Embed Size (px)
Citation preview

Reductive electrochemical study of Ni(II) complexes with N2O2 Schiffbase complexes and spectroscopic characterisation of the reduced
species. Reactivity towards CO
Fernando Azevedo, Cristina Freire *, Baltazar de Castro
CEQUP/Departamento de Quımica, Faculdade de Ciencias, Universidade do Porto, Rua do Campo Alegre, 687, 4169-007 Porto, Portugal
Received 20 November 2001; accepted 16 April 2002
Abstract
Reductive electrochemical properties of series of nickel(II) complexes with salen ligands, which have different diimine bridges and
substituents in the aldehyde moiety have been studied in several solvents (CH3CN, dmf and (CH3)2SO). In order to assess the
relative importance of the Ni(I) and Ni(II) anion radical species, the reduced species have been characterised by combining EPR and
UV�/Vis spectroscopy. The results have shown that complexes with aliphatic diimine bridges are reduced to four-coordinate Ni(I)
species with a B1 g (dxy )1 ground state, whereas those with aromatic diimine bridges are reduced to square�/planar Ni(II) anion
radical species that rapidly dimerise. None of the reduced species was found to bind pyridine, imidazole and triphenylphosphine, but
in the presence of the stronger p-acceptor ligand CO, new Ni(I) species were formed that, and on the basis of EPR data, can be
formulated as five-coordinate complexes with a B1g (dxy )1 ground state, [NiL �/CO]�. These new species are more stable than the
parent complexes as confirmed by the more positive E1/2 values as a consequence of the extensive p delocalisation M0/CO. # 2002
Elsevier Science Ltd. All rights reserved.
Keywords: Salen ligands; Ni(I) complexes; Nickel(II) complexes; EPR
1. Introduction
The chemistry of polydentate nickel(I) complexes has
attracted attention since they can act as powerful
catalysts on chemical or electrochemical reduction of
electrophiles, such as alkyl and aryl halides [1�/3] and
carbon dioxide [4]. The ability of the starting nickel(II)
complex to form upon one-electron transfer a nickel(I)
complex, rather than the anion radical of the ligand,
appears as a key point for obtaining an efficient
catalysis. Nickel(I) complexes are expected to react
with electrophiles by transfer of their metal centred
unpaired electron in an inner-sphere fashion, being more
efficient and selective than Ni(II) anion radicals, which
function as an outer-sphere electron donor due to the
delocalised nature of the ligand based unpaired electron.
Salen ligands can easily stabilise low and high
oxidation states of nickel and reduced [Ni(salen)] is
used in the electro-reduction of alkyl and aryl halides
[1a,1c,1d]. By introducing substituents in the ligand it is
possible to modulate the potential at which the reduc-
tion occurs, and to control concomitantly the catalytic
properties of the complexes. As salen has the desirable
characteristic of being readily subject to systematic
modification of its electronic and steric properties by
synthetic approaches, we have prepared a series of
nickel(II) complexes with salen derivatives that have
different diimine bridges and substituents in the alde-
hyde moieties (Scheme 1). The reductive behaviour of
the resulting complexes was studied in several solvents
and the reduced species characterised by combining
electrochemical, EPR and UV�/Vis spectroscopy in
order to assess the relative importance of the Ni(I) and
Ni(II) anion radical species. As our goal is to use these
complexes as catalysts, we report also the reactivity of
the reduced species towards p acceptor Lewis bases.
Some of the complexes have already been prepared and
* Corresponding authors. Tel.: �/351-22-6082-890; fax: �/351-22-
6082-959
E-mail address: [email protected] (C. Freire).
Polyhedron 21 (2002) 1695�/1705
www.elsevier.com/locate/poly
0277-5387/02/$ - see front matter # 2002 Elsevier Science Ltd. All rights reserved.
PII: S 0 2 7 7 - 5 3 8 7 ( 0 2 ) 0 1 0 2 5 - 2

characterised in some of the solvents used, [1a,1c,1d,5]
but they are included to provide a coherent framework
for the overall study reported.
2. Results and discussion
2.1. Cyclic voltammetry of nickel(II) complexes
The complexes studied can be divided in two groups,
based on their electrochemical response (Table 1).
Complexes with non-aromatic imine bridges (1�/7;
group A) show, in the potential range used, one
reduction process in all solvents used. With increasing
scan rates (v ), a linear dependence between ip and v1/2 is
observed (similar slopes for the ipc vs. v1/2 and ipa vs. v1/2
plots), and the cathodic�/anodic peak potential separa-
tions are similar to those observed for the couple Fc��/
Fc, and the ratios ipa/ipc are solvent independent,
constant and equal to 1 for all the scan rates used.
Electrochemical data for complexes of group A are thus
consistent with one electron, diffusion-controlled, rever-
sible reduction process [6].
Complexes with aromatic imine bridges (8 and 9;group B) present, in all solvents and for scan rates in the
range 0.02�/0.5 V s�1, cyclic voltammograms with one
cathodic wave at 1.210 V B/�/EpcB/1.378 V and one
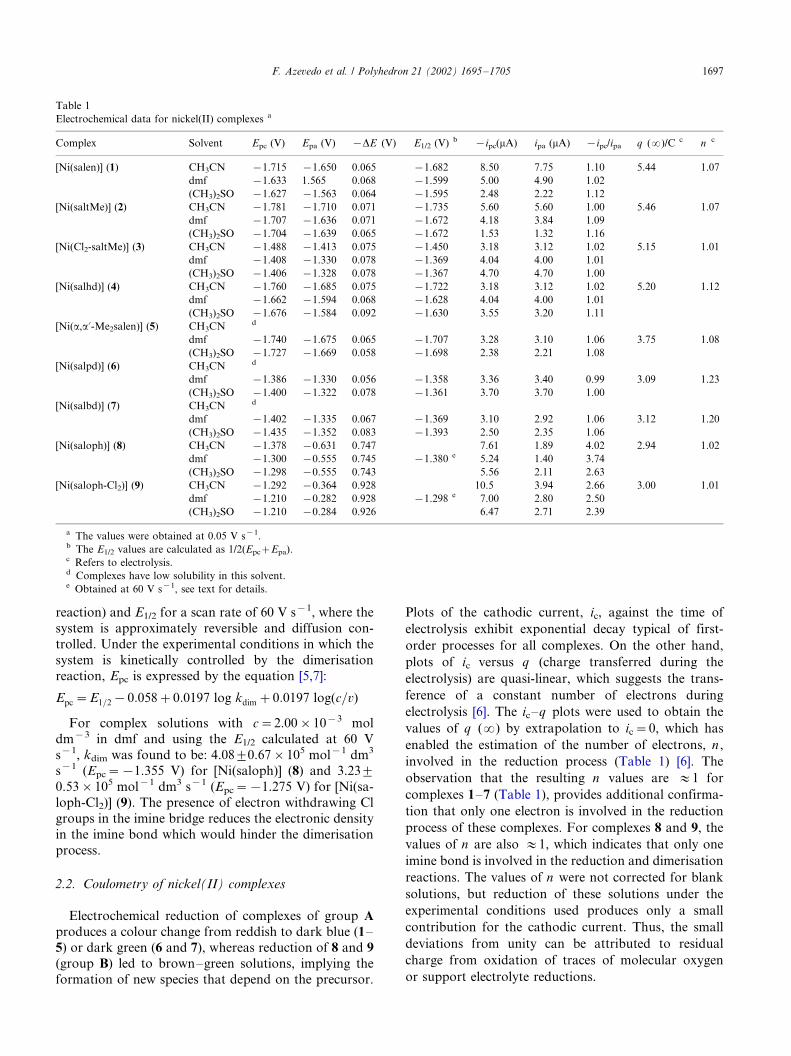
anodic wave at 0.282 V B/�/EpaB/0.630 V (Fig. 1(a)).
