proveer un total de 0,45 mg de amonio por litro como nitrógeno. Si
el agua de dilución no ha sido almacenada por el mejoramiento de la
calidad, agregar suficiente material de siembra para producir un
consumo de OD de 0,05 a 0,1 mg/L en 5 días a 20 °C. Incubar botellas
de DBO llenas de agua de dilución durante 5 días a 20 °C. Determinar
el oxígeno disuelto inicial y final. El oxígeno disuelto consumido durante
los 5 días a 20 °C no debería de ser más de 0,2 mg /L y preferiblemente
no más de 0,1 mg/L.
7.3. Chequeo con glucosa-ácido glutámico:
Debido a que la prueba de DBO es un bioensayo sus resultados pueden
estar influenciados grandemente por la presencia de tóxico o por el uso
de un material de siembra muy pobre. Las aguas destiladas con
frecuencia están contaminadas con cobre; algunos inóculos de aguas
residuales son relativamente inactivos. Bajos resultados siempre son
obtenidos con tales inóculos y aguas. Periódicamente chequear la
calidad del agua de dilución, la efectividad de la siembra, y la técnica
analítica haciendo mediciones de DBO en compuestos orgánicos puros
y en muestras con adiciones conocidas. En general, para las
determinaciones de DBO no se requiere de un inóculo adaptado, usar la
solución de glucosa-ácido glutámico como una solución estándar de
chequeo. La glucosa tiene excepcionalmente un alto y variable rango de
oxidación, pero cuando es usada con ácido glutámico, el rango de
oxidación es estabilizado y es similar al que se obtiene con muchos
desechos municipales. Alternativamente, si un agua residual contiene

56
un constituyente mayor que contribuya al DBO, usar este compuesto en
lugar de glucosa-ácido glutámico. Determinar el DBO a los 5 días a 20
°C de una dilución al 2% de solución estándar de ch equeo de glucosa-
ácido glutámico.
7.4. Inoculación
7.4.1. Fuente de inóculo
Es necesario tener presente una población de microorganismos
capaces de oxidar la materia orgánica biodegradable en la
muestra. Algunas aguas superficiales no contienen suficiente
población microbiana; para tales muestras sembrar el agua de
dilución por medio de la adición de poblaciones de
microorganismos. El inóculo preferido es el efluente procedente de
los sistemas de tratamiento biológico para desechos. En donde no
es posible, usar el sobrenadante procedente de las aguas
residuales domésticas después de sedimentarlas a temperatura
ambiental por lo menos durante 1 hora pero no más de 36 horas.
Cuando el efluente procedente de tratamiento biológico es usado,
se recomienda la inhibición de la nitrificación.
7.4.2. Control del inóculo
Determinar el DBO del material inoculado como para cualquier otra
muestra. Del valor del control del inóculo y del conocimiento de la
dilución del material de siembra (en el agua de dilución) determinar
el consumo de oxígeno disuelto. Idénticamente hacer diluciones
del inóculo tal como la mayor cantidad de resultados en por lo
menos 50% del consumo de oxígeno. Un gráfico del consumo de
oxígeno disuelto, en mg/L, versus mL de inóculo deberían de
presentar una línea recta, para la cual la pendiente indica el
consumo de oxígeno disuelto por mL de inóculo. El intercepto de

57
eje del oxígeno disuelto, es el consumo de oxígeno causado por el
agua de dilución y debe ser menor que 0,1 mg/L. Para determinar
el consumo de oxígeno disuelto de una muestra restar el consumo
de oxígeno disuelto del inóculo del total de oxígeno disuelto
consumido. El oxígeno disuelto consumido por el agua de dilución
inoculada debería estar entre 0,6 y 1,0 mg/L.
7.5. Pretratamiento de las muestras
7.5.1. En muestras conteniendo alcalinidad cáustica o acidez,
neutralizarlas a pH de 6.5 a 7.5 con solución de ácido sulfúrico ó de
hidróxido de sodio, de tal forma que la cantidad de reactivo no diluya
la muestra por más de 0,5%. El pH del agua de dilución inoculada no
debe de ser afectada por las diluciones mas bajas de la muestra.
7.5.2. Muestras conteniendo compuestos de cloro residual: si la muestra
ha sido clorada y el cloro residual no es detectable inocular la
muestra con el agua de dilución. Para muestras en las cuales el cloro
residual no se disipa en un tiempo razonable destruirlo por medio de
la adición de solución de sulfito de sodio. Determinar el volumen
requerido de sulfito se sodio en una porción de muestra neutralizada
de 100 a 1000 mL por medio de la adición de 10 mL de ácido
acético (1:1) o ácido sulfúrico (1:50), 10 mL de solución de yoduro
de potasio (10g/100 mL) por porciones de 1000 mL. Titular con sulfito
de sodio hasta el punto de finalización utilizando almidón como
indicador. Agregar a la muestra neutralizada el volumen de sulfito de
sodio obtenido en el procedimiento anterior, mezclar y dejar reposar
de 10 a 20 minutos y verificar la presencia de cloro residual en la
muestra.

58
Nota: el exceso de sulfito de sodio reacciona lentamente con
compuestos de cloraminas orgánicas
7.5.3. Muestras conteniendo otras sustancias tóxicas. Ciertos desechos
industriales, por ejemplo, desechos de industrias metalúrgicas,
contienen metales tóxicos. Tales muestras a menudo requieren
especial estudio y tratamiento.
7.5.4. Muestras supersaturadas con oxígeno disuelto. Muestras
conteniendo más de 9 mg/L de oxígeno disuelto a 20 °C pueden ser
encontradas en las aguas frías o en aguas donde ocurre la
fotosíntesis. Para prevenir la pérdida de oxígeno durante la
incubación de tales muestras, reducir el oxígeno disuelto a la
saturación a 20 °C poniendo la muestra a una temper atura cercana a
20 °C en un frasco parcialmente lleno y agitando vi gorosamente o
por medio de aireación con aire comprimido limpio y filtrado.
7.5.5. Ajuste de la temperatura de la muestra. Llevar las muestras a 20 ±
1 °C antes de hacer las diluciones.
7.5.6. Inhibición de la nitrificación. Si la inhibición de la nitrificación es
deseada, agregar 3 mg de 2 cloro-6-(triclorometil) piridina (TCMP) a
cada botella de 300 mL antes de taparlas o de agregar suficiente
cantidad de agua de dilución para hacer su concentración final de 10
mg/L. (Nota: El TCMP puro puede disolverse lentamente y puede
flotar en la parte superior de la muestra. Algunas formulaciones
comerciales se disuelven más fácilmente, pero no en un 100%,
ajustar las dosis). Las muestras pueden requerir inhibición de la
nitrificación y esta no solo incluye a los efluentes biológicamente
tratados, sino también a muestras inoculadas con efluentes tratados

59
biológicamente y aguas de ríos. Anotar el uso de inhibidor de
nitrógeno en los resultados.
7.5.7. Técnica de dilución. Las diluciones que resultan con un oxígeno
disuelto residual de por lo menos 1 mg/L y un consumo de oxígeno
disuelto de por lo menos 2 mg/L después de los 5 días de incubación
produce los resultados más confiables. Hacer varias diluciones de
muestra preparada para obtener un consumo de oxígeno dentro de
este rango. Se recomienda el uso de cinco diluciones. La experiencia
con muestras particulares permitirá el uso de un menor número de
diluciones. Un análisis más rápido, tal como el DQO, puede
correlacionarse aproximadamente con el DBO y servir como guía en
la selección de las diluciones. En ausencia de conocimiento previo,
usar las siguientes diluciones:
Rango de % de dilución Tipo de muestra
0,0 a 1 para desechos industriales fuertes
1 a 5 para aguas crudas y aguas de desecho sedimentadas
5 a 25 para efluentes tratados biológicamente
25 a 100 para aguas de ríos contaminados
Preparar cada dilución en probetas y luego transferirlas a las botellas de
DBO, ó prepararlas directamente en las botellas de DBO. Cada método
de dilución puede ser combinado con cualquier técnica de medición de
oxígeno disuelto. El número de botellas preparadas para cada dilución
depende de la técnica de oxígeno disuelto y el número de duplicados
deseados.

60
Cuando se utilizan probetas para preparar las diluciones, y cuando la
inoculación es necesaria, agregar el inóculo directamente al agua de
dilución ó a cada probeta individualmente antes de la dilución.
• Preparación de la dilución directamente en las botellas de DBO.
Utilizando una pipeta volumétrica, agregar el volumen deseado de
muestra individualmente a las botellas de DBO de capacidad
conocida. Agregar cantidades adecuadas de material de inoculación
a las botellas individuales de DBO ó al agua de dilución. Llenar las
botellas con suficiente agua de dilución, inoculada si es necesario,
de tal manera que el tapón sellador desplace todo el aire sin dejar
burbujas. Para diluciones mayores a 1:100 hacer una dilución
primaria en una probeta antes de hacer la dilución final en la botella.
Cuando se utiliza el método de titulación para mediciones de oxígeno
disuelto, preparar dos botellas de cada dilución. Determinar el oxígeno
disuelto inicial en una botella. Tapar fuertemente la segunda botella,
hacer sello de agua, e incubar por 5 días a 20 °C. Si el método del
electrodo de membrana es utilizado para la medición del oxígeno
disuelto inicial, preparar solamente una botella para cada dilución.
Determinar el oxígeno disuelto inicial y reemplazar el contenido
desplazado con agua de dilución para llenar la botella. Tapar
fuertemente, hacer sello de agua, e incubar por 5 días a 20 °C. Lavar el
electrodo de oxígeno disuelto entre cada determinación para prevenir la
contaminación cruzada de cada una de las muestras.
7.5.8. Determinación del oxígeno disuelto inicial. Si las muestras
contienen material que reacciona rápidamente con el oxígeno
disuelto, determinar el oxígeno disuelto inicial inmediatamente
después de llenar la botella con la muestra diluida. Si el consumo
rápido de oxígeno disuelto inicial es insignificante, el período de
tiempo entre la preparación de la dilución y la medición del oxígeno
disuelto inicial no es crítico, pero no debe exceder los 30 minutos. La

61
determinación de oxígeno disuelto puede ser realizado según las
metodologías señaladas anteriormente (3. Principio del método)
7.5.9. Blanco del agua de dilución. Usar un blanco del agua de dilución
como un control de calidad aproximado en el agua de dilución no
inoculada. El consumo de oxígeno disuelto no debe de ser mayor de
0.2 mg/L y preferiblemente no más de 0.1 mg/L. Descartar toda el
agua de dilución que tenga un oxígeno disuelto mayor a 0.2 mg/L .
7.5.10. Incubación. Incubar a 20 ± 1 °C las botella s de DBO conteniendo
las diluciones deseadas, controles de inoculación, blancos del agua
de dilución, y chequeos con glucosa-ácido glutámico. Hacerles sello
de agua a las botellas.
7.5.11. Determinación del oxígeno disuelto final: Después de 5 días de
incubación determinar el oxígeno disuelto en las diluciones de las
muestras, blancos y chequear como en 7.5.8.
8. CALCULOS Y EXPRESION DE RESULTADOS
8.1. Cuando el agua de dilución no está inoculada:
P
DDL/mg,DBO 21
5
−=
8.2. Cuando el agua de dilución está inoculada:
P
f)BB()DD(L/mg,DBO 2121
5
−−−=
donde;
D1 = OD de la muestra diluida inmediatamente después de la preparación,
mg/L
D2 = OD de la muestra diluida después de 5 días de incubación a 20 °C, mg/L

62
P = fracción volumétrica decimal de muestra usada,
B1 = OD del control del inóculo antes de la incubación, mg/L
B2 = OD del control del inóculo después de la incubación, mg/L
f = Cantidad de inóculo en la muestra diluida para sembrar el control del
inóculo
inóculodecontroleleninóculode
diluidamuestralaeninóculodef
%
%=
Si el material de inoculación es agregado directamente en las botellas de la
muestra ó en la de control del inóculo:
inóculodecontroleleninóculodevolumen
diluidamuestralaeninóculodevolumenf =
Reportar los resultados como DBO5 Carbonáceo si se ha inhibido la
nitrificación.
Si más de una dilución de la muestra cumple con el criterio de un oxígeno
disuelto residual de por lo menos 1 mg/L y el consumo de oxígeno disuelto es
de por lo menos 2 mg/L y no hay evidencia de toxicidad a concentraciones
mayores de muestra ó la existencia de una anormalidad obvia, promediar los
resultados en un rango aceptable.
En esos cálculos, no hacer correcciones para el consumo de oxígeno disuelto
por la dilución del blanco del agua de dilución durante la incubación. Esta
corrección es innecesaria si el agua de dilución cumple con el criterio del
blanco estipulado arriba. Si el agua de dilución no cumple con ese criterio, las
correcciones apropiadas son difíciles y los resultados pueden volverse
dudosos.
8.3. PRECISIÓN Y SESGO DE LOS RESULTADOS

63
No existen mediciones para establecer el sesgo del procedimiento de
DBO. El chequeo con la glucosa-ácido glutámico tiene por objetivo ser un
punto de referencia para la evaluación de la calidad del agua de dilución,
efectividad del inóculo, y de la técnica analítica.
8.3.1. Límites de control
Cada laboratorio, puede establecer su límite de control por medio de
la realización de un mínimo de 25 chequeos con glucosa-ácido
glutámico sobre un período de varias semanas o meses y calculando
el promedio y la desviación estándar. Usar la media y ± 3
desviaciones estándar como límite de control para futuros chequeos
de glucosa-ácido glutámico. Si los límites de control están fuera del
rango de 198 ± 30.5, reevaluar los límites de control e investigar la
fuente del problema. Si el DBO medido por el chequeo de glucosa-
ácido glutámico está fuera del rango de control aceptado, rechazar
las pruebas hechas con ese inóculo y con esa agua de dilución.
8.3.2. Rango de trabajo y límite de detección
El rango de trabajo es igual a la diferencia entre el oxígeno disuelto
inicial (de 7 a 9 mg/L) y el oxígeno disuelto residual mínimo de 1
mg/L multiplicado por el factor de dilución. El límite de detección más
bajo de 2 mg/L es establecido por los requerimientos de un mínimo
de disminución de oxígeno disuelto de 2 mg/L
9. REFERENCIAS
[1] Standard Methods for the Examination of Water and Wastewater. Method
5210 B 20th Edition. 2000. American Public Health Association, American Water
Works Association, Water Environment Federation.

64
[2] NMX-AA-028-SCFI-2001 - Secretaría de la Economía de DGN de
los Estados Unidos Mexicanos, Análisis de aguas-Determinación de la
demanda bioquímica de oxígeno en aguas naturales, residuales
(DBO5) y residuales tratadas.

65
2.3.1.4. Determinación de amonio (NH4+). Método tit ulométrico
previa destilación
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales”
Código del procedimiento: PT-FQ-02
Nombre del procedimiento: Determinación de amonio (NH4
+). Método titulométrico previa destilación.
Fecha de aprobación: 2008-05-02
Fecha de revisión: 2008-05-02
1. OBJETIVO
Describir el procedimiento para cuantificar el contenido de amonio utilizando
el método titulométrico previa destilación.
2. ALCANCE
Este procedimiento es aplicable para el análisis de muestras de aguas
superficiales. Es usado especialmente para concentraciones de NH3-N
mayores de 5 mg/L.
3. PRINCIPIO DEL METODO
El pH de la muestra se ajusta a 9,5 con buffer borato para evitar la hidrólisis
de los cianatos y los compuestos orgánicos de nitrógeno. El destilado es
recogido en ácido bórico para ser valorado posteriormente con solución de
ácido sulfúrico.
4. EQUIPOS
Debe utilizarse el equipo normal de laboratorio, además de:
4.1. Aparato de destilación
Utilizar un balón de destilación de borosilicato de 800-2000 mL de
capacidad, acoplado a un condensador. Colocar en el extremo de éste
una tubuladura lateral que debe quedar sumergida por debajo de la
superficie del recipiente que contiene la solución de ácido bórico.
4.2. Medidor de pH.
5. REACTIVOS Y MATERIALES
5.1. REACTIVOS

66
Utilizar únicamente reactivos de grado analítico reconocido y agua libre
de amonio para preparar todas las soluciones, para el lavado de la
cristalería y la dilución de la muestra.
• Tetraborato de de sodio. Na2B4O7.10H20
• Hidróxido de sodio. NaOH
• Tiosulfato de sodio. Na2S2O3.5H2O
• Indicador rojo de metilo.
• Azul de metileno. C6H18CIN3S.H2O
• Acido bórico. H3BO3
• Acido sulfúrico. H2SO4
5.2. SOLUCIONES
• Solución de tertraborato de sodio 0,025 M. Disolver 9,5 g de Na2B4O7.
10 H2O en un litro de agua.
• Solución amortiguadora de borato. Adicionar 88 mL de NaOH 0,1 N a
aproximadamente 500 mL de solución de borato de sodio 0,025 M y
diluir a 1L.
• Hidróxido de sodio 6N. Disolver lentamente 240 g de NaOH en 1 L de
agua. CUIDADO: Usar equipo de protección personal.
• Reactivo para eliminar el cloro. Disolver 3,5 g de tiosulfato de sodio en
agua y diluir a 1 litro. Preparar semanalmente. Use 1 mL de reactivo
para remover 1mg/L de cloro residual en 500 mL de muestra.
• Mezcla de indicadores: Disolver 200 mg del indicador rojo de metilo en
100 mL de alcohol etílico o isopropílico al 95 %. Disolver 100 mg de
azul de metileno en 50 mL de alcohol etílico o isopropílico al 95 %,
mezclar ambas disoluciones. Preparar mensualmente.

67
• Solución indicadora de ácido bórico. Disolver 20 g de H3BO3 en agua
y agregar 10 mL de la mezcla de indicadores y diluir a 1L. Preparar
mensualmente.
• Solución estándar de ácido sulfúrico 0,02 N.
5.3. MATERIALES
Debe emplearse cristalería normal de laboratorio.
6. ACCIONES PREVIAS
No corresponde
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Acondicionamiento del sistema de destilación.
Colocar 500 mL de agua libre de amonio en un beaker y adicionar 20
mL de la solución amortiguadora de borato. Ajustar el pH a 9,5 con
solución de hidróxido de sodio 6 N y trasvasar al balón de destilación.
Adicionar unas cuentas de vidrio o perlas de ebullición e iniciar el
proceso de destilación. Desechar la solución obtenida.
7.2. Preparación de la muestra.
Se toman 500 mL de muestra libre de cloro o una porción diluida de la
muestra. Si la concentración de amonio es menor a 100 µg/L, usar un
volumen de muestra de 1000 mL. Si es necesario, neutralizar a pH 7
con ácido o base diluidos usando el pH metro.
Adicionar 25 mL de la solución amortiguadora de borato y ajustar el pH
a 9,5 con solución de hidróxido de sodio 6 N usando el pH metro.
Transferir la solución al balón de destilación y añadir unas cuentas de
vidrio o perlas de ebullición. Conectar éste al aparato de destilación.
7.3. Destilación.
Destilar la muestra, cuidando que la temperatura del condensador no
pase de 29 °C. Recolectar el condensado en un erlen meyer de 500 mL

68
con la punta de la tubuladura lateral sumergida en 50 mL de solución
indicadora de ácido bórico.
La destilación se completa cuando se hayan recolectado 300 mL de
destilado aproximadamente, incluyendo los 50 mL de la solución
indicadora de ácido bórico.
Retirar el erlenmeyer de la tubuladura lateral.
Preparar un blanco con 500 mL de agua y darle el mismo tratamiento
que a la muestra.
7.4. Determinación de amonio.
Titular con solución de ácido sulfúrico 0,02 N, hasta que la solución vire
de un verde esmeralda a morado.
8. CALCULOS Y EXPRESION DE RESULTADOS
)(
280)(/3
mLmuestra
BAL�mg�H
×−=−
donde;
A = Volumen (mL) de H2SO4 empleado en la valoración de la muestra.
B = Volumen (mL) de H2SO4 empleado en la valoración del blanco.
9. REFERENCIAS
[1] Standard Methods for the Examination of Water and Wastewater, 20th.
Edition. 2000. Method 4500-NH3 B. American Public Health Association,
American Water Works Association, Water Environment Federation.

69
2.3.1.5. Determinación de amonio (NH 4+). Método fenato
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales”
Código del procedimiento: PT-FQ-03
Nombre del procedimiento: Determinación de amonio (NH4+).
Método fenato.
Fecha de aprobación: 2008-05-02
Fecha de revisión: 2008-05-02
1. OBJETIVO
Describir el procedimiento para cuantificar el contenido de amonio utilizando
el método de fenato.
2. ALCANCE
Se aplica para el análisis de muestras de aguas superficiales y es lineal hasta
0,6 mg NH3-N/L.
3. PRINCIPIO DEL METODO
La reacción entre el amonio, el hipoclorito y el fenol produce un compuesto de
color azul intenso llamado indofenol, el cual absorbe a 640 nm con un
recorrido de luz de 1 cm. La reacción es catalizada por el nitroprusiato de
sodio.
3.1. INTERFERENCIAS:
Las interferencias causadas por el calcio y el magnesio se eliminan
acomplejándolos con citrato, lo que evita que precipiten a valores de pH
alto. Si existe sulfuro de hidrógeno (H2S) se remueve acidificando la
muestra con ácido clorhídrico diluido a pH 3 y agitando vigorosamente,
hasta que el olor no se detecte.
4. EQUIPOS
Debe utilizarse el equipo normal de laboratorio, además de:
4.1. Espectrofotómetro de rango visible para medir a 640 nm.
5. REACTIVOS Y MATERIALES

70
5.1. REACTIVOS
Utilizar únicamente reactivos de grado analítico reconocido y agua libre de
amonio para preparar todas las soluciones, para el lavado de la cristalería y la
dilución de la muestra.
• Fenol. C6H6O
• Nitroprusiato de sodio. C5FeN6Na2O . 2H2O
• Citrato trisódico. C6H5Na3O7.2H2O
• Hipoclorito de sodio NaClO
• Cloruro de amonio. NH4Cl
• Etanol. C2H6OH
• Hidróxido de sodio. NaOH
5.2. SOLUCIONES
• Solución de fenol. Mezclar 11,1 mL de fenol líquido (≥ 89 %) con etanol al
95 % hasta llegar a un volumen final de 100 mL. CUIDADO: Preparar en
un lugar con la ventilación apropiada para evitar sobreexposición y con
equipo de protección personal.
• Solución de nitroprusiato de sodio, 0,5%. Disolver 0,5 g de nitroprusiato
de sodio en 100 mL de agua desionizada. Almacenar en botella ámbar
máximo por un mes.
• Solución de citrato alcalino: Disolver 200 g de citrato trisódico y 10 g de
hidróxido de sodio en agua desionizada. Diluir a 1L.
• Hipoclorito de sodio, solución comercial al 5 % aproximadamente: Esta
solución se descompone lentamente después de haber roto el sello de la
botella. Reemplazar cada dos meses.
• Solución oxidante: Mezclar 100 mL de la solución de citrato alcalino con
25 mL de la solución de hipoclorito de sodio. Preparar inmediatamente
antes de utilizar.

71
• Solución patrón de Amonio: Disolver 3,819 g de NH4Cl anhidro
(previamente desecado a 100 ° C por 2 h) en agua y diluir a 1 L. 1 mL = 1
mg N = 1,22 mg NH3.
• Solución estándar de amonio: Usar la solución patrón de amonio y el
agua para preparar la curva de calibración en el intervalo apropiado de la
muestra.
5.3. MATERIALES
Debe emplearse cristalería normal de laboratorio a demás de:
• Cubetas de vidrio o cuarzo de 1 cm.
6. ACCIONES PREVIAS
No corresponde.
7. DESCRIPCION DEL PROCEDIMIENTO
Tomar 25 mL de la muestra y colocar en un erlenmeyer de 50 mL. Agregar en el
siguiente orden y agitar después de cada adición: 1 mL de la solución de fenol,
1 mL de la solución de nitroprusiato de sodio y 2,5 mL de la solución oxidante.
Cubrir la boca del erlenmeyer con papel parafina y dejar que el color se
desarrolle a temperatura ambiente, en un lugar con luz tenue como mínimo 1 h.
El color es estable por 24 h.
Medir la absorbancia de las muestras por duplicado a 640 nm.
Preparar un blanco y al menos 5 estándares a partir de la solución stock de
amonio. Realizarles el mismo tratamiento que las muestras.
8. CALCULOS Y EXPRESION DE RESULTADOS
Realizar una curva de calibración graficando los valores de absorbancia
versus concentración de los estándares.
Calcular las concentraciones de las muestras interpolando los valores de
absorbancia en la curva de calibración.

72
9. REFERENCIAS
[1] Standard Methods for the Examination of Water and Wastewater, Method
4500-NH3 F. 20th Edition 2000. American Public Health Association, American
Water Works Association, Water Environment Federation.

73
2.3.1.6. Determinación de coliformes fecales. (Méto do de
Filtración por Membrana)
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales”
Código del procedimiento: PT-FQ-04
Nombre del procedimiento: Determinación de coliformes fecales (Método de Filtración por Membrana)
Fecha de aprobación: 2008-05-02
Fecha de revisión: 2008-05-02
1. OBJETIVO
Detectar y enumerar organismos coliformes fecales y Escherichia coli
presuntiva en agua, después de una filtración a través de una membrana
celulósica, su subsecuente cultivo en un medio diferencial lactosado y el
cálculo de sus números en la muestra.
2. ALCANCE
Se aplica a todo tipo de agua superficial, exceptuando aguas salinas con altos
contenidos de diatomeas, aguas con alta turbidez, aguas cloradas o cuando
números grandes de otros organismos puedan interferir con el crecimiento.
La selección de las pruebas empleadas en la detección y confirmación de los
grupos de organismos coliformes, incluyendo Escherichia coli puede verse
como parte de una secuencia continua. El grado de confirmación en una
muestra en particular depende parcialmente de la naturaleza del agua y de las
razones para llevar a cabo el examen. En la práctica, la detección en agua de
E. coli presuntiva da usualmente una indicación satisfactoria de
contaminación fecal.
3. PRINCIPIO DEL METODO
El método se basa en la filtración de una muestra directa o una alícuota de la
muestra a través de una membrana de celulosa que retiene los organismos,
colocando la membrana ya sea en un medio de cultivo selectivo o en un
cojinete absorbente saturado con un medio líquido. La membrana se incuba
durante 24 h a 44 ± 1°C para la presencia de organ ismos coliformes fecales.

74
Se lleva a cabo la cuenta directa de las colonias características desarrolladas
sobre la membrana, y algunas de estas colonias se resiembran para pruebas
confirmativas para producción de gas e indol. Finalmente, se hace el cálculo
del número de organismos coliformes fecales que pueden estar presentes en
100 mL de muestra.
4. EQUIPOS
4.1 Autoclave.
4.2 Bomba o sistema de vacío.
4.3 Balanza analítica.
4.4 Campana de Flujo Laminar (Opcional).
4.5 Equipo para filtración con membrana.
4.6 Horno de aire caliente para esterilización con calor seco.
4.7 Incubador o baño de agua, controlado termostáticamente a 35 °C – 37 °C.
4.8 Incubador o baño de agua, controlado termostáticamente a 44,0 °C ± 0,1
°C.
4.9 Medidor de pH.
5. REACTIVOS Y MATERIALES
5.1 REACTIVOS
• Cepas para los controles bacterianos positivo (Escherichia coli ATCC
25952) y negativo (Enterobacter aerogenes ATCC 13048).
• Ácido Rosólico, (C19
H14
O3)
• Agua destilada.
• Benzal al 1 %
• Cloruro de sodio (NaCl)
• Etanol 95%.
• Fosfato monobásico de potasio, (KH2PO4)
• Cloruro de magnesio, (MgCl2.6H2O).
• Hidróxido de sodio (NaOH).

75
• Caldo soya tripticaseína (TSB)
• Agar soya tripticaseína (TSA)
• Reactivo de kovacs
• Caldo laurel triptosa manitol con triptofano
• Medio m-FC. (Composición para 1 L.)
o Triptosa 10,0 g
o Peptona proteosa No. 3 o polipeptona 5,0 g
o Extracto de levadura 3,0 g
o Cloruro de sodio (NaCl) 5,0 g
o Lactosa 12,5 g
o Sales biliares No. 3 o mezcla de sales biliares 1,5 g
o Azul anilina 0,1 g
o Agua para llevar a 1 000 mL
Rehidratar en agua conteniendo 10 mL de ácido rosólico (aurina) (C19H14O3)
al 1 % en NaOH 0,2 N. Calentar el medio hasta su punto de ebullición, quitar
inmediatamente del calor enfriar y distribuir en volúmenes convenientes en
cajas de Petri. No esterilizar en autoclave. El pH final debe ser 7,4 ± 0,2.
El medio debe almacenarse entre 2 y 10°C y cualquie r porción no utilizada
debe desecharse después de 96 h.
Si dispone de la presentación comercial del medio de cultivo (lo mas
recomendable), rehidratar el medio de cultivo de acuerdo a las indicaciones
del proveedor.
NOTAS:
− Este medio puede solidificarse mediante la adición de 1,2 % al 5 % de agar
(m/m) antes de la ebullición.
− El reactivo de ácido rosólico (C19
H14
O3) se descompondrá si se esteriliza en
autoclave. La solución patrón debe almacenarse en la oscuridad entre 2 y

76
10°C y debe desecharse después de 2 semanas, o ante s si su color cambia de
rojo oscuro o café oscuro
5.2 SOLUCIONES
• Agua destilada.
• Solución de fosfato: Disolver 34 g de fosfato monobásico de potasio
en 500 mL de agua. Ajustar a pH 7,2 ± 0,5 con solución de hidróxido
de sodio 1 M y aforar a 1000 mL con agua.
• Solución de cloruro de magnesio: Disolver 81,1 g de MgCl2.6H2O en
1000 mL de agua.
• Solución amortiguadora de fosfato: Añadir 1,25 mL de solución de
fosfato y 5,0 mL de solución de cloruro de magnesio a 1000 mL de
agua. Distribuir en volúmenes convenientes y esterilizar en autoclave
a 121 ± 1 °C durante 15 min a una presión manométri ca de 0,098 066
MPa. (15 lb/inch2, a nivel del mar).
5.3 MATERIALES
• Erlenmeyer de 250 ml para la preparación de medios de cultivo
• Pipetas graduadas de 1, 2 y 10 ml
• Placas de Petri de 48 mm de diámetro
• Filtros de nitrocelulosa cuadriculados estériles de 0,45 ± 0,02 µm de
diámetro de poro.
• Pads estériles de 47 mm de diámetro
• Pinzas de punta plana.
• Cajas petri estériles
• Mechero Bunsen
6 ACCIONES PREVIAS
6.1 Los reactivos deben ser de grado analítico.
6.2 El material de vidrio deberá ser de uso exclusivo para determinaciones
microbiológicas y será lavado con una solución de Extran neutro al 2 %.

77
6.3 Las soluciones, material de vidrio y equipo que se utilicen para el análisis,
debe ser esterilizado en un autoclave a 121°C, dura nte 15 minutos a una
presión manométrica de 0,098 066 MPa (15 lb/pulg2, a nivel del mar).
7 DESCRIPCION DEL PROCEDIMIENTO
7.1 Preparación de las diluciones de las cepas control
Efectuar diluciones de las cepas control tomando 1 mL de la suspensión
bacteriana en TSB, y transferirlo a un tubo de ensayo que contenga 10
mL de solución amortiguadora de fosfato-cloruro de magnesio estéril (esta
dilución corresponde a la 1x10-1). Proseguir con las diluciones,
homogenizando cada vez la suspensión con la ayuda del agitador vortex
hasta las diluciones 1x10-8 para E. Coli y 1x10-5 para E. aerogenes.
7.2 Esterilización de equipos y del área de trabajo
Realizar la esterilización del área de trabajo y de los equipos de
laboratorio del área de microbiología de acuerdo con las normas internas
de trabajo.
7.3 Siembra de cepas control
Un día antes del análisis sembrar una asada de las cepas control en 10
mL de caldo TSB, contenido en un tubo de ensayo. Incubar de 18 a 24 h
a 35 + 1 ºC.
7.4 Preparación de cajas con caldos de cultivo
Preparar el caldo m-FC, dejarlo enfriar antes de verterlo en las cajas
Petri, para evitar la formación de gotas de condensación.
Colocar los pads en las cajas estériles en condiciones asépticas (cerca
del mechero o en la campana de flujo laminar); posteriormente dosificar 2
mL de caldo m-FC.

78
7.5 Preparación de las diluciones de las cepas control.
Efectuar diluciones de las cepas control tomando 1 mL de la suspensión
bacteriana en TSB, y transferirlo a un tubo de ensayo que contenga 10
mL de solución amortiguadora de fosfato-cloruro de magnesio estéril (esta
dilución corresponde a la 1x10-1).
Proseguir con las diluciones, homogenizando cada vez la suspensión con
la ayuda del agitador vortex, hasta las diluciones 1x10-5.
7.6 Preparación de diluciones para la muestra.
Si la muestra está muy turbia, proviene de un cuerpo de agua sin
tratamiento o de los pasos iniciales de un tratamiento, preparar diluciones
para la muestra de la misma manera que para las cepas control.
Las diluciones que se siembran son la 1x10 -1, 1x10 -2 y 1x10 -3 (estas
diluciones pueden variar, va a depender de la carga bacteriana que se
sospeche haya en la muestra). Para las muestras con un alto contenido
de materia en suspensión, filtrar utilizando un prefiltro de 0,8 a 1 µm.
Si la muestra por el contrario es casi transparente o proviene de los pasos
finales de tratamiento, sembrar directamente 100 mL. Cuando se espere
un contenido alto de bacterias, puede tomarse una muestra más pequeña
(20 mL).
NOTA:
− El volumen de muestra o la dilución elegida para la siembra es aquella en que
se sospecha que se tendrá un número de colonias que podrán ser contadas y
no se superponen entre unas y otras (entre 10 y 100 colonias). Para muestras
muy contaminadas usar la técnica de tubos múltiples de fermentación.
7.7 Filtración de muestras, controles y blancos

79
Armar el equipo de filtración en ambiente estéril (mecheros encendidos).
Colocar la membrana estéril con ayuda de las pinzas de punta plana
flameada. La cuadrícula de la membrana debe quedar visible.
Colocar el embudo con cuidado y sujetarlo con las pinzas.
Colocar aproximadamente 20 mL de solución amortiguadora de fosfatos-
cloruro de magnesio estéril dentro del vaso del equipo de filtración y filtrar
con ayuda de vacío. Este es el blanco antes de iniciar la filtración de
muestras.
Apagar la bomba de vacío, retirar la membrana y colocarla en la caja Petri
con el medio según sea el caso.
Agitar vigorosamente la muestra y verter 100 mL en el embudo, o bien
tomar 1 mL de la dilución elegida y colocarlo en 20 mL de la solución
amortiguadora estéril. Filtrar con ayuda del vacío, enjuagar el embudo con
la membrana aún en su lugar con una porción de 20 mL de la solución
amortiguadora estéril y filtrar totalmente hasta sequedad.
Retirar la membrana con la ayuda de unas pinzas y ponerla en una caja
Petri que contenga un pad estéril saturado previamente con caldo m-FC.
Tener cuidado de que no se formen burbujas de aire entre la membrana y
el pad, si esto sucede, golpear suavemente la caja contra una superficie.
Para evitar cualquier crecimiento confluente, verter cualquier exceso de
medio antes de colocar la membrana en el pad.
Enjuagar el embudo con 200 mL de solución amortiguadora y filtrar, antes
de trabajar con otra dilución o muestra. También se puede desinfectar el
equipo de filtración pasando una torunda de algodón impregnada con
etanol al 70 % en la superficie interna del embudo y flamear al mechero

80
con cuidado. Durante la filtración, no es necesario desinfectar el porta
membranas a menos que se contamine.
No alternar la filtración de muestras contaminadas con muestras de agua
tratada en el mismo equipo, a menos que se desinfecte. Primero filtrar
todas las muestras de agua cloradas y aquella de las que se esperan
resultados negativos y después las muestras contaminadas. Se sugiere
utilizar un equipo de filtración para todas las muestras cloradas y otro para
las muestras contaminadas.
Para los controles bacterianos positivo (Escherichia coli ATCC 25952) y
negativo (Enterobacter aerogenes ATCC 13048), filtrarlos por duplicado al
final del procedimiento, antes del último blanco. Seguir los mismos pasos
que para la filtración de las muestras.
Colocar posteriormente las membranas con E. coli en caldo m-FC (Control
positivo para coliformes fecales).
Por último, filtrar el blanco final después de haber desinfectado o
enjuagado el vaso con 200 mL de solución amortiguadora.
7.8 Incubación de las membranas
Incubar las membranas que contienen Caldo m-FC a 44,0 ± 0,2 °C entre 15
y 24 h para organismos coliformes fecales.
7.9 Examen de las membranas
Las membranas deben examinarse inmediatamente después de la
incubación. Contar como organismos coliformes fecales presuntivos todas
las colonias independientemente del tamaño, que muestren un color azul
marino después de su incubación a 44 °C en caldo m- FC.

81
NOTAS:
− Los blancos de reactivos no deben presentar contaminación, y la morfología
colonial o inhibición del crecimiento deben corresponden a las cepas control
positivo y negativo en el medio de cultivo respectivo.
− Las colonias que presentan las características mencionadas en Caldo m-FC
indican resultados presuntivos de Coliformes Fecales; dado que no se detecta
la producción de gas. Esto puede ser suficiente para agua cruda, agua residual
o parcialmente tratada, pero para agua potable o cuando el cliente así lo
indique por algún estudio especial se llevarán a cabo las pruebas
confirmativas.
7.10 Pruebas confirmativas
7.10.1 Producción de gas e indol.
Para confirmar los resultados, elegir una colonia de las aisladas en
medio mFC y sembrar en una placa con TSA, Incubar de 18 a 24
horas a 35± 1 °C.
Resembrarla en Caldo lauril triptosa manitol con triptofano
contenido en tubos de ensayo que tengan campanas de
fermentación Durham invertidas e incubar a a 44,0 ± 0,2 °C por 24
± 2 h.
Examinar los tubos para determinar la presencia o ausencia de
gas.
La producción de gas confirma la presencia de colif ormes
fecales.
Realizar la prueba de indol colocando a cada tubo en donde se
haya detectado la producción de gas, dos o tres gotas del reactivo
de Kovacs.

82
La formación de un anillo rojo en la superficie del cultivo
confirma la presencia de E. coli presuntiva .
7.11 Prueba de oxidasa.
De las colonias positivas para la producción de gas, tomar una asada
del cultivo en TSA y realizar la prueba de la oxidasa.
Las bacterias clasificadas dentro de los grupos coliformes totales y
fecales son oxidasa negativas.
8 CALCULOS Y EXPRESION DE RESULTADOS
A partir del número de colonias características contadas en las membranas y
tomando en cuenta los resultados de las pruebas confirmativas, calcular el
número de coliformes fecales presentes en la muestra, expresando el
resultado como unidades formadoras de colonias (UFC) por 100 mL de la
muestra de acuerdo con la siguiente fórmula:
Sin dilución:
muestradefiltradoVolumen
mLporcontadascoliformesColonias 100
Con dilución:
s
nnn
i
s VFVnFVnFVn
�C *
)(...)()( 222111 ++++= ∑

83
donde;
Cs es el número de unidades formadoras de colonias en el volumen de
referencia Vs de la muestra.
ΣNi
es la suma de las colonias en todas las cajas o membranas
contadas filtradas/siembras de dilución F1.
n1 es el número de cajas contadas para una dilución particular F1.
V1 es el volumen de dilución de la muestra F1 en la placa i.
F1 es la dilución usada para la porción de muestra V1
Vs es el volumen de referencia seleccionado para expresar la
concentración de los microorganismos en la muestra.
Nota
− La cuenta final así obtenida es el promedio ponderado de las cuentas de
cada una de las placas.
9 REFERENCIAS
[1] Standard Methods for the Examination of Water and Wastewater. Method
9222 B. 20th Edition. 2000. American Public Health Association, American
Water Works Association, Water Environment Federation.
[2] EPA 821-R-02-02. Method 1603: Escherichia coli(E. coli) in Water by
Membrane Filtration Using Modified membrane – Thermotolerant
Escherichia coli Agar (Modified mTEC). Septiembre 2002.
[3] NMX-AA-102-SCFI-2006 Calidad del Agua – Detección y Enumeración de
Organismos Coliformes, Organismos Coliformes Termolerantes y y
Escherichia coli Presuntiva – Método de Filtración en Membrana.
[4] L.A. Teixeira & Filhos S/C Litda. PA-AG 014 02: Determinação de
coliformes totais e fecais em águas. 2004.

84
[5] Instituto Tecnológico de Toluca, Laboratorio de Investigación en
Ingeniería Ambiental. LIIA-PT-16: Procedimiento para la Detección y
Enumeración de Organismos Coliformes, Organismos Coliformes
Termolerantes y y Escherichia coli Presuntiva – Método de Filtración en
Membrana. 2007.

