Embed Size (px)
Citation preview

Repetitive and Duplicitous Structure of Genomes
Jeff BaileyS5-432

Human Genome Structure
Hetrochromatic Sequence (tandem satellite repeats) Centromeric alpha-satellite, telomere CAGGG, acrocentric rRNA and beta-
satellite Euchromatic sequence ~3.1 gigabases
Genes (35%) ~25,000 Exons (1%) (transcription more ubiquitous ENCODE) Repetitive Sequences
3% Simple Sequence Repeats (poly A runs, dinucleotide and trinucleotide repeats)
45% Interspersed Repetitive Elements Repetitive Element Size Copies Fraction LINE elements (retrotransposon) up to 8 kb 850,000 21% Alu elements (retrotransposon) 300 bp 1,500,000 13% LTR-retrovirus-like 6-11 kb 450,000 8% DNA transposons 1-3 kb 300,000 3%
(International Human Genome Sequencing Consortium. Science 2001
Vast majority of sequence is non-coding and repetitive.Vast majority of sequence is non-coding and repetitive.
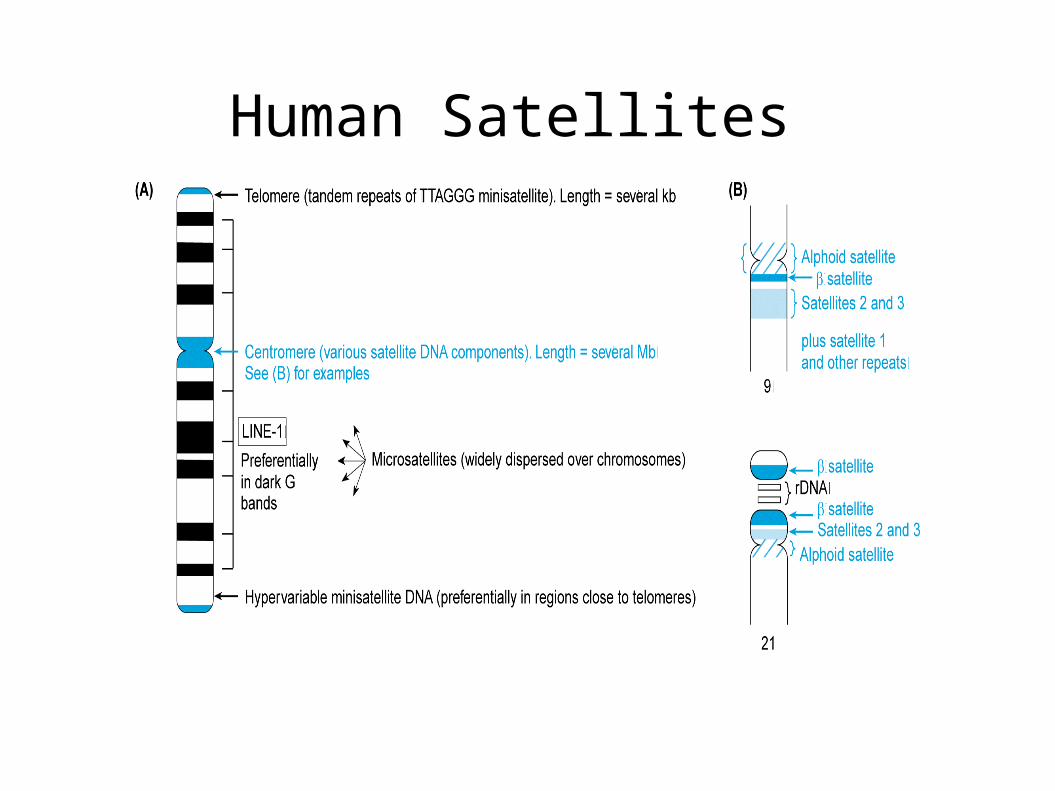
Human Satellites

Centromeric Sequence Human:
171 bp alpha-satellite in array of 2-5 Mb
higher order structure (only in Great Apes) 4-20
4-30 k-mer (A-B-C-D-A-B-C-D-A-B-C-D) A-B-C-D to A-B-C-D (2-5%) A-D- 20-40% Further flanked by other satellites (beta satellite)
Mouse:
234 bp major satellite (6 Mb) an 120 bp (600 kb) minor satellite at centromeric constriction
Arabibdopsis
178 bp satellite in 3 Mb array
Drosophilia:
5 bp simple arrays of AATAT and AAGAG
C. elegans:
Holocentric – entire chromosome acts as centromere
Yeast:
CEN3 1-2 kb of 83 bp repeat
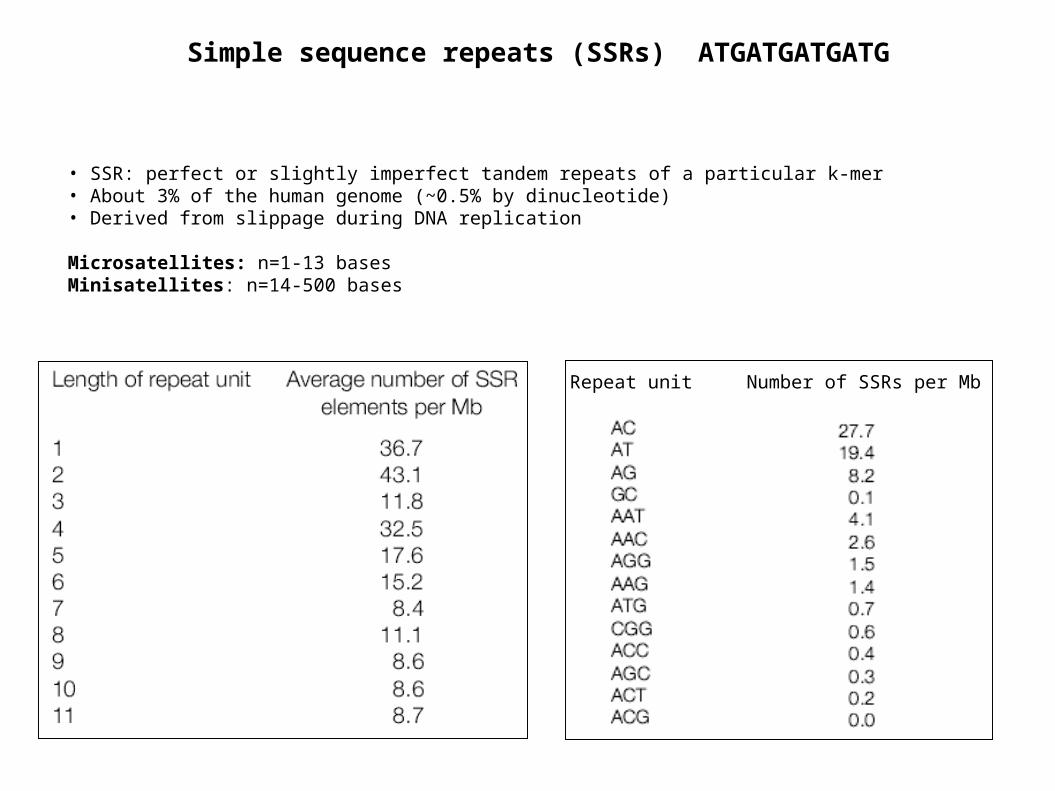
Simple sequence repeats (SSRs) ATGATGATGATG
• SSR: perfect or slightly imperfect tandem repeats of a particular k-mer• About 3% of the human genome (~0.5% by dinucleotide)• Derived from slippage during DNA replication
Microsatellites: n=1-13 basesMinisatellites: n=14-500 bases
Repeat unit Number of SSRs per Mb

