Embed Size (px)
Citation preview

Research ArticleDNAseq Workflow in a Diagnostic Context andan Example of a User Friendly Implementation
Beat Wolf12 Pierre Kuonen1 Thomas Dandekar2 and David Atlan3
1University of Applied Sciences and Arts of Western Switzerland Perolles 80 1700 Fribourg Switzerland2University of Wurzburg Am Hubland 97074 Wurzburg Germany3Phenosystems SA 137 Rue de Tubize 1440 Braine le Chateau Belgium
Correspondence should be addressed to Beat Wolf beatwolfhefrch
Received 13 March 2015 Revised 10 May 2015 Accepted 18 May 2015
Academic Editor Hong Lu
Copyright copy 2015 Beat Wolf et alThis is an open access article distributed under theCreativeCommonsAttribution License whichpermits unrestricted use distribution and reproduction in any medium provided the original work is properly cited
Over recent years next generation sequencing (NGS) technologies evolved from costly tools used by very few to a much moreaccessible and economically viable technology Through this recently gained popularity its use-cases expanded from researchenvironments into clinical settings But the technical know-how and infrastructure required to analyze the data remain an obstaclefor a wider adoption of this technology especially in smaller laboratories We present GensearchNGS a commercial DNAseqsoftware suite distributed by Phenosystems SAThe focus of GensearchNGS is the optimal usage of already existing infrastructurewhile keeping its use simpleThis is achieved through the integration of existing tools in a comprehensive software environment aswell as custom algorithms developedwith the restrictions of limited infrastructures inmindThis includes the possibility to connectmultiple computers to speed up computing intensive parts of the analysis such as sequence alignmentsWe present a typical DNAseqworkflow for NGS data analysis and the approach GensearchNGS takes to implement it The presented workflow goes from rawdata quality control to the final variant report This includes features such as gene panels and the integration of online databaseslike Ensembl for annotations or Cafe Variome for variant sharing
1 Introduction
Next generation sequencing (NGS) technologies saw a rapidprogression over recent years and improved in many aspectssince their introduction The data quality the length of thesequences and the speed at which the data is generatedimproved massively all while decreasing the costs associ-ated with the technology [1] Cost reductions and qualityimprovements now allow the technology to be used ina diagnostic setting replacing older technologies such asSanger sequencing [2] An increasing number of laboratoriesare either considering or already implementing NGS forroutine diagnostics procedures Next generation sequencinghas many use-cases in diagnostics and depending on theanalysis different types of information are searched for inthe sequencing data A very common type of analysis isthe search for SNPs (single-nucleotide polymorphisms) andsmall indels (insertions and deletions) in patient DNA With
the increasing number of documented variations in well-known disease related genes this type of analysis becamevery common Increasingly other types of analyses are alsoused in diagnostics such as the detection of structuralvariations which became possible with the increased qualityof sequencing data The sequencing data which can comefrom various sequencing technologies can be either tar-geted such as targeted gene panel sequencing and wholeexome sequencing (WES) or nontargeted like whole genomesequencing (WGS) Those different approaches to sequencethe patientrsquos genome produce a varying amount of data [3]and are suited for different types of analyses The complexityof the analysis increases with the amount of data to processboth from a computational point of view and from a humanresources point of view The general tendency today is tomove away from targeted sequencing towards WGS as thisremoves the need for targeted amplicon libraries as well asmaking it easier to reanalyse a sample at a later date without
Hindawi Publishing CorporationBioMed Research InternationalVolume 2015 Article ID 403497 11 pageshttpdxdoiorg1011552015403497
2 BioMed Research International
the need to resequence it This tendency to increase theamount of sequenced data creates increasing challenges forsmaller diagnostics laboratories They often do not have therequired computing infrastructure or technical know-how tohandle this type of data This is especially true in emergingmarkets which do not always have the required financial andhuman resources as well as the extensive know-how requiredto set up their own analysis pipeline
Today many types of NGS data analysis exist all ofthem answering different sorts of questions about a sampleOne example being copy number variation (CNV) analy-sis suited to replace traditional MLPA (multiplex ligation-dependent probe amplification) analysis Another one is denovo sequence alignment to detect large changes in thegenome for example in cancer patients But ultimately thedetection of SNPs and small indels in a sample based ona standard reference sequence remains the most prevalenttype of analysis This is why in the context of this paper welimit our study to this type of analysis the detection of SNPsand small indels in targeted or nontargeted resequencingAlthough this type of analysis can be approached in variousways it can be described as a set of generic steps that remaincommon to most approaches The next chapter describesthe commonly used steps in the analysis process providingexamples of existing tools that can be used to perform them
NGS Data Analysis This chapter describes a common NGSdata analysis workflow used in diagnostics Figure 1 showsan overview of the described workflow using a UnifiedModeling Language (UML) diagram
(A) QCFiltering The analysis starts with the quality controlof the raw sequencing data to determine if the sequencingprocess adheres to the defined quality requirements Differentmetrics such as the amount of reads sequenced the lengthdistribution of the sequenced reads and their quality areverified and compared to the expected values of the specificsequencer technology that has been used If the quality ofthe sequenced data does not match the required quality thesample is usually resequenced To verify the quality of the rawdata one commonly used tool is FastQC [4] which generatesa comprehensive list of statistics of the raw data Duringthis initial quality control stage the raw sequencing data isoften filtered discarding reads that do not match the definedquality criteria This raw data filtering is commonly donewith the FASTX toolkit [5] which allows removing reads thatdo not match the required quality by removing low qualityreads or reads that are too short Simultaneously to filter theraw data based on its quality it is increasingly common topreprocess the data for example to remove barcodes attachedto the sequenced reads Barcodes are added to the sequencedreads when multiple samples are sequenced together andneed to be separated again before the analysis This splittingbased on barcodes can also be done using a tool like FASTX
(B) Alignment The verified raw sequencing data is thenaligned against the human reference genome During thisprocess every sequenced read is placed at the most fitting
position on the reference genomeWhat themost fitting posi-tion will depend on the algorithm and parameters used Thisprocess is specially complicated when aligning sequencesfrom repeated regions or containing insertions and deletionsThe alignment process can be done by using one of manyavailable tools [6] Many of those tools are actively developedand possess their individual qualities depending on the use-case and sequence data to be aligned Some of the morecommonly used tools for sequence alignment are BWA [7]Bowtie 2 [8] or BLAST [9] This list is not exhaustive andfurther information on the subject can be found in [6]
(C) Realignment In addition to the alignment tools exist toimprove the quality of an existing alignment for examplewith local indel realignment Tools like GATK [10] and theComplete Genomics realigner [11] provide this functionalityThis and the previous Alignment analysis step are some ofthe heaviest in terms of computing power and require agood computing infrastructure to be performed in a shorttime frame This problem is accentuated with the increasingamount of produced raw data and expected qualities of thealignment notably for indels
(D) QC Another round of quality control is usually per-formed after the final alignment has been created It is usedto verify that the sample has been sequenced correctly inparticular in the regions of interest like the genes associatedwith the patients phenotype Several tools allow the extrac-tion of the information needed for this step such as samtools[12] or bedtools 2 [13] They allow the user to get metricssuch as the number of aligned reads and the coverage of theregions of interest out of the alignment file If the quality ofthe alignment is not sufficient at this stage it can be decided toresequence the sample This step of quality control is criticalin a diagnostic setting where it is necessary to show howwell the regions of interest for a specific sample have beencovered It is especially important for cases where no variantsof interest can be found in the specified zones and thus anegative findings report needs to be justified
(E) Variant Calling After the raw sequencing data has beenaligned and its quality verified SNPs and indels can be calledThe process of variant calling compares the aligned sequencesto the reference sequence in order to create a list of variantspresent in the sample This list can be filtered on differentcriteria such as minimum coverage at the variant position orthe frequency of the variant allele in the sample Commonlyused tools are GATK [10] samtools [12] and Varscan 2 [14]Different variant callers follow different underlying modelsresulting in differences in the called variants [15] Variantcalling usually yields a large amount of variants even whenrestricting the calling to only output variantswith aminimumquality and probability not to be a sequencing artifact
(F) Annotation This is why in the next step the list of thevariants is annotated with more information that is pertinentto the analysis being done on the sample This includesthe detection of variants present in public databases suchas dbSNP [16] or the information about which genes are
BioMed Research International 3
FastQCFASTX
BWABowtie 2BLASTand so forth
GATKcomplete genomicsrealigner
Data preparation
(A) QCfiltering
(B) Alignment
Data analysis
(D) QC
(E) Variant calling
(F) Annotation
(G) Variant filtering
Samtoolsbedtools 2
GATKsamtoolsVarscan 2
dbSNPPhenomizerVEPand so forth
Samtools
Validation and reporting
IGVTabletgenome viewUCSCbrowser and so forth
(I) Sanger validation
(J) Report generation
(H) Visualization
(C) Realignment
Figure 1 UML activity diagram of a NGS diagnostics workflow separated into different steps A collection of the available tools for everystep is mentioned
affected and their predicted effects on those genes Varioustools exist to annotate variants either locally or through aweb service Some of the more popular ones are the VariantEffect Predictor (VEP) from Ensembl [17] and Phenomizer[18] that add clinically relevant information to the detectedvariants Once the annotated list of variants has been createdthe phenotype relevant variant has to be identified
(G) Variant Filtering Analysis specific filters allow the userto filter the number of variants down to a smaller numberOne example is to only keep variants affecting genes withphenotypes matching the ones of the patient and that have apredicted consequence on the gene function The exact typeof filters to reduce the amount of candidate variants variesfrom case to case depending on the available informationThe filtering is often done using bcftools which is partof the samtools project [12] or vcftools [19] If during thisstep no clinically relevant variant related to the currentanalysis can be found depending on the analysis protocolthe filters may be relaxed This allows the user to includefor example variants on genes where the relation with thecurrent phenotype is less certain or variants with uncertainconsequences on a known disease gene
(H) Visualization Once a list of candidate variants has beenidentified it is common to visually verify them in a genomebrowser The genome browser displays the sequenced readsaround the variant and allows the user to verify the qualityof the data around that position allowing the user to identifyand discard variants originating from sequencing errorsThisis done using one of the various genome browsers installedlocally such as IGV [20] Tablet [21] or Genome view [22]There also exist web-based genome browsers such as theUCSC [23] or Ensembl [24] genome browsers
(I) Sanger Validation Before submitting the final findingsto the clinician it is still common practice to validatethe identified variants through Sanger sequencing Whilefuture advances in NGS technologies might make this stepredundant it is still considered the gold standard for variantvalidation
(J) Report Generation After visually inspecting and validatingthe variants the identified variants or the lack thereof aredocumented in a final report that is sent off to the clinicianto allow him to take decisions on the case
The described process can be done by taking severalapproaches A common approach is to use a selection ofthe previously described tools to perform the individualanalysis steps The different tools are usually connectedtogether through a series of automated scripts performingthe required analysis The creation of the resulting pipelineis a complex technical task as it requires insight in theworking of the different tools and the ability to chain themtogether This is a nontrivial task as not all tools follow thesame standards in terms of data file formats and handlingNonetheless this is a very common approach which resultedin a variety of custom made NGS analysis pipelines eitherprivate or public [25]
Complete software packages like CLCBio [26]NextGENe [27] or theweb-basedGalaxy [28] also existTheyallow the user to combine many of the mentioned analysistools without having to worry too much on how to connectthe different tools to perform the analysis Those toolsare focused on giving the user the possibility to automatethe analysis process as much as possible abstracting thecomplexity of the analysis GensearchNGS belongs to thisfamily of tools with its own approach and focus on theissues of smaller laboratories It focuses less on the possibility
4 BioMed Research International
to generate a custom automated pipeline but more on anintuitive interface to handle the data-files to perform theanalysis in a predefined pipeline optimised for diagnostics
The features of GensearchNGS are limited to thoseneeded to perform the presented NGS diagnostics workflowThis means the overall feature set is smaller than that of othercomplete software packages especially CLCBio or Galaxywhich both offer a wide variety of functionalities Havinga limited feature set allows for a cleaner and more focuseduser interface one of the major goals of GensearchNGSsomething that is highly appreciated by current users ofGensearchNGS
Aswill be discussed in the following chapters Gensearch-NGS uses either external applications through plugins orcustom implementations of standard algorithms for the dif-ferent analysis stepsThe resulting analysis quality is thereforecomparable to the one observed when using those toolsseparately by hand or through a software package like Galaxy
Approach The issues faced by small laboratories when usingthe previously mentioned tools which are often limitedin terms of computing and human resources are variousBased on our experience the biggest issue faced by smallerlaboratories is the lack of technical know-how and dedicatedbioinformaticians The usage and configuration of many ofthe described data analysis tools require a dedicated employeewhich not available in every laboratoryThis is especially truein emergingmarketswheremanymonetary restrictions are inplace The approach of solutions like Galaxy and CLCBio tothis problem is to abstract the usage of the underlying analysistools and provide a way for the user to automate the analysisprocessWhile the abstraction of the underlying tools reducesthe technical complexity of the analysis process this is oftennot enough tomake the analysis process more accessibleThecited tools can be used for many other types of analyses thanthe ones looked at in this paper which leads to a much morecomplex user interface and often overwhelms the user withmany options The cited tools also focus more on the abilityto create automated analysis pipelines leading to very littleinteractivity between the user and the data
The second issue often encountered is the lack of a suitablecomputing infrastructure to perform the computationallyintensive parts of the analysis Sequence alignment in par-ticular is very demanding in that regard Even though manylaboratories outsource the sequencing and alignment processthe possibility to realign the raw sequencing data is oftenrequired To perform the described analysis steps and moreGensearchNGS provides a complete software suite focusingon optimized algorithms and usage of distributed computingfor optimal usage of the existing infrastructure All theanalysis steps make usage of multiprocessor systems to speedup the analysis whereas only sequence alignment using thecustomGensearchNGS alignment algorithm uses distributedcomputing The entire functionality is provided throughan intuitive user interface to lower the technical know-how needed to perform NGS data analysis GensearchNGSprovides its own way on how the analysis is approachedby providing the user with a patient centric user interface
and favoring an interactive analysis approach instead ofa fully automated analysis pipeline The user interface isfocused on providing the functionality required to performthe previously described analysis without overwhelming theuser with rarely used features
2 Materials and Methods
GensearchNGS has been developed as a multiplatform appli-cation running onWindows Linux and OSX using Java 6+The application is a locally installed software and is not webbased In order to perform NGS analysis steps Gensearch-NGS includes several existing external tools through pluginsThose tools are mainly used during the sequence alignmentphase giving the user the choice between different alignmentalgorithms which may have specific advantages related tothe data to be analysed The external alignment algorithmssupported through plugins are BWA [7] and Bowtie 1 + 2[8] as well as Stampy [29] GensearchNGS can either uselocally installed versions of those external tools or downloadand update them automatically If the tools are installedby the user he can update them when desired otherwisethe same update mechanism used to update GensearchNGSitself is used to update the external tools For sequencingdata GensearchNGS supports various input and output fileformats For raw sequencing data the standard FASTQformat as well as FASTA SFF and FNA is supported All ofthem are converted to the standard FASTQ file format duringthe data import phase As FASTQ files require a quality scorefor every nucleotide in the sequenced reads a default qualityscore is assumed for all the nucleotides if the source formatlike FASTA is missing this information For aligned databoth SAM and BAM files are supported whereas SAM filesare converted to BAM during import BAM and SAM fileare accessed through the HTSJDK library HTSJDK is anopen source Java library developed as part of the samtoolsproject [12] It provides a standardized API to access variousNGS data file formats The library can be downloaded athttpsamtoolsgithubiohtsjdk Annotations can be pro-vided through the BED WIG and GFF files and the VCF fileformat is used to import or export variants
To benefit from the knowledge stored in various onlinedatabases GensearchNGS has been connected to several ofthem to annotate the NGS data on various levels from thegenes and their transcripts down to the individual variantsFor most of the annotation data GensearchNGS accessesthe Ensembl [24] web service through its biomart APIgiving the application access to gene and variant informationEnsembl provides a unified way to access several other onlinedatabases like dbSNP [16] or OMIM [30] through a singleonline service In addition to Ensembl GensearchNGS inte-grates other online services for additional data and featuresCafe Variome [31] provides a standardized way to sharevariants between various laboratories UCSC [32] providesaccess to the human reference sequence and HPO [18] pro-vides information about phenotypes associatedwith genes Insituations with a direct conflict in the information betweenthe different databases where possible the information withthe most severe clinical impact is displayed An example of
BioMed Research International 5
this is contradictory SIFT [33] and PolyPhen 2 [34] scores forvariants By default the most severe will be shown to the userwith the possibility to manually get more information whenrequested GensearchNGS also integrates the possibility totalk to other software installed by the user such as Alamut[35] allowing the user to easily open a variant found byGensearchNGS inside a local Alamut installation
3 Results
This chapter discusses how GensearchNGS implements thedifferent analysis steps and how it tries to lower as much aspossible the technical know-how and computing infrastruc-ture required for NGS data analysis
31 Software Suite GensearchNGS provides the user with thepossibility to create different projects to group the analysis ofvarious patients based on their type or their relation Whilemany other NGS analysis tools provide the possibility toorganize the data in projects GensearchNGS puts the notionof patients and their associatedmetainformation in the centerof the project management Every project contains one ormore patient and each can have several raw sequencing dataassociated The raw sequencing data can in turn have severalalignments associated for example using different alignersor alignment configurations Project specific settings such asspecific regions of interests to be used during the analysis canalso be configured Those regions can be described by BEDfiles to for example limit the analysis to regions relevant tothe type of analysis performed in the current project ThoseBED files can either be provided by the user or automaticallygenerated based on a list of gene names Projects alsoassociate the specific reference sequence to be used duringthe analysis The user can automatically download themfrom UCSC which provides reference sequences of the mostcommon human reference assemblies hg18 hg19 and hg38The usage of custom references sequences is also supportedThe patient centric database integrated into GensearchNGSallows the user to associate variants with patients a featurevery important in diagnostics The variants associated withpatients are shared between projects allowing the user toidentify variants found in different projects on previouslyanalysed patients
To perform the NGS data analysis GensearchNGS usesthe genes described in Ensembl as the gene model In a pro-cess transparent to the user the genemodel corresponding tothe reference sequence defined for the analysis is downloadedautomatically from Ensembl Additional metainformation isattached to the genes such as the associated phenotypesas described by HPO [18] or OMIM [30] Figure 2 showsthe main interface of GensearchNGS with the patient liston the left and the information about the currently selectedpatient in the center The central area of the image showsthe information about the currently selected patient Thisincludes his name internal ID associated phenotypes andall variants which have manually been confirmed by the userWhen validating variants the user can classify themmanuallyinto different categories The categories are based on the
Figure 2 Main interface of GensearchNGS showing a patient withits validated variants
ACMG variant classification guidelines [36] with the userrequested categories Artefact and False reference added Thesaved variants are used for any future variant detection in newpatients immediately telling the user if a variant has alreadybeen found in a different projectThe addition of the Artefactand False reference categories allows the user to quickly filterout common sequencing errors in his particular sequencingsetupThe hierarchical organization of the data is intended toguide the user through the analysis process while keeping itclear where the data comes from An example of this is thesequence alignment It is displayed as a child element of theraw sequencing data making the relation between the rawdata and the alignment immediately obvious
GensearchNGS does not provide the user with a singlebutton which goes from the raw sequencing data to the finalreport Instead of fully automating the pipeline Gensearch-NGS leads the user from one processing step to the nextBefore each step the user is given the choice to changeparameters or use the previously used (or standard) parame-ters After the initial import of the raw sequencing data theuser no longer has to handle the data files GensearchNGSperforms the required data file handling and conversionsbetween the processing steps
While there is no fully automated way to go from theraw sequencing data to the final variant report there is stilla way to automate a big part of the analysis When importingraw sequencing data the user has the possibility to performsequence alignment and variant calling automatically Thosetwo additional analysis steps are performed using eitherpreviously used configuration parameters or the default onesOnly the final variant validation has then to be performedby the user to create the final report Further details of thisprocess follow in the next chapter
32 Data Analysis To perform the initial quality control ofthe raw sequencing data GensearchNGS integrates its owntools allowing the user to import the raw data availablein different file formats The compatible data formats aredescribed in Section 2 The various formats are converted inthe standard FASTQ format during the data import step For
6 BioMed Research International
Figure 3 Overview of various statistics that the user can inspect to verify the data quality
the quality control of the raw sequencing data the user ispresented with various statistics as seen in Figure 3
The statistics allow the user to judge the quality of thesequencing data This includes statistics such as the lengthaverage quality andGC content distribution of the sequencedreads as well as the distribution of the sequenced basesand their quality for every read position and detection of
overrepresented K-mers at the start and the end of thesequenced reads
This raw data import process replaces existing tools likeFastQC to check the quality FASTX to filter the data andthe various utilities that transform different sequencing fileformats into the standard FASTQ file format Transparentlyfor the user the custom implementations of those tools
BioMed Research International 7
which produce the same results have been integrated into asingle processing stepThis includes the possibility to removeadapter sequences and split the raw sequencing data intomultiple files if they were produced through multiplexedsequencing Based on the reported quality information theuser can choose to filter the raw data following several filterssuch as maximum and minimum length of the sequencesand their minimal average quality He can also trim the 51015840and 31015840 parts of all reads or trim their excess length if theyare longer as a user defined length The trimming is done inaddition to the adapter and primer removalThe possibility tosplit the raw sequencing data based on barcodes in the readallowing the import of raw data from multiple samples thatwere sequenced at the same time is also integrated in theimport process
To align the sequenced data against a reference assemblyGensearchNGS provides the user with many options Severalexisting alignment algorithms are integrated into the applica-tion through plugins allowing their usage without the needof advanced technical know-how The currently integratedalignment algorithms are BWA [7] Bowtie 1 + 2 [8] andStampy [29]
Most of those alignment algorithms are developed in away that do not allow them to be used on multiple platformsIn fact most of them only run on Linux which is problematicfor a multiplatform application like GensearchNGS For thisreason a custom alignment algorithm written in Java hasbeen integrated directly into GensearchNGS This allows theuser to perform the full NGS data analysis even on operatingsystems likeWindows not supported bymost bioinformaticstools
The aligner supports paired end reads and gapped align-ment and has been optimized to use less than 5GB ofRAM to perform a full genome alignment This makes itsuitable for most of todayrsquos workspace desktops It is fullymultithreaded to support all cores of the machine and isable to offload calculations as described in Section 34 tomultiple computers The aligner uses a hash based indexa concept made popular through the BLAST aligner [9]with a custom heuristic based candidate alignment positionsselection [37] The final alignment is being done using theGotoh [38] algorithm a dynamic programming algorithmused for gapped alignment
The custom alignment algorithm being implemented inJava does not achieve quite the same performance as nativelyrunning aligners like BWA For WGS datasets it is generally50 slower The advantage of the GensearchNGS alignerover external aligners is that it is available on all supportedplatforms allowing the user to perform the complete NGSdata analysis on all platforms But when running on aplatform which supports the external aligners and speed isa deciding factor the users are encouraged to use the alignersavailable as plugins
The quality of resulting alignment can be verified throughvarious metrics by the user He can easily verify how manysequences have been discarded during the alignment pro-cess and how many sequences have been aligned to thedifferent chromosomes in the reference sequence Advancedstatistics similar to the ones created by bedtools 2 can also
be inspected giving detailed coverage information of thepreviously defined regions of interest The collected qualitycontrol information can be exported as a report for archivingpurposes using one of various formats such as PDF HTMLor PNG
While initially GensearchNGS integrated Varscan 2 [14]as its variant caller a replacement was created to reduce theexternal dependencies and to improve the performance ofvariant calling A variant scanner based on the model ofVarscan 2 has been implemented and is in the process of beingpublished as a separate tool called GNATY available for freefor noncommercial usage (httpgnatyphenosystemscom)Our custom variant caller shows important performanceadvantages over the existing Varscan 2 implementation of thesame variant calling model while producing the same analy-sis results Indeed GensearchNGS shows speed improvementof up to 18 times for variant calling compared to Varscan2 providing a similar range of features such as filters thatexclude sequencing artifacts
To be able to further filter down the detected variantsbased on their relevancy to the samplersquos phenotype severalsources are used to annotate them with various informationThis includes the prediction of the variant effect on thedifferent transcripts it affects a feature closely modeledafter the Variant Effect Predictor (VEP) from Ensembl [17]GensearchNGS implements its own version of the the VEPalgorithm to be able to perform the effect prediction in afraction of the time compared to the online VEP serviceThisis is mainly achieved by doing all calculations locally withouthaving to transfer the data to a remote server Predictionssuch as the addition or removal of a stop codon in an affectedtranscript if a splice site is affected and many others canbe made with this algorithm Additional information suchas if the variant is known in any public database associatedclinical significance and SIFT [33] and PolyPhen 2 [34]predictions are retrieved through the Ensembl web serviceOther services like the Human Phenotype Ontology [18]provide the information about the phenotype associated witha specific gene a crucial information in diagnostics
To filter the annotated variants GensearchNGS takesan interactive approach which allows the user to modifydifferent basic filters such as minimum or maximum samplefrequency coverage read balance or quality There are alsoadvanced filters such as the predicted consequence of thevariant as determined by the annotation process or if acertain phenotype is associated with the affected genes Thisincludes the possibility to filter by population frequency ofa variant The filtering by population frequency does not yettake into account the ethnicity of the patient Figure 4 showsan example of this list with several user selected interactivefilters applied
Additional features are available to perform a more in-depth analysis of variants such as family analysis where thevariants of multiple patients can be compared to find thoseonly present in a certain group of patients The usage ofpedigree information is of growing importance in NGS dataanalysis as it allows the user to filter sequencing artifactsand noncausative variants This is a very important featureespecially when usingWGSwhere the amount of variants can
8 BioMed Research International
Figure 4 Integrated variants list with several interactive filtersapplied
be very large Thanks to the usage of standard file formatslike VCF external tools like exome walker [39] can be usedto perform more advanced family analysis for features notcovered in GensearchNGS In contrast to many other NGSdata analysis software packages GensearchNGS does notfocus on the automation of every aspect of the analysisWhilecertain parts can be automated such as performing sequencealignment and variant calling on raw sequencing data thephilosophy of our software is to let the user to control thedifferent analysis steps This gives him enough flexibility toadapt to the data he is currently analysing without slowinghim down by having project specific analysis options whichare preselected for every analysis step This makes it possibleto perform most types of analysis with the project specificdefault options through a simple press of a button
33 Genome Browser A central tool of GensearchNGS isthe integrated genome browser to visualize the alignedsequencing data The genome browser as shown in Figure 5allows to interactively browse the sequence alignments Itis possible to display the aligned sequences and additionalgenomic data such as the gene locations and the encodedproteins by different transcripts
The genome browser allows the user to display sev-eral tracks containing information which helps to analysethe sequencing data The predefined tracks which can beactivated or deactivated allow the user to display coverageinformation GC content of the reference sequence thequality of the sequenced reads or custom annotation files instandard file formats such as BED WIG and GFF The focuswas put on optimizing the display of the data allowing theuser to visualize large datafiles without requiring a powerfulcomputerThis is achieved by streaming the data on demandonly loading the parts of the data currently needed forthe visualization The information about which variants arepresent in the alignment is available to the user in the sameform as the variant list in the main application It can be seenin Figure 4 allowing the user to quickly jump to the variantsof interest The genome browser like all of GensearchNGSis heavily connected with external data sources (EnsembldbSNP Entrez OMIM Blast and HPO) providing the user
Figure 5 Example view of the genome browser showing severalavailable tracks
with all relevant informationwhenneededTheuser can openexternal resources linked from many features in the visual-ized data such as genes transcripts or variants to retrieveadditional informationThe genome browser takes advantageof the integrated data sources like the Ensembl gene modelto display and interactively update encoded proteins basedon the variants present in the visualized data The genomebrowser is tightly integrated into GensearchNGS allowingthe user to easily visualize the alignment data belonging tofeatures of the sample
34 Distributed Computing The lack of computing powerto perform NGS data analysis is a very common issue inmany laboratories To address this problem GensearchNGSdoes not only optimize the algorithms for speed andmemoryusage but also try to exploit the already existing computinginfrastructure One way to do this is to use distributedcomputing a technique which allows the user to combinemultiple computers to perform computing intensive tasksThose computers can be in the same local network or at aremote location One of the most demanding steps in NGSdata analysis is sequence alignment For this reason it is thefirst feature to take advantage of this technique in Gensearch-NGS allowing the user to joinmultiple computers in the samenetwork to perform sequence alignment The approach usedby GensearchNGS is to transparently on request by the useruse all computers within the same networkwhereGensearch-NGS is installed Additionally the user has the possibility tostart up GensearchNGS instances in a cloud Currently onlythe amazon cloud is supportedThis process works regardlessof the operating system used by the different installationsof GensearchNGS By using an automatic service discoveryprotocol every installation of GensearchNGS located in thesame network is discovered without the need for manualconfigurationThis allows the user to use multiple computersat the same time or to completely offload calculations to adifferent computer making it possible to use computers notsuited for full genome alignments as an analysis workstationThe distributed sequence aligner used in GensearchNGS
