Embed Size (px)
Citation preview

An agency of the European Union
Registers and Observational Drug Studies Regulatory Perspective
June M Raine Chair, PRAC
EULAR RODS Meeting
14-15 November 2013

Outline of presentation
Objective of regulation
Regulatory role of registers
New EU pharmacovigilance legislation
Moving forward – challenges and opportunities

What is the objective of regulation?
Promote public health
Availability of innovative treatments and technologies without unnecessary delay
Protect public health
Active vigilance based on best evidence and prompt risk management
Support individual health decisions
As much information to patients and healthcare professionals as possible on benefits and risks

4
Regulators must…
Assess changing risk benefit of medicines in clinical use
Reach prompt decisions which take into account the therapeutic context
Implement proportionate risk minimising action
Constantly seek to improve & strengthen methodologies
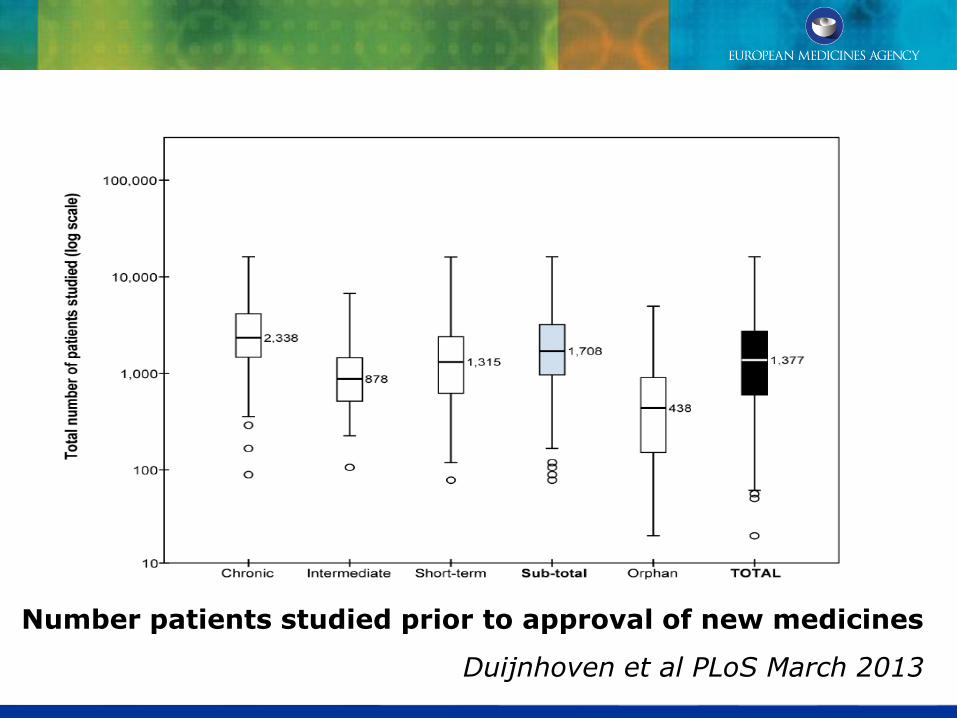
Number patients studied prior to approval of new medicines
Duijnhoven et al PLoS March 2013

Regulatory challenges
Access to robust evidence in post authorisation phase
- on emerging risk
- On rare risks, especially in long term use
- on background rates in exposed population
- on how medicine is being used in practice
- on real world efficacy and clinical outcomes
Effectiveness of risk minimisation
Time dimension – evolving evidence