Despite their large separation (:/0.75 V), the two waves
are related, as no anodic wave could be detected when
the potential is cycled from 0 to �/1.0 V.
With increasing scan rate a linear dependence betweenip and v1/2 is also observed, but with quite different
slopes for the ipc versus v1/2 and ipa versus v1/2 plots,
which implies different diffusion coefficients for the
reduced and oxidised species, and suggests significant
structural changes upon reduction.
Peak potential dependence with complex concentra-
tion at a constant scan rate in dmf was also studied. For8 and 9, the values of Epa are independent of complex
concentration, whereas those of Epc are shifted to more
negative potentials when the complex concentration is
reduced. For a scan rate of 200 mV s�1, Epc is shifted by
21 mV for [Ni(saloph)] (8) and 20 mV for [Ni(saloph-
Cl2)] (9), when the complex concentration is tenfold
reduced.
Similar results have been described in literature fordmf solutions of complex 8 [5,7] and have been
interpreted as a one-electron reversible reduction pro-
cess followed by a fast chemical reaction leading to the
formation of a stable new product, which is oxidised to
the original complex.
Floriani et al. [8,9] have isolated the product of
sodium reduction in the solution of [Ni(saloph)]. X-ray
and magnetic data show that the product, formulated as[Ni(saloph)]2Na2, is a diamagnetic Ni(II) dimeric com-
pound which is composed of two monomeric units
linked by one of the carbon imine atoms that has been
reduced to an amide carbon. Vianello et al. [5], based on
the similarity between the cyclic voltamograms of
[Ni(saloph)]2Na2 and [Ni(saloph)] in dmf, suggested
that the electro-reduction of [Ni(saloph)] in this solvent
produces in a first step a nickel(II) anion radical due toreduction of the imine bond to an amide bond, followed
by a fast dimerisation reaction to yield the Ni(II) dimeric
species, [Ni(saloph)]22�. These species are then reversi-
bly oxidised to the parent complex by cleavage of the
bonding between the two monomer units. We presup-
pose the same mechanism for reduction of complexes 8
and 9 in all solvents used, and experimental evidence for
the formation of the radical intermediate species havebeen obtained by EPR (see below).
Cyclic voltammograms recorded in dmf using very
high scan rates, typically in the 1�/60 V s�1 range exhibit
in the reverse scan a new anodic wave at potentials
similar to those of the cathodic wave, whereas the
anodic wave at more positive potentials looses intensity
as the scan rate is increased (Fig. 1(b)). When the scan
rate reaches 60 V s�1, the cyclic voltammograms, in allsolvents, resemble those of a reversible type system with
E1/2�/1/2(Epa�/Epc)�/�/1.380 for 8 and E1/2�/�/1.298
V for 9, and the ratio ipa/ipc reaches 1. The new anodic
wave is attributed to the oxidation of the reduced
monomeric species, in the absence of dimerisation; by
using very fast scan rates the monomeric reduced species
are oxidised before the dimerisation reaction takes
place.An estimate of the dimerisation rate constant, kdim,
can be obtained from the difference between Epc
(obtained for a scan rate of 50 mV s�1, where the
system is kinetically controlled by the dimerisation
Scheme 1. Structure of nickel(II) complexes.
F. Azevedo et al. / Polyhedron 21 (2002) 1695�/17051696
reaction) and E1/2 for a scan rate of 60 V s�1, where the
system is approximately reversible and diffusion con-
trolled. Under the experimental conditions in which the
system is kinetically controlled by the dimerisation
reaction, Epc is expressed by the equation [5,7]:
Epc�E1=2�0:058�0:0197 log kdim�0:0197 log(c=v)
For complex solutions with c�/2.00�/10�3 mol
dm�3 in dmf and using the E1/2 calculated at 60 V
s�1, kdim was found to be: 4.089/0.67�/105 mol�1 dm3
s�1 (Epc�/�/1.355 V) for [Ni(saloph)] (8) and 3.239/
0.53�/105 mol�1 dm3 s�1 (Epc�/�/1.275 V) for [Ni(sa-
loph-Cl2)] (9). The presence of electron withdrawing Cl
groups in the imine bridge reduces the electronic density
in the imine bond which would hinder the dimerisation
process.
2.2. Coulometry of nickel(II) complexes
Electrochemical reduction of complexes of group A
produces a colour change from reddish to dark blue (1�/
5) or dark green (6 and 7), whereas reduction of 8 and 9
(group B) led to brown�/green solutions, implying the
formation of new species that depend on the precursor.
Plots of the cathodic current, ic, against the time of
electrolysis exhibit exponential decay typical of first-
order processes for all complexes. On the other hand,
plots of ic versus q (charge transferred during the
electrolysis) are quasi-linear, which suggests the trans-
ference of a constant number of electrons during
electrolysis [6]. The ic�/q plots were used to obtain the
values of q (�) by extrapolation to ic�/0, which has
enabled the estimation of the number of electrons, n ,
involved in the reduction process (Table 1) [6]. The
observation that the resulting n values are :/1 for
complexes 1�/7 (Table 1), provides additional confirma-
tion that only one electron is involved in the reduction
process of these complexes. For complexes 8 and 9, the
values of n are also :/1, which indicates that only one
imine bond is involved in the reduction and dimerisation
reactions. The values of n were not corrected for blank
solutions, but reduction of these solutions under the
experimental conditions used produces only a small
contribution for the cathodic current. Thus, the small
deviations from unity can be attributed to residual
charge from oxidation of traces of molecular oxygen
or support electrolyte reductions.
