Embed Size (px)
Citation preview

CLINICOPATHOLOGIC CONFERENCE
Renal Failure, Dyspnea and Anemia in a 57 Year Old Woman
Stenographic reports of weekly clinicopathologic conferences held in Barnes and Wohl Hospitals are published in each issue of the Journal. Members of the Departments of Internal Medicine, Radi- ology, and Pathology of the Washington University School of Medi- cine participate jointly in these conferences. Kenneth M. Ludmerer, M.D., and John M. Kissane, M.D., are the editors of this feature.
A 57 year old black female was admitted to Jewish Hospital of St. Louis
February 13, 1980, with dyspnea and anemia. For many years, the patient had had severe hypertension. Six years
prior to admission, administration of guadethidine and high doses of
hydralazine and alpha-methyldopa was begun for this problem. Despite
treatment, symptoms of congestive heart failure developed, and di-
goxin therapy was initiated. Two years prior to admission, arthralgias
developed, particularly in the shoulders, elbows and hands. The
erythrocyte sedimentation rate (ESR) was normal, and antinuclear
antibodies (ANA) were absent. Naproxen, 200 mg four times a day,
was prescribed but did not relieve her symptoms. Six months prior to
admission, during a routine outpatient visit, her hemoglobin level was
11.5 g/d1 and her serum creatinine 1.2 mg/dl. Results of urinalysis at
that time were normal. Two weeks prior to admission, she noted
lightheadedness and dyspnea on exertion. She was seen again by her
physician, who found her hemoglobin level to be 5.5 g/dl, and hospi-
talization was promptly arranged. She denied overt blood loss, easy bruising, fever, sweats, chest pain, alopecia or photophobia.
Physical examination on admission revealed a temperature of
37.5’C, a blood pressure of 120/70 mm Hg without orthostasis, a pulse rate of 76/min and a respiratory rate of lG/min. The conjunctivae
were pale and the sclerae nonicteric. No retinal hemorrhages were
seen. The lung examination was noncontributory. The cardiac apical
impulse was enlarged and displaced to the left. The first and second
heart sounds were normal; a prominent fourth heart sound was heard;
and a grade 316 systolic murmur was present along the left sternal
border. The spleen was palpable 2 cm below the left costal margin. Rectal, pelvic, joint and neurologic examinations were unremarkable. No occult blood could be detected in the stool.
Laboratory work on admission was remarkable for a hemoglobin level of 5.0 g/dl, a hematocrit value of 16.1 percent, a mean corpus-
cular volume of 72 p3, a white blood cell count of 7,000/mm3, a re- ticulocyte count of 6.2 percent and a platelet count of 248,000/mm3. The urinalysis revealed a pH of 5, 2+ protein, moderate blood and red cell casts. The serum electrolytes were within normal limits. Other
test results included a glucose level of 108 mg/dl, creatinine level of
878 November 1981 The American Journal of Medicine Volume 71
CLINICOPATHOLOGIC CONFERENCE
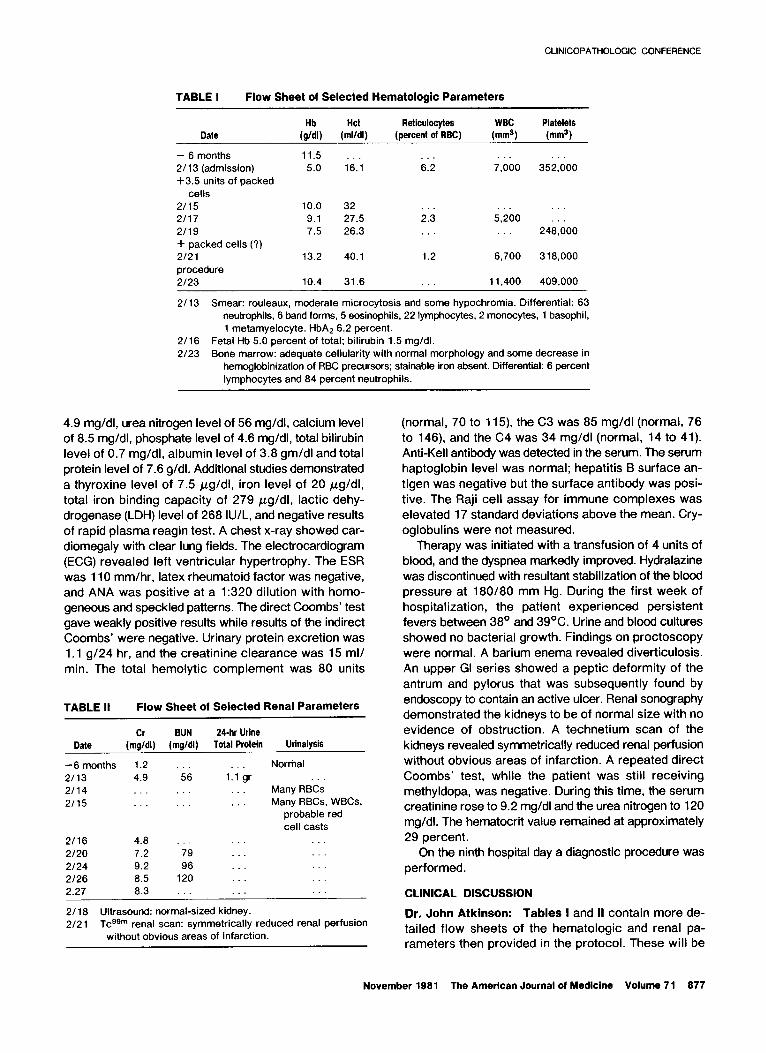
TABLE I Flow Sheet of Selecied Hematologic Parameters
Reticulocytes WBC Platelets Date (;I) (rzl) (percent of RBC) (mm3) (mm3)
- 6 months 11.5 2713 (admission) 5.0 l6.; 62 7,000 352:;oo +3.5 units of packed
cells 2115 10.0 32 2117 9.1 27.5 23 5,&o : : : 2/19 7.5 26.3 248,000 i- packed cells (?) 2/21 13.2 40.1 1.2 6,700 318,000 procedure 2123 10.4 31.6 11,400 409,000
2/13 Smear: rouleaux, moderate microcytosis and some hypochromia. Differential: 63 neutrophils, 6 band forms, 5 eosinophils, 22 lymphocytes, 2 monocytes, 1 basophil, 1 metamyelocyte. HbAs 6.2 percent.
2/16 Fetal Hb 5.0 percent of total; bilirubin 1.5 mg/dl. 2/23 Bone marrow: adequate cellularity with normal morphology and some decrease in
hemoglobinization of RBC precursors: stainable iron absent. Differential: 6 percent lymphocytes and 84 percent neutrophils.
