Embed Size (px)
Citation preview

REACH and the expectations in terms of data and risk assessment
Sharon Munn
European Chemicals Bureau, DG JRC / IHCP
Chairs of Scientific Committees for Risk Assessment , Brussels 24 October 2006

WHY REACH?
• Data gaps: 86% of HPVCs have less than base set data
• The process takes (too much) time• Burden of proof on public authorities• Administrative burden for new chemicals (low
volume) prevents innovation

The basic goal of REACH
“to ensure a high level of protection of human health and the environment as well as the free circulation of substances on the internal market while enhancing competitiveness and innovation”(Art 1(1)), “testing on vertebrate animals for the purpose of REACH shall be undertaken only as a last resort…necessary to take measures limiting duplication of other tests (Art 25.1)

Information requirements under REACHIntrinsic Properties
• Phys-chem properties (e.g. solubility, vapour pressure)
• Toxicity properties (e.g. acute toxicity, irritation, mutagenicity, carcinogenicity)
• Fate properties (e.g. (bio)degradation, partition coefficients)
• Ecotoxicity properties (e.g. toxicity to aquatic or terrestrial organisms)

Information requirements under REACH
• Annex VI of REACH proposal• Annex VII to X: staggered tonnage triggered approach
– Standard information in Technical Dossier depends on volume:≥ 1 tonne/y: Annex VII (~20,000 subst)≥ 10 tonne/y: Annex VIII≥ 100 tonne/y: Annex IX≥ 1000 tonne/y: Annex X (2,500 subst)
(~7,500 subst)

Adaptation of Information requirements
• Column 2; Specific adaptations “Study need not be conducted if…” lead to no/earlier/later testing
• Annex XI:– Annex XI(1): Testing is not scientifically necessary– Annex XI(2): Testing is technically not possible– Annex XI(3): Substance-tailored exposure-driven testing
Adequate justification and documentation needed

Use of information on intrinsic properties of substances in a regulatory context
• Information needs to be adequate for Classification and Labelling and the Chemical Safety Assessment
• Industries’ responsibility to decide and justify which information they consider necessary (starting from a minimum data set)

Annex XI: general rules for adaptation
• Testing not scientifically necessary– Use of existing data (not GLP/ non standard tests)– Historical Human data– (Q)SAR– Grouping of substances and read-across approach– In vitro methods– Weight of evidence

Annex XI: general rules for adaptation
• Testing is technically not possible
• Exposure-driven testing (only for tests in Annex VII and VIII i.e. ≥ 100 tons/year)

Testing does not appear scientifically necessary, cf. Annex XI(1)
• Non-testing methods (QSAR, read-across, categories), in vitro testing
– Scientific validation, domain of applicability– Adequate and reliable documentation of method– Results adequate for C&L and/or risk assessment

