Embed Size (px)
Citation preview

© The Author 2013. Published by Oxford University Press. All rights reserved. For Permissions, please email: [email protected]
Carcinogenesis vol.00 no.00 p.1 of 8, 2013 doi:10.1093/carcin/bgt043Advance Access publication February 6, 2013
Regulation of proteolytic cleavage of retinoid X receptor-α by GSK-3β
Weiwei Gao1,†, Jie Liu1,†, Mengjie Hu1, Mingfeng Huang1, Sisi Cai1, Zhiping Zeng1, Bingzhen Lin2, Xihua Cao2, Jiebo Chen2, Jin-zhang Zeng1, Hu Zhou1,2,* and Xiao-kun Zhang1,2,* 1School of Pharmaceutical Science, Xiamen University, Xiamen, Fujian 361102, China and 2Cancer Center, Sanford-Burnham Medical Research Institute, 10901 N. Torrey Pines Road, La Jolla, CA 92037, USA
*To whom correspondence should be addressed: Tel: +86 592 218 2552; Fax: +86 592 218 2552; Email: [email protected] may also be addressed to Xiao-kun Zhang. Tel: +86 592 218 1851; Fax: +86 592 218 1879; Email: [email protected]
We recently reported that an N-terminally truncated retinoid X receptor-α (tRXRα) produced in cancer cells acts to promote can-cer cell growth and survival through AKT activation. However, how RXRα is cleaved and how the cleavage is regulated in can-cer cells remain undefined. In this study, we demonstrated that calpain II could cleave RXRα protein in vitro, generating two truncated RXRα products. The cleavage sites in RXRα were mapped by Edman N-terminal sequencing to Gly90↓Ser91 and Lys118↓Val119. Transfection of the resulting cleavage product RXRα/∆90, but not RXRα/∆118, resulted in activation of AKT in cancer cells, similar to the effect of tRXRα. In support of the role of calpain II in cancer cells, transfection of calpain II expres-sion vector or its activation by ionomycin enhanced the produc-tion of tRXRα, whereas treatment of cells with calpain inhibitors reduced the levels of tRXRα. Co-immunoprecipitation assays also showed an interaction between calpain II and RXRα. In studying the regulation of tRXRα production, we observed that treatment of cells with lithium chloride or knockdown of glycogen synthase kinase-3β (GSK-3β) significantly increased the production of tRXRα. Conversely, overexpression of GSK-3β reduced tRXRα expression. Furthermore, we found that the inhibitory effect of GSK-3β on tRXRα production was due to its suppression of cal-pain II expression. Taken together, our data demonstrate that GSK-3β plays an important role in regulating tRXRα produc-tion by calpain II in cancer cells, providing new insights into the development of new strategies and agents for the prevention and treatment of tRXRα-related cancers.
Introduction
Retinoid X receptor α (RXRα), a unique member of the nuclear receptor superfamily (1), has pleiotropic functions ranging from cell proliferation to apoptosis through its action in both nucleus and cytoplasm (2,3). Like other nuclear receptors, RXRα consists of three main functional domains: (i) the non-conserved N-terminal A/B domain with an autonomous ligand-independent transcriptional activation function, (ii) the central DNA-binding domain respon-sible for DNA binding and (iii) the multifunctional C-terminal ligand-binding domain containing regions for receptor dimeriza-tion, ligand-binding and ligand-dependent transactivation (4). As a nuclear receptor, RXRα forms homodimers or heterodimers with
other nuclear receptors to bind to its cognate sequence in the pro-moters of target genes, leading to the activation or suppression of target gene transcription (4). Recently, the cytoplasmic localiza-tion of RXRα and its non-genomic functions have been extensively studied (3). In PC12 pheochromocytoma cells, RXRα together with Nur77 migrate from the nucleus to the cytoplasm in response to nerve growth factor, a process that is critical for nerve growth factor-induced PC12 cell differentiation (5). We also found that apoptotic stimuli such as retinoid-derived molecules induce nuclear export of RXRα and Nur77, which then interact with Bcl-2, resulting in Bcl-2 transformation from an apoptosis inhibitor to a promoter (6–8). Cytoplasmic localization of RXRα has been also implicated in inflammation (9).
Regulated proteolysis is a key step in a number of different signal-ing pathways that respond to developmental cues or external stimuli (10). We recently reported that an N-terminally truncated RXRα (tRXRα) resulted from proteolytic cleavage of RXRα was detected in different types of cancer cells and tumor tissues, but not in the cor-responding normal tissues (11). Unlike the full-length RXRα known to predominantly reside in the nucleus, tRXRα is localized mainly in the cytoplasm, where it interacts with the p85α regulatory subunit of phosphatidylinositol-3-OH kinase (PI3K), leading to activation of the PI3K/AKT signaling pathway and cancer cell growth in vitro and in animals (11,12). Thus, tRXRα acquires tumor-promoting function, which is different from the full-length RXRα. However, how RXRα is cleaved and how tRXRα production are regulated in cancer cells remains largely unknown.
Calpains are a large family of Ca2+-activated proteases involved in the regulation of cell adhesion, migration and death through the limited proteolysis of specific target proteins (13). The most well-characterized calpain isoforms are calpain I (µ-calpain) and calpain II (m-calpain), which are heterodimeric proteases composed of a 80 kDa catalytic subunit and a 30 kDa common regulatory subunit calpain 4 (14). The ubiquitously expressed protein calpastatin is an endogenous inhibitor of calpains (14). In addition, various kinases including mitogen-activated protein kinase kinase kinase 1/extracel-lular signal-regulated kinase and protein kinase A regulate calpain activity by phosphorylation (15–17). Calpain has been shown to account for limited proteolytic cleavage of several nuclear recep-tors. Cleavage of the androgen receptor (AR) by calpain II produces a truncated receptor that acts as a ligand-insensitive, constitutively active transcription factor, which may play a role in the development of androgen-independent prostate cancer (18,19). Calpain II also cleaves RXRα, suggesting its role in controlling the functions and activities of RXRα (20). However, whether calpain II cleavage of RXRα leads to production of tRXRα capable of activating AKT is currently unknown.