Interspersed Repeats
DNA transposons “extinct” in primate lineage (~40 mya). Quiescent in mammalian lineages.
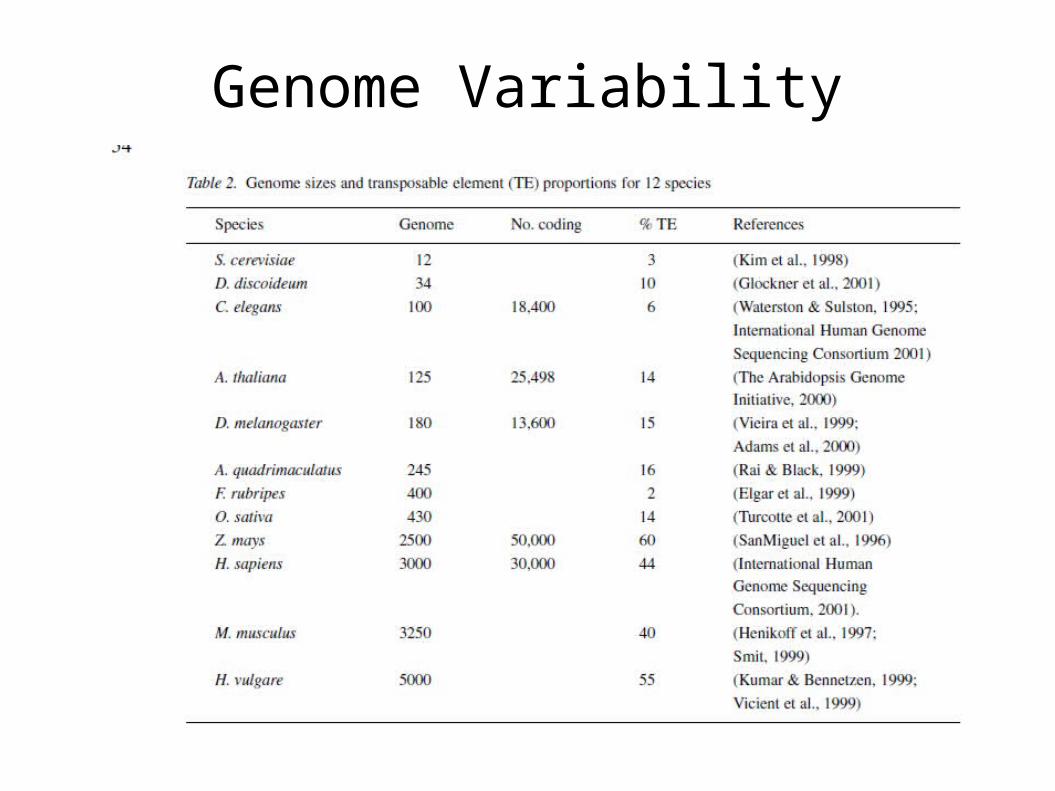
Genome Variability
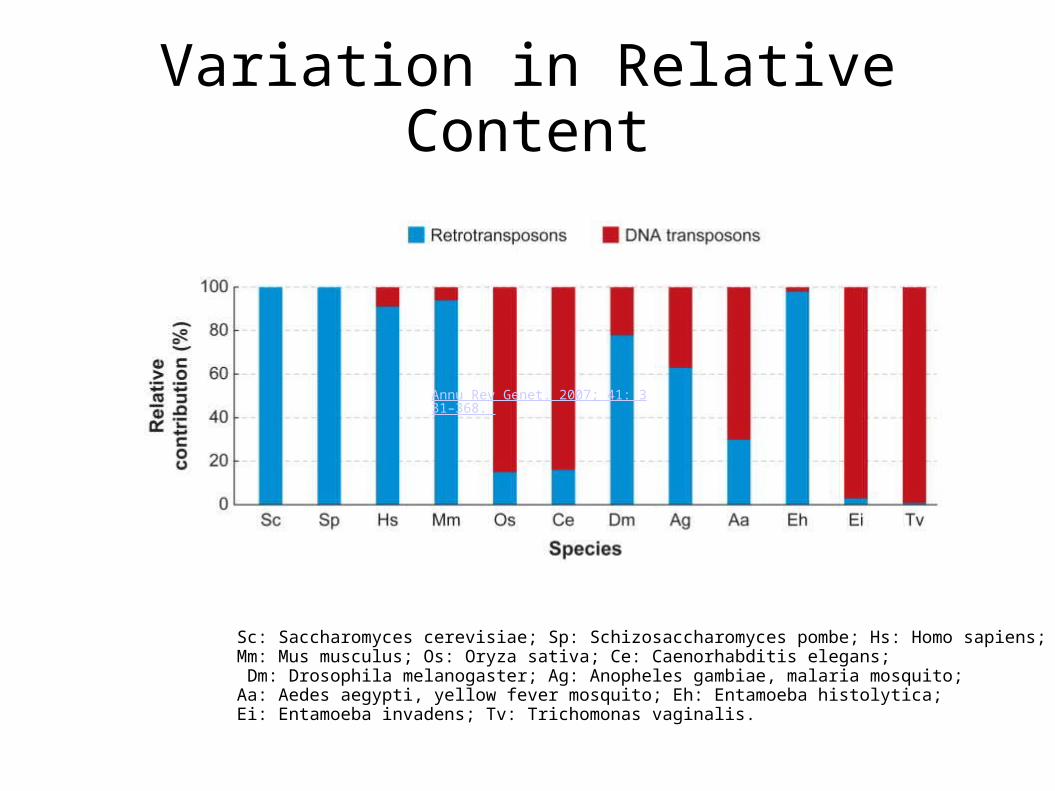
Annu Rev Genet. 2007; 41: 331–368.
Sc: Saccharomyces cerevisiae; Sp: Schizosaccharomyces pombe; Hs: Homo sapiens; Mm: Mus musculus; Os: Oryza sativa; Ce: Caenorhabditis elegans; Dm: Drosophila melanogaster; Ag: Anopheles gambiae, malaria mosquito; Aa: Aedes aegypti, yellow fever mosquito; Eh: Entamoeba histolytica; Ei: Entamoeba invadens; Tv: Trichomonas vaginalis.
Variation in Relative Content
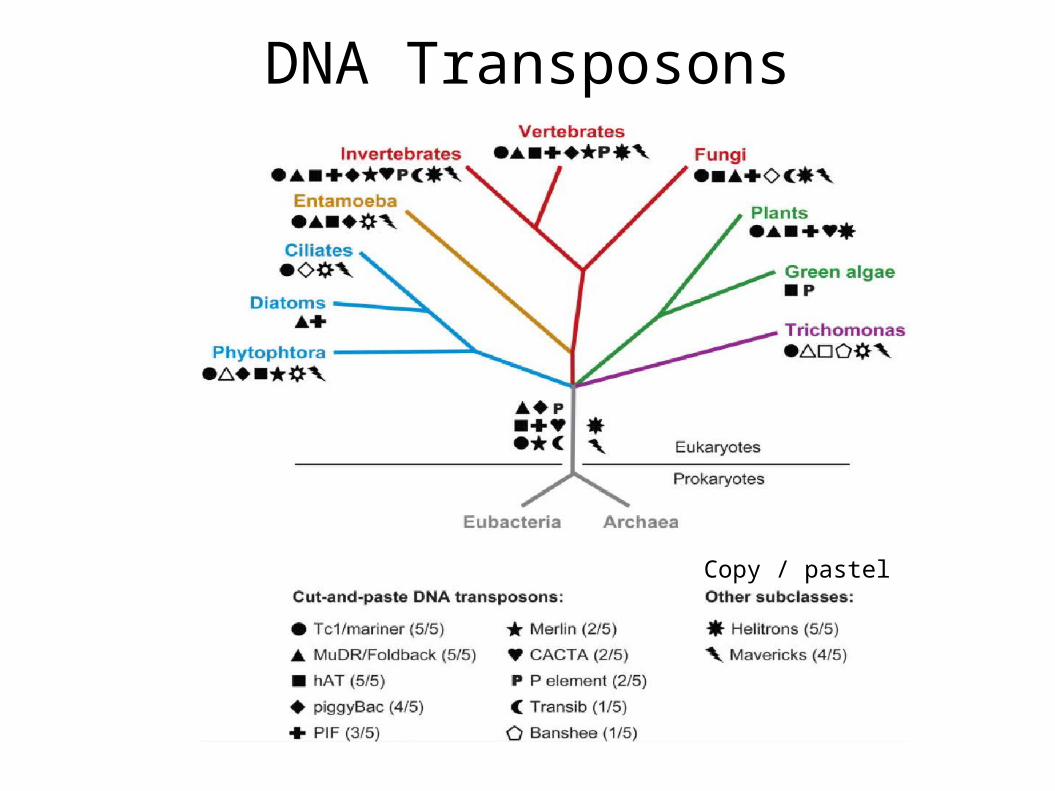
DNA Transposons
Copy / pastel
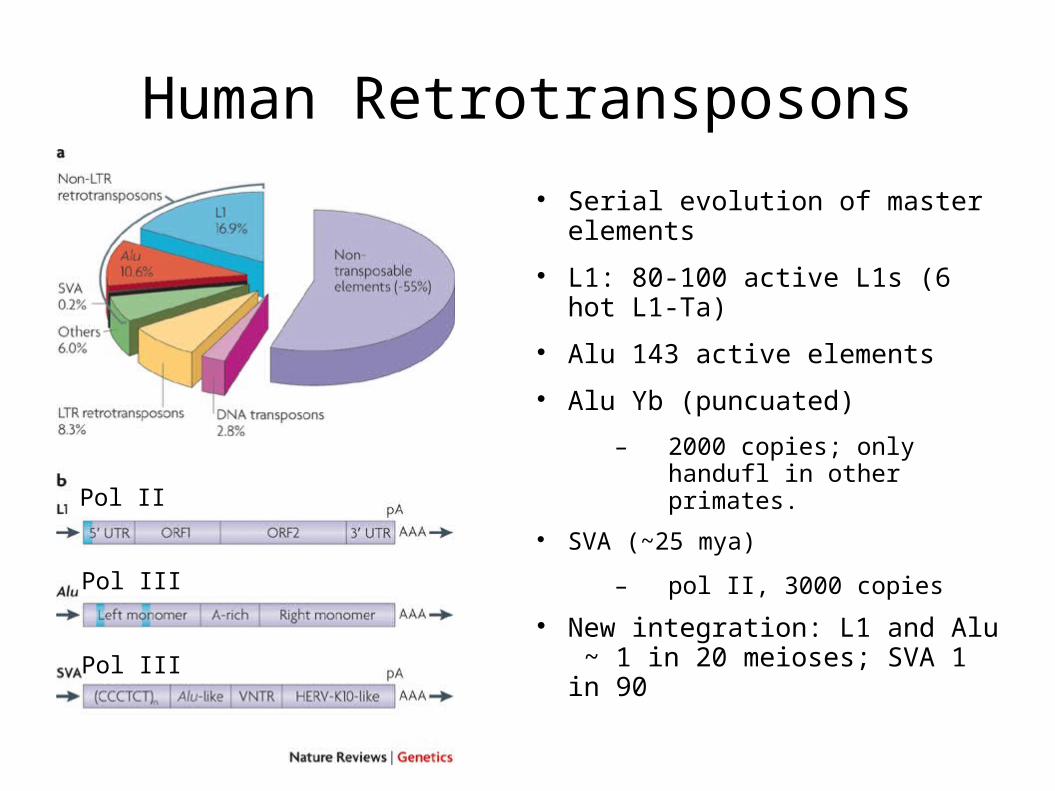
Human Retrotransposons
Serial evolution of master elements
L1: 80-100 active L1s (6 hot L1-Ta)
Alu 143 active elements
Alu Yb (puncuated)
– 2000 copies; only handufl in other primates.
SVA (~25 mya)
– pol II, 3000 copies
New integration: L1 and Alu ~ 1 in 20 meioses; SVA 1 in 90
Pol II
Pol III
Pol III