Substance-tailored exposure-driven testing, cf. Annex XI(3)
• General adaptation criteria (not endpoint specific)
• Testing may be omitted based on Exposure Scenarios (Annex VIII, 8.6 & 8.7; Annex IX, Annex X)
• Adequate justification is required based on exposure assessment, cf. Annex I(5) – COM to adopt criteria on adequate justification within 18 after EiF
• Specific conditions of use must be communicated through the supply chain (SDS or article 32)
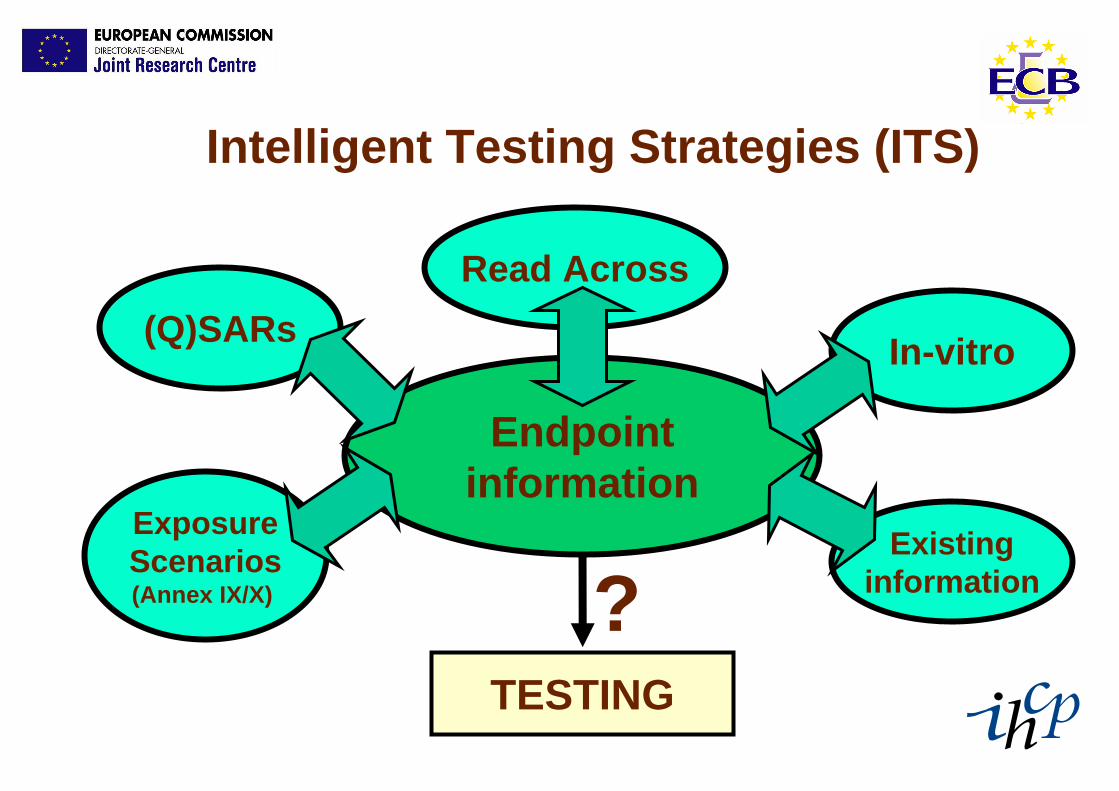
Intelligent Testing Strategies (ITS)
Endpointinformation
(Q)SARsRead Across
In-vitro
ExposureScenarios(Annex IX/X)
Existinginformation
TESTING
?

CONCLUSION – 1Intelligent (Integrated) Testing Strategies
1. Legislative text (Annex XI in particular) + GUIDANCE should limit use of animals and prevent box-ticking
2. A paradigm shift is needed from extensive animal testing to efficient, focussed animal testing
3. Impetus to refine current in vivo methods, and further develop non-test and in vitro test methods to be used in a regulatory context.
4. Further scientific work (2007 onwards) and regulatory implementation is needed.