Glycogen synthase kinase-3β (GSK-3β) is a highly conserved ser-ine/threonine protein kinase ubiquitously distributed in eukaryotes and plays a central role in many cellular functions by phosphorylating numerous target proteins (21). Unlike most kinases, GSK-3β is active in resting cells, and stimulation of cells by mitogens or growth fac-tors leads to its inactivation (22). The activity of GSK-3β is regulated by phosphorylations, of which the Tyr216 phosphorylation enhances GSK-3β activity, whereas the Ser9 phosphorylation inhibits its activ-ity (23). Dysfunction of GSK-3β leads to many diseases including cancers (22,24).
In this study, we investigated the role and regulation of calpain II in the production of tRXRα and tRXRα-mediated AKT activation. We report that calpain II could cleave RXRα at its N-terminal A/B region in vitro and in vivo. We also found that one of the resulting tRXRα products, which lack 90 N-terminal amino acids, could activate AKT when overexpressed in cancer cells. Furthermore, we showed that GSK-3β is a negative regulator of tRXRα production through its ability to inhibit calpain II expression.
Abbreviations: AR, androgen receptor; GSK-3β, glycogen synthase kinase-3β; GST, glutathione-S-transferase; IP, immunoprecipitated; LiCl, lithium chloride; PI3K, phosphatidylinositol-3-OH kinase; RXRα, retinoid X receptor α; SDS–PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; siRNA, small interfering RNA; TBST, Tris-buffered saline and Tween 20; tRXRα, truncated retinoid X receptor-α; WB, western blotting.
†These authors contributed equally to this work.
Page 1 of 8
Carcinogenesis Advance Access published March 5, 2013 at U
niversity of Alabam
a at Birm
ingham on M
arch 5, 2013http://carcin.oxfordjournals.org/
Dow
nloaded from
W.Gao et al.
Materials and methods
Antibodies and reagentsMouse monoclonal anti-Flag and anti-β-actin antibodies and lithium chloride (LiCl; Sigma–Aldrich), rabbit polyclonal anti-RXRα antibodies (D20 and ∆N197) and mouse monoclonal anti-Myc and anti-GFP antibodies (Santa Cruz Biotechnology), rabbit polyclonal anti-calpain II and anti-pAKT anti-bodies (Cell Signaling Technology), rabbit monoclonal anti-GSK-3β (phopho S9) and anti-glyceraldehyde 3-phosphate dehydrogenase antibodies (Abcam), mouse monoclonal anti-GSK-3β antibody (BD Biosciences), Lipofectamine 2000 transfection regent (Invitrogen), calpeptin (Millipore) and recombinant calpain II and ionomycin (Calbiochem) were used in this study.
Cell cultureHEK293T human embryonic kidney cells, Raw264.7 mouse monocyte mac-rophage, HaCat human skin keratinocyte cells, HepG2 human hepatocellular carcinoma cells and MCF-7 human breast adenocarcinoma cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. PC3 human prostate cancer cells, SW480 human colon cancer cells and QGY-7701 human hepatocellular carcinoma cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum.
TransfectionCalcium–phosphate precipitation and Lipofectamine 2000 were used to trans-fect cells as described previously (11,25). Calcium–phosphate precipitation
was used to transfect DNA into HEK293T cells. Briefly, cells were seeded in six-well plates 24 h before transfection. Plasmids (1 µg) in 50 µl of 0.25 M CaCl2 was mixed with 50 µl of 2× N,N-bis[2-hydroxyethyl] -2-aminoethanesulfonic acid-buffered saline (50 mM BES, 280 mM NaCl, 1.5 mM Na2HPO4·2H2O, pH 7.0), and the mixture solution was allowed to stand for 5 min at room tempera-ture. The mixture was added to the medium and incubated for 12 h. Other cell lines were transfected by Lipofectamine 2000 according to the manufacturer’s instruction. Briefly, Lipofectamine 2000 transfection reagent was mixed with DNA diluted in Opti-MEM I Reduced Serum Medium. After 20 min incuba-tion at room temperature, the complexes were applied to cells and incubated for 24–36 h.
Western blottingThe experiments were performed as described previously (25). Cell lysates were boiled in sodium dodecyl sulfate (SDS) sample loading buffer, resolved by 10% SDS–polyacrylamide gel electrophoresis (SDS–PAGE) and trans-ferred to nitrocellulose. The membranes were blocked in 5% milk in Tris-buffered saline and Tween 20 (TBST; 10 mM Tris–HCl [pH 8.0], 150 mM NaCl, 0.05% Tween 20) for 1 h at room temperature. After washing twice with TBST, the membranes were incubated with appropriate primary antibodies in TBST for 1 h and then washed twice, probed with horseradish peroxide-linked anti-immunoglobulin for 1 h at room temperature. After three washes with TBST, immunoreactive products were visualized using enhanced chemilumi-nescence reagents and autoradiography.
