Embed Size (px)
Citation preview

7 Westferry Circus ● Canary Wharf ● London E14 4HB ● United Kingdom Telephone +44 (0)20 7418 8400 Facsimile +44 (0)20 7523 7040 E-mail [email protected] Website www.ema.europa.eu An agency of the European Union
© European Medicines Agency, 2010. Reproduction is authorised provided the source is acknowledged.
27 April 2010 EMA/50813/2009 Human Medicines Development and Evaluation
Report to the European Commission Companies and products that have benefited from any of the rewards and incentives in the paediatric regulation and the companies that have failed to comply with any of the obligations in this regulation covering the years 2007 to 2009
Prepared by the Section Paediatric Medicines Human Medicines Special Areas Sector European Medicines Agency

Report to the European Commission EMA/50813/2009 Page 2/34
Table of contents
1. INTRODUCTION..................................................................................... 4 1.1. Scope of the report...............................................................................................4 1.2. Data collection .....................................................................................................4 1.3. Overview of the implementation of the Paediatric Regulation .....................................5
2. COMPANIES AND PRODUCTS THAT HAVE BENEFITED FROM ANY OF THE REWARDS AND INCENTIVES IN THE REGULATION ...................................... 6 2.1. Scientific advice ...................................................................................................6 2.1.1. Advice from the Agency......................................................................................6 2.1.2. Advice from the National Competent Authorities ....................................................7 2.2. Paediatric Investigation Plans – Waiver ...................................................................7 2.3. Compliance statement included in a marketing authorisation .....................................9 2.3.1. Compliance statement for centrally-authorised medicinal products...........................9 2.3.2. Compliance statement for medicinal products authorised through national/decentralised/mutual recognition procedure, including those subject to Article 29 of the Paediatric Regulation .............................................................................................9 2.4. Extension of the Supplementary Protection Certificate/Market Exclusivity ..................10 2.5. Marketing authorisation granted or varied with mention of the waiver or deferral in the Summary of Product Characteristics ............................................................................10 2.6. Price/reimbursement benefits ..............................................................................12 2.7. Research incentives ............................................................................................12 2.7.1. EU Framework Programme ...............................................................................12 2.7.2. European Network of Paediatric Research at the European Medicines Agency ..........12 2.7.3. National initiatives ...........................................................................................13 2.8. Authorisation of paediatric clinical trials.................................................................15 2.9. Procedures for marketing authorisation .................................................................15 2.10. Article 45/46 of the Paediatric Regulation ............................................................17 2.10.1. Article 45......................................................................................................17 2.10.2. Article 46......................................................................................................17
3. FAILURE TO COMPLY WITH THE OBLIGATIONS SET IN THE PAEDIATRIC REGULATION ............................................................................................. 19 3.1. Submissions of the PIP/waiver application to the PDCO ...........................................19 3.2. Validation of application for marketing authorisation/extension ................................19 3.3. Compliance with the paediatric requirements and rewards.......................................20 3.4. Mention of the Decision on waivers or deferrals in the product information ................20 3.5. Annual reports on deferrals .................................................................................20

Report to the European Commission EMA/50813/2009 Page 3/34
4. CONCLUSION......................................................................................... 20
Annex 1 - Questions sent to the Member States ........................................ 22
Annex 2 - Compliance statement included in a marketing authorisation for products authorised through national/decentralised/mutual recognition procedure.................................................................................................. 24
Annex 3 - 6-months extension of the supplementary protection certificate (SPC) granted by the National Patent Office ............................................. 28
Annex 4 – List of projects on off-patent medicines funded by the European Commission through the EU Framework Programme................................. 31
Annex 5 - List of products and resulting amendment of the SmPC further to submission of data through article 45 and 46............................................ 32

Report to the European Commission EMA/50813/2009 Page 4/34
1. INTRODUCTION
1.1. Scope of the report
Regulation (EC) No. 1901/2006 of the European Parliament and of the Council on medicinal products
for paediatric use and amending Regulation (EEC) No 1768/92, Directive 2001/20/EC, Directive
2001/83/EC and Regulation (EC) No 726/2004 (hereinafter 'the Paediatric Regulation') was adopted on
12th December 2006. It was published in the Official Journal of the European Communities on 27th
December 2006 and entered into force on 27th January 2007.
Article 50(1) states: “On the basis of a report from the Agency, and at least on an annual basis, the
Commission shall make public a list of the companies and of the products that have benefited from any
of the rewards and incentives in this Regulation and the companies that have failed to comply with any
of the obligations in this Regulation. The Member States shall provide this information to the Agency.”
Considering the early days of the implementation of the Paediatric Regulation and the lack of internal
resources, the European Medicines Agency (hereafter “the Agency”) was not able to produce such a
report for the years 2007 and 2008. This report therefore covers the whole period from the entry into
force of the Paediatric Regulation, i.e. 26 January 2007 to 31 December 2009.
As explicitly requested, this report lists the companies and products that have benefited from any of
the rewards and incentives defined in this Regulation both at the European Union and at national level.
The report examines also the situation where companies would have failed to comply with any of the
obligations set in this Regulation. The figures reported correspond to procedures with the start date
within the year.
Incentives available at EU level are supported by complementary national initiatives, particularly in
areas such as fiscal incentives and national research projects. Pursuant to Article 39(2) of the
Paediatric Regulation, the European Commission published such inventory on national measures based
on information received from 18 of 27 Member States on 30 July 2008.
1.2. Data collection
The Agency sent a letter on 10 December 2009 to all Member States to require contributions to
prepare this report (Letter sent to Permanent Representatives of the Member States of the European
Union with a response requested by 31 January 2010). The letter contained a list of information to be
provided (Annex 1).
We also contacted the DG Research to obtain information on the projects funded through the
framework programme in the context of the Paediatric Regulation.
The Agency also sent a copy of this letter to the Heads of Agencies and informed the Coordination
Group for Mutual Recognition and Decentralised Procedure – human, accordingly. In order to obtain
the highest return rate, a reminder was sent on 11 February 2010.
The Agency received feedback from 22 Member States:
Austria- Belgium - Bulgaria – Czech Republic - Cyprus – Denmark - Estonia – France - Finland –
Germany – Hungary - Ireland – Italy – Latvia – Lithuania – Luxembourg – Malta - The Netherlands -
Romania - Slovak Republic - Slovenia – Sweden - United Kingdom.
No answer has been received from Greece – Poland - Portugal – Spain.
Information was also received from EFTA States. The Paediatric Regulation is not yet part of the EEA
Agreement and therefore is not yet implemented in EFTA States (Iceland, Liechtenstein and Norway).

Report to the European Commission EMA/50813/2009 Page 5/34
Nonetheless Iceland and Norway have actively contributed to the work of the Paediatric Committee
since the beginning. In addition they requested the submission of paediatric data according to Article
45 and 46 of the Paediatric Regulation and are participating in the worksharing for assessment of these
data.
This report is not comprehensive. The limit of the methodology used is that some data was not
provided by all Member States. For instance some of the requested information may only be available
from certain bodies of the Member States such as the National Patent Office, the Competent Authority
for Medicinal Products, or the Ministry of Health. Therefore depending on the process chosen by the
Member State to collect such data, the feedback received varied substantially.
Some Member States provided additional information on some measures taken at national level. These
have been annexed to this report for completeness.
According to the Paediatric Regulation, the Agency should produce such report annually. Based on this
year’s experience, the Agency will reflect on how to improve the method to help Member States
collecting the data.
1.3. Overview of the implementation of the Paediatric Regulation
Three years have elapsed since the entry into force of the Paediatric Regulation whose objectives are:
i) to increase the availability of medicines intended for children, ii) to make information on those
medicines widely available and iii) to stimulate high quality paediatric research. This Regulation
includes a set of obligations and rewards/incentives for industry to compensate the investment in
paediatric development.
One of the pillars of the Regulation is the Paediatric Committee (PDCO) which is primarily responsible
for reviewing and agreeing applications for paediatric investigation plans (PIPs) including deferrals,
and/or waivers. Since its first meeting on 4 July 2007, the PDCO has performed strongly as shown by
the figures presented later in the report. This has been possible thanks to the preparation and
motivation of Committee members, supported for most of them by the National Competent Authorities,
as all of them have actively participated in the review process, as well as to the Agency secretariat,
namely the Paediatric Medicines section of the Human Special Areas sector, which supports the
Committee in its activities.
The Paediatric Regulation has created a series of new tasks for the Agency and for the Member States
which had an impact on the resources at Agency as well as at the National Competent Authorities level.
Most of the tasks have been dealt with, and the legal deadlines have been met with success. Again this
has been possible thanks to the cooperation of all partners and stakeholders, in particular learned
societies and industry.
Further information on the status of the implementation of the Paediatric Regulation with respect to
the various topics covered can be found on the Agency website (Medicines for Children
http://www.ema.europa.eu/htms/human/paediatrics/introduction.htm).

