Embed Size (px)
Citation preview

REACTIVOS ORGANOMETÀLICOS. FUENTES DE CARBONO NUCLEOFÍLICO PARA LA SÌNTESIS
DE ALCOHOLES

COMPUESTOS ORGANOMETÁLICOSLa reducciòn de aldehidos y cetonas con hidruros es una forma ùtil de sintetizaralcoholes. Èsta metodologìa serìa aún mas poderosa, si en lugar de hidrurosg p gpudièsemos utilizar una fuente de carbono nucleofílico. El ataque por un carbononucleofílico sobre un grupo carbonilo daría un alcohol y formarìa,simultaneamente un nuevo enlace Carbono-carbono. Habríamos construido, así,un producto con mas àtomos de carbono y, por tanto, con mas complejidad quela presente en el sustrato de partida
Para lograr tal transformaciòn, necesitamos un modo de hacer nucleòfilos basados en el carbono. Estudiaremos en esta sección como alcanzar tal objetivo.L l i l Li i M i i l h l dLos metales, particularmente Litio y Magnesio, reaccionan con los haluros dealquilo para generar un nuevo tipo de compuestos, denominados reactivosorganometálicos, en los cuales un àtomo de carbono de un compuestoorgánico está enlazado a un metal. Esas especies son bases muy fuertes ybuenos nucleòfilos y, como tales, son extremadamente ùtiles en sìntesisorgànica.

Preparación de Reactivos Alquillitio y Alquilmagnesio a partir de haloalcanos
Cl > Br > IOrden de reactividad : Cl > Br > IOrden de reactividad :
Los organometàlicos RMgX son denominados Reactivos de Grignard
En RLi y RMgX el metal es altamente deficiente de electrones y actùa como un àcidoEn RLi y RMgX el metal es altamente deficiente de electrones y actùa como un àcidode Lewis que se coordina facilmente con moléculas del solvente
Dado el carácter fuertemente básico ynucleofílico del àtomo de carbono de losorganometálicos, èstos deben prepararseen atmòsfera inerte, libres de O2 , CO2, yhumedad, en solventes tambiénperfectamente secos.
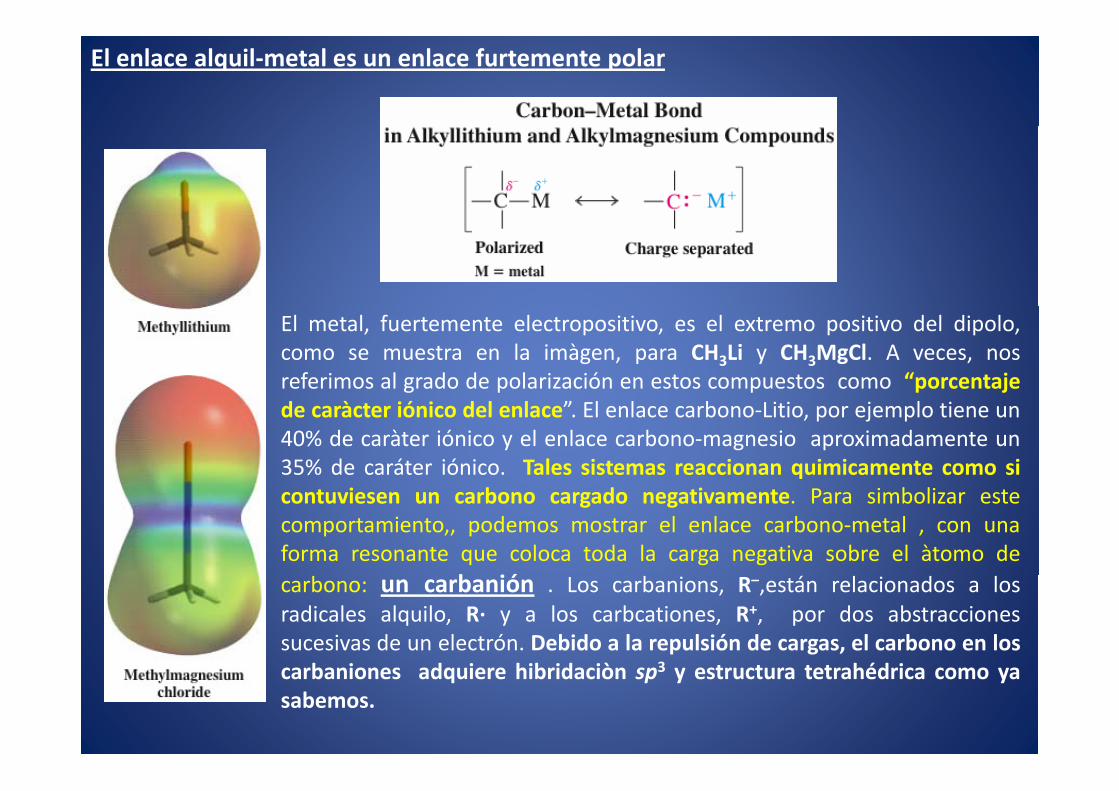
El enlace alquil-metal es un enlace furtemente polar
El metal, fuertemente electropositivo, es el extremo positivo del dipolo,como se muestra en la imàgen, para CH3Li y CH3MgCl. A veces, nosreferimos al grado de polarización en estos compuestos como “porcentajede caràcter iónico del enlace”. El enlace carbono-Litio, por ejemplo tiene un40% de caràter iónico y el enlace carbono-magnesio aproximadamente un35% de caráter iónico. Tales sistemas reaccionan quimicamente como sicontuviesen un carbono cargado negativamente. Para simbolizar estecomportamiento,, podemos mostrar el enlace carbono-metal , con unaforma resonante que coloca toda la carga negativa sobre el àtomo de
b iócarbono: un carbanión . Los carbanions, R–,están relacionados a losradicales alquilo, R· y a los carbcationes, R+, por dos abstraccionessucesivas de un electrón. Debido a la repulsión de cargas, el carbono en los
b i d i hib id iò 3 héd icarbaniones adquiere hibridaciòn sp3 y estructura tetrahédrica como yasabemos.

