Embed Size (px)
Citation preview

BRIEF COMMUNICATION www.jasn.org
Renal Fanconi Syndrome and Hypophosphatemic Ricketsin the Absence of Xenotropic and Polytropic RetroviralReceptor in the Nephron
Camille Ansermet,* Matthias B. Moor,* Gabriel Centeno,* Muriel Auberson,*Dorothy Zhang Hu,† Roland Baron,† Svetlana Nikolaeva,*‡ Barbara Haenzi,*Natalya Katanaeva,* Ivan Gautschi,* Vladimir Katanaev,*§ Samuel Rotman,| Robert Koesters,¶
Laurent Schild,* Sylvain Pradervand,** Olivier Bonny,*†† and Dmitri Firsov*
*Department of Pharmacology and Toxicology and **Genomic Technologies Facility, University of Lausanne, Lausanne,Switzerland; †Department of Oral Medicine, Infection, and Immunity, Harvard School of Dental Medicine, Boston,Massachusetts; ‡Institute of Evolutionary Physiology and Biochemistry, St. Petersburg, Russia; §School of Biomedicine,Far Eastern Federal University, Vladivostok, Russia; |Services of Pathology and ††Nephrology, Department of Medicine,University Hospital of Lausanne, Lausanne, Switzerland; and ¶Université Pierre et Marie Curie, Paris, France
ABSTRACTTight control of extracellular and intracellular inorganic phosphate (Pi) levels is crit-ical tomost biochemical and physiologic processes. Urinary Pi is freely filtered at thekidney glomerulus and is reabsorbed in the renal tubule by the action of the apicalsodium-dependent phosphate transporters, NaPi-IIa/NaPi-IIc/Pit2. However, themolecular identity of the protein(s) participating in the basolateral Pi efflux remainsunknown. Evidence has suggested that xenotropic and polytropic retroviral recep-tor 1 (XPR1) might be involved in this process. Here, we show that conditional in-activation of Xpr1 in the renal tubule in mice resulted in impaired renal Pireabsorption. Analysis of Pi transport in primary cultures of proximal tubular cellsor in freshly isolated renal tubules revealed that this Xpr1 deficiency significantlyaffected Pi efflux. Further, mice with conditional inactivation of Xpr1 in the renaltubule exhibited generalized proximal tubular dysfunction indicative of Fanconisyndrome, characterized by glycosuria, aminoaciduria, calciuria, and albuminuria.Dramatic alterations in the renal transcriptome, including a significant reduction inNaPi-IIa/NaPi-IIc expression, accompanied these functional changes. Additionally,Xpr1-deficient mice developed hypophosphatemic rickets secondary to renal dys-function. These results identify XPR1 as a major regulator of Pi homeostasis and as apotential therapeutic target in bone and kidney disorders.
J Am Soc Nephrol 28: ccc–ccc, 2016. doi: 10.1681/ASN.2016070726
The xenotropic and polytropic retrovirusreceptor1(XPR1)has longbeenconsideredas a candidate component of the inorganicphosphate (Pi) efflux mechanism becauseof its high degree of homology with PHO1protein in plants, which has been shownto mediate Pi transport from roots toshoots.1,2 However, evidence has only re-cently emerged supporting a role of XPR1in Pi transport. Battini and colleagues have
shown in vitro that XPR1 depletion or in-hibition results in a marked decrease in Piefflux.3 They also demonstrated thatXBRD, a XPR1 ligand derived from theX-MLV envelope glycoprotein, could effi-ciently inhibit Pi efflux, thereby providingevidence on the direct role of XPR1 in Pitransport. Wege and Poirier have demon-strated that ectopically expressed mouseXPR1 mediates Pi efflux in tobacco
leaves.4 Most recently, Legati et al. haveshown an association between geneticpolymorphisms in Xpr1 and primary fa-milial brain calcification disorder.5 How-ever, the role of XPR1 in the maintenanceof Pi homeostasis remains unknown.Here,we addressed this issue inmice deficient forXpr1 in the nephron.
Because Xpr1-null mice exhibit em-bryonic lethality (viable pups: wildtype, 254; heterozygous, 384; null, 0),we generated mice with a doxycycline(DOX)-inducible, Pax8-rtTA–driven,6
Received July 6, 2016. Accepted September 5,2016.
C.A., M.B.M., G.C., and M.A. contributed equally tothis work.
Present address: Natalya Katanaeva, Swiss FederalInstitute of Technology, Lausanne, Switzerland.
Present address: Dr. Barbara Haenzi, CambridgeCentre for Brain Repair, University of Cambridge,Cambridge, UK.
Published online ahead of print. Publication dateavailable at www.jasn.org.