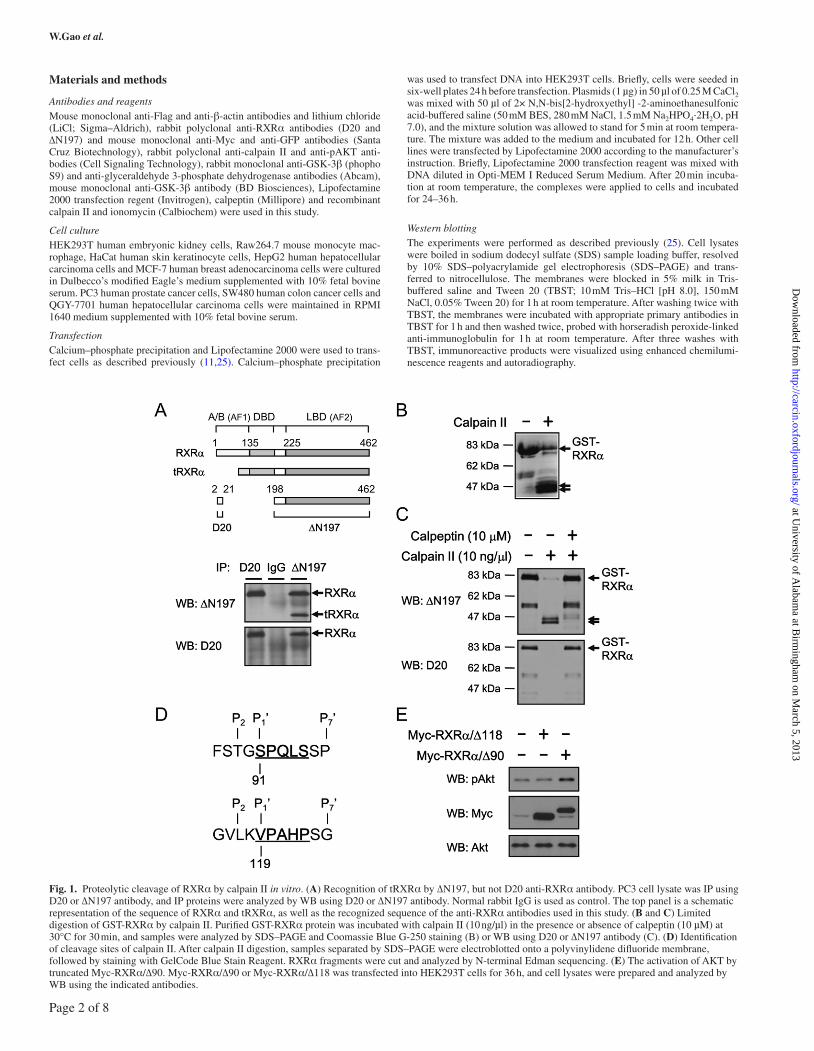
Fig. 1. Proteolytic cleavage of RXRα by calpain II in vitro. (A) Recognition of tRXRα by ∆N197, but not D20 anti-RXRα antibody. PC3 cell lysate was IP using D20 or ∆N197 antibody, and IP proteins were analyzed by WB using D20 or ∆N197 antibody. Normal rabbit IgG is used as control. The top panel is a schematic representation of the sequence of RXRα and tRXRα, as well as the recognized sequence of the anti-RXRα antibodies used in this study. (B and C) Limited digestion of GST-RXRα by calpain II. Purified GST-RXRα protein was incubated with calpain II (10 ng/µl) in the presence or absence of calpeptin (10 µM) at 30°C for 30 min, and samples were analyzed by SDS–PAGE and Coomassie Blue G-250 staining (B) or WB using D20 or ∆N197 antibody (C). (D) Identification of cleavage sites of calpain II. After calpain II digestion, samples separated by SDS–PAGE were electroblotted onto a polyvinylidene difluoride membrane, followed by staining with GelCode Blue Stain Reagent. RXRα fragments were cut and analyzed by N-terminal Edman sequencing. (E) The activation of AKT by truncated Myc-RXRα/∆90. Myc-RXRα/∆90 or Myc-RXRα/∆118 was transfected into HEK293T cells for 36 h, and cell lysates were prepared and analyzed by WB using the indicated antibodies.
Page 2 of 8
at University of A
labama at B
irmingham
on March 5, 2013
http://carcin.oxfordjournals.org/D
ownloaded from
Modulation of tRXRα production by calpain II and GSK-3β
Co-immunoprecipitationCells were harvested in lysis buffer (10 mM Tris [pH 7.4], 150 mM NaCl, 1% Triton X-100, 5 mM ethylenediaminetetraacetic acid, containing pro-tease inhibitors). Lysate was incubated with 1 µg antibody at 4°C for 2 h. Immunocomplexes were then precipitated with 30 µl of protein A/G-sepharose. After an extensive washing with lysis buffer, the beads were boiled in SDS sample loading buffer and assessed by western blotting (WB).
In vitro proteolysis assayPurified glutathione-S-transferase (GST)-RXRα protein was incubated with 10 ng/µl recombinant rat calpain II in a reaction buffer containing 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (pH 7.5), 5 mM CaCl2, 50 mM KCl, 2 mM MgCl2 and 1 mM dithiothreitol at 30°C for 30 min in the presence or absence of 10 µM calpeptin. The reactions were stopped by an addition of SDS–PAGE sample loading buffer, and the reaction mixtures were then loaded on a 10% SDS–PAGE gel. The cleaved fragments of RXRα were examined by Coomassie Blue staining and WB.
Edman N-terminal sequencingN-terminal Edman sequencing was performed on an ABI Procise 492 protein sequencer using standard procedure. GST-RXRα fragments generated by cal-pain II cleavage in vitro were resolved by 10% SDS–PAGE gel, followed by electroblotting to a polyvinylidene difluoride membrane. The membrane was stained with GelCode Blue Stain Reagent (Thermo Scientific), and the two bands were cut and subjected to Edman degradation.
RNA interferenceSmall interfering RNA (siRNA; 50 pmol) was transfected into cells grown in 12-well plate using Lipofectamine 2000 reagent according to the manufacturer’s recommendations. Briefly, 1 day before transfection, cells were plated in 12-well plates at appropriate concentration in order to reach 30–50% confluent at the time of transfection. Lipofectamine 2000 (2.5 µ l) and siRNA (50 pmol) were gently mixed with 125 µl Opti-MEM I Reduced Serum Medium, respectively, and in 5 min, the diluted siRNA and Lipofectamine 2000 were combined and mixed thoroughly. After 20 min of incubation, the siRNA/Lipofectamine complexes were applied to cells for transfection. Cells were treated with LiCl for 36 h and then harvested for immunoblotting assay. The siRNA sequences (Sigma) against human GSK-3β and non-targeting sequence were as follows: GUAUUGCAGGACAAGAGAUdTdT and UUCUCCGAACGUGUCACGUTT.