BioMed Research International 9
currently only supports the GensearchNGS aligner algorithmand not external aligners such as BWA or Bowtie 2 It uses astream based approach where the client machine streams thedata to be aligned to all computers involved in the alignmentprocess This includes the client machine which receives thealigned sequences back and combines them into the finalalignment file The process of distributing the alignmentsover multiple computers and the cloud is transparent to theuser but it is optional because the usage of an externalcloud is not compatible with the security guidelines of somelaboratories GensearchNGS is not the onlyNGSdata analysissoftware using distributed sequence alignment It is thisability to combine the computing resources of the localcomputer multiple computers in the same network as well asthe cloud whichmakes its approach unique and very flexible
4 Discussion
GensearchNGS addresses problems that smaller laboratoriesare facing in various ways The issue of the technical com-plexity of the analysis is approached through an intuitive userinterface The user interface abstracts the complexity of theunderlying NGS data analysis toolchain which is a majorusability improvement over the usage of individual toolsNevertheless it gives the user full control over the individualanalysis steps instead of focusing on the creation of auto-mated analysis pipelines It also provides an original approachby providing a patient centric user interface This userfriendly interface allows users unfamiliar with the underlyingsoftware tools to fine tune the analysis parameters withoutoverwhelming themwith features or rarely used optionsThisincreases the number of people able to perform NGS dataanalysis This is particularly interesting for geneticists thatmight not have special bioinformatics training The issue ofincreasing infrastructure requirements for NGS data analysisis being addressed twofold First more efficient algorithmsare being developed like the heavily optimized variant callingalgorithm included in GensearchNGS Secondly the abilityto distribute heavy calculations over several computers oreven a cloud service has been developed GensearchNGShas a unique approach to this problem by enabling the userto combine the various distribution methods to performsequence alignment It provides the flexibility needed tochoose the best compromise between data privacy andalignment speed depending on the laboratory policies andneeds The resulting software enabled various laboratoriesto perform NGS data analysis and publish their results invarious fields The topics range from muscular dystrophies[40 41] to hearing loss [42] andmalignant hyperthermia [43]but also yet unpublished work about CNV analysis and thedetection of somatic mutations in cancer screening
5 Conclusion
In this paper we reviewed the workflow used to analyzeNGS data in a diagnostics environment The reviewedworkflow includes the detection of SNPs and short indelsfrom panel WES or WGS DNAseq data We also pre-sented issues faced by smaller laboratories trying to setup a
NGS data analysis pipeline We then presented Gensearch-NGS a software suite integrating various algorithms toperform the discussed workflow GensearchNGS is a com-mercially available software distributed by PhenosystemsSA A demo version of GensearchNGS is freely availableat httpwwwphenosystemscom The two main issues thetechnical complexity of the analysis and the computinginfrastructure requirements have been addressed as follows
To reduce the technical complexity of the analysis theuser is provided with an intuitive user interface which guideshim through the process of the data analysis He is providedwith the required tools to go from raw sequencing data the thefinal visualization and report generation for the sequencedsamples without having to learn complex bioinformaticstools The interface abstracts the technical complexity fromthe user while keeping enough flexibility to adapt to differentanalysis protocols By lowering the technical complexity ofNGS data analysis compared to existing individual analysistools GensearchNGS expands the potential user base of thesoftware
To address the issue of the computing infrastructurerequirements GensearchNGS focuses on the optimal usageof existing computing resources This is primarily achievedthrough the development of analysis algorithms that runon a minimal amount of resources while producing thesame analysis results as standard tools An example has beendescribed with the variant calling algorithm which producesthe same results as Varscan 2 but is 18 times faster Thesecond approach is the integration of distributed computingallowing the distribution of computing intensive tasks likealignment to be speed up by the usage of multiple computers
With a solid framework built for DNAseq analysisGensearchNGS will expand into further types of analysis inthe futureOne example is the integration of RNAseq analysisas the combination of OMICS data for a single sample isof increasing importance to understand rare diseases [44]In fact many parts of GensearchNGS such as the genomebrowser have already been adapted to support RNAseq dataThe integration of multiple data sources will not only allowthe user to integrate different types of OMICS data but alsoallow multiple views on the same sample One example ofthis is the integration of Sanger sequencing to NGS dataa very useful feature for diagnostics where it is commonpractice to validate variants identified through NGS withSanger sequencing
Further optimizations of the algorithms used in theanalysis will also be evaluated including optimization for theunderlying libraries One example is HTSJDK where severalperformance patches have been submitted and includedimproving NGS data analysis performance not only for usersof GensearchNGS but also for the complete bioinformaticscommunity
Disclosure
Pierre Kuonen has a nonpaid advisory position at Phenosys-tems SA David Atlan is working full time for PhenosystemsSA
10 BioMed Research International
Conflict of Interests
Thomas Dandekar has no conflict of interests regarding thepublication of this paper
Acknowledgments
GensearchNGS is a commercially available software dis-tributed by Phenosystems SA Beat Wolf rsquos PhD thesis ispartially financially supported by Phenosystems SA
References
[1] E L van Dijk H Auger Y Jaszczyszyn and C ThermesldquoTen years of next-generation sequencing technologyrdquo Trendsin Genetics vol 30 no 9 pp 418ndash426 2014
[2] B Sikkema-Raddatz L F Johansson E N de Boer et al ldquoTar-geted next-generation sequencing can replace sanger sequenc-ing in clinical diagnosticsrdquo Human Mutation vol 34 no 7 pp1035ndash1042 2013
[3] H P J Buermans and J T den Dunnen ldquoNext generationsequencing technology advances and applicationsrdquo Biochimicaet Biophysica ActamdashMolecular Basis of Disease vol 1842 no 10pp 1932ndash1941 2014
[4] FastQC ldquoA quality control tool for high throughput sequencedatardquo Babraham Bioinformatics Website httpwwwbioinfor-maticsbabrahamacukprojectsfastqc
[5] W R Pearson T Wood Z Zhang andWMiller ldquoComparisonof DNA sequences with protein sequencesrdquo Genomics vol 46no 1 pp 24ndash36 1997
[6] H Li and N Homer ldquoA survey of sequence alignment algo-rithms for next-generation sequencingrdquo Briefings in Bioinfor-matics vol 11 no 5 pp 473ndash483 2010
[7] H Li and R Durbin ldquoFast and accurate long-read alignmentwith Burrows-Wheeler transformrdquo Bioinformatics vol 26 no5 pp 589ndash595 2010
[8] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[9] S F Altschul T L Madden A A Schaffer et al ldquoGappedBLAST and PSI-BLAST a new generation of protein databasesearch programsrdquo Nucleic Acids Research vol 25 no 17 pp3389ndash3402 1997
[10] M A DePristo E Banks R Poplin et al ldquoA framework forvariation discovery and genotyping using next-generationDNAsequencing datardquo Nature Genetics vol 43 no 5 pp 491ndash4982011
[11] P Carnevali J Baccash A L Halpern et al ldquoComputationaltechniques for human genome resequencing using matedgapped readsrdquo Journal of Computational Biology vol 19 no 3pp 279ndash292 2012
[12] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[13] A R Quinlan and I M Hall ldquoBEDTools a flexible suite ofutilities for comparing genomic featuresrdquo Bioinformatics vol26 no 6 pp 841ndash842 2010
[14] D C Koboldt Q Zhang D E Larson et al ldquoVarScan 2 somaticmutation and copy number alteration discovery in cancer byexome sequencingrdquo Genome Research vol 22 no 3 pp 568ndash576 2012
[15] J OrsquoRawe T Jiang G Sun et al ldquoLow concordance of multiplevariant-calling pipelines practical implications for exome andgenome sequencingrdquo Genome Medicine vol 5 no 3 article 282013
[16] S T SherryM-HWardM Kholodov et al ldquodbSNP the NCBIdatabase of genetic variationrdquoNucleic Acids Research vol 29 pp308ndash311 2001
[17] W McLaren B Pritchard D Rios Y Chen P Flicek and FCunningham ldquoDeriving the consequences of genomic variantswith the EnsemblAPI and SNPEffect PredictorrdquoBioinformaticsvol 26 no 16 Article ID btq330 pp 2069ndash2070 2010
[18] P N Robinson and S Mundlos ldquoThe human phenotypeontologyrdquo Clinical Genetics vol 77 no 6 pp 525ndash534 2010
[19] P Danecek A Auton G Abecasis et al ldquoThe variant call formatand VCFtoolsrdquo Bioinformatics vol 27 no 15 pp 2156ndash21582011
[20] J T Robinson HThorvaldsdottir W Winckler et al ldquoIntegra-tive genomics viewerrdquo Nature Biotechnology vol 29 no 1 pp24ndash26 2011
[21] I Milne G Stephen M Bayer et al ldquoUsing tablet for visualexploration of second-generation sequencing datardquo Briefings inBioinformatics vol 14 no 2 Article ID bbs012 pp 193ndash2022013
[22] T Abeel T van Parys Y Saeys J Galagan and Y van de PeerldquoGenomeView a next-generation genome browserrdquo NucleicAcids Research vol 40 no 2 article e12 2012
[23] W J Kent C W Sugnet T S Furey et al ldquoThe human genomebrowser at UCSCrdquo Genome Research vol 12 no 6 pp 996ndash1006 2002
[24] F Cunningham M Ridwan Amode D Barrell et al ldquoEnsembl2015rdquo Nucleic Acids Research vol 43 no 1 pp D662ndashD6692015
[25] NGS Data Analysis Software List SEQanswers 2012 httpseqanswerscomwikiSoftwarelist
[26] CLC Genomics Workbench CLC Bio Aarhus Denmark 2014httpwwwclcbiocom
[27] NextGENe Softgenetics State College Pa USA httpwwwsoftgeneticscom
[28] J Goecks A Nekrutenko J Taylor et al ldquoGalaxy a com-prehensive approach for supporting accessible reproducibleand transparent computational research in the life sciencesrdquoGenome Biology vol 11 no 8 article R86 2010
[29] G Lunter and M Goodson ldquoStampy a statistical algorithm forsensitive and fast mapping of Illumina sequence readsrdquoGenomeResearch vol 21 no 6 pp 936ndash939 2011
[30] Online Mendelian Inheritance in Man OMIM McKusick-Nathans Institute of Genetic Medicine Johns Hopkins Univer-sity Baltimore Md USA 2015 httpomimorg
[31] Cafe Variome httpwwwcafevariomeorg[32] The Genome Sequencing Consortium ldquoInitial sequencing and
analysis of the human genomerdquo Nature vol 409 pp 860ndash9212001
[33] P Kumar S Henikoff and P C Ng ldquoPredicting the effects ofcoding non-synonymous variants on protein function using theSIFT algorithmrdquo Nature Protocols vol 4 no 7 pp 1073ndash10822009
[34] I A Adzhubei S Schmidt L Peshkin et al ldquoA method andserver for predicting damaging missense mutationsrdquo NatureMethods vol 7 no 4 pp 248ndash249 2010
[35] Alamut Visual Interactive Biosoftware Rouen France httpwwwinteractivebiosoftwarecomalamut-visual
BioMed Research International 11
[36] S Richards N Aziz S Bale et al ldquoStandards and guidelinesfor the interpretation of sequence variants a joint consensusrecommendation of the American College of Medical Geneticsand Genomics and the Association for Molecular PathologyrdquoGenetics in Medicine vol 17 no 5 pp 405ndash423 2015
[37] BWolf and P Kuonen ldquoA novel approach for heuristic pairwiseDNA sequence alignmentrdquo in Proceedings of the InternationalConference on Bioinformatics amp Computational Biology (BIO-COMP rsquo13) Las Vegas Nev USA July 2013
[38] O Gotoh ldquoAn improved algorithm for matching biologicalsequencesrdquo Journal ofMolecular Biology vol 162 no 3 pp 705ndash708 1982
[39] D Smedley S Kohler J C Czeschik et al ldquoWalking theinteractome for candidate prioritization in exome sequencingstudies of Mendelian diseasesrdquo Bioinformatics vol 30 no 22pp 3215ndash3222 2014
[40] A L Semmler S Sacconi J Bach et al ldquoUnusual multisys-temic involvement and a novel BAG3 mutation revealed byNGS screening in a large cohort of myofibrillar myopathiesrdquoOrphanet Journal of Rare Diseases vol 9 no 1 p 121 2014
[41] M Larsen S Rost N El Hajj et al ldquoDiagnostic approach forFSHD revisited SMCHD1 mutations cause FSHD2 and act asmodifiers of disease severity in FSHD1rdquo European Journal ofHuman Genetics vol 23 no 6 pp 808ndash816 2015
[42] S Rost E Bach C Neuner et al ldquoNovel form of X-linkednonsyndromic hearing loss with cochlear malformation causedby a mutation in the type IV collagen gene COL4A6rdquo EuropeanJournal of Human Genetics vol 22 no 2 pp 208ndash215 2014
[43] M Broman I Kleinschnitz J E Bach S Rost G Islander andC R Muller ldquoNext-generation DNA sequencing of a Swedishmalignant hyperthermia cohortrdquo Clinical Genetics 2014
[44] T Low S vanHeesch H van den Toorn et al ldquoQuantitative andqualitative proteome characteristics extracted from in-depthintegrated genomics and proteomics analysisrdquo Cell Reports vol5 no 5 pp 1469ndash1478 2013
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

2 BioMed Research International
the need to resequence it This tendency to increase theamount of sequenced data creates increasing challenges forsmaller diagnostics laboratories They often do not have therequired computing infrastructure or technical know-how tohandle this type of data This is especially true in emergingmarkets which do not always have the required financial andhuman resources as well as the extensive know-how requiredto set up their own analysis pipeline
Today many types of NGS data analysis exist all ofthem answering different sorts of questions about a sampleOne example being copy number variation (CNV) analy-sis suited to replace traditional MLPA (multiplex ligation-dependent probe amplification) analysis Another one is denovo sequence alignment to detect large changes in thegenome for example in cancer patients But ultimately thedetection of SNPs and small indels in a sample based ona standard reference sequence remains the most prevalenttype of analysis This is why in the context of this paper welimit our study to this type of analysis the detection of SNPsand small indels in targeted or nontargeted resequencingAlthough this type of analysis can be approached in variousways it can be described as a set of generic steps that remaincommon to most approaches The next chapter describesthe commonly used steps in the analysis process providingexamples of existing tools that can be used to perform them
NGS Data Analysis This chapter describes a common NGSdata analysis workflow used in diagnostics Figure 1 showsan overview of the described workflow using a UnifiedModeling Language (UML) diagram
(A) QCFiltering The analysis starts with the quality controlof the raw sequencing data to determine if the sequencingprocess adheres to the defined quality requirements Differentmetrics such as the amount of reads sequenced the lengthdistribution of the sequenced reads and their quality areverified and compared to the expected values of the specificsequencer technology that has been used If the quality ofthe sequenced data does not match the required quality thesample is usually resequenced To verify the quality of the rawdata one commonly used tool is FastQC [4] which generatesa comprehensive list of statistics of the raw data Duringthis initial quality control stage the raw sequencing data isoften filtered discarding reads that do not match the definedquality criteria This raw data filtering is commonly donewith the FASTX toolkit [5] which allows removing reads thatdo not match the required quality by removing low qualityreads or reads that are too short Simultaneously to filter theraw data based on its quality it is increasingly common topreprocess the data for example to remove barcodes attachedto the sequenced reads Barcodes are added to the sequencedreads when multiple samples are sequenced together andneed to be separated again before the analysis This splittingbased on barcodes can also be done using a tool like FASTX
(B) Alignment The verified raw sequencing data is thenaligned against the human reference genome During thisprocess every sequenced read is placed at the most fitting
position on the reference genomeWhat themost fitting posi-tion will depend on the algorithm and parameters used Thisprocess is specially complicated when aligning sequencesfrom repeated regions or containing insertions and deletionsThe alignment process can be done by using one of manyavailable tools [6] Many of those tools are actively developedand possess their individual qualities depending on the use-case and sequence data to be aligned Some of the morecommonly used tools for sequence alignment are BWA [7]Bowtie 2 [8] or BLAST [9] This list is not exhaustive andfurther information on the subject can be found in [6]
(C) Realignment In addition to the alignment tools exist toimprove the quality of an existing alignment for examplewith local indel realignment Tools like GATK [10] and theComplete Genomics realigner [11] provide this functionalityThis and the previous Alignment analysis step are some ofthe heaviest in terms of computing power and require agood computing infrastructure to be performed in a shorttime frame This problem is accentuated with the increasingamount of produced raw data and expected qualities of thealignment notably for indels
(D) QC Another round of quality control is usually per-formed after the final alignment has been created It is usedto verify that the sample has been sequenced correctly inparticular in the regions of interest like the genes associatedwith the patients phenotype Several tools allow the extrac-tion of the information needed for this step such as samtools[12] or bedtools 2 [13] They allow the user to get metricssuch as the number of aligned reads and the coverage of theregions of interest out of the alignment file If the quality ofthe alignment is not sufficient at this stage it can be decided toresequence the sample This step of quality control is criticalin a diagnostic setting where it is necessary to show howwell the regions of interest for a specific sample have beencovered It is especially important for cases where no variantsof interest can be found in the specified zones and thus anegative findings report needs to be justified
(E) Variant Calling After the raw sequencing data has beenaligned and its quality verified SNPs and indels can be calledThe process of variant calling compares the aligned sequencesto the reference sequence in order to create a list of variantspresent in the sample This list can be filtered on differentcriteria such as minimum coverage at the variant position orthe frequency of the variant allele in the sample Commonlyused tools are GATK [10] samtools [12] and Varscan 2 [14]Different variant callers follow different underlying modelsresulting in differences in the called variants [15] Variantcalling usually yields a large amount of variants even whenrestricting the calling to only output variantswith aminimumquality and probability not to be a sequencing artifact
(F) Annotation This is why in the next step the list of thevariants is annotated with more information that is pertinentto the analysis being done on the sample This includesthe detection of variants present in public databases suchas dbSNP [16] or the information about which genes are
BioMed Research International 3
FastQCFASTX
BWABowtie 2BLASTand so forth
GATKcomplete genomicsrealigner
Data preparation
(A) QCfiltering
(B) Alignment
Data analysis
(D) QC
(E) Variant calling
(F) Annotation
(G) Variant filtering
Samtoolsbedtools 2
GATKsamtoolsVarscan 2
dbSNPPhenomizerVEPand so forth
Samtools
Validation and reporting
IGVTabletgenome viewUCSCbrowser and so forth
(I) Sanger validation
(J) Report generation
(H) Visualization
(C) Realignment
Figure 1 UML activity diagram of a NGS diagnostics workflow separated into different steps A collection of the available tools for everystep is mentioned
affected and their predicted effects on those genes Varioustools exist to annotate variants either locally or through aweb service Some of the more popular ones are the VariantEffect Predictor (VEP) from Ensembl [17] and Phenomizer[18] that add clinically relevant information to the detectedvariants Once the annotated list of variants has been createdthe phenotype relevant variant has to be identified
(G) Variant Filtering Analysis specific filters allow the userto filter the number of variants down to a smaller numberOne example is to only keep variants affecting genes withphenotypes matching the ones of the patient and that have apredicted consequence on the gene function The exact typeof filters to reduce the amount of candidate variants variesfrom case to case depending on the available informationThe filtering is often done using bcftools which is partof the samtools project [12] or vcftools [19] If during thisstep no clinically relevant variant related to the currentanalysis can be found depending on the analysis protocolthe filters may be relaxed This allows the user to includefor example variants on genes where the relation with thecurrent phenotype is less certain or variants with uncertainconsequences on a known disease gene
(H) Visualization Once a list of candidate variants has beenidentified it is common to visually verify them in a genomebrowser The genome browser displays the sequenced readsaround the variant and allows the user to verify the qualityof the data around that position allowing the user to identifyand discard variants originating from sequencing errorsThisis done using one of the various genome browsers installedlocally such as IGV [20] Tablet [21] or Genome view [22]There also exist web-based genome browsers such as theUCSC [23] or Ensembl [24] genome browsers
(I) Sanger Validation Before submitting the final findingsto the clinician it is still common practice to validatethe identified variants through Sanger sequencing Whilefuture advances in NGS technologies might make this stepredundant it is still considered the gold standard for variantvalidation
(J) Report Generation After visually inspecting and validatingthe variants the identified variants or the lack thereof aredocumented in a final report that is sent off to the clinicianto allow him to take decisions on the case
The described process can be done by taking severalapproaches A common approach is to use a selection ofthe previously described tools to perform the individualanalysis steps The different tools are usually connectedtogether through a series of automated scripts performingthe required analysis The creation of the resulting pipelineis a complex technical task as it requires insight in theworking of the different tools and the ability to chain themtogether This is a nontrivial task as not all tools follow thesame standards in terms of data file formats and handlingNonetheless this is a very common approach which resultedin a variety of custom made NGS analysis pipelines eitherprivate or public [25]
Complete software packages like CLCBio [26]NextGENe [27] or theweb-basedGalaxy [28] also existTheyallow the user to combine many of the mentioned analysistools without having to worry too much on how to connectthe different tools to perform the analysis Those toolsare focused on giving the user the possibility to automatethe analysis process as much as possible abstracting thecomplexity of the analysis GensearchNGS belongs to thisfamily of tools with its own approach and focus on theissues of smaller laboratories It focuses less on the possibility
4 BioMed Research International
to generate a custom automated pipeline but more on anintuitive interface to handle the data-files to perform theanalysis in a predefined pipeline optimised for diagnostics
The features of GensearchNGS are limited to thoseneeded to perform the presented NGS diagnostics workflowThis means the overall feature set is smaller than that of othercomplete software packages especially CLCBio or Galaxywhich both offer a wide variety of functionalities Havinga limited feature set allows for a cleaner and more focuseduser interface one of the major goals of GensearchNGSsomething that is highly appreciated by current users ofGensearchNGS
Aswill be discussed in the following chapters Gensearch-NGS uses either external applications through plugins orcustom implementations of standard algorithms for the dif-ferent analysis stepsThe resulting analysis quality is thereforecomparable to the one observed when using those toolsseparately by hand or through a software package like Galaxy
Approach The issues faced by small laboratories when usingthe previously mentioned tools which are often limitedin terms of computing and human resources are variousBased on our experience the biggest issue faced by smallerlaboratories is the lack of technical know-how and dedicatedbioinformaticians The usage and configuration of many ofthe described data analysis tools require a dedicated employeewhich not available in every laboratoryThis is especially truein emergingmarketswheremanymonetary restrictions are inplace The approach of solutions like Galaxy and CLCBio tothis problem is to abstract the usage of the underlying analysistools and provide a way for the user to automate the analysisprocessWhile the abstraction of the underlying tools reducesthe technical complexity of the analysis process this is oftennot enough tomake the analysis process more accessibleThecited tools can be used for many other types of analyses thanthe ones looked at in this paper which leads to a much morecomplex user interface and often overwhelms the user withmany options The cited tools also focus more on the abilityto create automated analysis pipelines leading to very littleinteractivity between the user and the data
The second issue often encountered is the lack of a suitablecomputing infrastructure to perform the computationallyintensive parts of the analysis Sequence alignment in par-ticular is very demanding in that regard Even though manylaboratories outsource the sequencing and alignment processthe possibility to realign the raw sequencing data is oftenrequired To perform the described analysis steps and moreGensearchNGS provides a complete software suite focusingon optimized algorithms and usage of distributed computingfor optimal usage of the existing infrastructure All theanalysis steps make usage of multiprocessor systems to speedup the analysis whereas only sequence alignment using thecustomGensearchNGS alignment algorithm uses distributedcomputing The entire functionality is provided throughan intuitive user interface to lower the technical know-how needed to perform NGS data analysis GensearchNGSprovides its own way on how the analysis is approachedby providing the user with a patient centric user interface
and favoring an interactive analysis approach instead ofa fully automated analysis pipeline The user interface isfocused on providing the functionality required to performthe previously described analysis without overwhelming theuser with rarely used features
2 Materials and Methods
GensearchNGS has been developed as a multiplatform appli-cation running onWindows Linux and OSX using Java 6+The application is a locally installed software and is not webbased In order to perform NGS analysis steps Gensearch-NGS includes several existing external tools through pluginsThose tools are mainly used during the sequence alignmentphase giving the user the choice between different alignmentalgorithms which may have specific advantages related tothe data to be analysed The external alignment algorithmssupported through plugins are BWA [7] and Bowtie 1 + 2[8] as well as Stampy [29] GensearchNGS can either uselocally installed versions of those external tools or downloadand update them automatically If the tools are installedby the user he can update them when desired otherwisethe same update mechanism used to update GensearchNGSitself is used to update the external tools For sequencingdata GensearchNGS supports various input and output fileformats For raw sequencing data the standard FASTQformat as well as FASTA SFF and FNA is supported All ofthem are converted to the standard FASTQ file format duringthe data import phase As FASTQ files require a quality scorefor every nucleotide in the sequenced reads a default qualityscore is assumed for all the nucleotides if the source formatlike FASTA is missing this information For aligned databoth SAM and BAM files are supported whereas SAM filesare converted to BAM during import BAM and SAM fileare accessed through the HTSJDK library HTSJDK is anopen source Java library developed as part of the samtoolsproject [12] It provides a standardized API to access variousNGS data file formats The library can be downloaded athttpsamtoolsgithubiohtsjdk Annotations can be pro-vided through the BED WIG and GFF files and the VCF fileformat is used to import or export variants
To benefit from the knowledge stored in various onlinedatabases GensearchNGS has been connected to several ofthem to annotate the NGS data on various levels from thegenes and their transcripts down to the individual variantsFor most of the annotation data GensearchNGS accessesthe Ensembl [24] web service through its biomart APIgiving the application access to gene and variant informationEnsembl provides a unified way to access several other onlinedatabases like dbSNP [16] or OMIM [30] through a singleonline service In addition to Ensembl GensearchNGS inte-grates other online services for additional data and featuresCafe Variome [31] provides a standardized way to sharevariants between various laboratories UCSC [32] providesaccess to the human reference sequence and HPO [18] pro-vides information about phenotypes associatedwith genes Insituations with a direct conflict in the information betweenthe different databases where possible the information withthe most severe clinical impact is displayed An example of
BioMed Research International 5
this is contradictory SIFT [33] and PolyPhen 2 [34] scores forvariants By default the most severe will be shown to the userwith the possibility to manually get more information whenrequested GensearchNGS also integrates the possibility totalk to other software installed by the user such as Alamut[35] allowing the user to easily open a variant found byGensearchNGS inside a local Alamut installation
3 Results
This chapter discusses how GensearchNGS implements thedifferent analysis steps and how it tries to lower as much aspossible the technical know-how and computing infrastruc-ture required for NGS data analysis
31 Software Suite GensearchNGS provides the user with thepossibility to create different projects to group the analysis ofvarious patients based on their type or their relation Whilemany other NGS analysis tools provide the possibility toorganize the data in projects GensearchNGS puts the notionof patients and their associatedmetainformation in the centerof the project management Every project contains one ormore patient and each can have several raw sequencing dataassociated The raw sequencing data can in turn have severalalignments associated for example using different alignersor alignment configurations Project specific settings such asspecific regions of interests to be used during the analysis canalso be configured Those regions can be described by BEDfiles to for example limit the analysis to regions relevant tothe type of analysis performed in the current project ThoseBED files can either be provided by the user or automaticallygenerated based on a list of gene names Projects alsoassociate the specific reference sequence to be used duringthe analysis The user can automatically download themfrom UCSC which provides reference sequences of the mostcommon human reference assemblies hg18 hg19 and hg38The usage of custom references sequences is also supportedThe patient centric database integrated into GensearchNGSallows the user to associate variants with patients a featurevery important in diagnostics The variants associated withpatients are shared between projects allowing the user toidentify variants found in different projects on previouslyanalysed patients
To perform the NGS data analysis GensearchNGS usesthe genes described in Ensembl as the gene model In a pro-cess transparent to the user the genemodel corresponding tothe reference sequence defined for the analysis is downloadedautomatically from Ensembl Additional metainformation isattached to the genes such as the associated phenotypesas described by HPO [18] or OMIM [30] Figure 2 showsthe main interface of GensearchNGS with the patient liston the left and the information about the currently selectedpatient in the center The central area of the image showsthe information about the currently selected patient Thisincludes his name internal ID associated phenotypes andall variants which have manually been confirmed by the userWhen validating variants the user can classify themmanuallyinto different categories The categories are based on the
Figure 2 Main interface of GensearchNGS showing a patient withits validated variants
ACMG variant classification guidelines [36] with the userrequested categories Artefact and False reference added Thesaved variants are used for any future variant detection in newpatients immediately telling the user if a variant has alreadybeen found in a different projectThe addition of the Artefactand False reference categories allows the user to quickly filterout common sequencing errors in his particular sequencingsetupThe hierarchical organization of the data is intended toguide the user through the analysis process while keeping itclear where the data comes from An example of this is thesequence alignment It is displayed as a child element of theraw sequencing data making the relation between the rawdata and the alignment immediately obvious
GensearchNGS does not provide the user with a singlebutton which goes from the raw sequencing data to the finalreport Instead of fully automating the pipeline Gensearch-NGS leads the user from one processing step to the nextBefore each step the user is given the choice to changeparameters or use the previously used (or standard) parame-ters After the initial import of the raw sequencing data theuser no longer has to handle the data files GensearchNGSperforms the required data file handling and conversionsbetween the processing steps
While there is no fully automated way to go from theraw sequencing data to the final variant report there is stilla way to automate a big part of the analysis When importingraw sequencing data the user has the possibility to performsequence alignment and variant calling automatically Thosetwo additional analysis steps are performed using eitherpreviously used configuration parameters or the default onesOnly the final variant validation has then to be performedby the user to create the final report Further details of thisprocess follow in the next chapter
32 Data Analysis To perform the initial quality control ofthe raw sequencing data GensearchNGS integrates its owntools allowing the user to import the raw data availablein different file formats The compatible data formats aredescribed in Section 2 The various formats are converted inthe standard FASTQ format during the data import step For
6 BioMed Research International
Figure 3 Overview of various statistics that the user can inspect to verify the data quality
the quality control of the raw sequencing data the user ispresented with various statistics as seen in Figure 3
The statistics allow the user to judge the quality of thesequencing data This includes statistics such as the lengthaverage quality andGC content distribution of the sequencedreads as well as the distribution of the sequenced basesand their quality for every read position and detection of
overrepresented K-mers at the start and the end of thesequenced reads
This raw data import process replaces existing tools likeFastQC to check the quality FASTX to filter the data andthe various utilities that transform different sequencing fileformats into the standard FASTQ file format Transparentlyfor the user the custom implementations of those tools
BioMed Research International 7
which produce the same results have been integrated into asingle processing stepThis includes the possibility to removeadapter sequences and split the raw sequencing data intomultiple files if they were produced through multiplexedsequencing Based on the reported quality information theuser can choose to filter the raw data following several filterssuch as maximum and minimum length of the sequencesand their minimal average quality He can also trim the 51015840and 31015840 parts of all reads or trim their excess length if theyare longer as a user defined length The trimming is done inaddition to the adapter and primer removalThe possibility tosplit the raw sequencing data based on barcodes in the readallowing the import of raw data from multiple samples thatwere sequenced at the same time is also integrated in theimport process