Report to the European Commission EMA/50813/2009 Page 6/34
2. COMPANIES AND PRODUCTS THAT HAVE BENEFITED FROM ANY OF THE REWARDS AND INCENTIVES IN THE REGULATION
2.1. Scientific advice
2.1.1. Advice from the Agency
In accordance with Article 26 of the Regulation, the Agency provides free scientific advice for any
paediatric questions. Scientific advice and protocol assistance (the special form of scientific advice
available for the development of designated medicines for 'orphan' or rare diseases) may be given to
companies on the design and conduct of trials necessary to demonstrate the quality, safety and
efficacy of the medicinal product in the paediatric population. The advice is provided by the Scientific
Advice Working Party of the Committee for Medicinal Products for Human Use (CHMP) and is adopted
by the CHMP. For the paediatric ones, members of the PDCO are routinely involved in the procedure as
experts. This is part of the collaboration established under the Executive Director’s responsibility
(article 3(3) of the Paediatric Regulation).
Applicants may choose to request scientific advice before submitting an application for a PIP to help
them to prepare such plan, or after the Agency Decision to discuss, for example, combined adult and
paediatric development in light of the PIP requirements. Some companies have submitted
simultaneous applications for a PIP and request for a scientific advice, but this is discouraged as the
procedures overlap creating unnecessary duplication of work. In addition despite active collaboration
between the PDCO and the Scientific Advice Working Party, a risk of divergence cannot be ruled out as
the two groups may not have the possibility to discuss all details of the applications.
At the difference of the decision on a PIP, scientific advice/protocol assistance received from the
Agency is not binding, either on the Agency, CHMP or on the sponsor, with regard to any future
marketing-authorisation application for the product concerned.
Since the entry into force of the Paediatric Regulation the number of such requests has increased
steadily (table 1).
Table 1. Number of requests for paediatric scientific advice(SA)/protocol assistance (PA) and follow-ups
Year 2007 Year 2008 Year 2009
Total SA requests 213 264 311
Total PA requests 68 56 77
Paediatric scientific advice 14 13 14
Paediatric follow-up SA 4 5 9
Paediatric protocol assistance - 5 4
Paediatric follow-up PA 3 - 3
Total paediatric SA+PA 21 23 30
As the information on scientific advice is considered commercially confidential, the list of the
companies and products which have benefited from this incentive is not included in this report but can
be found as a separate document.
A high number of mixed scientific advice/protocol assistance requests for adult and paediatric
development have also been submitted for which members of the PDCO may also be involved (see
Report to the European Commission EMA/50813/2009 Page 7/34
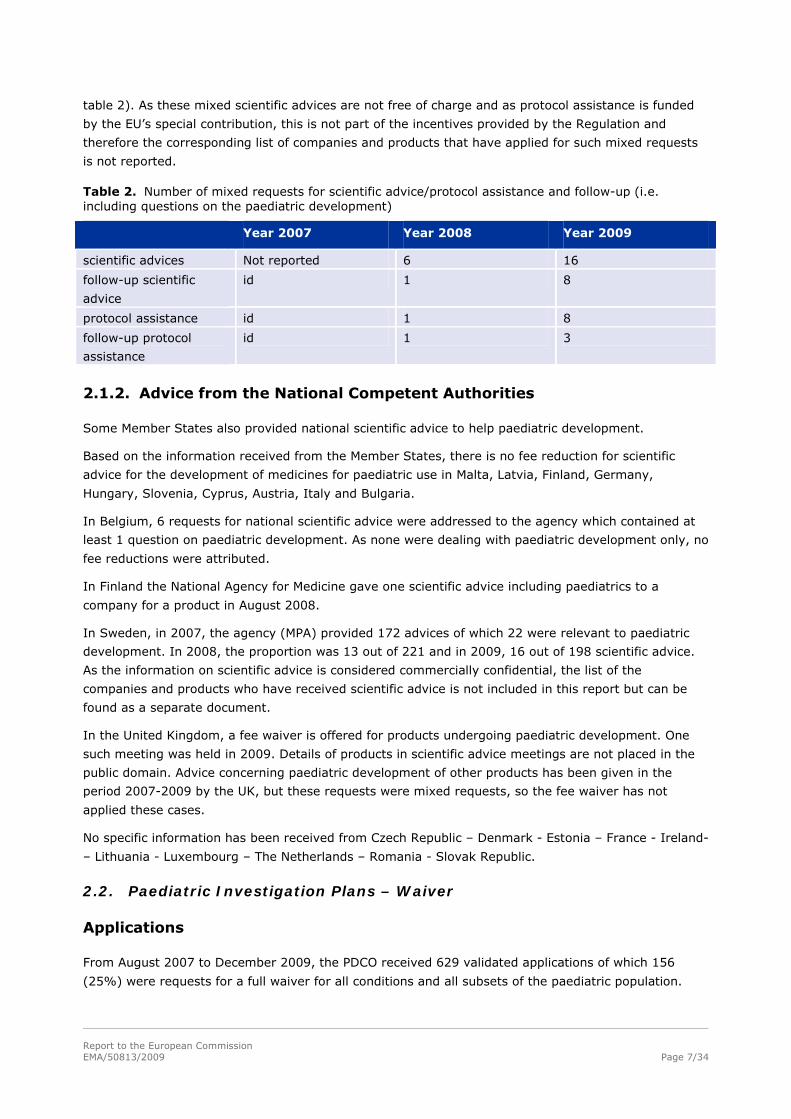
table 2). As these mixed scientific advices are not free of charge and as protocol assistance is funded
by the EU’s special contribution, this is not part of the incentives provided by the Regulation and
therefore the corresponding list of companies and products that have applied for such mixed requests
is not reported.
Table 2. Number of mixed requests for scientific advice/protocol assistance and follow-up (i.e. including questions on the paediatric development)
Year 2007 Year 2008 Year 2009
scientific advices Not reported 6 16
follow-up scientific
advice
id 1 8
protocol assistance id 1 8
follow-up protocol
assistance
id 1 3
2.1.2. Advice from the National Competent Authorities
Some Member States also provided national scientific advice to help paediatric development.
Based on the information received from the Member States, there is no fee reduction for scientific
advice for the development of medicines for paediatric use in Malta, Latvia, Finland, Germany,
Hungary, Slovenia, Cyprus, Austria, Italy and Bulgaria.
In Belgium, 6 requests for national scientific advice were addressed to the agency which contained at
least 1 question on paediatric development. As none were dealing with paediatric development only, no
fee reductions were attributed.
In Finland the National Agency for Medicine gave one scientific advice including paediatrics to a
company for a product in August 2008.
In Sweden, in 2007, the agency (MPA) provided 172 advices of which 22 were relevant to paediatric
development. In 2008, the proportion was 13 out of 221 and in 2009, 16 out of 198 scientific advice.
As the information on scientific advice is considered commercially confidential, the list of the
companies and products who have received scientific advice is not included in this report but can be
found as a separate document.
In the United Kingdom, a fee waiver is offered for products undergoing paediatric development. One
such meeting was held in 2009. Details of products in scientific advice meetings are not placed in the
public domain. Advice concerning paediatric development of other products has been given in the
period 2007-2009 by the UK, but these requests were mixed requests, so the fee waiver has not
applied these cases.
No specific information has been received from Czech Republic – Denmark - Estonia – France - Ireland-
– Lithuania - Luxembourg – The Netherlands – Romania - Slovak Republic.
2.2. Paediatric Investigation Plans – Waiver
Applications
From August 2007 to December 2009, the PDCO received 629 validated applications of which 156
(25%) were requests for a full waiver for all conditions and all subsets of the paediatric population.

Report to the European Commission EMA/50813/2009 Page 8/34
Of the 629 validated applications which covered 961 indications (on average 1.5 indications per
product):
• 416 applications (66%) referred to medicinal products not yet authorised in the EU at the time of
the entry into force of the Regulation (so called “Article 7 applications”).
• 192 applications (31%) referred to products already authorised, still under patent or
supplementary Protection certificate, in view of a submission of a variation/extension for a new
indication, pharmaceutical form or route of administration (so called “Article 8 applications”).
• 21 Applications (3%) referred to an off-patent product developed specifically for children with an
age-appropriate formulation (so called “Article 30 applications” with a view to submit a Paediatric
Use Marketing authorisation or PUMA).
In the infancy of the implementation of the Regulation, most of the applications were “Article 8
applications”. After about a year, the balance changed with a higher proportion of “Article 7
applications”.
Opinions
As a result of the assessment of these applications, the PDCO has since its first meeting in July 2007
adopted:
• 125 positive opinions on product-specific waivers (36%).
• 205 positive opinions on a PIP (59%). An opinion on a PIP may also contain a deferral and/or
waiver of the obligation to gather clinical trial data in certain age groups of children.
• 17 negative opinions (5%).
Since 2008 which saw the first Decision, the details of the decisions issued by the Agency on the PDCO
opinions are published in a summarised form and can be found on the following webpage:
http://www.ema.europa.eu/htms/human/paediatrics/decisions.htm.
Class Waivers
To effectively manage the anticipated high level of applications and in accordance with the Regulation,
very early on, the PDCO adopted a list of conditions that occur only in adult populations for which all
classes of medicinal products intended to treat these conditions would be exempt from the requirement
for a PIP and/or from the submission of an application for a product-specific waiver. The list is updated
at least once a year. In addition, the PDCO has adopted an opinion on a class of products (in a
condition) for which, similarly, there is an exemption.
The Agency decisions on the PDCO opinions on the class waivers can be found on the following
webpage: http://www.ema.europa.eu/htms/human/paediatrics/decisions.htm.
Modifications of agreed PIPs
Although it is too early for a prediction, it is anticipated that the number of requests for modifications
of an agreed PIP will increase exponentially over the coming years. Indeed in order to establish an
early dialogue between the sponsor and the PDCO, the Regulation sets a deadline for the submission of
the application for a PIP and/or waiver at the early stage of the development of the medicinal product.
As the development of medicinal products is a dynamic process depending on the results of ongoing
studies, it is estimated that 3 to 5 modifications will be submitted per agreed PIPs. As of December
2009, the PDCO already adopted 59 positive opinions on modifications of an agreed PIP.

