Embed Size (px)
Citation preview

THE JOURNAL OF BIOLOGICAL CHEMISTRY 0 1993 by The American Society for Biochemistry and Molecular Biology, Inc
Vol 268. No. 5. Issue of February 15. pp. 3161-3167,1993 Printed in U. S. A.
Purification and Analysis of Streptococcal NADH Peroxidase Expressed in Escherichia coli*
(Received for publication, July 29, 1992)
Derek Parsonage$, Holly Miller, R. Paul Ross, and A1 Claibornes From the Department of Biochemistry, Wake Forest University Medical Center, Winston-Salem, North Carolina 27157-1016
Using the T7 RNA polymerase expression system, a modified plasmid vector has been developed which gives reliable, high level expression in Escherichia coli of the gene encoding streptococcal NADH peroxidase. The recombinant enzyme has been purified to homo- geneity using a revised protocol which yields over 35 mg of pure protein per liter of culture. Recombinant NADH peroxidase is fully active and exhibits spectro- scopic and redox properties identical to those for the enzyme purified from Streptococcus faecalis 1OC 1. Re- ductive titrations and thiol analyses confirm the pres- ence of the unusual cysteine-sulfenic acid (Cys-SOH) redox center identified previously. N-terminal se- quence analysis, analytical gel filtration, and prelimi- nary x-ray diffraction data all confirm the structural identity of the recombinant and S. faecalis enzymes. Steady-state kinetic analysis of the peroxidase, cou- pled with results from static titration experiments is consistent with a limiting type of ternary complex mechanism and allows the determination of many of the corresponding kinetic constants. In addition, pre- liminary ‘H NMR spectra of the enzyme at millimolar concentrations show good dispersion in the amide re- gion and indicate that the recombinant peroxidase is suitable for one-dimensional NMR work with labeled amino acids.
The streptococcal flavoprotein NADH peroxidase catalyzes the reduction of HzOZ + 2H20 with reducing equivalents derived from the pyridine nucleotide substrate; as such, the enzyme represents one of two known flavin-linked peroxide reductases (1,2). In addition to FAD, the peroxidase contains a stabilized cysteine-sulfenic acid (Cys-SOH) redox center which cycles catalytically between Cys-SOH and Cys-SH states (3, 4). The npr gene encoding the pero3idase has been cloned and sequenced (5), and a refined 2.16-A structure has been determined for the enzyme purified from Streptococcus (now Enterococcus) faecalis lOCl (6). While the sequence reveals rather limited homology to glutathione reductase and the flavoprotein disulfide reductases, the crystal structure is strongly suggestive of an evolutionary relationship, given the similarities in overall chain folds and domain structures. The
* This work was supported by National Institutes of Health Grant GM-35394 and by a grant-in-aid from the American Heart Associa- tion. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
$ To whom correspondence should be addressed: Dept. of Biochem- istry, Wake Forest University Medical Ctr., Medical Center Blvd., Winston-Salem, NC 27157-1016. Tel.: 919-716-2575; Fax: 919-716- 7671.
J Established Investigator of the American Heart Association.
redox-active Cys4’ of the peroxidase, for example, is super- imposable with the charge-transfer of human glutathi- one reductase, and the respective FAD coenzymes are bound in similar regions and in similar conformations.
Two key areas for future emphasis with the peroxidase involve the kinetic and chemical mechanism and further structural analysis of the Cys-SOH redox center. Stoll and Blanchard (7), in work with the commercially available per- oxidase from Streptococcus fuecium (now Enterococcus hirae, ATCC 8043), concluded that the enzyme followed a ping pong kinetic mechanism, on the basis of initial velocity measure- ments and dead-end inhibition analyses. In a departure from the classical ping pong sequence, however, the binary EH2. NADH’ complex was proposed to play a central catalytic role. And, while the refined crystal structure of the S. faecalis enzyme is consistent with an active-site Cys-SOH redox cen- ter in the native state, the structure actually shows further oxidation of the Cys4* side chain, yielding the non-native Cys- S03H derivative.
Three particularly promising approaches to these remaining questions include rapid-reaction kinetics, solution NMR, and resonance Raman analyses, all of which require large amounts of protein. In this report we describe a protocol which allows optimized, large-scale expression and purification of the re- combinant peroxidase. In addition, we provide a revised steady-state kinetic mechanism for the S. faecalis lOCl en- zyme and demonstrate that the recombinant protein is suit- able for solution NMR studies.
EXPERIMENTAL PROCEDURES
Materials-IPTG was purchased from 5Prime -+ 3Prime, Inc., agarose from FMC Bioproducts, and streptomycin sulfate from Sigma. All other chemicals, as purchased from sources previously described, were of the best grades available. pBluescript was pur- chased from Stratagene, pSP73 from Promega, and PET-11 was from Novagen. DNA modification and restriction enzymes were obtained from Promega and were used as recommended by the manufacturer.
Bacterial Strains-Escherichia coli XL1-Blue (Stratagene) and JM109DE3 (8) were used as hosts. Cultures were grown with shaking at 37 “C in Luria-Bertani broth, 2xTY or TYP (16 g of Bacto Tryptone, 16 g of Bacto Yeast Extract, 5 g of NaC1, and 2.5 g of KzHPO, per liter) media. 25 pg/ml of chloramphenicol and 50 Kg/m1 of ampicillin were used as required. For expression experiments, TYP medium was supplemented with 30 mM glucose. DNA isolation and manipulation followed protocols previously described (5, 9).
