Embed Size (px)
Citation preview

PTEN PROMOTER IS METHYLATED IN A PROPORTION OF INVASIVEBREAST CANCERSSalma KHAN
1, Takashi KUMAGAI2,3, Jaimini VORA
1, Namrata BOSE1, Indu SEHGAL
1, Phillip H. KOEFFLER2,3 and Shikha BOSE
1,3
1Department of Pathology, Cedars Sinai Medical Center, Los Angeles, CA, USA2Department of Hematology/Oncology, Cedars Sinai Medical Center, Los Angeles, CA, USA3David Geffen School of Medicine, University of California, Los Angeles, CA
The PTEN protein is a negative regulator of the Akt path-way, leading to suppression of apoptosis and increased cellsurvival. Its role as a tumor-suppressor gene has been ade-quately substantiated, and homozygous mutations have beendemonstrated in familial and sporadic cancers. In breast can-cers, expression of PTEN protein is lost/reduced in 38% ofcases. Somatic mutations are, however, rarely found. Ourstudy was therefore designed to determine if differentialmethylation of the PTEN promoter region has a role in thetranscriptional inactivation of the gene in invasive breastcarcinomas. A total of 44 samples of invasive human breastcancer, 5 breast cancer cell lines and 16 samples of normalhuman breast tissue from young and elderly women werestudied for methylation of the PTEN promoter by methyla-tion-specific PCR and PTEN protein expression by immuno-histochemistry. PTEN methylation occurred in 34% of breastcancers, and 60% of these samples were associated with lossof PTEN protein. Analyzed from a different perspective, 34%of breast cancers had reduced expression of PTEN and 60%had a methylated PTEN promoter. None of the breast cancercell lines and normal breast tissues showed methylation. Insummary, methylation of the PTEN promoter leads to PTENinactivation in a subset of human breast cancers.© 2004 Wiley-Liss, Inc.
Key words: PTEN; methylation; breast cancer
PTEN (MMAC1/TEP1) is a tumor-suppressor gene located onchromosomal subband 10q23.3.1 It has been implicated in a num-ber of familial and sporadic cancers.2 Germline mutations havebeen reported in Cowden syndrome and Bannayan-Zonana syn-drome, 2 dominantly inherited conditions causing predisposition toa variety of cancers including breast cancer.3–5 Homozygous so-matic mutations have also been recorded in a high percentage ofhuman tumors, common among which are glioblastoma multi-forme6–8 and endometrial cancer.9–11 Genetic, functional and an-imal modeling studies have substantiated the tumor-suppressorfunction of PTEN. The gene encodes a PIP3 phosphatase andregulates negatively the PIP3–Akt pathway, leading to suppressionof apoptosis and increased cell survival.12–15
PTEN is involved in breast cancers. We have previously dem-onstrated that expression of PTEN protein is lost/reduced in 38%of invasive breast cancers, a feature that is seen with highestfrequency in high-grade carcinomas.16 Although breast cancersalso show 38% LOH at the PTEN locus,17 somatic mutations haverarely been documented.17–19 Thus, the events leading to loss ofprotein expression remain unclear. The discrepancy noted in thehigh incidence of LOH and/or protein loss and low mutation ratespoints to the possibility of epigenetic mechanisms in the inactiva-tion of PTEN.
The importance of epigenetic changes in human cancers is onlynow being recognized. Of primary interest is aberrant promoter hy-permethylation that leads to inappropriate gene silencing. Hypermeth-ylation of a growing number of genes (Rb, p16INK4a, VHL, E-cad-herin, APC and BRCA1, to name a few) occurs in promoter CpGislands in tumor cells and is being revealed as one of the most frequentmechanisms of loss of gene function.20,21 Epigenetic changes arebelieved to occur at a higher frequency than genetic changes. ThePTEN promoter region has been isolated,22 and preliminary studieshave identified methylation as a mechanism for PTEN inactivation in
lung carcinomas.23 Our study was therefore designed to determine ifepigenetic regulation by differential methylation has a role in thetranscriptional inactivation of the PTEN gene in invasive breast car-cinomas. Normal breast tissue was also tested for methylation of thePTEN promoter and its relationship, if any, to aging.
