Embed Size (px)
Citation preview

RBCs Disorders 1
Dr. Nabila Hamdi
MD, PhD

ILOs • Discuss the classification of anemia into hypochromic-microcytic, normochromic-
normocytic and macrocytic.
• Categorize laboratory test procedures used in the diagnosis of anemia, outlining the basic workup of a patient who presents with anemia.
• Understand the utilization of peripheral blood and bone marrow smears to assess the deviations from normal marrow response which occur in different types of anemia.
• Compare and contrast anemia secondary to acute vs. chronic blood loss.
• Discuss the different types of hemolytic anemia in terms of: genetics - molecular changes, etiology, pathogenesis, morphology, laboratory diagnosis and clinical features and course.
• Compare and contrast warm vs. cold antibody immunohemolytic anemias.
• Compare and contrast intravascular vs. extravascular hemolysis.
• Discuss and contrast the different types of anemia of diminished erythropoesis in terms of etiology and pathogenesis, marrow and peripheral blood morphology, laboratory diagnostic criteria and clinical features and course.

Outline
I. OVERVIEW
II. ANEMIA OF BLOOD LOSS: HEMORRHAGE
III. HEMOLYTIC ANEMIAS 1. Hereditary Spherocytosis
2. Sickle Cell Anemia
3. Thalassemia
4. Glucose-6-Phosphate Dehydrogenase Deficiency
5. Immunohemolytic Anemia
6. Mechanical Trauma to Red Cells
IV. ANEMIAS OF DIMINISHED ERYTHROPOIESIS 1. Iron Deficiency Anemia
2. Megaloblastic Anemias
3. Aplastic Anemia
3

Overview
4

Causes
Anemia
Bleeding Increased RBC destruction
Decreased red cell production
Acute: trauma Chronic: GIT lesions, gynecologic disturbances
(Hemolytic Anemias) Intrinsic (Intracorpuscular) Extrinsic (Extracorpuscular)
Proliferation and differentiation of stem cells Disturbed proliferation and maturation of erythroblasts Marrow replacement and infiltration

RBCs reference ranges
6
Average volume per RBC
Average mass of hemoglobin
Average Hb concentration in a given volume of packed RBCs
Coefficient of variation of RBC volume
Proportion of the blood that consists of packed RBCs
%

7
Microcytic hypochromic (iron deficiency, thalassemia)
Macrocytic anemia (Folate or vitamin B12 deficiency)
Normal Erythrocyte
Size: 6 - 9 μm, MCV: 80 - 100 fL
Size: < 6 μm, MCV: <80 fL Size: > 9μm, MCV: >100 fL
https://www.studyblue.com/online-flashcards
Normocytic normochromic (Hemolytic Anemia …)
Morphology
Peripheral smears

Clinical Manifestations
Acute: shortness of breath, organ failure, shock
Chronic
• Pallor, fatigue, lethargy
• With hemolysis: jaundice and pigment gallstones
• With ineffective erythropoiesis: iron overload, heart and endocrine failure (secondary hemochromatosis)
• If severe and congenital: growth retardation, bone deformities due to reactive marrow hyperplasia
8

ANEMIA OF BLOOD LOSS
With acute blood loss: If exceeding 20% of blood volume, the immediate threat is
hypovolemic shock rather than anemia.
The anemia is normocytic and normochromic
Rise in the erythropoietin level
Reticulocytosis within a period of 5 to 7 days
With chronic blood loss: Iron stores are gradually depleted
Iron deficiency anemia
9