Correspondence: Dr. Dmitri Firsov or Dr. OlivierBonny, Department of Pharmacology and Toxicol-ogy, University of Lausanne, 27 rue du Bugnon,1011 Lausanne, Switzerland. Email: [email protected] or [email protected]
Copyright © 2016 by the American Society ofNephrology
J Am Soc Nephrol 28: ccc–ccc, 2016 ISSN : 1046-6673/2804-ccc 1
conditional deletion of Xpr1 in the renaltubule (Xpr1lox/lox/Pax8-rtTA/LC1 mice,hereafter referred to as conditional
knockout [cKO] mice). LittermateXpr1lox/lox mice treated with DOX wereused as controls. Males and females were
investigated separately to assess possiblesex differences. As shown in Supplemen-tal Figure 1, DOX treatment resulted in a
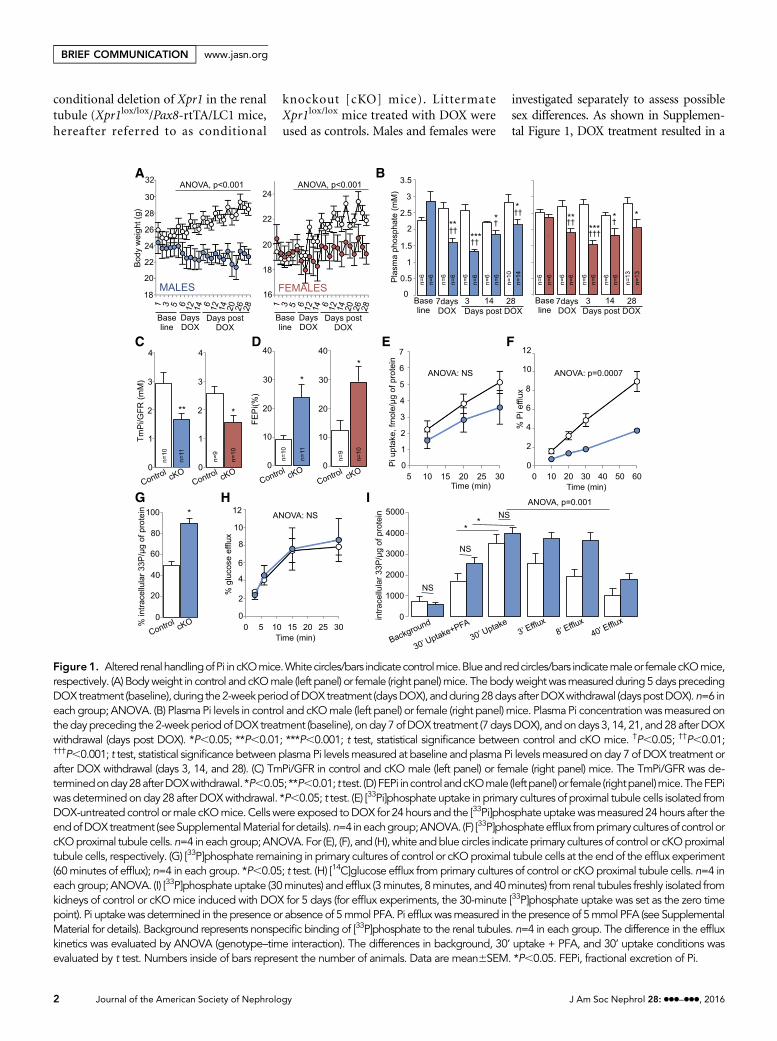
Figure 1. Altered renalhandlingofPi incKOmice.Whitecircles/bars indicatecontrolmice.Blueandredcircles/bars indicatemaleor femalecKOmice,respectively. (A) Bodyweight in control and cKOmale (left panel) or female (right panel)mice. The bodyweightwasmeasuredduring 5days precedingDOXtreatment (baseline),during the2-weekperiodofDOXtreatment (daysDOX), andduring28days afterDOXwithdrawal (dayspostDOX).n=6 ineach group; ANOVA. (B) Plasma Pi levels in control and cKOmale (left panel) or female (right panel) mice. Plasma Pi concentrationwasmeasured onthedaypreceding the2-weekperiodofDOX treatment (baseline), onday7ofDOX treatment (7daysDOX), andondays 3, 14, 21, and28afterDOXwithdrawal (days post DOX). *P,0.05; **P,0.01; ***P,0.001; t test, statistical significance between control and cKO mice. †P,0.05; ††P,0.01;†††P,0.001; t test, statistical significance between plasma Pi levelsmeasured at baseline and plasma Pi levelsmeasured on day 7 of DOX treatment orafter DOX withdrawal (days 3, 14, and 28). (C) TmPi/GFR in control and cKO male (left panel) or female (right panel) mice. The TmPi/GFR was de-terminedonday28afterDOXwithdrawal. *P,0.05; **P,0.01; t test. (D)FEPi incontrolandcKOmale (leftpanel)or female (rightpanel)mice.TheFEPiwas determinedonday 28 after DOXwithdrawal. *P,0.05; t test. (E) [33Pi]phosphate uptake in primary cultures of proximal tubule cells isolated fromDOX-untreated control ormale cKOmice. Cells were exposed toDOX for 24 hours and the [33Pi]phosphate uptakewasmeasured 24hours after theendofDOXtreatment (seeSupplementalMaterial fordetails).n=4 ineachgroup;ANOVA. (F) [33P]phosphateefflux fromprimaryculturesof controlorcKOproximal tubule cells.n=4 in eachgroup;ANOVA. For (E), (F), and (H), white andblue circles indicateprimary cultures of control or cKOproximaltubule cells, respectively. (G) [33P]phosphate remaining in primary cultures of control or cKOproximal tubule cells at the end of the efflux experiment(60minutes of efflux); n=4 in each group. *P,0.05; t test. (H) [14C]glucose efflux fromprimary cultures of control or cKOproximal tubule cells. n=4 ineachgroup; ANOVA. (I) [33P]phosphate uptake (30minutes) and efflux (3minutes, 8minutes, and 40minutes) from renal tubules freshly isolated fromkidneys of control or cKOmice induced with DOX for 5 days (for efflux experiments, the 