Table 1
Electrochemical data for nickel(II) complexes a
Complex Solvent Epc (V) Epa (V) �DE (V) E1/2 (V) b �ipc(mA) ipa (mA) �ipc/ipa q (�)/C c n c
[Ni(salen)] (1) CH3CN �1.715 �1.650 0.065 �1.682 8.50 7.75 1.10 5.44 1.07
dmf �1.633 1.565 0.068 �1.599 5.00 4.90 1.02
(CH3)2SO �1.627 �1.563 0.064 �1.595 2.48 2.22 1.12
[Ni(saltMe)] (2) CH3CN �1.781 �1.710 0.071 �1.735 5.60 5.60 1.00 5.46 1.07
dmf �1.707 �1.636 0.071 �1.672 4.18 3.84 1.09
(CH3)2SO �1.704 �1.639 0.065 �1.672 1.53 1.32 1.16
[Ni(Cl2-saltMe)] (3) CH3CN �1.488 �1.413 0.075 �1.450 3.18 3.12 1.02 5.15 1.01
dmf �1.408 �1.330 0.078 �1.369 4.04 4.00 1.01
(CH3)2SO �1.406 �1.328 0.078 �1.367 4.70 4.70 1.00
[Ni(salhd)] (4) CH3CN �1.760 �1.685 0.075 �1.722 3.18 3.12 1.02 5.20 1.12
dmf �1.662 �1.594 0.068 �1.628 4.04 4.00 1.01
(CH3)2SO �1.676 �1.584 0.092 �1.630 3.55 3.20 1.11
[Ni(a,a?-Me2salen)] (5) CH3CN d
dmf �1.740 �1.675 0.065 �1.707 3.28 3.10 1.06 3.75 1.08
(CH3)2SO �1.727 �1.669 0.058 �1.698 2.38 2.21 1.08
[Ni(salpd)] (6) CH3CN d
dmf �1.386 �1.330 0.056 �1.358 3.36 3.40 0.99 3.09 1.23
(CH3)2SO �1.400 �1.322 0.078 �1.361 3.70 3.70 1.00
[Ni(salbd)] (7) CH3CN d
dmf �1.402 �1.335 0.067 �1.369 3.10 2.92 1.06 3.12 1.20
(CH3)2SO �1.435 �1.352 0.083 �1.393 2.50 2.35 1.06
[Ni(saloph)] (8) CH3CN �1.378 �0.631 0.747 7.61 1.89 4.02 2.94 1.02
dmf �1.300 �0.555 0.745 �1.380 e 5.24 1.40 3.74
(CH3)2SO �1.298 �0.555 0.743 5.56 2.11 2.63
[Ni(saloph-Cl2)] (9) CH3CN �1.292 �0.364 0.928 10.5 3.94 2.66 3.00 1.01
dmf �1.210 �0.282 0.928 �1.298 e 7.00 2.80 2.50
(CH3)2SO �1.210 �0.284 0.926 6.47 2.71 2.39
a The values were obtained at 0.05 V s�1.b The E1/2 values are calculated as 1/2(Epc�Epa).c Refers to electrolysis.d Complexes have low solubility in this solvent.e Obtained at 60 V s�1, see text for details.
F. Azevedo et al. / Polyhedron 21 (2002) 1695�/1705 1697
2.3. EPR and electronic spectroscopy of reduced species
Analysis of electrochemical data has shown that all
complexes of group A and B are reduced by an electron
charge transfer process, but that different species are
produced by complexes with aromatic and non-aromatic
imine bridges, a consequence of different reduction
processes. Coupling EPR and electronic spectroscopy
provides further characterisation of the reduced species.
It must be pointed out that all complexes were also
reduced with Na(Hg) and in the same solvents used for
coulometric reductions. As the resulting solutions have
the same colours and identical frozen solution EPR
spectra it is possible to conclude that the same reduced
species are formed by chemical and electrochemical
reduction. The following description is thus applicable
to chemically and electrochemically reduced nickel(II)
complexes.
Frozen solution EPR spectra of reduced species from
group A in any of the solvents are very similar, and
show large g tensor anisotropy with gav:/2.12�/2.15,
typical of metal centred reduced species, in this case
nickel(I) complexes. The spectra of reduced solutions of
1�/5 are rhombic and exhibit hyperfine splittings in the
two high magnetic field regions (a :/0.6�/1.0 mT) due to
the interaction of the unpaired electron with two
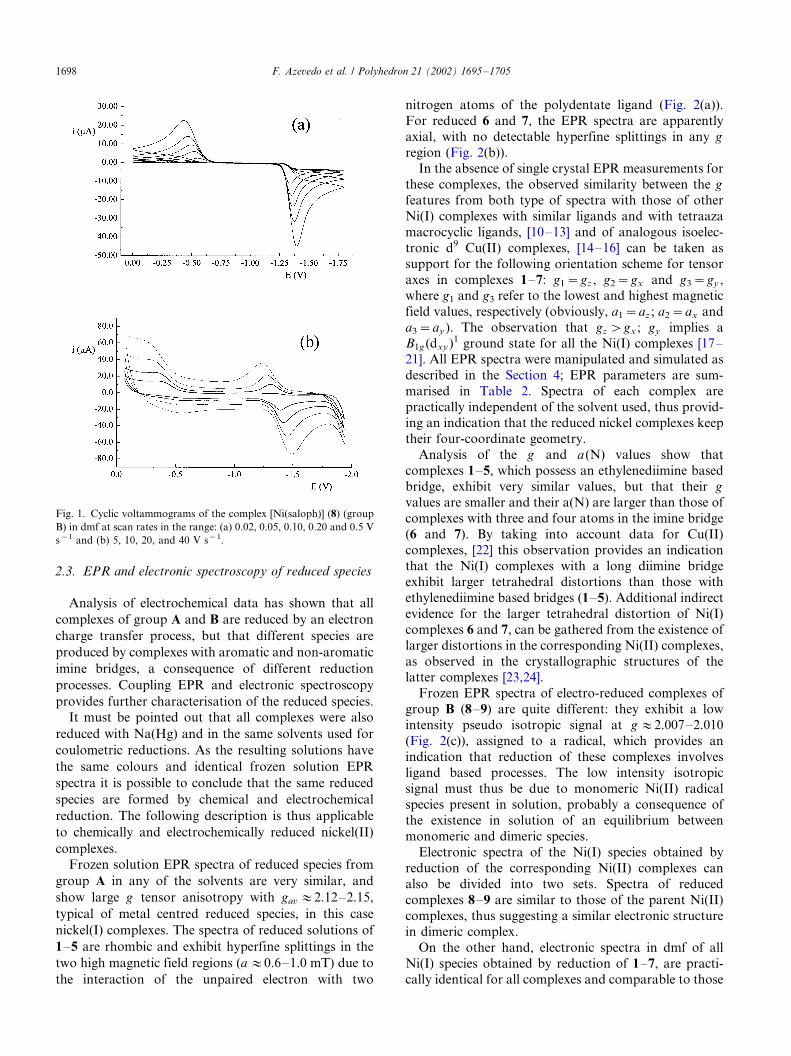
nitrogen atoms of the polydentate ligand (Fig. 2(a)).
For reduced 6 and 7, the EPR spectra are apparently
axial, with no detectable hyperfine splittings in any g
region (Fig. 2(b)).In the absence of single crystal EPR measurements for
these complexes, the observed similarity between the g
features from both type of spectra with those of other
Ni(I) complexes with similar ligands and with tetraaza
macrocyclic ligands, [10�/13] and of analogous isoelec-
tronic d9 Cu(II) complexes, [14�/16] can be taken as
support for the following orientation scheme for tensor
axes in complexes 1�/7: g1�/gz , g2�/gx and g3�/gy ,where g1 and g3 refer to the lowest and highest magnetic
field values, respectively (obviously, a1�/az ; a2�/ax and
a3�/ay ). The observation that gz �/gx ; gy implies a
B1g(dxy)1 ground state for all the Ni(I) complexes [17�/
21]. All EPR spectra were manipulated and simulated as
described in the Section 4; EPR parameters are sum-
marised in Table 2. Spectra of each complex are
practically independent of the solvent used, thus provid-ing an indication that the reduced nickel complexes keep
their four-coordinate geometry.
Analysis of the g and a (N) values show that
complexes 1�/5, which possess an ethylenediimine based
bridge, exhibit very similar values, but that their g
values are smaller and their a(N) are larger than those of
complexes with three and four atoms in the imine bridge
(6 and 7). By taking into account data for Cu(II)complexes, [22] this observation provides an indication
that the Ni(I) complexes with a long diimine bridge
exhibit larger tetrahedral distortions than those with
ethylenediimine based bridges (1�/5). Additional indirect
evidence for the larger tetrahedral distortion of Ni(I)
complexes 6 and 7, can be gathered from the existence of
larger distortions in the corresponding Ni(II) complexes,
as observed in the crystallographic structures of thelatter complexes [23,24].