Results
Calpain II cleaves RXRα in vitroTo determine whether calpain II could cleave RXRα to gener-ate tRXRα known to activate the AKT signaling pathway (11), we performed the in vitro protease assay by using purified GST-RXRα fusion protein. Two anti-RXRα antibodies, D20 and ∆N197, were used for this study (Figure 1A). The D20 anti-RXRα antibody recog-nizes the N-terminal 2–20 amino acids of RXRα, whereas the ∆N197 anti-RXRα antibody recognizes the C-terminal 198–462 sequence of RXRα. As tRXRα, which activates the PI3K/AKT pathway, is a
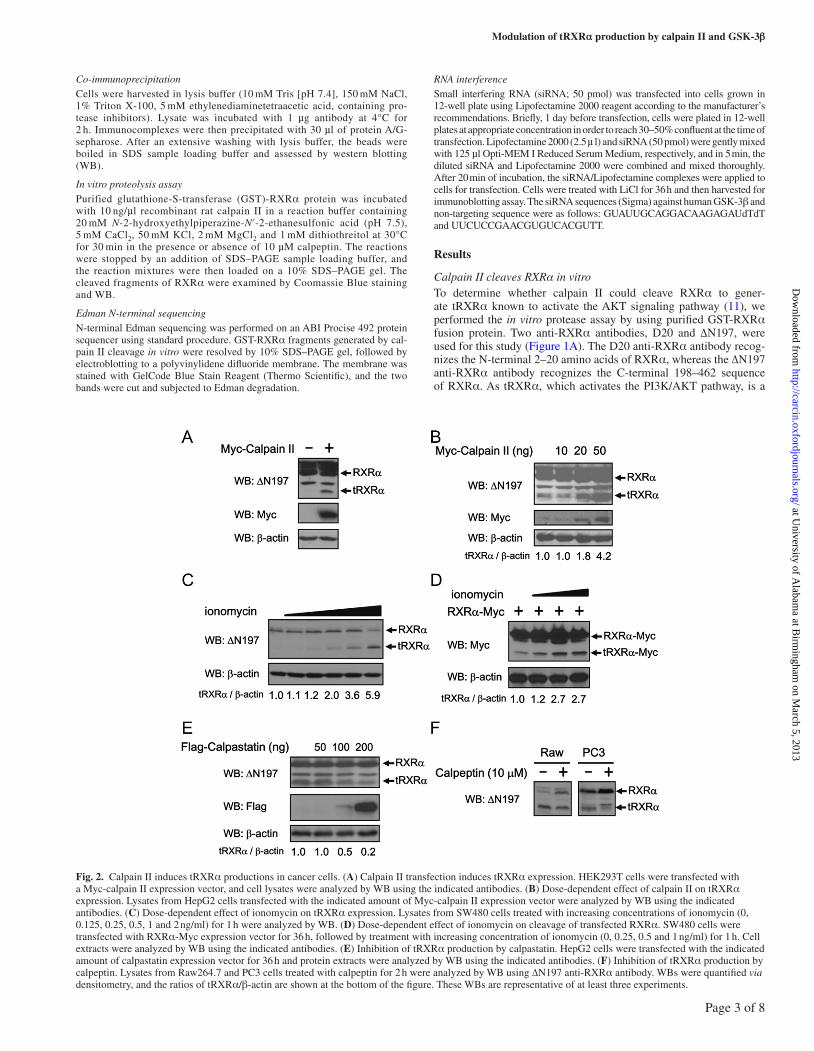
Fig. 2. Calpain II induces tRXRα productions in cancer cells. (A) Calpain II transfection induces tRXRα expression. HEK293T cells were transfected with a Myc-calpain II expression vector, and cell lysates were analyzed by WB using the indicated antibodies. (B) Dose-dependent effect of calpain II on tRXRα expression. Lysates from HepG2 cells transfected with the indicated amount of Myc-calpain II expression vector were analyzed by WB using the indicated antibodies. (C) Dose-dependent effect of ionomycin on tRXRα expression. Lysates from SW480 cells treated with increasing concentrations of ionomycin (0, 0.125, 0.25, 0.5, 1 and 2 ng/ml) for 1 h were analyzed by WB. (D) Dose-dependent effect of ionomycin on cleavage of transfected RXRα. SW480 cells were transfected with RXRα-Myc expression vector for 36 h, followed by treatment with increasing concentration of ionomycin (0, 0.25, 0.5 and 1 ng/ml) for 1 h. Cell extracts were analyzed by WB using the indicated antibodies. (E) Inhibition of tRXRα production by calpastatin. HepG2 cells were transfected with the indicated amount of calpastatin expression vector for 36 h and protein extracts were analyzed by WB using the indicated antibodies. (F) Inhibition of tRXRα production by calpeptin. Lysates from Raw264.7 and PC3 cells treated with calpeptin for 2 h were analyzed by WB using ∆N197 anti-RXRα antibody. WBs were quantified via densitometry, and the ratios of tRXRα/β-actin are shown at the bottom of the figure. These WBs are representative of at least three experiments.
Page 3 of 8
at University of A
labama at B
irmingham
on March 5, 2013
http://carcin.oxfordjournals.org/D
ownloaded from

W.Gao et al.
C-terminal RXRα product, it could be detected by ∆N197, but not by D20 anti-RXRα antibody (11). Figure 1A showed that ∆N197, but not D20 anti-RXRα antibody, recognized tRXRα produced in PC3 prostate cancer cells, confirming the specificity of the antibod-ies. When GST-RXRα purified from Escherichia coli, with molecu-lar weight of ~80 kDa, was incubated with recombinant calpain II, it was cleaved, generating two proteolytic fragments with the apparent molecular weight of ~47 and ~45 kDa, as revealed by SDS–PAGE and Coomassie Blue staining (Figure 1B). Because the fragments were recognized by ∆N197, but not D20 anti-RXRα antibody (Figure 1C), they likely represented tRXRα lacking N-terminal sequences. The cleavage of GST-RXRα by calpain II was specific as its effect was completely inhibited by calpeptin, the selective calpain II inhibitor (Figure 1C). These results are consistent with previous observation (20), demonstrating that calpain II is capable of cleaving RXRα at its N-terminal region.
