Embed Size (px)
Citation preview

Vol. 173, No. 6JOURNAL OF BACTERIOLOGY, Mar. 1991, p. 1894-19010021-9193/91/061894-08$02.00/0Copyright C) 1991, American Society for Microbiology
Purification and Properties of Tryptophan-Sensitive 3-Deoxy-D-arabino-Heptulosonate-7-Phosphate Synthase from Escherichia coli
JILL M. RAYt AND RONALD BAUERLE*Department of Biology and the Molecular Biology Institute, University of Virginia, Charlottesville, Virginia 22901
Received 31 October 1990/Accepted 9 January 1991
The aroH gene of Escherichia coli, which encodes the tryptophan-sensitive 3-deoxy-n-arabino-heptulosonate-7-phosphate synthase isoenzyme of the common aromatic biosynthetic pathway, was cloned behind the tacpromoter in expression plasmid pKK223-3. The enzyme was overexpressed, purified to homogeneity, andcharacterized. The native enzyme was found to be a dimeric metalloprotein containing 0.3 mol of iron per molof subunit and variable amounts of zinc. The activity of the native enzyme was stimulated two- to threefoldwhen assayed in the presence of Fe2+ ions. Pretreatment of the enzyme with Fe2+ also resulted in activation,accompanied by an equivalent increase in iron content. Treatment of the enzyme with chelating agents led toinactivation, which was fully reversed by the presence of Fe2+ in the assay mixture. The native enzymeexhibited a unique absorption profile, having a shoulder of absorbance on the aromatic band with a maximumaround 350 nm and a broad, weak band with a maximum around 500 nm. Treatment of the enzyme with Fe2tenhanced the absorbance at 350 nm and eliminated the band at 500 nm. Treatment with reducing agents causedthe disappearance of both bands and destabilized the enzyme. Feedback regulation of the activity of the enzymewas specific for tryptophan, with maximum inhibition at about 70%.
In bacteria, a common aromatic pathway of seven stepsleads to the synthesis of chorismic acid, the branch pointprecursor of the aromatic amino acids tryptophan, tyrosine,and phenylalanine, as well as of folic acid, ubiquinone,menaquinone, and enterochelin. The first committed step ofthis pathway is the condensation of erythrose-4-phosphate(E4P) and phosphoenolpyruvate (PEP), yielding the phos-phorylated 7-carbon keto sugar acid 3-deoxy-D-arabino-heptulosonate-7-phosphate (DAHP). The enzyme that cata-lyzes this reaction is DAHP synthase (EC 4.1.2.15).
In Escherichia coli there are three DAHP synthase isoen-zymes, each specifically feedback regulated by one of thethree aromatic amino acids (6). The Phe-sensitive [DAHP-S(Phe)] and Tyr-sensitive [DAHPS(Tyr)] isoenzymes, en-coded by aroG and aroF, respectively, are responsible foressentially all of the DAHP synthase activity in the wild-typecell; the Trp-sensitive isoenzyme [DAHPS(Trp)], encodedby aroH, contributes only about 1% of the total activity (29).Molecular cloning and DNA sequencing of the aroF, aroG,and aroH genes (8, 21, 26) have revealed that the threepolypeptides are nearly identical in size and have a highdegree of sequence similarity. On this basis, it has beenconcluded that the three are homologs with a commonevolutionary origin (19, 21). DAHPS(Tyr) (1, 24) and DAHP-S(Phe) (17) from E. coli have been purified to homogeneityand extensively characterized; however, only a preliminarystudy of a partially purified preparation of DAHPS(Trp) hasbeen reported (7).DAHPS(Phe) from E. coli is tetrameric in structure, while
DAHPS(Trp) and DAHPS(Tyr) are dimeric. Chelation stud-ies have indicated that DAHPS(Phe) and DAHPS(Tyr) aremetalloenzymes (1, 10, 16, 24); however, there are disparateresults concerning the nature and stoichiometry of the acti-vating metal in the native enzymes. DAHPS(Phe) was found
* Corresponding author.t Present address: Department of Plant Biology, Carnegie Insti-
tution of Washington, Stanford, CA 94305-1297.
to be irreversibly inactivated by the iron chelator o-phenan-throline and to contain 1 mol of iron per enzyme tetramer(16). On the other hand, in one study, purified DAHPS(Tyr)was found to contain 1 mol each of copper, iron, manganese,and zinc per enzyme dimer (13), while in another evidencewas presented that Cu2" is the activating metal in the nativeenzyme (1).Here we describe methods for the overexpression and
purification of DAHPS(Trp) from E. coli and report theresults of an analysis of the homogeneous enzyme withrespect to its stability, metal composition, absorbance prop-erties, and feedback sensitivity.
MATERIALS AND METHODS
Chemicals and enzymes. Amino acids, PEP (monocyclo-hexyl-ammonium salt), E4P (sodium salt), 1,3-bis[tris(hy-droxymethyl)-methylamino]propane (BTP), 3-(cyclohexy-lamino)-2-hydroxy-1-propanesulfonic acid, isopropyl-P-D-thiogalactoside (IPTG), dithiothreitol, dithioerythritol(DTE), o-phenanthroline, and ampicillin were from SigmaChemical Co. All enzymes used for the construction andmanipulation of recombinant DNA molecules were obtainedcommercially and used as recommended by the varioussuppliers. All other chemicals were analytical grade.
Bacterial strains and plasmids. E. coli AB3248 (aroF363aroG365 aroH367 proA2 argE3 his4 thi lac gal-2 tsx-358)(31) and CB198 (W3110 lacIq tnaA2 trpR) were obtainedfrom C. Yanofsky, Stanford University. Plasmid pAHH1 isa derivative of plasmid pBR322 carrying the E. coli aroHgene under the control of its natural promoter and terminator(21). Plasmid pCHR40, constructed as described below, is aderivative of plasmid pKK223-3 (5) in which the aroH geneis expressed under the control of the tac promoter.
Construction of expression plasmid pCHR40. The 1,332-bpDdeI fragment containing the aroH coding region togetherwith its upstream ribosome-binding site was excised fromplasmid pAHH1 and, after end filling with Klenow polymer-ase, was ligated into the unique SmaI site of expression
1894
on Septem
ber 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

TRYPTOPHAN-SENSITIVE DAHP SYNTHASE 1895
A.
Sail
B.
SD
....AGGAACAGAATTCCC To CTTGTAATGAACAGAACTGAC...t
Sma/DdeJunction
FIG. 1. Physical map and upstream junction sequence of aroHexpression vector pCHR40. (A) ptac, tac promoter; t', aroH tran-scription terminator; tt, rrnB tandem transcription terminator. (B)Shaded nucleotides indicate the Shine-Dalgarno (SD) sequence andthe translation initiation codon of aroH.
plasmid pKK223-3, using T4 DNA ligase. The ligated DNAwas transformed into E. coli AB3248, an aromatic auxotrophrequiring phenylalanine, tryptophan, and tyrosine forgrowth, with selection for ampicillin resistance and aromaticamino acid prototrophy on minimal agar plus ampicillin.Plasmid DNA was isolated from clones purified by using thesame selective medium, and a construct with an intact,properly oriented aroH insert was identified by the patternsof digestion by appropriate combinations of restriction en-
donucleases. The derived plasmid, designated pCHR40 (Fig.1), was transformed into E. coli host CB198 where it was
stably maintained. The nucleotide sequence of the-upstreamvector-insert junction of pCHR40 (Fig. 1) was verified bydideoxy sequencing (23) of both strands of the 240-bpEcoRI-PstI fragment of pCHR40 which had been subclonedinto the EcoRI-PstI sites of bacteriophages M13mpl8 andM13mpl9. All media and methods used in the constructionand characterization of pCHR40 were as described before(22).