4.9 mg/dl, urea nitrogen level of 56 mg/dl, calcium level
of 8.5 mg/dl, phosphate level of 4.6 mg/dl, total bilirubin level of 0.7 mg/dl, albumin level of 3.8 gm/dl and total
protein level of 7.6 g/dl. Additional studies demonstrated
a thyroxine level of 7.5 pgldl, iron level of 20 pg/dl,
total iron binding capacity of 279 pg/dl, lactic dehy-
drogenase (LDH) level of 268 IWL, and negative results
of rapid plasma reagin test. A chest x-ray showed car-
diomegaly with clear lung fields. The electrocardiogram
(ECG) revealed left ventricular hypertrophy. The ESR
was 110 mm/hr, latex rheumatoid factor was negative, and ANA was positive at a 1:320 dilution with homo-
geneous and speckled patterns. The direct Coombs’ test
gave weakly positive results while results of the indirect
Coombs’ were negative. Urinary protein excretion was
1.1 g/24 hr, and the creatinine clearance was 15 ml/
min. The total hemolytic complement was 80 units
TABLE II Flow Sheet of Selected Renal Parameters
Date 24-hr Urine
Total Protein Urinalysis
-6 months 2113 2114 2115
2116 2/20 2124 2126
1.2 Norrhal 4.9 ‘56 “. 1.1 gr
Many RBCs Many RBCs, WBCs,
probable red cell casts
4.8 7.2 ‘79 9.2 96 8.5 120
2.27 8.3 . CLINICAL DISCUSSION
2/ 18 Ultrasound: normal-sized kidney. 2/21 Tcggm renal scan: symmetrically reduced renal perfusion
without obvious areas of infarction.
(normal, 70 to 115) the C3 was 85 mg/dl (normal, 76
to 146) and the C4 was 34 mg/dl (normal, 14 to 41).
Anti-Kell antibody was detected in the serum. The serum
haptoglobin level was normal; hepatitis B surface an-
tigen was negative but the surface antibody was posi-
tive. The Raji cell assay for immune complexes was
elevated 17 standard deviations above the mean. Cry-
oglobulins were not measured.
Therapy was initiated with a transfusion of 4 units of
blood, and the dyspnea markedly improved. Hydralazine
was discontinued with resultant stabilization of the blood
pressure at 180180 mm Hg. During the first week of
hospitalization, the patient experienced persistent
fevers between 38’ and 39OC. Urine and blood cultures
showed no bacterial growth. Findings on proctoscopy
were normal. A barium enema revealed diverticulosis.
An upper GI series showed a peptic deformity of the
antrum and pylorus that was subsequently found by
endoscopy to contain an active ulcer. Renal sonography demonstrated the kidneys to be of normal size with no
evidence of obstruction. A technetium scan of the
kidneys revealed symmetrically reduced renal perfusion
without obvious areas of infarction. A repeated direct
Coombs’ test, while the patient was still receiving
methyldopa, was negative. During this time, the serum creatinine rose to 9.2 mg/dl and the urea nitrogen to 120
mg/dl. The hematocrit value remained at approximately
29 percent. On the ninth hospital day a diagnostic procedure was
performed.
Dr. John Atkinson: Tables I and II contain more de-
tailed flow sheets of the hematologic and renal pa- rameters then provided in the protocol. These will be
November 1981 The American Journal of Medicine Volume 71 877

RENAL FAILURE, DYSPNEA AND ANEMIA
useful to us later. Right now, I would like to discuss
certain aspects of the patient’s history.
First, she had had severe high blood pressure for six
years. On admission, her antihypertensive medications
consisted of guanethidine (15 mg four times a day),
hydralazine (100 mg four times a day), alpha-methyl-
dopa (500 mg four times a day) and propranolol(l0 mg
four times a day). We have no record of her blood
pressure readings during this time, but it should be noted
that she was under a physician’s care and that the hy-
pertension was carefully monitored and treated.
Second, congestive heart failure developed in 1978,
and digoxin and a diuretic were administered. We do not
know the basis for this diagnosis. I suspect that her
physician believed she had hypertensive cardiovascular
disease. Since subsequently we will be entertaining the
diagnosis of systemic lupus erythematosus (SLE), the possibility of pleural-pericardial and myocardial in-
volvement secondary to this disease becomes an al-
ternative explanation of her heart failure.
Third, arthralgias in her shoulders, elbows and hands
developed two years before admission. The location of
her symptoms differed from that of the usual “aches and
pains” syndrome seen commonly in outpatient prac-
tice-a syndrome that much more often involves the
low back, knees and hips. Therefore, her complaints,
together with the knowledge she was taking hydralazine,
raise the specter of an underlying rheumatic disease.
Her physician apparently had similar thoughts and or-
dered, in my opinion, the two best screening tests in this situation: an ESR and an ANA test. The results of these
tests were both normal, and at the time it was appro-
priate not to undertake further evaluation. Despite
medication, however, her arthralgias persisted.
Fourth, she was lightheaded for at least two weeks
prior to seeing her physician. This delay in seeking
medical consultation may explain the apparent absence
of blood in her stool. After a major upper gastrointestinal
bleed, blood can be detected in the stool for no longer
than one week.
Fifth, some of her physical findings on admission are
surprising. Despite a hematocrit value of 16 percent,
her blood pressure was normal and there was no or- thostatic fall. In addition, on repeated examinations her
pulse rate was only 76/min, her respiratory rate lG/min,
and her temperature 37.5’C. The propranolol may have
slowed her pulse rate, but it should not have prevented an orthostatic fall. Thus, the patient’s cardiovascular
status was surprisingly well compensated, especially in view of her prior history of cardiomegaly and con- gestive heart failure.
Sixth, the absence of retinal hemorrhages represents an important negative finding. If present, such hemor-
rhages could have resulted from either the severe anemia or hypertension. Since the fundus contained no
findings compatible with long-standing hypertension,
it becomes much less likely that high blood pressure
caused her renal and cardiac problems.
Seventh, a cardiologist who examined the patient the
morning after admission noted the presence of a loud
S-3 gallop, a left ventricular heave, a wide pulse pres-
sure, and Quincke’s and Corrigan’s pulses. Accordingly, he suggested that she had high output failure from se-
vere anemia superimposed upon probable hypertensive
heart disease.
Eighth, in addition to the exceedingly low hematocrit
level that prompted hospitalization, the patient’s serum
creatinine was also notably abnormal on admission. Six
months previously, her creatinine level had been 1.2
mg/dl and results of urinalysis normal. The deterioration
in renal function, too, demands explanation.
Ninth, I reviewed previous chest radiographs as well as her ECGs. In 1977, the cardiac silhouette was nor-
mal. In February 1980, the chest x-ray revealed car-
diomegaly with left ventricle predominance. The lung
fields at that time were clear. Although her admission
ECG met the Estes criteria for left ventricular hyper-
trophy, an echocardiogrm that hospitalization was within
normal limits.
Last, although her temperature was normal on ad- mission, she was febrile throughout the remainder of
her lo-day hospitalization. Appropriate cultures were
negative.