Engineering of the Gene for NADH Peroxidase-Plasmid pNPR4, containing the gene for streptococcal NADH peroxidase (5), was digested with Sal1 and Sac1 to release a 1.87-kb fragment containing the npr gene. This fragment was ligated into pSP73C, a derivative of
The abbreviations used are: EH2, two-electron reduced enzyme; E, oxidized enzyme; IPTG, isopropyl-P-D-thiogalactopyranoside; TY, tryptone-yeast extract medium; TYP, tryptone-yeast extract-phos- phate medium; kb, kilobase(s); bp, base pair(s); MES, 2-(4-morpho- 1ino)ethanesulfonic acid MOPS, 3-(4-morpholino)propanesulfonic acid.
3161

3162 Characterization of Recombinant S. faecalis NADH Peroxidase
pSP73 in which the 8-lactamase gene had been replaced by the cat gene from pACYC184 (10). The resultant plasmid was designated pNPX8 (Fig. 1A). During construction of pSP73C the HindIII, PstI, and SphI restriction sites were removed by digesting with Pstl and XhoI, then blunt ends were created by a 5-min incubation with T4 DNA polymerase before adding dNTPs to fill in any ragged ends prior to religation. In this way the XhoI site was recreated in the vector at the site of ligation.
In order to remove the npr promoter sequence from pNPX8, the 402-bp Ssp1 fragment from pNPR4 was isolated and ligated into pBluescript I1 SK-, which had been digested with XhoI, made blunt- ended with the T4 DNA polymerase and dNTPs, and dephosphoryl- ated using calf intestinal alkaline phosphatase. The resultant plas- mids were screened by restriction digestion, and a clone (pNPR5) containing the insert in the correct orientation was then digested with Sal1 and SphI to produce a fragment containing only the npr ribosome-binding site and the N-terminal sequence of the npr struc- tural gene. This Sall-SphI fragment was then used to replace the equivalent fragment in pNPX8 to produce plasmid pNPX9, in which the ribosome-binding site of the npr gene is positioned 36 bp down- stream of the T7 promoter (Fig. 1B).
Construction of a T7 Expression Vector, pOX04”In order to re- move the EcoRI site from the cat gene, the 1.63-kb HincII-AccI fragment from pACYC184 was made blunt-ended by treatment with T 4 DNA polymerase and dNTPs and then ligated with the 1.95-kb BspHI fragment (filled in with T4 DNA polymerase and dNTPs) from pBluescriptII SK-. After transformation of the ligated DNA into competent XL1-Blue cells, plasmid DNA from chloramphenicol- resistant colonies was prepared and checked by restriction digests to ensure that the construction was correct. Single-stranded phagemid DNA was prepared by infection with helper phage M13K07 and then used as template for oligonucleotide-directed mutagenesis. The oli-
gonucleotide 5’-CATCCGGAGTTCCGTATGG-3’ was synthesized to cause the silent mutation GAA + GAG specifying glutamate, hence removing the EcoRI recognition sequence. Oligonucleotide- directed mutagenesis was carried out using the kit from Amersham except that T7 DNA polymerase (New England Biolabs) was used for the initial elongation of the mutagenic primer. Annealing of the phosphorylated primer to the single-stranded template was carried out in 5 pl of 104 mM Tris-HC1, pH 7.5, containing 26 mM MgC12 and 130 mM NaCI, by heating to 65 “C for 2 min followed by slow cooling to room temperature. The annealed primer-template mix was then supplemented with 1 p1 of 0.1 M dithiothreitol, 4.75 p1 of nucleotide mix 1 from the Amersham kit, 5 units of T7 DNA polymerase, and 1.5 units T4 DNA ligase, and the volume was made up to 13 p1 with 0.1 mg/ml bovine serum albumin. Elongation and ligation were car- ried out a t 37 “C for 3 h. The remaining steps of the mutagenesis protocol were continued as per the manufacturer’s instructions, with appropriate reduction in all reagent volumes. Successfully mutated plasmids were identified by restriction digests, and one was used as the source of a 1.17-kb HhaI fragment carrying the mutated cat gene to construct a new vector (pOXO3) by ligation with the 1.35-kb BspHI fragment of pSP73, after both fragments were made blunt- ended using T4 DNA polymerase and dNTPs. The T7 transcription terminator T@ sequence from PET-11 was isolated with BamHI and BglII and ligated into BglII-digested and dephosphorylated pOXO3 to generate the expression vector, pOXO4. The T@ termination sequence limits transcription from the T7 promoter solely to se- quences cloned into the multiple-cloning site of pOXO4. The struc- tural gene and ribosome-binding site for npr were excised from pNPX9 with XhoI and EcoRI and ligated into pOXO4 between the T7 Dromoter and termination seauences. The resultant expression plasmid pNPX12 (Fig. 1C) was used in this study.
Expression of Recombinant NADH Peroxidase-The
EmR 10.02 0.03 “I
plasmid
1.51
FIG. 1. Construction of expres- sion plasmids for NADH peroxidase. A , pNPX8 was constructed by ligating a 1.87-kb SalI-Sac1 fragment of pNPR4 to similarly cleaved pSP73C. B, pNPX9 was constructed from pNPR4 and B pNPX8 as described under “Experimen- s s p I fragment of pNPR4 tal Procedures.” C , pNPX12 was con- structed from pNPX9 and the general
S O C I 0.03
expression the T7 termination plasmid pOXO4. sequence T@ refers cloned to + pNPR5 7 from PET-11, and the cat gene lacks the EcoRI site found in pNPX8 and pNPX9.