To determine the cleavage sites of calpain II, two calpain II-generated proteolytic products of RXRα were purified. Edman N-terminal sequencing analysis revealed two peptide sequences of SPQLS and VPAHP, which correspond to two cleavage sites STG90↓S91PQLS and VLK118↓V119PAHP in RXRα, respectively (Figure 1D). Although there is no consensus sequence for calpain cleavage sites, there are residue preferences around the cleavage sites from P4′ to P7′ (26), of which the preferred residues at the P2 position are Leu, Thr and Val, and the P1 position are Lys, Tyr and Arg as well as the P1′ position are Ser, Thr and Ala. The sequences surrounding the cleavage sites of calpain II in RXRα partially follow the rule, with Thr89 and Leu117 meeting with the P2 preferences, Lys118 meeting with the P1 prefer-ences and Ser91 meeting with the P1′ preferences (Figure 1D).
To study whether calpain II-cleaved RXRα fragments, RXRα/∆90 and RXRα/∆118, could activate the AKT signaling pathway, the cor-responding tRXRα complementary DNAs were cloned into an expres-sion vector containing Myc epitope and transfected into HEK293T cells. Immunoblotting demonstrated that transfection of RXRα/∆90, but not RXRα/∆118, resulted in enhanced expression of phospho-AKT, but not total AKT (Figure 1E). Thus, RXRα/∆90 could activate AKT in cells, suggesting that calpain II cleavage of RXRα contributes to production of tRXRα for regulating AKT signaling pathway.
Regulation of tRXRα production by calpain II in cellsTo study the role of calpain II in tRXRα production in cells, we determined whether altered expression of calpain II could regulate tRXRα production. The level of tRXRα was undetectable in HEK293T cells under conditions used. However, transfection of calpain II strongly enhanced tRXRα expression (Figure 2A). In HepG2 hepatocellular carcinoma cells, transfection of calpain II expression vector also dose dependently increased the level of tRXRα protein (Figure 2B). Thus, tRXRα production is correlated with calpain II expression. We next examined the effect of calpain II activation on tRXRα production. The calcium ionophore ionomycin is known to activate endogenous calpain by raising the intracellular level of calcium (27,28). Treatment of cancer cells with ionomycin led to an increase of endogenous tRXRα level in a dose-dependent manner (Figure 2C). Proteolytic cleavage of transfected RXRα was also enhanced by ionomycin (Figure 2D). The production of tRXRα was inhibited by transfection of calpastatin, the endogenous calpain II inhibitor (14) (Figure 2E), and by treatment of cells with the selective calpain II inhibitor calpeptin (Figure 2F). Thus, the expression and activation of calpain II play an important role in tRXRα production in cancer cells.
To further study the role of calpain II, we examined whether there was an interaction between calpain II and RXRα by co-immunopre-cipitation assays. Thus, Flag-RXRα expression vector was co-trans-fected with Myc-calpain I or Myc-calpain II expression vectors into HepG2 cells. Flag-RXRα was immunoprecipitated (IP) by anti-Flag antibody, and its interaction with calpain I or calpain II was exam-ined by immunoblotting using anti-Myc antibody. Our co-immuno-precipitation assays showed that Myc-calpain II, but not Myc-calpain
I, could interact with Flag-RXRα (Figure 3A). Conversely, trans-fected GFP-RXRα was co-immunoprecipitated by anti-Myc antibody when it was co-transfected with Myc-calpain II into HEK293T cells (Figure 3B). Transfection of RXRα expression vector into PC3 cells also resulted in its co-immunoprecipitation of endogenous calpain II (Figure 3C). Together, these results demonstrate that calpain II inter-acts with RXRα, further supporting its role in RXRα cleavage and tRXRα production.
GSK-3β negatively regulates tRXRα productionIn studying the regulation of tRXRα production in cancer cells, we evaluated the effect of chemical inhibitors of several protein kinases including Jun N-terminal kinase, extracellular signal-regulated kinase, p38, PI3K and GSK-3β. Our results showed that LiCl, an inhibitor of GSK-3β (29), could upregulate tRXRα productions. In HepG2 cells, the level of tRXRα protein gradually increased with the treatment of 20 mM LiCl in a time-dependent manner, with the highest tRXRα level occurring at 24 h post-treatment (Figure 4A). The optimal concentration of LiCl required for induction of tRXRα production was about 20 mM (Figure 4B), which is the concentration widely used for inhibiting GSK-3β (30,31). This inducing effect of LiCl on tRXRα production was also observed in QGY-7701 liver cancer cells (Figure 4C) and several other cancer cell lines including HaCat keratinocytes, MCF-7 breast cancer and SW480 colon cancer cells (Figure 4D). As expected, exposure of cancer cells to LiCl resulted in inhibition of GSK-3β activation, revealed by induction of GSK-3β serine 9 phosphorylation by LiCl (Figure 4B and D). To further address the role of GSK-3β on tRXRα production, we transfected
Fig. 3. Interaction of calpain II with RXRα. (A) HepG2 cells were transfected with Flag-RXRα expression vector alone or together with Myc-calpain I or Myc-calpain II expression vector for 36 h. Cell lysates prepared were IP by anti-Flag antibody, and immunoprecipitates were analyzed by WB using anti-Myc or anti-Flag antibody. (B) HEK293T cells were transfected with GFP-RXRα alone or together with Myc-calpain II expression vector. Cell lysates were IP with anti-Myc antibody, and the immunoprecipitates were analyzed by WB using anti-GFP or anti-Myc antibody. (C) PC3 cells were transfected with RXRα-Myc, and cell extracts were IP using anti-Myc antibody followed by WB using anti-calpain II antibody.
Page 4 of 8
at University of A
labama at B
irmingham
on March 5, 2013
http://carcin.oxfordjournals.org/D
ownloaded from

Modulation of tRXRα production by calpain II and GSK-3β
GSK-3β expression vector into HepG2 cells. Immunoblotting showed that transfection of GSK-3β potently inhibited the production of tRXRα in a dose-dependent manner (Figure 4E) and the inducing effect of LiCl on tRXRα production (Figure 4F). We also examined the effects of GSK-3β downregulation on tRXRα production by siRNA approach. Transfection of GSK-3β siRNA into HepG2 cells, which reduced the level of GSK-3β, enhanced the expression of tRXRα (Figure 4G). In addition, transfection of GSK-3β siRNA, but not control siRNA, impaired the effect of LiCl on inducing tRXRα production (Figure 4G). Thus, GSK-3β activation negatively regulates the expression of tRXRα protein.