Preparation of extracts. Cultures of CB198/pCHR40 were
grown at 37`C in minimal salts medium (2) containing caseinhydrolysate (U.S. Biochemical Corp.) (1 g/liter), glucose (5g/liter), ampicillin (30 mg/liter), tyrosine (50 mg/liter), phen-ylalanine (50 mg/liter), and tryptophan (25 mg/liter). Whenthe A550 of the culture reached 0.5, IPTG was added to 0.5mM to induce expression of the tac-promoted aroH gene.After 3 h of induction, the cells were harvested, washed in 1volume of cold 0.9% saline, and resuspended in 4 ml of coldBPP buffer (10 mM BTP, 100 p.M PEP, pH 6.7) for each gramof wet cells. All subsequent steps were carried out at 0 to40C. The cells were disrupted in 6-ml batches by sonification
for 35 s, using a Branson model W185 Sonifier operated at anoutput of 70 W. Cell debris was removed by centrifugation at48,000 x g for 20 min.
Assay for DAHP synthase activity. DAHP synthase activitywas assayed as described previously (21) by a modificationof the continuous spectrophotometric method of Schonerand Herrmann (24). The standard assay mixture contained100 ,uM PEP, 300 ,uM E4P, 10 mM BTP (pH 7.0), and, unlessindicated otherwise, 10 ,uM FeSO4 in a final volume of 1 ml.The reaction was initiated by the addition of enzyme to aquartz cuvette containing the reaction mixture which hadbeen equilibrated to 25°C. The rate of disappearance of PEPwas measured at 232 nm in a Varian DMS200 UV-visibledouble-beam 'spectrophotometer equipped with a thermo-statted cell holder and kinetics software. One unit of enzymeactivity is defined as the disappearance of 1 ,umol of PEP permin at 25°C.
Purification of DAHPS(Trp). Homogeneous enzyme wasprepared from crude extracts of IPTG-induced cultures ofCB198/pCHR40, using the purification protocol outlinedbelow. All procedures were carried out on ice or at 4°Cunless noted otherwise.
(i) Treatment with streptomycin sulfate. A one-tenth vol-ume of cold 20% streptomycin sulfate prepared in BPP bufferwas added' dropwise with slow stirring to the crude cellextract. After 15 min, the nucleic acid precipitate wasremoved by centrifugation at 48,000 x g for 20 min and thesupernatant was recovered.
(ii) Acetone precipitation. Ice-cold acetone was addeddropwise with stirring to the streptomycin-treated extract,held in an ice bath, to a final concentration of 25%. Midwaythrough the addition, NaCl was' added to the ice bath todecrease the temperature to -5 to -10°C. After 15 min withstirring, the preparation was centrifuged at 12,000 x g for 15min. The supernatant was decanted and the protein pelletwas dissolved in BPP buffer containing 250 mM KCl (4ml/liter of original culture). The preparation was then dilutedwith 1.5 volumes of BPP buffer, reducing the KCl concen-tration to 100 mM.
(iui) Anion-exchange fast protein liquid chromatography(FPLC). The preparation from step ii was clarified by pas-sage through a 0.22-,um nylon filter cartridge and thenchromatographed over a Pharmacia Mono Q HR 5/5 anion-exchange column (Pharmacia LKB Biotechnology, Inc.),using a Beckman high-performance liquid chromatography(HPLC) system. The column was prepared for each chro-matographic cycle by successive 0.5-ml injections of 2 MNaCl-2 M NaOH-75% acetic acid-2 M NaCl at a flow rate of0.5 ml/min followed by equilibration with 10 mM BTP (pH6.7) at 1.0 ml/min. Two-milliliter aliquots of the enzymepreparation (15 to 20 mg of total protein) were injected foreach cycle. The column was developed at a flow rate of 1ml/min with the following discontinuous KCl gradient, pre-pared in 10 mM BTP (pH 6.7): (a) phase 1-buffer wash for4 min; (b) phase 2-linear gradient of 0 to 140 mM KCl in 2min, dwell at 140 mM KCl for 5 min; (c) phase 3-lineargradient of 140 to 190 mM KCl in 8 min, dwell at 190 mM for5 min; (d) phase 4-190 to 500 mM KCl in 2 min, dwell at 500mM for 15 min. Fractions of 0.5 ml were collected. TheDAHP synthase activity was eluted from the column at 160mM KCI (12 to 13 min into the run). The peak fractions fromeach chromatographic cycle were combined and PEP, whichgreatly increases the stability of the enzyme, was immedi-ately added to a concentration of 100 p.M. The preparationwas then dialyzed exhaustively against 10 mM BPP (pH 7.0)before use.
VOL. 173, 1991
on Septem
ber 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

1896 RAY AND BAUERLE
Determination of relative and absolute protein concentra-tions. The relative protein content of crude and purifiedenzyme preparations was estimated by the method of Brad-ford (4), using the Bio-Rad protein reagent (Bio-Rad Labo-ratories) and bovine serum albumin as the standard. For thecalculation of metal-enzyme stoichiometries, relative con-centrations of purified DAHPS(Trp) were converted to ac-tual concentrations by the application of a correction factorthat compensates for the overestimation of the enzyme bythe Bio-Rad reagent. The correction factor was determinedfrom the results of the quantitative amino acid analysis ofacid hydrolysates of homogeneous DAHPS(Trp). A 200-pmol amount of purified enzyme, as estimated with theBio-Rad reagent, was hydrolyzed in triplicate, and theamount of each amino acid in the hydrolysates was deter-mined by using precolumn phenylthiohydantoin derivatiza-tion and reversed-phase HPLC (3). The ratio of the meandetermined value for each amino acid to that expected fromthe amino acid sequence of the AroH polypeptide, as de-rived from the nucleotide sequence of the aroH gene (21),was calculated for all amino acids except tryptophan, cyste-ine, and proline (glutamine and asparagine were determinedas glutamate and aspartate, respectively). An identical anal-ysis of crystalline bovine serum albumin was used to assessand correct for loss in the experiment, which averaged 15%.This procedure revealed that the Bradford method overesti-mates the content of pure preparations of DAHPS(Trp) by afactor of 1.64.PAGE. Sodium dodecyl sulfate-polyacrylamide gel elec-
trophoresis (SDS-PAGE) of proteins (14) was carried out in10% acrylamide minigels. Protein bands were fixed with 20%methanol-7% acetic acid and stained with 0.1% Coomassiebrilliant blue R-250 in the same solvent.