MATERIAL AND METHODS
Archived tissue blocks from surgical specimens of 50 patientswith invasive breast cancer and 16 with normal breast tissueremoved for reduction mammoplasties and other benign conditionswere retrieved from the surgical pathology files of Cedars SinaiMedical Center after appropriate approval from the InstitutionalReview Board. Cases were randomly selected. Cancer patientsranged in age from 36 to 94 years (median 56). Tumor sizes ranged0.8–15 cm, and 28 tumors were �2 cm in size. Pathologic clas-sification of invasive cancers was ductal (41 cases), lobular (1case), inflammatory (1case) and medullary (1 case). Tumorsranged in differentiation from well to poor. ER was positive in77%, PR was positive in 68% and Her2/neu overexpression wasobserved in 24% of invasive cancers, 75% of which had Her2/neugene amplification. As determined by MIB-1 IHC, 36% of tumorshad a low, 32% had an intermediate and 32% had a high prolif-eration index. Normal breast tissue was selected from both youngand older women. Six women were less than 20 years of age,whereas the remaining 10 were older than 50 years.
Invasive cancers and normal tissues were tested for the presenceof methylated CpG islands in the PTEN promoter region usingMSP24 after bisulfite modification of tumor DNA.25 The findingswere correlated with PTEN protein expression as determined byIHC. Additionally, 5 breast cancer cell lines (MCF-7, MDA-MB-231, MCF-12A, BT474 and BT549) obtained from the ATCC(Manassas, VA) were analyzed for the presence of promoter meth-ylation by MSP.
MSPGenomic DNA was isolated from 10-�-thick tissue sections
after manual microdissection of invasive cancer cases, to enrich
Abbreviations: ER, estrogen receptor; IHC, immunohistochemistry;LOH, loss of heterozygosity; MSP, methylation-specific PCR; NSCLC,non-small cell lung cancer; PIP3, phosphatidylinositol 3-phosphate; PR,progesterone receptor.
Grant sponsor: California Breast Cancer Research Program; Grant num-ber: 8KB-0011.
*Correspondence to: Department of Pathology, South Tower 8732,Cedars Sinai Medical Center, 8400 Beverly Blvd., Los Angeles, CA 90048,USA. Fax: �310-423-0122. E-mail: [email protected]
Received 2 March 2004; Accepted 12 May 2004
DOI 10.1002/ijc.20447Published online 29 June 2004 in Wiley InterScience (www.interscience.
wiley.com).
Int. J. Cancer: 112, 407–410 (2004)© 2004 Wiley-Liss, Inc.
Publication of the International Union Against Cancer
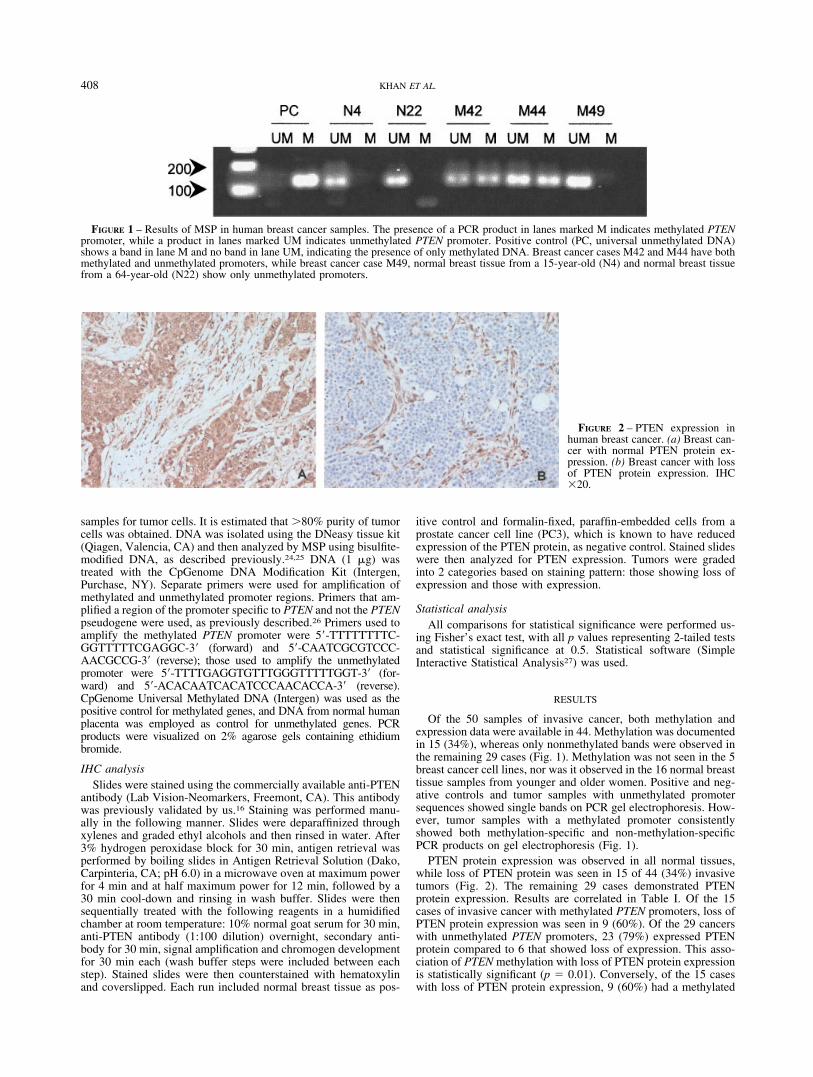
samples for tumor cells. It is estimated that �80% purity of tumorcells was obtained. DNA was isolated using the DNeasy tissue kit(Qiagen, Valencia, CA) and then analyzed by MSP using bisulfite-modified DNA, as described previously.24,25 DNA (1 �g) wastreated with the CpGenome DNA Modification Kit (Intergen,Purchase, NY). Separate primers were used for amplification ofmethylated and unmethylated promoter regions. Primers that am-plified a region of the promoter specific to PTEN and