EU strategy for pharmacovigilance
Shift from responding reactively to new safety issues, to proactively investigating drug safety in clinical use
Generate robust data, including on long term use, rare adverse reactions Monitor the effectiveness of risk minimisation and evaluating meaningful outcomes
Engage with healthcare professionals and patients
Better communications and greater transparency
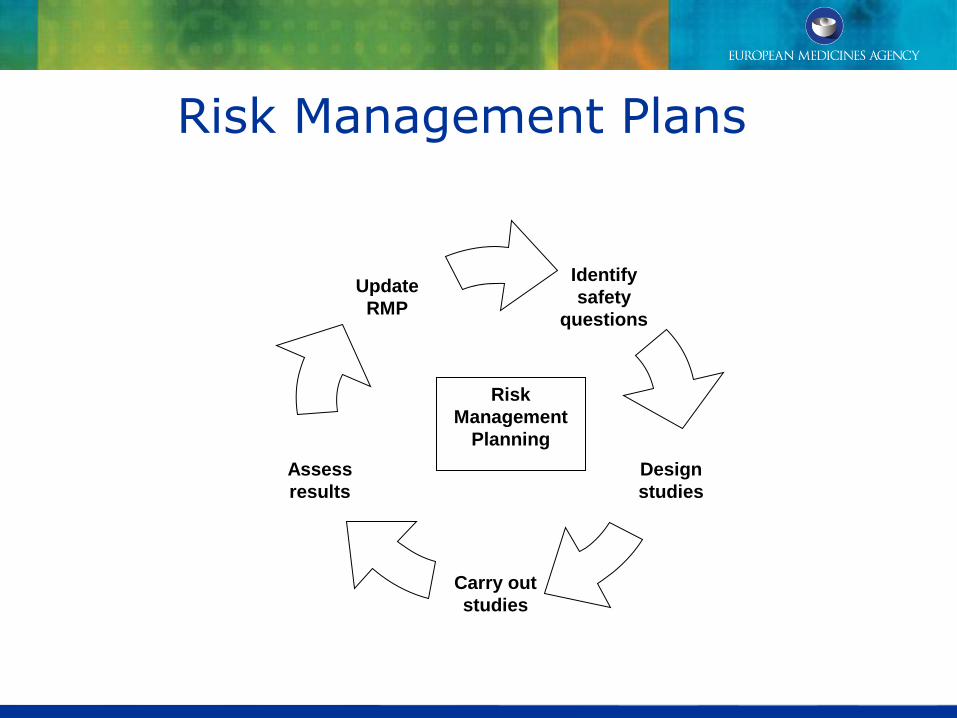
Risk Management Plans
Identify
safety
questions
Assess
results
Carry out
studies
Design
studies
Update
RMP
Risk
Management
Planning

What is currently required in RMPs?
For all new MA with RMP 1/1/2010 to 31/12/2012:
How many of the RMP for new MA mention a registry?
• Of the 123 RMPs identified, 30 had a registry (24%)
• All of these were additional PhV activities
Number of RMPs by risk Identified Risk
Potential risk
Missing information
Number of RMPs with registry
11
13
24

Regulatory view of registries
Definition of registry
An organised system that uses observational methods to collect uniform data on specified outcomes in a population defined by a particular disease, condition or exposure (Good Vigilance Practice 2012)
Types of registry
• By geography, population or coverage
• By therapeutics - product, substance, class, disease
- - -

Regulatory view of registries
Patient registries are key instruments for:
• Pooling scarce data without bias for epidemiological and/or
clinical research
• Assessing the feasibility of clinical trials, and to facilitate
the planning of appropriate clinical trials
• Gathering of evidence on effectiveness of the treatment &
on its possible side effects
• Development of clinical research, specially in field of rare
diseases
• Improvement of patient care and healthcare planning
• Study of social, economic & quality of life outcomes

Important regulatory RODS
Biologics in rheumatoid arthritis
HAART in HIV/AIDS
Factor VIII, IX in haemophilia
Anti-epileptics, SSRIs in pregnancy
Betaferons, natalizumab in Multiple Sclerosis

Contributions of registries to understanding of safety of biopharmaceuticals
Serious infections, particularly tuberculosis and opportunistic infections
Malignancies in general
Lymphoma, melanoma and keratinocyte cancers
Cardiovascular events
Immunogenicity

Data Collection on Adverse Events on Anti-HIV Drugs
Collaborative, observational study
11 established cohorts of HIV-1-infected persons prospectively
followed at 212 clinics in Europe, the US and Australia
– Cohort I: 23,437 patients enrolled 1999-March 2001
– Cohort II: 10,021 patients enrolled 2001-2004
– Cohort III: 5,000 enrolled after 2004
Estimated to have accumulated 295,000-310,000 person-
years until the 13th annual data merger on 31/01/2012
Important findings on cardiovascular risk, non-AIDs
malignancies