Determination of native molecular weight. The native mo-lecular weight of DAHPS(Trp) was determined by sizeexclusion chromatography over a Superose-12 HR 10/30column (Pharmacia LKB Biotechnology, Inc.). The columnwas equilibrated and developed with 50 mM potassiumphosphate-150 mM NaCl (pH 7.0) at a flow rate of 0.4ml/min, using a Beckman HPLC system. The followingstandard proteins were used to calibrate the fractionation:yeast alcohol dehydrogenase (150/37.5 kDa, tetramer/mono-mer), horse liver alcohol dehydrogenase (84/42 kDa, dimer/monomer), chymotrypsin (22.5 kDa), and cytochrome c(12.4 kDa). The void volume (VO) of the column was deter-mined with blue dextran 2000 (Pharmacia LKB Technology,Inc.). DAHPS(Trp) was chromatographed both by itself andtogether with a mixture of the standard proteins, excludingchymotrypsin. The relative molecular weight of DAHP-S(Trp) was estimated from a plot of the ratio of elutionvolume to void volume (VeIVo) for each standard proteinversus the logarithm of its molecular weight.
Amino-terminal sequencing. Samples containing 200 to 500pmol of purified enzyme were fractionated by SDS-PAGE ina 10% polyacrylamide minigel at 15-W constant power. TheLaemmli (14) buffer system was used except that sodiumthioglycolate (250 ,ug/liter) was added to the tank buffer toreduce the formation of free radicals during electrophoresis.Protein bands were transferred onto polyvinylidine difluo-ride membrane (Amicon Corp.) by electroblotting for 12 h at130 mA in a transblot apparatus (Hoefer Scientific, Inc.).The transfer buffer was 10 mM 3-(cyclohexylamino)-2-hy-droxy-1-propanesulfonic acid in 10% methanol, pH 11. Themembrane was washed with water, stained with 0.1%Coomassie brilliant blue R-250 in 50% methanol for 5 min,destained in 50% methanol-10% acetic acid for 10 min,
washed with water, and air dried. Segments of the mem-brane containing the bands of interest were excised and useddirectly for automated amino-terminal sequencing (15) in anApplied Biosystems model 470A Sequenator equipped withan on-line phenylthiohydantoin analyzer.
Metal saturation and analysis. Freshly prepared FeSO4was added at 20 ,uM to 5 ,uM enzyme monomer (40 ,ug ofenzyme in 200 ,ld of BPP buffer, pH 7.0) at 20°C. Thepreparation was then passed through a 4-ml Sephadex G-25Fine gel filtration column equilibrated with BPP buffer toseparate unbound iron from the protein. An untreated en-zyme sample was processed in the same manner. Proteincontent and enzymatic activity were assayed in each prepa-ration, and the metal content was analyzed by atomicabsorption analysis. Metal concentrations were determinedin duplicate with a Perkin-Elmer model 5100 atomic absorp-tion spectrometer, using either a graphite furnace (for iron,cobalt, and manganese) or a flame analyzer (for zinc andmagnesium). Values for each metal in the enzyme prepara-tions were corrected for the small amount of each metaldetected in the BPP buffer. Metal-enzyme stoichiometrieswere calculated by using actual protein concentrations de-rived from the concentrations determined colorimetrically,as described above.
Absorption spectrophotometry. Absorption spectra wererecorded with a Varian DMS200 UV-visible double-beamspectrophotometer operated at wavelength slit of 1 nm andscanning speed of 100 nm/min.
RESULTS
Expression system for the DAHPS(Trp) isoenzyme. Be-cause expression of the aroH gene, which encodes theDAHPS(Trp) isoenzyme, is very inefficient under the controlof its own promoter (32), the aroH coding region was clonedbehind the strong, IPTG-inducible tac promoter (9) inexpression plasmid pKK223-3. The derived plasmid,pCHR40, was stably maintained in the lacIq host, CB198. Avariety of conditions were assessed for the expression ofaroH in CB198/pCHR40. It was found that maximum induc-tion was achieved 3 h after the addition of 0.5 mM IPTG tomid-log cultures grown in minimal salts medium supple-mented with 0.5% glucose, 0.1% Casamino Acids, and 30 ,ugof ampicillin per ml. Under these conditions, the specificactivity of DAHPS(Trp) in crude extracts of strain CB198/pCHR40 was found to be 60-fold higher than that obtainedby indoleacrylic acid derepression of a strain with a singlechromosomal aroH gene and 5-fold higher than that of anindoleacrylic acid-derepressed multicopy strain (AB3248/pAHH1) (21) in which the aroH gene was carried on plasmidpBR322 under the control of its own promoter.
Purification and preliminary characterization of DAHP-S(Trp). The DAHPS(Trp) isoenzyme was purified fromcrude extracts of IPTG-induced cultures of CB198/pCHR40following the protocol described in Materials and Methods.The results of a representative purification are shown inTable 1 and Fig. 2. Generally, the overall procedure gave afive- to sixfold purification with about 50% recovery andyielded 12 to 15 mg of enzyme per liter of culture. Thus, theCB198/pCHR40 expression system elevated the enzyme toabout 18 to 20% of the total soluble protein in crude extracts.SDS-PAGE analysis revealed that the purified enzyme
was essentially homogeneous, containing one major poly-peptide with an estimated molecular weight of 39,000 (Fig.2). This was in excellent agreement with the value predicted(38,590) from the sequence of the aroH gene (21). In addi-
J. BACTERIOL.
on Septem
ber 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

TRYPTOPHAN-SENSITIVE DAHP SYNTHASE 1897
TABLE 1. Purification of DAHPS(Trp)a
Vol Total Total Sp act RecoveryPrepn (ml) activity protein (U/mg)(U) (Mg)b mg ()
Crude extract 35 604 256 2.4 100Streptomycin supernatant 34 580 191 3.1 96Acetone precipitate 10 506 88 5.8 84Mono Q peak 16 313 24 13.1 52
a From the cells of four 500-ml cultures of CB198(pCHR40) grown andinduced as described in Materials and Methods.
b Relative concentrations determined colorimetrically as described in Ma-terials and Methods.
tion, one minor polypeptide with a molecular weight of about32,000, representing no more than a few percent of the totalprotein, was consistently observed. The native enzymefractionated as a single component during analytical sizeexclusion FPLC with a relative molecular weight of 69,000,indicating that the holoenzyme is a homodimer.
Analysis of the minor polypeptide. A number of differentchromatographic strategies were used in an attempt toremove the minor 32,000-molecular-weight (32K) polypep-tide present in purified DAHPS(Trp) preparations. Theseincluded size exclusion and hydrophobic interaction FPLC,reversed-phase HPLC, and manual chromatography overhydroxylapatite and procion red agarose. None was effectivein resolving the minor polypeptide from the DAHPS(Trp)holoenzyme, in spite of the distinctly different mechanismsof fractionation of the chromatographic media. These resultssuggested an avid association between the 32K protein andthe native enzyme, rather than their chance cofractionation.A likely possibility was that the 32K protein was a fragmentof DAHPS(Trp) arising by the in vivo and/or in vitroproteolytic "nicking" of one or possibly both of the AroHsubunits in a small fraction of the holoenzyme molecules,without leading to disassembly.To test this idea, the intact AroH polypeptide and the
minor 32K polypeptide were separated by preparative SDS-PAGE, recovered by electroblotting, and analyzed by ami-no-terminal sequencing. The results of five cycles of auto-mated Edman degradation revealed that the amino-terminalsequences of the two polypeptides were identical, namely,
1 2 3 4 5 6
AI.