To align the sequenced data against a reference assemblyGensearchNGS provides the user with many options Severalexisting alignment algorithms are integrated into the applica-tion through plugins allowing their usage without the needof advanced technical know-how The currently integratedalignment algorithms are BWA [7] Bowtie 1 + 2 [8] andStampy [29]
Most of those alignment algorithms are developed in away that do not allow them to be used on multiple platformsIn fact most of them only run on Linux which is problematicfor a multiplatform application like GensearchNGS For thisreason a custom alignment algorithm written in Java hasbeen integrated directly into GensearchNGS This allows theuser to perform the full NGS data analysis even on operatingsystems likeWindows not supported bymost bioinformaticstools
The aligner supports paired end reads and gapped align-ment and has been optimized to use less than 5GB ofRAM to perform a full genome alignment This makes itsuitable for most of todayrsquos workspace desktops It is fullymultithreaded to support all cores of the machine and isable to offload calculations as described in Section 34 tomultiple computers The aligner uses a hash based indexa concept made popular through the BLAST aligner [9]with a custom heuristic based candidate alignment positionsselection [37] The final alignment is being done using theGotoh [38] algorithm a dynamic programming algorithmused for gapped alignment
The custom alignment algorithm being implemented inJava does not achieve quite the same performance as nativelyrunning aligners like BWA For WGS datasets it is generally50 slower The advantage of the GensearchNGS alignerover external aligners is that it is available on all supportedplatforms allowing the user to perform the complete NGSdata analysis on all platforms But when running on aplatform which supports the external aligners and speed isa deciding factor the users are encouraged to use the alignersavailable as plugins
The quality of resulting alignment can be verified throughvarious metrics by the user He can easily verify how manysequences have been discarded during the alignment pro-cess and how many sequences have been aligned to thedifferent chromosomes in the reference sequence Advancedstatistics similar to the ones created by bedtools 2 can also
be inspected giving detailed coverage information of thepreviously defined regions of interest The collected qualitycontrol information can be exported as a report for archivingpurposes using one of various formats such as PDF HTMLor PNG
While initially GensearchNGS integrated Varscan 2 [14]as its variant caller a replacement was created to reduce theexternal dependencies and to improve the performance ofvariant calling A variant scanner based on the model ofVarscan 2 has been implemented and is in the process of beingpublished as a separate tool called GNATY available for freefor noncommercial usage (httpgnatyphenosystemscom)Our custom variant caller shows important performanceadvantages over the existing Varscan 2 implementation of thesame variant calling model while producing the same analy-sis results Indeed GensearchNGS shows speed improvementof up to 18 times for variant calling compared to Varscan2 providing a similar range of features such as filters thatexclude sequencing artifacts
To be able to further filter down the detected variantsbased on their relevancy to the samplersquos phenotype severalsources are used to annotate them with various informationThis includes the prediction of the variant effect on thedifferent transcripts it affects a feature closely modeledafter the Variant Effect Predictor (VEP) from Ensembl [17]GensearchNGS implements its own version of the the VEPalgorithm to be able to perform the effect prediction in afraction of the time compared to the online VEP serviceThisis is mainly achieved by doing all calculations locally withouthaving to transfer the data to a remote server Predictionssuch as the addition or removal of a stop codon in an affectedtranscript if a splice site is affected and many others canbe made with this algorithm Additional information suchas if the variant is known in any public database associatedclinical significance and SIFT [33] and PolyPhen 2 [34]predictions are retrieved through the Ensembl web serviceOther services like the Human Phenotype Ontology [18]provide the information about the phenotype associated witha specific gene a crucial information in diagnostics
To filter the annotated variants GensearchNGS takesan interactive approach which allows the user to modifydifferent basic filters such as minimum or maximum samplefrequency coverage read balance or quality There are alsoadvanced filters such as the predicted consequence of thevariant as determined by the annotation process or if acertain phenotype is associated with the affected genes Thisincludes the possibility to filter by population frequency ofa variant The filtering by population frequency does not yettake into account the ethnicity of the patient Figure 4 showsan example of this list with several user selected interactivefilters applied
Additional features are available to perform a more in-depth analysis of variants such as family analysis where thevariants of multiple patients can be compared to find thoseonly present in a certain group of patients The usage ofpedigree information is of growing importance in NGS dataanalysis as it allows the user to filter sequencing artifactsand noncausative variants This is a very important featureespecially when usingWGSwhere the amount of variants can
8 BioMed Research International
Figure 4 Integrated variants list with several interactive filtersapplied
be very large Thanks to the usage of standard file formatslike VCF external tools like exome walker [39] can be usedto perform more advanced family analysis for features notcovered in GensearchNGS In contrast to many other NGSdata analysis software packages GensearchNGS does notfocus on the automation of every aspect of the analysisWhilecertain parts can be automated such as performing sequencealignment and variant calling on raw sequencing data thephilosophy of our software is to let the user to control thedifferent analysis steps This gives him enough flexibility toadapt to the data he is currently analysing without slowinghim down by having project specific analysis options whichare preselected for every analysis step This makes it possibleto perform most types of analysis with the project specificdefault options through a simple press of a button
33 Genome Browser A central tool of GensearchNGS isthe integrated genome browser to visualize the alignedsequencing data The genome browser as shown in Figure 5allows to interactively browse the sequence alignments Itis possible to display the aligned sequences and additionalgenomic data such as the gene locations and the encodedproteins by different transcripts
The genome browser allows the user to display sev-eral tracks containing information which helps to analysethe sequencing data The predefined tracks which can beactivated or deactivated allow the user to display coverageinformation GC content of the reference sequence thequality of the sequenced reads or custom annotation files instandard file formats such as BED WIG and GFF The focuswas put on optimizing the display of the data allowing theuser to visualize large datafiles without requiring a powerfulcomputerThis is achieved by streaming the data on demandonly loading the parts of the data currently needed forthe visualization The information about which variants arepresent in the alignment is available to the user in the sameform as the variant list in the main application It can be seenin Figure 4 allowing the user to quickly jump to the variantsof interest The genome browser like all of GensearchNGSis heavily connected with external data sources (EnsembldbSNP Entrez OMIM Blast and HPO) providing the user
Figure 5 Example view of the genome browser showing severalavailable tracks
with all relevant informationwhenneededTheuser can openexternal resources linked from many features in the visual-ized data such as genes transcripts or variants to retrieveadditional informationThe genome browser takes advantageof the integrated data sources like the Ensembl gene modelto display and interactively update encoded proteins basedon the variants present in the visualized data The genomebrowser is tightly integrated into GensearchNGS allowingthe user to easily visualize the alignment data belonging tofeatures of the sample
34 Distributed Computing The lack of computing powerto perform NGS data analysis is a very common issue inmany laboratories To address this problem GensearchNGSdoes not only optimize the algorithms for speed andmemoryusage but also try to exploit the already existing computinginfrastructure One way to do this is to use distributedcomputing a technique which allows the user to combinemultiple computers to perform computing intensive tasksThose computers can be in the same local network or at aremote location One of the most demanding steps in NGSdata analysis is sequence alignment For this reason it is thefirst feature to take advantage of this technique in Gensearch-NGS allowing the user to joinmultiple computers in the samenetwork to perform sequence alignment The approach usedby GensearchNGS is to transparently on request by the useruse all computers within the same networkwhereGensearch-NGS is installed Additionally the user has the possibility tostart up GensearchNGS instances in a cloud Currently onlythe amazon cloud is supportedThis process works regardlessof the operating system used by the different installationsof GensearchNGS By using an automatic service discoveryprotocol every installation of GensearchNGS located in thesame network is discovered without the need for manualconfigurationThis allows the user to use multiple computersat the same time or to completely offload calculations to adifferent computer making it possible to use computers notsuited for full genome alignments as an analysis workstationThe distributed sequence aligner used in GensearchNGS
BioMed Research International 9
currently only supports the GensearchNGS aligner algorithmand not external aligners such as BWA or Bowtie 2 It uses astream based approach where the client machine streams thedata to be aligned to all computers involved in the alignmentprocess This includes the client machine which receives thealigned sequences back and combines them into the finalalignment file The process of distributing the alignmentsover multiple computers and the cloud is transparent to theuser but it is optional because the usage of an externalcloud is not compatible with the security guidelines of somelaboratories GensearchNGS is not the onlyNGSdata analysissoftware using distributed sequence alignment It is thisability to combine the computing resources of the localcomputer multiple computers in the same network as well asthe cloud whichmakes its approach unique and very flexible
4 Discussion
GensearchNGS addresses problems that smaller laboratoriesare facing in various ways The issue of the technical com-plexity of the analysis is approached through an intuitive userinterface The user interface abstracts the complexity of theunderlying NGS data analysis toolchain which is a majorusability improvement over the usage of individual toolsNevertheless it gives the user full control over the individualanalysis steps instead of focusing on the creation of auto-mated analysis pipelines It also provides an original approachby providing a patient centric user interface This userfriendly interface allows users unfamiliar with the underlyingsoftware tools to fine tune the analysis parameters withoutoverwhelming themwith features or rarely used optionsThisincreases the number of people able to perform NGS dataanalysis This is particularly interesting for geneticists thatmight not have special bioinformatics training The issue ofincreasing infrastructure requirements for NGS data analysisis being addressed twofold First more efficient algorithmsare being developed like the heavily optimized variant callingalgorithm included in GensearchNGS Secondly the abilityto distribute heavy calculations over several computers oreven a cloud service has been developed GensearchNGShas a unique approach to this problem by enabling the userto combine the various distribution methods to performsequence alignment It provides the flexibility needed tochoose the best compromise between data privacy andalignment speed depending on the laboratory policies andneeds The resulting software enabled various laboratoriesto perform NGS data analysis and publish their results invarious fields The topics range from muscular dystrophies[40 41] to hearing loss [42] andmalignant hyperthermia [43]but also yet unpublished work about CNV analysis and thedetection of somatic mutations in cancer screening
5 Conclusion
In this paper we reviewed the workflow used to analyzeNGS data in a diagnostics environment The reviewedworkflow includes the detection of SNPs and short indelsfrom panel WES or WGS DNAseq data We also pre-sented issues faced by smaller laboratories trying to setup a
NGS data analysis pipeline We then presented Gensearch-NGS a software suite integrating various algorithms toperform the discussed workflow GensearchNGS is a com-mercially available software distributed by PhenosystemsSA A demo version of GensearchNGS is freely availableat httpwwwphenosystemscom The two main issues thetechnical complexity of the analysis and the computinginfrastructure requirements have been addressed as follows
To reduce the technical complexity of the analysis theuser is provided with an intuitive user interface which guideshim through the process of the data analysis He is providedwith the required tools to go from raw sequencing data the thefinal visualization and report generation for the sequencedsamples without having to learn complex bioinformaticstools The interface abstracts the technical complexity fromthe user while keeping enough flexibility to adapt to differentanalysis protocols By lowering the technical complexity ofNGS data analysis compared to existing individual analysistools GensearchNGS expands the potential user base of thesoftware
To address the issue of the computing infrastructurerequirements GensearchNGS focuses on the optimal usageof existing computing resources This is primarily achievedthrough the development of analysis algorithms that runon a minimal amount of resources while producing thesame analysis results as standard tools An example has beendescribed with the variant calling algorithm which producesthe same results as Varscan 2 but is 18 times faster Thesecond approach is the integration of distributed computingallowing the distribution of computing intensive tasks likealignment to be speed up by the usage of multiple computers
With a solid framework built for DNAseq analysisGensearchNGS will expand into further types of analysis inthe futureOne example is the integration of RNAseq analysisas the combination of OMICS data for a single sample isof increasing importance to understand rare diseases [44]In fact many parts of GensearchNGS such as the genomebrowser have already been adapted to support RNAseq dataThe integration of multiple data sources will not only allowthe user to integrate different types of OMICS data but alsoallow multiple views on the same sample One example ofthis is the integration of Sanger sequencing to NGS dataa very useful feature for diagnostics where it is commonpractice to validate variants identified through NGS withSanger sequencing
Further optimizations of the algorithms used in theanalysis will also be evaluated including optimization for theunderlying libraries One example is HTSJDK where severalperformance patches have been submitted and includedimproving NGS data analysis performance not only for usersof GensearchNGS but also for the complete bioinformaticscommunity
Disclosure
Pierre Kuonen has a nonpaid advisory position at Phenosys-tems SA David Atlan is working full time for PhenosystemsSA
10 BioMed Research International
Conflict of Interests
Thomas Dandekar has no conflict of interests regarding thepublication of this paper
Acknowledgments
GensearchNGS is a commercially available software dis-tributed by Phenosystems SA Beat Wolf rsquos PhD thesis ispartially financially supported by Phenosystems SA
References
[1] E L van Dijk H Auger Y Jaszczyszyn and C ThermesldquoTen years of next-generation sequencing technologyrdquo Trendsin Genetics vol 30 no 9 pp 418ndash426 2014
[2] B Sikkema-Raddatz L F Johansson E N de Boer et al ldquoTar-geted next-generation sequencing can replace sanger sequenc-ing in clinical diagnosticsrdquo Human Mutation vol 34 no 7 pp1035ndash1042 2013
[3] H P J Buermans and J T den Dunnen ldquoNext generationsequencing technology advances and applicationsrdquo Biochimicaet Biophysica ActamdashMolecular Basis of Disease vol 1842 no 10pp 1932ndash1941 2014
[4] FastQC ldquoA quality control tool for high throughput sequencedatardquo Babraham Bioinformatics Website httpwwwbioinfor-maticsbabrahamacukprojectsfastqc
[5] W R Pearson T Wood Z Zhang andWMiller ldquoComparisonof DNA sequences with protein sequencesrdquo Genomics vol 46no 1 pp 24ndash36 1997
[6] H Li and N Homer ldquoA survey of sequence alignment algo-rithms for next-generation sequencingrdquo Briefings in Bioinfor-matics vol 11 no 5 pp 473ndash483 2010
[7] H Li and R Durbin ldquoFast and accurate long-read alignmentwith Burrows-Wheeler transformrdquo Bioinformatics vol 26 no5 pp 589ndash595 2010
[8] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[9] S F Altschul T L Madden A A Schaffer et al ldquoGappedBLAST and PSI-BLAST a new generation of protein databasesearch programsrdquo Nucleic Acids Research vol 25 no 17 pp3389ndash3402 1997
[10] M A DePristo E Banks R Poplin et al ldquoA framework forvariation discovery and genotyping using next-generationDNAsequencing datardquo Nature Genetics vol 43 no 5 pp 491ndash4982011
[11] P Carnevali J Baccash A L Halpern et al ldquoComputationaltechniques for human genome resequencing using matedgapped readsrdquo Journal of Computational Biology vol 19 no 3pp 279ndash292 2012
[12] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[13] A R Quinlan and I M Hall ldquoBEDTools a flexible suite ofutilities for comparing genomic featuresrdquo Bioinformatics vol26 no 6 pp 841ndash842 2010
[14] D C Koboldt Q Zhang D E Larson et al ldquoVarScan 2 somaticmutation and copy number alteration discovery in cancer byexome sequencingrdquo Genome Research vol 22 no 3 pp 568ndash576 2012
[15] J OrsquoRawe T Jiang G Sun et al ldquoLow concordance of multiplevariant-calling pipelines practical implications for exome andgenome sequencingrdquo Genome Medicine vol 5 no 3 article 282013
[16] S T SherryM-HWardM Kholodov et al ldquodbSNP the NCBIdatabase of genetic variationrdquoNucleic Acids Research vol 29 pp308ndash311 2001
[17] W McLaren B Pritchard D Rios Y Chen P Flicek and FCunningham ldquoDeriving the consequences of genomic variantswith the EnsemblAPI and SNPEffect PredictorrdquoBioinformaticsvol 26 no 16 Article ID btq330 pp 2069ndash2070 2010
[18] P N Robinson and S Mundlos ldquoThe human phenotypeontologyrdquo Clinical Genetics vol 77 no 6 pp 525ndash534 2010
[19] P Danecek A Auton G Abecasis et al ldquoThe variant call formatand VCFtoolsrdquo Bioinformatics vol 27 no 15 pp 2156ndash21582011
[20] J T Robinson HThorvaldsdottir W Winckler et al ldquoIntegra-tive genomics viewerrdquo Nature Biotechnology vol 29 no 1 pp24ndash26 2011
[21] I Milne G Stephen M Bayer et al ldquoUsing tablet for visualexploration of second-generation sequencing datardquo Briefings inBioinformatics vol 14 no 2 Article ID bbs012 pp 193ndash2022013
[22] T Abeel T van Parys Y Saeys J Galagan and Y van de PeerldquoGenomeView a next-generation genome browserrdquo NucleicAcids Research vol 40 no 2 article e12 2012
[23] W J Kent C W Sugnet T S Furey et al ldquoThe human genomebrowser at UCSCrdquo Genome Research vol 12 no 6 pp 996ndash1006 2002
[24] F Cunningham M Ridwan Amode D Barrell et al ldquoEnsembl2015rdquo Nucleic Acids Research vol 43 no 1 pp D662ndashD6692015
[25] NGS Data Analysis Software List SEQanswers 2012 httpseqanswerscomwikiSoftwarelist
[26] CLC Genomics Workbench CLC Bio Aarhus Denmark 2014httpwwwclcbiocom
[27] NextGENe Softgenetics State College Pa USA httpwwwsoftgeneticscom
[28] J Goecks A Nekrutenko J Taylor et al ldquoGalaxy a com-prehensive approach for supporting accessible reproducibleand transparent computational research in the life sciencesrdquoGenome Biology vol 11 no 8 article R86 2010
[29] G Lunter and M Goodson ldquoStampy a statistical algorithm forsensitive and fast mapping of Illumina sequence readsrdquoGenomeResearch vol 21 no 6 pp 936ndash939 2011
[30] Online Mendelian Inheritance in Man OMIM McKusick-Nathans Institute of Genetic Medicine Johns Hopkins Univer-sity Baltimore Md USA 2015 httpomimorg
[31] Cafe Variome httpwwwcafevariomeorg[32] The Genome Sequencing Consortium ldquoInitial sequencing and
analysis of the human genomerdquo Nature vol 409 pp 860ndash9212001
[33] P Kumar S Henikoff and P C Ng ldquoPredicting the effects ofcoding non-synonymous variants on protein function using theSIFT algorithmrdquo Nature Protocols vol 4 no 7 pp 1073ndash10822009
[34] I A Adzhubei S Schmidt L Peshkin et al ldquoA method andserver for predicting damaging missense mutationsrdquo NatureMethods vol 7 no 4 pp 248ndash249 2010
[35] Alamut Visual Interactive Biosoftware Rouen France httpwwwinteractivebiosoftwarecomalamut-visual
BioMed Research International 11
[36] S Richards N Aziz S Bale et al ldquoStandards and guidelinesfor the interpretation of sequence variants a joint consensusrecommendation of the American College of Medical Geneticsand Genomics and the Association for Molecular PathologyrdquoGenetics in Medicine vol 17 no 5 pp 405ndash423 2015
[37] BWolf and P Kuonen ldquoA novel approach for heuristic pairwiseDNA sequence alignmentrdquo in Proceedings of the InternationalConference on Bioinformatics amp Computational Biology (BIO-COMP rsquo13) Las Vegas Nev USA July 2013
[38] O Gotoh ldquoAn improved algorithm for matching biologicalsequencesrdquo Journal ofMolecular Biology vol 162 no 3 pp 705ndash708 1982
[39] D Smedley S Kohler J C Czeschik et al ldquoWalking theinteractome for candidate prioritization in exome sequencingstudies of Mendelian diseasesrdquo Bioinformatics vol 30 no 22pp 3215ndash3222 2014
[40] A L Semmler S Sacconi J Bach et al ldquoUnusual multisys-temic involvement and a novel BAG3 mutation revealed byNGS screening in a large cohort of myofibrillar myopathiesrdquoOrphanet Journal of Rare Diseases vol 9 no 1 p 121 2014
[41] M Larsen S Rost N El Hajj et al ldquoDiagnostic approach forFSHD revisited SMCHD1 mutations cause FSHD2 and act asmodifiers of disease severity in FSHD1rdquo European Journal ofHuman Genetics vol 23 no 6 pp 808ndash816 2015
[42] S Rost E Bach C Neuner et al ldquoNovel form of X-linkednonsyndromic hearing loss with cochlear malformation causedby a mutation in the type IV collagen gene COL4A6rdquo EuropeanJournal of Human Genetics vol 22 no 2 pp 208ndash215 2014
[43] M Broman I Kleinschnitz J E Bach S Rost G Islander andC R Muller ldquoNext-generation DNA sequencing of a Swedishmalignant hyperthermia cohortrdquo Clinical Genetics 2014
[44] T Low S vanHeesch H van den Toorn et al ldquoQuantitative andqualitative proteome characteristics extracted from in-depthintegrated genomics and proteomics analysisrdquo Cell Reports vol5 no 5 pp 1469ndash1478 2013
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

BioMed Research International 3
FastQCFASTX
BWABowtie 2BLASTand so forth
GATKcomplete genomicsrealigner
Data preparation
(A) QCfiltering
(B) Alignment
Data analysis
(D) QC
(E) Variant calling
(F) Annotation
(G) Variant filtering
Samtoolsbedtools 2
GATKsamtoolsVarscan 2
dbSNPPhenomizerVEPand so forth
Samtools
Validation and reporting
IGVTabletgenome viewUCSCbrowser and so forth
(I) Sanger validation
(J) Report generation
(H) Visualization
(C) Realignment
Figure 1 UML activity diagram of a NGS diagnostics workflow separated into different steps A collection of the available tools for everystep is mentioned
affected and their predicted effects on those genes Varioustools exist to annotate variants either locally or through aweb service Some of the more popular ones are the VariantEffect Predictor (VEP) from Ensembl [17] and Phenomizer[18] that add clinically relevant information to the detectedvariants Once the annotated list of variants has been createdthe phenotype relevant variant has to be identified
(G) Variant Filtering Analysis specific filters allow the userto filter the number of variants down to a smaller numberOne example is to only keep variants affecting genes withphenotypes matching the ones of the patient and that have apredicted consequence on the gene function The exact typeof filters to reduce the amount of candidate variants variesfrom case to case depending on the available informationThe filtering is often done using bcftools which is partof the samtools project [12] or vcftools [19] If during thisstep no clinically relevant variant related to the currentanalysis can be found depending on the analysis protocolthe filters may be relaxed This allows the user to includefor example variants on genes where the relation with thecurrent phenotype is less certain or variants with uncertainconsequences on a known disease gene
(H) Visualization Once a list of candidate variants has beenidentified it is common to visually verify them in a genomebrowser The genome browser displays the sequenced readsaround the variant and allows the user to verify the qualityof the data around that position allowing the user to identifyand discard variants originating from sequencing errorsThisis done using one of the various genome browsers installedlocally such as IGV [20] Tablet [21] or Genome view [22]There also exist web-based genome browsers such as theUCSC [23] or Ensembl [24] genome browsers
(I) Sanger Validation Before submitting the final findingsto the clinician it is still common practice to validatethe identified variants through Sanger sequencing Whilefuture advances in NGS technologies might make this stepredundant it is still considered the gold standard for variantvalidation
(J) Report Generation After visually inspecting and validatingthe variants the identified variants or the lack thereof aredocumented in a final report that is sent off to the clinicianto allow him to take decisions on the case
The described process can be done by taking severalapproaches A common approach is to use a selection ofthe previously described tools to perform the individualanalysis steps The different tools are usually connectedtogether through a series of automated scripts performingthe required analysis The creation of the resulting pipelineis a complex technical task as it requires insight in theworking of the different tools and the ability to chain themtogether This is a nontrivial task as not all tools follow thesame standards in terms of data file formats and handlingNonetheless this is a very common approach which resultedin a variety of custom made NGS analysis pipelines eitherprivate or public [25]
Complete software packages like CLCBio [26]NextGENe [27] or theweb-basedGalaxy [28] also existTheyallow the user to combine many of the mentioned analysistools without having to worry too much on how to connectthe different tools to perform the analysis Those toolsare focused on giving the user the possibility to automatethe analysis process as much as possible abstracting thecomplexity of the analysis GensearchNGS belongs to thisfamily of tools with its own approach and focus on theissues of smaller laboratories It focuses less on the possibility
4 BioMed Research International
to generate a custom automated pipeline but more on anintuitive interface to handle the data-files to perform theanalysis in a predefined pipeline optimised for diagnostics
The features of GensearchNGS are limited to thoseneeded to perform the presented NGS diagnostics workflowThis means the overall feature set is smaller than that of othercomplete software packages especially CLCBio or Galaxywhich both offer a wide variety of functionalities Havinga limited feature set allows for a cleaner and more focuseduser interface one of the major goals of GensearchNGSsomething that is highly appreciated by current users ofGensearchNGS
Aswill be discussed in the following chapters Gensearch-NGS uses either external applications through plugins orcustom implementations of standard algorithms for the dif-ferent analysis stepsThe resulting analysis quality is thereforecomparable to the one observed when using those toolsseparately by hand or through a software package like Galaxy
Approach The issues faced by small laboratories when usingthe previously mentioned tools which are often limitedin terms of computing and human resources are variousBased on our experience the biggest issue faced by smallerlaboratories is the lack of technical know-how and dedicatedbioinformaticians The usage and configuration of many ofthe described data analysis tools require a dedicated employeewhich not available in every laboratoryThis is especially truein emergingmarketswheremanymonetary restrictions are inplace The approach of solutions like Galaxy and CLCBio tothis problem is to abstract the usage of the underlying analysistools and provide a way for the user to automate the analysisprocessWhile the abstraction of the underlying tools reducesthe technical complexity of the analysis process this is oftennot enough tomake the analysis process more accessibleThecited tools can be used for many other types of analyses thanthe ones looked at in this paper which leads to a much morecomplex user interface and often overwhelms the user withmany options The cited tools also focus more on the abilityto create automated analysis pipelines leading to very littleinteractivity between the user and the data
The second issue often encountered is the lack of a suitablecomputing infrastructure to perform the computationallyintensive parts of the analysis Sequence alignment in par-ticular is very demanding in that regard Even though manylaboratories outsource the sequencing and alignment processthe possibility to realign the raw sequencing data is oftenrequired To perform the described analysis steps and moreGensearchNGS provides a complete software suite focusingon optimized algorithms and usage of distributed computingfor optimal usage of the existing infrastructure All theanalysis steps make usage of multiprocessor systems to speedup the analysis whereas only sequence alignment using thecustomGensearchNGS alignment algorithm uses distributedcomputing The entire functionality is provided throughan intuitive user interface to lower the technical know-how needed to perform NGS data analysis GensearchNGSprovides its own way on how the analysis is approachedby providing the user with a patient centric user interface
and favoring an interactive analysis approach instead ofa fully automated analysis pipeline The user interface isfocused on providing the functionality required to performthe previously described analysis without overwhelming theuser with rarely used features
2 Materials and Methods
GensearchNGS has been developed as a multiplatform appli-cation running onWindows Linux and OSX using Java 6+The application is a locally installed software and is not webbased In order to perform NGS analysis steps Gensearch-NGS includes several existing external tools through pluginsThose tools are mainly used during the sequence alignmentphase giving the user the choice between different alignmentalgorithms which may have specific advantages related tothe data to be analysed The external alignment algorithmssupported through plugins are BWA [7] and Bowtie 1 + 2[8] as well as Stampy [29] GensearchNGS can either uselocally installed versions of those external tools or downloadand update them automatically If the tools are installedby the user he can update them when desired otherwisethe same update mechanism used to update GensearchNGSitself is used to update the external tools For sequencingdata GensearchNGS supports various input and output fileformats For raw sequencing data the standard FASTQformat as well as FASTA SFF and FNA is supported All ofthem are converted to the standard FASTQ file format duringthe data import phase As FASTQ files require a quality scorefor every nucleotide in the sequenced reads a default qualityscore is assumed for all the nucleotides if the source formatlike FASTA is missing this information For aligned databoth SAM and BAM files are supported whereas SAM filesare converted to BAM during import BAM and SAM fileare accessed through the HTSJDK library HTSJDK is anopen source Java library developed as part of the samtoolsproject [12] It provides a standardized API to access variousNGS data file formats The library can be downloaded athttpsamtoolsgithubiohtsjdk Annotations can be pro-vided through the BED WIG and GFF files and the VCF fileformat is used to import or export variants
To benefit from the knowledge stored in various onlinedatabases GensearchNGS has been connected to several ofthem to annotate the NGS data on various levels from thegenes and their transcripts down to the individual variantsFor most of the annotation data GensearchNGS accessesthe Ensembl [24] web service through its biomart APIgiving the application access to gene and variant informationEnsembl provides a unified way to access several other onlinedatabases like dbSNP [16] or OMIM [30] through a singleonline service In addition to Ensembl GensearchNGS inte-grates other online services for additional data and featuresCafe Variome [31] provides a standardized way to sharevariants between various laboratories UCSC [32] providesaccess to the human reference sequence and HPO [18] pro-vides information about phenotypes associatedwith genes Insituations with a direct conflict in the information betweenthe different databases where possible the information withthe most severe clinical impact is displayed An example of
BioMed Research International 5
this is contradictory SIFT [33] and PolyPhen 2 [34] scores forvariants By default the most severe will be shown to the userwith the possibility to manually get more information whenrequested GensearchNGS also integrates the possibility totalk to other software installed by the user such as Alamut[35] allowing the user to easily open a variant found byGensearchNGS inside a local Alamut installation
3 Results
This chapter discusses how GensearchNGS implements thedifferent analysis steps and how it tries to lower as much aspossible the technical know-how and computing infrastruc-ture required for NGS data analysis
31 Software Suite GensearchNGS provides the user with thepossibility to create different projects to group the analysis ofvarious patients based on their type or their relation Whilemany other NGS analysis tools provide the possibility toorganize the data in projects GensearchNGS puts the notionof patients and their associatedmetainformation in the centerof the project management Every project contains one ormore patient and each can have several raw sequencing dataassociated The raw sequencing data can in turn have severalalignments associated for example using different alignersor alignment configurations Project specific settings such asspecific regions of interests to be used during the analysis canalso be configured Those regions can be described by BEDfiles to for example limit the analysis to regions relevant tothe type of analysis performed in the current project ThoseBED files can either be provided by the user or automaticallygenerated based on a list of gene names Projects alsoassociate the specific reference sequence to be used duringthe analysis The user can automatically download themfrom UCSC which provides reference sequences of the mostcommon human reference assemblies hg18 hg19 and hg38The usage of custom references sequences is also supportedThe patient centric database integrated into GensearchNGSallows the user to associate variants with patients a featurevery important in diagnostics The variants associated withpatients are shared between projects allowing the user toidentify variants found in different projects on previouslyanalysed patients
To perform the NGS data analysis GensearchNGS usesthe genes described in Ensembl as the gene model In a pro-cess transparent to the user the genemodel corresponding tothe reference sequence defined for the analysis is downloadedautomatically from Ensembl Additional metainformation isattached to the genes such as the associated phenotypesas described by HPO [18] or OMIM [30] Figure 2 showsthe main interface of GensearchNGS with the patient liston the left and the information about the currently selectedpatient in the center The central area of the image showsthe information about the currently selected patient Thisincludes his name internal ID associated phenotypes andall variants which have manually been confirmed by the userWhen validating variants the user can classify themmanuallyinto different categories The categories are based on the
Figure 2 Main interface of GensearchNGS showing a patient withits validated variants