30-minute [33P]phosphate uptake was set as the zero timepoint). Pi uptakewas determined in the presence or absence of 5mmol PFA. Pi effluxwasmeasured in the presence of 5mmol PFA (see SupplementalMaterial for details). Background represents nonspecific binding of [33P]phosphate to the renal tubules. n=4 in each group. The difference in the effluxkinetics was evaluated by ANOVA (genotype–time interaction). The differences in background, 30’ uptake + PFA, and 30’ uptake conditions wasevaluated by t test. Numbers inside of bars represent the number of animals. Data are mean6SEM. *P,0.05. FEPi, fractional excretion of Pi.
2 Journal of the American Society of Nephrology J Am Soc Nephrol 28: ccc–ccc, 2016
BRIEF COMMUNICATION www.jasn.org
significant reduction in Xpr1 mRNA andprotein levels in whole kidneys and in mi-crodissected proximal tubules of cKOmice. The decrease in renal XPR1
expression was accompanied by a progres-sively increasing difference in body weightbetween control and cKO mice thatreached 220.4% (cKO males) and
212.1% (cKO females) 28 days after theend ofDOX treatment (Figure 1A). Assess-ment of renal Pi handling revealed thatcKO mice exhibited hypophosphatemia
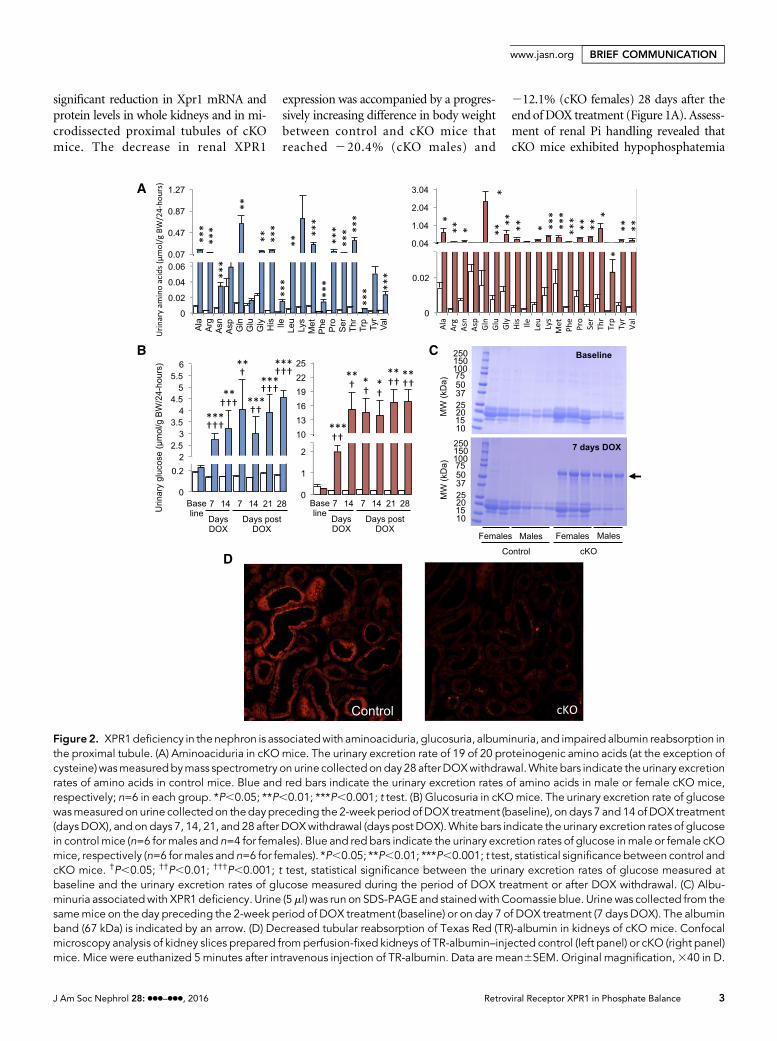
Figure2. XPR1deficiency in the nephron is associatedwith aminoaciduria, glucosuria, albuminuria, and impaired albumin reabsorption inthe proximal tubule. (A) Aminoaciduria in cKOmice. The urinary excretion rate of 19 of 20 proteinogenic amino acids (at the exception ofcysteine)wasmeasuredbymass spectrometry on urine collectedonday 28 afterDOXwithdrawal.White bars indicate the urinary excretionrates of amino acids in control mice. Blue and red bars indicate the urinary excretion rates of amino acids in male or female cKO mice,respectively; n=6 in each group. *P,0.05; **P,0.01; ***P,0.001; t test. (B) Glucosuria in cKOmice. The urinary excretion rate of glucosewasmeasured on urine collectedon thedaypreceding the 2-week period of DOX treatment (baseline), on days 7 and14 of DOX treatment(daysDOX), and on days 7, 14, 21, and 28 after DOXwithdrawal (days post DOX).White bars indicate the urinary excretion rates of glucosein control mice (n=6 for males and n=4 for females). Blue and red bars indicate the urinary excretion rates of glucose inmale or female cKOmice, respectively (n=6 formales and n=6 for females). *P,0.05; **P,0.01; ***P,0.001; t test, statistical significance between control andcKO mice. †P,0.05; ††P,0.01; †††P,0.001; t test, statistical significance between the urinary excretion rates of glucose measured atbaseline and the urinary excretion rates of glucose measured during the period of DOX treatment or after DOX withdrawal. (C) Albu-minuria associatedwith XPR1 deficiency. Urine (5ml) was run on SDS-PAGE and stainedwith Coomassie blue. Urinewas collected from thesamemice on the day preceding the 2-week period of DOX treatment (baseline) or on day 7 of DOX treatment (7 days DOX). The albuminband (67 kDa) is indicated by an arrow. (D) Decreased tubular reabsorption of Texas Red (TR)-albumin in kidneys of cKO mice. Confocalmicroscopy analysis of kidney slices prepared fromperfusion-fixed kidneys of TR-albumin–injected control (left panel) or cKO (right panel)mice. Mice were euthanized 5 minutes after intravenous injection of TR-albumin. Data are mean6SEM. Original magnification,340 in D.
