Embed Size (px)
Citation preview

of April 11, 2018.This information is current as
CytokinesParticulate Antigen Modulates Murine Lung Repeated Intratracheal Challenge with
Seitzman, Bin Hu and Jeffrey L. CurtisJill Todt, Joanne Sonstein, Timothy Polak, Gerami D.
http://www.jimmunol.org/content/164/8/4037doi: 10.4049/jimmunol.164.8.4037
2000; 164:4037-4047; ;J Immunol
Referenceshttp://www.jimmunol.org/content/164/8/4037.full#ref-list-1
, 27 of which you can access for free at: cites 76 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2000 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on April 11, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on April 11, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Repeated Intratracheal Challenge with Particulate AntigenModulates Murine Lung Cytokines1,2
Jill Todt,* Joanne Sonstein,* Timothy Polak,* Gerami D. Seitzman,* Bin Hu,* andJeffrey L. Curtis 3*†
When lungs of experimental animals are repeatedly challenged with Ag, pulmonary inflammation wanes via unknown mecha-nisms. We hypothesized that changes in the balance of lung cytokines are responsible for immune down-regulation to repeated Agchallenge. We used intratracheal (IT) challenge of primed C57BL/6 mice with SRBC and on various days after single (1IT) ortriple (3IT) challenge counted lung inflammatory cells and measured whole-lung cytokine mRNA and protein concentrations usingRT-PCR and ELISA. We found that lung lymphocyte numbers and parenchymal lung inflammation decreased significantly atdays 6 and 9 after final Ag challenge in 3IT mice compared with 1IT mice. Lungs of 3IT mice showed the following changes inrelative mRNA expression: an earlier peak in IL-10, decreased IL-1b, and a change from a Th2 response in 1IT mice to a Th1response in 3IT mice (with pronounced increases in IL-12, IL-18, and IFN-g and decreased IL-4, IL-13, and IL-5). Similar typesof changes were seen in whole-lung protein concentrations for TNF-a, IL-10, IL-12 p40, IFN- g, and IL-4. Additionally, mRNAexpression of the endothelial selectins CD62E and CD62P decreased and lung lymphocyte apoptosis increased in the 3IT group.Thus, physiologic down-regulation of the pulmonary immune response to repeated Ag exposure is characterized by increased anti-and decreased proinflammatory cytokines that accompanies Th1 polarization. Similar mechanisms may act to minimize chroniclung inflammation in the majority of normal humans who do not develop progressive lung pathology when repeatedly exposed toinhaled or aspirated environmental Ags. The Journal of Immunology,2000, 164: 4037–4047.
T he pathogenesis of common immunologic lung diseases isknown or suspected to involve inappropriately regulatedpulmonary immune responses to inhaled Ags. Because
many of the causative Ags are ubiquitous or unidentified, Agavoidance cannot be relied upon as the sole therapeutic strategy.Current therapies for immunologic lung diseases are only partiallyeffective and in many cases are toxic and expensive. To developnovel immunomodulatory therapies, the mechanisms that success-fully regulate physiologic pulmonary immune responses must bedefined in greater detail. Definition of these mechanisms cannotrely solely on extrapolation from in vitro studies but instead ulti-mately requires analysis of immune responses in the lungs of intactanimals or patients.
To this purpose, we and others have analyzed the response ofAg-primed animals of a variety of species to intratracheal (IT)4
challenge with the prototypic particulate T cell Ag, the SRBC (1).The most prevalent model system uses inbred mice, predominantlyof the C57BL/6 or A/J strains, which are high responders to SRBC(2–4). This experimental model system has been used to analyzethe anatomic and molecular mechanisms of lymphocyte recruit-ment to lung parenchyma (5–7), the cytokine requirements for air-ways hyperresponsiveness (AHR) (8), and the role of neuropep-tides in development of lung inflammation (9). Lung inflammationin this model system depends crucially on recruitment of func-tional CD41 cells, which enter the lung via both VLA-4-dependentand selectin-dependent pathways (10, 11). Net lung lymphocyteaccumulation depends minimally on in situ lymphocyte prolifera-tion, but is associated with prominent lung eosinophilia (12). Wehave observed previously that inflammatory cell recruitment is ac-companied by transient angiitis, alveolar macrophage activation,and in situ maturation of specific Ab-secreting cells; however fi-brosis is not seen (5, 13). Thus, this experimental model systemshares many of the characteristics of relevant human immunologiclung diseases. Importantly, however, the response is self-limitingwithin 3 wk (3, 5).
A key question in pulmonary immunology is why a minority ofindividuals develop progressive lung inflammation to ubiquitousinhaled agents that are ignored by the immune systems of mostexposed individuals. One potential clue to this puzzle comes fromanimal models that indicate that the pulmonary immune responsetypically decreases spontaneously during repeated pulmonary Agchallenge of normal animals (14–17). Thus, normal animals ap-pear to mimic the natural response of most healthy humans toinhalation of noninjurious Ags. The molecular basis for this wan-ing response during continued Ag exposure remains unknown de-spite considerable study. Evidence against a variety of mechanismshas been provided. The normally antiproliferative effect of alveolarmacrophages on activated lymphocytes in vitro is not decreased on
*Division of Pulmonary and Critical Care Medicine, Department of Internal Medi-cine, University of Michigan Medical School, and†Pulmonary and Critical CareMedicine Section, Medical Service, Department of Veterans Affairs Medical Center,Ann Arbor, MI 48105
Received for publication June 28, 1999. Accepted for publication February 3, 2000.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby markedadvertisementin accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by research funds and a REAP center from the Departmentof Veterans Affairs, Grant RO1 HL56309 from the U.S. Public Health Service, and bythe University of Michigan Medical School Student Biomedical Research Fund (toG.D.S.). J.L.C. is a Career Investigator of the American Lung Association ofMichigan.2 Portions of these data were presented at the International Scientific Conference ofthe American Thoracic Society (San Diego, CA; April 27, 1999) and have beenpublished in abstract form (1999 Am. J. Respir. Crit. Care Med. 159:A660).3 Address correspondence and reprint requests to Dr. Jeffrey L. Curtis, Pulmonary andCritical Care Medicine Section (111G), Department of Veterans Affairs Medical Cen-ter, 2215 Fuller Road, Ann Arbor, MI 48105-2303. E-mail address: [email protected] Abbreviations used in this paper: IT, intratracheal; 1IT, group of mice which re-ceived a single Ag challenge; 3IT, group of mice which received three Ag challenges
at weekly intervals; AHR, airways hyperresponsiveness; BAL, bronchoalveolarlavage.
Copyright © 2000 by The American Association of Immunologists 0022-1767/00/$02.00
by guest on April 11, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

repeated challenge of rabbits or mice withMicropolyspora faeni(18, 19). Peripheral lymphocyte tolerance does not occur usingmultiple IT challenges ofM. faeni in rabbits orThermoactinomy-ces vulgarisAg in mice (20, 21), and T suppresser activity is notenhanced on repeated challenge of mice withT. vulgarisAg (21).However, because T cells can differentiate in vivo into immune orsuppressor cells depending upon the nature and dosage of Ag (22–24), oral tolerance cannot be totally eliminated as an explanationfor the waning response to multiple challenge.
We have previously found that termination of the pulmonaryimmune response to a single IT SRBC challenge does not requireCD81 T cells, but does involve considerable lymphocyte apoptosiswithin the alveolar space and lung interstitium (13, 25). We alsofound that, at the single time-point tested, the magnitude of lunginflammation and lymphocyte accumulation after three IT Ag chal-lenges is markedly less than after a single IT Ag challenge (25).Previous experiments showed that multiple i.p. priming exposuresto SRBC did not appreciably alter lung lymphocyte numbers aftersingle IT challenge (12), excluding the trivial explanation of ex-haustion of the systemic immune response or systemic tolerance.These findings suggested that a variation of our well-characterizedmodel system could be studied to understand the mechanism ofthis previously described physiologic down-regulation seen inmany animal models of pulmonary hypersensitivity. The goal ofthis study was to further characterize the down-regulation of lunginflammation and of lymphocyte accumulation on repeated IT Agchallenge, which we hypothesized would result from decreasedproduction of proinflammatory cytokines within the lungs. Wefound that repeated Ag challenge was associated with up-regula-tion of antiinflammatory IL-10 as well as a down-regulation of E-and P-selectin mRNA. These changes were accompanied by a shiftfrom a predominantly Th2 cytokine response to a distinct Th1response.
Materials and MethodsAnimals
Pathogen-free female C57BL/6J mice were obtained at 7–8 wks of agefrom Charles River Laboratory (Wilmington, MA). Mice were housed inthe Animal Care Facility at the Ann Arbor Veterans Affairs Medical Cen-ter, which is fully accredited by the American Association for Accredita-tion of Laboratory Animal Care. This study complied with the NationalInstitutes of Health “Guide for the Care and Use of Laboratory Animals”(Department of Health, Education, and Welfare Publication No. (NationalInsitutes of Health) 80-23). Mice were fed standard animal chow (RodentLab Chow 5001; Purina, St. Louis, MO) and chlorinated tap water adlibitum. Mice were used at 8–14 wks of age.
Monoclonal Abs
The following mAbs were purchased from PharMingen (San Diego, CA):RM4-4 (anti-murine CD4; rat IgG2b), 53-6.72 (anti-murine CD8; ratIgG2b), 1D3 (anti CD19), RM2-5 (anti CD2), 7D4 (anti-murine CD25, ratIgM), 145-2C-11 (hamster anti-murine CD3), and H1.2F3 (anti-murineCD69, hamster IgG). Monoclonal Abs were either directly FITC conju-gated or biotinylated; in the latter case staining was visualized usingstreptavidin-PE (PharMingen). Isotype-matched irrelevant control Abs(107.3 (anti-TNP control mouse IgG1,k), anti-TNP hamster IgG, and ratIgM, k (PharMingen)) were tested simultaneously to exclude nonspecificstaining.