New EU pharmacovigilance legislation
Clear legal framework for post-authorisation monitoring Implementation and delivery of risk management plans Regulatory oversight of post-authorisation studies Good Vigilance Practice standards Greater transparency, patient and public involvement
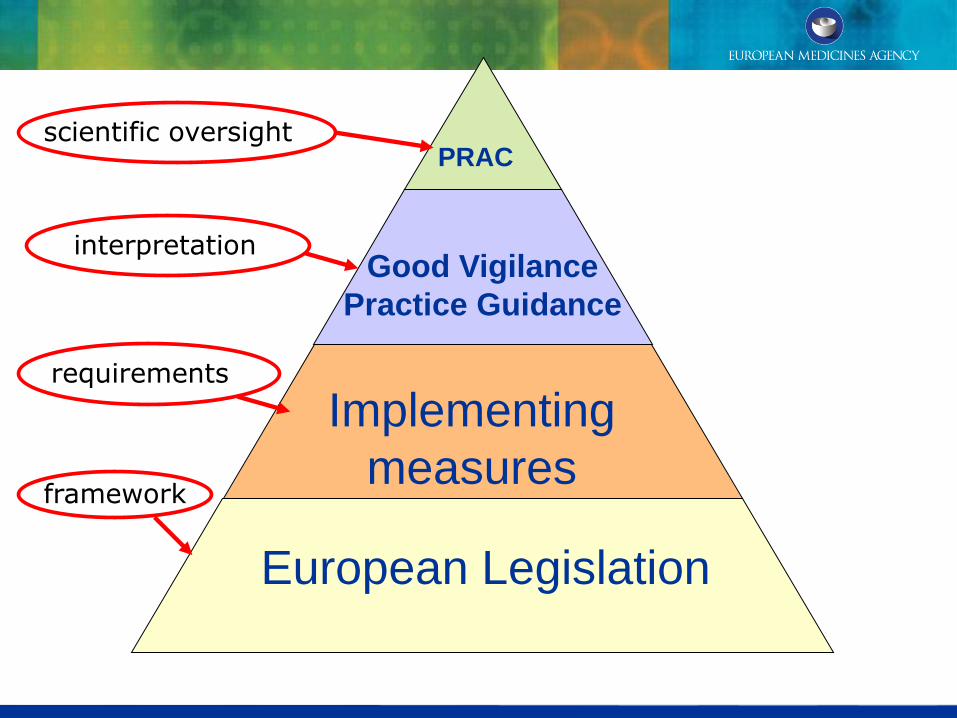
European Legislation
Implementing
measures
Good Vigilance
Practice Guidance
PRAC
requirements
interpretation
scientific oversight
framework

Pharmacovigilance Risk Assessment Committee - Mandate
All aspects of the risk management of the use of medicinal products including the detection, assessment, minimisation and communication relating to the risk of adverse reactions, having
due regard to the therapeutic effect of the medicinal product, the design and evaluation of
post-authorisation safety studies and pharmacovigilance audit