Frozen EPR spectra of electro-reduced complexes of
group B (8�/9) are quite different: they exhibit a low
intensity pseudo isotropic signal at g :/2.007�/2.010
(Fig. 2(c)), assigned to a radical, which provides an
indication that reduction of these complexes involves
ligand based processes. The low intensity isotropic
signal must thus be due to monomeric Ni(II) radicalspecies present in solution, probably a consequence of
the existence in solution of an equilibrium between
monomeric and dimeric species.
Electronic spectra of the Ni(I) species obtained by
reduction of the corresponding Ni(II) complexes can
also be divided into two sets. Spectra of reduced
complexes 8�/9 are similar to those of the parent Ni(II)
complexes, thus suggesting a similar electronic structurein dimeric complex.
On the other hand, electronic spectra in dmf of all
Ni(I) species obtained by reduction of 1�/7, are practi-
cally identical for all complexes and comparable to those
Fig. 1. Cyclic voltammograms of the complex [Ni(saloph)] (8) (group
B) in dmf at scan rates in the range: (a) 0.02, 0.05, 0.10, 0.20 and 0.5 V
s�1 and (b) 5, 10, 20, and 40 V s�1.
F. Azevedo et al. / Polyhedron 21 (2002) 1695�/17051698
of similar Ni(I) compounds described in literature
[13,25�/29]; they show a medium intensity band at l�/
800�/830 nm (o�/1100�/1800 dm�3 mol�1 cm�1) with a
shoulder at higher energies l�/637�/760 nm (o�/3200�/
5100 dm�3 mol�1 cm�1), and high-intensity bands in
the UV region lB/400 (o�/10 000 dm�3 mol�1 cm�1)
(Fig. 3(a) and Table 2). On the basis of their extinction
coefficients the low energy band can be tentatively
assigned to a d�/d transition, albeit with a large
contribution from ligand orbitals, and the shoulder at
higher energy to charge transfer bands. The wavelength
of the d�/d band in spectra of reduced species is
bathochromic shifted by about 30% relative to the d�/d
transitions of the precursor Ni(II) complexes, which
occur in the range l�/550�/500 nm (Table 2) [13,25�/29].
This indicates a weakening in the ligand field caused by
the decrease in effective nuclear charge of the metal
centre concomitant with its reduction. The extinction
coefficients for Ni(I) complexes are usually much larger
than those observed for similar Ni(II) complexes which
is attributed to a large degree of covalency in the metal�/
ligand bonds (see below) [13,25�/29].Using the splitting scheme proposed by Nishida et al.
[30,31] for the d-orbitals in isoelectronic square�/planar
copper(II) complexes with di-negatively charged tetra-
dentate ligands, dx2�/y2B/dz2B/dxz 5/dyz B/dxy , and as-
suming the same d-splitting for the isoelectronic Ni(I)
complexes used in this work, the low energy band in
each spectrum can be assigned to the transition dxz :/
dyz 1/dx2.
Confirmation for this assignment is provided by EPR
data. Ni(I) complexes are at best of D2h symmetry and
using the g equations deduced for d9 systems with B1g
ground state, the values of s and p-out-of plane
covalency parameters, a2 and d ?2, respectively, can be
estimated from the gx and gy values [18�/21]. This
calculation was made using the methodology outlined in
[13], with the further assumption that the low-energy
electronic band corresponds to the transition dxz :/
dyz 1/dz2. The values of these parameters, summarised
Fig. 2. Frozen solution EPR spectra of the reduced species in dmf (�/120 8C): (a) [Ni(a,a?-Me2salen)]�; (b) [Ni(salbd)]� and (c) [Ni(saloph)]22� in
N2 atmosphere and (a?) [Ni(a,a?-Me2salen)]� in CO atmosphere.
F. Azevedo et al. / Polyhedron 21 (2002) 1695�/1705 1699
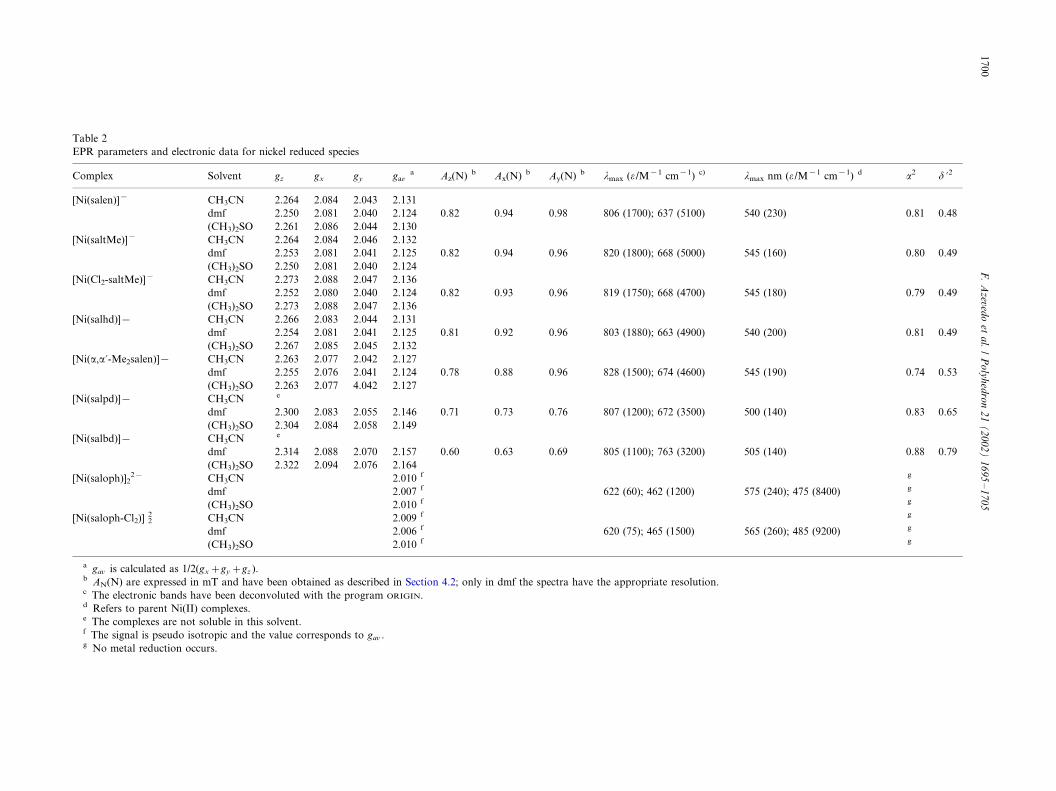
Table 2
EPR parameters and electronic data for nickel reduced species
Complex Solvent gz gx gy gava Az(N) b Ax(N) b Ay(N) b lmax (o /M�1 cm�1) c) lmax nm (o /M�1 cm�1) d a2 d ?2
[Ni(salen)]� CH3CN 2.264 2.084 2.043 2.131
dmf 2.250 2.081 2.040 2.124 0.82 0.94 0.98 806 (1700); 637 (5100) 540 (230) 0.81 0.48
(CH3)2SO 2.261 2.086 2.044 2.130
[Ni(saltMe)]� CH3CN 2.264 2.084 2.046 2.132
dmf 2.253 2.081 2.041 2.125 0.82 0.94 0.96 820 (1800); 668 (5000) 545 (160) 0.80 0.49
(CH3)2SO 2.250 2.081 2.040 2.124
[Ni(Cl2-saltMe)]� CH3CN 2.273 2.088 2.047 2.136
dmf 2.252 2.080 2.040 2.124 0.82 0.93 0.96 819 (1750); 668 (4700) 545 (180) 0.79 0.49
(CH3)2SO 2.273 2.088 2.047 2.136
[Ni(salhd)]� CH3CN 2.266 2.083 2.044 2.131
dmf 2.254 2.081 2.041 2.125 0.81 0.92 0.96 803 (1880); 663 (4900) 540 (200) 0.81 0.49
(CH3)2SO 2.267 2.085 2.045 2.132
[Ni(a,a?-Me2salen)]� CH3CN 2.263 2.077 2.042 2.127
dmf 2.255 2.076 2.041 2.124 0.78 0.88 0.96 828 (1500); 674 (4600) 545 (190) 0.74 0.53
(CH3)2SO 2.263 2.077 4.042 2.127
[Ni(salpd)]� CH3CN e
dmf 2.300 2.083 2.055 2.146 0.71 0.73 0.76 807 (1200); 672 (3500) 500 (140) 0.83 0.65
(CH3)2SO 2.304 2.084 2.058 2.149
[Ni(salbd)]� CH3CN e
dmf 2.314 2.088 2.070 2.157 0.60 0.63 0.69 805 (1100); 763 (3200) 505 (140) 0.88 0.79
(CH3)2SO 2.322 2.094 2.076 2.164
[Ni(saloph)]22� CH3CN 2.010 f g
dmf 2.007 f 622 (60); 462 (1200) 575 (240); 475 (8400) g
(CH3)2SO 2.010 f g
[Ni(saloph-Cl2)] 22 CH3CN 2.009 f g
dmf 2.006 f 620 (75); 465 (1500) 565 (260); 485 (9200) g
(CH3)2SO 2.010 f g
a gav is calculated as 1/2(gx�gy�gz ).b AN(N) are expressed in mT and have been obtained as described in Section 4.2; only in dmf the spectra have the appropriate resolution.c The electronic bands have been deconvoluted with the program ORIGIN.d Refers to parent Ni(II) complexes.e The complexes are not soluble in this solvent.f The signal is pseudo isotropic and the value corresponds to gav .g No metal reduction occurs.