Induction of pulmonary immune response
SRBC (sheep 4151) (Colorado Serum, Denver, CO) were washed threetimes in normal saline before use. Mice were Ag primed by i.p. injectionwith 1 3 108 SRBC in 0.5 ml normal saline. Beginning 2 wks later, micewere IT challenged with 53 108 SRBC in 50ml normal saline as previ-ously described (7) either once (1IT group) or three times with 1 wk be-tween challenges (3IT group) (Fig. 1). Two control experiments were per-formed (Fig. 1). In control A, 4 wk elapsed between i.p. priming and ITchallenge. This experimental group was used to control for the differencebetween the 1IT and 3IT groups in the time from initial priming to final IT
challenge. In control B, the first two of the 3IT challenges used the samevolume of normal saline rather than SRBC. This experimental group wasused to control for the repeated immunization procedures to which the 3ITgroup was subjected. In a single additional control experiment not depictedin Fig. 1, SRBC-primed mice were IT challenged a single time with 50mlsaline containing India ink (Pelikan, Hannover, Germany), and lungs wereharvested 6 days later. This group was used to analyze the effect of theimmunization procedure at the site of Ag deposition.
Sample collection
At various times from 0–14 d after the final IT Ag challenge, mice weredeeply anesthetized with pentobarbital (80 mg/kg i.p.) and killed by ex-sanguination and induction of bilateral pneumothoraces. Lungs were thenprocessed in one of three ways. First, to obtain lung inflammatory cells,some lungs were perfused via the right heart using normal saline, and thenbronchoalveolar lavage (BAL) and total and differential cell counts wereperformed as previously described (3). Aliquots of these cells were stainedwith mAbs and analyzed on a FACScanflow cytometer (Becton Dickinson,Mountain View, CA) as previously described (26), except that CellQuestsoftware (Becton Dickinson) was used. Second, for histologic analysissome lungs were inflated using 1 ml 10% neutral-buffered formalin, andthen the entire thoracic contents were dissected free and fixed by immer-sion in 10% formalin in PBS for 18–24 h. Parasagittal sections throughthese fixed lungs were then cut, embedded in paraffin, and sectioned at 5mm thickness. The slides, each consisting of sections of both lungs from anindividual mouse, were stained with hematoxylin and eosin and with Mas-son’s trichrome stains. Third, for samples to be analyzed for total lungcytokine mRNA or protein production, lungs that were perfused but notlavaged were carefully dissected to exclude any extrapleural lymphatictissue. These lungs, separated from the mainstem bronchi at the medialpleural surface, were snap frozen in liquid nitrogen and stored at270°Cuntil processed as described below.
FIGURE 1. Experimental protocols. The horizontal axis denotes time indays, synchronized such that the day of the final IT challenge is day zerofor each group. Arrows above the line denote Ag administration by theintraperitoneal (IP) or intratracheal (IT) route, whereas arrows below theline indicate days on which groups of mice were euthanized for assay.
4038 LUNG CYTOKINES IN REPEATED PULMONARY Ag CHALLENGE
by guest on April 11, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
Apoptosis measurement
Apoptotic lymphocytes were measured using the Apoptosis Detection Kit(R&D Systems, Minneapolis, MN) according to the manufacturer’s in-structions. This method uses flow cytometry to identify apoptotic cells asthose that bind annexin V (27) and exclude propidium iodide.
Isolation of RNA
Lungs were homogenized in 2 ml of TRIzol reagent (Life Technologies,Grand Island, NY), and RNA was isolated as described in the TRIzolprotocol. RNA was quantitated spectrophotometrically. The integrity ofindividual RNA samples was confirmed by electrophoresing aliquots on a2% agarose gel containing 0.5mg/ml ethidium bromide and observing 28Sand 18S rRNA bands. RNA samples were stored at270°C.
RT-PCR detection of cytokine mRNA
Isolated RNA was reverse transcribed to DNA as follows. To 20mg RNAwas added 24.4ml of mix A (403 ml RNase inhibitor (Life Technologies;10 U/ml) and 280ml of random primers (Life Technologies, 3mg/ml)),which was heated at 70°C for 2 min in a programmable thermocycler(Perkin-Elmer 9600, Norwalk, CT). The temperature was reduced to 4°Cand 68.9ml of mix B (812 ml of double distilled water, 300ml of 0.1 MDTT, 75ml of 20 mM each dNTP, 602ml of 53 first-strand buffer, and 140ml of 200 U/ml Moloney murine leukemia virus reverse transcriptase (allfrom Life Technologies)) was added. RNA was transcribed in this mixturefor 1 h at 42°C, and then the reaction was stopped by a 5-min incubationat 95°C.
The DNA was subjected to PCR as follows. In a thin-walled PCR tube,5 ml of sample was added to 20ml of PCR buffer containing 5 nM of eachdNTP, 50 nM MgCl2, 1.0 U of TaqDNA polymerase (all from Life Tech-nologies), and 0.15 ng each of sense and anti-sense primers. Amplificationwas then performed in a thermocycler as follows: 5 min at 95°C, followedby up to 35 cycles of 15 s each at 95°C, 20 s at 58°C, 30 s at 72°C. Aftercycling, there was a DNA extension period of 6 min at 72°C. The amplifiedDNA was analyzed by electrophoresis on 1.5% agarose containing 0.5mg/ml ethidium bromide.
The primer sequences used are defined in Table I. Primers were de-signed based upon nucleotide sequences downloaded from the National
Center for Biotechnology Information data bank, using primer design soft-ware (Primer 2; Scientific & Educational Software, State Line, PA). Allprimers were prepared by the University of Michigan DNA Core Facility(Ann Arbor, MI). PCR conditions and cycle number were defined for eachcytokine primer pair such that a linear relationship between input RNA andfinal PCR product (defined as OD signal) was obtained. Positive and neg-ative controls were included in each assay to establish that only cDNAPCR products were detected and that none of the reagents were contami-nated with cDNA or extraneous PCR products. Authenticity of reactionproducts was verified by Southern analysis using a chemiluminescent de-tection system (Amersham Life Science, Little Chalfont, U.K.).
To quantitate cDNA bands, the ethidium bromide-stained agarose gelswere photographed using Polaroid 667 film, scanned using a Scan Jet IIcx
FIGURE 2. Kinetics of lung lymphocyte accumulation. Lung lympho-cytes were collected by BAL of SRBC-primed C57BL/6 mice at variousdays after final IT SRBC challenge. 1IT,f; 3IT, L. Absolute lymphocytenumbers were determined as the product of total BAL cell count (by he-mocytometer) and lymphocyte percentage on differential cell count of he-matoxylin and eosin-stained preparations. Data represent mean6 SEM ofat least three mice assayed in two to five experiments per time point;p, p ,0.05, unpaired Studentt test.
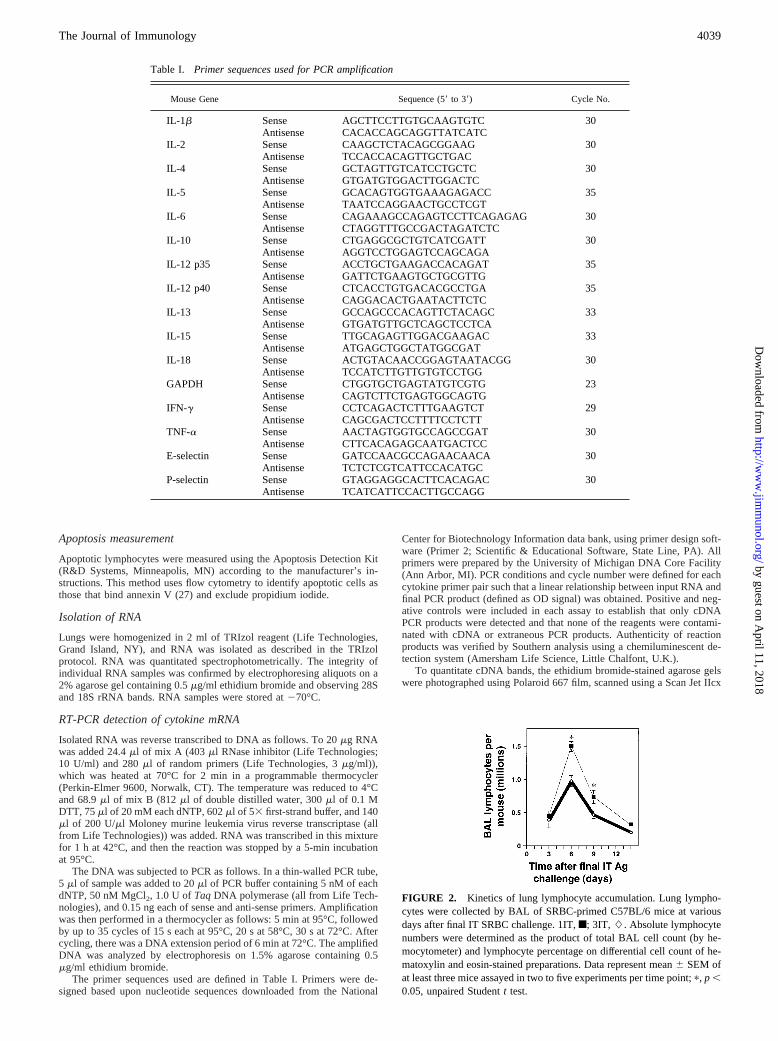
Table I. Primer sequences used for PCR amplification
Mouse Gene Sequence (59to 39) Cycle No.