85
2.3.1.7. Determinación de coniformes fecales. (Méto do del
número más probable)
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales”
Código del procedimiento: PT-FQ-05
Nombre del procedimiento: Determinación de coniformes fecales (Método del número más probable)
Fecha de aprobación: 2008-05-02
Fecha de revisión: 2008-05-02
1 OBJETIVO
Este procedimiento describe un método de análisis microbiológico para la
determinación cuantitativa de bacterias coliformes fecales, empleando el
método de tubos múltiples de fermentación (NMP) en todo tipo de agua.
2 ALCANCE
Se aplica a todo tipo de agua. La selección de las pruebas empleadas en la
detección y confirmación de los grupos de organismos coliformes puede verse
como parte de una secuencia continua. El grado de confirmación en una
muestra en particular depende parcialmente de la naturaleza del agua y de las
razones para llevar a cabo el examen.
3 PRINCIPIO DEL METODO
El método se basa en el enriquecimiento de las muestras de agua con caldo
lactosado y EC incubados entre 44,5 °C ± 0,2 °C durante 24 h.
4 EQUIPOS
4.1 Autoclave
4.2 Campana de Flujo Laminar (Opcional)
4.3 Horno de aire caliente para esterilización con calor seco
4.4 Incubador o baño de agua, controlado termostáticamente
4.5 Medidor de pH

86
5 REACTIVOS Y MATERIALES
5.1 REACTIVOS
Los reactivos deben ser grado analítico.
• Fosfato monobásico de potasio. KH2PO4
• Sulfato de magnesio. MgSO4.7H2O.
• NaOH
• Caldo lactosado.
• Caldo EC
5.2 SOLUCIONES
• Agua de dilución.
• Solución de Fosfato: Disolver 34 g de fosfato monobásico de potasio
en 1000 mL de agua. Ajustar a pH 7,2 ± 0,5 con solución de hidróxido
de sodio 1 mol/L y aforar a 1 000 mL con agua Tipo 1.
• Solución de sulfato de magnesio heptahidratado: Pesar 50,00 g de
MgSO4.7H2O y disolver a 1 L con agua Tipo 1.
• Mezclar 1,25 mL de la solución de fosfato y 5 mL de la solución de
sulfato de magnesio heptahidratado y aforar a 1 L con agua Tipo 1.
Esterilizar a 121 °C por 15 minutos.
• Caldo lactosado sencillo: Rehidratar el medio de acuerdo a las
indicaciones del fabricante.
• Caldo EC: Rehidratar el medio de acuerdo a las indicaciones del
fabricante.
• Hidróxido de sodio 1mol/L.
5.3 MATERIALES
• Asas de metal con un mínimo de 3mm de diámetro.
• Gradilla para tubo de ensayo de 180 mm x 20 mm.
• Pipetas bacteriológicas esterilizadas de 1 mL y 10 mL.
• Tubos de ensayo de 180 x 20 mm.

87
• Tubos de fermentación Durham
6 ACCIONES PREVIAS
El material de vidrio deberá ser de uso exclusivo para determinaciones
microbiológicas y será lavado con una disolución de Extran neutro al 2 %.
Las soluciones, material de vidrio y equipo que se utilicen para el análisis,
deben ser esterilizados en un autoclave a 121°C, a 15 psi durante 15 min.
7 DESCRIPCION DEL PROCEDIMIENTO
7.1 Esterilización de equipos y del área de trabajo.
Realizar la esterilización del área de trabajo y de los equipos de
laboratorio del área de microbiología de acuerdo con las normas internas
de trabajo.
7.2 Preparación de diluciones para la muestra.
Si la muestra es muy turbia, proviene de un cuerpo de agua sin
tratamiento o de los pasos iniciales de un tratamiento, preparar diluciones
para la muestra. Las diluciones que se siembran son la 1x10-1, 1x10-2 y
1x10-3 (estas diluciones pueden variar, va a depender de la carga
bacteriana que se sospeche haya en la muestra). Para las muestras con
un alto contenido de materia en suspensión, filtrar utilizando un prefiltro
de 0,8 µm a 1 µm.
Si la muestra por el contrario es casi transparente o proviene de los pasos
finales de tratamiento, sembrar directamente 10 mL. Cuando se espere
un contenido alto de bacterias, puede tomarse una muestra más
pequeña.
7.3 Determinación del número más probable de coliformes fecales.
7.3.1 Prueba presuntiva
De la muestra de ensayo de agua tratada, se toman 5 porciones de
10 mL cada una empleando pipetas estériles graduadas de 10 mL y

88
se transfieren a todos los tubos de ensayo que contengan 10 mL de
caldo lactosado (con tubos invertidos onifo en su interior) y se
mezcla el contenido. Cuando se analiza agua no tratada, se inoculan
porciones de la muestra y sus diluciones respectivas.
Se incuban los tubos entre 35 ± 0,5°C y 37 °C durante 24 ± 2 h . Al
cabo de 24 h se examina cada tubo inoculado y si no se ha
producido gas, se reincuban y se examinan nuevamente al cabo de
48 ± 3 h. La formación de gas dentro de las 48 h constituyen una
prueba presuntiva positiva y la ausencia de gas constituye una
prueba negativa.
7.3.2 Prueba confirmativa
A partir de cada uno de los tubos gas positivo en caldo lactosado (en
la prueba presuntiva), se inoculan, con una asa, los tubos que
contienen el caldo EC. Los tubos de caldo EC se incuban en baño
de agua a temperatura de 44,5 ± 0,2 °C durante 24 ± 2 h.
Los tubos sembrados se colocaran en baño de agua dentro de los
30 minutos después de la inoculación y se mantendrán sumergidos
a un nivel por encima del medio de cultivo. La producción de gas en
un tubo de fermentación dentro de 24 h o menos se considera una
reacción positiva indicando coliformes de origen fecal. Cuando no se
produce gas la reacción se considera negativa.
8 CALCULOS Y EXPRESION DE RESULTADOS
Los tubos considerados como positivos en la prueba confirmativa se llevan a
la tabla del NMP que corresponda (según el numero de tubos utilizados) los
resultados de NMP de coliformes fecales se expresan por 100 mL.
9 REFERENCIAS

89
[1] Standard Methods for the Examination of Water and Wastewater. Method
9221 E. 20th. Edition. 2000. American Public Health Association, American
Water Works Association, Water Environment Federation.
[2] Norma Cubana 93-01-128. 1998. Determinación del Número Más
Probable de Coliformes totales y fecales.

90
2.3.1.8. Análisis de Turbiedad o Turbidez
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales”
Código del procedimiento: PT-FQ-06
Nombre del procedimiento: Análisis de Turbiedad o Turbidez
Fecha de aprobación: 2008-05-02
Fecha de revisión: 2008-05-02
1 OBJETIVO
Describir el procedimiento para determinar la turbiedad o turbidez utilizando el
método nefelométrico.
2 ALCANCE
Se limita al análisis de muestras de aguas superficiales.
3 PRINCIPIO DEL METODO
La turbiedad en el agua es causada por la materia coloidal o suspendida
(materia orgánica, materia inorgánica, organismos microscópicos, etc.). Es
una expresión de la propiedad óptica que causa que la luz sea dispersada y
absorbida, en lugar de ser transmitida en una línea recta a través de la
muestra.
Las lecturas son realizadas empleando un turbidímetro, calibrado con un
patrón de referencia de formazina.
3.1 INTERFERENCIAS
La presencia de residuos flotantes y materia fina de rápida sedimentación.
Burbujas de aire, suciedad en los recipientes de vidrio y las celdas de
medición, así como efectos de vibración que alteran la visibilidad
superficial de la muestra pueden conducir a resultados falsos.
“El color verdadero” por ejemplo, el color del agua debido a las sustancias
disueltas que absorben luz puede causar medidas de turbidez baja,
aunque este efecto generalmente no es significativo en aguas tratadas.

91
4 EQUIPOS
Debe utilizarse el equipo normal de laboratorio, además de:
4.1 Turbidímetro
El equipo debe cumplir como mínimo las siguientes características:
• Angulo de la luz aceptada por el detector.- Centrado a 900 al paso de
luz incidente y no exceda ± 30° de los 90°.
• Detector y sistema de filtro. - El detector y el sistema de filtro, si es
usado, debe tener una respuesta del pico espectral entre 400 nm y 600
nm.
• Distancia atravesada por la luz incidnte y la luz dispersada dentro del
tubo de muestra. - En total no debe exceder los 10 cm.
• Fuente de luz. - Lámpara de tungsteno que opera a una temperatura
entre 2200 ° K y 3000 ° K.
4.2 Balanza analítica con una precisión menor de 0,1 mg.
5 REACTIVOS Y MATERIALES
5.1 REACTIVOS
Utilizar únicamente reactivos de grado analítico reconocido.
• Agua libre de turbiedad: Pasar agua destilada y/o desionizada a través
de un filtro de membrana de tamaño de poro de 0,45µm.
• Sulfato de hidracina (NH2)2 H2SO4
• Hexametilentetramina (CH2)6N4
5.2 SOLUCIONES
• Solución estándar primaria de formazina (4000 UNT).

92
Solución I: Disolver 1,000 g de sulfato de hidracina en agua y llevar a
100 mL en un matraz aforado. Cuidado : El sulfato de hidracina es
cancerígeno, evite inhalación, ingestión y contacto con la piel. La
suspensión de formazina puede contener sulfato de hidracina residual.
Solución II: Disolver 10,00 g de hexametilentetramina en agua y llevar
a 100 mL en un matraz aforado.
Mezclar 5 mL de la Solución I y 5 mL de la Solución II. Dejar en reposo
por 24 h a 25 ± 3 °C. Tranferir esta solución a un frasco ámbar para su
almacenamiento. Esta solución es estable hasta 1 año cuando es
almacenada apropiadamente.
• Juego de soluciones estándar de turbidez.
A partir de la solución estándar primaria de 4000 UNT, obtener las
soluciones de turbidez en el rango de interés. Deben prepararse
diariamente.
5.3 MATERIALES
Debe emplearse cristalería normal de laboratorio, así como:
• Celdas de vidrio o plástico incoloro, transparente y limpias.
6 ACCIONES PREVIAS
Las muestras deben de analizarse lo antes posible después del muestreo y
en un periodo no mayor de 24 h, mientras permanezcan en el laboratorio
deben conservarse refrigeradas a 4 ºC.
7 DESCRIPCION DEL PROCEDIMIENTO

93
Si la muestra se encuentra refrigerada esperar que alcance la temperatura
ambiente antes de que se realice el ensayo.
Seguir las instrucciones del manual de operación del equipo para la
calibración del mismo.
Agite la muestra. Espere que desaparezcan las burbujas de aire y coloque la
muestra en la celda cuidadosamente.
Leer la turbidez de la muestra. Se recomienda tomar 3 lecturas,
homogeneizando entre cada una de ellas.
8 CALCULOS Y EXPRESION DE RESULTADOS
Leer la turbiedad directamente del instrumento en Unidades Nefelométricas
de Turbiedad (UNT).
8.1 CONTROL DE CALIDAD
La diferencia entre las réplicas no debe ser mayor que ± 2% para lecturas
menores a 100 UNT y ± 3% para lecturas mayores 100 UNT.
9 REFERENCIAS
[1] Standard Methods for the Examination of Water and Wastewater. Method
2130 B. 20th. Edition. 2000. American Public Health Association, American
Water Works Association, Water Environment Federation.
[2] U.S. Environmental Protection Agency. Método 180.1: Determination of
Turbidity by Nephelometry. 1993
[3] NMX-AA-038-SCFI-2001: Análisis de Agua – Determinación de Turbiedad
en aguas naturales, residuales y residuales tratadas. 2001.
[4] L.A. Teixeira & Filhos S/C Litda. PA-AG 014 02: Determinação de Turbidez em
águas. 2008

94
2.3.1.9. Determinación de Nitratos (NO 3-) previa reducción con
cadmio o hidracina
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales”
Código del procedimiento: PT-FQ-07
Nombre del procedimiento: Determinación de Nitratos (NO3-) previa reducción con cadmio o hidracina.
Fecha de aprobación: 2008-05-08
Fecha de revisión: 2008-05-08
1 OBJETIVO
Determinar la concentración de nitratos en agua por espectrofotometría,
utilizando dos métodos de reducción: reducción con cadmio cuperizado, o con
sulfato de hidracina.
2 ALCANCE
Este procedimiento se utiliza para el análisis de muestras de aguas
superficiales.
El método de reducción con cadmio cuperizado es recomendable para la
cuantificación de nitratos en muestras que contienen entre 0,01 y 1 mg de
N/L.
El método de reducción con sulfato de hidracina se recomienda para
determinar concentraciones entre 0,01 y 10 mg N/L.
3 PRINCIPIO DEL METODO
3.1 Reducción con cadmio cuperizado
El ión nitrato se reduce cuantitativamente a nitrito (NO2-) en presencia de
cadmio granulado, tratado con sulfato de cobre y empacado en una
columna de vidrio.

95
3.2 Reducción con sulfato de hidracina
El ión nitrato (NO3-) es reducido espontáneamente a nitrito (NO2
-) con
sulfato de hidracina.
3.3 Determinación de nitrito por espectrofotometría.
El nitrito obtenido por cualquiera de los dos métodos de reducción
descritos anteriormente es determinado espectrofotométricamente por
diazotización con sulfanilamida y posterior copulación con N (1 – Naftol)
etilendiamina dihidrocloruro, para formar un compuesto coloreado cuya
intensidad es proporcional a la concentración de nitrato.
3.4 Interferencias
Las muestras cuyo color absorben en el rango fotométrico utilizado
pueden interferir. Las concentraciones del ión sulfito menores de 10 mg/l
causa variaciones en la concentración de nitratos y nitritos en un 10%
aproximadamente.
4 EQUIPOS
Debe utilizarse el equipo normal de laboratorio, además de:
4.1 Espectrofotómetro ultravioleta –visible.
4.2 Balanza analítica.
4.3 Estufa.
4.4 Medidor de pH.
5 REACTIVOS Y MATERIALES
5.1 REACTIVOS

96
Utilizar únicamente reactivos de grado analítico reconocido y agua
desionizada o de pureza equivalente.
5.1.1 Método de reducción con cadmio cuperizado
• Gránulos de cadmio cuperizado
• Ácido clorhídrico concentrado (HCl)
• Ácido fosfórico (H3PO4)
• Sulfanilamida (NH2C6H4SO2NH2)
• N (1 – Naftol) etilendiamina dihidrocloruro (C10H7NHCH2NH2. 2HCl)
• Cloruro de amonio (NH4Cl)
• Sal sódica del ácido etilendiaminotetracético (EDTANa2)
• Sulfato de cobre pentahidratado (CuSO4.5H2O)
• Hidróxido de amonio concentrado (NH3OH)
• Nitrito de sodio (NaNO2)
• Nitrato de sodio (NaNO3)
• Cloroformo (CHCl3)
5.1.2 Método de reducción con sulfato de hidracina
• Ácido fosfórico concentrardo (H3PO4)
• Etilendiamina dihidrocloruro (1- naftol) (C10H7NHCH2NH2.2HCl)
• Hidróxido de sodio (NaOH)
• Nitrato de potasio (KNO3)
• Sulfanilamida (NH2C6H4SO2NH2)
• Sulfato de cobre pentahidratado (CuSO4.5H2O)
• Sulfato de hidracina (N2H4H2SO4.H20)
• Nitrito de sodio (NaNO2)
• Nitrato de sodio (NaNO3)

97
• Cloroformo (CHCl3)
5.2 SOLUCIONES
5.2.1 Método de reducción con cadmio cuperizado
Gránulos de cadmio cuperizado: Lavar 25 g de gránulos de
cadmio, de un tamaño entre 20 y 100 mesh con ácido clorhídrico 6
N y enjuagarlos con agua. Colocar el cadmio en 100 mL de la
solución de sulfato de cobre al 2 % por 5 minutos o hasta que
palidezca parcialmente el color azul de la solución. Decantar y
repetir la operación con sulfato de cobre fresco hasta iniciar el
desarrollo de un precipitado coloidal de color café. Lavar con agua
y retirar todo el cobre precipitado.
Reactivo de color: Agregar a 800 mL de agua desionizada 100 mL
de ácido fosfórico concentrado al 85% v/v y 10 g de sulfanilamida.
Después de la disolución completa de la sulfanilamida, añadir 1 g
de etilendiamina dihidrocloruro N (1- Naftol). Diluir con agua hasta
1 litro. Envasar en botella ámbar y refrigerar. Esta solución es
estable por 1 mes. Si la solución se colorea, se debe descartar. La
presencia de coloración rosada indica contaminación.
Solución cloruro de amonio-EDTA: Disolver 13 g de cloruro de
amonio y 1,7g de EDTANa2 en un volumen de aproximadamente
900 mL. Ajustar a pH 8,5 con amoníaco, enrasar a un litro en
matraz aforado.
Solución de sulfato de cobre: Disolver 20 g de sulfato de cobre en
500 mL de agua y completar el volumen a 1 litro en matraz
aforado.

98
Solución patrón de nitrato. 100 mg NO3--N/L: Secar nitrato de
potasio a 105° C por 24 horas. Disolver 0,7218 g de la sal en agua
y diluir a 1 litro. Preservar con 2 mL de cloroformo
1mL = 100 µg NO3—N.
Solución intermedia de nitrato. 10 mg/L: Diluir 100 mL de solución
estándar de nitrato a 1 litro con agua. Preservar con 2 mL de
cloroformo.
1mL = 10 µg NO3--N.
Solución patrón de nitritos. 250 mg N /L: Pesar 1,232 g de nitrito de
sodio secados previamente a 105 °C durante 24 horas , y disolver
en aproximadamente 800 mL de agua, añadir 1 mL de cloroformo.
Enrasar a 1 litro y mezclar. Refrigerar.
1mL = 250 µg N.
Solución intermedia de nitrito. 50 mg N /L: Diluir 50 mL de la
solución estándar de nitrito a 250 mL con agua en matraz aforado.
Preparar cuando vaya a ser usada.
1 mL = 50 µg N.
Solución de trabajo de nitrito: Diluir 10 mL de la solución intermedia
de nitritos a 1 L con agua en matraz aforado. Preparar cuando
vaya a ser usada.
1 mL = 0,50 µg N.
5.2.2 Método de reducción con sulfato de hidracina
Reactivo formador de color: Agregar a 500 mL de agua 200 mL de
ácido fosfórico concentrado al 85% v/v y 10 g de sulfanilamida.
Después de la disolución completa de la sulfanilamida, añadir 0.8 g

99
de etilendiamina dihidrocloruro N (1- Naftol). Diluir con agua hasta
1 litro. Envasar en botella ámbar y refrigerar. Esta solución es
estable por 1 mes.
Solución patrón de nitrato, 100 mg NO3--N/L: Secar nitrato de
potasio a 105° C por 24 horas. Disolver 0,7218 g de la sal en agua
desionizada y diluir a 1 litro. Preservar con 2 mL de cloroformo
1mL = 100 µg NO3--N.
Solución intermedia de nitrato.10 mg NO3--N /L: Prepararla a partir
de la solución patrón de nitrato.
1mL = 10 µg NO3—N
Solución patrón de hidróxido de sodio 10 N: Disolver 400 g de
hidróxido de sodio en 750 mL de agua y diluir a 1 litro con agua.
Solución intermedia de hidróxido de sodio 1,0 N: Tomar un
volumen de 100 mL de la solución patrón de hidróxido de sodio y
llevar a 1 litro con agua.
Solución patrón de sulfato de cobre: Disolver 2,5 g de sulfato de
cobre pentahidratado en agua y diluir hasta 1 litro.
Solución diluida de sulfato de cobre: Tomar 20 mL de la solución
patrón de sulfato de cobre y diluir con agua hasta 2 litros.
Solución patrón de sulfato de hidracina: Disolver 27.5 g de sulfato
de hidracina en 400 mL de agua y diluir hasta 1 litro. Esta solución
es estable por 6 meses. PRECAUCION: Su ingestión es tóxica.
5.2.3 MATERIALES

100
Debe emplearse cristalería normal de laboratorio, así como Celdas
de vidrio o cuarzo de 1 cm de paso óptico.
6 ACCIONES PREVIAS
Si el almacenamiento es necesario, guardar las muestras por 24 horas a 4°
C. Para almacenajes mas prolongados conservar con 2 ml de H2SO4
concentrado y a 4° C.
NOTA
- Cuando la muestra es conservada con ácido, el nitrato y el nitrito no se
pueden determinar como especies individuales.
7 DESCRIPCION DEL PROCEDIMIENTO
7.1 Reducción con cadmio cuperizado
7.1.1 Preparación de la columna de reducción
Insertar un tapón de lana de vidrio o algodón en la base de la
columna de reducción y llenar con agua.
Añadir suficientes gránulos de cadmio cuperizado para empacar
una columna de 18,5 cm. Mantener el nivel de agua por encima de
los gránulos de cadmio cuperizado para evitar burbujas de aire.
Lavar la columna con 200 mL de solución de cloruro de amonio-
EDTA. Activar la columna haciéndole pasar, a velocidad de 7 a 10
mL/min. 100 mL o más de una disolución compuesta por 25% de
estándar de 1,0 mg de N-NO3--/L y 75% de solución de cloruro de
amonio-EDTA.
7.1.2 Tratamiento de la muestra

101
Ajustar el pH (si fuera necesario) entre 7 y 9 usando pHmetro y
ácido clorhídrico o hidróxido de sodio diluidos. Esto asegura un pH
de 8,5, después de añadir la solución de cloruro de amonio-EDTA.
7.1.3 Reducción de la muestra
A 25 mL de muestra o una porción diluida previamente llevada a 25
mL, adicionar 75 mL de la solución de cloruro de amonio-EDTA y
mezclar.
Verter la muestra mezclada en la columna y ajustar el flujo a una
velocidad entre 7 mL/min. a 10 mL/min. Descartar los primeros 25
mL y colectar el resto en el matraz de la muestra original.
No es necesario lavar la columna entre muestras y muestra; pero si
no va a ser reutilizada durante varias horas o por algún tiempo,
verter 50 mL de solución diluida de cloruro de amonio-EDTA en la
columna y dejarla pasar a través del sistema. Almacenar la
columna de cadmio cuperizado en esta solución y no permitir que
se seque.
7.1.4 Desarrollo del color y medición
Tan pronto como sea posible y no más de 15 min después de la
reducción, añadir 2,0 mL de reactivo de color a 50 mL de la
muestra reducida y mezclar. Entre 10 min y antes de las 2 h medir
la absorbancia a 543 nm contra un blanco de reactivos.
NOTA: Si la concentración de NO3- excede el intervalo de la curva
(aproximadamente 1 mg NO3- N- / L), usar el sobrante de muestra reducida
para hacer una dilución apropiada y analizar nuevamente.
7.1.5 Preparación de la curva de calibración

102
Utilizando la solución intermedia de NO3--N, preparar los
estándares en el rango entre 0,05 a 1,0 mg NO3--N/L en
volumétricos de 100 mL. Proceda de igual forma que la muestra
(reducción de la misma).
Comparar un estándar de nitrito de igual concentración a uno de
nitrato reducido y verifique la eficiencia de la columna. Reactivar
los gránulos cadmio cuperizado cuando la eficiencia de reducción
esté aproximadamente por debajo del 75 %.
7.2 Método de reducción con sulfato de hidracina
Tomar 500 µl de la muestra y adicionar 1 ml de NaOH, 1 mL de agua
desionizada, 500 µl de sulfato de cobre y 500 µl de hidrazina. Dejar
reposar la muestra por ½ hora y luego agregar 500 µl de colorante y 500
µl de agua. Los patrones deben ser preparados a partir de la solución
intermedia de nitrato en un rango de 1 a 7 mg/L. Leer los espectros de
cada muestra en el espectrofotómetro uv- visible a una longitud de onda
de 520 nm.
8 CALCULOS Y EXPRESION DE RESULTADOS
Para ambos métodos:
Obtener una curva de calibración graficando la absorbancia contra la
concentración de N-NO3 de los estándares.
Calcular la concentración de la muestra directamente de la curva de
calibración.
Reportar como miligramos de N por litro (la suma de N-NO3- más N-NO2
-) a
menos que la concentración de N-NO2- se determine y reste separadamente.
Hacer una gráfica con los valores de la curva de calibración y asegurarse de
obtener el coeficiente de correlación aceptable.

103
Calcular la concentración de la muestra por medio de la ecuación de la recta
obtenida de la curva de calibración representada por la siguiente ecuación:
Y = mX + b
Donde:
m = pendiente
b = ordenada al origen
Y = absorbancia
X = concentración (mg N-NO3-/L).
Reportar mg N-NO3-/L con la precisión correspondiente.
9 REFERENCIAS
[1] Standard Methods for the Examination of Water and Wastewater. Method
4500 NO3 E y G. 20 th Edition. 2000. American Public Health Association,
American Water Works Association, Water Environment Federation.
[2] Secretaría de la Economía de DGN de los Estados Unidos Mexicanos,
Análisis de aguas- Determinación de nitratos en aguas naturales, potables,
residuales y residuales tratadas. NMX-AA-079-SCFI-200.

104
2.3.1.10. Determinación de nitratos (NO 3-). Método del electrodo
de nitrato
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales”
Código del procedimiento: PT-FQ-08
Nombre del procedimiento: Determinación de nitratos (NO3-). Método del electrodo de nitrato.
Fecha de aprobación: 2008-05-08
Fecha de revisión: 2008-05-08
1 OBJETIVO
Determinar la concentración de nitratos en agua, en el rango de 0,14 a 1400
mg NO3-- N /L, utilizando el método del electrodo de nitrato.
2 ALCANCE:
Se aplica para el análisis de muestras de aguas superficiales in situ o llevadas
al laboratorio.
3 PRINCIPIO DEL METODO
El electrodo de ión NO3- es un sensor selectivo que desarrolla un potencial a
través de una membrana delgada, porosa, inerte, que se mantiene en
posición en un intercambiador iónico en un líquido inmiscible con agua. El
electrodo responde a la actividad del ión NO3- entre aproximadamente 10-5 y
10-1 M (0,14 a 1400 mg NO3-- N/L).
3.1 INTERFERENCIAS
Los iones cloruros y bicarbonato interfieren cuando su proporción en peso
frente a NO3-- N es > 10 o > 5, respectivamente. Los iones potencialmente
interferentes pero que no se encuentran en niveles significativos en las
aguas potables son: NO2-, CN-, S2
-, Br-, I-, ClO3- y ClO4
-. Se han
observado respuestas erráticas del electrodo cuando el pH no se
mantiene constante; se recomienda que esté en un rango entre 3 y 9 para

105
el funcionamiento óptimo del mismo. Dado que el electrodo responde a la
actividad de NO3- más que a su concentración, la fuerza iónica debe ser
constante en todas las muestras y patrones.
Para minimizar estos problemas se utiliza una solución buffer que
contenga:
• Ag2SO4 , que remueve los iones Cl-, Br-, I-, S-2 y CN-
• Al2(SO4)3, que forma complejos con los ácidos orgánicos presentes
• Acido sulfámico, que remueve el NO2-
• pH=3 que elimina el HCO3-, mantiene el pH y la fuerza iónica constante.
4 EQUIPOS
Debe utilizarse el equipo normal de laboratorio, además de:
4.1 Electrodo selectivo para determinación de nitrato
4.2 Medidor de pH/mV, (resolución de 0,1 mV) o multímetro compatible con el
electrodo.
4.3 Agitador magnético.
5 REACTIVOS Y MATERIALES
5.1 REACTIVOS
Utilizar únicamente reactivos de grado analítico reconocido y agua
desionizada o de pureza equivalente.
• Nitrato de sodio. NaNO3 o Nitrato de potasio. KNO3
• Sulfato de amonio. (NH4)2SO4
• Sulfato de aluminio. Al2(SO4)3.18H2O
• Sulfato de plata. Ag2SO4

106
• Ácido bórico. H3BO3
• Ácido sulfámico H2NSO3H
• Hidróxido de sodio. NaOH
• Cloroformo CHCl3
5.2 SOLUCIONES
• Agua exenta de nitratos: Utilizar agua bidestilada o destilada,
desionizada de la máxima pureza para preparar todas las soluciones y
diluciones.
• Solución estándar de nitrato, 1000 mg/L NO3-: Secar NaNO3 o KNO3
en una estufa a 105 °C durante 24 h. Disolver 1.37 g de NaNO3 o 0.7218
g de KNO3 en agua y diluir a 1000 mL en matraz aforado. Preservar la
solución de KNO3 con 2 mL de CHCl3/L. La solución es estable por un
período de 6 meses
• Solución buffer: Disolver 17,32 g de Al2(SO4)3.18H2O, 3,43 g de
Ag2SO4, 1,28 g de H3BO3 y 2,52 g de H2NSO3H, en aproximadamente
800 mL de agua. Ajustar el pH a 3 agregando lentamente solución de
NaOH 0,1 N. Diluir a 1000 mL y guardar en botella de vidrio color ámbar.
• Solución de NaOH 0,1N: Disolver 4 g de NaOH en un vaso de
precipitado, con aproximadamente 500 mL de agua, posteriormente
transferir cuantitativamente la solución a un matraz aforado de 1000 mL y
completar el volumen con agua.
• Solución de llenado del electrodo de referencia: Disolver 0.53 g de
(NH4)2SO4 en agua y diluir a 100 mL.
5.3 MATERIALES
Debe emplearse cristalería normal de laboratorio, así como:
• Pastilla magnética (magnetos)
• Termómetro.

107
6 ACCIONES PREVIAS
Si la muestra va a ser llevada al laboratorio, se recomienda recolectarla en
envase de plástico (polietileno o equivalente) o vidrio de un volumen mínimo
de 100 mL, sin cámara de aire y cerrar herméticamente. Analizar lo antes
posible o refrigerar. El tiempo máximo de conservación recomendado es de
48 h.
7 DESCRIPCION DEL PROCEDIMIENTO
7.1 Preparación del electrodo
Llenar el electrodo con la solución de relleno hasta justo debajo del orificio
de llenado. Agitarlo suavemente hacia abajo (como un termómetro) para
eliminar las burbujas de aire. Previo al primer uso, o luego de un período
largo de almacenamiento, sumergir la membrana de nitrato en un
estándar de nitrato durante 30 minutos.
7.2 Preparación de la curva de calibración
Por dilución seriada del estándar de nitrato de 1000 mg/L, preparar los
estándares de 100 y 10 mg/L. Agregar 10 mL de solución buffer por cada
10 mL de estándar. Preparar estándares de composición similar a la
muestra, si estas tienen una fuerza iónica superior a 0,1 M.
Colocar la solución más diluida (10 mg/L) en el agitador magnético y
agitar a velocidad constante. Colocar el electrodo en la solución (con el
medidor en el modo de mV) y cuando se estabiliza la lectura registrarla.
Colocar la solución intermedia (100 mg/L) en el agitador magnético y
agitar a velocidad constante. Introducir el electrodo en la solución (con el
medidor en el modo de mV) y cuando se estabiliza la lectura registrarla.
Poner la solución más concentrada (1000 mg/L) en el agitador magnético
y agitar a velocidad constante. Colocar el electrodo en la solución (con el
medidor en el modo de mV) y cuando se estabiliza la lectura registrarla.

108
Graficar las lecturas en mV vs el logaritmo de las concentraciones.
Extrapolar la curva hasta 1.0 mg/L.
Chequear la calibración cada dos horas. Asumiendo que no hubo
cambios en la temperatura ambiente, sumergir el electrodo en el estándar
intermedio. Luego que la lectura se estabiliza, se compara con la lectura
original registrada de la solución intermedia. Si la diferencia es mayor a
0.5 mV o la temperatura ambiente cambió hacer nuevamente la curva de
calibración.
Preparar la curva de calibración diariamente.
7.3 Medición de la muestra
Agregar en un vaso limpio y seco de 150 mL, 10 mL de la muestra y 10
mL de solución Buffer, colocar en el agitador magnético. Luego de
enjuagar el electrodo en agua exenta de nitratos, secar y sumergirlo en la
solución. Cuando se estabiliza, registrar la lectura en mV.
8 CALCULOS Y EXPRESION DE RESULTADOS
Determinar la concentración directamente de la curva de calibración.
Los resultados se expresan en mg NO3-N/L
8.1 Precisión
Dentro del rango del método se espera una precisión de ±0,4 mV, que
corresponde al 2,5% en concentración.
9 REFERENCIAS
[1] Standard Methods for the Examination of Water and Wastewater. Method
4500 D. 20 th Edition, 2000. American Public Health Association, American
Water Works Association, Water Environment Federation

109
2.3.1.11. Determinación de oxígeno disuelto por el método
iodométrico (modificación azida)
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales”
Código del procedimiento: PT-FQ-09
Nombre del procedimiento: Determinación de oxígeno disuelto por el método iodométrico (modificación azida)
Fecha de aprobación: 2008-05-08
Fecha de revisión: 2008-05-08
1 OBJETIVO
Determinar el oxígeno disuelto (OD) por el método iodométrico (modificación
azida)
2 ALCANCE
Se aplica para analizar el oxígeno disuelto en aguas superficiales.
3 PRINCIPIO DEL METODO
El método yodométrico es el método más preciso y confiable de los análisis
titulométricos para la determinación de OD. Está basado en la adición de una
solución de manganeso divalente, seguida de la adición de un álcali fuerte a
la muestra en una botella de vidrio tapada. El OD oxida rápidamente una
cantidad equivalente del precipitado de hidróxido manganoso divalente
disperso a un hidróxido con estado de valencia mayor. En presencia de iones
yoduro en una solución ácida, el manganeso oxidado se revierte al estado
divalente, con liberación de yoduro equivalente al contenido original de OD.
El yoduro es entonces titulado con una solución estándar de tiosulfato.
El punto final de la titulación puede ser detectado visualmente, con un
indicador de almidón.
Los análisis experimentales pueden alcanzar una precisión de ±50 µg /L con

110
una detección visual del punto final y una precisión de ±5 µg/L con la
detección electrométrica del punto final.
La modificación de azida es usada para muestras de agua superficiales,
especialmente si las muestras contienen mas de 50 µg NO2--N/L y no mas de
1 mg de hierro ferroso/L. Otros materiales oxidantes y reductores podrían
estar ausentes. Si 1mL de solución de KF es adicionado antes de que la
muestra sea acidificada y no haya retraso en la titulación, el método es
aplicable en presencia de 100 a 200 mg hierro férrico/L.
La modificación de azida, remueve efectivamente la interferencia causada
por los nitritos, que son muy comunes en efluentes tratados biológicamente,
4 EQUIPOS
Debe utilizarse el equipo normal de laboratorio, además de:
4.1 Balanza analítica
5 REACTIVOS Y MATERIALES
5.1 REACTIVOS
Utilizar únicamente reactivos de grado analítico reconocido y agua
desionizada o de pureza equivalente.
• Sulfato de manganeso. MnSO4.4H2O, MnSO4.2H2O, MnSO4.H2O
• Yoduro de sodio o Yoduro de potasio. NaI o KI
• Azida de sodio. NaN3
• Ácido sulfúrico concentrado. H2SO4
• Almidón soluble. (C6H10O5)n
• Ácido salicílico C7H6O3
• Tiosulfato de sodio. Na2S2O3.H2O

111
• Biyodato de potasio. KH(IO3)2
• Hidróxido de sodio o Hidróxido de potasio NaOH o KOH
5.2 SOLUCIONES:
• Solución sulfato manganoso tetrahidratado: Disolver 480 g de MnSO4
.4 H2O, o 400 g de MnSO4.2 H2O ó 364 g MnSO4 x. H2O en agua
destilada, filtrar y diluir a 1 L.
La solución de sulfato manganoso no deberá dar color con el almidón
cuando es adicionado a una solución acidificada de KI.
• Solución álcali-yoduro-azida:
Para muestras saturadas o de menor saturación . Disolver 500 g de
NaOH (ó 700 g de KOH), 135 g de NaI (ó 150 g KI) en agua destilada y
diluir a un litro. Añadir 10 g de NaN3 disueltos en 40 mL de agua
destilada. Las sales de sodio y potasio pueden ser utilizadas en forma
intercambiada. Este reactivo no debe dar color con el almidón cuando es
diluida y acidificada.
Para muestras sobresaturadas: Disolver 10 g de NaN3 en 500 ml de
agua destilada. Añadir 480 g de NaOH y 750 g de NaI, y agitar hasta
disolución. Se producirá una turbidez blanca debida al carbonato de sodio
pero que no es perjudicial.
• Acido sulfúrico concentrado: Un mililitro es equivalente a
aproximadamente 3 mL de reactivo álcali-yoduro-azida.
• Solución de almidón: Disolver 2 g de almidón soluble y 0,2 g de ácido
salicílico como preservante en 100 mL de agua destilada caliente.

112
NOTA
- Se ha probado que sin preservante y guardándolo a la refrigeradora
da buenos resultados para que no se descomponga tan rápidamente.
• Solución estándar de Tiosulfato de Sodio 0,025 M: Disolver 6.205 g de
Na2S2O3.5 H2O en agua destilada. Añadir 1,5 mL de NaOH 6N ó 0,4 g
de NaOH sólido y diluir a 1 litro. Estandarizar con solución de biyodato
de potasio 0,0021 M.
• Solución estándar de biyodato de potasio 0,0021 M. Disolver 812,4 mg
de KH(IO3)2 en agua destilada y diluir a 1 litro.
Para la estandarización:
Disolver aproximadamente 2 g de KI libre de yodato en un frasco
erlenmeyer con 100 a 150 mL de agua destilada, añadir 1 mL de H2SO4
6N ó unas pocas gotas de H2SO4 concentrado y 20 mL de solución
estándar de biyodato. Diluir a 200 mL y titular el yodo liberado con la
solución estándar de tiosulfato, adicionando almidón cerca del punto final
de la titulación cuando un color amarillo pálido es alcanzado. Cuando las
soluciones tienen igual fuerza, se requieren 20 mL de tiosulfato de sodio
0,025 M . Si no es así, ajustar la solución de Na2S2O3 a 0,025 M.
5.3 MATERIALES
Debe emplearse cristalería normal de laboratorio, así como:
• Frascos Winkler
6 ACCIONES PREVIAS

113
Se debe evitar durante la toma de muestra la incorporación de burbujas de
aire en la botella y la presencia de sustancias oxidantes o reductoras.
7 DESCRIPCION DEL PROCEDIMIENTO
A la muestra colectada en un frasco de 250 a 300 mL, añada 1 mL de
MnSO4, seguido por 1 mL de solución álcali-yoduro-azida. Si la pipeta es
sumergida dentro de la muestra enjuagarla antes de regresarla al frasco del
reactivo. Alternativamente mantenga la punta de la pipeta justo arriba de la
superficie del líquido cuando se estén añadiendo los reactivos. Tapar
cuidadosamente para evitar burbujas de aire y mezclar invirtiendo el frasco
varias veces. Cuando el precipitado haya sedimentado lo suficiente
(aproximadamente la mitad del volumen del frasco) dejar que el
sobrenadante aclare arriba del floculo de MnSO4, añadir 1 mL de H2SO4
concentrado. Tapar nuevamente y mezclar por inversión hasta que la
solución esté completa. Titular un volumen correspondiente a 200 mL de la
muestra original después de la corrección por pérdida de muestra por
desplazamiento de los reactivos. Así para un total de 2 mL (1 mL de cada
reactivo) en un frasco de 300 mL, titular 200 x 300/(300-2) = 201 mL.
Titular con solución de Na2S2O3 0,025 M hasta la aparición de un color
amarillo pálido. Añadir unas pocas gotas de solución de almidón y continuar
la titulación hasta la primera desaparición del color azul. Si el punto final es
sobrepasado titule de retroceso con solución de biyodato 0,0021 M añadido
gota a gota ó por la adición de un volumen medido de muestra tratada.
Corregir la cantidad de solución de biyodato o de muestra. No tomar en
cuenta las recoloraciones siguientes que aparecen por efecto catalítico de
nitritos o trazas de sales férricas que no fueron complejadas con el fluoruro.
8 CALCULOS Y EXPRESION DE RESULTADOS
Para 200 mL de muestra, 1 mL de Na2S2O3 0,025 M es = 1 mg de OD/L.

114
8.1 PRECISIÓN Y SESGO DE LOS RESULTADOS
El OD puede ser determinado con una precisión, expresada como una
desviación estándar, de cerca de 20 µg/L en agua destilada y cerca de 60
µg/L en aguas de desecho y en efluentes secundarios. En presencia de
interferentes apreciables hasta con las modificaciones apropiadas, la
desviación estándar puede ser tan alta como 100 µg/L.
9 REFERENCIAS
[1] Standard Methods for the Examination of Water and Wastewater 20th
Edition. 4500-B. 2000. American Public Health Association, American Water
Works Association, Water Environment Federation.
[2] Secretaria de la Economía. Análisis de agua-NMX-AA-012-SCFI-2001–
Determinación de oxígeno disuelto en aguas naturales, residuales y residuales
tratadas-México.

115
2.3.1.12. Determinación de fósforo total. (Método d el ácido
ascórbico)
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales” Código del procedimiento:
PT-FQ-10 Nombre del procedimiento:
Determinación de fósforo total (Método del ácido ascórbico)
Fecha de aprobación: 2008-05-02
Fecha de revisión: 2008-05-02
1 OBJETIVO
Describir el procedimiento para cuantificar los niveles de fósforo total
utilizando el método colorimétrico del ácido ascórbico.
2 ALCANCE
Este procedimiento se limita al análisis de muestras de aguas superficiales.
La concentración mínima detectable es de aproximadamente 10 µg/L.
3 PRINCIPIO DEL METODO
El ortofosfato reacciona con molibdato de amonio, el tartrato potásico de
antimonio y produce fosfomolibdato de antimonio, este se reduce con ácido
ascórbico a un complejo de color azul. Los complejos presentan absorción de
radiación a la cual se le puede aplicar la Ley de Beer para realizar la
determinación del fósforo total. La intensidad del color azul en el intervalo de
trabajo es directamente proporcional a la concentración de fósforo total.
3.1 INTERFERENCIAS
A partir de 0,1 mg/L de arsénico hay interferencia en la determinación. El
cromo hexavalente y el nitrito interfieren.