Report to the European Commission EMA/50813/2009 Page 9/34
2.3. Compliance statement included in a marketing authorisation
Once the PIP is completed, there is a need to check compliance, i.e. to verify that the measures set out
in the Agency decision have been carried out in accordance with the agreed PIP, including its timelines.
This is done at the time of validation either of applications for marketing authorisation, or of
variation/extensions, or prior to the submission of the application, on request from the applicant to the
PDCO.
Until now and as a pilot phase, the National Competent Authorities have requested the PDCO to check
the compliance check on their behalf.
Opinions of the PDCO on compliance are provided once the PIP is completed. As of December 2009,
the number of PDCO positive opinions on compliance with an agreed PIP reached 13. The PDCO also
adopted 1 negative opinion on compliance.
2.3.1. Compliance statement for centrally-authorised medicinal products
For 3 centrally authorised medicinal products, 2 companies have already provided the results of all
studies performed in compliance with an agreed PIP as part of the submission of an application for
authorisation of new indications, including paediatric indications, new pharmaceutical forms and new
routes of administration. This has resulted in a compliance statement included in the marketing
authorisation by the European Commission (Table 3).
Table 3. List of companies and products with a compliance statement (centrally approved)
Companies Products: invented name (international non-proprietary name)
Year
2008
Merck Sharp
and Dohme
Cancidas (caspofungin)
http://www.ema.europa.eu/humandocs/Humans/EPAR/cancidas/cancidasM2.ht
m
Year
2009
Schering-
Plough
Europe
Schering-
Plough
Europe
Rebetol (ribavirin)
http://www.ema.europa.eu/humandocs/Humans/EPAR/rebetol/rebetolM2.htm
PegIntron/Viraferon peg (peginterferon alfa-2b)
http://www.ema.europa.eu/humandocs/Humans/EPAR/pegintron/pegintronM2.h
tm
http://www.ema.europa.eu/humandocs/Humans/EPAR/viraferonpeg/viraferonpe
gM2.htm
2.3.2. Compliance statement for medicinal products authorised through national/decentralised/mutual recognition procedure, including those subject to Article 29 of the Paediatric Regulation
The list of companies and products which have benefited from the inclusion of the compliance
statement is presented in Annex 2. So far this list includes only the products which have been subject
to an “Article 29” procedure of the Paediatric Regulation. This procedure allows companies to submit
an application to the Agency for a new indication (including in children), pharmaceutical form or route
of administration for medicines that are already authorised at the level of the Member States. Data
supporting such applications have to be generated in accordance with an agreed paediatric
investigation plan (PIP). This results in the adoption of an EU harmonised decision on the use of a
medicinal product in the paediatric population. Once the Commission Decision is adopted, the Member
States are required to vary the terms of an existing marketing authorisation as necessary, to comply
with the Decision within 30 days of its notification.

Report to the European Commission EMA/50813/2009 Page 10/34
Table 4. Commission Decisions for medicinal products for human use, pursuant to Article 29 of Regulation (EC) No 1901/2006, with a compliance statement
Companies Products: invented name (international non-proprietary name)
Year
2008
Merck Sharp
and Dohme BV
Cozaar and associated names (losartan)
Commission Decision C(2009)488
http://www.ema.europa.eu/pdfs/human/press/pr/55020608en.pdf
Year
2009
AstraZeneca
AB
Novartis
Pharma AG
Arimidex and associated names (anastrazole) Commission Decision
C(2009)8750
http://www.ema.europa.eu/pdfs/human/press/pr/55020608en.pdf
Diovan and associated names (valsartan) –(CHMP Opinion adopted in
December 2009)
http://www.ema.europa.eu/pdfs/human/press/pr/83310409en.pdf
2.4. Extension of the Supplementary Protection Certificate/Market Exclusivity
In order to be eligible for an extension of the Supplementary Protection Certificate (SPC) by 6 months,
several conditions need to be met (assuming that the SPC extension application is made in due time
and complies with the provisions of Regulation (EC) No 469/2009):
i) he compliance statement with the agreed PIP is included in the marketing authorisation;
ii) there is an authorisation of the medicinal product in all Member States;
iii) there is an update of the Summary of Product Characteristics (SmPC) with results of the studies
conducted in compliance with the agreed PIP. This applies even if the results fail to lead to the
authorisation of a paediatric indication.
• Extensions of the supplementary protection certificate are granted by the National Patent Offices at
national level. Therefore the companies will have to file for an SPC extension with the NPO of each
and every Member State where the active substance used for the medicinal product is protected by
a basic patent or an SPC.
Annex 3 compiles the information received from Member States on those products which have
fulfilled the paediatric requirements and therefore have been granted by the National Patent Offices
a 6-month extension of the Supplementary Protection Certificate.
• For orphan medicinal products, the reward is a 2-year extension of the market exclusivity. So far
no orphan medicinal product has benefited from this reward. Orphan medicines represent
approximately 20% of the applications for PIPs and waivers.
As no PUMA has yet been authorised, no company has benefited from the defined data and
marketing protections periods.
2.5. Marketing authorisation granted or varied with mention of the waiver or deferral in the Summary of Product Characteristics
According to Article 28(2) of the Paediatric Regulation, any Agency decision on a waiver or deferral is
recorded in the Summary of Product Characteristics (SmPC) and if appropriate in the package leaflet of
the product concerned when the initial marketing authorisation is granted or when the marketing
authorisation is varied to include a new indication, including paediatric indication, new pharmaceutical
form or new route of administration.
Report to the European Commission EMA/50813/2009 Page 11/34
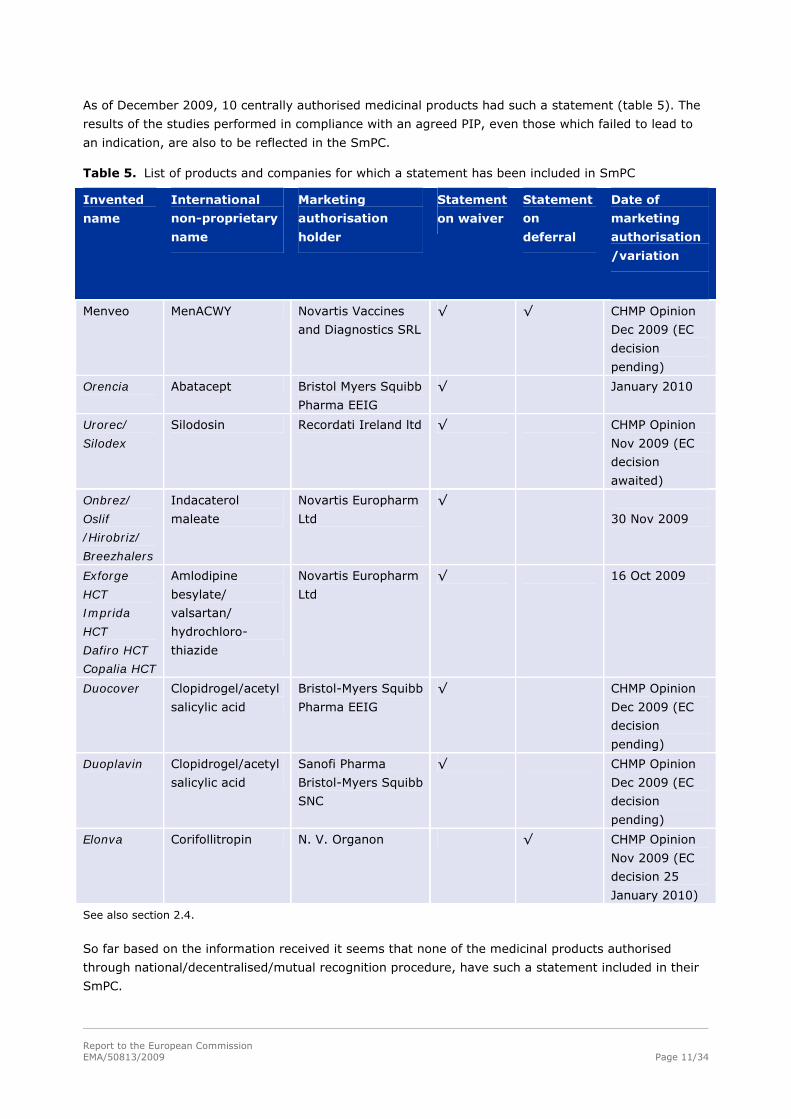
As of December 2009, 10 centrally authorised medicinal products had such a statement (table 5). The
results of the studies performed in compliance with an agreed PIP, even those which failed to lead to
an indication, are also to be reflected in the SmPC.
Table 5. List of products and companies for which a statement has been included in SmPC
Invented
name
International
non-proprietary
name
Marketing
authorisation
holder
Statement
on waiver
Statement
on
deferral
Date of
marketing
authorisation
/variation
Menveo
MenACWY Novartis Vaccines
and Diagnostics SRL
√ √ CHMP Opinion
Dec 2009 (EC
decision
pending)
Orencia
Abatacept Bristol Myers Squibb
Pharma EEIG
√ January 2010
Urorec/
Silodex
Silodosin Recordati Ireland ltd √ CHMP Opinion
Nov 2009 (EC
decision
awaited)
Onbrez/
Oslif
/Hirobriz/
Breezhalers
Indacaterol
maleate
Novartis Europharm
Ltd
√
30 Nov 2009
Exforge
HCT
Imprida
HCT
Dafiro HCT
Copalia HCT
Amlodipine
besylate/
valsartan/
hydrochloro-
thiazide
Novartis Europharm
Ltd
√ 16 Oct 2009
Duocover Clopidrogel/acetyl
salicylic acid
Bristol-Myers Squibb
Pharma EEIG
√ CHMP Opinion
Dec 2009 (EC
decision
pending)
Duoplavin Clopidrogel/acetyl
salicylic acid
Sanofi Pharma
Bristol-Myers Squibb
SNC
√ CHMP Opinion
Dec 2009 (EC
decision
pending)
Elonva Corifollitropin N. V. Organon √ CHMP Opinion
Nov 2009 (EC
decision 25
January 2010)
See also section 2.4.
So far based on the information received it seems that none of the medicinal products authorised
through national/decentralised/mutual recognition procedure, have such a statement included in their
SmPC.