IL-1b Sense AGCTTCCTTGTGCAAGTGTC 30Antisense CACACCAGCAGGTTATCATC
IL-2 Sense CAAGCTCTACAGCGGAAG 30Antisense TCCACCACAGTTGCTGAC
IL-4 Sense GCTAGTTGTCATCCTGCTC 30Antisense GTGATGTGGACTTGGACTC
IL-5 Sense GCACAGTGGTGAAAGAGACC 35Antisense TAATCCAGGAACTGCCTCGT
IL-6 Sense CAGAAAGCCAGAGTCCTTCAGAGAG 30Antisense CTAGGTTTGCCGACTAGATCTC
IL-10 Sense CTGAGGCGCTGTCATCGATT 30Antisense AGGTCCTGGAGTCCAGCAGA
IL-12 p35 Sense ACCTGCTGAAGACCACAGAT 35Antisense GATTCTGAAGTGCTGCGTTG
IL-12 p40 Sense CTCACCTGTGACACGCCTGA 35Antisense CAGGACACTGAATACTTCTC
IL-13 Sense GCCAGCCCACAGTTCTACAGC 33Antisense GTGATGTTGCTCAGCTCCTCA
IL-15 Sense TTGCAGAGTTGGACGAAGAC 33Antisense ATGAGCTGGCTATGGCGAT
IL-18 Sense ACTGTACAACCGGAGTAATACGG 30Antisense TCCATCTTGTTGTGTCCTGG
GAPDH Sense CTGGTGCTGAGTATGTCGTG 23Antisense CAGTCTTCTGAGTGGCAGTG
IFN-g Sense CCTCAGACTCTTTGAAGTCT 29Antisense CAGCGACTCCTTTTCCTCTT
TNF-a Sense AACTAGTGGTGCCAGCCGAT 30Antisense CTTCACAGAGCAATGACTCC
E-selectin Sense GATCCAACGCCAGAACAACA 30Antisense TCTCTCGTCATTCCACATGC
P-selectin Sense GTAGGAGGCACTTCACAGAC 30Antisense TCATCATTCCACTTGCCAGG
4039The Journal of Immunology
by guest on April 11, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
(Hewlett Packard, Palo Alto, CA), and analyzed on a Macintosh 8100/80AV computer using the public domain software program NIH Image(version 1.60; developed at the National Institutes of Health and availableon the Internet at http://rsb.info.nih.gov/nih-image/). Results were ex-pressed as a ratio of OD signal for a given cytokine to that for GAPDH, andfold increases were calculated relative to unchallenged mouse lung.
ELISA
Lungs frozen at280°C were homogenized in 1 ml of PBS and centrifugedat 1500 rpm. The supernatant was aliquoted and stored at280°C. Thesesamples were analyzed for IFN-g, IL-4, TNF-a, IL-12 p40, and IL-10using PharMingen kits according the manufacturer’s instructions. OD read-ings of samples were converted to picograms per milliliter using standardcurves generated with varying concentrations of recombinant cytokine sup-plied with the kit. The limit of detection was 31.2 pg/ml for each assay.
Statistical analysis
Data were expressed as mean6 SEM. Statistical calculations were per-formed using Statview (Abacus Concepts, Berkeley, CA) on a MacintoshPowerPC G3 computer. Differences between the groups of mice in con-tinuous ratio scale data at individual time points after final IT challenge
were evaluated using the unpaired Studentt test (28). Differences in cyto-kine levels between the groups of mice were evaluated using Mann-Whit-ney nonparametric unpaired analysis. Significant differences were definedasp , 0.05.
ResultsLung inflammation wanes on repeated IT Ag challenge
Lung lymphocyte numbers peaked at day 6 after single Ag chal-lenge and then progressively decreased, in agreement with previ-ous data in this model system. Compared with the 1IT group, lym-phocyte numbers were significantly decreased in the 3IT group ondays 6 and 9 after final IT challenge, but not during early inflam-mation (day 3) or on day 14, when inflammation in both groupswas largely resolved (Fig. 2). Histologic analysis showed a de-crease in the extent but no discernible change in the anatomicdistribution of inflammatory cell accumulation after 3IT (Fig. 3,A–D). Inflammatory cell accumulation in both groups was chieflyassociated with the bronchovascular bundles and pulmonary veins,
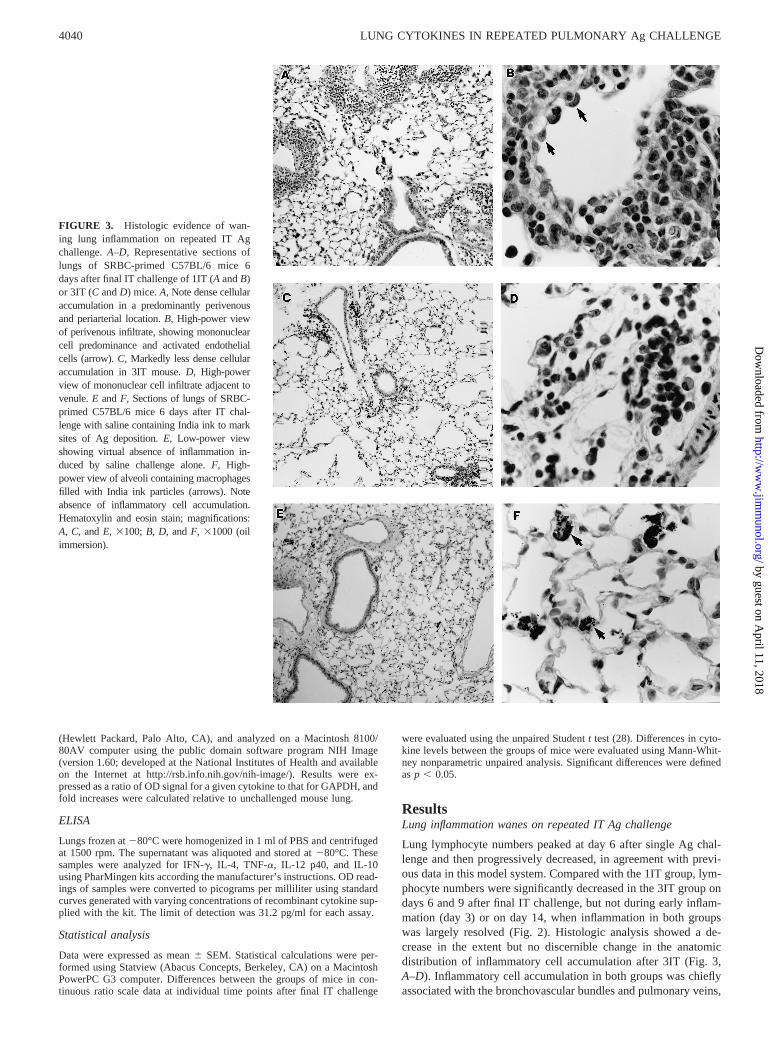
FIGURE 3. Histologic evidence of wan-ing lung inflammation on repeated IT Agchallenge.A–D, Representative sections oflungs of SRBC-primed C57BL/6 mice 6days after final IT challenge of 1IT (AandB)or 3IT (C andD) mice.A, Note dense cellularaccumulation in a predominantly perivenousand periarterial location.B, High-power viewof perivenous infiltrate, showing mononuclearcell predominance and activated endothelialcells (arrow).C, Markedly less dense cellularaccumulation in 3IT mouse.D, High-powerview of mononuclear cell infiltrate adjacent tovenule.E andF, Sections of lungs of SRBC-primed C57BL/6 mice 6 days after IT chal-lenge with saline containing India ink to marksites of Ag deposition.E, Low-power viewshowing virtual absence of inflammation in-duced by saline challenge alone.F, High-power view of alveoli containing macrophagesfilled with India ink particles (arrows). Noteabsence of inflammatory cell accumulation.Hematoxylin and eosin stain; magnifications:A, C, andE, 3100; B, D, andF, 31000 (oilimmersion).
4040 LUNG CYTOKINES IN REPEATED PULMONARY Ag CHALLENGE
by guest on April 11, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

as previously reported (5, 7). Examination of trichrome-stainedsections did not reveal any evidence of fibrosis. Histologic analysisof the lungs of mice challenged solely with saline containing Indiaink to detect the site of Ag deposition did not disclose inflamma-tory cell accumulation (Fig. 3,E and F). Thus, kinetic analysisindicated that, in this model system as in other models (14–18, 20,21), repeated intrapulmonary Ag deposition led to a diminution ofthe lung inflammatory cell response.
In control experiments that compared timing of the initial i.p.Ag priming at 2 wk (Fig. 1, 1 IT) or 4 wk (Fig. 1, Control A)before IT SRBC injection, there were no significant differences innumbers of total BAL inflammatory cells, macrophages, or lym-phocytes (data not shown). There were also no differences in BALcell numbers or types when comparing experiments with 1IT chal-lenge and 3IT challenge where the first two injections were saline(Fig. 1, Control B) (data not shown). These results indicate thatmultiple surgeries or variations in timing of i.p. injections were notthe causes of differences between 1IT- and 3IT-challenged mice inlung lymphocyte numbers.
Lung lymphocyte phenotypes change minimally after repeatedAg challenge
Flow cytometric analysis of lung lymphocyte phenotypes showedsignificant differences only at day 9 after final IT challenge, when3IT mice had a decreased percentage of CD191 cells and increasedpercentage of CD81 cells (Fig. 4). Overall, there was a trend to-ward an increasing fraction of CD41 and toward a decreasing frac-tion of CD191 cells in the 3IT group, both with time after chal-lenge and relative to the 1IT group, but these differences did notattain statistical significance. We and others have previously re-ported that a large fraction of lymphocytes in the lungs of immu-nized mice are negative for most subpopulation markers (3, 29). Inthe current study, the percentage of such null cells (defined asCD21, CD42, CD82, and CD192 cells within light-scatter gatescharacteristic of lymphocytes) did not differ between the twogroups. Thus, no systematic change in the distribution of lunglymphocyte subsets between the two groups was consistently seenover the course of the immune response.
To investigate the degree of T cell activation in the two treat-ment groups, in some experiments CD41 and CD81 cells weredouble stained with mAb against CD25 or CD69 (Table II). In allbut two of the comparisons, a greater percentage of both CD41
cells and CD81 cells were positive for these activation receptors inthe 3IT group, although the difference was statistically significantfor only five of the 12 conditions tested.
Down-regulation of lung inflammation on repeated Ag challengeis characterized by a decrease in proinflammatory cytokines andan increase in IL-10
To determine which cytokines may be involved in down-regula-tion of the immune response in 3IT-challenged mice, we deter-mined relative levels of whole-lung cytokine mRNA by RT-PCRand of selected whole-lung cytokine protein concentrations byELISA. In the remainder of this paper, significant differences be-tween 1IT and 3IT mice are only mentioned if results of 1IT aswell as control A and control B mice are similarly significantlydifferent from those of 3IT mice. In the 1IT groups, relative mRNAlevels of the proinflammatory cytokine IL-1b was actually ele-vated at the time of initial IT challenge, perhaps reflecting residualchanges from previous i.p. priming (Fig. 5). IL-1b levels weresignificantly higher in the 1IT and control groups relative to 3IT atday 1. TNF-a mRNA levels were greater in 1IT and control groupsthan 3IT levels at early time points but did not reach significance.Relative mRNA levels of the antiinflammatory cytokine IL-10peaked earlier in 3IT mice, at day 1 compared with days 3–4 in
FIGURE 4. Lung lymphocyte phenotypes. Phenotypes of BAL lympho-cytes, collected as described in Fig. 2, were determined by direct immu-nofluorescence staining and flow cytometry. 1IT, dark hatching, 3IT, lighthatching. Data are expressed as the percentage of each phenotype at var-ious days after final IT challenge. Note differences in scales. Each pointrepresents the mean and SEM of four to eight individual mice assayed intwo experiments for days 6 and 9 or three samples of pooled cells from atleast six mice each for day 14.p, p , 0.05, unpaired Studentt test.