HEMOLYTIC ANEMIAS
10

HEMOLYTIC ANEMIAS
Features shared by hemolytic anemias: A decreased RBC life span.
Erythroid hyperplasia in the marrow and reticulocytosis.
In severe hemolytic anemias, extramedullary hematopoiesis may appear in the liver, spleen, and lymph nodes.
Because the pathways for the excretion of excess iron are limited, this often causes iron to accumulate, giving rise to systemic hemosiderosis or, in very severe cases, secondary hemochromatosis.
11
12
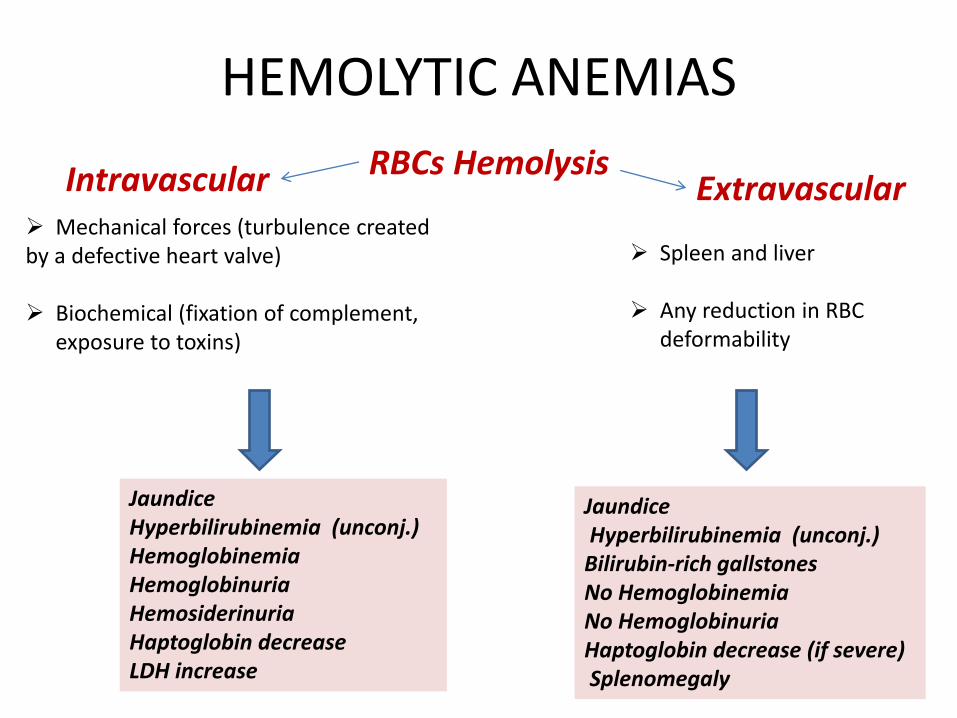
HEMOLYTIC ANEMIAS
RBCs Hemolysis Intravascular Extravascular Mechanical forces (turbulence created by a defective heart valve) Biochemical (fixation of complement,
exposure to toxins)
Jaundice Hyperbilirubinemia (unconj.) Hemoglobinemia Hemoglobinuria Hemosiderinuria Haptoglobin decrease LDH increase
Spleen and liver
Any reduction in RBC deformability
Jaundice Hyperbilirubinemia (unconj.) Bilirubin-rich gallstones No Hemoglobinemia No Hemoglobinuria Haptoglobin decrease (if severe) Splenomegaly

Hereditary Spherocytosis
13
The horizontal spectrin-spectrin and vertical spectrin-intrinsic membrane protein interactions serve to stabilize the membrane and are responsible for
the normal shape, strength, and flexibility of the red cell.
Hereditary Spherocytosis
14
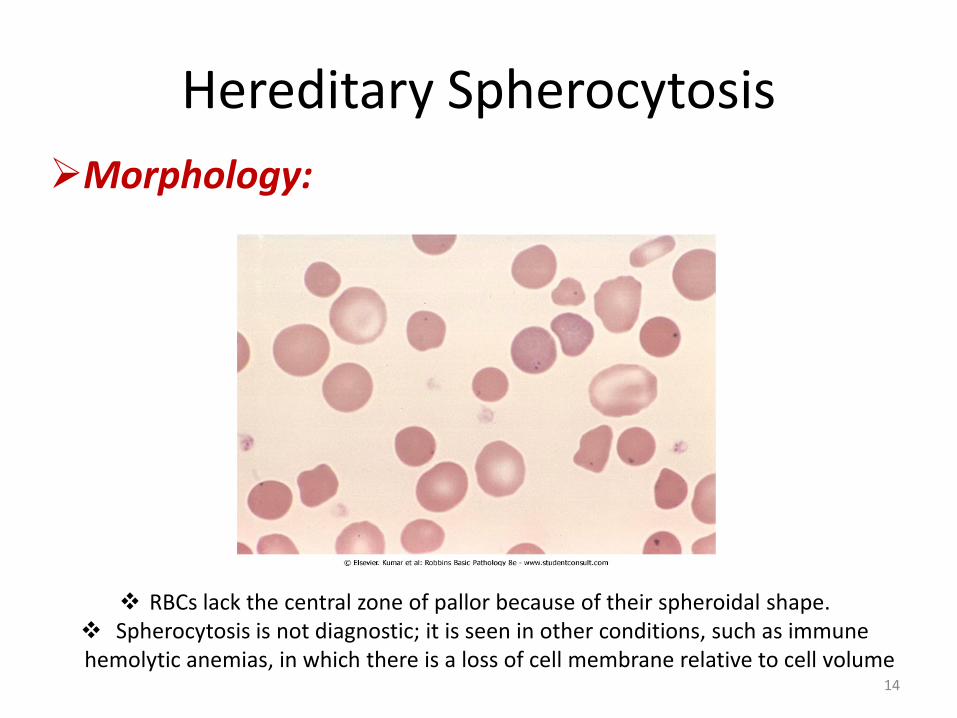
Morphology:
RBCs lack the central zone of pallor because of their spheroidal shape. Spherocytosis is not diagnostic; it is seen in other conditions, such as immune hemolytic anemias, in which there is a loss of cell membrane relative to cell volume