Xho I - fi l led
pBluescript II SK (-) pNPX8
C %I-EcoRI
pNPX9
pOXO4 X h l 1.82

Characterization of Recombinant S. faecalis NADH Peroxidase 3163
pNPX12 was transformed into E. coli JM109DE3, which contains a chromosomal copy of the T7 RNA polymerase gene under the control of a lacUV5 promoter (8). Freshly transformed cells were grown in 50 ml of TYP medium containing 30 rg/ml chloramphenicol and 30 mM glucose. After reaching late-log phase, 20 ml of these starter cultures were used to inoculate 2.8-1 Fernbach flasks containing 500 ml of the same medium. Flasks were shaken vigorously at 37 "C until an A m of 1.3 was reached, and expression was induced by addition of 0.4 mM IPTG (final concentration). Four h later cells were harvested, washed with 50 mM phosphate, pH 7.0, plus 0.6 mM EDTA, and frozen at -80 "C.
Purification of Recombinant NADH Peroxidase-All buffers used for purification contained 0.6 mM EDTA, all steps were carried out a t 4 "C, and all ammonium sulfate concentrations refer to percent saturation at 25 'C. For purification of the enzyme, 15 g of cells (wet weight; from 3 liters of culture) were thawed and resuspended in 90 ml of 50 mM potassium phosphate, pH 7.0. Equal volumes of cell suspension and wet glass beads were placed in the 85-ml chamber of a Bead-Beater (Biospec Products); breaking was done in two batches. The chamber, which had been precooled at -20 "C for 15 min, was surrounded by an ice/salt bath to maintain the temperature at or below 4 "C. Cells were disrupted with three 1-min pulses with inter- vening 1-min cooling periods. After decanting the extract, the beads were rinsed with 45 ml of phosphate buffer. The combined extract was centrifuged to remove cell debris. Streptomycin sulfate was then added to a concentration of 2.5% (w/v), and the suspension was stirred for 1 h to precipitate nucleic acids. The supernatant from a 20-min (30,000 X g) spin was brought to 50% saturation with solid ammonium sulfate and stirred for 30 min. Precipitated proteins were removed by centrifugation, and the supernatant was applied to a 20- ml Phenyl Sepharose 6 Fast Flow (Pharmacia LKB Biotechnology Inc.) column equilibrated with 52% saturated ammonium sulfate in 50 mM potassium phosphate, pH 7.0. Column chromatography was carried out using a Pharmacia fast-protein liquid chromatography system. The column was washed with the equilibration buffer until the AZm of the eluate was less than 0.1. A 150-ml gradient from 52 to 0% saturated ammonium sulfate was used to elute the peroxidase. Peak fractions (4 ml each) with 450-nm absorbance greater than 0.2 were pooled and dialyzed twice against 4 liters of 0.2 M NaCl in 50 mM potassium phosphate, pH 7.0. The dialyzed protein was loaded onto a 50-ml Q Sepharose Fast Flow column equilibrated with the same buffer as that used for dialysis. After extensive washing, the major yellow protein was eluted from the column by applying a 250- ml gradient from 0.2 to 0.4 M NaCl in 50 mM potassium phosphate, pH 7.0. Peak fractions with A280/A450 < 9 were pooled and dialyzed against 50 mM potassium phosphate, pH 7.0, buffer and then concen- trated to -9 mg/ml by ultrafiltration over a YM-30 membrane (Amicon). Concentrated enzyme was stored in aliquots at -20 "C and retained full activity for at least 6 months.
Steady-State Kinetics-Initial velocity studies with purified NADH peroxidase followed assay procedures described previously (11). The kinetic parameters at pH 5.5 were obtained by varying both NADH and Hz02 concentrations in an assay buffer of 0.1 M potassium acetate/O.l M MES, plus 0.3 mM EDTA, at 25 "C. Data were analyzed as described by Cornish-Bowden (12) as follows.
u = Vab
K i a + KAb + ab
where u is the initial velocity, superscripts A and B represent NADH and H202, respectively, a and b represent the corresponding concen- trations, and V represents V,,,,,. Primary plots of a/u uersus a intersect on the y-axis in the case of a substituted-enzyme mechanism, and the intercept equals KA/V. A secondary plot of (slope X b ) uersus b gives a y-axis intercept of Ki/V and has a slope equal to 1/V. Apparent kinetic parameters were determined at pH 7.5 in a buffer of 0.1 M potassium acetate/O.l M MOPS, plus 0.3 mM EDTA, by varying [NADH] at 1.3 mM H202 and by varying [Hz021 at 0.16 mM NADH. When the fixed substrate concentration is much greater than its K,,,, the slope of the primary plot equals 1/V, and the x-axis intercept equals -K, for the varied substrate. NAD+ inhibition of NADH peroxidase turnover, with the much poorer substrate NADPH replac- ing NADH, was analyzed at pH 5.5 and 7.5, essentially as described by Stoll and Blanchard (7).
Proton NMR Methods-One-dimensional proton NMR spectra were obtained on a Varian Unity 600 spectrometer at the Duke University NMR Spectroscopy Center. 40 ml of freshly prepared NADH peroxidase (-1.6 mg/ml) was dialyzed exhaustively against
25 mM potassium phosphate, pH 7.0, without EDTA, prior to concen- tration with an Amicon YM-30 ultrafiltration membrane to a volume of 5-10 ml. The enzyme was then taken to a volume of -0.5 ml in successive applications with a single CM-30 microconcentrator (Ami- con). Prior to analysis, -10% D20 was added to provide the lock signal and the final volume was brought to -0.55 ml. Spectra were recorded at 28 "C using a precision 5-mm round-bottom NMR tube (Wilmad). The actual protein concentration was 2.1 mM (FAD).