GSK-3β inhibits tRXRα production by downregulation of calpain IIThe inhibition of tRXRα production by GSK-3β prompted us to investigate whether its inhibitory effect involved calpain II. We first examined the effect of LiCl on calpain II expression. Our immunoblotting assay showed that treatment of HepG2 cells with LiCl induced the expression of calpain II (Figure 5A). The expression level of calpastatin was not altered in the presence or absence of LiCl, demonstrating the specific effect of LiCl on cal-pain II expression (Figure 5A). To address the role of GSK-3β, cells were transfected with GSK-3β expression vector and exam-ined for calpain II expression. Figure 5B showed that transfection of GSK-3β expression vector inhibited the expression of calpain
II in a dose-dependent manner. Knockdown of GSK-3β by GSK-3β siRNA transfection also increased the expression of calpain II and production of tRXRα (Figure 5C). Thus, GSK-3β plays a critical role in the regulation of calpain II expression in cancer cells. We also examined the effect of LiCl on AKT activation and found that treatment of cells with LiCl induced AKT activation in a dose-dependent manner, which was closely correlated with decreased GSK-3β activation, increased calpain II expression and enhanced tRXRα production (Figure 5D). Together, our results suggest a GSK-3β/calpain II/tRXRα cascade for AKT activation in cancer cells.
Discussion
Studying proteases responsible for RXRα cleavage and signaling pathways regulating tRXRα production is critical for understanding the newly identified tRXRα-dependent survival pathway in cancer cells. Here, we provide evidence that calpain II could cleave RXRα, resulting in the production of tRXRα in vitro and in vivo. We also identified its cleavage sites in RXRα and showed that one of the cleavage products represented tRXRα protein with its ability to activate AKT in cancer cells. Moreover, we found that GSK-3β negatively regulates tRXRα production by inhibiting the expression of calpain II. These results, therefore, identified a GSK-3β/calpain
Fig. 4. GSK-3β inhibits tRXRα production. (A) Time-course effect of LiCl on tRXRα production. Lysates from HepG2 cells treated with LiCl for the indicated time were analyzed by WB using the indicated antibodies. (B–D) Dose-dependent effect of LiCl on tRXRα expression in different cell types. HepG2 (B), QGY-7701 (C) and HaCat, MCF-7, SW480 (D) cells were treated with LiCl for the indicated concentrations for 24 h. Lysates were examined by WB using the indicated antibodies. (E) Inhibition of tRXRα production by GSK-3β. Lysates from HepG2 cells transfected with the indicated amount of Flag-GSK-3β expression vector were analyzed by WB using the indicated antibodies. (F) GSK-3β antagonizes the effect of LiCl on tRXRα productions. HepG2 cells were transfected with GSK-3β expression vector, and 24 h later, cells were treated with LiCl for 24 h. WB was performed to examine the protein expression using the indicated antibodies. (G) Induction of tRXRα production by knocking down GSK-3β. HepG2 cells were transfected with control siRNA or GSK-3β siRNA for 36 h, and cells were then treated with LiCl for 24 h. WB was performed using the indicated antibodies. The bands were quantified by densitometry, and the ratios of tRXRα to β-actin or glyceraldehyde 3-phosphate dehydrogenase are indicated. These WBs are representative of at least three experiments.
Page 5 of 8
at University of A
labama at B
irmingham
on March 5, 2013
http://carcin.oxfordjournals.org/D
ownloaded from

W.Gao et al.
II/tRXRα axis for regulating the PI3K/AKT signaling pathway in cancer cells.
Previous studies showed that calpain II was involved in the proteo-lytic cleavage of RXRα. Results obtained in this study confirm that calpain II could cleave RXRα, resulting in the production of tRXRα in vitro and in vivo. Cleavage of RXRα by calpain II in vitro generates two C-terminal fragments of RXRα with apparent molecular masses of ~47 and ~44 kDa (20). However, the cleavage sites of calpain II in RXRα were not determined, and whether calpain II cleaves RXRα in cells and whether the proteolytic products of RXRα are functional remained unknown. Our in vitro cleavage assay showed that cal-pain II could cleave E. coli expressed GST-RXRα to generate two C-terminal RXRα fragments with molecular weight ~47 and ~44 kDa (Figure 1B and C). By using Edman N-terminal sequencing, we iden-tified the cleavage sites of calpain II in RXRα, which are located at ThrGly90↓Ser91Pro and LeuLys118↓Val119Pro in the A/B region of RXRα (Figure 1D). When we evaluated the effect of the resulting C-terminal RXRα products, RXRα/∆90 and RXRα/∆118, for their activation of AKT, we found that RXRα/∆90, but not RXRα/∆118, could activate AKT in cancer cells (Figure 1E). These results are consistent with our previous finding that RXRα/∆80 lacking 80 N-terminal amino acids, but not RXRα/∆100 lacking 100 N-terminal amino acids, was able to activate AKT in cancer cells (11). Thus, tRXRα product generated by calpain II cleavage of RXRα at Gly90↓Ser91 likely represents tRXRα that we reported previously (11). Interestingly, calpain II cleavage of AR results in expression of several truncated AR molecules and one of them acts as an androgen-independent isoform, promoting hor-mone-independent growth of prostate tumor cells (19). Thus, calpain II-mediated proteolysis likely represents an important mechanism that regulates the activities of nuclear receptors.
The notion that calpain II activation contributes to the genera-tion of oncogenic tRXRα protein was further supported by our data showing that calpain II expression and activation were critical for the production of tRXRα in various cancer cell lines. Overexpression of calpain II increased the production of tRXRα in different cell types (Figure 2A and B), and the activation of calpain II by iono-mycin led to an enhanced expression of tRXRα (Figure 2C and D). Consistently, inhibition of calpain II activation by calpain inhibitors calpastatin and calpeptin reduced tRXRα production (Figure 2E and F). Moreover, our co-immunoprecipitation assays showed that calpain II, but not calpain I, could interact with RXRα in cells (Figure 3). These results convincingly demonstrate that calpain II is one of the proteases responsible for production of tRXRα, which in turn acts non-genomically to activate the PI3K/AKT signaling pathway in can-cer cells. Calpain expression and activity are often altered during tum-origenesis (14,32). Overexpression of calpain II and downregulation of calpastatin are often observed in different types of cancer cells, and calpain inhibitors have shown promising anticancer effects in vitro and in animals (14,33,34). Calpain-mediated proteolytic cleavages of various substrates including AR, inhibitors of nuclear factor-κB, focal adhesion protein and several proto-oncogenes have been implicated in tumor pathogenesis (16,28,35). Our present finding that calpain II is involved in the generation of tRXRα together with our previous observation that tRXRα could promote cancel cell growth in vitro and in animals by activating TNFα-dependent PI3K/AKT signaling path-way suggest a new molecular event contributing to the tumorigenic effect of the altered calpain system in cancer.