Principio de Polarizaciòn reversaLa preparación de alquilmetales a partir de haloalcanos, ilustra un principioi t t í i á i i téti l l i iò Eimportante en química orgánica sintética: la polarizaciòn reversa. En unhaloalcano, la presencia de un halógeno electronegativo hace del carbono un centroelectrofílico. Por tratamiento con un metal, la unidad Cδ+–Xδ– , es convertida en Cδ––Mδ+ E t l b i i t l di iò d l i iò L ióMδ+ . En otras palabras, se invierte la direcciòn de polarizaciòn. ¡La reacción conun metal (metalación) ha transformado un carbono electrofílico en uncentro nucleofílico!.
El grupo alquilo de los alquilmetales es fuertemente básicoLos carbaniones son bases muy fuertes. De hecho, los alquilmetales son mucho masbásicos que las aminas ó los alcóxidos, ya que el carbono es considerable menoselectronegativo que el nitrógeno ó el oxígeno y mucho menos capaz de soportar unacarga negativa. Recuerda que los alcanos son ácidos extremadamente débiles. El pKa
N í d t lde metano es aproximadamente 50. No es, así, sorprendente que loscarbaniones sean bases tan fuertes: son, después de todo las basesconjugadas de los alcanos . Su basicidad hace que los reactivos organometálicossean muy sensibles a la humedad e incompatibles con el grupo funcional OH ógrupos funcionales acídicos similares. En presencia de agua, ocurre hidrólisis, amenudo violenta, con formación de hidróxido del metal y un alcano. El resultado deésta transformación es predecible sobre consideraciones puramente electrostáticas.

La secuencia metalación-hidrólisis, convierte un haloalcano en un alcano
Una manera mas directa de lograr el mismo objetivo es la reacción de unhaloalcano con el poderoso donador de hidruro LiAlH4 , un desplazamiento SN2 delhaloalcano con el poderoso donador de hidruro LiAlH4 , un desplazamiento SN2 delhaluro por el anión hidruro. NaBH4 , mucho menos reactivo , es incapaz de producirésta sustitución.
Otra aplicación útil de la secuencia metalación-hidrólisis es la introducción deOtra aplicación útil de la secuencia metalación hidrólisis es la introducción de isótopos de hidrógeno

Reactivos Organometálicos en la Síntesis de Alcoholes Entre las aplicaciones mas útiles de los organometálicos de Magnesio y Litio están
ll l l l il l i d ti t iaquellas en las que el grupo alquilo, polarizado negativamente, reacciona comonucleófilo. Como los hidruros, esos reactivos pueden atacar al grupo carbonilo de unaldehído ó cetona para producir un alcohol ( después de verter la mezcla de reacción
b ) L dif i d á l fsobre agua). La diferencia es que, además, en el proceso se forma un nuevoenlace carbono-carbono