PRAC review - CVS safety of diclofenac
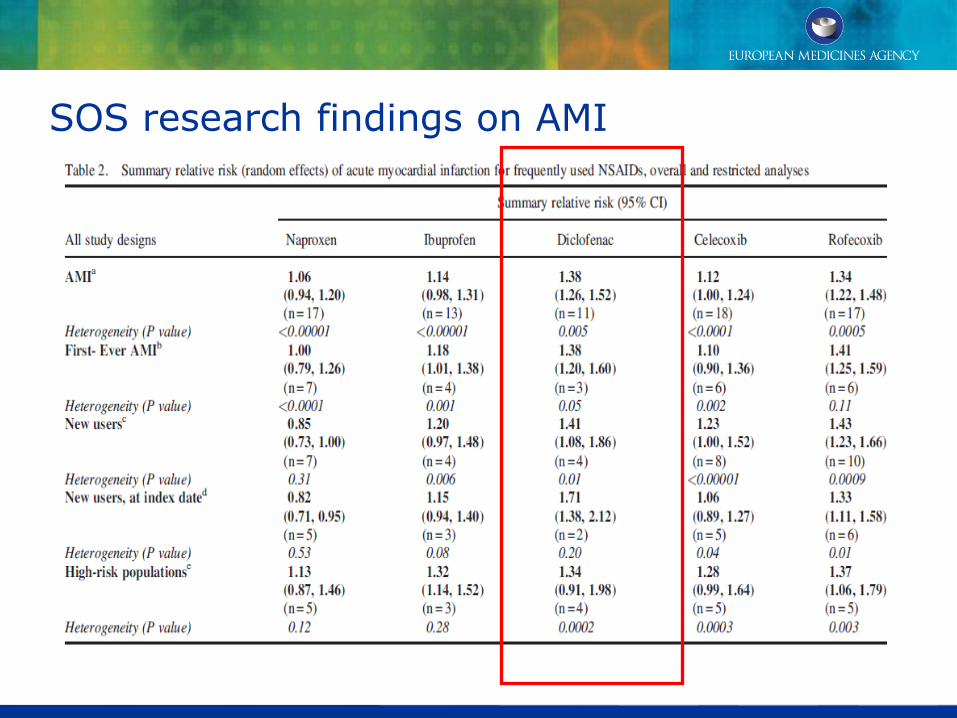
SOS research findings on AMI

PRAC advice - benefit risk of strontium ranelate
Periodic safety update report identified increased risk of cardiac disorders including MI
PRAC advised safety restrictions
CHMP referral under Art 31
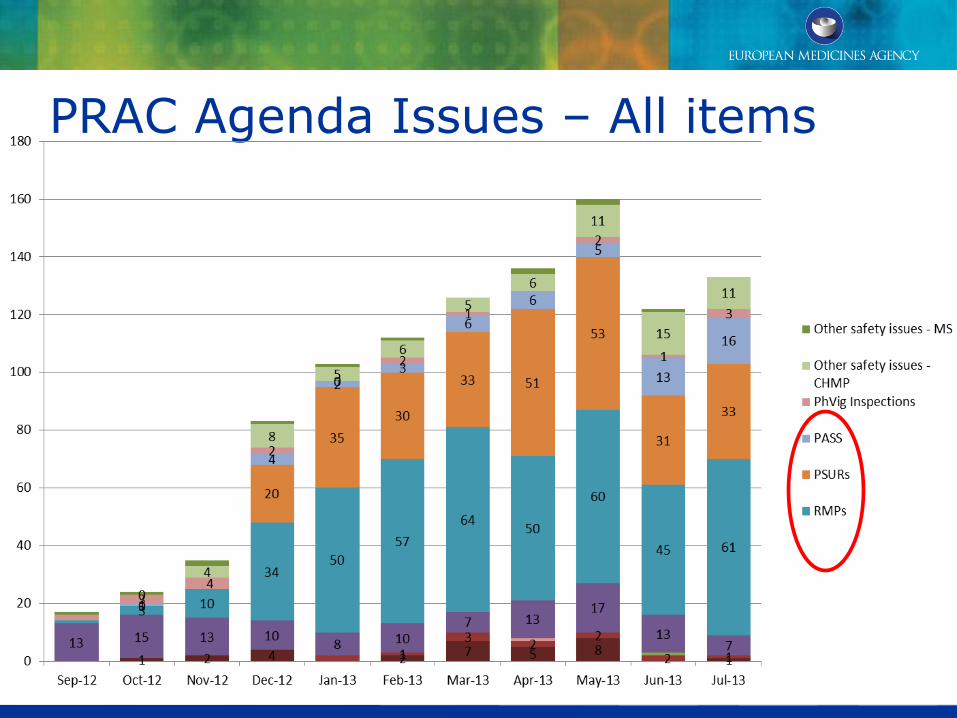
PRAC Agenda Issues – All items