J Am Soc Nephrol 28: ccc–ccc, 2016 Retroviral Receptor XPR1 in Phosphate Balance 3
www.jasn.org BRIEF COMMUNICATION

(Figure 1B), phosphaturia (transient inmales, Supplemental Figure 2), inappro-priately low maximal tubular reabsorptionof Pi per volume of glomerular filtrate(TmPi/GFR) (Figure 1C), and significantlyincreased fractional excretion of Pi (Figure1D). Furthermore, we assessed the role ofXPR1 in Pi efflux in primary cultures ofproximal tubular cells isolated from kid-neys of DOX-untreated control and cKOmice. Xpr1 deficiency was induced ex vivoby 24 hours of DOXexposure. Twenty fourhours after the end of DOX treatment, theXpr1 mRNA expression was significantlydecreased in the proximal tubular cells iso-lated from cKOmice, as assessed by quan-titative PCR (Xpr1 mRNA expression incKO versus control cells: 18.967.3%;n=3; P=0.01, t test). As shown in Figure1E, proximal tubular cells from cKO micehad a nonsignificant trend toward lower[33Pi]phosphate uptake. In contrast,[33P]phosphate effluxwas strongly affectedby XPR1 deficiency (Figure 1F). The lat-ter correlated with higher percentage of[33P]phosphate remaining in the proximaltubularcells fromcKOmiceafter60minutesof efflux (Figure 1G). Importantly, effluxof[14C]glucose was not different betweenproximal tubular cells isolated from kid-neys of control or cKO mice (Figure 1H),indicating that the short-term ex-vivoXpr1deficiency did not result in the overall de-pression of efflux transport activity. The Piefflux was also evaluated in renal tubules
freshly isolated from kidneys of control orcKO mice treated with DOX for 5 days.As shown in Figure 1I, the 30-minute[33P]phosphate uptake was similar in bothgenotypes, and was significantly reducedin the presence of phosphonoformic acid(PFA), a low potency competitive inhibi-tor of apical Na+/Pi cotransporters. Thepersistent Pi uptake in the presence ofPFA likely results from partial inhibitionof apical Pi transport, but importantly, thefraction of PFA-sensitive Pi uptake wasnot different between genotypes. At theend of the 30-minute uptake period,the [33P]phosphate was removed fromthe bath and Pi efflux was measured. Intubules isolated from kidneys of cKOmice, the Pi efflux was significantlyslower compared with control mice,providing further evidence for anXPR1-mediated Pi efflux. Pi efflux isgenerally considered to occur throughthe basolateral membrane; indeed,an apical Pi efflux is very unlikely be-cause intracellular Pi concentrationremains far below the thermodynamicequilibrium for Na+-dependent Pitransporters.7 Collectively, these exper-iments demonstrate a critical role ofXPR1 in Pi efflux from renal tubularcells, and suggest Xpr1 deficiency asthe primary cause of phosphaturia incKO mice.