Report to the European Commission EMA/50813/2009 Page 12/34
2.6. Price/reimbursement benefits
On the basis of the information provided by some Member States, it appears that none of those having
answered had introduced any concession for medicinal products for paediatric use in connection with
the fixing of prices and reimbursement. Similarly, it seems that no specific measures have been
introduced to favour a priority review for determining the price and reimbursement for medicinal
products for paediatric use.
The feedback received from Lithuania specifically indicates that no special price/reimbursement
benefits for paediatric medicinal products are foreseen in the legislation. However special conditions for
reimbursement of medicinal products intended for treatment of children exist. That means that all
medicinal products included into positive lists, (i.e. the list of diseases and the reimbursed medicinal
products used for their treatment and the list of reimbursed medicinal products) may be prescribed for
children until 18 years by applying 100% compensation of a basic price.
The United Kingdom explained that, under the Pharmaceutical Price Regulation Scheme (PPRS),
companies may benefit from the variable rate for paediatrics element of the R&D allowance. However,
the amount is small (a maximum of 3% of a company's sales of branded medicines to the NHS) and in
practice only one or two companies have claimed the paediatrics element. Companies have not
benefited materially as the amount was insufficient to generate a price increase for the company or to
reduce the amount of excess profits repayable under the PPRS and so NHS list prices were unchanged.
2.7. Research incentives
2.7.1. EU Framework Programme
Thanks to the Paediatric Regulation (Article 40), funding of studies into off-patent medicinal products
(i.e. those not covered by a patent or supplementary protection certificate) is available. This funding,
provided through the EU Framework Programmes for Research and Technological Development, covers
the development of off-patent medicinal products with a view to the submission of an application for a
PUMA. In order to ensure that funds are directed into research of medicinal products with the highest
need in the paediatric population and in agreement with DG Research, the PDCO has adopted a priority
list of off-patent products for which studies are required in advance of each call.
The European Commission has already launched four calls within the 7th Framework Programme. Six
projects have been funded in response to the second (first paediatric) call in 2007 with a budget of
about €22 million for 3 years (maximum of €6 million per project), and three projects have been
funded in response to the third call with a budget of about €18 million. The procedure for selecting the
projects in response to the fourth call is ongoing. This year the PDCO will adopt a new revised list in
advance of the next call. The fourth call with a total budget available of 40 million Euros was issued in
September 2009. The selection process is ongoing and three projects have been proposed to be
funded. For this call, the Commission, with the involvement of the Agency has initiated a greater
collaboration with the US (FDA/NIH) in order to have a close co-operation for research in paediatrics
and thereby avoiding any unnecessary duplication of studies. Further details of these projects can be
found in Annex 4 as provided by DG Research.
2.7.2. European Network of Paediatric Research at the European Medicines Agency
The Paediatric Regulation requires the setting up of a European network of existing national and
European networks, investigators and centers with specific expertise in the performance of studies in
the paediatric population coordinated by the Agency. Further to the adoption of the implementing

Report to the European Commission EMA/50813/2009 Page 13/34
strategy for the network by the Agency Management Board on 15 January 2008 the network has been
set up. Although the network will not fund studies or research per se, its objectives are to coordinate
studies relating to paediatric medicinal products, to build up the necessary scientific and administrative
competences at European level, in order to avoid duplication of studies and testing in children.
Companies will therefore benefit for having such a network as a tool for the development of medicinal
products in children. Up to 60 networks have been identified and work is ongoing to implement the
recognition criteria and the Coordinating Group.
2.7.3. National initiatives
Some initiatives have been taken for funding the development of medicinal products for paediatric use
at national level. The following feedback from the following Member States has been received.
Belgium
The Belgian agency launched the Belgian Paediatric Research Network in collaboration with the Belgian
Paediatric Association.
Germany
There is a survey on off-label use “Off-Label-Use von Arzneimitteln bei Kindern und Jugendlichen in
Deutschland – Ergebnisse von KiGGS (Kinder-und Jugendgesundheitssurvey)” (Off-label use of
medicinal products in children and adolescents in Germany - Results from the KiGGS (Children and
Adolescent Health Survey), Budget Euros 28,520.00.
Hungary
There are some measures of general scope which may promote the research and development of
paediatric medicines.
In July 2009, an Action Plan for the Pharmaceutical Industry and Biotechnology was adopted which
provides for the deduction of R&D costs from certain payment obligations on the pharmaceutical
companies. Currently the deduction rate is 20% of the R&D investment, but from 2011 the total
amount invested shall be taken into account. Although so far this incentive has not been explicitly used
for paediatrics medicines R&D, it is hoped that pharmaceutical companies will extend their innovation
activities to this direction as well.
Italy
Independent research is funded by AIFA. The promotion of independent research on drugs represents
one of the strategic tasks assigned to AIFA by Italian Legislation. The general aim of the program is to
support clinical research on drugs in areas of interest for the National Health Service (NHS) and where
commercial support is normally insufficient. The target populations are patients normally excluded by
clinical studies on efficacy and safety, such as children, pregnant women and the elderly. The focusing
research issues are those less explored in commercial research, such as clinically relevant end points,
relative efficacy of drugs (including the assessment of multimodal strategies) and long term follow up
on efficacy and safety of therapies.
AIFA set up the program on independent research in 2005, and five calls for proposals (2005 2006-
2006-2007-2008-2009) have already been launched. The call for proposals is aimed at investigators
working in public (e.g. national health service [SSN], universities, etc.) or non-profit organisations
(e.g. scientific foundations, patient associations, etc.).