not the PTENpseudogene were used, as previously described.26 Primers used toamplify the methylated PTEN promoter were 5�-TTTTTTTTC-GGTTTTTCGAGGC-3� (forward) and 5�-CAATCGCGTCCC-AACGCCG-3� (reverse); those used to amplify the unmethylatedpromoter were 5�-TTTTGAGGTGTTTGGGTTTTTGGT-3� (for-ward) and 5�-ACACAATCACATCCCAACACCA-3� (reverse).CpGenome Universal Methylated DNA (Intergen) was used as thepositive control for methylated genes, and DNA from normal humanplacenta was employed as control for unmethylated genes. PCRproducts were visualized on 2% agarose gels containing ethidiumbromide.
IHC analysisSlides were stained using the commercially available anti-PTEN
antibody (Lab Vision-Neomarkers, Freemont, CA). This antibodywas previously validated by us.16 Staining was performed manu-ally in the following manner. Slides were deparaffinized throughxylenes and graded ethyl alcohols and then rinsed in water. After3% hydrogen peroxidase block for 30 min, antigen retrieval wasperformed by boiling slides in Antigen Retrieval Solution (Dako,Carpinteria, CA; pH 6.0) in a microwave oven at maximum powerfor 4 min and at half maximum power for 12 min, followed by a30 min cool-down and rinsing in wash buffer. Slides were thensequentially treated with the following reagents in a humidifiedchamber at room temperature: 10% normal goat serum for 30 min,anti-PTEN antibody (1:100 dilution) overnight, secondary anti-body for 30 min, signal amplification and chromogen developmentfor 30 min each (wash buffer steps were included between eachstep). Stained slides were then counterstained with hematoxylinand coverslipped. Each run included normal breast tissue as pos-
itive control and formalin-fixed, paraffin-embedded cells from aprostate cancer cell line (PC3), which is known to have reducedexpression of the PTEN protein, as negative control. Stained slideswere then analyzed for PTEN expression. Tumors were gradedinto 2 categories based on staining pattern: those showing loss ofexpression and those with expression.
Statistical analysisAll comparisons for statistical significance were performed us-
ing Fisher’s exact test, with all p values representing 2-tailed testsand statistical significance at 0.5. Statistical software (SimpleInteractive Statistical Analysis27) was used.
RESULTS
Of the 50 samples of invasive cancer, both methylation andexpression data were available in 44. Methylation was documentedin 15 (34%), whereas only nonmethylated bands were observed inthe remaining 29 cases (Fig. 1). Methylation was not seen in the 5breast cancer cell lines, nor was it observed in the 16 normal breasttissue samples from younger and older women. Positive and neg-ative controls and tumor samples with unmethylated promotersequences showed single bands on PCR gel electrophoresis. How-ever, tumor samples with a methylated promoter consistentlyshowed both methylation-specific and non-methylation-specificPCR products on gel electrophoresis (Fig. 1).