F.
Azeved
oet
al.
/P
oly
hed
ron
21
(2
00
2)
16
95�
/17
05
17
00

in Table 2, compare with those observed for other Ni(I)
complexes with similar ligands [11�/13,32,33] which
provides indirect support for the assignment. The values
indicate that covalency is greater for the p-out-of plane
bonding, suggesting large p delocalisation between theligand and the metal centre. The lower planarity of the
Ni(I) complexes with a long aliphatic diimine bridge (6,
7) is also evidenced by the larger values calculated for
d ?2.
2.4. Reactivity of reduced species towards Lewis bases
No changes in the frozen EPR spectra or electronic
spectra were observed upon addition of pyridine,
imidazole or triphenylphosphine (in stoichiometric or
excess molar ratio) to nickel reduced solutions, either
freshly electrolysed or chemically reduced, an indication
that neither these Ni(I) species interact with the strong
Lewis bases, nor do the nickel(II) dimeric species.Probably the monodentate N and P donors ligands act
as weaker p acceptors than the tetradentate ligand in
stabilising the Ni(I) species.
2.5. Reactivity of reduced species towards carbon
monoxide
The binding of CO, a stronger p acceptor ligand, with
complexes 1�/9 was assessed by studying the reduction
process by cyclic voltammetry and coulometry under a
CO atmosphere, and the reduced species were charac-
terised by EPR and electronic spectroscopy.
2.6. Cyclic voltammetry and electrolysis
No changes were observed in the cyclic voltammo-
grams of complexes of group B (8, 9) obtained under a
CO atmosphere when compared with those under a
nitrogen atmosphere. This suggests no interactionbetween CO and the nickel species, and corroborates
further that reduction of group B complexes does not
involve the metal centre. Contrastingly, the voltammo-
grams of group A complexes are slightly different from
those in a nitrogen atmosphere: a reduction process is
always observed, but (i) the DE values are larger; (ii) the
values of DE increase significantly with increasing scanrates; and (iii) the ipa/ipc ratios are smaller than 1, which
suggests a slow reaction between the nickel species and
CO [6,7].
In order to get further information on the interaction
of CO with reduced complexes of 1�/7, electrolysis of the
Ni(II) complexes were made in a CO atmosphere.
During the electrolysis a change in colour was observed
from red to crimson, which differs from the dark blue�/
green colour of the reduced species in a nitrogen
atmosphere, indicating the presence of different species
in solution. Cyclic voltammograms obtained after
electrolysis show one reduction process with DE�/60�/
70 mV at n�/50 mV s�1, and with ipa/ipc�/1, but with
E1/2 at slightly more positive potentials than those
observed in a N2 atmosphere. In Table 3 are summarised
the electrochemical data for the complexes in COatmosphere. The new electrochemical process corre-
sponds to reduction of nickel species with axial coordi-
nated CO; the more positive E1/2 values imply that
coordination of CO originates more stable Ni(I) species.
The difference between E1/2(N2) and E1/2(CO) can be
taken as a measure of the extent of CO coordination.
From data in Tables 1 and 3 it is possible to infer that
complexes with large tetrahedral distortions (6 and 7)show the weaker interactions with CO, and that com-
plexes with ethylenic bridges, except those with axial
substituents (2, 3 and 4), exhibit the strongest interac-
tions. These results suggest that the planarity of the
coordination sphere [(NiI)N2O2]� plays an important
role in the CO interaction with the nickel centre.
2.7. EPR and electronic spectroscopy of reduced species
with bound CO
Frozen EPR spectra of nickel species with bound CO
have g features similar to those obtained in a N2
atmosphere, but with larger anisotropy and rhombicity
(gav:/2.15, Dxy :/0.06�/0.07). EPR data are summarised
in Table 4, and an analysis of data in this table shows
that the reduced species are still Ni(I) complexes with aB1g(dxy)1 ground state, as gz �/gx , gy . Furthermore, the
observation that gz and gx have been shifted to lower
magnetic field values and that no hyperfine splitting
could be detected in any of the g regions (Fig. 2(a))
suggests that only one CO molecule is bound per nickel
centre, leading to a square pyramidal geometry for the
Ni(I) complexes, as a six-coordinate complex would
have the gz and gx shifted to higher magnetic fields[14,22]. EPR spectra of the reduced species that interact
less extensively with CO (6,7) show a sobreposition of
the parent Ni(I) complex and the one with bound CO.
Fig. 3. UV�/Vis spectra in dmf of [Ni(salen)]� (1 mmol dm�3) in: (a)
N2 and (b) CO atmospheres.
F. Azevedo et al. / Polyhedron 21 (2002) 1695�/1705 1701
Electronic spectra of these CO bound Ni(I) complexesare quite different from those of their parent complexes
and show one intense electronic band at lmax�/546 nm
(o�/5000�/6900 dm�3 mol�1 cm�1) and several high
intense bands in the UV region (Fig. 3). The new band in
the visible region occurs at the same energy for all the
complexes and taking in account their extinction
coefficient they can be assigned to a M0/CO charge
transfer band and is indicative of the high p acceptorcapacity of CO. The non-observation of any d�/d band
can be accounted for by a combination of two factors.