Síntesis de alcoholes secundarios:
Síntesis de alcoholes terciarios

Reacciones de acoplamiento cruzadas catalizadas por metales de transición
L ió l d l i t d h l l ti bLa reacción general de acoplamiento de un haloalcano, que contiene un carbonopolarizado positivamente, con un alquilmetal, que contiene un átomo de carbonopolarizado negativamente, es totalmente exotérmica.
Sin embargo, en el caso de Li y Mg, esos acoplamiento ó son demasiado lentos atemperatura ambiente ó dan lugar a mezclas de productos cuando se suministracalor. Por otra parte, tales procesos constituyen algunas de las reacciones masf d l l f ió d l b b ífundamentales para la formación de enlaces carbono-carbono; así, no essorprendente que los químicos sintéticos hayan dedicado un esfuerzo considerablepara solucionar el problema, un esfuerzo que aún continúa. Una primera solución se
t ó l d l d b t li d C b lencontró en el uso de sales de cobre como catalizadores. Como ya sabemos, loscatalizadores hacen posible que las reacciones transcurran mucho mas rápidamente,a través de mecanismos y estados de transición de energía mas baja

Por ejemplo, el método ha sido aplicado a escala industrial en la fabricación de muscalure, la feromona de la mosca común.
El mecanismo de esas reacciones procede a través de la formación de especiesorganocobre, denominadas también organocupratos, que pueden obtenerse yusarse estequiométricamente a partir, por ejemplo, de reactivos alquillitio
Mas recientemente, se han explorado numerosas variantes de ésta metodología,con M = Zn, Sn, Al, y otros, empleando catalizadores basados sobre Ni, Pd, Fe, y, , , y , p , , , yRh, para nombrar unos pocos. El objetivo es, no solamente mejorar la eficacia,sino también la tolerancia por otros grupos funcionales. Por ejemplo, y adiferencia de los alquillitio y los reactivos de Grignard, los correspondientescompuestos de Zn, no atacan a la funciónes carbonílicas.
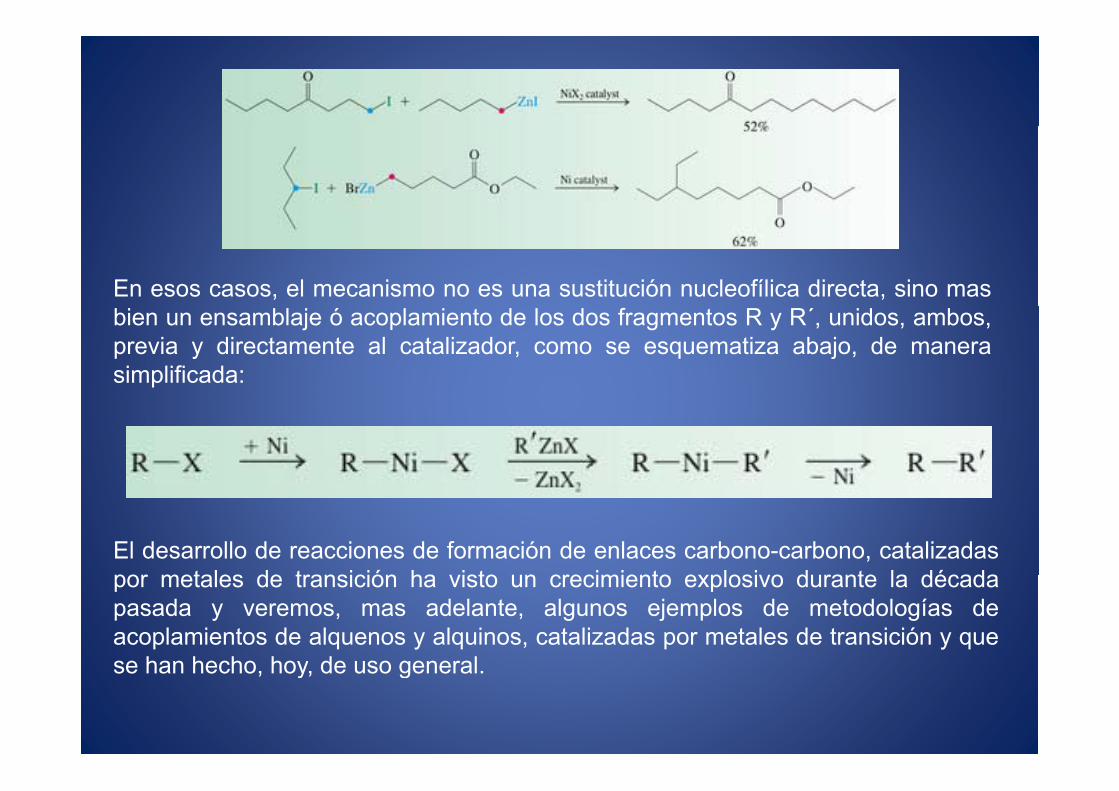
En esos casos, el mecanismo no es una sustitución nucleofílica directa, sino masbien un ensamblaje ó acoplamiento de los dos fragmentos R y R´, unidos, ambos,previa y directamente al catalizador, como se esquematiza abajo, de manerasimplificada:
El desarrollo de reacciones de formación de enlaces carbono-carbono, catalizadaspor metales de transición ha visto un crecimiento explosivo durante la décadapor metales de transición ha visto un crecimiento explosivo durante la décadapasada y veremos, mas adelante, algunos ejemplos de metodologías deacoplamientos de alquenos y alquinos, catalizadas por metales de transición y quese han hecho hoy de uso generalse han hecho, hoy, de uso general.