L1 “master” elements
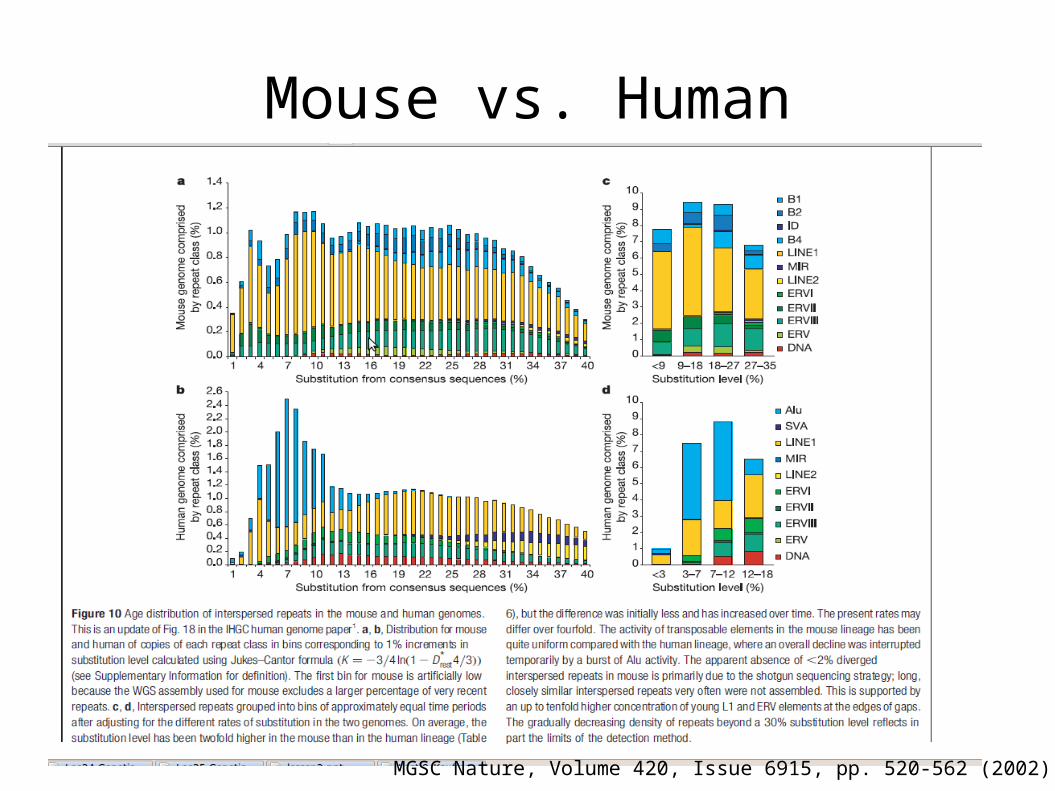
Mouse vs. Human
MGSC Nature, Volume 420, Issue 6915, pp. 520-562 (2002).
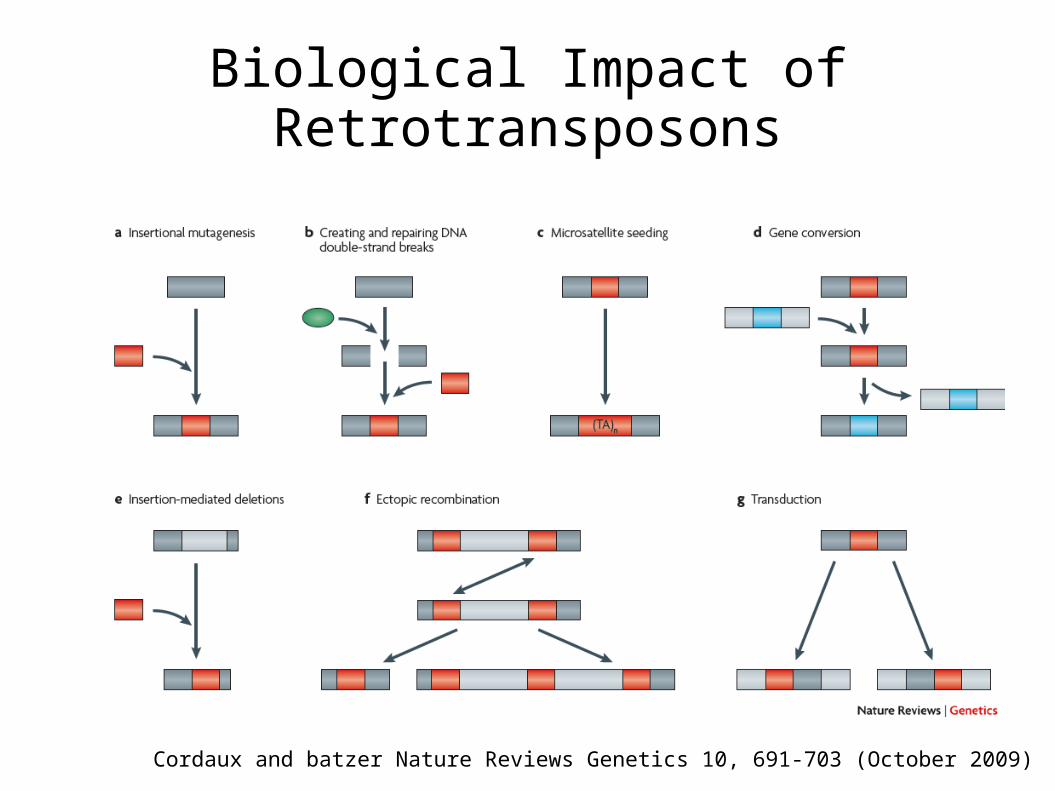
Biological Impact of Retrotransposons
Cordaux and batzer Nature Reviews Genetics 10, 691-703 (October 2009)

Biological Importance (cont.)
Boundary / Insulator Elements Alternative splicing / novel
exons / novel genes Role in suppression of poly II
transcription in cellular stress What accounts for long-
term maintenance?

Human Genome Structure
Hetrochromatic Sequence (tandem satellite repeats) Centromeric alpha-satellite, telomere CAGGG, acrocentric rRNA and beta-
satellite Euchromatic sequence ~3.1 gigabases
Genes (35%) ~25,000 Exons (1%) (transcription more ubiquitous ENCODE) Repetitive Sequences
3% Simple Sequence Repeats (poly A runs, dinucleotide and trinucleotide repeats)
45% Interspersed Repetitive Elements Repetitive Element Size Copies Fraction LINE elements (retrotransposon) up to 8 kb 850,000 21% Alu elements (retrotransposon) 300 bp 1,500,000 13% LTR-retrovirus-like 6-11 kb 450,000 8% DNA transposons 1-3 kb 300,000 3%
(International Human Genome Sequencing Consortium. Science 2001
Vast majority of sequence is non-coding and repetitive.Vast majority of sequence is non-coding and repetitive.