RISK ASSESSMENT UNDER REACH
Chemical Safety Assessment
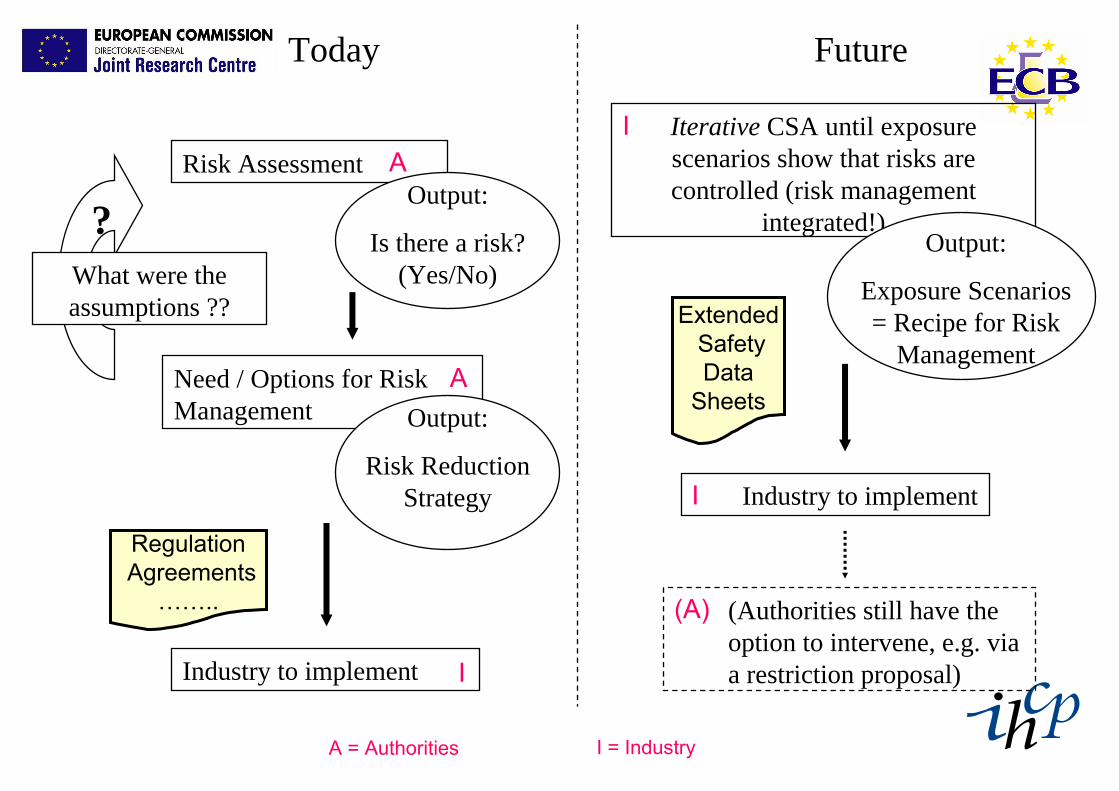
Today Future
What were the assumptions ??
?
Industry to implement I
I = IndustryA = Authorities
Iterative CSA until exposure scenarios show that risks are controlled (risk management
integrated!)
I
(Authorities still have the option to intervene, e.g. via a restriction proposal)
(A)
Industry to implementI
Output:
Exposure Scenarios = Recipe for Risk
Management
Risk Assessment A
ExtendedSafetyData
Sheets
RegulationAgreements
……..
Need / Options for Risk Management
A
Output:
Is there a risk? (Yes/No)
Output:
Risk Reduction Strategy

Core tools for Industry under REACH• The Chemical Safety Assessment (CSA) is the
tool used to determine• The Chemicals Safety Report (CSR) is the tool
used to record/document• The Safety Data Sheet (SDS) is the tool used to
communicate
Conditions for use (for sufficiently protecting human health and the environment):
– risk management measures
– operational conditions
Exposure Exposure ScenarioScenario

When is a CSA needed ?Part of Registration requirements, when:• ≥ 10 tonne per year• If the substance is dangerous, PBT or vPvB
exposure assessment, including exposure scenarios and risk characterisation to be included in the CSR
• Registrant to perform a CSA for :–Manufacture–Own use–All identified uses–All life cycle stages resulting from the manufacture, own use and identified uses
• Downstream User to perform a CSA for its uses (and all life cycle resulting from these) non identified by its supplier
Part of application for Authorisation

Exposure scenarios Hazard identification
Classification and labelling
Exposure assessment
Hazard assessment
Risk characterisation
Existing knowledge/data
YES
Are risks adequately controlled?
Revise assumed RMM and/or operational
conditions
ESs with RMM and operational conditions to adequately control the risks, to be:Documented in the Chemical Safety Report (CSR)
Communicated to users (to downstream users via an SDS annex)
NO NO

3.1: Preparing theregistration dossier3.1: Preparing the
registration dossier
3.2: Preparing the CSR
3.2: Preparing the CSR
3.3: Information requirements
3.3: Information requirements
3.10: Guidance on substance ID
3.10: Guidance on substance ID
3.5: Guidance for downstream users3.5: Guidance for downstream users
3.8: Requirementsfor articles
3.8: Requirementsfor articles
3.6: Guidance onC&L under GHS
3.6: Guidance onC&L under GHS
3.7: Guidance on applications for
authorisation
3.7: Guidance on applications for
authorisation
3.9: Guidance on SEA
3.9: Guidance on SEA
3.4: Guidance on data-sharing
3.4: Guidance on data-sharing
RIP-3Guidance for
Industry

4.1: Guidance on dossier evaluation4.1: Guidance on dossier evaluation
4.2: Guidance on substance evaluation
4.2: Guidance on substance evaluation
4.3: Inclusion of substances in
Annex XIII
4.3: Inclusion of substances in
Annex XIII
4.4: Preparation of Annex XV dossiers
4.4: Preparation of Annex XV dossiers
4.5: Priority settingfor evaluation
4.5: Priority settingfor evaluation
RIP-4Guidance for
Authorities