Alcoholes complejos: Una introducciòn a la estrategia sintèticaLas reacciones introducidas hasta ahora forman parte del “vocabulario” de la químicaorgánica; a menos que conzcamos ése vocabulario, no podemos hablar el lenguaje deg q p g jla química orgánica. Esas reacciones nos permiten manipular las moléculas einterconvertir grupos funcionales; así, es importante familiarizarse con esastransformaciones, sus tipos, los reactivos usados, las condiciones bajo las cualesocurren (en especial cundo las condiciones son cruciales para el èxito del proceso) ylas limitaciones de cada tipo.Esta tarea puede parecer monumental y demandante de mucha memorización: Pero sehace mucho mas fácil si se comprenden los mecanismos de reacción. Ya sabemos quela reactividad puede predecirse a partir de un pequeño nùmero de factores, tales comoelectronegatividad, fuerzas culómbicas y fortalezas de enlace. Vamos a ver aquí, unpoco de, como los químicos orgánicos aplicamos èsta comprensión de los mecanismospara diseñar estrategias sintéticas útiles; es decir, secuencias de reacciones quepermitan la construcción de una molécula objetivo deseada, en el mínimo
ú d d lt di i tnúmero de pasos de alto rendimiento.Empezaremos con unos pocos ejemplos, en los que predeciremos la reactividadsobre fundamentos mecanísticos. Luego volveremos a la síntesis— la construcción del lé l C d ll l í i ét d i tétilas moléculas. ¿ Como desarrollamos los químicos nuevos métodos sintéticos y comopodemos construir una molécula “objetivo” de la forma mas eficaz posible? Ambostópicos están estrechamente relacionados. El segundo, conocido como síntesis total,req iere s almente na serie de reacciones Al est diar esas tareas estaremsrequiere usualmente una serie de reacciones. Al estudiar esas tareas estaremstambién, por tanto, revisando muchas de las reacciones consideradas hasta ahora

Los mecanismos ayudan a predecir el resultado de una reacciònRecordemos primero como predecimos el resultado de una reacción. ¿Cuales son losf h i i l f i ? V lfactores que hacen que un mecanismo en particular funcione?. Veamos algunos ejemplos:
Explicaciòn: El bromuro es mejor grupo saliente que el fluorurop j g p q
Explicación: El carbono carbonílico polarizado positivamente forma un enlace alExplicación: El carbono carbonílico polarizado positivamente forma un enlace algrupo alquilo, polarizado negativamente, del reactivo organometálico,.
Explicación: El enlace C-H terciario es más débil que los enlaces C-H primarios ó secundarios y el Br2 es muy selectivo en las halogenaciones.