116
El sulfuro y el silicato no interfieren a concentraciones entre 1 y 10 mg
P/L. Altas concentraciones de hierro causan precipitaciones y pérdidas de
fósforo.
4 EQUIPOS
Debe utilizarse el equipo normal de laboratorio, además de:
4.1 Placa calefactora.
4.2 Espectrofotómetro rango visible.
4.3 Balanza Analítica con una precisión menor de 0,1 mg.
4.4 Equipo de filtración.
4.5 Microondas.
4.6 Cronómetro.
5 REACTIVOS Y MATERIALES
5.1 REACTIVOS
Utilizar únicamente reactivos de grado analítico reconocido y agua
desionizada o de pureza equivalente.
• Molibdato de amonio (NH4)6Mo7O24
• Ácido ascórbico C6H8O6
• Tartrato potásico de antimonio K(SbO)C4H4O6
• Fosfato monobásico de potasio KH2PO4
• Fenolftaleína C20H14O4
• Hidróxido de sodio libre de fosfatos. NaOH
• Ácido sulfúrico concentrado H2SO4
• Ácido nítrico concentrado HNO3

117
• Ácido clorhídrico concentrado HCl
• Persulfato de amonio (NH4)2S2O8 o Persulfato de potasio K2S2O8
5.2 SOLUCIONES
• Ácido sulfúrico 2,5 M: Se diluye 70 mL de ácido sulfúrico concentrado
con agua desionizada a un volumen final de 500 mL.
• Solución de tartrato potásico de antimonio: Se disuelve 1,3715 g de
tartrato potásico de antimonio en 400 mL de agua desionizada. Se lleva
a un volumen final de 500 mL en matraz aforado. Se almacena en un
frasco de vidrio.
• Solución de molibdato de amonio: Se disuelve 20 g de molibdato de
amonio en 500 mL de agua desionizada (requiere calentamiento). Se
almacena en un frasco de vidrio.
• Solución de ácido ascórbico 0,1 M: Se disuelve 1,76 g de ácido
ascórbico en 100 mL de agua desionizada, esta solución es estable por
una semana almacenada a 4 °C.
• Reactivo combinado: Se mezcla las siguientes soluciones para
preparar 100 mL del reactivo combinado:
o 50 mL de ácido sulfúrico 2,5 M
o 5 mL de solución de tartrato potásico de antimonio
o 15 mL de solución de molibdato de amonio
o 30 mL de ácido ascórbico
NOTA
- Se mezcla después de agregar cada reactivo. Se permite a los
reactivos alcanzar la temperatura ambiente antes de mezclarse. Se
deben mezclar en el orden dado. Si se forma turbidez mezclar y se deja

118
hasta que la turbidez desaparezca. El reactivo tiene una estabilidad de
4 horas.
• Solución estándar de fósforo 50 mg P/L: Se disuelve 219,5 mg de
fosfato monobásico de potasio anhidro (previamente secado por 2 h en
estufa a 105 °C) en agua desionizada. Se lleva a 1 L. Se con serva en
una botella de plástico oscuro y se refrigera.
• Solución de trabajo de 2,5 mg P / L: Se diluye 50 mL de la solución
estándar de fósforo a un volumen final de 1 L con agua desionizada.
• Solución de fenolftaleína: Se pesa 0,05 gramos de fenolftaleína y se
diluye a 100 mL con etanol.
• Hidróxido de sodio 10 M.
• Hidróxido de sodio 1 M
• Ácido sulfúrico 1 M
5.3 MATERIALES
Debe emplearse cristalería normal de laboratorio, así como:
• Filtros Whatman número 42 o equivalente
• Embudo de espiga larga
• Cubetas de vidrio o cuarzo de 1 cm.
6 ACCIONES PREVIAS
La muestra se recolecta en botella plástica o de vidrio. El equipo se debe
lavar con detergente libre de fosfatos. La muestra se debe mantener a 4 °C.
Si no se puede realizar el análisis inmediatamente, conserve con 1 mL de
ácido clorhídrico concentrado o ácido sulfúrico concentrado, para mantener el
pH < 2.
La cristalería se debe lavar con ácido nítrico 1 + 1 o con ácido clorhídrico
diluido caliente, y enjuagado con agua bidestilada o desionizada.

119
7 DESCRIPCION DEL PROCEDIMIENTO
7.1 Digestión tradicional
Se toman 50 mL de muestra y se colocan en un erlenmeyer. Se adiciona
1 mL de ácido sulfúrico 5,5 M y 0,4 g de persulfato de amonio o 0,5 g de
persulfato de potasio. Se digiere la muestra sobre una placa de
calentamiento aproximadamente por un período de 30 a 40 min hasta un
volumen de 10 mL. Si hay presencia de compuestos organofosforados, el
tiempo de digestión puede incrementarse hasta por 2 h.
Si hay presencia de sólidos después de la digestión la muestra se debe
filtrar en papel filtro Whatman número 42 o equivalente. Se trasvasa la
muestra a un matraz aforado de 50 mL, (nota: se utilizan
aproximadamente 25 mL de agua desionizada para hacer el trasvase y el
lavado).
Se agregan 2 gotas de solución de fenolftaleína, posteriormente se
agrega hidróxido de sodio 10 M hasta que cambie a un color rosa. En
esta etapa es posible que aparezca la presencia de un precipitado que no
se debe filtrar debido a que puede indicar presencia de fosfato de calcio.
Se adiciona ácido sulfúrico 1 M gota a gota hasta la desaparición del color
rosado (alcanzar la neutralidad). Se continúa con la etapa de desarrollo
de color respectivo.
Se adicionan 8 mL del reactivo combinado y se afora a 50 mL, se agita
vigorosamente y se mide entre 10 y 30 min después de mezclado y se lee
en espectrofotómetro visible a una longitud de onda 880 nm. Si la muestra
presenta color o turbiedad se utiliza la misma muestra como blanco
agregando 8 mL del reactivo combinado. La absorbancia del blanco de la
muestra se debe sustraer de la absorbancia de la muestra.

120
Según la capacidad del equipo, se preparan patrones en un intervalo de
concentraciones de 0,15 mg P / L hasta 1,30 mg P / L. El número de
patrones dependerá de la decisión de cada laboratorio.
Para ello, se mide el volumen apropiado del patrón de la solución de
trabajo de 2.5 mg/L y se lleva a un volumen final de aproximadamente 50
mL con agua desionizada. Se aplica el proceso de digestión apropiado,
desarrollo de color y lectura.
NOTAS:
-Tanto las muestras como los patrones deben ser analizadas con el mismo
tiempo de desarrollo de color y medición.
- Es necesario tener más de un blanco de reactivos si el tiempo de operación así
lo requiere.
8 CALCULOS Y EXPRESION DE RESULTADOS
Se determina la curva de calibración Absorbancia vs concentración. Se
interpola la absorbancia de la muestra. Se debe ajustar la concentración por
dilución con la siguiente fórmula:
mg P/L = f x Cn
donde;
mg P/L = Concentración de fósforo de la muestra
f = factor de dilución
Cn = ¿???
8.1 CONTROL DE CALIDAD

121
Se deben preparar enriquecidos de muestras por cada lote, se considera
el análisis en control cuando el porcentaje de recuperación está entre 90 y
110%.
9 REFERENCIAS
[1] EPA 365-1 Determination of Phosphorus by Semi-Automated Colorimetric
[2] Standard Methods for the Examination of Water and Wastewater. Method
4500 P E. 19a. Edition. 1995. American Public Health Association, American
Water Works Association, Water Environment Federation.

122
2.3.1.13. Determinación de fósforo total. (Método d e molibdato de
amonio y cloruro de estaño II)
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales” Código del procedimiento: PT-FQ-11
Nombre del procedimiento: Determinación de fósforo total (Método de molibdato de amonio y cloruro de estaño II)
Fecha de aprobación: 2008-05-02
Fecha de revisión: 2008-05-02
1 OBJETIVO
Describir el procedimiento para cuantificar los niveles de fósforo total
utilizando el método de reducción con cloruro de estaño mediante la
determinación espectrofotométrica.
2 ALCANCE
Este procedimiento se limita al análisis de muestras de aguas superficiales.
La concentración mínima detectable es de aproximadamente 3 µg P/L.
3 PRINCIPIO DEL METODO
El ortofosfato se compleja con molibdato de amonio. El método se basa en la
formación de ácido molibdofosfórico que es reducido por el cloruro de estaño
II para formar un compuesto de molibdeno de coloración azul intensa. Los
complejos presentan absorción de radiación a la cual se le puede aplicar la
Ley de Beer para realizar la determinación del fósforo total. La intensidad del
color azul entre los rangos de trabajo es directamente proporcional a la
concentración de fósforo total.
3.1 INTERFERENCIAS
La presencia de sílice, arsenato, fluoruro, torio, bismuto, sulfuro,
tiosulfato, tiocianato o exceso de molibdato, así como concentraciones

123
superiores a 100 mg /L de hierro II. La interferencia de sulfuro se puede
eliminar al adicionar agua de bromo. La dureza cálcica no es una
interferencia para concentraciones inferiores a 1000 mg/L al igual que los
iones de aluminio, hierro III, magnesio, bario, estroncio II, litio, sodio,
potasio, amonio, cadmio, manganeso, plomo, mercurio I y II, estaño II,
cobre, níquel, plata, uranio IV, circonio IV, bromuro, carbonato, clorato,
hipoclorito, cianuro, yodato, silicato, nitrato, nitrito, sulfato, sulfito,
tetraborato, selenato. Si se utiliza ácido nítrico los cloruros interfieren a
una concentración superior a 75 mg/ L.
4 EQUIPOS
Debe utilizarse el equipo normal de laboratorio, además de:
4.1 Equipo para calentamiento.
4.2 Espectrofotómetro de rango visible para leer a 690 nm, si el equipo no es
capaz de leer a esa longitud de onda, utilizar 650 nm para soluciones
acuosas, con algo de reducción de la sensibilidad y precisión.
4.3 Balanza analítica con una precisión menor de 0.1 mg.
4.4 Equipo de filtración.
4.5 Microondas (si se realizara digestión de muestra por este método).
4.6 Cronómetro.
4.7 Baño de agua.
5 REACTIVOS Y MATERIALES
5.1 REACTIVOS
Utilizar únicamente reactivos de grado analítico reconocido y agua
desionizada o de pureza equivalente.
• Molibdato de Amonio. (NH4)6Mo7O24.4H2O

124
• Cloruro de Estaño II dihidratado. SnCl2.2H2O
• Gricerol.
• Fosfato monobásico de potasio. KH2PO4
• Fenolftaleína. C20H14O4
• Etanol. C2H5OH
• Hidróxido de sodio libre de fosfatos. NaOH
• Ácido Sulfúrico concentrado. H2SO4
• Ácido Nítrico concentrado. HNO3
• Ácido Clorhídrico concentrado. HCl
5.2 SOLUCIONES
• Solución de molibdato de amonio (Reactivo I):
o Se disuelve 12,5 gramos de (NH4)6Mo7O24 x 4H2O en 90 mL de
agua desionizada (requiere calentamiento para su disolución
completa).
o Se adiciona lentamente a 200 mL de agua desionizada 140 mL de
H2SO4 concentrado (CUIDADO: se adiciona lentamente para evitar
un sobrecalentamiento peligroso).
Se permite deja enfriar ambas soluciones y luego se mezclan. La
mezcla se lleva a un volumen final de 500 mL. Se almacena en botella
de plástico oscuro y se refrigera para su conservación.
• Solución de cloruro de estaño (Reactivo II). Se disuelven 2,5 gramos
de SnCl2. 2H2O en 100 mL de glicerol. Se calienta la mezcla en un
baño de agua y se agita para apresurar la disolución. Se almacena en
botella de plástico y se refrigera para su conservación.
• Solución intermedia de 150,0 mg P/L: Se disuelven 329,2 mg de
KH2PO4 anhidro (se deseca la sal dos horas a 105 °C) en a gua
desionizada. Se lleva a 500 mL. Se conserva en una botella de plástico
oscuro y se refrigera.

125
• Solución de trabajo de 15,0 mg P/L: Se mide 5 mL de la solución
intermedia de fosfato y se agrega en un matraz aforado de 50 mL, se
lleva a volumen con agua desionizada. Se almacena en una botella de
plástico oscuro bajo refrigeración por un tiempo no superior a un mes.
• Solución de fenolftaleína: Se pesa 0,05 gramos de fenolftaleína y se
diluye a 100 mL con etanol.
• Hidróxido de sodio 10 molar.
• Hidróxido de sodio 1 molar
• Ácido sulfúrico 1 molar
5.3 MATERIALES
Debe emplearse cristalería normal de laboratorio, así como:
• Cubetas de vidrio o cuarzo de 1 cm.
• Filtros Whatman Nº 42 o equivalente
6 ACCIONES PREVIAS
La muestra se recoge en botella plástica o de vidrio previamente lavados con
detergente libre de fosfatos. La muestra se debe mantener a 4 °C. Si no se
puede realizar el análisis inmediatamente, conserve con 1 mL de ácido
clorhídrico concentrado o ácido sulfúrico concentrado, para mantener el pH <
2.
La cristalería se debe lavar con ácido nítrico 1 + 1 (o con ácido clorhídrico
diluido caliente) y enjuagar con agua bidestilada o desionizada.
7 DESCRIPCION DEL PROCEDIMIENTO
7.1 Digestión tradicional:

126
Se toman 50 mL de muestra y se coloca en un erlenmeyer. Se adiciona 1
mL de ácido sulfúrico concentrado y 5 mL de ácido nítrico concentrado.
Se digiere la muestra sobre una placa de calentamiento hasta un volumen
de 1 mL y se continúa hasta la decoloración (indica la remoción de NO2
del HNO3).
Después de digerida la muestra, se permite que alcance la temperatura
ambiente. Se trasvasa cuantitativamente a matraces aforados de 100 mL,
si hay presencia de sólidos después de la digestión la muestra se debe
filtrar en papel filtro cualitativo Whatman Nº 42 o equivalente. Se debe
procurar no utilizar un exceso de agua de lavado en el trasvase. Se
agregan 2 gotas de solución de fenolftaleína, posteriormente se agrega
hidróxido de sodio 10 M hasta que cambie a un color rosa, en esta etapa
es posible que aparezca la presencia de un precipitado que no se debe
filtrar debido a que puede indicar presencia de fosfato de calcio. Se
adiciona ácido sulfúrico 1 M gota a gota hasta la desaparición del color
rosado (alcanzar la neutralidad).
7.2 Digestión por microondas
Se toman 50 mL de muestra. Se adiciona 5 mL de ácido nítrico
concentrado, se programa la temperatura del microondas con
incrementos constantes por 25 minutos hasta un máximo de 150 °C, la
presión y la temperatura de seguridad se selecciona según las
instrucciones del fabricante.
Después de digerida la muestra, se permite que alcance la temperatura
ambiente. Se trasvasa cuantitativamente a matraces aforados de 100 mL,
si hay presencia de sólidos después de la digestión la muestra se debe
filtrar en papel filtro cualitativo Whatman Nº 42 o equivalente. Se debe
procurar no utilizar un exceso de agua de lavado en el trasvase. Se
agregan 2 gotas de solución de fenolftaleína, posteriormente se agrega
hidróxido de sodio 10 M hasta que cambie a un color rosa, en esta etapa

127
es posible que aparezca la presencia de un precipitado que no se debe
filtrar debido a que puede indicar presencia de fosfato de calcio. Se
adiciona ácido sulfúrico 1 M gota a gota hasta la desaparición del color
rosado (alcanzar la neutralidad).
Una vez que la muestra alcance la temperatura ambiente, se agregan en
el siguiente orden, con rapidez y con agitación 4 mL de la solución de
molibdato de amonio y 10 gotas de solución de cloruro de estaño II, se
agita vigorosamente y se afora a volumen final. La lectura de la muestra
se realiza después de 10 minutos, pero antes de los 12 minutos en
espectrofotómetro visible a una longitud de onda de 690 nm.
Según la capacidad del equipo se preparan patrones en un intervalo de
concentraciones de 0,003 mg P / L hasta 1 mg P / L. El número de
patrones dependerá de la decisión de cada laboratorio.
Para ello, se mide el volumen del patrón de la solución de trabajo de 15
mg P/L y se lleva a un volumen final de aproximadamente 50 mL con
agua desionizada. Se aplica el proceso de digestión apropiado, desarrollo
de color y lectura.
NOTAS
- Tanto las muestras como los patrones deben ser analizadas con el mismo
tiempo de desarrollo de color y medición.
- Es necesario tener más de un blanco de reactivos si el tiempo de
operación así lo requiere
8 CALCULOS Y EXPRESION DE RESULTADOS
Se determina la curva de calibración absorbancia vs concentración. Se
interpola la absorbancia de la muestra, se debe ajustar la concentración por
dilución con la siguiente fórmula:

128
muestrademL
VfxCn:L/PmgmuestraladeiónConcentrac
Donde:
Cn = Concentración de la muestra leída en la curva de calibración (mg/L)
Vf = Volumen final de la muestra después de la digestión realizada (100 mL)
mL de muestral = Alícuota de muestra utilizada para la digestión (50 mL)
8.1 CONTROL DE CALIDAD
Se deben preparar enriquecidos de muestras por cada lote, se considera el
análisis en control cuando el porcentaje de recuperación está entre 90 y
110%.
9 REFERENCIAS
[1] EPA 365-1 Determination of Phosphorus by Semi-Automated Colorimetric.
[2] Norma Mexicana NMX-AA-029-SCFI-2001. Determinación de Fósforo Total
en aguas naturales, residuales y residuales tratadas. Método de Prueba.
[3] Standard Methods for the Examination of Water and Wastewater. Method
4500 P C. 20th. Edition. 2000. American Public Health Association, American
Water Works Association, Water Environment Federation.
[4] MAQA-1. Determinación de Fósforo Total. Centro de Investigación en
Contaminación Ambiental, UCR. Costa Rica. Versión 9.

129
2.3.1.14. Determinación de fósforo total. (Método d e molibdato de
amonio y cloruro de estaño II)
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales” Código del procedimiento:
PT-FQ-12 Nombre del procedimiento:
Determinación de fósforo total (Método del molibdovanadato de amonio)
Fecha de aprobación: 2008-05-02
Fecha de revisión: 2008-05-02
1 OBJETIVO
Describir el procedimiento para cuantificar los niveles de fósforo total
utilizando el método colorimétrico del molibdovanadato de amonio.
2 ALCANCE
Este procedimiento se limita al análisis de muestras de aguas superficiales.
La concentración mínima detectable es de aproximadamente 200 µg/L
utilizando celdas de 1 cm.
3 PRINCIPIO DEL METODO
El ortofosfato en presencia de molibdato de amonio y vanadato forma el ácido
vanadomolibdofosfórico. Los complejos presentan absorción de radiación a la
cual se le puede aplicar la Ley de Beer para realizar la determinación del
fósforo total. La intensidad del color amarillo entre los rangos de trabajo es
directamente proporcional a la concentración de fósforo total.
3.1 INTERFERENCIAS
La presencia de sílice, arsenato, fluoruro, torio, bismuto, sulfuro, tiosulfato,
tiocianato o exceso de molibdato. Concentraciones superiores a 100 mg /L de
hierro II. La interferencia de sulfuro se puede eliminar al adicionar agua de

130
bromo. La dureza cálcica no es una interferencia para concentraciones
inferiores a 1000 mg/L al igual que los iones de aluminio, hierro III, magnesio,
bario, estroncio II, litio, sodio, potasio, amonio, cadmio, manganeso, plomo,
mercurio I y II, estaño II, cobre, níquel, plata, uranio IV, circonio IV, bromo,
carbonato, clorato, hipoclorito, cianuro, yodato, silicato, nitrato, nitrito, sulfato,
sulfito, tetraborato, selenato. Si se utiliza ácido nítrico los cloruros interfieren a
una concentración superior a 75 mg / L.
4 EQUIPOS
Debe utilizarse el equipo normal de laboratorio, además de:
4.1 Equipo de calentamiento (placa calefactora o hot plate)
4.2 Espectrofotómetro de rango visible
4.3 Balanza analítica con una precisión menor de 0,1 mg
4.4 Equipo de filtración
4.5 Microondas
4.6 Cronómetro
5 REACTIVOS Y MATERIALES
5.1 REACTIVOS
Utilizar únicamente reactivos de grado analítico reconocido y agua
desionizada o de pureza equivalente.
• Molibdato de amonio (NH4)6Mo7O24.4H2O
• Metavanadato de amonio NH4VO3
• Fosfato monobásico de potasio KH2PO4
• Fenolftaleína C20H14O4
• Etanol C2H5OH
• Hidróxido de sodio libre de fosfatos. NaOH

131
• Ácido sulfúrico concentrado H2SO4
• Ácido nítrico concentrado HNO3
• Ácido clorhídrico concentrado HCl
5.2 SOLUCIONES
• Reactivo molibdo-vanadato: Este reactivo se prepara mezclando la
Solución A con la Solución B y diluyendo a 1 L.
Solución A: Se disuelve 25 gramos de molibdato de amonio en 300 mL
de agua desionizada (requiere calentamiento).
Solución B: Se disuelve 1,25 gramos de metavanadato de amonio en
300 mL de agua (se debe calentar). Se enfría la solución B a
temperatura ambiente, se agrega 330 mL de ácido clorhídrico
concentrado. Se enfría a temperatura ambiente.
• Solución estándar de fósforo 50 mg P/L: Se disuelve 219,5 mg de
fosfato monobásico de potasio anhidro (previamente secada a 105 oC)
en agua desionizada. Se lleva a 1 L. Se conserva en una botella de
plástico oscuro y se refrigera.
• Solución de fenolftaleína: Se pesa 0,05 gramos de fenolftaleína y se
diluye a 100 mL con etanol.
• Hidróxido de sodio 10 molar.
• Hidróxido de sodio 1 molar
• Ácido sulfúrico 1 molar
5.3 MATERIALES
Debe emplearse cristalería normal de laboratorio, así como
• Cubetas de vidrio o cuarzo de 1 cm.

132
• Filtros Whatman Número 42 o equivalente
6 ACCIONES PREVIAS
La muestra se recoge en botella plástica o de vidrio previamente lavados con
detergente libre de fosfatos. La muestra se debe mantener a 4 °C. Si no se
puede realizar el análisis inmediatamente, conserve con 1 mL de ácido
clorhídrico concentrado o ácido sulfúrico concentrado, para mantener el pH <
2.
La cristalería se debe lavar con ácido nítrico 1 + 1 (o con ácido clorhídrico
diluido caliente) y enjuagar con agua desionizada o de pureza equivalente.
7 DESCRIPCION DEL PROCEDIMIENTO
7.1 Digestión tradicional:
Se toman 50 mL de muestra y se colocan en un erlenmeyer. Se adiciona
1 mL de ácido sulfúrico concentrado y 5 mL de ácido nítrico concentrado.
Se digiere la muestra sobre una placa calefactora hasta un volumen de 1
mL y se continúa hasta la decoloración (indica la remoción de NO2 del
HNO3).
Después de digerida la muestra, se permite que alcance la temperatura
ambiente. Se trasvasa cuantitativamente a matraces aforados de 100 mL,
si hay presencia de sólidos después de la digestión la muestra se debe
filtrar en papel filtro Whatman número 42 o equivalente. Se debe procurar
no utilizar un exceso de agua de lavado en el trasvase. Se agregan 2
gotas de solución de fenolftaleína, posteriormente se agrega hidróxido de
sodio 10 M hasta que cambie a un color rosa, en esta etapa es posible
que aparezca la presencia de un precipitado que no se debe filtrar debido
a que puede indicar presencia de fosfato de calcio.

133
7.2 Digestión por microondas
Se toman 50 mL de muestra. Se adiciona 5 mL de ácido nítrico
concentrado, se programa la temperatura con incrementos constantes por
25 minutos hasta un máximo de 150 °C, la presión y la temperatura de
seguridad se selecciona según las instrucciones del fabricante.
Después de digerida la muestra, se permite que alcance la temperatura
ambiente. Se trasvasa cuantitativamente a matraces aforados de 100 mL,
si hay presencia de sólidos después de la digestión la muestra se debe
filtrar en papel filtro Whatman número 42 o equivalente. Se debe procurar
no utilizar un exceso de agua de lavado en el trasvase. Se agregan 2
gotas de solución de fenolftaleína, posteriormente se agrega hidróxido de
sodio 10 M hasta que cambie a un color rosa, en esta etapa es posible
que aparezca la presencia de un precipitado que no se debe filtrar debido
a que puede indicar presencia de fosfato de calcio.
Una vez que se permite que la muestra alcance la temperatura ambiente,
se afora la muestra al volumen final de 100 mL.
Se toman 25 mL de la muestra y se vierten en un matraz aforado de 50
mL. Se adiciona 10 mL del reactivo vanadato-molibdato y se lleva a la
marca de aforo. La lectura de la muestra se puede realizar después de 10
minutos debido a que el complejo es estable durante varios días. La
lectura se realiza en espectrofotómetro visible ó equivalente.
Dependiendo de la concentración de las muestras se selecciona la
longitud de onda como se presenta en la Tabla 1.
Tabla 1. Selección de longitud de onda según rango de trabajo
Rango de trabajo
(mg P/L)
Longitud de onda
(nm)
1 – 5 400
2 – 10 420

134
4 – 18 470
Según las concentraciones de las muestras se preparan patrones como
se indica en la tabla I. El número de patrones dependerá de la decisión de
cada laboratorio.
Para ello, se mide el volumen del patrón de la solución de trabajo de 50
mg P /L y se lleva a un volumen final de aproximadamente 50 mL con
agua desionizada. Se aplica el proceso de digestión apropiado, desarrollo
de color y lectura.
NOTAS
- Tanto las muestras como los patrones deben ser analizadas con el mismo
tiempo de desarrollo de color y medición.
- Es necesario tener más de un blanco de reactivos si el tiempo de operación
así lo requiere.
8 CALCULOS Y EXPRESION DE RESULTADOS
Se determina la curva de calibración absorbancia vs concentración. Se
interpola la absorbancia de la muestra, se debe ajustar la concentración por
dilución con la siguiente fórmula:
muestrademL
VfxCn:L/PmgmuestraladeiónConcentrac
Donde:
Cn = Concentración de la muestra leída en la Curva de calibración (mg/L)
Vf = Volumen final de la muestra después de la digestión realizada
mL de muestral = Alícuota de muestra utilizada para la digestión

135
8.1 CONTROL DE CALIDAD
Se deben preparar enriquecidos de muestras por cada lote, se considera
el análisis en control cuando el porcentaje de recuperación está entre 90 y
110%.
9 REFERENCIAS
[1] Norma Mexicana NMX-AA-029-SCFI-2001. Determinación de Fósforo Total
en aguas naturales, residuales y residuales tratadas. Método de Prueba.
[2] Standard Methods for the Examination of Water and Wastewater. Method
4500 P C. 20th. Edition 2000.American Public Health Association, American
Water Works Association, Water Environment Federation.

136
2.3.1.15. Determinación de sólidos totales disuelto s secados a
180 °C
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales”
Código del procedimiento: PT-FQ-13
Nombre del procedimiento: Determinación de sólidos totales disueltos secados a 1800 C
Fecha de aprobación: 2008-05-02
Fecha de revisión: 2008-05-02
1 OBJETIVO
Cuantificar los niveles de sólidos totales disueltos mediante la determinación
gravimétrica.
2 ALCANCE
Se aplica al análisis de muestras de aguas superficiales.
3 PRINCIPIO DEL METODO
La muestra de agua homogenizada (por agitación) se filtra a través de un filtro
de fibra de vidrio. El filtrado se evapora a sequedad en un recipiente de peso
conocido y el residuo es secado a 180 ± 2 ºC hasta conseguir un peso
constante. El incremento en el peso del recipiente representa los sólidos
disueltos totales.
3.1 INTERFERENCIAS
Las aguas con alta dureza y alcalinidad pueden requerir secado prolongado y
rápido pesado debido a las propiedades higroscópicas.
Si parte de la muestra se adhiere en las paredes del recipiente de recolección
o en los materiales de análisis, se debe indicar en el reporte de resultados.
4 EQUIPOS

137
Debe utilizarse el equipo normal de laboratorio, además de:
4.1 Estufa que opere entre los 180 ± 2 °C.
4.2 Sistema de filtración al vacío.
4.3 Balanza analítica con una precisión menor de 0,1 mg
4.4 Agitador magnético
5 REACTIVOS Y MATERIALES
5.1 REACTIVOS
Utilizar únicamente agua desionizada o de pureza equivalente.
5.2 MATERIALES
Debe emplearse cristalería normal de laboratorio, así como:
• Filtros de fibra de vidrio de 47 mm de diámetro, Whatman grado
934AH, Gelman tipo A/E, Millipore tipo AP40, E-D Scientific
Specialities grado 161 o productos equivalentes.
• Embudo con filtro de fibra de vidrio o Embudo Gooch.
• Kitasato
• Cápsulas de porcelana con capacidad de 100 a 150 mL.
• Pinzas
• Desecador
6 ACCIONES PREVIAS
La muestra se debe recolectar en un recipiente de plástico resistente
(polietileno) o vidrio. El tiempo máximo de almacenamiento de la muestra es
de 7 días, por lo que se debe procurar realizar el análisis dentro de las 24
horas después del muestreo.

138
Se debe mantener en refrigeración una temperatura de 4 °C.
Antes de realizar el análisis la muestra se debe llevar a temperatura ambiente
y homogenizar con agitación.
Se debe controlar el buen estado del desecante.
7 DESCRIPCION DEL PROCEDIMIENTO
7.1 Preparación de las cápsulas de porcelana
Lavar con agua destilada o desionizada y rotular antes de ingresarlas a la
estufa.
Secar en la estufa durante 1 hora a 180º C.
Retirar las cápsulas de la estufa y colocarlas en un desecador hasta que
sea necesario. Se pesan inmediatamente antes de usar.
7.2 Selección del filtro y el tamaño de la muestra
El volumen de la muestra deberá contener entre 2,5 y 200 mg de residuo
seco. Si la filtración completa de la muestra requiere más de 10 minutos,
incremente el tamaño del filtro o disminuya el volumen de muestra.
7.3 Análisis de la muestra.
Agitar la muestra en agitador magnético y tomar un volumen de muestra
dado y aplicar vacío. Lavar sucesivamente el filtro tres veces con 10 mL
de agua. Desechar los lavados y continuar la succión durante 3 minutos.
Después de haber hecho la filtración completa, se procura eliminar la
máxima cantidad de agua posible.
Transferir cuantitativamente el filtrado a la cápsula de evaporación
previamente pesada y colocarla en una estufa o en un baño de vapor
hasta la evaporación de la muestra. Si es necesario, adicionar sucesivas

139
porciones de la muestra en la cápsula de porcelana. La muestra
evaporada es secada por al menos 1 hora a 180 °C.
Retirar la cápsula y colocarla en el desecador hasta alcanzar la
temperatura ambiente. Pesar.
Repetir el ciclo de secado, enfriado, desecado y pesado hasta que se
obtenga un peso constante, haya un cambio de peso de al menos el 4%
del valor inicial o hasta que la pérdida de peso sea inferior a 0,5 mg.
8 CALCULOS Y EXPRESION DE RESULTADOS
V
BALTotalesDisueltosSólidosmg
1000)(/
×−=
donde;
A = Peso del residuo seco + cápsula en mg.
B= Peso de la cápsula en mg
V = volumen de la muestra en mL
8.1 PRECISIÓN Y SESGO DE LOS RESULTADOS
Las determinaciones de duplicados no deben diferir en más del 5 % del
peso promedio.
9 REFERENCIAS

140
[1] Standard Methods for the Examination of Water and Wastewater. Method
2540 C. 20th Edition 2000. American Public Health Association, American Water
Works Association, Water Environment Federation
[2] Norma Mexicana. NMX-AA-034-SCFI-2001

141
2.3.1.16. Determinación de sólidos totales suspendi dos
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales”
Código del procedimiento: PT-FQ-14
Nombre del procedimiento: Determinación de sólidos totales suspendidos
Fecha de aprobación: 2008-05-02
Fecha de revisión: 2008-05-02
1 OBJETIVO
Cuantificar los niveles de sólidos totales suspendidos mediante la
determinación gravimétrica.
2 ALCANCE
Se aplica al análisis de muestras de aguas superficiales.
3 PRINCIPIO DEL METODO
La muestra de agua homogénea y representativa se hace pasar a través de
un filtro de fibra de vidrio previamente pesado. El filtro y el residuo retenido se
secan en una estufa entre 103 a 105°C hasta peso co nstante. El incremento
en el peso del filtro representa los sólidos totales suspendidos.
3.1 INTERFERENCIAS
Las aguas con alta dureza pueden requerir secado prolongado y rápido
pesado debido a las propiedades higroscópicas.
Cualquier partícula o aglomerados de material no homogéneo que no se
quiera incluir en el resultado final, se deben descartar.
4 EQUIPOS

142
Debe utilizarse el equipo normal de laboratorio, además de:
4.1 Estufa que opere entre los 103 - 105 °C.
4.2 Sistema de filtración al vacío.
4.3 Balanza analítica con una precisión menor de 0,1 mg.
4.4 Licuadora.
4.5 Agitador magnético.
5 REACTIVOS Y MATERIALES
5.1 REACTIVOS
Utilizar únicamente agua desionizada o de pureza equivalente.
5.2 MATERIALES
Debe emplearse cristalería normal de laboratorio, así como:
• Filtros de fibra de vidrio de 47 mm de diámetro, Whatman grado
934AH, Gelman tipo A/E, Millipore tipo AP40, E-D Scientific
Specialities grado 161 o productos equivalentes.
• Embudo con filtro de vidrio o Embudo Gooch.
• Kitasato
• Desecador
6 ACCIONES PREVIAS
La muestra se debe recolectar en un recipiente de plástico resistente
(polietileno) o vidrio.
Si la muestra contiene grasas y aceites, dispersar con una licuadora antes de
realizar el análisis.

143
El tiempo máximo de almacenamiento de la muestra es de 7 días, se debe
procurar realizar el análisis dentro de las 24 horas después del muestreo.
Se debe mantener en refrigeración a una temperatura de 4 °C.
La muestra se debe llevar a temperatura ambiente antes del análisis.
Si parte de la muestra se adhiere en la superficie del recipiente de recolección
o en los materiales de análisis, se debe indicar en el reporte de resultados.
Se debe controlar el buen estado del desecante.
7 DESCRIPCION DEL PROCEDIMIENTO
7.1 Preparación del filtro
Colocar el filtro en el embudo. Aplicar vacío y lavar el mismo con tres
porciones sucesivas de 20 mL de agua. Continuar la succión para que se
seque parcialmente.
Se remueve el filtro y se coloca en un soporte (cápsula de porcelana o
vidrio reloj) y se introduce en una estufa a 103-105 °C durante al menos 1
hora.
Retirar el filtro de la estufa y colocarlo (junto con el soporte) en el
desecador por un período de 2 horas hasta que se enfríen.
Repetir el ciclo de secado, enfriado, desecado y pesado hasta que se
obtenga un peso constante, haya un cambio de peso de al menos el 4%
del valor inicial o hasta que la pérdida de peso sea inferior a 0, 5 mg.
7.2 Selección del filtro y el tamaño de la muestra
El volumen de la muestra a analizar deberá contener entre 2 y 200 mg de
residuo seco. Si el volumen filtrado cubre una porción mínima del filtro,
incrementar el volumen de muestra hasta 1 litro. Si la filtración completa

144
de la muestra requiere más de 10 minutos, incrementar el tamaño del
filtro o disminuir el volumen de muestra.
7.3 Análisis de la muestra
Montar el equipo de filtración y comenzar a succionar. Humedecer el filtro
con un pequeño volumen de agua.
Se aplica agitación magnética constante a un volumen dado de muestra y
se filtra a través del embudo (la toma del volumen debe realizarse en la
parte media entre la pared del recipiente y el remolino).
Lavar el filtro tres veces con porciones de 10 mL de agua y continuar
succionando después que se ha terminado de filtrar para secar
parcialmente el filtro.
Retirar cuidadosamente el filtro para evitar la pérdida de muestra y
conservar la integridad del filtro. Se coloca sobre el soporte.
Colocarlo en la estufa a 103-105 0C durante 1 hora. Enfriar en un
desecador para alcanzar un balance de temperatura y pesar. Repetir el
ciclo de secado, enfriado, desecado y pesado hasta que se obtenga un
peso constante, haya un cambio de peso de al menos el 4% del valor
inicial o hasta que la pérdida de peso sea inferior a 0,5 mg.
8 CALCULOS Y EXPRESION DE RESULTADOS
V
BALTotalessSuspendidoSólidosmg
1000)(/
×−=
donde;
A = Peso del filtro + residuo seco en mg

145
B = Peso del filtro en mg
V = Volumen de muestra en mL
8.1 PRECISIÓN Y SESGO DE LOS RESULTADOS
Las determinaciones de duplicados no deben diferir en más del 5 % del
peso promedio.
9 REFERENCIAS
[1] Standard Methods for the Examination of Water and Wastewater. Method
2540 D. 20th Edition 2000. American Public Health Association, American Water
Works Association, Water Environment Federation
[2] Norma Mexicana. NMX-AA-034-SCFI-2001

146
2.3.1.17. Nitratos por cromatografía iónica
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales”
Código del procedimiento: PT-FQ-15
Nombre del procedimiento: Nitratos por cromatografía iónica
Fecha de aprobación: 2008-05-02
Fecha de revisión: 2008-08-06
1 OBJETIVO
Cuantificar el contenido de nitratos mediante cromatografía iónica.
2 ALCANCE
Se aplica para el análisis de muestras de aguas superficiales.
3 PRINCIPIO DEL METODO
Al pasar a través de una columna de intercambio aniónico, utilizando una fase
móvil, los aniones se separan por diferencia de carga, peso molecular y por
su afinidad con la columna, y son detectados generalmente mediante un
detector de conductividad la diferencia producida entre la fase móvil y los
analitos que se analizan. Este método tiene la ventaja de necesitar poca
cantidad de muestra.
3.1 INTERFERENCIAS:
4 EQUIPOS
Debe utilizarse el equipo normal de laboratorio, además de:
4.1 Balanza analítica.
4.2 Baño ultrasónico.
4.3 Bomba de vacío.

147
4.4 Cromatógrafo Iónico.
4.5 Equipo de filtración.
5 REACTIVOS Y MATERIALES
5.1 REACTIVOS
Utilizar únicamente reactivos de grado analítico reconocido y agua
desionizada o de pureza equivalente.
• Nitrato de sodio (NaNO3).
• Reactivos para preparar la fase móvil de acuerdo al equipo que se use.
5.2 SOLUCIONES
• Solución estándar de nitrato, 1000 mg/L NO3-: Secar NaNO3 en una
estufa a 105 °C durante 24 h. Disolver 1.37 g de Na NO3 en agua y diluir a
1000 mL en matraz aforado. Preservar la solución de KNO3 con 2 mL de
CHCl3/L. La solución es estable por un período de 6 meses.
• Solución intermedia de nitrato. 150 mg/L: Diluir 150 mL de solución
estándar de nitrato a 1 litro con agua. Preservar con 2 mL de cloroformo.
• Prepare una curva de calibración de al menos siete patrones
(incluyendo el blanco) en el ámbito de linealidad del método, a partir de la
disolución intermedia de nitrato. El ámbito de linealidad del método
depende del equipo que se emplee y de las condiciones propias de cada
laboratorio.
5.3 MATERIALES
Debe emplearse cristalería normal de laboratorio, así como:

148
• Filtros de acetato de celulosa, diámetro de poro de 0,20 µm y 0,45 µm,
ambos de 47 mm de diámetro.
6 ACCIONES PREVIAS
La muestra debe analizarse lo antes posible después del muestreo.
Almacenar a una temperatura superior al punto de congelación, pero inferior
a 4 °C.
La muestra debe filtrarse para evitar la formación de tapones en las líneas de
flujo, válvulas de chequeo, columna y detector.
Contar siempre con un mínimo de 400 mL de fase móvil en el reservorio para
evitar la entrada de aire al sistema y por tanto el deterioro de la columna.
7 DESCRIPCION DEL PROCEDIMIENTO
Preparar la fase móvil y fíltrela a través de un filtro millipore o similar de 0,20
µm ó 0,45 µm.
Desgasificar la fase móvil por 60 min utilizando un baño ultrasónico.
Estabilizar el cromatógrafo de intercambio iónico de acuerdo con el
procedimiento del equipo.
Preparar la curva de calibración.
Filtrar las muestras en un filtro millipore o similar de 0,20 µm ó 0,45 µm.
Si la línea base se ha logrado estabilizar, proceder a inyectar en primer lugar
el blanco, luego el resto de los patrones y finalmente las muestras y controles
respectivos.
8 CALCULOS Y EXPRESION DE RESULTADOS
Se determina la curva de calibración Área vs concentración, mediante
mínimos cuadrados simples. Se interpola el área de la muestra, se debe
ajustar la concentración por dilución, con la siguiente fórmula:

149
Área = m × C + b
Donde;
m = pendiente de la curva
C = concentración en mg/L
b = intercepto
8.1 CONTROL DE CALIDAD
El control de calidad consiste en analizar una muestra enriquecida o
fortificada por cada lote de 10 muestras que se analicen. El
enriquecimiento se realiza con la disolución patrón intermedio preparada.
Una vez que la muestra se ha enriquecido se procede a analizarla, luego
se calcula el porcentaje de recuperación y se evalúa este valor en un
gráfico de control. En caso de que este valor incumpla con los límites
especificados para el análisis, repita el control y si la situación prevalece
será necesario repetir el lote completo de análisis junto con los controles
pertinentes. Es importante revisar también las tendencias de los datos
registrados en los gráficos control.
9 REFERENCIAS:
[1] Standard Methods for the Examination of Water and Wastewater. Método
4110 C. 21a. Edición. 2005. American Public Health Association, American Water
Works Association, Water Environment Federation.