ACMG variant classification guidelines [36] with the userrequested categories Artefact and False reference added Thesaved variants are used for any future variant detection in newpatients immediately telling the user if a variant has alreadybeen found in a different projectThe addition of the Artefactand False reference categories allows the user to quickly filterout common sequencing errors in his particular sequencingsetupThe hierarchical organization of the data is intended toguide the user through the analysis process while keeping itclear where the data comes from An example of this is thesequence alignment It is displayed as a child element of theraw sequencing data making the relation between the rawdata and the alignment immediately obvious
GensearchNGS does not provide the user with a singlebutton which goes from the raw sequencing data to the finalreport Instead of fully automating the pipeline Gensearch-NGS leads the user from one processing step to the nextBefore each step the user is given the choice to changeparameters or use the previously used (or standard) parame-ters After the initial import of the raw sequencing data theuser no longer has to handle the data files GensearchNGSperforms the required data file handling and conversionsbetween the processing steps
While there is no fully automated way to go from theraw sequencing data to the final variant report there is stilla way to automate a big part of the analysis When importingraw sequencing data the user has the possibility to performsequence alignment and variant calling automatically Thosetwo additional analysis steps are performed using eitherpreviously used configuration parameters or the default onesOnly the final variant validation has then to be performedby the user to create the final report Further details of thisprocess follow in the next chapter
32 Data Analysis To perform the initial quality control ofthe raw sequencing data GensearchNGS integrates its owntools allowing the user to import the raw data availablein different file formats The compatible data formats aredescribed in Section 2 The various formats are converted inthe standard FASTQ format during the data import step For
6 BioMed Research International
Figure 3 Overview of various statistics that the user can inspect to verify the data quality
the quality control of the raw sequencing data the user ispresented with various statistics as seen in Figure 3
The statistics allow the user to judge the quality of thesequencing data This includes statistics such as the lengthaverage quality andGC content distribution of the sequencedreads as well as the distribution of the sequenced basesand their quality for every read position and detection of
overrepresented K-mers at the start and the end of thesequenced reads
This raw data import process replaces existing tools likeFastQC to check the quality FASTX to filter the data andthe various utilities that transform different sequencing fileformats into the standard FASTQ file format Transparentlyfor the user the custom implementations of those tools
BioMed Research International 7
which produce the same results have been integrated into asingle processing stepThis includes the possibility to removeadapter sequences and split the raw sequencing data intomultiple files if they were produced through multiplexedsequencing Based on the reported quality information theuser can choose to filter the raw data following several filterssuch as maximum and minimum length of the sequencesand their minimal average quality He can also trim the 51015840and 31015840 parts of all reads or trim their excess length if theyare longer as a user defined length The trimming is done inaddition to the adapter and primer removalThe possibility tosplit the raw sequencing data based on barcodes in the readallowing the import of raw data from multiple samples thatwere sequenced at the same time is also integrated in theimport process
To align the sequenced data against a reference assemblyGensearchNGS provides the user with many options Severalexisting alignment algorithms are integrated into the applica-tion through plugins allowing their usage without the needof advanced technical know-how The currently integratedalignment algorithms are BWA [7] Bowtie 1 + 2 [8] andStampy [29]
Most of those alignment algorithms are developed in away that do not allow them to be used on multiple platformsIn fact most of them only run on Linux which is problematicfor a multiplatform application like GensearchNGS For thisreason a custom alignment algorithm written in Java hasbeen integrated directly into GensearchNGS This allows theuser to perform the full NGS data analysis even on operatingsystems likeWindows not supported bymost bioinformaticstools
The aligner supports paired end reads and gapped align-ment and has been optimized to use less than 5GB ofRAM to perform a full genome alignment This makes itsuitable for most of todayrsquos workspace desktops It is fullymultithreaded to support all cores of the machine and isable to offload calculations as described in Section 34 tomultiple computers The aligner uses a hash based indexa concept made popular through the BLAST aligner [9]with a custom heuristic based candidate alignment positionsselection [37] The final alignment is being done using theGotoh [38] algorithm a dynamic programming algorithmused for gapped alignment
The custom alignment algorithm being implemented inJava does not achieve quite the same performance as nativelyrunning aligners like BWA For WGS datasets it is generally50 slower The advantage of the GensearchNGS alignerover external aligners is that it is available on all supportedplatforms allowing the user to perform the complete NGSdata analysis on all platforms But when running on aplatform which supports the external aligners and speed isa deciding factor the users are encouraged to use the alignersavailable as plugins
The quality of resulting alignment can be verified throughvarious metrics by the user He can easily verify how manysequences have been discarded during the alignment pro-cess and how many sequences have been aligned to thedifferent chromosomes in the reference sequence Advancedstatistics similar to the ones created by bedtools 2 can also
be inspected giving detailed coverage information of thepreviously defined regions of interest The collected qualitycontrol information can be exported as a report for archivingpurposes using one of various formats such as PDF HTMLor PNG
While initially GensearchNGS integrated Varscan 2 [14]as its variant caller a replacement was created to reduce theexternal dependencies and to improve the performance ofvariant calling A variant scanner based on the model ofVarscan 2 has been implemented and is in the process of beingpublished as a separate tool called GNATY available for freefor noncommercial usage (httpgnatyphenosystemscom)Our custom variant caller shows important performanceadvantages over the existing Varscan 2 implementation of thesame variant calling model while producing the same analy-sis results Indeed GensearchNGS shows speed improvementof up to 18 times for variant calling compared to Varscan2 providing a similar range of features such as filters thatexclude sequencing artifacts
To be able to further filter down the detected variantsbased on their relevancy to the samplersquos phenotype severalsources are used to annotate them with various informationThis includes the prediction of the variant effect on thedifferent transcripts it affects a feature closely modeledafter the Variant Effect Predictor (VEP) from Ensembl [17]GensearchNGS implements its own version of the the VEPalgorithm to be able to perform the effect prediction in afraction of the time compared to the online VEP serviceThisis is mainly achieved by doing all calculations locally withouthaving to transfer the data to a remote server Predictionssuch as the addition or removal of a stop codon in an affectedtranscript if a splice site is affected and many others canbe made with this algorithm Additional information suchas if the variant is known in any public database associatedclinical significance and SIFT [33] and PolyPhen 2 [34]predictions are retrieved through the Ensembl web serviceOther services like the Human Phenotype Ontology [18]provide the information about the phenotype associated witha specific gene a crucial information in diagnostics
To filter the annotated variants GensearchNGS takesan interactive approach which allows the user to modifydifferent basic filters such as minimum or maximum samplefrequency coverage read balance or quality There are alsoadvanced filters such as the predicted consequence of thevariant as determined by the annotation process or if acertain phenotype is associated with the affected genes Thisincludes the possibility to filter by population frequency ofa variant The filtering by population frequency does not yettake into account the ethnicity of the patient Figure 4 showsan example of this list with several user selected interactivefilters applied
Additional features are available to perform a more in-depth analysis of variants such as family analysis where thevariants of multiple patients can be compared to find thoseonly present in a certain group of patients The usage ofpedigree information is of growing importance in NGS dataanalysis as it allows the user to filter sequencing artifactsand noncausative variants This is a very important featureespecially when usingWGSwhere the amount of variants can
8 BioMed Research International
Figure 4 Integrated variants list with several interactive filtersapplied
be very large Thanks to the usage of standard file formatslike VCF external tools like exome walker [39] can be usedto perform more advanced family analysis for features notcovered in GensearchNGS In contrast to many other NGSdata analysis software packages GensearchNGS does notfocus on the automation of every aspect of the analysisWhilecertain parts can be automated such as performing sequencealignment and variant calling on raw sequencing data thephilosophy of our software is to let the user to control thedifferent analysis steps This gives him enough flexibility toadapt to the data he is currently analysing without slowinghim down by having project specific analysis options whichare preselected for every analysis step This makes it possibleto perform most types of analysis with the project specificdefault options through a simple press of a button
33 Genome Browser A central tool of GensearchNGS isthe integrated genome browser to visualize the alignedsequencing data The genome browser as shown in Figure 5allows to interactively browse the sequence alignments Itis possible to display the aligned sequences and additionalgenomic data such as the gene locations and the encodedproteins by different transcripts
The genome browser allows the user to display sev-eral tracks containing information which helps to analysethe sequencing data The predefined tracks which can beactivated or deactivated allow the user to display coverageinformation GC content of the reference sequence thequality of the sequenced reads or custom annotation files instandard file formats such as BED WIG and GFF The focuswas put on optimizing the display of the data allowing theuser to visualize large datafiles without requiring a powerfulcomputerThis is achieved by streaming the data on demandonly loading the parts of the data currently needed forthe visualization The information about which variants arepresent in the alignment is available to the user in the sameform as the variant list in the main application It can be seenin Figure 4 allowing the user to quickly jump to the variantsof interest The genome browser like all of GensearchNGSis heavily connected with external data sources (EnsembldbSNP Entrez OMIM Blast and HPO) providing the user
Figure 5 Example view of the genome browser showing severalavailable tracks
with all relevant informationwhenneededTheuser can openexternal resources linked from many features in the visual-ized data such as genes transcripts or variants to retrieveadditional informationThe genome browser takes advantageof the integrated data sources like the Ensembl gene modelto display and interactively update encoded proteins basedon the variants present in the visualized data The genomebrowser is tightly integrated into GensearchNGS allowingthe user to easily visualize the alignment data belonging tofeatures of the sample
34 Distributed Computing The lack of computing powerto perform NGS data analysis is a very common issue inmany laboratories To address this problem GensearchNGSdoes not only optimize the algorithms for speed andmemoryusage but also try to exploit the already existing computinginfrastructure One way to do this is to use distributedcomputing a technique which allows the user to combinemultiple computers to perform computing intensive tasksThose computers can be in the same local network or at aremote location One of the most demanding steps in NGSdata analysis is sequence alignment For this reason it is thefirst feature to take advantage of this technique in Gensearch-NGS allowing the user to joinmultiple computers in the samenetwork to perform sequence alignment The approach usedby GensearchNGS is to transparently on request by the useruse all computers within the same networkwhereGensearch-NGS is installed Additionally the user has the possibility tostart up GensearchNGS instances in a cloud Currently onlythe amazon cloud is supportedThis process works regardlessof the operating system used by the different installationsof GensearchNGS By using an automatic service discoveryprotocol every installation of GensearchNGS located in thesame network is discovered without the need for manualconfigurationThis allows the user to use multiple computersat the same time or to completely offload calculations to adifferent computer making it possible to use computers notsuited for full genome alignments as an analysis workstationThe distributed sequence aligner used in GensearchNGS
BioMed Research International 9
currently only supports the GensearchNGS aligner algorithmand not external aligners such as BWA or Bowtie 2 It uses astream based approach where the client machine streams thedata to be aligned to all computers involved in the alignmentprocess This includes the client machine which receives thealigned sequences back and combines them into the finalalignment file The process of distributing the alignmentsover multiple computers and the cloud is transparent to theuser but it is optional because the usage of an externalcloud is not compatible with the security guidelines of somelaboratories GensearchNGS is not the onlyNGSdata analysissoftware using distributed sequence alignment It is thisability to combine the computing resources of the localcomputer multiple computers in the same network as well asthe cloud whichmakes its approach unique and very flexible
4 Discussion
GensearchNGS addresses problems that smaller laboratoriesare facing in various ways The issue of the technical com-plexity of the analysis is approached through an intuitive userinterface The user interface abstracts the complexity of theunderlying NGS data analysis toolchain which is a majorusability improvement over the usage of individual toolsNevertheless it gives the user full control over the individualanalysis steps instead of focusing on the creation of auto-mated analysis pipelines It also provides an original approachby providing a patient centric user interface This userfriendly interface allows users unfamiliar with the underlyingsoftware tools to fine tune the analysis parameters withoutoverwhelming themwith features or rarely used optionsThisincreases the number of people able to perform NGS dataanalysis This is particularly interesting for geneticists thatmight not have special bioinformatics training The issue ofincreasing infrastructure requirements for NGS data analysisis being addressed twofold First more efficient algorithmsare being developed like the heavily optimized variant callingalgorithm included in GensearchNGS Secondly the abilityto distribute heavy calculations over several computers oreven a cloud service has been developed GensearchNGShas a unique approach to this problem by enabling the userto combine the various distribution methods to performsequence alignment It provides the flexibility needed tochoose the best compromise between data privacy andalignment speed depending on the laboratory policies andneeds The resulting software enabled various laboratoriesto perform NGS data analysis and publish their results invarious fields The topics range from muscular dystrophies[40 41] to hearing loss [42] andmalignant hyperthermia [43]but also yet unpublished work about CNV analysis and thedetection of somatic mutations in cancer screening
5 Conclusion
In this paper we reviewed the workflow used to analyzeNGS data in a diagnostics environment The reviewedworkflow includes the detection of SNPs and short indelsfrom panel WES or WGS DNAseq data We also pre-sented issues faced by smaller laboratories trying to setup a
NGS data analysis pipeline We then presented Gensearch-NGS a software suite integrating various algorithms toperform the discussed workflow GensearchNGS is a com-mercially available software distributed by PhenosystemsSA A demo version of GensearchNGS is freely availableat httpwwwphenosystemscom The two main issues thetechnical complexity of the analysis and the computinginfrastructure requirements have been addressed as follows
To reduce the technical complexity of the analysis theuser is provided with an intuitive user interface which guideshim through the process of the data analysis He is providedwith the required tools to go from raw sequencing data the thefinal visualization and report generation for the sequencedsamples without having to learn complex bioinformaticstools The interface abstracts the technical complexity fromthe user while keeping enough flexibility to adapt to differentanalysis protocols By lowering the technical complexity ofNGS data analysis compared to existing individual analysistools GensearchNGS expands the potential user base of thesoftware
To address the issue of the computing infrastructurerequirements GensearchNGS focuses on the optimal usageof existing computing resources This is primarily achievedthrough the development of analysis algorithms that runon a minimal amount of resources while producing thesame analysis results as standard tools An example has beendescribed with the variant calling algorithm which producesthe same results as Varscan 2 but is 18 times faster Thesecond approach is the integration of distributed computingallowing the distribution of computing intensive tasks likealignment to be speed up by the usage of multiple computers
With a solid framework built for DNAseq analysisGensearchNGS will expand into further types of analysis inthe futureOne example is the integration of RNAseq analysisas the combination of OMICS data for a single sample isof increasing importance to understand rare diseases [44]In fact many parts of GensearchNGS such as the genomebrowser have already been adapted to support RNAseq dataThe integration of multiple data sources will not only allowthe user to integrate different types of OMICS data but alsoallow multiple views on the same sample One example ofthis is the integration of Sanger sequencing to NGS dataa very useful feature for diagnostics where it is commonpractice to validate variants identified through NGS withSanger sequencing
Further optimizations of the algorithms used in theanalysis will also be evaluated including optimization for theunderlying libraries One example is HTSJDK where severalperformance patches have been submitted and includedimproving NGS data analysis performance not only for usersof GensearchNGS but also for the complete bioinformaticscommunity
Disclosure
Pierre Kuonen has a nonpaid advisory position at Phenosys-tems SA David Atlan is working full time for PhenosystemsSA
10 BioMed Research International
Conflict of Interests
Thomas Dandekar has no conflict of interests regarding thepublication of this paper
Acknowledgments
GensearchNGS is a commercially available software dis-tributed by Phenosystems SA Beat Wolf rsquos PhD thesis ispartially financially supported by Phenosystems SA
References
[1] E L van Dijk H Auger Y Jaszczyszyn and C ThermesldquoTen years of next-generation sequencing technologyrdquo Trendsin Genetics vol 30 no 9 pp 418ndash426 2014
[2] B Sikkema-Raddatz L F Johansson E N de Boer et al ldquoTar-geted next-generation sequencing can replace sanger sequenc-ing in clinical diagnosticsrdquo Human Mutation vol 34 no 7 pp1035ndash1042 2013
[3] H P J Buermans and J T den Dunnen ldquoNext generationsequencing technology advances and applicationsrdquo Biochimicaet Biophysica ActamdashMolecular Basis of Disease vol 1842 no 10pp 1932ndash1941 2014
[4] FastQC ldquoA quality control tool for high throughput sequencedatardquo Babraham Bioinformatics Website httpwwwbioinfor-maticsbabrahamacukprojectsfastqc
[5] W R Pearson T Wood Z Zhang andWMiller ldquoComparisonof DNA sequences with protein sequencesrdquo Genomics vol 46no 1 pp 24ndash36 1997
[6] H Li and N Homer ldquoA survey of sequence alignment algo-rithms for next-generation sequencingrdquo Briefings in Bioinfor-matics vol 11 no 5 pp 473ndash483 2010
[7] H Li and R Durbin ldquoFast and accurate long-read alignmentwith Burrows-Wheeler transformrdquo Bioinformatics vol 26 no5 pp 589ndash595 2010
[8] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[9] S F Altschul T L Madden A A Schaffer et al ldquoGappedBLAST and PSI-BLAST a new generation of protein databasesearch programsrdquo Nucleic Acids Research vol 25 no 17 pp3389ndash3402 1997
[10] M A DePristo E Banks R Poplin et al ldquoA framework forvariation discovery and genotyping using next-generationDNAsequencing datardquo Nature Genetics vol 43 no 5 pp 491ndash4982011
[11] P Carnevali J Baccash A L Halpern et al ldquoComputationaltechniques for human genome resequencing using matedgapped readsrdquo Journal of Computational Biology vol 19 no 3pp 279ndash292 2012
[12] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[13] A R Quinlan and I M Hall ldquoBEDTools a flexible suite ofutilities for comparing genomic featuresrdquo Bioinformatics vol26 no 6 pp 841ndash842 2010
[14] D C Koboldt Q Zhang D E Larson et al ldquoVarScan 2 somaticmutation and copy number alteration discovery in cancer byexome sequencingrdquo Genome Research vol 22 no 3 pp 568ndash576 2012
[15] J OrsquoRawe T Jiang G Sun et al ldquoLow concordance of multiplevariant-calling pipelines practical implications for exome andgenome sequencingrdquo Genome Medicine vol 5 no 3 article 282013
[16] S T SherryM-HWardM Kholodov et al ldquodbSNP the NCBIdatabase of genetic variationrdquoNucleic Acids Research vol 29 pp308ndash311 2001
[17] W McLaren B Pritchard D Rios Y Chen P Flicek and FCunningham ldquoDeriving the consequences of genomic variantswith the EnsemblAPI and SNPEffect PredictorrdquoBioinformaticsvol 26 no 16 Article ID btq330 pp 2069ndash2070 2010
[18] P N Robinson and S Mundlos ldquoThe human phenotypeontologyrdquo Clinical Genetics vol 77 no 6 pp 525ndash534 2010
[19] P Danecek A Auton G Abecasis et al ldquoThe variant call formatand VCFtoolsrdquo Bioinformatics vol 27 no 15 pp 2156ndash21582011
[20] J T Robinson HThorvaldsdottir W Winckler et al ldquoIntegra-tive genomics viewerrdquo Nature Biotechnology vol 29 no 1 pp24ndash26 2011
[21] I Milne G Stephen M Bayer et al ldquoUsing tablet for visualexploration of second-generation sequencing datardquo Briefings inBioinformatics vol 14 no 2 Article ID bbs012 pp 193ndash2022013
[22] T Abeel T van Parys Y Saeys J Galagan and Y van de PeerldquoGenomeView a next-generation genome browserrdquo NucleicAcids Research vol 40 no 2 article e12 2012
[23] W J Kent C W Sugnet T S Furey et al ldquoThe human genomebrowser at UCSCrdquo Genome Research vol 12 no 6 pp 996ndash1006 2002
[24] F Cunningham M Ridwan Amode D Barrell et al ldquoEnsembl2015rdquo Nucleic Acids Research vol 43 no 1 pp D662ndashD6692015
[25] NGS Data Analysis Software List SEQanswers 2012 httpseqanswerscomwikiSoftwarelist
[26] CLC Genomics Workbench CLC Bio Aarhus Denmark 2014httpwwwclcbiocom
[27] NextGENe Softgenetics State College Pa USA httpwwwsoftgeneticscom
[28] J Goecks A Nekrutenko J Taylor et al ldquoGalaxy a com-prehensive approach for supporting accessible reproducibleand transparent computational research in the life sciencesrdquoGenome Biology vol 11 no 8 article R86 2010
[29] G Lunter and M Goodson ldquoStampy a statistical algorithm forsensitive and fast mapping of Illumina sequence readsrdquoGenomeResearch vol 21 no 6 pp 936ndash939 2011
[30] Online Mendelian Inheritance in Man OMIM McKusick-Nathans Institute of Genetic Medicine Johns Hopkins Univer-sity Baltimore Md USA 2015 httpomimorg
[31] Cafe Variome httpwwwcafevariomeorg[32] The Genome Sequencing Consortium ldquoInitial sequencing and
analysis of the human genomerdquo Nature vol 409 pp 860ndash9212001
[33] P Kumar S Henikoff and P C Ng ldquoPredicting the effects ofcoding non-synonymous variants on protein function using theSIFT algorithmrdquo Nature Protocols vol 4 no 7 pp 1073ndash10822009
[34] I A Adzhubei S Schmidt L Peshkin et al ldquoA method andserver for predicting damaging missense mutationsrdquo NatureMethods vol 7 no 4 pp 248ndash249 2010
[35] Alamut Visual Interactive Biosoftware Rouen France httpwwwinteractivebiosoftwarecomalamut-visual
BioMed Research International 11
[36] S Richards N Aziz S Bale et al ldquoStandards and guidelinesfor the interpretation of sequence variants a joint consensusrecommendation of the American College of Medical Geneticsand Genomics and the Association for Molecular PathologyrdquoGenetics in Medicine vol 17 no 5 pp 405ndash423 2015
[37] BWolf and P Kuonen ldquoA novel approach for heuristic pairwiseDNA sequence alignmentrdquo in Proceedings of the InternationalConference on Bioinformatics amp Computational Biology (BIO-COMP rsquo13) Las Vegas Nev USA July 2013
[38] O Gotoh ldquoAn improved algorithm for matching biologicalsequencesrdquo Journal ofMolecular Biology vol 162 no 3 pp 705ndash708 1982
[39] D Smedley S Kohler J C Czeschik et al ldquoWalking theinteractome for candidate prioritization in exome sequencingstudies of Mendelian diseasesrdquo Bioinformatics vol 30 no 22pp 3215ndash3222 2014
[40] A L Semmler S Sacconi J Bach et al ldquoUnusual multisys-temic involvement and a novel BAG3 mutation revealed byNGS screening in a large cohort of myofibrillar myopathiesrdquoOrphanet Journal of Rare Diseases vol 9 no 1 p 121 2014
[41] M Larsen S Rost N El Hajj et al ldquoDiagnostic approach forFSHD revisited SMCHD1 mutations cause FSHD2 and act asmodifiers of disease severity in FSHD1rdquo European Journal ofHuman Genetics vol 23 no 6 pp 808ndash816 2015
[42] S Rost E Bach C Neuner et al ldquoNovel form of X-linkednonsyndromic hearing loss with cochlear malformation causedby a mutation in the type IV collagen gene COL4A6rdquo EuropeanJournal of Human Genetics vol 22 no 2 pp 208ndash215 2014
[43] M Broman I Kleinschnitz J E Bach S Rost G Islander andC R Muller ldquoNext-generation DNA sequencing of a Swedishmalignant hyperthermia cohortrdquo Clinical Genetics 2014
[44] T Low S vanHeesch H van den Toorn et al ldquoQuantitative andqualitative proteome characteristics extracted from in-depthintegrated genomics and proteomics analysisrdquo Cell Reports vol5 no 5 pp 1469ndash1478 2013
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

4 BioMed Research International
to generate a custom automated pipeline but more on anintuitive interface to handle the data-files to perform theanalysis in a predefined pipeline optimised for diagnostics
The features of GensearchNGS are limited to thoseneeded to perform the presented NGS diagnostics workflowThis means the overall feature set is smaller than that of othercomplete software packages especially CLCBio or Galaxywhich both offer a wide variety of functionalities Havinga limited feature set allows for a cleaner and more focuseduser interface one of the major goals of GensearchNGSsomething that is highly appreciated by current users ofGensearchNGS
Aswill be discussed in the following chapters Gensearch-NGS uses either external applications through plugins orcustom implementations of standard algorithms for the dif-ferent analysis stepsThe resulting analysis quality is thereforecomparable to the one observed when using those toolsseparately by hand or through a software package like Galaxy
Approach The issues faced by small laboratories when usingthe previously mentioned tools which are often limitedin terms of computing and human resources are variousBased on our experience the biggest issue faced by smallerlaboratories is the lack of technical know-how and dedicatedbioinformaticians The usage and configuration of many ofthe described data analysis tools require a dedicated employeewhich not available in every laboratoryThis is especially truein emergingmarketswheremanymonetary restrictions are inplace The approach of solutions like Galaxy and CLCBio tothis problem is to abstract the usage of the underlying analysistools and provide a way for the user to automate the analysisprocessWhile the abstraction of the underlying tools reducesthe technical complexity of the analysis process this is oftennot enough tomake the analysis process more accessibleThecited tools can be used for many other types of analyses thanthe ones looked at in this paper which leads to a much morecomplex user interface and often overwhelms the user withmany options The cited tools also focus more on the abilityto create automated analysis pipelines leading to very littleinteractivity between the user and the data
The second issue often encountered is the lack of a suitablecomputing infrastructure to perform the computationallyintensive parts of the analysis Sequence alignment in par-ticular is very demanding in that regard Even though manylaboratories outsource the sequencing and alignment processthe possibility to realign the raw sequencing data is oftenrequired To perform the described analysis steps and moreGensearchNGS provides a complete software suite focusingon optimized algorithms and usage of distributed computingfor optimal usage of the existing infrastructure All theanalysis steps make usage of multiprocessor systems to speedup the analysis whereas only sequence alignment using thecustomGensearchNGS alignment algorithm uses distributedcomputing The entire functionality is provided throughan intuitive user interface to lower the technical know-how needed to perform NGS data analysis GensearchNGSprovides its own way on how the analysis is approachedby providing the user with a patient centric user interface
and favoring an interactive analysis approach instead ofa fully automated analysis pipeline The user interface isfocused on providing the functionality required to performthe previously described analysis without overwhelming theuser with rarely used features
2 Materials and Methods
GensearchNGS has been developed as a multiplatform appli-cation running onWindows Linux and OSX using Java 6+The application is a locally installed software and is not webbased In order to perform NGS analysis steps Gensearch-NGS includes several existing external tools through pluginsThose tools are mainly used during the sequence alignmentphase giving the user the choice between different alignmentalgorithms which may have specific advantages related tothe data to be analysed The external alignment algorithmssupported through plugins are BWA [7] and Bowtie 1 + 2[8] as well as Stampy [29] GensearchNGS can either uselocally installed versions of those external tools or downloadand update them automatically If the tools are installedby the user he can update them when desired otherwisethe same update mechanism used to update GensearchNGSitself is used to update the external tools For sequencingdata GensearchNGS supports various input and output fileformats For raw sequencing data the standard FASTQformat as well as FASTA SFF and FNA is supported All ofthem are converted to the standard FASTQ file format duringthe data import phase As FASTQ files require a quality scorefor every nucleotide in the sequenced reads a default qualityscore is assumed for all the nucleotides if the source formatlike FASTA is missing this information For aligned databoth SAM and BAM files are supported whereas SAM filesare converted to BAM during import BAM and SAM fileare accessed through the HTSJDK library HTSJDK is anopen source Java library developed as part of the samtoolsproject [12] It provides a standardized API to access variousNGS data file formats The library can be downloaded athttpsamtoolsgithubiohtsjdk Annotations can be pro-vided through the BED WIG and GFF files and the VCF fileformat is used to import or export variants
To benefit from the knowledge stored in various onlinedatabases GensearchNGS has been connected to several ofthem to annotate the NGS data on various levels from thegenes and their transcripts down to the individual variantsFor most of the annotation data GensearchNGS accessesthe Ensembl [24] web service through its biomart APIgiving the application access to gene and variant informationEnsembl provides a unified way to access several other onlinedatabases like dbSNP [16] or OMIM [30] through a singleonline service In addition to Ensembl GensearchNGS inte-grates other online services for additional data and featuresCafe Variome [31] provides a standardized way to sharevariants between various laboratories UCSC [32] providesaccess to the human reference sequence and HPO [18] pro-vides information about phenotypes associatedwith genes Insituations with a direct conflict in the information betweenthe different databases where possible the information withthe most severe clinical impact is displayed An example of
BioMed Research International 5
this is contradictory SIFT [33] and PolyPhen 2 [34] scores forvariants By default the most severe will be shown to the userwith the possibility to manually get more information whenrequested GensearchNGS also integrates the possibility totalk to other software installed by the user such as Alamut[35] allowing the user to easily open a variant found byGensearchNGS inside a local Alamut installation
3 Results
This chapter discusses how GensearchNGS implements thedifferent analysis steps and how it tries to lower as much aspossible the technical know-how and computing infrastruc-ture required for NGS data analysis
31 Software Suite GensearchNGS provides the user with thepossibility to create different projects to group the analysis ofvarious patients based on their type or their relation Whilemany other NGS analysis tools provide the possibility toorganize the data in projects GensearchNGS puts the notionof patients and their associatedmetainformation in the centerof the project management Every project contains one ormore patient and each can have several raw sequencing dataassociated The raw sequencing data can in turn have severalalignments associated for example using different alignersor alignment configurations Project specific settings such asspecific regions of interests to be used during the analysis canalso be configured Those regions can be described by BEDfiles to for example limit the analysis to regions relevant tothe type of analysis performed in the current project ThoseBED files can either be provided by the user or automaticallygenerated based on a list of gene names Projects alsoassociate the specific reference sequence to be used duringthe analysis The user can automatically download themfrom UCSC which provides reference sequences of the mostcommon human reference assemblies hg18 hg19 and hg38The usage of custom references sequences is also supportedThe patient centric database integrated into GensearchNGSallows the user to associate variants with patients a featurevery important in diagnostics The variants associated withpatients are shared between projects allowing the user toidentify variants found in different projects on previouslyanalysed patients
To perform the NGS data analysis GensearchNGS usesthe genes described in Ensembl as the gene model In a pro-cess transparent to the user the genemodel corresponding tothe reference sequence defined for the analysis is downloadedautomatically from Ensembl Additional metainformation isattached to the genes such as the associated phenotypesas described by HPO [18] or OMIM [30] Figure 2 showsthe main interface of GensearchNGS with the patient liston the left and the information about the currently selectedpatient in the center The central area of the image showsthe information about the currently selected patient Thisincludes his name internal ID associated phenotypes andall variants which have manually been confirmed by the userWhen validating variants the user can classify themmanuallyinto different categories The categories are based on the
Figure 2 Main interface of GensearchNGS showing a patient withits validated variants
ACMG variant classification guidelines [36] with the userrequested categories Artefact and False reference added Thesaved variants are used for any future variant detection in newpatients immediately telling the user if a variant has alreadybeen found in a different projectThe addition of the Artefactand False reference categories allows the user to quickly filterout common sequencing errors in his particular sequencingsetupThe hierarchical organization of the data is intended toguide the user through the analysis process while keeping itclear where the data comes from An example of this is thesequence alignment It is displayed as a child element of theraw sequencing data making the relation between the rawdata and the alignment immediately obvious