General Procedures-Anaerobic titrations, thiol determinations, and other analytical methods followed protocols previously described (11).
RESULTS
Expression Vector for NADH Peroxidase-Initial attempts to express the recombinant peroxidase in E. coli, using the plasmid pNPR4 described previously, were not successful. This was attributed to the loss of the selective pressure (ampicillin-resistance) required for stable maintenance of pNPR4, because of secretion of p-lactamase into the culture medium. Therefore, the NADH peroxidase gene was cloned into pSP73C, a derivative of pSP73 (Fig. 1A) in which the p- lactamase gene had been replaced by the chloramphenicol acetyltransferase gene from pACYC184. The resulting plas- mid (pNPX8) gave consistent IPTG-induced expression of NADH peroxidase, as confirmed by both SDS-polyacrylamide gel electrophoresis and by enzyme assays of crude E. coli extracts. However, it was noticed in the SDS-polyacrylamide gel electrophoresis analyses that some expression of NADH peroxidase frequently preceded addition of IPTG. In order to eliminate any transcriptional activity of those native strep- tococcal promoter sequences upstream of the structural gene, sequences preceding the ribosome-binding site were removed (Fig. 1B), as described under "Experimental Procedures." The resulting expression vector (pNPX9) gave very little, if any, peroxidase protein before IPTG induction, when transformed into JM109DE3.
Finally, T7 termination sequences were introduced down- stream of the peroxidase structural gene in order to restrict the mRNA transcript to the minimum necessary length. The resulting plasmid (pNPX12; Fig. IC) contains a T7 termina- tion sequence derived from PET-11, located approximately 360 bp beyond the npr stop codon, and an EcoRI-resistant cat gene, prepared as described under "Experimental Proce- dures." A series of experiments was then undertaken to opti- mize expression of NADH peroxidase from pNPXl2 in 50-ml cultures. Maximal expression, as analyzed by SDS-polyacryl- amide gel electrophoresis, was found with TYP medium after 4 h of IPTG induction. Optimal induction occurred when IPTG was added at a culture Asoo of 1.3.
Purification of Recombinant NADH Peroxidase-Crude ex- tracts of recombinant E. coli, harboring pNPX12 and induced following the optimized protocol described above, gave a per- oxidase specific activity of 35-40 units/mg, which represents a 50- t o 60-fold increase over levels in S. faecalis l0Cl extracts (11). Recombinant NADH peroxidase accounts for over 20% of the total protein in these E. coli extracts. Since nonrecom- binant E. coli do not have NADH peroxidase activity the enzyme was purified in a modified two-column protocol di- rectly from JM109DE3. Possible interference in the enzyme assay (1.3 mM H202) from endogenous catalase did not ma- terialize, possibly due to the low p H (5.4) employed in the standard assay. The purification scheme is outlined under "Experimental Procedures" and generally yields -110 mg of pure enzyme from three liters of E . coli in 3 days. The purified peroxidase is fully active, with a specific activity of 115-125 units/mg. SDS-polyacrylamide gel electrophoresis analysis (Fig. 2) gives a molecular weight of 49,000 for the monomer,

3 164 Characterization of Recombinant S. faecalis NADH Peroxidase
identical to that observed for the enzyme purified from S. fuecalis (1 1).
Structural Analyses of Recombinant Peroxidase-The N- terminal amino acid sequence of recombinant NADH perox- idase is shown in Table I and is compared with that predicted from the npr gene sequence and with that originally reported for the enzyme purified from S. faecalis lOCl (5, 13). While the recombinant protein sequence is identical to that pre- dicted from the gene sequence, residue 29 in the earlier protein sequence was incorrectly given as asparagine. Re-evaluation of the original N-terminal sequence data confirms the identity of residue 29 as glutamine, and residue 31 is unambiguously identified as tyrosine in the recombinant protein sequence.
NADH peroxidase differs from NADH oxidase and the flavoprotein disulfide reductases by virtue of its tetrameric quaternary structure, as determined by sedimentation equilib- rium analysis. Analytical gel filtration gives an estimated Stokes radius of 5.3 nm on Sephacryl S-300; the peroxidase tetramer ( M , = 201,300) elutes just slightly ahead of catalase (13). The recombinant enzyme showed very similar behavior when analyzed on a 1.6 X 51-cm column of Superose 12 (prep grade; Pharmacia) calibrated with ferritin, catalase, bovine serum albumin, and horseradish peroxidase. We conclude that recombinant NADH peroxidase displays a tetrameric quater- nary structure identical to the enzyme purified from S. faecalis 1oc1.
In addition, recombinant peroxidase was crystallized as previously reported for the S. faecalis enzyme and analyzed by x-ray diffraction. Crystals 9f the recombinant enzyme diffracted to a resolution of 2.4 A and were of the same space group and unit cell dimensions as reported for the enzyme
1 2 3 4 5 6 7 8 9 FIG. 2. Expression of NADH peroxidase in E. coli and pu-
rification. A 10% SDS-polyacrylamide gel was run with samples pretreated by heating a t 100 "C for 5 min in sample buffer containing 2% SDS and 0.1 M dithiothreitol. Lanes 4-8 contain samples from the crude E. coli extract (10 pg), the streptomycin sulfate supernatant (10 p g ) , the 50% ammonium sulfate supernatant (10 pg) , the Phenyl Sepharose pool (10 pa) , and the final Q Sepharose pool (5 pg). Lanes 2 and 3 contain samples from whole cells taken before and 4 h after induction with IPTG, respectively. Lanes I and 9 contain low range protein standards (Rio-Rad).
from S. faecalis.' A difference Fourier map confirmed the identity in structures for the native and recombinant NADH peroxidase.