150
2.3.1.18. Determinación de pH por medición potencio métrica
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales”
Código del procedimiento: PT-FQ-16
Nombre del procedimiento: Determinación de pH por medición potenciométrica
Fecha de aprobación: 2008-05-08
Fecha de revisión: 2008-05-08
1. OBJETIVO
Determinar la actividad de los iones hidrógeno (H+) en cuerpos de agua por
medición potenciométrica, haciendo uso de un electrodo de vidrio y un
electrodo de referencia ó un electrodo combinado, previa calibración del
instrumento con soluciones estándares de pH.
2. ALCANCE
Este método de ensayo establece la metodología para el análisis de pH en
cuerpos de agua medida en campo.
3. PRINCIPIO DEL METODO
La base del principio electrométrico en la medición de pH es la determinación
de la actividad del ión hidrógeno por mediciones potenciométricas usando
un electrodo estándar de hidrógeno y un electrodo de referencia. La
determinación del pH en el agua es una medida de la tendencia de la acidez
o de la alcalinidad. No mide el valor de la acidez o alcalinidad
3.1. INTERFERENCIAS:
El electrodo de vidrio está relativamente libre de interferencia de color,
turbiedad, materia coloidal, oxidantes, reductores o elevada salinidad a
excepción del error de sodio a pH>10. En caso de requerirse, este error

151
puede ser reducido utilizando un electrodo especial con bajo error de
sodio
Lugar monitoreado en presencia de aceites o material particulado puede
causar interferencia en la lectura. Esta interferencia puede ser removida
lavando el electrodo con detergente suave y enjuagando varias veces con
agua destilada y/o aplicando un lavado con HCl + agua (1+9) de ser
necesario.
El problema más frecuente que puede causar un conjunto combinado de
electrodos es una obstrucción en el diafragma. Si esto ocurre, como lo
indica una desviación persistente se debe remojar el electrodo en
solución de hidróxido de amonio.
4. EQUIPOS
4.1. pH –metro portátil o de campo
4.2. Electrodo de vidrio
4.3. Electrodo de referencia de calomelano, plata cloruro de plata u otro
electrodo de referencia con un potencial constante.
4.4. Termómetro o sensor de temperatura con compensación automática.
5. REACTIVOS Y MATERIALES
5.1. REACTIVOS
Durante el análisis se deben utilizar solamente reactivos de calidad para
análisis (p.a.), y agua destilada o desionizada.
• Tampón (Buffer) pH 4.00.
• Tampón (Buffer) pH 7.00.
• Tampón (Buffer) pH 10.00.
5.2. MATERIALES
Debe emplearse cristalería normal de laboratorio, tales como: probeta,
pizeta o frasco lavador

152
6. ACCIONES PREVIAS
6.1. Si los electrodos se han almacenado secos durante un largo período, la
membrana de vidrio debe remojarse en una solución de 3 M de KCl
durante 12 a 24 horas antes de usarse.
6.2. El ajuste y calibración del instrumento a utilizar deberá realizarse según
el manual del equipo.
6.2.1. Verificar la calibración del instrumento en el laboratorio y siempre
antes de cada uso en el campo (Tabla 1).
6.2.2. Seleccionar los reactivos (tampón o buffers), cuidando que se
encuentren entre 1 y 2 unidades respecto del pH a medir en las
muestras.
6.2.3. Confirmar la compensación de temperatura de estándares y
muestras.
6.2.4. Al verificar la calibración del instrumento la lectura obtenida debe
estar dentro de ±0,1 unidades de pH
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Repetir en el campo la verificación de la calibración del equipo según
item 6.
7.2. Enjuagar los electrodos con agua para análisis grado reactivo, secar con
un papel suave e introducir al cuerpo de agua superficialmente
previniendo los lugares o sectores con turbulencia.
7.3. Registrar el valor de pH entregado por el instrumento, cuando se alcance
una lectura estable.
7.4. Registrar la temperatura a la que se efectuó la medición.
7.5. Si la temperatura difiere ±2ºC de la solución buffer, la medición de pH
debe ser corregida mediante un ajuste electrónico (automático o manual)
y compensar la diferencia de temperatura. Remitirse a las instrucciones
del manual de operaciones.

153
7.6. Para el control de calidad analítica por cada punto de muestreo realizar la
lectura por duplicado (precisión mínima 90%, es decir un CV de 10%)
7.7. Si no se puede medir el pH en la corriente, deberá medirse en el
recipiente utilizado para tomar muestras de agua. Las precauciones que
deben tomarse cuando se usa un recipiente para hacer mediciones de
pH en el terreno son las mismas que las especificadas en los item 7.1 al
7.6
8. CALCULOS Y EXPRESION DE RESULTADOS
El resultado se lee directamente en el equipo y debe expresarse como pH a
25ºC. Para medidas que no se hayan llevado a cabo a 25,0 ± 0,1ºC debe
indicarse el método de corrección junto con la temperatura real medida.
9. REFERENCIAS:
[1] Standard Methods For The Examination Of Water And Wastewater. APHA.
AWWA. WEF. 21th Edition 2005, Part. 4500H+-B Electrometric Method.
[2] Environmental Protection Agency. Methods For Chemicals Analisys Of
Water And Wastewater. 2th ed. Cincinnati, EPA 1983, 150.1
10. ANEXO
154
Tabla 1: Variación del pH en soluciones tampón (Buffer)
pH 4,0 pH 7,0 pH 10,0 Temp. (ºC)
pH Temp. (ºC)
pH Temp. (ºC)
pH
0 4,01 0 7,10 0 9,24 5 4,00 5 7,07 5 9,17
10 4,00 10 7,04 10 9,10 15 4,00 15 7,02 15 9,05 20 4,00 20 7,00 20 9,00 25 4,00 25 6,98 25 8,98 30 4,01 30 6,97 30 8,91 35 4,02 35 6,96 35 8,88 40 4,03 40 6,96 40 8,84 45 4,04 45 6,95 45 8,81 50 4,06 50 6,95 50 8,79 55 4,08 55 6,95 55 8,76 60 4,10 60 6,96 60 8,74 70 4,12 70 6,96 70 8,70 80 4,16 80 6,98 80 8,66 90 4,21 90 7,00 90 8,63

155
2.3.1.19. Determinación de oxígeno disuelto por el método
electrométrico
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales” Código del procedimiento:
PT-FQ-17 Nombre del procedimiento:
Determinación de oxígeno disuelto por el método electrométrico
Fecha de aprobación: 2008-05-08
Fecha de revisión: 2008-05-08
1. OBJETIVO:
Determinar la cantidad de oxígeno que está disuelta en el agua y ser un
indicador de cuán contaminada está el agua y cuán bien puede dar soporte
esta agua a la vida vegetal y animal.
2. ALCANCE:
Este procedimiento es válido para analizar el oxígeno disuelto (OD) en
cuerpos de agua mediante electrodo de membrana.
3. PRINCIPIO DEL METODO
En el método electrométrico los electrodos de membrana sensible al oxígeno,
ya sean galvánicos o polarizados están constituidos por dos electrodos de
metal en contacto con un electrolito soporte, separado de la disolución de
muestra por medio de una membrana selectiva. En la celda del oxímetro, en
el cátodo, ocurre la reducción del oxígeno, mientras que en el ánodo ocurre
la oxidación del metal. La diferencia básica entre el sistema galvánico y el
polarizado es que en el primero la reacción en el electrodo ocurre
espontáneamente, mientras que en el segundo es necesario aplicar un
potencial externo para polarizar el electrodo indicador.
Generalmente se utilizan membranas de polietileno y fluorocarbón que son
permeables al oxígeno molecular y relativamente rugosas.

156
3.1. INTERFERENCIAS
La entrada de aire atmosférico dentro de las muestras puede causar
medidas erróneas del instrumento.
Las pruebas basadas en difusión están sujetas a errores negativos por la
formación de capas como los óxidos de hierro, los cuales impiden la
difusión del oxígeno.
La hidrazina, aminas, ácido sulfhídrico e hidrógeno, que pasen a través
de la membrana causan errores negativos.
4. EQUIPOS
4.1. Medidor de oxígeno disuelto con electrodo de membrana sensitiva al
oxígeno, de tipo galvánico o polarizado (oxímetro)
4.2. Balanza analítica ±0,001g
5. REACTIVOS Y MATERIALES
5.1. REACTIVOS
5.1.1. Cloruro de cobalto (CoCl2)
5.1.2. Sulfito de sodio (Na2SO3)
5.2. SOLUCIONES
5.2.1. Disolución saturada de cloruro de cobalto. Pesar aproximadamente
4,500±0,001 g de cloruro de cobalto y disolver en 10 mL de agua.
5.2.2. Disolución estándar de concentración nula de oxígeno disuelto
(OD). Pesar 5,000 ± 0,001 g de sulfito de sodio, aforar a 100 mL con
agua y añadir 2 gotas de la disolución saturada de cloruro de cobalto
(item 5.2.1).
5.3. MATERIALES
Debe emplearse cristalería normal de laboratorio, tales como: probeta, pizeta
o frasco lavador

157
6. ACCIONES PREVIAS
6.1. Verificar la calibración del medidor de oxígeno disuelto según
especificaciones del fabricante.
6.2. La estandarización cero de OD consiste en la inmersión del electrodo en
la disolución estándar de concentración nula de oxígeno (item 5.2.2). La
lectura debe estar en cero mg/L de OD, de no ser así, ajustar el
instrumento a cero.
6.3. Ajustar el instrumento a la presión atmosférica.
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Repetir en campo la verificación de la calibración del equipo según item
6.
7.2. Enjuagar la celda del oxímetro con agua para análisis grado reactivo e
introducir al cuerpo de agua, tomando en cuenta la profundidad y el lugar
de lectura de campo previniendo efectuar en lugares o sectores con
turbulencia.
7.3. Registrar el valor de OD entregado por el instrumento, cuando se alcance
una lectura estable y el orificio de la celda esté sumergido en el cuerpo
de agua.
7.4. Registrar la temperatura a la que se efectuó la medición. El Porcentaje
de Saturación del Oxígeno Disuelto depende de la temperatura del agua
y la elevación del sitio donde se toma la muestra de agua. Determine la
altitud (elevación) o la presión atmosférica haciendo uso de la Tabla 1,
remitirse al item 10, para determinar el factor de corrección.
7.5. Si la temperatura difiere ±2ºC de la solución estándar, la medición de CE
debe ser corregida mediante un ajuste electrónico automático (remitirse a
las instrucciones del manual de operaciones) ó manualmente (según
Tabla1) y compensar la diferencia de temperatura.
7.6. Para el control de calidad analítica por cada punto de muestreo realizar la
lectura por triplicado (precisión mínima 90%, es decir un CV de 10%)

158
8. CALCULOS Y EXPRESION DE RESULTADOS
El resultado se lee directamente en el equipo y el OD debe expresarse en
mg/L a 25ºC. Para medidas que no se hayan llevado a cabo a 25,0 ± 0,1ºC
debe indicarse el método de corrección junto con la temperatura real medida.
9. REFERENCIAS:
[1] Standard Methods For The Examination Of Water And Wastewater.
APHA. AWWA. WEF. 21th Edition 2005, Part. 4500-OG
[2] Environmental Protection Agency. Methods For Chemicals Análisis Of
Water And Waste Water. 2th ed. Cincinnati, EPA, 360.1
10. ANEXO
Tabla 1: Determinación del factor de corrección para OD en función de la presión
atmosférica (mm de Hg) o altitud (m)
Presión Atmosférica (mmHg)
Altitud Equivalente (m) Factor de Corrección
775 165 1.02 760 0 1.00 745 166 0.98 730 334 0.96 714 516 0.94 699 695 0.92 684 875 0.90 669 1059 0.88 654 1247 0.86 638 1453 0.84 623 1651 0.82 608 1853 0.80 593 2061 0.78 578 2273 0.76 562 2507 0.74 547 2731 0.72

159
532 2962 0.70 517 3200 0.68
NOTA
El Pocentaje de Saturación es la cantidad de oxígeno disuelto en la muestra
de agua comparada con la cantidad máxima que podría estar presente a la
misma temperatura. Por ejemplo, se dice que el agua está saturada en un
100% si contiene la cantidad máxima de oxígeno a esa temperatura. Una
muestra de agua que está saturada en un 50% solamente tiene la mitad de la
cantidad de oxígeno que potencialmente podría tener a esa temperatura. A
veces, el agua se supersatura con oxígeno debido a que el agua se mueve
rápidamente. Esto generalmente dura un período corto de tiempo, pero puede
ser dañino para los peces y otros organismos acuáticos. Los valores del
Porcentaje de Saturación del OD de 80-120% se consideran excelentes y los
valores menores al 60% o superiores a 125% se consideran malos.

160
2.3.1.20. Determinación de solutos ionizables
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales”
Código del procedimiento: PT-FQ-18
Nombre del procedimiento: Determinación de solutos ionizables
Fecha de aprobación: 2008-05-08
Fecha de revisión: 2008-05-08
1. OBJETIVO
Determinar la concentración de solutos ionizables presentes en cuerpos de
agua.
2. ALCANCE
Este método de ensayo establece la metodología para el análisis de
conductividad (CE) en cuerpos de agua medida en campo.
3. PRINCIPIO DEL METODO
La conductividad eléctrica es una medida de la corriente conducida por los
iones presentes en el agua y depende de:
- concentración de los iones
- naturaleza de los iones
- temperatura de la solución
- viscocidad de la solución
3.1. INTERFERENCIAS
Las lecturas repetibles requieren que la celda del conductímetro esté
protegida de la suciedad, movimientos bruscos y de temperaturas de
congelación.

161
Los valores medidos de CE pueden verse afectados por contaminación
de la muestra en presencia de materias en suspensión de tamaño
considerable, de aceites o grasas
4. EQUIPOS
4.1. Conductímetro portátil o de campo
4.2. Celda de conductividad
4.3. Termómetro o sensor de temperatura con compensación automática.
5. REACTIVOS Y MATERIALES
5.1. REACTIVOS
Durante el análisis se deben utilizar solamente reactivos de calidad para
análisis (p.a.), y agua destilada o desionizada (≤0,1µS/cm). Para calibrar
el equipo puede hacer uso del item 5.1.1 ó 5.1.2.
5.1.1. Solución estándar de KCl 0.01M
Disolver 0.7456 ± 0,001 g de cloruro de potasio (KCl), secado
previamente 2 horas a 105ºC, en agua calidad reactivo y aforar a 1L a
25ºC ó 77ºF. Esta solución estándar de referencia a 25ºC ó 77ºF tiene
una conductividad de 1412 ± 1 µs/cm. Preservar dicha solución en
frasco de vidrio de borosilicato.
5.1.2. Solución comercial de calibración de conductividad 1413 µS/cm a
25ºC ó 77ºF.
5.2. MATERIALES
Debe emplearse cristalería normal de laboratorio, tales como: probeta,
pizeta o frasco lavador
6. ACCIONES PREVIAS

162
6.1. El ajuste y calibración del instrumento a utilizar deberá realizarse según
el manual del equipo.
6.2. Verificar la calibración del instrumento en el laboratorio y siempre antes
de cada uso en campo.
6.3. La verificación de calibración de un (1) punto deberá realizarse haciendo
uso de solución de KCl de conductividad conocida (p.ej. 1413 uS/cm) ó
en el orden de la medición a efectuarse a una temperatura dada (Tabla
1).
6.4. La limpieza de la celda de conductividad se lleva a cabo con agua
destilada o desionizada (≤0,1µS/cm).
6.5. Se debe confirmar la compensación de temperatura de estándares y
muestras.
6.6. Al verificar la calibración del instrumento la lectura obtenida debe estar
dentro de ±10% de error relativo.
7. DESCRIPCION DEL PROCEDIMIENTO:
7.1. Repetir en campo la verificación de la calibración del equipo según item
6.
7.2. Enjuagar la celda del conductímetro con agua para análisis grado
reactivo, secar con un papel suave e introducir al cuerpo de agua
superficial previniendo los lugares o sectores con turbulencia.
7.3. Registrar el valor de CE entregado por el instrumento, cuando se alcance
una lectura estable y el orificio de la celda esté sumergido en el cuerpo
de agua.
7.4. Registrar la temperatura a la que se efectuó la medición.
7.5. Si la temperatura difiere ±2ºC de la solución estándar, la medición de CE
debe ser corregida mediante un ajuste electrónico automático (remitirse a
las instrucciones del manual de operaciones) ó manual (Tabla1, Anexo) y
compensar la diferencia de temperatura.
7.6. Para el control de calidad analítica por cada punto de muestreo realizar la
lectura por duplicado (precisión mínima 90%, es decir un CV de 10%).

163
8. CALCULOS Y EXPRESION DE RESULTADOS
El resultado se lee directamente en el equipo y la CE debe expresarse en
µS/cm a 25ºC. Para medidas que no se hayan llevado a cabo a 25,0 ± 0,1ºC
debe indicarse el método de corrección junto con la temperatura real medida.
8.1. Ejemplos:
8.1.1. Ejemplo 1:
CE (25ºC) = 25,2 µS/cm
Temperatura de medida 25,0ºC
8.1.2. Ejemplo 2:
CE (25ºC) = 1413 µS/cm
Temperatura de medida 11,5ºC
Corrección según Tabla 1: Conductividad Electrica 0,01M KCl (EPA,
120.1)
8.1.3. Ejemplo 3
CE (25ºC) = 480 µS/cm
Temperatura de medida 12,1ºC
Corrección mediante un sistema de compensación de temperatura
9. REFERENCIAS:
[1] Standard Methods For The Examination Of Water And Wastewater.
APHA. AWWA. WEF. 21th Edition 2005, Part. 2510-B
[2] Environmental Protection Agency. Methods For Chemicals Analisys Of
Water And Waste Water. 2th ed. Cincinnati, EPA, 120.1
10. ANEXO
164
Tabla 1.-Conductividad eléctrica 0.01 M KCl
°C ºF µS/cm 0 32.0 776 5 41.0 896 10 50.0 1020 15 59.0 1147 16 60.8 1173 17 62.6 1199 18 64.4 1225 19 64.4 1225 20 68.0 1278 21 69.8 1305 22 71.6 1332 23 73.4 1359 24 75.2 1386 25 77.0 1413 26 78.8 1440 27 80.6 1467 28 82.4 1494 29 84.2 1521 30 86.0 1548 31 87.8 1575

165
Tabla 2: Calidad del agua en función de la concentración (mg/L) de OD
Nivel de OD (mg/L) Calidad del Agua
0,0 - 4,0 Mala Algunas poblaciones de peces y macroinvertebrados
empezarán a bajar.
4,1 - 7,9 Aceptable
8,0 - 12,0 Buena
>12,0 Repita la prueba. El agua puede airearse artificialmente.
A 20º C (temperatura ambiente) y presión atmosférica estándar (nivel del
mar), la cantidad máxima de oxígeno que puede disolverse en agua dulce es
9 mg/L. Si la temperatura del agua está por debajo de 20º C, puede haber
más oxígeno disuelto en la muestra. En general, un nivel de oxígeno disuelto
de 9-10 mg/L se considera muy bueno.

166
2.3.1.21. Digestión ácida de aguas para análisis de metales
recuperables totales o metales disueltos por GFAAS
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales
Código del procedimiento: PT-DA-02
Nombre del procedimiento: Digestión ácida de aguas para análisis de metales recuperables totales
o metales disueltos por GFAAS
Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Es la digestión ácida de muestras de aguas superficiales y subterráneas para
su posterior análisis por espectrometría de absorción atómica por horno de
grafito (GFAAS).
2. ALCANCE
A las muestras preparadas por este procedimiento, se les puede determinar
los siguientes metales:
Cadmio – Plomo – Cromo - Cobre
3. PRINCIPIO DEL METODO
El método es aplicable a metales recuperables totales y para metales
disueltos (ver Procedimiento PT-DA-01).
4. EQUIPOS
4.1. Plancha calefactora o equivalente, ajustable y capaz de mantener una
temperatura de 90-95°C .
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Vasos de precipitación de tamaños adecuados o equivalentes.
5.1.2. Vidrios de reloj o equivalentes.
5.1.3. Membranas filtrantes de acetato de celulosa de 0,45 µm de tamaño
de poro.
5.1.4. Papel de filtro y embudos de filtración.
5.1.5. Probetas graduadas o equivalentes.

167
5.2. REACTIVOS
Se deben emplear reactivos de grado analítico en todos los ensayos. A
menos que se especifique lo contrario, se debe lograr que todos los
reactivos cumplan con lo dispuesto en las especificaciones del Comité de
Reactivos Analíticos de la American Chemical Society (ACS). Se pueden
usar otros grados de reactivos en la medida que sean de suficiente
pureza para permitir su uso sin perder la exactitud de las
determinaciones.
5.2.1. Agua grado reactivo.
5.2.2. Solución de ácido nítrico concentrado. Se debe analizar el ácido
para determinar el grado de pureza. Si el valor del blanco de
reactivos es menor al límite de detección del método, puede usarse
el ácido correspondiente.
6. ACCIONES PREVIAS
6.1. Todas las muestras se deben haber colectado de acuerdo al
procedimiento de muestreo del presente manual.
6.2. Todos los contenedores de las muestras deben haber sido prelavados
con detergentes, ácidos y agua.
6.3. Para el muestreo de metales recuperables totales, todas las muestras
deben acidificarse en el momento de la calefacción con solución de ácido
nítrico concentrado (5 mL/litro de muestra).
6.4. Para el muestreo de metales recuperables totales o disueltos, ver
Procedimiento PT-DA-01.
7. DESCRIPCION DEL PROCEDIMIENTO (para metales recuperables totales)
7.1. Transferir una alícuota de 100 mL de una muestra bien mezclada a un
vaso de precipitación.
7.2. Agregar 3 mL de solución de ácido nítrico concentrado. Cubrir el vaso
con un vidrio de reloj o equivalente.
7.3. Calentar la muestra sobre plancha calefactora o equivalente en forma
cuidadosa y suave hasta reducir el volumen a 5 mL, tomando la
precaución que la muestra no llegue a sequedad.

168
7.4. Retirar el vaso y dejar enfriar. Agregar 3 mL de solución de ácido nítrico
concentrado.
7.5. Aumentar la temperatura de la plancha para que comience el reflujo de la
muestra. Continuar hasta que el digesto sea de color suave y que no
cambie con el tiempo.
7.6. Cuando la digestión se complete, evaporar hasta un volumen de 3 mL,
no permitiendo que ninguna porción del fondo se seque.
7.7. Agregar 10 mL de agua reactivo para permitir la solubilización de
cualquier residuo que pudiera haberse producido.
7.8. Retirar el vaso de la plancha y lavar las paredes del mismo y el vidrio de
reloj con agua reactivo. Si es necesario, filtrar o centrifugar la muestra
para eliminar cualquier residuo que pueda permanecer y que interfiera
con la determinación posterior.
Nota: acondicionar el papel de filtro y el embudo con ácido nítrico para
eliminar posibles impurezas.
7.9. Ajustar el volumen final a 100 mL con agua reactivo, quedando la
muestra ya preparada para su análisis.
8. CALCULOS Y EXPRESION DE RESULTADOS
9. REFERENCIAS
[1] Norma EPA 3020.
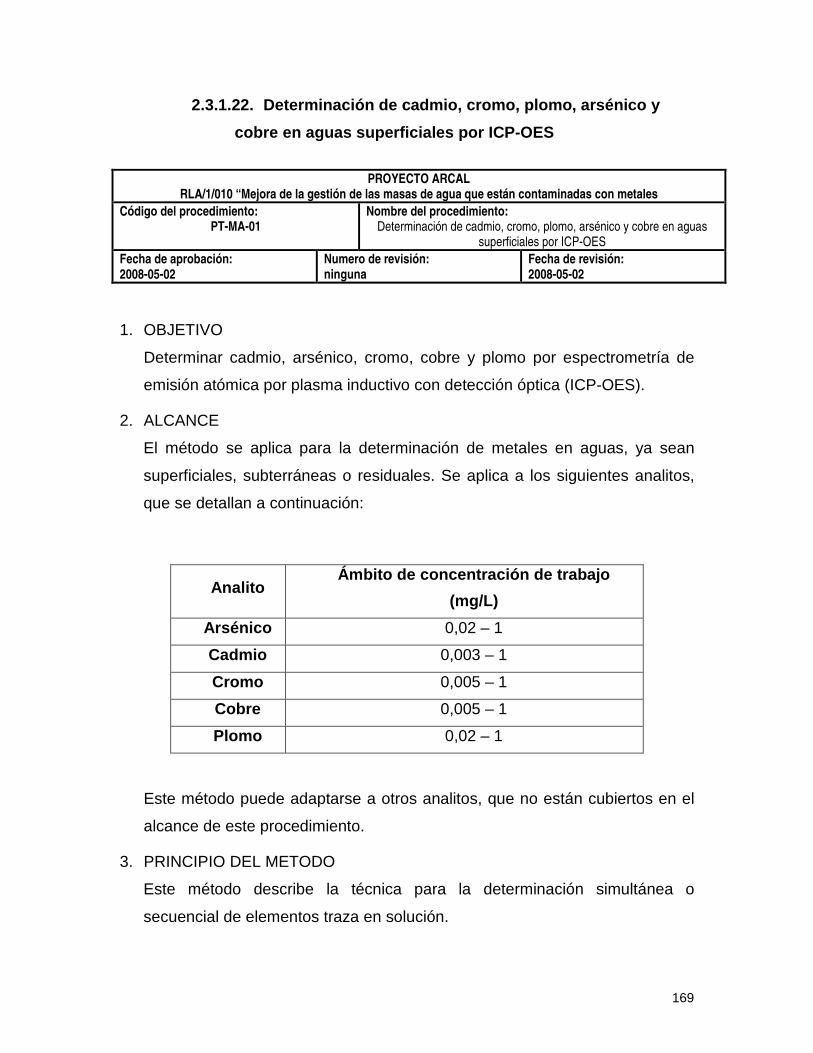
169
2.3.1.22. Determinación de cadmio, cromo, plomo, ar sénico y
cobre en aguas superficiales por ICP-OES
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales Código del procedimiento:
PT-MA-01
Nombre del procedimiento: Determinación de cadmio, cromo, plomo, arsénico y cobre en aguas
superficiales por ICP-OES
Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determinar cadmio, arsénico, cromo, cobre y plomo por espectrometría de
emisión atómica por plasma inductivo con detección óptica (ICP-OES).
2. ALCANCE
El método se aplica para la determinación de metales en aguas, ya sean
superficiales, subterráneas o residuales. Se aplica a los siguientes analitos,
que se detallan a continuación:
Analito Ámbito de concentración de trabajo
(mg/L)
Arsénico 0,02 – 1
Cadmio 0,003 – 1
Cromo 0,005 – 1
Cobre 0,005 – 1
Plomo 0,02 – 1
Este método puede adaptarse a otros analitos, que no están cubiertos en el
alcance de este procedimiento.
3. PRINCIPIO DEL METODO
Este método describe la técnica para la determinación simultánea o
secuencial de elementos traza en solución.

170
La base es la medida de la emisión atómica por una técnica espectroscópica
óptica.
Se nebulizan las muestras y se transporta el aerosol producido a la antorcha
del plasma donde ocurre la excitación.
Se debe emplear una técnica de corrección de fondo para compensar la
contribución del fondo variable para la determinación de elementos traza.
Esta contribución de fondo debe medirse en forma adyacente a las líneas del
analito durante el análisis.
La posición que se selecciona para la medición del fondo, a ambos lados de
la línea analítica, se determina por la complejidad del espectro adyacente a la
línea del analito.
La posición debe estar libre de interferencias espectrales y debe reflejar el
mismo cambio en la intensidad de fondo que ocurre a la longitud de onda de
medición.
Las interferencias pueden caracterizarse en dos tipos; interferencias
espectrales e interferencias no espectrales.
Interferencias espectrales Incluyen la emisión de luz desde otras fuentes espectrales diferentes al
analito de interés. Incluyen la superposición de líneas espectrales directas, el
ensanchamiento de líneas espectrales distintas, los efectos de la
recombinación ion-átomo, la emisión de bandas moleculares y luz espúrea
proveniente de la emisión de elementos a altas concentraciones. Estos
inconvenientes se evitan o eliminan seleccionando longitudes de onda
analíticas. Además se debe hacer uso de la corrección de fondo provista por
el “software”” del instrumento y de los factores de corrección interelementos
cuando se requiera.

171
Interferencias no espectrales Pueden ser provocadas por las propiedades físicas de la muestra. Los
cambios en la viscosidad y la tensión superficial pueden ocasionar errores
significativos. Esto sucede frecuentemente cuando las muestras contienen
más de un 10 % en volumen de ácido o más de 1500 mg/L de sólidos
disueltos y se analizan con soluciones estándar de calibración que contienen
menos del 5 % en volumen de ácido. Estos factores se compensan
habitualmente por dilución de la muestra. Las interferencias químicas
causadas por otros motivos pueden provocar errores. Esto debe tenerse en
cuenta por una selección muy adecuada de las líneas analíticas y de la
corrección de fondo.
Para más detalle, se debe consultar la norma 200.7 de la EPA.
4. EQUIPOS
4.1. Espectrómetro de emisión atómica por plasma inductivo.
4.2. Equipo refrigerador para el espectrómetro.
4.3. Equipo purificador de agua.
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Matraces aforados, clase A, de 50, 100 y 1000 mL.
5.1.2. Pipetas automáticas de punta descartable, de volumen variable
entre 10 y 100 µL, de volumen variable entre 100 y 1000 µL, de
volumen variable entre 50 y 250 µL y de volumen variable entre 1000
y 5000 µL.
5.1.3. Botellas de plástico de 125 mL.
5.1.4. Jeringas de filtración.
5.1.5. Filtros de acetato de celulosa de 0,45 µm para usar con jeringas.
5.1.6. Tubos descartables de plástico de 15 y 50 mL.
5.1.7. Botellas de plástico de alta densidad de 125 mL.
5.2. REACTIVOS

172
5.2.1. Agua grado reactivo.
5.2.2. Solución estándar de arsénico de 1000 mg/L, calidad ultrapura.
5.2.3. Solución estándar de cadmio de 1000 mg/L, calidad ultrapura.
5.2.4. Solución estándar de cromo de 1000 mg/L, calidad ultrapura.
5.2.5. Solución estándar de cobre de 1000 mg/L, calidad ultrapura.
5.2.6. Solución estándar de plomo de 1000 mg/L, calidad ultrapura.
5.2.7. Solución de ácido nítrico concentrado, calidad ultrapuro.
5.2.8. Solución de ácido nítrico (0,2 % V/V). Preparar con 2 mL de
solución de ácido nítrico concentrado (5.2.7.) diluidos a 1000 mL con
agua grado reactivo (5.2.1.).
5.2.9. Argón, calidad UAP, (ultra alta pureza).
5.2.10. Nitrógeno, calidad UAP, (ultra alta pureza).
5.2.11. Soluciones estándar de control de procedimiento: material
certificado. Provisto por una empresa acreditada. Posee fecha de
vencimiento.
5.2.12. Materiales de referencia certificados, provistos por el National
Water Research Institute (NWRI), el NIST, o de calidad similar.
Soluciones estándar mixtas
Se utilizan las siguientes combinaciones recomendadas por la EPA
5.2.13. Solución estándar mixta I: cadmio y plomo.
5.2.14. Solución estándar mixta II: cobre.
5.2.15. Solución estándar mixta III: arsénico.
5.2.16. Solución estándar mixta IV: cromo.
5.2.17. Solución estándar mixta I de 50 mg/L: Pipetear 5,00 mL de cada
una de las siguientes soluciones estándar de 1000 mg/L: cadmio y
plomo en un matraz aforado de 100 mL, y diluir hasta el aforo con
solución de ácido nítrico 0,2 % V/V. Almacenar en botella de plástico
de 125 mL. Preparar mensualmente.

173
5.2.18. Solución estándar mixta II de 50 mg/L: Pipetear 5,00 mL de cada
una de las siguientes soluciones estándar de 1000 mg/L: cobre en un
matraz aforado de 100 mL, y diluir hasta el aforo con solución de
ácido nítrico 0,2 % V/V. Almacenar en botella de plástico de 125 mL.
Preparar mensualmente.
5.2.19. Solución estándar mixta III de 50 mg/L: Pipetear 5,00 mL de cada
una de las siguientes soluciones estándar de 1000 mg/L: arsénico en
un matraz aforado de 100 mL, y diluir hasta el aforo con solución de
ácido nítrico 0,2 % V/V. Almacenar en botella de plástico de 125 mL.
Preparar mensualmente.
5.2.20. Solución estándar mixta II de 50 mg/L: Pipetear 5,00 mL de cada
una de las siguientes soluciones estándar de 1000 mg/L: cromo en
un matraz aforado de 100 mL, y diluir hasta el aforo con solución de
ácido nítrico 0,2 % V/V. Almacenar en botella de plástico de 125 mL.
Preparar mensualmente.
Soluciones estándar de calibración
5.2.21. Estándar de calibración de 0,03 mg/L: Pipetear 30 µL de cada
una de las cuatro soluciones estándares mixtas de 50 mg/L en un
matraz aforado de 50 mL, y diluir hasta el aforo con solución de ácido
nítrico 0,2 % V/V. Almacenar en tubo de polipropileno de 50 mL.
Preparar mensualmente.
5.2.22. Estándar de calibración de 0,10 mg/L: Pipetear 100 µL de cada
una de las cuatro soluciones estándares mixtas de 50 mg/L en un
matraz aforado de 50 mL, y diluir hasta el aforo con solución de ácido
nítrico 0,2 % V/V. Almacenar en tubo de polipropileno de 50 mL.
Preparar mensualmente.
5.2.23. Estándar de calibración de 0,20 mg/L: Pipetear 200 µL de cada
una de las cuatro soluciones estándares mixtas de 50 mg/L en un
matraz aforado de 50 mL, y diluir hasta el aforo con solución de ácido

174
nítrico 0,2 % V/V. Almacenar en tubo de polipropileno de 50 mL.
Preparar mensualmente.
5.2.24. Estándar de calibración de 0,40 mg/L: Pipetear 400 µL de cada
una de las cuatro soluciones estándares mixtas de 50 mg/L en un
matraz aforado de 50 mL, y diluir hasta el aforo con solución de ácido
nítrico 0,2 % V/V. Almacenar en tubo de polipropileno de 50 mL.
Preparar mensualmente.
5.2.25. Estándar de calibración de 0,70 mg/L: Pipetear 700 µL de cada
una de las cuatro soluciones estándares mixtas de 50 mg/L en un
matraz aforado de 50 mL, y diluir hasta el aforo con solución de ácido
nítrico 0,2 % V/V. Almacenar en tubo de polipropileno de 50 mL.
Preparar mensualmente.
5.2.26. Estándar de calibración de 1,00 mg/L: Pipetear 1000 µL de cada
una de las cuatro soluciones estándares mixtas de 50 mg/L en un
matraz aforado de 50 mL, y diluir hasta el aforo con solución de ácido
nítrico 0,2 % V/V. Almacenar en tubo de polipropileno de 50 mL.
Preparar mensualmente.
Blanco de calibración
5.2.27. Solución 5.2.8.
Blanco de filtro
5.2.28. Es agua grado reactivo que se hace pasar por el filtro de acetato
de celulosa. Usar sólo si se necesitó el uso de los filtros para la
muestra.
6. ACCIONES PREVIAS
6.1. Las muestras deben estar libres de sólidos en suspensión previo a la
aspiración.

175
6.2. Las muestras deben tratarse antes del análisis, si se sospecha la
presencia de sólidos en suspensión o sedimentados.
6.3. Las muestras se filtran por medio de filtros con jeringa.
6.4. Las muestras deben conservarse a bajas temperaturas (4° C).
6.5. Las soluciones estándar de control de performance se utilizan tal cuál las
entrega el proveedor.
7. DESCRIPCION DEL PROCEDIMIENTO
Operación del equipo
7.1. Preparar el equipo de ICP de acuerdo a las instrucciones que figuran
en el manual de instrucciones del instrumento.
7.2. La siguiente tabla describe los parámetros operativos para cada uno
de los analitos.
Analito Longitud de onda (nm)
Arsénico 188,98
Cadmio 228,80
Cromo 267,72
Cobre 324,75
Plomo 220,35
Estas longitudes de onda son recomendadas por su sensibilidad y
aceptación. Se pueden sustituir por otras longitudes de onda, si se
considera que hay interferencias espectrales.
7.3. Antes de comenzar el uso del instrumento, proceder a estabilizar el
plasma durante 20 minutos.
7.4. Llevar a cabo una alineación óptica empleando la lámpara de Hg
provista con el equipo.
7.5. Verificar la alineación de la antorcha del plasma y de la ranura de
entrada del espectrómetro, si se llevó a cabo un mantenimiento del
sistema de introducción de las muestras.

176
7.6. En el muestreador automático, se colocan los tubos de plástico de 50
mL, conteniendo el blanco de calibración y las soluciones estándares
de calibración, comenzando por la solución más diluida. Se programa
el equipo y se lleva a cabo la calibración correspondiente.
7.7. Purgar la cañería con cada estándar o blanco a una velocidad de 4
mL/min durante un mínimo de 15 segundos. Luego dejar estabilizar
durante otro mínimo de 15 segundos.
7.8. Enjuagar con agua reactivo durante 30 segundos antes de cada
estándar para minimizar cualquier efecto de memoria del estándar
previo. Emplear tres integraciones de los estándares o muestras para
reducir el error aleatorio.
7.9. Todas estas operaciones, una vez programadas, el equipo las lleva a
cabo en forma automática.
7.10. A continuación del estándar más concentrado, aspirar agua reactivo
(limpieza), luego un blanco de calibración, y a posteriori dos
soluciones estándar de control de procedimiento.
7.11. Si la calibración y los valores de las muestras de control de
procedimiento están dentro de los límites aceptables, continuar con el
análisis de las muestras. Si no lo están, proceder a tomar las acciones
correctivas.
7.12. Las muestras se cargan en el muestreador automático.
7.13. El blanco de filtración se incluye como una muestra para verificar la
ausencia de contaminantes por el proceso de filtración. Asegurar que
la lista de muestras en el “software” coincida con la posición correcta
en el “tray” del muestreador.
7.14. El equipo prepara automáticamente la curva de calibración, y en base
a esta curva de calibración, el equipo efectúa los cálculos
correspondientes a las muestras ingresadas.
7.15. Se ingresan por computadora los factores de dilución de cada muestra
y el instrumento automáticamente calcula la concentración correcta.

177
7.16. En el caso que la curva de calibración que calcula el equipo no sea
satisfactoria (mal coeficiente de correlación por alguna lectura errónea
de un estándar de calibración), el analista la puede construir con un
paquete estadístico en forma manual.
8. CALCULOS Y EXPRESION DE RESULTADOS
Expresar el resultado de cada analito en µg/L en base a la curva de
calibración.
Control de Calidad:
Medir duplicados en el 10 % de los casos, como mínimo. Los resultados
deben estar dentro del valor previamente calculado para cada analito, que a
su vez depende de la zona de la curva de calibración en la cuál se encuentra
su valor. El valor no es el mismo a bajas concentraciones como a altas
concentraciones. Verificar el cumplimiento del gráfico de control para
duplicados.
Introducir en el análisis cada 10 muestras, como mínimo, un estándar de
calibración de 0,70 mg/L, para determinar si se produjo alguna variación
instrumental. El resultado debe estar dentro del ± 5% del valor esperado. Si
esto no se produce, terminar el análisis de las muestras, corregir el problema
y recalibrar el instrumento.
Antes de finalizar el análisis, reanalizar una o más muestras. Los resultados
deben coincidir entre sí dentro del ± 5%. Si esto no se produce, hay que
reanalizar todas las muestras analizadas después del último control
aceptable del estándar de calibración de 0,7 mg/L.
Hay que analizar los dos estándares de control de procedimiento con cada
corrida. Esto permite verificar la exactitud.
Los resultados deben estar dentro del 95 % del límite de confianza que viene
suministrado por el proveedor para las muestras estándar de control de
procedimiento.

178
Verificar el cumplimiento del gráfico de control para las muestras estándar de
control.
Si los resultados de las muestras estándar de control de performance no
están comprendidos dentro del 95 % del límite de confianza, se repite el
análisis para los estándares en cuestión. Si este procedimiento falla, se
deben preparar nuevamente los estándares de calibración y reanalizarse.
Si todavía los resultados caen fuera del límite, se debe controlar que se
hayan seguido en forma adecuada las condiciones analíticas, y se deben
efectuar las correcciones y ajustes necesarios.
Si todavía existe una falla, se chequea el instrumento por la eventualidad de
algún daño. Deben analizarse nuevamente los estándares de calibración.
Si esta solución falla, el método se considera inválido y no se informan los
resultados hasta que la situación se clarifique. Hay que llamar al soporte
técnico del instrumento.
Los límites de detección, los estudios de repetibilidad y de recuperación se
deben encontrar documentados en el laboratorio.
9. REFERENCIAS
[1] Norma EPA 200.7.