=-
...
kDa
-97- 66
;" 45
._" -31
FIG. 2. SDS-PAGE analysis of DAHPS(Trp) preparations dur-ing purification. Lane 1, Crude extract; lane 2, streptomycin sulfatesupernatant; lane 3, acetone supematant; lane 4, acetone precipi-tate; lane 5, Mono Q FPLC peak; lane 6, molecular weight stan-dards. Approximately 5 jig of total protein was loaded per lane.Arrowhead indicates the DAHPS(Trp) band.
Met-Asn-Xxx-Thr-Asp... (the third residue was not identi-fied in either of the degradations), and matched that pre-dicted from the DNA sequence of the aroH gene (i.e.,Met-Asn-Arg-Thr-Asp... .). Thus, it was concluded that the32K polypeptide was an amino-terminal proteolytic fragmentof the intact AroH polypeptide.
Stimulation of enzymatic activity by metals. The activity ofthe purified enzyme was assayed in the presence of a varietyof metals. It was found that activity was significantly en-hanced by the addition of ferrous ions. Maximum activation,which varied from two- to threefold between preparations,was attained at concentrations as low as 10 p.M and wasequivalent with FeSO4, Fe(NH4)2(SO4)2, and FeCl2. Mn2+and Zn2+ were marginally effective, with a maximum stim-ulation of 20 to 30% at 100 p.M. Ca2+, Co2+, Cu2+, Cu3+,Fe3", and Mg2+ had no significant effect on enzymaticactivity. Over the course of many experiments, it wasobserved that the magnitude of the response to Fe2+ wasmaximal in freshly purified enzyme preparations and de-clined during extended storage of the enzyme at 0°C as wellas when frozen at -20 or -75°C. Higher concentrations (50to 100 ,uM) of Fe2+ in the assay did not compensate for thisloss of activation.Metal content of purified DAHPS(Trp). One explanation of
the observed metal activation is that the enzyme requiresFe2+ for activity and that the added Fe2+ stimulated activityby saturating unoccupied metal binding sites in the purifiedenzyme. Alternatively, the activation might have been theresult of the action of Fe2+ as a reducing agent. To distin-guish these possibilities, the metal content of the enzymebefore and after in vitro treatment with a fourfold molarexcess of FeSO4 was determined by atomic absorptionanalysis. In one preparation it was found that the untreatedenzyme contained 0.3 mol of iron per mol of enzymemonomer and that treatment of the enzyme with FeSO4increased this to 0.75 mol/mol. Similar results were obtainedwith a second enzyme preparation that had been purified atanother time. This 2.5-fold increase in iron content wasaccompanied by a similar increase in activity in the FeSO4-treated enzyme. Thus, it appeared that the activation byFe2+ was a direct result of the binding of additional ironatoms to the enzyme. However, the Fe2+-treated enzymewas apparently not fully activated since its activity increasedan additional 20% when assayed in the presence of Fe2+.Assuming that this was due to unoccupied metal bindingsites, the iron content of fully saturated DAHPS(Trp) wasextrapolated to be 0.9 mol/mol of enzyme monomer.Atomic absorption analysis also showed that the untreated
enzyme contained only background levels of cobalt, manga-nese, and magnesium but did have substantial, althoughhighly variable, amounts of zinc. In the two enzyme prepa-rations described above, where the iron content was essen-tially equivalent (0.3 mol/mol of enzyme monomer), the zinccontents were 1.8 and 4.4 mol/mol of enzyme monomer.Such variable results raise the possibility that some or all ofthe zinc, a ubiquitous metal, was bound nonspecificallyduring the preparation of the enzyme.Metal chelation studies. The possible role of Fe2+ and Zn2+
in DAHPS(Trp) was investigated further by analyzing theeffect of metal chelators on the activity of the enzyme.Treatment of the native enzyme with 1 mM o-phenanthrolineled to a time-dependent inactivation of the enzyme whenactivity was assayed in the absence of Fe2+ (Fig. 3). Treat-ment with 100 p.M EDTA caused partial inactivation (Fig. 3);higher concentrations enhanced the inactivation but at 1 mMEDTA was still not as effective as o-phenanthroline (not
VOL. 173, 1991
on Septem
ber 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from
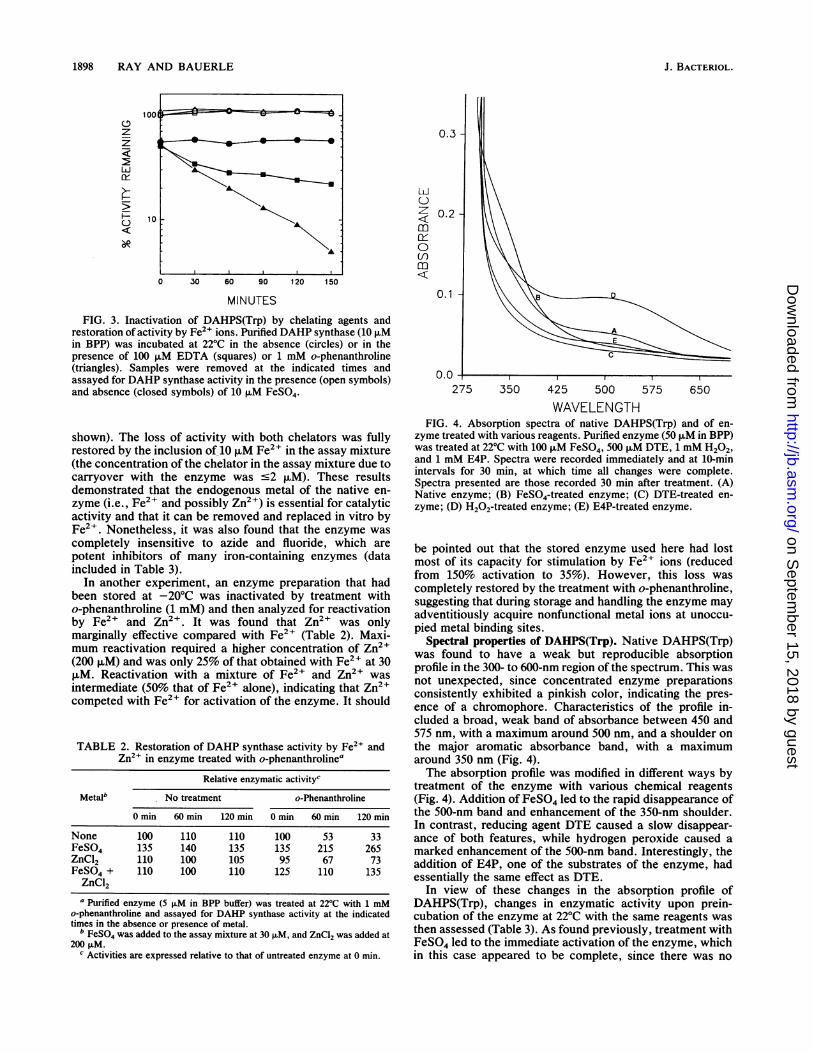
1898 RAY AND BAUERLE
cLzz
L,J
10
0 30 60 90 120 150
MINUTESFIG. 3. Inactivation of DAHPS(Trp) by chelating agents and
restoration of activity by Fe2" ions. Purified DAHP synthase (10 ,uMin BPP) was incubated at 22°C in the absence (circles) or in thepresence of 100 ,uM EDTA (squares) or 1 mM o-phenanthroline(triangles). Samples were removed at the indicated times andassayed for DAHP synthase activity in the presence (open symbols)and absence (closed symbols) of 10 ,uM FeSO4.