• Whole Genome Duplication– Ancient 4N 2N
• Segmental Duplications– Tandem– Interspersed
• Interchromosomal• intrachromosomal
Types of Duplications
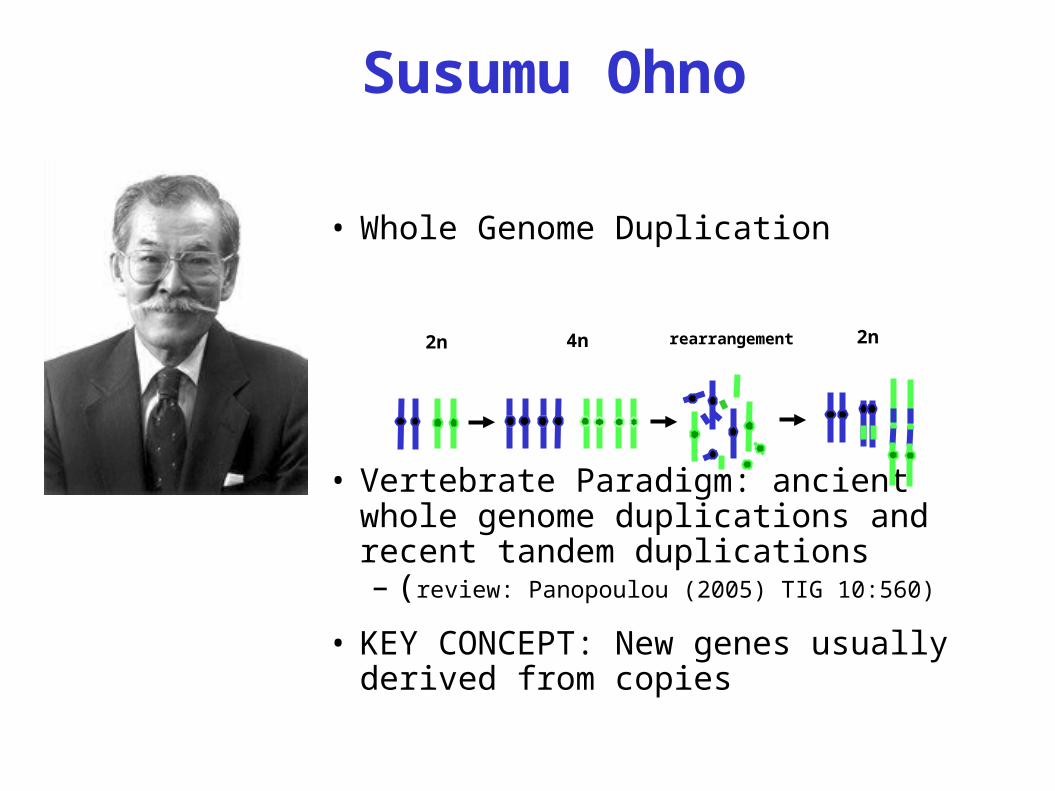
Susumu Ohno
• Whole Genome Duplication
• Vertebrate Paradigm: ancient whole genome duplications and recent tandem duplications– (review: Panopoulou (2005) TIG 10:560)
• KEY CONCEPT: New genes usually derived from copies
2n 4n rearrangement 2n
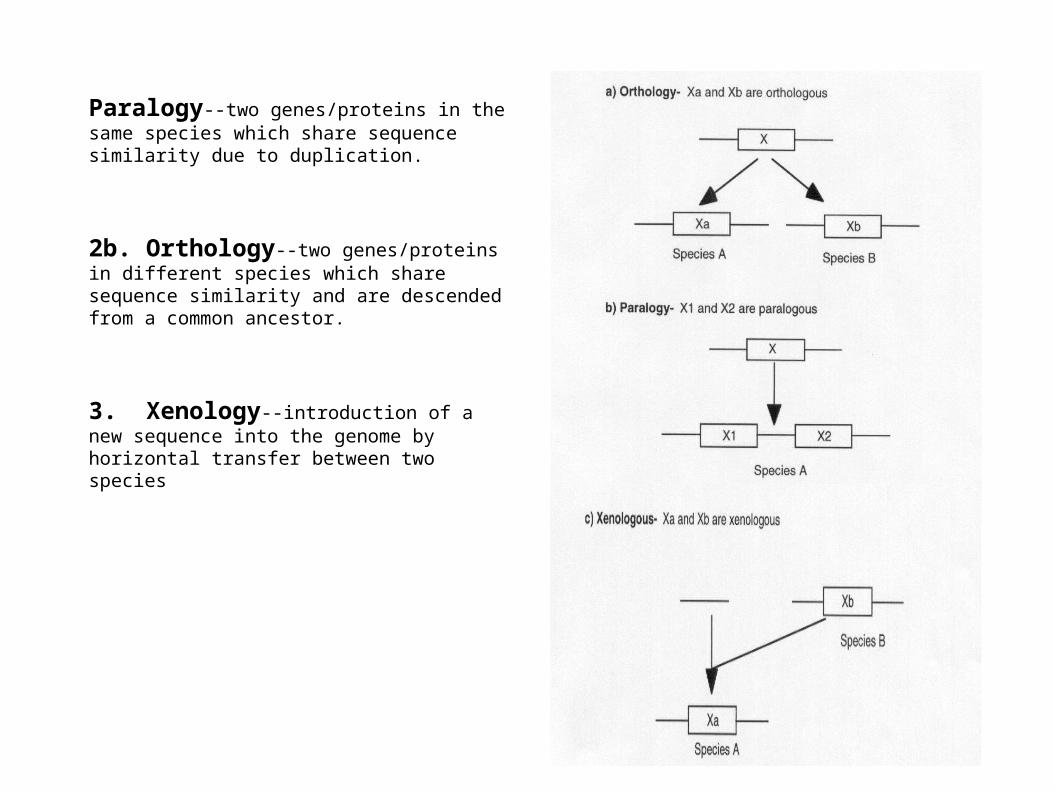
Paralogy--two genes/proteins in the same species which share sequence similarity due to duplication.
2b. Orthology--two genes/proteins in different species which share sequence similarity and are descended from a common ancestor.
3. Xenology--introduction of a new sequence into the genome by horizontal transfer between two species
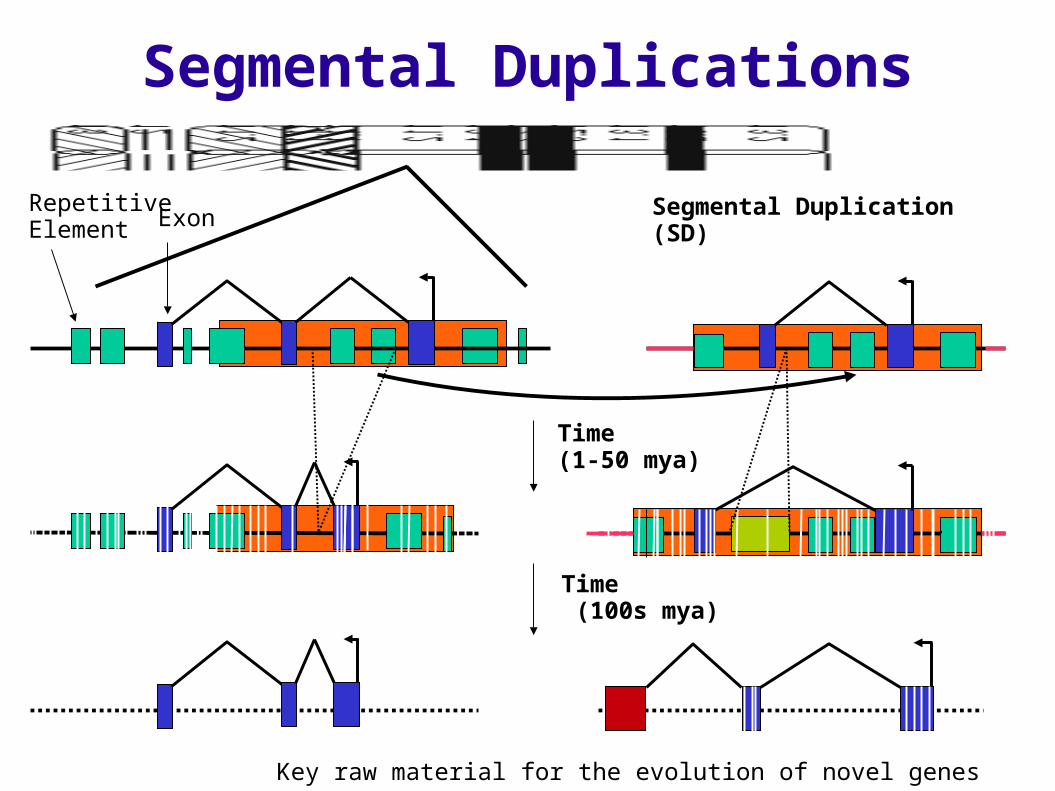
Segmental Duplication (SD)
Segmental Duplications
Repetitive Element Exon
Time (100s mya)
Key raw material for the evolution of novel genes
Time (1-50 mya)
`
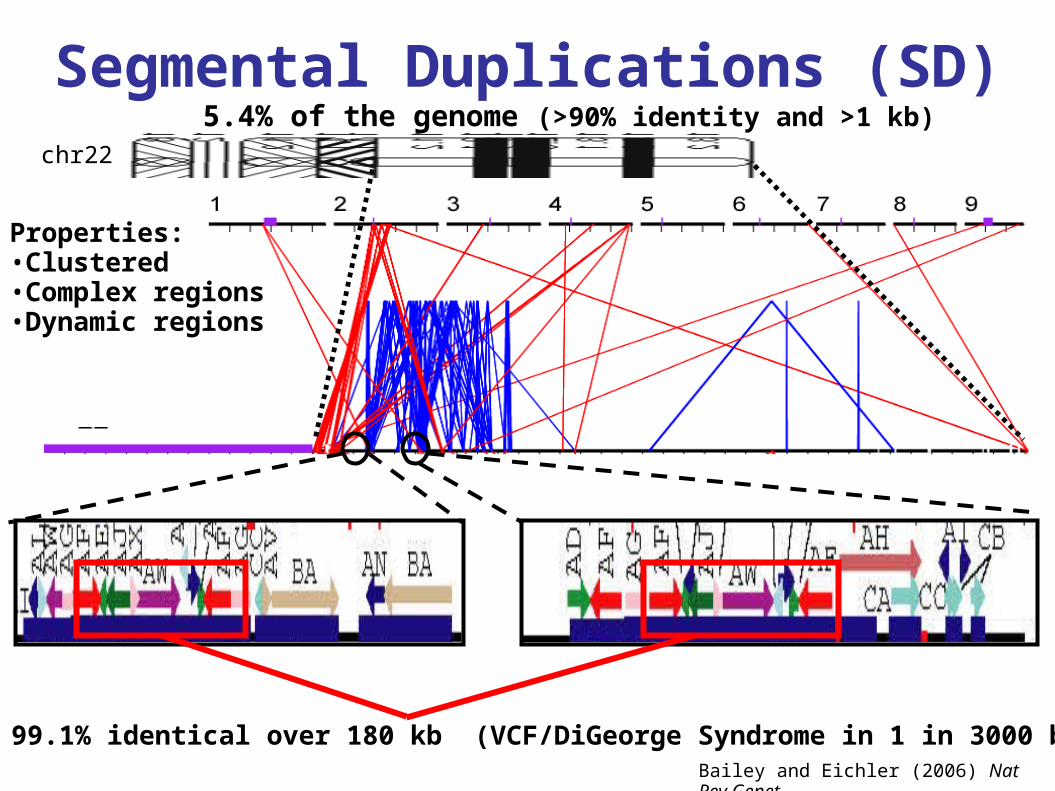
Segmental Duplications (SD)
Bailey and Eichler (2006) Nat Rev Genet
Properties:•Clustered•Complex regions•Dynamic regions
99.1% identical over 180 kb (VCF/DiGeorge Syndrome in 1 in 3000 births)
5.4% of the genome (>90% identity and >1 kb)chr22
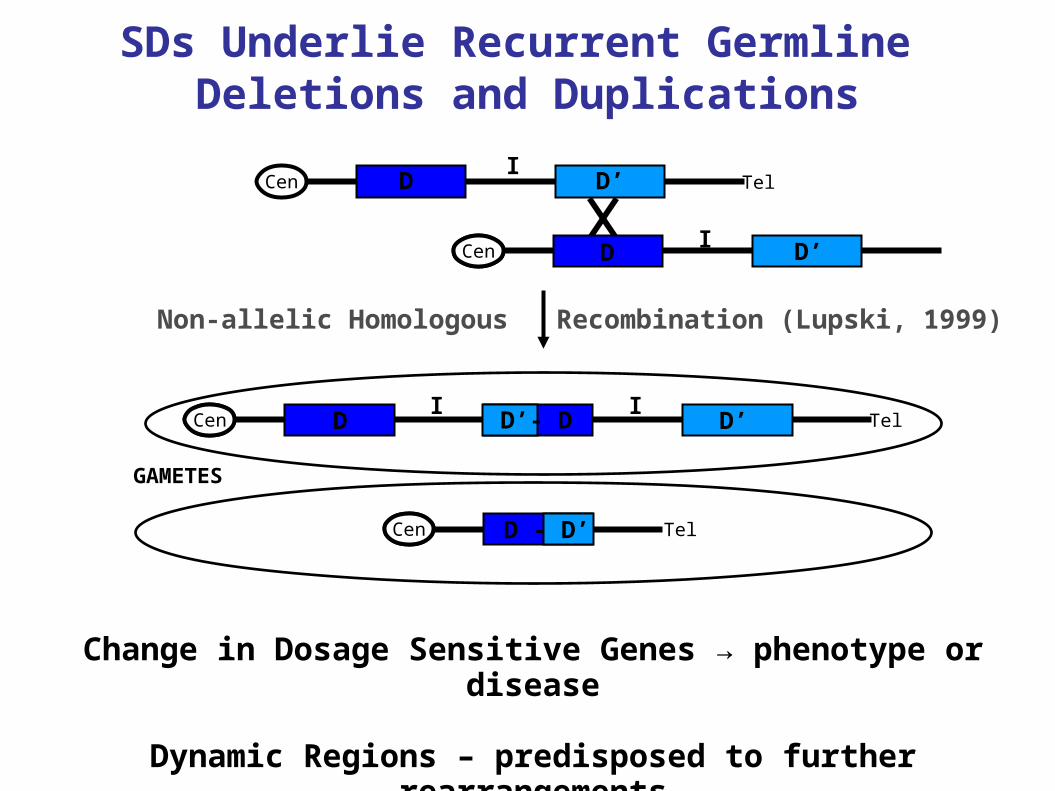
SDs Underlie Recurrent Germline Deletions and Duplications
Cen TelI
D D’
CenI D’D
Tel
Tel
Cen
Cen
GAMETES
D D’I I
Change in Dosage Sensitive Genes → phenotype or disease
Dynamic Regions – predisposed to further rearrangements
Non-allelic Homologous Recombination (Lupski, 1999)
D’- D
D - D’
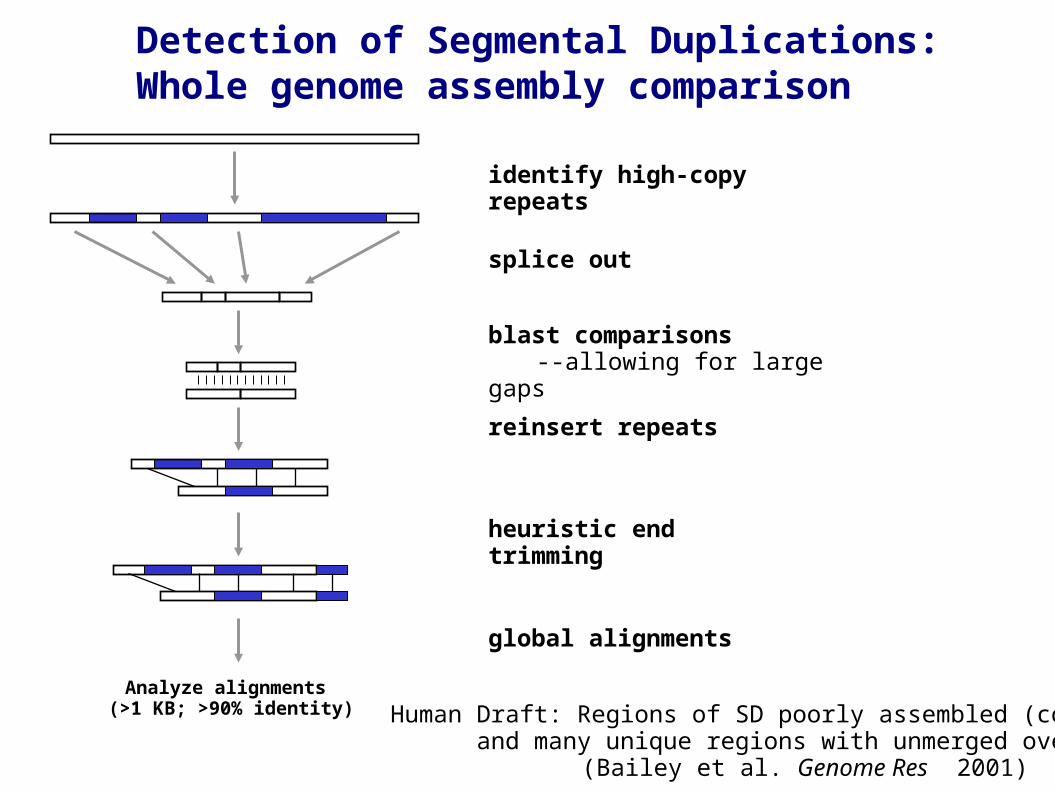
Figure 1identify high-copy repeats
splice out
Analyze alignments (>1 KB; >90% identity)
blast comparisons--allowing for large gaps
reinsert repeats
heuristic end trimming
global alignments
Detection of Segmental Duplications:Whole genome assembly comparison
Human Draft: Regions of SD poorly assembled (collapsed) and many unique regions with unmerged overlaps (allelic) (Bailey et al. Genome Res 2001)