Spectral and Redox Properties-As purified, recombinant NADH peroxidase has a visible absorption spectrum very similar to that previously reported, but with higher absorb- ance in the 540-nm region (11). The possibility that this spectral difference could be due to a small amount of the air- stable EH2 species was confirmed by the finding that addition of 0.25 equivalents of H202/FAD generated the spectrum of the fully oxidized enzyme. Subsequent anaerobic titration with NADH (Fig. 3) displayed the characteristic spectral changes observed with the enzyme from S. faecalis. During the first phase of the titration, reducing equivalents from NADH are transferred via FAD to the active-site cysteine- sulfenic acid (Cys-SOH), producing the two-electron reduced EHz species. The second phase of the titration leads to the formation of the EH,.NADH complex; the overall titration exhibits endpoints of 0.75 and 1.63 equivalents of NADH/ FAD. Dithionite titration of the enzyme led to full reduction to the EH, species in a biphasic process with endpoints of 0.75 and 1.85 equivalents/FAD, respectively.
As reported previously for the S. faecalis enzyme, thiol assays under anaerobic denaturing conditions (4 M guanidine- HCl) gave no reaction ((0.01 thiols/FAD). Analysis of the NADH-reduced (one equivalent of NADH/FAD), denatured peroxidase, however, gave 0.83 thiols/FAD. This determina- tion is identical to that originally reported for reduced per- oxidase and is consistent with the presence of a single redox- active Cys-SOH per subunit (3).
Steady-State Kinetic Mechanism-Earlier initial velocity studies with the commercially available NADH peroxidase from S. faecium (now Enterococcus hirae; ATCC 8043) gave parallel double-reciprocal plots when either NADH or Hz02 was varied a t fixed concentrations of the second substrate (7). The finding that NAD' was unable to compete for NADH binding to EH, in product inhibition studies, however, in combination with the results of direct spectral titrations of the oxidized enzyme with NADH, suggested that the EHz. NADH complex plays a central catalytic role. This behavior furthermore indicates that NADH peroxidase may follow a limiting type of ternary complex mechanism, characterized by parallel double-reciprocal plots (as described in detail under "Discussion").
The pH optimum for S. faecalis lOCl NADH peroxidase is 5.4, and acetate ion stimulates activity. Recently, Stoll and Blanchard (14) have reported a similar pH optimum for the S. faecium enzyme; however, their original steady-state analy- sis was performed at pH 7.5 where VmaX is only 30% that at pH 5.4. We have now repeated this work with the S. faecalis peroxidase in buffers containing 0.1 M acetate a t pH 5.5 and 7.5. As discussed by Cornish-Bowden (12), plots of [H202]/v versus [H202] at fixed concentrations of NADH intersect on the y-axis (Fig. 4), consistent with a classical ping pong
* T. Stehle, personal communication.
TABLE I N-terminal seuuences determined for NADH Deroxidase
Source Sequence
Translated DNA sequence" MKVIVLGSSH GGYEAVEELL NLHPDAEIQW YEKGDFISFL Recombinant peroxidase MKVIVLGSSH GGYEAVEELL NLHPDAEIQW YEKGDFISFL Peroxidase isolated from S. faecalis 10Clh MKVIVLGSSH GGYEAVEELL NLHPDAEINW XEKGDFISFL
Ref. 5. Ref. 13.