As I evaluate the case, we have several tasks. Most
importantly, we need to determine the cause of the anemia and renal failure. In addition, we must construct
a diagnosis that accounts for all patient’s problems.
Finally, we need to predict the procedure that was
performed and the pathologic findings that were noted. We will address these issues in order.
Before turning the discussion over to Dr. Chaplin, I would like to make a few points concerning her anemia.
We know that six months prior to admission she had a
hemoglobin level of 11.5 g/dl; however, by February
1980, severe anemia had developed. On admission, the
differential diagnosis was principally between hemolysis
and blood loss. On the basis of the relatively low retic-
ulocyte count, the normal bilirubin level, a blood smear inconsistent with ongoing hemolysis and normal LDH
and haptoglobin levels, the house staff correctly as-
sumed that the picture was incompatible with acute
hemolysis. Within 24 hours of admission she had re-
ceived 3.5 units of packed red blood cells, with sub- sequent improvement in her sense of well-being and
with the anticipated rise in hematocrit level. However, over the next three days, without evidence of blood loss,
the hemoglobin level dropped from 10 to 7.5 g/dl. I would like Dr. Chaplin to address the problem of the
post-transfusion drop in hemoglobin as well as to dis- cuss the cause or causes of the anemia that was
present on admission. Dr. Hugh Chaplin: The hematologic problems of this
878 November 1981 The American Journal of Medicine Volume 71

case are difficult and controversial. Because time is
limited, I would like to focus on the four major questions
posed by our patient: (1) Does our patient have an
underlying chronic, possibly inherited, hematologic
abnormality? (2) Are her hematologic findings secon-
dary to any of the drugs she was taking? (3) What was
the primary cause of her severe anemia on admission?
(4) Could she have had a delayed hemolytic transfusion reaction?
Regarding the possibility of an underlying chronic
inherited hemolytic anemia, several points are pertinent:
the moderate anemia six months prior to admission; the
microcytic, hypochromic indices at this admission; a
hemoglobin A2 value of 6 percent: and a fetal hemo- globin value of 5 percent on one occasion prior to
transfusion. In addition, she had mild splenomegaly. I
believe these findings suggest the diagnosis of thalas-
semia minor, and I believe she had this condition
throughout her life, presumably without having suffered
any significant consequences. I believe our patient also
had superimposed iron deficiency on the basis of sev- eral findings: her low serum iron level, the slightly ele-
vated total iron binding capacity and the absence of iron
in the bone marrow. It is interesting that the elevated
A2 hemoglobin value characteristic of thalassemia
minor may fall to normal if superimposed iron deficiency
develops. In today’s case, however, the elevated A2
hemoglobin persisted, a phenomenon that occurs in at
least half the patients with thalassemia minor in whom
iron deficiency develops.
Next, we must consider the possible role of drugs in
the development of her anemia. The three most likely
pharmacologic culprits are methyldopa, hydralazine and naproxen. Methyldopa must always be considered when
severe anemia develops in patients taking the drug, but in this case I believe it can be totally exonerated. In
cases of methyldopa-induced hemolytic anemia, the
Coombs’ test result invariably is very strongly positive
and remains so for months after the drug is discontin-
ued. In today’s case, the Coombs’ test on admission
gave only weakly positive results, a fact that eliminates
methyldopa as an offending agent. The weakly positive
Coombs’ test result might have been related to the hy-
dralazine because this phenomenon occurs as part of the lupus-like syndrome associated with the drug. In
such cases, the positive Coombs’ result can be shown to be caused by the binding of the third component of
complement to the red blood cells. However, it is highly
unlikely that hydralazine caused our patient’s severe
anemia. The lupus-like syndrome associated with hy-
dralazine and other drugs is much more often charac- terized by a weakly positive Coombs’ test result than
by frank, brisk hemolysis. In those rare cases when hydralazine produces significant hemolysis, the he-
molysis results from a drug/anti-drug immune complex mechanism. As a consequence, the Coombs’ result is
CLINICOPATHOLOGIC CONFERENCE
strongly positive for complement, which was not found
in today’s case. Furthermore, without good evidence
for hemolysis-her admission bilirubin and haptoglobin
levels were normal-the likelihood that either methyl-
dopa or hydralazine is implicated decreases even more. Naproxen causes an abnormality in platelet aggregation
and a modest elevation in bleeding time, both of which
may facilitate hemorrhage, especially when underlying
gastrointestinal disease is present. Therefore, I think
the naproxen may have contributed to blood loss from
our patient’s gastrointestinal tract, which I believe was
the primary cause of her very low hemoglobin level on
admission. The hemorrhage almost certainly occurred
from the channel ulcer that was demonstrated radio-
graphically. The bleeding had stopped by the time of
admission, and her reticulocyte response was subop-
timal for a hemoglobin level of 5 g/dl. However, such
a poor reticulocyte response is readily explainable on
the basis of iron deficiency, chronic illness, fever and
rapidly progressive renal failure.
Finally, we must consider the possibility of a delayed
transfusion reaction. Initially, this woman’s serum was
not difficult to cross-match, and she received the first
3 of her 4 units of blood without incident. During the
subsequent 96 hours, however, the hemoglobin level
dropped from 10 g/dl to 7.5 g/dl; and 48 hours after her
transfusions had ended, her bilirubin level had risen to 1.5 mg/dl. Following transfusion, the bilirubin can rise
simply from the destruction of nonviable cells that are
always present to some extent in transfused blood.
However, this mechanism occurs within the first 8 hours
after the cessation of the transfusion and hence does
not explain an elevation of the bilirubin 48 hours later. Thus, I believe the patient had a mild delayed transfusion
reaction. Enhancing this possibility is that anti-Kell
antibody, which had not been demonstrable earlier, was
subsequently clearly evident in her serum. These ob-
servations suggest the stimulation of alloantibody in a
previously sensitized woman. Our patient was para-IX,
gravida-IX; it is very likely that an earlier pregnancy
caused her initial anti-Kell sensitization. Since Kell-
positivity is found in approximately 1 of 10 units of
blood, it is quite possible that at least 1 of the 4 units she
received contained that antigen. In summary, I think our patient had underlying tha-
lassemia minor. Although she was taking certain drugs
that can cause hemolytic anemia, I do not believe they were responsible for the severe anemia in her case.
Rather, I believe her anemia arose from gastrointestinal
blood loss that was possibly aggravated by the bleeding diathesis produced by naproxen. I think she also had a
mild hemolytic transfusion reaction secondary to re- stimulation of anti-Kell antibodies which would have
originally been provoked during one or more of her
pregnancies. Dr. Atkinson: Thank you. A likely source of the gas-
November 1981 The American Journal of Medicine Volume 71 879

RENAL FAILURE, DYSPNEA AND ANEMIA
trointestinal bleeding was found, and no further bleeding
occurred in the hospital. Thus, despite a rather complex
hematologic picture on admission, a successful eval-
uation was completed within a few days. Hence, let us
now analyze the renal failure (Table II).