179
2.3.1.23. Determinación de cadmio, cobre, cromo y p lomo total
en agua superficial mediante GFAAS
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales Código del procedimiento:
PT-MA-02
Nombre del procedimiento: Determinación de cadmio, cobre, cromo y plomo total en agua
superficial mediante GFAAS
Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determinar la concentración total de cadmio, cobre, cromo y plomo en
muestras de agua superficial, utilizando espectrometría de absorción atómica
con horno de grafito (GFAAS).
2. ALCANCE
Este método se aplica a muestras de agua superficial y a determinaciones
de Cd, Cr y Pb cuando se requieran límites de cuantificación, por debajo de
0,030 mg/L y de Cu por debajo de 0,050 mg/L.
Dichas muestras han tenido el tratamiento preliminar de digestión ácida
siguiendo el Procedimiento PT-DA-02.
3. PRINCIPIO DEL METODO
El principio se basa en la absorción de luz por átomos a una específica
longitud de onda. La espectrometría de absorción atómica electrotérmica
utiliza para la atomización un horno de grafito.
Un volumen de la muestra se deposita en un atomizador (horno de grafito) y
se incrementa la temperatura controlada antes de la atomización, para
remover el solvente y concomitantes. La alícuota introducida en el tubo de
grafito se atomiza dentro de un tiempo corto, dando lugar a una señal cuya
área (absorbancia integrada) es proporcional a la masa del analito en la
solución medida.
El uso de fuentes de luz especiales y de la selección de la longitud de onda
permite la determinación específica de los elementos individuales.
Interferencias

180
Las interferencias pueden deberse a la absorción molecular cuando los
componentes de la matriz se volatilizan durante la atomización, dando lugar a
una banda ancha de absorción.
Otra interferencia proviene de los efectos de la matriz y químicos.
Algunas interferencias específicas son:
Cadmio: Absorción severa no específica y scattering de la luz en la
atomización, causada por los componentes de la matriz. El exceso de cloruro
puede causar volatilización prematura de cadmio.
Cromo: Bajas concentraciones de calcio y/o fosfato pueden causar
interferencias. A concentraciones por debajo de 200 mg/L, el efecto del calcio
es constante y elimina el efecto del fosfato.
El uso del corrector de fondo y de los modificadores de matriz, minimizan las
interferencias en la determinación de estos analitos.
4. EQUIPOS
4.1. Espectrómetro de absorción atómica, provisto de un corrector de fondo.
4.2. Horno de grafito capaz de alcanzar la temperatura especificada y
requerida. Seguir las instrucciones del fabricante para considerar los
tiempos y temperaturas de secado, pirólisis y atomización.
4.3. Lámparas de cátodo hueco o de descarga eléctrica del elemento a
determinarse.
4.4. Muestreador automático (opcional)
4.5. Es recomendable, un sistema de registro de datos, de tal manera que se
tenga un registro permanente del proceso y que, cualquier problema con
el análisis (incompleta atomización, perdidas durante la pirólisis, cambios
en la sensibilidad, forma del pico obtenido, etc), pueda ser fácilmente
reconocido.
4.6. Balanza analítica, de sensibilidad igual o mayor a 0,1 mg.
5. REACTIVOS Y MATERIALES

181
5.1. MATERIALES
5.1.1. Pipetas calibradas clase A para medición de volúmenes iguales o
mayores a 1 mL.
5.1.2. Micropipetas calibradas de los rangos de volumen necesarios, para
mediciones de volumen menores de 1 mL, con puntas de buena
calidad y de un solo uso.
5.1.3. Material de uso habitual en el laboratorio: matraces aforados,
vasos de precipitados, puntas de micropipeta, etc, limpio, lavado con
solución de HNO3 1:1, enjuagado con agua grado reactivo.
5.2. REACTIVOS
5.2.1. Agua grado reactivo.
5.2.2. Solución de ácido nítrico concentrado, calidad ultrapuro.
5.2.3. Aire comprimido.
5.2.4. Argón, calidad UAP, (ultra alta pureza).
Soluciones estándar primarias
Las soluciones estándar primarias puede ser preparadas de metales de
alta pureza, óxidos o sales no higróscópicas usando agua pura. Estas
soluciones son preparadas con una concentración de 1000 mg/L.
Es posible utilizar soluciones estándar comerciales de 1000 mg/L.
Cuando se usa metales para la preparación, es necesaria la limpieza del
metal (especialmente alambre) para la preparación de los estándares.
Algunas formas de preparar las soluciones estándar primarias son:
5.2.5. Solución estándar de cadmio: Disolver 1.000 g de cadmio metálico
(mínimo 99,99 %) en 20 mL de HNO3 1:1 y diluir a 1 L con agua
grado reactivo.
5.2.6. Solución estándar de cobre: Disolver 1.000 g de cobre electrolítico
(mínimo 99,99 %) en 20 mL de HNO3 1:1 y diluir a 1 L con agua
grado reactivo.

182
5.2.7. Solución estándar de cromo: Disolver 1.923 g de trióxido de Cr,
CrO3 (mínimo 99,99 %) en agua grado reactivo, acidificar con
solución de ácido nítrico ultrapuro, y diluir a 1 L con agua grado
reactivo.
5.2.8. Solución estándar de plomo: Disolver 1.599 g de nitrato de plomo
Pb(NO3)2 (mínimo 99.99 %) en agua grado reactivo, acidificar con
10 mL de solución de ácido nítrico ultrapuro, y diluir a 1 L con agua
grado reactivo.
Soluciones estándar de calibración
Se preparan diluyendo la solución estándar primaria del metal con los
mismos ácidos y concentración que la muestra. Los estándares de
calibración deben ser frescos y preparados cada vez que se realiza el
análisis de un lote de muestras.
Modificadores de matriz
5.2.9. El uso de Pd y Mg(NO3)2 como modificadores de matriz, se
recomienda para la determinación de Cd y Cu: Se disuelven 300 mg
de paladio en polvo, en solución de ácido nítrico concentrado (1 mL
de HNO3 y si fuera necesario, agregar 0.1 mL de HCl concentrado).
Disolver 200 mg de Mg(NO3)2 en agua pura. Verter las dos
soluciones juntas y diluir a 100 mL con agua grado reactivo.
5.2.10. El uso de NH4H2PO4 y Mg(NO3)2 como modificadores de matriz
se recomienda para la determinación de Cd y Pb: La solución de
fosfato de amonio, NH4H2PO4 al 40%, se prepara disolviendo 40 g
del reactivo en agua grado reactivo y diluir a 100 mL.
5.2.11. El uso de Mg(NO3)2 como modificador de matriz , se recomienda
para la determinación de Cr.
5.2.12. Es posible también el uso de soluciones estándar primarias
comerciales de dichos modificadores de matriz.
Blancos

183
5.2.13. Se requiere el uso de dos tipos de blancos: El blanco de
calibración para establecer la curva analítica y el blanco reactivo
utilizado para identificar posible contaminación de los reactivos, o del
material utilizado durante la preparación de la muestra.
5.2.14. El blanco de calibración se prepara acidificando el agua grado
reactivo a la misma concentración de los ácidos utilizados para los
estándares de calibración y muestras.
5.3. El blanco reactivo, debe contener todos los reactivos en el mismo
volumen como se usó en la preparación de las muestras.
6. ACCIONES PREVIAS
6.1. Se requiere digestión ácida antes del análisis debido a que algunas
muestras pueden ser complejas y contener material suspendido que
debe ser sometido a solubilización.
6.2. Los bajos límites de cuantificación que se alcancen con este método
dependerán del equipo (tipo de espectrómetro y horno de grafito, la
fuente de energía, el grado de expansión eléctrica de la señal) y de la
matriz de la muestra.
6.3. Referirse al manual del equipo para detalles de operación optima.
6.4. Tomar en cuenta los efectos de posibles interferencias por el uso de
diluciones, modificadores de matriz o la aplicación del método de
adición estándar.
6.5. Trabajar en un laboratorio limpio para prevenir la contaminación de las
muestras.
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Obtener la curva de calibración respectiva utilizando el blanco de
calibración y los estándares, acidificados adecuadamente y
conteniendo los modificadores de matriz respectivos en las cantidades
siguientes: 5 µg de Pd + 3 µg de Mg(NO3)2 para la determinación de
Cd y Cu; 15 µg de Mg(NO3)2 para la determinación de Cr y 50 µg de
NH4H2PO4 + 3 µg de Mg(NO3)2 para la determinación de Cd y Pb.
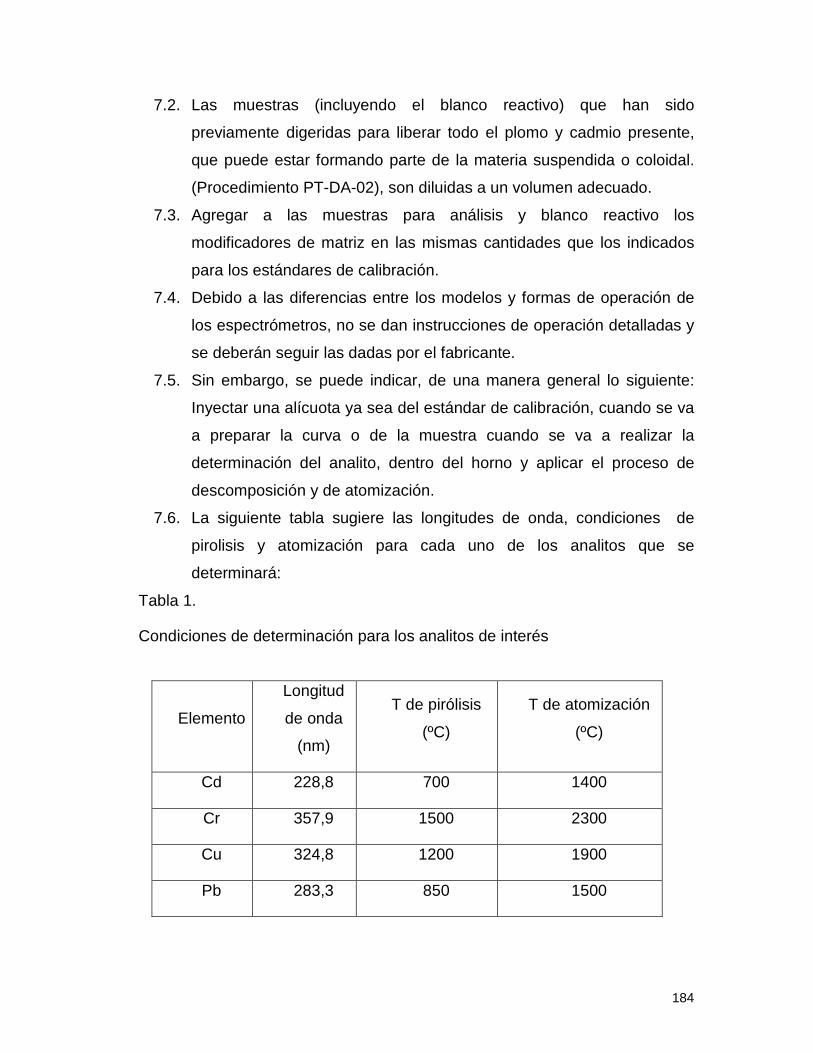
184
7.2. Las muestras (incluyendo el blanco reactivo) que han sido
previamente digeridas para liberar todo el plomo y cadmio presente,
que puede estar formando parte de la materia suspendida o coloidal.
(Procedimiento PT-DA-02), son diluidas a un volumen adecuado.
7.3. Agregar a las muestras para análisis y blanco reactivo los
modificadores de matriz en las mismas cantidades que los indicados
para los estándares de calibración.
7.4. Debido a las diferencias entre los modelos y formas de operación de
los espectrómetros, no se dan instrucciones de operación detalladas y
se deberán seguir las dadas por el fabricante.
7.5. Sin embargo, se puede indicar, de una manera general lo siguiente:
Inyectar una alícuota ya sea del estándar de calibración, cuando se va
a preparar la curva o de la muestra cuando se va a realizar la
determinación del analito, dentro del horno y aplicar el proceso de
descomposición y de atomización.
7.6. La siguiente tabla sugiere las longitudes de onda, condiciones de
pirolisis y atomización para cada uno de los analitos que se
determinará:
Tabla 1.
Condiciones de determinación para los analitos de interés
Elemento
Longitud
de onda
(nm)
T de pirólisis
(ºC)
T de atomización
(ºC)
Cd 228,8 700 1400
Cr 357,9 1500 2300
Cu 324,8 1200 1900
Pb 283,3 850 1500

185
7.7. Si se observa que la concentración de la muestra es mayor que la
concentración del mayor estándar, ésta deberá ser diluida con la
misma matriz ácida y reanalizada.
8. CALCULOS Y EXPRESION DE RESULTADOS
8.1. Cálculos para la preparación de la soluciones de calibración a partir de
un estándar primario comercial:
est
dldl
estC
VCV
⋅= mL
8.2. Cálculos para determinar la concentración del analito (µg/L).
dle FCC ⋅=
donde;
Ce Concentración del elemento en la muestra original (g/L),
C Concentración del elemento en la solución de la muestra (µg/L),
leída directamente o usando la curva de calibración.
Fdi Factor de dilución: Volumen de aforo de la solución de la muestra
(mL) / volumen de la alícuota tomada para dilución (mL).
Cdl Concentración a la que se quiere diluir el estándar (µg/L)
Vdl Volumen de aforo de la dilución (mL)
Cest Concentración del estándar (µg/L)
8.3. Expresión del resultado
Los resultados serán expresados en mg/L o µg/L acompañado de la
incertidumbre expandida a un nivel de confianza del 95%
aproximadamente.
Control de Calidad:
Establecer y mantener un sistema de registro para documentar la calidad de
los datos generados.

186
Establecer los límites de detección para evaluar el nivel de ruido del
instrumento y los cambios de respuesta en el tiempo para cada analito,
analizando una serie de blancos reactivos.
Estos límites en µg/L, se estiman calculando el promedio de las desviaciones
estándar de tres corridas y en días no consecutivos del blanco reactivo con 7
mediciones por día.
Esta medición debe realizarse al menos cada tres meses.
Con cada lote de muestras, se deben analizar por lo menos un blanco.
Realizar el análisis por lo menos por triplicado.
Con cada lote de muestras, se debe analizar por lo menos un material de
control, que pueden ser muestras fortificadas con un volumen conocido del
analito en análisis o un material de referencia certificado.
Es necesario establecer un criterio de aceptación de los resultados
obtenidos. Para muestras fortificadas el límite debe ser ± 20% del valor
fortificado.
9. REFERENCIAS
[1] Standard Method for the examination of water and wastewater “American
Health Association” Ed. 21 th 2005 3113 A
[2] Standard Method for the examination of water and wastewater “American
Health Association” Ed. 21 th 2005 3113 B
[3] Environmental Protection Agency, EPA Método 7010 – 2. SW-845.
[4] Bernhard Welz, Michael Sperlling. Atomic Absorption Spectrometry. 3era.
Edición. WILEY-VCH.

187
2.3.1.24. Determinación de As, Co, Cr, Cs, Fe, Rb, Sb, Sr, U, Zn en
muestras de agua mediante el análisis por activació n
neutrónica, método del k subcero
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales Código del procedimiento:
PT-MA-04
Nombre del procedimiento: Determinación de As, Co, Cr, Cs, Fe, Rb, Sb, Sr, U, Zn en muestras de
agua mediante el análisis por activación neutrónica, método del k
subcero
Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determinar la concentración multielemental en muestras de agua superficial,
utilizando el análisis por activación neutrónica, basado en el método del
ksubcero (k0- NAA).
2. ALCANCE
Este método se aplica a muestras de agua superficial y a la determinación de
As, Co, Cr, Cs, Fe, Rb, Sb, Sr, U, Zn.
3. PRINCIPIO DEL METODO
El análisis por activación neutrónica instrumental (AANI) es una técnica
analítica nuclear, que utiliza el proceso de transformación de un nucleido
cualquiera en otro artificialmente radiactivo, mediante la captura de un
neutrón y la posterior medición de la actividad inducida, para el análisis
multielemental cualitativo y cuantitativo de elementos mayores, menores y
traza en muestras geológicas, biológicas, arqueológicas, ambientales, etc.
El método de análisis por activación comparativo ha sido el más utilizado por
varias décadas. Este consiste en la irradiación simultánea y en la misma
posición de la muestra y un comparador que contiene cantidades
exactamente conocidas de cada uno de los elementos que interesa analizar.
El método del k0, es un método paramétrico, que utiliza un solo comparador
para la determinación multielemental en la muestra y requiere un
conocimiento exacto de los parámetros que caracterizan la facilidad de
irradiación.

188
4. EQUIPOS
4.1. Fuente de neutrones con flujos térmicos del orden de 1013 n.cm-2. s-1.
4.2. Sistema de Espectrometría Gamma de alta resolución constituído por:
Detector semiconductor de Ge-Hiperpuro, con preamplificador incorporado.
Fuente de alta Tensión
Amplificador adecuado
Convertidor analógico digital.
4.3. Sistema intercambiador de medición de muestras
4.4. Balanza analítica de sensibilidad igual o mayor a 0.1 mg
4.5. Polietileno (PE) de buena calidad para irradiación.
4.6. Cápsulas de PE para envío de muestras a irradiación
4.7. Fuente patrón de 152Eu
4.8. Contenedor de Pb
4.9. Material de laboratorio limpio, guantes de polietileno.
4.10. Horno digestor de microondas.
4.11. Prensa hidráulica
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Micropipeta calibrada y puntas de un solo uso
5.2. REACTIVOS
Se deben emplear reactivos de grado analítico en todos los ensayos. A
menos que se especifique lo contrario, se debe lograr que todos los
reactivos cumplan con lo dispuesto en las especificaciones del Comité de
Reactivos Analíticos de la ACS. Se pueden usar otros grados de reactivos
en la medida que sean de suficiente pureza para permitir su uso sin
perder la exactitud de las determinaciones.
5.2.1. Agua grado reactivo.
5.2.2. Solución estándar de Na de 10000 mg/L.
5.2.3. Solución estándar de Co de 1000 mg/L.
5.2.4. Solución estándar de Mo de 1000 mg/L.

189
5.2.5. Solución estándar de Au de 500 mg/L.
5.2.6. Materiales de referencia certificados conteniendo los analitos de
interés.
5.2.7. Papel de filtro Whatman 42 de 12.5 cm.
6. ACCIONES PREVIAS
6.1. La manipulación de muestras y material se realiza con guantes y en
laboratorio limpio.
6.2. Preparación de los comparadores de Na
Se depositan aproximadamente con micropipeta, 1000 µg de solución
estándar primaria de Na , en discos de papel filtro y se pesan.
Se evapora el disco de papel filtro, hasta secado bajo lámpara IR a
temperatura controlada.
Se prensa el disco seco, se empaca en bolsitas de PE previamente
lavado con HNO3 al 10%.
6.3. Preparación de monitores de flujo
De igual manera que en la preparación del comparador de Na, se
depositan con micropipeta, 15 µg de solución de Au, 350 µg de solución
de Mo y 120 µg de solución de Co.
Se evapora el disco de papel filtro, hasta secado bajo lámpara IR a
temperatura controlada
Se prensa el disco seco, se empaca en bolsitas de PE previamente
lavado con HNO3 al 10%.
6.4. Calibración del sistema de espectrometría gamma
Se utiliza una fuente de 152Eu u otra adecuada para la calibración. Esta
se coloca sobre el detector que se usara para las mediciones.
Se mide la fuente por 30 minutos y se calibra utilizando las energías de
122 keV, 344.3 keV, 778.9 keV y 1408 keV, en caso de utilizarse 152Eu.
7. DESCRIPCION DEL PROCEDIMIENTO

190
7.1. Las muestras se preparan pesando aproximadamente 100 mL de
muestra en vaso Pirex para ser pre-concentrados en un horno digestor
de microondas a 85 o C y luego se toman 200 µL de muestra pre-
concentrada y se depositan en papel de filtro Whatman 42 para ser
evaporado en lámpara infrarroja a temperatura controlada.
7.2. Los filtros secos son prensados como pastillas de 13 mm de diámetro,
luego es embolsado, codificado y acondicionado en viales de polietileno
para irradiación.
7.3. Se acondicionan en las cápsulas para irradiación alternadamente
comparadores, muestras, monitores de flujo, blancos y materiales de
referencia preparados de igual forma que las muestras.
7.4. Se envía la cápsula a irradiar por 5 h, a una potencia de 10 MW y un flujo
de neutrones térmicos de aprox. 3.0 · 1013 n. cm2· s-1.
7.5. Medir y registrar la tasa de dosis de la cápsula irradiada
aproximadamente a 25 cm y colocarla en el tacho de plomo anexo a la
estación de envío. En caso superar los 0.5 mSv transportar en
contenedor de plomo a la campana radioquímica.
7.6. Abrir la cápsula dentro de la campana y retirar los envases del estándar y
la muestra verificando la integridad de los mismos.
7.7. Transportar en el contenedor de plomo, las muestras, comparadores al
laboratorio de mediciones de espectrometría gamma.
7.8. Medir las muestras, materiales de referencia y comparadores de sodio
utilizando el detector de Ge (HP), de eficiencia relativa adecuada y con
una buena resolución para el pico de la fuente utilizada en la calibración
del espectrómetro gamma.
7.9. Evaluar los espectros.
7.10. Calcular la concentración de los elementos de interés.
7.11. Documentar la información en formatos preparados para ello.
8. CALCULOS Y EXPRESION DE RESULTADOS
8.1. Cálculo de la concentración en µg/L

191
Se emplea el área de los siguientes radionúclidos:

192
56As 559 keV 60Co 1173 y 1332.5 keV 51Cr 320 keV 134Cs 796 keV 59Fe 1099 y 1291 kev 239Np (U) 277 keV 86Rb 1076 keV 124Sb 1690 keV 88mSr 388 keV 95Zn 1115 keV
Calcular la concentración ρi del elemento “i” en una muestra x aplicando
el método de k0 según la convención de Högdahl:
)(
)(
,0
,0
,
,
,
,
αα
ρx
p
x
p
xo
po
pesp
xesp
iQF
QF
K
K
T
T
++
∈∈
=
Donde “x “ y “p” denota la muestra y el estándar respectivamente y Tesp
es igual:
)(
,
),(gwS
HCDCT
in
cxesp ⋅⋅⋅⋅
=
dteD⋅= λ
( )iteC ⋅−−
= λλ
1
l
r
t
tH =

193
Donde; ti, td son los tiempos de irradiación, decaimiento
respectivamente, λ es la constante de decaimiento = 0.693/periodo de
semidesintegración; є es la eficiencia del detector para la energía
medida, Q(α) es la razón de la integral de resonancia a la sección eficaz
como función de α y F es la relación de flujo térmico sobre flujo
epitérmico.
8.2. Expresión de Resultados.
La concentración calculada se expresa en términos de µg.L-1 con la
incertidumbre expandida.
9. CONTROL DE CALIDAD
9.1. Las muestras se analizan en duplicado
9.2. Por cada corrida de muestras se analiza como mínimo un material de
referencia certificado.
9.3. Por cada corrida de muestra se analiza un blanco de reactivos.
9.4. Participar en ensayos de aptitud o rondas de intercomparación en matriz
agua por lo menos una vez al año
10. REFERENCIAS
10.1. Knoll G.F. Radiation Detection and Measurement 2ndEdition 1989.
10.2. G. Erdtmann Nuclear Activation and Radioisotope Methods of Analyses.
10.3. IAEA-TECDOC-564 Practical Aspects of Operating a Neutron Activation
Analyses Laboratory. Documento Técnico del Taller Regional. Tópicos
selectos sobre Aplicaciones del Método de k-sub cero y otros métodos
Paramétricos en Análisis por Activación Neutrónica.
10.4. Frans De Corte. The k0-Standardization Method, 1987.

194
2.3.1.25. Determinación de Cadmio por el Método de
Espectrometría de Absorción Atómica con Aspiración
Directa
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales
Código del procedimiento: PT-MA-05
Nombre del procedimiento: Determinación de Cadmio por el Método de Espectrometría de
Absorción Atómica con Aspiración Directa
Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determinar cadmio total por espectrometría de absorción atómica.
2. ALCANCE
Se aplica a cualquier agua superficial y corresponde a la concentración de
metales determinados en una muestra no filtrada.
3. PRINCIPIO DEL METODO
3.1. Una fracción de muestra previamente digerida, es aspirada hacia una
llama Aire-Acetileno posicionada en el paso óptico de un espectrómetro
de absorción atómica. La muestra es atomizada bajo estas condiciones.
3.2 La población de átomos al estado elemental absorbe radiación
característica proveniente una fuente de emisión de líneas atómicas de
cadmio, la relación entre potencia incidente y potencia transmitida es una
medida de la concentración del elemento en la muestra.
4. EQUIPOS
4.1. Balanza Analítica, de sensibilidad igual o mayor a 0.1 mg.
4.2. Campana, con sistema de extracción de gases.
4.3. Espectrómetro de absorción atómica, dotado con quemador aire-
acetileno y corrector de deuterio (o correctores de acuerdo a equipo
utilizado).
4.4. Fuente de emisión de líneas atómicas de cadmio.

195
4.5. Plancha calefactora con regulación de temperatura o digestor de micro
ondas (opcional).
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Material de uso habitual en laboratorio (ej. vasos de precipitado,
pipetas, matraces y otros).
5.2. REACTIVOS
5.2.1. Agua grado reactivo.
5.2.2. Solución de ácido clorhídrico concentrado, calidad ultrapuro.
5.2.3. Solución de ácido nítrico concentrado, calidad ultrapuro.
5.2.4. Solución estándar de cadmio de 1000 mg/L.
5.2.5. Aire comprimido.
5.2.6. Acetileno, calidad UAP, (ultra alta pureza).
Soluciones estándares de calibración
5.2.7. Preparar una serie de soluciones de calibración en el rango óptimo,
a partir de diluciones apropiadas de la solución estándar de cadmio
de 1000 mg/L.
5.2.8. Almacenar en envases de polietileno de alta densidad
debidamente etiquetados. La condición de acidez final para los
estándares es solución 1% HNO3 V/V. Se exige para la curva de
calibración un r≥0.995.
5.2.9. Solución blanco término cero: la solución término cero debe ser
preparada con agua grado reactivo y en un medio 1% HNO3 V/V.
6. ACCIONES PREVIAS
6.1. Las muestras que contienen material particulado o materia orgánica,
generalmente requieren pretratamiento antes del análisis
espectroscópico. Estas se deben someter a un pretratamiento previo de
digestión para liberar todo el cadmio presente, que puede estar formando
parte de la materia suspendida o coloidal. Se recomienda utilizar el
procedimiento PT-DA-01.

196
6.2. La aplicación de la variante metodológica de omitir la digestión en
muestras con turbiedad < 1UNT, no es aplicable en la determinación de
Cd por el método de aspiración directa, debido al nivel exigido de LDM
para este parámetro. Por tanto, para la determinación de Cd en agua
potable utilizando la metodología propuesta, es imprescindible realizar el
tratamiento de preconcentración.
Preconcentración por evaporación
6.3. Transferir 250 mL de muestra previamente digerida a un vaso de
precipitación de tamaño adecuado.
6.4. Adicionar 5ml de solución de HNO3 concentrado, calentar en plancha
calefactora hasta desprendimiento de vapores rojizos. Continuar el
calentamiento a temperatura controlada hasta asegurarse el
desprendimiento de exceso de HNO3 [estado pastoso]. Evitar que la
muestra se seque.
6.5. Enfriar y disgregar las sales con 2,5ml de solución de HCl concentrado.
6.6. Transferir cuantitativamente a un matraz aforado de 25ml y aforar con
agua p.a. grado reactivo exenta de Cd.
6.7. Llevar junto a las muestras, por cada set de análisis, un blanco de
reactivo con la cantidad de ácidos y agua utilizados en al análisis de las
muestras.
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Encender el espectrómetro y verificar las presiones de aire y acetileno,
ajustando si procede.
7.2. Verificar que el extractor de gases se encuentra en correcto
funcionamiento.
7.3. Posicionar y encender la fuente de emisión de líneas atómicas de
cadmio; esperar a que la fuente se estabilice
7.4. Ajustar la energía de la fuente respecto del detector de acuerdo al
manual del equipo.
7.5. Ajustar la longitud de onda principal y abertura del monocromador de
acuerdo a las especificaciones establecidas en el manual de

197
operaciones del equipo. Longitud de onda principal 228 nm y abertura
del slit 0,7 nm).
7.6. Continuar con el ajuste y calibración, de acuerdo a instrucciones del
fabricante del equipo.
7.7. Una vez ajustadas las condiciones, calibrar el instrumento
procediendo como sigue:
7.8. Realizar autozero aspirando el blanco termino cero de la curva de
calibración.
7.9. Calibrar empleando un número adecuado de estándares como mínimo
blanco y tres concentraciones en el rango de trabajo,
recomendándose 5 puntos.
7.10. Verificar la calibración después de la lectura de cada seis muestras. Si
la calibración permanece constante continuar con la lectura de las
siguientes muestras. Si la calibración ha variado, recalibrar y chequear
la lectura de las muestras anteriores.
Lectura de las muestras
7.11. Una vez calibrado el instrumento, determinar la concentración de las
muestras de acuerdo al siguiente protocolo:
7.12. Aspirar el blanco reactivo del set de análisis que ha sido sometido al
mismo proceso de las muestras y registrar la concentración de los
analitos.
7.13. Aspirar las muestras en estudio en secuencia de seis en seis,
verificando la calibración y la estabilidad del cero entre lotes de
muestra.
7.14. Repetir las lecturas en caso de que sea necesario.
7.15. Aspirar agua grado reactivo entre muestra y muestra para evitar
contaminación cruzada.
Control de calidad analítica
7.16. Por cada set de análisis, llevar de forma paralela a las muestras, los
siguientes controles:

198
7.17. Un blanco reactivo preparado a partir de agua para análisis grado
reactivo exenta de cadmio.
7.18. Un ensayo duplicado de una muestra elegida al azar.
7.19. Un ensayo de un estándar de concentración conocida cercana al valor
normado.
8. CALCULOS Y EXPRESION DE RESULTADOS
Cd (mg/L) = (Lect - Bco) x D
donde:
Cd (mg/L) = concentración de Cd, expresada en mg de Cd por litro de
agua
Lect = lectura de la muestra en mg/L
Bco = lectura blanco reactivo sometido al mismo proceso de las muestras
D = factor de dilución o concentración
9. REFERENCIAS
[1] Standard Methods For The Examination Of Water And Wastewater.
APHA. AWWA. WEF. 21th Edition 2005, Part. 3111-B; Direct Air –Acetylene
Flame Method .

199
2.3.1.26. Determinación de Cromo por el Método de
Espectrometría de Absorción Atómica con Aspiración
Directa
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agu a que están contaminadas con metales Código del procedimiento:
PT-MA-06
Nombre del procedimiento: Determinación de Cromo por el Método de Espectrometría
de Absorción Atómica con Aspiración Directa Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determinar cromo total por espectrometría de absorción atómica.
2. ALCANCE
Se aplica a cualquier agua superficial y corresponde a la concentración de
metales determinados en una muestra no filtrada.
3. PRINCIPIO DEL METODO
3.1. Una fracción de muestra previamente digerida, es aspirada hacia una
llama Aire-Acetileno posicionada en el paso óptico de un espectrómetro
de absorción atómica. La muestra es atomizada bajo estas condiciones.
3.2 La población de átomos al estado elemental absorbe radiación
característica proveniente una fuente de emisión de líneas atómicas de
cromo, la relación entre potencia incidente y potencia transmitida es una
medida de la concentración del elemento en la muestra.
4. EQUIPOS
4.1. Balanza Analítica, de sensibilidad igual o mayor a 0.1 mg.
4.2. Campana, con sistema de extracción de gases.
4.3. Espectrómetro de absorción atómica, dotado con quemador aire-
acetileno y corrector de deuterio (o correctores de acuerdo a equipo
utilizado).
4.4. Fuente de emisión de líneas atómicas de cromo.

200
4.5. Plancha calefactora con regulación de temperatura o digestor de micro
ondas (opcional).
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Material de uso habitual en laboratorio (ej. vasos de precipitado,
pipetas, matraces y otros).
5.2. REACTIVOS
5.2.1. Agua grado reactivo.
5.2.2. Solución de ácido clorhídrico concentrado, calidad ultrapuro.
5.2.3. Solución de ácido nítrico concentrado, calidad ultrapuro.
5.2.4. Solución estándar de cromo de 100 mg/L. Disolver 0,1923 g de
CrO3 en agua, acidificar con 10 mL de solución de HNO3
concentrado y diluir a 1000 mL con agua destilada 1,00 mL = 100
microgramos Cr.
5.2.5. Oxido nitroso.
5.2.6. Acetileno, calidad UAP, (ultra alta pureza).
Soluciones estándares de calibración
5.2.7. Preparar una serie de soluciones de calibración en el rango de 0,2
a 3,0 mg/L, a partir de diluciones apropiadas de la solución estándar
de cromo de 100 mg/L.
5.2.8. Almacenar en envases de polietileno de alta densidad
debidamente etiquetados. La condición de acidez final para los
estándares es solución 1% HNO3 V/V. Se exige para la curva de
calibración un r≥0.995.
5.2.9. Solución blanco término cero: la solución término cero debe ser
preparada con agua grado reactivo y en un medio 1% HNO3 V/V.
6. ACCIONES PREVIAS
6.1. ver Procedimiento PT-MA-05.
Preconcentración por evaporación
6.2. Transferir 250 mL de muestra previamente digerida a vaso precipitado y
calentar en plancha de calentamiento para disminuir el volumen de la

201
solución, evitando que se seque la muestra. Debe evitarse la ebullición
violenta que puede provocar pérdidas del analito.
6.3. Enfriar y diluir a 25 mL con agua.
6.4. En caso de aparecer sales, disgregar primero con 2,5 mL de solución de
HCl concentrado y después diluir a 25 mL.
6.5. Llevar junto a las muestras, por cada set de análisis, un blanco de
reactivo con la cantidad de ácidos y agua utilizados en al análisis de las
muestras.
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Encender el espectrómetro y verificar las presiones de aire y acetileno,
ajustando si procede.
7.2. Verificar que el extractor de gases se encuentra en correcto
funcionamiento.
7.3. Posicionar y encender la fuente de emisión de líneas atómicas de
cobre; esperar a que la fuente se estabilice
7.4. Ajustar la energía de la fuente respecto del detector de acuerdo al
manual del equipo.
7.5. Ajustar la longitud de onda principal y abertura del monocromador de
acuerdo a las especificaciones establecidas en el manual de
operaciones del equipo. Longitud de onda principal 357,87 nm.
7.6. Continuar con el ajuste y calibración, de acuerdo a instrucciones del
fabricante del equipo.
7.7. Una vez ajustadas las condiciones, calibrar el instrumento
procediendo como sigue:
7.8. Realizar autozero aspirando el blanco termino cero de la curva de
calibración.
7.9. Calibrar empleando un número adecuado de estándares como mínimo
blanco y tres concentraciones en el rango de trabajo,
recomendándose 5 puntos.
7.10. Verificar la calibración después de la lectura de cada seis muestras. Si
la calibración permanece constante continuar con la lectura de las

202
siguientes muestras. Si la calibración ha variado, recalibrar y chequear
la lectura de las muestras anteriores.
Lectura de las muestras
7.11. Una vez calibrado el instrumento, determinar la concentración de las
muestras de acuerdo al siguiente protocolo:
7.12. Aspirar el blanco reactivo del set de análisis que ha sido sometido al
mismo proceso de las muestras y registrar la concentración de los
analitos.
7.13. Aspirar las muestras en estudio en secuencia de seis en seis,
verificando la calibración y la estabilidad del cero entre lotes de
muestra.
7.14. Repetir las lecturas en caso de que sea necesario.
7.15. Aspirar agua grado reactivo entre muestra y muestra para evitar
contaminación cruzada.
Control de calidad analítica
7.16. Por cada set de análisis, llevar de forma paralela a las muestras, los
siguientes controles:
7.17. Un blanco reactivo preparado a partir de agua para análisis grado
reactivo exenta de cobre.
7.18. Un ensayo duplicado de una muestra elegida al azar.
7.19. Un ensayo de un estándar de concentración conocida cercana al valor
normado.
8. CALCULOS Y EXPRESION DE RESULTADOS
Cr (mg/L) = (Lect - Bco) x D
donde:
Cr (mg/L) = concentración de Cr, expresada en mg de Cr por litro de agua
Lect = lectura de la muestra en mg/L
Bco = lectura blanco reactivo sometido al mismo proceso de las muestras

203
D = factor de dilución o concentración
9. REFERENCIAS
[1] Standard Methods For The Examination Of Water And Wastewater.
APHA. AWWA. WEF. 21th Edition 2005, Part. 3111-B; Direct Air –Acetylene
Flame Method.

204
2.3.1.27. Determinación de Cobre por el Método de
Espectrometría de Absorción Atómica con Aspiración
Directa
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales
Código del procedimiento: PT-MA-07
Nombre del procedimiento: Determinación de Cobre por el Método de Espectrometría de Absorción
Atómica con Aspiración Directa
Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determinar cobre total por espectrometría de absorción atómica.
2. ALCANCE
Se aplica a cualquier agua superficial y corresponde a la concentración de
metales determinados en una muestra no filtrada.
3. PRINCIPIO DEL METODO
Ver Procedimiento PT-MA-05.
4. EQUIPOS
4.1. Balanza Analítica, de sensibilidad igual o mayor a 0.1 mg.
4.2. Campana, con sistema de extracción de gases.
4.3. Espectrómetro de absorción atómica, dotado con quemador aire-
acetileno y corrector de deuterio (o correctores de acuerdo a equipo
utilizado).
4.4. Fuente de emisión de líneas atómicas de cobre.
4.5. Plancha calefactora con regulación de temperatura o digestor de micro
ondas (opcional).
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Material de uso habitual en laboratorio (ej. vasos de precipitado,
pipetas, matraces y otros).
5.2. REACTIVOS

205
5.2.1. Agua grado reactivo.
5.2.2. Solución de ácido clorhídrico concentrado, calidad ultrapuro.
5.2.3. Solución de ácido nítrico concentrado, calidad ultrapuro.
5.2.4. Solución estándar de cobre de 100 mg/L. Disolver 0,100 g de cobre
metálico en 2 ml de solución de HNO3 concentrado, añadir 10 mL de
solución de HNO3 concentrado y diluir a 1000 mL con agua.
5.2.5. Aire comprimido.
5.2.6. Acetileno, calidad UAP, (ultra alta pureza).
Soluciones estándares de calibración
5.2.7. Preparar una serie de soluciones de calibración en el rango óptimo
de 1 a 5 mg/L, a partir de diluciones apropiadas de la solución
estándar de cobre de 100 mg/L.
5.2.8. Almacenar en envases de polietileno de alta densidad
debidamente etiquetados. La condición de acidez final para los
estándares es solución 1% HNO3 V/V. Se exige para la curva de
calibración un r≥0.995.
5.2.9. Solución blanco término cero: la solución término cero debe ser
preparada con agua grado reactivo y en un medio 1% HNO3 V/V.
6. ACCIONES PREVIAS
6.1. Ver Procedimiento PT-MA-05.
6.2. Para muestras con bajo contenido de cobre se hace necesario realizar
una etapa de preconcentración. Pueden aplicarse la concentración por
evaporación y mediante extracción con APDC.
Preconcentración por evaporación
6.3. Transferir 250 mL de muestra previamente digerida a un vaso de
precipitación, y calentar en plancha calefactora para disminuir el volumen
de la solución, evitando que se seque la muestra. Debe evitarse la
ebullición violenta que puede provocar pérdidas del analito.
6.4. Enfriar y aforar a 25 mL con agua.
6.5. En caso de aparecer sales, disgregar primero con 2,5 mL de solución de
HCl concentrado y aforar a 25 mL.

206
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Encender el espectrómetro y verificar las presiones de aire y acetileno,
ajustando si procede.
7.2. Verificar que el extractor de gases se encuentra en correcto
funcionamiento.
7.3. Posicionar y encender la fuente de emisión de líneas atómicas de
cobre; esperar a que la fuente se estabilice
7.4. Ajustar la energía de la fuente respecto del detector de acuerdo al
manual del equipo.
7.5. Ajustar la longitud de onda principal y abertura del monocromador de
acuerdo a las especificaciones establecidas en el manual de
operaciones del equipo. Longitud de onda principal 324,8 nm.
7.6. Continuar con el ajuste y calibración, de acuerdo a instrucciones del
fabricante del equipo.
7.7. Una vez ajustadas las condiciones, calibrar el instrumento
procediendo como sigue:
7.8. Realizar autozero aspirando el blanco termino cero de la curva de
calibración.
7.9. Calibrar empleando un número adecuado de estándares como mínimo
blanco y tres concentraciones en el rango de trabajo,
recomendándose 5 puntos.
7.10. Verificar la calibración después de la lectura de cada seis muestras. Si
la calibración permanece constante continuar con la lectura de las
siguientes muestras. Si la calibración ha variado, recalibrar y chequear
la lectura de las muestras anteriores.
Lectura de las muestras
7.11. Una vez calibrado el instrumento, determinar la concentración de las
muestras de acuerdo al siguiente protocolo:
7.12. Aspirar el blanco reactivo del set de análisis que ha sido sometido al
mismo proceso de las muestras y registrar la concentración de los
analitos.

207
7.13. Aspirar las muestras en estudio en secuencia de seis en seis,
verificando la calibración y la estabilidad del cero entre lotes de
muestra.
7.14. Repetir las lecturas en caso de que sea necesario.
7.15. Aspirar agua grado reactivo entre muestra y muestra para evitar
contaminación cruzada.
Control de calidad analítica
7.16. Por cada set de análisis, llevar de forma paralela a las muestras, los
siguientes controles:
7.17. Un blanco reactivo preparado a partir de agua para análisis grado
reactivo exenta de cobre.
7.18. Un ensayo duplicado de una muestra elegida al azar.
7.19. Un ensayo de un estándar de concentración conocida cercana al valor
normado.
8. CALCULOS Y EXPRESION DE RESULTADOS
Cu (mg/L) = (Lect - Bco) x D
donde:
Cu (mg/L) = concentración de Cu, expresada en mg de Cu por litro de
agua
Lect = lectura de la muestra en mg/L
Bco = lectura blanco reactivo sometido al mismo proceso de las muestras
D = factor de dilución o concentración
9. REFERENCIAS
[1] Standard Methods For The Examination Of Water And Wastewater.
APHA. AWWA. WEF. 21th Edition 2005, Part. 3111-B; Direct Air –Acetylene
Flame Method.

208
2.3.1.28. Determinación de Plomo por el Método de
Espectrometría de Absorción Atómica con Aspiración
Directa
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agu a que están contaminadas con metales
Código del procedimiento: PT-MA-08
Nombre del procedimiento: Determinación de Plomo por el Método de Espectrometría
de Absorción Atómica con Aspiración Directa Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determina plomo total por espectrometría de absorción atómica.
2. ALCANCE
Este método de ensayo es aplicable a cualquier agua superficial y
corresponde a la concentración de metales determinados en una muestra no
filtrada.
3. PRINCIPIO DEL METODO
Ver Procedimiento PT-MA-05.
4. EQUIPOS
4.1. Balanza Analítica, de sensibilidad igual o mayor a 0.1 mg.
4.2. Campana, con sistema de extracción de gases.
4.3. Espectrómetro de absorción atómica, dotado con quemador aire-
acetileno y corrector de deuterio (o correctores de acuerdo a equipo
utilizado).
4.4. Fuente de emisión de líneas atómicas de plomo.
4.5. Plancha calefactora con regulación de temperatura o digestor de micro
ondas (opcional).
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Material de uso habitual en laboratorio (ej. vasos de precipitado,
pipetas, matraces y otros).