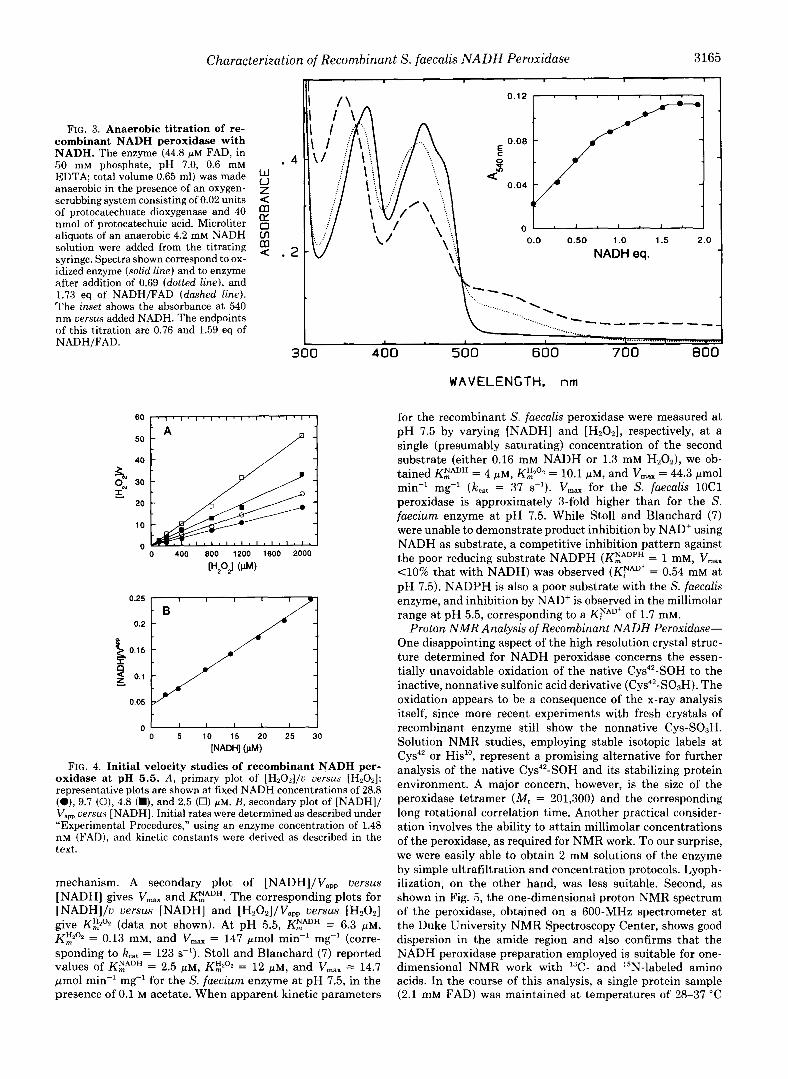
Characterization of Recombinant S. faecalis NADH Peroxidase 3165 -,
FIG. 3. Anaerobic titration of re- combinant NADH peroxidase with NADH. The enzyme (44.8 FM FAD, in 50 mM phosphate, pH 7.0, 0.6 mM EDTA; total volume 0.65 ml) was made anaerobic in the presence of an oxygen- scrubbing system consisting of 0.02 units < of protocatechuate dioxygenase and 40 nmol of protocatechuic acid. Microliter 0 IY
aliquots of an anaerobic 4.2 mM NADH Ln solution were added from the titrating $ . syringe. Spectra shown correspond to ox- NADH eq. idized enzyme (solid line) and to enzyme after addition of 0.69 (dotted line), and 1.73 eq of NADH/FAD (dashed line). The inset shows the absorbance at 540 nm uersus added NADH. The endpoints %
of this titration are 0.76 and 1.59 eq of NADH/FAD.
-
1 . 1 . 1 .
0.0 0.50 1.0 1.5 2.0
..__ .... ..._ -""" ""
300 400 500 600 700 800
-.._..,
WAVELENGTH. nrn
% Ow 30 3
20
10
n 0 400 800 1200 1600 2000
[H202] ( W
0.25 . I I I I
- B 0.2 -
0.05
I I I I I I 0 5 10 15 20 25 30
[NADH] (PM)
FIG. 4. Initial velocity studies of recombinant NADH per- oxidase at pH 5.5. A , primary plot of [H,Oz]/u uersus [HzOz]; representative plots are shown at fixed NADH concentrations of 28.8 (O), 9.7 (0), 4.8 (H), and 2.5 (0) g ~ . B, secondary plot of [NADH]/ Vapp uersus [NADH]. Initial rates were determined as described under "Experimental Procedures," using an enzyme concentration of 1.48 nM (FAD), and kinetic constants were derived as described in the text.
mechanism. A secondary plot of [NADH]/V,,, uersus [NADH] gives V,,, and KEADH. The corresponding plots for [NADH]/u uersus [NADH] and [H2O2]/VaP, uersus [H202] give K3'2 (data not shown). At pH 5.5, KZADH = 6.3 p ~ , Kf;i202 = 0.13 mM, and Vmax = 147 pmol min" mg-' (corre- sponding to kc,, = 123 s-'). Stoll and Blanchard (7) reported values of KZADH = 2.5 PM, K?', = 12 p ~ , and V,,, = 14.7 pmol min-' mg" for the S. faecium enzyme at pH 7.5, in the presence of 0.1 M acetate. When apparent kinetic parameters
for the recombinant S. faecalis peroxidase were measured at pH 7.5 by varying [NADH] and [Hz02], respectively, at a single (presumably saturating) concentration of the second substrate (either 0.16 mM NADH or 1.3 mM Hz02), we ob- tained K:""" = 4 FM, K3'2 = 10.1 pM, and V,,, = 44.3 pmol min" mg-' (k,,, = 37 s-'). V,,, for the S. faecalis lOCl peroxidase is approximately 3-fold higher than for the S. faecium enzyme at pH 7.5. While Stoll and Blanchard (7) were unable to demonstrate product inhibition by NAD' using NADH as substrate, a competitive inhibition pattern against the poor reducing substrate NADPH (KZADPH = 1 mM, V,,, - 4 0 % that with NADH) was observed (KNAD' = 0.54 mM a t pH 7.5). NADPH is also a poor substrate with the S. faecalis enzyme, and inhibition by NAD+ is observed in the millimolar range at pH 5.5, corresponding to a KYAD' of 1.7 mM.