shown). The loss of activity with both chelators was fullyrestored by the inclusion of 10 ,uM Fe2+ in the assay mixture(the concentration of the chelator in the assay mixture due tocarryover with the enzyme was s2 ,uM). These resultsdemonstrated that the endogenous metal of the native en-zyme (i.e., Fe2+ and possibly Zn2+) is essential for catalyticactivity and that it can be removed and replaced in vitro byFe2+. Nonetheless, it was also found that the enzyme wascompletely insensitive to azide and fluoride, which arepotent inhibitors of many iron-containing enzymes (dataincluded in Table 3).
In another experiment, an enzyme preparation that hadbeen stored at -20°C was inactivated by treatment witho-phenanthroline (1 mM) and then analyzed for reactivationby Fe2+ and Zn2+. It was found that Zn2+ was onlymarginally effective compared with Fe2+ (Table 2). Maxi-mum reactivation required a higher concentration of Zn2+(200 ,uM) and was only 25% of that obtained with Fe2' at 30,uM. Reactivation with a mixture of Fe2+ and Zn2+ wasintermediate (50% that of Fe2+ alone), indicating that Zn2+competed with Fe2+ for activation of the enzyme. It should
TABLE 2. Restoration of DAHP synthase activity by Fe2' andZn2+ in enzyme treated with o-phenanthrolinea
Relative enzymatic activityc
Metalb No treatment o-Phenanthroline
0 min 60 min 120 min 0 min 60 min 120 min
None 100 110 110 100 53 33FeSO4 135 140 135 135 215 265ZnCl2 110 100 105 95 67 73FeSO4 + 110 100 110 125 110 135ZnCl2a Purified enzyme (5 ,uM in BPP buffer) was treated at 22°C with 1 mM
o-phenanthroline and assayed for DAHP synthase activity at the indicatedtimes in the absence or presence of metal.
b FeSO4 was added to the assay mixture at 30 F.M, and ZnC12 was added at200 ,uM.
c Activities are expressed relative to that of untreated enzyme at 0 min.
0.3 -
Z0.2-
0
m
0.1
0.0
275 350 425 500 575 650
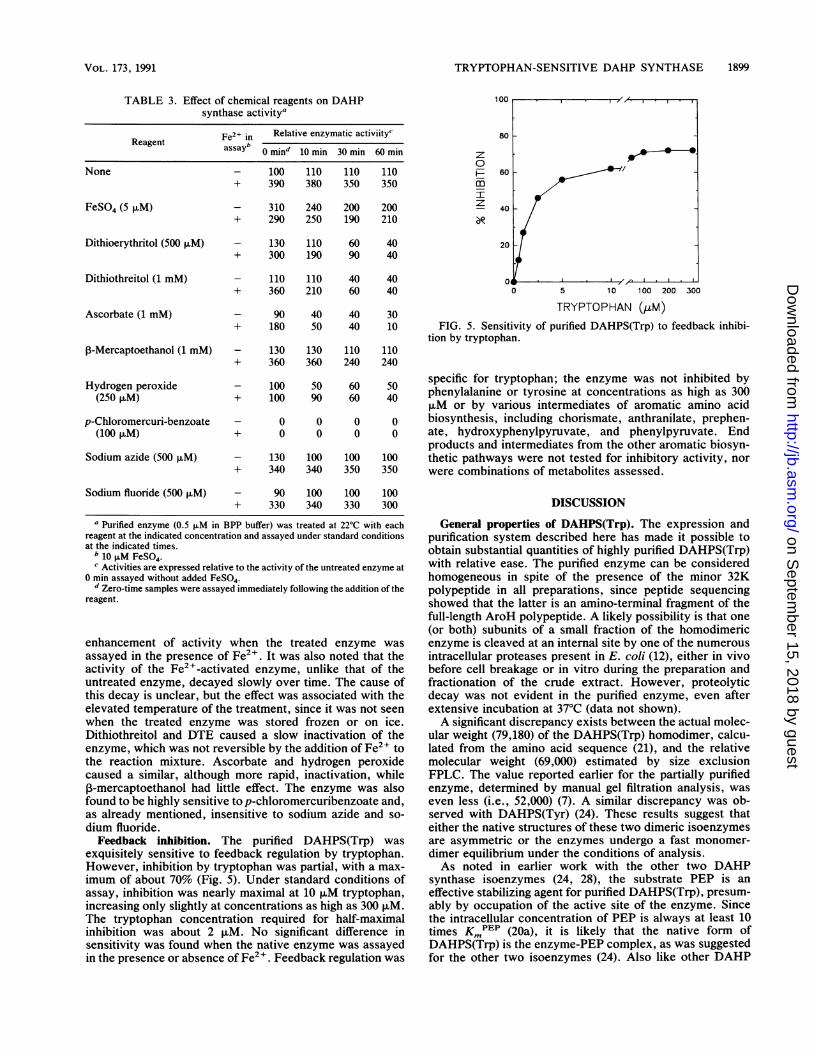
WAVELENGTHFIG. 4. Absorption spectra of native DAHPS(Trp) and of en-
zyme treated with various reagents. Purified enzyme (50 ,uM in BPP)was treated at 22°C with 100 ,uM FeSO4, 500 ,uM DTE, 1 mM H202,and 1 mM E4P. Spectra were recorded immediately and at 10-minintervals for 30 min, at which time all changes were complete.Spectra presented are those recorded 30 min after treatment. (A)Native enzyme; (B) FeSO4-treated enzyme; (C) DTE-treated en-zyme; (D) H202-treated enzyme; (E) E4P-treated enzyme.
be pointed out that the stored enzyme used here had lostmost of its capacity for stimulation by Fe2+ ions (reducedfrom 150% activation to 35%). However, this loss wascompletely restored by the treatment with o-phenanthroline,suggesting that during storage and handling the enzyme mayadventitiously acquire nonfunctional metal ions at unoccu-pied metal binding sites.