209
5.2. REACTIVOS
5.2.1. Agua grado reactivo.
5.2.2. Solución de ácido clorhídrico concentrado, calidad ultrapuro.
5.2.3. Solución de ácido nítrico concentrado, calidad ultrapuro.
5.2.4. Solución estándar de plomo de 100 mg/L. Disolver 0,100 g de
plomo metálico en 2 ml de solución de HNO3 concentrado, añadir 10
mL de solución de HNO3 concentrado y diluir a 1000 mL con agua.
5.2.5. Aire comprimido.
5.2.6. Acetileno, calidad UAP, (ultra alta pureza).
Soluciones estándares de calibración
5.2.7. Preparar una serie de soluciones de calibración en el rango óptimo
de 2 a 10 mg/L, a partir de diluciones apropiadas de la solución
estándar de plomo de 100 mg/L.
5.2.8. Almacenar en envases de polietileno de alta densidad
debidamente etiquetados. La condición de acidez final para los
estándares es solución 1% HNO3 V/V. Se exige para la curva de
calibración un r≥0.995.
5.2.9. Solución blanco término cero: la solución término cero debe ser
preparada con agua grado reactivo y en un medio 1% HNO3 V/V.
6. ACCIONES PREVIAS
6.1. Ver Procedimiento PT-MA-05.
6.2. Para muestras con bajo contenido de plomo se hace necesario realizar
una etapa de preconcentración. Pueden aplicarse la concentración por
evaporación y mediante extracción con APDC.
Preconcentración por evaporación
6.3. Transferir 250 mL de muestra previamente digerida a un vaso de
precipitación, y calentar en plancha calefactora para disminuir el volumen
de la solución, evitando que se seque la muestra. Debe evitarse la
ebullición violenta que puede provocar pérdidas del analito.
6.4. Enfriar y aforar a 25 mL con agua.

210
6.5. En caso de aparecer sales, disgregar primero con 1,5 mL de solución de
HNO3 concentrado y aforar a 25 mL.
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Encender el espectrómetro y verificar las presiones de aire y acetileno,
ajustando si procede.
7.2. Verificar que el extractor de gases se encuentra en correcto
funcionamiento.
7.3. Posicionar y encender la fuente de emisión de líneas atómicas de
plomo; esperar a que la fuente se estabilice.
7.4. Ajustar la energía de la fuente respecto del detector de acuerdo al
manual del equipo.
7.5. Ajustar la longitud de onda principal y abertura del monocromador de
acuerdo a las especificaciones establecidas en el manual de
operaciones del equipo. Longitud de onda principal 217,0 nm.
7.6. Continuar con el ajuste y calibración, de acuerdo a instrucciones del
fabricante del equipo.
7.7. Una vez ajustadas las condiciones, calibrar el instrumento
procediendo como sigue:
7.8. Realizar autozero aspirando el blanco termino cero de la curva de
calibración.
7.9. Calibrar empleando un número adecuado de estándares como mínimo
blanco y tres concentraciones en el rango de trabajo,
recomendándose 5 puntos.
7.10. Verificar la calibración después de la lectura de cada seis muestras. Si
la calibración permanece constante continuar con la lectura de las
siguientes muestras. Si la calibración ha variado, recalibrar y chequear
la lectura de las muestras anteriores.
Lectura de las muestras
7.11. Una vez calibrado el instrumento, determinar la concentración de las
muestras de acuerdo al siguiente protocolo:

211
7.12. Aspirar el blanco reactivo del set de análisis que ha sido sometido al
mismo proceso de las muestras y registrar la concentración de los
analitos.
7.13. Aspirar las muestras en estudio en secuencia de seis en seis,
verificando la calibración y la estabilidad del cero entre lotes de
muestra.
7.14. Repetir las lecturas en caso de que sea necesario.
7.15. Aspirar agua grado reactivo entre muestra y muestra para evitar
contaminación cruzada.
Control de calidad analítica
7.16. Por cada set de análisis, llevar de forma paralela a las muestras, los
siguientes controles:
7.17. Un blanco reactivo preparado a partir de agua para análisis grado
reactivo exenta de cobre.
7.18. Un ensayo duplicado de una muestra elegida al azar.
7.19. Un ensayo de un estándar de concentración conocida cercana al valor
normado.
8. CALCULOS Y EXPRESION DE RESULTADOS
Pb (mg/L) = (Lect - Bco) x D
donde:
Pb (mg/L) = concentración de Pb, expresada en mg de Pb por litro de
agua
Lect = lectura de la muestra en mg/L
Bco = lectura blanco reactivo sometido al mismo proceso de las muestras
D = factor de dilución o concentración
9. REFERENCIAS

212
[1] Standard Methods For The Examination Of Water And Wastewater.
APHA. AWWA. WEF. 21th Edition 2005, Part. 3111-B; Direct Air –Acetylene
Flame Method.

213
2.3.1.29. Determinación de Arsénico Total por el Mé todo de
Espectrometría de Absorción Atómica con Generación de
Hidruros
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agu a que están contaminadas con metales
Código del procedimiento: PT-MA-09
Nombre del procedimiento: Determinación de Arsénico Total por el Método de
Espectrometría de Absorción Atómica con Generación de Hidruros
Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determinar arsénico total mediante espectrometría de absorción atómica con
generación de hidruros.
2. ALCANCE
Se aplica a toda agua superficial y corresponde a la concentración de
arsénico en una muestra ya digerida.
3. PRINCIPIO DEL METODO
3.1 El arsénico presente en una fracción de muestra previamente digerida,
es reducido a arsina mediante reacción con borohidruro de sodio en
medio ácido. La arsina generada es conducida por un gas de arrastre
hacia una celda de cuarzo posicionada en el paso óptico de un
espectrómetro de absorción atómica. La celda se encuentra a una
temperatura aproximada de 1000ºC. La arsina, en estas condiciones, y a
través de mecanismos combinados [descomposición térmica y
reacciones con radicales hidrógeno] es atomizada.
3.2 La población de átomos al estado elemental absorbe radiación
característica proveniente de una fuente de emisión de líneas atómicas
de arsénico, la relación entre potencia incidente y potencia transmitida es
una medida de la concentración del elemento en la muestra.
4. EQUIPOS

214
4.1. Balanza Analítica, de sensibilidad igual o mayor a 0.1 mg.
4.2. Campana, con sistema de extracción de gases.
4.3. Espectrómetro de absorción atómica, dotado con quemador y con
generador de hidruros y corrector de deuterio [u otro corrector de
acuerdo a equipo utilizado).
4.4. Fuente de emisión de líneas atómicas de arsénico.
4.5. Sistema generador de hidruros continuo o discontinuo.
4.6. Celda de cuarzo y soporte de celda.
4.7. Plancha calefactora con regulación de temperatura o digestor de micro
ondas (opcional).
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Material de uso habitual en laboratorio (ej. vasos de precipitado,
pipetas, matraces y otros).
5.2. REACTIVOS
5.2.1. Agua grado reactivo.
5.2.2. Solución de ácido clorhídrico concentrado, calidad ultrapuro.
5.2.3. Solución de ácido nítrico concentrado, calidad ultrapuro.
5.2.4. Solución estándar comercial de arsénico de 1000 mg/L.
5.2.5. Aire comprimido.
5.2.6. Argón o nitrógeno
5.2.7. Borohidruro de sodio (NaBH4)
5.2.8. Yoduro de potasio (KI)
5.2.9. Hidróxido de sodio
5.2.10. Indicador fenolftaleína
5.2.11. Solución de ácido sulfúrico concentrado, calidad ultrapuro.
5.2.12. Solución de NaBH4 1 – 4%; la concentración dependerá del
sistema de generación empleado. Pesar la cantidad requerida para
1L de solución (el reactivo es corrosivo en contacto con la piel).
Disolver con solución de NaOH (0,5 -1% m/v). Una vez disuelto,
filtrar a través de filtro rápido de alta pureza. Descartar el filtro y

215
transferir el filtrado a un matraz aforado de 1000 ml. Aforar con
solución de NaOH (0,5 -1% m/v).
5.2.13. Solución de KI 20% m/v: Pesar 20g. de KI, disolver y diluir a 100
mL con agua grado reactivo exenta de arsénico. Proteger de la
oxidación por efecto de la luz.
Soluciones estándares de calibración
5.2.14. Preparar una serie de soluciones de calibración en el rango
óptimo de trabajo, a partir de diluciones apropiadas del estándar de
1000 mg As (III)/L 1% v/v H2SO4.
5.2.15. Por cada 100 ml de solución adicionar 10 ml de solución de KI
(20% m/v). Agitar y dejar reposar aproximadamente 30 minutos.
5.2.16. Cubrir debidamente el matraz para evitar la oxidación por efecto
de la luz. La condición de acidez final para estándares es 20% v/v
HCl. Preparar diariamente. Se exige para la curva de calibración un
r≥0.995.
5.2.17. Solución blanco término cero: la solución término cero debe ser
preparada con agua grado reactivo exenta de arsénico y en un medio
20 % HCl v/v, adicionando 10 ml de solución de KI (20% m/v).
Proteger de la oxidación por efecto de la luz. Preparar diariamente.
6. ACCIONES PREVIAS
Ensayo de las muestras:
6.1. Las muestras que contienen material particulado o materia orgánica,
generalmente requieren pretratamiento antes del análisis
espectroscópico. Estas se deben someter a un pretratamiento previo de
digestión para liberar todo el arsénico presente que puede estar
formando parte de la materia suspendida o coloidal. Para muestras de
agua potable, transparentes, incoloras e inodoras, con turbiedad < 1
UNT, puede obviarse el pretratamiento de digestión para analizar por
FAAS con lectura directa. Para esta condición, luego que la muestra se
recolecta, se acidifica a pH<2 con solución de HNO3 concentrado [1,5ml
HNO3/L es usualmente adecuado para agua potable] y se analiza sin

216
necesidad de digestión. Antes de aplicar esa modalidad como rutina, y
sobre todo en muestras sin historial, es necesario analizarlas con o sin
digestión, se manera que haya evidencia que los resultados obtenidos
sean comparables.
6.2. El procedimiento analítico para la modalidad sin digestión solo permite
obviar esta operación, por tanto, deberá considerarse cada uno de los
pasos descritos en la preparación de las muestras para la lectura final.
La aplicación de esta variante metodológica está supeditada a lograr,
bajo las condiciones operativas y disposición de instrumental adecuado,
llegar a los niveles exigidos de LDM para este parámetro. Si ello no es
posible, aún cuando la muestra tenga turbiedad < 1 UNT, se deberá
aplicar el tratamiento de preconcentración de muestras descrito más
adelante.
6.3. Si se requiere digestión de la muestra, proceder por alguna de las
alternativas que siguen para luego continuar con el procedimiento.
6.4. Los volúmenes de muestra a ensayar dependen de la concentración
esperada para el analito, por lo que para su definición se deberá tener
en consideración esta variable.
Digestión en plancha calefactora. Sin preconcentrac ión.
6.5. Transferir el volumen seleccionado de la muestra preservada a un vaso
de precipitado de tamaño adecuado.
6.6. Adicionar 5 mL de solución de HNO3 concentrado y calentar en plancha
calefactora hasta desprendimiento de vapores rojizos. Continuar el
calentamiento a temperatura controlada hasta asegurarse el
desprendimiento del exceso de HNO3 [estado pastoso]. Evitar que la
muestra se seque.
6.7. Enfriar y disgregar las sales con 10 mL de solución de HCl concentrado.
6.8. Transferir cuantitativamente a un matraz aforado de 100 mL.
6.9. Adicionar 10ml de solución de KI [prerreducción de As(V) a As(III)].

217
6.10. Aforar y mezclar con agua p.a. grado reactivo exenta de arsénico.
6.11. Proteger de la oxidación por efecto de la luz. 6.12. Llevar junto a las muestras, por cada set de análisis, un blanco de
reactivo con la cantidad de ácidos, KI y agua utilizados en al análisis de
las muestras.
6.13. Llevar, por cada set de análisis, un estándar a través toda la
marcha analítica a fin de evaluar la recuperabilidad en el procedimiento.
Digestión en plancha calefactora. Con preconcentrac ión inicial.
6.14. Transferir los 250 mL de la muestra preservada a un vaso de
precipitados de tamaño adecuado.
6.15. Adicionar 5ml de solución de HNO3 concentrado, calentar en
plancha calefactora hasta desprendimiento de vapores rojizos. Continuar
el calentamiento a temperatura controlada hasta asegurarse el
desprendimiento de exceso de HNO3 [estado pastoso]. Evitar que la
muestra se seque.
6.16. Enfriar y disgregar las sales con 5ml de HCl.
6.17. Transferir cuantitativamente a un matraz aforado de 50ml.
6.18. Adicionar 10 mL de solución de KI [prerreducción de As(V) a
As(III)].
6.19. Aforar y mezclar con agua grado reactivo exenta de arsénico.
6.20. Proteger de la oxidación por efecto de la luz.
6.21. Llevar junto a las muestras, por cada set de análisis, un blanco de
reactivo con la cantidad de ácidos, KI y agua utilizados en al análisis de
las muestras.
6.22. Llevar, por cada set de análisis, un estándar a través toda la
marcha analítica a fin de evaluar la recuperabilidad en el procedimiento.
Digestión con Microondas:
6.23. Ajustar el programa de acuerdo a las condiciones de la matriz de
la muestra.

218
6.24. Transferir al recipiente de digestión el volumen adecuado de
muestra, teniendo presente el efecto de las diluciones sobre el LDM.
6.25. Aplicar el programa.
6.26. Ajustar la muestra de acuerdo a las condiciones finales descritas
en los procedimientos descriptos anteriormente, dependiendo si se
requiere o no preconcentración inicial.
7. DESCRIPCION DEL PROCEDIMIENTO
Ajuste y calibración del instrumento:
7.1. Encender el espectrómetro y verificar las presiones de aire, acetileno y
nitrógeno o argón, ajustando si procede.
7.2. Verificar que el extractor de gases se encuentre en correcto
funcionamiento.
7.3. Chequear con una fuente de emisión que emita luz visible [Cu, Mn],
que el haz esté pasando central a al ranura del quemador.
7.4. Posicionar el soporte de la celda de cuarzo en el paso óptico del
espectrómetro y ajustar que el haz pase por el interior de la celda,
siguiendo las instrucciones proporcionadas en el manual del
instrumento [generador de hidruros].
7.5. Posicionar y encender la fuente de emisión de líneas atómicas de As.
Esperar a que la fuente se estabilice.
7.6. Ajustar la longitud de onda principal [193,7nm] y la abertura del
monocromador de acuerdo a las especificaciones establecidas en el
manual de operaciones del equipo.
7.7. Ajustar la energía de la fuente respecto al detector de acuerdo a las
instrucciones descritas en el manual del equipo.
7.8. Ajustar los flujos de encendido y encender el instrumento.
Sistema manual o automático de generación de hidru ros [generación
discontinua].

219
7.9. De acuerdo las instrucciones indicadas en el manual de operaciones
del generador, adicionar el volumen de estándar requerido y aplicar el
programa.
7.10. Ajustar el flujo de gas portador [Ar o N2].
7.11. Encender el corrector de absorción no específica.
7.12. Verificar la sensibilidad analítica empleando el estándar de calibración
recomendado en el manual de operaciones del equipo.
7.13. Construir la curva de calibración empleando un numero adecuado de
estándares, como mínimo blanco y tres concentraciones del rango de
trabajo, recomendándose 5 puntos.
7.14. Verificar la calibración después de la lectura de cada 6 muestras. Si la
calibración permanece constante, continuar con las lecturas de las
siguientes muestras, si la calibración ha variado, recalibrar y chequear
las lecturas de las muestras anteriores.
7.15. Una vez calibrado el instrumento, determinar la concentración de
muestras de acuerdo al siguiente protocolo:
7.16. Aspirar el blanco y realizar auto -cero
7.17. Aspirar las muestras en estudio, en secuencia de 6 en 6, verificando la
calibración y la estabilidad del cero, entre lotes de muestras. Repetir
las lecturas en caso que sea necesario.
7.18. Aspirar agua para análisis grado reactivo exenta de As entre muestra
y muestra con el propósito de evitar contaminación cruzada.
7.19. Para el control de calidad analítica por cada set de análisis llevar en
forma paralela a las muestras, los siguientes controles:
7.19.1. Un blanco reactivo preparado a partir de agua para análisis
grado reactivo exenta de As.
7.19.2. Un ensayo duplicado de una muestra elegida al azar
(precisión mínima 80%; RSD ≤ 20%)
7.19.3. Un ensayo de un estándar de concentración conocida,
cercana al valor medido (exactitud mínima 85-115% como
recuperación del estándar).

220
Sistema de Generación Continua de Hidruros:
7.20. Encender y ajustar el equipo de acuerdo a las instrucciones descritas
en el manual del equipo y ajustar las condiciones (longitud de onda
principal 193,7 nm y abertura del monocromador slit 0,7 nm).
7.21. Ajustar el flujo de gas (Ar o N2) en el sistema de generación continua
(FIAS)
7.22. Ajustar las velocidades de flujo del reactivo reductor y muestra de
acuerdo a las especificaciones indicadas en el manual del generador
de hidruros (FIAS)
7.23. Verificar la sensibilidad analítica empleando el estándar de calibración
recomendado en el manual de operaciones del equipo.
7.24. Construir la curva de calibración empleando un numero adecuado de
estándares, como mínimo blanco y tres concentraciones del rango de
trabajo, recomendándose 5 puntos.
7.25. Verificar la calibración después de la lectura de cada 6 muestras. Si la
calibración permanece constante, continuar con las lecturas de las
siguientes muestras, si la calibración ha variado, recalibrar y chequear
las lecturas de las muestras anteriores.
7.26. Una vez calibrado el instrumento, determinar la concentración de
muestras de acuerdo al siguiente protocolo:
7.26.1. Aspirar el blanco y realizar auto -cero
7.26.2. Aspirar las muestras en estudio, en secuencia de 6 en 6,
verificando la calibración y la estabilidad del cero, entre lotes de
muestras. Repetir las lecturas en caso que sea necesario.
7.26.3. Aspirar agua grado reactivo exenta de As entre muestra y
muestra con el propósito de evitar contaminación cruzada.
7.27. Para el control de calidad analítica por cada set de análisis llevar en
forma paralela a las muestras, los siguientes controles:
7.27.1. Un blanco reactivo preparado a partir de agua para análisis
grado reactivo exenta de As

221
7.27.2. Un ensayo duplicado de una muestra elegida al azar
(precisión mínima 80%; RSD≤ 20%)
7.27.3. Un ensayo de un estándar de concentración conocida,
cercana al valor medido (exactitud mínima 85-115% como
recuperación del estándar).
8. CALCULOS Y EXPRESION DE RESULTADOS
As (mg/L) = (Lect - Bco) x D
donde:
As (mg/L) = concentración de As, expresada en mg de As por litro de
agua
Lect = lectura de la muestra en mg/L
Bco = lectura blanco reactivo sometido al mismo proceso de las muestras
D = factor de dilución o concentración
8.1. Si la muestra no acusa presencia de As, se debe informar como el LDM
<0,001 mg/L.
8.2. La posible presencia de materia orgánica constituye un interferente, que
se remueve por digestión de la muestra.
Interferencias:
8.3. No se puede establecer un mecanismo único de eliminación de
interferentes, no obstante se recomienda aplicar los siguientes criterios:
8.3.1. Identificar la naturaleza de la interferencia

222
8.3.2. La posible presencia de materia orgánica constituye un
interferente, que se remueve por digestión de la muestra.
8.3.3. Si la interferencia es de carácter físico y se manifiesta en el
transporte del hidruro, revisar mangueras a fin de evitar fugas,
reacciones de hidrólisis o cualquier efecto que pudiese producirse
debido a la naturaleza del medio de transporte.
8.3.4. Si la interferencia es de carácter físico y se manifiesta durante la
atomización, verificar la limpieza de la celda de cuarzo. Verificar los
niveles de los otros elementos susceptibles de generar hidruros
volátiles y realizar el análisis con adición estándar.
9. REFERENCIAS
9.1. Standard Methods For The Examination Of Water And Wastewater.
APHA. AWWA. WEF. 21th Edition 2005, Part. 3114-B; Arsenic and
Selenium by Hydride Generation/Atomic Absorption Spectrometry.
9.2. Taller Regional Para La Elaboración del Manual de protocolos
Armonizados y Evaluados Para La Toma de Muestras y Análisis de
Aguas y Sedimentos. ARCAL RLA /1/10. El Salvador, 5 – 9 de Mayo del
2008.

223
2.3.1.30. Determinación de Mercurio total por el mé todo de
Espectrometría de Absorción Atómica con Generación de
Vapor Atómico de Mercurio (Vapor Frío)
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agu a que están contaminadas con metales Código del procedimiento:
PT-MA-10
Nombre del procedimiento: Determinación de Mercurio total por el método de
Espectrometría de Absorción Atómica con Generación de Vapor Atómico de Mercurio (Vapor Frío)
Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determinar mercurio total mediante espectrometría de absorción atómica con
generación de Vapor Atómico de Hg (Vapor Frio).
2. ALCANCE
Este método de ensayo es aplicable a cualquier agua superficial y
corresponde a la concentración de mercurio determinada en una muestra no
filtrada.
3. PRINCIPIO DEL METODO
El mercurio presente en una fracción de muestra previamente digerida, es
reducido a vapor atómico de mercurio, mediante reacción con cloruro estanoso
en medio ácido, o aplicando el procedimiento de generación continua usando
solución de boro hidruro de sodio como agente reductor.
El vapor atómico generado es conducido por un gas de arrastre hacia una celda
de cuarzo posicionada en el paso óptico de un espectrofotómetro de absorción
atómica. La celda se encuentra a temperatura ambiente, la relación entre
potencia incidente y potencia transmitida es una medida de la concentración del
elemento en la muestra.
4. EQUIPOS
4.1. Balanza analítica, de sensibilidad igual o mayor a 0,1mg.

224
4.2. Campana, con sistema de extracción de gases.
4.3. Espectrómetro de absorción atómica, provisto de generador de vapor frio,
celdas con ventanas de cuarzo y accesorios necesarios.
4.4. Fuente de ignición, de líneas atómicas de mercurio.
4.5. Sistema Generador de Vapor Frío.
4.6. Plancha calefactora o equivalente, con regulación de temperatura o baño
termoregulado.
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Material de uso habitual de laboratorio [ej. vasos de precipitado,
matraces, pipetas, y otros].
5.1.2. Recipiente de reacción, matraz erlenmeyer de 250ml, incorporando
tubo de aireación.
5.2. REACTIVOS
5.2.1. Durante el análisis se deben utilizar solamente reactivos de calidad
para análisis (p.a.), y con agua para análisis grado reactivo.
5.2.2. Solución de ácido clorhídrico concentrado, calidad ultrapuro.
5.2.3. Solución de ácido nítrico concentrado, calidad ultrapuro.
5.2.4. Estándar comercial de Hg de 1000 mg/L
5.2.5. Argón o nitrógeno
5.2.6. Acetileno
5.2.7. Borohidruro de sodio
5.2.8. Hidróxido de sodio
5.2.9. Soluciones estándares de calibración: preparar una serie de
soluciones de calibración en el rango óptimo de trabajo, a partir de
diluciones apropiadas del estándar de 1000 mg Hg/L 10% v/v HCl.
Preparar diariamente. Se exige para la curva de calibración un r ≥
0.995.
5.2.10. Solución blanco término cero: la solución término cero debe
ser preparada con agua para análisis grado reactivo exenta de
mercurio y en un medio 10 % HCl v/v. Preparar diariamente.

225
5.2.11. Como agente reductor se utiliza una solución acuosa de
NaBH4 al 0.2% en NaOH al 0.05%, debiéndose preparar cada vez
que es utilizado.
5.2.12. Se pesan 0.5 gr de NaOH disolviéndolo en agua Mili Q para
luego añadir 2 gr de NaBH4, posteriormente se filtra y se afora a 1 L.
6. ACCIONES PREVIAS
6.1. Para la toma, transporte y almacenamiento de las muestras se
recomienda utilizar recipientes de vidrio en lugar de plástico si la muestra
se almacenará por más de 15 días.
6.2. En vista que el mercurio se puede perder rápidamente desde las
muestras, se recomienda utilizar HNO3 como preservante y así mantener
el pH < 2,0.
Ensayo de las muestras:
6.3. Las muestras que contienen material particulado o materia orgánica,
generalmente requieren pretratamiento antes del análisis
espectroscópico. Estas se deben someter a un pretratamiento previo de
digestión para liberar todo el mercurio presente que puede estar
formando parte de la materia suspendida o coloidal. Para muestras de
agua potable, transparentes, incoloras e inodoras, con turbiedad < 1
UNT, puede obviarse el pretratamiento de digestión para analizar por
FAAS con lectura directa. Para esta condición, luego que la muestra se
recolecta, se acidifica a pH<2 con HNO3 65% m/v [1,5ml HNO3/L es
usualmente adecuado para agua potable] y se analiza sin necesidad de
digestión. Antes de aplicar esa modalidad como rutina, y sobre todo en
muestras sin historial, es necesario analizarlas con o sin digestión, de
manera que haya evidencia que los resultados obtenidos sean
comparables. El procedimiento analítico para la modalidad sin digestión
solo permite obviar esta operación, por tanto, deberá considerarse cada
uno de los pasos descritos en la preparación de las muestras para la
lectura final. La aplicación de esta variante metodológica está supeditada

226
a lograr, bajo las condiciones operativas y disposición de instrumental
adecuado, llegar a los niveles exigidos de LDM para este parámetro.
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Encender el computador, el espectrómetro de absorción atómica y el
sistema de análisis de inyección de flujo FIAS, de acuerdo a las
referencias descritas en el manual de operaciones de su equipo.
7.2. Alinear la posición de la celda de cuarzo y optimizar la intensidad de la
señal ajustando la posición de la lámpara y la longitud de onda (253.7
nm).
7.3. Abrir el programa que pone a disposición el equipo utiizado, encender la
lámpara de mercurio y dejar estabilizar por un tiempo aproximado de 30
minutos para aumentar su energía, encender luego la celda (100ºC).
7.4. Encender el extractor de aire.
7.5. Ajustar las mangueras y dejar por un tiempo prudente el funcionamiento
de las bombas para el lavado con agua de calidad para análisis.
7.6. Llenar los contenedores con la solución de ácido clorhídrico al 10% y
solución de boro hidruro (0.2% en 0.05% de NaOH).
7.7. Ajustar la presión del argón entre 320 y 400 kpa.
7.8. Ajustar la velocidad de flujo sobre 100 ml/min
7.9. Calibrar el equipo con las soluciones estándares de concentración de 10
y 20 µg/L de mercurio. Construir la curva de calibración empleando un
numero adecuado de estándares, como mínimo blanco y tres
concentraciones del rango de trabajo, recomendándose 5 puntos.
7.10. Una vez calibrado el instrumento, determinar la concentración de
muestras de acuerdo al siguiente protocolo:
7.10.1. Aspirar el blanco y realizar auto –cero
7.10.2. Aspirar las muestras en estudio, en secuencia de 6 en 6,
verificando la calibración y la estabilidad del cero, entre lotes de
muestras. Repetir las lecturas en caso que sea necesario.

227
7.10.3. Aspirar agua para análisis grado reactivo exenta de Hg entre
muestra y muestra con el propósito de evitar contaminación cruzada.
7.10.4. Para el control de calidad analítica por cada set de análisis
llevar en forma paralela a las muestras, los siguientes controles:
7.10.4.1. Un blanco reactivo preparado a partir de agua para análisis
grado reactivo exenta de Hg.
7.10.4.2. Un ensayo duplicado de una muestra elegida al azar
(precisión mínima 80%; RSD≤ 20%).
7.10.4.3. Un ensayo de un estándar de concentración conocida,
cercana al valor medido (exactitud mínima 85-115% como
recuperación del estándar).
7.11. Interferencias
No se puede establecer un mecanismo único de eliminación de
interferentes, no obstante se recomienda aplicar los siguientes
criterios:
7.11.1. Identificar la naturaleza de la interferencia.
7.11.2. La posible presencia de materia orgánica constituye un
interferente, que se remueve por digestión de la muestra.
7.11.3. Si la interferencia es de carácter físico y se manifiesta en el
transporte del vapor atómico de mercurio, revisar mangueras a fin de
evitar fugas, reacciones de hidrólisis o cualquier efecto que pudiese
producirse debido a la naturaleza del medio de transporte.
7.11.4. Si la interferencia es de carácter físico y se manifiesta durante la
atomización, verificar la limpieza de la celda de cuarzo. Si existe
interferencia de matriz, realizar el análisis con adición estándar.
8. CALCULOS Y EXPRESION DE RESULTADOS
mg Hg/L = (Lect - Bco) x D
donde:

228
mg Hg/L = concentración de Hg, expresada en mg de Hg por litro de
agua.
Lect = lectura de la muestra en mg/L.
Bco = lectura blanco reactivo sometido al mismo proceso de las muestras.
D = factor de dilución o concentración
8.1. Si la muestra no acusa presencia de Hg, se debe informar como el LDM
<0,001 mg/L.
8.2. La posible presencia de materia orgánica constituye un interferente, que
se remueve por digestión de la muestra.
9. REFERENCIAS
9.1. Standard Methods for the Examination of Water and Wastewater. APHA.
AWWA. WEF. 21TH Edition 2005, Part 3000. Cold-Vapor Atomic
Absorption Spectrometry Method 3112 B.
9.2. Taller Regional Para La Elaboración del Manual de protocolos
Armonizados y Evaluados Para La Toma de Muestras y Análisis de
Aguas y Sedimentos. ARCAL RLA /1/10. El Salvador, 5 – 9 de Mayo del
2008.

229
2.3.1.31. Pretratamiento y Preparación de Muestras para análisis de Sedimentos
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales
Código del procedimiento: PT-PS-01
Nombre del procedimiento: Pretratamiento y Preparación de Muestras para análisis de Sedimentos
Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Pretratamiento y preparación de las muestras de sedimentos para el análisis
químico.
2. ALCANCE
Se aplica a todas las muestras de sedimentos que serán analizados y
comprende todas las etapas desde la recepción de las muestras hasta su
digestión ácida, sin incluir esta etapa.
3. PRINCIPIO DEL METODO
Comprende las etapas de secado y preparación física de las muestras de
sedimentos.
4. EQUIPOS
4.1. Balanza de aproximadamente 200 kg de capacidad.
4.2. Estufa de secado de muestras con convección de aire.
5. REACTIVOS Y MATERIALES
6. ACCIONES PREVIAS
6.1. Todas las muestras se deben haber colectado de acuerdo al
procedimiento de muestreo del presente manual.
7. DESCRIPCION DEL PROCEDIMIENTO
Secado de muestras
7.1. Las técnicas de secado deben ser apropiadas para todos los análisis
subsiguientes que se realizarán.

230
7.2. Es importante la etapa de secado porque las concentraciones de los
contaminantes se informan sobre base seca.
7.3. La etapa de secado proporcionará una manera de calcular el contenido
de agua por pesada de la muestra antes y después del secado.
7.4. Esta etapa de secado se hará en estufa provista de un sistema interior
que minimice la contaminación de las muestras durante este proceso.
7.5. La temperatura de secado se fijará en 50 °C y esta etapa finalizará
cuando el peso se mantenga constante.
Fraccionamiento de muestras
7.6. Los sedimentos constituyen una mezcla de partículas de diferente
tamaño. Por tal motivo, es crítico poder maximizar la homogeneidad de
una muestra seleccionada para el análisis.
7.7. Se debe comenzar con una muestra representativa que sea lo
suficientemente grande para poder incorporar todo el ámbito de tamaños
de partículas presentes en el punto de muestreo.
7.8. Esto es importante ya que el proceso de homogeneización está enfocada
en una mezcla exhaustiva de la muestra. Toda la etapa de mezclado
debe realizarse en contenedores especiales y con herramientas no
contaminantes. Se puede emplear, si se dispone de ello, de
mezcladores comerciales para tamaño de muestras muy grande para
ser manipuladas manualmente.
7.9. A continuación, se procede a realizar el submuestreo de las muestras.
7.10. Para muestras secas, se emplea la técnica de cuarteo que produce
submuestras de menor volumen a partir de un volumen inicial grande.
7.11. La muestra se apila en forma de cono y se va separando en cuartos.
Se separan dos cuartos y el resto se recombina, se vuelve a formar la
pila y se cuartea nuevamente. Se repite este procedimiento hasta lograr
una masa adecuada de submuestra.
Preparación de las muestras

231
Este proceso es de gran importancia para lograr resultados
experimentales bien representativos.
El objetivo es preparar una muestra de tal manera que se preserve la
distribución original de los analitos al momento del muestreo y que se
prevenga la introducción de elementos extraños y que puedan contaminar
las muestras.
Una homogeneización adecuada implica que todas las partículas
minerales presentes en el sedimento, estén igualmente representadas en
la submuestras.
De acuerdo a la masa de submuestra requerida para el análisis, se
obtiene un tamaño de partícula adecuado para el trabajo.
Generalmente se puede trabajar con tamaños de partícula menores que
malla #150 o menores que 0,10 mm.
Este proceso de preparación requiere de equipos provistos con elementos
de molienda que no aporten ningún tipo de contaminación posterior.
Hay que realizar al finalizar este procedimiento un test de homogeneidad
para garantizar que toda la submuestra que llega al laboratorio químico
sea homogénea.
Preparación de las muestras
7.12. Este proceso es de gran importancia para lograr resultados
experimentales bien representativos.
7.13. El objetivo es preparar una muestra de tal manera que se preserve la
distribución original de los analitos al momento del muestreo y que se
prevenga la introducción de elementos extraños y que puedan
contaminar las muestras.
7.14. Una homogeneización adecuada implica que todas las partículas
minerales presentes en el sedimento, estén igualmente representadas
en la submuestras. De acuerdo a la masa de submuestra requerida para
el análisis, se obtiene un tamaño de partícula adecuado para el trabajo.
7.15. Generalmente se puede trabajar con tamaños de partícula menores
que malla #150 o menores que 0,10 mm. Este proceso de preparación

232
requiere de equipos provistos con elementos de molienda que no
aporten ningún tipo de contaminación posterior.
7.16. Hay que realizar al finalizar este procedimiento un test de
homogeneidad para garantizar que toda la submuestra que llega al
laboratorio químico sea homogénea.
8. CALCULOS Y EXPRESION DE RESULTADOS
9. REFERENCIAS
IAEA-TECDOC-1360

233
2.3.1.32. Digestión ácida total de sedimentos utili zando microondas
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales Código del procedimiento:
PT-DS-01
Nombre del procedimiento: Digestión ácida total de sedimentos utilizando microondas
Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Es la digestión ácida de sedimentos con horno de microondas para análisis
de metales por técnicas espectroscópicas.
2. ALCANCE
Este método se aplica a la digestión ácida asistida con horno de microondas
de muestras de sedimentos para los siguientes analitos:
Arsénico – Cromo – Cadmio – Cobre – Plomo – Mercurio
3. PRINCIPIO DEL METODO
Este método se basa en la descomposición ácida de muestras de sedimento
con horno de microondas y se considera un método muy rápido y
multielemental. Los digestos producidos por este método son adecuados
para analizar elementos por espectrometría de absorción atómica por llama
(FAAS), por espectrometría de absorción atómica por vapor frío (CVAAS),
por espectrometría de absorción atómica por horno de grafito (GFAAS) y por
espectrometría de emisión atómica por excitación por plasma inductivo (ICP-
OES). Permite valores altos de recuperación para los analitos citados.
4. EQUIPOS
4.1. Balanza analítica de 160 g de capacidad con precisión de 0,0001 g.
4.2. Horno de microondas con tubos de Teflón con sensores de temperatura
con exactitud de ± 2°C a la temperatura final de 180 °C.
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Pipetas graduadas de tamaños adecuados.

234
5.1.2. Matraces aforados de polipropileno de 50 mL.
5.1.3. Papel de filtro cualitativo o equivalente.
5.1.4. Embudos de polipropileno.
5.2. REACTIVOS
Se deben emplear reactivos de grado analítico en todos los ensayos. A
menos que se especifique lo contrario, se debe lograr que todos los
reactivos cumplan con lo dispuesto en las especificaciones del Comité de
Reactivos Analíticos de la ACS. Se pueden usar otros grados de reactivos
en la medida que sean de suficiente pureza para permitir su uso sin
perder la exactitud de las determinaciones.
5.2.3. Agua grado reactivo.
5.2.4. Solución de ácido nítrico concentrado. Se debe analizar el ácido
para determinar el grado de pureza. Si el valor del blanco de
reactivos es menor al límite de detección del método, puede usarse
el ácido correspondiente.
5.2.5. Solución de ácido fluorhídrico concentrado. Se debe analizar el
ácido para determinar el grado de pureza. Si el valor del blanco de
reactivos es menor al límite de detección del método, puede usarse
el ácido correspondiente.
5.2.6. Peróxido de hidrógeno. Se debe analizar para determinar el grado
de pureza. Si el valor del blanco de reactivos es menor al límite de
detección del método, puede usarse el ácido correspondiente.
6. ACCIONES PREVIAS
6.1. Todas las muestras se deben haber colectado de acuerdo al
procedimiento de muestreo del presente manual.
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Pesar alrededor de 0,5000 g de muestra con aproximación de 0,0001 g y
transferir al recipiente de Teflón del horno de microondas.
7.2. Agregar 9,0 mL de solución de ácido nítrico concentrado y 3,0 mL de
solución de ácido fluorhídrico concentrado. Esta operación debe hacerse
bajo campana de extracción de humos.

235
Nota: se puede agregar hasta 0,5 mL de peróxido de hidrógeno antes de
colocar el tubo en el horno a microondas, tomando la precaución de
adicionarlo en porciones pequeñas, ya que pueden producirse reacciones
violentas.
7.3. Cerrar herméticamente el vaso y colocarlo en el horno a microondas, de
acuerdo a los manuales de cada instrumento.
7.4. La temperatura de la muestra debería alcanzar los 180 °C ± 5°C en
aproximadamente 5 minutos y permanecer a esa temperatura durante 10
minutos.
7.5. Al finalizar el ciclo del horno, dejar enfriar los recipientes durante 5
minutos antes de retirarlos del horno. Verificar que no haya ocurrido
pérdidas de peso durante el proceso, considerando aceptable una
pérdida de hasta el 1 % del peso de la muestra original más los reactivos.
7.6. Si el digesto presenta partículas que pueden interferir con las
determinaciones analíticas, se pueden centrifugar o filtrar.
7.7. Diluir a volumen en un matraz de polipropileno de 50 mL usando solución
de ácido nítrico 0,28 M.
8. CALCULOS Y EXPRESION DE RESULTADOS
9. REFERENCIAS
Norma EPA 3052.

236
2.3.1.33. Digestión ácida total de sedimentos en si stema abierto
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales
Código del procedimiento: PT-DS-02
Nombre del procedimiento: Digestión ácida total de sedimentos en sistema abierto
Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Es la digestión ácida de sedimentos a sistema abierto para análisis de
metales por ICP-OES.
2. ALCANCE
Este método se aplica a la digestión ácida total de muestras de sedimentos
para los siguientes analitos a ser determinados por ICP-OES:
Arsénico – Cromo – Cadmio – Cobre – Plomo
3. PRINCIPIO DEL METODO
Este método se basa en la descomposición total de la muestra con una
combinación de ácidos que permite valores altos de recuperación para los
analitos citados.
4. EQUIPOS
4.1. Balanza analítica de 160 g de capacidad con precisión de 0,0001 g.
4.2. Plancha calefactora o equivalente, ajustable y con posibilidad de
termostatizar.
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Vasos de precipitación de Teflón adecuados o equivalentes.
5.1.2. Tapa de vasos de teflón o equivalentes.
5.1.3. Membranas filtrantes de acetato de celulosa de 0,45 µm de tamaño
de poro.
5.1.4. Pipetas graduadas de tamaños adecuados.
5.1.5. Matraces aforados de polipropileno de 50 mL.
5.2. REACTIVOS

237
Se deben emplear reactivos de grado analítico en todos los ensayos. A
menos que se especifique lo contrario, se debe lograr que todos los
reactivos cumplan con lo dispuesto en las especificaciones del Comité de
Reactivos Analíticos de la ACS. Se pueden usar otros grados de reactivos
en la medida que sean de suficiente pureza para permitir su uso sin
perder la exactitud de las determinaciones.
5.2.1. Agua grado reactivo.
5.2.2. Solución de ácido nítrico concentrado. Se debe analizar el ácido
para determinar el grado de pureza. Si el valor del blanco de
reactivos es menor al límite de detección del método, puede usarse
el ácido correspondiente.
5.2.3. Solución de ácido clorhídrico concentrado. Se debe analizar el
ácido para determinar el grado de pureza. Si el valor del blanco de
reactivos es menor al límite de detección del método, puede usarse
el ácido correspondiente.
5.2.4. Solución de ácido perclórico concentrado. Se debe analizar el
ácido para determinar el grado de pureza. Si el valor del blanco de
reactivos es menor al límite de detección del método, puede usarse
el ácido correspondiente.
5.2.5. Solución de ácido fluorhídrico concentrado. Se debe analizar el
ácido para determinar el grado de pureza. Si el valor del blanco de
reactivos es menor al límite de detección del método, puede usarse
el ácido correspondiente.
6. ACCIONES PREVIAS
6.1. Todas las muestras se deben haber colectado de acuerdo al
procedimiento de muestreo del presente manual.
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Pesar alrededor de 0,5000 g de muestra con aproximación de 0,0001 g y
transferir al recipiente de Teflón del horno de microondas.
7.2. Agregar 10 mL de solución de ácido nítrico concentrado, cubrir el vaso
con la cubierta adecuada y calentar la solución a 70°C durante 3 horas.

238
7.3. Agregar 5 mL de solución de ácido perclórico concentrado y calentar la
muestra a 90°C durante 15 horas.
7.4. Agregar 5 mL de ácido nítrico concentrado, 5 mL de solución de ácido
perclórico concentrado y 5 mL de solución de ácido fluorhídrico
concentrado y calentar a 70 °C para llegar casi a s equedad.
7.5. Disolver el residuo con 5 mL de solución de ácido clorhídrico concentrado
y 5 mL de solución de ácido fluorhídrico concentrado y calentar a 70 °C
durante 15 horas.
7.6. Llevar la solución casi a sequedad 3 veces y redisolver cada vez con 10
mL de solución de ácido nítrico 0,28 M.
7.7. Una vez terminada esta etapa, transferir a un matraz de polipropileno de
50 mL y diluir con solución de ácido nítrico 0,28 M.
8. CALCULOS Y EXPRESION DE RESULTADOS
9. REFERENCIAS
Goossens, Jan; Moens, Luc; Dams, Richard: Inductively coupled plasma
mass spectrometric determination of heavy metals in soils and sludge
candidate reference materials; Analytica Chimica Acta 304 (1995). 307-315.