Proton N M R Analysis of Recombinant NADH Peroxidase- One disappointing aspect of the high resolution crystal struc- ture determined for NADH peroxidase concerns the essen- tially unavoidable oxidation of the native Cys4'-SOH to the inactive, nonnative sulfonic acid derivative (Cys4'-S03H). The oxidation appears to be a consequence of the x-ray analysis itself, since more recent experiments with fresh crystals of recombinant enzyme still show the nonnative Cys-SOsH. Solution NMR studies, employing stable isotopic labels at Cys4' or His'', represent a promising alternative for further analysis of the native Cys4*-SOH and its stabilizing protein environment. A major concern, however, is the size of the peroxidase tetramer ( M r = 201,300) and the corresponding long rotational correlation time. Another practical consider- ation involves the ability to attain millimolar concentrations of the peroxidase, as required for NMR work. To our surprise, we were easily able to obtain 2 mM solutions of the enzyme by simple ultrafiltration and concentration protocols. Lyoph- ilization, on the other hand, was less suitable. Second, as shown in Fig. 5, the one-dimensional proton NMR spectrum of the peroxidase, obtained on a 600-MHz spectrometer at the Duke University NMR Spectroscopy Center, shows good dispersion in the amide region and also confirms that the NADH peroxidase preparation employed is suitable for one- dimensional NMR work with 13C- and "N-labeled amino acids. In the course of this analysis, a single protein sample (2.1 mM FAD) was maintained at temperatures of 28-37 "C

3166 Characterization of Recombinant S. faecalis NADH Peroxidase
1 0 8 8 4 2 0
'H (ppm)
FIG. 5. 600-MHz 'H NMR spectrum of recombinant NADH peroxidase. The sample contains 2.1 mM protein (FAD) in 25 mM phosphate buffer, pH 7.0, with 10% D20 added as an internal lock. A total of 256 scans of 8192 data points were collected at 28 "C.
for approximately 5 h. More importantly, subsequent analysis showed the enzyme retained full activity, with no change in spectral properties. We are indebted to Drs. Len Spicer and Ron Venters for their assistance with this NMR analysis.
Resonance Raman Analysis of NADH Peroxidase-Another potential spectroscopic probe of the C ~ S ~ ~ - S O H redox center of the native peroxidase involves resonance Raman identifi- cation of the characteristic S-0 stretching vibration. In collaboration with Dr. David Dooley, Amherst College, at- tempts were made to obtain resonance Raman spectra of the peroxidase. However, repeated attempts to acquire these data (using 457.9-, 488.0-, or 514.5-nm excitation) invariably led to FAD dissociation, presumably due to either localized heat- ing or photochemical effects induced during exposure to the laser excitation source.
DISCUSSION
We have developed a reliable system for high level expres- sion of streptococcal NADH peroxidase in E. coli. Three significant improvements over our previous expression vector include 1) replacing the ampicillin-resistance (/I-lactamase) marker with the chloramphenicol acetyltransferase gene, thereby maintaining selective pressure favoring transform- ants harboring the pNPX12 plasmid, 2) addition of a T7 RNA polymerase terminator sequence to limit transcription to the npr gene, and 3) removal of the native npr promoter to allow tighter control over heterologous expression and, thus, to optimize growth of the culture before IPTG induction. The general purpose expression plasmid, pOXO4, which results from modifications 1 and 2 contains a multiple-cloning site between the T7 promoter and the T@ transcription terminator and was specifically engineered to have wide applicability to other systems.
Application of this expression system to the streptococcal NADH peroxidase allows the purification of 35 mg of protein per liter of culture in a 90-95% overall yield. The recombinant peroxidase is fully active and appears to be identical to the enzyme originally isolated from S. faecalis 10Cl in all respects. The observation of a moderate amount (-25%) of the reduced EH, form of the peroxidase in the purified enzyme is consist- ent with the highly reducing cytosolic environment of E. coli, the short purification time required with the overexpressed protein, and the very slow rate of aerobic autoxidation re-
ported for EHz in the absence of Hz02. Furthermore, since the only "posttranslational" event involved in generating the functional Cys-SOH redox center is oxidation of Cys-SH by intracellular H202, expression of the fully active peroxidase in the heterologous host is not hindered. In addition, we have shown that the recombinant enzyme is well suited for more protein-intensive applications such as x-ray crystallography and one-dimensional NMR analyses. Mutagenesis studies already underway have demonstrated both the general appli- cability of the expression and purification protocol and the suitability of the corresponding protein product(s) for these types of studies.
Before applying rapid-reaction approaches to the study of the chemical and kinetic mechanism, it is essential to perform a complete analysis of the steady-state mechanism. Earlier studies by Stoll and Blanchard (7, 14) have focused on the commercial enzyme preparation from S. faecium (ATCC 8043). We have shown that the latter peroxidase is antigeni- cally similar to the enzyme purified from S. faecalis (ATCC 9790), but that neither enzyme cross-reacts with antibodies to the S. faecalis lOCl (ATCC 11700) protein (15). Further- more, preliminary sequence analysis of a PCR fragment cor- responding to the ATCC 9790 npr gene indicates that overall amino acid sequence identity with the ATCC 11700 protein may be only about 40%.3 This suggests that similar sequence differences may exist for the ATCC 8043 enzyme when com- pared with that from ATCC 11700. And, at the present time, the single complete npr sequence and the three-dimensional protein structure correspond to the S. faecalis lOCl peroxi- dase.
Stoll and Blanchard (7) first suggested the central role of an enzyme- NADH. H20z ternary complex in the catalytic mechanism. However, no attempt has been made to reconcile the parallel double-reciprocal pattern observed in their initial velocity studies. As pointed out by Palmer and Massey (16) and by others (17), there are cases (e.g. D-amino acid oxidase, lactate oxidase) where ternary complex mechanisms still yield parallel double-reciprocal plots. Stoll and Blanchard (7), in agreement with previous work from this laboratory (4, ll), have suggested that an initial priming step in the peroxidase mechanism reduces oxidized enzyme ( E ) to EHz at the ex- pense of one NADH and that, in the steady-state, the perox- idase cycles between EH2 and EHz.NADH forms. On the basis of the data available, and using the Dalziel type II(i) ternary complex mechanism as a guide (181, we propose the following kinetic scheme for NADH peroxidase.
