Embed Size (px)
Citation preview

ANALYTICAL BIOCHEMISTRY 47, 132-138 (1972)
Rapid Assay for Purine Phosphoribosyltransferases
WOLF GUTENSOHN AND GORDON GUROFF
Section on Intermediary Metabolism, Laboratory of Biomedical Sciences, National Institute of Child Health and Human Development,
National Institutes of Health, Bethesda, Maryland SOOl/,
Received August 11, 1971
Purine phosphoribosyltransferases, especially hypoxanthine-guanine phosphoribosyltransferase (EC 2.4.2.8, 1MP:pyrophosphate phosphoribo- syltransferase) (HGPRTase) , have been intensively studied in recent years because of the relationship of the latter enzyme to the Lesch- Nyhan syndrome, a condition of children characterized by compulsive biting of fingers and lips (for review see reference 1). The most thor- oughly investigated enzyme is that from human erythrocytes (24), but the enzyme has also been found and characterized in a number of other human and mammalian tissues including tumor cells (5-11) and cells in tissue culture (12-13), as well as in microorganisms (14).
In most of the previous work the enzyme was assayed by incubating the 14C-labeled purine bases with PRPP,l Mg++ ions, buffer, and the enzyme preparation. Separation of product and substrate was achieved by paper chromatography, thin-layer chromatography, or high-voltage paper electrophoresis. These procedures, though accurate and repro- ducible, are time consuming and are difficult to adjust to the processing of large numbers of samples, as needed for an enzyme purification.
We will describe an assay procedure requiring only small amounts of protein (5 mpg to 25 pg) in a total volume of 20 ~1 by which many samples can be assayed in a short time. The method used by Murray et al: (lOJ5) in which the nucleotide formed is adsorbed on DEAE- cellulose paper discs might be equally rapid but uses more protein in bigger incubation volumes. Also, there is no indication in Murray’s papers as to the absolute counts of the blanks in the assay. A compari- son with the assay recently described by Chow et al. (16) using chelat- ing columns (Chelex 100 loaded with a copper-ammonia complex) will be given below.
1 Abbreviations: PRPP, 5-phosphbribosyl-l-pyrophosphate. TTP, thymidine triphosphate.
132 @ 1972 by Academic Press, Inc.

PRTase ASSAY 133
METHODS
Incubation
Incubation mixture: Tris-HCI buffer, pH 7.4 70 mM PRPP (di Mg salt) 0.9 mM Mg” 4mM TTP (where indicated) 3.3 mM
Purine : Adenine-8-14C (3.9 mCi/mmole) 0.38 mM Hypoxanthine-8-Y (4.0 mCi/mmole) 0.37 mM Guanine-8-Y? (3.4 mCi/mmole) 0.14 mM
(The lower concentration of G was taken because of the low solubility of this purine.j Enzyme (for concentrations see below) in 0.05 M Tris- HCl, pH 7.4, was added to a total volume of 20 ~1.
Reactions were started by addition of either enzyme or radioactive purine base. (The initial burst of activity described by others (3) when the enzyme was preincubated with PRPP was not observed under our conditions.) Samples were incubated in small test tubes at 37°C with shaking. The reaction was stopped by adding 50 ~1 of 0.1 M EDTA and immediate immersion of the test tube in a dry ice-acetone bath. Stopping the reactions by boiling the mixtures gave less reproducible result’s,
Separation
Our separation procedure is actually an adaptation of an already existing method (17) to a micro scale. Dowex 1-X8 (200-400 mesh) is brought to the formate form as described by Rembold and Busch- mann (18). A stock. suspension of 2 parts of wet resin and 5 parts of water is prepared. Disposable Pasteur pipets are plugged with a piece of cotton wool at the narrow end and are filled with 1 ml of the vigor- ously stirred resin-suspension yielding a resin bed of about 1 cm height. Each column is washed with about 5 ml of water before use.