Nuevas Reacciones dan lugar a Nuevos Mètodos de Síntesis
Cuando se descubre una nueva reacciòn (ya sea por diseño ò por accidente) esCuando se descubre una nueva reacciòn (ya sea por diseño ò por accidente), esimportante mostrar su alcance y sus limitaciones. Para ello se prueba la reacción conmuchos sustratos diferentes, se anotan las reacciones secundarias (si existen), sesometen otros grupos funcionales a las condiciones de la reacciòn y se llevan a cabosometen otros grupos funcionales a las condiciones de la reacciòn y se llevan a caboestudios mecanísticos de la reacción. Si esas investigaciones probasen que la nuevareacción es de aplicabilidad general, se añade, como un nuevo método de síntesis alarsenal de los químicos orgánicosarsenal de los químicos orgánicos.
Puesto que una reacciòn conduce a un cambio muy específico en una molécula, confrecuencia es ùtil poner énfasis en la naturaleza general de ésta “alteracióngmolecular” . Un ejemplo simple es la adición de un reactivo de Grignard aformaldehído ; ¿Cual es el cambio estructural en esta transformación? . Se añadeuna nueva unidad carbonada a un grupo alquilo. El método es valioso porquepermite, de forma directa, extender la cadena carbonada en un carbono más unproceso tambien denominado homologación.
Aunque , en esta etapa, nuestro vocabulario sintético es aún relativamente limitadoya conocemos, y tenemos a nuestra disoposición, un cierto número de alteracionesmoleculares. Por ejemplo, los bromoalcanos son excelentes puntos de partida para
t f inumerosas transformaciones
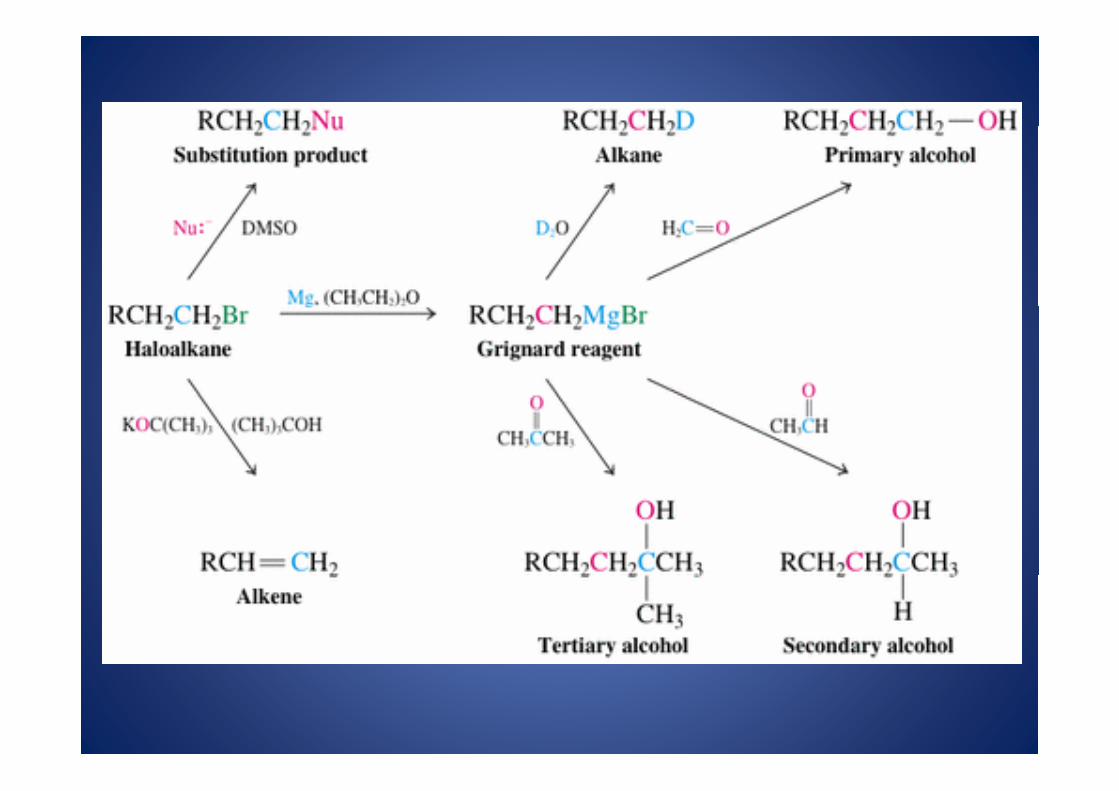