Report to the European Commission EMA/50813/2009 Page 14/34
The way of funding this independent research derives from an ad hoc fund set up, requiring
pharmaceutical companies to contribute 5% of their yearly expenditure devoted to promotional
initiatives.
Regarding the period of interest of this Report, years 2007-2009, AIFA has selected several protocols
to be funded (program 2007 and program 2008). Program 2009 has just been launched and the
preliminary results will be available in April 2010.
See separate document (Additional information provided by Member States) ‘Paediatric Studies funded
by AIFA 2007-2008’ for the paediatric protocols funded in 2007 and 2008.
Framework agreements
The Italian Medicines Agency, through the Framework Agreements on Research & Development,
promotes at national level investment in production, research and development in the pharmaceutical
sector, according to the ten-year European Union Programme of renewal and encouragement of the
economic and social environment, as defined by the Lisbon European Council. Framework Agreements
are intended for pharmaceutical companies operating in the Italian territory, which are selected for
admission to the funding through a specific call for proposals that AIFA launched in 2008. Investment,
amounting to a total of €100 million, is destined to:
• The promotion of phase I and II clinical trials in Italy.
• The establishment or expansion of production sites (including feasibility studies, land, buildings,
machinery).
• The opening or improvement of research laboratories.
• The hiring of new permanent staff involved in production.
• The hiring of new permanent staff assigned to R&D activities.
In 2008 52 companies submitted proposals for 114 projects. An appointed Commission assessed the
most interesting projects to be funded: 29 projects are focusing on pre-clinical research; 15 on clinical
trials and 15 on production sites. Projects involving paediatrics are listed in the document “Framework
Agreements by AIFA in 2008 (Projects in Paediatric Field)” (Additional information provided by Member
States).
The Netherlands
There is a grant for development via ZonMw: the Netherlands organisation for health research and
development. The Ministry of Health has given ZonMw the assignment to start a programme on the
promotion of pharmaceuticals for children. The programme started in 2009 and lasts till 2017 with a
budget of 14.3 million Euros.
The Medicines for Children Research Network (MCRN) is a network of Dutch paediatricians of university
paediatric hospitals. The MCRN receives its money from a starting grant (2008-2011) of the
Netherlands Federation of University Medical Centres (NFU), Nefarma and the Ministry of Health. The
MCRN needs to generate its own income from 2012 onwards and be financially independent.
Lithuania
There are possibilities to get financial support available through available financial programmes
coordinated by the Ministry of Economy. So far, domestic companies involved in research and
development of medicinal product did not submit any applications to take part in tendering processes.

Report to the European Commission EMA/50813/2009 Page 15/34
Malta
Research on medicinal products inclusive for paediatric use can be funded under the National Research
and Innovation Programme set up by the Malta Council for Science and Technology. However there is
no specific incentive in place for developing paediatric medicines.
United Kingdom
The UK Government provides support for the Medicines for Children Research Network which provides
infrastructure to encourage and support paediatric studies although not direct funding. The work of this
Network is described in a separate document (Additional information provided by Member States).
Grants for particular products intended for use in children may have been awarded under a number
of other general research programmes, but this information is not readily available. In addition to the
research network, the UK NHS provides experts who advise the UK Licensing Authority on the quality,
safety and efficacy of paediatric medicines in the context of the Paediatric Regulation, through the
Paediatric Medicines Expert Advisory Group of the Commission on Human Medicines. This work is not
directly remunerated and is therefore supported by the UK Government.
2.8. Authorisation of paediatric clinical trials
The European Medicines Agency has no responsibility in the authorisation of clinical trials in the EU.
This is under the responsibility of the Member States according to Directive 2001/20/EC.
For those Member States who have answered the question, no fee reduction/fee waiver or priority
review has been introduced with respect to paediatric products.
Collaboration of the PDCO with the Clinical Trials Facilitation Group (CTFG) has been initiated to resolve
potential issues of divergences between PDCO and national competent authorities regarding the trials
from the PIP that the Competent Authorities are supervising. The CTFG will have direct access to the
paediatric database and PIP evaluation, thereby allowing immediate retrieval of information regarding
the clinical trials included in the PIP opinions.
The Slovak Republic and Sweden took the initiative to send a list of paediatric trials that have been
authorised by them. This list is not included in this report but can be found as a separate document.
It has to be kept in mind that one of the provisions of the Paediatric Regulation is to make public the
information on paediatric trials entered into the EU Database on Clinical Trials (EudraCT). The Agency
with its PDCO have been working with the European Commission to produce guidance on the protocol-
related information and on the results concerning paediatric clinical trials to be entered as well as the
information to be made public. This year will see the first roll-out of publicly available protocol-related
information. Results-related information will follow.
2.9. Procedures for marketing authorisation
The existing procedures for the granting of a marketing authorisation of medicinal products and for
extension of the marketing authorisation to add a new indication, pharmaceutical form and/or route of
administration have not been changed.
The Paediatric Regulation has however introduced a new type of marketing authorisation: the
paediatric-use marketing authorisation (PUMA); it may be requested for a medicine which is already
authorised or not, but in all cases no longer covered by intellectual property rights (patent,
supplementary protection certificate), and which will be exclusively developed for use in children in
compliance with an agreed PIP. This type of marketing authorisation will cover the indication(s) and

Report to the European Commission EMA/50813/2009 Page 16/34
age-appropriate formulation for the paediatric population. A paediatric-use marketing authorisation will
benefit from 10 years of market protection as an incentive to the development in children. The
submission of an application for a PUMA is automatically eligible to the centralised procedure but it
may also be made through the national/decentralised/mutual recognition procedures.
At the European Medicines Agency level there are no specific provisions to either prioritise or
accelerate the review of medicinal products intended for use in children, including for PUMA. However
the CHMP may consider shortening the review time for such products, in accordance with the
accelerated assessment procedure.
With respect to the fees, the European Medicines Agency has decided not to introduce any fee
reduction for centralised procedure for medicinal products indicated in children or for extension of the
marketing authorisation to add a new paediatric indication, pharmaceutical form and/or route of
administration relevant for paediatric use. However, with respect to PUMA and in line with Article 47 of
the Regulation, the Agency is granting a partial exemption from the payment of the fees laid down in
the fee regulation for paediatric use marketing authorisation applications submitted under Article 30 of
Regulation (EC) No 1901/2006 on medicinal products for paediatric use. So far there has been no
application for PUMA and therefore no companies have benefited from this.
At national level some initiatives have also been taken. The following feedback from Member States
has been received.
Belgium
A policy for priority review of the applications for variations was introduced, but it has not been used
because no case occurred where it could have been applied.
Hungary
The National Institute of Pharmacy is considering giving priority to paediatrics related dossiers in the
near future. These measures may include either more rapid processing of applications for marketing
authorisations or provision of scientific advice.
Slovak Republic
Fees for authorisation of medicines in the Slovak Republic are far below average EU fees. Therefore in
2008 the Slovak agency has proposed a new fee scheme in this respect which is still in progress in the
Ministry of Health. This proposed new scheme for fees would allow introducing differences in fees in
this respect.
United Kingdom
A fee waiver applies in certain cases for products being developed specifically for paediatric use, for
example a new paediatric formulation or extension of indications into the paediatric population. One
such application (which has not needed to comply with a PIP) has been received but is pending
approval.
The United Kingdom has a system of priority reviews which would include paediatric medicines but is
not specific to that category. UK has approved one such request but an application has not yet been
submitted.
For the other Member States, either no such initiatives have been taken or no information was
received.

Report to the European Commission EMA/50813/2009 Page 17/34
2.10. Article 45/46 of the Paediatric Regulation
2.10.1. Article 45
In accordance with Article 45 of the Paediatric Regulation, marketing authorisation holders were
required to submit to the competent authorities all paediatric studies completed by the date of entry
into force of the Regulation. These studies were to be submitted by 26 January 2008. Upon
assessment of these data the competent authority may update the SmPC and package leaflet and may
vary the marketing authorisation.
• For centrally authorised medicinal products, data have been submitted for approximately 60
products. The assessment of the data resulted to an update of the SmPC for only 4 medicinal
products. For these 4 cases there was no update of the package leaflet was amended. There are
still a couple of procedures ongoing.
The figures may be underestimated as in order to streamline the process, when regulatory action
was deemed necessary (i.e. where amendments to SmPC, labelling and/or PL were identified by
the MAH) MAHs were advised to straightaway submit a variation containing the Article 45
paediatric studies.
• For products authorised through national/decentralised/mutual recognition procedure the extent of
information received has been enormous. Information has been received for approximately 1000
active substances, with several documents for each of them (some may relate to the same study).
To cope with the workload, there is an ongoing work-sharing exercise between the Member States
and the assessment is being performed in stages. Four waves have been agreed, and the
assessment has already been completed for 11 medicines, for which Paediatric Assessment reports
have been published on the Head of Agencies website.
Both at the level of CHMP and of the Member States it was considered that the data assessed
were, in most cases, either of insufficient quality and too limited to lead to a change in the SmPC
of the information related to the use in children.
2.10.2. Article 46
In accordance with Article 46 of the Paediatric Regulation the MAH has the obligation to submit to the
Competent Authority any MAH-sponsored studies involving the use in the paediatric population of an
authorised medicinal product, whether or not they are part of a PIP, within 6 months of completion.
• For centrally authorised products, studies have so far been submitted for about 24 products, and
this has led to an update of the SmPC in only 2 occasions. The CHMP has been reminded of the
importance of systematically including information into the SmPC. As for Article 45, the figures are
approximate as similarly, when regulatory action is necessary (i.e. in case amendments to SmPC,
labelling and/or PL are identified by the MAH) MAHs are advised to straightaway submit a variation
containing the Article 46 paediatric study(ies). In some cases it was agreed that the assessment of
the data would be postponed as the MAHs intended to submit a variation procedure within a short
period of time. It is therefore difficult to conclude on how many cases the submission of these data
has truly led to a change of the Summary of Products Characteristics.
• For nationally authorised medicinal products and those authorised through mutual recognition, or
decentralised procedures, studies have been received for 70 medicinal products.
While Article 45 was a retrospective exercise and therefore the data were submitted at about the same
time, the obligation set by Article 46 is prospective. As such the number of studies to be submitted is
anticipated to increase steadily over the years.