PTEN protein expression was observed in all normal tissues,while loss of PTEN protein was seen in 15 of 44 (34%) invasivetumors (Fig. 2). The remaining 29 cases demonstrated PTENprotein expression. Results are correlated in Table I. Of the 15cases of invasive cancer with methylated PTEN promoters, loss ofPTEN protein expression was seen in 9 (60%). Of the 29 cancerswith unmethylated PTEN promoters, 23 (79%) expressed PTENprotein compared to 6 that showed loss of expression. This asso-ciation of PTEN methylation with loss of PTEN protein expressionis statistically significant (p � 0.01). Conversely, of the 15 caseswith loss of PTEN protein expression, 9 (60%) had a methylated
FIGURE 1 – Results of MSP in human breast cancer samples. The presence of a PCR product in lanes marked M indicates methylated PTENpromoter, while a product in lanes marked UM indicates unmethylated PTEN promoter. Positive control (PC, universal unmethylated DNA)shows a band in lane M and no band in lane UM, indicating the presence of only methylated DNA. Breast cancer cases M42 and M44 have bothmethylated and unmethylated promoters, while breast cancer case M49, normal breast tissue from a 15-year-old (N4) and normal breast tissuefrom a 64-year-old (N22) show only unmethylated promoters.
FIGURE 2 – PTEN expression inhuman breast cancer. (a) Breast can-cer with normal PTEN protein ex-pression. (b) Breast cancer with lossof PTEN protein expression. IHC�20.
408 KHAN ET AL.

PTEN; while of the 29 cases showing PTEN expression, 6 (21%)were methylated.
No significant correlations were observed in tumor characteris-tics (size, histologic grade, mitotic rate, proliferation index, pres-ence of axillary metastasis, presence of ERs and PRs and overex-pression/amplification of Her2/neu gene) between cancers withmethylated promoters and cancers with unmethylated promoters.However, when the above parameters were compared in protein-expressing and -nonexpressing breast cancers with methylatedpromoters, some differences were observed (Table II). All of the 6PTEN-expressing cancers expressed both ER and PR. Of the 9PTEN-nonexpressing cancers with methylated promoter, 4 werenegative for ER and 6 were negative for PR. Values for ER did notreach statistical significance (p � 0.09); however, the findings forPR were statistically significant (p � 0.016).
DISCUSSION
A detailed analysis of the PTEN promoter region has not beenperformed in breast cancer. Studies of the PTEN promoterregion have been restricted due to the strong homology with ahighly conserved PTEN processed pseudogene (GenBank ac-cession AF040103, PTEN processed pseudogene AF029308,Homo sapiens chromosome 9 duplication of the T-cell receptor� locus and trypsinogen gene families), which shares over 98%homology with the coding region of functional PTEN. Thisgene is located on chromosome 9p21.28 Examination of the 2sequences indicates that the identity extends – 841 nucleotidesupstream of the PTEN translation start site. This high degree ofhomology has made analysis of the methylation status of thePTEN promoter quite challenging, and careful consideration ofcritical nucleotide differences is required for the design ofappropriate primers for analysis. Initial studies were performedusing primer sequences located in the region of homology.29,30
These revealed methylation of invasive tumors; however, in astudy of the methylation profile of the PTEN promoter inendometrial, colon and breast cancer cell lines, Zysman et al.26
showed that it is the PTEN pseudogene, and not the PTEN gene,that is predominantly methylated in cell lines. In their study, 2of the 3 breast cancer cell lines that were positive for methyl-ation with primers common to both PTEN and PTEN pseudo-gene were unmethylated with primers specific for PTEN. Sim-ilarly, Sato et al.31 demonstrated that gastric cancers, whichwere previously reported to show methylation of the PTENpromoter,30 did not in fact contain a methylated PTEN pro-moter. However, using primers specific for the PTEN gene, thepromoter has been found to be methylated in lung cancers. Soriaet al.23 analyzed 30 cases of NSCLC for PTEN promotermethylation and concomitant PTEN expression by IHC. PTENexpression was lost in 24% of NSCLCs. PTEN methylation wasdetected in 7 (35%) of the 20 PTEN-nonexpressing carcinomasand 0 of the 10 PTEN-positive samples. Furthermore, PTENmethylation was observed in 11 (69%) of the 16 NSCLC celllines. Also, levels of PTEN mRNA increased in an NSCLC cellline (NCI-H1299) cultured with 5-aza-2�-deoxycytidine, a dem-ethylating agent. Our data in breast cancer samples are similar.