Cada uno de los productos en el esquema anterior, pueden, a su vez, sufrirtransformaciones posteriores dando así lugar a productos mas complicados.Cuando nos preguntamos “ ¿hasta que punto es buena esta reacción?, ¿Que clasede estructuras podemos fabricar mediante su aplicación?”, estamos apuntando a unproblema de metodología sintética. Supongamos, por otra parte que queremos
lé l bj ti t h l i i t tpreparar una molécula objetivo concreta y nos hacemos las siguientes preguntas:“¿Como hacemos para divisar una ruta de preparación eficiente para ella?, ¿cualesserían los materiales de partida adecuados?”, el problema con el que estamostratando ahora es de Sintesis Total
Los químicos orgánicos podemos desear fabricar moléculas complejas con diversospropósitos específicos Por ejemplo ciertos compuestos pueden tener propiedades
tratando ahora es de Sintesis Total.
propósitos específicos. Por ejemplo, ciertos compuestos pueden tener propiedadesmedicinales valiosas pero no son de fácil acceso desde fuentes naturales. Losbioquímicos pueden necesitar una molécula particular marcada isotopicamente ,para trazar rutas metabólicas. Los Químicos orgánicos-físicos diseñan conpara trazar rutas metabólicas. Los Químicos orgánicos físicos diseñan confrecuencia nuevas estructuras para su estudios espectroscópicos, electrónicos, etc.Hay, en fin, muchas posibles razones para la síntesis total de moléculas orgánicas.
Cualesquiera que sea el objetivo final, una síntesis exitosa debe caracterizarsepor su brevedad y alto rendimiento global. Los materiales de partida deberían deser facilmente accesibles, con preferencia, comerciales y baratos. Más aún,p ypreocupaciones medioambientales y de seguridad, demandan que, idealmente,losreactivos usados sean relativamente poco tóxicos y seguros de manejar.

El Análisis Retrosintético Simplifica Los Problemas de SíntesisMuchos compuestos disponibles comercialmete y baratos, son también pequeños ycontienen seis o algunos más átomos de carbono Así la tarea mas frecuente con lacontienen seis o algunos más átomos de carbono. Así, la tarea mas frecuente con laque se enfrenta el químico orgánico sintético es la de construir una molécula masgrande y mas complicada, a partir de fragmentos simples más pequeños. La mejoraproximación a la preparación de la molécula objetivo es trabajar su síntesis “haciaaproximación a la preparación de la molécula objetivo, es trabajar su síntesis haciaatrás”, sobre el papel, una aproximación a la que se denomina análisisretrosintético. En éste análisis, enlaces carbono-carbono estratégicos de la moléculaobjetivo son rotos ó “desconectados” en puntos donde la formación del enlace pareceobjetivo son rotos ó desconectados en puntos donde la formación del enlace pareceposible y mas adecuada. Este modo de pensar “hacia atrás”, puede parecerte extraño alprincipio, porque estás acostumbrado a aprender las reaciones en su sentido “haciaadelante”; p ej “A +B da C” La retrosíntesis requiere que pienses en el proceso deadelante ; p.ej. A +B da C . La retrosíntesis requiere que pienses en el proceso demanera inversa, p. ej. “ C deriva de A+B”.
¿Porqué retrosíntesis? La respuesta es que en cualquier “construcción” de una¿Porqué retrosíntesis? La respuesta es que, en cualquier construcción de unaestructura compleja a partir de ladrillos simples, el número de posibilidades de añadirlas piezas incrementa drásticamente a medida que avanzamos e incluye miles deopciones que constituyen puntos muertos. En contraste, trabajando hacia atrás, laopciones que constituyen puntos muertos. En contraste, trabajando hacia atrás, lacomplejidad disminuye y se minimizan las soluciones inviables. Una analogía simplepodría ser un puzzle: obviamente, es mas fácil desmantelarlo paso a paso queensamblarlo. Por ejemplo, un análisis retrosintético de la síntesis del 3-hexanol a partirj p , pde unidades de tres carbonos sugiere su formación a partir de un organometálico depropilo y propanal:

La doble flecha se usa para indicar una desconección estratégica . Hemosreconocido que el enlace entre C3 y C4 del producto, desconectado en éste análisis,puede ser construido usando una transformación que conocemos: la reacción de unp qreactivo organometálico con un aldehído. En éste caso, sólo se necesita una reacciónpara lograr la conección; en otros puede que se requieran varios pasos.
P é l i t ibl d i C C j t C4 C5? E¿Porqué no elegir otras posibles desconecciones C-C, p. ej. entre C4 y C5?. Ennuestro ejemplo esta estrategia es perfectamente razonable y requeriría comomateriales de partida : butanal y bromuro de etil magnesio . Sin embargo, en general,las desconecciones retrosintéticos deben hacerse de tal forma que suministrenlas desconecciones retrosintéticos deben hacerse de tal forma, que suministrenpiezas moleculares lo más iguales en tamaño posible. En éste sentido, nuestro primeranálisis es mejor.