Hereditary Spherocytosis
Pathogenesis: • An inherited (intrinsic) defect in the red cell membrane that renders the
cells spheroidal, less deformable, and vulnerable to splenic sequestration and destruction.
• Hereditary spherocytosis (HS) is transmitted most commonly as an autosomal dominant trait.
• Approximately 25% of patients have a more severe autosomal recessive form of the disease.
• The mutations most frequently involve ankyrin, band 3, and spectrin.
• RBCs have reduced membrane stability and consequently lose membrane fragments after their release into the periphery, while retaining most of their volume.
• Spherocytes are sequestered in the splenic cords and eventually destroyed by macrophages.
15

Hereditary Spherocytosis
Clinical Features: • Anemia: most commonly it is of moderate degree
• Splenomegaly: is greater and more common in HS than in any other form of
hemolytic anemia.
• Jaundice
• The clinical course often is stable but may be punctuated by aplastic crises, due to infection and destruction of erythroblasts in the bone marrow by parvovirus B19.
• Such episodes are self-limited, but some patients need supportive blood transfusions during the period of red cell aplasia.
RBCs have increased osmotic fragility when placed in hypotonic salt solutions.
16

Hereditary Spherocytosis Treatment: • There is no specific treatment for hereditary spherocytosis.
• Splenectomy provides relief for symptomatic patients.
• The benefits of splenectomy must be weighed against the risk of increased susceptibility to infections, particularly in children.
• Partial splenectomy is gaining favor, because this approach may produce hematologic improvement while maintaining protection against sepsis.
17

Sickle Cell Anemia
18
http://www.chem.purdue.edu/courses/chm333/
Normal hemoglobin is a tetramers composed of two pairs of similar chains.
On average, the normal adult red cell contains 96% HbA (α2β2) 3% HbA2 (α2δ2) 1% fetal Hb (HbF, α2γ2)
Normal Hb:

Sickle Cell Anemia
19
Sickle cell anemia is the most common
familial hemolytic anemia in the world.
In parts of Africa where malaria is endemic, the gene frequency approaches 30% as a result of a small but significant protective effect of HbS against Plasmodium falciparum malaria.
In the US, approximately 8% of blacks are
heterozygous for HbS, and about 1 in 600 have sickle cell anemia.
http://www.mun.ca/biology/scarr/Hb_Val_substitution.html
Epidemiology:
Sickle Cell Anemia
20
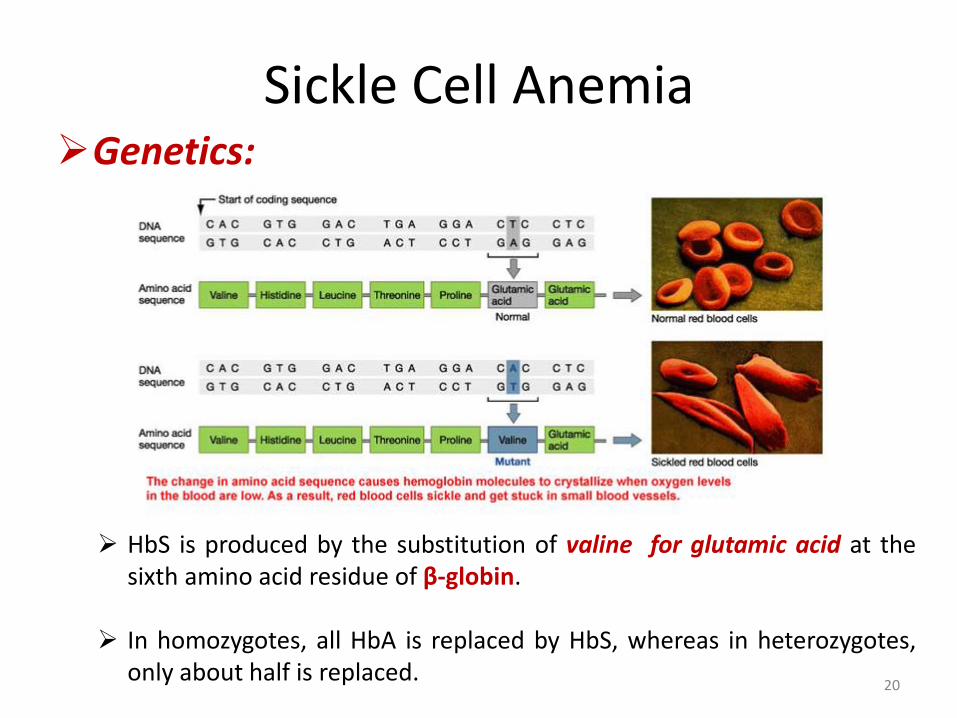
HbS is produced by the substitution of valine for glutamic acid at the sixth amino acid residue of β-globin.
In homozygotes, all HbA is replaced by HbS, whereas in heterozygotes,
only about half is replaced.
Genetics:

21
Missense mutations Position 6 on β-globin
Valine Glutamic acid

Sickle Cell Anemia
Pathogenesis: • On deoxygenation, HbS molecules form long polymers by means of
intermolecular contacts that involve the abnormal valine residue at position 6.
• These polymers distort the red cell, which assumes an elongated crescentic, or sickle, shape.
• The sickling of red cells initially is reversible upon reoxygenation. However, the distortion of the membrane that is produced by each sickling episode leads to an influx of calcium, which causes the loss of potassium and water and also damages the membrane skeleton.
• Over time, this cumulative damage creates irreversibly sickled cells, which are rapidly hemolyzed.
22

Sickle Cell Anemia
23
Sickle cell anemia—peripheral blood smear. A, Low magnification shows sickle cells B, Higher magnification shows an irreversibly sickled cell in the center. (Courtesy of Dr. Robert W. McKenna, Department of Pathology, University of Texas Southwestern Medical School, Dallas, Texas.)

Sickle Cell Anemia Factors that influence RBCs sickling: 1. The presence of hemoglobins other than HbS: • Sickle cell trait: in heterozygotes 40% of Hb is HbS and the remainder
is HbA, which interacts only weakly with deoxygenated HbS. RBCs have little tendency to sickle in vivo.
• HbF interacts weakly with HbS, so newborns with sickle cell anemia
do not manifest the disease until HbF falls to adult levels, generally around the age of 5 to 6 months.
2. The intracellular concentration of HbS: • RBC dehydration facilitates sickling. • Coexistence of α-thalassemia (decreased Hb concentration) reduces
sickling. • Absence of sickling in heterozygotes with sickle cell trait.
24

Sickle Cell Anemia
3. The transit time for RBCs through the microvasculature:
• Sickling in microvascular beds is confined to areas of the body in which blood flow is sluggish (spleen and bone marrow)
• Inflammation slows the flow of blood by increasing the adhesion of leukocytes and red cells to endothelium.
25