Spectral properties of DAHPS(Trp). Native DAHPS(Trp)was found to have a weak but reproducible absorptionprofile in the 300- to 600-nm region of the spectrum. This wasnot unexpected, since concentrated enzyme preparationsconsistently exhibited a pinkish color, indicating the pres-ence of a chromophore. Characteristics of the profile in-cluded a broad, weak band of absorbance between 450 and575 nm, with a maximum around 500 nm, and a shoulder onthe major aromatic absorbance band, with a maximumaround 350 nm (Fig. 4).The absorption profile was modified in different ways by
treatment of the enzyme with various chemical reagents(Fig. 4). Addition of FeSO4 led to the rapid disappearance ofthe 500-nm band and enhancement of the 350-nm shoulder.In contrast, reducing agent DTE caused a slow disappear-ance of both features, while hydrogen peroxide caused amarked enhancement of the 500-nm band. Interestingly, theaddition of E4P, one of the substrates of the enzyme, hadessentially the same effect as DTE.
In view of these changes in the absorption profile ofDAHPS(Trp), changes in enzymatic activity upon prein-cubation of the enzyme at 22°C with the same reagents wasthen assessed (Table 3). As found previously, treatment withFeSO4 led to the immediate activation of the enzyme, whichin this case appeared to be complete, since there was no
__ _r~~~~~~~~~~ * * *
I.
i nn
J. BACTERIOL.
on Septem
ber 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from
TRYPTOPHAN-SENSITIVE DAHP SYNTHASE 1899
TABLE 3. Effect of chemical reagents on DAHPsynthase activity'
Fe2" in Relative enzymatic activiitycReagent assayb 0 mind 10 min 30 min 60 min
None - 100 110 110 110+ 390 380 350 350
FeSO4 (5 jiM) - 310 240 200 200+ 290 250 190 210
Dithioerythritol (500 ,uM) - 130 110 60 40+ 300 190 90 40
Dithiothreitol (1 mM) - 110 110 40 40+ 360 210 60 40
Ascorbate (1 mM) - 90 40 40 30+ 180 50 40 10
P-Mercaptoethanol (1 mM) - 130 130 110 110+ 360 360 240 240
Hydrogen peroxide - 100 50 60 50(250 ,uM) + 100 90 60 40
p-Chloromercuri-benzoate - 0 0 0 0(100 ,uM) + 0 0 0 0
Sodium azide (500 ,uM) - 130 100 100 100+ 340 340 350 350
Sodium fluoride (500 ,uM) - 90 100 100 100+ 330 340 330 300
a Purified enzyme (0.5 ,uM in BPP buffer) was treated at 22°C with eachreagent at the indicated concentration and assayed under standard conditionsat the indicated times.
b 10 ,uM FeSO4.c Activities are expressed relative to the activity of the untreated enzyme at
0 min assayed without added FeSO4.d Zero-time samples were assayed immediately following the addition of the
reagent.
enhancement of activity when the treated enzyme wasassayed in the presence of Fe2+. It was also noted that theactivity of the Fe2+-activated enzyme, unlike that of theuntreated enzyme, decayed slowly over time. The cause ofthis decay is unclear, but the effect was associated with theelevated temperature of the treatment, since it was not seenwhen the treated enzyme was stored frozen or on ice.Dithiothreitol and DTE caused a slow inactivation of theenzyme, which was not reversible by the addition of Fe2+ tothe reaction mixture. Ascorbate and hydrogen peroxidecaused a similar, although more rapid, inactivation, whileP-mercaptoethanol had little effect. The enzyme was alsofound to be highly sensitive to p-chloromercuribenzoate and,as already mentioned, insensitive to sodium azide and so-dium fluoride.Feedback inhibition. The purified DAHPS(Trp) was
exquisitely sensitive to feedback regulation by tryptophan.However, inhibition by tryptophan was partial, with a max-imum of about 70% (Fig. 5). Under standard conditions ofassay, inhibition was nearly maximal at 10 ,uM tryptophan,increasing only slightly at concentrations as high as 300 ,uM.The tryptophan concentration required for half-maximalinhibition was about 2 ,uM. No significant difference insensitivity was found when the native enzyme was assayedin the presence or absence of Fe2+. Feedback regulation was
100
80
z0
m
z
60
40
20
0 5 1 0 100 200 300
TRYPTOPHAN (FaM)FIG. 5. Sensitivity of purified DAHPS(Trp) to feedback inhibi-
tion by tryptophan.
specific for tryptophan; the enzyme was not inhibited byphenylalanine or tyrosine at concentrations as high as 300pFM or by various intermediates of aromatic amino acidbiosynthesis, including chorismate, anthranilate, prephen-ate, hydroxyphenylpyruvate, and phenylpyruvate. Endproducts and intermediates from the other aromatic biosyn-thetic pathways were not tested for inhibitory activity, norwere combinations of metabolites assessed.
DISCUSSION
General properties of DAHPS(Trp). The expression andpurification system described here has made it possible toobtain substantial quantities of highly purified DAHPS(Trp)with relative ease. The purified enzyme can be consideredhomogeneous in spite of the presence of the minor 32Kpolypeptide in all preparations, since peptide sequencingshowed that the latter is an amino-terminal fragment of thefull-length AroH polypeptide. A likely possibility is that one(or both) subunits of a small fraction of the homodimericenzyme is cleaved at an internal site by one of the numerousintracellular proteases present in E. coli (12), either in vivobefore cell breakage or in vitro during the preparation andfractionation of the crude extract. However, proteolyticdecay was not evident in the purified enzyme, even afterextensive incubation at 37°C (data not shown).A significant discrepancy exists between the actual molec-
ular weight (79,180) of the DAHPS(Trp) homodimer, calcu-lated from the amino acid sequence (21), and the relativemolecular weight (69,000) estimated by size exclusionFPLC. The value reported earlier for the partially purifiedenzyme, determined by manual gel filtration analysis, waseven less (i.e., 52,000) (7). A similar discrepancy was ob-served with DAHPS(Tyr) (24). These results suggest thateither the native structures of these two dimeric isoenzymesare asymmetric or the enzymes undergo a fast monomer-dimer equilibrium under the conditions of analysis.As noted in earlier work with the other two DAHP
synthase isoenzymes (24, 28), the substrate PEP is aneffective stabilizing agent for purified DAHPS(Trp), presum-ably by occupation of the active site of the enzyme. Sincethe intracellular concentration of PEP is always at least 10times KmPEP (20a), it is likely that the native form ofDAHPS(Trp) is the enzyme-PEP complex, as was suggestedfor the other two isoenzymes (24). Also like other DAHP
VOL. 173, 1991
on Septem
ber 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

1900 RAY AND BAUERLE
synthases, DAHPS(Trp) was markedly sensitive to p-chlo-romercuribenzoate, which suggests a role for an essentialcysteine(s) in the structure or activity of the enzyme. Therecovery of an inactivating mutation at residue Cys-326,which is invariant in the three E. coli isoenzymes, afterrandom mutagenesis of the aroH gene (21) supports thisconclusion. In view of this, it is significant that the enzymewas destabilized by reducing agents such as dithiothreitoland DTE (Table 3). This could occur if a cysteine sulfhydrylof the enzyme, perhaps Cys-326, were directly involved inPEP binding at the active site. If so, strong reducing agentsmight act to reverse the binding, thereby destabilizing theenzyme over time, as observed. A reaction mechanism forDAHP synthase that involves such an enzyme-phosphoe-nolpyruvyl intermediate coupled by a covalent thiol linkagehas been proposed (11).DAHPS(Trp) as a metalloenzyme. The results reported