The thawed incubation mixture is t’ransferred quantitatively to the top of the resin bed by use of a long-tipped Pasteur pipet; the column is filled with 0.1% HCOOH and connected by rubber tubing to a reser- voir containing 0.1% HCOOH. The column is eluted with 10 to 15 ml of 0.1% HCOOH. An exact measurement of this elution volume is not crucial, but large variations might affect the reproducibility. Groups of columns can be run at the same time. After completion of the 0.1% HCOOH elution the columns are allowed to run dry. Counting vials

134 GUTENSOHN AND GUROFF
are placed under the columns and the nucleotides are now eluted with 2 ml of either 4 N or 8 N HCOOH. 10 ml of Bray solution (19) is added and the samples are counted.
For the electrophoretic separation the reaction was stopped by add- ing 2 ,~l of 42% perchloric acid; 2 ~1 of 0.5 M Tris-HCl, pH 7.0, and 2 ~1 of 4.4111 KOH were added and the mixture was centrifuged. Then 15 ~1 of the supernatant fraction was applied to 3MM Whatman paper with appropriate standards. Electrophoresis was run for 30 min in 0.02 M sodium lactate, pH 3.6, at 5000V and 30 mA. The areas containing the nucleotides were inspected under UV light, cut out, and counted in 10 ml of Bray solution.
In contrast to a Dowex l-X8 formate column, a Chelex 100 column of the same dimensions loaded with a copper-ammonia complex (16) holds the purine bases, whereas the mononucleotides are immediately eluted with water. 10 ml of water is needed to complete elution, only 2 ml of which is actually counted; therefore the sensitivity of the method is only 20% of the theoretical value.
Sensitivity
Samples counted in 4 N or 8 N HCOOH show higher quenching com- pared with water. 14C is counted in our system with 58% efficiency in water, 35% efficiency in 4N HCOOH, and 22% efficiency in 8N HCOOH, i.e., the yield in 4N HCOOH is about 60% and in 8 N HCOOH about 40% of that in water.
Columns were calibrated with I%-labeled purine bases, the corre- sponding mononucleotides, and mixtures of both. The amounts of 14C-purines (G, H, and A) used in the assay are almost quantitatively eluted with 10 ml of 0.1% HCOOH. The subsequent elution with 4N or 8 N HCOOH yields only 20-50 cpm above background, which corresponds to only 0.05-0.15% of the total substrate.
The commerically available mononucleotides are well held by the column during the 0.1% HCOOH elution, except for a certain percentage of impurities as indicated by the supplier. This, however, is no impair- ment to the procedure, since in the actual assay the mononucleotides are enzymically produced.
Since, as shown above, HCOOH concentration has an influence on quenching, it is desirable to make the concentration as low as possible. On the other hand to achieve a relatively quantit,ative elution of the mononucleotides we had to choose 4 N HCOOH for GMP and 8 N HCOOH for AMP and IMP. Under these conditions 95% of GMP, 94% of AMP, and 88% of IMP are eluted with 2 ml of the acid. The use of the Dowex column in the chloride instead of the formate form or
PRTase ASSAY 135
of HCl instead of HCOOH as eluant does not bring any advantage in this respect.
These figures together with the quench given above have an influence on the sensitivity of the method. Compared with the theoretical value measured in water, only 56% of the counts for GMP and only 38% of the counts for AMP and IMP are actually measured.
RESULTS
We have applied our method to the determination of PRTase activ- ities in a crude extract, in a partially purified enzyme preparation from rat brain, and in an eryt;hrocyte lysate from a normal human subject prepared according to Krenitsky et al. (2).