Otras dos posibles alternativas retrosintéticas para 3-hexanol podrían ser las siguientes:
Ninguna de ellas es tan buena como la primera, puesto que no simplificansignificativamente la estructura de la molécula objetivo: no hay desconección designificativamente la estructura de la molécula objetivo: no hay desconección deenlaces carbono-carbono.
El análisis retrosintético ayuda en la construcción de alcoholes
Apliquemos el análisis retrosintético a la preparación de un alcohol terciario: 4-etil-4-nonanol. Debido a su naturaleza impedida estericamente e hidrofóbica, éstealcohol y sus homólogos, tienen aplicaciones industriales importantes comoy g p pcosolventes y aditivos en ciertos procesos de polimerización. Hay dos pasos aseguir en cada etapa del proceso. Primero tenemos que identificar todas lasposibles desconecciones estratégicas, “rompiendo” todos los enlaces que puedanvolver a formarse mediante reacciones que conocemos. Segundo, evaluamos losméritos relativos de esas desconecciones , buscando aquellas que simplifiquenmejor la estructura de la molécula objetivo. Los enlaces estratégicos en 4-ethyl-4-
fnonanol son aquellos que rodean al grupo funcional. Hay tres desconecciones quedan los precursores mas simples:

La ruta a desconecta el grupo etilo desde C4, sugiriendo como materiales de partidapara su construcción, bromuro de etilmagnesio y 4-nonanona. La ruta b es unaposibilidad alternativa que da un reactivo de Grignard propílico y 3-octanona comoprecursores . Finalmente, la desconección c revela una tercera ruta sintética derivadade la adición de bromuro de pentilmagnesio a 3-hexanone.
Evaluación revela que la ruta c es la mejor : Los materiales de partida, son casi igualesen tamaño con cinco y seis átomos de carbono; así ésta desconección suministra laen tamaño, con cinco y seis átomos de carbono; así, ésta desconección suministra lasimplificación mayor de la estructura.

¿Podemos someter a alguno de los fragmentos obtenidos por desconección a través dela ruta c ,anterior, a posterior desconección, para llegar a materiales de partida aún massencillos? . La respuesta es Sí; recuerda que las cetonas pueden obtenerse porsencillos? . La respuesta es Sí; recuerda que las cetonas pueden obtenerse poroxidación de alcoholes secundarios por reactivos de Cr(VI) . Podemos, así, considerarla preparación de 3-hexanona a partir del correspondiente alcohol,.
Puesto que al comienzo del análisis retrosintético, identificamos una desconeccióneficiente de 3-hexanol en dos fragmentos de tres átomos de carbono, estamos ahoraen condiciones de presentar nuestro esquema sintético al completo:
Este ejemplo ilustra una secuencia general muy poderosa para la construcción dealcoholes complejos : primero adición de un reactivo de Grignard ó de un organolitio aalcoholes complejos : primero, adición de un reactivo de Grignard ó de un organolitio aun aldehído para dar un alcohol secundario; segundo, oxidación a cetona y, finalmente,adición de otro reactivo organometálico para dar un alcohol terciario.

Aspectos a tener en cuenta al planificar una síntesis
A la hora de planificar una síntesis hay varias consideraciones a tener en cuenta, queayudarán a evitar diseños poco exitosos ó aproximaciones con bajos rendimientos a
lé l bj ti Primero t t d i i i l ú t t l duna molécula objetivo. Primero, tratar de minimizar el número total detransformaciones requeridas para convertir el material inicial de partida en elproducto deseado. Este punto es tan importante que, en algunos casos, espreferible aceptar un paso de bajo rendimiento si con ello vamos a conseguir unpreferible aceptar un paso de bajo rendimiento, si con ello vamos a conseguir unacortamiento de la secuencia sintética. Por ejemplo (y asumiendo que todos losmateriales de partida tienen coste comparable), una síntesis en siete pasos en la quetodos ellos dan un rendimiento del 85% es inferior a una síntesis en cuatro pasostodos ellos dan un rendimiento del 85%, es inferior a una síntesis en cuatro pasos,en la que tres de ellos tienen un rendimiento del 95% y, el restante un rendimientodel 45%. La eficacia global en la primera secuencia sería: (0.85 x 0.85 x 0.85 x 0.85x 0 85 x 0 85 x 0 85) x 100 = 32% mientras que la seguna síntesis además de serx 0.85 x 0.85 x 0.85) x 100 32%, mientras que la seguna síntesis, además de sertres pasos mas corta, tendría un rendimiento global de: (0.95 x 0.95 x 0.95 x 0.45) x100 = 39%.