Of the 44 invasive ductal carcinomas of the breast, PTENpromoter methylation occurred in 34%, of which 60% wereassociated with loss of PTEN protein expression. Of the 29cases without PTEN promoter methylation, 23 (79%) expressedPTEN protein. Promoter methylation, therefore, correlates sig-nificantly with loss of PTEN protein expression (p � 0.01). Sixcases had an unmethylated PTEN promoter but also had loss ofPTEN protein expression, indicating that other inactivatingmechanisms are also at play. These include LOH associatedwith inactivating mutation of the second allele, although thishas been demonstrated in only a minority of breast cancers(6%).17 Subsequent studies in animal models have suggestedthat haploinsufficiency (i.e., loss of a single PTEN allele) alonemay be sufficient for inducing protein loss and tumor progres-sion.32,33 Although definitive studies are lacking in breast can-cer, that this may occur in humans is suggested by the fact thatheterozygous inactivation of PTEN results in 3 dominantlyinherited disorders: Cowden syndrome (associated with a pre-disposition to thyroid and breast cancers), Bannayan-Zonanasyndrome and Lhermitte-Duclos syndrome.3–5 Of course, thismore likely suggests that it is easier to mutate the second alleleonce the first allele is lost.
Interestingly, 6 of the 15 cases with methylated promoters(40%) expressed PTEN protein. We hypothesize that this occursas a result of incomplete methylation at the promoter site. MSPis an extremely sensitive method for the study of CpG islandhypermethylation from small amounts of genomic DNA. Her-man et al.24 were consistently able to detect 0.1% of methylatedDNA in an otherwise unmethylated sample. Thus, perhapstumor cells need to achieve a certain level of methylation priorto translational inactivation. This hypothesis is in concordancewith the view proposed by Turker34 that gene silencing is not asingle event but instead a series of events that begins with amarked drop in transcription potential and ends with its com-plete cessation. This transition is portrayed as a chaotic processthat ensues when transcription levels drop and DNA methyl-ation begins, spreading haltingly toward the diminished pro-moter. According to this view, silencing is stabilized when thepromoter region is “captured” by the spread of DNA methyl-ation near to or into its transcription factor binding sites. Thepresence of incomplete PTEN promoter methylation may alsoexplain the band of unmethylated product in MSP in tumorsamples with methylated bands. However, this band may rep-resent contamination of the breast cancer sample by normalcells. Our finding of loss of ERs and PRs in PTEN-nonexpress-ing breast cancers with methylated promoters also supports theabove hypothesis. Loss of ERs and PRs occurs with tumorprogression, and this is observed in methylated tumors showingloss of PTEN protein expression as opposed to methylatedcancers without loss of PTEN protein expression.
We did not observe methylation in any of the normal samples,including those from older women. This indicates that methylation ofthe PTEN promoter is not related to aging but occurs specificallyduring carcinogenesis. Moreover, our study also did not reveal meth-ylation in 5 breast cancer cell lines. Two of these (MCF-7 andMDA-MB-231) have been previously analyzed with similar results.26
Although our sample size of cell lines is small and may not berepresentative of all breast cancer cell lines, it is also possible that
TABLE II – CORRELATION OF ER AND PR STATUS AND LOSS OF PTENPROTEIN EXPRESSION IN INVASIVE BREAST CANCERS WITH
METHYLATED PTEN PROMOTER
Breast cancers with methylatedPTEN promoter
Receptor status in invasivebreast cancer
ER� ER� PR� PR�
PTEN protein expressionPresent (6) 6 0 6 0Absent (9) 5 4 3 6
p value (Fisher’s exact test) 0.09 0.016
TABLE I – CORRELATION OF PTEN PROMOTER METHYLATION STATUSWITH PROTEIN EXPRESSION IN INVASIVE BREAST CANCER
PTEN promoterPTEN protein expression
Total (%)Present Absent
Unmethylated 23 6 29 (66)Methylated 6 91 15 (34)Total (%) 29 (66) 15 (34) 44 (100)1Of PTEN methylated cases, 9/15 (60%) were associated with
PTEN protein loss compared to 23/29 (79%) PTEN unmethylatedcases that expressed PTEN protein. Results are statistically significant(p � 0.01).