239
2.3.1.34. Digestión ácida total de sólidos en suspe nsión
PROYECTO ARCAL RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales Código del procedimiento: PT-DS-03
Nombre del procedimiento: Digestión ácida total de sólidos en suspensión.
Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Es la digestión ácida de sólidos en suspensión para análisis de metales por
ICP-OES.
2. ALCANCE
Este método se aplica a la digestión ácida total de de sólidos en suspensión
para los siguientes analitos a ser determinados por ICP-OES:
Arsénico – Cromo – Cadmio – Cobre – Plomo
3. PRINCIPIO DEL METODO
Está basado en la descomposición total de la muestra con una combinación
de ácidos que permite valores altos de recuperación para los analitos citados.
Es aplicable a metales en suspensión.
Metales en suspensión: son los metales retenidos por un filtro de membrana
de 0,45 µm de poro. Se debe filtrar la muestra a través del filtro en el
momento de la colección de la muestra, separar la fracción líquida y
preservar los filtros con los sólidos en suspensión.
4. EQUIPOS
4.1. Balanza analítica de 160 g de capacidad con precisión de 0,0001 g.
4.2. Plancha calefactora o equivalente, ajustable y con posibilidad de
termostatizar.
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Vasos de precipitación de Teflón adecuados o equivalentes.
5.1.2. Vidrios de reloj o equivalentes.

240
5.1.3. Membranas filtrantes de acetato de celulosa de 0,45 µm de tamaño
de poro.
5.1.4. Papel de filtro y embudos de filtración.
5.1.5. Probetas graduadas o equivalentes.
5.2. REACTIVOS
Se deben emplear reactivos de grado analítico en todos los ensayos. A
menos que se especifique lo contrario, se debe lograr que todos los
reactivos cumplan con lo dispuesto en las especificaciones del Comité de
Reactivos Analíticos de la ACS. Se pueden usar otros grados de reactivos
en la medida que sean de suficiente pureza para permitir su uso sin
perder la exactitud de las determinaciones.
5.2.1. Agua grado reactivo.
5.2.2. Solución de ácido nítrico concentrado. Se debe analizar el ácido
para determinar el grado de pureza. Si el valor del blanco de
reactivos es menor al límite de detección del método, puede usarse
el ácido correspondiente.
5.2.3. Solución de ácido clorhídrico concentrado. Se debe analizar el
ácido para determinar el grado de pureza. Si el valor del blanco de
reactivos es menor al límite de detección del método, puede usarse
el ácido correspondiente.
6. ACCIONES PREVIAS
6.1. Todas las muestras se deben haber colectado de acuerdo al
procedimiento de muestreo del presente manual.
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Transferir el filtro de membrana que contiene el material insoluble a un
vaso de precipitación de 150 mL y agregar 4 mL de solución de ácido
nítrico concentrado.
7.2. Cubrir el vaso con un vidrio de reloj y calentar suavemente sobre plancha
calefactora. El ácido disolverá la membrana muy rápidamente.
7.3. Aumentar la temperatura de la plancha y digerir el material particulado.

241
7.4. Cuando el ácido se haya evaporado, enfriar el vaso y agregar 3 mL
adicionales de solución de ácido nítrico. Cubrir y seguir el calentamiento
hasta que la digestión sea completa, lo que generalmente se indica por
un color muy débil del digesto.
7.5. Evaporar casi a sequedad (2 mL), enfriar, agregar 10 mL de solución de
ácido clorhídrico (1+1) y 15 mL de agua grado reactivo y calentar
suavemente durante 15 minutos para disolver cualquier precipitado o
material insoluble.
7.6. Dejar enfriar, lavar bien las paredes del vaso y el vidrio de reloj con agua
grado reactivo y filtrar la muestra para remover cualquier partícula que
puede obturar el nebulizador.
7.7. Ajustar el volumen a 50 mL con agua grado reactivo.
La muestra está preparada para su posterior análisis.
8. CALCULOS Y EXPRESION DE RESULTADOS
9. REFERENCIAS
EPA 200.7
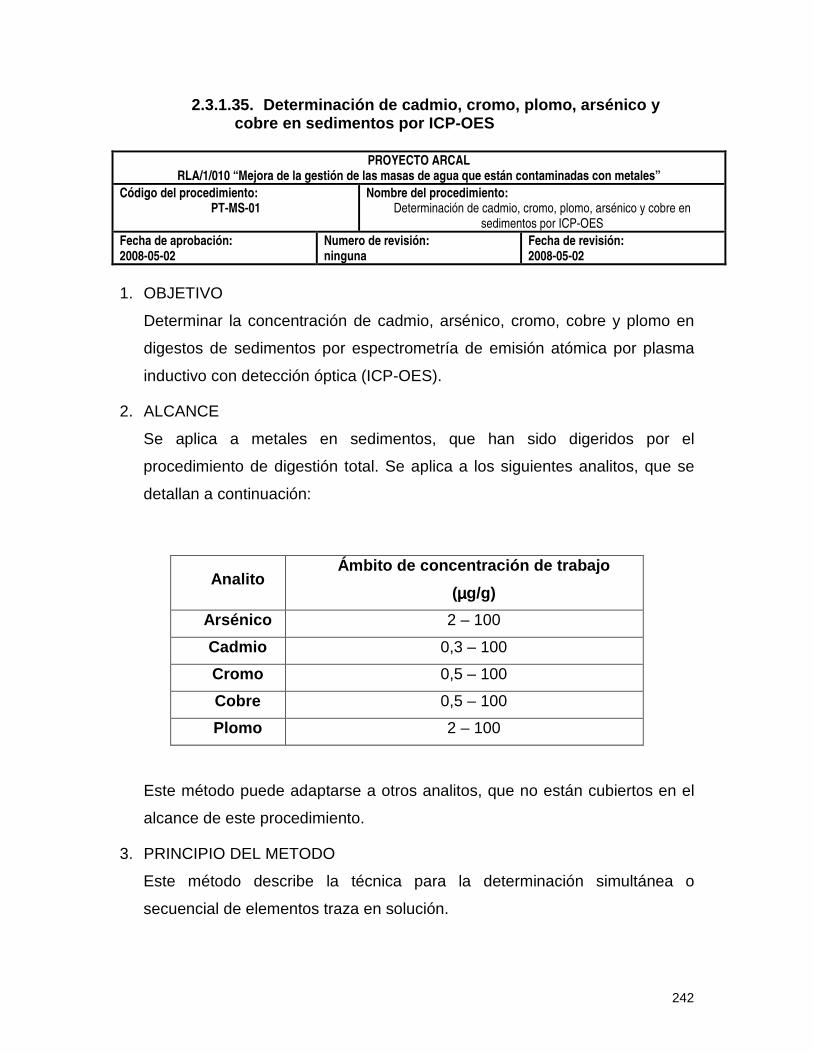
242
2.3.1.35. Determinación de cadmio, cromo, plomo, ar sénico y cobre en sedimentos por ICP-OES
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales” Código del procedimiento:
PT-MS-01
Nombre del procedimiento: Determinación de cadmio, cromo, plomo, arsénico y cobre en
sedimentos por ICP-OES
Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determinar la concentración de cadmio, arsénico, cromo, cobre y plomo en
digestos de sedimentos por espectrometría de emisión atómica por plasma
inductivo con detección óptica (ICP-OES).
2. ALCANCE
Se aplica a metales en sedimentos, que han sido digeridos por el
procedimiento de digestión total. Se aplica a los siguientes analitos, que se
detallan a continuación:
Analito Ámbito de concentración de trabajo
(µµµµg/g)
Arsénico 2 – 100
Cadmio 0,3 – 100
Cromo 0,5 – 100
Cobre 0,5 – 100
Plomo 2 – 100
Este método puede adaptarse a otros analitos, que no están cubiertos en el
alcance de este procedimiento.
3. PRINCIPIO DEL METODO
Este método describe la técnica para la determinación simultánea o
secuencial de elementos traza en solución.

243
La base es la medida de la emisión atómica por una técnica espectroscópica
óptica.
Se nebulizan los digestos y se transporta el aerosol producido a la antorcha
del plasma donde ocurre la excitación.
Se debe emplear una técnica de corrección de fondo para compensar la
contribución del fondo variable para la determinación de elementos traza.
Esta contribución de fondo debe medirse en forma adyacente a las líneas del
analito durante el análisis.
La posición que se selecciona para la medición del fondo, a ambos lados de
la línea analítica, se determina por la complejidad del espectro adyacente a la
línea del analito.
La posición debe estar libre de interferencias espectrales y debe reflejar el
mismo cambio en la intensidad de fondo que ocurre a la longitud de onda de
medición.
Las interferencias pueden caracterizarse en dos tipos; interferencias
espectrales e interferencias no espectrales. (ver Procedimiento PT-MA-01.)
4. EQUIPOS
4.1. Espectrómetro de emisión atómica por plasma inductivo.
4.2. Equipo refrigerador para el espectrómetro.
4.3. Equipo purificador de agua.
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Matraces aforados, clase A, de 50, 100 y 1000 mL.
5.1.2. Pipetas automáticas de punta descartable, de volumen variable
entre 10 y 100 µL, de volumen variable entre 100 y 1000 µL, de
volumen variable entre 50 y 250 µL y de volumen variable entre 1000
y 5000 µL.
5.1.3. Botellas de plástico de 125 mL.
5.1.4. Jeringas de filtración.
5.1.5. Filtros de acetato de celulosa de 0,45 µm para usar con jeringas.

244
5.1.6. Tubos descartables de plástico de 15 y 50 mL.
5.2. REACTIVOS
5.2.1. Agua grado reactivo.
5.2.2. Solución estándar de arsénico de 1000 mg/L, calidad ultrapura.
5.2.3. Solución estándar de cadmio de 1000 mg/L, calidad ultrapura.
5.2.4. Solución estándar de cromo de 1000 mg/L, calidad ultrapura.
5.2.5. Solución estándar de cobre de 1000 mg/L, calidad ultrapura.
5.2.6. Solución estándar de plomo de 1000 mg/L, calidad ultrapura.
5.2.7. Solución de ácido nítrico concentrado, calidad ultrapuro.
5.2.8. Solución de ácido nítrico (0,2 % V/V). Preparar con 2 mL de
solución de ácido nítrico concentrado (5.2.7.) diluidos a 1000 mL con
agua grado reactivo (5.2.1.).
5.2.9. Argón, calidad UAP, (ultra alta pureza).
5.2.10. Nitrógeno, calidad UAP, (ultra alta pureza).
5.2.11. Soluciones estándar de control de procedimiento: material
certificado. Provisto por una empresa acreditada. Posee fecha de
vencimiento.
5.2.12. Materiales de referencia certificados, provistos por el National
Water Research Institute (NWRI), el NIST, o de calidad similar. Estas
muestras sólidas se deben digerir de acuerdo al procedimiento para
digestión de muestras de sedimentos. Se sugiere digerir tres
muestras de sedimentos certificados para poder validar la
metodología analítica.
Soluciones estándar mixtas
Se utilizan las siguientes combinaciones recomendadas por la EPA
5.2.13. Solución estándar mixta I: cadmio y plomo.
5.2.14. Solución estándar mixta II: cobre.
5.2.15. Solución estándar mixta III: arsénico.
5.2.16. Solución estándar mixta IV: cromo.
5.2.17. Solución estándar mixta I de 50 mg/L: Pipetear 5,00 mL de cada
una de las siguientes soluciones estándar de 1000 mg/L: cadmio y

245
plomo en un matraz aforado de 100 mL, y diluir hasta el aforo con
solución de ácido nítrico 0,2 % V/V. Almacenar en botella de plástico
de 125 mL. Preparar mensualmente.
5.2.18. Solución estándar mixta II de 50 mg/L: Pipetear 5,00 mL de cada
una de las siguientes soluciones estándar de 1000 mg/L: cobre en un
matraz aforado de 100 mL, y diluir hasta el aforo con solución de
ácido nítrico 0,2 % V/V. Almacenar en botella de plástico de 125 mL.
Preparar mensualmente.
5.2.19. Solución estándar mixta III de 50 mg/L: Pipetear 5,00 mL de cada
una de las siguientes soluciones estándar de 1000 mg/L: arsénico en
un matraz aforado de 100 mL, y diluir hasta el aforo con solución de
ácido nítrico 0,2 % V/V. Almacenar en botella de plástico de 125 mL.
Preparar mensualmente.
5.2.20. Solución estándar mixta II de 50 mg/L: Pipetear 5,00 mL de cada
una de las siguientes soluciones estándar de 1000 mg/L: cromo en
un matraz aforado de 100 mL, y diluir hasta el aforo con solución de
ácido nítrico 0,2 % V/V. Almacenar en botella de plástico de 125 mL.
Preparar mensualmente.
Soluciones estándar de calibración
5.2.21. Estándar de calibración de 0,03 mg/L: Pipetear 30 µL de cada
una de las cuatro soluciones estándares mixtas de 50 mg/L en un
matraz aforado de 50 mL, y diluir hasta el aforo con solución de ácido
nítrico 0,2 % V/V. Almacenar en tubo de polipropileno de 50 mL.
Preparar mensualmente.
5.2.22. Estándar de calibración de 0,10 mg/L: Pipetear 100 µL de cada
una de las cuatro soluciones estándares mixtas de 50 mg/L en un
matraz aforado de 50 mL, y diluir hasta el aforo con solución de ácido
nítrico 0,2 % V/V. Almacenar en tubo de polipropileno de 50 mL.
Preparar mensualmente.
5.2.23. Estándar de calibración de 0,20 mg/L: Pipetear 200 µL de cada
una de las cuatro soluciones estándares mixtas de 50 mg/L en un

246
matraz aforado de 50 mL, y diluir hasta el aforo con solución de ácido
nítrico 0,2 % V/V. Almacenar en tubo de polipropileno de 50 mL.
Preparar mensualmente.
5.2.24. Estándar de calibración de 0,40 mg/L: Pipetear 400 µL de cada
una de las cuatro soluciones estándares mixtas de 50 mg/L en un
matraz aforado de 50 mL, y diluir hasta el aforo con solución de ácido
nítrico 0,2 % V/V. Almacenar en tubo de polipropileno de 50 mL.
Preparar mensualmente.
5.2.25. Estándar de calibración de 0,70 mg/L: Pipetear 700 µL de cada
una de las cuatro soluciones estándares mixtas de 50 mg/L en un
matraz aforado de 50 mL, y diluir hasta el aforo con solución de ácido
nítrico 0,2 % V/V. Almacenar en tubo de polipropileno de 50 mL.
Preparar mensualmente.
5.2.26. Estándar de calibración de 1,00 mg/L: Pipetear 1000 µL de cada
una de las cuatro soluciones estándares mixtas de 50 mg/L en un
matraz aforado de 50 mL, y diluir hasta el aforo con solución de ácido
nítrico 0,2 % V/V. Almacenar en tubo de polipropileno de 50 mL.
Preparar mensualmente.
Blanco de calibración
5.2.27. Solución 5.2.8.
Blanco de filtro
5.2.28. Es agua grado reactivo que se hace pasar por el filtro de acetato
de celulosa. Usar sólo si se necesitó el uso de los filtros para la
muestra.
6. ACCIONES PREVIAS
6.1. Las muestras deben estar libres de sólidos en suspensión previo a la
aspiración.
6.2. Las muestras deben tratarse antes del análisis, si se sospecha la
presencia de sólidos en suspensión o sedimentados.
6.3. Las muestras se filtran por medio de filtros con jeringa.

247
6.4. Las muestras deben conservarse a bajas temperaturas (4° C).
6.5. Las soluciones estándar de control de performance se utilizan tal cuál las
entrega el proveedor.
7. DESCRIPCION DEL PROCEDIMIENTO
Operación del equipo
7.1. Preparar el equipo de ICP de acuerdo a las instrucciones que figuran
en el manual de instrucciones del instrumento.
7.2. La siguiente tabla describe los parámetros operativos para cada uno
de los analitos.
Analito Longitud de onda
(nm)
Arsénico 188,98
Cadmio 228,80
Cromo 267,72
Cobre 324,75
Plomo 220,35
Estas longitudes de onda son recomendadas por su sensibilidad y
aceptación. Se pueden sustituir por otras longitudes de onda, si se
considera que hay interferencias espectrales.
7.3. Antes de comenzar el uso del instrumento, proceder a estabilizar el
plasma durante 20 minutos.
7.4. Llevar a cabo una alineación óptica empleando la lámpara de Hg
provista con el equipo.
7.5. Verificar la alineación de la antorcha del plasma y de la ranura de
entrada del espectrómetro, si se llevó a cabo un mantenimiento del
sistema de introducción de las muestras.
7.6. En el muestreador automático, se colocan los tubos de plástico de 50
mL, conteniendo el blanco de calibración y las soluciones estándares

248
de calibración, comenzando por la solución más diluida. Se programa
el equipo y se lleva a cabo la calibración correspondiente.
7.7. Purgar la cañería con cada estándar o blanco a una velocidad de 4
mL/min durante un mínimo de 15 segundos. Luego dejar estabilizar
durante otro mínimo de 15 segundos.
7.8. Enjuagar con agua reactivo durante 30 segundos antes de cada
estándar para minimizar cualquier efecto de memoria del estándar
previo. Emplear tres integraciones de los estándares o muestras para
reducir el error aleatorio.
7.9. Todas estas operaciones, una vez programadas, el equipo las lleva a
cabo en forma automática.
7.10. A continuación del estándar más concentrado, aspirar agua reactivo
(limpieza), luego un blanco de calibración, y a posteriori dos
soluciones estándar de control de procedimiento.
7.11. Si la calibración y los valores de las muestras de control de
procedimiento están dentro de los límites aceptables, continuar con el
análisis de las muestras. Si no lo están, proceder a tomar las acciones
correctivas.
7.12. Las muestras se cargan en el muestreador automático.
7.13. El blanco de filtración se incluye como una muestra para verificar la
ausencia de contaminantes por el proceso de filtración. Asegurar que
la lista de muestras en el “software” coincida con la posición correcta
en el “tray” del muestreador.
7.14. El equipo prepara automáticamente la curva de calibración, y en base
a esta curva de calibración, el equipo efectúa los cálculos
correspondientes a las muestras ingresadas.
7.15. Se ingresan por computadora los factores de dilución de cada muestra
y el instrumento automáticamente calcula la concentración correcta.
7.16. En el caso que la curva de calibración que calcula el equipo no sea
satisfactoria (mal coeficiente de correlación por alguna lectura errónea

249
de un estándar de calibración), el analista la puede construir con un
paquete estadístico en forma manual.
8. CALCULOS Y EXPRESION DE RESULTADOS
Expresar el resultado de cada analito en µg/L en base a la curva de
calibración.
Control de Calidad:
Medir duplicados en el 10 % de los casos, como mínimo. Los resultados
deben estar dentro del valor previamente calculado para cada analito, que a
su vez depende de la zona de la curva de calibración en la cuál se encuentra
su valor. El valor no es el mismo a bajas concentraciones como a altas
concentraciones. Verificar el cumplimiento del gráfico de control para
duplicados.
Introducir en el análisis cada 10 muestras, como mínimo, un estándar de
calibración de 0,70 mg/L, para determinar si se produjo alguna variación
instrumental. El resultado debe estar dentro del ± 5% del valor esperado. Si
esto no se produce, terminar el análisis de las muestras, corregir el problema
y recalibrar el instrumento.
Antes de finalizar el análisis, reanalizar una o más muestras. Los resultados
deben coincidir entre sí dentro del ± 5%. Si esto no se produce, hay que
reanalizar todas las muestras analizadas después del último control
aceptable del estándar de calibración de 0,7 mg/L.
Hay que analizar los dos estándares de control de procedimiento con cada
corrida. Esto permite verificar la exactitud.
Los resultados deben estar dentro del 95 % del límite de confianza que viene
suministrado por el proveedor para las muestras estándar de control de
procedimiento.
Verificar el cumplimiento del gráfico de control para las muestras estándar de
control.

250
Si los resultados de las muestras estándar de control de performance no
están comprendidos dentro del 95 % del límite de confianza, se repite el
análisis para los estándares en cuestión. Si este procedimiento falla, se
deben preparar nuevamente los estándares de calibración y reanalizarse.
Si todavía los resultados caen fuera del límite, se debe controlar que se
hayan seguido en forma adecuada las condiciones analíticas, y se deben
efectuar las correcciones y ajustes necesarios.
Si todavía existe una falla, se chequea el instrumento por la eventualidad de
algún daño. Deben analizarse nuevamente los estándares de calibración.
Si esta solución falla, el método se considera inválido y no se informan los
resultados hasta que la situación se clarifique. Hay que llamar al soporte
técnico del instrumento.
Los límites de detección, los estudios de repetibilidad y de recuperación se
deben encontrar documentados en el laboratorio.
9. REFERENCIAS
9.1 Norma EPA 200.7.

251
2.3.1.36. Determinación de cadmio, cobre y plomo to tal en sedimentos mediante GFAAS
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales Código del procedimiento:
PT-MS-02
Nombre del procedimiento: Determinación de cadmio, cobre y plomo total en sedimentos mediante
GFAAS
Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determinar la concentración total de cadmio, cobre y plomo en muestras de
sedimentos, utilizando espectrometría de absorción atómica con horno de
grafito (GFAAS).
2. ALCANCE
Se aplica a muestras de sedimentos y a determinaciones de Cd, Cu y Pb
cuando se requieran límites de cuantificación, por debajo de 0,030 mg/L y de
Cu por debajo de 0,050 mg/L.
Dichas muestras han tenido el tratamiento preliminar de digestión ácida
siguiendo el PT-DS-01.
3. PRINCIPIO DEL METODO
El principio se basa en la absorción de luz por átomos a una específica
longitud de onda. La espectrometría de absorción atómica electrotérmica
utiliza para la atomización un horno de grafito.
Un volumen de la muestra se deposita en un atomizador (horno de grafito) y
se incrementa la temperatura controlada antes de la atomización, para
remover el solvente y concomitantes. La alícuota introducida en el tubo de
grafito se atomiza dentro de un tiempo corto, dando lugar a una señal cuya
área (absorbancia integrada) es proporcional a la masa del analito en la
solución medida.
El uso de fuentes de luz especiales y de la selección de la longitud de onda
permite la determinación específica de los elementos individuales.

252
Interferencias
Las interferencias pueden deberse a la absorción molecular cuando los
componentes de la matriz se volatilizan durante la atomización, dando lugar a
una banda ancha de absorción.
Otra interferencia proviene de los efectos de la matriz y químicos.
Algunas interferencias específicas son:
Cadmio: Absorción severa no específica y scattering de la luz en la
atomización, causada por los componentes de la matriz. El exceso de cloruro
puede causar volatilización prematura de cadmio.
El uso del corrector de fondo y de los modificadores de matriz, minimizan las
interferencias en la determinación de estos analitos.
4. EQUIPOS
4.1. Espectrómetro de absorción atómica, provisto de un corrector de fondo.
4.2. Horno de grafito capaz de alcanzar la temperatura especificada y
requerida. Seguir las instrucciones del fabricante para considerar los
tiempos y temperaturas de secado, pirólisis y atomización.
4.3. Lámparas de cátodo hueco o de descarga eléctrica del elemento a
determinarse.
4.4. Muestreador automático (opcional)
4.5. Es recomendable, un sistema de registro de datos, de tal manera que se
tenga un registro permanente del proceso y que, cualquier problema con
el análisis (incompleta atomización, perdidas durante la pirólisis, cambios
en la sensibilidad, forma del pico obtenido, etc), pueda ser fácilmente
reconocido.
4.6. Balanza analítica, de sensibilidad igual o mayor a 0,1 mg.
5. REACTIVOS Y MATERIALES
5.1. MATERIALES

253
5.1.1. Pipetas calibradas clase A para medición de volúmenes iguales o
mayores a 1 mL.
5.1.2. Micropipetas calibradas de los rangos de volumen necesarios, para
mediciones de volumen menores de 1 mL, con puntas de buena
calidad y de un solo uso.
5.1.3. Material de uso habitual en el laboratorio: matraces aforados,
vasos de precipitados, puntas de micropipeta, etc, limpio, lavado con
solución de HNO3 1:1, enjuagado con agua grado reactivo.
5.2. REACTIVOS
5.2.1. Agua grado reactivo.
5.2.2. Solución de ácido nítrico concentrado, calidad ultrapuro.
5.2.3. Aire comprimido.
5.2.4. Argón, calidad UAP, (ultra alta pureza).
Soluciones estándar primarias
Las soluciones estándar primarias puede ser preparadas de metales de
alta pureza, óxidos o sales no higroscópicas usando agua pura. Estas
soluciones son preparadas con una concentración de 1000 mg/L.
Es posible utilizar soluciones estándar comerciales de 1000 mg/L.
Cuando se usa metales para la preparación, es necesaria la limpieza del
metal (especialmente alambre) para la preparación de los estándares.
Algunas formas de preparar las soluciones estándar primarias son:
5.2.5. Solución estándar de cadmio: Disolver 1.000 g de cadmio metálico
(mínimo 99,99 %) en 20 mL de HNO3 1:1 y diluir a 1 L con agua
grado reactivo.
5.2.6. Solución estándar de cobre: Disolver 1.000 g de cobre electrolítico
(mínimo 99,99 %) en 20 mL de HNO3 1:1 y diluir a 1 L con agua
grado reactivo.

254
5.2.7. Solución estándar de plomo: Disolver 1.599 g de nitrato de plomo
Pb(NO3)2 (mínimo 99.99 %) en agua grado reactivo, acidificar con
10 mL de solución de ácido nítrico ultrapuro, y diluir a 1 L con agua
grado reactivo.
Soluciones estándar de calibración
Se preparan diluyendo la solución estándar primaria del metal con los
mismos ácidos y concentración que la muestra. Los estándares de
calibración deben ser frescos y preparados cada vez que se realiza el
análisis de un lote de muestras.
Modificadores de matriz
5.2.8. El uso de Pd y Mg(NO3)2 como modificadores de matriz, se
recomienda para la determinación de Cd y Cu: Se disuelven 300 mg
de paladio en polvo, en solución de ácido nítrico concentrado (1 mL
de HNO3 y si fuera necesario, agregar 0.1 mL de HCl concentrado).
Disolver 200 mg de Mg(NO3)2 en agua pura. Verter las dos
soluciones juntas y diluir a 100 mL con agua grado reactivo.
5.2.9. El uso de NH4H2PO4 y Mg(NO3)2 como modificadores de matriz
se recomienda para la determinación de Cd y Pb: La solución de
fosfato de amonio, NH4H2PO4 al 40%, se prepara disolviendo 40 g
del reactivo en agua grado reactivo y diluir a 100 mL.
5.2.10. Es posible también el uso de soluciones estándar primarias
comerciales de dichos modificadores de matriz.
Blancos
5.2.11. Se requiere el uso de dos tipos de blancos: El blanco de
calibración para establecer la curva analítica y el blanco reactivo

255
utilizado para identificar posible contaminación de los reactivos, o del
material utilizado durante la preparación de la muestra.
5.2.12. El blanco de calibración se prepara acidificando el agua grado
reactivo a la misma concentración de los ácidos utilizados para los
estándares de calibración y muestras.
5.3. El blanco reactivo, debe contener todos los reactivos en el mismo
volumen como se usó en la preparación de las muestras.
6. ACCIONES PREVIAS
6.1. Los bajos límites de cuantificación que se alcancen con este método
dependerán del equipo (tipo de espectrómetro y horno de grafito, la
fuente de energía, el grado de expansión eléctrica de la señal) y de la
matriz de la muestra.
6.2. Referirse al manual del equipo para detalles de operación optima.
6.3. Tomar en cuenta los efectos de posibles interferencias por el uso de
diluciones, modificadores de matriz o la aplicación del método de
adición estándar.
6.4. Trabajar en un laboratorio limpio para prevenir la contaminación de las
muestras.
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Obtener la curva de calibración respectiva utilizando el blanco de
calibración y los estándares, acidificados adecuadamente y
conteniendo los modificadores de matriz respectivos en las cantidades
siguientes: 5 µg de Pd + 3 µg de Mg(NO3)2 para la determinación de
Cd y Cu y 50 µg de NH4H2PO4 + 3 µg de Mg(NO3)2 para la
determinación de Cd y Pb.
7.2. Las muestras (incluyendo el blanco reactivo) que han sido
previamente digeridas para liberar todo el plomo y cadmio presente,
que puede estar formando parte de la materia suspendida o coloidal.
(Procedimiento PT-DA-02), son diluidas a un volumen adecuado.

256
7.3. Agregar a las muestras para análisis y blanco reactivo los
modificadores de matriz en las mismas cantidades que los indicados
para los estándares de calibración.
7.4. Debido a las diferencias entre los modelos y formas de operación de
los espectrómetros, no se dan instrucciones de operación detalladas y
se deberán seguir las dadas por el fabricante.
7.5. Sin embargo, se puede indicar, de una manera general lo siguiente:
Inyectar una alícuota ya sea del estándar de calibración, cuando se va
a preparar la curva o de la muestra cuando se va a realizar la
determinación del analito, dentro del horno y aplicar el proceso de
descomposición y de atomización.
7.6. La siguiente tabla sugiere las longitudes de onda, condiciones de
pirolisis y atomización para cada uno de los analitos que se
determinará:
Tabla 1. Condiciones de determinación para los analitos de interés
7.7. Si se observa que la concentración de la muestra es mayor que la
concentración del mayor estándar, ésta deberá ser diluida con la
misma matriz ácida y reanalizada.
8. CALCULOS Y EXPRESION DE RESULTADOS
8.1. Cálculos para la preparación de la soluciones de calibración a partir de
un estándar primario comercial:
est
dldl
estC
VCV
⋅= mL
Elemento Longitud de onda (nm)
T de pirólisis
(ºC)
T de atomización
(ºC)
Cd 228,8 700 1400
Cu 324,8 1200 1900
Pb 283,3 850 1500

257
8.2. Cálculos para determinar la concentración del analito (µg/g).
W
FVCC dl
e
∗∗=
donde;
Ce = Concentración del elemento en la muestra original (µg/g).
C = Concentración del elemento en la solución de la muestra (µg/mL),
leída directamente o usando la curva de calibración.
V = Volumen de la solución de aforo de la muestra (mL).
W = masa de muestra (g)
Fdi = Factor de dilución: Volumen de aforo de la solución de la muestra
(mL) / volumen de la alícuota tomada para dilución (mL).
8.3. Expresión del resultado
Los resultados serán expresados en µg/g acompañado de la
incertidumbre expandida a un nivel de confianza del 95%
aproximadamente.
Control de Calidad:
Establecer y mantener un sistema de registro para documentar la calidad de
los datos generados.
Establecer los límites de detección para evaluar el nivel de ruido del
instrumento y los cambios de respuesta en el tiempo para cada analito,
analizando una serie de blancos reactivos.
Estos límites en µg/g, se estiman calculando el promedio de las desviaciones
estándar de tres corridas y en días no consecutivos del blanco reactivo con 7
mediciones por día.
Esta medición debe realizarse al menos cada tres meses.

258
Con cada lote de muestras, se deben analizar por lo menos un blanco.
Realizar el análisis por lo menos por triplicado.
Con cada lote de muestras, se debe analizar por lo menos un material de
control, que pueden ser muestras fortificadas con un volumen conocido del
analito en análisis o un material de referencia certificado.
Es necesario establecer un criterio de aceptación de los resultados
obtenidos. Para muestras fortificadas el límite debe ser ± 20% del valor
fortificado.
9. REFERENCIAS
[1] Standard Method for the examination of water and wastewater “American
Health Association” Ed. 21th 2005 3113 A
[2] Standard Method for the examination of water and wastewater “American
Health Association” Ed. 21 th 2005 3113 B
[3] Environmental Protection Agency, EPA Método 7010 – 2. SW-845.
[4] Bernhard Welz, Michael Sperlling. Atomic Absorption Spectrometry. 3era.
Edición. WILEY-VCH.

259
2.3.1.37. Determinación de As, Co, Cr, Cs, Fe, Rb, Sb, Sr, U, Zn en muestras de sedimentos mediante el análisis por act ivación neutrónica, método del k subcero
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agu a que están contaminadas con metales Código del procedimiento:
PT-MS-04
Nombre del procedimiento: Determinación de As, Co, Cr, Cs, Fe, Rb, Sb, Sr, U, Zn en muestras de sedimentos mediante el análisis por activación
neutrónica, método del k subcero Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determinar la concentración multielemental en muestras de sedimentos,
utilizando el análisis por activación neutrónica, basado en el método del
ksubcero (k0- NAA).
2. ALCANCE
Se aplica a muestras de sedimentos y a la determinación de As, Co, Cr, Cs,
Fe, Rb, Sb, Sr, U, Zn.
3. PRINCIPIO DEL METODO
El análisis por activación neutrónica instrumental (AANI) es una técnica
analítica nuclear, que utiliza el proceso de transformación de un nucleido
cualquiera en otro artificialmente radiactivo, mediante la captura de un
neutrón y la posterior medición de la actividad inducida, para el análisis
multielemental cualitativo y cuantitativo de elementos mayores, menores y
traza en muestras geológicas, biológicas, arqueológicas, ambientales, etc.
El método de análisis por activación comparativo ha sido el más utilizado por
varias décadas. Este consiste en la irradiación simultánea y en la misma
posición de la muestra y un comparador que contiene cantidades
exactamente conocidas de cada uno de los elementos que interesa analizar.
El método del k0, es un método paramétrico, que utiliza un solo comparador
para la determinación multielemental en la muestra y requiere un
conocimiento exacto de los parámetros que caracterizan la facilidad de
irradiación.

260
4. EQUIPOS
4.1. Fuente de neutrones con flujos térmicos del orden de 1013 n.cm-2. s-1.
4.2. Sistema de Espectrometría Gamma de alta resolución constituído por:
4.3. Detector semiconductor de Ge-Hiperpuro, con preamplificador
incorporado.
4.4. Fuente de alta Tensión
4.5. Amplificador adecuado
4.6. Convertidor analógico digital.
4.7. Balanza analítica de sensibilidad igual o mayor a 0,1 mg
4.8. Sistema neumático de transferencia de muestras
4.9. Polietileno (PE) de buena calidad para irradiación.
4.10. Cápsulas de PE para envío de muestras a irradiación
4.11. Fuente patrón de 152Eu
4.12. Contenedor de Pb
4.13. Material de laboratorio limpio, guantes de polietileno.
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Micropipeta calibrada y puntas de un solo uso
5.2. REACTIVOS
Se deben emplear reactivos de grado analítico en todos los ensayos. A
menos que se especifique lo contrario, se debe lograr que todos los
reactivos cumplan con lo dispuesto en las especificaciones del Comité de
Reactivos Analíticos de la ACS. Se pueden usar otros grados de reactivos
en la medida que sean de suficiente pureza para permitir su uso sin
perder la exactitud de las determinaciones.
5.2.1. Agua grado reactivo.
5.2.2. Solución estándar de Na de 10000 mg/L.
5.2.3. Solución estándar de Co de 1000 mg/L.
5.2.4. Solución estándar de Mo de 1000 mg/L.
5.2.5. Solución estándar de Au de 500 mg/L.

261
5.2.6. Materiales de referencia certificados conteniendo los analitos de
interés.
5.2.7. Celulosa
6. ACCIONES PREVIAS
6.1. La manipulación de muestras y material se realiza con guantes y en
laboratorio limpio.
6.2. Preparación de los comparadores de Na
Se pesa aproximadamente 50 mg de celulosa en los viales de irradiación
previamente lavados con HNO3 al 10%.
Se depositan con micropipeta calibrada 0,250 mL de solución estándar
de sodio sobre la celulosa y se pesa.
Los viales conteniendo el comparador de Na, se secan bajo lámpara IR,
se tapan y sellan para ser acondicionados en las cápsulas de irradiación.
6.3. Preparación de monitores de flujo
De igual manera que en la preparación del comparador de Na, se
depositan en viales de irradiación conteniendo celulosa 15 µg de solución
de Au, 350 µg de solución de Mo y 120 µg de solución de Co.De igual
manera que en la preparación del comparador de Na, se depositan con
micropipeta, 15 µg de solución de Au, 350 µg de solución de Mo y 120 µg
de solución de Co.
6.4. Calibración del sistema de espectrometría gamma
Se utiliza una fuente de 152Eu u otra adecuada para la calibración. Esta
se coloca sobre el detector que se usara para las mediciones.
Se mide la fuente por 30 minutos y se calibra utilizando las energías de
122 keV, 344.3 keV, 778.9 keV y 1408 keV, en caso de utilizarse 152Eu.
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Las muestras y materiales de referencia se preparan pesando
aproximadamente 200 mg de muestra en viales de PE para irradiación,
previamente lavados en solución de HNO3 al 10% y rotulados.

262
7.2. Se sellan los viales
7.3. Se acondicionan en las cápsulas para irradiación alternadamente
comparadores, muestras, monitores de flujo, blancos y materiales de
referencia preparados.
7.4. Se envía la cápsula a irradiar por 1200 segundos, usando el sistema
neumático de transferencia de muestras a una potencia de 10 MW y un
flujo de neutrones térmicos de aprox. 3.0 · 1013 n. cm2· s-1.
7.5. Medir y registrar la tasa de dosis de la cápsula irradiada
aproximadamente a 25 cm y colocarla en el tacho de plomo anexo a la
estación de envío. En caso superar los 0,5 mSv transportar en
contenedor de plomo a la campana radioquímica.
7.6. Abrir la cápsula dentro de la campana y retirar los envases del estándar y
la muestra verificando la integridad de los mismos.
7.7. Transportar en el contenedor de plomo, las muestras, comparadores al
laboratorio de mediciones de espectrometría gamma.
7.8. Medir las muestras, materiales de referencia y comparadores de sodio
utilizando el detector de Ge (HP), de eficiencia relativa adecuada y con
una buena resolución para el pico de la fuente utilizada en la calibración
del espectrómetro gamma.
7.9. Evaluar los espectros
7.10. Calcular la concentración de los elementos tomando como base las
formulas expresadas en el punto 8.
7.11. Documentar la información en los formatos
8. CALCULOS Y EXPRESION DE RESULTADOS
8.1. Cálculo de la concentración en µg/g
Se emplea el área de los fotopicos de los siguientes radionúclidos:

263
56As 559 keV 60Co 1173 y 1332.5 keV 51Cr 320 keV 134Cs 796 keV 59Fe 1099 y 1291 kev 239Np (U) 277 keV 86Rb 1076 keV 124Sb 1690 keV 88mSr 388 keV 95Zn 1115 keV
Calcular la concentración ρi del elemento “i” en una muestra x aplicando
el método de k0 según la convención de Högdahl:
)(
)(
,0
,0
,
,
,
,
αα
ρx
p
x
p
xo
po
pesp
xesp
iQF
QF
K
K
T
T
++
∈∈
=
Donde “x “ y “p” denota la muestra y el estándar respectivamente y Tesp
es igual:
)(
,
),(gwS
HCDCT
in
cxesp ⋅⋅⋅⋅
=
dteD⋅= λ
( )iteC ⋅−−
= λλ
1
l
r
t
tH =
Donde; ti, td son los tiempos de irradiación, decaimiento
respectivamente, λ es la constante de decaimiento = 0.693/periodo de
semidesintegración; є es la eficiencia del detector para la energía

264
medida, Q(α) es la razón de la integral de resonancia a la sección eficaz
como función de α y F es la relación de flujo térmico sobre flujo
epitérmico.
8.2. Expresión de Resultados
La concentración calculada se expresa en términos de µg.g-1 con la
incertidumbre expandida.
9. CONTROL DE CALIDAD
9.1. Las muestras se analizan en duplicado
9.2. Por cada corrida de muestras se analiza como mínimo un material de
referencia certificado.
9.3. Por cada corrida de muestra se analiza un blanco de reactivos.
9.4. Participar en ensayos de aptitud o rondas de intercomparación en matriz
agua por lo menos una vez al año
10. REFERENCIAS
10.1. Knoll G.F. Radiation Detection and Measurement 2ndEdition 1989.
10.2. G. Erdtmann Nuclear Activation and Radioisotope Methods of Analyses.
10.3. IAEA-TECDOC-564 Practical Aspects of Operating a Neutron Activation
Analyses Laboratory. Documento Técnico del Taller Regional. Tópicos
selectos sobre Aplicaciones del Método de k-sub cero y otros métodos
Paramétricos en Análisis por Activación Neutrónica.
10.4. Frans De Corte. The k0-Standardization Method, 1987.