Analysis of urine samples revealedthat 1 week after beginning DOX treat-
ment, cKO mice developed generalizedproximal tubule dysfunction, or renalFanconi syndrome, characterized byaminoaciduria (Figure 2A), glycosuria(Figure 2B), albuminuria (Figure 2C),magnesuria (Supplemental Figure 3A),calciuria (Supplemental Figure 3B),lower urinary pH (Supplemental Figure4A), polyuria (Supplemental Figure 4B),and decreased urine osmolality (Supple-mental Figure 4C). Transcriptome analysis(GSE87450) of kidneys from control andcKO mice (males) revealed dramaticchanges in expression levels of RNAs en-coding proteins involved in apical Pi reab-sorption (NaPi-IIa [Slc34a1]: 27.19-fold;NaPi-IIc [Slc34a3]: 225.37-fold), glucosereabsorption (SGLT2 [Slc5a2]: 22.88-fold; GLUT2 [Slc2a2]:22.87-fold), aminoacid transport (LAT2 [Slc7a8]: 24.59-fold; BAT1 [Slc7a9]: 24.24-fold; LAT1[Slc7a7]: 23.36-fold; 4F2hc [Slc3a2]:21.83-fold), and in endocytic receptorsrequired for reuptake of filtered albuminin the proximal tubule (megalin [Lrp2]:23.52-fold; cubilin [Cubn]: 23.40-fold)(Supplemental Table 1). The impairmentin tubular albumin reabsorption was as-sessed functionally by confocal micros-copy analysis of kidney slices preparedfrom kidneys of mice intravenously in-jected with fluorescent albumin (TexasRed albumin). As shown in Figure 2D,Texas Red albumin was abundantly pre-sent in the subapical region of the
Table 1. Plasma chemistry and GFR in control and cKO mice euthanized on day 28 after DOX withdrawal
PlasmaMales Females
Control cKO P Value Control cKO P Value
Osmolality, mosm/kg H2O 325.661.1 (6) 324.665.6 (6) 0.86 324.462.2 (4) 323.862.1 (6) 0.86Ca2+, mM 2.1260.01 (4) 2.1860.02 (5) 0.03 2.1160.02 (5) 2.4160.06 (4) 0.001Na+, mM 156.562.2 (6) 159.964.2 (6) 0.44 158.661.2 (5) 160.962.89 (4) 0.58K+, mM 4.0460.16 (6) 3.6460.20 (3) 0.16 3.7460.29 (5) 3.1960.09 (4) 0.09Creatinine, mM 11.0361.03 (16) 21.2461.68 (15) 1.2631025 14.0361.69 (16) 20.1661.37 (16) 0.01GFR (inulin), ml/min 235613 (5) 147624 (5) 0.01Aldosterone, pg/ml 202.46665.27 (8) 408.25656.76 (8) 0.03 222.02654.06 (9) 506.85678.97 (8) 0.01FGF23, pg/ml 122.95615.57 (5) ,30 (4) 0.001 142.61620.37 (4) ,30 (5) 9.331025
CTX1, pg/ml 5.9560.87 (4) 17.7162.34 (4) 0.003 6.5660.30 (4) 30.31612.85 (4) 0.111.25(OH)2-D3 nmol/ml 88.97613.41 (5) 66.0465.97 (5) 0.16 56.5364.57 (5) 55.8666.13 (5) 0.93PTH, pg/ml 20.2967.30 (6) 34.05611.34 (6) 0.33 39.25612.75 (6) 15.1166.83 (6) 0.12ALP activity, U/L 3.8761.81 (4) 3.7760.75 (4) 0.96 9.0664.43 (4) 7.4863.45 (4) 0.79TRAP, ng/ml 124.13613.02 (4) 92.54612.13 (4) 0.13 98.07611.79 (4) 62.1266.89 (4) 0.03Osteocalcin, ng/ml 66.13618.61 (6) 210.79659.18 (4) 0.03 104.59623.77 (4) 145.92614.79 (3) 0.23Data are means6SEM (n). P values calculated using unpaired t test. CTX1, C-terminal telopeptides of type I collagen; PTH, parathyroid hormone; ALP, alkalinephosphatase; TRAP, tartrate-resistant acid phosphatase.