First, the energy of the d�/d bands is increased due to the
high ligand field of CO; second, that of the CT bands is
decreased due to the high p acceptor capacity of CO. As
a consequence the CT bands that occur in the same
region mask less intense d�/d bands.
3. Concluding remarks
Species generated by one-electron electrochemical
reduction of the nickel(II) Schiff base complexes re-
ported in this work were characterised by combining
EPR and UV�/Vis�/NIR spectroscopy and the results
obtained allow for unambiguous identification of the
electron transfer site. Reduction of nickel complexes was
found to be solvent independent, but on the other hand
the final reduction product depends largely on the
ligand diimine bridge. Complexes with aromatic diimine
bridges, (Group B), yielded ligand-centred reduced
species, whereas those with aliphatic imine bridges,
(group A), exhibit metal-centred reduction processes.
Furthermore, the observation that the E1/2 values for all
complexes are practically solvent independent implies
that the solvent does not participate in the charge
transfer process, and indicates that the reduced nickel
species must keep their tetracoordinate geometry. This
result must be contrasted with the oxidation process of
the same complexes, for which the solvent has a marked
influence on oxidation site (metal vs. ligand), with good
donor solvents favouring formation of Ni(III) com-
plexes [34�/39].
As referred above the extent of unsaturation not only
is critical in controlling the ultimate reduction site, but
influences also the E1/2 values, as complexes with greater
unsaturation exhibit the more positive values. Further-
more, the size of the imine bridge and the substituents in
the ligand were also found to influence E1/2 values.
There is a pronounced positive shift in E1/2 values when
the number of carbon atoms in the aliphatic chain of the
Table 3
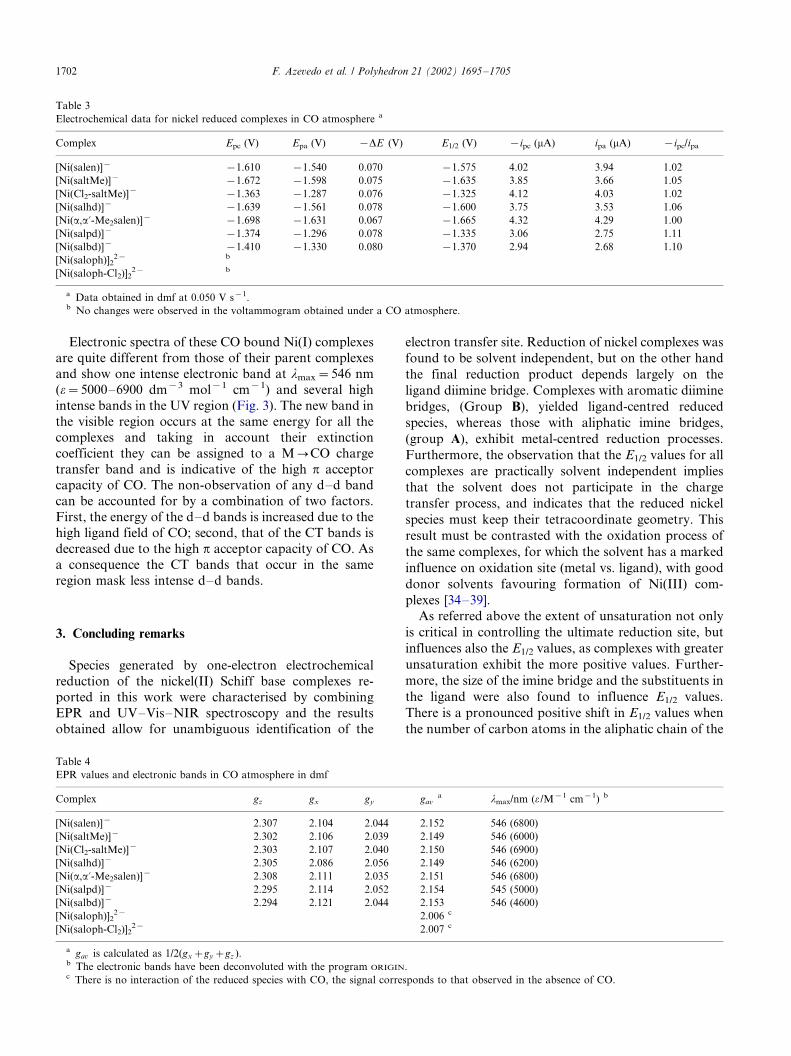
Electrochemical data for nickel reduced complexes in CO atmosphere a
Complex Epc (V) Epa (V) �DE (V) E1/2 (V) �ipc (mA) ipa (mA) �ipc/ipa
[Ni(salen)]� �1.610 �1.540 0.070 �1.575 4.02 3.94 1.02
[Ni(saltMe)]� �1.672 �1.598 0.075 �1.635 3.85 3.66 1.05
[Ni(Cl2-saltMe)]� �1.363 �1.287 0.076 �1.325 4.12 4.03 1.02
[Ni(salhd)]� �1.639 �1.561 0.078 �1.600 3.75 3.53 1.06
[Ni(a,a?-Me2salen)]� �1.698 �1.631 0.067 �1.665 4.32 4.29 1.00
[Ni(salpd)]� �1.374 �1.296 0.078 �1.335 3.06 2.75 1.11
[Ni(salbd)]� �1.410 �1.330 0.080 �1.370 2.94 2.68 1.10
[Ni(saloph)]22� b
[Ni(saloph-Cl2)]22� b
a Data obtained in dmf at 0.050 V s�1.b No changes were observed in the voltammogram obtained under a CO atmosphere.
Table 4
EPR values and electronic bands in CO atmosphere in dmf
Complex gz gx gy gava lmax/nm (o /M�1 cm�1) b
[Ni(salen)]� 2.307 2.104 2.044 2.152 546 (6800)
[Ni(saltMe)]� 2.302 2.106 2.039 2.149 546 (6000)
[Ni(Cl2-saltMe)]� 2.303 2.107 2.040 2.150 546 (6900)
[Ni(salhd)]� 2.305 2.086 2.056 2.149 546 (6200)
[Ni(a,a?-Me2salen)]� 2.308 2.111 2.035 2.151 546 (6800)
[Ni(salpd)]� 2.295 2.114 2.052 2.154 545 (5000)
[Ni(salbd)]� 2.294 2.121 2.044 2.153 546 (4600)
[Ni(saloph)]22� 2.006 c
[Ni(saloph-Cl2)]22� 2.007 c
a gav is calculated as 1/2(gx�gy�gz ).b The electronic bands have been deconvoluted with the program ORIGIN.c There is no interaction of the reduced species with CO, the signal corresponds to that observed in the absence of CO.
F. Azevedo et al. / Polyhedron 21 (2002) 1695�/17051702

imine bridge is increased. The larger hole sizes and
larger tetrahedral distortions of [Ni(salpd)] (6) and
[Ni(salbd)] (7), [23,24] make it easier to accommodate
the size increase expected upon reduction of the metaland thus the more positive E1/2 values.
The number, ring position and electronic character-
istics of the substituents also influence the reductive
properties of the complexes: introduction of chloride
atoms increases the stability of the reduced state
(comparison between 2 and 3), whereas the introduction
of methyl groups decreases the stability of reduced
complexes (comparison of 1 and 2 with 7). Thisbehaviour is attributed to changes in the electronic
density in the metal centre induced by the substituents.