Table II. Lung T cell activation receptor expression increases on repeated Ag challenge
Subset Receptor Daya 1IT Group 3IT Group Significanceb
CD4 CD25 6 36.26 1.6c 42.96 2.8 NS9 36.76 1.7 47.36 1.5 p , 0.05
14 17.16 1.8 19.76 2.7 NSCD4 CD69 6 39.16 3.8 38.66 1.3 NS
9 43.16 3.5 54.96 2.8 NS14 19.96 2.3 39.66 1.1 p , 0.05
CD8 CD25 6 17.36 1.4 29.46 2.3 NS9 10.86 1.4 40.36 3.8 p , 0.05
14 5.86 0.1 5.36 0.2 NSCD8 CD69 6 35.66 0.0 52.16 3.3 p , 0.05
9 62.76 0.1 68.86 2.5 NS14 48.46 0.1 70.46 4.0 p , 0.05
a Day after final IT Ag challenge.b As determined by unpaired Studentt test.c Percentage positive (mean6 SEM) as determined by immunofluorescent staining and flow cytometry.
4041The Journal of Immunology
by guest on April 11, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
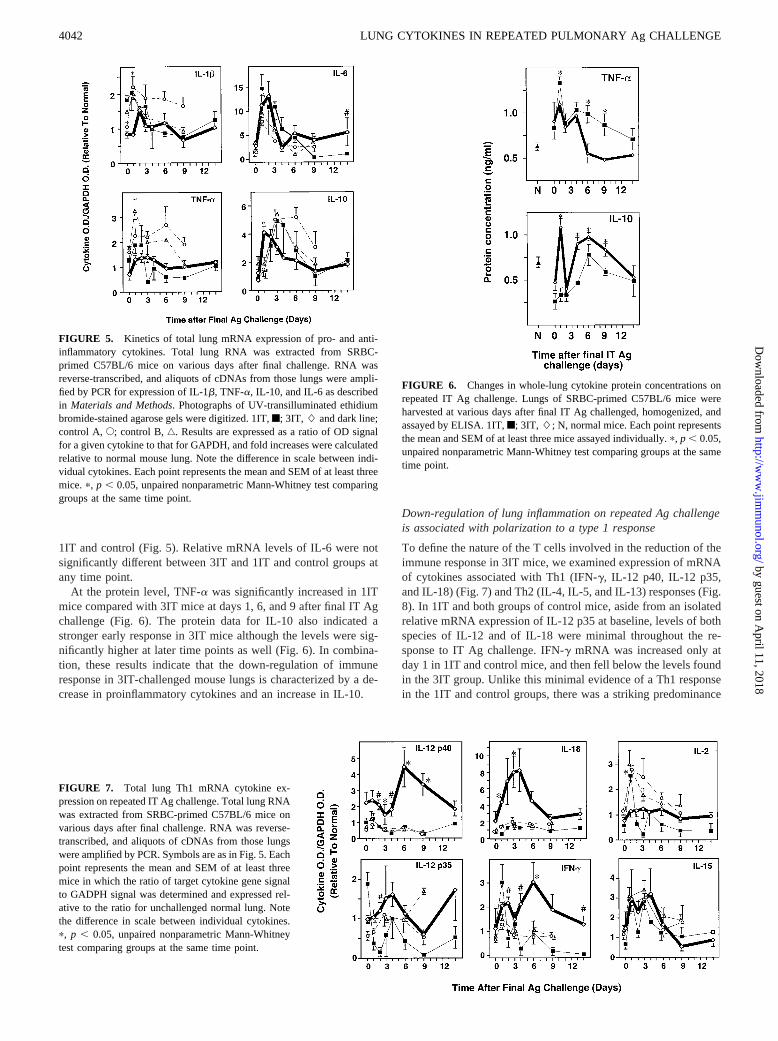
1IT and control (Fig. 5). Relative mRNA levels of IL-6 were notsignificantly different between 3IT and 1IT and control groups atany time point.
At the protein level, TNF-a was significantly increased in 1ITmice compared with 3IT mice at days 1, 6, and 9 after final IT Agchallenge (Fig. 6). The protein data for IL-10 also indicated astronger early response in 3IT mice although the levels were sig-nificantly higher at later time points as well (Fig. 6). In combina-tion, these results indicate that the down-regulation of immuneresponse in 3IT-challenged mouse lungs is characterized by a de-crease in proinflammatory cytokines and an increase in IL-10.
Down-regulation of lung inflammation on repeated Ag challengeis associated with polarization to a type 1 response
To define the nature of the T cells involved in the reduction of theimmune response in 3IT mice, we examined expression of mRNAof cytokines associated with Th1 (IFN-g, IL-12 p40, IL-12 p35,and IL-18) (Fig. 7) and Th2 (IL-4, IL-5, and IL-13) responses (Fig.8). In 1IT and both groups of control mice, aside from an isolatedrelative mRNA expression of IL-12 p35 at baseline, levels of bothspecies of IL-12 and of IL-18 were minimal throughout the re-sponse to IT Ag challenge. IFN-g mRNA was increased only atday 1 in 1IT and control mice, and then fell below the levels foundin the 3IT group. Unlike this minimal evidence of a Th1 responsein the 1IT and control groups, there was a striking predominance
FIGURE 6. Changes in whole-lung cytokine protein concentrations onrepeated IT Ag challenge. Lungs of SRBC-primed C57BL/6 mice wereharvested at various days after final IT Ag challenged, homogenized, andassayed by ELISA. 1IT,f; 3IT, L; N, normal mice. Each point representsthe mean and SEM of at least three mice assayed individually.p, p , 0.05,unpaired nonparametric Mann-Whitney test comparing groups at the sametime point.
FIGURE 5. Kinetics of total lung mRNA expression of pro- and anti-inflammatory cytokines. Total lung RNA was extracted from SRBC-primed C57BL/6 mice on various days after final challenge. RNA wasreverse-transcribed, and aliquots of cDNAs from those lungs were ampli-fied by PCR for expression of IL-1b, TNF-a, IL-10, and IL-6 as describedin Materials and Methods. Photographs of UV-transilluminated ethidiumbromide-stained agarose gels were digitized. 1IT,f; 3IT, L and dark line;control A,E; control B,‚. Results are expressed as a ratio of OD signalfor a given cytokine to that for GAPDH, and fold increases were calculatedrelative to normal mouse lung. Note the difference in scale between indi-vidual cytokines. Each point represents the mean and SEM of at least threemice.p, p , 0.05, unpaired nonparametric Mann-Whitney test comparinggroups at the same time point.
FIGURE 7. Total lung Th1 mRNA cytokine ex-pression on repeated IT Ag challenge. Total lung RNAwas extracted from SRBC-primed C57BL/6 mice onvarious days after final challenge. RNA was reverse-transcribed, and aliquots of cDNAs from those lungswere amplified by PCR. Symbols are as in Fig. 5. Eachpoint represents the mean and SEM of at least threemice in which the ratio of target cytokine gene signalto GADPH signal was determined and expressed rel-ative to the ratio for unchallenged normal lung. Notethe difference in scale between individual cytokines.p, p , 0.05, unpaired nonparametric Mann-Whitneytest comparing groups at the same time point.
4042 LUNG CYTOKINES IN REPEATED PULMONARY Ag CHALLENGE
by guest on April 11, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
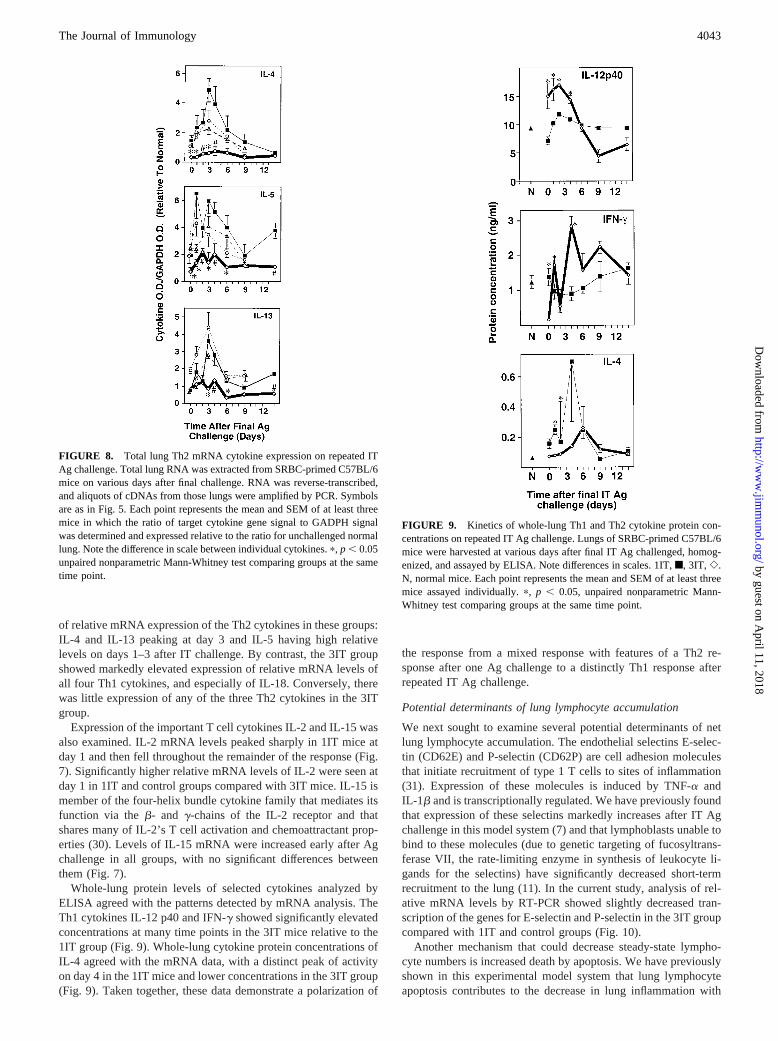
of relative mRNA expression of the Th2 cytokines in these groups:IL-4 and IL-13 peaking at day 3 and IL-5 having high relativelevels on days 1–3 after IT challenge. By contrast, the 3IT groupshowed markedly elevated expression of relative mRNA levels ofall four Th1 cytokines, and especially of IL-18. Conversely, therewas little expression of any of the three Th2 cytokines in the 3ITgroup.