here indicate that, like other DAHP synthases, DAHPS(Trp)has a metal as an essential cofactor. It appears likely that theendogenous metal is ferrous iron. The activity of the purifiedenzyme was stimulated two- to threefold by Fe2+ but not toany significant degree by other metals tested. The enzymewas inactivated by chelation with o-phenanthroline andEDTA and was fully reactivated by Fe2+ ions (Fig. 2).Finally, the native enzyme contained 0.3 mol of iron permonomeric subunit and when activated by treatment withFe2+ gained additional iron such that the content of fullyactivated enzyme was extrapolated to be close to stoichio-metric. Since the activity of the enzyme in crude extractswas stimulated to about the same extent as the purifiedenzyme (data not shown), it appears that, under the condi-tions of expression and purification used here, the enzyme isnot fully saturated with metal when released from the cell.The large variation in the amount of zinc in different prepa-rations of the native enzyme argue that this ubiquitous metalmay have been acquired in vitro by adventitious bindingduring the purification and handling of the enzyme (30).Nonetheless, the weak reactivation of chelated enzyme byZn2+ indicates that it can also interact specifically with theenzyme as a metal activator.These results differ in some respects with those of other
studies. It has been reported that a homogeneous prepara-tion of the tetrameric DAHPS(Phe) from E. coli contained0.25 mol of iron per mol of enzyme monomer and wasirreversibly inactivated by chelation with o-phenanthroline(16). Similarly, the monomeric DAHPS(Phe) from Saccha-romyces cerevisiae was found to contain 0.6 mol of iron permol of enzyme and to be inactivated by EDTA; however, itwas readily reactivated not only by Fe2 , but also by Mn2 ,Co2+, and Zn2+ (18). In contrast, the dimeric DAHPS(Tyr)isoenzyme from E. coli was recently reported to contain 0.5mol of copper per enzyme monomer and no significantamount of iron. Further, it was inactivated by the copperbinding reagent cyanide and was reactivated to differentextents by numerous divalent metals, including Cu2+, Mn2+,Zn2+, and Cd2+, but not by Fe2+. Thus, it appears that themetal requirement of different DAHP synthases can besatisfied in vitro by different groups of metals, each with adifferent catalytic efficiency. It is possible that the samesituation obtains in vivo, although metal utilization wouldnecessarily be limited by intracellular availability, whichmay vary between strains as well as with the culture condi-tions used (1).The spectral properties of DAHPS(Trp) (Fig. 4) are un-
doubtedly a manifestation of its nature as a metalloprotein.The shoulder of absorbance centered around 350 nm is
similar to the well-defined, copper-dependent absorbancepeak at 350 nm seen with DAHPS(Tyr) (1). This feature ofDAHPS(Tyr), and perhaps that of DAHPS(Trp) as well, isindicative of a ligand-to-metal charge transfer transition (27).On the basis of the absorbance behavior of the DAHPS(Trp)isoenzyme (Fig. 4), we suggest that the 350-nm band isdependent on the presence of PEP bound at the active site ofthe enzyme. That activation of the native enzyme by Fe2 ,which apparently binds to enzyme molecules with unoccu-pied metal binding sites, caused an enhancement of the350-nm shoulder of absorbance is consistent with this idea.The bleaching of the absorbance at 350 nm by treatment ofthe enzyme with reducing agents could result from thereduction of the proposed thiol linkage of the enzyme-PEPcomplex, leading to the release of the bound PEP and metaland to the eventual destabilization of the enzyme. Treatmentwith E4P should have a similar effect, as was observed, bydepleting the enzyme of PEP by its conversion to DAHP.The weak absorbance band centered at 500 nm may be due
to a fraction of enzyme molecules whose bound iron hasbeen oxidized to the ferric form. The observed elimination ofthis band by treatment of the enzyme with Fe2+ ions, whichcould act to reduce the bound Fe3" to Fe2+, and the markedenhancement of the band by treatment of the enzyme withH202, which would act to oxidize Fe2+ to Fe3+, are consis-tent with this idea. Work is now under way to analyzefurther the role of metals in the catalytic mechanism of the E.coli DAHP synthases.Feedback inhibition of DAHPS(Trp). Feedback sensitivity
of the homogeneous enzyme prepared here was significantlygreater than that detected with crude or partially purifiedpreparations, with which maximum inhibition was only 30 to50% (compared with 70%) (13, 20, 21). Such partial inhibi-tion has been consistently observed with DAHPS(Trp) fromE. coli. This character is typical of enzymes when bothsubstrate and inhibitor can bind simultaneously, presumablyoccupying separate sites, and when the enzyme-substratecomplex and the enzyme-substrate-inhibitor complex bothyield product (25). Thus, as observed here with DAHP-S(Trp), at a fixed concentration of substrate the velocity ofthe reaction can never be driven to zero no matter how highthe concentration of inhibitor. The existence of separateinhibitor and substrate binding sites in DAHPS(Trp) isindicated by the kinetic properties of a group of mutantenzymes with altered feedback sensitivity (20a).
ACKNOWLEDGMENTS
This work was supported by Public Health Service research grantGM35889 and training grant DMB8703685 from the National Insti-tutes of Health.We thank Charles Yanofsky for bacterial strains, Wilson Mclvor
for technical assistance, and Craig Stephens, Bruce Averill, andMichael Dunn for helpful discussions.
REFERENCES
1. Baasov, T., and J. R. Knowles. 1989. Is the first enzyme of theshikimate pathway, 3-deoxy-D-arabino-heptulosonate-7-phos-phate synthase (tyrosine sensitive), a copper metalloenzyme? J.Bacteriol. 171:6155-6160.
2. Bauerle, R., J. Hess, and S. French. 1987. Anthranilate syn-thase-anthranilate-5'-phosphoribosyl-1-pyrophosphate phos-phoribosyltransferase complex and subunits of Salmonella ty-phimurium. Methods Enzymol. 142:366-386.
3. Bidlingmeyer, B. A., S. A. Cohen, and T. L. Tarvin. 1984. Rapidanalysis of amino acids using pre-column derivatization. J.Chem. 336:93-104.
J. BACTERIOL.
on Septem
ber 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

TRYPTOPHAN-SENSITIVE DAHP SYNTHASE 1901
4. Bradford, M. 1976. A rapid and sensitive method for thequantitation of microgram amounts of protein utilizing theprinciple of protein-dye binding. Anal. Biochem. 72:248-254.
5. Brosius, J., and A. Holy. 1984. Regulation of ribosomal RNApromoters with a synthetic lac operator. Proc. Natl. Acad. Sci.USA 81:6929-6933.