Report to the European Commission EMA/50813/2009 Page 18/34
The list of products and resulting amendments of the SmPCs further to submission of data through
Article 45 and 46 are presented in Annex 5.

Report to the European Commission EMA/50813/2009 Page 19/34
3. FAILURE TO COMPLY WITH THE OBLIGATIONS SET IN THE PAEDIATRIC REGULATION
3.1. Submissions of the PIP/waiver application to the PDCO
Article 16 of the Paediatric Regulation requires companies to submit applications for a PIP or a waiver
for agreement no later than upon completion of the human pharmacokinetic studies in adults except
where duly justified. It was agreed that this corresponds approximately to the end of phase 1.
In the early days of the entry into force of the Regulation, the companies submitted their application as
soon as it was possible, but for the majority the overall development of the product was already
beyond that stage and often reaching confirmatory (phase III) clinical trials in adults, to comply with
the new requirements for products already in development.
However, it is clear that late submissions of PIP/Waivers applications have occurred in a number of
cases, causing a delay in the submission or the validation of the application for the marketing
authorisation in adults, as the applicant did not have an Agency Decision on a PIP including a deferral
and/or on a waiver. This also forced the PDCO to evaluate PIP applications with proposed trials and
studies that were already ongoing or completed; in many cases the proposal of the applicant was not
considered satisfactory but could not be modified. The PDCO considered at the same time that
requesting new studies would have led to unnecessary studies in children. It is acknowledged that
some learning process had to take place but this situation is repeating itself, including with
experienced companies.
The Agency will therefore monitor closely the compliance with this requirement of the Regulation and
will present the outcome in the next report.
3.2. Validation of application for marketing authorisation/extension
As set out in Article 7 of the Paediatric Regulation, applications concerning a medicinal product not
authorised in the EEA on 26 July 2008, must include one of the following in order to be considered
‘valid’:
• The results of all studies performed and details of all information collected in compliance with an
agreed Paediatric Investigation Plan (PIP).
This means that the application will have to include the PIP decision but also the results in accordance
with the agreed PIP.
• A decision of the EMEA on a PIP including the granting of a deferral.
This means that the application will have to include the PIP decision including the deferral granted.
• A decision of the EMEA granting a product-specific waiver.
• A decision of the EMEA granting a class waiver on condition.
The same requirements as set out in Article 8 of the Paediatric Regulation, apply to applications
submitted from 26 January 2009, for new indication(s), new pharmaceutical form(s) and/or new
route(s) of administration concerning an authorised medicinal product protected either by a
supplementary protection certificate or by a patent which qualifies for the granting of such a
certificate:
• So far it seems that no application falling under Article 7 or 8 has been validated without having
complied with these requirements at the Agency level.

Report to the European Commission EMA/50813/2009 Page 20/34
• For the centralised procedure, despite early identification of applicants, and reminders (letters were
sent to future applicants in 2007) a few companies have submitted their application for marketing
authorisation or application to add a new indication, new pharmaceutical form, new route of
administration without having either a Decision on a PIP or on a waiver, or without having
requested prior to the submission an opinion on the compliance check from the PDCO. This has
resulted in a couple of delays for the start of the procedure.
3.3. Compliance with the paediatric requirements and rewards
So far there is no indication that a company has benefited from the reward without having complied
with the paediatric requirements set in the Regulation.
3.4. Mention of the Decision on waivers or deferrals in the product information
There is a requirement set in the Regulation (Article 28(2)) to include in the marketing authorisation
granted or varied a statement on the waiver or deferral. However, the Guideline on the Summary of
Product Characteristics was only revised in September 2009 and applies as of 1st May 2010. The
guideline provides guidance on how to word statements on waivers and deferrals. Obviously,
submissions may also be made on the basis of this guidance, prior to the implementation date.
For the centralised procedure, only a few products were identified for which this statement has been
omitted. It is planned to correct the situation at the next regulatory procedure involving an
amendment of the Summary of Product Characteristics and a Procedural announcement was made
through the October 2009 CHMP meeting report.
Based on the information received from the Member States, it appears that no marketing authorisation
has been granted or varied for medicinal products for which an Agency decision on a waiver or deferral
has been issued to date.
3.5. Annual reports on deferrals
Article 34.4 of the Paediatric Regulation states that “in the case of a deferral, the marketing
authorisation holder shall submit an annual report to the Agency providing an update on progress with
paediatric studies in accordance with the decision of the Agency agreeing the paediatric investigation
plan and granting a deferral”.
The Agency had published guidance and a form for the electronic submission of the reports, and has
already received 13 reports. The Agency will analyse the adherence with such requirements and will
aim to report on this in the next report.
4. CONCLUSION
After three years of entry into force of the Paediatric Regulation, companies have already benefited
from incentives and rewards, and although still few, some companies have benefited from an extension
of the Supplementary Protection Certificate in some Member States. So far no companies have
benefited from 2-year extension of market exclusivity for an orphan medicinal product, nor from the
data and marketing protection periods granted for PUMA.
To date the Agency has not received any report identifying any failure to comply with any of the
obligations set in the Paediatric Regulation. However not all Member States have provided information
on this point. It has to be noted that the Commission Regulation on financial penalties has not been

Report to the European Commission EMA/50813/2009 Page 21/34
revised as requested in Article 49(3) to make possible financial penalties linked to infringement of the
Paediatric Regulation.
In a wider context, it is already clear that the Regulation has created real awareness of the need for
conducting appropriate paediatric clinical trials and for developing suitable formulations for use in
children. It has become the norm to include the development of a medicinal product in children within
the development in adults, where relevant.
The Regulation is stimulating the conduct of high-quality ethical research, in particular through the
European network, and is promoting information with the transparency of agreed PIPs and waivers,
and the access to clinical trial data generated by research.

Report to the European Commission EMA/50813/2009 Page 22/34
Annex 1 - Questions sent to the Member States
In addition to the Community rewards/incentives, and in accordance to Article 39 of Paediatric
Regulation, Member States may have introduced other measures that companies may benefit from
when developing medicinal products for paediatric use. The below list encompasses measures taken by
various Member States on the basis of the report published by the European Commission on 30 July
2008. For all the below items, if applicable to your Member State, please list the companies and
products that have benefited in 2007, 2008 and 2009, separated by year from:
1) Benefits
• Compliance statement included in a marketing authorisation for those products
approved nationally (number of statements, name of products and of marketing authorisation
holders).
• 6 months extension of the Supplementary of Protection Certificate granted by your National
Patent Office (number of extensions, name of products and patent holders).
This applies to products who have complied with Article 36 of the Paediatric Regulation.
• Price/reimbursement benefits (name of products marketing authorisation holders, type and
amount of concessions).
Your Member State may have introduced concessions granted for medicinal products for paediatric use
in connection with the fixing of prices and reimbursement, including priority review for this process.
• Grants for development (name of products, beneficiaries and type of support).
Your Member State may have provisions for funding support to the development of medicinal products
for paediatric use.
• Clinical trials, Marketing Authorisation.
Your Member State may have introduced fee reduction/fee waiver for:
• Authorisation of paediatric clinical trials (name of products, sponsors, type and amount of fee
reduction/waiver).
• Request for national scientific advice to help companies wishing to develop medicinal products for
paediatric use (number of advice, name of products and of marketing authorisation holders).
• Granting of and/or extension of the marketing authorisation, including for Paediatric Use Marketing
Authorisation (PUMA) (type and amount of fee reductions, name of products and marketing
authorisation applicant/holder).
• Your Member State may have introduced policy for priority review of the applications for marketing
authorisation for products for paediatric use (number of priority reviews, PUMA, name of products
and marketing authorisation holder).
• Paediatric Use Marketing Authorisation (PUMA) granted nationally* (number, name of products and
marketing authorisation holder).
• National marketing Authorisation/extension granted with statement on deferral/waiver in the
product information (number, products, marketing authorisation holder).

Report to the European Commission EMA/50813/2009 Page 23/34
2) Infringement
• Application for marketing authorisation/extension submitted and validated without compliance with
Article 7 or 8 of the Paediatric Regulation (number, product, date of marketing authorisation,
marketing authorisation holder).
• Reward obtained without inclusion on the product information of the paediatric data (products,
marketing authorisation holder).
• Marketing authorisation granted or varied without any mention of the waiver or deferral in the
Summary of Product Characteristics (products, marketing authorisation holder).
* For those products submitted under mutual recognition or decentralised products the European Medicines Agency will contact the Coordination Group (CMD(h)) directly.