Expression of the important T cell cytokines IL-2 and IL-15 wasalso examined. IL-2 mRNA levels peaked sharply in 1IT mice atday 1 and then fell throughout the remainder of the response (Fig.7). Significantly higher relative mRNA levels of IL-2 were seen atday 1 in 1IT and control groups compared with 3IT mice. IL-15 ismember of the four-helix bundle cytokine family that mediates itsfunction via theb- and g-chains of the IL-2 receptor and thatshares many of IL-2’s T cell activation and chemoattractant prop-erties (30). Levels of IL-15 mRNA were increased early after Agchallenge in all groups, with no significant differences betweenthem (Fig. 7).
Whole-lung protein levels of selected cytokines analyzed byELISA agreed with the patterns detected by mRNA analysis. TheTh1 cytokines IL-12 p40 and IFN-g showed significantly elevatedconcentrations at many time points in the 3IT mice relative to the1IT group (Fig. 9). Whole-lung cytokine protein concentrations ofIL-4 agreed with the mRNA data, with a distinct peak of activityon day 4 in the 1IT mice and lower concentrations in the 3IT group(Fig. 9). Taken together, these data demonstrate a polarization of
the response from a mixed response with features of a Th2 re-sponse after one Ag challenge to a distinctly Th1 response afterrepeated IT Ag challenge.
Potential determinants of lung lymphocyte accumulation
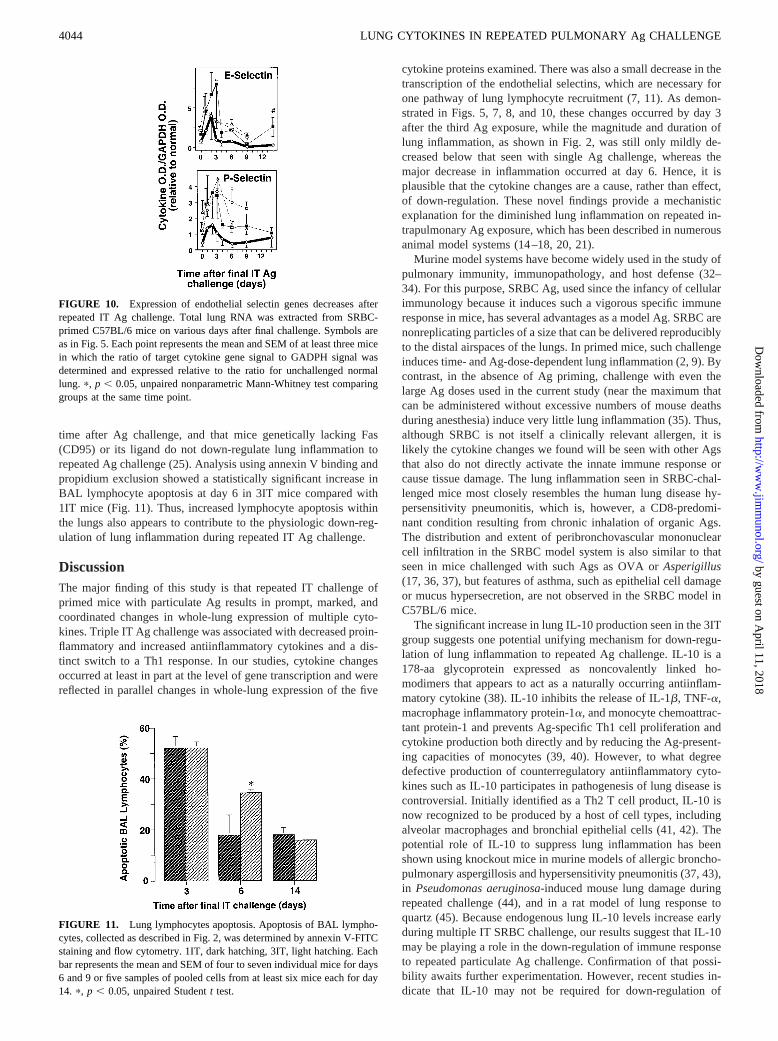
We next sought to examine several potential determinants of netlung lymphocyte accumulation. The endothelial selectins E-selec-tin (CD62E) and P-selectin (CD62P) are cell adhesion moleculesthat initiate recruitment of type 1 T cells to sites of inflammation(31). Expression of these molecules is induced by TNF-a andIL-1b and is transcriptionally regulated. We have previously foundthat expression of these selectins markedly increases after IT Agchallenge in this model system (7) and that lymphoblasts unable tobind to these molecules (due to genetic targeting of fucosyltrans-ferase VII, the rate-limiting enzyme in synthesis of leukocyte li-gands for the selectins) have significantly decreased short-termrecruitment to the lung (11). In the current study, analysis of rel-ative mRNA levels by RT-PCR showed slightly decreased tran-scription of the genes for E-selectin and P-selectin in the 3IT groupcompared with 1IT and control groups (Fig. 10).
Another mechanism that could decrease steady-state lympho-cyte numbers is increased death by apoptosis. We have previouslyshown in this experimental model system that lung lymphocyteapoptosis contributes to the decrease in lung inflammation with
FIGURE 8. Total lung Th2 mRNA cytokine expression on repeated ITAg challenge. Total lung RNA was extracted from SRBC-primed C57BL/6mice on various days after final challenge. RNA was reverse-transcribed,and aliquots of cDNAs from those lungs were amplified by PCR. Symbolsare as in Fig. 5. Each point represents the mean and SEM of at least threemice in which the ratio of target cytokine gene signal to GADPH signalwas determined and expressed relative to the ratio for unchallenged normallung. Note the difference in scale between individual cytokines.p, p , 0.05unpaired nonparametric Mann-Whitney test comparing groups at the sametime point.
FIGURE 9. Kinetics of whole-lung Th1 and Th2 cytokine protein con-centrations on repeated IT Ag challenge. Lungs of SRBC-primed C57BL/6mice were harvested at various days after final IT Ag challenged, homog-enized, and assayed by ELISA. Note differences in scales. 1IT,f, 3IT, e.N, normal mice. Each point represents the mean and SEM of at least threemice assayed individually.p, p , 0.05, unpaired nonparametric Mann-Whitney test comparing groups at the same time point.
4043The Journal of Immunology
by guest on April 11, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
time after Ag challenge, and that mice genetically lacking Fas(CD95) or its ligand do not down-regulate lung inflammation torepeated Ag challenge (25). Analysis using annexin V binding andpropidium exclusion showed a statistically significant increase inBAL lymphocyte apoptosis at day 6 in 3IT mice compared with1IT mice (Fig. 11). Thus, increased lymphocyte apoptosis withinthe lungs also appears to contribute to the physiologic down-reg-ulation of lung inflammation during repeated IT Ag challenge.
DiscussionThe major finding of this study is that repeated IT challenge ofprimed mice with particulate Ag results in prompt, marked, andcoordinated changes in whole-lung expression of multiple cyto-kines. Triple IT Ag challenge was associated with decreased proin-flammatory and increased antiinflammatory cytokines and a dis-tinct switch to a Th1 response. In our studies, cytokine changesoccurred at least in part at the level of gene transcription and werereflected in parallel changes in whole-lung expression of the five
cytokine proteins examined. There was also a small decrease in thetranscription of the endothelial selectins, which are necessary forone pathway of lung lymphocyte recruitment (7, 11). As demon-strated in Figs. 5, 7, 8, and 10, these changes occurred by day 3after the third Ag exposure, while the magnitude and duration oflung inflammation, as shown in Fig. 2, was still only mildly de-creased below that seen with single Ag challenge, whereas themajor decrease in inflammation occurred at day 6. Hence, it isplausible that the cytokine changes are a cause, rather than effect,of down-regulation. These novel findings provide a mechanisticexplanation for the diminished lung inflammation on repeated in-trapulmonary Ag exposure, which has been described in numerousanimal model systems (14–18, 20, 21).
Murine model systems have become widely used in the study ofpulmonary immunity, immunopathology, and host defense (32–34). For this purpose, SRBC Ag, used since the infancy of cellularimmunology because it induces such a vigorous specific immuneresponse in mice, has several advantages as a model Ag. SRBC arenonreplicating particles of a size that can be delivered reproduciblyto the distal airspaces of the lungs. In primed mice, such challengeinduces time- and Ag-dose-dependent lung inflammation (2, 9). Bycontrast, in the absence of Ag priming, challenge with even thelarge Ag doses used in the current study (near the maximum thatcan be administered without excessive numbers of mouse deathsduring anesthesia) induce very little lung inflammation (35). Thus,although SRBC is not itself a clinically relevant allergen, it islikely the cytokine changes we found will be seen with other Agsthat also do not directly activate the innate immune response orcause tissue damage. The lung inflammation seen in SRBC-chal-lenged mice most closely resembles the human lung disease hy-persensitivity pneumonitis, which is, however, a CD8-predomi-nant condition resulting from chronic inhalation of organic Ags.The distribution and extent of peribronchovascular mononuclearcell infiltration in the SRBC model system is also similar to thatseen in mice challenged with such Ags as OVA orAsperigillus(17, 36, 37), but features of asthma, such as epithelial cell damageor mucus hypersecretion, are not observed in the SRBC model inC57BL/6 mice.