4 Journal of the American Society of Nephrology J Am Soc Nephrol 28: ccc–ccc, 2016
BRIEF COMMUNICATION www.jasn.org
proximal tubular cells in kidneys of controlmice, whereas the fluorescence intensitywas significantly lower in kidneys of cKOmice.
The kidneys of cKO mice exhibitedreduced expression of genes encodingmitochondrially located proteins (Sup-plemental Figure 5, A and B) despitenormal mitochondrial biogenesis (Sup-plemental Figure 5C) and apparentlynormal mitochondria, as examined byelectron microscopy (Supplemental Fig-ure 5E). TheNAD+/NADH ratiowas sig-nificantly reduced in kidneys of cKOmice, suggesting a shift in the metabolicstatus resulting from the XPR1 defi-ciency (Supplemental Figure 5D).
The GFR was significantly decreasedinmale cKOmice, along with an increasein plasma creatinine levels in cKOmice ofboth sexes (Table 1). The cKO mice ex-hibited slightly higher calcemia, whereasplasma levels of glucose, sodium, and po-tassium, and plasma osmolality were notdifferent from controls (Table 1). Plasmaaldosterone levels were significantly in-creased, suggesting extracellular volumecontraction in cKO mice (Table 1).
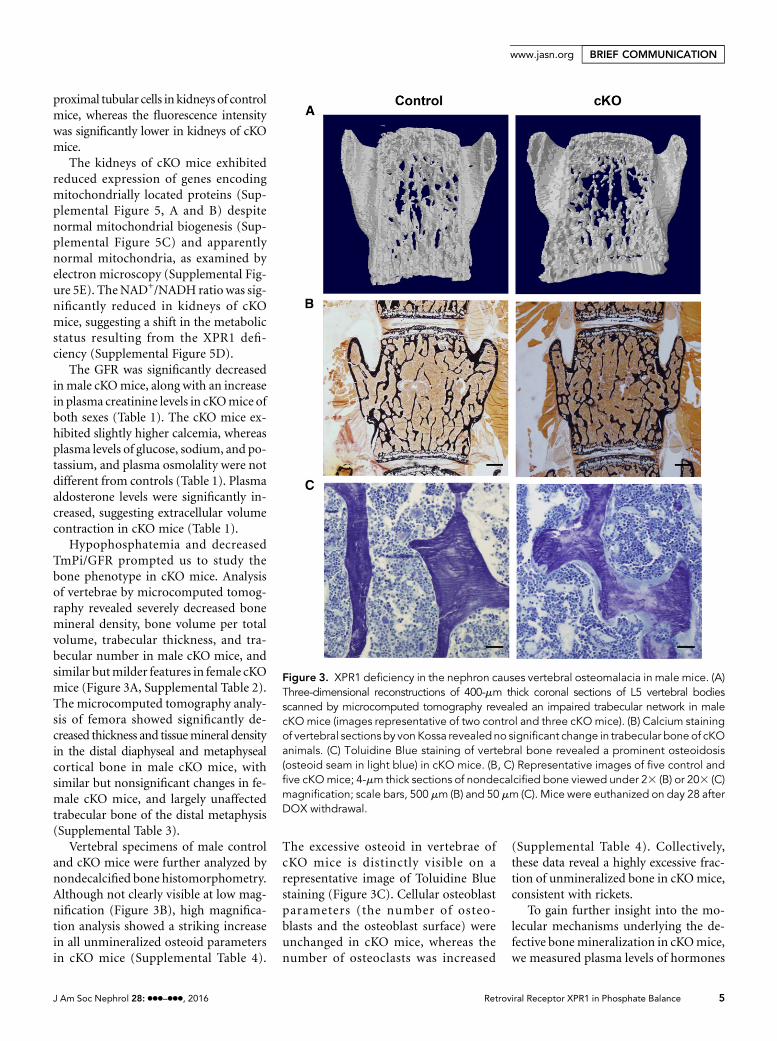
Hypophosphatemia and decreasedTmPi/GFR prompted us to study thebone phenotype in cKO mice. Analysisof vertebrae by microcomputed tomog-raphy revealed severely decreased bonemineral density, bone volume per totalvolume, trabecular thickness, and tra-becular number in male cKO mice, andsimilar butmilder features in female cKOmice (Figure 3A, Supplemental Table 2).The microcomputed tomography analy-sis of femora showed significantly de-creased thickness and tissuemineral densityin the distal diaphyseal and metaphysealcortical bone in male cKO mice, withsimilar but nonsignificant changes in fe-male cKO mice, and largely unaffectedtrabecular bone of the distal metaphysis(Supplemental Table 3).