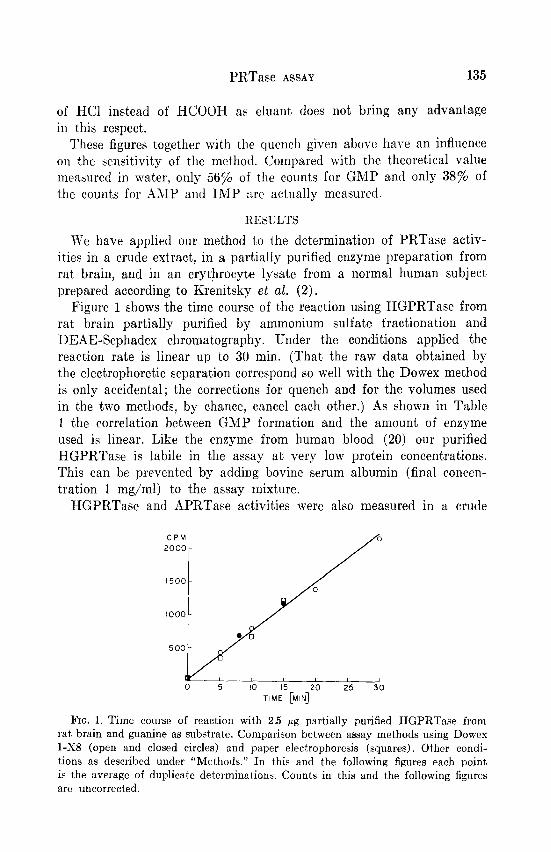
Figure 1 shows the time course of the reaction using HGPRTase from rat brain partially purified by ammonium sulfate fractionation and DEAE-Sephadex chromatography. Under the conditions applied the reaction rate is linear up to 30 min. (That the raw data obtained by the electrophoretic separation correspond so well with the Dowex method is only accidental; the corrections for quench and for the volumes used in the two methods, by chance, cancel each other.) As shown in Table 1 the correlation between GMP formation and the amount, of enzyme used is linear. Like the enzyme from human blood (20) our purified HGPRTase is labile in the assay at very low protein concentrations. This can be prevented by adding bovine serum albumin (final concen- tration 1 mg/ml) to the assay mixture.
HGPRTase and APRTase activit’ies were also measured in a crude
CPM 2000 -
FIG. 1. Time course of reaction with 2.5 pg partially purified HGPRTase from rat brain and guanine as substrate. Comparison between assay methods using Dowes l-X8 (open and closed circles) and paper electrophoresis (squares). Other condi- tions as described under “Methods.” In this and the following figures each point is the average of duplicate determinations. Counts in this and the following figures are uncorrected.
136 GUTENSOHN AND GUROFF
TABLE 1
FormaCon of GMP as Function of Enzyme Concent,ration in Partially Purified Preparation from Bat Brain”
(other conditions given under “Methods”)
Protein, mpg/assay
cpm minus background, av. of duplicate detns.
5 min incubated 10 min incubated
4 209 436 12 604 1246
20 998 2181
a The enzyme used in these experiments was somewhat more purified by ammonium sulfate fractionation, a heat step, and DEAE-cellulose chromatography.
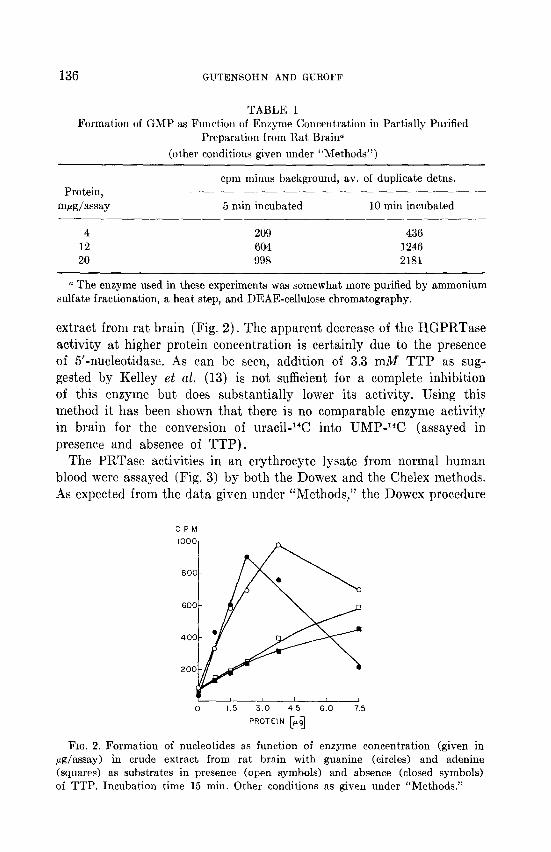
extract from rat brain (Fig. 2). The apparent decrease of the HGPRTase activity at higher protein concentration is certainly due to the presence of 5’-nucleotidase. As can be seen, addition of 3.3 mM TTP as sug- gested by Kelley et al. (13) is not sufficient for a complete inhibition of this enzyme but does substantially lower its activity. Using this method it has been shown that there is no comparable enzyme activity in brain for the conversion of uracil-14C into UMP-W (assayed in presence and absence of TTP).