The significant increase in lung IL-10 production seen in the 3ITgroup suggests one potential unifying mechanism for down-regu-lation of lung inflammation to repeated Ag challenge. IL-10 is a178-aa glycoprotein expressed as noncovalently linked ho-modimers that appears to act as a naturally occurring antiinflam-matory cytokine (38). IL-10 inhibits the release of IL-1b, TNF-a,macrophage inflammatory protein-1a, and monocyte chemoattrac-tant protein-1 and prevents Ag-specific Th1 cell proliferation andcytokine production both directly and by reducing the Ag-present-ing capacities of monocytes (39, 40). However, to what degreedefective production of counterregulatory antiinflammatory cyto-kines such as IL-10 participates in pathogenesis of lung disease iscontroversial. Initially identified as a Th2 T cell product, IL-10 isnow recognized to be produced by a host of cell types, includingalveolar macrophages and bronchial epithelial cells (41, 42). Thepotential role of IL-10 to suppress lung inflammation has beenshown using knockout mice in murine models of allergic broncho-pulmonary aspergillosis and hypersensitivity pneumonitis (37, 43),in Pseudomonas aeruginosa-induced mouse lung damage duringrepeated challenge (44), and in a rat model of lung response toquartz (45). Because endogenous lung IL-10 levels increase earlyduring multiple IT SRBC challenge, our results suggest that IL-10may be playing a role in the down-regulation of immune responseto repeated particulate Ag challenge. Confirmation of that possi-bility awaits further experimentation. However, recent studies in-dicate that IL-10 may not be required for down-regulation of
FIGURE 10. Expression of endothelial selectin genes decreases afterrepeated IT Ag challenge. Total lung RNA was extracted from SRBC-primed C57BL/6 mice on various days after final challenge. Symbols areas in Fig. 5. Each point represents the mean and SEM of at least three micein which the ratio of target cytokine gene signal to GADPH signal wasdetermined and expressed relative to the ratio for unchallenged normallung. p, p , 0.05, unpaired nonparametric Mann-Whitney test comparinggroups at the same time point.
FIGURE 11. Lung lymphocytes apoptosis. Apoptosis of BAL lympho-cytes, collected as described in Fig. 2, was determined by annexin V-FITCstaining and flow cytometry. 1IT, dark hatching, 3IT, light hatching. Eachbar represents the mean and SEM of four to seven individual mice for days6 and 9 or five samples of pooled cells from at least six mice each for day14. p, p , 0.05, unpaired Studentt test.
4044 LUNG CYTOKINES IN REPEATED PULMONARY Ag CHALLENGE
by guest on April 11, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

mouse lung inflammation associated with CpG DNA treatment ofLPS-induced injury (46), with blockade of the chemokine C10 inexperimental allergic bronchopulmonary aspergillosis (47), or dur-ing resolution of replicativeLegionella pneumophilainfection(48). Hence, the importance of IL-10 in down-regulation of lunginflammation appears to vary with the experimental model system.
The finding that IL-12 p40 mRNA and protein expression in-creased on repeated Ag challenge is significant because IL-12plays an important role in both innate resistance and Ag-specificimmunity to intracellular bacterial, fungal, viral, and protozoanpathogens. Moreover, a variety of protocols that down-regulateallergic manifestations against soluble Ags, including infectionswith Mycobacterium tuberculosisor adenovirus, administration ofunmethylated CpG oligodeoxynucleotides, and allergen-injectionimmunotherapy, both up-regulate IL-12 and switch T cells towarda Th1 phenotype (49–52). Endogenous production of IL-12 is es-sential for the resistance of some murine strains to OVA-inducedAHR (53). Low-dose intranasal IL-12 decreased Ag-induced AHRand lung eosinophilia in an OVA model of asthma in BALB/cmice (54). Interestingly, Hofstra and colleagues found in anotherOVA model in BALB/c mice that IL-12 plus IL-18, but not IL-12alone, was required to significantly inhibit Ag-induced lung eo-sinophilia, AHR, and elevated serum IgE (55). Therefore, lunginflammation involving predominance of Th2 cytokines followedby recovery associated with up-regulation of Th1 cytokines ap-pears to be well established in response to soluble Ags such asOVA. However, many Ags of clinical significance are of a partic-ulate nature, and with these results are not as consistent. For ex-ample, human and animal studies of hypersensitivity pneumonitis,a granulomatous inflammatory lung disease caused by inhalationof particulate Ag (most commonly thermophilic actinomycetes)indicate that Th1 cytokines predominate during infection (56, 57).In contrast, particulateAsperigillus fumigatusAgs elicited a Th2response in BALB/c mice (58, 59). Single IT SRBC challenge ofprimed A/J mice induced AHR and lung eosinophilia in a CD4 Tcell-dependent fashion reminiscent of human asthma (4). In thatmodel system, repeated systemic administration of exogenousIL-12 at the time of a second IT SRBC challenge abolished AHRand lung eosinophilia, increased IFN-g, and decreased IL-4 andIL-5 expression; these effects of IL-12 were only partially depen-dent on IFN-g (8). The current study extends these results byshowing spontaneous production within the lungs of IL-12, IL-18,and IFN-g in response to repeated particulate Ag challenge.
The dose of SRBC used in this study, although seemingly large,is in the same range used by others measuring immune response toparticulate cellular Ag (4, 35, 60, 61). In contrast to soluble Ags,generation of pulmonary immune responses to particulates typi-cally requires large Ag doses, presumably reflecting evolutionarypressure for alveolar macrophages to eliminate particles withoutinvoking lung inflammation. Nevertheless, the immune response toparticulates merits investigation due to the large quantities of par-ticulates to which even normal individuals are repeatedly exposedfocally through aspiration (62) and more insidiously by inhalation(63). With regard to the effect of dose on Th2/Th1 polarization,published results are contradictory. Some studies have indicatedthat low-dose soluble Ag preferentially supports IL-4 over IFN-gproduction (64–69). A recent study using transgenic T cells bear-ing a single TCR indicated that over the long term, high doses ofhigh-affinity peptides led selectively to IFN-g-secreting cellswhereas IL-4- and IL-5-secreting cells predominated with moder-ate doses (70). In contrast, with intact organisms such asLeish-mania major,Trichuris muris, andSchistosoma mansoni,a greaterdegree of Ag stimulation induced a Th2 response, whereas singleimmunizations or low-level infection favored Th1 cytokines (71–
73). The cytokine patterns seen in our 1IT and control groups areconsistent with results of a recent study that found that a high doseof SRBC (43 108 cells) given i.v. to naive CBA/J mice induceseither a mixed Th1/Th2 or a predominantly Th2 response, in con-trast to the Th1 response seen with lower doses (43 105 or 4 3106 cells) (61). Because Th1/Th2 polarization can also be influ-enced by other variables including strength of TCR signal (65, 74),amount of costimulation (75), nature of cytokines present (76), andT cell responsiveness to particular cytokines (77), it is currentlydifficult to predict accurately the effect of a given immunizationprotocol. The results of the current study are important becausethey provide additional information about the pulmonary immuneresponse to nonreplicating particulate Ag, and because they implythat route of Ag administration should also be considered.
There are probably multiple causes for the observed decrease inlung lymphocyte accumulation in the 3IT group. Based on previ-ous findings in this model system, decreased expression of endo-thelial selectins and increased lymphocyte apoptosis would both beanticipated to have major effects on steady-state lymphocyte ac-cumulation and hence the severity and duration of lung inflamma-tion. It is likely that lymphocyte retention within lung parenchymais also decreased after repeated Ag challenge; however, we havenot yet measured this parameter. We also did not measure locallymphocyte proliferation within the lungs, reasoning that it wouldbe difficult to account for significant net changes between thegroups based on further reduction of the already very low rate ofproliferation previously seen in the 1IT protocol (12).
Several points about our results merit discussion in relationshipto previous studies using this experimental model system. Kaltre-ider and colleagues demonstrated expression of cytokine proteins(IL-2, IL-4, IL-6, and IFN-g) as a function of time after a single ITSRBC challenge (9). Because that study analyzed concentratedBAL fluid, whereas ours analyzed total lung digests, the results arenot directly comparable. However, both studies found peak IL-4production at 3–4 days after challenge. Our finding of decreasedlymphocyte and total leukocyte numbers in the 3IT group differfrom results of Denis and Bisson (35). In addition, they foundstriking fibrosis in mice challenged three times, which was notobserved in our studies. There are several methodological differ-ences between our study and theirs, including control groups (theycompared 3IT only to primed mice without IT challenge and tounprimed mice after single IT challenge), interval between IT chal-lenges (10-day rather than 7-day intervals as is our studies), andmethod of cytokine analysis (cytokines were analyzed in the su-pernatants of purified BAL T cells stimulated in vitro with SRBC,whereas we measured protein levels in unstimulated lung cell di-gests). Despite these methodological differences, both the Denisand Bisson and our study found that protein concentrations ofIFN-g increased on repeated Ag challenge, consistent with evolu-tion of a Th1 response. In agreement with our findings, most otheranimal models of repeated stimulation using exogenous Ag havefound both decreased inflammation and distinct difficulty in induc-ing fibrosis (14–16, 18, 20, 21).
In summary, this study demonstrates that, relative to single Agchallenge, repeated IT Ag challenge of primed mice is associatedwith significantly decreased lymphocyte accumulation and signif-icant changes in total lung production of mRNA and proteins.These changes are down-regulation of inflammatory cytokines, up-regulation of the antiinflammatory cytokine IL-10, as well as strik-ing Th1 polarization. These cytokine changes are associated withdecreased transcription of the genes for the inducible endothelialselectins and by increased lung lymphocyte apoptosis. These re-sults are important because similar mechanisms may act to mini-mize chronic lung inflammation in the majority of normal humans
4045The Journal of Immunology
by guest on April 11, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

who do not develop progressive lung pathology when repeatedlyexposed to inhaled or aspirated particulate environmental allergensand Ags.
AcknowledgmentsWe thank Dr. Richard E. Goodman (Monsanto, St. Louis, MO) for theprimer and probe sequences for GAPDH, GM-CSF, IL-2, IL-4, IFN-g, andIL-12 p35; Dr. Lloyd Stoolman for recombinant human IL-2; Angela Pres-ton, Kelly Warmington and Drs. James M. Beck, Paul Christensen, GaryHuffnagle, and Bethany Moore for helpful suggestions; Joyce O’Brien forsecretarial support; Michael Hormuth and Carolyn White for assistancewith the photomicrographs; and Drs. Beck and Moore for critiquing themanuscript.