GensearchNGS does not provide the user with a singlebutton which goes from the raw sequencing data to the finalreport Instead of fully automating the pipeline Gensearch-NGS leads the user from one processing step to the nextBefore each step the user is given the choice to changeparameters or use the previously used (or standard) parame-ters After the initial import of the raw sequencing data theuser no longer has to handle the data files GensearchNGSperforms the required data file handling and conversionsbetween the processing steps
While there is no fully automated way to go from theraw sequencing data to the final variant report there is stilla way to automate a big part of the analysis When importingraw sequencing data the user has the possibility to performsequence alignment and variant calling automatically Thosetwo additional analysis steps are performed using eitherpreviously used configuration parameters or the default onesOnly the final variant validation has then to be performedby the user to create the final report Further details of thisprocess follow in the next chapter
32 Data Analysis To perform the initial quality control ofthe raw sequencing data GensearchNGS integrates its owntools allowing the user to import the raw data availablein different file formats The compatible data formats aredescribed in Section 2 The various formats are converted inthe standard FASTQ format during the data import step For
6 BioMed Research International
Figure 3 Overview of various statistics that the user can inspect to verify the data quality
the quality control of the raw sequencing data the user ispresented with various statistics as seen in Figure 3
The statistics allow the user to judge the quality of thesequencing data This includes statistics such as the lengthaverage quality andGC content distribution of the sequencedreads as well as the distribution of the sequenced basesand their quality for every read position and detection of
overrepresented K-mers at the start and the end of thesequenced reads
This raw data import process replaces existing tools likeFastQC to check the quality FASTX to filter the data andthe various utilities that transform different sequencing fileformats into the standard FASTQ file format Transparentlyfor the user the custom implementations of those tools
BioMed Research International 7
which produce the same results have been integrated into asingle processing stepThis includes the possibility to removeadapter sequences and split the raw sequencing data intomultiple files if they were produced through multiplexedsequencing Based on the reported quality information theuser can choose to filter the raw data following several filterssuch as maximum and minimum length of the sequencesand their minimal average quality He can also trim the 51015840and 31015840 parts of all reads or trim their excess length if theyare longer as a user defined length The trimming is done inaddition to the adapter and primer removalThe possibility tosplit the raw sequencing data based on barcodes in the readallowing the import of raw data from multiple samples thatwere sequenced at the same time is also integrated in theimport process
To align the sequenced data against a reference assemblyGensearchNGS provides the user with many options Severalexisting alignment algorithms are integrated into the applica-tion through plugins allowing their usage without the needof advanced technical know-how The currently integratedalignment algorithms are BWA [7] Bowtie 1 + 2 [8] andStampy [29]
Most of those alignment algorithms are developed in away that do not allow them to be used on multiple platformsIn fact most of them only run on Linux which is problematicfor a multiplatform application like GensearchNGS For thisreason a custom alignment algorithm written in Java hasbeen integrated directly into GensearchNGS This allows theuser to perform the full NGS data analysis even on operatingsystems likeWindows not supported bymost bioinformaticstools
The aligner supports paired end reads and gapped align-ment and has been optimized to use less than 5GB ofRAM to perform a full genome alignment This makes itsuitable for most of todayrsquos workspace desktops It is fullymultithreaded to support all cores of the machine and isable to offload calculations as described in Section 34 tomultiple computers The aligner uses a hash based indexa concept made popular through the BLAST aligner [9]with a custom heuristic based candidate alignment positionsselection [37] The final alignment is being done using theGotoh [38] algorithm a dynamic programming algorithmused for gapped alignment
The custom alignment algorithm being implemented inJava does not achieve quite the same performance as nativelyrunning aligners like BWA For WGS datasets it is generally50 slower The advantage of the GensearchNGS alignerover external aligners is that it is available on all supportedplatforms allowing the user to perform the complete NGSdata analysis on all platforms But when running on aplatform which supports the external aligners and speed isa deciding factor the users are encouraged to use the alignersavailable as plugins
The quality of resulting alignment can be verified throughvarious metrics by the user He can easily verify how manysequences have been discarded during the alignment pro-cess and how many sequences have been aligned to thedifferent chromosomes in the reference sequence Advancedstatistics similar to the ones created by bedtools 2 can also
be inspected giving detailed coverage information of thepreviously defined regions of interest The collected qualitycontrol information can be exported as a report for archivingpurposes using one of various formats such as PDF HTMLor PNG
While initially GensearchNGS integrated Varscan 2 [14]as its variant caller a replacement was created to reduce theexternal dependencies and to improve the performance ofvariant calling A variant scanner based on the model ofVarscan 2 has been implemented and is in the process of beingpublished as a separate tool called GNATY available for freefor noncommercial usage (httpgnatyphenosystemscom)Our custom variant caller shows important performanceadvantages over the existing Varscan 2 implementation of thesame variant calling model while producing the same analy-sis results Indeed GensearchNGS shows speed improvementof up to 18 times for variant calling compared to Varscan2 providing a similar range of features such as filters thatexclude sequencing artifacts
To be able to further filter down the detected variantsbased on their relevancy to the samplersquos phenotype severalsources are used to annotate them with various informationThis includes the prediction of the variant effect on thedifferent transcripts it affects a feature closely modeledafter the Variant Effect Predictor (VEP) from Ensembl [17]GensearchNGS implements its own version of the the VEPalgorithm to be able to perform the effect prediction in afraction of the time compared to the online VEP serviceThisis is mainly achieved by doing all calculations locally withouthaving to transfer the data to a remote server Predictionssuch as the addition or removal of a stop codon in an affectedtranscript if a splice site is affected and many others canbe made with this algorithm Additional information suchas if the variant is known in any public database associatedclinical significance and SIFT [33] and PolyPhen 2 [34]predictions are retrieved through the Ensembl web serviceOther services like the Human Phenotype Ontology [18]provide the information about the phenotype associated witha specific gene a crucial information in diagnostics
To filter the annotated variants GensearchNGS takesan interactive approach which allows the user to modifydifferent basic filters such as minimum or maximum samplefrequency coverage read balance or quality There are alsoadvanced filters such as the predicted consequence of thevariant as determined by the annotation process or if acertain phenotype is associated with the affected genes Thisincludes the possibility to filter by population frequency ofa variant The filtering by population frequency does not yettake into account the ethnicity of the patient Figure 4 showsan example of this list with several user selected interactivefilters applied
Additional features are available to perform a more in-depth analysis of variants such as family analysis where thevariants of multiple patients can be compared to find thoseonly present in a certain group of patients The usage ofpedigree information is of growing importance in NGS dataanalysis as it allows the user to filter sequencing artifactsand noncausative variants This is a very important featureespecially when usingWGSwhere the amount of variants can
8 BioMed Research International
Figure 4 Integrated variants list with several interactive filtersapplied
be very large Thanks to the usage of standard file formatslike VCF external tools like exome walker [39] can be usedto perform more advanced family analysis for features notcovered in GensearchNGS In contrast to many other NGSdata analysis software packages GensearchNGS does notfocus on the automation of every aspect of the analysisWhilecertain parts can be automated such as performing sequencealignment and variant calling on raw sequencing data thephilosophy of our software is to let the user to control thedifferent analysis steps This gives him enough flexibility toadapt to the data he is currently analysing without slowinghim down by having project specific analysis options whichare preselected for every analysis step This makes it possibleto perform most types of analysis with the project specificdefault options through a simple press of a button
33 Genome Browser A central tool of GensearchNGS isthe integrated genome browser to visualize the alignedsequencing data The genome browser as shown in Figure 5allows to interactively browse the sequence alignments Itis possible to display the aligned sequences and additionalgenomic data such as the gene locations and the encodedproteins by different transcripts
The genome browser allows the user to display sev-eral tracks containing information which helps to analysethe sequencing data The predefined tracks which can beactivated or deactivated allow the user to display coverageinformation GC content of the reference sequence thequality of the sequenced reads or custom annotation files instandard file formats such as BED WIG and GFF The focuswas put on optimizing the display of the data allowing theuser to visualize large datafiles without requiring a powerfulcomputerThis is achieved by streaming the data on demandonly loading the parts of the data currently needed forthe visualization The information about which variants arepresent in the alignment is available to the user in the sameform as the variant list in the main application It can be seenin Figure 4 allowing the user to quickly jump to the variantsof interest The genome browser like all of GensearchNGSis heavily connected with external data sources (EnsembldbSNP Entrez OMIM Blast and HPO) providing the user
Figure 5 Example view of the genome browser showing severalavailable tracks
with all relevant informationwhenneededTheuser can openexternal resources linked from many features in the visual-ized data such as genes transcripts or variants to retrieveadditional informationThe genome browser takes advantageof the integrated data sources like the Ensembl gene modelto display and interactively update encoded proteins basedon the variants present in the visualized data The genomebrowser is tightly integrated into GensearchNGS allowingthe user to easily visualize the alignment data belonging tofeatures of the sample
34 Distributed Computing The lack of computing powerto perform NGS data analysis is a very common issue inmany laboratories To address this problem GensearchNGSdoes not only optimize the algorithms for speed andmemoryusage but also try to exploit the already existing computinginfrastructure One way to do this is to use distributedcomputing a technique which allows the user to combinemultiple computers to perform computing intensive tasksThose computers can be in the same local network or at aremote location One of the most demanding steps in NGSdata analysis is sequence alignment For this reason it is thefirst feature to take advantage of this technique in Gensearch-NGS allowing the user to joinmultiple computers in the samenetwork to perform sequence alignment The approach usedby GensearchNGS is to transparently on request by the useruse all computers within the same networkwhereGensearch-NGS is installed Additionally the user has the possibility tostart up GensearchNGS instances in a cloud Currently onlythe amazon cloud is supportedThis process works regardlessof the operating system used by the different installationsof GensearchNGS By using an automatic service discoveryprotocol every installation of GensearchNGS located in thesame network is discovered without the need for manualconfigurationThis allows the user to use multiple computersat the same time or to completely offload calculations to adifferent computer making it possible to use computers notsuited for full genome alignments as an analysis workstationThe distributed sequence aligner used in GensearchNGS
BioMed Research International 9
currently only supports the GensearchNGS aligner algorithmand not external aligners such as BWA or Bowtie 2 It uses astream based approach where the client machine streams thedata to be aligned to all computers involved in the alignmentprocess This includes the client machine which receives thealigned sequences back and combines them into the finalalignment file The process of distributing the alignmentsover multiple computers and the cloud is transparent to theuser but it is optional because the usage of an externalcloud is not compatible with the security guidelines of somelaboratories GensearchNGS is not the onlyNGSdata analysissoftware using distributed sequence alignment It is thisability to combine the computing resources of the localcomputer multiple computers in the same network as well asthe cloud whichmakes its approach unique and very flexible
4 Discussion
GensearchNGS addresses problems that smaller laboratoriesare facing in various ways The issue of the technical com-plexity of the analysis is approached through an intuitive userinterface The user interface abstracts the complexity of theunderlying NGS data analysis toolchain which is a majorusability improvement over the usage of individual toolsNevertheless it gives the user full control over the individualanalysis steps instead of focusing on the creation of auto-mated analysis pipelines It also provides an original approachby providing a patient centric user interface This userfriendly interface allows users unfamiliar with the underlyingsoftware tools to fine tune the analysis parameters withoutoverwhelming themwith features or rarely used optionsThisincreases the number of people able to perform NGS dataanalysis This is particularly interesting for geneticists thatmight not have special bioinformatics training The issue ofincreasing infrastructure requirements for NGS data analysisis being addressed twofold First more efficient algorithmsare being developed like the heavily optimized variant callingalgorithm included in GensearchNGS Secondly the abilityto distribute heavy calculations over several computers oreven a cloud service has been developed GensearchNGShas a unique approach to this problem by enabling the userto combine the various distribution methods to performsequence alignment It provides the flexibility needed tochoose the best compromise between data privacy andalignment speed depending on the laboratory policies andneeds The resulting software enabled various laboratoriesto perform NGS data analysis and publish their results invarious fields The topics range from muscular dystrophies[40 41] to hearing loss [42] andmalignant hyperthermia [43]but also yet unpublished work about CNV analysis and thedetection of somatic mutations in cancer screening
5 Conclusion
In this paper we reviewed the workflow used to analyzeNGS data in a diagnostics environment The reviewedworkflow includes the detection of SNPs and short indelsfrom panel WES or WGS DNAseq data We also pre-sented issues faced by smaller laboratories trying to setup a
NGS data analysis pipeline We then presented Gensearch-NGS a software suite integrating various algorithms toperform the discussed workflow GensearchNGS is a com-mercially available software distributed by PhenosystemsSA A demo version of GensearchNGS is freely availableat httpwwwphenosystemscom The two main issues thetechnical complexity of the analysis and the computinginfrastructure requirements have been addressed as follows
To reduce the technical complexity of the analysis theuser is provided with an intuitive user interface which guideshim through the process of the data analysis He is providedwith the required tools to go from raw sequencing data the thefinal visualization and report generation for the sequencedsamples without having to learn complex bioinformaticstools The interface abstracts the technical complexity fromthe user while keeping enough flexibility to adapt to differentanalysis protocols By lowering the technical complexity ofNGS data analysis compared to existing individual analysistools GensearchNGS expands the potential user base of thesoftware
To address the issue of the computing infrastructurerequirements GensearchNGS focuses on the optimal usageof existing computing resources This is primarily achievedthrough the development of analysis algorithms that runon a minimal amount of resources while producing thesame analysis results as standard tools An example has beendescribed with the variant calling algorithm which producesthe same results as Varscan 2 but is 18 times faster Thesecond approach is the integration of distributed computingallowing the distribution of computing intensive tasks likealignment to be speed up by the usage of multiple computers
With a solid framework built for DNAseq analysisGensearchNGS will expand into further types of analysis inthe futureOne example is the integration of RNAseq analysisas the combination of OMICS data for a single sample isof increasing importance to understand rare diseases [44]In fact many parts of GensearchNGS such as the genomebrowser have already been adapted to support RNAseq dataThe integration of multiple data sources will not only allowthe user to integrate different types of OMICS data but alsoallow multiple views on the same sample One example ofthis is the integration of Sanger sequencing to NGS dataa very useful feature for diagnostics where it is commonpractice to validate variants identified through NGS withSanger sequencing
Further optimizations of the algorithms used in theanalysis will also be evaluated including optimization for theunderlying libraries One example is HTSJDK where severalperformance patches have been submitted and includedimproving NGS data analysis performance not only for usersof GensearchNGS but also for the complete bioinformaticscommunity
Disclosure
Pierre Kuonen has a nonpaid advisory position at Phenosys-tems SA David Atlan is working full time for PhenosystemsSA
10 BioMed Research International
Conflict of Interests
Thomas Dandekar has no conflict of interests regarding thepublication of this paper
Acknowledgments
GensearchNGS is a commercially available software dis-tributed by Phenosystems SA Beat Wolf rsquos PhD thesis ispartially financially supported by Phenosystems SA
References
[1] E L van Dijk H Auger Y Jaszczyszyn and C ThermesldquoTen years of next-generation sequencing technologyrdquo Trendsin Genetics vol 30 no 9 pp 418ndash426 2014
[2] B Sikkema-Raddatz L F Johansson E N de Boer et al ldquoTar-geted next-generation sequencing can replace sanger sequenc-ing in clinical diagnosticsrdquo Human Mutation vol 34 no 7 pp1035ndash1042 2013
[3] H P J Buermans and J T den Dunnen ldquoNext generationsequencing technology advances and applicationsrdquo Biochimicaet Biophysica ActamdashMolecular Basis of Disease vol 1842 no 10pp 1932ndash1941 2014
[4] FastQC ldquoA quality control tool for high throughput sequencedatardquo Babraham Bioinformatics Website httpwwwbioinfor-maticsbabrahamacukprojectsfastqc
[5] W R Pearson T Wood Z Zhang andWMiller ldquoComparisonof DNA sequences with protein sequencesrdquo Genomics vol 46no 1 pp 24ndash36 1997
[6] H Li and N Homer ldquoA survey of sequence alignment algo-rithms for next-generation sequencingrdquo Briefings in Bioinfor-matics vol 11 no 5 pp 473ndash483 2010
[7] H Li and R Durbin ldquoFast and accurate long-read alignmentwith Burrows-Wheeler transformrdquo Bioinformatics vol 26 no5 pp 589ndash595 2010
[8] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[9] S F Altschul T L Madden A A Schaffer et al ldquoGappedBLAST and PSI-BLAST a new generation of protein databasesearch programsrdquo Nucleic Acids Research vol 25 no 17 pp3389ndash3402 1997
[10] M A DePristo E Banks R Poplin et al ldquoA framework forvariation discovery and genotyping using next-generationDNAsequencing datardquo Nature Genetics vol 43 no 5 pp 491ndash4982011
[11] P Carnevali J Baccash A L Halpern et al ldquoComputationaltechniques for human genome resequencing using matedgapped readsrdquo Journal of Computational Biology vol 19 no 3pp 279ndash292 2012
[12] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[13] A R Quinlan and I M Hall ldquoBEDTools a flexible suite ofutilities for comparing genomic featuresrdquo Bioinformatics vol26 no 6 pp 841ndash842 2010
[14] D C Koboldt Q Zhang D E Larson et al ldquoVarScan 2 somaticmutation and copy number alteration discovery in cancer byexome sequencingrdquo Genome Research vol 22 no 3 pp 568ndash576 2012
[15] J OrsquoRawe T Jiang G Sun et al ldquoLow concordance of multiplevariant-calling pipelines practical implications for exome andgenome sequencingrdquo Genome Medicine vol 5 no 3 article 282013
[16] S T SherryM-HWardM Kholodov et al ldquodbSNP the NCBIdatabase of genetic variationrdquoNucleic Acids Research vol 29 pp308ndash311 2001
[17] W McLaren B Pritchard D Rios Y Chen P Flicek and FCunningham ldquoDeriving the consequences of genomic variantswith the EnsemblAPI and SNPEffect PredictorrdquoBioinformaticsvol 26 no 16 Article ID btq330 pp 2069ndash2070 2010
[18] P N Robinson and S Mundlos ldquoThe human phenotypeontologyrdquo Clinical Genetics vol 77 no 6 pp 525ndash534 2010
[19] P Danecek A Auton G Abecasis et al ldquoThe variant call formatand VCFtoolsrdquo Bioinformatics vol 27 no 15 pp 2156ndash21582011
[20] J T Robinson HThorvaldsdottir W Winckler et al ldquoIntegra-tive genomics viewerrdquo Nature Biotechnology vol 29 no 1 pp24ndash26 2011
[21] I Milne G Stephen M Bayer et al ldquoUsing tablet for visualexploration of second-generation sequencing datardquo Briefings inBioinformatics vol 14 no 2 Article ID bbs012 pp 193ndash2022013
[22] T Abeel T van Parys Y Saeys J Galagan and Y van de PeerldquoGenomeView a next-generation genome browserrdquo NucleicAcids Research vol 40 no 2 article e12 2012
[23] W J Kent C W Sugnet T S Furey et al ldquoThe human genomebrowser at UCSCrdquo Genome Research vol 12 no 6 pp 996ndash1006 2002
[24] F Cunningham M Ridwan Amode D Barrell et al ldquoEnsembl2015rdquo Nucleic Acids Research vol 43 no 1 pp D662ndashD6692015
[25] NGS Data Analysis Software List SEQanswers 2012 httpseqanswerscomwikiSoftwarelist
[26] CLC Genomics Workbench CLC Bio Aarhus Denmark 2014httpwwwclcbiocom
[27] NextGENe Softgenetics State College Pa USA httpwwwsoftgeneticscom
[28] J Goecks A Nekrutenko J Taylor et al ldquoGalaxy a com-prehensive approach for supporting accessible reproducibleand transparent computational research in the life sciencesrdquoGenome Biology vol 11 no 8 article R86 2010
[29] G Lunter and M Goodson ldquoStampy a statistical algorithm forsensitive and fast mapping of Illumina sequence readsrdquoGenomeResearch vol 21 no 6 pp 936ndash939 2011
[30] Online Mendelian Inheritance in Man OMIM McKusick-Nathans Institute of Genetic Medicine Johns Hopkins Univer-sity Baltimore Md USA 2015 httpomimorg
[31] Cafe Variome httpwwwcafevariomeorg[32] The Genome Sequencing Consortium ldquoInitial sequencing and
analysis of the human genomerdquo Nature vol 409 pp 860ndash9212001
[33] P Kumar S Henikoff and P C Ng ldquoPredicting the effects ofcoding non-synonymous variants on protein function using theSIFT algorithmrdquo Nature Protocols vol 4 no 7 pp 1073ndash10822009
[34] I A Adzhubei S Schmidt L Peshkin et al ldquoA method andserver for predicting damaging missense mutationsrdquo NatureMethods vol 7 no 4 pp 248ndash249 2010
[35] Alamut Visual Interactive Biosoftware Rouen France httpwwwinteractivebiosoftwarecomalamut-visual
BioMed Research International 11
[36] S Richards N Aziz S Bale et al ldquoStandards and guidelinesfor the interpretation of sequence variants a joint consensusrecommendation of the American College of Medical Geneticsand Genomics and the Association for Molecular PathologyrdquoGenetics in Medicine vol 17 no 5 pp 405ndash423 2015
[37] BWolf and P Kuonen ldquoA novel approach for heuristic pairwiseDNA sequence alignmentrdquo in Proceedings of the InternationalConference on Bioinformatics amp Computational Biology (BIO-COMP rsquo13) Las Vegas Nev USA July 2013
[38] O Gotoh ldquoAn improved algorithm for matching biologicalsequencesrdquo Journal ofMolecular Biology vol 162 no 3 pp 705ndash708 1982
[39] D Smedley S Kohler J C Czeschik et al ldquoWalking theinteractome for candidate prioritization in exome sequencingstudies of Mendelian diseasesrdquo Bioinformatics vol 30 no 22pp 3215ndash3222 2014
[40] A L Semmler S Sacconi J Bach et al ldquoUnusual multisys-temic involvement and a novel BAG3 mutation revealed byNGS screening in a large cohort of myofibrillar myopathiesrdquoOrphanet Journal of Rare Diseases vol 9 no 1 p 121 2014
[41] M Larsen S Rost N El Hajj et al ldquoDiagnostic approach forFSHD revisited SMCHD1 mutations cause FSHD2 and act asmodifiers of disease severity in FSHD1rdquo European Journal ofHuman Genetics vol 23 no 6 pp 808ndash816 2015
[42] S Rost E Bach C Neuner et al ldquoNovel form of X-linkednonsyndromic hearing loss with cochlear malformation causedby a mutation in the type IV collagen gene COL4A6rdquo EuropeanJournal of Human Genetics vol 22 no 2 pp 208ndash215 2014
[43] M Broman I Kleinschnitz J E Bach S Rost G Islander andC R Muller ldquoNext-generation DNA sequencing of a Swedishmalignant hyperthermia cohortrdquo Clinical Genetics 2014
[44] T Low S vanHeesch H van den Toorn et al ldquoQuantitative andqualitative proteome characteristics extracted from in-depthintegrated genomics and proteomics analysisrdquo Cell Reports vol5 no 5 pp 1469ndash1478 2013
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

BioMed Research International 5
this is contradictory SIFT [33] and PolyPhen 2 [34] scores forvariants By default the most severe will be shown to the userwith the possibility to manually get more information whenrequested GensearchNGS also integrates the possibility totalk to other software installed by the user such as Alamut[35] allowing the user to easily open a variant found byGensearchNGS inside a local Alamut installation
3 Results
This chapter discusses how GensearchNGS implements thedifferent analysis steps and how it tries to lower as much aspossible the technical know-how and computing infrastruc-ture required for NGS data analysis
31 Software Suite GensearchNGS provides the user with thepossibility to create different projects to group the analysis ofvarious patients based on their type or their relation Whilemany other NGS analysis tools provide the possibility toorganize the data in projects GensearchNGS puts the notionof patients and their associatedmetainformation in the centerof the project management Every project contains one ormore patient and each can have several raw sequencing dataassociated The raw sequencing data can in turn have severalalignments associated for example using different alignersor alignment configurations Project specific settings such asspecific regions of interests to be used during the analysis canalso be configured Those regions can be described by BEDfiles to for example limit the analysis to regions relevant tothe type of analysis performed in the current project ThoseBED files can either be provided by the user or automaticallygenerated based on a list of gene names Projects alsoassociate the specific reference sequence to be used duringthe analysis The user can automatically download themfrom UCSC which provides reference sequences of the mostcommon human reference assemblies hg18 hg19 and hg38The usage of custom references sequences is also supportedThe patient centric database integrated into GensearchNGSallows the user to associate variants with patients a featurevery important in diagnostics The variants associated withpatients are shared between projects allowing the user toidentify variants found in different projects on previouslyanalysed patients
To perform the NGS data analysis GensearchNGS usesthe genes described in Ensembl as the gene model In a pro-cess transparent to the user the genemodel corresponding tothe reference sequence defined for the analysis is downloadedautomatically from Ensembl Additional metainformation isattached to the genes such as the associated phenotypesas described by HPO [18] or OMIM [30] Figure 2 showsthe main interface of GensearchNGS with the patient liston the left and the information about the currently selectedpatient in the center The central area of the image showsthe information about the currently selected patient Thisincludes his name internal ID associated phenotypes andall variants which have manually been confirmed by the userWhen validating variants the user can classify themmanuallyinto different categories The categories are based on the
Figure 2 Main interface of GensearchNGS showing a patient withits validated variants
ACMG variant classification guidelines [36] with the userrequested categories Artefact and False reference added Thesaved variants are used for any future variant detection in newpatients immediately telling the user if a variant has alreadybeen found in a different projectThe addition of the Artefactand False reference categories allows the user to quickly filterout common sequencing errors in his particular sequencingsetupThe hierarchical organization of the data is intended toguide the user through the analysis process while keeping itclear where the data comes from An example of this is thesequence alignment It is displayed as a child element of theraw sequencing data making the relation between the rawdata and the alignment immediately obvious
GensearchNGS does not provide the user with a singlebutton which goes from the raw sequencing data to the finalreport Instead of fully automating the pipeline Gensearch-NGS leads the user from one processing step to the nextBefore each step the user is given the choice to changeparameters or use the previously used (or standard) parame-ters After the initial import of the raw sequencing data theuser no longer has to handle the data files GensearchNGSperforms the required data file handling and conversionsbetween the processing steps
While there is no fully automated way to go from theraw sequencing data to the final variant report there is stilla way to automate a big part of the analysis When importingraw sequencing data the user has the possibility to performsequence alignment and variant calling automatically Thosetwo additional analysis steps are performed using eitherpreviously used configuration parameters or the default onesOnly the final variant validation has then to be performedby the user to create the final report Further details of thisprocess follow in the next chapter
32 Data Analysis To perform the initial quality control ofthe raw sequencing data GensearchNGS integrates its owntools allowing the user to import the raw data availablein different file formats The compatible data formats aredescribed in Section 2 The various formats are converted inthe standard FASTQ format during the data import step For
6 BioMed Research International
Figure 3 Overview of various statistics that the user can inspect to verify the data quality
the quality control of the raw sequencing data the user ispresented with various statistics as seen in Figure 3
The statistics allow the user to judge the quality of thesequencing data This includes statistics such as the lengthaverage quality andGC content distribution of the sequencedreads as well as the distribution of the sequenced basesand their quality for every read position and detection of
overrepresented K-mers at the start and the end of thesequenced reads
This raw data import process replaces existing tools likeFastQC to check the quality FASTX to filter the data andthe various utilities that transform different sequencing fileformats into the standard FASTQ file format Transparentlyfor the user the custom implementations of those tools
BioMed Research International 7
which produce the same results have been integrated into asingle processing stepThis includes the possibility to removeadapter sequences and split the raw sequencing data intomultiple files if they were produced through multiplexedsequencing Based on the reported quality information theuser can choose to filter the raw data following several filterssuch as maximum and minimum length of the sequencesand their minimal average quality He can also trim the 51015840and 31015840 parts of all reads or trim their excess length if theyare longer as a user defined length The trimming is done inaddition to the adapter and primer removalThe possibility tosplit the raw sequencing data based on barcodes in the readallowing the import of raw data from multiple samples thatwere sequenced at the same time is also integrated in theimport process
To align the sequenced data against a reference assemblyGensearchNGS provides the user with many options Severalexisting alignment algorithms are integrated into the applica-tion through plugins allowing their usage without the needof advanced technical know-how The currently integratedalignment algorithms are BWA [7] Bowtie 1 + 2 [8] andStampy [29]
Most of those alignment algorithms are developed in away that do not allow them to be used on multiple platformsIn fact most of them only run on Linux which is problematicfor a multiplatform application like GensearchNGS For thisreason a custom alignment algorithm written in Java hasbeen integrated directly into GensearchNGS This allows theuser to perform the full NGS data analysis even on operatingsystems likeWindows not supported bymost bioinformaticstools
The aligner supports paired end reads and gapped align-ment and has been optimized to use less than 5GB ofRAM to perform a full genome alignment This makes itsuitable for most of todayrsquos workspace desktops It is fullymultithreaded to support all cores of the machine and isable to offload calculations as described in Section 34 tomultiple computers The aligner uses a hash based indexa concept made popular through the BLAST aligner [9]with a custom heuristic based candidate alignment positionsselection [37] The final alignment is being done using theGotoh [38] algorithm a dynamic programming algorithmused for gapped alignment
The custom alignment algorithm being implemented inJava does not achieve quite the same performance as nativelyrunning aligners like BWA For WGS datasets it is generally50 slower The advantage of the GensearchNGS alignerover external aligners is that it is available on all supportedplatforms allowing the user to perform the complete NGSdata analysis on all platforms But when running on aplatform which supports the external aligners and speed isa deciding factor the users are encouraged to use the alignersavailable as plugins
The quality of resulting alignment can be verified throughvarious metrics by the user He can easily verify how manysequences have been discarded during the alignment pro-cess and how many sequences have been aligned to thedifferent chromosomes in the reference sequence Advancedstatistics similar to the ones created by bedtools 2 can also
be inspected giving detailed coverage information of thepreviously defined regions of interest The collected qualitycontrol information can be exported as a report for archivingpurposes using one of various formats such as PDF HTMLor PNG
While initially GensearchNGS integrated Varscan 2 [14]as its variant caller a replacement was created to reduce theexternal dependencies and to improve the performance ofvariant calling A variant scanner based on the model ofVarscan 2 has been implemented and is in the process of beingpublished as a separate tool called GNATY available for freefor noncommercial usage (httpgnatyphenosystemscom)Our custom variant caller shows important performanceadvantages over the existing Varscan 2 implementation of thesame variant calling model while producing the same analy-sis results Indeed GensearchNGS shows speed improvementof up to 18 times for variant calling compared to Varscan2 providing a similar range of features such as filters thatexclude sequencing artifacts