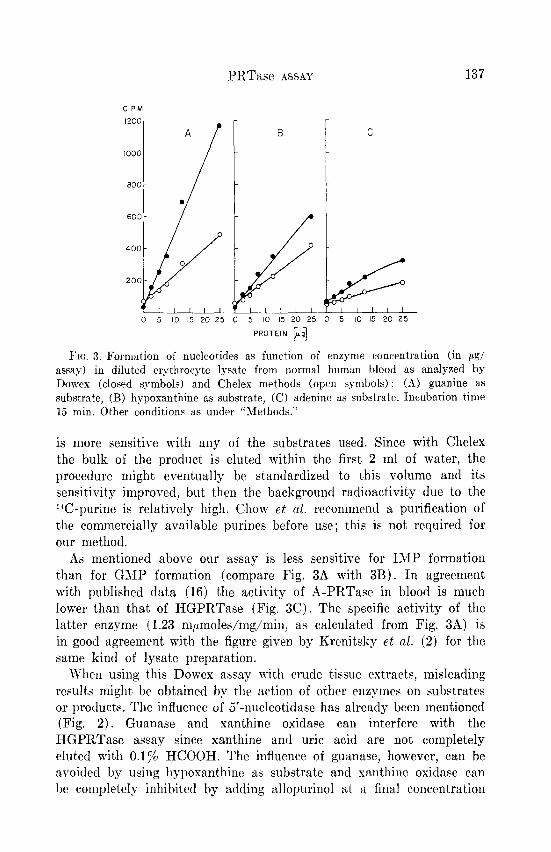
The PRTase activities in an erythrocyte lysate from normal human blood were assayed (Fig. 3) by both the Dowex and the Chelex methods. As expected from the data given under “Methods,” the Dowex procedure
CPM
0 15 30 45 6.0 75
PROTEIN [+g
FIG. 2. Formation of nucleotides as function of enzyme concentration (given in ag/assay) in crude extract from rat brain with guanine (circles) and adenine (squares) as substrates in presence (open symbols) and absence (closed symbols) of TTP. Incubation time 15 min. Other conditions as given under “Methods.”
PRTasc ASSAY
C PM
IZOO-
1000
SOOL
6OOL
400
A
/
.
B
/ 2oo[ k 0 5 10 15 20 25 0 5 IO I5 20 25
$ 0
C
$6 5 IO 15 20 25
Fro. 3. Formation of nucleotides as function of enzyme concentration (in pg/ assay) in diluted erythrocyte lysate from normal human blood as analyzed by Dowes (closed symbols) and Cheles methods (open symbols) : (A) guanine as substrate, (B) hypoxanthine as substrate, (C) adenine as substrate. Incubation time 15 min. Other conditions as under “Methods.”
is more sensitive with any of the substrates used. Since with Chelex the bulk of the product is eluted within the first 2 ml of water, the procedure might eventually be standardized to t’his volume and its sensitivity improved, but, then the background radioactivity due to the *%-purine is relatively high. Chow et al. recommend a purification of the commercially available purines before use; this is not required for our method.
As mentioned above our assay is less sensitive for IMP formation than for GMP formation (compare Fig. 3A with 3B). In agreement with published data (16) the activity of A-PRTase in blood is much lower than that of HGPRTase (Fig. 3C). The specific activity of the latter enzyme (1.23 m~moles/mg/min, as calculated from Fig. 3A) is in good agreement with the figure given by Krenitsky et al. (2) for the same kind of lysate preparation.