Report to the European Commission EMA/50813/2009 Page 24/34
Annex 2 - Compliance statement included in a marketing authorisation for products authorised through national/decentralised/mutual recognition procedure
Austria
• Cosaar 12,5 mg Filmtabletten, NL/H/1457/001, MAH = Merck Sharp & Dohme
GmbH: 1 compliance statement (04.05.2009)
• Cosaar 50 mg Filmtabletten, NL/H/1457/002, MAH = Merck Sharp & Dohme
GmbH: 1 compliance statement (04.05.2009)
• Cosaar 100 mg Filmtabletten, NL/H/1457/003, MAH = Merck Sharp & Dohme
GmbH: 1 compliance statement (04.05.2009)
• Cosaar 2,5 mg Pulver und Lösungsmittel zur Herstellung einer Suspension, NL/H/1457/004, MAH =
Merck Sharp & Dohme GmbH: 1 compliance statement (13.11.2009)
• Arimidex 1 mg Filmtabletten, UK/H/0111/001, MAH = AstraZeneca Österreich
GmbH: national implementation is ongoing
(Variation to introduce data an paediatric use in accordance with the European Commission
Decision issued in Nov. 2009 was approved on 18.12.2009)
There were no compliance statements introduced in marketing authorisation in 2007 or 2008.
Belgium
No information provided (although information has been provided on the 6-month extension of the
Supplementary Protection Certificated).
Bulgaria
Arimidex 1 mg film-coated tablet, anastrozole, AstraZeneca Pharmaceuticals AB.
Czech Republic
None.
Cyprus
None.
Denmark
• Cozaar (losartan potassium) Merck Sharp & Dohme BV, The Netherlands.
• Arimidex (anastrozol) from AstraZeneca A/S, Denmark.
There were no compliance statements introduced in marketing authorisation in 2007 or 2008.
Estonia
• Cozaar (losartan potassium) Merck Sharp & Dohme.

Report to the European Commission EMA/50813/2009 Page 25/34
• Arimidex (anastrozol) from AstraZeneca UK Ltd.
There were no compliance statements introduced in marketing authorisation in 2007 or 2008.
Finland
• Arimidex 1 mg tabletti (MAnr12097 Hnr1 193367/2009) AstraZeneca Oy (Type II variation
application approved 16.12.2009).
• Cozaar 12,5 mg tabletti kalvopäällysteinen (MAnr 12916 Hnr 172362/2008
NL/H/1457/01/II/02) Merck Sharp & Dohme B.V (type II variation approved 8-05-2009) PIP
compliance statement.
• Cozaar 50 mg tabletti kalvopäällysteinen (MAnr 11637 Hnr 172363/2008
NL/H/1457/01/II/02) Merck Sharp & Dohme B.V (type II variation approved 8-05-2009) PIP
compliance statement.
• Cozaar 100 mg tabletti kalvopäällysteinen (MAnr 16602 Hnr 172364/2008 NL/H/1457/01/II/02
Merck Sharp & Dohme B.V - (type II variation approved 8-05-2009) PIP compliance statement.
• Cozaar 2,5 mg/ml jauhe ja liuotin oraalisuspensiota varten (MAnr 25172 Hnr 161675/2009),
Merck Sharp & Dohme B.V – (line extension approved 26-02-2009).
• Cozaar 2,5 mg/ml jauhe ja liuotin oraalisuspensiota varten (MAnr 25172 Hnr 180745/2009
NL/H/1457/01/II/02), Merck Sharp & Dohme B.V – (type II variation approved 23-09-2009) PIP
compliance statement.
France
No information provided (although information has been provided on the 6-month extension of the
Supplementary Protection Certificated)
Germany
• Arimidex (anastrozol) from AstraZeneca.
• Cosaar (losartan).
There were no compliance statements introduced in marketing authorisation in 2007 or 2008.
Hungary
There were no compliance statements introduced in marketing authorisation in 2007, 2008 or 2009.
Ireland
• Cozaar (losartan) 2.5mg powder and solvent for oral solution (PA 0970/003/001): Marketing
Authorisation Holder: Merck Sharp and Dohme.
• Arimidex 1mg film coated tablets (PA 1286/004/004): Marketing Authorisation Holder: Astra
Zeneca UK Limited.
There were no compliance statements introduced in marketing authorisation in 2007 or 2008.

Report to the European Commission EMA/50813/2009 Page 26/34
Italy
• Lortaan (losartan potassium) (MSD) NL/H/1457/01-04.
• Neo-Lotan (losartan potassium) NL/H/1457/01-04.
• Losaprex (losartan potassium) NL/H/1457/01-04.
• Arimidex (AstraZeneca) UK/H/111/01.
Latvia
There were no compliance statements introduced in marketing authorisation in 2007, 2008 or 2009.
Lithuania
There were no compliance statements introduced in marketing authorisation in 2007, 2008 or 2009.
Luxembourg
There were no compliance statements introduced in marketing authorisation in 2007, 2008 or 2009.
The Netherlands
There were 2 products for which a compliance statement has been introduced in the marketing
authorisation.
Malta
There were no compliance statements introduced in marketing authorisation in 2007, 2008 or 2009.
Romania
2009: COZAAR 2,5 mg/ml powder and solvent for oral suspension Merck Sharpe & Dohme Romania
SRL.
Slovakia
There were no compliance statements introduced in marketing authorisation in 2007, 2008 or 2009.
Slovenia
• Arimidex 1 mg film-coated tablet, anastrozole, AstraZeneca UK Limited.
• Cozaar 12,5 mg film-coated tablets, Merck Sharp & Dohme inovativna zdravila d.o.o - Cozaar 50
mg film-coated tablets, Merck Sharp & Dohme inovativna zdravila d.o.o - Cozaar 100 mg film-
coated tablets, Merck Sharp & Dohme inovativna zdravila d.o.o.
• Cozaar 2,5 mg/ml powder and vehicle for oral suspension, Merck Sharp & Dohme inovativna
zdravila d.o.o.
Sweden
• Cozaar film coated tablet and powder and solvent for oral suspension, MAH is Merck Sharp &
Dohme BV, The Netherlands and the representative is Merck Sharp & Dohme AB, Sweden.

Report to the European Commission EMA/50813/2009 Page 27/34
• Arimidex film coated tablet, MAH is AstraZeneca AB, Södertälje, Sweden.
United Kingdom
• Cozaar (Merck, Sharp and Dohme Ltd) compliance statement included in UK MA on 6/5/2009.
• Arimidex (Astra Zeneca) compliance statement included in UK MA on 18/12/2009.
There were no compliance statements introduced in marketing authorisation in 2007 or 2008.

Report to the European Commission EMA/50813/2009 Page 28/34
Annex 3 - 6-months extension of the supplementary protection certificate (SPC) granted by the National Patent Office
Austria
No information provided.
Belgium
• Coozar (losartan potassium) Merck Sharp & Dohme Inc.
• Request for Caspofungine under evaluation Merck Sharp & Dohme.
Bulgaria
There are no SPCs granted during 2009.
Czech Republic
No extension has been granted yet.
Cyprus
No extension has been granted yet.
Denmark
In 2009 granted 2 extensions of the Supplementary Protection Certificate for losartan potassium
(Cozaar), patent holder E.I. du Pont de Nemours and Company and Caspofunginacetat (Cancidas)
patent holder Merck & Co.
Udstedte forlængelser af supplerende beskyttelsescertifikat (Issued extensions of SPCs):
Indl.dag: 2009-02-24
Certifikat ans.nr: CA 2004 00003
Udløbsdag (expire): 2010-03-02
Grundpatent nr (Patent no): PR 174700
Ansøger (Applicant): E.I. DU PONT DE NEMOURS AND COMPANY, 1007 Market Street, Wilmington,
Delaware 19898, USA
Fuldmægtig: Zacco Denmark A/S, Hans Bekkevolds Allé 7, 2900 Hellerup, Danmark
DK markedsførelsestilladelse nr (MA no): MT 15844
Dato for samme: 1994-09-26
Produkt (product): Losartankalium
Første markedsføringstilladelse i EU (First MA in EU): 12209
Dato for samme (Date for first MA in EU): 1994-09-02
Benævnelse: Substituerede imidazoler samt farmaceutisk præparat
indeholdende dem
Indl.dag: 2002-04-19
Certifikat ans.nr: CA 2002 00007
Udløbsdag: 2017-04-24
Grundpatent nr: DK/EP 0620232