Genome Wide Detection
Assembly % finished 90-98% >98%July 2000 20% 3.6% 12.9%
January 2001 23% 3.6% 10.6%August 2001 44% 4.1% 15.3%
Problem:
Allelic/True Overlap vs.
Duplication
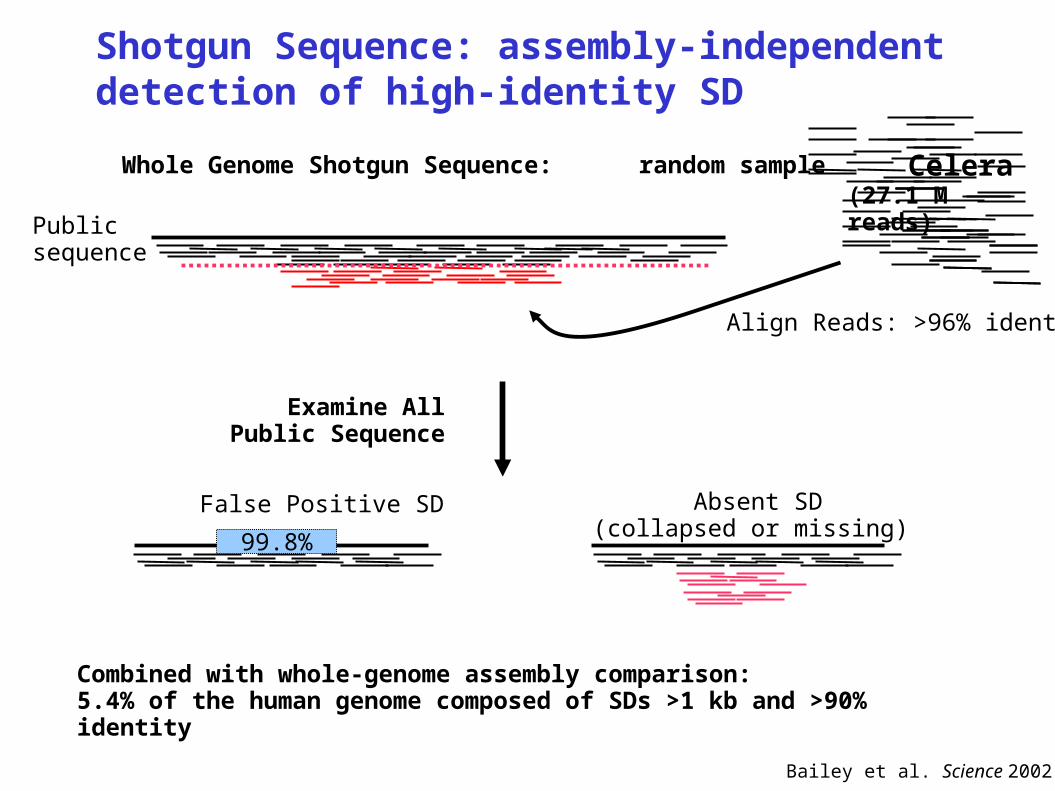
Shotgun Sequence: assembly-independentdetection of high-identity SD
Whole Genome Shotgun Sequence: random sample
Bailey et al. Science 2002
Combined with whole-genome assembly comparison:5.4% of the human genome composed of SDs >1 kb and >90% identity
99.8%
False Positive SD Absent SD (collapsed or missing)
Examine All Public Sequence
Publicsequence
Align Reads: >96% identity
Celera(27.1 M reads)
REPEATS
47
100
200
# R
ead
s / 5
kb
Public
Celera
223
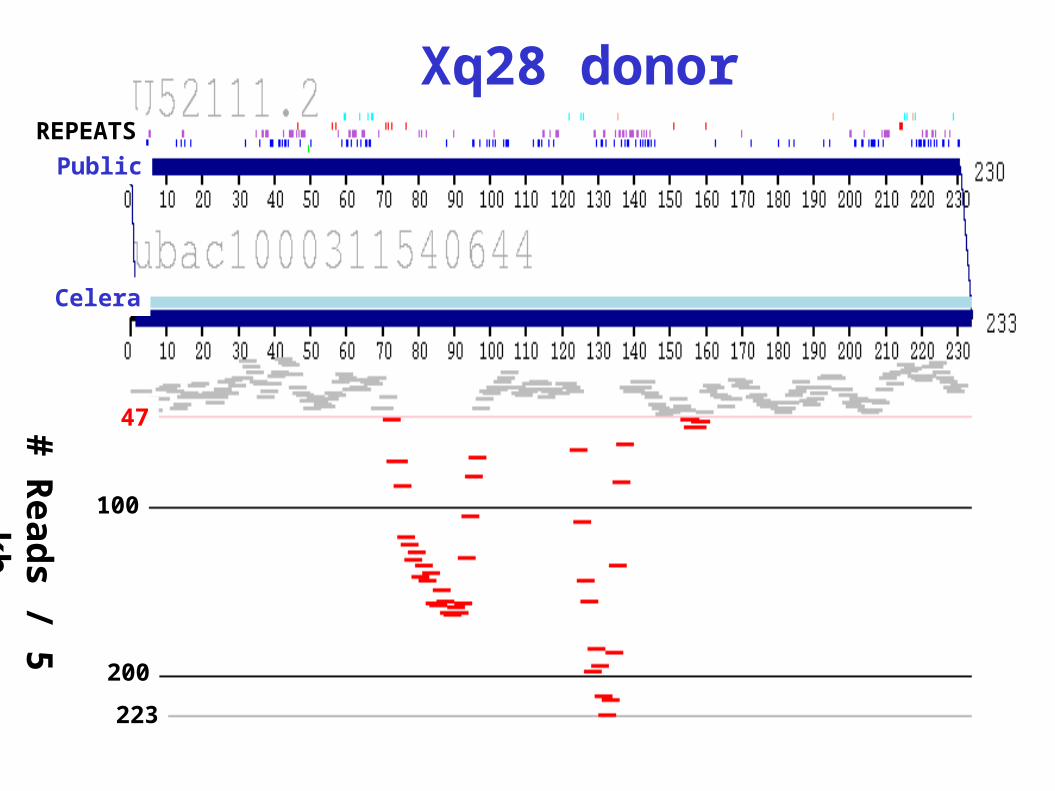
Xq28 donor
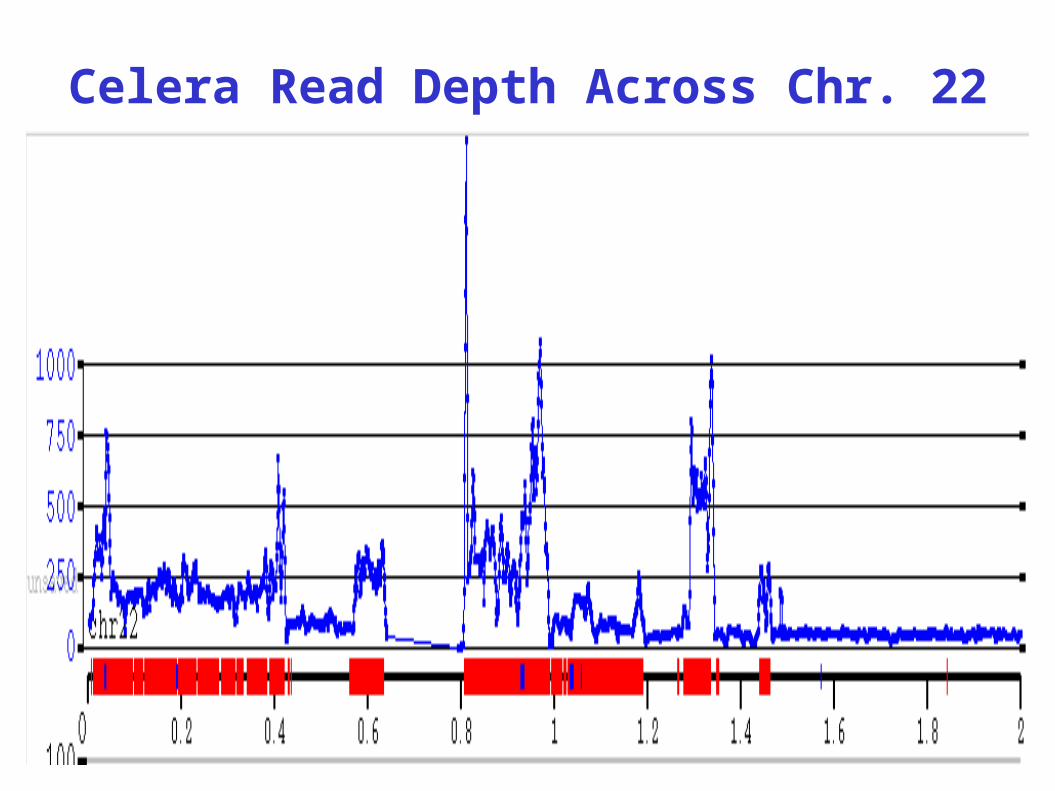
Celera Read Depth Across Chr. 22
Covera
ge
Nu
mb
er o
f Read
s/5
kb
w
ind
ow
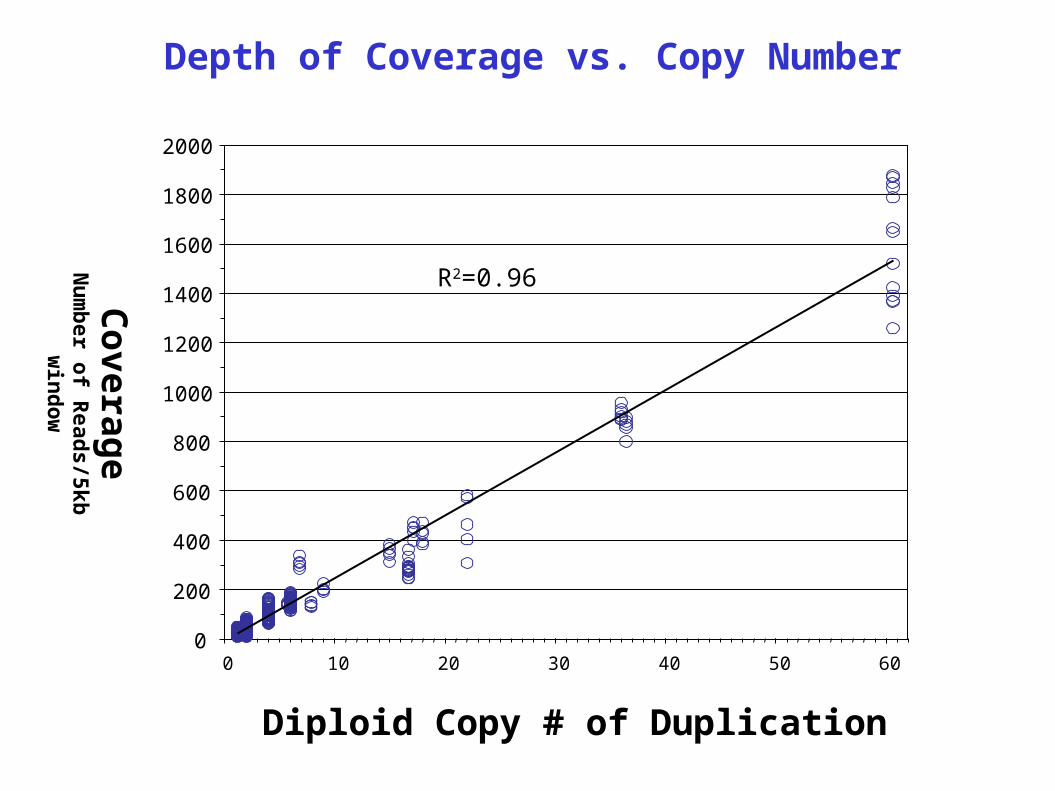
Diploid Copy # of Duplication
Depth of Coverage vs. Copy Number
R2=0.96
0
200
400
600
800
1000
1200
1400
1600
1800
2000
0 10 20 30 40 50 60
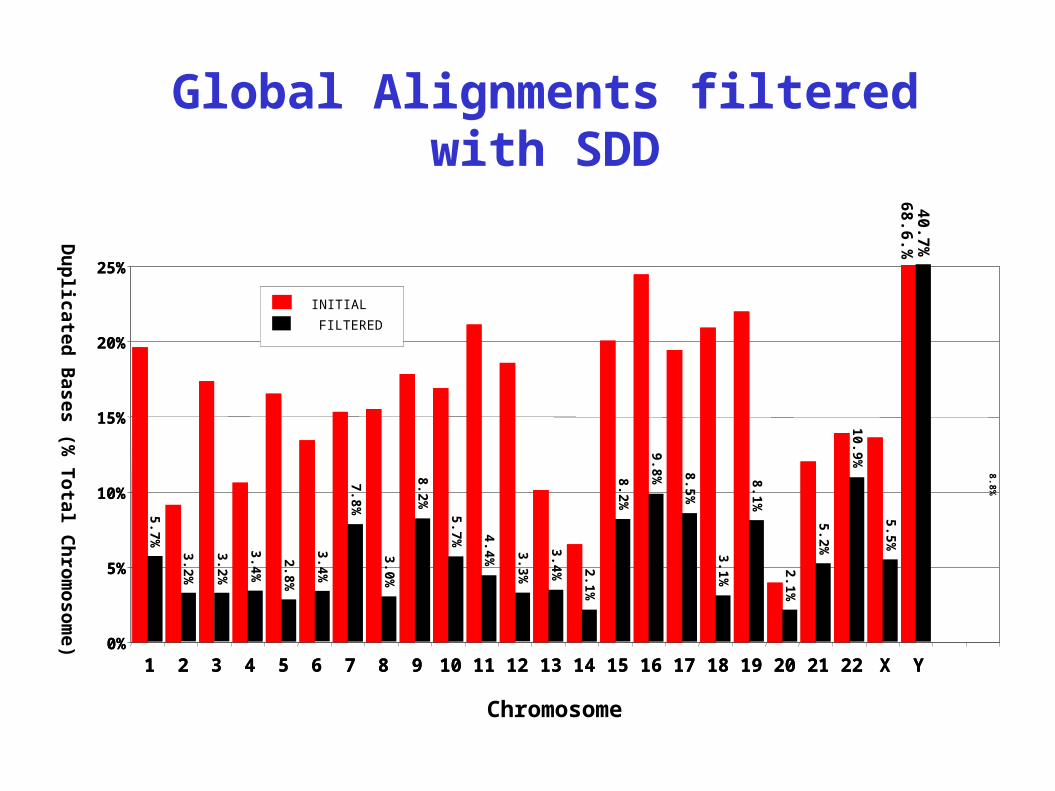
Global Alignments filtered with SDD
5.7%
3.2%
3.2%
3.4%
2.8%
3.4%
7.8%
3.0%
8.2%
5.7% 4.4% 3.3%
3.4% 2.1%
8.2%
9.8% 8.5%
3.1%
8.1%
2.1%
5.2%
10.9%
5.5%
8.8%
40
.7%
0%
5%
10%
15%
20%
25%
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X Y
Chromosome
INITIAL
FILTERED
68
.6.%
0%
5%
10%
15%
20%
25%
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X Y
Du
plicated
Bases (%
To
tal Ch
rom
oso
me)
INITIAL
FILTERED