On admission, the patient displayed elevated urea
nitrogen and creatinine levels. Since she gave no history
of previous renal disease but was known to be hyper-
tensive, her physicians initially made the reasonable
assumption that her renal impairment resulted from
nephrosclerosis. However, several factors militated
against the diagnosis of hypertensive renal disease: a
normal serum creatinine six months prior to admission,
a well-controlled blood pressure during the previous six
months and the absence of end organ damage in the
eye grounds. The initial results of urinalysis appeared on the chart
the evening of February 14 or the morning of February
15. The urinary sediment contained protein and nu-
merous red blood cells. No casts were seen. On the
evening of February 15, a temperature of 392OC de-
veloped. At that time, an intern carefully evaluated the
possible causes of her fever. His urinalysis documented
a sediment laden with numerous red blood cells and, in
all likelihood, red cell casts. Since the creatinine level
on February 16 had not changed from that on admission,
the focus of her evaluation remained on the anemia. However, five or six days later, her renal function began
to deteriorate rapidly, and the direction of the work-up turned toward the kidneys. A renal scan and ultrasound
study both demonstrated normal kidney size. The renal scan also showed symmetrically reduced function in
both kidneys. Dr. Martin will now present his analysis
of the renal failure.
Dr. Kelvin Martin: In order to evaluate a patient with
new renal failure, it is useful to consider the main ne-
phrologic syndromes [I]. Acute nephritis denotes an
acute inflammatory process in the kidney, usually in the
gfomeruli, that is manifested by renal failure, proteinuria
and the presence of blood or red cell casts in the urinary
sediment. The presentation of today’s patient clearly
fits this syndrome. An additional consideration in our patient is hypertensive renal disease, which was orig-
inally believed to be the cause of her renal failure. Al-
though hypertensive nephrosclerosis commonly pro-
duces renal failure and urinary findings similar to those
seen in today’s patient, the normal retinal examination makes hypertension highly unlikely as the cause of her
rapidly progressive renal failure. Since this patient’s renal failure developed over six
months, the major consideration is subacute nephritis. Since casts may disappear with storage, I wish to em-
phasize the importance of frequently examining the sediment in freshly voided urine rather than sending
specimens to the laboratory. Renal biopsy is indicated
in such cases, and this presumably was the procedure
performed on our patient. I would anticipate that biopsy
revealed proliferative glomerulonephritis. Dr. Atkinson: We now need to construct a differential
diagnosis. The most likely possibility by far is SLE. A
more difficult issue to resolve is whether the SLE is
idiopathic or drug-related. Another possibility, quite
remote, is that she had idiopathic rapidly progressing
glomerulonephritis of an immune complex type with the
positive serologic studies occurring incidentally sec- ondary to the hydralazine. Although I cannot rule out this
possibility, I am going to devote the rest of my discus-
sion to SLE.
In complicated cases such as this one, it is useful to
list points for and against the diagnosis of SLE. Several
features in the case favor that diagnosis: (1) a two-year
history of arthralgias of the upper extremities unre- sponsive to nonsteroidal anti-inflammatory drugs; (2)
an unexplained fever; (3) splenomegaly (although, as
Dr. Chaplin suggested, this could have resulted from the
heterozygous ,&thalassemia); (4) rapidly progressive nonobstructive renal failure with an active sediment,
which strongly suggests glomerulonephritis; (5) a pos-
itive ANA result, which admittedly could have been an
incidental finding secondary to the hydralazine; (6) a
positive test for circulating immune complexes, as detected by the Raji assay, with very high levels that are
frequently seen in SLE but only occasionally in other
diseases: (7) a diffuse polyclonal increase in IgG; and
(8) a very high ESR. Although none of these findings
alone is definitive, together they argue strongly for an
immune complex type of illness-probably SLE, either
of drug-induced or idiopathic type.
A few points weigh against SLE. The patient had no
involvement of the skin or mucous membranes, a
finding present in about 90 percent of patients with that
illness. Indeed, dermatologic findings account for the
majority of the 14 criteria used by the American
Rheumatism Association to diagnose SLE clinically. She
had normal serum complement levels, whereas ap-
proximately two-thirds of patients with renal involve-
ment in SLE have reduced levels. She had no rheuma-
toid factor and a normal partial thromboplastin time, but
these are abnormal in only approximately one-third of patients with SLE. She also had no thrombocytopenia
or leukopenia, both common hematologic manifesta-
tions of SLE. Obviously, in the appropriate clinical setting, none of these points rules out the diagnosis of SLE, and I still believe she had this disease. Assuming
this to be the case, we must next consider whether she
had drug-induced SLE secondary to hydralazine or id- iopathic SLE.
Our knowledge of hydralazine-induced SLE largely results from studies performed at Washington University under the direction of Dr. Mitchell Perry. We are fortu-
880 November 1981 The American Journal of Medicine Volume 71

nate to have Dr. Perry with us today, and later in the
discussion he will present his analysis of the present
case. Dr. Perry, as director of the Washington University
Hypertension Unit, began treating hypertensive patients
with hydralazine in 1952 when this agent first became
available. At that time hydralazine was one of the few
effective and reasonably well tolerated drugs for high
blood pressure. In 1953, he and his co-workers pub- lished the initial description of the “hydralazine syn-
drome” [ 21. Several of his patients had development
of a clinical syndrome consisting of such constitutional
symptoms as fatigue, fever, malaise, asthenia and ar- thralgias and arthritis and pleurisy. These patients also
had characteristic laboratory abnormalities, including
elevated gamma globulin levels, a high ESR and a positive LE preparation. At that time, SLE was consid-
ered a rare disease, and serologic tests to assist with
the diagnosis were just becoming available. Several
other reports around the same time confirmed these features of the illness [3,4]. Dr. Perry then initiated a
long-term study of these patients, and I will summarize
four of his most important publications [5-81.
In 1965, he and his co-workers described a number
of additional clinical and serologic features of the hy-
dralazine syndrome [ 51. The syndrome developed in
about one-fifth of the patients who had taken more than
200 mg of hydralazine per day for several years. Anti- nuclear antibodies could be found in approximately 60
percent of these patients. The illness rarely occurred
in blacks, although blacks who had the syndrome also had antinuclear antibodies. Clinically, the syndrome
could not be distinguished from idiopathic SLE. How-
ever, hydralazine-induced SLE only rarely involved the
central nervous or renal systems. In addition, the hy-
dralazine syndrome was generally characterized by
absent cryoglobulins and normal complement deter-
minations. Severe high blood pressure prompted re-
institution of the drug in several patients, and the syn-
drome again developed in about two-thirds of these patients.