Our results demonstrate that GSK-3β could serve as a nega-tive regulator of tRXRα production in cancer cells by inhibiting calpain II expression. Several lines of evidence were presented to
Fig. 5. GSK-3β downregulates calpain II expression. (A) Induction of calpain II expression by LiCl. Lysates from HepG2 cells treated with 5 or 10 mM LiCl for 24 h were analyzed by WB using the indicated antibodies. (B) Inhibition of calpain II expression by GSK-3β. HepG2 cells were transfected with the indicated amount of Flag-GSK-3β expression vector for 36 h, and lysates were analyzed by WB using the indicated antibodies. (C) Induction of calpain II expression by GSK-3β siRNA. HepG2 cells were transfected with control siRNA or GSK-3β siRNA, and 36 h later, cells were treated with LiCl for 24 h. WB was performed to examine the protein expression using the indicated antibodies. (D) AKT activation by LiCl. Lysates from HepG2 cells treated with 10 or 20 mM LiCl for 24 h were analyzed by WB using the indicated antibodies. The bands were quantified by densitometry, and the ratios of tRXRα to β-actin were calculated and indicated. All WBs are representative of at least three independent experiments.
Page 6 of 8
at University of A
labama at B
irmingham
on March 5, 2013
http://carcin.oxfordjournals.org/D
ownloaded from

Modulation of tRXRα production by calpain II and GSK-3β
illustrate the role of GSK-3β in tRXRα production. First, treat-ment of cancer cells with LiCl, an inhibitor of GSK-3β (29), induced expression of tRXRα (Figure 4A–D). Second, transfection of GSK-3β could inhibit tRXRα production (Figure 4E), whereas knocking down GSK-3β by siRNA approach induced tRXRα expression (Figure 4G). Furthermore, overexpression of GSK-3β could antagonize the inducing effect of LiCl on tRXRα produc-tion (Figure 4F). In studying the possible mechanism by which GSK-3β inhibited tRXRα production, we provided evidence that its effect was due to its inhibition of calpain II expression. Thus, LiCl treatment (Figure 5A) and transfection of GSK-3β siRNA (Figure 5C) resulted in an increase of calpain II expression. In contrast, transfection of GSK-3β dose dependently inhibited the expression of calpain II (Figure 5B). The molecular mechanism for downregulating calpain II expression by GSK-3β is currently unknown, but may involve its phosphorylation of calpain II lead-ing to its ubiquitination and degradation, which is currently under investigation.
GSK-3β is a multifunctional serine/threonine kinase, which unlike most protein kinases is constitutively active in resting cells and undergoes a rapid and transient inhibition in response to a number of external signals (31,36). Dysregulation of GSK-3β has been implicated in a range of human pathologies including diabetes (37), cardiovascular disease (38) and neurodegenerative diseases (23), although its role in tumorigenesis remains controversial (24,39). GSK-3β regulates diverse substrates and signaling pathways. One of the most well-known substrates of GSK-3β is β-catenin, which is targeted for ubiquitin-mediated degradation by GSK-3β leading to inhibition of Wnt/β-catenin signaling (40). In addition to β-catenin, activities of p53, AP-1 and NF-κB are also regulated by GSK-3β (24). Our results identify RXRα as another downstream effector of GSK-3β action in cancer cells. The fact that GSK-3β activation inhibited the production of tRXRα known to activate the PI3K/AKT signaling pathway (11) suggests that inhibition of RXRα cleavage by GSK-3β may account for the tumor suppressive effect of GSK-3β. Interestingly, our results showed that inhibition of GSK-3β by LiCl could profoundly activate AKT (Figure 5D), which is known to inactivate GSK-3β by phosphorylating Ser9of GSK-3β (41). These results suggest a GSK-3β-tRXRα axis for amplifying AKT signaling in cancer cells.
Funding
Fundamental Research Funds for the Central Universities (2010111081); the National Natural Science Foundation of China (NSFC-91129302 and NSFC- 31271453); the Tobacco-Related Disease Research Program (15FT-0243); the National Institutes of Health (CA140980 and GM089927) and the United States Army Medical Research and Material Command (W81XWH-11-1-0677).
Conflict of Interest Statement: None declared.
References
1. Ahuja,H.S. et al. (2003) The retinoid X receptor and its ligands: versatile regulators of metabolic function, cell differentiation and cell death. J. Biol. Regul. Homeost. Agents, 17, 29–45.
2. Szanto,A. et al. (2004) Retinoid X receptors: X-ploring their (patho)physi-ological functions. Cell Death Differ., 11 (suppl. 2), S126–S143.
3. Moll,U.M. et al. (2006) p53 and Nur77/TR3 - transcription factors that directly target mitochondria for cell death induction. Oncogene, 25, 4725–4743.
4. Lefebvre,P. et al. (2010) Retinoid X receptors: common heterodimeri-zation partners with distinct functions. Trends Endocrinol. Metab., 21, 676–683.
5. Katagiri,Y. et al. (2000) Modulation of retinoid signalling through NGF-induced nuclear export of NGFI-B. Nat. Cell Biol., 2, 435–440.
6. Li,H. et al. (2000) Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3. Science, 289, 1159–1164.
7. Cao,X. et al. (2004) Retinoid X receptor regulates Nur77/TR3-dependent apoptosis [corrected] by modulating its nuclear export and mitochondrial targeting. Mol. Cell. Biol., 24, 9705–9725.