•130 candidate regions (298 Mb) •23 associated with genetic disease
SD “Hotspot”Map of Human
Genome
Bailey et al. Science 2002
Interrogation of these regions has lead to detection of 16 additional pathogenic rearrangements including new microdeletions on 1q21.1, 15q13, 15q24 and 17q12. (Sharp et al. Nat Genet 2006; Mefford et al. Am J Hum Genet 2007; Mefford et al. N Engl J Med 2008)
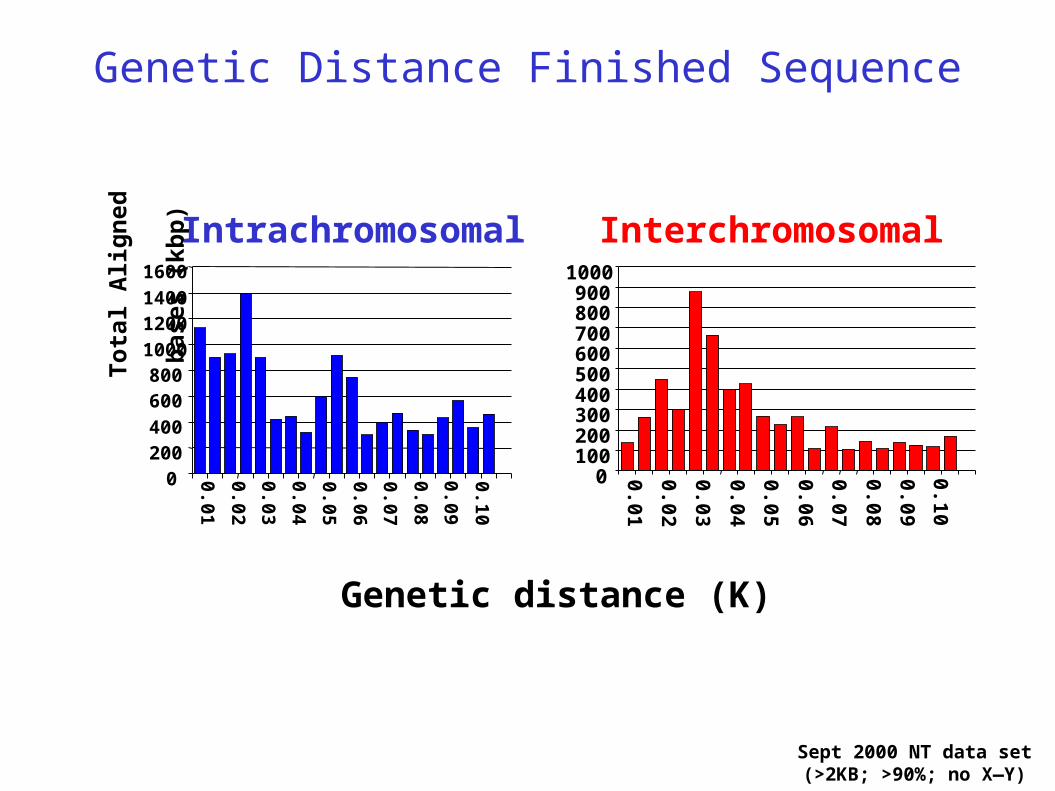
Genetic Distance Finished Sequence
Sept 2000 NT data set(>2KB; >90%; no X—Y)
0200400
600800
1000
12001400
1600
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0.09
0.10
0100200300400500600700800900
1000
0.0
1
0.0
2
0.0
3
0.0
4
0.0
5
0.0
6
0.0
7
0.0
8
0.0
9
0.1
0Tota
l A
lig
ned
b
ases (
kb
p)
Genetic distance (K)
Intrachromosomal Interchromosomal
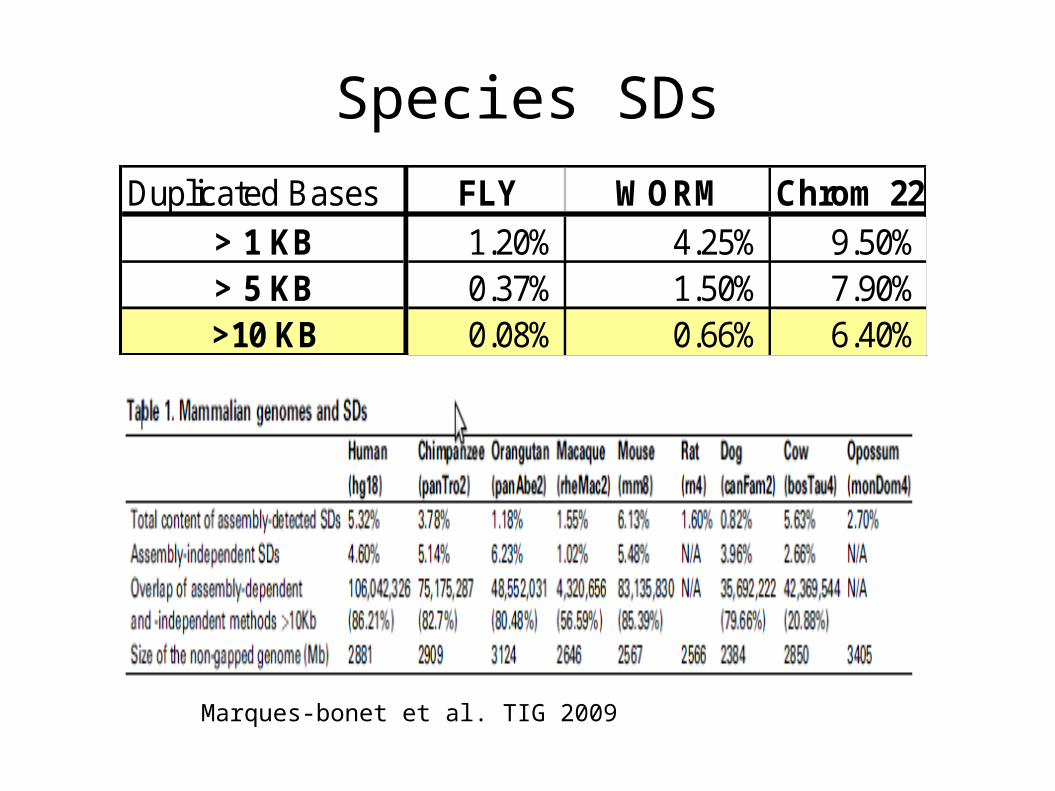
Species SDs
Marques-bonet et al. TIG 2009
Duplicated Bases FLY WORM Chrom 22> 1 KB 1.20% 4.25% 9.50%> 5 KB 0.37% 1.50% 7.90%>10 KB 0.08% 0.66% 6.40%
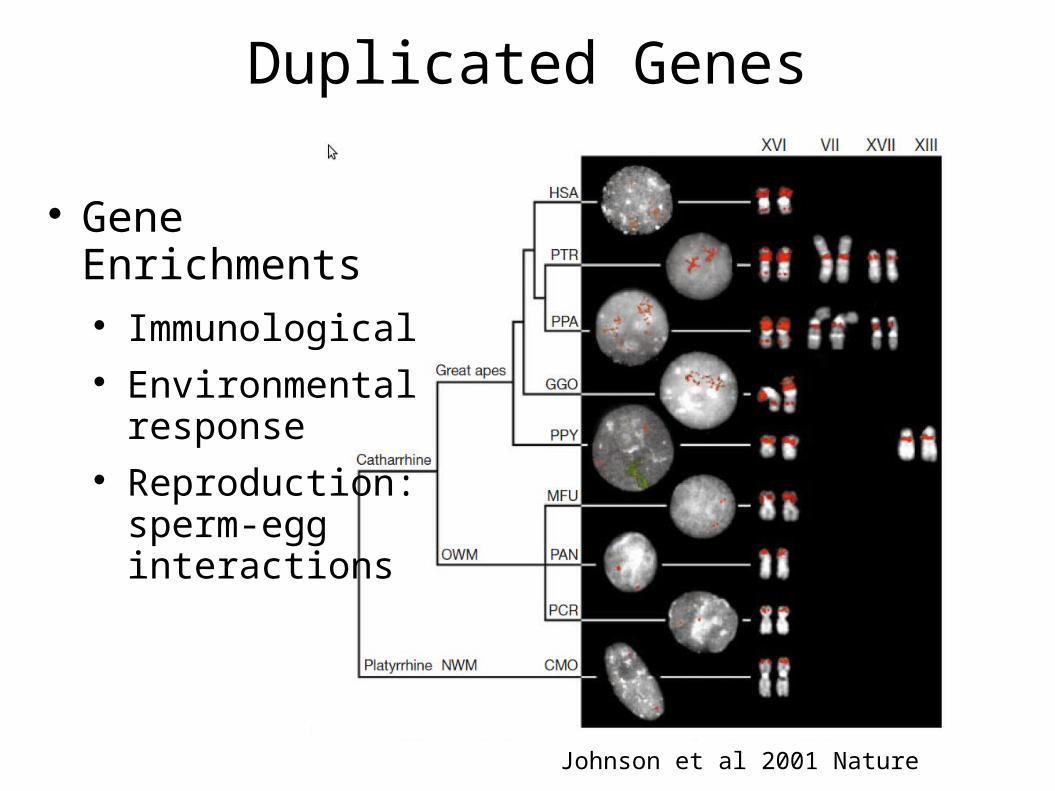
Duplicated Genes
Johnson et al 2001 Nature
Gene Enrichments Immunological Environmental
response Reproduction:
sperm-egg interactions
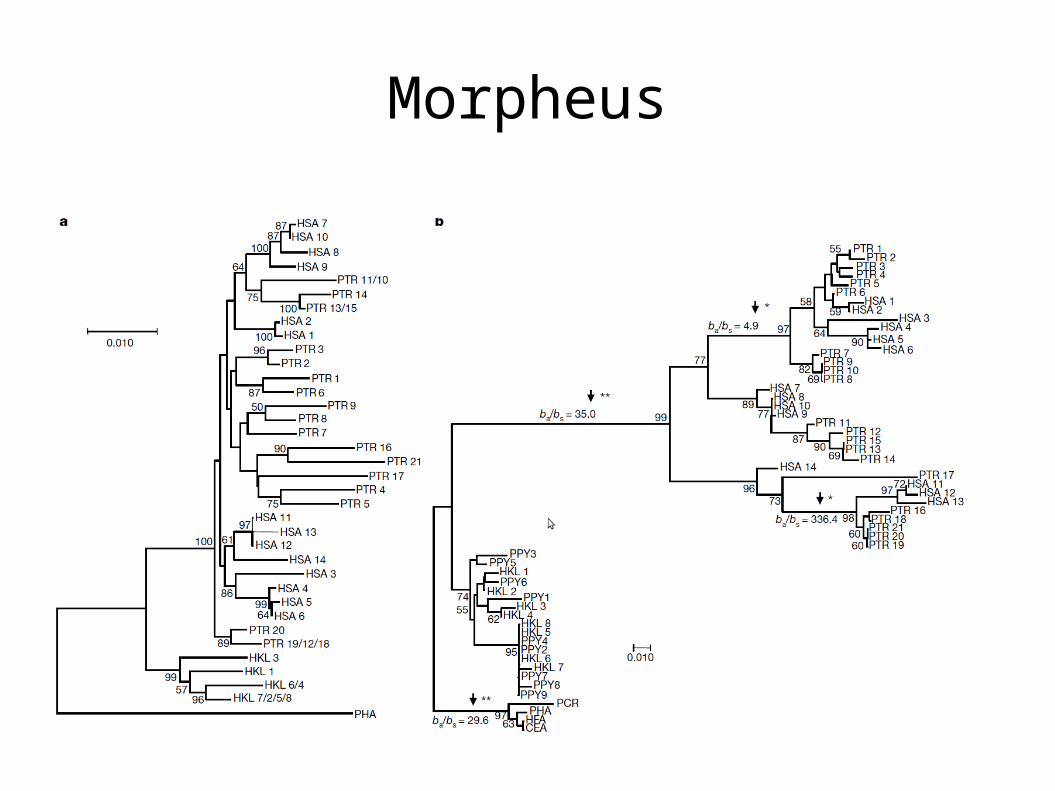
Morpheus