In 1970, Dr. Perry made another very interesting
observation [6]. Dr. Perry and his colleagues examined
the ability of 57 patients on maintenance hydraiazine
therapy to acetylate drugs. Acetyl transferase-N is a
hepatic enzyme important in the catabolism of a number
of drugs, including procainamide and hydralazine. The
rate of hepatic acetylation can be determined by mea- suring urinary concentations of a sulfonamide following
intravenous administration of the drug. About 50 percent
of both normotensive and hypertensive persons are fast
acetylators. Dr. Perry evaluated the acetylation status of 57 patients who had been taking hydralazine at a minimal dose of 200 mg a day for a period of one to
fifteen years. Thirty-one of 57 patients were ANA-
positive. Of the slow acetylators, more than half were
cLlNtc0PATH0L00i~ CONFERENCE
ANA-positive; of the fast acetylators, nine of 24 became ANA-positive. Dr. Perry also showed that ANA was
found in the slow acetylators sooner than in the fast
acetylators. The most important point in the study was
the observation that of the 12 persons in whom SLE
developed, all were slow acetylators and ANA-positive. Thus, hydralazine-related SLE usually occurs in patients
on long-standing, high doses of the drug who have
positive ANA and who are slow acetylators.
A few years later Dr. Bevra Hahn, in collaboration
with Dr. Perry and other colleagues, examined the im-
munologic response of these patients to hydralazioe
[7]. They showed that patients with this syndrome
produced antibodies specific for both hydralazine and
certain nuclear constituents. In addition, they showed that the lymphocytes of these persons underwent
modest but definite transformation in response to hy-
dralazine. Patients with idiopathic SLE, in contrast, did
not have antihydralazine antibodies. More recently,
another group demonstrated that ANA in drug-induced
SLE were quite specific, being primarily directed to
histones, the basic nuclear proteins associated with
DNA [9]. In idiopathic SLE, only one-third of the patients
had antibodies to histones. In contrast, 23 of 23 patients
with drug-induced SLE possessed such antibodies.
Moreover, patients with the drug-induced syndrome did
not have antibodies to native DNA. Some data in the literature have suggested that hydralazine, a highly re-
active molecule, may form physicochemical bonds with
DNA-histones [9]. In this way, hydralazine could be
acting like a hapten. This could explain why persons
with the drug-induced syndrome have antibodies to
hydralazine, histones, and to a hydralazine-histone
complex. In summary, the initial description and most
of the advances in our understanding of this syndrome
have resulted from. Dr. Perry’s work at Washington
University. His 20-year experience with hydralazine-
inducedSLE has been reviewed in this journal [8].
Drug-induced SLE commonly occurs in two principal clinical set$ngs. One is that of today’s patient-hya
pert&nsive persons who have been taking hydralazine
for several years. The illness begins insidiously with arthralgias, followed by arthritis and constitutional
symptoms. The second setting involves patients re-
ceiving procainamide for control of ventricular pre-
mature beats. After several months, they can have a syndrome of arthralgias and arthritis (particularly in the
shoulder girdle), fever and pleural-pericardial mani-
festations that easily mimic other conditions such as the
postmyocardial infarction syndrome, pulmonary em-
bolus, congestive heart failure and recurrent coronary pain. In both forms of drug-induced lupus, central ner-
vous system and renal involvement are almost invari-
ably absent. Serologic studies in both situations gen- erally demonstrate ANA, but normal complement levels,
November 1981 The American Journal of Medicine Volume 71 881

RENAL FAILURE, DYSPNEA AND ANEMIA
no cryoglobulins and no antibodies to native DNA. In
both cases, the disease usually remits within a few
months after the drug is stopped.
To reinforce some of these comparisons, I would like
to mention a review by Dubois et al. [lo]. They studied
33 patients with procainamide-induced lupus, 50 pa-
tients with hydralazine-induced lupus, and 500 patients
with idiopathic lupus. All three groups demonstrated a
high incidence of fever, arthralgias and arthritis. Pleuritis
and pericarditis occurred frequently in the patients taking procainamide. In the two groups with drug-in-
duced lupus, skin manifestations occurred less com-
monly and central nervous system and renal involve-
ment were distinctly unusual. Dubois also found a low
incidence of adenopathy and gastrointestinal complaints
in the patients with the drug-induced syndrome, but I
have not found these latter items useful in distinguishing
idiopathic from drug-related disease.
With this background, we can now summarize the
information for and against drug-induced SLE in today’s
patient. Strongly against the diagnosis is this patient’s
race. As mentioned, only in rare instances has hydral- azine-induced SLE developed in black persons. Renal
failure, although described, is also quite unusual in hy-
dralazine-induced lupus. For example, in the Wash-
ington University experience, renal involvement pos-
sibly occurred in only two patients [8]. Of note, a con-
trary report from the Mayo Clinic suggested that renal
disease develops in 10 to 25 percent of patients with
hydralazine-induced lupus [ 111. However, this series
must be taken lightly because it was not clearly estab-
lished that the subjects had hydralazine-induced SLE,
in contrast to the alternative possibility that they had
already contracted lupus and were taking hydralazine
for secondary hypertension. Unfortunately, we do not have certain laboratory in-
formation that would help us distinguish between hy-
dralazine-induced and idiopathic lupus. For example,
the presence of antibodies to native DNA would militate
against drug-induced SLE. If we knew our patient were
a fast acetylator, the diagnosis of drug-induced SLE would again be unlikely. (In this case, however, testing
her acetylator staus by the “sulfa method” during the
last admission would have been impossible because
of her renal failure.) The most helpful factor, however,
would be knowledge of the clinical outcome after the
drug was stopped. If, after hydrajazine was discontinued, the clinical syndrome resolved, the ANA titer progres- sively decreased and no further steroid therapy were required, then I would assume that the clinical picture
was drug-induced. Of course, such a sequence of
events would not eliminate the possibility that the patient
had idiopathic SLE that was exacerbated or induced by hydralazine [4,8,10-131.
Next, we need to predict the procedure performed and the likely pathologic findings. Kidney biopsy was
undoubtedly the procedure undertaken since she had
an active urinary sediment with rapidly progressive renal
failure of unknown cause. The creatinine level reached 9.2 mg/dl on the seventh day of hospitalization. In view
of this clinical course, I would have probably begun
pulse therapy with corticosteroids even before biopsy
was performed in the hope of stopping or reversing the
process. While reviewing the chart, I found some ad-
ditional reasons why I think kidney biopsy was per-
formed. First, by the time of the procedure, her hema-
tologic work-up had been completed, and her anemia
had been treated with transfusions. In addition, around the time of the procedure, lymphocytopenia and gran-
ulocytosis developed and she became afebrile-all of
which could have been secondary to the administration
of corticosteroids. Taken together, these observations
make me believe that she underwent kidney biopsy on
the ninth hospital day and probably received a steroid
pulse about the same time.
Dr. David McLain, a former fellow in the Washington
University Rheumatology Division, two years ago began
to study the serologic features of drug-induced lupus.
He has accumulated data on 12 patients and has shared his findings with me. Even in this small series, he has
found patients with drug-induced SLE who have had
cryoglobulins [ 141, immune complexes by Raji assay,
typical lupus anticoagulant and low complement levels.