In the absence of steric effects, electron-withdrawing
substituents decrease electron density on the metal
making it easier to reduce, whereas electron donating
groups will have the opposite effect. The position of the
methyl groups in the ligand is also important, as
introduction of two methyl groups in the imine carbonfragment, �/C�/N, shifts the E1/2 to more negative values
than four methyl groups in the aliphatic imine bridge.
Due to the large p delocalisation between the Ni(I)
and thepolydentate ligands (as quantified by s and p-
out-of plane covalency parameters, a2 and d ?2), no
interaction with monodentate N and P donor Lewis
bases could be observed. However, with a stronger pacceptor like CO, a strong interaction was observed andmore stable Ni(I) species were formed as proved by their
more positive E1/2 values and lower energy charge
transfer bands.
4. Experimental
4.1. Reagents, solvents and nickel(II) complexes
The solvents for syntheses were of reagent grade
(Merck), and those for spectroscopic and electrochemi-
cal measurements were of analytical grade (Merck, pro
analysi); all were used as received. Tetra-n-butylammo-
nium perchlorate (TBAP) was prepared by published
procedures from tetrabutylammonium chloride (Al-
drich) and perchloric acid (Merck, p.a.) and recrystal-lised twice from ethanol [40] (CAUTION: perchlorates
are hazardous and may explode). Carbon monoxide gas
was purchased from Praxair.
The nickel(II) complexes [Ni(salen)] (1), [Ni(saltMe)]
(2), [Ni(salhd)] (4), [Ni(salpd)] (5) and [Ni(a,a?-Me2sa-
len)] (7), [Ni(saloph)] (8), [Ni(saloph-Cl2)] (9) have been
prepared and fully characterised in [35,36]. The com-
plexes [Ni(Cl4saltMe)] (3) and [Ni(salbd)] (6) have beenprepared by standard methods [35,36] and characterised
by elemental analysis: [Ni(Cl4saltMe)], bis (3,5-tetra-
chlorosalicylaldehyde)tetramethylethylenediamine:
NiC16H18N2O2Cl4. Anal . Calc.: C, 40.8; H, 3.9; N, 5.9.
Found: C, 40.2; H, 3.8; N, 5.5%.
[Ni(salbd)], bis (salicylaldehyde)butylenediamine:
NiC18H18N2O2. Anal . Calc.: C, 61.2; H, 5.1; N, 7.9.Found: C, 61.1; H, 5.0; N, 8.1%.
4.2. Physical measurements
Electrochemical measurements were performed using
an EG&G PAR 273 potentiostat�/galvanostat using
solutions 1 mmol dm�3 in nickel(II) complex and 0.1
mol dm�3 TBAP. Cyclic voltammograms were obtained
in a standard one-compartment electrochemical cellusing a platinum-disc electrode (area of 0.0314 cm2) as
working electrode, a platinum gauze as the counter
electrode and an Ag�/AgCl (1 mol dm�3 NaCl)
reference electrode. All voltammograms were obtained
without IR feedback compensation and measured
potentials were not corrected for liquid junction poten-
tials. All potentials are reported relative to that of the
Ag�/AgCl (1 mol dm�3 NaCl) reference electrode and toE1/2 of the ferrocenium�/ferrocene couple. Cyclic vol-
tammetry was performed in Me2SO, dmf and MeCN in
the potential range �/1.0�/�/2.0 V using scan rates 0.02�/
0.5 V s�1; for complexes of group B scan rates in the
range 5�/60 V s�1 were also used. Under the experi-
mental conditions used and for scan rate 0.05 V s�1,
E1/2 (DE ) for Fc��/Fc couple are 0.50 V (0.09 V) in
Me2SO, 0.50 V (0.10 V) in dmf, and E1/2�/0.48 V (0.085V) in MeCN.
Controlled potential electrolysis was carried out
under a N2 or a CO atmosphere at a potential 0.1 V
higher than the cathodic peak potentials. The electro-
chemical cell has two compartments: one for the
platinum-gauze working electrode and the other for
the platinum-gauze counter electrode, which are sepa-
rated by a silica frit. A Ag�/AgCl (1 mol dm�3 NaCl)reference electrode was used.
Room-temperature (r.t.) electronic spectra of
nickel(II)�/(I) complexes were recorded with a Schi-
madzu UV/3101PC spectrometer in the range 1500�/200
nm. Solutions of nickel(II)�/(I) complexes, under an
atmosphere of N2 or CO, were typically 1 mmol dm�3.
The EPR spectra were obtained with an X-band
Bruker ESP 300E spectrometer equipped with BrukerB-VT 2000 temperature accessory controller, in a dual
cavity and using diphenylpicrylhydrazyl (dpph; g�/
2.0037) as standard; the magnetic field was calibrated
by use of Mn2� in MgO. Typical experimental condi-
tions used were: microwave power 10�/15 mW, modula-
tion amplitude 0.5 mT; modulation frequency 100 kHz.
Spectra were recorded at �/120 8C, using sealed quartz
tubes and the reported EPR parameters were obtainedby computer simulation, [41] using a rhombic g tensor.
As the spectra in Me2SO and CH3CN have lower
resolution than in dmf, the values of superhyperfine
F. Azevedo et al. / Polyhedron 21 (2002) 1695�/1705 1703

splittings obtained for spectra in dmf were used to
simulate spectra in the former solvents. Even for spectra
in dmf the superhyperfine couplings with the ligand
nitrogen atoms had to be obtained from second-derivative EPR spectra, after a Fourier transformation
and the subsequent elimination of all high-frequency
components with a cut off filter just above the typical
frequency components of nitrogen hyperfine coupling.
After back-transformation to the magnetic field space,
the noise at high frequencies introduced by differentia-
tion is eliminated.
4.3. Reduction of Ni(II) complexes
Nickel(I) species were generated in-situ by electrolysis
or chemical reduction of 1 mmol dm�3 solutions of the
corresponding nickel(II) complexes under strict anaero-
bic conditions. Electrochemical reduction was per-formed in all solvents used the procedure outlined
above. Chemical reduction was performed in dimethyl-
formamide using an excess of 5% sodium mercury
amalgam. The solution�/amalgam was stirred for 15
min, and a fast solution colour change was observed.
The progress of the reduction was monitored by UV�/
Vis and EPR spectroscopy and was assumed to be
complete (1 mmol dm�3 in Ni(I) species, at approxi-mately 15 min), when no changes in intensity of the EPR
signals and in the absorbance of electronic bands were
detected.
Manipulations of nickel(I) species were carried out in
a nitrogen atmosphere, by use of standard Schlenk
techniques and/or of a glove box. Addition of pyridine,
imidazole and triphenylphosphine to freshly electrolysed
or chemically reduced solutions was performed underanaerobic conditions at r.t. The resulting solutions were
immediately transferred to EPR tubes and frozen in
liquid nitrogen. Adducts with CO were prepared by
making the reduction (electrochemically and chemically)
under a CO atmosphere.
Acknowledgements
This work was partially supported by the ‘Fundacao
para a Ciencia e Tecnologia’, Lisboa, Portugal, through
Project POCTI/32831/QUI/2000.