265
2.3.1.38. Determinación de Cadmio total en muestras de sedimentos por el Método de Espectrometría de Absor ción Atómica
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agu a que están contaminadas con metales Código del procedimiento:
PT-MS-06
Nombre del procedimiento: Determinación de Cadmio total en muestras de sedimentos
por el Método de Espectrometría de Absorción Atómica Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determinar la concentración de cadmio total por espectrometría de absorción
atómica.
2. ALCANCE
Se aplica a cualquier sedimento y corresponde a la concentración de metales
determinados en una muestra previamente digerida.
3. PRINCIPIO DEL METODO
3.1. Una fracción de muestra previamente digerida, es aspirada hacia una
llama Aire-Acetileno posicionada en el paso óptico de un espectrómetro
de absorción atómica. La muestra es atomizada bajo estas condiciones.
3.2. La población de átomos al estado elemental absorbe radiación
característica proveniente una fuente de emisión de líneas atómicas de
cadmio, la relación entre potencia incidente y potencia transmitida es una
medida de la concentración del elemento en la muestra.
4. EQUIPOS
4.1. Balanza Analítica, de sensibilidad igual o mayor a 0,1 mg.
4.2. Campana, con sistema de extracción de gases.
4.3. Espectrómetro de absorción atómica, dotado con quemador aire-
acetileno y corrector de deuterio (o correctores de acuerdo a equipo
utilizado).
4.4. Fuente de emisión de líneas atómicas de cadmio.
5. REACTIVOS Y MATERIALES
5.1. MATERIALES

266
5.1.1. Material de uso habitual en laboratorio (ej. vasos de precipitado,
pipetas, matraces y otros).
5.2. REACTIVOS
5.2.1. Agua grado reactivo.
5.2.2. Solución de ácido nítrico concentrado, calidad ultrapuro.
5.2.3. Solución estándar de cadmio de 1000 mg/L.
5.2.4. Aire comprimido.
5.2.5. Acetileno, calidad UAP, (ultra alta pureza).
Soluciones estándares de calibración
5.2.6. Preparar una serie de soluciones de calibración en el rango óptimo,
a partir de diluciones apropiadas de la solución estándar de cadmio
de 1000 mg/L.
5.2.7. Almacenar en envases de polietileno de alta densidad
debidamente etiquetados. La condición de acidez final para los
estándares es solución 1% HNO3 V/V. Se exige para la curva de
calibración un r≥0.995.
5.2.8. Solución blanco término cero: la solución término cero debe ser
preparada con agua grado reactivo y en un medio 1% HNO3 V/V.
6. ACCIONES PREVIAS
6.1. Las muestras deben ser digeridas como se indica en el procedimiento
PT-DS-01.
6.2. Aforar la muestra digerida a volumen predeterminado.
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Encender el espectrómetro y verificar las presiones de aire y acetileno,
ajustando si procede.
7.2. Verificar que el extractor de gases se encuentra en correcto
funcionamiento.
7.3. Posicionar y encender la fuente de emisión de líneas atómicas de
cadmio; esperar a que la fuente se estabilice

267
7.4. Ajustar la energía de la fuente respecto del detector de acuerdo al
manual del equipo.
7.5. Ajustar la longitud de onda principal y abertura del monocromador de
acuerdo a las especificaciones establecidas en el manual de
operaciones del equipo. Longitud de onda principal 228 nm y abertura
del slit 0,7 nm).
7.6. Continuar con el ajuste y calibración, de acuerdo a instrucciones del
fabricante del equipo.
7.7. Una vez ajustadas las condiciones, calibrar el instrumento
procediendo como sigue:
7.8. Realizar autozero aspirando el blanco termino cero de la curva de
calibración.
7.9. Calibrar empleando un número adecuado de estándares como mínimo
blanco y tres concentraciones en el rango de trabajo,
recomendándose 5 puntos.
7.10. Verificar la calibración después de la lectura de cada seis muestras. Si
la calibración permanece constante continuar con la lectura de las
siguientes muestras. Si la calibración ha variado, recalibrar y chequear
la lectura de las muestras anteriores.
Lectura de las muestras
7.11. Una vez calibrado el instrumento, determinar la concentración de las
muestras de acuerdo al siguiente protocolo:
7.12. Aspirar el blanco reactivo del set de análisis que ha sido sometido al
mismo proceso de las muestras y registrar la concentración de los
analitos.
7.13. Aspirar las muestras en estudio en secuencia de seis en seis,
verificando la calibración y la estabilidad del cero entre lotes de
muestra.
7.14. Repetir las lecturas en caso de que sea necesario.
7.15. Aspirar agua grado reactivo entre muestra y muestra para evitar
contaminación cruzada.

268
Control de calidad analítica
7.16. Por cada set de análisis, llevar de forma paralela a las muestras, los
siguientes controles:
7.17. Un blanco reactivo preparado a partir de agua para análisis grado
reactivo exenta de cobre.
7.18. Un ensayo duplicado de una muestra elegida al azar.
7.19. Un ensayo de un estándar de concentración conocida cercana al valor
normado.
8. CALCULOS Y EXPRESION DE RESULTADOS
Cd (µµµµg/g) = ((Lect - Bco) x D) / m
donde:
Cd (µg/g) = concentración de Cd, expresada en µg de Cd por gramo de
sedimento
Lect = lectura de la muestra en mg/L
Bco = lectura blanco reactivo sometido al mismo proceso de las muestras
D = factor de dilución o concentración
m = masa de muestra analizada (en gramos)
9. REFERENCIAS
[1] Standard Methods For The Examination Of Water And Wastewater.
APHA. AWWA. WEF. 21th Edition 2005, Part. 3111-B; Direct Air –Acetylene
Flame Method.

269
2.3.1.39. Determinación de Cromo en sedimentos por el Método de Espectrometría de Absorción Atómica
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con metales” Código del procedimiento:
PT-MS-07
Nombre del procedimiento: Determinación de Cromo en sedimentos por el Método de
Espectrometría de Absorción Atómica
Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determinar cromo total en muestras de sedimentos por espectrometría de
absorción atómica por llama.
2. ALCANCE
Este procedimiento es valido para la determinación de cromo en muestras de
sedimentos procesadas según PT-DS-01.
3. PRINCIPIO DEL METODO
3.1. Una fracción de muestra previamente digerida, es aspirada hacia una
llama Aire-Acetileno posicionada en el paso óptico de un espectrómetro
de absorción atómica. La muestra es atomizada bajo estas condiciones.
3.2. La población de átomos al estado elemental absorbe radiación
característica proveniente una fuente de emisión de líneas atómicas de
cromo, la relación entre potencia incidente y potencia transmitida es una
medida de la concentración del elemento en la muestra.
4. EQUIPOS
4.1. Balanza Analítica, de sensibilidad igual o mayor a 0,1 mg.
4.2. Campana, con sistema de extracción de gases.
4.3. Espectrómetro de absorción atómica, dotado con quemador aire-
acetileno y corrector de deuterio (o correctores de acuerdo a equipo
utilizado).
4.4. Fuente de emisión de líneas atómicas de cromo.
4.5. Plancha calefactora con regulación de temperatura o digestor de micro
ondas (opcional).
5. REACTIVOS Y MATERIALES

270
5.1. MATERIALES
5.1.1. Material de uso habitual en laboratorio (ej. vasos de precipitado,
pipetas, matraces aforados y otros).
5.2. REACTIVOS
5.2.1. Agua grado reactivo.
5.2.2. Solución de ácido clorhídrico concentrado, calidad ultrapuro.
5.2.3. Solución de ácido nítrico concentrado, calidad ultrapuro.
5.2.4. Solución estándar de cromo de 100 mg/L. Disolver 0,1923 g de
CrO3 en agua, acidificar con 10 mL de solución de HNO3
concentrado y diluir a 1000 mL con agua destilada 1,00 mL = 100
microgramos Cr.
5.2.5. Oxido nitroso.
5.2.6. Acetileno, calidad UAP, (ultra alta pureza).
Soluciones estándares de calibración
5.2.7. Preparar una serie de soluciones de calibración en el rango de 0,2
a 3,0 mg/L, a partir de diluciones apropiadas de la solución
estándar de cromo de 100 mg/L.
5.2.8. Almacenar en envases de polietileno de alta densidad
debidamente etiquetados. La condición de acidez final para los
estándares es solución 1% HNO3 V/V. Se exige para la curva de
calibración un r≥0.995.
5.2.9. Solución blanco término cero: la solución término cero debe ser
preparada con agua grado reactivo y en un medio 1% HNO3 V/V.
6. ACCIONES PREVIAS
6.1. El material empleado en el análisis debe ser lavado con detergente
neutro y agua corriente. Enjuagar con agua destilada. Lavar después con
una mezcla 1:1 de ácido nítrico y agua destilada.
6.2. Las muestras de sedimento deben someterse a un tratamiento previo de
digestión para liberar todo el analito presente. Para el procesamiento de
las muestras y blanco referirse al procedimiento PT-DS-01.

271
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Encender el espectrómetro y verificar las presiones de óxido nitroso y
acetileno, ajustando si procede.
7.2. Verificar que el extractor de gases se encuentra en correcto
funcionamiento.
7.3. Posicionar y encender la fuente de emisión de líneas atómicas de
cobre; esperar a que la fuente se estabilice
7.4. Ajustar la energía de la fuente respecto del detector de acuerdo al
manual del equipo.
7.5. Ajustar la longitud de onda principal y abertura del monocromador de
acuerdo a las especificaciones establecidas en el manual de
operaciones del equipo. Longitud de onda principal 357,87 nm.
7.6. Continuar con el ajuste y calibración, de acuerdo a instrucciones del
fabricante del equipo.
7.7. Una vez ajustadas las condiciones, calibrar el instrumento
procediendo como sigue:
7.8. Realizar autozero aspirando el blanco termino cero de la curva de
calibración.
7.9. Calibrar empleando un número adecuado de estándares como mínimo
blanco y tres concentraciones en el rango de trabajo,
recomendándose 5 puntos.
7.10. Verificar la calibración después de la lectura de cada seis muestras. Si
la calibración permanece constante continuar con la lectura de las
siguientes muestras. Si la calibración ha variado, recalibrar y chequear
la lectura de las muestras anteriores.
Lectura de las muestras
7.11. Una vez calibrado el instrumento, determinar la concentración de las
muestras de acuerdo al siguiente protocolo:

272
7.12. Aspirar el blanco reactivo del set de análisis que ha sido sometido al
mismo proceso de las muestras y registrar la concentración de los
analitos.
7.13. Aspirar las muestras en estudio en secuencia de seis en seis,
verificando la calibración y la estabilidad del cero entre lotes de
muestra.
7.14. Repetir las lecturas en caso de que sea necesario.
7.15. Aspirar agua grado reactivo entre muestra y muestra para evitar
contaminación cruzada.
Control de calidad analítica
7.16. Por cada set de análisis, llevar de forma paralela a las muestras, los
siguientes controles:
7.17. Un blanco reactivo preparado a partir de agua para análisis grado
reactivo exenta de cobre.
7.18. Un ensayo duplicado de una muestra elegida al azar.
7.19. Un ensayo de un estándar de concentración conocida cercana al valor
normado.
8. CALCULOS Y EXPRESION DE RESULTADOS
Cr (µµµµg/g) = ((Lect - Bco) x D) / m
donde:
Cr (µg/g) = concentración de Cr, expresada en µg de Cr por gramo de
muestra.
Lect = lectura de la muestra en µg/mL
Bco = lectura blanco reactivo sometido al mismo proceso de las muestras
D = factor de dilución o concentración

273
m = masa de muestra (en gramos)
9. REFERENCIAS
[1] Standard Methods For The Examination Of Water And Wastewater.
APHA. AWWA. WEF. 21th Edition 2005, Part. 3111-B; Direct Air –Acetylene
Flame Method.

274
2.3.1.40. Determinación de Cobre en sedimentos por el Método de Espectrometría de Absorción Atómica
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agu a que están contaminadas con metales Código del procedimiento:
PT-MS-08
Nombre del procedimiento: Determinación de Cobre en sedimentos por el Método de
Espectrometría de Absorción Atómica Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determinar la concentración de cobre total en sedimentos por espectrometría
de absorción atómica por llama.
2. ALCANCE
Este método de ensayo es aplicable a cualquier muestra de sedimentos
procesados por el procedimiento PT-DS-01.
3. PRINCIPIO DEL METODO
Una fracción de muestra, directamente o previamente digerida, es aspirada
hacia una llama de aire-acetileno, posicionada en el paso óptico de un
espectrómetro de absorción atómica. La muestra en estas condiciones es
atomizada.
La población de átomos al estado elemental absorbe radiación característica
proveniente de una fuente de emisión de líneas atómicas de cobre; la
relación entre potencia incidente y potencia transmitida es una medida de
concentración del elemento en muestra.
4. EQUIPOS
4.1. Balanza Analítica, de sensibilidad igual o mayor a 0,1 mg.
4.2. Campana, con sistema de extracción de gases.
4.3. Espectrómetro de absorción atómica, dotado con quemador aire-
acetileno y corrector de deuterio (o correctores de acuerdo a equipo
utilizado).
4.4. Fuente de emisión de líneas atómicas de cobre.

275
4.5. Plancha calefactora con regulación de temperatura o digestor de micro
ondas (opcional).
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Material de uso habitual en laboratorio (ej. vasos de precipitado,
pipetas, matraces y otros).
5.2. REACTIVOS
5.2.1. Agua grado reactivo.
5.2.2. Solución de ácido clorhídrico concentrado, calidad ultrapuro.
5.2.3. Solución de ácido nítrico concentrado, calidad ultrapuro.
5.2.4. Solución estándar de cobre de 100 mg/L. Disolver 0,100 g de cobre
metálico en 2 ml de solución de HNO3 concentrado, añadir 10 mL de
solución de HNO3 concentrado y diluir a 1000 mL con agua.
5.2.5. Aire comprimido.
5.2.6. Acetileno, calidad UAP, (ultra alta pureza).
Soluciones estándares de calibración
5.2.7. Preparar una serie de soluciones de calibración en el rango óptimo
de 1 a 5 mg/L, a partir de diluciones apropiadas de la solución
estándar de cobre de 100 mg/L.
5.2.8. Almacenar en envases de polietileno de alta densidad
debidamente etiquetados. La condición de acidez final para los
estándares es solución 1% HNO3 V/V. Se exige para la curva de
calibración un r≥0,995.
5.2.9. Solución blanco término cero: la solución término cero debe ser
preparada con agua grado reactivo y en un medio 1% HNO3 V/V.
6. ACCIONES PREVIAS
6.1. Las muestras de sedimento deben someterse a un tratamiento previo de
digestión para liberar todo el analito presente. Para el procesamiento de
las muestras y blanco referirse al procedimiento PT-DS-01.
7. DESCRIPCION DEL PROCEDIMIENTO

276
7.1. Encender el espectrómetro y verificar las presiones de aire y acetileno,
ajustando si procede.
7.2. Verificar que el extractor de gases se encuentra en correcto
funcionamiento.
7.3. Posicionar y encender la fuente de emisión de líneas atómicas de
cobre; esperar a que la fuente se estabilice
7.4. Ajustar la energía de la fuente respecto del detector de acuerdo al
manual del equipo.
7.5. Ajustar la longitud de onda principal y abertura del monocromador de
acuerdo a las especificaciones establecidas en el manual de
operaciones del equipo. Longitud de onda principal 324,8 nm.
7.6. Continuar con el ajuste y calibración, de acuerdo a instrucciones del
fabricante del equipo.
7.7. Una vez ajustadas las condiciones, calibrar el instrumento
procediendo como sigue:
7.8. Realizar autozero aspirando el blanco termino cero de la curva de
calibración.
7.9. Calibrar empleando un número adecuado de estándares como mínimo
blanco y tres concentraciones en el rango de trabajo,
recomendándose 5 puntos.
7.10. Verificar la calibración después de la lectura de cada seis muestras. Si
la calibración permanece constante continuar con la lectura de las
siguientes muestras. Si la calibración ha variado, recalibrar y chequear
la lectura de las muestras anteriores.
Lectura de las muestras
7.11. Una vez calibrado el instrumento, determinar la concentración de las
muestras de acuerdo al siguiente protocolo:
7.12. Aspirar el blanco reactivo del set de análisis que ha sido sometido al
mismo proceso de las muestras y registrar la concentración de los
analitos.

277
7.13. Aspirar las muestras en estudio en secuencia de seis en seis,
verificando la calibración y la estabilidad del cero entre lotes de
muestra.
7.14. Repetir las lecturas en caso de que sea necesario.
7.15. Aspirar agua grado reactivo entre muestra y muestra para evitar
contaminación cruzada.
Control de calidad analítica
7.16. Por cada set de análisis, llevar de forma paralela a las muestras, los
siguientes controles:
7.17. Un blanco reactivo preparado a partir de agua para análisis grado
reactivo exenta de cobre.
7.18. Un ensayo duplicado de una muestra elegida al azar.
7.19. Un ensayo de un estándar de concentración conocida cercana al valor
normado.
8. CALCULOS Y EXPRESION DE RESULTADOS
Cu (µµµµg/g) = ((Lect - Bco) x D) / m
donde:
Cu (µg/g) = concentración de Cu, expresada en µg de Cu por gramo de
muestra
Lect = lectura de la muestra en µg/mL
Bco = lectura blanco reactivo sometido al mismo proceso de las muestras
D = factor de dilución o concentración
m =masa de muestra (en gramos)
9. REFERENCIAS

278
[1] Standard Methods For The Examination Of Water And Wastewater.
APHA. AWWA. WEF. 21th Edition 2005, Part. 3111-B; Direct Air –Acetylene
Flame Method.

279
2.3.1.41. Determinación de Plomo en sedimentos por el Método de Espectrometría de Absorción Atómica
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agu a que están contaminadas con metales Código del procedimiento:
PT-MS-09
Nombre del procedimiento: Determinación de Plomo en sedimentos por el Método de
Espectrometría de Absorción Atómica Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determinar plomo en muestras de sedimentos por espectrometría de
absorción atómica por llama.
2. ALCANCE
Se aplica a la determinación de Pb en muestras de sedimentos procesadas
según el procedimiento PT-DS-01.
3. PRINCIPIO DEL METODO
Una fracción de muestra, directamente o previamente digerida, es aspirada
hacia una llama de aire-acetileno, posicionada en el paso óptico de un
espectrómetro de absorción atómica. La muestra en estas condiciones es
atomizada.
La población de átomos al estado elemental absorbe radiación característica
proveniente de una fuente de emisión de líneas atómicas de plomo; la
relación entre potencia incidente y potencia transmitida es una medida de
concentración del elemento en muestra.
4. EQUIPOS
4.1. Balanza Analítica, de sensibilidad igual o mayor a 0,1 mg.
4.2. Campana, con sistema de extracción de gases.
4.3. Espectrómetro de absorción atómica, dotado con quemador aire-
acetileno y corrector de deuterio (o correctores de acuerdo a equipo
utilizado).
4.4. Fuente de emisión de líneas atómicas de plomo.

280
4.5. Plancha calefactora con regulación de temperatura o digestor de micro
ondas (opcional).
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Material de uso habitual en laboratorio (ej. vasos de precipitado,
pipetas, matraces y otros).
5.2. REACTIVOS
5.2.1. Agua grado reactivo.
5.2.2. Solución de ácido clorhídrico concentrado, calidad ultrapuro.
5.2.3. Solución de ácido nítrico concentrado, calidad ultrapuro.
5.2.4. Solución estándar de plomo de 100 mg/L. Disolver 0,100 g de
plomo metálico en 2 ml de solución de HNO3 concentrado, añadir 10
mL de solución de HNO3 concentrado y diluir a 1000 mL con agua.
5.2.5. Aire comprimido.
5.2.6. Acetileno, calidad UAP, (ultra alta pureza).
Soluciones estándares de calibración
5.2.7. Preparar una serie de soluciones de calibración en el rango óptimo
de 2 a 10 mg/L, a partir de diluciones apropiadas de la solución
estándar de plomo de 100 mg/L.
5.2.8. Almacenar en envases de polietileno de alta densidad
debidamente etiquetados. La condición de acidez final para los
estándares es solución 1% HNO3 V/V. Se exige para la curva de
calibración un r≥0,995.
5.2.9. Solución blanco término cero: la solución término cero debe ser
preparada con agua grado reactivo y en un medio 1% HNO3 V/V.
6. ACCIONES PREVIAS
6.1. El material empleado en el análisis debe ser lavado con detergente
neutro y agua corriente. Enjuagar con agua destilada. Lavar después con
una mezcla 1:1 de ácido nítrico y agua destilada.

281
6.2. Las muestras de sedimento deben someterse a un tratamiento previo de
digestion para liberar todo el analito presente. Para el procesamiento de
las muestras y blanco referirse al procedimiento PT-DS-01.
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Encender el espectrómetro y verificar las presiones de aire y acetileno,
ajustando si procede.
7.2. Verificar que el extractor de gases se encuentra en correcto
funcionamiento.
7.3. Posicionar y encender la fuente de emisión de líneas atómicas de
plomo; esperar a que la fuente se estabilice.
7.4. Ajustar la energía de la fuente respecto del detector de acuerdo al
manual del equipo.
7.5. Ajustar la longitud de onda principal y abertura del monocromador de
acuerdo a las especificaciones establecidas en el manual de
operaciones del equipo. Longitud de onda principal 217,0 nm.
7.6. Continuar con el ajuste y calibración, de acuerdo a instrucciones del
fabricante del equipo.
7.7. Una vez ajustadas las condiciones, calibrar el instrumento
procediendo como sigue:
7.8. Realizar autozero aspirando el blanco termino cero de la curva de
calibración.
7.9. Calibrar empleando un número adecuado de estándares como mínimo
blanco y tres concentraciones en el rango de trabajo,
recomendándose 5 puntos.
7.10. Verificar la calibración después de la lectura de cada seis muestras. Si
la calibración permanece constante continuar con la lectura de las
siguientes muestras. Si la calibración ha variado, recalibrar y chequear
la lectura de las muestras anteriores.
Lectura de las muestras

282
7.11. Una vez calibrado el instrumento, determinar la concentración de las
muestras de acuerdo al siguiente protocolo:
7.12. Aspirar el blanco reactivo del set de análisis que ha sido sometido al
mismo proceso de las muestras y registrar la concentración de los
analitos.
7.13. Aspirar las muestras en estudio en secuencia de seis en seis,
verificando la calibración y la estabilidad del cero entre lotes de
muestra.
7.14. Repetir las lecturas en caso de que sea necesario.
7.15. Aspirar agua grado reactivo entre muestra y muestra para evitar
contaminación cruzada.
Control de calidad analítica
7.16. Por cada set de análisis, llevar de forma paralela a las muestras, los
siguientes controles:
7.17. Un blanco reactivo preparado a partir de agua para análisis grado
reactivo exenta de cobre.
7.18. Un ensayo duplicado de una muestra elegida al azar.
7.19. Un ensayo de un estándar de concentración conocida cercana al valor
normado.
8. CALCULOS Y EXPRESION DE RESULTADOS
Pb (µµµµg/g) = ((Lect - Bco) x D) / m
donde:
Pb (µg/g) = concentración de Pb, expresada en µg de Pb por gramo de
muestra
Lect = lectura de la muestra en mg/L
Bco = lectura blanco reactivo sometido al mismo proceso de las muestras

283
D = factor de dilución o concentración
m =masa de muestra (en gramos)
9. REFERENCIAS
[1] Standard Methods For The Examination Of Water And Wastewater.
APHA. AWWA. WEF. 21th Edition 2005, Part. 3111-B; Direct Air –Acetylene
Flame Method.

284
2.3.1.42. Determinación de Arsénico Total en sedime ntos por el Método de Espectrometría de Absorción Atómica con Generación de Hidruros
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agu a que están contaminadas con metales Código del procedimiento:
PT-MS-10
Nombre del procedimiento: Determinación de Arsénico Total en sedimentos por el Método de Espectrometría de Absorción Atómica con
Generación de Hidruros Fecha de aprobación: 2008-05-02
Numero de revisión: ninguna
Fecha de revisión: 2008-05-02
1. OBJETIVO
Determinar arsénico total mediante espectrometría de absorción atómica con
generación de hidruros.
2. ALCANCE
Se aplica a todo sedimento y corresponde a la concentración de arsénico en
una muestra previamente digerida.
3. PRINCIPIO DEL METODO
3.1. El arsénico presente en una fracción de muestra previamente digerida,
es reducido a arsina mediante reacción con borohidruro de sodio en
medio ácido. La arsina generada es conducida por un gas de arrastre
hacia una celda de cuarzo posicionada en el paso óptico de un
espectrómetro de absorción atómica. La celda se encuentra a una
temperatura aproximada de 1000ºC. La arsina, en estas condiciones, y a
través de mecanismos combinados [descomposición térmica y
reacciones con radicales hidrógeno] es atomizada.
3.2. La población de átomos al estado elemental absorbe radiación
característica proveniente de una fuente de emisión de líneas atómicas
de arsénico, la relación entre potencia incidente y potencia transmitida es
una medida de la concentración del elemento en la muestra.
4. EQUIPOS
4.1. Balanza Analítica, de sensibilidad igual o mayor a 0,1 mg.
4.2. Campana, con sistema de extracción de gases.

285
4.3. Espectrómetro de absorción atómica, dotado con quemador y con
generador de hidruros y corrector de deuterio [u otro corrector de
acuerdo a equipo utilizado).
4.4. Fuente de emisión de líneas atómicas de arsénico.
4.5. Sistemas generador de hidruros continuo o discontinuo.
4.6. Celda de cuarzo y soporte de celda.
4.7. Plancha calefactora con regulación de temperatura o digestor de micro
ondas (opcional).
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Material de uso habitual en laboratorio (ej. vasos de precipitado,
pipetas, matraces y otros).
5.2. REACTIVOS
5.2.1. Agua grado reactivo.
5.2.2. Solución de ácido clorhídrico concentrado, calidad ultrapuro.
5.2.3. Solución de ácido nítrico concentrado, calidad ultrapuro.
5.2.4. Solución estándar comercial de arsénico de 1000 mg/L.
5.2.5. Aire comprimido.
5.2.6. Argón o nitrógeno
5.2.7. Borohidruro de sodio (NaBH4)
5.2.8. Yoduro de potasio (KI)
5.2.9. Hidróxido de sodio
5.2.10. Indicador fenolftaleína
5.2.11. Solución de ácido sulfúrico concentrado, calidad ultrapuro.
5.2.12. Solución de NaBH4 1 – 4%; la concentración dependerá del
sistema de generación empleado. Pesar la cantidad requerida para
1L de solución (el reactivo es corrosivo en contacto con la piel).
Disolver con solución de NaOH (0,5 -1% m/v). Una vez disuelto,
filtrar a través de filtro rápido de alta pureza. Descartar el filtro y
transferir el filtrado a un matraz aforado de 1000 ml. Aforar con
solución de NaOH (0,5 -1% m/v).

286
5.2.13. Solución de KI 20% m/v: Pesar 20g. de KI, disolver y diluir a 100
mL con agua grado reactivo exenta de arsénico. Proteger de la
oxidación por efecto de la luz.
Soluciones estándares de calibración
5.2.14. Preparar una serie de soluciones de calibración en el rango
óptimo de trabajo, a partir de diluciones apropiadas del estándar de
1000 mg As (III)/L 1% v/v H2SO4.
5.2.15. Por cada 100 ml de solución adicionar 10 ml de solución de KI
(20% m/v). Agitar y dejar reposar aproximadamente 30 minutos.
5.2.16. Cubrir debidamente el matraz para evitar la oxidación por efecto
de la luz. La condición de acidez final para estándares es 20% v/v
HCl. Preparar diariamente. Se exige para la curva de calibración un
r≥0,995.
5.2.17. Solución blanco término cero: la solución término cero debe ser
preparada con agua grado reactivo exenta de arsénico y en un medio
20 % HCl v/v, adicionando 10 ml de solución de KI (20% m/v).
Proteger de la oxidación por efecto de la luz. Preparar diariamente.
6. ACCIONES PREVIAS
6.1. Para la toma, transporte y almacenamiento de las muestras se
recomienda utilizar recipientes de plástico.
6.2. Las muestras deben ser digeridas como se indica en el procedimiento
PT-DS-01
7. DESCRIPCION DEL PROCEDIMIENTO
Ajuste y calibración del instrumento:
7.1. Encender el espectrómetro y verificar las presiones de aire, acetileno y
nitrógeno o argón, ajustando si procede.
7.2. Verificar que el extractor de gases se encuentre en correcto
funcionamiento.

287
7.3. Chequear con una fuente de emisión que emita luz visible [Cu, Mn],
que el haz esté pasando central a al ranura del quemador.
7.4. Posicionar el soporte de la celda de cuarzo en el paso óptico del
espectrómetro y ajustar que el haz pase por el interior de la celda,
siguiendo las instrucciones proporcionadas en el manual del
instrumento [generador de hidruros].
7.5. Posicionar y encender la fuente de emisión de líneas atómicas de As.
Esperar a que la fuente se estabilice.
7.6. Ajustar la longitud de onda principal [193,7nm] y la abertura del
monocromador de acuerdo a las especificaciones establecidas en el
manual de operaciones del equipo.
7.7. Ajustar la energía de la fuente respecto al detector de acuerdo a las
instrucciones descritas en el manual del equipo.
7.8. Ajustar los flujos de encendido y encender el instrumento.
Sistema manual o automático de generación de hidru ros [generación
discontinua].
7.9. De acuerdo las instrucciones indicadas en el manual de operaciones
del generador, adicionar el volumen de estándar requerido y aplicar el
programa.
7.10. Ajustar el flujo de gas portador [Ar o N2].
7.11. Encender el corrector de absorción no específica.
7.12. Verificar la sensibilidad analítica empleando el estándar de calibración
recomendado en el manual de operaciones del equipo.
7.13. Construir la curva de calibración empleando un numero adecuado de
estándares, como mínimo blanco y tres concentraciones del rango de
trabajo, recomendándose 5 puntos.
7.14. Verificar la calibración después de la lectura de cada 6 muestras. Si la
calibración permanece constante, continuar con las lecturas de las

288
siguientes muestras, si la calibración ha variado, recalibrar y chequear
las lecturas de las muestras anteriores.
7.15. Una vez calibrado el instrumento, determinar la concentración de
muestras de acuerdo al siguiente protocolo:
7.16. Aspirar el blanco y realizar auto -cero
7.17. Aspirar las muestras en estudio, en secuencia de 6 en 6, verificando la
calibración y la estabilidad del cero, entre lotes de muestras. Repetir
las lecturas en caso que sea necesario.
7.18. Aspirar agua para análisis grado reactivo exenta de As entre muestra
y muestra con el propósito de evitar contaminación cruzada.
7.19. Para el control de calidad analítica por cada set de análisis llevar en
forma paralela a las muestras, los siguientes controles:
7.19.1. Un blanco reactivo preparado a partir de agua para análisis
grado reactivo exenta de As.
7.19.2. Un ensayo duplicado de una muestra elegida al azar (precisión
mínima 80%; RSD ≤ 20%)
7.19.3. Un ensayo de un estándar de concentración conocida, cercana al
valor medido (exactitud mínima 85-115% como recuperación del
estándar).
Sistema de Generación Continua de Hidruros:
7.20. Encender y ajustar el equipo de acuerdo a las instrucciones descritas
en el manual del equipo y ajustar las condiciones (longitud de onda
principal 193,7 nm y abertura del monocromador slit 0,7 nm).
7.21. Ajustar el flujo de gas (Ar o N2) en el sistema de generación continua
(FIAS)
7.22. Ajustar las velocidades de flujo del reactivo reductor y muestra de
acuerdo a las especificaciones indicadas en el manual del generador
de hidruros (FIAS)
7.23. Verificar la sensibilidad analítica empleando el estándar de calibración
recomendado en el manual de operaciones del equipo.

289
7.24. Construir la curva de calibración empleando un numero adecuado de
estándares, como mínimo blanco y tres concentraciones del rango de
trabajo, recomendándose 5 puntos.
7.25. Verificar la calibración después de la lectura de cada 6 muestras. Si la
calibración permanece constante, continuar con las lecturas de las
siguientes muestras, si la calibración ha variado, recalibrar y chequear
las lecturas de las muestras anteriores.
7.26. Una vez calibrado el instrumento, determinar la concentración de
muestras de acuerdo al siguiente protocolo:
7.26.1. Aspirar el blanco y realizar auto -cero
7.26.2. Aspirar las muestras en estudio, en secuencia de 6 en 6,
verificando la calibración y la estabilidad del cero, entre lotes de
muestras. Repetir las lecturas en caso que sea necesario.
7.26.3. Aspirar agua grado reactivo exenta de As entre muestra y
muestra con el propósito de evitar contaminación cruzada.
7.27. Para el control de calidad analítica por cada set de análisis llevar en
forma paralela a las muestras, los siguientes controles:
7.27.1. Un blanco reactivo preparado a partir de agua para análisis
grado reactivo exenta de As
7.27.2. Un ensayo duplicado de una muestra elegida al azar (precisión
mínima 80%; RSD≤ 20%)
7.27.3. Un ensayo de un estándar de concentración conocida, cercana al
valor medido (exactitud mínima 85-115% como recuperación del
estándar).
8. CALCULOS Y EXPRESION DE RESULTADOS
As ( µµµµg/g) = ((Lect - Bco) x D) / m
donde:

290
As (µg/g) = concentración de As, expresada en µg de As por gramo de
muestra
Lect = lectura de la muestra en µg/mL
Bco = lectura blanco reactivo sometido al mismo proceso de las muestras
D = factor de dilución o concentración
m =masa de muestra (en gramos)
8.1. Si la muestra no acusa presencia de As, se debe informar como el LDM
<0,1 µg/g.
8.2. La posible presencia de materia orgánica constituye un interferente, que
se remueve por digestión de la muestra.
Interferencias:
8.3. No se puede establecer un mecanismo único de eliminación de
interferentes, no obstante se recomienda aplicar los siguientes criterios:
8.3.1. Identificar la naturaleza de la interferencia
8.3.2. La posible presencia de materia orgánica constituye un
interferente, que se remueve por digestión de la muestra.
8.3.3. Si la interferencia es de carácter físico y se manifiesta en el
transporte del hidruro, revisar mangueras a fin de evitar fugas,
reacciones de hidrólisis o cualquier efecto que pudiese producirse
debido a la naturaleza del medio de transporte.
8.3.4. Si la interferencia es de carácter físico y se manifiesta durante la
atomización, verificar la limpieza de la celda de cuarzo. Verificar los
niveles de los otros elementos susceptibles de generar hidruros
volátiles y realizar el análisis con adición estándar.

291
9. REFERENCIAS
9.1. Standard Methods For The Examination Of Water And Wastewater.
APHA. AWWA. WEF. 21th Edition 2005, Part. 3114-B; Arsenic and
Selenium by Hydride Generation/Atomic Absorption Spectrometry.
9.2. Taller Regional Para La Elaboración del Manual de protocolos
Armonizados y Evaluados Para La Toma de Muestras y Análisis de
Aguas y Sedimentos. ARCAL RLA /1/10. El Salvador, 5 – 9 de Mayo del
2008.

292
2.3.1.43. Determinación de Mercurio en sedimentos p or el método de Espectrometría de Absorción Atómica con Generación de Vapor Atómico de Mercurio (Vapor Frío )
PROYECTO ARCAL
RLA/1/010 “Mejora de la gestión de las masas de agua que están contaminadas con
metales”
Código del procedimiento:
PT-MS-10
%ombre del procedimiento: Determinación de Mercurio en sedimentos por el
método de Espectrometría de Absorción Atómica con
Generación de Vapor Atómico de Mercurio (Vapor
Frío)
Fecha de aprobación:
2008-05-02
%umero de revisión:
ninguna
Fecha de revisión:
2008-05-02
1. OBJETIVO
Determinar mercurio total mediante espectrometría de absorción atómica con
generación de Vapor Atómico de Hg (Vapor Frio).
2. ALCANCE
Se aplica a cualquier sedimento y corresponde a la concentración de
mercurio determinada en una muestra previamente digerida.
3. PRINCIPIO DEL METODO
El mercurio presente en una francción de muestra previamente digerida, es
reducido a vapor atómico de mercurio, mediante reacción con cloruro
estannoso en medio ácido, o aplicando el procedimiento de generación
contínua usando solución de boro hidruro de sodio como agente reductor.
El vapor atómico generado es conducido por un gas de arrastre hacia una
celda de cuarzo posicionada en el paso óptico de un espectrofotómetro de
absorción atómica. La celda se encuentra a temperatura ambiente, la
relación entre potencia incidente y potencia transmitida es una medida de la
concentración del elemento en la muestra.
4. EQUIPOS
4.1. Balanza analítica, de sensibilidad igual o mayor a 0,1mg.
4.2. Campana, con sistema de extracción de gases.

293
4.3. Espectrómetro de absorción atómica, provisto de generador de vapor frio,
celdas con ventanas de cuarzo y accesorios necesarios.
4.4. Fuente de ignición, de líneas atómicas de mercurio.
4.5. Sistema Generador de Vapor Frío.
4.6. Plancha calefactora o equivalente, con regulación de temperatura o baño
termoregulado.
5. REACTIVOS Y MATERIALES
5.1. MATERIALES
5.1.1. Material de uso habitual de laboratorio [ej. vasos de precipitado,
matraces, pipetas, y otros].
5.1.2. Recipiente de reacción, matraz erlenmeyer de 250ml, incorporando
tubo de aireación.
5.2. REACTIVOS
5.2.1. Durante el análisis se deben utilizar solamente reactivos de calidad
para análisis (p.a.), y con agua para análisis grado reactivo.
5.2.2. Solución de ácido clorhídrico concentrado, calidad ultrapuro.
5.2.3. Solución de ácido nítrico concentrado, calidad ultrapuro.
5.2.4. Estándar comercial de Hg de 1000 mg/L
5.2.5. Argón o nitrógeno
5.2.6. Acetileno
5.2.7. Borohidruro de sodio
5.2.8. Hidróxido de sodio
5.2.9. Soluciones estándares de calibración: preparar una serie de
soluciones de calibración en el rango óptimo de trabajo, a partir de
diluciones apropiadas del estándar de 1000 mg Hg/L 10% v/v HCl.
Preparar diariamente. Se exige para la curva de calibración un r ≥
0,995.
5.2.10. Solución blanco término cero: la solución término cero debe ser
preparada con agua para análisis grado reactivo exenta de mercurio
y en un medio 10 % HCl v/v. Preparar diariamente.

294
5.2.11. Como agente reductor se utiliza una solución acuosa de NaBH4
al 0,2% en NaOH al 0,05%, debiéndose preparar cada vez que es
utilizado.
5.2.12. Se pesan 0,5 gr de NaOH disolviéndolo en agua Mili Q para
luego añadir 2 gr de NaBH4, posteriormente se filtra y se afora a 1 L.
6. ACCIONES PREVIAS
6.1. Para la toma, transporte y almacenamiento de las muestras se
recomienda utilizar recipientes de plástico.
6.2. Las muestras deben ser digeridas como se indica en el procedimiento
PT-DS-01.
7. DESCRIPCION DEL PROCEDIMIENTO
7.1. Encender el computador, el espectrómetro de absorción atómica y el
sistema de análisis de inyección de flujo FIAS, de acuerdo a las
referencias descritas en el manual de operaciones de su equipo.
7.2. Alinear la posición de la celda de cuarzo y optimizar la intensidad de la
señal ajustando la posición de la lámpara y la longitud de onda (253.7
nm).
7.3. Abrir el programa que pone a disposición el equipo utiizado, encender la
lámpara de mercurio y dejar estabilizar por un tiempo aproximado de 30
minutos para aumentar su energía, encender luego la celda (100ºC).
7.4. Encender el extractor de aire.
7.5. Ajustar las mangueras y dejar por un tiempo prudente el funcionamiento
de las bombas para el lavado con agua de calidad para análisis.
7.6. Llenar los contenedores con la solución de ácido clorhídrico al 10% y
solución de boro hidruro (0.2% en 0.05% de NaOH).
7.7. Ajustar la presión del argón entre 320 y 400 kpa.
7.8. Ajustar la velocidad de flujo sobre 100 ml/min
7.9. Calibrar el equipo con las soluciones estándares de concentración de 10
y 20 µg/L de mercurio. Construir la curva de calibración empleando un

295
numero adecuado de estándares, como mínimo blanco y tres
concentraciones del rango de trabajo, recomendándose 5 puntos.
7.10. Una vez calibrado el instrumento, determinar la concentración de
muestras de acuerdo al siguiente protocolo:
7.10.1. Aspirar el blanco y realizar auto -cero
7.10.2. Aspirar las muestras en estudio, en secuencia de 6 en 6,
verificando la calibración y la estabilidad del cero, entre lotes de
muestras. Repetir las lecturas en caso que sea necesario.
7.10.3. Aspirar agua para análisis grado reactivo exenta de Hg entre
muestra y muestra con el propósito de evitar contaminación cruzada.
7.11. Para el control de calidad analítica por cada set de análisis llevar en
forma paralela a las muestras, los siguientes controles:
7.11.1. Un blanco reactivo preparado a partir de agua para análisis
grado reactivo exenta de Hg.
7.11.2. Un ensayo duplicado de una muestra elegida al azar (precisión
mínima 80%; RSD≤ 20%).
7.11.3. Un ensayo de un estándar de concentración conocida, cercana al
valor medido (exactitud mínima 85-115% como recuperación del
estándar).
7.12. Interferencias:
No se puede establecer un mecanismo único de eliminación de
interferentes, no obstante se recomienda aplicar los siguientes
criterios:
7.12.1. Identificar la naturaleza de la interferencia.
7.12.2. La posible presencia de materia orgánica constituye un
interferente, que se remueve por digestión de la muestra.
7.12.3. Si la interferencia es de carácter físico y se manifiesta en el
transporte del vapor atómico de mercurio, revisar mangueras a fin de
evitar fugas, reacciones de hidrólisis o cualquier efecto que pudiese
producirse debido a la naturaleza del medio de transporte.

296
7.12.4. Si la interferencia es de carácter físico y se manifiesta durante la
atomización, verificar la limpieza de la celda de cuarzo. Si existe
interferencia de matriz, realizar el análisis con adición estándar.
8. CALCULOS Y EXPRESION DE RESULTADOS
Hg( µµµµg/g) = ((Lect - Bco) x D) / m
donde:
µg Hg/g = concentración de Hg, expresada en µg de Hg por gramo de
muestra.
Lect = lectura de la muestra en mg/L.
Bco = lectura blanco reactivo sometido al mismo proceso de las muestras.
D = factor de dilución o concentración
m = masa de muestra (en gramos)
8.1. Si la muestra no acusa presencia de Hg, se debe informar como el LDM
<0,001 mg/L.
8.2. La posible presencia de materia orgánica constituye un interferente, que
se remueve por digestión de la muestra.
9. REFERENCIAS
9.1. Standard Methods for the Examination of Water and Wastewater. APHA.
AWWA. WEF. 21TH Edition 2005, Part 3000. Cold-Vapor Atomic
Absorption Spectrometry Methd 3112 B.
9.2. Taller Regional Para La Elaboración del Manual de protocolos
Armonizados y Evaluados Para La Toma de Muestras y Análisis de

297
Aguas y Sedimentos. ARCAL RLA /1/10. El Salvador, 5 – 9 de Mayo del
2008.





