EHZ + NADH + EH2.NADH kl
k2 k3 k
k4 k'
(Es. 2)
EH2.NADH + Hz02 EHz.NADH.Hz02 G= E.NADH + Hz0
k4 I
k3 ' k2'
12,'
E.NADH + H+ + EHz.NAD+ + H20
EH2.NAD' + EH2 + NAD'
The general form of the corresponding initial rate equation is:
The condition for obtaining parallel double-reciprocal plots is
' R. P. Ross and A. Claiborne, unpublished observations.

Characterization of Recombinant S. faecalis NADH Peroxidase 3167
TABLE I1 Kinetic constants for streptococcal NADH peroxidase
Values were obtained as described in the text, using the kinetic coefficients defined by Dalziel for the type II(i) mechanism (Ref. 18). All kinetic constants refer to 0.1 M acetate/O.l M MES, pH 5.5, at 25 "C. The KYADH was determined in 0.1 M phosphate, pH 7.0, a t 25 "C.
k2 KdNADH
2 X lo7 M" S-l
6 s-' 0.3 p M 9.1 x lo5 ~ - 1 s-ln NDb ND 0' ND Od ND ND 1.7 mM 123 s-l 6.3 p M 0.13 mM
If k (forward rate of H,02 reduction) >> k4 (dissociation rate of
H202 from the ternary complex), then kS = -. 1
$2
ND, not determined. Chemical reduction of H202 is irreversible. NAD' cannot oxidize EH2 to E.
$2 = (k4k4' + k4k' + kh'k)
knk4'k (Eq. 5)
Therefore,
The ratio k2 /k l equals the dissociation constant for NADH binding to EH2, which has been measured in static titrations
at pH 7.0 (Fig. 3; also Ref. 11) to be 4 0.3 WM. Also, at pH 5.5,
Therefore, $12 = (3 X M) (1.1 X M .s) = 3.3 X 10"" M'.s, which when divided by the product of [NADH] and [Hz02], represents a negligibly small term in the initial rate equation. Double-reciprocal plots, as initially observed by Stoll and Blanchard ( 7 ) , will be parallel. Table I1 summarizes our values for the kinetic constants corresponding to the Dalziel type II(i) mechanism, as determined from both steady- state kinetic and static titration methods. The large amounts of purified NADH peroxidase now available will greatly facil- itate direct testing of this mechanism by rapid reaction tech- niques.
Acknowledgments-We thank Dr. Ron Venters and Dr. Len Spicer for their help with NMR spectroscopy and Dr. David Dooley for performing the resonance Raman trials. We thank Dr. David Porter for helpful discussions concerning the kinetic mechanism. N-terminal protein sequence analysis was performed by the Protein Analysis Core Facility of the Comprehensive Cancer Center of Wake Forest University. The Duke University Magnetic Resonance Spectroscopy Center was established with grants from the National Institutes of Health, the National Science Foundation, and the North Carolina Biotechnology Center.
REFERENCES 1. D o h M. I. (1982) in Ex eriences in Biochemical Perception (Omston, L.
N.,'and Sligar, S. G., e&) p 293-307, Academic Press, New York 2. Claiborne, A., Ross, R. P., anx'parsonage, D. (1992) Trends Biochem. Sci.
17 , 183-186 3. Poole, L. B., and Claiborne, A. (1988) Biochem. Biophys. Res. Commun.
4. Poole L. B. and Claiborne A. (1989) J . Biol. Chem. 2 6 4 12330-12338 153,261-266
5. Ross 'R. P.,'and Claiborne,'A.,(1991) J. Mol. Biol. 221, 8,!7-871 6. Stehie, T., Ahmed, S. A., Clalborne, A., and Schulz, G. E. (1991) J . Mol.
7. Stoll, V. S., and Blancbard, J. S. (1988) Arch. Biochem. Biophys. 260,752-
8. Studier F. W. and Moffatt B. A. (1986) J. Mol. Biol. 169 113-130 9. Ross R . P. a i d Claiborne 'A. (1992) J. Mol. Biol. 227 656-671
10. Chan'g A. k. Y. and Coheh S. N. (1978) J . Bacteriol. i34 1141-1156 11. Poole 'L. B. anh Claiborne'A. (1986) J. Biol. Chem. 261 i4525-14533 12. CornGh-Bohden, A. (1979) k u n d a m e n t a k of Enzyme Kine'tics, Butterworth
Biol. 221, 1325-1344
762
13. Poole L. B. and Claiborne A. (1989) J. Biol. Chem. 264 12322-12329 14. Stoll 'V. S. h d Blanchard'J. S. (1991) Biochemist 30 '942-948 15. Mill&, H., Poole L. B., and'claiborne, A. (1990) J . gal. &em. 265,9857-
L Co., London
OYG? 16. Pai;;;kyr, G., and Massey, V. (1968) in Biological Oxidations (Singer, T. P.,
17. Lockrl ge, 0 , Massey, q, and Sullivan, P. A. (1972) J. B h . Chem. 247, ed) PI . 263,-300, Wile Interscience, John Wiley & Sons New York
~ W R I 06 18. Dalziel, K.-<1957) Acta Chem. Scand. 1 1 , 1706-1723





