Post-authorisation studies
“It is necessary from a public health perspective to
complement the data available at the time of
authorisation with additional data about the safety
and, in certain cases, efficacy of medicines
Competent authorities should therefore be
empowered to impose on the MAH the obligation to
conduct post-authorisation studies on safety and on
efficacy”
Such studies may be aimed at collecting data to
enable the assessment of safety or efficacy… in
everyday medical practice”

Good Vigilance Practice Module V
Guideline on Risk Management Systems
Refers to registries
Further module specifically on biologics under development

Article 22a of Directive 2010/84/EU
“If the same concerns apply to more than one medicinal product, the national competent authority shall, after consultation with the PRAC, encourage the marketing authorisation holders concerned to conduct a joint post-authorisation safety study”

Article 22a of Directive 2010/84/EU
“The national competent authority may impose an obligation to conduct a post-authorisation efficacy study when the understanding of the disease or the clinical methodology indicate the previous efficacy evaluations may have to be revised significantly”
EMA workshop 24-25 October 2013

Further legislation…

Moving forward
Strengthening the science base - building an EU “infrastructure”
Anticipating the scientific challenges
Developing new methodologies – adaptive licensing
Engaging with companies, academia, health professionals and patients
A clear vision of success

Regulatory Science
“The areas of science used in assessment
of quality, safety & efficacy of medicines
throughout their lifespan, as well as the
scientific areas used in regulatory
decision-making
…..It encompasses basic and applied
medicinal science and social sciences
and contributes to the development of
regulatory standards and tools”

Strengthening the science base

European network to promote access to European pharmacoepidemiological resources
Improve research standards
Increase independence and transparency in research
Stimulate collaboration and exchange of information and experience

Pharmepi and Drug Safety
2012; 20: 690-696

ENCePP Centres with experience in registries
Centres distribution by geographical origin
UK – 13 Spain – 11 Germany – 10 Less than 10 in descending order – Italy, France, the Netherlands, Denmark, Finland and
Portugal, Sweden and Greece and Belgium, Ireland, Slovakia, Switzerland

Of 77 centres in EU listed in ENCePP that mention ‘Registry’
51 centres have drug registry/ies 51 centres have disease registry/ies 34 have other registry/ies (e.g. from observational studies,
family database, mortality registry, adverse events registry, etc.)
Centres distribution by organisation/affiliation
University based (44) Hospital based (23) Government based (13) Charity of non-for-profit organisation (16) For profit organisation (15) Other (not listed specifically under the categories above) (8)
ENCePP Centres with experience in registries

Scientific challenges
New risks
Traceability
Biosimilars
Children
Rare diseases, orphans
Early access

Stakeholder engagement - how to encourage
collaboration?
By imposing on several different companies the obligation to
create a registry at the same time?
Not imposing registry but bringing companies together to
discuss creating of a disease registry?
Identify and approach a suitable academic/patient
organization and suggest collaboration with relevant
companies?
Bring together relevant companies and relevant ENCePP
centres to start the collaboration?

A clear vision of ideal registry
• Built around a specific disease or a defined group of diseases
• Number of individuals on the registry must be adequate to provide sufficient power in relation to defined questions
• Optimal geographic scope
• Direct reporting by healthcare professionals as well as patients to improve completeness and robustness of data collection
• Measures for quality assurance/control, requirements for updates
• Linked with data and biological specimens when possible
• Robust and appropriate case definition
• Accurate and as complete as possible data to guarantee scientific integrity, internal/external validity
• Interoperable, harmonized format and standards
• Transparent, impartial governance inclusive of all stakeholders, complying with data protection and confidentiality legislation

Conclusions
Registries and observational studies have played a critical role in understanding risk benefit in real world use of medicines
New European legislation has placed observational studies at the centre of new strengthened regime
Scientific innovations in therapeutics require new regulatory approaches and methodologies
From risk to benefit:risk life-cycle monitoring opens opportunities for new models of regulation
Collaboration between all stakeholders is key to ensuring we achieve optimum value from RODS





