Other investigators have also reported patients with
drug-induced lupus in which these serologic test results
were positive or abnormal [ 15-181. In most such pa-
tients, however, these serologic tests give normal re-
sults. Indeed, a patient very similar to the one under
discussion today was described in Dr. McLain’s file, and
Dr. McLain had concluded that this person did not have
drug-induced lupus.
Assuming renal biopsy was performed, we must predict the likely pathologic findings. The marked and
rapid deterioration of renal function argues against a
focal proliferative glomerulonephritis, although the
modest proteinuria is compatible with such a histologic
finding. More likely, in view of the rapid course of her
renal disease, she had diffuse proliferative glomerulo-
nephritis, although such patients generally have low
complement levels and detectable antibodies to native
DNA.
In summary, I believe this patient had high blood
pressure, probably idiopathic or essential. She may also have had cardiovascular disease secondary to the hy- pertension. In view of the negative ANA results in 1978,
I do not believe that smoldering SLE of several years’
duration caused the hypertension. I believe SLE then
developed, which produced her renal failure. I cannot
be sure whether her SLE was hydralazine-induced or idiopathic, but I favor the latter. She also had a pyloric channel ulcer that probably bled several weeks prior to admission and thus contributed substantially to the
882 November 1981 The American Journal of Medicine Volume 71

anemia. Other factors contributing to the anemia in- cluded a chronic inflammatory disease (lupus), renal failure, iron deficiency and fl-thalassemia minor. Dr. Perry, would you please comment. Dr. H. Mitchell Perry: I would disagree with only one of your points. I believe you have overstated the frequency of renal dysfunction as a complication of hydralazine- related lupus. We have seen only one patient in whom acute renal failure could be unequivocally related to hydralazine-related lupus [ 191. In none of our other patients has any causal link been demonstrated be- tween hydralazine-related lupus and renal dysfunc- tion-despite the fact that many hydralazine-treated patients have renal failure either from hypertension or from the underlying renal disease that presumably caused their hypertension. I know of only one other reported case in which hydralazine-related lupus seemed definitely to be implicated as a cause of renal failure [20].
Another feature would make the diagnosis of hy- dralazine-related lupus unusual in this case: the patient’s race. Of our first 44 cases of hydralazine-related lupus, two occurred in black patients [8], and other investi- gators have also reported the syndrome in blacks. De- spite these occasional instances, however, drug- related lupus occurs much less frequently in blacks than in whites. In one study, we observed that the risk was al- most five times greater in white patients [ 211. Our two black patients with hydralazine-related lupus both had atypical presentations. Each was a woman with “ma- lignant” hypertension, and both women were receiving 750 mg of hydralazine daily. A clinical syndrome con- sistent with hepatitis developed in the first patient after three months of exposure. In the second, whose very complicated case has been reported [22], marked asthenia developed after 23 months of exposure.
While I have the floor, I would like to make several additional comments about drug-related lupus. First, the protocol indicates that this patient originally had “severe hypertension,” although it does not specify how high the blood pressure. Most of our early patients with hy- dralazine-related lupus had truly severe hypertension, with “malignant” hypertension in more than half [8].
Second, the protocol described this patient’s hy- dralazine dosage as “high,” and I have learned that at one time she ingested 400 mg daily. This is a significant dosage; 75 percent of our early patients with hydrala- zine-related lupus took at least 400 mg of the drug for six months [8]. According to the information provided, this patient had been taking hydralazine for four years when arthralgias developed and for six years when the renal failure appeared. In only one of our 44 patients with hydralazine-related lupus did symptoms develop after more than four years of exposure to the drug. Thus, it is unlikely that her symptoms resulted from hydrala-
CLINICOPATHOLOGIC CONFERENCE
zine-related lupus. Our only patient with documented renal failure secondary to hydralazine-related lupus was unusual because of the extreme severity of her hyper- tension and the high dosages of hydralazine (up to 1 g per day) she received for therapy. That case has been reported elsewhere [ 191.
Third, the protocol indicates that the patient was normotensive at the time renal failure became apparent. In our experience, the blood pressure almost always falls to normal before symptoms of hydralazine-related lupus appear. In addition, the patients generally remain normotensive for at least as long as the hydralazine- related lupus persists [8].
All our patients with hydralazine-related lupus had positive ANA, as did today’s patient; in each case, the positive ANA antedated the development of clinical symptoms [6]. A positive ANA result. however, appears much more frequently than does symptomatic drug- induced lupus in hydralazine-treated patients. For ex- ample, hydralazine often induces positive ANA reults in blacks and white “fast acetylators,” although neither group stands much risk of development of hydrala- zine-related lupus. “Fast acetylators” may require up to 1,200 mg of hydralazine for development of positive ANA, versus the 400 mg that will produce a positive ANA result in 50 percent of “slow acetylators” [6]. Thus, the appearance of a positive ANA does not war- rant the discontinuation of hydralazine, although white “slow acetylators” should receive closer follow-up observation.
Finally, although our patient’s anemia probably does not relate to hydralazine, it is noteworthy that patients receiving large doses of hydralazine initially experience a drop of hemoglobin averaging almost 2 gm/dl without development of hydralazine-related lupus [23]. With severe hydralazine-related lupus, the anemia is even greater, with an average decrease in hemoglobin of 4.5 gm/dl, and we have seen patients with severe hydral- azine-related lupus in whom the hemoglobin decreased as much as 7.4 gm/dl. These anemic patients had di- minished red blood cell volume, low serum iron levels and erythroid hyperplasia of the marrow-all suggesting iron deficiency anemia [8].
In summary, I am impressed that renal failure only rarely results from hydralazine-related lupus. The one case we have seen represented an unusual situation in which we were attempting to control severe hyper- tension with large doses of hydralazine as the sole drug. Thus, I think it unlikely that today’s patient had renal failure secondary to hydralazine. Dr. Atkinson: I appreciate your comments and agree with your analysis of the hydralazine data. The one published study suggesting that renal failure develops in 20 percent of the patients with hydralazine-induced SLE must be interpreted with caution because of the likelihood that many of the patients, in fact, had idio-
November 1981 The American Journal of Medicine Volume 71 883
RENAL FAILURE. DYSPNEA AND ANEMIA
pathic SLE with hypertension [ IO]. Hypertension occurs
in more than one-third of patients with SLE. Am I cor-
rect, Dr. Perry, in assuming that you believe today’s patient had idiopathic SLE, with hydralazine being either
unrelated to or, at most, an inducing factor in her
SLE?
Dr. Perry: Yes.
PATHOLOGIC DISCUSSION
Dr. John Meyer: Since I do not know the follow-up in
this patient after hydralazine was discontinued, I can
approach the diagnostic problem with some objectivity.