REACH IMPLEMENTATION PROJECTS
• RIP 3.3: TGD on Information Requirements– Integrated Testing or Information Strategy, i.e. how best to
obtain the information that is required, use of alternative data (in vitro, QSAR, etc)
• RIP 3.2: TGD on conducting a Chemical Safety Assessment– Includes how to develop ES and estimate exposure and
how to characterise potential risks

RIP 3.3-1 (scoping study) Information Requirements
• State of the Art reports:– Testing– Non-testing– Exposure considerations
• General framework Integrated/Intelligent Testing Strategies
• Draft of testing strategies for 4 endpoints: – irritancy-corrosivity– reprotoxicity– aquatic toxicity– degradation

Main conclusions / Recommendations from RIP 3.3-1
• Good starting point but not yet guidance (scoping study only)
• Need for flexibility and updating: Final guidance to be presented in a web-based tool.
• Apply a tiered approach, with general guidance that would assist the fairly skilled industry registrant, and indications of when more expert help should be sought.
• Weight-of-evidence (over and across endpoints) approaches should be developed further.
• Categorisation/read-across promising: Further guidance needed!
• How can in vitro, (Q)SAR /read across be better integrated into testing strategy?

RIP 3.3-2 (Main Study)Information requirements
• testing strategies assigned to specific Endpoints Working Groups (EWG)– Physicochemical properties– Irritation/corrrosivity– Sensitisation– Acute toxicity– Repeated dose toxicity– Mutagenicity/carcinogenicity– Reproductive and developmental toxicity– Degradation– Aquatic toxicity– Bioconcentration/Bioaccumulation– Terrestrial organisms
• cross cutting issues as toxicokinetics, guidance on the use of categories, weight of evidence, (Q)SAR, exposure-based waiving
It is foreseen that the project will be completed in April 2007.

ECB Project on computational toxicology (http://ecb.jrc.it/QSAR)
1. Research on the development, validation and regulatory application of (Q)SARs, and in emerging areas(e.g. computational nanotoxicology)
2. Making computational tools available from ECB website3. Coordinating the activities of the EU QSAR Working Group4. Contributing to the activities of the OECD ad hoc QSAR Group5. Providing Training on computational methods
Experimental log BCF
Pred
icte
d lo
g BC
F
6543210-1
6
5
4
3
2
1
0
-1
Training setExternal test
LogBCF = 1.06 + 0.64 LogKow - 0.11 DMax_max - 0.20 ELUMO_min