Sickle Cell Anemia
Two major consequences of RBCs sickling: Chronic hemolytic anemia: • RBC membrane damage and dehydration are caused by repeated episodes
of sickling • The mean life span of red cells in sickle cell anemia is only 20 days • There is a compensatory hyperplasia of erythroid progenitors in the
marrow. • As with the other hemolytic anemias, hemosiderosis and gallstones are
common.
Microvascular obstructions: • Result in ischemic tissue damage and pain crises commonly in the bone
marrow (sluggish blood flow), where it often progresses to infarction. • Vaso-occlusion is enhanced by infection, inflammation, dehydration, and
acidosis that enhance the sickling of reversibly sickled cells. • Infarction and autosplenectomy, is complete by adulthood.
26
27
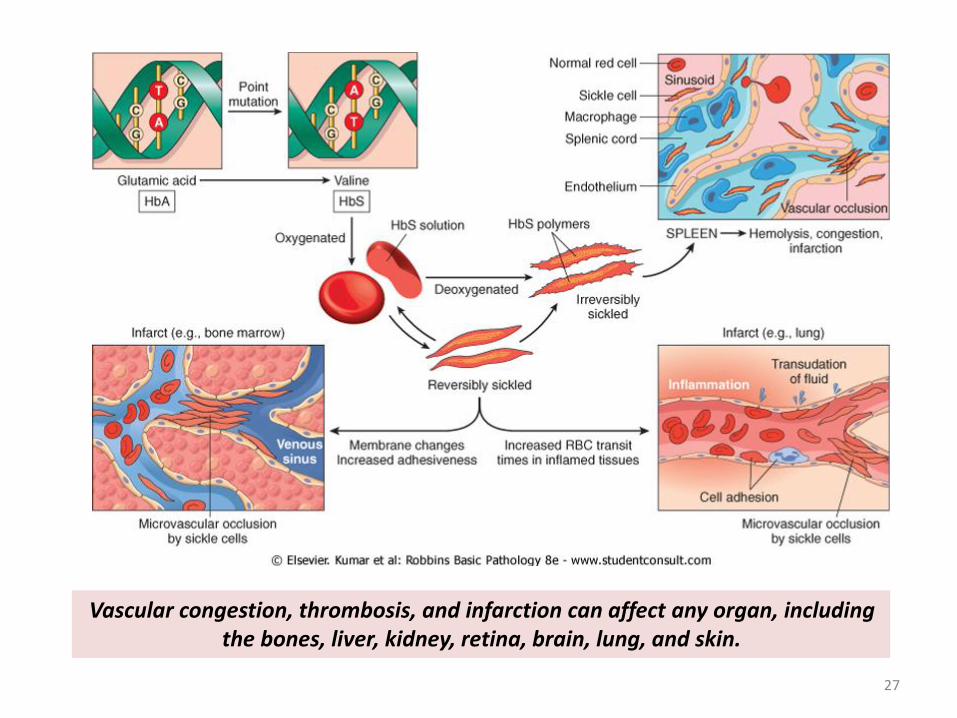
Vascular congestion, thrombosis, and infarction can affect any organ, including the bones, liver, kidney, retina, brain, lung, and skin.

Sickle Cell Anemia
Clinical course: • From its onset, the disease runs an unremitting course punctuated
by sudden crises.
• The vaso-occlusion in these episodes can involve many sites but occurs most commonly in the bone marrow (BM), where it often progresses to infarction.
• Acute chest syndrome, which can be triggered by pulmonary infections or fat emboli/BM embolism from infarcted marrow.
• The acute chest syndrome and stroke are the two leading causes of ischemia-related death.
• Aplastic crisis, is caused by a sudden decrease in red cell production, usually is triggered by the infection of erythroblasts by parvovirus B19 and, while severe, is self-limited.
28

Sickle Cell Anemia
Clinical course: • Both children and adults with sickle cell disease are functionally
asplenic, making them susceptible to infections • In adults the basis for “hyposplenism” is autoinfarction. • Even children with enlarged spleens are at risk for development of
fatal septicemia. • Patients with sickle cell disease also are predisposed to Salmonella
osteomyelitis • In homozygous sickle cell disease, irreversibly sickled red cells are
seen in routine peripheral blood smears. • In sickle cell trait, sickling can be induced in vitro by exposing cells to
marked hypoxia. • The diagnosis is confirmed by electrophoretic demonstration of HbS. • Prenatal diagnosis can be performed by analyzing fetal DNA
obtained by amniocentesis or biopsy of chorionic villi.
29

Sickle Cell Anemia
Clinical course: The clinical course is highly variable.
Of particular importance is prophylactic treatment with penicillin to prevent pneumococcal infections.
Approximately 50% of patients survive beyond the fifth decade.
sickle cell trait causes symptoms rarely and only under extreme conditions, such as after vigorous exertion at high altitudes.
A mainstay of therapy is hydroxyurea by:
• increasing RBC levels of HbF
• an anti-inflammatory effect due to the inhibition of white cell production
• increasing in red cell size (decreasing Hb concentration)
30

References
ROBBINS Basic Pathology 9th Edition Source of the cover: http://kidney2.blogspot.com/2012_07_01_archive.html
31

Thank you…
32















![ERYTHROCYTES [RBCs]](https://img.pdfslide.net/doc/110x75/56812e48550346895d93dd1e/erythrocytes-rbcs.jpg)






![ERYTHROCYTES [RBCs]](https://img.pdfslide.net/doc/110x75/56813dc0550346895da78963/erythrocytes-rbcs-56ea22b2e2743.jpg)