Duplicon Structure Chr 22

Organizing the MESS
Jiang et al. 2007 Nat Gen:39:1361-8
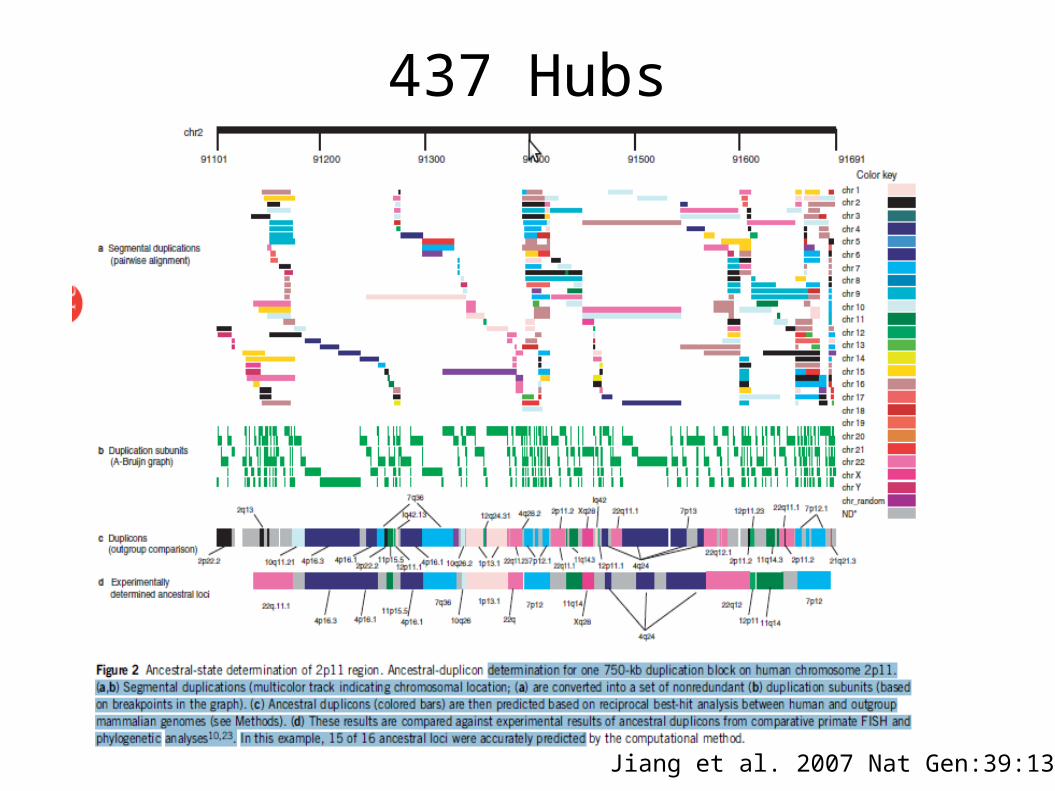
437 Hubs
Jiang et al. 2007 Nat Gen:39:1361-8