8. Lin,B. et al. (2004) Conversion of Bcl-2 from protector to killer by interac-tion with nuclear orphan receptor Nur77/TR3. Cell, 116, 527–540.
9. Mey,J. et al. (2007) Effects of inflammatory cytokines IL-1beta, IL-6, and TNFalpha on the intracellular localization of retinoid receptors in Schwann cells. Glia, 55, 152–164.
10. Urban,S. et al. (2002) Intramembrane proteolysis controls diverse sig-nalling pathways throughout evolution. Curr. Opin. Genet. Dev., 12, 512–518.
11. Zhou,H. et al. (2010) NSAID sulindac and its analog bind RXRalpha and inhibit RXRalpha-dependent AKT signaling. Cancer Cell, 17, 560–573.
12. Lu,N. et al. (2012) Antagonist effect of triptolide on AKT activation by truncated retinoid X receptor-alpha. PLoS ONE, 7, e35722.
13. Sorimachi,H. et al. (2001) The structure of calpain. J. Biochem., 129, 653–664.
14. Storr,S.J. et al. (2011) The calpain system and cancer. Nat. Rev. Cancer, 11, 364–374.
15. Glading,A. et al. (2004) Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol. Cell. Biol., 24, 2499–2512.
16. Chen,H. et al. (2010) ERK regulates calpain 2-induced androgen receptor proteolysis in CWR22 relapsed prostate tumor cell lines. J. Biol. Chem., 285, 2368–2374.
17. Shiraha,H. et al. (2002) Activation of m-calpain (calpain II) by epidermal growth factor is limited by protein kinase A phosphorylation of m-calpain. Mol. Cell. Biol., 22, 2716–2727.
18. Pelley,R.P. et al. (2006) Calmodulin-androgen receptor (AR) interaction: calcium-dependent, calpain-mediated breakdown of AR in LNCaP prostate cancer cells. Cancer Res., 66, 11754–11762.
19. Libertini,S.J. et al. (2007) Evidence for calpain-mediated androgen recep-tor cleavage as a mechanism for androgen independence. Cancer Res., 67, 9001–9005.
20. Matsushima-Nishiwaki,R. et al. (1996) Limited degradation of retinoid X receptor by calpain. Biochem. Biophys. Res. Commun., 225, 946–951.
21. Forde,J.E. et al. (2007) Glycogen synthase kinase 3: a key regulator of cel-lular fate. Cell. Mol. Life Sci., 64, 1930–1944.
22. Manoukian,A.S. et al. (2002) Role of glycogen synthase kinase-3 in can-cer: regulation by Wnts and other signaling pathways. Adv. Cancer Res., 84, 203–229.
23. Doble,B.W. et al. (2003) GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell. Sci., 116(Pt 7), 1175–1186.
24. Luo,J. (2009) Glycogen synthase kinase 3beta (GSK3beta) in tumorigen-esis and cancer chemotherapy. Cancer Lett., 273, 194–200.
25. Zhang,L. et al. (2004) Zebrafish Dpr2 inhibits mesoderm induction by pro-moting degradation of nodal receptors. Science, 306, 114–117.
26. Tompa,P. et al. (2004) On the sequential determinants of calpain cleavage. J. Biol. Chem., 279, 20775–20785.
27. Rios-Doria,J. et al. (2003) The role of calpain in the proteolytic cleavage of E-cadherin in prostate and mammary epithelial cells. J. Biol. Chem., 278, 1372–1379.
28. Chan,K.T. et al. (2010) Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK). J. Biol. Chem., 285, 11418–11426.
29. Oreña,S.J. et al. (2000) Inhibition of glycogen-synthase kinase 3 stimulates glycogen synthase and glucose transport by distinct mechanisms in 3T3-L1 adipocytes. J. Biol. Chem., 275, 15765–15772.
30. Beurel,E. et al. (2008) Differential regulation of STAT family members by glycogen synthase kinase-3. J. Biol. Chem., 283, 21934–21944.
31. Frame,S. et al. (2001) GSK3 takes centre stage more than 20 years after its discovery. Biochem. J., 359(Pt 1), 1–16.
32. Liu,X. et al. (2004) The role of calpain in oncotic cell death. Annu. Rev. Pharmacol. Toxicol., 44, 349–370.
33. Mataga,M.A. et al. (2012) Anti-breast cancer effects of histone deacetylase inhibitors and calpain inhibitor. Anticancer Res., 32, 2523–2529.
34. Niapour,M. et al. (2008) Regulation of calpain activity by c-Myc through calpastatin and promotion of transformation in c-Myc-negative cells by calpastatin suppression. J. Biol. Chem., 283, 21371–21381.
35. Han,Y. et al. (1999) Tumor necrosis factor-alpha-inducible IkappaBalpha proteolysis mediated by cytosolic m-calpain. A mechanism parallel to the ubiquitin-proteasome pathway for nuclear factor-kappab activation. J. Biol. Chem., 274, 787–794.
36. Grimes,C.A. et al. (2001) The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol., 65, 391–426.
Page 7 of 8
at University of A
labama at B
irmingham
on March 5, 2013
http://carcin.oxfordjournals.org/D
ownloaded from

W.Gao et al.
37. Henriksen,E.J. et al. (2006) Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr. Drug Targets, 7, 1435–1441.
38. Oudit,G.Y. et al. (2004) The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J. Mol. Cell. Cardiol., 37, 449–471.
39. Kim,L. et al. (2000) GSK3, a master switch regulating cell-fate specifica-tion and tumorigenesis. Curr. Opin. Genet. Dev., 10, 508–514.
40. Wu,D. et al. (2010) GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem. Sci., 35, 161–168.
41. Rössig,L. et al. (2002) Glycogen synthase kinase-3 couples AKT-dependent signaling to the regulation of p21Cip1 degradation. J. Biol. Chem., 277, 9684–9689.
Received October 28, 2012; revised January 11, 2013; accepted January 26, 2013
Page 8 of 8
at University of A
labama at B
irmingham
on March 5, 2013
http://carcin.oxfordjournals.org/D
ownloaded from





