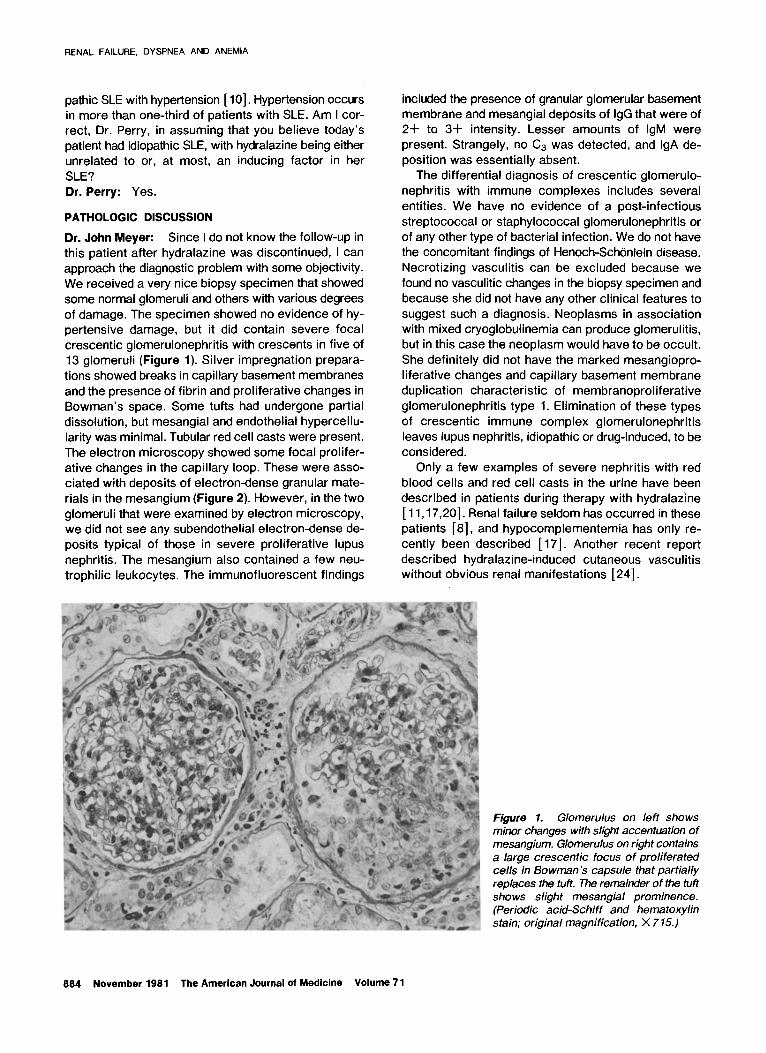
We received a very nice biopsy specimen that showed
some normal glomeruli and others with various degrees
of damage. The specimen showed no evidence of hy-
pertensive damage, but it did contain severe focal crescentic glomerulonephritis with crescents in five of
13 glomeruli (Figure 1). Silver impregnation prepara-
tions showed breaks in capillary basement membranes
and the presence of fibrin and proliferative changes in
Bowman’s space. Some tufts had undergone partial
dissolution, but mesangial and endothelial hypercellu-
larity was minimal. Tubular red cell casts were present.
The electron microscopy showed some focal prolifer-
ative changes in the capillary loop. These were asso- ciated with deposits of electron-dense granular mate-
rials in the mesangium (Figure 2). However, in the two
glomeruli that were examined by electron microscopy, we did not see any subendothelial electron-dense de-
posits typical of those in severe proliferative lupus
nephritis. The mesangium also contained a few neu-
trophilic leukocytes. The immunofluorescent findings
included the presence of granular glomerular basement membrane and mesangial deposits of IgG that were of
2+ to 3+ intensity. Lesser amounts of IgM were
present. Strangely, no C3 was detected, and IgA de-
position was essentially absent.
The differential diagnosis of crescentic glomerulo-
nephritis with immune complexes includes several
entities. We have no evidence of a post-infectious
streptococcal or staphylococcal glomerulonephritis or
of any other type of bacterial infection. We do not have
the concomitant findings of Henoch-Schonlein disease.
Necrotizing vasculitis can be excluded because we
found no vasculitic changes in the biopsy specimen and
because she did not have any other clinical features to
suggest such a diagnosis. Neoplasms in association
with mixed cryoglobulinemia can produce glomerulitis,
but in this case the neoplasm would have to be occult.
She definitely did not have the marked mesangiopro-
liferative changes and capillary basement membrane
duplication characteristic of membranoproliferative
glomerulonephritis type 1. Elimination of these types
of crescentic immune complex glomerulonephritis
leaves lupus nephritis, idiopathic or drug-induced, to be
considered.
Only a few examples of severe nephritis with red
blood cells and red cell casts in the urine have been
described in patients during therapy with hydralazine
[ 11,17,20]. Renal failure seldom has occurred in these
patients [8], and hypocomplementemia has only re-
cently been described [ 171. Another recent report
described hydralazine-induced cutaneous vasculitis without obvious renal manifestations [24].
Figure 1. Glomerulus on left shows minor changes with slight accentuation of mesangium. Glomerulus on right contains a large crescent/c focus of proliferated ceils in Bowman’s capsule that partially replaces the tuft. The remainder of the tuft shows slight mesangial prominence. (Periodic acid-Schiff and hematoxylin stain; original magnification, X 7 15.)
884 November 1981 The American Journal of Medicine Volume 71

CLINICOPATHOLOGIC CONFERENCE
Figure 2. Electron-dense deposits typ- ical of immune complexes can be seen in the electron micrograph of the glomerular mesangium. (&any/ acetate and lead ci- trate stain; original magnification, X 8,200.)
The renal biopsy findings in our patient are consistent
with idiopathic systemic lupus nephritis, but the long
history of therapy with high-dose hydraiazine certainly
raises the question of drug-induced immune complex
disease. Unfortunately, the literature provides no in- formation on how to differentiate the two conditions. We
have not been able to find any reports of renal biopsy
studies in patients with the hydraiazine toxicity syn-
drome.
Several features of our patient’s case are notable.
First, she received hydraiazine for an unusually long
time after the appearance of symptoms that may in
retrospect have represented drug toxicity, and the
symptoms were treated with naproxen rather than with
discontinuation of the hydraiazine. Second, several
features of the biopsy specimen are unusual for severe
idiopathic lupus nephritis: the absence of endotheiiai
and mesangiai proliferative responses, the lack of
abundant subendotheliai immune complexes in electron
micrographs of the giomeruii and the lack of immuno-
reactive IgA in the glomeruiar deposits. Of note, in this
medical center we have seen mesangiai deposits by
electron microscopy in the renal biopsy specimens of
two other patients with presumed hydraiazine-induced
lupus, and neither showed subendothelial deposits. The current patient, however, is the only one of the three in
whom biopsy demonstrated a destructive giomeruiitis. With these considerations in mind, I believe that we very possibly could be seeing a case of hydraiazine-induced
crescentic immune complex giomerulitis. Of course,
the definitive test of this concept would be observation
of the patient’s course after discontinuation of hydral-
azine. in addition, determination of the patient’s N-acetyi
transferase level would be helpful since the hydraiazine toxicity syndrome appears to be confined to slow
acetyiators [8,25]. Dr. Victor Meltzer: I have some follow-up information.
