Embed Size (px)
Citation preview

Repetitive DNA Detection and Classification
Vijay Krishnan
Masters Student
Computer Science Department

2
Repetitive DNA Refers to substrings of the genome that
repeat multiple times. Different instances of the repeat element can
have slightly different patterns Highly prevalent in eukaryotes (organisms
with a visible nucleus and cell structure, as opposed to bacteria)• About 50% of the human genome is repetitive
DNA.

3
Why detect repetitive DNA? Repeats Drive Evolution in Diverse Ways
(Kazazian, 2004). Repetitive DNA are generally not found to
have any function. Homology searches need repeat masking.
• To avoid explosion of unnecessary results.
Repeats also contain information about parentage.

4
Hit Defined as a local alignment between two
regions Q and T. Q and T are called images of the hit. Q = partner(T) with respect to the hit. Completely defined by the endpoint
coordinates of Q and T. Endpoints of Q referred to as start(Q) and
end(Q).
5
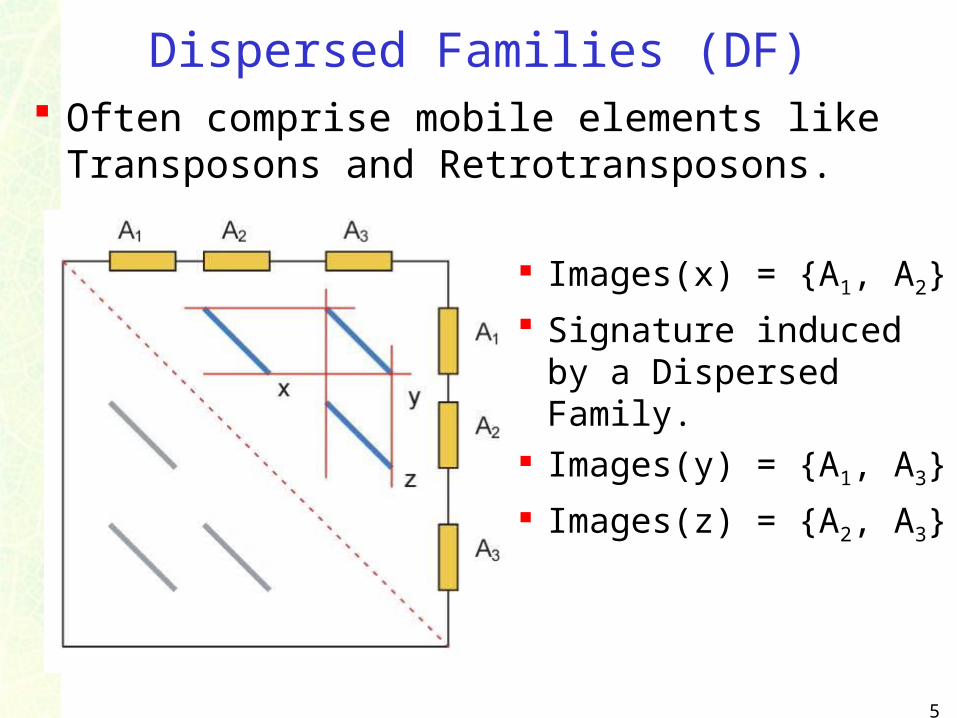
Dispersed Families (DF) Often comprise mobile elements like
Transposons and Retrotransposons.
Images(x) = {A1, A2}
Signature induced by a Dispersed Family.
Images(y) = {A1, A3}
Images(z) = {A2, A3}
6
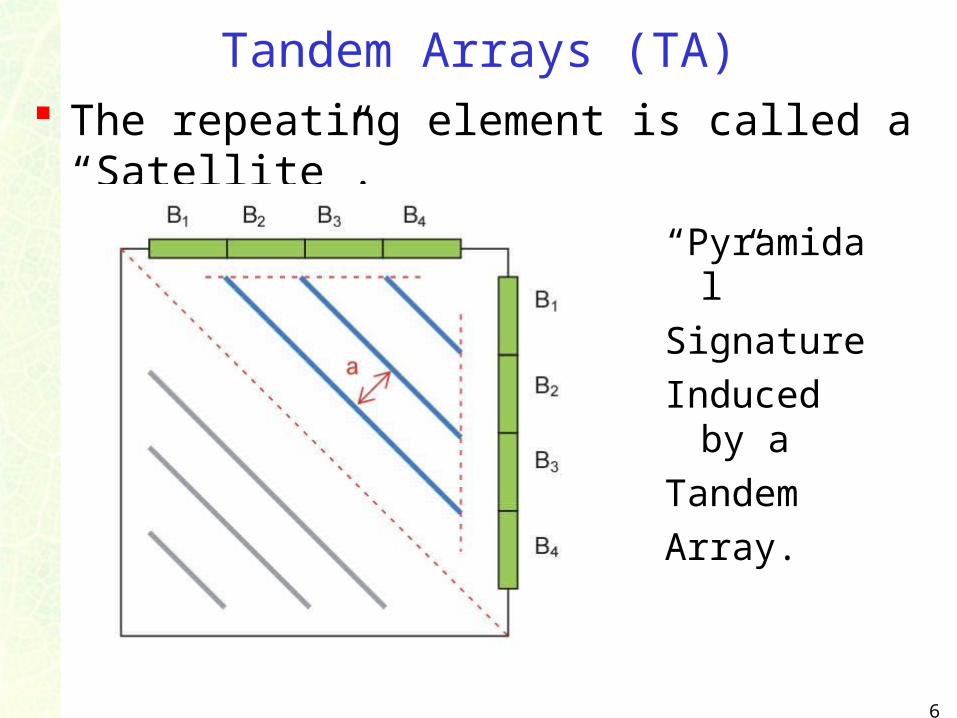
Tandem Arrays (TA) The repeating element is called a “Satellite”.
“Pyramidal”
Signature
Induced by a
Tandem
Array.