Vertebral specimens of male controland cKO mice were further analyzed bynondecalcified bone histomorphometry.Although not clearly visible at low mag-nification (Figure 3B), high magnifica-tion analysis showed a striking increasein all unmineralized osteoid parametersin cKO mice (Supplemental Table 4).
The excessive osteoid in vertebrae ofcKO mice is distinctly visible on arepresentative image of Toluidine Bluestaining (Figure 3C). Cellular osteoblastparameters (the number of osteo-blasts and the osteoblast surface) wereunchanged in cKO mice, whereas thenumber of osteoclasts was increased
(Supplemental Table 4). Collectively,these data reveal a highly excessive frac-tion of unmineralized bone in cKOmice,consistent with rickets.
To gain further insight into the mo-lecular mechanisms underlying the de-fective bonemineralization in cKOmice,we measured plasma levels of hormones
Figure 3. XPR1 deficiency in the nephron causes vertebral osteomalacia in male mice. (A)Three-dimensional reconstructions of 400-mm thick coronal sections of L5 vertebral bodiesscanned by microcomputed tomography revealed an impaired trabecular network in malecKOmice (images representative of two control and three cKOmice). (B) Calcium stainingof vertebral sections by von Kossa revealed no significant change in trabecular bone of cKOanimals. (C) Toluidine Blue staining of vertebral bone revealed a prominent osteoidosis(osteoid seam in light blue) in cKO mice. (B, C) Representative images of five control andfive cKOmice; 4-mm thick sections of nondecalcified bone viewed under 23 (B) or 203 (C)magnification; scale bars, 500 mm (B) and 50 mm (C). Mice were euthanized on day 28 afterDOX withdrawal.
J Am Soc Nephrol 28: ccc–ccc, 2016 Retroviral Receptor XPR1 in Phosphate Balance 5
www.jasn.org BRIEF COMMUNICATION

involved in calcium/phosphate homeo-stasis andbone turnover biomarkers (Ta-ble 1). Most strikingly, fibroblast growthfactor 23 (FGF23) levels were undetect-able in cKO mice of both sexes, whereas1,25-dihydroxyvitaminD3 [1,25(OH)2-D3,or calcitriol] and parathyroid hormonelevels were unchanged. Collagen degra-dation product CTX1 was significantlyincreased in male cKO mice, and a non-significant trend in the same direction wasfound in female cKO mice, suggesting anincrease in bone resorption consistentwith the increased osteoclast numbersobserved. However, alkaline phosphataseactivity was unchanged. The levels of theosteoblast-produced hormone osteocalcinwere increased in male cKO mice. To sum-marize, distinct signs of overall altered boneturnover were present in cKO mice.
To conclude, mice deficient for Xpr1 inthe renal tubule develop complete Fanconisyndrome and hypophosphatemic rickets.The severity of renal dysfunction was sim-ilar in cKOmice of both sexes, whereas thebone phenotype was more prominent inmales compared with females, an observa-tion that has been made in human pa-tients.8 Hypophosphatemic ricketsrepresents a heterogeneous entity thatcan be further divided into conditions as-sociated with high FGF23 levels and sup-pressed 1,25(OH)2-D3, such as X-linkedhypophosphatemic rickets and autosomalrecessive hypophosphatemic rickets, orwith low FGF23 and high 1,25(OH)2-D3
levels, found when defects of renal phos-phate transport are present. Indeed, mu-tations of NaPi-IIa and NaPi-IIc, the twosodium-phosphate cotransporters presentin the brush border of the proximal tubule,lead to hereditary hypophosphatemicrickets with hypercalciuria.9,10 Here, weprovide evidence for involvement ofXPR1 in hypophosphatemic rickets asso-ciated with low FGF23 levels and normal1,25(OH)2-D3 levels, reminiscent of he-reditary hypophosphatemic rickets withhypercalciuria. Furthermore, we showthat renal XPR1 is essential for phosphate
homeostasis and bone physiology, andopen new avenues for treatment options.
CONCISE METHODS
Detailed methods are described in the Sup-
plemental Material.
ACKNOWLEDGMENTS
The authors thank Drs. Jean-Luc Battini and
Yves Poirier for helpful discussions, Dr.
Florence Morgenthaler (Cellular Imaging
Facility, University of Lausanne, Lausanne,
Switzerland) for help with microcomputed
tomography analysis, and the Lausanne
Genomic Technologies Facility for tran-
scriptome analysis.
This work was supported by the Swiss
National Science Foundation Research grants
31003A-149440 (to D.F.) and 310030-163340
(to O.B.).
DISCLOSURESNone.