When using this Dowex assay with crude tissue extracts, misleading results might be obtained by the action of other enzymes on substrates or products. The influence of 5’-nucleotidase has already been mentioned (Fig. 2). Guanase and xanthine oxidase can interfere with the HGPRTase assay since xanthine and uric acid are not completely eluted with 0.1% HCOOH. The influence of guanase, however, can be avoided by using hypoxanthine as substrate and xanthine oxidase can be completely inhibited by adding allopurinol at a final concentration

138 GUTEh-SOHS AND GVROFF
of 1 mM, which (as we could show) does not interfere with HGPRTase. In principle the Dowex method is also applicable to tritiated purine
substrates. There is no tritium exchange during the separation pro- cedure. Since, however, the quench for tritium counts in water and formic acid is very high, considerable errors might be brought in by the necessary correction factors.
SUMMARY
A micro assay for purine phosphoribosyltransferases has been de- veloped using small Dowex l-X8 columns. This method compares favorably in accuracy, sensitivity, and reproducibility with many estab- lished procedures. It seems to be most suitable for enzyme purifications,
which require many assays tjo be performed in a reasonable time.
REFERENCES
1. KELLEY, W. N.. Ann. Intern. Med. 70, 155 (1969). 2. KRENITSKY, T. rZ., PAPAIOANWXJ, R., AND ELION, G. B., J. Biol. Chem. 244, 1263
(1969). 3. KRENITSKY, T. A., AND PAPAIOANNOU, R., J. Biol. Chem. 244, 1271 (1969). 4. HENDERSON, J. F.. BRONX, L. W., KELLEY, W. N., ROSENBLOOM, F. M., AND
SEEGMILLER, J. E., J. Biol. Chem. 243, 2514 (1968). 5. RLISEXBLOOM, F. M., KELLEY, W. IT., MILLER, J., HENDERSON, J. F., .~ND
SEEGMILLER, J. E., J. Amer. Med. Ass. 202, 175 (1967). 6. GARTLER, S. M., SCOTT, R. C., GOLDSTEIN, J. C., AND CAMPBELL, B., Science 172,
572 (1971) 7. KRENITSKY, T. A., NEIL, S. M., ELION, G. B., AND HITCHINGS, G. H., J. Biol.
Chem. 244, 4779 (1969). 8. KRENITSFCY, T. A., Biochim. Biophys. Acfn 179, 506 (1969). 9. SANTOS. J. N., HEMPSTEAD. K. W., KOPP, L. E., AXD MIECH, R. P., J. NezLro-
them. 15, 367 (1968). 10. MURRAY, A. W., Biochem. J. 104 664 (1966). 11. MURRAY, A. W.. Biochem. J. 104, 675 (1967). 12. DEMARS, R., Fed. Proc. 30, 944 (1971). 13. KELLEY, W. N., ASD MEADE, J. C., J. Biol. Chem. 246, 2953 (1971). 14. KRENITSKY. T. A., NEIL, S. H., AND MILLER, R. L., J. Biol. Chem. 245, 2605
( 1970). 15. ATKINSON, M. R., AND MURRAY, 9. W., Riochem. J. 94, 64 (1965). 16. CHOW, D. C., K.4w.4H.4RA. F. S., SAUNDERS, T., AND SORENSEN, L. B., 1. Lab.
Clin. Med. 76, 733 (1970). 17. FLAKS, J. G.. its “Methods in Enzymology” (Colowick, S. P.. and Kaplan, N. O.,
vds.), Vol. VI. 11. 136. Academic Press, New York. 1963. 18. &MBOLD, H., AND BuScHMANN, L., Hoppe-Seyler’s 2. Physiol. Chem. 330, 132
(1962). 19. BRAY, G. A., Anal. &o&em. 1, 279 (1960). 20. RUBIN, C. S.. DANCIS, J., YIP, L. C., NOWINSKI. R. C.. BALIS. M. E., Proc. Nat.
AC&. Sci. U. S. 68, 1461 (1971).





