RIP 3.2 (CSR)
• Guidance for preparing the Chemical Safety Assessment (CSA) and Chemical Safety Report (CSR)– Scoping study: RIP 3.2-1 (Jan-July 2005)– Second phase: RIP 3.2-2: To be finalised 2007
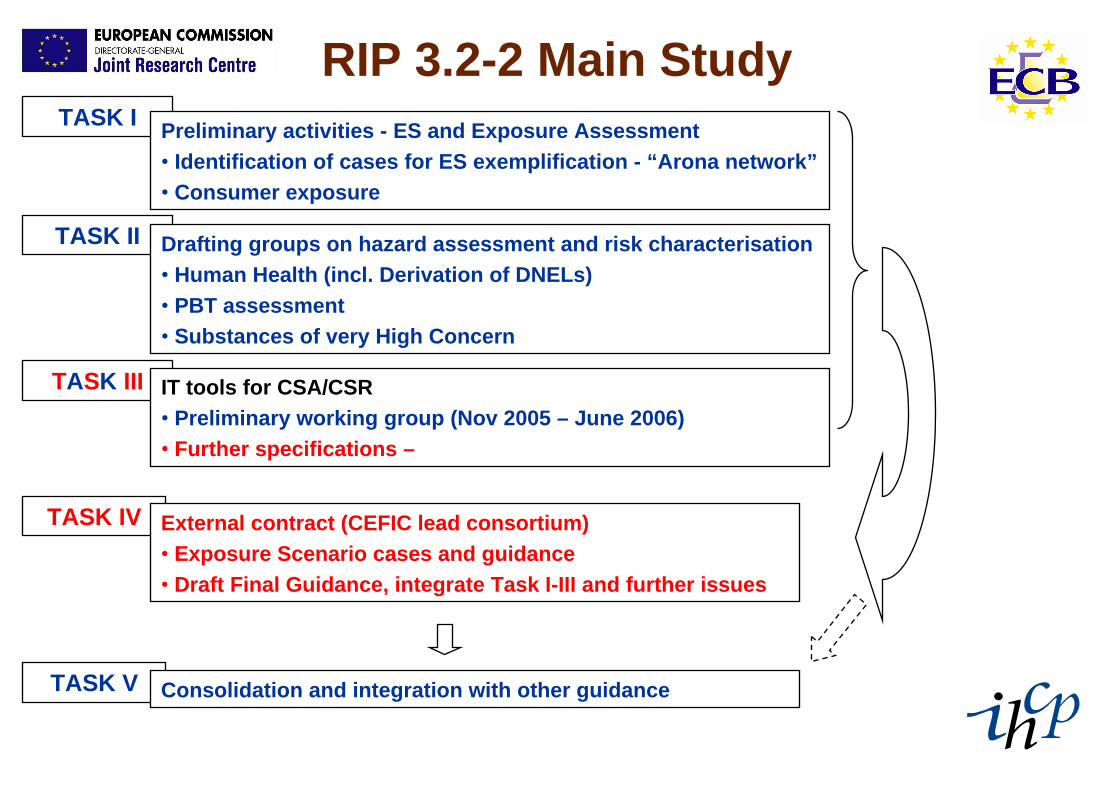
RIP 3.2-2 Main StudyTASK I Preliminary activities - ES and Exposure Assessment
• Identification of cases for ES exemplification - “Arona network”• Consumer exposure
TASK II Drafting groups on hazard assessment and risk characterisation• Human Health (incl. Derivation of DNELs)• PBT assessment• Substances of very High Concern
TASK III IT tools for CSA/CSR• Preliminary working group (Nov 2005 – June 2006)• Further specifications –
TASK V Consolidation and integration with other guidance
TASK IV External contract (CEFIC lead consortium)• Exposure Scenario cases and guidance• Draft Final Guidance, integrate Task I-III and further issues

Annex I – Human Health Hazard Assessment
• REACH ( Annex I, 1.0.1 ) defines the Derived No-Effect Level (DNEL), i.e. the level of exposure above which humans should not be exposed.
• In the risk characterisation, the exposure of each human population known to be or likely to be exposed is compared with the appropriate DNEL. The risk to humans can be considered to be adequately controlled if the exposure levels estimated do not exceed the appropriate DNEL.

RIP 3.2-2 TASKS
• Establish the detailed ‘mechanisms’ for deriving a DNEL (‘translation’ of current ‘MOS’ TGD on risk characterisation into REACH ‘DNEL’ guidance)
• Establish guidance for assessment of non-threshold effects and substances of very high concern

DNEL versus MOS
MOS =NOAEL or NOAECExposure
If MOS > Overall Assessment Factor → No concernIf MOS < Overall Assessment Factor → Concern
DNEL = NOAEL or NOAEC Overall Assessment Factor
If Exposure < DNEL → Risk is adequately controlledIf Exposure > DNEL → Risk is NOT adequately controlled

Critical DNEL
• The critical (lowest) DNEL across all endpoints should be determined for each relevant exposure route and population
• DNEL is exposure driven – i.e. only the critical DNEL taken to the risk characterisation for each relevant combination of population/exposure route/exposure duration/local-systemic effects