On the ninth hospital day the patient underwent renal
biopsy. Two days later, she began receiving methyi-
prednisoione, 1 g daily for three days. in addition, she
received cyciophosphamide, 100 mg per day,
throughout the remainder of her hospitalization. At the
time of discharge 12 days later, her serum creatinine
level had fallen to 3.6 mg/di. Prior to therapy, her anti-
DNA antibody level was 63 U/ml; on the day prior to
discharge, it was 44 U/ml. Normally, this value does not exceed 15 U/ml; values greater than 25 U/ml occur
largely in patients with lupus. As an out-patient, she
began receiving prednisone, and the cyciophosphamide
was eventually stopped. Six months later, she was readmitted with pneumo-
nia. At that time, still without hydralazine, antinuclear
antibodies were still present, and her serum creatinine level had decreased further to 2.6 mg/dl. Subsequently, she has continued to take alternate-day, low-dose
prednisone; and her most recent serum creatinine level, 12 months after the original hospitalization, was 2.5
mg/di. Without hydraiazine. her blood pressure has
November 1981 The American Journal of Medicine Volume 71 885

RENAL FAILURE, DYSPNEA AND ANEMIA
been difficult to control, requiring institution of minoxidil and propranolol. Dr. Atkinson: The elevated level of anti-DNA anti-
bodies provides more evidence in favor of idiopathic
SLE. The gratifying response to therapy does not help
resolve the diagnostic dilemma of idiopathic versus
drug-induced SLE. Since she now is much improved on
low-dose steroids, one could still argue that she had a
drug-induced syndrome. However, I do not believe she
has undergone sufficient follow-up to be certain.
In summary, despite the unusual histologic features
indicated by Dr. Meyer, I still favor the diagnosis of id-
iopathic SLE in this case. However, I would accept the
diagnosis of drug-induced lupus if, over the next two
years, the clinical syndrome and serologic abnormalities
were to remit without further prednisone therapy.
Final pathologic diagnosis: Immune complex cres-
centic glomerulonephritis, possibly hydralazine-in-
duced.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
a.
9.
10.
11.
12.
13.
Coe FL: Clinical and laboratory assessment of the patient with renal disease. In, Brenner BB, Rector FC, eds: The kidney, 2nd ed. Philadelphia: WB Saunders, 1981; 1135-l 180.
Morrow JD, Schroeder HA, Perry HM Jr: Studies on the control of hypertension by Hyphex. II. Toxic reactions and side effects. Circulation 1953; a: 829-839.
Reinhardt DJ, Waldron JM: Lupus erythematosus-like syn- drome complicating hydralazine (Apresolinie) therapy. JAMA 1954; 155: 1491-1492.
Lee SL, Chase PH: Drug-induced systemic lupus erythema- tosus: a critical review. Semin Arthritis Rheum 1975; 5: 83-103.
Condemi JJ, Moore-Jones D, Vaughan JH, Perry HM Jr: Anti- nuclear antibodies following hydralazine toxicity. N Engl J Med 1965; 276: 486-491.
Perry HM Jr, Tan EM, Carmody S, Sakamoto A: Relationship of acetyl transferase activity to antinuclear antibodies and toxic symptoms in hypertensive patients treated with hy- dralazine. J Lab Clin Med 1970; 76: 114-125.
Hahn BH, Sharp GC, Irvin WS, et al.: immune responses to hydralazine and nuclear antigens in hydralazine-induced lupus erythematosus. Ann Intern Med 1972; 76: 365- 374.
Perry HM Jr: Late toxicity to hydralazine resembling systemic lupus erythematosus or rheumatoid arthritis. Am J Med 1973; 54: 58-72.
Fritzler MJ, Tan EM: Antibodies to histones in drug-induced and idiopathic lupus erythematosus. J Clin Invest 1978; 64: 560-567.
Dubois EL, Tallman E, Wonka RA: Chlorpromazine-induced systemic lupus erythematosus. Case report and review of the literature. JAMA 1972; 221: 595-596.
Alarcon-Segovia D, Wakim KG, Worthington JW, Ward LE: Clinical and experimental studies on the hydralazine syn- drome and its relationship to systemic lupus erythematosus. Medicine (Baltimore) 1967; 46: l-33.
Alarcon-Segovia D: Drug-induced lupus syndromes. Mayo Clin Proc 1969; 44: 664-681.
Blomgren SE: Drug-induced lupus erythematosus. Semin
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
Hematol 1973; 10: 345-349. McLain DA, Hahn BH: Cryoglobulins in the procainamide-
induced lupus syndrome. Arthritis Rheum 1979; 22: 305-307.
Utsinger PD, Zvaifler NJ, Bluestein HG: Hypocomplementemia in proceinamide-associated systemic lupus erythematosus. Ann Intern Med 1976; 84: 293-296.
Bell WR, Boss CR, Wolfson JS: Circulating anticoagulant in the procainamide-induced lupus syndrome. Arch Intern Med 1977; 137: 1471-1473.
Weinstein J: Hypocomplementemia in hydralazine-associated systemic lupus erythematosus. Am J Med 1978; 65: 553-556.
Becker M, Klajman A, Moalem T, Yaretzky A, Ben-Efraim S: Circulating immune complexes in sera from patients re- ceiving procainamide. Clin lmmunol lmmunopathol 1979; 12: 220-227.
Perry HM Jr: Hydralazine-induced lupus. Pharmacological and clinical aspects of hydralazine (Apresoline). In, Today’s treatment of hypertension. Oslo, Norway: Hassle-Ciba- Geigy, 1978; 124.
Dammin GJ, Nora JR, Reardan JB: Hydralazine reaction: case with LE cells antemortem and postmortem, and pulmonary, renal, splenic, and muscular lesions of disseminated lupus erythematosus. J Lab Clin Med 1955; 46: 806.
Perry HM Jr: Possible mechanisms of the hydralazine-induced lupus-like syndrome. Arthritis Rheum 1981; 24: 1093.
Perry HM Jr, O’Neal R, Thomas WA: Pulmonary disease fol- lowing chronic chemical ganglionic blockade: a clinical and pathological study. Am J Med 1957; 22: 37.
Perry HM Jr, Schroeder HA, Comens P: Abnormalities of cir- culating cells and proteins in hydralazine-treated patients without toxic symptoms. Am J Med Sci 1962; 244: 82.
Bernstein RM, Egerton-Vernon J, Webster J: Hydralazine- induced cutaneous vasculitis. Br Med J 1980; 280: 156- 157.
Strandberg I, Boman G, Hassler L, Sjoqvist F: Acetylator phenotype in patients with hydralazine-induced lupoid syndrome. Acta Med Stand 1976; 200; 367-371.
888 November 1981 The American Journal of Medicine Volume 71





