References1. Bice, D. E., and B. A. Muggenburg. 1988. Localized immune memory in the
lung. Am. Rev. Respir. Dis. 138:565.2. Kaltreider, H. B., J. L. Curtis, and S. M. Arraj. 1987. The mechanism of appear-
ance of specific antibody-forming cells in lungs of inbred mice after intratrachealimmunization with sheep erythrocytes. II. Dose-dependence and kinetics. Am.Rev. Respir. Dis. 135:87.
3. Curtis, J. L., and H. B. Kaltreider. 1989. Characterization of bronchoalveolarlymphocytes during a specific antibody-forming cell response in the lungs ofmice.Am. Rev. Respir. Dis. 139:393.
4. Gavett, S. H., X. Chen, F. Finkelman, and M. Wills-Karp. 1994. Depletion ofmurine CD41 T lymphocytes prevents antigen-induced airway hyperreactivityand pulmonary eosinophilia.Am. J. Respir. Cell. Mol. Biol. 10:587.
5. Curtis, J. L., M. L. Warnock, S. M. Arraj, and H. B. Kaltreider. 1990. Histologicanalysis of an immune response in the lung parenchyma of mice: angiopathyaccompanies inflammatory cell influx.Am. J. Pathol. 137:689.
6. Ichikawa, S., Y. Goto, S. Uchino, H. B. Kaltreider, E. J. Goetzl, andS. P. Sreedharan. 1996. Changes in adhesion molecule expression during distinctpatterns of immune cell migration in the inflamed lung.Arch. Histol. Cytol.59:443.
7. Wolber, F. M., J. L. Curtis, A. M. Milik, G. D. Seitzman, K. L. Fields, K.-M.Kim, S. Kim, J. Sonstein, and L. M. Stoolman. 1997. Lymphocyte recruitmentand the kinetics of adhesion receptor expression during the pulmonary immuneresponse to particulate antigen.Am. J. Pathol. 151:1715.
8. Gavett, S. H., D. J. O’Hearn, X. Li, S. K. Huang, F. D. Finkelman, andM. Wills-Karp. 1995. Interleukin 12 inhibits antigen-induced airway hyperre-sponsiveness, inflammation, and Th2 cytokine expression in mice.J. Exp. Med.182:1527.
9. Kaltreider, H. B., S. Ichikawa, P. K. Byrd, D. A. Ingram, J. L. Kishiyama,S. P. Sreedharan, M. L. Warnock, J. M. Beck, and E. J. Goetzl. 1997. Upregu-lation of neuropeptides and neuropeptide receptors in a murine model of immuneinflammation in lung parenchyma.Am. J. Respir. Cell. Mol. Biol. 16:133.
10. Curtis, J. L., P. K. Byrd, M. L. Warnock, and H. B. Kaltreider. 1991. Requirementof CD41 T cells for cellular recruitment to the lungs of mice in response tointratracheal antigen.J. Clin. Invest. 88:1244.
11. Wolber, F. M., J. L. Curtis, J. Lowe, P. Maly, P. Smith, T. A. Yednock,R. J. Kelly, and L. M. Stoolman. 1998. Endothelial selectins anda4 integrinregulate independent pathways of T lymphocyte recruitment in pulmonary in-flammation.J. Immunol. 161:4396.
12. Seitzman, G. D., J. Sonstein, W. Choy, S. Kim, and J. L. Curtis. 1998. Lunglymphocytes proliferate minimally in the murine pulmonary immune response tointratracheal sheep erythrocytes.Am. J. Respir. Cell. Mol. Biol. 18:800.
13. Curtis, J. L., P. K. Byrd, M. L. Warnock, J. M. Beck, and H. B. Kaltreider. 1993.Pulmonary lymphocyte recruitment: Depletion of CD81 T cells does not impairthe pulmonary immune response to intratracheal antigen.Am. J. Respir. Cell.Mol. Biol. 9:90.
14. Richerson, H. B., J. J. Seidenfeld, H. V. Ratjczak, and D. W. Richards. 1978.Chronic experimental interstitial pneumonitis in the rabbit.Am. Rev. Respir. Dis.117:5.
15. Ratajczak, H. V., D. W. Richards, and H. B. Richerson. 1980. Systemic and locallymphocyte responses in experimental hypersensitivity pneumonitis.Am. Rev.Respir. Dis. 122:761.
16. Schuyler, M. R., J. Kleinerman, J. R. Pensky, C. Brandt, and D. Schmitt. 1983.Pulmonary response to repeated exposure toMicropolyspora faeni. Am. Rev.Respir. Dis. 128:1071.
17. Yiamouyiannis, C. A., C. M. Schramm, L. Puddington, P. Stengel,E. Baradaran-Hosseini, W. W. Wolyniec, H. E. Whiteley, and R. S. Thrall. 1999.Shifts in lung lymphocyte profiles correlate with the sequential development ofacute allergic and chronic tolerant stages in a murine asthma model.Am. J. Pathol. 154:1911.
18. Kopp, W. C., M. T. Suelzer, and H. B. Richerson. 1988. Alveolar macrophageimmunosuppression is maintained in rabbit models of hypersensitivity pneumo-nitis. J. Allergy Clin. Immunol. 82:204.
19. Denis, M., D. Bisson, and E. Ghadirian. 1993. Cellular and cytokine profiles inspontaneous regression phase of hypersensitivity pneumonitis.Exp. Lung Res.19:257.
20. Schuyler, M. R., and D. Schmitt. 1984. Experimental hypersensitivity pneumo-nitis: lack of tolerance.Am. Rev. Respir. Dis. 130:772.
21. Takizawa, H., M. Suko, N. Kobayashi, K. Ohta, S. Shoji, T. Horiuchi,H. Okudaira, T. Miyamoto, and J. Shiga. 1989. Spontaneous regression in murinehypersensitivity pneumonitis: lack of immunological tolerance.Int. Arch. AllergyAppl. Immunol. 89:173.
22. Friedman, A., and H. L. Weiner. 1994. Induction of anergy or active suppressionfollowing oral tolerance is determined by antigen dosage.Proc. Natl. Acad. Sci.USA 91:6688.
23. Chen, Y., J. Inobe, V. K. Kuchroo, J. L. Baron, C. A. Janeway, Jr., andH. L. Weiner. 1996. Oral tolerance in myelin basic protein T-cell receptor trans-genic mice: suppression of autoimmune encephalomyelitis and dose-dependentinduction of regulatory cells.Proc. Natl. Acad. Sci. USA 93:388.
24. Chung, Y., S. Y. Chang, and C. Y. Kang. 1999. Kinetic analysis of oral tolerance:memory lymphocytes are refractory to oral tolerance.J. Immunol. 163:3692.
25. Milik, A. M., V. A. Beuchner-Maxwell, S. Kim, J. Sonstein, G. D. Seitzman,T. F. Beals, and J. L. Curtis. 1997. Lung lymphocyte elimination by apoptosis inthe murine response to intratracheal particulate antigen.J. Clin. Invest. 99:1082.
26. Curtis, J. L., S. Kim, P. J. Scott, and V. A. Buechner-Maxwell. 1995. Adhesionreceptor phenotypes of murine lung CD41 T cells during a pulmonary immuneresponse to sheep erythrocytes.Am. J. Respir. Cell. Mol. Biol. 12:520.
27. Vermes, I., C. Haanen, H. Steffens-Nakken, and C. Reutelingsperger. 1995. Anovel assay for apoptosis. Flow cytometric detection of phosphatidylserine ex-pression on early apoptotic cells using fluorescein labelled Annexin V.J. Immu-nol. Methods 184:39.
28. Zar, J. H. 1974.Biostatistical Analysis. Prentice-Hall, Englewood Cliffs, NJ,p. 718.
29. Chang, J. C. C., L. Zhang, S. G. Distler, G. Ziang, and A. M. Kaplan. 1992.Characterization and function of CD31 CD42 CD82 TcR-ab bearing cells in-filtrating the lung during the immune response.Reg. Immunol. 4:25.
30. Tagaya, Y., R. N. Bamford, A. P. DeFilippis, and T. A. Waldmann. 1996. IL-15:a pleiotropic cytokine with diverse receptor/signaling pathways whose expressionis controlled at multiple levels.Immunity 4:329.
31. Austrup, F., D. Vestweber, E. Borges, M. Lohning, R. Brauer, U. Herz, H. Renz,R. Hallman, A. Scheffold, A. Radbruch, and A. Hamann. 1997. P- and E-selectinmediate recruitment of T-helper-1 but not T-helper-2 cells into inflammed tissues.Nature 385:81.
32. Perez Arellano, J. L., N. M. Barrios Gonzalez, T. Martin Dominguez,M. L. Sanchez Benitez de Soto, and A. Jimenez Lopez. 1992. Experimentalmodels of hypersensitivity pneumonitis.J. Invest. Allerg. Clin. Immunol. 2:219.
33. Kumar, R. K. 1995. Experimental models in pulmonary pathology.Pathology27:130.
34. Wills-Karp, M., and S. L. Ewart. 1997. The genetics of allergen-induced airwayhyperresponsiveness in mice.Am. J. Respir. Crit. Care Med. 156:S89.
35. Denis, M., and D. Bisson. 1995. Antigen-induced alveolitis: cytokine productionin a mouse model.Inflammation 19:157.
36. Wilder, J. A., D. D. Collie, B. S. Wilson, D. E. Bice, C. R. Lyons, andM. F. Lipscomb. 1999. Dissociation of airway hyperresponsiveness from immu-noglobulin E and airway eosinophilia in a murine model of allergic asthma.Am. J. Respir. Cell Mol. Biol. 20:1326.
37. Grunig, G., D. B. Corry, M. W. Leach, B. W. Seymour, V. P. Kurup, andD. M. Rennick. 1997. Interleukin-10 is a natural suppressor of cytokine produc-tion and inflammation in a murine model of allergic bronchopulmonary aspergil-losis.J. Exp. Med. 185:1089.
38. Moore, K. W., A. O’Garra, R. de Waal Malefyt, P. Vieira, and T. R. Mosmann.1993. Interleukin-10.Annu. Rev. Immunol. 11:165.
39. de Waal-Malefyt, R., J. Haanen, H. Spits, M. G. Roncarolo, T. V. A., C. Figdor,K. Johnson, R. Kastelein, H. Yssel, and J. E. de Vries. 1991. Interleukin 10(IL-10) and viral IL-10 strongly reduce antigen-specific human T cell prolifera-tion by diminishing the antigen-presenting capacity of monocytes via downregu-lation of class II major histocompatibility complex expression.J. Exp. Med. 174:915.