To be able to further filter down the detected variantsbased on their relevancy to the samplersquos phenotype severalsources are used to annotate them with various informationThis includes the prediction of the variant effect on thedifferent transcripts it affects a feature closely modeledafter the Variant Effect Predictor (VEP) from Ensembl [17]GensearchNGS implements its own version of the the VEPalgorithm to be able to perform the effect prediction in afraction of the time compared to the online VEP serviceThisis is mainly achieved by doing all calculations locally withouthaving to transfer the data to a remote server Predictionssuch as the addition or removal of a stop codon in an affectedtranscript if a splice site is affected and many others canbe made with this algorithm Additional information suchas if the variant is known in any public database associatedclinical significance and SIFT [33] and PolyPhen 2 [34]predictions are retrieved through the Ensembl web serviceOther services like the Human Phenotype Ontology [18]provide the information about the phenotype associated witha specific gene a crucial information in diagnostics
To filter the annotated variants GensearchNGS takesan interactive approach which allows the user to modifydifferent basic filters such as minimum or maximum samplefrequency coverage read balance or quality There are alsoadvanced filters such as the predicted consequence of thevariant as determined by the annotation process or if acertain phenotype is associated with the affected genes Thisincludes the possibility to filter by population frequency ofa variant The filtering by population frequency does not yettake into account the ethnicity of the patient Figure 4 showsan example of this list with several user selected interactivefilters applied
Additional features are available to perform a more in-depth analysis of variants such as family analysis where thevariants of multiple patients can be compared to find thoseonly present in a certain group of patients The usage ofpedigree information is of growing importance in NGS dataanalysis as it allows the user to filter sequencing artifactsand noncausative variants This is a very important featureespecially when usingWGSwhere the amount of variants can
8 BioMed Research International
Figure 4 Integrated variants list with several interactive filtersapplied
be very large Thanks to the usage of standard file formatslike VCF external tools like exome walker [39] can be usedto perform more advanced family analysis for features notcovered in GensearchNGS In contrast to many other NGSdata analysis software packages GensearchNGS does notfocus on the automation of every aspect of the analysisWhilecertain parts can be automated such as performing sequencealignment and variant calling on raw sequencing data thephilosophy of our software is to let the user to control thedifferent analysis steps This gives him enough flexibility toadapt to the data he is currently analysing without slowinghim down by having project specific analysis options whichare preselected for every analysis step This makes it possibleto perform most types of analysis with the project specificdefault options through a simple press of a button
33 Genome Browser A central tool of GensearchNGS isthe integrated genome browser to visualize the alignedsequencing data The genome browser as shown in Figure 5allows to interactively browse the sequence alignments Itis possible to display the aligned sequences and additionalgenomic data such as the gene locations and the encodedproteins by different transcripts
The genome browser allows the user to display sev-eral tracks containing information which helps to analysethe sequencing data The predefined tracks which can beactivated or deactivated allow the user to display coverageinformation GC content of the reference sequence thequality of the sequenced reads or custom annotation files instandard file formats such as BED WIG and GFF The focuswas put on optimizing the display of the data allowing theuser to visualize large datafiles without requiring a powerfulcomputerThis is achieved by streaming the data on demandonly loading the parts of the data currently needed forthe visualization The information about which variants arepresent in the alignment is available to the user in the sameform as the variant list in the main application It can be seenin Figure 4 allowing the user to quickly jump to the variantsof interest The genome browser like all of GensearchNGSis heavily connected with external data sources (EnsembldbSNP Entrez OMIM Blast and HPO) providing the user
Figure 5 Example view of the genome browser showing severalavailable tracks
with all relevant informationwhenneededTheuser can openexternal resources linked from many features in the visual-ized data such as genes transcripts or variants to retrieveadditional informationThe genome browser takes advantageof the integrated data sources like the Ensembl gene modelto display and interactively update encoded proteins basedon the variants present in the visualized data The genomebrowser is tightly integrated into GensearchNGS allowingthe user to easily visualize the alignment data belonging tofeatures of the sample
34 Distributed Computing The lack of computing powerto perform NGS data analysis is a very common issue inmany laboratories To address this problem GensearchNGSdoes not only optimize the algorithms for speed andmemoryusage but also try to exploit the already existing computinginfrastructure One way to do this is to use distributedcomputing a technique which allows the user to combinemultiple computers to perform computing intensive tasksThose computers can be in the same local network or at aremote location One of the most demanding steps in NGSdata analysis is sequence alignment For this reason it is thefirst feature to take advantage of this technique in Gensearch-NGS allowing the user to joinmultiple computers in the samenetwork to perform sequence alignment The approach usedby GensearchNGS is to transparently on request by the useruse all computers within the same networkwhereGensearch-NGS is installed Additionally the user has the possibility tostart up GensearchNGS instances in a cloud Currently onlythe amazon cloud is supportedThis process works regardlessof the operating system used by the different installationsof GensearchNGS By using an automatic service discoveryprotocol every installation of GensearchNGS located in thesame network is discovered without the need for manualconfigurationThis allows the user to use multiple computersat the same time or to completely offload calculations to adifferent computer making it possible to use computers notsuited for full genome alignments as an analysis workstationThe distributed sequence aligner used in GensearchNGS
BioMed Research International 9
currently only supports the GensearchNGS aligner algorithmand not external aligners such as BWA or Bowtie 2 It uses astream based approach where the client machine streams thedata to be aligned to all computers involved in the alignmentprocess This includes the client machine which receives thealigned sequences back and combines them into the finalalignment file The process of distributing the alignmentsover multiple computers and the cloud is transparent to theuser but it is optional because the usage of an externalcloud is not compatible with the security guidelines of somelaboratories GensearchNGS is not the onlyNGSdata analysissoftware using distributed sequence alignment It is thisability to combine the computing resources of the localcomputer multiple computers in the same network as well asthe cloud whichmakes its approach unique and very flexible
4 Discussion
GensearchNGS addresses problems that smaller laboratoriesare facing in various ways The issue of the technical com-plexity of the analysis is approached through an intuitive userinterface The user interface abstracts the complexity of theunderlying NGS data analysis toolchain which is a majorusability improvement over the usage of individual toolsNevertheless it gives the user full control over the individualanalysis steps instead of focusing on the creation of auto-mated analysis pipelines It also provides an original approachby providing a patient centric user interface This userfriendly interface allows users unfamiliar with the underlyingsoftware tools to fine tune the analysis parameters withoutoverwhelming themwith features or rarely used optionsThisincreases the number of people able to perform NGS dataanalysis This is particularly interesting for geneticists thatmight not have special bioinformatics training The issue ofincreasing infrastructure requirements for NGS data analysisis being addressed twofold First more efficient algorithmsare being developed like the heavily optimized variant callingalgorithm included in GensearchNGS Secondly the abilityto distribute heavy calculations over several computers oreven a cloud service has been developed GensearchNGShas a unique approach to this problem by enabling the userto combine the various distribution methods to performsequence alignment It provides the flexibility needed tochoose the best compromise between data privacy andalignment speed depending on the laboratory policies andneeds The resulting software enabled various laboratoriesto perform NGS data analysis and publish their results invarious fields The topics range from muscular dystrophies[40 41] to hearing loss [42] andmalignant hyperthermia [43]but also yet unpublished work about CNV analysis and thedetection of somatic mutations in cancer screening
5 Conclusion
In this paper we reviewed the workflow used to analyzeNGS data in a diagnostics environment The reviewedworkflow includes the detection of SNPs and short indelsfrom panel WES or WGS DNAseq data We also pre-sented issues faced by smaller laboratories trying to setup a
NGS data analysis pipeline We then presented Gensearch-NGS a software suite integrating various algorithms toperform the discussed workflow GensearchNGS is a com-mercially available software distributed by PhenosystemsSA A demo version of GensearchNGS is freely availableat httpwwwphenosystemscom The two main issues thetechnical complexity of the analysis and the computinginfrastructure requirements have been addressed as follows
To reduce the technical complexity of the analysis theuser is provided with an intuitive user interface which guideshim through the process of the data analysis He is providedwith the required tools to go from raw sequencing data the thefinal visualization and report generation for the sequencedsamples without having to learn complex bioinformaticstools The interface abstracts the technical complexity fromthe user while keeping enough flexibility to adapt to differentanalysis protocols By lowering the technical complexity ofNGS data analysis compared to existing individual analysistools GensearchNGS expands the potential user base of thesoftware
To address the issue of the computing infrastructurerequirements GensearchNGS focuses on the optimal usageof existing computing resources This is primarily achievedthrough the development of analysis algorithms that runon a minimal amount of resources while producing thesame analysis results as standard tools An example has beendescribed with the variant calling algorithm which producesthe same results as Varscan 2 but is 18 times faster Thesecond approach is the integration of distributed computingallowing the distribution of computing intensive tasks likealignment to be speed up by the usage of multiple computers
With a solid framework built for DNAseq analysisGensearchNGS will expand into further types of analysis inthe futureOne example is the integration of RNAseq analysisas the combination of OMICS data for a single sample isof increasing importance to understand rare diseases [44]In fact many parts of GensearchNGS such as the genomebrowser have already been adapted to support RNAseq dataThe integration of multiple data sources will not only allowthe user to integrate different types of OMICS data but alsoallow multiple views on the same sample One example ofthis is the integration of Sanger sequencing to NGS dataa very useful feature for diagnostics where it is commonpractice to validate variants identified through NGS withSanger sequencing
Further optimizations of the algorithms used in theanalysis will also be evaluated including optimization for theunderlying libraries One example is HTSJDK where severalperformance patches have been submitted and includedimproving NGS data analysis performance not only for usersof GensearchNGS but also for the complete bioinformaticscommunity
Disclosure
Pierre Kuonen has a nonpaid advisory position at Phenosys-tems SA David Atlan is working full time for PhenosystemsSA
10 BioMed Research International
Conflict of Interests
Thomas Dandekar has no conflict of interests regarding thepublication of this paper
Acknowledgments
GensearchNGS is a commercially available software dis-tributed by Phenosystems SA Beat Wolf rsquos PhD thesis ispartially financially supported by Phenosystems SA
References
[1] E L van Dijk H Auger Y Jaszczyszyn and C ThermesldquoTen years of next-generation sequencing technologyrdquo Trendsin Genetics vol 30 no 9 pp 418ndash426 2014
[2] B Sikkema-Raddatz L F Johansson E N de Boer et al ldquoTar-geted next-generation sequencing can replace sanger sequenc-ing in clinical diagnosticsrdquo Human Mutation vol 34 no 7 pp1035ndash1042 2013
[3] H P J Buermans and J T den Dunnen ldquoNext generationsequencing technology advances and applicationsrdquo Biochimicaet Biophysica ActamdashMolecular Basis of Disease vol 1842 no 10pp 1932ndash1941 2014
[4] FastQC ldquoA quality control tool for high throughput sequencedatardquo Babraham Bioinformatics Website httpwwwbioinfor-maticsbabrahamacukprojectsfastqc
[5] W R Pearson T Wood Z Zhang andWMiller ldquoComparisonof DNA sequences with protein sequencesrdquo Genomics vol 46no 1 pp 24ndash36 1997
[6] H Li and N Homer ldquoA survey of sequence alignment algo-rithms for next-generation sequencingrdquo Briefings in Bioinfor-matics vol 11 no 5 pp 473ndash483 2010
[7] H Li and R Durbin ldquoFast and accurate long-read alignmentwith Burrows-Wheeler transformrdquo Bioinformatics vol 26 no5 pp 589ndash595 2010
[8] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[9] S F Altschul T L Madden A A Schaffer et al ldquoGappedBLAST and PSI-BLAST a new generation of protein databasesearch programsrdquo Nucleic Acids Research vol 25 no 17 pp3389ndash3402 1997
[10] M A DePristo E Banks R Poplin et al ldquoA framework forvariation discovery and genotyping using next-generationDNAsequencing datardquo Nature Genetics vol 43 no 5 pp 491ndash4982011
[11] P Carnevali J Baccash A L Halpern et al ldquoComputationaltechniques for human genome resequencing using matedgapped readsrdquo Journal of Computational Biology vol 19 no 3pp 279ndash292 2012
[12] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[13] A R Quinlan and I M Hall ldquoBEDTools a flexible suite ofutilities for comparing genomic featuresrdquo Bioinformatics vol26 no 6 pp 841ndash842 2010
[14] D C Koboldt Q Zhang D E Larson et al ldquoVarScan 2 somaticmutation and copy number alteration discovery in cancer byexome sequencingrdquo Genome Research vol 22 no 3 pp 568ndash576 2012
[15] J OrsquoRawe T Jiang G Sun et al ldquoLow concordance of multiplevariant-calling pipelines practical implications for exome andgenome sequencingrdquo Genome Medicine vol 5 no 3 article 282013
[16] S T SherryM-HWardM Kholodov et al ldquodbSNP the NCBIdatabase of genetic variationrdquoNucleic Acids Research vol 29 pp308ndash311 2001
[17] W McLaren B Pritchard D Rios Y Chen P Flicek and FCunningham ldquoDeriving the consequences of genomic variantswith the EnsemblAPI and SNPEffect PredictorrdquoBioinformaticsvol 26 no 16 Article ID btq330 pp 2069ndash2070 2010
[18] P N Robinson and S Mundlos ldquoThe human phenotypeontologyrdquo Clinical Genetics vol 77 no 6 pp 525ndash534 2010
[19] P Danecek A Auton G Abecasis et al ldquoThe variant call formatand VCFtoolsrdquo Bioinformatics vol 27 no 15 pp 2156ndash21582011
[20] J T Robinson HThorvaldsdottir W Winckler et al ldquoIntegra-tive genomics viewerrdquo Nature Biotechnology vol 29 no 1 pp24ndash26 2011
[21] I Milne G Stephen M Bayer et al ldquoUsing tablet for visualexploration of second-generation sequencing datardquo Briefings inBioinformatics vol 14 no 2 Article ID bbs012 pp 193ndash2022013
[22] T Abeel T van Parys Y Saeys J Galagan and Y van de PeerldquoGenomeView a next-generation genome browserrdquo NucleicAcids Research vol 40 no 2 article e12 2012
[23] W J Kent C W Sugnet T S Furey et al ldquoThe human genomebrowser at UCSCrdquo Genome Research vol 12 no 6 pp 996ndash1006 2002
[24] F Cunningham M Ridwan Amode D Barrell et al ldquoEnsembl2015rdquo Nucleic Acids Research vol 43 no 1 pp D662ndashD6692015
[25] NGS Data Analysis Software List SEQanswers 2012 httpseqanswerscomwikiSoftwarelist
[26] CLC Genomics Workbench CLC Bio Aarhus Denmark 2014httpwwwclcbiocom
[27] NextGENe Softgenetics State College Pa USA httpwwwsoftgeneticscom
[28] J Goecks A Nekrutenko J Taylor et al ldquoGalaxy a com-prehensive approach for supporting accessible reproducibleand transparent computational research in the life sciencesrdquoGenome Biology vol 11 no 8 article R86 2010
[29] G Lunter and M Goodson ldquoStampy a statistical algorithm forsensitive and fast mapping of Illumina sequence readsrdquoGenomeResearch vol 21 no 6 pp 936ndash939 2011
[30] Online Mendelian Inheritance in Man OMIM McKusick-Nathans Institute of Genetic Medicine Johns Hopkins Univer-sity Baltimore Md USA 2015 httpomimorg
[31] Cafe Variome httpwwwcafevariomeorg[32] The Genome Sequencing Consortium ldquoInitial sequencing and
analysis of the human genomerdquo Nature vol 409 pp 860ndash9212001
[33] P Kumar S Henikoff and P C Ng ldquoPredicting the effects ofcoding non-synonymous variants on protein function using theSIFT algorithmrdquo Nature Protocols vol 4 no 7 pp 1073ndash10822009
[34] I A Adzhubei S Schmidt L Peshkin et al ldquoA method andserver for predicting damaging missense mutationsrdquo NatureMethods vol 7 no 4 pp 248ndash249 2010
[35] Alamut Visual Interactive Biosoftware Rouen France httpwwwinteractivebiosoftwarecomalamut-visual
BioMed Research International 11
[36] S Richards N Aziz S Bale et al ldquoStandards and guidelinesfor the interpretation of sequence variants a joint consensusrecommendation of the American College of Medical Geneticsand Genomics and the Association for Molecular PathologyrdquoGenetics in Medicine vol 17 no 5 pp 405ndash423 2015
[37] BWolf and P Kuonen ldquoA novel approach for heuristic pairwiseDNA sequence alignmentrdquo in Proceedings of the InternationalConference on Bioinformatics amp Computational Biology (BIO-COMP rsquo13) Las Vegas Nev USA July 2013
[38] O Gotoh ldquoAn improved algorithm for matching biologicalsequencesrdquo Journal ofMolecular Biology vol 162 no 3 pp 705ndash708 1982
[39] D Smedley S Kohler J C Czeschik et al ldquoWalking theinteractome for candidate prioritization in exome sequencingstudies of Mendelian diseasesrdquo Bioinformatics vol 30 no 22pp 3215ndash3222 2014
[40] A L Semmler S Sacconi J Bach et al ldquoUnusual multisys-temic involvement and a novel BAG3 mutation revealed byNGS screening in a large cohort of myofibrillar myopathiesrdquoOrphanet Journal of Rare Diseases vol 9 no 1 p 121 2014
[41] M Larsen S Rost N El Hajj et al ldquoDiagnostic approach forFSHD revisited SMCHD1 mutations cause FSHD2 and act asmodifiers of disease severity in FSHD1rdquo European Journal ofHuman Genetics vol 23 no 6 pp 808ndash816 2015
[42] S Rost E Bach C Neuner et al ldquoNovel form of X-linkednonsyndromic hearing loss with cochlear malformation causedby a mutation in the type IV collagen gene COL4A6rdquo EuropeanJournal of Human Genetics vol 22 no 2 pp 208ndash215 2014
[43] M Broman I Kleinschnitz J E Bach S Rost G Islander andC R Muller ldquoNext-generation DNA sequencing of a Swedishmalignant hyperthermia cohortrdquo Clinical Genetics 2014
[44] T Low S vanHeesch H van den Toorn et al ldquoQuantitative andqualitative proteome characteristics extracted from in-depthintegrated genomics and proteomics analysisrdquo Cell Reports vol5 no 5 pp 1469ndash1478 2013
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology
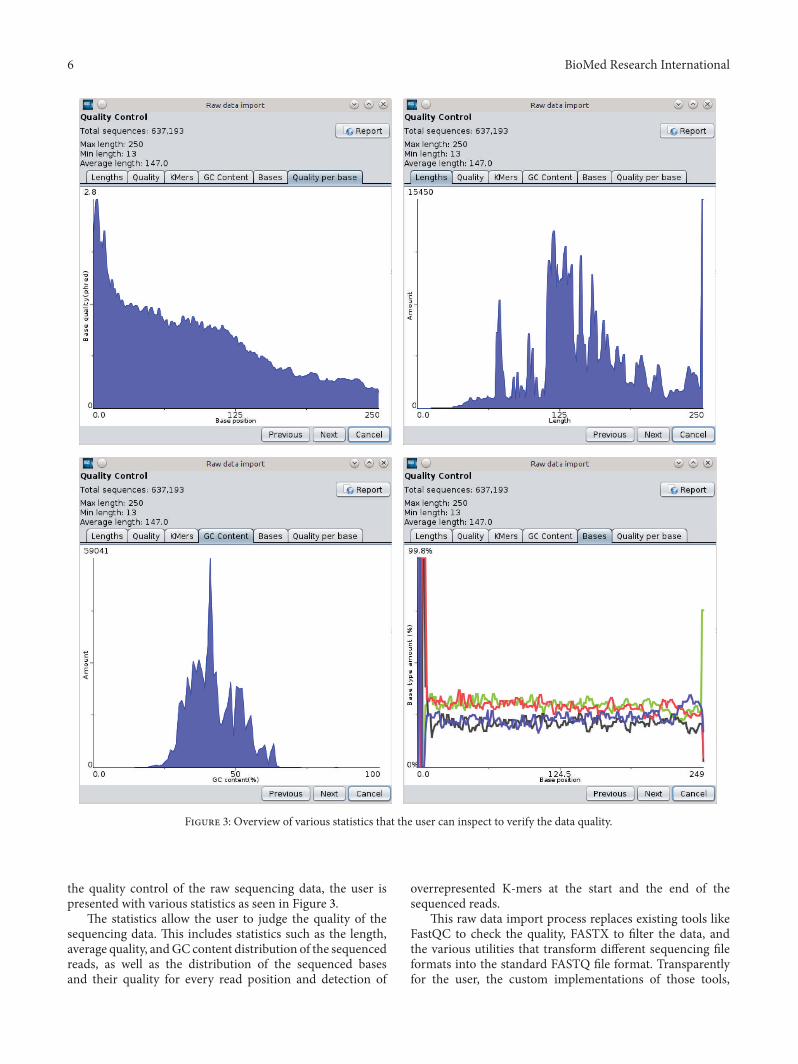
6 BioMed Research International
Figure 3 Overview of various statistics that the user can inspect to verify the data quality
the quality control of the raw sequencing data the user ispresented with various statistics as seen in Figure 3
The statistics allow the user to judge the quality of thesequencing data This includes statistics such as the lengthaverage quality andGC content distribution of the sequencedreads as well as the distribution of the sequenced basesand their quality for every read position and detection of
overrepresented K-mers at the start and the end of thesequenced reads
This raw data import process replaces existing tools likeFastQC to check the quality FASTX to filter the data andthe various utilities that transform different sequencing fileformats into the standard FASTQ file format Transparentlyfor the user the custom implementations of those tools
BioMed Research International 7
which produce the same results have been integrated into asingle processing stepThis includes the possibility to removeadapter sequences and split the raw sequencing data intomultiple files if they were produced through multiplexedsequencing Based on the reported quality information theuser can choose to filter the raw data following several filterssuch as maximum and minimum length of the sequencesand their minimal average quality He can also trim the 51015840and 31015840 parts of all reads or trim their excess length if theyare longer as a user defined length The trimming is done inaddition to the adapter and primer removalThe possibility tosplit the raw sequencing data based on barcodes in the readallowing the import of raw data from multiple samples thatwere sequenced at the same time is also integrated in theimport process
To align the sequenced data against a reference assemblyGensearchNGS provides the user with many options Severalexisting alignment algorithms are integrated into the applica-tion through plugins allowing their usage without the needof advanced technical know-how The currently integratedalignment algorithms are BWA [7] Bowtie 1 + 2 [8] andStampy [29]
Most of those alignment algorithms are developed in away that do not allow them to be used on multiple platformsIn fact most of them only run on Linux which is problematicfor a multiplatform application like GensearchNGS For thisreason a custom alignment algorithm written in Java hasbeen integrated directly into GensearchNGS This allows theuser to perform the full NGS data analysis even on operatingsystems likeWindows not supported bymost bioinformaticstools
The aligner supports paired end reads and gapped align-ment and has been optimized to use less than 5GB ofRAM to perform a full genome alignment This makes itsuitable for most of todayrsquos workspace desktops It is fullymultithreaded to support all cores of the machine and isable to offload calculations as described in Section 34 tomultiple computers The aligner uses a hash based indexa concept made popular through the BLAST aligner [9]with a custom heuristic based candidate alignment positionsselection [37] The final alignment is being done using theGotoh [38] algorithm a dynamic programming algorithmused for gapped alignment
The custom alignment algorithm being implemented inJava does not achieve quite the same performance as nativelyrunning aligners like BWA For WGS datasets it is generally50 slower The advantage of the GensearchNGS alignerover external aligners is that it is available on all supportedplatforms allowing the user to perform the complete NGSdata analysis on all platforms But when running on aplatform which supports the external aligners and speed isa deciding factor the users are encouraged to use the alignersavailable as plugins
The quality of resulting alignment can be verified throughvarious metrics by the user He can easily verify how manysequences have been discarded during the alignment pro-cess and how many sequences have been aligned to thedifferent chromosomes in the reference sequence Advancedstatistics similar to the ones created by bedtools 2 can also
be inspected giving detailed coverage information of thepreviously defined regions of interest The collected qualitycontrol information can be exported as a report for archivingpurposes using one of various formats such as PDF HTMLor PNG
While initially GensearchNGS integrated Varscan 2 [14]as its variant caller a replacement was created to reduce theexternal dependencies and to improve the performance ofvariant calling A variant scanner based on the model ofVarscan 2 has been implemented and is in the process of beingpublished as a separate tool called GNATY available for freefor noncommercial usage (httpgnatyphenosystemscom)Our custom variant caller shows important performanceadvantages over the existing Varscan 2 implementation of thesame variant calling model while producing the same analy-sis results Indeed GensearchNGS shows speed improvementof up to 18 times for variant calling compared to Varscan2 providing a similar range of features such as filters thatexclude sequencing artifacts
To be able to further filter down the detected variantsbased on their relevancy to the samplersquos phenotype severalsources are used to annotate them with various informationThis includes the prediction of the variant effect on thedifferent transcripts it affects a feature closely modeledafter the Variant Effect Predictor (VEP) from Ensembl [17]GensearchNGS implements its own version of the the VEPalgorithm to be able to perform the effect prediction in afraction of the time compared to the online VEP serviceThisis is mainly achieved by doing all calculations locally withouthaving to transfer the data to a remote server Predictionssuch as the addition or removal of a stop codon in an affectedtranscript if a splice site is affected and many others canbe made with this algorithm Additional information suchas if the variant is known in any public database associatedclinical significance and SIFT [33] and PolyPhen 2 [34]predictions are retrieved through the Ensembl web serviceOther services like the Human Phenotype Ontology [18]provide the information about the phenotype associated witha specific gene a crucial information in diagnostics
To filter the annotated variants GensearchNGS takesan interactive approach which allows the user to modifydifferent basic filters such as minimum or maximum samplefrequency coverage read balance or quality There are alsoadvanced filters such as the predicted consequence of thevariant as determined by the annotation process or if acertain phenotype is associated with the affected genes Thisincludes the possibility to filter by population frequency ofa variant The filtering by population frequency does not yettake into account the ethnicity of the patient Figure 4 showsan example of this list with several user selected interactivefilters applied
Additional features are available to perform a more in-depth analysis of variants such as family analysis where thevariants of multiple patients can be compared to find thoseonly present in a certain group of patients The usage ofpedigree information is of growing importance in NGS dataanalysis as it allows the user to filter sequencing artifactsand noncausative variants This is a very important featureespecially when usingWGSwhere the amount of variants can
8 BioMed Research International
Figure 4 Integrated variants list with several interactive filtersapplied
be very large Thanks to the usage of standard file formatslike VCF external tools like exome walker [39] can be usedto perform more advanced family analysis for features notcovered in GensearchNGS In contrast to many other NGSdata analysis software packages GensearchNGS does notfocus on the automation of every aspect of the analysisWhilecertain parts can be automated such as performing sequencealignment and variant calling on raw sequencing data thephilosophy of our software is to let the user to control thedifferent analysis steps This gives him enough flexibility toadapt to the data he is currently analysing without slowinghim down by having project specific analysis options whichare preselected for every analysis step This makes it possibleto perform most types of analysis with the project specificdefault options through a simple press of a button
33 Genome Browser A central tool of GensearchNGS isthe integrated genome browser to visualize the alignedsequencing data The genome browser as shown in Figure 5allows to interactively browse the sequence alignments Itis possible to display the aligned sequences and additionalgenomic data such as the gene locations and the encodedproteins by different transcripts
The genome browser allows the user to display sev-eral tracks containing information which helps to analysethe sequencing data The predefined tracks which can beactivated or deactivated allow the user to display coverageinformation GC content of the reference sequence thequality of the sequenced reads or custom annotation files instandard file formats such as BED WIG and GFF The focuswas put on optimizing the display of the data allowing theuser to visualize large datafiles without requiring a powerfulcomputerThis is achieved by streaming the data on demandonly loading the parts of the data currently needed forthe visualization The information about which variants arepresent in the alignment is available to the user in the sameform as the variant list in the main application It can be seenin Figure 4 allowing the user to quickly jump to the variantsof interest The genome browser like all of GensearchNGSis heavily connected with external data sources (EnsembldbSNP Entrez OMIM Blast and HPO) providing the user
Figure 5 Example view of the genome browser showing severalavailable tracks
with all relevant informationwhenneededTheuser can openexternal resources linked from many features in the visual-ized data such as genes transcripts or variants to retrieveadditional informationThe genome browser takes advantageof the integrated data sources like the Ensembl gene modelto display and interactively update encoded proteins basedon the variants present in the visualized data The genomebrowser is tightly integrated into GensearchNGS allowingthe user to easily visualize the alignment data belonging tofeatures of the sample
34 Distributed Computing The lack of computing powerto perform NGS data analysis is a very common issue inmany laboratories To address this problem GensearchNGSdoes not only optimize the algorithms for speed andmemoryusage but also try to exploit the already existing computinginfrastructure One way to do this is to use distributedcomputing a technique which allows the user to combinemultiple computers to perform computing intensive tasksThose computers can be in the same local network or at aremote location One of the most demanding steps in NGSdata analysis is sequence alignment For this reason it is thefirst feature to take advantage of this technique in Gensearch-NGS allowing the user to joinmultiple computers in the samenetwork to perform sequence alignment The approach usedby GensearchNGS is to transparently on request by the useruse all computers within the same networkwhereGensearch-NGS is installed Additionally the user has the possibility tostart up GensearchNGS instances in a cloud Currently onlythe amazon cloud is supportedThis process works regardlessof the operating system used by the different installationsof GensearchNGS By using an automatic service discoveryprotocol every installation of GensearchNGS located in thesame network is discovered without the need for manualconfigurationThis allows the user to use multiple computersat the same time or to completely offload calculations to adifferent computer making it possible to use computers notsuited for full genome alignments as an analysis workstationThe distributed sequence aligner used in GensearchNGS
BioMed Research International 9
currently only supports the GensearchNGS aligner algorithmand not external aligners such as BWA or Bowtie 2 It uses astream based approach where the client machine streams thedata to be aligned to all computers involved in the alignmentprocess This includes the client machine which receives thealigned sequences back and combines them into the finalalignment file The process of distributing the alignmentsover multiple computers and the cloud is transparent to theuser but it is optional because the usage of an externalcloud is not compatible with the security guidelines of somelaboratories GensearchNGS is not the onlyNGSdata analysissoftware using distributed sequence alignment It is thisability to combine the computing resources of the localcomputer multiple computers in the same network as well asthe cloud whichmakes its approach unique and very flexible
4 Discussion
GensearchNGS addresses problems that smaller laboratoriesare facing in various ways The issue of the technical com-plexity of the analysis is approached through an intuitive userinterface The user interface abstracts the complexity of theunderlying NGS data analysis toolchain which is a majorusability improvement over the usage of individual toolsNevertheless it gives the user full control over the individualanalysis steps instead of focusing on the creation of auto-mated analysis pipelines It also provides an original approachby providing a patient centric user interface This userfriendly interface allows users unfamiliar with the underlyingsoftware tools to fine tune the analysis parameters withoutoverwhelming themwith features or rarely used optionsThisincreases the number of people able to perform NGS dataanalysis This is particularly interesting for geneticists thatmight not have special bioinformatics training The issue ofincreasing infrastructure requirements for NGS data analysisis being addressed twofold First more efficient algorithmsare being developed like the heavily optimized variant callingalgorithm included in GensearchNGS Secondly the abilityto distribute heavy calculations over several computers oreven a cloud service has been developed GensearchNGShas a unique approach to this problem by enabling the userto combine the various distribution methods to performsequence alignment It provides the flexibility needed tochoose the best compromise between data privacy andalignment speed depending on the laboratory policies andneeds The resulting software enabled various laboratoriesto perform NGS data analysis and publish their results invarious fields The topics range from muscular dystrophies[40 41] to hearing loss [42] andmalignant hyperthermia [43]but also yet unpublished work about CNV analysis and thedetection of somatic mutations in cancer screening
5 Conclusion
In this paper we reviewed the workflow used to analyzeNGS data in a diagnostics environment The reviewedworkflow includes the detection of SNPs and short indelsfrom panel WES or WGS DNAseq data We also pre-sented issues faced by smaller laboratories trying to setup a
NGS data analysis pipeline We then presented Gensearch-NGS a software suite integrating various algorithms toperform the discussed workflow GensearchNGS is a com-mercially available software distributed by PhenosystemsSA A demo version of GensearchNGS is freely availableat httpwwwphenosystemscom The two main issues thetechnical complexity of the analysis and the computinginfrastructure requirements have been addressed as follows
To reduce the technical complexity of the analysis theuser is provided with an intuitive user interface which guideshim through the process of the data analysis He is providedwith the required tools to go from raw sequencing data the thefinal visualization and report generation for the sequencedsamples without having to learn complex bioinformaticstools The interface abstracts the technical complexity fromthe user while keeping enough flexibility to adapt to differentanalysis protocols By lowering the technical complexity ofNGS data analysis compared to existing individual analysistools GensearchNGS expands the potential user base of thesoftware
To address the issue of the computing infrastructurerequirements GensearchNGS focuses on the optimal usageof existing computing resources This is primarily achievedthrough the development of analysis algorithms that runon a minimal amount of resources while producing thesame analysis results as standard tools An example has beendescribed with the variant calling algorithm which producesthe same results as Varscan 2 but is 18 times faster Thesecond approach is the integration of distributed computingallowing the distribution of computing intensive tasks likealignment to be speed up by the usage of multiple computers
With a solid framework built for DNAseq analysisGensearchNGS will expand into further types of analysis inthe futureOne example is the integration of RNAseq analysisas the combination of OMICS data for a single sample isof increasing importance to understand rare diseases [44]In fact many parts of GensearchNGS such as the genomebrowser have already been adapted to support RNAseq dataThe integration of multiple data sources will not only allowthe user to integrate different types of OMICS data but alsoallow multiple views on the same sample One example ofthis is the integration of Sanger sequencing to NGS dataa very useful feature for diagnostics where it is commonpractice to validate variants identified through NGS withSanger sequencing
Further optimizations of the algorithms used in theanalysis will also be evaluated including optimization for theunderlying libraries One example is HTSJDK where severalperformance patches have been submitted and includedimproving NGS data analysis performance not only for usersof GensearchNGS but also for the complete bioinformaticscommunity