REFERENCES
1. Hamburger D, Rezzonico E, MacDonald-Comber Petétot J, Somerville C, Poirier Y:Identification and characterization of theArabidopsis PHO1 gene involved in phos-phate loading to the xylem. Plant Cell 14:889–902, 2002
2. Stefanovic A, Arpat AB, Bligny R, Gout E,Vidoudez C, Bensimon M, Poirier Y: Over-expression of PHO1 in Arabidopsis leavesreveals its role inmediating phosphate efflux.Plant J 66: 689–699, 2011
3. Giovannini D, Touhami J, Charnet P, SitbonM, Battini JL: Inorganic phosphate export bythe retrovirus receptor XPR1 in metazoans.Cell Reports 3: 1866–1873, 2013
4. Wege S, Poirier Y: Expression of the mam-malian Xenotropic Polytropic Virus Receptor1 (XPR1) in tobacco leaves leads to phos-phate export. FEBS Lett 588: 482–489, 2014
5. Legati A, Giovannini D, Nicolas G, López-Sánchez U, Quintáns B, Oliveira JR, Sears RL,
Ramos EM, Spiteri E, Sobrido MJ, CarracedoÁ, Castro-Fernández C, Cubizolle S, FogelBL, Goizet C, Jen JC, Kirdlarp S, Lang AE,Miedzybrodzka Z, Mitarnun W, Paucar M,Paulson H, Pariente J, Richard AC, Salins NS,Simpson SA, Striano P, Svenningsson P,Tison F, Unni VK, Vanakker O, Wessels MW,Wetchaphanphesat S, Yang M, Boller F,Campion D, Hannequin D, Sitbon M,Geschwind DH, Battini JL, Coppola G: Mu-tations in XPR1 cause primary familial braincalcification associated with altered phos-phate export. Nat Genet 47: 579–581,2015
6. Traykova-Brauch M, Schönig K, Greiner O,Miloud T, Jauch A, Bode M, Felsher DW,Glick AB, Kwiatkowski DJ, Bujard H, Horst J,von Knebel Doeberitz M, Niggli FK, Kriz W,Gröne HJ, Koesters R: An efficient and ver-satile system for acute and chronic modula-tion of renal tubular function in transgenicmice. Nat Med 14: 979–984, 2008
7. Freeman D, Bartlett S, Radda G, Ross B: En-ergetics of sodium transport in the kidney.Saturation transfer 31P-NMR. Biochim Bio-phys Acta 762: 325–336, 1983
8. Beck-Nielsen SS, Brusgaard K, RasmussenLM, BrixenK, Brock-JacobsenB, PoulsenMR,Vestergaard P, Ralston SH, Albagha OM,Poulsen S, Haubek D, Gjørup H, Hintze H,Andersen MG, Heickendorff L, Hjelmborg J,Gram J: Phenotype presentation of hypo-phosphatemic rickets in adults. Calcif TissueInt 87: 108–119, 2010
9. Lorenz-Depiereux B, Benet-Pages A, EcksteinG, Tenenbaum-Rakover Y, Wagenstaller J,Tiosano D, Gershoni-Baruch R, Albers N,Lichtner P, Schnabel D, Hochberg Z, StromTM: Hereditary hypophosphatemic ricketswith hypercalciuria is caused by mutations inthe sodium-phosphate cotransporter geneSLC34A3. Am J Hum Genet 78: 193–201,2006
10. Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, FrappierD, Burkett K, Carpenter TO, Anderson D,Garabedian M, Sermet I, Fujiwara TM,Morgan K, Tenenhouse HS, Juppner H:SLC34A3 mutations in patients with he-reditary hypophosphatemic rickets withhypercalciuria predict a key role for thesodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeosta-sis. Am J Hum Genet 78: 179–192, 2006
This article contains supplemental material onlineat http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2016070726/-/DCSupplemental.
6 Journal of the American Society of Nephrology J Am Soc Nephrol 28: ccc–ccc, 2016
BRIEF COMMUNICATION www.jasn.org
![The Difficult Diagnosis of Hypophosphatemic Rickets-A Review of …article.clinicalmed.org/pdf/10.11648.j.cmr.20200905.11.pdf · D-resistant rickets/osteomalacia [5]. 2. Material](https://img.pdfslide.net/doc/110x75/60407e73e4edd922d0572ba6/the-difficult-diagnosis-of-hypophosphatemic-rickets-a-review-of-d-resistant-ricketsosteomalacia.jpg)




![y roi d Dis Thyroid Disorders Ther 2012, 1:2 d T h ers l n ...€¦ · may be the late manifestation of hypophosphatemic rickets in adults [7-10]. These can be excluded as etiology](https://img.pdfslide.net/doc/110x75/6068ebbefc79904c7e27223b/y-roi-d-dis-thyroid-disorders-ther-2012-12-d-t-h-ers-l-n-may-be-the-late-manifestation.jpg)