Mechanism: Junction Content
Control +/- 1 kb
Junction (50 bp)
•Duplications >95% and < 99.5%•Only finished sequence•Enrichment for Alu elements
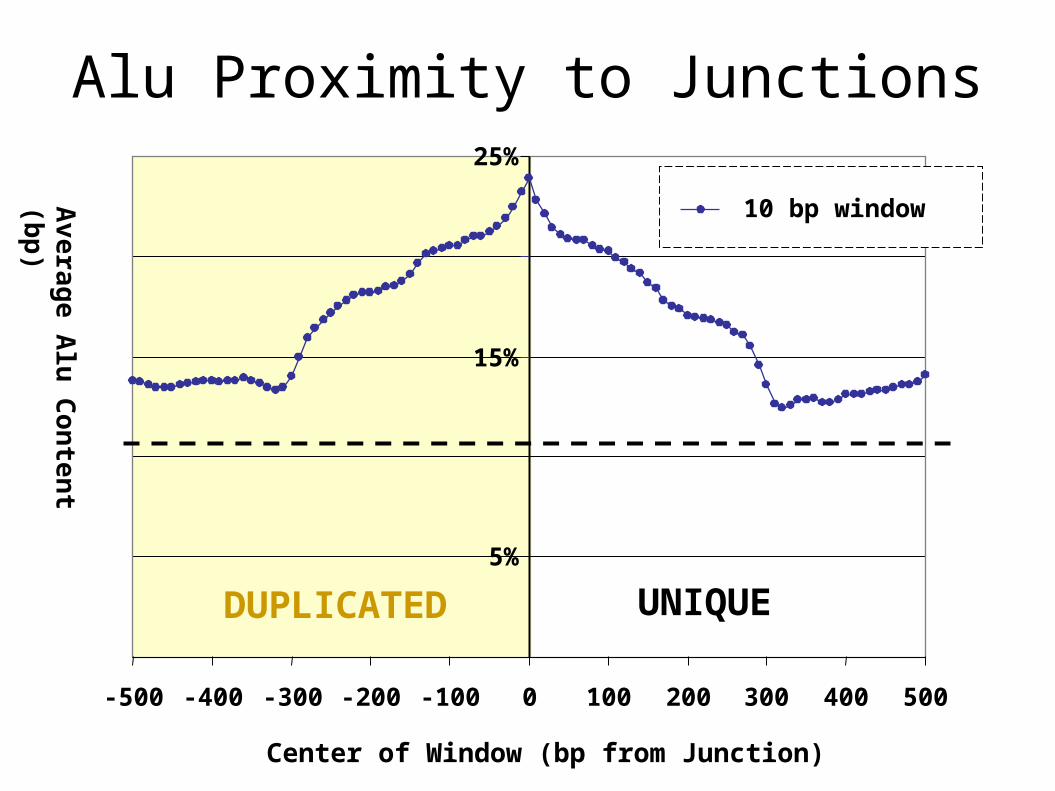
Alu Proximity to Junctions
5%
15%
25%
-500 -400 -300 -200 -100 0 100 200 300 400 500
10 bp window
DUPLICATED UNIQUE
Center of Window (bp from Junction)
Av
era
ge
Alu
Co
nte
nt
(bp
)
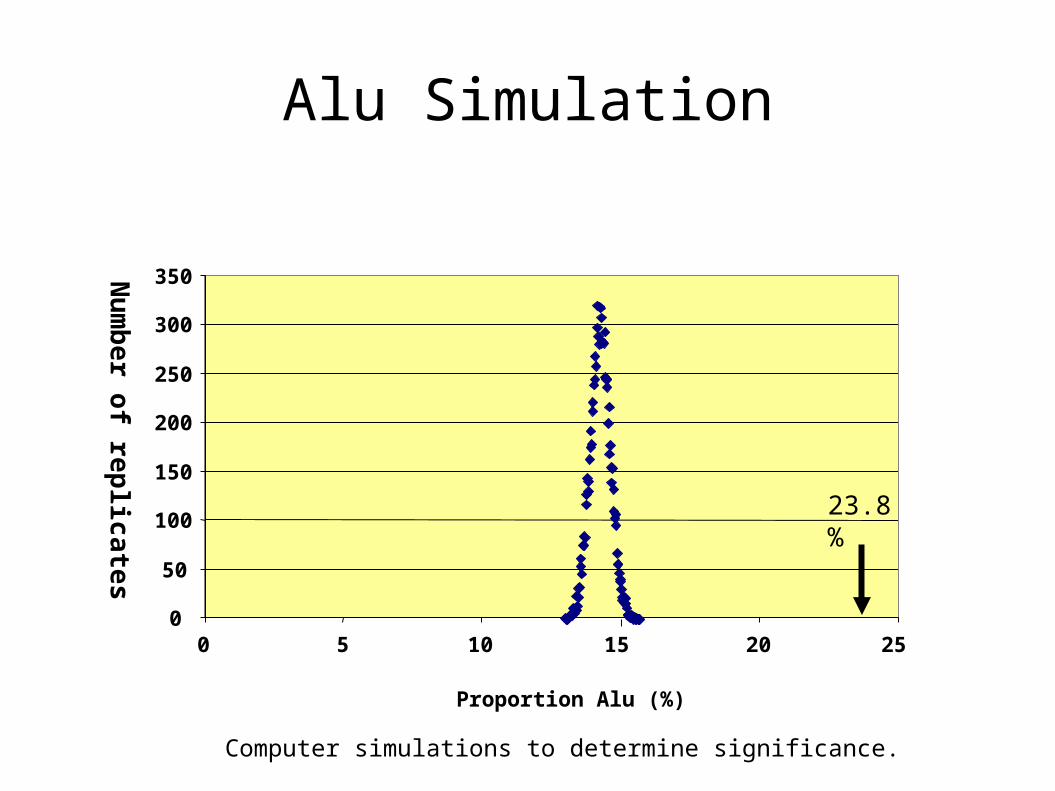
Alu Simulation
0
50
100
150
200
250
300
350
0 5 10 15 20 25
Proportion Alu (%)
Nu
mb
er o
f replica
tes
23.8%
Computer simulations to determine significance.
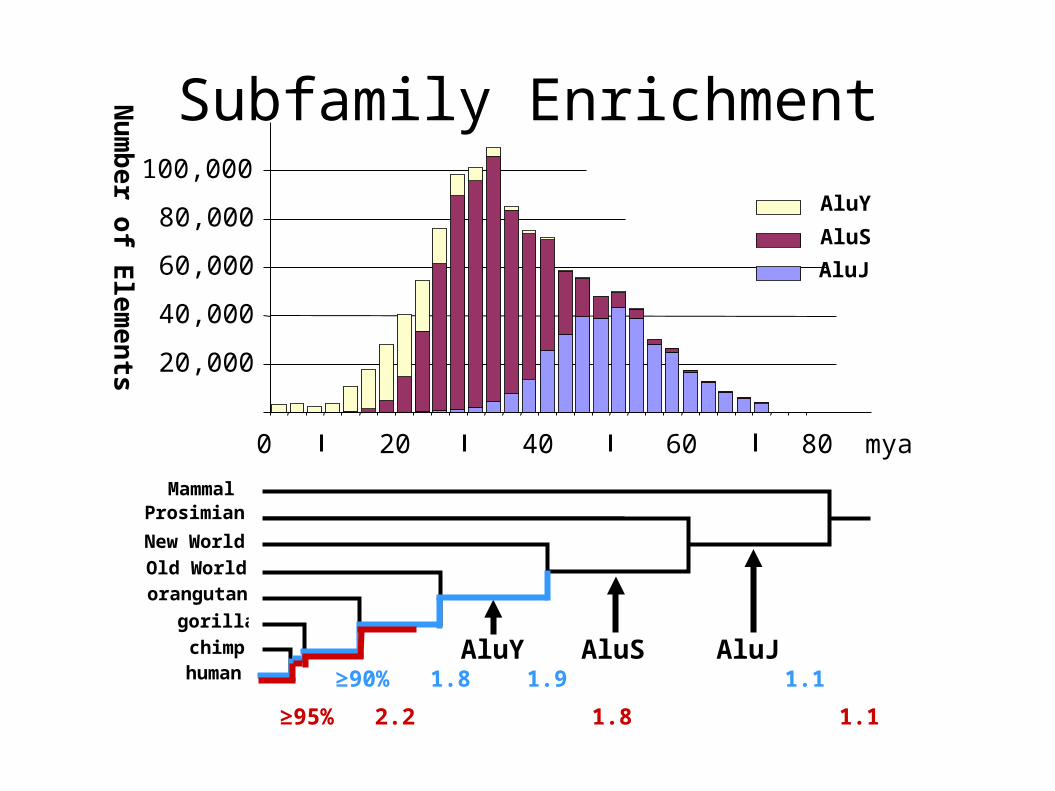
Subfamily Enrichment
20,000
40,000
60,000
80,000
100,000AluY
AluS
AluJ
20
humanchimp
orangutanOld World
New World
ProsimianMammal
gorilla
AluJAluSAluY
40 60 80 mya
≥90% 1.8 1.9 1.1
≥95% 2.2 1.8 1.1
0
Nu
mb
er o
f Ele
me
nts
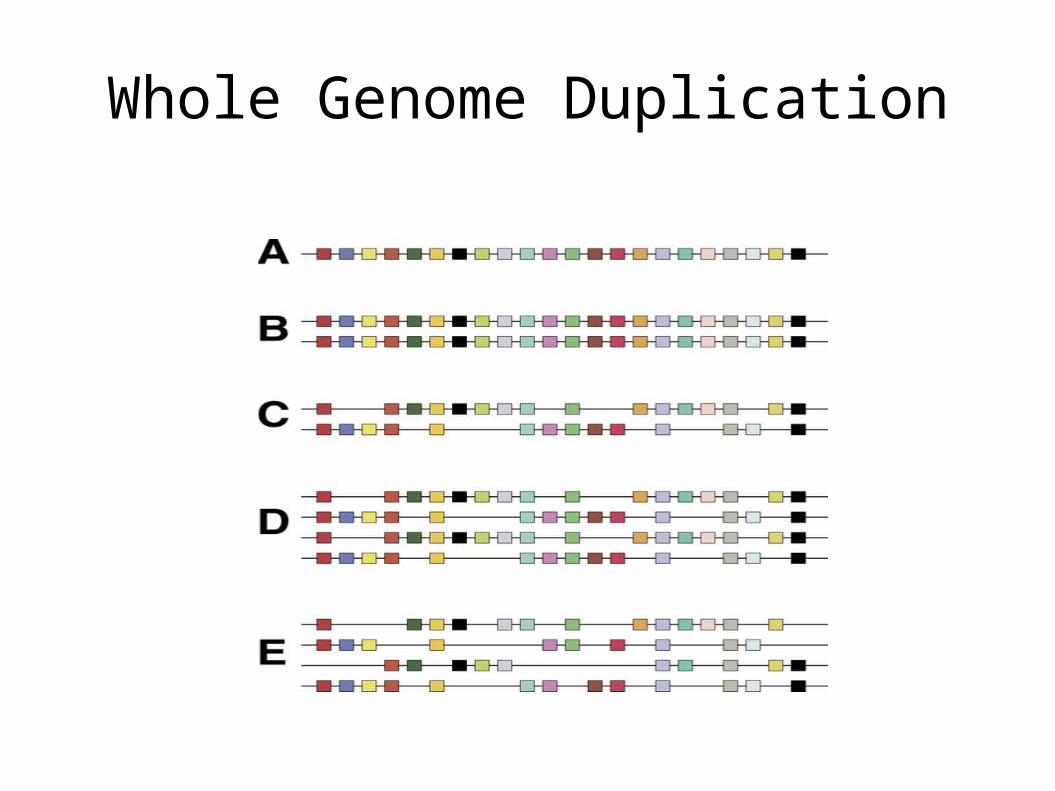
Whole Genome Duplication

Whole Genome Duplication Yeast
Kellis and Lander (Nature 428:617-24 2004)

Explore Resources
REMINDER OF CLASSExercises for analysis of repetitive elements and segmental duplications





