40. Fiorentino, D. F., A. Zlotnik, T. R. Mosmann, M. Howard, and A. O’Garra. 1991.IL-10 inhibits cytokine production by activated macrophages.J. Immunol. 147:3815.
41. Xing, Z., M. Jordana, H. Kirpalani, K. E. Driscoll, T. J. Schall, and J. Gauldie.1994. Cytokine expression by neutrophils and macrophages in vivo: endotoxininduces tumor necrosis factor-a, macrophage inflammatory protein- 2, interleu-kin-1b, and interleukin-6 but not RANTES or transforming growth factor-b 1mRNA expression in acute lung inflammation.Am. J. Respir. Cell Mol. Biol.10:148.
42. Bonfield, T. L., M. W. Konstan, P. Burfeind, J. R. Panuska, J. B. Hilliard, andM. Berger. 1995. Normal bronchial epithelial cells constitutively produce theanti-inflammatory cytokine interleukin-10, which is downregulated in cystic fi-brosis.Am. J. Respir. Cell Mol. Biol. 13:257.
43. Gudmundsson, G., A. Bosch, B. L. Davidson, D. J. Berg, and G. W. Hunning-hake. 1998. Interleukin-10 modulates the severity of hypersensitivity pneumonitisin mice.Am. J. Respir. Cell Mol. Biol. 19:812.
44. Yu, H., M. Hanes, C. E. Chrisp, J. C. Boucher, and V. Deretic. 1998. Microbialpathogenesis in cystic fibrosis: pulmonary clearance of mucoidPseudomonasaeruginosaand inflammation in a mouse model of repeated respiratory challenge.Infect. Immun. 66:280.
45. Driscoll, K. E., J. M. Carter, B. W. Howard, D. Hassenbein, M. Burdick,S. L. Kunkel, and R. M. Strieter. 1998. Interleukin-10 regulates quartz-inducedpulmonary inflammation in rats.Am. J. Physiol. 275:L887.
46. Schwartz, D. A., C. L. Wohlford-Lenane, T. J. Quinn, and A. M. Krieg. 1999.Bacterial DNA or oligonucleotides containing unmethylated CpG motifs canminimize lipopolysaccharide-induced inflammation in the lower respiratory tractthrough an IL-12-dependent pathway.J. Immunol. 163:224.
4046 LUNG CYTOKINES IN REPEATED PULMONARY Ag CHALLENGE
by guest on April 11, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

47. Hogaboam, C. M., C. S. Gallinat, D. D. Taub, R. M. Strieter, S. L. Kunkel, andN. W. Lukacs. 1999. Immunomodulatory role of C10 chemokine in a murinemodel of allergic bronchopulmonary aspergillosis.J. Immunol. 162:6071.
48. Brieland, J. K., D. G. Remick, M. L. Legendre, N. C. Engleberg, andJ. C. Fantone. 1998. In vivo regulation of replicativeLegionella pneumophilalung infection by endogenous interleukin-12.Infect. Immun. 66:65.
49. Sano, K., K. Haneda, G. Tamura, and K. Shirato. 1999. Ovalbumin (OVA) andMycobacterium tuberculosisbacilli cooperatively polarize anti-OVA T-helper(Th) cells toward a Th1-dominant phenotype and ameliorate murine trachealeosinophilia.Am. J. Respir. Cell Mol. Biol. 20:1260.
50. Stampfli, M. R., S. A. Ritz, G. S. Neigh, P. J. Sime, X. F. Lei, Z. Xing,K. Croitoru, and M. Jordana. 1998. Adenoviral infection inhibits allergic airwaysinflammation in mice.Clin. Exp. Allergy. 28:1581.
51. Sur, S., J. S. Wild, B. K. Choudhury, N. Sur, R. Alam, and D. M. Klinman. 1999.Long term prevention of allergic lung inflammation in a mouse model of asthmaby CpG oligodeoxynucleotides.J. Immunol. 162:6284.
52. Durham, S. R., A. B. Kay, and Q. Hamid. 1995. Changes in allergic inflammationassociated with successful immunotherapy.Int. Arch. Allergy Immunol. 107:282.
53. Keane-Myers, A., M. Wysocka, G. Trinchieri, and M. Wills-Karp. 1998. Resis-tance to antigen-induced airway hyperresponsiveness requires endogenous pro-duction of IL-12.J. Immunol. 161:919.
54. Schwarze, J., E. Hamelmann, G. Cieslewicz, A. Tomkinson, A. Joetham,K. Bradley, and E. W. Gelfand. 1998. Local treatment with IL-12 is an effectiveinhibitor of airway hyperresponsiveness and lung eosinophilia after airway chal-lenge in sensitized mice.J. Allergy Clin. Immunol. 102:86.
55. Hofstra, C. L., I. Van Ark, G. Hofman, M. Kool, F. P. Nijkamp, andA. J. Van Oosterhout. 1998. Prevention of Th2-like cell responses by coadmin-istration of IL-12 and IL-18 is associated with inhibition of antigen-induced air-way hyperresponsiveness, eosinophilia, and serum IgE levels.J. Immunol. 161:5054.
56. Yamasaki, H., M. Ando, W. Brazer, D. M. Center, and W. W. Cruikshank. 1999.Polarized type 1 cytokine profile in bronchoalveolar lavage T cells of patientswith hypersensitivity pneumonitis.J. Immunol. 163:3516.
57. Gudmundsson, G., M. M. Monick, and G. W. Hunninghake. 1998. IL-12 mod-ulates expression of hypersensitivity pneumonitis.J. Immunol. 161:991.
58. Kurup, V. P., B. W. Seymour, H. Choi, and R. L. Coffman. 1994. ParticulateAsperigillus fumigatusantigens elicit a TH2 response in BALB/c mice.J. Allergy Clin. Immunol. 93:1013.
59. Kurup, V. P., J. Q. Xia, D. A. Rickaby, C. A. Dawson, H. Choi, and J. N. Fink.1999.Asperigillus fumigatusantigen exposure results in pulmonary airway re-sistance in wild-type but not in IL-4 knockout mice.Clin. Immunol. 90:404.
60. Menon, J. N., and P. A. Bretscher. 1998. Parasite dose determines the Th1/Th2nature of the response toLeishmania majorindependently of infection route andstrain of host or parasite.Eur. J. Immunol. 28:4020.
61. Ismail, N., and P. A. Bretscher. 1999. The Th1/Th2 nature of concurrent immuneresponses to unrelated antigens can be independent.J. Immunol. 163:4842.
62. Huxley, E. J., J. Viroslav, W. R. Gray, and A. K. Pierce. 1978. Pharyngealaspiration in normal adults and patients with depressed consciousness.Am. J. Med. 64:564.
63. Brunekreef, B. 1999. All but quiet on the particulate front.Am. J. Respir. Crit.Care Med. 159:354.
64. Kumar, V., V. Bhardwaj, L. Soares, J. Alexander, A. Sette, and E. Sercarz. 1995.Major histocompatibility complex binding affinity of an antigenic determinant iscrucial for the differential secretion of interleukin 4/5 or interferong by T cells.Proc. Natl. Acad. Sci. USA 92:9510.
65. Constant, S., C. Pfeiffer, A. Woodard, T. Pasqualini, and K. Bottomly. 1995.Extent of T cell receptor ligation can determine the functional differentiation ofnaive CD41 T cells.J. Exp. Med. 182:1591.
66. Secrist, H., R. H. DeKruyff, and D. T. Umetsu. 1995. Interleukin 4 production byCD41 T cells from allergic individuals is modulated by antigen concentration andantigen-presenting cell type.J. Exp. Med. 181:1081.
67. Tao, X., S. Constant, P. Jorritsma, and K. Bottomly. 1997. Strength of TCR signaldetermines the costimulatory requirements for Th1 and Th2 CD41 T cell differ-entiation.J. Immunol. 159:5956.
68. Schountz, T., J. P. Kasselman, F. A. Martinson, L. Brown, and J. S. Murray.1996. MHC genotype controls the capacity of ligand density to switch T helper(Th)-1/Th-2 priming in vivo.J. Immunol. 157:3893.
69. Sakai, K., A. Yokoyama, N. Kohno, and K. Hiwada. 1999. Effect of differentsensitizing doses of antigen in a murine model of atopic asthma.Clin. Exp.Immunol. 118:9.
70. Rogers, P. R., and M. Croft. 1999. Peptide dose, affinity, and time of differen-tiation can contribute to the Th1/Th2 cytokine balance.J. Immunol. 163:1205.
71. Caulada-Benedetti, Z., F. al-Zamel, A. Sher, and S. James. 1991. Comparison ofTh1- and Th2-associated immune reactivities stimulated by single versus multiplevaccination of mice with irradiatedSchistosoma mansonicercariae.J. Immunol.146:1655.
72. Bretscher, P. A., G. Wei, J. N. Menon, and H. Bielefeldt-Ohmann. 1992. Estab-lishment of stable, cell-mediated immunity makes “susceptible” mice resistant toLeishmania major. Science 257:539.
73. Bancroft, A. J., K. J. Else, and R. K. Grencis. 1994. Low-level infection withTrichuris murissignificantly affects the polarization of the CD4 response.Eur.J. Immunol. 24:3113.
74. Constant, S. L., and K. Bottomly. 1997. Induction of Th1 and Th2 CD41 T cellresponses: the alternative approaches.Annu. Rev. Immunol. 15:297.
75. Bluestone, J. A. 1995. New perspectives of CD28–B7-mediated T cell costimu-lation. Immunity 2:555.
76. Abbas, A. K., K. M. Murphy, and A. Sher. 1996. Functional diversity of helperT lymphocytes.Nature 383:787.
77. Guler, M. L., J. D. Gorham, C. S. Hsieh, A. J. Mackey, R. G. Steen,W. F. Dietrich, and K. M. Murphy. 1996. Genetic susceptibility toLeishmania:IL-12 responsiveness in TH1 cell development.Science 271:984.
4047The Journal of Immunology
by guest on April 11, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from





