En el ejemplo visto, todos los pasos de síntesis se han llevado a cabo de maneraconsecutiva; es lo que se suele denominar una Síntesis lineal. En general, paramoléculas objetivo complejas es mejor una aproximación sintética que conlleve dos ómoléculas objetivo complejas, es mejor una aproximación sintética que conlleve dos ómas rutas concurrentes, siempre y cuando el número de pasos sea el mismo; talestrategia se define como una Síntesis convergente. Aunque para una estrategiaconvergente no es posible un cálculo simple del rendimoento global basta paraconvergente, no es posible un cálculo simple del rendimoento global, basta paraconvencernos de su mejor eficacia, el comparar las cantidades reales de de materialesde partida requeridas por cada una de las dos aproximaciones para obtener la mismacantidad de producto deseado. En el siguiente ejemplo, se preparan primero 10 g dep g j p , p p p gun producto H mediante una secuencia lineal en tres pasos A → B → C → H ( conrendimiento de 50% en cada paso) y, después, por una síntesis convergente,partiendo de D y F, a través de E y G respectivamente. Si asumimos (para mayorp y y p (p ysimpliciadad) que los pesos moleculares de todos los compuestos son los mismos, laprimera preparación requiere 80 g de material de partida y la segunda sólo 40 g (enconjunto).

Segundo , no usar reactivos cuyas moléculas tengan grupos funcionales quepuedan interferir con la reacción deseada. Por ejemplo, el tratamiento de unhidroxialdehído con un reactivo de Grignard produce una reacción ácido-base quehidroxialdehído con un reactivo de Grignard , produce una reacción ácido base quedestruye el reactivo organometálico, y no la formación del nuevo enlace carbono-carbono deseada.
Una solución posible para este problema, sería añadir dos equivalentes de reactivode Grignard: uno para reaccionar con el hidrógeno ácido del grupo alcohólico, comoindicado arriba, el otro para lograr la adición deseada al grupo carbonilo. Otra
l ió “ ” l f ió hid i f d ésolución es “proteger” la función hidroxi en forma de un éter , como veremos masadelante.No trate de fabricar un reactivo de Grignard a partir de una bromocetona. Tal reactivo
í t bl d t i í t t f ióno sería estable ya que se destruiría tan pronto como se forma, por reacción con supropio grupo carbonilo (en la misma ó en otra molécula). Aprenderemos, masadelante, como proteger una función carbonilo.

Tercero, tener en cuenta cualquier restricción mecanística ó estructural queafecte a las reacciones bajo consideración. Por ejemplo, las bromacionesradicalarias son mas selectivas que las cloraciones Tener en mente las limitacionesradicalarias son mas selectivas que las cloraciones. Tener en mente las limitacionesestructurales de las reacciones nucleofílicas, y no olvidar la falta de reactividad de2,2-dimetil-1-halopropanos. Aunque no fáciles de reconocer, algunas veces, muchoshaloalcanos tienen tales estructuras estericamente impedidas y son de análogahaloalcanos tienen tales estructuras estericamente impedidas y son, de análogamanera, no reactivos . No obstante, tales sistemas forman reactivos organometálicosque, así, pueden funcionalizarse posteriormente. Por ejemplo, tratamiento delreactivo de Grignard obtenido a partir de 1-bromo-2 2-dimetilpropano conreactivo de Grignard obtenido a partir de 1 bromo 2,2 dimetilpropano conformaldehído, dá el alcohol correspondiente.
Haloalcanos terciarios, cuando incorporados en estructuras mas complejas, son,algunas veces, también difíciles de reconocer. Recuerda, que los haluros terciariosno dan reacciones SN2 , síno eliminaciones, en presencia de bases.

La experiencia en síntesis, como en otros muchos aspectos de la química orgánica,se desarrolla, en gran mediada, con la práctica. La planificación de síntesis paramoléculas complejas requiere una revisión de las reacciones y mecanismos quemoléculas complejas requiere una revisión de las reacciones y mecanismos quehemos visto hasta ahora. Los conocimientos adquiridos así pueden, entonces,aplicarse a la solución de problemas sintéticos.






