7
Other Repeat Families Pseudo-Satellites: Intermediate between
Satellites and Dispersed Families. Tandem Repeat:
• Often defined to be the same as TA.• The PILER paper defines it as images with size
50-2000 bases, separated by 50 to 15000 bases.

8
De novo identification of repeat families Input: The Genome sequence Output: The repeat families and the positions
where they occur in the Genome.

PILER: identification and classification of genomic repeats
Robert C. Edgar
and
Eugene W. Myers

10
Finding Local Alignments (Hits) Pairwise Alignment of Local Sequences
(PALS) software used as a black box. Used to find local alignments of minimum
length(λ) and minimum identity(μ). Additional optimizations for banded search
for alignments.• Finding regions separated by maximum distance
β.

11
Pile Suppose we are given a list of N hits. This corresponds to 2N images (intervals). A pile is a list of all images covering a
maximal contiguous region.• “Merge” overlapping images and “erase” the
boundaries between adjacent images.
Let images = { [1,3], [2,4], [3,6], [8,9], [9,13] }• Pile boundaries = { [1,6], [8,13] }.• Pile Images = { {[1,3], [2,4], [3,6]}, {[8,9], [9,13]} }

12
Construction of Piles (Example) Images = { [1,3], [2,4], [6,7] }
Index 1 2 3 4 5 6 7
Value 0 0 0 0 0 0 0
Index 1 2 3 4 5 6 7
Value 1 1 1 0 0 0 0
Index 1 2 3 4 5 6 7
Value 1 2 2 1 0 0 0
Index 1 2 3 4 5 6 7
Value 1 2 2 1 0 1 1
Index 1 2 3 4 5 6 7
Value 1 1 1 1 0 2 2

13
PILER-DF Let G be a graph with one node for each pile, and
no edges. is-global-image(Q) is true if:
• #bases in Q >= g * (#bases in pile(Q))
For each pile p in P:• For each image Q in p:
• Let T = partner(Q)• if is-global-image(Q) and is-global-image(T ):
– Add edge p−pile(T ) to G
Find connected components of G of order ≥ t. t >= 3 to avoid segmental duplication. Each Connected Component is a DF.

14
PILER-PS Similar to the problem of finding DFs, except
that PSs are typically closer to one another. Algorithm identical to PILER-DF except for
banded search to identify hits. Banded Search:
• Ensures that the PSs are clustered.• Allows a faster and more sensitive search for
hits.

15
PILER-TA TAs have pyramids as signatures. We can avoid comparing every pair of hits
since:• Hits in a pyramid belong to the same pile.• The images should be separated by at most
distance β (banded search).
Define first(h) = image in h with smaller start coordinate.
Define last(h) = image in h with larger start coordinate.

16
PILER-TA For each pile p:
• Create an empty graph G with allhits in the pile• For each pair of hits (h1, h2) in p:
• Set shorter_length = min(|h1|, |h2|)• Set longer_length = max(|h1|, |h2|)• Set Q1 = first(h1) … here (B1,B2,B3)• Set T1 = last(h1) … here (B2,B3,B4)• Set Q2= first(h2) … here (B1,B2)• Set T2 = last(h2) … here (B3,B4)• Set dS = (start(Q2) − start(Q1)) / shorter_length … here 0-0=0• Set dE = (end(T2) − end(T1)) / shorter_length …. Here 4-4 = 0• if shorter_length / longer_length > 0.5 and
– |dS| < m and |dT | < m:– Add edge h1 − h2 to G
Each connected components of G is a TA. 0 <=m <= 1. By default m= 0.05.

17
PILER-TR
Identify and mask Satellites and PSs. Two pass method:
• Pass1: perform banded search for TR candidates.
• Pass2: Find hits that align TR pairs to each other.

18
Library Construction Use MUSCLE (Edgar, 2004a,b)
• Create multiple alignments of family members found by PILER.
• Use these to find consensus sequences.
This library can be used by BLAST or RepeatMasker to find intact and partial instances.