6. Brown, K. D., and C. H. Doy. 1966. Control of three isoenzymic7-phospho-2-oxo-3-deoxy-D-arabino-heptonate-D-erythrose-4-phosphate lyases of Escherichia coli W and derived mutants byrepressive and "inductive" effects of the aromatic amino acids.Biochim. Biophys. Acta 118:157-172.
7. Camakaris, J., and J. Pittard. 1974. Purification and propertiesof 3-deoxy-D-arabino-heptulosonic acid-7-phosphate synthetase(trp) from Escherichia coli. J. Bacteriol. 120:590-597.
8. Davies, W. D., and B. E. Davidson. 1982. The nucleotidesequence of aroG, the gene for 3-deoxy-D-arabino heptu-losonate-7-phosphate synthetase (phe) in Escherichia coli K-12.Nucleic Acids Res. 10:4045-4058.
9. deBoer, H. A., L. J. Comstock, and M. Vasser. 1983. The tacpromoter: a functional hybrid derived from trp and lac promot-ers. Proc. Natl. Acad. Sci. USA 78:21-25.
10. DeLeo, A. B., J. Dayan, and D. B. Sprinson. 1973. Purificationand kinetics of tyrosine-sensitive 3-deoxy-D-arabino-heptu-losonic acid 7-phosphate synthetase from Salmonella typhimu-rium. J. Biol. Chem. 248:2344-2353.
11. Ganem, B. 1978. From glucose to aromatics: recent develop-ments in natural products of the shikimate pathway. Tetrahe-dron 34:3353-3383.
12. Gottesman, S. 1989. Genetics of proteolysis in Escherichia coli.Annu. Rev. Genet. 23:163-198.
13. Herrmann, K. M. 1983. The common aromatic biosyntheticpathway, p. 301-318. In K. M. Herrmann and R. L. Somerville(ed.), Amino acids: biosynthesis and genetic regulation. Addi-son-Wesley Publishing Co., Reading, Mass.
14. Laemmli, U. K. 1970. Cleavage of structural proteins during theassembly of the head of bacteriophage T4. Nature (London)227:680-685.
15. Matsudaira, P. 1987. Sequence from picomole quantities ofproteins electroblotted onto polyvinylidene difluoride mem-branes. J. Biol. Chem. 262:10035-10038.
16. McCandliss, R. J., and K. M. Herrmann. 1978. Iron, an essentialelement for biosynthesis of aromatic compounds. Proc. Natl.Acad. Sci. USA 75:4810-4813.
17. McCandliss, R. J., M. D. Poling, and K. M. Herrmann. 1978.3-Deoxy-D-arabino-heptulosonate-7-phosphate synthase. Puri-fication and molecular characterization of the phenylalanine-sensitive isoenzyme from Escherichia coli. J. Biol. Chem.253:4259-4265.
18. Paravicini, G., T. Schmidheini, and G. Braus. 1989. Purificationand properties of the 3-deoxy-D-arabino-heptulosonate-7-phos-phate synthase (phenylalanine-inhibitable) of Saccharomycescerevisiae. Eur. J. Biochem. 186:361-366.
19. Pittard, J. 1987. Biosynthesis of the aromatic amino acids, p.
368-394. In J. L. Ingraham, L. B. Low, B. Magasanik, M.Schaechter, and H. E. Umbarger (ed.), Escherichia coli andSalmonella typhimurium: cellular and molecular biology, vol. 1.American Society for Microbiology, Washington, D.C.
20. Pittard, J., J. Camakaris, and B. J. Wallace. 1969. Inhibition of3-deoxy-D-arabino-heptulosonic acid-7-phosphate synthetase(trp) in Escherichia coli. J. Bacteriol. 97:1242-1247.
20a.Ray, J. M., and R. Bauerle. Unpublished data.21. Ray, J. M., C. Yanofsky, and R. Bauerle. 1988. Mutational
analysis of the catalytic and feedback sites of the tryptophan-sensitive 3-deoxy-D-arabino-heptulosonate-7-phosphate syn-thase of Escherichia coli. J. Bacteriol. 170:5500-5506.
22. Sambrook, J., E. Fritsch, and T. Maniatis. 1989. Molecularcloning: a laboratory manual. Cold Spring Harbor Laboratory,Cold Spring Harbor, N.Y.
23. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequenc-ing with chain-terminating inhibitors. Proc. Natl. Acad. Sci.USA 74:5436-5467.
24. Schoner, R., and K. M. Herrmann. 1976. 3-Deoxy-D-arabino-heptulosonate 7-phosphate synthase. Purification, properties,and kinetics of the tyrosine-sensitive isoenzyme from Esche-richia coli. J. Biol. Chem. 251:5440-5447.
25. Segel, I. 1975. Enzyme kinetics, p. 161-166. John Wiley & Sons,Inc., New York.
26. Shultz, J., M. A. Hermodson, C. C. Garner, and K. M. Her-rmann. 1984. The nucleotide sequence of the aroF gene ofEscherichia coli and the amino acid sequence of the encodedprotein, the tyrosine-sensitive 3-deoxy-D-arabino-heptu-losonate-7-phosphate synthase. J. Biol. Chem. 259:9655-9661.
27. Solomon, E. I., K. W. Penfield, and D. E. Wilcox. 1983. Activesites in copper proteins: an electronic structure overview, p.1-57. In M. J. Clarke et al., (ed.), Structure and bonding, (vol.53.) Springer-Verlag, Berlin.
28. Staub, M., and G. Denes. 1969. Purification and properties of the3-deoxy-D-arabino-heptulosonate-7-phosphate synthase (phen-ylalanine-sensitive) of Escherichia coli K-12. Biochim. Biophys.Acta 178:599-608.
29. Tribe, D. E., H. Camakaris, and J. Pittard. 1976. Constitutiveand repressible enzymes of the common pathway of aromaticbiosynthesis in Escherichia coli K-12: regulation of enzymesynthesis at different growth rates. J. Bacteriol. 127:1085-1097.
30. Wagner, F. W. 1988. Preparation of metal-free enzymes. Meth-ods Enzymol. 158:21-32.
31. Wallace, B. J., and J. Pittard. 1967. Genetic and biochemicalanalysis of the isozymes concerned in the first reaction ofaromatic biosynthesis in Escherichia coli. J. Bacteriol. 93:234-244.
32. Zurawski, G., R. P. Gunsalus, K. D. Brown, and C. Yanofsky.1981. Structure and regulation of aroH, the structural gene forthe tryptophan repressible 3-deoxy-D-arabino-heptulosonic ac-id-7-phosphate synthetase of Escherichia coli. J. Mol. Biol.145:47-73.
VOL. 173, 1991
on Septem
ber 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from












![Purification andProperties of Undecyl Acetate Esterase ... · UNDECYLACETATEESTERASEFROMP. CEPACIA.I. suring hydrolysis of undecyl [2- 4C]acetate into 1-undecanol and [1-_4C]acetate](https://img.pdfslide.net/doc/110x75/5e10065c5061b41b950418f8/purification-andproperties-of-undecyl-acetate-esterase-undecylacetateesterasefromp.jpg)
