409PTEN PROMOTER METHYLATION IN BREAST CANCER

PTEN promoter methylation in breast cancer is an in vivo process; andthis may highlight a difference between in vivo and in vitro tumorgrowth patterns. A situation similar to this was observed by Esteller etal.35 Hypermethylation of the BRCA-1 promoter was found in 13% ofunselected breast cancer samples in their study. However, none of the21 breast cancer cell lines as well as samples of normal breast tissuerevealed abnormal methylation of the BRCA-1 gene.
In summary, our data show that methylation of the PTENpromoter in a subset of breast carcinomas is associated with aprominent reduction of PTEN protein levels in these samples.Some breast cancer samples do not express PTEN, but the pro-moter of the gene coding for this protein is not methylated,suggesting that other genetic/epigenetic events may have a role ingene inactivation.
REFERENCES
1. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang S, Puc J,Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, etal. PTEN, a putative protein tyrosine phosphatase gene mutated inhuman brain, breast, and prostate cancer. Science 1997;275:1943–7.
2. Waite KA, Eng C. Protean PTEN: form and function. Am J HumGenet 2002;70:829–44.
3. Liaw D, Marsh DJ, Li J, Dahia PLM, Wang SI, Zheng Z, Bose S, CallKM, Tsou HC, Peacocke M, Eng C, Parsons R. PTEN gene inCowden disease, an inherited breast and thyroid cancer syndrome. NatGenet 1997;16:64–7.
4. Marsh DJ, Coulon V, Lunetta KL, Rocca-Serra P, Dahia PL, Zheng Z,Liaw D, Caron S, Duboue B, Lin AY, Richardson AL, BonnetblancJM, et al. Mutation spectrum and genotype–phenotype analyses inCowden disease and Bannayan-Zonana syndrome, two hamartomasyndromes with germline PTEN mutation. Hum Mol Genet 1998;7:507–15.
5. Eng C, Peacocke M. PTEN and inherited hamartoma-cancer syn-dromes. Nat Genet 1998;19:223.
6. Durr E-M, Rollbrocker B, Hayashi Y, Peters N, Meyer-Puttlitz B,Louis DN, Schramm J, Wiestler OD, Parsons R, Eng C, von DeimlingA. PTEN mutations in gliomas and glioneuronal tumors. Oncogene1998;16:2259–64.
7. Rasheed BKA, Stenzel TT, McLendon RE, Parsons R, Friedman AH,Friedman HS, Bigner DD, Bigner SH. PTEN gene mutations are seenin high-grade but not in low-grade gliomas. Cancer Res 1997;37:4187–90.
8. Wang SI, Puc J, Li J, Bruce JN, Cairns P, Sidransky D, Parsons R.Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res1997;57:4183–6.
9. Tashiro H, Blazes MS, Wu R, Cho KR, Bose S, Wang SL, Si J,Parsons R, Ellenson LH. Mutations in PTEN are frequent in endome-trial carcinoma but are rare in other common gynecological malig-nancies. Cancer Res 1997;57:3935–40.
10. Bussaglia E, del Rio E, Matias-Guiu X, Prat J. PTEN mutations inendometrial carcinomas: a molecular and clinicopathologic analysisof 38 cases. Hum Pathol 2000;31:312–7.
11. Ali IU. Gatekeeper for endometrium: the PTEN tumor suppressorgene. J Natl Cancer Inst 2000;92:861–3.
12. Maehama T, Dixon JE. PTEN: a tumour suppressor that functions asa phospholipid phosphatase. Trends Cell Biol 1999;9:125–8.
13. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, de-phosphorylates the lipid second messenger, phosphatidylinositol3,4,5-trisphosphate. J Biol Chem 1998;273:13375–8.
14. Lu Y, Lin YZ, LaPushin R, Cuevas B, Fang X, Yu SX, Davies MA,Khan H, Furui T, Mao M, Zinner R, Hung MC, et al. The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and in-duces apoptosis and anoikis in breast cancer cells. Oncogene 1999;18:7034–45.
15. Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C,Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negativeregulation of PKB/Akt-dependent cell survival by the tumor suppres-sor PTEN. Cell 1998;95:29–39.
16. Bose S, Crane A, Hibshoosh H, Mansukhani M, Sandweis L, ParsonsR. Reduced expression of PTEN correlates with breast cancer pro-gression. Hum Pathol 2002;33:405–9.
17. Bose S, Wang SI, Terry MB, Hibshoosh H, Parsons R. Allelic loss ofchromosome 10q23 is associated with invasion in breast carcinomas.Oncogene 1998;17:123–7.
18. Rhei E, Kang L, Bogomolinly F, Federici MG, Borgen PI, Boyd J.Mutation analysis of the putative tumor suppressor gene PTEN/MMAC1 in primary breast carcinomas. Cancer Res 1997;57:3657–9.
19. Freihoff D, Kempe A, Beste B, Wappenschmidt B, Kreyer E, HayashiY, Meindl A, Krebs D, Wiestler OD, von Deimling A, SchmutzlerRK. Exclusion of a major role for the PTEN tumour-suppressor genein breast carcinomas. Br J Cancer 1999;79:754–8.
20. Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis:epigenetics joins genetics. Trends Genet 2000;16:168–74.
21. Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet1999;21:163–7.
22. Sheng X, Koul D, Liu JL, Liu TJ, Yung WK. Promoter analysis oftumor suppressor gene PTEN: identification of minimum promoterregion. Biochem Biophys Res Commun 2002;292:422–6.
23. Soria JC, Lee HY, Lee JI, Wang L, Issa JP, Kemp BL, Liu DD, KurieJM, Mao L, Khuri FR. Lack of PTEN expression in non-small celllung cancer could be related to promoter methylation. Clin Cancer Res2002;8:1178–84.
24. Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methy-lation-specific PCR: a novel PCR assay for methylation status of CpGislands. Proc Natl Acad Sci USA 1996;93:9821–6.
25. Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mappingof methylated cytosines. Nucleic Acids Res 1994;22:2990–7.
26. Zysman MA, Chapman WB, Bapat B. Considerations when analyzingthe methylation status of PTEN tumor suppressor gene. Am J Pathol2002;160:795–800.
27. Uitenbroek DG. SISA-binomial. 1997. (http://homeclaranet/sisa/bino-mialhtm) Accessed 1 January 2002.
28. Dahia PL, FitzGerald MG, Zhang X, Marsh DJ, Zheng Z, Pietsch T,von Deimling A, Haluska FG, Haber DA, Eng C. A highly conservedprocessed PTEN pseudogene is located on chromosome band 9p21.Oncogene 1998;16:2403–6.
29. Salvesen HB, MacDonald N, Ryan A, Jacobs IJ, Lynch ED, AkslenLA, Das S. PTEN methylation is associated with advanced stage andmicrosatellite instability in endometrial carcinoma. Int J Cancer 2001;91:22–6.
30. Kang YH, Lee HS, Kim WH. Promoter methylation and silencing ofPTEN in gastric carcinoma. Lab Invest 2002;82:285–91.
31. Sato K, Tamura G, Tsuchiya T, Endoh Y, Sakata K, Motoyama T,Usuba O, Kimura W, Terashima M, Nishizuka S, Zou T, Meltzer SJ.Analysis of genetic and epigenetic alterations of the PTEN gene ingastric cancer. Virchows Arch 2002;440:160–5.
32. Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB,Pandolfi PP. Impaired Fas response and autoimmunity in Pten�/�
mice. Science 1999;285:2122–5.33. Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada
KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R. Mutation ofPten/Mmac1 in mice causes neoplasia in multiple organ systems. ProcNatl Acad Sci USA 1999;96:1563–8.
34. Turker MS. Gene silencing in mammalian cells and the spread ofDNA methylation. Oncogene 2002;21:5388–93.
35. Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, LermaE, Bussaglia E, Prat J, Harkes IC, Repasky EA, Gabrielson E, SchutteM, et al. Promoter hypermethylation and BRCA1 inactivation insporadic breast and ovarian tumors. J Natl Cancer Inst 2000;92:564–9.
410 KHAN ET AL.





