Disclosure
Pierre Kuonen has a nonpaid advisory position at Phenosys-tems SA David Atlan is working full time for PhenosystemsSA
10 BioMed Research International
Conflict of Interests
Thomas Dandekar has no conflict of interests regarding thepublication of this paper
Acknowledgments
GensearchNGS is a commercially available software dis-tributed by Phenosystems SA Beat Wolf rsquos PhD thesis ispartially financially supported by Phenosystems SA
References
[1] E L van Dijk H Auger Y Jaszczyszyn and C ThermesldquoTen years of next-generation sequencing technologyrdquo Trendsin Genetics vol 30 no 9 pp 418ndash426 2014
[2] B Sikkema-Raddatz L F Johansson E N de Boer et al ldquoTar-geted next-generation sequencing can replace sanger sequenc-ing in clinical diagnosticsrdquo Human Mutation vol 34 no 7 pp1035ndash1042 2013
[3] H P J Buermans and J T den Dunnen ldquoNext generationsequencing technology advances and applicationsrdquo Biochimicaet Biophysica ActamdashMolecular Basis of Disease vol 1842 no 10pp 1932ndash1941 2014
[4] FastQC ldquoA quality control tool for high throughput sequencedatardquo Babraham Bioinformatics Website httpwwwbioinfor-maticsbabrahamacukprojectsfastqc
[5] W R Pearson T Wood Z Zhang andWMiller ldquoComparisonof DNA sequences with protein sequencesrdquo Genomics vol 46no 1 pp 24ndash36 1997
[6] H Li and N Homer ldquoA survey of sequence alignment algo-rithms for next-generation sequencingrdquo Briefings in Bioinfor-matics vol 11 no 5 pp 473ndash483 2010
[7] H Li and R Durbin ldquoFast and accurate long-read alignmentwith Burrows-Wheeler transformrdquo Bioinformatics vol 26 no5 pp 589ndash595 2010
[8] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[9] S F Altschul T L Madden A A Schaffer et al ldquoGappedBLAST and PSI-BLAST a new generation of protein databasesearch programsrdquo Nucleic Acids Research vol 25 no 17 pp3389ndash3402 1997
[10] M A DePristo E Banks R Poplin et al ldquoA framework forvariation discovery and genotyping using next-generationDNAsequencing datardquo Nature Genetics vol 43 no 5 pp 491ndash4982011
[11] P Carnevali J Baccash A L Halpern et al ldquoComputationaltechniques for human genome resequencing using matedgapped readsrdquo Journal of Computational Biology vol 19 no 3pp 279ndash292 2012
[12] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[13] A R Quinlan and I M Hall ldquoBEDTools a flexible suite ofutilities for comparing genomic featuresrdquo Bioinformatics vol26 no 6 pp 841ndash842 2010
[14] D C Koboldt Q Zhang D E Larson et al ldquoVarScan 2 somaticmutation and copy number alteration discovery in cancer byexome sequencingrdquo Genome Research vol 22 no 3 pp 568ndash576 2012
[15] J OrsquoRawe T Jiang G Sun et al ldquoLow concordance of multiplevariant-calling pipelines practical implications for exome andgenome sequencingrdquo Genome Medicine vol 5 no 3 article 282013
[16] S T SherryM-HWardM Kholodov et al ldquodbSNP the NCBIdatabase of genetic variationrdquoNucleic Acids Research vol 29 pp308ndash311 2001
[17] W McLaren B Pritchard D Rios Y Chen P Flicek and FCunningham ldquoDeriving the consequences of genomic variantswith the EnsemblAPI and SNPEffect PredictorrdquoBioinformaticsvol 26 no 16 Article ID btq330 pp 2069ndash2070 2010
[18] P N Robinson and S Mundlos ldquoThe human phenotypeontologyrdquo Clinical Genetics vol 77 no 6 pp 525ndash534 2010
[19] P Danecek A Auton G Abecasis et al ldquoThe variant call formatand VCFtoolsrdquo Bioinformatics vol 27 no 15 pp 2156ndash21582011
[20] J T Robinson HThorvaldsdottir W Winckler et al ldquoIntegra-tive genomics viewerrdquo Nature Biotechnology vol 29 no 1 pp24ndash26 2011
[21] I Milne G Stephen M Bayer et al ldquoUsing tablet for visualexploration of second-generation sequencing datardquo Briefings inBioinformatics vol 14 no 2 Article ID bbs012 pp 193ndash2022013
[22] T Abeel T van Parys Y Saeys J Galagan and Y van de PeerldquoGenomeView a next-generation genome browserrdquo NucleicAcids Research vol 40 no 2 article e12 2012
[23] W J Kent C W Sugnet T S Furey et al ldquoThe human genomebrowser at UCSCrdquo Genome Research vol 12 no 6 pp 996ndash1006 2002
[24] F Cunningham M Ridwan Amode D Barrell et al ldquoEnsembl2015rdquo Nucleic Acids Research vol 43 no 1 pp D662ndashD6692015
[25] NGS Data Analysis Software List SEQanswers 2012 httpseqanswerscomwikiSoftwarelist
[26] CLC Genomics Workbench CLC Bio Aarhus Denmark 2014httpwwwclcbiocom
[27] NextGENe Softgenetics State College Pa USA httpwwwsoftgeneticscom
[28] J Goecks A Nekrutenko J Taylor et al ldquoGalaxy a com-prehensive approach for supporting accessible reproducibleand transparent computational research in the life sciencesrdquoGenome Biology vol 11 no 8 article R86 2010
[29] G Lunter and M Goodson ldquoStampy a statistical algorithm forsensitive and fast mapping of Illumina sequence readsrdquoGenomeResearch vol 21 no 6 pp 936ndash939 2011
[30] Online Mendelian Inheritance in Man OMIM McKusick-Nathans Institute of Genetic Medicine Johns Hopkins Univer-sity Baltimore Md USA 2015 httpomimorg
[31] Cafe Variome httpwwwcafevariomeorg[32] The Genome Sequencing Consortium ldquoInitial sequencing and
analysis of the human genomerdquo Nature vol 409 pp 860ndash9212001
[33] P Kumar S Henikoff and P C Ng ldquoPredicting the effects ofcoding non-synonymous variants on protein function using theSIFT algorithmrdquo Nature Protocols vol 4 no 7 pp 1073ndash10822009
[34] I A Adzhubei S Schmidt L Peshkin et al ldquoA method andserver for predicting damaging missense mutationsrdquo NatureMethods vol 7 no 4 pp 248ndash249 2010
[35] Alamut Visual Interactive Biosoftware Rouen France httpwwwinteractivebiosoftwarecomalamut-visual
BioMed Research International 11
[36] S Richards N Aziz S Bale et al ldquoStandards and guidelinesfor the interpretation of sequence variants a joint consensusrecommendation of the American College of Medical Geneticsand Genomics and the Association for Molecular PathologyrdquoGenetics in Medicine vol 17 no 5 pp 405ndash423 2015
[37] BWolf and P Kuonen ldquoA novel approach for heuristic pairwiseDNA sequence alignmentrdquo in Proceedings of the InternationalConference on Bioinformatics amp Computational Biology (BIO-COMP rsquo13) Las Vegas Nev USA July 2013
[38] O Gotoh ldquoAn improved algorithm for matching biologicalsequencesrdquo Journal ofMolecular Biology vol 162 no 3 pp 705ndash708 1982
[39] D Smedley S Kohler J C Czeschik et al ldquoWalking theinteractome for candidate prioritization in exome sequencingstudies of Mendelian diseasesrdquo Bioinformatics vol 30 no 22pp 3215ndash3222 2014
[40] A L Semmler S Sacconi J Bach et al ldquoUnusual multisys-temic involvement and a novel BAG3 mutation revealed byNGS screening in a large cohort of myofibrillar myopathiesrdquoOrphanet Journal of Rare Diseases vol 9 no 1 p 121 2014
[41] M Larsen S Rost N El Hajj et al ldquoDiagnostic approach forFSHD revisited SMCHD1 mutations cause FSHD2 and act asmodifiers of disease severity in FSHD1rdquo European Journal ofHuman Genetics vol 23 no 6 pp 808ndash816 2015
[42] S Rost E Bach C Neuner et al ldquoNovel form of X-linkednonsyndromic hearing loss with cochlear malformation causedby a mutation in the type IV collagen gene COL4A6rdquo EuropeanJournal of Human Genetics vol 22 no 2 pp 208ndash215 2014
[43] M Broman I Kleinschnitz J E Bach S Rost G Islander andC R Muller ldquoNext-generation DNA sequencing of a Swedishmalignant hyperthermia cohortrdquo Clinical Genetics 2014
[44] T Low S vanHeesch H van den Toorn et al ldquoQuantitative andqualitative proteome characteristics extracted from in-depthintegrated genomics and proteomics analysisrdquo Cell Reports vol5 no 5 pp 1469ndash1478 2013
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

BioMed Research International 7
which produce the same results have been integrated into asingle processing stepThis includes the possibility to removeadapter sequences and split the raw sequencing data intomultiple files if they were produced through multiplexedsequencing Based on the reported quality information theuser can choose to filter the raw data following several filterssuch as maximum and minimum length of the sequencesand their minimal average quality He can also trim the 51015840and 31015840 parts of all reads or trim their excess length if theyare longer as a user defined length The trimming is done inaddition to the adapter and primer removalThe possibility tosplit the raw sequencing data based on barcodes in the readallowing the import of raw data from multiple samples thatwere sequenced at the same time is also integrated in theimport process
To align the sequenced data against a reference assemblyGensearchNGS provides the user with many options Severalexisting alignment algorithms are integrated into the applica-tion through plugins allowing their usage without the needof advanced technical know-how The currently integratedalignment algorithms are BWA [7] Bowtie 1 + 2 [8] andStampy [29]
Most of those alignment algorithms are developed in away that do not allow them to be used on multiple platformsIn fact most of them only run on Linux which is problematicfor a multiplatform application like GensearchNGS For thisreason a custom alignment algorithm written in Java hasbeen integrated directly into GensearchNGS This allows theuser to perform the full NGS data analysis even on operatingsystems likeWindows not supported bymost bioinformaticstools
The aligner supports paired end reads and gapped align-ment and has been optimized to use less than 5GB ofRAM to perform a full genome alignment This makes itsuitable for most of todayrsquos workspace desktops It is fullymultithreaded to support all cores of the machine and isable to offload calculations as described in Section 34 tomultiple computers The aligner uses a hash based indexa concept made popular through the BLAST aligner [9]with a custom heuristic based candidate alignment positionsselection [37] The final alignment is being done using theGotoh [38] algorithm a dynamic programming algorithmused for gapped alignment
The custom alignment algorithm being implemented inJava does not achieve quite the same performance as nativelyrunning aligners like BWA For WGS datasets it is generally50 slower The advantage of the GensearchNGS alignerover external aligners is that it is available on all supportedplatforms allowing the user to perform the complete NGSdata analysis on all platforms But when running on aplatform which supports the external aligners and speed isa deciding factor the users are encouraged to use the alignersavailable as plugins
The quality of resulting alignment can be verified throughvarious metrics by the user He can easily verify how manysequences have been discarded during the alignment pro-cess and how many sequences have been aligned to thedifferent chromosomes in the reference sequence Advancedstatistics similar to the ones created by bedtools 2 can also
be inspected giving detailed coverage information of thepreviously defined regions of interest The collected qualitycontrol information can be exported as a report for archivingpurposes using one of various formats such as PDF HTMLor PNG
While initially GensearchNGS integrated Varscan 2 [14]as its variant caller a replacement was created to reduce theexternal dependencies and to improve the performance ofvariant calling A variant scanner based on the model ofVarscan 2 has been implemented and is in the process of beingpublished as a separate tool called GNATY available for freefor noncommercial usage (httpgnatyphenosystemscom)Our custom variant caller shows important performanceadvantages over the existing Varscan 2 implementation of thesame variant calling model while producing the same analy-sis results Indeed GensearchNGS shows speed improvementof up to 18 times for variant calling compared to Varscan2 providing a similar range of features such as filters thatexclude sequencing artifacts
To be able to further filter down the detected variantsbased on their relevancy to the samplersquos phenotype severalsources are used to annotate them with various informationThis includes the prediction of the variant effect on thedifferent transcripts it affects a feature closely modeledafter the Variant Effect Predictor (VEP) from Ensembl [17]GensearchNGS implements its own version of the the VEPalgorithm to be able to perform the effect prediction in afraction of the time compared to the online VEP serviceThisis is mainly achieved by doing all calculations locally withouthaving to transfer the data to a remote server Predictionssuch as the addition or removal of a stop codon in an affectedtranscript if a splice site is affected and many others canbe made with this algorithm Additional information suchas if the variant is known in any public database associatedclinical significance and SIFT [33] and PolyPhen 2 [34]predictions are retrieved through the Ensembl web serviceOther services like the Human Phenotype Ontology [18]provide the information about the phenotype associated witha specific gene a crucial information in diagnostics
To filter the annotated variants GensearchNGS takesan interactive approach which allows the user to modifydifferent basic filters such as minimum or maximum samplefrequency coverage read balance or quality There are alsoadvanced filters such as the predicted consequence of thevariant as determined by the annotation process or if acertain phenotype is associated with the affected genes Thisincludes the possibility to filter by population frequency ofa variant The filtering by population frequency does not yettake into account the ethnicity of the patient Figure 4 showsan example of this list with several user selected interactivefilters applied
Additional features are available to perform a more in-depth analysis of variants such as family analysis where thevariants of multiple patients can be compared to find thoseonly present in a certain group of patients The usage ofpedigree information is of growing importance in NGS dataanalysis as it allows the user to filter sequencing artifactsand noncausative variants This is a very important featureespecially when usingWGSwhere the amount of variants can
8 BioMed Research International
Figure 4 Integrated variants list with several interactive filtersapplied
be very large Thanks to the usage of standard file formatslike VCF external tools like exome walker [39] can be usedto perform more advanced family analysis for features notcovered in GensearchNGS In contrast to many other NGSdata analysis software packages GensearchNGS does notfocus on the automation of every aspect of the analysisWhilecertain parts can be automated such as performing sequencealignment and variant calling on raw sequencing data thephilosophy of our software is to let the user to control thedifferent analysis steps This gives him enough flexibility toadapt to the data he is currently analysing without slowinghim down by having project specific analysis options whichare preselected for every analysis step This makes it possibleto perform most types of analysis with the project specificdefault options through a simple press of a button
33 Genome Browser A central tool of GensearchNGS isthe integrated genome browser to visualize the alignedsequencing data The genome browser as shown in Figure 5allows to interactively browse the sequence alignments Itis possible to display the aligned sequences and additionalgenomic data such as the gene locations and the encodedproteins by different transcripts
The genome browser allows the user to display sev-eral tracks containing information which helps to analysethe sequencing data The predefined tracks which can beactivated or deactivated allow the user to display coverageinformation GC content of the reference sequence thequality of the sequenced reads or custom annotation files instandard file formats such as BED WIG and GFF The focuswas put on optimizing the display of the data allowing theuser to visualize large datafiles without requiring a powerfulcomputerThis is achieved by streaming the data on demandonly loading the parts of the data currently needed forthe visualization The information about which variants arepresent in the alignment is available to the user in the sameform as the variant list in the main application It can be seenin Figure 4 allowing the user to quickly jump to the variantsof interest The genome browser like all of GensearchNGSis heavily connected with external data sources (EnsembldbSNP Entrez OMIM Blast and HPO) providing the user
Figure 5 Example view of the genome browser showing severalavailable tracks
with all relevant informationwhenneededTheuser can openexternal resources linked from many features in the visual-ized data such as genes transcripts or variants to retrieveadditional informationThe genome browser takes advantageof the integrated data sources like the Ensembl gene modelto display and interactively update encoded proteins basedon the variants present in the visualized data The genomebrowser is tightly integrated into GensearchNGS allowingthe user to easily visualize the alignment data belonging tofeatures of the sample
34 Distributed Computing The lack of computing powerto perform NGS data analysis is a very common issue inmany laboratories To address this problem GensearchNGSdoes not only optimize the algorithms for speed andmemoryusage but also try to exploit the already existing computinginfrastructure One way to do this is to use distributedcomputing a technique which allows the user to combinemultiple computers to perform computing intensive tasksThose computers can be in the same local network or at aremote location One of the most demanding steps in NGSdata analysis is sequence alignment For this reason it is thefirst feature to take advantage of this technique in Gensearch-NGS allowing the user to joinmultiple computers in the samenetwork to perform sequence alignment The approach usedby GensearchNGS is to transparently on request by the useruse all computers within the same networkwhereGensearch-NGS is installed Additionally the user has the possibility tostart up GensearchNGS instances in a cloud Currently onlythe amazon cloud is supportedThis process works regardlessof the operating system used by the different installationsof GensearchNGS By using an automatic service discoveryprotocol every installation of GensearchNGS located in thesame network is discovered without the need for manualconfigurationThis allows the user to use multiple computersat the same time or to completely offload calculations to adifferent computer making it possible to use computers notsuited for full genome alignments as an analysis workstationThe distributed sequence aligner used in GensearchNGS
BioMed Research International 9
currently only supports the GensearchNGS aligner algorithmand not external aligners such as BWA or Bowtie 2 It uses astream based approach where the client machine streams thedata to be aligned to all computers involved in the alignmentprocess This includes the client machine which receives thealigned sequences back and combines them into the finalalignment file The process of distributing the alignmentsover multiple computers and the cloud is transparent to theuser but it is optional because the usage of an externalcloud is not compatible with the security guidelines of somelaboratories GensearchNGS is not the onlyNGSdata analysissoftware using distributed sequence alignment It is thisability to combine the computing resources of the localcomputer multiple computers in the same network as well asthe cloud whichmakes its approach unique and very flexible
4 Discussion
GensearchNGS addresses problems that smaller laboratoriesare facing in various ways The issue of the technical com-plexity of the analysis is approached through an intuitive userinterface The user interface abstracts the complexity of theunderlying NGS data analysis toolchain which is a majorusability improvement over the usage of individual toolsNevertheless it gives the user full control over the individualanalysis steps instead of focusing on the creation of auto-mated analysis pipelines It also provides an original approachby providing a patient centric user interface This userfriendly interface allows users unfamiliar with the underlyingsoftware tools to fine tune the analysis parameters withoutoverwhelming themwith features or rarely used optionsThisincreases the number of people able to perform NGS dataanalysis This is particularly interesting for geneticists thatmight not have special bioinformatics training The issue ofincreasing infrastructure requirements for NGS data analysisis being addressed twofold First more efficient algorithmsare being developed like the heavily optimized variant callingalgorithm included in GensearchNGS Secondly the abilityto distribute heavy calculations over several computers oreven a cloud service has been developed GensearchNGShas a unique approach to this problem by enabling the userto combine the various distribution methods to performsequence alignment It provides the flexibility needed tochoose the best compromise between data privacy andalignment speed depending on the laboratory policies andneeds The resulting software enabled various laboratoriesto perform NGS data analysis and publish their results invarious fields The topics range from muscular dystrophies[40 41] to hearing loss [42] andmalignant hyperthermia [43]but also yet unpublished work about CNV analysis and thedetection of somatic mutations in cancer screening
5 Conclusion
In this paper we reviewed the workflow used to analyzeNGS data in a diagnostics environment The reviewedworkflow includes the detection of SNPs and short indelsfrom panel WES or WGS DNAseq data We also pre-sented issues faced by smaller laboratories trying to setup a
NGS data analysis pipeline We then presented Gensearch-NGS a software suite integrating various algorithms toperform the discussed workflow GensearchNGS is a com-mercially available software distributed by PhenosystemsSA A demo version of GensearchNGS is freely availableat httpwwwphenosystemscom The two main issues thetechnical complexity of the analysis and the computinginfrastructure requirements have been addressed as follows
To reduce the technical complexity of the analysis theuser is provided with an intuitive user interface which guideshim through the process of the data analysis He is providedwith the required tools to go from raw sequencing data the thefinal visualization and report generation for the sequencedsamples without having to learn complex bioinformaticstools The interface abstracts the technical complexity fromthe user while keeping enough flexibility to adapt to differentanalysis protocols By lowering the technical complexity ofNGS data analysis compared to existing individual analysistools GensearchNGS expands the potential user base of thesoftware
To address the issue of the computing infrastructurerequirements GensearchNGS focuses on the optimal usageof existing computing resources This is primarily achievedthrough the development of analysis algorithms that runon a minimal amount of resources while producing thesame analysis results as standard tools An example has beendescribed with the variant calling algorithm which producesthe same results as Varscan 2 but is 18 times faster Thesecond approach is the integration of distributed computingallowing the distribution of computing intensive tasks likealignment to be speed up by the usage of multiple computers
With a solid framework built for DNAseq analysisGensearchNGS will expand into further types of analysis inthe futureOne example is the integration of RNAseq analysisas the combination of OMICS data for a single sample isof increasing importance to understand rare diseases [44]In fact many parts of GensearchNGS such as the genomebrowser have already been adapted to support RNAseq dataThe integration of multiple data sources will not only allowthe user to integrate different types of OMICS data but alsoallow multiple views on the same sample One example ofthis is the integration of Sanger sequencing to NGS dataa very useful feature for diagnostics where it is commonpractice to validate variants identified through NGS withSanger sequencing
Further optimizations of the algorithms used in theanalysis will also be evaluated including optimization for theunderlying libraries One example is HTSJDK where severalperformance patches have been submitted and includedimproving NGS data analysis performance not only for usersof GensearchNGS but also for the complete bioinformaticscommunity
Disclosure
Pierre Kuonen has a nonpaid advisory position at Phenosys-tems SA David Atlan is working full time for PhenosystemsSA
10 BioMed Research International
Conflict of Interests
Thomas Dandekar has no conflict of interests regarding thepublication of this paper
Acknowledgments
GensearchNGS is a commercially available software dis-tributed by Phenosystems SA Beat Wolf rsquos PhD thesis ispartially financially supported by Phenosystems SA
References
[1] E L van Dijk H Auger Y Jaszczyszyn and C ThermesldquoTen years of next-generation sequencing technologyrdquo Trendsin Genetics vol 30 no 9 pp 418ndash426 2014
[2] B Sikkema-Raddatz L F Johansson E N de Boer et al ldquoTar-geted next-generation sequencing can replace sanger sequenc-ing in clinical diagnosticsrdquo Human Mutation vol 34 no 7 pp1035ndash1042 2013
[3] H P J Buermans and J T den Dunnen ldquoNext generationsequencing technology advances and applicationsrdquo Biochimicaet Biophysica ActamdashMolecular Basis of Disease vol 1842 no 10pp 1932ndash1941 2014
[4] FastQC ldquoA quality control tool for high throughput sequencedatardquo Babraham Bioinformatics Website httpwwwbioinfor-maticsbabrahamacukprojectsfastqc
[5] W R Pearson T Wood Z Zhang andWMiller ldquoComparisonof DNA sequences with protein sequencesrdquo Genomics vol 46no 1 pp 24ndash36 1997
[6] H Li and N Homer ldquoA survey of sequence alignment algo-rithms for next-generation sequencingrdquo Briefings in Bioinfor-matics vol 11 no 5 pp 473ndash483 2010
[7] H Li and R Durbin ldquoFast and accurate long-read alignmentwith Burrows-Wheeler transformrdquo Bioinformatics vol 26 no5 pp 589ndash595 2010
[8] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[9] S F Altschul T L Madden A A Schaffer et al ldquoGappedBLAST and PSI-BLAST a new generation of protein databasesearch programsrdquo Nucleic Acids Research vol 25 no 17 pp3389ndash3402 1997
[10] M A DePristo E Banks R Poplin et al ldquoA framework forvariation discovery and genotyping using next-generationDNAsequencing datardquo Nature Genetics vol 43 no 5 pp 491ndash4982011
[11] P Carnevali J Baccash A L Halpern et al ldquoComputationaltechniques for human genome resequencing using matedgapped readsrdquo Journal of Computational Biology vol 19 no 3pp 279ndash292 2012
[12] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[13] A R Quinlan and I M Hall ldquoBEDTools a flexible suite ofutilities for comparing genomic featuresrdquo Bioinformatics vol26 no 6 pp 841ndash842 2010
[14] D C Koboldt Q Zhang D E Larson et al ldquoVarScan 2 somaticmutation and copy number alteration discovery in cancer byexome sequencingrdquo Genome Research vol 22 no 3 pp 568ndash576 2012
[15] J OrsquoRawe T Jiang G Sun et al ldquoLow concordance of multiplevariant-calling pipelines practical implications for exome andgenome sequencingrdquo Genome Medicine vol 5 no 3 article 282013
[16] S T SherryM-HWardM Kholodov et al ldquodbSNP the NCBIdatabase of genetic variationrdquoNucleic Acids Research vol 29 pp308ndash311 2001
[17] W McLaren B Pritchard D Rios Y Chen P Flicek and FCunningham ldquoDeriving the consequences of genomic variantswith the EnsemblAPI and SNPEffect PredictorrdquoBioinformaticsvol 26 no 16 Article ID btq330 pp 2069ndash2070 2010
[18] P N Robinson and S Mundlos ldquoThe human phenotypeontologyrdquo Clinical Genetics vol 77 no 6 pp 525ndash534 2010
[19] P Danecek A Auton G Abecasis et al ldquoThe variant call formatand VCFtoolsrdquo Bioinformatics vol 27 no 15 pp 2156ndash21582011
[20] J T Robinson HThorvaldsdottir W Winckler et al ldquoIntegra-tive genomics viewerrdquo Nature Biotechnology vol 29 no 1 pp24ndash26 2011
[21] I Milne G Stephen M Bayer et al ldquoUsing tablet for visualexploration of second-generation sequencing datardquo Briefings inBioinformatics vol 14 no 2 Article ID bbs012 pp 193ndash2022013
[22] T Abeel T van Parys Y Saeys J Galagan and Y van de PeerldquoGenomeView a next-generation genome browserrdquo NucleicAcids Research vol 40 no 2 article e12 2012
[23] W J Kent C W Sugnet T S Furey et al ldquoThe human genomebrowser at UCSCrdquo Genome Research vol 12 no 6 pp 996ndash1006 2002
[24] F Cunningham M Ridwan Amode D Barrell et al ldquoEnsembl2015rdquo Nucleic Acids Research vol 43 no 1 pp D662ndashD6692015
[25] NGS Data Analysis Software List SEQanswers 2012 httpseqanswerscomwikiSoftwarelist
[26] CLC Genomics Workbench CLC Bio Aarhus Denmark 2014httpwwwclcbiocom
[27] NextGENe Softgenetics State College Pa USA httpwwwsoftgeneticscom
[28] J Goecks A Nekrutenko J Taylor et al ldquoGalaxy a com-prehensive approach for supporting accessible reproducibleand transparent computational research in the life sciencesrdquoGenome Biology vol 11 no 8 article R86 2010
[29] G Lunter and M Goodson ldquoStampy a statistical algorithm forsensitive and fast mapping of Illumina sequence readsrdquoGenomeResearch vol 21 no 6 pp 936ndash939 2011
[30] Online Mendelian Inheritance in Man OMIM McKusick-Nathans Institute of Genetic Medicine Johns Hopkins Univer-sity Baltimore Md USA 2015 httpomimorg
[31] Cafe Variome httpwwwcafevariomeorg[32] The Genome Sequencing Consortium ldquoInitial sequencing and
analysis of the human genomerdquo Nature vol 409 pp 860ndash9212001
[33] P Kumar S Henikoff and P C Ng ldquoPredicting the effects ofcoding non-synonymous variants on protein function using theSIFT algorithmrdquo Nature Protocols vol 4 no 7 pp 1073ndash10822009
[34] I A Adzhubei S Schmidt L Peshkin et al ldquoA method andserver for predicting damaging missense mutationsrdquo NatureMethods vol 7 no 4 pp 248ndash249 2010
[35] Alamut Visual Interactive Biosoftware Rouen France httpwwwinteractivebiosoftwarecomalamut-visual
BioMed Research International 11
[36] S Richards N Aziz S Bale et al ldquoStandards and guidelinesfor the interpretation of sequence variants a joint consensusrecommendation of the American College of Medical Geneticsand Genomics and the Association for Molecular PathologyrdquoGenetics in Medicine vol 17 no 5 pp 405ndash423 2015
[37] BWolf and P Kuonen ldquoA novel approach for heuristic pairwiseDNA sequence alignmentrdquo in Proceedings of the InternationalConference on Bioinformatics amp Computational Biology (BIO-COMP rsquo13) Las Vegas Nev USA July 2013
[38] O Gotoh ldquoAn improved algorithm for matching biologicalsequencesrdquo Journal ofMolecular Biology vol 162 no 3 pp 705ndash708 1982
[39] D Smedley S Kohler J C Czeschik et al ldquoWalking theinteractome for candidate prioritization in exome sequencingstudies of Mendelian diseasesrdquo Bioinformatics vol 30 no 22pp 3215ndash3222 2014
[40] A L Semmler S Sacconi J Bach et al ldquoUnusual multisys-temic involvement and a novel BAG3 mutation revealed byNGS screening in a large cohort of myofibrillar myopathiesrdquoOrphanet Journal of Rare Diseases vol 9 no 1 p 121 2014
[41] M Larsen S Rost N El Hajj et al ldquoDiagnostic approach forFSHD revisited SMCHD1 mutations cause FSHD2 and act asmodifiers of disease severity in FSHD1rdquo European Journal ofHuman Genetics vol 23 no 6 pp 808ndash816 2015
[42] S Rost E Bach C Neuner et al ldquoNovel form of X-linkednonsyndromic hearing loss with cochlear malformation causedby a mutation in the type IV collagen gene COL4A6rdquo EuropeanJournal of Human Genetics vol 22 no 2 pp 208ndash215 2014
[43] M Broman I Kleinschnitz J E Bach S Rost G Islander andC R Muller ldquoNext-generation DNA sequencing of a Swedishmalignant hyperthermia cohortrdquo Clinical Genetics 2014
[44] T Low S vanHeesch H van den Toorn et al ldquoQuantitative andqualitative proteome characteristics extracted from in-depthintegrated genomics and proteomics analysisrdquo Cell Reports vol5 no 5 pp 1469ndash1478 2013
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

8 BioMed Research International
Figure 4 Integrated variants list with several interactive filtersapplied
be very large Thanks to the usage of standard file formatslike VCF external tools like exome walker [39] can be usedto perform more advanced family analysis for features notcovered in GensearchNGS In contrast to many other NGSdata analysis software packages GensearchNGS does notfocus on the automation of every aspect of the analysisWhilecertain parts can be automated such as performing sequencealignment and variant calling on raw sequencing data thephilosophy of our software is to let the user to control thedifferent analysis steps This gives him enough flexibility toadapt to the data he is currently analysing without slowinghim down by having project specific analysis options whichare preselected for every analysis step This makes it possibleto perform most types of analysis with the project specificdefault options through a simple press of a button
33 Genome Browser A central tool of GensearchNGS isthe integrated genome browser to visualize the alignedsequencing data The genome browser as shown in Figure 5allows to interactively browse the sequence alignments Itis possible to display the aligned sequences and additionalgenomic data such as the gene locations and the encodedproteins by different transcripts
The genome browser allows the user to display sev-eral tracks containing information which helps to analysethe sequencing data The predefined tracks which can beactivated or deactivated allow the user to display coverageinformation GC content of the reference sequence thequality of the sequenced reads or custom annotation files instandard file formats such as BED WIG and GFF The focuswas put on optimizing the display of the data allowing theuser to visualize large datafiles without requiring a powerfulcomputerThis is achieved by streaming the data on demandonly loading the parts of the data currently needed forthe visualization The information about which variants arepresent in the alignment is available to the user in the sameform as the variant list in the main application It can be seenin Figure 4 allowing the user to quickly jump to the variantsof interest The genome browser like all of GensearchNGSis heavily connected with external data sources (EnsembldbSNP Entrez OMIM Blast and HPO) providing the user
Figure 5 Example view of the genome browser showing severalavailable tracks
with all relevant informationwhenneededTheuser can openexternal resources linked from many features in the visual-ized data such as genes transcripts or variants to retrieveadditional informationThe genome browser takes advantageof the integrated data sources like the Ensembl gene modelto display and interactively update encoded proteins basedon the variants present in the visualized data The genomebrowser is tightly integrated into GensearchNGS allowingthe user to easily visualize the alignment data belonging tofeatures of the sample
34 Distributed Computing The lack of computing powerto perform NGS data analysis is a very common issue inmany laboratories To address this problem GensearchNGSdoes not only optimize the algorithms for speed andmemoryusage but also try to exploit the already existing computinginfrastructure One way to do this is to use distributedcomputing a technique which allows the user to combinemultiple computers to perform computing intensive tasksThose computers can be in the same local network or at aremote location One of the most demanding steps in NGSdata analysis is sequence alignment For this reason it is thefirst feature to take advantage of this technique in Gensearch-NGS allowing the user to joinmultiple computers in the samenetwork to perform sequence alignment The approach usedby GensearchNGS is to transparently on request by the useruse all computers within the same networkwhereGensearch-NGS is installed Additionally the user has the possibility tostart up GensearchNGS instances in a cloud Currently onlythe amazon cloud is supportedThis process works regardlessof the operating system used by the different installationsof GensearchNGS By using an automatic service discoveryprotocol every installation of GensearchNGS located in thesame network is discovered without the need for manualconfigurationThis allows the user to use multiple computersat the same time or to completely offload calculations to adifferent computer making it possible to use computers notsuited for full genome alignments as an analysis workstationThe distributed sequence aligner used in GensearchNGS
BioMed Research International 9
currently only supports the GensearchNGS aligner algorithmand not external aligners such as BWA or Bowtie 2 It uses astream based approach where the client machine streams thedata to be aligned to all computers involved in the alignmentprocess This includes the client machine which receives thealigned sequences back and combines them into the finalalignment file The process of distributing the alignmentsover multiple computers and the cloud is transparent to theuser but it is optional because the usage of an externalcloud is not compatible with the security guidelines of somelaboratories GensearchNGS is not the onlyNGSdata analysissoftware using distributed sequence alignment It is thisability to combine the computing resources of the localcomputer multiple computers in the same network as well asthe cloud whichmakes its approach unique and very flexible