References
[1] (a) C. Gosden, K.P. Healy, D. Pletcher, J. Chem Soc. Dalton
Trans. (1978) 972;
(b) C. Gosden, D. Pletcher, J. Organomet. Chem. 186 (1980) 401;
(c) K.P. Healey, D. Pletcher, J. Organomet. Chem. 161 (1978)
109;
(d) C. Gosden, J.B. Kerr, D. Pletcher, R. Rosas, J. Electroanal.
Chem. 117 (1981) 101;
(e) J.Y. Becker, J.B. Kerr, D. Pletcher, R. Rosas, J. Electroanal.
Chem. Interf. Electrochem. 117 (1981) 87.
[2] (a) A. Bakac, J.H. Espenson, J. Am. Chem. Soc. 108 (1986) 713;
(b) A. Bakac, J.H. Espenson, J. Am. Chem. Soc. 108 (1986) 719;
(c) A. Bakac, J.H. Espenson, J. Am. Chem. Soc. 108 (1986) 5353;
(d) M.S. Ram, A. Bakac, J.H. Espenson, Inorg. Chem. 25 (1986)
3267;
(e) N. Sadler, S.L. Scott, A. Bakac, J.H. Espenson, Inorg. Chem.
28 (1989) 3951.
[3] (a) G.K. Lahiri, L.J. Schussel, A.M. Stolzenberg, Inorg. Chem. 31
(1992) 4991;
(b) K.M. Kadish, M.M. Franzen, B.C. Han, C. Araullo-McA-
dams, D. Sazou, Inorg. Chem. 31 (1992) 4399.
[4] (a) B. Fischer, R. Eisenberg, J. Am. Chem. Soc. 102 (1980) 7361;
(b) M. Beley, J.P. Collin, R. Rupert, J.P. Sauvage, J. Am. Chem.
Soc. 108 (1986) 7641;
(c) J.B. Collin, A. Jouaiti, J.P. Sauvage, Inorg. Chem. 27 (1988)
1986.
[5] A.A. Isse, A. Gennaro, E. Vianello, Electrochim. Acta 37 (1992)
113.
[6] A.J. Bard, L.R. Faulkner, Electrochemical Methods. Fundamen-
tals and Applications, 2nd ed., Wiley, New York, 2001.
[7] R.S. Nickolson, Anal. Chem. 37 (1965) 178.
[8] S. Gambarotta, C. Floriani, A. Chiesi-Villa, C. Guastini, J. Chem.
Soc., Chem. Commun. (1982) 756.
[9] S. Gambarotta, F. Urso, C. Floriani, A. Chiesi-Villa, C. Guastini,
Inorg. Chem. 22 (1983) 3966.
[10] F. Azevedo, M.A.A.F. De, C.T. Carrondo, M. Coney, B. de
Castro, D. Domingues, M.T. Duarte, C. Freire, K. Nielsen, I.
Santos, Inorg. Chim. Acta 112 (1994) 1634.
[11] G.A. Bowmarker, P.D.W. Boyd, G.K. Campbell, J.H. Hop,
Inorg. Chem. 21 (1982) 1152.
[12] G.A. Bowmaker, G.A. Boyd, G.K. Campbell, M. Zvagulis, J.
Chem. Soc., Dalton Trans. (1986) 1065.
[13] M. Valente, C. Freire, B. de Castro, J. Chem. Soc., Dalton Trans.
(1998) 1557.
[14] J. Maroney, N.J. Rose, Inorg. Chem. 23 (1984) 2252.
[15] A.D. Toy, M.D. Hobday, P.D.W. Boyd, J. Chem. Soc., Dalton
Trans. (1973) 1259.
[16] M. Chikira, H. Yokoi, T. Isobe, Bull. Chem. Soc. Jpn. 47 (1974)
2208.
[17] A.H. Maki, N. Edelstein, A. Davison, R.H. Holm, J. Am. Chem.
Soc. 86 (1964) 4580.
[18] A.H. Maki, B.R. McGarvey, J. Chem. Phys. 29 (1958) 31.
[19] A.H. Maki, B.R. McGarvey, J. Chem. Phys. 29 (1958) 39.
[20] R. Neiman, D. Kivelson, J. Chem. Phys. 35 (1962) 149.
[21] H.G. Gersmann, J.D. Swalen, J. Chem. Phys. 36 (1962) 3221.
[22] U. Sakaguchi, A.W. Addison, J. Chem. Soc. (1979) 600.
[23] M.G.B. Drew, R.N. Prasad, R.P. Sharma, Acta Crystallogr. Sect.
C 41 (1985) 1755.
[24] M. Calligaris, G. Nardin, L. Randaccio, Coord. Chem. Rev. 7
(1972) 385.
[25] A.M. Stolzenberg, M.T. Stershic, J. Am. Chem. Soc. 110 (1988)
5397.
[26] L.R. Furenlid, M.W. Renner, K.M. Smith, J. Fajer, J. Am. Chem.
Soc. 112 (1990) 1634.
[27] M.P. Sugh, I.S. Kim, S.J. Cho, W. Shin, J. Chem. Soc., Dalton
Trans. (1994) 2765.
[28] M.P. Suh, Adv. Inorg. Chem. 44 (1996) 93.
[29] D.J. Szalda, E. Fijita, R. Sanzenbacher, H. Paulus, H. Elias,
Inorg. Chem. 33 (1994) 5855.
[30] Y. Nishida, K. Hayashida, A. Sumita, S. Kida, Inorg. Chim. Acta
31 (1978) 19.
[31] Y. Nishida, K. Hayashida, S. Kida, J. Coord. Chem. 10 (1980)
101.
[32] M.C.R. Symmons, D.X. West, J. Chem. Soc., Dalton Trans.
(1985) 379.
F. Azevedo et al. / Polyhedron 21 (2002) 1695�/17051704

[33] P.J. Chmielewski, L. Latos-Grazynski, E. Paholska, Inorg. Chem.
33 (1994) 1992.
[34] M. Vilas Boas, C. Freire, B. de Castro, P.A. Christensen, A.R.
Hillman, Inorg. Chem. 36 (1997) 4919.
[35] C. Freire, B. de Castro, J. Chem. Soc., Dalton Trans. (1998) 1491.
[36] C. Freire, B. de Castro, Polyhedron 17 (1998) 4227.
[37] M. Vilas-Boas, C. Freire, B. de Castro, A.R. Hillman, J. Phys.
Chem. B 102 (1998) 8533.
[38] M. Vilas Boas, M.J. Henderson, C. Freire, A.R. Hillman, E. Vieil,
Chem. Eur. J. 6 (2000) 1160.
[39] I.C. Santos, M. Vilas-Boas, F. Piedade, C. Freire, M. Teresa
Duarte, B. de Castro, Polyhedron 19 (2000) 655.
[40] S.T. Donald, R.L. Julien, Experimental Electrochemistry for
Chemistry, Wiley, New York, 1974.
[41] J.R. Pilbrow, M.E. Winfield, Mol. Phys. 25 (1973)
1073.
F. Azevedo et al. / Polyhedron 21 (2002) 1695�/1705 1705











![Hetero-binuclear Schiff Base Complexes of Copper(II ...nopr.niscair.res.in/bitstream/123456789/49062/1... · Hetero-binuclear schiff base complexes of the types [Cu Ll \1X1] and ["liL'\1X1J](https://img.pdfslide.net/doc/110x75/5fab3d32647cdd491452f6fc/hetero-binuclear-schiff-base-complexes-of-copperii-nopr-hetero-binuclear-schiff.jpg)

