19
Satellites and PSs in A.thalania

De novo identification of repeat families in large genomes
Alkes L. Price
Neil C. Jones
Pavel A. Pevzner

21
The RepeatScout Algorithm Improves on the RECON algorithm (Bao and
Eddy, 2002). Builds repeat families using high-frequency
L-mers as seeds. Input: DNA Sequences S1,…..,Sn each of
which contains a similar repeat element and extends past the repeat element on either side.
Output: Substrings R1,…..,Rn that give the repeat element boundaries, and consensus sequence Q.

22
RepeatScout (contd) Q is defined to be the sequence that
maximizes:
A(Q;S1,...,Sk) = [ ∑k max{a(Q,Sk),0}] -c|Q|,
Where a(Q,Sk) can be any reasonable sequence alignment score.
The penalty factor c|Q| discourages long Qs,• c can be thought of as the minimum number of
repeat elements that must align with each given position of Q.

23
Choice of a(Q,S) Local Alignment Score:
Fit Alignment Score (Waterman, 1995)• Boundaries of Q shared by all segments.• Strict constraint on Q.

24
Fit-Preferred Alignment Score

25
Comparison of Alignment Scores

26
Optimizing A(Q; S1, . . . , Sn) Even dynamic Programming for the optimal
solution is intractable.• The problem would be n-dimensional.• Both time and space requirements are
exponential in n.
Greedy Heuristic:• Suppose L is the high freqency lmer and S1, . . . ,
Sn surround its exact matches.
• Initialize Q0 to L and greedily extend Q.

27
Optimizing A(Q; S1, . . . , Sn) N Є {A, C, G, T} Choose Qt+1 =Qt .N where N maximizes:
• A(Qt .N; S1, . . . , Sn)
We can re-use alignment scores from the previous iteration while computing alignment scores for the (t+1)th iteration.
Terminate after a certain no. of iterations gives no improvement.
Use this procedure for extending to the right, and then to the left.

28
Optimizing A(Q; S1, . . . , Sn) Prevent redundancy in finding consensus
sequences. After identifying Q, locate its occurrences
and reduce the counts of L-mers corresponding to those locations.
Algorithm terminates when we have no L-mers with effective count of at least m.
Refine Q after the optimal alignment boundaries are determined.
More details of parameter settings in the paper.

29
Results

30
Results
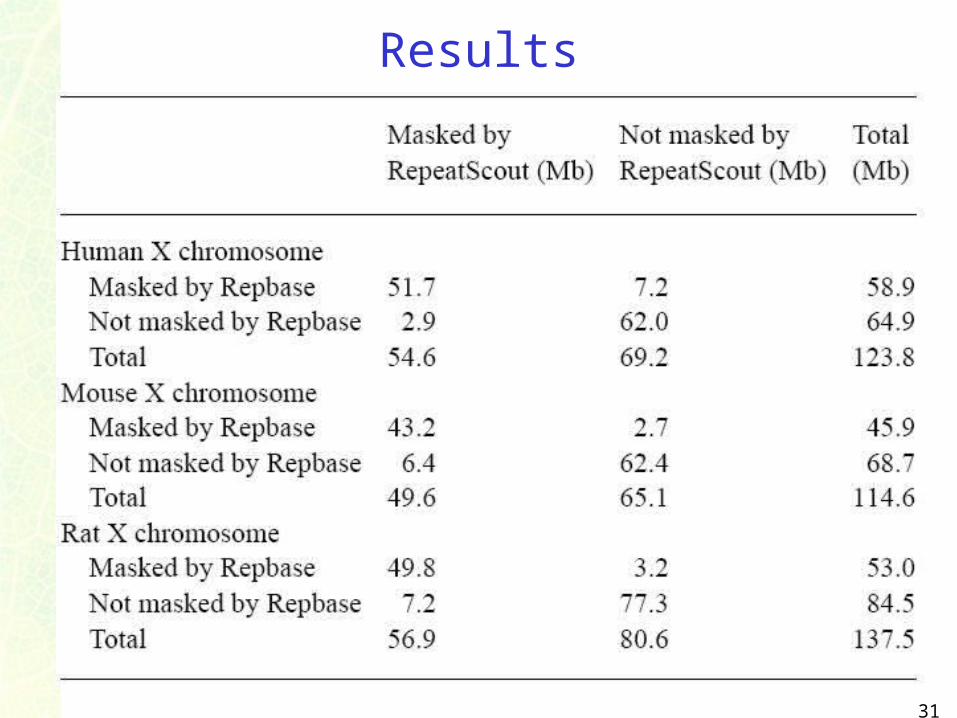
31
Results

32
Conclusions Both PILER and RepeatScout address DNA
repeats. PILER focuses more on finding diverse kinds
of repeat families and uses MUSCLE to find the consensus sequences
RepeatScout focuses more on finding the consensus sequence given members of a repeat family.

33
Thank You!
Questions?





