4 Discussion
GensearchNGS addresses problems that smaller laboratoriesare facing in various ways The issue of the technical com-plexity of the analysis is approached through an intuitive userinterface The user interface abstracts the complexity of theunderlying NGS data analysis toolchain which is a majorusability improvement over the usage of individual toolsNevertheless it gives the user full control over the individualanalysis steps instead of focusing on the creation of auto-mated analysis pipelines It also provides an original approachby providing a patient centric user interface This userfriendly interface allows users unfamiliar with the underlyingsoftware tools to fine tune the analysis parameters withoutoverwhelming themwith features or rarely used optionsThisincreases the number of people able to perform NGS dataanalysis This is particularly interesting for geneticists thatmight not have special bioinformatics training The issue ofincreasing infrastructure requirements for NGS data analysisis being addressed twofold First more efficient algorithmsare being developed like the heavily optimized variant callingalgorithm included in GensearchNGS Secondly the abilityto distribute heavy calculations over several computers oreven a cloud service has been developed GensearchNGShas a unique approach to this problem by enabling the userto combine the various distribution methods to performsequence alignment It provides the flexibility needed tochoose the best compromise between data privacy andalignment speed depending on the laboratory policies andneeds The resulting software enabled various laboratoriesto perform NGS data analysis and publish their results invarious fields The topics range from muscular dystrophies[40 41] to hearing loss [42] andmalignant hyperthermia [43]but also yet unpublished work about CNV analysis and thedetection of somatic mutations in cancer screening
5 Conclusion
In this paper we reviewed the workflow used to analyzeNGS data in a diagnostics environment The reviewedworkflow includes the detection of SNPs and short indelsfrom panel WES or WGS DNAseq data We also pre-sented issues faced by smaller laboratories trying to setup a
NGS data analysis pipeline We then presented Gensearch-NGS a software suite integrating various algorithms toperform the discussed workflow GensearchNGS is a com-mercially available software distributed by PhenosystemsSA A demo version of GensearchNGS is freely availableat httpwwwphenosystemscom The two main issues thetechnical complexity of the analysis and the computinginfrastructure requirements have been addressed as follows
To reduce the technical complexity of the analysis theuser is provided with an intuitive user interface which guideshim through the process of the data analysis He is providedwith the required tools to go from raw sequencing data the thefinal visualization and report generation for the sequencedsamples without having to learn complex bioinformaticstools The interface abstracts the technical complexity fromthe user while keeping enough flexibility to adapt to differentanalysis protocols By lowering the technical complexity ofNGS data analysis compared to existing individual analysistools GensearchNGS expands the potential user base of thesoftware
To address the issue of the computing infrastructurerequirements GensearchNGS focuses on the optimal usageof existing computing resources This is primarily achievedthrough the development of analysis algorithms that runon a minimal amount of resources while producing thesame analysis results as standard tools An example has beendescribed with the variant calling algorithm which producesthe same results as Varscan 2 but is 18 times faster Thesecond approach is the integration of distributed computingallowing the distribution of computing intensive tasks likealignment to be speed up by the usage of multiple computers
With a solid framework built for DNAseq analysisGensearchNGS will expand into further types of analysis inthe futureOne example is the integration of RNAseq analysisas the combination of OMICS data for a single sample isof increasing importance to understand rare diseases [44]In fact many parts of GensearchNGS such as the genomebrowser have already been adapted to support RNAseq dataThe integration of multiple data sources will not only allowthe user to integrate different types of OMICS data but alsoallow multiple views on the same sample One example ofthis is the integration of Sanger sequencing to NGS dataa very useful feature for diagnostics where it is commonpractice to validate variants identified through NGS withSanger sequencing
Further optimizations of the algorithms used in theanalysis will also be evaluated including optimization for theunderlying libraries One example is HTSJDK where severalperformance patches have been submitted and includedimproving NGS data analysis performance not only for usersof GensearchNGS but also for the complete bioinformaticscommunity
Disclosure
Pierre Kuonen has a nonpaid advisory position at Phenosys-tems SA David Atlan is working full time for PhenosystemsSA
10 BioMed Research International
Conflict of Interests
Thomas Dandekar has no conflict of interests regarding thepublication of this paper
Acknowledgments
GensearchNGS is a commercially available software dis-tributed by Phenosystems SA Beat Wolf rsquos PhD thesis ispartially financially supported by Phenosystems SA
References
[1] E L van Dijk H Auger Y Jaszczyszyn and C ThermesldquoTen years of next-generation sequencing technologyrdquo Trendsin Genetics vol 30 no 9 pp 418ndash426 2014
[2] B Sikkema-Raddatz L F Johansson E N de Boer et al ldquoTar-geted next-generation sequencing can replace sanger sequenc-ing in clinical diagnosticsrdquo Human Mutation vol 34 no 7 pp1035ndash1042 2013
[3] H P J Buermans and J T den Dunnen ldquoNext generationsequencing technology advances and applicationsrdquo Biochimicaet Biophysica ActamdashMolecular Basis of Disease vol 1842 no 10pp 1932ndash1941 2014
[4] FastQC ldquoA quality control tool for high throughput sequencedatardquo Babraham Bioinformatics Website httpwwwbioinfor-maticsbabrahamacukprojectsfastqc
[5] W R Pearson T Wood Z Zhang andWMiller ldquoComparisonof DNA sequences with protein sequencesrdquo Genomics vol 46no 1 pp 24ndash36 1997
[6] H Li and N Homer ldquoA survey of sequence alignment algo-rithms for next-generation sequencingrdquo Briefings in Bioinfor-matics vol 11 no 5 pp 473ndash483 2010
[7] H Li and R Durbin ldquoFast and accurate long-read alignmentwith Burrows-Wheeler transformrdquo Bioinformatics vol 26 no5 pp 589ndash595 2010
[8] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[9] S F Altschul T L Madden A A Schaffer et al ldquoGappedBLAST and PSI-BLAST a new generation of protein databasesearch programsrdquo Nucleic Acids Research vol 25 no 17 pp3389ndash3402 1997
[10] M A DePristo E Banks R Poplin et al ldquoA framework forvariation discovery and genotyping using next-generationDNAsequencing datardquo Nature Genetics vol 43 no 5 pp 491ndash4982011
[11] P Carnevali J Baccash A L Halpern et al ldquoComputationaltechniques for human genome resequencing using matedgapped readsrdquo Journal of Computational Biology vol 19 no 3pp 279ndash292 2012
[12] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[13] A R Quinlan and I M Hall ldquoBEDTools a flexible suite ofutilities for comparing genomic featuresrdquo Bioinformatics vol26 no 6 pp 841ndash842 2010
[14] D C Koboldt Q Zhang D E Larson et al ldquoVarScan 2 somaticmutation and copy number alteration discovery in cancer byexome sequencingrdquo Genome Research vol 22 no 3 pp 568ndash576 2012
[15] J OrsquoRawe T Jiang G Sun et al ldquoLow concordance of multiplevariant-calling pipelines practical implications for exome andgenome sequencingrdquo Genome Medicine vol 5 no 3 article 282013
[16] S T SherryM-HWardM Kholodov et al ldquodbSNP the NCBIdatabase of genetic variationrdquoNucleic Acids Research vol 29 pp308ndash311 2001
[17] W McLaren B Pritchard D Rios Y Chen P Flicek and FCunningham ldquoDeriving the consequences of genomic variantswith the EnsemblAPI and SNPEffect PredictorrdquoBioinformaticsvol 26 no 16 Article ID btq330 pp 2069ndash2070 2010
[18] P N Robinson and S Mundlos ldquoThe human phenotypeontologyrdquo Clinical Genetics vol 77 no 6 pp 525ndash534 2010
[19] P Danecek A Auton G Abecasis et al ldquoThe variant call formatand VCFtoolsrdquo Bioinformatics vol 27 no 15 pp 2156ndash21582011
[20] J T Robinson HThorvaldsdottir W Winckler et al ldquoIntegra-tive genomics viewerrdquo Nature Biotechnology vol 29 no 1 pp24ndash26 2011
[21] I Milne G Stephen M Bayer et al ldquoUsing tablet for visualexploration of second-generation sequencing datardquo Briefings inBioinformatics vol 14 no 2 Article ID bbs012 pp 193ndash2022013
[22] T Abeel T van Parys Y Saeys J Galagan and Y van de PeerldquoGenomeView a next-generation genome browserrdquo NucleicAcids Research vol 40 no 2 article e12 2012
[23] W J Kent C W Sugnet T S Furey et al ldquoThe human genomebrowser at UCSCrdquo Genome Research vol 12 no 6 pp 996ndash1006 2002
[24] F Cunningham M Ridwan Amode D Barrell et al ldquoEnsembl2015rdquo Nucleic Acids Research vol 43 no 1 pp D662ndashD6692015
[25] NGS Data Analysis Software List SEQanswers 2012 httpseqanswerscomwikiSoftwarelist
[26] CLC Genomics Workbench CLC Bio Aarhus Denmark 2014httpwwwclcbiocom
[27] NextGENe Softgenetics State College Pa USA httpwwwsoftgeneticscom
[28] J Goecks A Nekrutenko J Taylor et al ldquoGalaxy a com-prehensive approach for supporting accessible reproducibleand transparent computational research in the life sciencesrdquoGenome Biology vol 11 no 8 article R86 2010
[29] G Lunter and M Goodson ldquoStampy a statistical algorithm forsensitive and fast mapping of Illumina sequence readsrdquoGenomeResearch vol 21 no 6 pp 936ndash939 2011
[30] Online Mendelian Inheritance in Man OMIM McKusick-Nathans Institute of Genetic Medicine Johns Hopkins Univer-sity Baltimore Md USA 2015 httpomimorg
[31] Cafe Variome httpwwwcafevariomeorg[32] The Genome Sequencing Consortium ldquoInitial sequencing and
analysis of the human genomerdquo Nature vol 409 pp 860ndash9212001
[33] P Kumar S Henikoff and P C Ng ldquoPredicting the effects ofcoding non-synonymous variants on protein function using theSIFT algorithmrdquo Nature Protocols vol 4 no 7 pp 1073ndash10822009
[34] I A Adzhubei S Schmidt L Peshkin et al ldquoA method andserver for predicting damaging missense mutationsrdquo NatureMethods vol 7 no 4 pp 248ndash249 2010
[35] Alamut Visual Interactive Biosoftware Rouen France httpwwwinteractivebiosoftwarecomalamut-visual
BioMed Research International 11
[36] S Richards N Aziz S Bale et al ldquoStandards and guidelinesfor the interpretation of sequence variants a joint consensusrecommendation of the American College of Medical Geneticsand Genomics and the Association for Molecular PathologyrdquoGenetics in Medicine vol 17 no 5 pp 405ndash423 2015
[37] BWolf and P Kuonen ldquoA novel approach for heuristic pairwiseDNA sequence alignmentrdquo in Proceedings of the InternationalConference on Bioinformatics amp Computational Biology (BIO-COMP rsquo13) Las Vegas Nev USA July 2013
[38] O Gotoh ldquoAn improved algorithm for matching biologicalsequencesrdquo Journal ofMolecular Biology vol 162 no 3 pp 705ndash708 1982
[39] D Smedley S Kohler J C Czeschik et al ldquoWalking theinteractome for candidate prioritization in exome sequencingstudies of Mendelian diseasesrdquo Bioinformatics vol 30 no 22pp 3215ndash3222 2014
[40] A L Semmler S Sacconi J Bach et al ldquoUnusual multisys-temic involvement and a novel BAG3 mutation revealed byNGS screening in a large cohort of myofibrillar myopathiesrdquoOrphanet Journal of Rare Diseases vol 9 no 1 p 121 2014
[41] M Larsen S Rost N El Hajj et al ldquoDiagnostic approach forFSHD revisited SMCHD1 mutations cause FSHD2 and act asmodifiers of disease severity in FSHD1rdquo European Journal ofHuman Genetics vol 23 no 6 pp 808ndash816 2015
[42] S Rost E Bach C Neuner et al ldquoNovel form of X-linkednonsyndromic hearing loss with cochlear malformation causedby a mutation in the type IV collagen gene COL4A6rdquo EuropeanJournal of Human Genetics vol 22 no 2 pp 208ndash215 2014
[43] M Broman I Kleinschnitz J E Bach S Rost G Islander andC R Muller ldquoNext-generation DNA sequencing of a Swedishmalignant hyperthermia cohortrdquo Clinical Genetics 2014
[44] T Low S vanHeesch H van den Toorn et al ldquoQuantitative andqualitative proteome characteristics extracted from in-depthintegrated genomics and proteomics analysisrdquo Cell Reports vol5 no 5 pp 1469ndash1478 2013
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

BioMed Research International 9
currently only supports the GensearchNGS aligner algorithmand not external aligners such as BWA or Bowtie 2 It uses astream based approach where the client machine streams thedata to be aligned to all computers involved in the alignmentprocess This includes the client machine which receives thealigned sequences back and combines them into the finalalignment file The process of distributing the alignmentsover multiple computers and the cloud is transparent to theuser but it is optional because the usage of an externalcloud is not compatible with the security guidelines of somelaboratories GensearchNGS is not the onlyNGSdata analysissoftware using distributed sequence alignment It is thisability to combine the computing resources of the localcomputer multiple computers in the same network as well asthe cloud whichmakes its approach unique and very flexible
4 Discussion
GensearchNGS addresses problems that smaller laboratoriesare facing in various ways The issue of the technical com-plexity of the analysis is approached through an intuitive userinterface The user interface abstracts the complexity of theunderlying NGS data analysis toolchain which is a majorusability improvement over the usage of individual toolsNevertheless it gives the user full control over the individualanalysis steps instead of focusing on the creation of auto-mated analysis pipelines It also provides an original approachby providing a patient centric user interface This userfriendly interface allows users unfamiliar with the underlyingsoftware tools to fine tune the analysis parameters withoutoverwhelming themwith features or rarely used optionsThisincreases the number of people able to perform NGS dataanalysis This is particularly interesting for geneticists thatmight not have special bioinformatics training The issue ofincreasing infrastructure requirements for NGS data analysisis being addressed twofold First more efficient algorithmsare being developed like the heavily optimized variant callingalgorithm included in GensearchNGS Secondly the abilityto distribute heavy calculations over several computers oreven a cloud service has been developed GensearchNGShas a unique approach to this problem by enabling the userto combine the various distribution methods to performsequence alignment It provides the flexibility needed tochoose the best compromise between data privacy andalignment speed depending on the laboratory policies andneeds The resulting software enabled various laboratoriesto perform NGS data analysis and publish their results invarious fields The topics range from muscular dystrophies[40 41] to hearing loss [42] andmalignant hyperthermia [43]but also yet unpublished work about CNV analysis and thedetection of somatic mutations in cancer screening
5 Conclusion
In this paper we reviewed the workflow used to analyzeNGS data in a diagnostics environment The reviewedworkflow includes the detection of SNPs and short indelsfrom panel WES or WGS DNAseq data We also pre-sented issues faced by smaller laboratories trying to setup a
NGS data analysis pipeline We then presented Gensearch-NGS a software suite integrating various algorithms toperform the discussed workflow GensearchNGS is a com-mercially available software distributed by PhenosystemsSA A demo version of GensearchNGS is freely availableat httpwwwphenosystemscom The two main issues thetechnical complexity of the analysis and the computinginfrastructure requirements have been addressed as follows
To reduce the technical complexity of the analysis theuser is provided with an intuitive user interface which guideshim through the process of the data analysis He is providedwith the required tools to go from raw sequencing data the thefinal visualization and report generation for the sequencedsamples without having to learn complex bioinformaticstools The interface abstracts the technical complexity fromthe user while keeping enough flexibility to adapt to differentanalysis protocols By lowering the technical complexity ofNGS data analysis compared to existing individual analysistools GensearchNGS expands the potential user base of thesoftware
To address the issue of the computing infrastructurerequirements GensearchNGS focuses on the optimal usageof existing computing resources This is primarily achievedthrough the development of analysis algorithms that runon a minimal amount of resources while producing thesame analysis results as standard tools An example has beendescribed with the variant calling algorithm which producesthe same results as Varscan 2 but is 18 times faster Thesecond approach is the integration of distributed computingallowing the distribution of computing intensive tasks likealignment to be speed up by the usage of multiple computers
With a solid framework built for DNAseq analysisGensearchNGS will expand into further types of analysis inthe futureOne example is the integration of RNAseq analysisas the combination of OMICS data for a single sample isof increasing importance to understand rare diseases [44]In fact many parts of GensearchNGS such as the genomebrowser have already been adapted to support RNAseq dataThe integration of multiple data sources will not only allowthe user to integrate different types of OMICS data but alsoallow multiple views on the same sample One example ofthis is the integration of Sanger sequencing to NGS dataa very useful feature for diagnostics where it is commonpractice to validate variants identified through NGS withSanger sequencing
Further optimizations of the algorithms used in theanalysis will also be evaluated including optimization for theunderlying libraries One example is HTSJDK where severalperformance patches have been submitted and includedimproving NGS data analysis performance not only for usersof GensearchNGS but also for the complete bioinformaticscommunity
Disclosure
Pierre Kuonen has a nonpaid advisory position at Phenosys-tems SA David Atlan is working full time for PhenosystemsSA
10 BioMed Research International
Conflict of Interests
Thomas Dandekar has no conflict of interests regarding thepublication of this paper
Acknowledgments
GensearchNGS is a commercially available software dis-tributed by Phenosystems SA Beat Wolf rsquos PhD thesis ispartially financially supported by Phenosystems SA
References
[1] E L van Dijk H Auger Y Jaszczyszyn and C ThermesldquoTen years of next-generation sequencing technologyrdquo Trendsin Genetics vol 30 no 9 pp 418ndash426 2014
[2] B Sikkema-Raddatz L F Johansson E N de Boer et al ldquoTar-geted next-generation sequencing can replace sanger sequenc-ing in clinical diagnosticsrdquo Human Mutation vol 34 no 7 pp1035ndash1042 2013
[3] H P J Buermans and J T den Dunnen ldquoNext generationsequencing technology advances and applicationsrdquo Biochimicaet Biophysica ActamdashMolecular Basis of Disease vol 1842 no 10pp 1932ndash1941 2014
[4] FastQC ldquoA quality control tool for high throughput sequencedatardquo Babraham Bioinformatics Website httpwwwbioinfor-maticsbabrahamacukprojectsfastqc
[5] W R Pearson T Wood Z Zhang andWMiller ldquoComparisonof DNA sequences with protein sequencesrdquo Genomics vol 46no 1 pp 24ndash36 1997
[6] H Li and N Homer ldquoA survey of sequence alignment algo-rithms for next-generation sequencingrdquo Briefings in Bioinfor-matics vol 11 no 5 pp 473ndash483 2010
[7] H Li and R Durbin ldquoFast and accurate long-read alignmentwith Burrows-Wheeler transformrdquo Bioinformatics vol 26 no5 pp 589ndash595 2010
[8] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[9] S F Altschul T L Madden A A Schaffer et al ldquoGappedBLAST and PSI-BLAST a new generation of protein databasesearch programsrdquo Nucleic Acids Research vol 25 no 17 pp3389ndash3402 1997
[10] M A DePristo E Banks R Poplin et al ldquoA framework forvariation discovery and genotyping using next-generationDNAsequencing datardquo Nature Genetics vol 43 no 5 pp 491ndash4982011
[11] P Carnevali J Baccash A L Halpern et al ldquoComputationaltechniques for human genome resequencing using matedgapped readsrdquo Journal of Computational Biology vol 19 no 3pp 279ndash292 2012
[12] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[13] A R Quinlan and I M Hall ldquoBEDTools a flexible suite ofutilities for comparing genomic featuresrdquo Bioinformatics vol26 no 6 pp 841ndash842 2010
[14] D C Koboldt Q Zhang D E Larson et al ldquoVarScan 2 somaticmutation and copy number alteration discovery in cancer byexome sequencingrdquo Genome Research vol 22 no 3 pp 568ndash576 2012
[15] J OrsquoRawe T Jiang G Sun et al ldquoLow concordance of multiplevariant-calling pipelines practical implications for exome andgenome sequencingrdquo Genome Medicine vol 5 no 3 article 282013
[16] S T SherryM-HWardM Kholodov et al ldquodbSNP the NCBIdatabase of genetic variationrdquoNucleic Acids Research vol 29 pp308ndash311 2001
[17] W McLaren B Pritchard D Rios Y Chen P Flicek and FCunningham ldquoDeriving the consequences of genomic variantswith the EnsemblAPI and SNPEffect PredictorrdquoBioinformaticsvol 26 no 16 Article ID btq330 pp 2069ndash2070 2010
[18] P N Robinson and S Mundlos ldquoThe human phenotypeontologyrdquo Clinical Genetics vol 77 no 6 pp 525ndash534 2010
[19] P Danecek A Auton G Abecasis et al ldquoThe variant call formatand VCFtoolsrdquo Bioinformatics vol 27 no 15 pp 2156ndash21582011
[20] J T Robinson HThorvaldsdottir W Winckler et al ldquoIntegra-tive genomics viewerrdquo Nature Biotechnology vol 29 no 1 pp24ndash26 2011
[21] I Milne G Stephen M Bayer et al ldquoUsing tablet for visualexploration of second-generation sequencing datardquo Briefings inBioinformatics vol 14 no 2 Article ID bbs012 pp 193ndash2022013
[22] T Abeel T van Parys Y Saeys J Galagan and Y van de PeerldquoGenomeView a next-generation genome browserrdquo NucleicAcids Research vol 40 no 2 article e12 2012
[23] W J Kent C W Sugnet T S Furey et al ldquoThe human genomebrowser at UCSCrdquo Genome Research vol 12 no 6 pp 996ndash1006 2002
[24] F Cunningham M Ridwan Amode D Barrell et al ldquoEnsembl2015rdquo Nucleic Acids Research vol 43 no 1 pp D662ndashD6692015
[25] NGS Data Analysis Software List SEQanswers 2012 httpseqanswerscomwikiSoftwarelist
[26] CLC Genomics Workbench CLC Bio Aarhus Denmark 2014httpwwwclcbiocom
[27] NextGENe Softgenetics State College Pa USA httpwwwsoftgeneticscom
[28] J Goecks A Nekrutenko J Taylor et al ldquoGalaxy a com-prehensive approach for supporting accessible reproducibleand transparent computational research in the life sciencesrdquoGenome Biology vol 11 no 8 article R86 2010
[29] G Lunter and M Goodson ldquoStampy a statistical algorithm forsensitive and fast mapping of Illumina sequence readsrdquoGenomeResearch vol 21 no 6 pp 936ndash939 2011
[30] Online Mendelian Inheritance in Man OMIM McKusick-Nathans Institute of Genetic Medicine Johns Hopkins Univer-sity Baltimore Md USA 2015 httpomimorg
[31] Cafe Variome httpwwwcafevariomeorg[32] The Genome Sequencing Consortium ldquoInitial sequencing and
analysis of the human genomerdquo Nature vol 409 pp 860ndash9212001
[33] P Kumar S Henikoff and P C Ng ldquoPredicting the effects ofcoding non-synonymous variants on protein function using theSIFT algorithmrdquo Nature Protocols vol 4 no 7 pp 1073ndash10822009
[34] I A Adzhubei S Schmidt L Peshkin et al ldquoA method andserver for predicting damaging missense mutationsrdquo NatureMethods vol 7 no 4 pp 248ndash249 2010
[35] Alamut Visual Interactive Biosoftware Rouen France httpwwwinteractivebiosoftwarecomalamut-visual
BioMed Research International 11
[36] S Richards N Aziz S Bale et al ldquoStandards and guidelinesfor the interpretation of sequence variants a joint consensusrecommendation of the American College of Medical Geneticsand Genomics and the Association for Molecular PathologyrdquoGenetics in Medicine vol 17 no 5 pp 405ndash423 2015
[37] BWolf and P Kuonen ldquoA novel approach for heuristic pairwiseDNA sequence alignmentrdquo in Proceedings of the InternationalConference on Bioinformatics amp Computational Biology (BIO-COMP rsquo13) Las Vegas Nev USA July 2013
[38] O Gotoh ldquoAn improved algorithm for matching biologicalsequencesrdquo Journal ofMolecular Biology vol 162 no 3 pp 705ndash708 1982
[39] D Smedley S Kohler J C Czeschik et al ldquoWalking theinteractome for candidate prioritization in exome sequencingstudies of Mendelian diseasesrdquo Bioinformatics vol 30 no 22pp 3215ndash3222 2014
[40] A L Semmler S Sacconi J Bach et al ldquoUnusual multisys-temic involvement and a novel BAG3 mutation revealed byNGS screening in a large cohort of myofibrillar myopathiesrdquoOrphanet Journal of Rare Diseases vol 9 no 1 p 121 2014
[41] M Larsen S Rost N El Hajj et al ldquoDiagnostic approach forFSHD revisited SMCHD1 mutations cause FSHD2 and act asmodifiers of disease severity in FSHD1rdquo European Journal ofHuman Genetics vol 23 no 6 pp 808ndash816 2015
[42] S Rost E Bach C Neuner et al ldquoNovel form of X-linkednonsyndromic hearing loss with cochlear malformation causedby a mutation in the type IV collagen gene COL4A6rdquo EuropeanJournal of Human Genetics vol 22 no 2 pp 208ndash215 2014
[43] M Broman I Kleinschnitz J E Bach S Rost G Islander andC R Muller ldquoNext-generation DNA sequencing of a Swedishmalignant hyperthermia cohortrdquo Clinical Genetics 2014
[44] T Low S vanHeesch H van den Toorn et al ldquoQuantitative andqualitative proteome characteristics extracted from in-depthintegrated genomics and proteomics analysisrdquo Cell Reports vol5 no 5 pp 1469ndash1478 2013
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

10 BioMed Research International
Conflict of Interests
Thomas Dandekar has no conflict of interests regarding thepublication of this paper
Acknowledgments
GensearchNGS is a commercially available software dis-tributed by Phenosystems SA Beat Wolf rsquos PhD thesis ispartially financially supported by Phenosystems SA
References
[1] E L van Dijk H Auger Y Jaszczyszyn and C ThermesldquoTen years of next-generation sequencing technologyrdquo Trendsin Genetics vol 30 no 9 pp 418ndash426 2014
[2] B Sikkema-Raddatz L F Johansson E N de Boer et al ldquoTar-geted next-generation sequencing can replace sanger sequenc-ing in clinical diagnosticsrdquo Human Mutation vol 34 no 7 pp1035ndash1042 2013
[3] H P J Buermans and J T den Dunnen ldquoNext generationsequencing technology advances and applicationsrdquo Biochimicaet Biophysica ActamdashMolecular Basis of Disease vol 1842 no 10pp 1932ndash1941 2014
[4] FastQC ldquoA quality control tool for high throughput sequencedatardquo Babraham Bioinformatics Website httpwwwbioinfor-maticsbabrahamacukprojectsfastqc
[5] W R Pearson T Wood Z Zhang andWMiller ldquoComparisonof DNA sequences with protein sequencesrdquo Genomics vol 46no 1 pp 24ndash36 1997
[6] H Li and N Homer ldquoA survey of sequence alignment algo-rithms for next-generation sequencingrdquo Briefings in Bioinfor-matics vol 11 no 5 pp 473ndash483 2010
[7] H Li and R Durbin ldquoFast and accurate long-read alignmentwith Burrows-Wheeler transformrdquo Bioinformatics vol 26 no5 pp 589ndash595 2010
[8] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[9] S F Altschul T L Madden A A Schaffer et al ldquoGappedBLAST and PSI-BLAST a new generation of protein databasesearch programsrdquo Nucleic Acids Research vol 25 no 17 pp3389ndash3402 1997
[10] M A DePristo E Banks R Poplin et al ldquoA framework forvariation discovery and genotyping using next-generationDNAsequencing datardquo Nature Genetics vol 43 no 5 pp 491ndash4982011
[11] P Carnevali J Baccash A L Halpern et al ldquoComputationaltechniques for human genome resequencing using matedgapped readsrdquo Journal of Computational Biology vol 19 no 3pp 279ndash292 2012
[12] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[13] A R Quinlan and I M Hall ldquoBEDTools a flexible suite ofutilities for comparing genomic featuresrdquo Bioinformatics vol26 no 6 pp 841ndash842 2010
[14] D C Koboldt Q Zhang D E Larson et al ldquoVarScan 2 somaticmutation and copy number alteration discovery in cancer byexome sequencingrdquo Genome Research vol 22 no 3 pp 568ndash576 2012
[15] J OrsquoRawe T Jiang G Sun et al ldquoLow concordance of multiplevariant-calling pipelines practical implications for exome andgenome sequencingrdquo Genome Medicine vol 5 no 3 article 282013
[16] S T SherryM-HWardM Kholodov et al ldquodbSNP the NCBIdatabase of genetic variationrdquoNucleic Acids Research vol 29 pp308ndash311 2001
[17] W McLaren B Pritchard D Rios Y Chen P Flicek and FCunningham ldquoDeriving the consequences of genomic variantswith the EnsemblAPI and SNPEffect PredictorrdquoBioinformaticsvol 26 no 16 Article ID btq330 pp 2069ndash2070 2010
[18] P N Robinson and S Mundlos ldquoThe human phenotypeontologyrdquo Clinical Genetics vol 77 no 6 pp 525ndash534 2010
[19] P Danecek A Auton G Abecasis et al ldquoThe variant call formatand VCFtoolsrdquo Bioinformatics vol 27 no 15 pp 2156ndash21582011
[20] J T Robinson HThorvaldsdottir W Winckler et al ldquoIntegra-tive genomics viewerrdquo Nature Biotechnology vol 29 no 1 pp24ndash26 2011
[21] I Milne G Stephen M Bayer et al ldquoUsing tablet for visualexploration of second-generation sequencing datardquo Briefings inBioinformatics vol 14 no 2 Article ID bbs012 pp 193ndash2022013
[22] T Abeel T van Parys Y Saeys J Galagan and Y van de PeerldquoGenomeView a next-generation genome browserrdquo NucleicAcids Research vol 40 no 2 article e12 2012
[23] W J Kent C W Sugnet T S Furey et al ldquoThe human genomebrowser at UCSCrdquo Genome Research vol 12 no 6 pp 996ndash1006 2002
[24] F Cunningham M Ridwan Amode D Barrell et al ldquoEnsembl2015rdquo Nucleic Acids Research vol 43 no 1 pp D662ndashD6692015
[25] NGS Data Analysis Software List SEQanswers 2012 httpseqanswerscomwikiSoftwarelist
[26] CLC Genomics Workbench CLC Bio Aarhus Denmark 2014httpwwwclcbiocom
[27] NextGENe Softgenetics State College Pa USA httpwwwsoftgeneticscom
[28] J Goecks A Nekrutenko J Taylor et al ldquoGalaxy a com-prehensive approach for supporting accessible reproducibleand transparent computational research in the life sciencesrdquoGenome Biology vol 11 no 8 article R86 2010
[29] G Lunter and M Goodson ldquoStampy a statistical algorithm forsensitive and fast mapping of Illumina sequence readsrdquoGenomeResearch vol 21 no 6 pp 936ndash939 2011
[30] Online Mendelian Inheritance in Man OMIM McKusick-Nathans Institute of Genetic Medicine Johns Hopkins Univer-sity Baltimore Md USA 2015 httpomimorg
[31] Cafe Variome httpwwwcafevariomeorg[32] The Genome Sequencing Consortium ldquoInitial sequencing and
analysis of the human genomerdquo Nature vol 409 pp 860ndash9212001
[33] P Kumar S Henikoff and P C Ng ldquoPredicting the effects ofcoding non-synonymous variants on protein function using theSIFT algorithmrdquo Nature Protocols vol 4 no 7 pp 1073ndash10822009
[34] I A Adzhubei S Schmidt L Peshkin et al ldquoA method andserver for predicting damaging missense mutationsrdquo NatureMethods vol 7 no 4 pp 248ndash249 2010
[35] Alamut Visual Interactive Biosoftware Rouen France httpwwwinteractivebiosoftwarecomalamut-visual
BioMed Research International 11
[36] S Richards N Aziz S Bale et al ldquoStandards and guidelinesfor the interpretation of sequence variants a joint consensusrecommendation of the American College of Medical Geneticsand Genomics and the Association for Molecular PathologyrdquoGenetics in Medicine vol 17 no 5 pp 405ndash423 2015
[37] BWolf and P Kuonen ldquoA novel approach for heuristic pairwiseDNA sequence alignmentrdquo in Proceedings of the InternationalConference on Bioinformatics amp Computational Biology (BIO-COMP rsquo13) Las Vegas Nev USA July 2013
[38] O Gotoh ldquoAn improved algorithm for matching biologicalsequencesrdquo Journal ofMolecular Biology vol 162 no 3 pp 705ndash708 1982
[39] D Smedley S Kohler J C Czeschik et al ldquoWalking theinteractome for candidate prioritization in exome sequencingstudies of Mendelian diseasesrdquo Bioinformatics vol 30 no 22pp 3215ndash3222 2014
[40] A L Semmler S Sacconi J Bach et al ldquoUnusual multisys-temic involvement and a novel BAG3 mutation revealed byNGS screening in a large cohort of myofibrillar myopathiesrdquoOrphanet Journal of Rare Diseases vol 9 no 1 p 121 2014
[41] M Larsen S Rost N El Hajj et al ldquoDiagnostic approach forFSHD revisited SMCHD1 mutations cause FSHD2 and act asmodifiers of disease severity in FSHD1rdquo European Journal ofHuman Genetics vol 23 no 6 pp 808ndash816 2015
[42] S Rost E Bach C Neuner et al ldquoNovel form of X-linkednonsyndromic hearing loss with cochlear malformation causedby a mutation in the type IV collagen gene COL4A6rdquo EuropeanJournal of Human Genetics vol 22 no 2 pp 208ndash215 2014
[43] M Broman I Kleinschnitz J E Bach S Rost G Islander andC R Muller ldquoNext-generation DNA sequencing of a Swedishmalignant hyperthermia cohortrdquo Clinical Genetics 2014
[44] T Low S vanHeesch H van den Toorn et al ldquoQuantitative andqualitative proteome characteristics extracted from in-depthintegrated genomics and proteomics analysisrdquo Cell Reports vol5 no 5 pp 1469ndash1478 2013
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

BioMed Research International 11
[36] S Richards N Aziz S Bale et al ldquoStandards and guidelinesfor the interpretation of sequence variants a joint consensusrecommendation of the American College of Medical Geneticsand Genomics and the Association for Molecular PathologyrdquoGenetics in Medicine vol 17 no 5 pp 405ndash423 2015
[37] BWolf and P Kuonen ldquoA novel approach for heuristic pairwiseDNA sequence alignmentrdquo in Proceedings of the InternationalConference on Bioinformatics amp Computational Biology (BIO-COMP rsquo13) Las Vegas Nev USA July 2013
[38] O Gotoh ldquoAn improved algorithm for matching biologicalsequencesrdquo Journal ofMolecular Biology vol 162 no 3 pp 705ndash708 1982
[39] D Smedley S Kohler J C Czeschik et al ldquoWalking theinteractome for candidate prioritization in exome sequencingstudies of Mendelian diseasesrdquo Bioinformatics vol 30 no 22pp 3215ndash3222 2014
[40] A L Semmler S Sacconi J Bach et al ldquoUnusual multisys-temic involvement and a novel BAG3 mutation revealed byNGS screening in a large cohort of myofibrillar myopathiesrdquoOrphanet Journal of Rare Diseases vol 9 no 1 p 121 2014
[41] M Larsen S Rost N El Hajj et al ldquoDiagnostic approach forFSHD revisited SMCHD1 mutations cause FSHD2 and act asmodifiers of disease severity in FSHD1rdquo European Journal ofHuman Genetics vol 23 no 6 pp 808ndash816 2015
[42] S Rost E Bach C Neuner et al ldquoNovel form of X-linkednonsyndromic hearing loss with cochlear malformation causedby a mutation in the type IV collagen gene COL4A6rdquo EuropeanJournal of Human Genetics vol 22 no 2 pp 208ndash215 2014
[43] M Broman I Kleinschnitz J E Bach S Rost G Islander andC R Muller ldquoNext-generation DNA sequencing of a Swedishmalignant hyperthermia cohortrdquo Clinical Genetics 2014
[44] T Low S vanHeesch H van den Toorn et al ldquoQuantitative andqualitative proteome characteristics extracted from in-depthintegrated genomics and proteomics analysisrdquo Cell Reports vol5 no 5 pp 1469ndash1478 2013
Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology

Submit your manuscripts athttpwwwhindawicom
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Anatomy Research International
PeptidesInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporation httpwwwhindawicom
International Journal of
Volume 2014
Zoology
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Molecular Biology International
GenomicsInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
The Scientific World JournalHindawi Publishing Corporation httpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioinformaticsAdvances in
Marine BiologyJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Signal TransductionJournal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
BioMed Research International
Evolutionary BiologyInternational Journal of
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Biochemistry Research International
ArchaeaHindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Genetics Research International
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Advances in
Virolog y
Hindawi Publishing Corporationhttpwwwhindawicom
Nucleic AcidsJournal of
Volume 2014
Stem CellsInternational
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
Enzyme Research
Hindawi Publishing Corporationhttpwwwhindawicom Volume 2014
International Journal of
Microbiology







![利用Ion半导体测序...序列质量调整、定位和计数 每个FASTQ文件经过最短序列长度的 过滤,并从每个读取的3'端读入,根 据每个碱基检出相关的质量值过滤。这是利用FASTX-工具箱实现的[8]。如](https://img.pdfslide.net/doc/110x75/5e2814495bd08a060f67b497/cion-eeefoee-fastqceoecec.jpg)


![Separation of breast cancer and organ …...classifying RNA from different species in PDX models. RNA-Seq pre-processing and quality control FastQC v.0.11.5 [19] was used for quality](https://img.pdfslide.net/doc/110x75/5f033fea7e708231d4084760/separation-of-breast-cancer-and-organ-classifying-rna-from-different-species.jpg)