When no DNEL can be derived
• Non-threshold mutagens/carcinogens– Further needs as compared to qualitative and quantitative approaches
described in current TGD? Other approaches e.g. EFSA
• Other end-points (non-threshold or no DNEL established)– Further needs as compared to current TGD?
• High Exposure Uncertainty– Await progress in the PBT drafting group
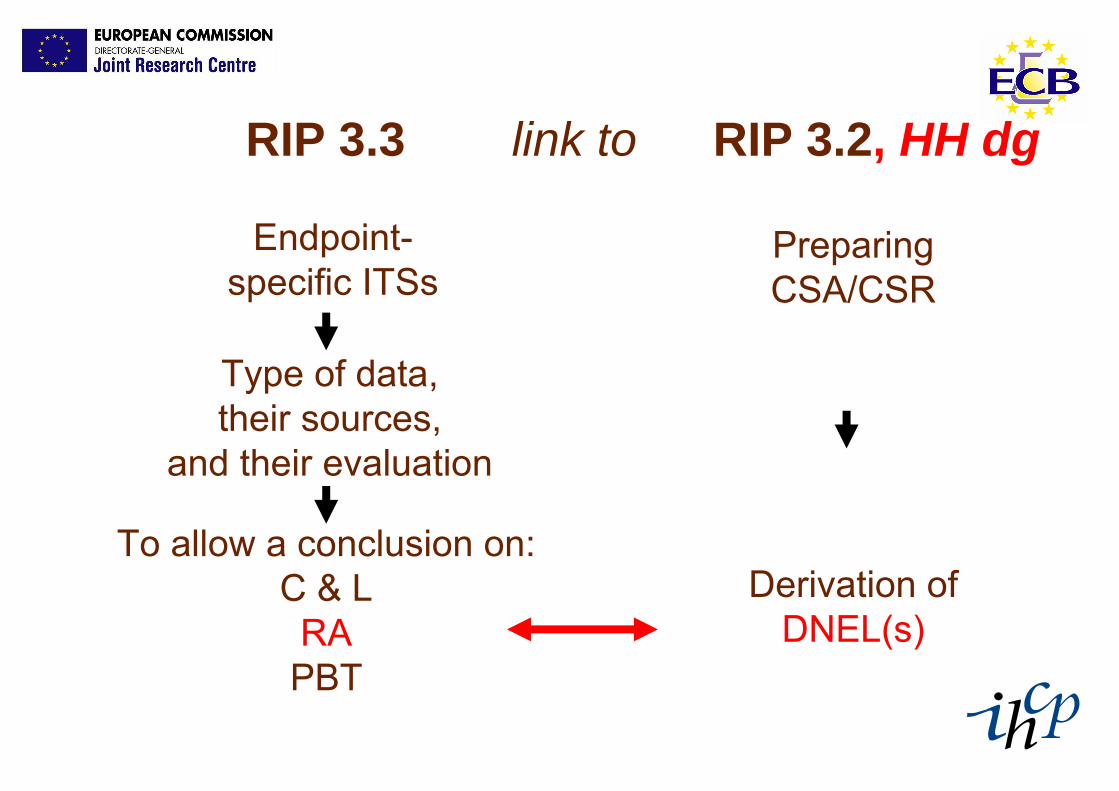
RIP 3.3 RIP 3.2, HH dglink to
Preparing CSA/CSR
Endpoint-specific ITSs
Type of data, their sources,
and their evaluation
To allow a conclusion on:C & LRAPBT
Derivation of DNEL(s)

RIP 3.3 RIP 3.2, HH dglink to
RA element:
Identify suitable dose-descriptors:e.g.NOAEL (or NOAEC),LOAELLD50T25 BMD10etc.
Convert these dose-descriptors
into DNEL(s)or
some non-threshold effect
level

Use of Alternative DataNeeds to be Adequate for C&L and risk assessment
• Qualitative• Potency – semi-quantitative• Quantitative (dose/response, dose-descriptor)

Use of Alternative Data
• Measure of Uncertainty around QSAR prediction, or in vitro test result
• Acceptance of additional uncertainty– Consequence of getting it wrong, seriousness of effect, vicinity to likely
exposures
• Additional Assessment Factor for Alternative Data
?

CONCLUSIONS - 2
• Information requirements under REACH– Flexible with adaptation possibilities– Adequate justification and documentation needed
• Guidance is needed– Input from experts and practitioners required

Thank you for your attention!

This paper was produced for a meeting organized by Health & Consumer Protection DG and represents the views of its author on thesubject. These views have not been adopted or in any way approved by the Commission and should not be relied upon as a statement of the Commission's or Health & Consumer Protection DG's views. The European Commission does not guarantee the accuracy of the dataincluded in this paper, nor does it accept responsibility for any use made thereof.













![Psychology of Marketing FINAL [Read-Only]...Companies Get Their Mojo From Maslow, Conley) + Hierarchy of Customer Needs Level 1—Reach Satisfaction by Meeting Expectations: Companies](https://img.pdfslide.net/doc/110x75/5ed02d1c2ce38e77c2543dbd/psychology-of-marketing-final-read-only-companies-get-their-mojo-from-maslow.jpg)