Report to the European Commission EMA/50813/2009 Page 29/34
Ansøger: Merck & Co., Inc., 126, East Lincoln Avenue, P.O. Box 2000, Rahway, New Jersey 07065-
0900, USA
Fuldmægtig: Zacco Denmark A/S, Hans Bekkevolds Allé 7, 2900 Hellerup, Danmark
DK markedsførelsestilladelse nr: EU/1/01/196/001-003
Dato for samme: 2001-10-24
Produkt: Caspofunginacetat
Første markedsføringstilladelse i EU: EU/1/01/196/001-003
Dato for samme: 2001-10-24
Benævnelse: Azacyclohexapeptid-forbindelser
Estonia
No information provided.
France
• Cozaar: laboratoires Merck-Sharp & Dohme-Chibret.
• Arimidex: laboratoires AstraZeneca.
Finland
No information provided apart from the link to the National Board of Patents and Registration of
Finland (NBPR) website http://www/prh.fi/en.html.
Germany
Certificate (Schutzzertifikat DE 196 75 001)
Handelsnamen (z.B) Cosaar, Cozaar oder Lorzaar
Wirkstoffname/Bezeichnung des Erzeugnisses: Losartan-Kalium
Schutzzertifikatsinhaber: EI. Du Pont de Nemours & Co
Certificate (Schutzzertifikat DE 102 99 013)
Handelsnamen (z.B) Cancidas
Wirkstoffname/Bezeichnung des Erzeugnisses: Caspofungin
Schutzzertifikatsinhaber: Merck & Co
Hungary
No information provided.
Ireland
2009: Merck Sharpe and Dohme Cozaar.
Italy
• Lortaan (MSD) NL/H/1457/04/II/04 N. 754 20/10/09

Report to the European Commission EMA/50813/2009 Page 30/34
• Neo-Lotan NL/H/1457/04/II/04 “N. 753 20/10/09
• Losaprex NL/H/1457/04/II/04.” N. 755 20/10/09
• Arimidex (AstraZeneca) UK/H/111/01/II/52 Provv 946 del 18/12/09
Latvia
No information provided.
Lithuania
No SPC has been granted.
Luxembourg
No SPC has been granted.
Malta
No SPC has been granted.
The Netherlands
No information provided.
Romania
No information provided.
Slovak Republic
No information provided.
Slovenia
No SPC has been granted.
Sweden
No information.
United Kingdom
2009
• Caspofungin (Merck & Co, Inc) SPC extension granted 30 July 2009
SPC/GB/02/002 extended until 23 April 2017
• losartan (EI du Pont Nemours & Co) SPC extension granted 24 August 2009
SPC/GB/95/010 extended until 1 March 2010

Report to the European Commission EMA/50813/2009 Page 31/34
Annex 4 – List of projects on off-patent medicines funded by the European Commission through the EU Framework Programme
2008
• LOULLA & PHILLA
Development of oral liquid formulations of Methotrexate and 6-Mercaptopurine for paediatric acute
lymphoblastic leukaemia (ALL).
• TINN
Aims to evaluate PK & PD of ciprofloxacin and fluconazole in neonates.
• O3K
Oral liquid formulations of Cyclophosphamide and Temozolomide.
• NEUROSIS
Efficacy of Budesonide (BS) in reducing bronchopulmonary dysplasia (BPD).
• EPOC
Aims to evaluate pharmacokinetics and pharmacodynamics of doxorubicin.
• NeoOpioid
Compares morphine and fentanyl in pain relief in pre-term infants.
2009
• NEMO
Evaluates the efficacy safety, PK, PD, mechanisms of action of bumetanide in neonatal seizures,
including the effect on neurodevelopment and to develop and adapt a bumetanide formulation
suitable for newborns in order to apply for a Paediatric Use Marketing Authorisation (PUMA).
• NeoMero
European multicentre network to evaluate pharmacokinetics, safety and efficacy of Meropenem in
neonatal sepsis and meningitis.
• PERS
Focuses on two indications, the use of risperidone in children and adolescents with conduct
disorder who are not mentally retarded, and the use of risperidone in adolescents with
schizophrenia.
Report to the European Commission EMA/50813/2009 Page 32/34
Annex 5 - List of products and resulting amendment of the SmPC further to submission of data through article 45 and 46
Article 45
Centrally authorised medicinal products
International
Non-
proprietary
name
Invented
name
Marketing
authorisation
holders
Change in the
Summary of
Product
Characteristics
Link to EPAR
Pegfilgrastim Neulasta Amgen
Europe B.V.
Section 4.2 and
introduction of new
information in
sections 4.8, 5.1
and 5.2
http://www.ema.europa.eu/human
docs/PDFs/EPAR/neulasta/296102e
n8b.pdf
Ritonavir Norvir Abbott
Laboratories
Limited
Section 5.1 http://www.ema.europa.eu/human
docs/PDFs/EPAR/Norvir/H-127-
en8b.pdf
Mangafodipir Teslascan GE
Healthcare
AS
Section 4.2
http://www.ema.europa.eu/human
docs/PDFs/EPAR/Teslascan/013097
en8b.pdf
Interferon
beta-1a
Avonex Biogen Idec
Limited
Sections 4.2, 4.8
and 5.1
CHMP opinion 2010
Medicinal products authorised through national/mutual recognition/decentralised procedure
Tranexamic
Acid
Exacyl,
Ugurol
Sanofi-
Aventis,
Rottapharm
Section 4.2 and
5.1 (+ other
sections not
specifically linked
to submission of
paediatric data)
http://www.hma.eu/fileadmin/datei
en/Human_Medicines/CMD_h_/Pae
diatric_Regulation/Assessment_Rep
orts/Article_45_work-
sharing/Tranexamic_acid_45PaedA
R.pdf
Amlodipine Pfizer Section 4.2 (dose
recommendation),
5.1 and 5.2
http://www.hma.eu/fileadmin/datei
en/Human_Medicines/CMD_h_/Pae
diatric_Regulation/Assessment_Rep
orts/Article_45_work-
sharing/Amlodipine_2009_11_45Pd
AR.pdf
Bisacodyl Dulcolax
Prepacol
Fenolax
Boehringer
Ingelheim
Guerbet
ICN Polfa
Rzeszow
Harmonisation of
section 4.2
http://www.hma.eu/fileadmin/datei
en/Human_Medicines/CMD_h_/Pae
diatric_Regulation/Assessment_Rep
orts/Article_45_work-
sharing/Bisacodyl_2009_12_45PdA
R.pdf
Felodipine
Renedil,
Prevex,
Plendil
Sanofi-
Aventis
Section 5.1 and
5.2
http://www.hma.eu/fileadmin/datei
en/Human_Medicines/CMD_h_/Pae
diatric_Regulation/Assessment_Rep
orts/Article_45_work-

Report to the European Commission EMA/50813/2009 Page 33/34
Centrally authorised medicinal products
sharing/Felodipine_2009_11_45PdA
R.pdf
Glucosamine
Donacom
and other
trade
names
Rottapharm
Ltd
Harmonisation of
section 4.2 and 4.4
http://www.hma.eu/fileadmin/datei
en/Human_Medicines/CMD_h_/Pae
diatric_Regulation/Assessment_Rep
orts/Article_45_work-
sharing/Glucosamine_2009_11_45P
dAR.pdf
Lisinopril Prinivil,
Zestril
Merck, Astra
Zeneca
4.2 (dose
recommendations)
4.8, 5.1 and 5.2
http://www.hma.eu/fileadmin/datei
en/Human_Medicines/CMD_h_/Pae
diatric_Regulation/Assessment_Rep
orts/Article_45_work-
sharing/Lisinopril_2009_11_45PdA
R.pdf
Adumbran Oxazepam Boehringer
Ingelheim
Section 4.4 http://www.hma.eu/fileadmin/datei
en/Human_Medicines/CMD_h_/Pae
diatric_Regulation/Assessment_Rep
orts/Article_45_work-
sharing/Oxazepam_2009_09_Paed
AR.pdf
Salmon
Calcitonin
Calsynar
Calcitonin
armour
Tonocalcin
Aventis
Pharma
Rorer
Pharma-
ceuticals
Alfa Biotech,
Alfa
Wasserman
Section 4.2 http://www.hma.eu/fileadmin/datei
en/Human_Medicines/CMD_h_/Pae
diatric_Regulation/Assessment_Rep
orts/Article_45_work-
sharing/Salmon_calcitonin_45Paed
PAR.pdf
Simvastatin Zocor Merck
Sharpe &
Dohme
GmbH
4.2 (dose
recommendations)
4.3, 4.4, 4.8, 5.1
and 5.2
http://www.hma.eu/fileadmin/datei
en/Human_Medicines/CMD_h_/Pae
diatric_Regulation/Assessment_Rep
orts/Article_45_work-
sharing/simvastatin_45PaedPAR_.p
df

Report to the European Commission EMA/50813/2009 Page 34/34
Article 46
Centrally authorised medicinal products
International
Non-
proprietary
name
Invented
name
Marketing
authorisation
holders
Change in the
Summary of
Product
Characteristics
Link to EPAR
Cinacalcet Mimpara Amgen Europe
B.V
Section 5.2 http://www.ema.europa.eu/humando
cs/PDFs/EPAR/mimpara/H-570-
en8.pdf
Telithromycin Ketek Aventis Pharma
S.A.
Section 4.2
and 5.2
http://www.ema.europa.eu/humando
cs/PDFs/EPAR/ketek/H-354-en8.pdf





























